Изобретение относится к новым производным 7-(замещенный алкокси) кумарина полезным, например, для лечения цереброваскулярных нарушений.
Соединения, которые в настоящее время известны в качестве лекарственных средств для лечения цереброваскулярных нарушений, могут быть отнесены к двум группам: одна группа состоит из соединений, способствующих улучшению мозгового кровообращения; а другая группа состоит из соединений, способствующих улучшению мозгового кровообращения; а другая группа состоит из соединений, активирующих церебральный метаболизм. Соединения, улучшающие мозговое кровообращение, такие, как никардипин и пентоксифиллин, повышают церебральный кровоток путем релаксации гладких мышц кровесных сосудов мозга или путем улучшения церебральной микроциркуляции, что приводит к улучшению в картине любой церебральной дисфункции. Соединения, активирующие церебральный метаболизм, такие, как тиаприд и инделоксазин, способствуют улучшению симптоматики психических расстройств путем моноаминовых соединений, действующих в качестве нейтротрансмиттеров, таких, как серотонин, норепинефрин и допамин. В настоящее время разрабатываются соединения, имеющие обе из указанных выше активностей, однако соединения, показывающие достаточно хорошую клиническую активность, пока еще не получены.
Производные 7-(замещенный алкокси) кумарина являются известными соединениями, которые, как было установлено, иеют до некоторой степени ограниченную терапевтическую активность, хотя до настоящее времени они никогда не использовались для лечения цереброваскулярных расстройств. Известные соединения этого класса раскрываются в работах [1-3] Примерами соединений, раскрытых в указанных документах, являются соединения, приведенные ниже и обозначенные как соединения А-F.
Соединение А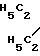 N-(CH2)2-O-
N-(CH2)2-O- 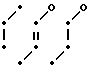
Соединение В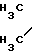 N-(CH2)3-O-
N-(CH2)3-O- 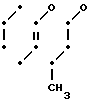
Соединение С N-(CH2)2-O-
N-(CH2)2-O- 
Соединение 1
H2N-(CH2)3-O-
Соединение Е
H )3-O
)3-O
Соединение F N-(CH2)2-O-
N-(CH2)2-O- 
Известны соединения, являющиеся ценными при лечении цереброваскулярных нарушений, и использование которых является частью изобретения. Однако ни одно из соединений, раскрытых в указанных работах, не обладает активностью, аналогичной активности соединений, рассматриваемых в изобретении.
В изобретении раскрывается серия производных 7-(замещенный алкокси)кумарина, обладающих способностью снижать повышенную вязкость крови, вызванную ишемией, и показывающих антирезерпиновую активность, и поэтому указанные соединения могут быть использованы при лечении пациентов, страдающих цереброваскулярными нарушениями, например, инфрактами миокарда и сенильной деменцией.
Целью изобретения является предоставление производных 7-(замещенный алкокси)кумарина, полезных для использования для лечения цереброваскулярных расстройств.
Соединения настоящего изобретения являются соединениями формулы (I) или их фармацевтически приемлемыми солями.
A-(CH2)m-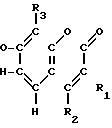 (I) где А является группой формулы (III)
(I) где А является группой формулы (III)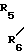 N-
N-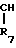 -
-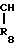 - (III) R1,R2 и R3 являются независимо выбранными из группы, содержащей; атомы водорода, алкильные группы, имеющие от 1 до 4 атомов углерода и атомы галогена;
- (III) R1,R2 и R3 являются независимо выбранными из группы, содержащей; атомы водорода, алкильные группы, имеющие от 1 до 4 атомов углерода и атомы галогена;
R5 и R6 являются независимо выбранными из группы, содержащей атомы водорода и алкильные группы, имеющие от 1 до 4 атомов углерода;
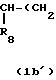
R7 и R8 являются выбранными из группы, содержащей атомы водорода и алкильные группы, имеющие от 1 до 4 атомов углерода; или R7 и R8 вместе представляют собой алкиленовую группу, имеющую 3 или 4 атома углерода; и m является 1 или 2; при условии, что R5 и R6 не являются одинаковыми, и не представляют собой либо метильную, либо этильную группу, когда R1,R3,R7 и R8 все являются атомами водорода, R2 является метильной группой, а m равно 1.
В соединениях настоящего изобретения, когда R1,R2,R3,R5,R6,R7 или R8 представляют собой алкильные группы, эти алкильные группы имеют от 1 до 4 атомов углерода и могут быть прямыми или разветвленными. Примерами указанных групп могут служить метил, этил, пропил, изопропил, бутил, изобутил, фтор-бутил, и т-бутил, где метильная группа является предпочтительной.
В случае, если R1, R2 или R3 являются атомами галогена, то указанными атомами могут быть атомы хлора, фтора, брома или йода, причем предпочтительными являются атомы фтора и хлора.
Когда R7 и R8 вместе представляют собой алкиленовую группу, имеющую 3 или 4 атома углерода, группа формулы (III) представляет, таким образом, циклопентил (С3-алкилен) или циклогексил (С4-алкилен), группу имеющую 2-аминозаместитель.
Из соединений формулы (I), предпочтительными являются следующие соединения:
А соединения, в которых R5 и R6 являются атомами водорода;
В соединения, в которых R5 является атомом водорода, а R6 является алкильной группой, содержащей от 1 до 4 атома углерода; более предпочтительными являются соединения, в которых R является атомом водорода, а R6 является метильной группой;
С соединения, в которых R5 и R6 являются одинаковыми или различными и каждый является метильной или этильной группой;
D соединения, в которых R1 является атомом водорода, атомом хлора или метильной группой;
Е соединения, в которых R2 является атомом водорода, или метильной группой;
F соединения, в которых R3 является атомом водорода или метильной группой.
Более предпочтительными соединениями формулы (I) являются
(G) соединения, в которых
R1,R2 и R3 все являются метильными группами;
R5 и R6 являются атомами водорода;
R7 и R8 независимо являются выбранными из группы, содержащей атомы водорода, метильные группы, этильные группы, и изопропильные группы.
(Н) соединения, в которых:
R1, R2 и R3 все являются метильными группами;
R5 является атомом водорода;
R6 является метильной группой;
R7 и R8 являются независимо выбранными из группы, содержащей атомы водорода, метильные группы, этильные группы и изопропильные группы.
(V) соединения, в которых:
R1,R2 и R3 все являются метильными группами;
R5 и R6 являются метильными группами;
R7 и R8 независимо являются выбранными из группы, содержащей атомы водорода, метильные группы, этильные группы и изопропильные группы.
Хотя соединения изобретения представлены в описании лишь одной формулой (I), однако указанные соединения могут существовать в виде различных изомеров, в зависимости от природы групп-заместителей. В частности, соединения изобретения могут сущестовать в виде двух или четырех форм оптических изомеров, например, в (R)-r форме и (S)-форме, благодаря ассиметрическому атому или атомам углерода, когда R7 и/или R8являются алкильной группой. Изобретение включает в себя оба отдельных изолированных изомера, а также их смеси, включая рацемические смеси (которые могут быть образованы из эквимолярных смесей реагентов). Специалистам хорошо известно, что иногда один из пары изомеров проявляет большую активность или другие более желательные свойства, чем другой изомер из этой пары, и в таких случаях, желательно использовать отдельный изомер. С другой стороны, если необходимо, то указанные соединения могут быть использованы в виде смеси двух или более изомеров, не оказывая при этом никаких неблагоприятных воздействий. Если требуются отдельные изомеры, то они могут быть образованы путем техники стереоспецифического синтеза, который будет описан и проиллюстрирован следующими примерами. Альтернативно могут быть образованы смеси соединений изобретения, а затем разделены с помощью стандартных способов, известных специалистам.
Соединения изобретения содержат основной атом азота и поэтому могут образовывать соли с соответствующими кислотами. В принципе, природа кислот, используемых для формирования таких солей, может быть любой. Однако, если указанная соль предназначена для терапевтического использования, то необходимо чтобы указанная соль была фармацевтически приемлемой, которая не должна иметь пониженную активность (или неприемлемо низкую активность) или повышенную токсичность (или неприемлемо повышенную токсичность) по сравнению со свободно основанными соединениями. Однако в случае, если соль используется в других целях, например в качестве промежуточного соединения при получении другого, и возможно, более активного соединения, эти ограничения могут не приниматься во внимание. Примерами подходящих кислот могут служить: неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота; органические карбоновые кислоты, такие как щавелевая кислота, молочная кислота, лимонная кислота, винная кислота, янтарная кислота, малеиновая кислота и муравьиная кислота; и органические сульфоновые кислоты, такие как метасульфоновая кислота.
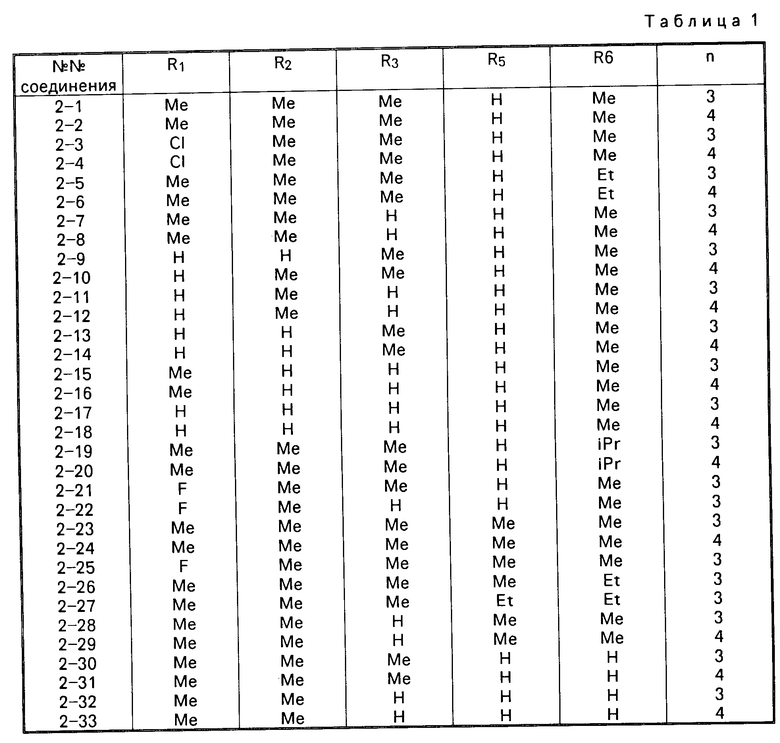
Примеры конкретных соединений изобретения приводятся ниже в виде формул (1-2)-(1-3), в которых заместители определены в соответствующих таблицах (т. е. табл.1 соотвествует формалам (1-2) и тал.2 соответсвует формула (1-3)).
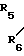 N-(CH2)n-
N-(CH2)n-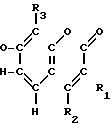 (1-2)
(1-2)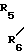 N-
N-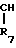 -
-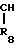 -CH2-
-CH2-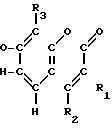 (1-3)
(1-3)
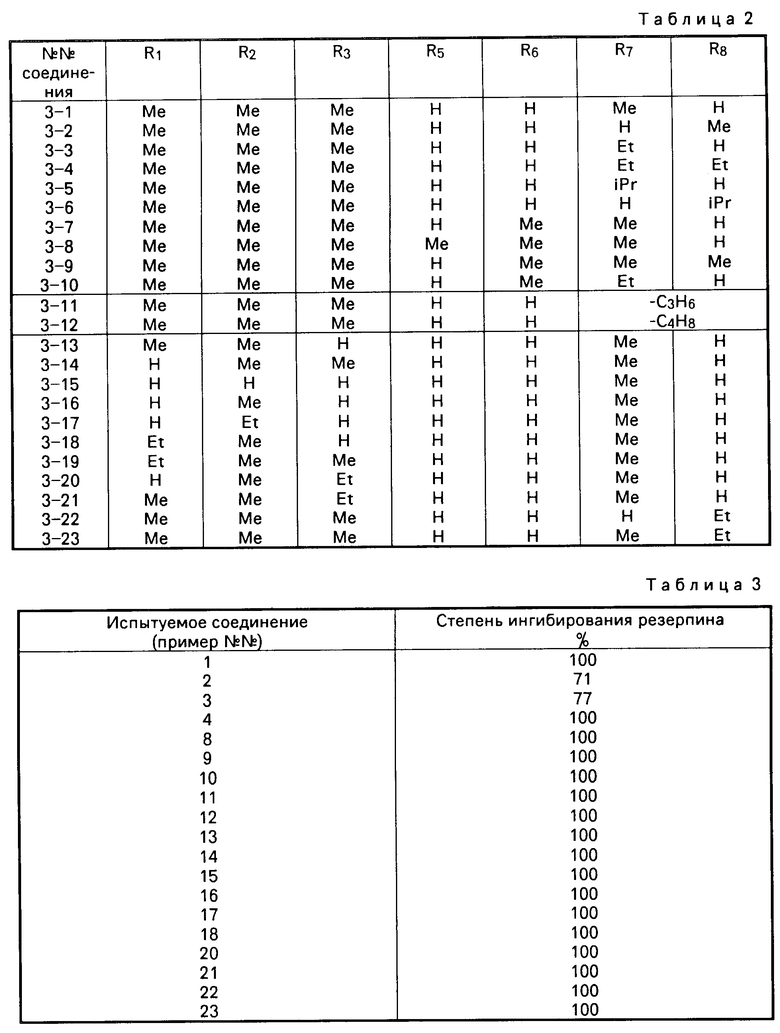
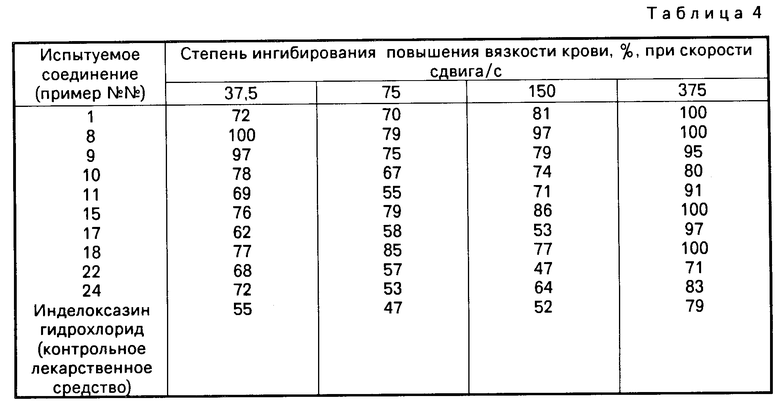
В табл. 2, в соединениях 3-11 и 3-12, R7 и R8 вместе являются либо триметиленовой, либо тетраметиленовой группой соответственно и образуют с атомом углерода, с которым они связаны, либо циклопентановое, либо циклогексановое кольцо.
В приведенных таблицах используются следующие сокращения: Вu бутил; Еt этил; Меt метил; iР изопропил.
Из перечисленных соединений предпочтительными являются следующие соединения, соединения: 2-1, 2-2, 2-5, 2-7, 2-23, 2-24, 2-28, 2-30, 2-31, 2-32, 3-1, 3-2, 3-5, 3-7, 3-8 и 3-11; а более предпочтительными являются следующие соединения: 2-1, 2-23, 2-30, 3-1, 3-5, 3-8 и 3-11.
Наиболее предпочтительными являются следующие соединения:
2-23. 7-3-(N,N-диметиламинопропокси)3,4,8-триметилкумарин;
2-30. 7-(3-аминопропокси)-3,4,8-триметилкумарин;
3-1. 7-(3-аминобутокси)-3,4,8-триметилкумарин.
Предпочтительными являются также фармацевтически приемлемые соли указанных соединений, особенно гидрохлориды и фумараты.
Соединения изобретения могут быть получены различными способами, обычно используемыми для получения такого типа соединений и хорошо известными специалистам.
Например, указанные соединения могут быть получены при помощи
(а) реакции взаимодействия соединения формулы (IV)
H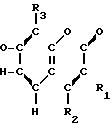 (IV) где R1, R2 и R3 определены выше с соединением формулы (V):
(IV) где R1, R2 и R3 определены выше с соединением формулы (V):
Y-(СН2)m-А, (V) где m определено выше; Y является атомом галогена, сульфонилоксильной группой или гидроксильной группой; а А является группой формулы (III'): N-
N- -
-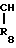 - (III) где R5,R7 и R8 определены выше: а R6 является аминозащитной группой или алкильной группой, имеющей от 1 до 4 атома углерода; и если требуется, при помощи одной из стадий (b) или (с), а именно:
- (III) где R5,R7 и R8 определены выше: а R6 является аминозащитной группой или алкильной группой, имеющей от 1 до 4 атома углерода; и если требуется, при помощи одной из стадий (b) или (с), а именно:
(b) если А является группой формулы (III'), а R6 является аминозащитной группой; осуществляют удаление аминозащитной группы;
(с) если продукт является соединением, в котором один или оба R5 и R6 являются атомами водорода, то алкилируют атома водорода в целях превращения его в алкильную группу, имеющую от 1 до 4 атомов углерода; и необязательно осуществляют реакцию образования солей полученного продукта.
Для большей наглядности, предпочтительные способы изобретения могут быть проиллюстрированы с помощью следующих реакционных схем
Реакционная схема С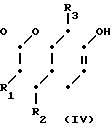 +
+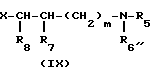
Основание
Стадия С1 ->> -
-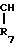 -
- )m-
)m-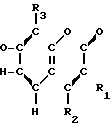
Реакционная схема D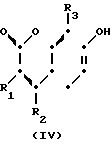 +
+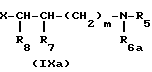
Основание
Стадия D1 ->>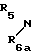 -
- -
- H2)m-
H2)m-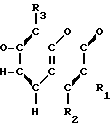
Разблокирование
Стадия D2 ->>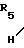 N-
N- -
- )m-
)m-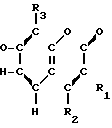
(Ib )
Реакционная схема Е +
+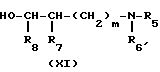
Дегидратирующий агент
Н2О ->>
Стадия Е1 N-
N- -
- -(CH2)
-(CH2)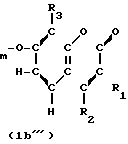
В приведенных формулах, R1,R2,R3, R5,R6,R7,R8, m и R6 определены выше; R6 является алкильной группой; R6а является аминозащитной группой; а Х является атомом галогена или сульфонилокси-группой.
Примерами атомов галогена, которые могут быть представлены Х и Y, являются атомы хлора, брома и йода. Примерами сульфонилокси-групп, которые могут быть представлены Х и Y, являются алкилсульфонилокси-группы, такие как метилсульфонилокси и этилсульфонилокси-группы; арилсульфонилокси-группы, такие как фенилсульфонилокси-группы, 4-метилфенилсульфонилокси-группы и 4-нитрофенилсульфокси-группы.
Аминозащитные группы, используемые в указанных реакциях, могут быть любыми по своей природе, так как эти группы в процессе реакции удаляются, и поэтому не влияют на конечный продукт. Отсюда следует, что может быть использована любая группа, которая способна защитить аминогруппу от нежелательного участия в реакции. Примерами аминозащитных групп, которые могут быть использованы в реакции, являются аралкильные группы, такие как бензильная, n-метоксибензильная и трифенилметильная группы; триалкилсилильные группы, такие, как триметилсилильные и т-бутилдиметилсилильные группы; ацильные группы, такие как формильные, ацетильные и трифторацетильные группы; и алококсикарбонильные и аралкилоксикарбонильные группы, такие как бензилоксикарбонильная, n-нитробензилоксикарбонильная, метоксикарбонильная, этоксикарбонильная и т-бутоксикарбонильная группы. Из указанных групп предпочтительными являются ацильная, алоксикарбонильная и аралкилоксикарбонильная группы.
Стадии С1 и D1 в реакционных схемах С и D соответственно включают в основном одни и те же реакции, в которых гидроксильное соединение формулы (IV) взаимодействует с галогенидом или сульфонатом формулы (IХ), или (IХа) соответственно в присутствии основания или инертного растворителя.
В указанных реакциях может быть использован любой растворитель, при условии, что он не оказывает неблагоприятного воздействия на реакцию или его реагенты. Примерами подходящих растворителей могут служить: углероводороды, в частности ароматические углеводороды, такие как бензол, или толуол; простые эфиры, такие как тетрагидрофуран и диоксан; кетоны, такие как ацетон и метилэтилкетон; спирты, такие как метанол, этанол и т-бутанол; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид и N-метил-2-пирролидон; и сульфоксиды, такие, как диметилсульфоксид. Предпочтительными являются кетоны и амиды.
В рассматриваемых реакциях могут быть использованы любые основания, которые обычно используются в подобного типа реакциях, при условии, что они не оказывают неблагоприятного воздействия на любые другие части молекул. Примерами таких оснований могут служить гидриды щелочных металлов, такие как гидрид лития и гидрид натрия; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия и т-бутоксид калия; карбонаты щелочных металлов, такие как карбонат натрия и карбонат калия; и бикарбонаты щелочных металлов, такие как бикарбонат натрия и бикарбонат калия. Предпочтительными являются гидриды и карбонаты щелочных металлов.
Указанные реакции могут протекать в широком диапазоне температур, поскольку для способа температура не является критическим параметром. Обычно реакцию проводят при температурах 0-120оС более предпочтителтьно 20-80оС. Время проведения реакции также может широко варьироваться в зависимости от многих факторов, например от температуры реакции и природы реагентов. В условиях предпочтительного осуществления изобретения этот период времени составляет 1-24 ч.
Полученное соединение формулы (Ib) или (Х) может по необходимости быть выделено из реакционной смеси стандартным способом, хорошо известным специалистам. Например, указанное соединение может быть выделено с помощью следующих процедур: экстрагирование органическим растворителем, таким как этилацетат; промывание экстракта водой; его сушка, например, в присутствии безводного сульфата натрия или магния; и наконец, отгонка растворителя. По желанию указанное соединение может быть затем очищено с помощью различных стандартных способов, таких как перекристаллизация или различного рода хроматографическая техника, например колоночная хроматография или препаративная тонкослойная хроматография. Альтернативно, рассматриваемое соединение может быть использовано в любой из перечисленных стадий без использования промежуточной очистки.
Исходное соединение формулы (IV), используемое в указанных стадиях, является либо известным соединением, либо может быть легко получено известными способами.
В стадии D2 аминозащитные группы при необходимости удаляют. Указанная реакция удаления может быть осуществлена с помощью стандартных способов и используемый способ зависит от природы удаляемой защитной группы. Например, аралкильная и аралкилоксикарбонильная группы, такие как бензильная и бензилоксикабонильная группы, могут быть удалены с помощью каталитического восстановления с использованием палладиевого катализатора; трифенилметильные группы, триалкилсилильные группы (такие как триметилсилил) и т-бутоксикаронильные группы могут быть удалены с использованием кислоты, такой как водная уксусная кислота, хлористый водород-диоксан или хлористый водород-этилацетат; ацильные группы (такие, как формильная, ацетильная и трифторацетильная группы), алкоксикарбонильные и аралкилоксикарбонильные группы (такие, как бензилоксикарбонильная группа) могут быть удалены с помощью щелочи, например метоксида натрия, этоксида натрия, гидроксида натрия или гидроксида калия; а триалкилсилильные группы, такие как т-бутилдиметилсилильная группа, могут быть удалены с помощью соединения, способного генерировать ионы фтора.
В указанных реакциях разблокирования (снятия защиты) может быть использован растворитель любой природы, если он не оказывает неблагоприятного воздействия на реакции и на ее реагенты. Примерами подходящих для этой цели растворителей могут служить вода, спирты, такие как метанол и этанол; низшие жирные кислоты, такие как уксусная кислота; простые эфиры, такие как тетрагидрофуран и диоксан; сложные эфиры, такие как этилацетат; и галогенированные углеводороды, в частности, галогенированные алифатические углеводороды, такие как метиленхлорид. Указанные реакции могут протекать в широких диапазонах температур, поскольку реакционная температура не является критическим параметром изобретения. В основном реакция протекает при температуре 0-100оС (более предпочтительно от комнатной температуры до температуры 80оС). Время прохождения указанных реакций также может варьироваться в широких пределах в зависимости от многих факторов, например от температуры реакции и природы реагентов. Однако в условиях предпочтительного осуществления изобретения этот период времени в основном составляет 30-24 ч (более предпочтительно 1-16 ч).
После завершения реакции реакционный продукт в органическом растворителе, таком как этилацетат или диэтиловый эфир, может быть выделен в виде соли соединения формулы (I) путем добавления кислоты, такой как гидрохлорид. Альтернативно реакционный продукт может быть выделен с помощью реакции щелочного раствора с последующим экстрагированием свободного соединения формулы (I) орагинческим растворителем, таким как этилацетат. По желанию указанное соединение может быть затем подвергнуто очистке различными стандартными способами, такими как перекристаллизация или хроматографическая техника, например хроматография на колонках или препаративная тонкослойная хроматография.
В стадии Е1 реакционной схеме Е, гидроксильное соединение формулы (IV) взаимодействует с α -амино- ω -гидроксисоединением формулы (ХI) в присутствии агента дегидратации. Обычно и предпочтительно реакция протекает в присутствии растворителя. В указанной реакции может быть использован растворитель любой природы, если только он не оказывает неблагоприятного воздействия на реакцию или ее реагенты. Примерами подходящих растворителей могут служить углеводороды, в частности ароматические углеводороды, такие как бензол и толуол; галогенированные углеводороды, в частности галогенированные алифатические углеводороды, такие как метиленхлорид и 1,2-дихлорэтан; кетоны, такие как ацетон и метилэтилкетон; и амиды, в частности амиды жирных кислот, такие как N,N-диметилформамид, N,N-диметилацетамид и N-метил-2-пирролидон, причем особенно предпочтительными являются галогенированные углеводороды и амиды.
Указанная реакция может протекать в широких диапазонах температур, поскольку реакционная температура не является критическим параметром изобретения. В основном реакция протекает при температуре 0-60оС, а более предпочтительно 10-30оС. Время прохождения указанной реакции также может варьироваться в широких пределах в зависимости от многих факторов, например от температуры реакции и природы реагентов. Однако в условиях предпочтительного осуществления изобретения этот период времени в основном составляет 1-24 ч.
Если R6 представляет собой аминозащитную группу, то она может быть удалена с помощью стандартных реакций, указанных в описании стадии D2.
Целевой продукт может быть выделен из реакционной смеси стандартными способами, например, путем простого концентрирования реакционной смеси. Полученное соединение может быть по желанию подвергнуто дальнейшей очистке стандартными способами, такими как перекристаллизация или хроматографическая техника различного типа, например колоночная хроматография или препаративная тонкослойная хроматография.
Как указано выше, соединения изобретения могут существовать в виде различных оптических изомеров, того, что в соединении формулы (Ib) присутствует ассиметрический атом или атомы, если R7 и/или R8 являются алкильной группой. Если полученное соединение представляет собой смесь оптических изомеров, то оптическое разделение может быть осуществлено с помощью реакции оптически активного соединения формулы (IХ) с соединением формулы (IV) на стадии С1 или указанное оптическое разделение может быть проведено после превращения соединения формулы (I) в соль путем реакции с оптически активной кислотой, в частности сульфоновой кислотой или карбоновой кислотой, такой как C- или D-камфорсульфоновая кислота, L- или D-винная кислота, L- или D-молочная кислота или с ацилированной L- или D-аминокислотой.
Соединения формулы (I) могут быть превращены в их фармацевтически приемлемые соли с помощью обработки кислотой стандартными способами. Например, соединение формулы (I) может быть подвергнуто растворению в органическом растворителе, таком как этилацетат или метиленхлорид, и при этом может быть добавлено эквимолярное количество или избыточное количество кислоты, такой как гидрохлорид диоксан. Растворитель можно отогнать, и в результате соединение формулы (I) может быть получено в виде соли путем кристаллизации или отверждения в органическом растворителе, таком как диэтиловый эфир или диизопропиловый эфир.
Соединение формулы (Ib), в котором один из радикалов R5 и R6является атомом водорода, а другой алкильной группой, может быть получено из соответствующего соединения, в котором оба радикала R5 и R6являются атомами водорода, т.е. соединения формулы (Iс)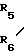 N-
N- -
- -(CH2)m-O-
-(CH2)m-O-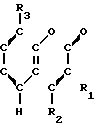 (1с) (где R1,R2,R3,R7,R8 и m определены выше) путем алкилирования указанного соединения. Подходящими способами алкилирования являются восстановительная реакция в присутствии карбонильного соединения формулы R9R10С=0 (ХII)= где R9 и R10 являются одинаковыми или различными и каждый является атомом водорода или алкильной группой, имеющей от 1 до 3 атомов углерода; и алкилирование с соединением формулы R6 Х (ХIII), где R6 и Х определены выше.
(1с) (где R1,R2,R3,R7,R8 и m определены выше) путем алкилирования указанного соединения. Подходящими способами алкилирования являются восстановительная реакция в присутствии карбонильного соединения формулы R9R10С=0 (ХII)= где R9 и R10 являются одинаковыми или различными и каждый является атомом водорода или алкильной группой, имеющей от 1 до 3 атомов углерода; и алкилирование с соединением формулы R6 Х (ХIII), где R6 и Х определены выше.
При восстановительной реакции алкилирования основания Шиффа получают с помощью реакции производного аминоалкоксикумарина формулы (1с) с карбонильным соединением формулы (ХII), а затем указанное основание Шиффа восстанавливают в присутствии восстанавливающего агента (такого, как цианоборогидрид натрия) или путем каталитического восстановления в атмосфере водорода и в присутствии восстановительного катализатора, такого как скелетный никелевый или платиновый катализатор гидрирования, предпочтительно в одной стадии. Таким образом предпочтительно если реакция протекает в присутствии подходящего растворителя (например, спирта, такого, как метанол или этанол) и в присутствии водорода предпочтительно при атмосферном или сверхатмосферном давлении, например, от 1 до 5 атм. или в присутствии восстановительного агента. Указанная реакция может протекать в широком диапазоне температур, поскольку выбираемая температура не является критическим параметром для данного изобретения. В основном, подходящей для данной реакции является температура от 0 до 50оС (а более предпочтительной от 0 до комнатной температуры). Аналогично время протекания реакции также может варьироваться в широких пределах в зависимости от многих факторов, например, от температуры реакции и природы реагентов. Однако в большинстве случаев этот период колеблется от 30 мин до 24 ч.
После завершения реакции, целевой продукт формулы (Ib ), где R5является атомом водорода, может быть выделен из реакционной смеси любым стандартным способом. Например, если реакцию проводят путем каталитического восстановления, то выделение соединения осуществляют путем отфильтровки катализатора с последующей конденсацией фильтрата; и альтернативно, при использовании восстанавливающего агента, подходящим способом выделения может служить конденсация реакционного раствора при пониженном давлении с последующим экстрагированием остатка органическим растворителем, таким, как метиленхлорид или этилацетат.
Альтернативно, реакцию алкилирования проводят с помощью реакции соединения формулы (Iс) с соединением формулы R6 -Х (ХIII), где R6 и Х определены выше, т.е. алкилгалогенидом или алкилсульфонатом. Предпочтительно, если указанная реакция протекает в присутствии инертного растворителя и основания. Условия реакции аналогичны условиям реакции соединения формулы (IV) с соединением формулы (ХI) или (ХIа). Для получения соединения формулы, (Ib), в котором только один из радикалов R5 и R6 является алкильной группой, а другой является атомом водорода, предпочтительно использовать около I эквивалента соединения формулы (ХIII) на I эквивалент соединения формулы (1с) или, во всяком случае, менее 2 эквивалентов; если используются 2 или более эквивалента соединения формулы (ХIII) на I эквивалент соединения формулы (1с), то полученный продукт будет, преимущественно, являться соединением формулы (Ib), в котором оба радикала R5 и R6 являются алкильной группой.
Биологическая активность.
Соединения изобретения обладают способностью ингибировать повышение вязкости крови, которая является результатом церебральной ишемии; а также они обладают сильной антирезерпиновой активностью. Поэтому следует ожидать, что соединения настоящего изобретения будут способствовать улучшению церебрального метаболизма и кровообращения в результате их моноамино-активирующего действия и действия, улучшающего микроциркуляцию.
В приведенных ниже экспериментах соединения настоящего изобретения обозначены числом, являющимся номером Примера, в котором описано его получение.
(I) Антирезерпиновое действие.
В эксперименте использовали самцов мыши в возрасте 4 недели и весом 22-27 г каждая. Животных разделили на две группы, по 3 мыши в каждой. Испытуемые соединения растворяли или суспендировали в физиологическом солевом растворе, в 0,5%-ном растворе СМС (карбоксиметилцеллюлозы) или в 1%-ном растворе диметилсульфоксида, затем 100 мг/кг образца полученного раствора перрорально вводили каждой мыши одной группы (обработанная группа). Мышам из другой группы вводили только раствор, не содержащей активное соединение (контрольная группа). Сразу после введения активного соединения и контрольного раствора, каждой мыши вводили подкожно 2 мг/кг резерпина. Через 90 мин определяли степень опущения века (степень блефароптоза). Для оценки результатов нормальную мышь, не имеющую птоза, определяли как 0; мышей, обнаруживающих птоз на 1/3-1/2, определяли как 1; мышь, обнаруживающую птоз на 2/3 и до состояния лишь чуть приоткрытого века, определяли как 2, а мышь с полностью закрытым веком определяли как 3. Степень ингибирования резерпина испытуемого образца рассчитывали по следующему уравнению: т
т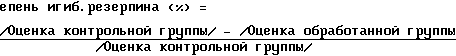
Результаты этих оценок представлены в табл.3.
(2) Ингибирующее действие на повышение вязкости крови, вызванной церебральной ишемией.
Взрослые крысы самцы штамма Wistar были разделены на 2 группы, каждая из которых содержала по 6 животных. Испытуемой образец растворяли или суспендировали в физиологическом солевом растворе или в 0,5%-ном растворе СМС, после чего каждой крысе из одной группы (обработанная группа) перрорально вводили 100 мг/кг образца полученного раствора. Крысам из другой группы вводили только раствор, не содержащий активное соединение (контрольная группа). Сразу после введния каждой крысе внутрибрюшинно вводили 40 мг/кг пентоборбитала для анестезии. Животное держали в положении супинации. Из яремной вены с одной стороны брали пробы крови (0,6 мл). Затем с помощью вискозиметра для измерения крови (Вiorheolyxer, Токио Кei ki) измеряли вязкость крови со скоростями сдвига 37,5/с, 75/с, 150/с, и 375/с. После чего крысам перевязывали общие сонные артерии с обеих сторон в течение одного часа в целях индуцирования неполной церебральной ишемии; и из яремной вены с другой стороны собирали 0,5 мл крови в целях измерения ее вязкости с помощью указанного выше прибора. Ингибирование повышения вязкости крови посредством испытуемого лекарственного средства определяли по приведенному ниже уравнению при каждой скорости сдвига.
Эксперименты проводили с использованием нескольких соединений изобретения, а также известного соединения, Инделоксазингидрохлорида.

Полученные результаты представлены в табл.4.
Приведенные выше результаты показывают, что соединения формулы (I) и его фармацетивческие приемлемые соли могут быть использованы для улучшения мозгового кровообращения и метаболизма. Для этой цели указанные соединения могут быть введены перрорально или перантерально в лекарственной форме, соответствующей пути введения. Например, для перрорального введения могут быть использованы такие препараты, как порошки, гранулы, таблетки или капсулы, а для парентерального введения такие препараты, как инъекции, суппозитории и пластыри. При этом соединение изоретения могут быть использованы в чистом виде или в смеси с любыми фармацевтически приемлемыми носителями, наполнителями или разбавителями. Предпочтительные дозы активного соединения могут варьироваться в завимости от природы заболевания пациента, его возраста, физического состояния и веса тела, а также от способа введения лекарственного средства; при этом суточная доза обычно составляет 1-1000 мг, а более предпочтительная доза составляет 0,5-30 мг (при внутривенном введении). Соединение изобретения может быть введено в виде одноразовой дозы или в виде разделенных доз один или три раза в день в зависимости от симптомов.
В качестве иллюстрации изобретения ниже приводятся примеры получения некоторых соединений изобретения.
П р и м е р 1. 7-(3-N-Метиламинопропокси)-3,4,8-триметилкумарина гидрохлорид(а) 7-[3-(N-т-бутоксикарбонил-N-метиламино) пропокси] -3,4,8-триметилкумарин.
9 г карбоната калия добавляли к 50 мл раствора диметилформамида, содержащего 3,0 г 7-гидрокси-3,4,8-триметилкумарина и 6,73 г 3-(N-т-бутоксикарбонил-N-метиламино)-пропил-р-толуол-сульфоната. Затем реакционную смесь перемешивали в течение 5 ч при 65оС, после чего в смесь добавляли этилацетат и воду. Слой этилацетата отделяли и осушали безводным сульфатом магния, а растворитель удаляли путем дистилляции при пониженном давлении. Полученный кристаллический осадок очищали с помощью хроматографии на колонках с силикагелем, элюируя смесью этилацетата и метиленхлорида (1:9), в результате чего получали 4,04 г целевого соединения в виде кристаллов с т.пл. 110оС.
ЯМР (CDCl3), δ млн.дол. 1,46 (9Н, с); 1,85-2,25 (2Н, м), 2,17 (3Н, с); 2,30 (3Н, с); 2,34 (3Н, с); 2,90 (3Н, с); 3,45 (2Н, т, I=7 Гц); 4,06 (2Н, т, I=7 Гц); 6,77 (1Н, д, I=8,4 Гц); 7,37 (1Н, д, I=8,5 Гц).
(b) 7-(3-N-метиламинопропокси)-3,4,8-триметилкумарина гидрохлорид.
30 мл 4н. раствора хлороводорода в этилацетате добавляли к 3,9 г 7-[3-N-т-бутоксикаронил-N-метиламино)пропокси]-3,4, 8-триметликумарина (полученного согласно описанию в стадии (а)), и полученную смесь перемешивали в течение 4 ч при комнатной температуре. По истечении этого времени студенистую реакционную смесь конденсировали путем выпаривания при пониженном давлении. К остатку добавляли 20 мл этанола и полученную смесь нагревали с обратным холодильником, а затем охлаждали. После чего добавляли 40 мл этилацетата и полученный осадок собирали фильтрацией, в результате получали 2,99 г целевого соединения, температура размягчения которого составляет 215оС, а т.пл. 253оС.
ЯМР (гексадейтерированной диметилсульфоксид), δ млн.дол. 2,08 (3Н, с); 2,21 (3Н, с); 2,05 -2,25 (2Н, м); 2,36 (3Н, с); 2,59 (3Н, с); 3,08 (2Н, т, I= 7,5 Гц); 4,21 (2Н, т, I=7,5 Гц); 7,04 (1Н, д, I=8,5 Гц); 7,63 (1Н, д. I= 8,5 Г).
П р и м е р 2. 7-(4-N-Метиламинобутокси)-3,4,8-триметилкумарина гидрохлорид (а) 7-[4-(N-т-бутоксикарбонил-N-метиламино)бу- токси]-3,4,8-триметилкумарин.
Повторяли процедуру, описанную в примере 1(а), за исключением того, что использовали 0,96 г 7-гидрокси-3,4,8-триметилкумарина, 2,19 г 4-N-т-бутоксикарбонил-N-метиламино)бу- тила р-нитробензолсульфоната и 2,6 г карбоната калия. Полученный в результате продукт очищали с помощью хроматографии на колонках с силикагелем, элюируя смесью этилацетата и гексана (1:4), и получали 1,80 г целевого соединения в виде кристаллов с т.пл.86оС.
ЯМР (СDCl3), δ млн.дол. 1,48 (9Н, с); 1,65-1,95 (4Н, м); 2,16 (3Н; с); 2,29 (3Н, с); 2,33 (3Н, с); 2,86 (3Н, с); 3,15-3,45 (2Н, м); 3,95-4,15 (2Н, м); 6,76 (1Н, д, I=8,5 Гц); 7,37 (1Н, д, I=8,5 Гц).
(b) 7-(4-N-метиламинобутокси)-3,4,8-триметилкумарина гидрохлорид.
Повторяли процедуру, описанную в примере 8 (b), за исключением того, что использовали 1,70 г 7-[4-(N-т-бутоксикарбонил-N-метиламино)бутокси]-3,4,8-триметил- кумарина (полученного согласно описанию в стадии (а)), в результате чего получали 1,30 г целевого соединения в виде кристаллов с т.пл. 197оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,75-1,95 (4Н, м); 2,09 (3Н, с); 2,22 (3Н, с); 2,36 (3Н, с); 2,55 (3Н, с); 2,85-3,10 (2Н, м); 4,00-4,20 (2Н, м); 7,03 (1Н, д, I=8,5 Гц); 7,59 (1Н, д, I=8,5 Гц).
П р и м е р 3. 3-Хлоро-4,8-диметил-7-(3-N-метиламинопропокси)кумарина гидрохлорид
(а) 7-[3-(N-т-бутоксикарбонил-N-метиламино)пропокси]-3-хлоро-4,8-диметилкума- рин.
Повторяли процедуру, описанную в примере 1(а), за исключением того, что использовали 0,65 г 3-хлоро-7-гидрокси-4,8-диметилкумарина, 1,44 г 3-(N-т-бутоксикарбонил-N-метиламино)пропила n-толуолсульфоната и 2,0 г карбоната калия, и полученное соединение очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и метиленхлорида (1:9), в результате чего по- лучали 0,93 г целевого соединения в виде кристаллов с т.пл. 161оС.
ЯМР (CDCl3), δ млн.дол. 1,41 (9Н, с); 2,06 (2Н, квинтет, I=7 Гц); 2,27 (3Н, с); 2,50 (3Н, с); 2,87 (3Н, с); 3,44 (2Н, т, I=7 Гц); 4,08 (2Н, т, I= 8,5); 6,86 (1Н, д, I=8,5 Гц); 7,44 (1Н, д, I=8,5 Гц).
(b) 3-Хлоро-4,8-диметил-7-(3-N-метиламинопропокси)кумарина гидрохлорид.
Повторяли процедуру, описанную в примере 1(b), за исключением того, что использовали 0,90 г 7-[3-(N-т-бутоксикарбонил-N-метиламино)пропокси]-3-хло- ро-4,8-диметилкумарина (полученного согласно описанию в стадии (а)), в результате чего получали 0,73 г целевого соединения в виде кристаллов, температура размягчения которого составляла 245оС, а т.пл. 273оС (с сокращением).
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 2,0-2,35 (2Н, м); 2,21 (3Н, с); 2,54 (6Н, с); 2,95-3,2 (2Н,м); 4,24 (2Н, т, I=7,5); 7,10 (1Н, д, I=8,5 Гц); 7,68 (1Н, д, I=8,5 Гц).
П р и м е р 4. 3,4-Диметил-7-(4-N-метиламинобутокси)кумарина гидрохлорид (а) 7-[4-(N-т-бутоксикарбонил-N-метиламино)бу- токси]-3,4-диметилкумарин.
Повторяли процедуру, описанную в примере 1(а), за исключением того, что использовали 1,00 г 3,4-диметил-7-гидроксикумарина, 2,4 г 4-N-т-бутоксикарбонил-N-метиламино бутила п-нитробензолсульфоната и 2,83 г карбоната калия, в результате чего получали 2,08 г целевого соединения в виде сиропа (после очистки с помощью колочной хроматографии на силикагеле с элюированием смесью этилацетата и гексана (1:2)).
ЯМР (СDCl3), δ млн.дол. 1,44 (9Н, с); 1,65-1,85 (4Н, м); 2,17 (3Н, с); 2,36 (3Н, с); 2,86 (3Н, с); 3,18-3,41 (2Н,м); 3,92-4,13 (2Н, м); 6,78-6,96 (2Н, м); 7,42-7,60 (1Н, м).
(b) 3,4-Диметил-7-(4-N-метиламинобутокси)кумарина гидрохлорид.
Повторяли процедуру, описанную в примере 1(b), за исключением того, что использовали 2,00 г 7-[4-N-т-бутоксикарбонил-N-метиламино)бутокси]-3,4-диметилку- марина (полученного согласно описанию в стадии (а)), в результате чего получали 1,24 г целевого соединения в виде кристаллов с т.пл. 196-198оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,8-1,9 (4Н, м); 2,8 (3Н, с); 2,37 (3Н, с); 2,54 (3Н, с); 2,8-3,05 (2Н, м); 4,0-4,2 (2Н, м); 6,85-7,05 (2Н, м); 7,65-7,75 (1Н, м).
П р и м е р 5. 7-(3-N-этиламинопропокси)-3,4,8-триметилкумарина гидрохлорид (а) 7-[3-(N-т-бутоксикарбонил-N-этиламино) пропокси]-3,4,8-триметилкумарина.
Повторяли процедуру, описанную в примере 1(а), за исключением того, что использовали 1,0 г 7-гидрокси-3,4,8-триметилакумарина, 2,3 г 3-(N-т-бутоксикарбонил-N-этиламино)пропила п-толуолсульфоната и 3,0 г карбоната калия, и после очистки с помощью хроматографии на колонках с силикагелем и элюированием смесью этилацетата и гексана (1:2), получали 1,20 г целевого соединения в виде кристаллов с т.пл. 107оС.
ЯМР (СDСl3), δ млн.дол. 1,11 (3Н, т, I=7 Гц); 1,43 (9Н, с); 1,85-2,25 (2Н, м); 2,17 (3Н, с); 2,29 (3Н, с); 2,34 (3Н, с); 3,25 (2Н, кв. I=7 Гц); 3,41 (2Н, т, I=7 Гц); 4,06 (2Н, т, I=7 Гц); 6,81 (1Н, д, I=10 Гц); 7,42 (1Н, д, I=10 Гц).
(b) 7-(3-N-этиламинопропокси)-3,4,8-триметилкумарина гидрохлорид.
Повторяли процедуру, описанную в примере 1 (b), за исключением того, что использовали 0,90 г [3-(N-т-бутоксикарбонил-N-этиламино)пропокси]-3,4,8-триметилкума- рина (полученного согласно описанию в стадии (а)), в результате чего получали 0,74 г целевого соединения в виде кристаллов с т.пл. 245оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,27 (3Н, т, I= 7 Гц); 1,9-2,4 (2Н, м); 2,08 (3Н, с); 2,20 (3Н, с); 2,33 (3Н, м); 2,7-3,3 (4Н, м); 4,20 (2Н, т, I=6 Гц); 6,98 (1Н, д, I=9,5 Гц); 7,55 (1Н, д, I=9,5 Гц); 9,30 (2Н, шир.с).
П р и м е р 6. 7-(3-N-изопропиламинопропокси)-3,4,8-триметилкумарина- гидрохлорид (а) 7-[3-(N-т-бутоксикарбонил -N-изопропиламино)пропокси] 3,4,8-триметилкумарин.
0,23 мл ацетона и 0,30 мл уксусной кислоты добавляли к 15 мл метанолового раствора, содержащего 642 мг 7-(3-амино-пропокси)-3,4,8-триметилкумарина (по- лученного согласно описанию в примере 10). Затем смесь помещали в ледяную баню и добавляли 157 винооборогидрида натрия. После чего смесь перемешивали при комнатной температуре в течение 2 ч и затем конденсировали путем выпаривания при пониженном давлении. Затем добавляли насыщенный водный раствор бикарбоната натрия и получали нужное свободное аминосоединение в виде осадка. Этот осадок собирали фильтрацией и суспендировали в этилацетате. К полученной суспензии добавляли 0,42 мл триэтиламина и 643 мг ди-т-бутилпирокарбоната, после чего реакционную смесь перемещали в течение 3 ч.
По истечение этого времени смесь конденсировали путем выпаривания при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и метиленхлорида (1:9), и получали 0,57 г 7-[3-(N-т-бутоксикарбонил-N-изопропила- мино)пропокси]-3,4,8-триметилкумарина в виде кристаллов с т.пл. 132оС.
ЯМР (СDCl3), δ млн.дол. 1,17 (6Н, д, I=7 Гц); 1,50 (9Н, с); 1,95-2,25 (2Н, м); 2,20 (3Н, с); 2,33 (3Н, с); 2,36 (3Н, с); 3,31 (2Н, дд, I=6 и 7, 5Гц); 4,08 (2Н, т, I=7 Гц); 4,0-4,8 (1Н, м); 6,81 (1Н, д, I=8,5 Гц); 7,42 (1Н, д, I=8,5 Гц).
(b) 7-(3-N-изопропиламинопропокси)-3,4,8-триметилкумарина гидрохлорид.
Повторяли процедуру, описанную в примере 1(b), за исключением того, что использовали что исользовали 0,52 г 7-[3-N-т-бутокси-карбонил-N-изопропиламино)пропокси] -3,4,7-триметилкумарина (полученного согласно описанию в стадии (а)), в результате чего получали 0,44 г целевого соединения в виде кристаллов с температурой размягчения 270оС, и т.пл. 286оС.
ЯМР (D2О), δ млн, дол. 1,89 (6Н, д, I=6,5 Гц); 2,19 (3Н, с); 2,05-2,4 (2Н, м); 2,33 (6Н, с); 3,7-3,9 (2Н, м); 3,85-4,2 (1Н, м); 4,45-4,6 (2Н, м); 7,07 (1Н, д, I=8,5 Гц); 7,43 (1Н, д, I=8,5 Гц).
П р и м е р 7. 7-(3-N-метиламинопропокси)кумарина фумарат.
Повторяли процедуру, описанную в примерах 1 (а) и (b), за исключением того, что использовали 1,3 и 7-гидроксикумарина, в результате чего получали 2,1 г гидрохлорида целевого соединения в виде гигроскопических кристаллов. Полученный гидрохлорид растворял в 10 мл воды. Полученный раствор нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия и затем экстрагировали метиленхлоридом. Экстракт метиленхлорида осушали безводным сульфатом магния, а растворитель удаляли путем дистилляции при пониженном давлении, в реузльтате чего получали 1,66 г производного свободного кумарина, которое является целевым соединением в виде смолы. Эту смолу обрабатывали 0,83 г фумаровой кислоты, и полученный продукт промывали сначала диэтиловым эфиром, а затем этилацетатом, в результате чего получали 2,30 г целевого соединения в виде кристаллов с т.пл. 108оС (с размягчением).
ЯМР (гексадейтерированный диметилсульфоксид) δ млн.дол. 1,9-2,35 (2Н, м); 2,56 (3Н, с); 3,05 (2Н, т, I=8 Гц; 4,18 (2Н, т, I=6 Гц; 6,28 (1Н, д. I= 9,5 Гц); 6,50 (2Н, с); 6,8-7,05 (2Н, м); 7,64 (1Н, д, I=9,5 Гц); 7,89 (1Н, д, I=9,5 Гц).
П р и м е р 8. 3,4-Диметил-7-(3-N-метиламинопропокси)кумарина гидрохлорид.
Повторяли процедуру, описанную в примерах 1(а) и (b), за исключением того, что использовали 2,88 г 3-(N-бутоксикарбонил-N-метиламино)пропил-n-толуолсульфоната и 1,2 г 7-гидрокси-3,4-диметилкумарина, в результате чего получали 1,69 г целевого соединения в виде кристаллов с т.пл. 180-182оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млнд.дол. 1,95-2,3 (2Н, м); 2,09 (3Н, с); 2,37 (3Н, с); 2,57 (3Н, с); 3,06 (2Н, т, I=7,5 Гц), 4,20 (2Н, т, I=6 Гц); 6,9-7,05 (2Н, м); 7,71 (1Н, д, I=9,5 Гц); 9,23 (2Н, шир.с).
П р и м е р 9. 7-(3-N,N-Диметиламинопропокси)-3,4,8-триметилкумарин и его гидрохлорид.
1,2 г 7-гидрокси-3,4,8-триметилкумарина, 1,0 г 3-(диметиламино)пропилхлорида-гидрохлорида и 1,5 г карбоната калия добавляли к 30 мл метилэтилкетона. Полученную смесь перемешивали, нагревая при этом с обратным холодильником в течение 9 ч. По истечении этого времени, реакционный раствор конденсировали путем выпаривания при пониженном давлении. Остаток растворяли в этилацетате и воде. Слой этилацетата отделяли и высушивали безводным сульфатом магния. Затем растворитель удаляли путем дистилляции при пониженном давлении, кристаллы отделяли и промывали диэтиловым эфиром, в результате чего получали целевое соединение (0,98 г) в виде кристаллов с т. пл. 86оС.
ЯМР (СDCl3), δ млнд.дол. 1,8-2,6 (4Н, м); 2,16 (3Н, с); 2,27 (3Н, с); 2,30 (3Н, с); 2,33 (3Н, с); 4,09 (2Н, т, I=6 Гц); 6,79 (1Н, д, I=9 Гц); 7,36 (1Н, д, I=9 Гц).
0,90 г полученных кристаллов подвергали взаимодействию с 4н раствором хлороводорода в этилацетате и получали 1,05 г гидрохлорида целевого соединения с температурой размягчения 220оС и т.пл. 252оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 2,09 (3Н, с); 2,05-2,35 (2Н, м); 2,23 (3Н, с); 2,37 (3Н, с); 2,77 (3Н, с); 2,83 (3Н, с); 3,1-3,4 (2Н, м); 4,21 (2Н, т, I=6 Гц); 7,05 (1Н, д, I=9 Гц); 7,62 (1Н, д, I= 9 Гц).
П р и м е р 10. 7-(3-Аминопропокси)-3,4,8-триметилкумарина гидрохлорид (а) 7-[3-(N-т-бутоксикарбониламино)пропокси] 3,4,8-триметилкумарин.
4 г карбоната калия добавляли к раствору 2,7 г 7-гидрокси-3,4,8-триметилкумарина и 3,8 г 3-(N-т-бутоксикарбониламино)пропилбромида в 50 мл диметилформамида. Полученную смесь перемешивали при 65оС в течение 5 ч, а затем добавляли этилацетат в воду. Слой этилацетата отделяли, высушивали безводным сульфатом магния, а затем концентрировали путем выпаривания в вакууме. Полученный остаток перекристаллизовывали из этанола, в результате чего получали 3,80 г целевого соединения в виде кристаллов с температурой пл.137оС.
ЯМР (СDCl3), δ млн. дол. 1,45 (9Н, с); 1,8-2,3 (2Н, м); 2,15 (3Н, с); 2,29 (3Н, с); 2,32 (3Н, с); 3,35 (2Н, кв. I=6 Гц); 4,10 (2Н, т, I=6 Гц); 4,95 (1Н, шир.с.); 6,78 (1Н, д, I=9 Гц); 7,35 (1Н, д, I=9 Гц).
(b) 7-(3-аминопропокси)-3,4,8-триметилкумарина гидрохлорид.
30 мл 4 н раствора хлороводорода в этилацетате добавляли в горячий раствор 7-[3-(N-т-бутоксикарбониламино)пропокси [-3,4,8-триметилкумарина (полученного согласно описанию в стадии (а)) в 40 мл этилацетата, и полученную смесь перемешивали при комнатной температуре в течение 4 часов. По истечении этого времени реакционную смесь концентрировали путем выпаривания в вакууме и получали целевое соединение в виде кристаллов. Полученный продукт перекристаллизовывали их 80% объем водного этанола, в результате чего получали 2,88 г целевого соединения в виде кристаллов с т.пл. 260оС.
ЯМР (гексадентерированный диметилсульфоксид), δ млн.дол. 1,95-2,35 (2Н, м); 2,08 (3Н, с); 2,21 (3Н, с); 2,37 (3Н, с); 2,8-3,15 (2Н, м); 4,22 (2Н, т, I=6 Гц); 7,03 (1Н, д, I=9 Гц); 7,62 (1Н, д, I=9 Гц); 8,27 (3Н, шир.с.).
П р и м е р 11. 7-4-Аминобутокси-3,4-диметилкумарина гидрохлорид.
Повторяли процедуру, описанную в примерах 1 (а) и (b), за исключением того, что использовали 3,12 г 4-(N-т-бутоксикарбониламино)бутил-n-толуолсульфоната и 1,3 г 7-гидрокси-3,4-диметилкумарина, в результате чего получали 1,87 г целевого соединения, температура размягчения которого составляет 190оС, а т.пл. 215оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,65-1,95 (4Н, м); 2,08 (3Н, с); 2,36 (3Н, с); 2,7-3,0 (2Н, м); 4,0-4,25 (2Н, м); 6,85-7,05 (2Н, м); 7,68 (1Н, д, I=9 Гц); 8,22 (3Н, шир.с).
П р и м е р 12. 7-(4-Аминобутокси)-3,4,8-триметилкумарина гидрохлорид.
Повторяли процедуру, аналогичную описанной в примерах 1(а) и 1(b), за исключением того, что использовали в 2,91 г 4-(N-т-бутоксикарбониламино)бутил-n-толуолсу- льфоната и 1,3 г 7-гидрокси-3,4,8-триметилкумарина, в результате чего получали 1,71 г целевого соединения с т.пл. 245-247оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,65-2,05 (4Н, м); 2,06 (3Н, с); 2,20 (3Н, с); 2,34 (3Н, с); 2,7-3,1 (2Н, м); 4,0-4,3 (2Н, м); 7,02 (1Н, д, I=9 Гц); 7,59 (1Н, д, I=9 Гц); 8,20 (3Н, шир.с).
П р и м е р 13. 7-[(RS)-3-аминобутокси]-3,4,8-триметилкумарина гидрохлорид (а) 7-[(RS)-3-(т-бутоксикарбониламино)бутокси] -3,4,8-триметилкумарин.
4,02 г карбоната калия добавляли к 20 мл раствора диметилформамида, содержащего 1,32 г 7-гидрокси-3,4,8-триметилкумарина и 2,67 г 3-(т-бутоксикарбониламино)бутил-n-толуолсульфоната. Реакционную смесь перемешивали при 80оС в течение 5 ч, после чего добавляли этилацетат и воду. Слой этилацетата отделяли и высушивали безводным сульфатом магния, а затем удаляли растворитель путем дистилляции при пониженном давлении. Кристаллический остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (2:5), в результате чего получали 1,87 г целевого соединения в виде кристаллов, т.пл. 132-134оС.
ЯМР (СDCl3), δ млн.дол. 1,23 (3Н, д, I=7 Гц); 1,43 (9Н, с); 1,8-2,3 (2Н, м); 2,16 (3Н, с); 2,30 (3Н, с) 2,34 (3Н, с); 3,55-4,3 (3Н, м); 4,65 (1Н, шир. д, I=7 Гц); 6,77 (1н, д, I=8,5 Гц); 7,36 (1Н, д, I=8,5 Гц).
(b) 7[(RS)-3-аминобутокси]-3,4,8-триметилкумарина гидрохлорид.
20 мл 4н. раствора хлороводорода в этилацетате добавляли к 1,87 7-[RS)-3-(т-бутоксикарбониламино)бутокси]-3,4,8-триметил- кумарина (полученного согласно описанию в стадии (а)). Полученную реакционную смесь перемешивали при комнатной температуре в течение 4 ч, после чего целевое соединение выделяли путем фильтрации и промывали смесью этилацетата и диэтилового эфира. После перекристаллизации из 90% об./об. водного этанола, получали 1,26 г целевого соединения, т.пл. 226-227оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,31 (3Н, д, I= 6 Гц); 1,9-2,3 (2Н, м); 2,06 (3Н, с); 2,18 (3Н, с); 2,34 (3Н, с); 3,3-3,65 (1Н, м); 4,21 (2Н, т, I=6 Гц); 7,03 (1Н, д, I=9 Гц); 7,61 (1Н, д, I=9 Гц); 8,30 (3Н, шир.с).
П р и м е р 14. 7-[RS)-3-Аминопентокси]-3,4,8-триметилкумарина гидрохлорид. (а) 7-[RS)-3-(т-бутоксикарбониламино)пентокси] 3,4,8-триметилкумарин.
2 мл раствора метиленхлорида, содержащего 2,26 г диэтилазодикарбоксилата по капле добавляли к 30 мл раствора метиленхлорида, содержащего 2,04 г 7-гидрокси-3,4,8-триметилкумарина, 2,58 г 3-(т-бутоксикарбониламино)пентанола и 3,40 г трифенилфосфина и помещенного в ледяную баню. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По истечении этого времени растворитель удаляли путем дистилляции при пониженном давлении. Осадок очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (2:3), в результате чего получали 3,85 г целевого соединения в виде кристаллов, т.пл. 148оС.
ЯМР (СDCl3), δ млн.дол. 0,95 (3Н, т, I=7 Гц); 1,39 (9Н, с); 1,4-2,1 (4Н, м); 2,14 (2Н, с); 2,28 (3Н, с); 2,32 (3Н, с); 3,5-3,9 (1Н, м); 4,12 (2Н, т, I=6 Гц); 6,80 (1Н, д, I=9 Гц); 7,38 (1Н, д, I=9 Гц).
(b) 7-[(RS)-3-Аминопентокси]-3,4,8-триметилкумарина гидрохлорид.
Повторяли процедуру, аналогичную описанной в примере 13 (а) за исключением того, что использовали 3,30 г 7-[(RS)-3(т-бутоксикарбониламино)пентокси] -3,4,8-три- метилкумарина (полученного согласно описанию в стадии (а)), в результате чего получали 2,04 г целевого соединения в виде кристаллов, т.пл. 242-245оС.
ЯМР (D2О), δ млн.дол. 1,54 (3Н, т, I= Гц); 2,13 (3Н, с); 2,25 (3Н, с); 2,30 (3Н, с); 2,25-2,45 (2Н, м); 2,5-2,8 (2Н, м); 3,85-4,1 (1Н, м); 4,4-4,7 (2Н, м); 7,08 (1Н, д, I=9 Гц); 7,41 (1Н, д, I=9 Гц).
П р и м е р ы 15-24. Повторяли процедуру, аналогичную описанной в примере 14, в результате чего получали соединения, приведенные ниже.
П р и м е р 15. 7-[(S)-3-Аминобутокси]-3,4,8-триметилкумарина гидрохлорид.
Температура плавления 232-234оС.
Оптическое вращение [α]D25 5,8о (с=1, Н2О).
ЯМР этого соединения аналогичен ЯМР соединения, полученного по описанию в примере 13 (b).
П р и м е р 16. 7-[(RS)-3-Аминобутокси]-3,4,8-триметилкумарина гидрохлорид.
Температура плавления 232-234оС.
Оптическое вращение [α]D25 5,5о (с=1, Н2О).
ЯМР этого соединения аналогичен ЯМР соединения, полученного по описанию в примере 13 (b).
П р и м е р 17. Температура размягчения 230оС. Температура разложения 238-241оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,12 (3Н, д, I= 7 Гц); 2,0-2,4 (1Н, м); 2,08 (3Н, с); 2,21 (3Н, с); 2,36 (3Н, с); 2,7-3,15 (2Н, м); 4,07 (2Н, д, I=6 Гц); 7,02 (1Н, д, I=9 Гц); 7,61 (1Н, д, I=9 Гц); 8,30 (3Н, шир.с).
П р и м е р 18. 7-[(RS)-3-Амино-4-метилпентокси]-3,4,8-триметилкумарина гидрохлорид.
Температура размягчения 240оС. Температура разложения 243-245оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,00 (6Н, д,I= 6,5 Гц); 1,85-2,20 (3Н, м); 2,09 (2Н, с); 2,21 (3Н, с); 2,36 (3Н, с); 3,05-3,40 (1Н, м); 4,28 (2Н, т, I=6 Гц); 7,05 (1Н, д, I=9 Гц); 7,62 (1Н, д, I=9 Гц); 8,23 (3Н, шир.с).
П р и м е р 19. 7-(RS)-транс-(2-аминоциклогексил)метокси]3,4,8-триметилкумарина гидрохлорид.
Температура плавления 293-296оС (с разложением).
ЯМР (гексадейтерированный диметилсульфоксид), млн.дол. 1,0-2,3 (9Н, м); 2,06 (3Н, с); 2,21 (3Н, с); 2,35 (3Н, с); 2,7-3,3 (1Н, м); 4,0-4,3 (2Н, м); 7,05 (1Н, д, I=9 Гц); 7,61 (1Н, д, I=9 Гц); 8,38 (3Н, шир.с).
П р и м е р 20. 7-[(RS)-цис-(2-аминоциклогексил)метокси]-3,4,8-триметилкумарина гидрохлорид.
Температура плавления 261-265оС (с разложением).
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,1-2,1 (9Н, м); 2,09 (3Н, с); 2,22 (3Н, с); 2,36 (3Н, с); 3,2-3,6 (1Н, м); 4,0-4,2 (2Н, м); 7,05 (1Н, д, I=9 Гц); 7,58 (1Н, д, I=9 Гц); 8,30 (3Н, шир.с).
П р и м е р 21. 7-[(RS)-транс или цис-(2-аминоциклопентил)метокси]-3,4,8-триме- тилкумарина гидрохлорид.
Температура пл.264оС (окрашивание), 265-269оС (разложение).
ЯМР (D2О), δ млн.дол. 2,16 (3Н, с); 2,28 (3Н, с); 2,33 (3Н, с); 2,1-3,2 (7Н, м); 4,0-4,25 (91Н, м); 4,46 (2Н, д, I= 6 Гц); 7,08 (1Н, д, I=9 Гц); 7,44 (1Н, д, I=9 Гц).
П р и м е р 22. 7-[(RS)-транс или цис-(2-аминоциклопентил)метокси]-3,4,8-триме- тилкумарина гидрохлорид.
Это соединение является диастереоизомером соединения, полученного по описанию в примере 21.
Т.пл. 260оС (с разложением).
ЯМР (D2О), δ млн.дол. 2,1-2,8 (6Н, м); 2,20 (3Н, с); 2,37 (3Н, с); 2,40 (3Н, с); 3,0-3,3 (1Н, м); 4,3-4,45 (1Н, м); 4,60 (2Н, д, I=6 Гц); 7,22 (1Н, д, I=9 Гц); 7,58 (1Н, д, I=9 Гц).
П р и м е р 23. 7-[(RS)-3-(метиламино)пентокси]-3,4,8-триметилкумарина гидрохлорид.
Температура пл. 218-220оС.
ЯМР (D2О), δ млн.дол. 1,52 (3Н, т, I=7 Гц); 2,15 (3Н, с); 2,2-2,45 (2Н, м); 2,26 (3Н, с); 2,32 (3Н, с); 2,55-2,8 (2Н, м); 3,7-3,95 (1Н, м); 4,4-4,65 (2Н, м); 8,09 (1Н, д, I=9 Гц); 7,42 (1Н, д, I=9 Гц).
П р и м е р 24. 7-[(RS)-3-(Диметиламино)бутокси]-3,4,8-триметилкумарина гидрохлорид.
Температура пл.220-222оС.
ЯМР (гексадейтерированный диметилсульфоксид), δ млн.дол. 1,34 (3Н, д, I= 7 Гц); 1,8-2,5 (3Н, м); 2,06 (3Н, с); 2,20 (3Н, с); 2,33 (3Н, с); 2,66 (3Н, с); 2,73 (3Н, с); 3,1-3,7 (1Н, м); 4,20 (2Н, т, I=6 Гц); 7,04 (1Н, д, I=9 Гц); 7,60 (1Н, д, I=9 Гц); 11,10 (1Н, шир.с).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2038354C1 |
| ПРОИЗВОДНЫЕ АЗЕТИДИНОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2047602C1 |
| ТИАЗОЛИДИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СПОСОБ СНИЖЕНИЯ СОДЕРЖАНИЯ САХАРА В КРОВИ У МЛЕКОПИТАЮЩИХ | 1992 |
|
RU2095354C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНОГО ПРОИЗВОДНОГО ИНДОЛОБЕНЗОХИНОЛИНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2045528C1 |
| ПРОИЗВОДНЫЕ 1-БИФЕНИЛМЕТИЛИМИДАЗОЛА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ ЭФИРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2092481C1 |
| ПРОИЗВОДНЫЕ ТИАЗОЛИДИН-2,4-ДИОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2103265C1 |
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2097383C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНКАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2091373C1 |
| ФУНГИЦИДНЫЕ ПРОИЗВОДНЫЕ ОКСЕТАНА И ИХ СОЛИ | 1992 |
|
RU2044736C1 |
Использование: в медицине для лечения цереброваскулярных нарушений. Сущность изобретения: продукт - производные кумарина ф-лы 1, где R1, R2, R3 одинаковые или различные и означают Н, алк C1-C4, На 1, R5 и R6 одинаковые или различные и означают Н, алк C1-C4, R7 и R8 одинаковые или различные и означают Н, алк- C1-C4, или R7 и R8 вместе означают алкилен C3-C4, m 1 или 2; при условии, что R5 и R6 не являются идентичными и представляют CH3 или C2H5, когда R1,R3,R7 и R8-H,R2-CH3 и n = 1, или их фармацевтически приемлемая соль. Условия процесса: взаимодействием гидроксильного производного ф-лы 2 с соответсвующим галогенидом или сульфонатом в присутствии основания или инертного растворителя. Структура соединений ф-л 1 и 2 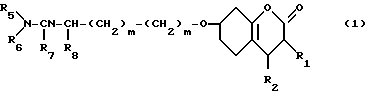
 8 з. п. ф-лы, 4 табл.
8 з. п. ф-лы, 4 табл.
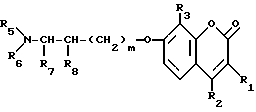
где R1, R2 и R3, одинаковые или разные, каждый - водород, С1-С4-алкильная группа или галоген;
R5 и R6, одинаковые или разные, каждый водород или С1-С4-алкильная группа;
R7 и R8, одинаковые или разные, каждый водород или С1-С4-алкильная группа или R7 и R8 вместе - С3-С4-алкиленовая группа;
m 1 или 2, при условии, что R5 и R6 не одинаковые и не представляют собой либо метильную, либо этильную группу; когда R1, R3, R7 и R8 водород, R2 метильная группа и m 1,
или их фармацевтически приемлемая соль.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
