Изобретение относится к ряду производных тиазолидина, которые характеризуются наличием наряду с другими гидрохиноновой группы или нафтогидрохиноновой группы в их молекулах. Эти соединения обладают ценными видами терапевтической и профилактической активности, включая противодиабетическую активность, и, следовательно, изобретение предоставляет также способы и композиции, использующие эти соединения для лечения и профилактики диабета и диабетических осложнений, как это описывается более подробно. Изобретение также предоставляет способы получения этих новых соединений.
Известен ряд соединений, в которых к 5-положению тиазолидин-2,4-дионовой группы присоединена замещенная алкоксибензильная группа. Эти соединения в общем виде могут быть представлены формулой (A):
Например, в европейской патентной публикации N 8203 раскрывается ряд соединений типа, показанного формулой (A), в которой Ra может быть алкильной или циклоалкильной группой. В европейской патентной публикации N 139421 раскрываются такие соединения, в которых группа, эквивалентная группа Ra в формуле (A), приведенной выше, представляет хромановую или аналогичную группу, а Y.Kawamatsu и др. Chem. Pharm. Bull. 30, 3580-3600 (1962)) раскрывают обширный ряд соединений формулы (A), в которой Ra может представлять различные фенильные, замещенные фенильные, алкиламино, циклоалкильные, терпенильную и гетероциклические группы.
Говорится, что все из известных производных тиазолидина, на которые дается ссылкае, обладают способностью понижать уровни глюкозы в крови и считается, что это достигается путем уменьшения стойкости к инсулину в периферической системе.
Однако в настоящее время считают, что соединения известного уровня техники, которые являются наиболее близкими к соединениям настоящего изобретения, раскрываются в европейской патентной публикации N 441605, так как они, как и соединения по изобретению могут содержать гидрохиноновую группу или нафтогидрохиноновую группу, хотя и присоединенную иным образом в алкиленовой группе формулы -(CH2)n.
В настоящее время обнаружен ряд новых соединений, которые в дополнение к способности уменьшать устойчивость к инсулину в периферических тканях (которая является единственной основой потиводиабетической активности большинства соединений известного уровня техники), также проявляют другие виды активности, например, как и соединения европейской патентной публикации N 441605, настоящие соединения обладают способностью подавлять гепатический глюконогенез в печени, который является одной из причин диабетов. Эти дополнительные виды активности в сочетании с низкой токсичностью означают, что соединения по изобретению являются эффективнее, чем известные соединения, и способны лечить более широкий круг различных нарушений. Соединения по изобретению, как это обнаружено, обладают гораздо лучшей активностью, чем известные соединения из европейской патентной публикации N 441605.
Краткая характеристика изобретения.
Таким образом, объектом изобретения является предоставление ряда новых тиазолидиновых соединений, имеющих бензогидрохинонильную или нафтогидрохинонильную группы.
Еще одной целью изобретения является предоставление таких соединений, которые имеют полезную терапевтическую активность, такую, как противодиабетическая активность.
Другие цели и преимущества станут очевидными по мере дальнейшего описания изобретения.
Соответственно соединения по изобретению представляют соединения формулы (I):
в которой:
R1 представляет алкильную группу, имеющую от 1 до 5 атомов углерода;
R2 и R3 являются одинаковыми или различными и каждый представляет алкильную группу, имеющую от 1 до 5 атомов углерода, или алкокси группу, имеющую от 1 до 5 атомов углерода, или
R2 и R3 вместе образуют бензольное кольцо, которое является незамещенным или которое замещено по крайней мере одним заместителем, выбранным из группы, состоящей из заместителей A, определенных ниже, и, когда R2 и R3 вместе образуют указанное бензольное кольцо, R1 представляет атом водорода, атом галогена или алкильную группу, имеющую от 1 до 5 атомов углерода;
R4 и R5 оба представляют атомы водорода, или R4 и R5 вместе представляют одинарную углерод-углеродную связь (образуя двойную связь между двумя углеродными атомами, к которым они присоединены, как это показано);
Y1 и Y2 являются одинаковыми друг с другом или различными и каждый представляет:
атом водорода,
алкильную группу, имеющую от 1 до 5 атомов углерода,
Алифатическую карбоциклическую ацильную группу, имеющую от 1 до 7 атомов углерода, или
бензоильную, нафтоильную, пиридинкарбонильную или хинолинкарбонильную группу, которая является незамещенной или замещенной по крайней мере одним заместителем, выбранным из группы, состоящей из заместителей A определенных ниже;
W представляет одинарную связь или алкиленовую группу, имеющую от 1 до 5 атомов углерода; и
Z представляет атом водорода или 1/x эквивалент катиона, где X является зарядом у катиона; и
заместители A выбираются из группы, состоящей из алкильных групп, имеющих от 1 до 5 атомов углерода, алкокси групп, имеющих от 1 до 5 атомов углерода, и атомов галогена.
Изобретение также представляет фармацевтическую композицию для лечения или профилактики диабетов или гиперлипемии, которая включает эффективное количество активного соединения в смеси с фармацевтически приемлемым носителем или разбавителем, в которой указанное активное соединение выбрано из группы, состоящей из соединений формулы (I), определенной выше.
Далее изобретение представляет способ лечения или профилактики диабетов или гиперлипемии у млекопитающих, которым может быть человек, который включает назначение для приема указанным млекопитающим эффективного количества активного соединения, и при котором активное соединение выбирается из группы, состоящей из соединений формулы (I), определенной выше.
Данное изобретение предоставляет также способы получения соединений по изобретению, которые описываются далее более подробно.
В соединениях по изобретению, когда R1, R2, R3, Y1, Y2 или заместитель A представляет алкильную группу, ею может быть алкильная группа с прямой или разветвленной цепью, имеющей от 1 до 5 атомов углерода, и примеры ее включают метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, петильную, неопентильную и изопентильную группы. Из них мы предпочитаем алкильные группы, имеющие от 1 до 4 атомов углерода, наиболее предпочтительно метильную группу.
Когда R2 и R3 вместе образуют бензольное кольцо (т.е. бензольное кольцо образует с кольцом, с которым оно сконденсировано, нафтогидрохиноновую группу), оно может быть незамещенным или может иметь на кольцевой части, представленной символами R2 и R3, один или более заместителей, выбранных из группы, состоящей из заместителей A, примеры которых приведены ниже. В дополнение к сказанному, в данном случае R1 может представлять атом водорода, галогена или одну из алкильных групп, примеры которых приведены выше. В данном случае заместители A могут быть также выбраны из группы, состоящей из алкильных групп, имеющих от 1 до 5 атомов углерода, таких, как группы, примеры которых приведены выше, алкокси групп, имеющих от 1 до 5 атомов углерода.
Когда получающееся в результате сконденсированное бензольное кольцо является замещенным в отношении числа заместителей нет никаких особых ограничений, за исключением таких, которые могут налагаться числом, способных к замещению положений или возможно пространственными (стерическими) сдерживающими факторами. Обычно возможно наличие от 1 до 4 заместителей, хотя предпочитается меньшее число, обычно более предпочтительно присутствие от 1 до 3, а еще более предпочтительно 1 или 2 заместителя. Мы больше всего предпочитаем данное сконденсированное бензольное кольцо без заместителей.
Когда R2, R3 или заместитель А представляет алкокси группу, ею может быть алкокси группа с прямой или разветвленной цепью, имеющая от 1 до 5 атомов углерода, и примеры ее включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, неопентилокси и изопентилокси группы. Из них мы предпочитаем алкокси группы, имеющие от 1 до 4 атомов углерода, наиболее предпочтительно метокси группу.
Когда R1 или заместитель А представляет атом галогена, им может быть, например, атом хлора, фтора или брома, предпочтительно атом хлора или фтора, и наиболее предпочтительно атом хлора.
Когда Y1 и/или Y2 представляет алифатическую карбоновую ацильную группу, имеющую от 1 до 7 атомов углерода, ею может быть группа с прямой или разветвленной цепью, и примеры таких ацильных групп включают формильную, ацетильную, пропионильную, бутирильную, изобутирильную, валерильную изовалерильную, пивалоильную, пентаноильную и гесаноильную группы. Из них мы предпочитаем алифатические карбоксильные ацильные группы с прямой или разветвленной цепью, имеющие от 2 до 4 атомов углерода, и наиболее всего предпочитаем ацетильную группу.
Когда Y1 и/или Y2 представляет обязательно замещенную бензоильную, нафтоильную, пиридинкарбонильную или хинолинкарбонильную группу, примеры таких групп включают бензоильную, альфа-нафтоильную, бета-нафтоильную, пиколиноильную, никотиноильную, изоникстиноильную, хинолин-2-карбонильную, хинолин-3-карбонильную и хинолин-4-карбонильную группу. Из них мы предпочитаем необязательно замещенную бензоильную группу или необязательно замещенную пиридинкарбонильную группу и больше всего предпочитаем никотинсильную группу.
W может представлять одинарную связь или алкиленовую группу. Когда W представляет алкиленовую группу, ею может быть алкиленовая группа с прямой или разветвленной цепью, имеющая от 1 до 5 атомов углерода. Связи алкиленовой группы, по которым она присоединяется с одной стороны к гидрохиноновой или нафтогидрохиноновой группе, а с другой стороны к атому кислорода, могут быть у одних и тех же атомов углерода или у различных атомов углерода. Когда связи находятся у одних и тех же углеродных атомов, эти группы иногда называют "алкалиденовыми группами". Однако обычным или общепринятым является использование общего термина "алкиленовая группа" для включения как тех групп, у которых связи находятся у одного и того же атома углерода, так и тех, у которых они находятся у различных углеродных атомов. Примеры таких групп включают метиленовую, этиленовую, триметиленовую, тетраметиленовую пентаметиленовую, метилметиленовую, 2,2-диметилтриметиленову, 2-этилтриметиленовую, 1-метилтетраметиленовую, 2-метилтетраметиленовую и 3-метилтетраметиленовую группы, из которых мы предпочитаем те алкиленовые группы (которые могут быть группами с прямой или разветвленной цепью), имеющие от 1 до 4 атомов углерода, и наиболее всего предпочитаем алкиленовые группы с прямой цепью, имеющие 2 или 3 атома углерода.
Z может представлять атом водорода или катион. Когда катион имеет множественный заряд, например 2+, тогда Z представляет число эквивалентов того катиона, который является соответствующим заряду. Например, когда Z представляет щелочной металл, примеры таких щелочных металлов включают литий, натрий, или калий, и зарядом, который несут эти металлы, является 1+, Z представляет для каждого эквивалента соединения формулы (1) один эквивалент металла. Когда Z представляет щелочно-земельный металл, примеры таких металлов включают кальций или барий, и заряд, несомый этими металлами, составляет 2+, Z представляет для каждого эквивалента соединения формулы (1) половину эквивалента металла. Когда Z представляет основную аминокислоту, примеры таких аминокислот включают лизинг или аргинин, и заряд, несомый этими кислотами, составляет 1+, Z представляет для каждого эквивалента соединения формулы (1), один эквивалент кислоты.
Предпочтительно Z представляет щелочной металл, половину эквивалента щелочно-земельного металла или основную аминокислоту.
Соединения по изобретению обязательно содержат по крайней мере один асимметричный углерод в 5-положении тиазолидинового кольца, и в зависимости от природы групп и атомов, представленных символами R1, R2, R3, Y1, Y2 и W, могут содержать несколько асимметричных атомов углерода в своих молекулах. Они могут также образовывать оптические изомеры. Они могут также образовывать таутомеры вследствие взаимопревращений имидной группы, образуемой оксо группами в 2- и 4- положениях тиазолидинового кольца, в группу формулы N= C/OH/-. Хотя эти оптические изомеры и таутомеры все представлены здесь одной молекулярной формулой, изобретение включает, как индивидуальные выделенные изомеры, так и смеси, включая их рацематы. Когда применяются стереоспецифические приемы синтеза или в качестве исходных материалов применяются оптически активные соединения, могут непосредственно получаться индивидуальные изомеры; с другой стороны, если получается смесь изомера, индивидуальные изомеры могут быть получены с помощью общепринятых приемов разделения.
Предпочтительным классом соединений по изобретению являются те соединения формулы (1), в которых:
R1 представляет алкильную группу, имеющую от 1 до 5 атомов углерода;
R2 и R3 являются одинаковыми или различными (особенно предпочтительно, одинаковыми), и каждый представляет алкильную группу, имеющую от 1 до 5 атомов углерода, или алкокси группу, имеющую от 1 до 5 атомов углерода, или R2 и R3 вместе образуют бензольное кольцо, которое является незамещенным или замещенным по крайней мере одним заместителем, выбранным из группы, состоящей из заместителей А, определенных выше, и, когда R2 и R3 вместе образуют указанное бензольное кольцо, R1 представляет атом водорода, атом галогена или алкильную группу, имеющую от 1 до 65 атомов углерода.
R4 и R5 каждый представляет атом водорода;
Y1 и Y2 является одинаковыми или различными и каждый представляет атом водорода, метильную группу, ацетильную группу, бензоильную группу или никотиноильную группу;
W представляет алкиленовую группу, имеющую от 1 до 5 атомов углерода; и
Z представляет атом водорода или атом натрия.
Более предпочтительным классом соединений данного изобретения являются те соединения формулы (1), в которых:
R1 представляет алкильную группу с 1-5 атомами углерода;
R2 и R3 являются одинаковыми и каждый представляет алкильную группу, имеющую от 1 до 5 атомов углерода, или R2 и R3 вместе образуют незамещенное бензольное кольцо, и, когда R2 и R3 вместе образуют указанное бензольное кольцо, R1 представляет атом водорода, метильную группу или атом хлора, более предпочтительно, атом водорода;
R4 и R5 каждый представляет атом водорода;
Y1 и Y2 являются одинаковыми или различными и каждый представляет атом водорода, метильную группу или ацетильную группу, более предпочтительно метильную или ацетильную группу;
W представляет алкиленовую группу, имеющую от 2 до 4 атомов углерода; и
Z представляет атом водорода или атом натрия.
Наиболее предпочтительным классом соединений данного изобретения являются соединения формулы (1), в которых:
R1, R2 и R3 каждый представляет метильную группу;
Y1 и Y2 являются одинаковыми и каждый представляет метильную или ацетильную группу;
W представляет этиленовую или триметиленовую группу; и
Z представляет атом водорода или атом натрия.
Конкретными примерами соединений изобретения являются соединения, имеющие формулы (1-1) (1-3) см. фиг. 1, в которых заместители имеют значения, определенные в отношении одной из табл.1-3, т.е. табл. 1 относится к формуле (1-1), табл. 2 к формуле (1-2) и табл.3 к формуле (1-3). В табл. 1-3 используются следующие сокращения для некоторых групп; в других отношениях для обозначения атомов используются стандартные международно признанные символы:
Ac ацетил Et этил
Boz бензоил Me метил
Bu бутил Nic никотиноил
Byr бутирил Ipr изопропил
Из соединений, перечисленных выше, предпочтительными являются соединения N:
1-7. 5- 4-[3-(2,5-Дигидрокси-3,4,6-триметилфенил)пропокси]бензил} тиазолидин-2,4-дион;
1-14. 5-[4-(2,5-диметокси-3,4,6-триметилбензилокси)(бензил] тиазолидин-2,4-дион-натриевая соль,
1-17. 5-{4-[3-(2,5-диметокси-3,4,6-триметилфенил)пропокси]бензил} тиазолидин-2,4-дион.
1-18. 5-{4-[3-(2,5-диметокси-3,4,6-триметилфенил)пропокси]бензил} тиазолидин-2,4-диона натриевая соль;
1-20. 5-{4-[4-(2,5-диметокси-3,4,6-триметилфенил)бутокси]бензил} тиазолидин-2,4-диона натриевая соль,
1-23. 5-[4-(2,5-диацетокси-3,4,6-триметилфенокси)бензил] тиазолидин-2,4-дион.
1-27. 5-{4-[2-(2,5диацетокси-3,4,6-триметилфенил)этокси]бензил} тиазолидин-2,4-дион.
1-30. Натриевая соль 5-{4-[2,5-диацетокси-3,4,6-триметилфенил) пропокси] бензил}тиазолидин-2,4-диона.
1-47. 5-{4-[2-(2,3,4,5-тетраметокси-6-метилфенил)-этокси]бензил} тиазолидин-2,4-диона натриевая соль.
1-49. Натриевая соль 5-{4-[3-(2,3,4,5-тетраметокси-6-метилфенил) пропокси]бензил}тиазолидин-2,4-диона,
1-50. 5-{4-[4-(2,3,4,5-тетраметокси -6- метилфенил)бутокси]бензил} тиазолидин-2,4-дион,
1-51. Натриевая соль 5-{4-[4-(2,3,4,5-тетраметокси-6-метилфенил) бутокси]бензил}тиазолидин-2,4-диона,
2-4. 5-[4-(2,7-диметоксинафтилметокси)бензил]тиазолидин-2,4-дион,
2-5. Натриевая соль 5 [4-(2,7-диметоксинафтилметокси)бензил]-тиазолидин-2,4-диона,
2-12. 5-[4(2,7-диметокси-8-метилнафтилметокси)бензил] тиазолидин-2,4-дион,
2-14. 5-{4-[2-(2,7-диметоксинафтил)этокси]бензил} тиазолидин-2,4-дион, и
2-15. Натриевая соль 5-{4- [2-(2,7-диметоксинафтил)этокси]бензил} тиазолидин-2,4-диона.
Из них предпочтительны соединения NN 1-18, 1-27, 2-4 и 2-14 и наиболее предпочтительны соединения NN 1-18 м 2-14.
Соединения по изобретению могут быть получены с помощью большого разнообразия процессов, известных для получения данного типа соединений. Например, они могут быть проиллюстрированы следующими методами A-H.
Метод A:
Метод A включает осуществление процедуры, описанной в европейской патентной публикации N 139421 (японская патентная заявка Кокаи N Sh 060-51189) японской патентной публикации N He i 2-31079), сведения из которой включены в описание для ссылки. Желаемое соединение формулы (I) могут быть получены с помощью взаимодействия соединения общей формулы (II):
(в которой R1, R2, R3, Y1, Y2 и W имеют значения, определенные выше, A представляет карбоксильную, алкоксикарбонильную или карбамоильную группу, или группу формулу -COOM; и X представляет атом галогена), которое может быть получено, как описано в указанном патенте в отношении альфа-галоидкарбоновых кислот, используемых в качестве исходных материалов и/или в "Ссылочных примерах" с тиомочевиной с получением промежуточного соединения формулы (III):
(в которой R1, R2, R3, Y1, Y2 и W имеют значения, определенные выше), а затем гидролиза соединения формулы (III), например, как описано в цитированном патенте.
Примеры алкоксикарбонильных групп, которые могут быть представлены символом A, включают метоксикрабонильную, этоксикарбонильную, изопропоксикарбонильную и бутоксикарбонильную группу. В группе формулы -COOM M представляет атом металла, например натрия, калия, кальция или алюминия, или эквивалентный катион, такой, как ион аммония. X представляет атом галогена, такой как атом хлора, брома или иода.
Реакция соединения формулы (II) с тиомочевиной обычно и предпочтительно проводится в присутствии растворителя. В отношении характера применяемого растворителя нет никаких особых ограничений при условии, что он не оказывает отрицательного влияния на реакцию или на реагенты, участвующие в реакции, и что он может растворять реагенты по крайней мере до некоторой степени. Примеры подходящих растворителей включают: спирты, такие как метанол, этанол, пропанол, бутанол или этиленгликольмонометиловый эфир; простые эфиры, такие как тетрагидрофуран или диоксан; кетоны, такие как ацетон; сульфоксиды, такие как диметилсульфоксид или сульфодан; и амиды, особенно амиды жирных кислот, такие как диметилформамид и диметилацетамид. Нет особых ограничений в отношении используемого молярного отношения соединения формулы II) к тиомочевине, но предпочтительно реакция проводится с использованием по крайней мере небольшого молярного избытка тиомочевины на моль соединения формулы (II). Более предпочтительно использовать от 1 до 2 молей тиомочевины на моль соединения формулы (II).
Реакция может проходить в широком интервале температур и точная температура реакции не является критической для изобретения и предпочтительная температура может варьировать в зависимости от природы исходного материала и используемого растворителя. Обычно мы находим удобным осуществлять реакцию при температуре кипения растворителя или при температуре от 80 до 150oC. Время, требуемое для реакции, также может широко варьировать в зависимости от многих факторов, а именно температуры реакции и природы применяемых реагентом и растворителя. Однако при условии, что реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным бывает период от 1 до нескольких десятков часов.
После этого соединение формул (III) может гидролизоваться с помощью нагревания его в соответствующем растворителе и в присутствии воды и органической кислоты, такой как уксусная кислота, или минеральной кислоты, такой как серная или соляная кислота, или минеральной кислоты, такой как серная или соляная кислота. Реакция обычно и предпочтительно проводится в присутствии растворителя. Нет особых ограничений относительно природы применяемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию и реагенты, участвующие в реакции и при условии, что он может по крайней мере до некоторой степени растворять реагенты. Примеры подходящих растворителей включают: сульфоксиды, такие как сульфолан; и спирты, такие как метанол, этанол и этиленгликольмонометиловый эфир. Количество используемой кислоты обычно и предпочтительно составляет от 0,1 до 10 молей, более предпочтительно от 0,2 до 3 молей, на моль соединения формулы (III). Вода или водный растворитель добавляется обычно в большом избытке относительно молярного количества соединения формулы (III).
Реакция может происходить в широком интервале температуры, и точная температура реакции не является существенной для изобретения. Обычно мы находим удобным осуществлять реакцию при температуре порядка 50-100oC. Время, требуемое для реакции, также может широко варьировать в зависимости от многих факторов, а именно температуры реакции и природы применяемых реагентов и растворителя. Однако при условии, что реакция осуществляется в предпочтительных условиях, описанных выше, обычно достаточным является период от нескольких часов до нескольких десятков часов.
После гидролиза Y1 и Y2 в соединении формулы (I) каждый обычно представляет атом водорода или соответствующую алкильную группу. Когда Y1 и Y2 каждый представляет ацильную группу, они могут оставаться незатронутыми в зависимости от выбора условий реакции.
Метод B:
Метод B предусматривает получение соединения формулы (I) с помощью процедуры, описанной в J. Med. Chem. 1538 (1991)), информация, о которой включена в описание для сведения. См. фиг. 2.
В приведенных выше формулах R1, R2, R3, Y1, Y2 и W имеют значения, определенные выше, и R6 представляет атом водорода или защитную группу.
В способе B спиртовое соединение формулы (IV), которое используется в качестве исходного материала, может получаться с помощью процедуры, описанной, например, в J. Am. Chem. Soc. 64, 440 (1942), J. Am. chem. soc. 94, 227 (1972)f, J. Chem. soc. Perkin Jrans. 1. 1591 (1 83), японской патентной заявке Кокаи N Sho 58-83698)= японская патентная публикация N Hei 1-33114), японской патентной заявке Кокаи N 58-174342)= японская патентная публикация N Hei 1-39411) или J. Iakeda Res. Zab. 45, N 3 и 4, 73 (1986) с последующими обычными реакциями превращения. Желаемое соединение формулы (VI) затем может получаться с помощью реакции дегидратационной конденсации, например, реакции, известной как реакция Мицунобу Fiesen Fieser, "Reagents for Grganic Synthesis" том 6, стр. 645, дубликация Вили-Интерсайенс, издания John Wiley and Санз), между соединением формулы (IV) и необязательно защищенным тиазолидиновым соединением формулы (V).
Реакция обычно и предпочтительно проводится в присутствии растворителя. Относительно природы применяемого растворителя нет каких-либо особых ограничений, единственным условием является то, чтобы он не оказывал отрицательного воздействия на реакцию и применяемые реагенты и мог растворять реагенты по крайней мере до некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол или толуол; алифатические углеводороды, такие как гексан или гептен; простые эфиры, такие как тетрагидрофуран или диоксан; галоидированные углеводороды, особенно галоидированные алифатические углеводороды, такие как метиленхлорид; и сульфоксиды, такие как диметилсульфоксид. Молярное отношение соединения формулы (Iv) к соединению формулы (V) не имеет особого значения, но предпочитается использовать от 1 до 3 молей соединения формулы (V) на моль соединения формулы (IV).
Реакция может протекать в широком интервале температур, но точная реакционная температура не является критической для реакции. Обычно мы находим удобным осуществлять реакцию при температуре от -20 до 150oC. Время, необходимое для реакции, также может широко варьировать в зависимости от многих факторов, а именно температуры реакции и природы применяемых реагентов и растворителя. Однако при условии, что реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным является период от 10 мин до нескольких десятков часов.
Когда соединение формулы (VI), полученное таким образом, имеет защитную группу, например тритильную группу, если необходимо может быть достигнуто снятие защиты с помощью обработки соединения формулы (VI) органической кислотой, такой как трифторуксусная кислота, давая соединение формулы (I).
Реакция обычно и предпочтительно проводится в присутствии растворителя. Нет каких-либо ограничений в отношении природы применяемого растворителя при условии, что он не оказывает отрицательного влияния на ход реакции или на реагенты, вовлеченные в реакцию, и может растворять по крайней мере до некоторой степени реагенты. Примеры подходящих растворителей включают простые эфиры, такие как тетрагидрофуран или диоксан; и галоидированные углеводороды, особенно галоидированные алифатические углеводороды, такие, как металенхлорид. Молярное отношение трифторуксусной кислоты к соединению формулы (VI) составляет предпочтительно от 0,5:1 до большого избытка.
Реакция может проходить в широком интервале температур, но точная температура реакции не является критической для изобретения, а предпочтительная температура может варьировать в зависимости от природы исходного материала и используемого растворителя. Обычно мы считаем удобным осуществлять реакцию при температуре от -20 до 40oC. Время, необходимое для реакции, также может широко изменяться в зависимости от многих реакторов, а именно от температуры реакции и природы применяемых реагентов и растворителя. Однако при условии, что реакция проводится в предпочтительных условиях, охарактеризованных выше, обычно достаточным является период от нескольких минут до нескольких десятков часов.
Метод C:
По способу C желаемое соединение формулы (I) может получаться с помощью превращения соединения формулы (IV), описанного в методе B, в активное сложно-эфирное производное или в галоидированное соединение и взаимодействия продукта с соединением формулы (V).
На первой стадии соединения формулы (II) превращается в активное сложно-эфирное соединение, такое как метансульфонат, бензолсульфонат или толуолсульфонат, с помощью обычных средств или в галоидированное соединение, такое, как хлорид, бромид или иодид, с помощью известных средств. Требуемое соединение формулы (I) может затем получаться по реакции активного сложно-эфирного соединения или галоидированного соединения, полученного таким образом, с соединением формулы (V).
Реакция активного сложно-эфирного соединения или галоидированного соединения с соединением формулы (V) обычно предпочтительно осуществляется в присутствии основания, например неорганического основания, такого как карбонат щелочного металла (например, карбонат натрия или карбонат калия), или гидрооксись щелочного металла (например, гидроокись натрия или гидроокись калия); алкоголят щелочного металла, такой как металат натрия, этилат или трет-бутилат калия; или гидрид металла, такой как гидрид натрия, гидрид калия или гидрид лития. Реакция обычно предпочтительно проводится в присутствии растворителя. В отношении применяемого растворителя нет каких-либо особых ограничений, при условии, что он не влияет отрицательно на реакцию или на участвующие в ней реагенты и может растворять реагенты по крайней мере до некоторой степени. Предпочтительный используемый растворитель варьирует в зависимости от природы используемого основания. Однако примеры подходящих растворителей включают ароматические углеводороды, такие, как бензол, толуол или ксилол; простые эфиры, такие, как диэтиловый эфир, тетрагидрофуран или диметилформамид или диметилацетамид; и органические соединения серы, такие, как диметилсульфоксид или сульфолан.
Из них предпочтительны амиды. Молярное отношение соединения формулы (V) к основанию обычно составляет от 0,5:1 до 5:1, более предпочтительно от 1:1 до 3:1. Молярное отношение соединения формулы (V) к активному сложно-эфиному или галоидированному соединению составляет обычно от 0,5:1 до 4,1, более предпочтительно от 1:1 до 3:1.
Реакция может протекать в широком интервале температур, и точная реакционная температура не является критической для изобретения, предпочитаемая используемая температура варьирует в зависимости от природы исходного материала, основания и растворителя, которые используются при реакции. Обычно мы находим удобным осуществлять реакцию при температуре 0-50oC, более предпочтительно 5-20oC. Время, необходимое для реакции также может варьировать широко в зависимости от многих факторов, а именно от реакционной температуры и природы применяемых реагентов и растворителя. Однако при условии, что реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным является период от нескольких минут до нескольких десятков часов.
Защитная группа может затем, если необходимо, удаляться с помощью процедуры, описанной при описании Метода B.
Метод D:
По данному способу соединение формулы (1) может получаться с помощью процедуры, описанной, например, в европейской патентной публикации N 306228 (Японская патентная публикация Кокан N Hei-1-131169) см. фиг. 3.
В приведенных формулах R1, R2, R3, Y1, Y2 и W имеют значения, определенные выше.
Согласно данной схеме реакции соединение формулы (I) может получаться с помощью реакции конденсации между альдегидным соединением формулы (VII), получаемым с помощью процедуры, описанной в указанном выше патенте, с тиазолидин-2,4-дионовым соединением формулы (VIII), давая соединение формулы (Ia), которое затем восстанавливается.
Альтернативно соединение формулы (Ia) может также получаться из соединения формулы (X) при соответствующем выборе условий реакции в Методе E, описанном далее. Так, соединение формулы (X) окисляется с помощью цериевого нитрата аммония (CAN), как описано в Методе E, давая бензилиденовое соединение формулы (XI), и данный продукт восстанавливается с использованием боргидрида натрия с помощью процедуры, описанной в Методике E, давая бензилиденовое соединение формулы (XII). Требуемое соединение формулы (Ia) может затем получаться путем ацилирования или алкилирования бензилиденового соединения формулы (XII), полученного выше, с помощью обычных средств, например, с помощью процедуры, описанной при описании Метода F.
Данная реакционная последовательность иллюстрируется в виде реакционной схемы D, приведенной на фиг. 4.
В указанной схеме R1, R2, R3 и W имеют значения, определенные выше, и Y3 и Y4, которые могут быть одинаковыми или отличаться друг от друга, каждый представляет алкильную группу, предпочтительно имеющую от 1 до 5 атомов углерода, например, также, как определены выше в отношении R1 предпочтительно, метильную группу.
Метод E:
Согласно данному соединение формулы (I), в которой Y1 и Y2 оба представляют атомы водорода, могут получаться с помощью реакционной схемы E, представленной ниже (см. фиг. 5).
В приведенных выше формулах R1, R2, R3, Y3, Y4 и W имеют значения, определенные выше.
На стадии EI данной схемы реакций соединение формулы (X), в которой Y3 и Y4 каждый представляет низшую алкильную группу, предпочтительно метильную группу, превращается с помощью окисления с использованием цериевого нитрата аммония в соединение формулы (XIII) с помощью процедуры, описанной в "Fieser u Fieser for Grganic Synthesis",том 7, с. 55, публикация Вилли-Интерсайенс из-во Джон Вилли энд Санз, информация из которой включена в данное описание для сведения. Реакция окисления с использованием цериевого нитрата аммония обычно и предпочтительно проводится в присутствии растворителя. В отношении природы применяемого растворителя нет особых ограничений при условии, что он не оказывает отрицательного воздействия на реакцию и участвующие в ней реагенты, и что он может растворять реагенты по крайней мере до некоторой степени. Примеры подходящих растворителей включают воду; нитрилы, такие как ацетонитрил; кетоны, такие как ацетон; и смеси любых двух или более из вышеуказанных растворителей. Количество используемого цериевого нитрата аммония особенно не является критическим, но предпочтительно использовать 1-10 молей цериевого нитрата аммония на моль соединения формулы (X). Реакция может протекать в широком интервале температур, не точная температура реакции не является критической, хотя предпочтительная температура может варьировать в зависимости от природы используемых исходного материала и растворителя. Обычно удобно осуществлять реакцию при температуре от -10 до 40oC. Время, требуемое для реакции, также может варьировать широко в зависимости от многих факторов, а именно от температуры реакции и природы применяемых реагентов и растворителя. Однако при условии, что реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным является период реакции от нескольких минут до нескольких десятков часов.
В последствие соединение формулы (XIY) может получаться из соединения формулы (XIII) с помощью восстановления, например каталитического восстановления, или с использованием восстанавливающего агента, такого как гидрид (например, боргидрид натрия) или металл (например цинк или железо).
Если необходимо, исходное соединение может подвергаться сначала окислению с использованием цериевого нитрата аммония, например, как показано на схеме реакции E:
(в которой
R1, R2, R3, Y3, Y4 и W имеют значения, определенные выше, и Br представляет бензильную или замешенную бензильную группу). Как пример данный метод применим к соединению формулы (XV), т.е. соединению формулы (IV), в котором гидроксильная группа защищена бензильной группой давая соединение формулы (XVI).
Метод F:
Данный метод состоит в получении соединения формулы (i), в которой Y1 и Y2 каждый представляет ациальную группу.
По данному способу хиноновое или нафтохиноновое соединение, например соединение формулы (XIV), которое может быть получено, как описано при описании Метода E, и после выделения из реакционной смеси или без выделения подвергается ацилированию с помощью обычных средств, давая соединение, эквивалентное соединение формулы (X), но в котором алкильные группы, представленные символами Y3 и Y4, заменены ацильными группами.
Данная реакция может при желании проводиться на стадии получения исходных материалов. Например, как показано на схеме реакций F, приведенной на фиг.6.
В приведенных формулах R1, R2, R3, Y3, Y4 и W имеют значения, определенные выше; X представляет атом галогена, такой как атом хлора, брома или иода; и Y5 и Y6 являются одинаковыми или различными и каждый представляет ацильную группу, как она определена для Y1 и Y2.
По данной схеме реакции соединение формулы (XVII), в которой Y3 и Y4 каждый представляет низшую алкильную группу (особенно, описанному в Методе E, давая соединение формулы (XVIII). Впоследствии диациальное соединение формулы (XX) может получаться для использования в качестве исходного материала с помощью восстановления соединения формулы (XVIII) с применением процедуры, описанной в методе E, давая соединение формулы (XIX), а затем ацилирования продукта, давая соединения формулы (XX).
Ацилирование и может осуществляться после выделения или без выделения соединения формулы (XIX). Когда ацилирование поведется без выделения соединения формулы (XIX), соединение формулы (XX) Может получаться путем восстановления соединения формулы (XVIII) с использованием металла, такого, как цинк или железо, в присутствии ацилирующего агента, такого как ангидрид кислоты (например, уксусной ангидрид), или галоидированное ацильное соединение (например, ацетилхлорид). Реакция обычно и предпочтительно проводится в присутствии растворителя. В отношении применяемого растворителя нет особых ограничений при условии, что он не оказывает вредного воздействия на реакцию и участвующие в ней реагенты и может растворять реагенты по крайней мере в некоторой степени. Примеры подходящих растворителей включают органические кислоты, такие как уксусная кислота или пропионовая кислота; и органические основания, такие как пиридин.
Метод G:
Согласно реакционной схеме G желаемое соединение формулы (I) например, где Z представляет атом натрия, может получаться в форме соли, т.е. путем замещения атома водорода имидной группы атомом металла с помощью взаимодействия соединения формулы (I), в котором Z представляет атом водорода, с подходящим основанием с помощью обычных средств. В отношении природы используемого основания нет никаких особых ограничений. Примеры таких оснований включают: гидроокись натрия, алкоголяты, такие, как метилат натрия или этилат натрия, и натриевые соли органических кислот, такие, как 2-этилгексаноат натрия. Реакция обычно и предпочтительно проводится в присутствии растворителя. В отношении характера применяемого растворителя нет особых ограничений при условии, что он не оказывает вредного влияния на реакцию и участвующие в ней реагенты, и может растворять реагенты по крайней мере до некоторой степени. Используемые предпочитаемые растворители могут изменяться в зависимости от природы используемого основания и примеры растворителей, которые могут использоваться, включают низшие спирты, такие как метанол или этанол: сложные эфиры, такие как этилацетат или пропилацетат; простые эфиры, такие как тетрагидрофуран или диоксан; воду; и смеси любых двух или более указанных выше растворителей.
Соли других металлов, например калия или кальция, или соответствующие соли основных аминокислот или других органических оснований могут приготавливаться по способу, аналогичному получению натриевых солей, описанному выше.
Метод H:
Данный способ может применяться для получения соединений формулы (T), в которой R2 и R3 вместе образуют бензольное кольцо, имеющее от 1 до 4 заместителей, выбранных из группы, состоящей из заместителей A, определенных и приведенных в качестве примеров вышей и W представляет одинарную связь, как показано на схеме реакции H; приведенной на фиг.7. (в которой R1, R2, R3 и X, имеют значения, определенные выше).
Реакция обычно и предпочтительно осуществляется в присутствии основания или с использованием соли щелочного металла (например, натриевой соли) 5-/4-гидроксибензил/тиазолидин-2,4-диона формулы (XXII). Используемое основание, используемый растворитель, температура реакция и время, необходимое для реакции, являются аналогичными таковым метода G.
Альтернативно соединение формулы (XXI) может подвергаться взаимодействию с 4-гидроксинитробензолом или с солью его, давая 3-галоид-2-/4-нитрофенокси/-1,4-нафтохиноновое) производное, а затем продукт превращается с соединение формулы (II) с помощью процедуры, описанной в литературе для Метода A. Впоследствии после процедуры Метода A соединение формулы (I) может получаться из соединения формулы (II). Реакция осуществляется в тех же условиях, что и для Метода A.
После завершения любой из описанных выше реакций необходимые соединения могут выделяться из реакционной смеси и, если необходимо, очищаться общепринятыми приемами, например, с помощью разнообразных приемов хроматографии, таких, как хроматография на колонке, или с помощью перекристаллизации, переосаждения или аналогичных. Пример такой процедуры включает добавление растворителя к реакционной смеси и затем отгонку растворителя из экстракта. Остаток, полученный таким образом, может очищаться с помощью хроматографии на колонке из силикагеля или аналогичным образом, давая желаемое соединение в чистом состоянии.
Кроме того, когда полученное соединение включает смесь различных изомеров, эти изомеры могут разделяться с помощью обычных приемов разделения на соответствующей стадии.
Биологическая активность
Тиазолидиновые соединения данного изобретения показали превосходную гипогликемическую активность и выдающееся ингибирующее действие против гепатического глюконогенеза в системе испытаний с использованием генетически диабетических животных. Соответственно предполагается, что соединения изобретения полезны для лечения и/или предотвращения диабетов, диабетических осложнений, гиперлипидемии, гиперлипопероксидемии, связанной с точностью или полнотой гипертензии, астеопороза и аналогичных.
Соединения по изобретению могут назначаться в разнообразных формах в зависимости от расстройства, подлежащего лечению, и состояния пациента, как это хорошо известно в данной области. Например, когда соединения предполагаются для орального назначения, они могут преобразовываться в форму таблеток, капсул, гранул, порошков или сиропов, для парэнтерального назначения они могут формироваться в виде инъекций (внутривенной, внутримышечной или подкожной), препаратов для капельного вливания или суппозиториев. Для приема по способу через офтальмическую мукозную мембрану они могут формироваться в виде глазных капель или мази для глаз. Эти препаративные формы могут приготавливаться с помощью обычных средств, и, если необходимо, активный ингредиент может смешиваться с любой общепринятой добавкой, такой как носитель, связующее, дезинтегратор, смазочный агент, корригент, солюбилизирующий агент, суспензионное вспомогательное средство, эмульгирующий агент или покрывающий агент. Хотя дозировка варьирует в зависимости от симптомов, возраста и веса тела пациента, характера и тяжести заболевания, подвергаемого лечению или предотвращению, способа назначения и формы лекарства, для лечения диабетов, диабетических осложнений и/или гиперлипемии. Для взрослого человека рекомендуется дневная доза от 1 до 1000 мг соединения, и эта доза может назначаться в виде единичной дозы или в виде раздельных доз.
Активность соединений настоящего изобретения иллюстрируется следующим экспериментом.
Эксперимент.
Гипогликемическая активность.
Используемыми опытными животными были самцы диабетических мышей штамма KK, каждое животное имело вес тела более 40 г. Каждому животному назначали орально 50 мг/кг испытываемого соединения, а затем им давали возможность питаться свободно в течение 18 ч. В конце данного времени из хвостовых вен без анестезии собиралась кровь. Определялся уровень глюкозы в крови с помощью анализатора глюкозы (GL 101, выпускаемого фирмой Мицубиси Касси Ко.
Степень понижения глюкозы в крови вычислялась с помощью следующего уравнения:
Степень понижения глюкозы в крови (5)=
[(BGLs-BGLt)/BGLs]•100
где:
BGLs уменьшение глюкозы в крови (BGL) в группе, получающей растворитель; и
BGLt-BGL в группе, которой назначалось для приема испытываемое соединение.
Результаты показаны ниже, где каждое соединение по изобретению идентифицируется по номеру соединения следующих ниже примеров, в которых иллюстрируется их получение.
В качестве контроля мы также использовали следующие испытываемые соединения:
5-{4-[2-метил-2-гидрокси-4-(3,5,6-триметил-1,4-бензохинон-2-ил) бутокси] бензил}тиазолидин-2,4-дион, который представляет собой соединение примера 1, описанного в европейской патентной публикации N 441605). Данное соединение обозначается как "Контроль 1"; и
5-{ 4-[-(2,5-дигидрокси-3,4,6-триметилфенил)-2-гидрокси-2-метилбутокси] бензил} тиазолидин-2,4-дион, который является соединением примера 3, описанного в европейской патентной публикации N 441605). Данное соединение идентифицируется как "Контроль 2".
Соединение Степень понижения BGL (%)
Соединение примера 7 24,0
Соединение примера 9 28,8
Соединение примера 10 46,0
Соединение примера 12 24,0
Соединение примера 16 20,2
Соединение примера 18 22,0
Соединение примера 19 26,6
Соединение примера 21 33,4
Соединение примера 23 24,4
Соединение примера 31 32,9
Соединение примера 33 28,4
Соединение примера 34 40,0
Контроль 1 -0,6
Контроль 2 10,4
Как можно видеть из приведенных результатов, соединения по изобретению показали гораздо большую активность, чем соединение известного уровня техники.
Получение соединений по изобретению далее иллюстрируется следующими неограничивающими примерами, а получение различных промежуточных продуктов, используемых в этих примерах, иллюстрируется в следующих получениях.
Пример 1. 5-[4-(2,4,5-Триметил-3,6-диметоксифенокси)бензил]тиазолидин-2, 4-дион (соединение N 1-11).
Смесь 5,7 г бутил 2-бром-3-[4-(2,4,5-триметил-3,6-диметокси-фенокси) фенил]пропионата (полученного, как описано в получении 1); 1,2 г тиомочевины и 10 мл сульфолана нагревалась при 120oC в течение 5 ч в атмосфере азота, а затем к получающейся смеси добавлялись 10 мл 2 н. водной соляной кислоты. Смесь затем нагревалась при 100oC в течение 5 ч, после чего реакционная смесь выливалась в воду, а затем экстрагировалась бензолом. Экстракт промывался водой и сушился над безводным сульфатом натрия. Растворитель удалялся из экстракта с помощью перегонки при пониженном давлении и остаток, полученный таким образом, очищался с помощью хроматографии на колонке при пропускании через силикагель с использованием 9:1 по объему смеси бензола и этилацетата в качестве элюента, давая 4,7 г названного в заголовке соединения в виде белого стеклянистого порошка, размягчающегося при 47-50oC.
Спектр ядерно-магнитного резонанса (гексадейтергированный диметилсульфоксид) δ млн. дол.
1,97 (3H, синглет)
2,11 (3H, стнглет), 2,15 (3H, синглет),
3,04 (1H, дублет дублетов, J=9 и 14 Гц),
3,32 (1H, дублет дублетов, J=4 и 14 Гц),
3,54 (3H, синглет, 3,61 (3H, синглет),
4,85 (1H, дублет дублетов, J=4 и 9 Гц),
6,70 (2H, дублет, J=8 Гц),
7,15 (2M, дублет, J=8 Гц).
Пример 2. 5-{4-[2(2,4,5-Триметил-3,6-диметоксифенил)этокси]бензил} тиазолидин-2,4-дион (соединение N 1-15).
3,2 г диэтилазодикарбоксилата добавлялись по каплям при охлаждении льдом и в атмосфере азота к раствору 3,5 г 2-/2,4,5-триметил-3,6-диметоксифенил/этанола, 7,3 г 5-/4-гидроксибензил/ -3-трифенилметилтиазолидин-2,4-диона (полученного, как описано в получении 23) и 4,9 г трифенилфосфина в 100 мл тетрагидрофурана, и получающаяся смесь перемешивалась при комнатной температуре в течение 5 ч. В конце данного периода времени реакционная смесь выливалась в воду, после чего она экстрагировалась этилацетатом. Экстракт промывался насыщенным водным раствором хлористого натрия и сушился над безводным сульфатом натрия. Растворитель затем удалялся из экстракта с помощью перегонки при пониженном давлении и получающийся остаток очищался с помощью хроматографии на колонке из силикагеля с использованием смеси 4:1 по объему гексана и эти6лацетата в качестве элюента, давая 5-{4-[2-(2,4,5-триметил-3,6-диметоксифенил)этокси] бензил}-3-трифенилметилтиазолидин-2,4-дион в виде маслянистого промежуточного вещества. Добавлялось 50 мл трифторуксусной кислоты при охлаждении льдом к 7,9 г промежуточного продукта и полученная смесь перемешивалась в течение 1 ч. В конце данного времени реакционная смесь разбавлялась водой, после чего она экстртагировалась этилацетатом. Экстракт промывался дважды, каждый раз насыщенным водой раствором бикарбоната натрия, затем удалялся с помощью перегонки при пониженном давлении и остаток, полученный таким образом, очищался с помощью хроматографии на колонке при пропускании через силикагель с использованием 3:1 по объему смеси гексана и этилацетата в качестве элюента, давая 3,6 г целевого соединения, размягчающегося при 44-45oC.
Пример 3. 5-{4-[3-(2,5-Диметокси-3,4,5-триметилфенил)пропокси]бензил}тиазолидин-2,4-дион (соединение N 1-17).
8,01 г 5-(4-гидроксибензил)тиазолидин-2,4-диона добавлялось небольшими количествами при охлаждении льдом к суспензии, приготовленной при добавлении 80 мл диметилформамида к 3,45 г гидрида натрия (в виде 55 мас.) дисперсии в минеральном масле, которая предварительно промывалась два раза сухим гексаном). Получающаяся смесь перемешивалась при той же температуре в течение 30 мин, после чего к раствору добавлялся по каплям раствор 13,73 г 3-/2,5-диметокси-3,4,5-триметилфенил/пропилиодида /полученного, как описано в получении 15/ в 20 мл диметилформамида. Смесь затем перемешивалась при комнатной температуре в течение 1,5 ч. В конце данного периода времени реакционная смесь выливалась в 300 мл льда и воды, после чего она экстрагировалась этилацетатом. Экстракт промывался два раза, каждый раз насыщенным водным раствором хлористого натрия, и сушился над безводным сульфатом натрия. Растворитель затем удалялся из экстракта с помощью перегонки при пониженном давлении, и остаток, полученный таким образом, очищался с помощью хроматографии на колонке при пропускании через силикагель с использованием метода градиентного элюирования с помощью смесей гексана и этилацетата с изменением их от 3: 1 до 2:1 по объему в качестве элюента, давая 6,7 г целевого соединения, плавящегося при 111-113oC.
Пример 4. 5-[4-(2,5-Дигидрокси-3,4,5-триметилфенокси)бензил] тиазолидин-2,4-дион (соединение N 1-1).
50 мг боргидрида натрия добавлялись при охлаждении льдом к смеси 480 мг 5-[4- 3,5,6-триметил-1,4-бензохиной-2-илокси)бензил] -тиазолидин-2,4-диона (полученного, как описано в получении 2) в 8 мл этанола, и получающаяся смесь перемешивалась при комнатной температуре в течение 30 мин. В конце данного периода времени реакционная смесь выливалась и охлажденную разбавленную водную соляную кислоту для осаждения кристаллов, которые собирались с помощью фильтрования, давая таким образом 470 мг целевого соединения, плавящегося при 124-130oC.
Пример 5. 5-[4-(2,4,5-Триметил-3,6-диметоксифенил)бензил]тиазолидин-2,4-диона натриевая соль (соединение N 1-12).
35 мг метилата натрия добавлялись к раствору 250 мг 5-[4-(2,4,5-триметил-3,6-диметоксифенокси)бензил] тиазолидин-2,4-диона (полученного, как описано в примере 1) в 2 мл метанола. В конце данного периода времени растворитель удалялся изреакционной смеси с помощью перегонки при пониженном давлении, давая 240 мг целевого соединения в виде бесцветного стеклянистого порошка, плавящегося при 120-125oC (точка размягчения).
Спектр ядерно-магнитного резонанса (гексадейтерированный диметилсульфоксид) d млн дол.
1,98 (3H, синглет), 2,11 '3H, синглет), 2,15 (3H, синглет)
2,63 (1H, дублет дублетов J 10 и 14 Гц),
3,33 (1H, дублет дублетов, J 3 и 14 Гц),
3,56 (3H, синглет), 3,61 (3H, синглет),
4,14 (1H, дублет дублетов, J 3 и 10 Гц),
6,64 (2H, дублет, J 8 Гц), 7,10 (2H, дублет, J 8 Гц).
Пример 6. 5-{ 4-[2-(2,4,5-Триметил-3,6-диметоксифенил)этокси] бензил} тиазолидин-2,4-диона натриевая соль (соединения N 1-16).
0,12 г 2-этилгексаноата натрия добавлялось к раствору 0,3 г 5-{ 4-[2-(2,4,5-триметил-3,6-диметоксифенил)этокси] бензил} тиазолидин-2,4-диона (полученного, как описано в примере 2) в 10 мл этилацетата, и получающаяся смесь перемешивалась при комнатной температуре в течение 17 ч. В конце данного периода растворитель удалялся из реакционной смеси с помощью перегонки при пониженном давлении. Получающийся в результате кристаллический остаток затем промывался 10 мл-ми гексана, 252 мг целевого соединения, плавящегося при 165-170oC.
Пример 7. 5-[4-(2,5-Диацетокси-3,4,6-триметилфенокси)бензил] тиазолидин-2,4-дион (соединение N 1-23).
0,4 г уксусного ангидрида и 0,3 г пиридина добавлялись к раствору 340 мг 5-[4-(2,5-дигидрокси-3,4,6-триметилфенокси)бензил] -тиазолидин-2,4-диона (полученного, как описано в примере 4) и 6 мл толуола, и получающаяся смесь перемешивалась при комнатной температуре в течение 3 дней. В конце данного времени реакционная смесь разбавлялась бензолом, и разбавленная смесь промывалась водой. Смесь сушилась над безводным сульфатом натрия, после чего растворитель удалялся с помощью перегонки при пониженном давлении. Получающийся в результате остаток очищался с помощью хроматография колонке при пропускании через силикагель с использованием 4:1 по объему смеси бензола и этилацетата в качестве элюента, давая 340 мг целевого соединения, плавящегося при 174-176oC.
Примеры 8-25.Следуя процедурам, аналогичным процедурам, описанным в примерах 1-7, приведенных выше, мы также получали соединения формулы (1-4):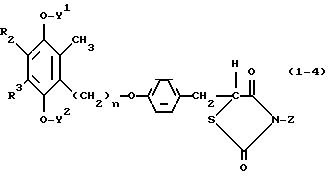
в которой R2, R3, W и Z имеют значения, определенные в табл. 4. В табл.4 столбец "Как в прим. N" показывает номер примера, процедура которого выполнялась.
В табл. 4 и 5 используются следующие сокращения:
Ac ацетил, Me метил,
MeO метокси, Nic никотиноил,
Cpd N Соединение N (из табл. 1-3)
(d) точка разложения; и
(S) точка размягчения
Пример 26-29. Следуя процедурам, аналогичным описанным в примерах 4, 6, и 7, приведенных выше, мы получили также соединения формулы (1-5):
в которой
Y1, Y2 n и Z имеют значения, определенные для табл. 5. В этой таблице столбец "Как в примере N" показывает номер примера, процедуре которого следовали, а сокращения расшифровываются, как определено в отношении табл. 4.
Примеры 30-39. Следуя процедурам, аналогичным описанным в примерах 2-7, приведенных выше, мы получили также соединения формулы (1-6);
в которой
R1, Y1, Y2, n и Z имеют значения, определенные в табл.6. В табл.6 столбец "Как в прим. N" показывает номер примера, процедуре которого следовали, а сокращения расшифровываются так, как определено в отношении табл. 4.
Ниже дано примечание к табл.6.
xСпектр ЯМР соединения примера 30 (дельта млн.дол. гексадейтерированный диметилсульфоксид): 2,21 (3H, синглет), 2,55 (3H, синглет), 3,10 (1Н, дублет дублетов, J 9 и 14 Гц), 3,3-3,4 (1Н, не определенный), 4,89 (1Н, дублет дублетов, J 4 и 9 Гц), 6,84 (2Н, дублет, J 8Гц), 7,21 (2Н, дублет, J 8 Гц), 7,21 (2Н, дублет, J 8 Гц), 7,67-7,75 (2Н, мультиплет), 7,95-8,1 (2Н, мультиплет), 12,03 (1Н, широкий синглет).
xСпектр ядерно-магнитного резонанса соединения примера 31 (дельта млн. дол. СДСI3): 3,12 (1Н, дублет дублетов, J 9 и 14 Гц), 3,46 (1Н, дублет дублетов, J 4 и 14 Гц), 3,94 (3Н, синглет), 3,98 (3Н, синглет), 4,51 (1Н, дубдет дублетов, J 4 и 9 Гц), 5,26 (2Н, синглет), 6,87 (1Н, синглет), 7,00 (2Н, дублет, J 9 Гц), 7,16 (2Н, дублет, J 9 Гц),7,45-7,60 (2Н, мультиплект), 8,08 (1Н, дублет, J 8 Гц), 8,16 (1Н, широкий синглет), 8,24 (1Н, дублет, J 9 Гц).
xСпектр ядерно-магнитного разноса соединения примера 32 ( дельта млн. дол. гексадейтерированный диметилсульфоксид): 2,71 (1Н, дублет дублетов, J 10 и 14 Гц), 3,33 (1Н, дублет дублетов, J 4 и 14 Гц), 3,87 (3Н, синглет), 3,95 (3Н, синглет), 4,22 (1Н, дублет дублетов, J 4 и 10 Гц), 5,20 (2Н, синглет), 6,97 (1Н, синглет), 7,00 (2Н, дублет, J 8 Гц), 7,15 (2Н, дублет, J 8 Гц), 7,55 (1Н, дублет, J 8 Гц), 7,61 (1Н, триплет, J 8 Гц), 8,04 (1Н, дублет, J 8 Гц), 8,16 (1Н, дублет, J 8 Гц).
xСпектр ЯМР соединения примера 33 (дельта млн.дол. СДCI3: 2,46 (3Н, синглет), 3,13 (1Н,дублет дублетов, J 9 и 14 Гц), 3,48 (1Н, дублет дублетов, J 4 и 14 Гц), 3,88 (3Н, синглет), 3,95 (3Н, синглет), 4,52 (1Н, дублет дублетов, J 4 и 9 Гц), 5,24 (2Н, синглет), 7,03 (2Н, дублет, J 9 Гц), 7,20 (2Н, дублет, J 9 Гц), 7,45-7,53 (2Н, мультиплет), 8,07-8,16 (2Н, мультиплет), 8,42 (1Н, широкий синглет).
xСпектр ЯМР соединения примера 34 (дельта млн.дол. СДCI3): 3,10 (1Н, дублет дублетов, J 14 и 9 Гц)), 3,28 (2Н, триплет, J 7 Гц), 3,44 (1Н, дублет дублетов, J 14 и 4 Гц), 3,93 (3Н, синглет), 3,98 (3Н, синглет), 4,25 (2Н, триплет, J 7 Гц), 4,49 (1Н, дублет дублетов, J 9 и 4 Гц), 6,71 (1Н, синглет), 6,88 (2Н, дублет, J 9 Гц), 7,13 (2Н, дублет, J 9 Гц), 7,42-7,58 (2Н, мультиплет), 7,99-8,12 (1Н, широкий синглет), 8,08 (1Н, дублет, J 8 Гц).
xСпектр ядерно-магнитного резонанса соединения примера 35 (дельта млн. дол. гексадейтерированный димметилсульфоксид): 2,63 (1Н, дублет дублетов, J 10 и 14 Гц),3,20 (2Н, триплет, J 7 Гц), 3,31 (1Н, дублет дублетов, J 4 и 14 Гц), 3,85 (3Н, синглет), 3,94 (3Н, синглет), 4,12 (1Н, булдет дублетов, J 4 и 14 Гц), 4,25 (2Н, триплет J 7 Гц), 6,88 (2Н, дублет, J 9 Гц), 6,95 (1Н, синглет), 7,10 (2Н, дублет, J 9 Гц), 7,48 (1Н триплет, J 8 Гц), 7,57 (1Н, триплет, J 8 Гц), 7,98 (1Н, дублет, J 8 Гц),8,11 (1Н, дублет, J 8Гц).
xСпектр ЯМР соединения примера 36 (дельта млн.дол.СДCI3): 2,12-2,25 (2Н, мультиплет), 2,99 (2Н, триплет, J 8 Гц), 3,10 (1Н, дублет дублетов, J 14 и 9Гц), 3,45 (1Н, дублет, дублетов, J 14 и 4 Гц), 3,88 (3Н, синглет), 3,90 (3Н, синглет), 4,01 (2Н, триплет, J 6 Гц), 4,50 (1Н, дублет дублетов, J 9 и 4 Гц), 6,61 (1Н синглет), 8,86 (2Н, дублет, J 9 Гц), 7,14 (2Н, дублет, J 9 Гц), 7,40-7,57 (2Н, мультиплет), 7,98-8,12 (1Н, широкий синглет), 8,02 (1Н, дублет, J Гц), 8,20 (1Н, дублет, J 9 Гц).
xСпектр ядерно-магнитного резонанса соединения примера 37 (дельта млн. дол. гексадейтерированный диметилсульфоксид): 2,05-2,14 (2Н, мультиплект), 2,63 (1Н, дублет дублетов, J 11 и 14 Гц), 2,91 (2Н, триплет, J 8Гц), 3,31 (1Н, дублет дублетов, J 4 и 14 Гц), 3,30 (3Н, синглет), 3,87 (3Н, синглет), 4,00 (2Н, триплет, J 6 Гц), 4,11 (1Н, дублет дублетов, J 4 и 11 Гц), 6,80 (1Н, синглет, 6,84 (2Н, дублет, J 9 Гц), 7,10 (2Н, дублет, J 9 Гц), 7,46 (1Н, триплет, J 8 Гц), 7,55 (1Н, триплет, J 8 Гц), 7,96 (1Н, дублет, J 8 Гц), 8,10 (1Н, дублет, J 8 Гц).
xСпектр ЯМР соединения примера 38 (дельта млн.дол. СДCI3): 1,84-1,93 (4Н, мультиплет), 2,82-2,92 (2Н, мультиплет), 3,10 (1Н, дублет дублетов, J 9 и 14 Гц), 3,44 (1Н, дублет дублетов, J 4 и 14 Гц), 3,57 (3Н, синглет), 3,97 (3Н, синглет), 3,95-4,04 (2Н, мультиплет), 4,50 (1Н, дублет дублетов, J 4 и 9 Гц), 6,63 (1Н, синглет), 6,84 (2Н,дублет, J 9 Гц), 7,12 (2Н, дублет, J 9 Гц), 7,41-7,55 (2Н, мультиплет), 7,88 (1Н,широкий синглет), 8,02 (1Н,дублет, J 9 Гц), 8,20 (1Н, дублет, J 9 Гц).
Получение 1.
Бутил 2-бром-3-[4-(2,4,5-тримеил-3,6-диметоксифенокси)фенил]пропионат.
1(a) 2,5-Диметокси-3,4,6-триметилфенол
Раствор 9,4 г м-хлорнадбензойной кислоты (70% чистоты) в 100 мл метиленхлорида добавляют по каплям при охлаждении льдом к раствору 4,6 г 1,4-диметокси-2,3,5-триметилбензола в 20 мл метиленхлорида, и получающаяся смесь перемешивалась при той же температуре в течение 30 мин, а затем при комнатной температуре в течение 5 ч. В конце данного периода времени реакционная смесь промывалась 5 мас./об. водным раствором бисульфита натрия, 5 мас. /об. водным раствором бикарбоната натрия и водой в указанном порядке, после чего она сушилась над безводным сульфатом натрия. Растворитель затем удалялся из реакционной смеси с помощью перегонки при пониженном давлении, и получающийся остаток очищался с помощью хроматографии на силикагеле с использованием бензола и 50:1 по объему смеси бензола и этилацетата в качестве элюентов, давая 1,3 г целевого соединения.
Спектр ядерно-магнитного резонанса (CDCl3) δ млн. дол.
2,12 (3H, синглет), 2,17 (6H, синглет), 3,65 (3H, синглет), 3,73 (3H, синглет), 5,59 (1H, синглет, исчезал при добавлении окиси дейтерия).
1(b) 2,5-дтметокси-3,4,6-триметил-1-(4-нитрофенокси/бензол.
5,8 г 2,5-Диметоки-3,4,6-триметилфенола (полученного, как описано в стадии (a) выше) в 10 мл диметилформамида добавлялось к суспензии 1,4 г гидрида натрия (в виде 55 мас./мас. дисперсии в минеральном масле) в 50 мл диметилформамида при охлаждении льдом, и смесь перемешивалась при комнатной температуре в течение 2 ч. В конце данного времени к смеси добавлялся раствор 4,6 г п-фторнитробензола в 10 мл диметилформамида при охлаждении льдом. Смесь затем перемешивалась при комнатной температуре в течение 1 ч, а затем при 80oC в течение 7 ч. В конце данного периода времени смесь выливалась в воду, и получающееся сырое масло экстрагировалось бензолом. Бензольный экстракт промывался водой и сушился над безводным сульфатом натрия. Растворитель затем удалялся с помощью перегонки при пониженном давлении, и получающееся масло очищалось с помощью хроматографии на колонке с пропусканием через силикагель, с использованием 4:1 по объему смеси бензола и гексана, а затем одного бензола в качестве элюента, давая 3,9 г указанного в заголовке соединения.
Спектр ядерно-магнитного резонанса (CDCl3) d млн. дол.
2,08 (3H, синглет), 2,19 (3H, синглет), 2,23 (3H, синглет), 3,65 (3H, синглет), 3,70 (3H, синглет), 6,89 (2H, дублет, J 9 Гц), 8,17 (2H, дублет, J 9 Гц).
1(c) 4-(2,5-Диметокси-3,4,6-триметилфенокси/анилин.
Смесь 4,8 г 2,5-диметокси-3,4,6-триметил-1-/4-нитрофеноксибензола (полученного, как описано на стадии (b) выше), 1,0 г 10 мас./мас. палладия на угле и 100 мл этанола перемешивалась в атмосфере азота при комнатной температуре в течение 3 ч. В конце данного периода времени катализатор отфильтровывался, и фильтрат концентрировался с помощью выпаривания при пониженном давлении, давая 3,9 г целевого соединения.
Спектр ядерно-магнитного резонанса (CDCl3) d млн. дол.
2,09 (3H, синглет), 2,17 (3H, синглет), 2,20 (3H, синглет), 3,4 (2H, широкий синглет), исчезал при добавлении окиси дейтерия), 3,674 (3H, синглет), 6,59 (2H, дублет, J 9 Гц), 6,65 (2H, дублет, J 9 Гц).
1(D) Бутил 2-бром-3-[2,4,6-триметил-3,6-диметоксифенокси)-фенил]пропионат.
7,7 г 47 мас./об. водного раствора бромистоводородной кислоты и раствор 1,3 г нитрита натрия в 3 мл воды добавлялись по каплям в указанном порядке к раствору 4,3 г 4-/2,5-диметокси-3,4,6-триметилфенокси/анилина/ полученного, как описано на стадии (c) выше) в 10 мл ацетона, после чего к смеси добавлялось 21 мл бутилакрилата. После этого постепенно добавлялось 0,3 г бромистой меди (2), и получающаяся смесь перемешивалась при комнатной температуре в течение 4 ч. В конце данного периода реакционная смесь выливалась в воду, после чего она экстрагировалась бензолом. Экстракт промывался водой и сушился над безводным сульфатом натрия. Растворитель удалялся с помощью перегонки при пониженном давлении из экстракта, и полученный таким образом остаток очищался с помощью хроматографии на колонке при пропускании через силикагель, с использованием 3:7 по объему смеси гексана и бензола в качестве элюента, давая 5,7 г целевого соединения.
Спектр ядерно-магнитного резонанса (CDCl3) d млн. дол.
0,87 (3H, синглет), 0,91 (3H, синглет), 0,93 (3H, синглет), 1,2-1,4 (2H, мультиплет), 1,5-1,65 (2H, мультиплет), 2,07 (3H, синглет), 2,17 (3H, синглет), 2,21 (3H, синглет), 3,16 (1H, дублет дублетов, J=7 и 10 Гц), 3,39 (1H, дублет дублетов, J=9 и 14 Гц)
3,65 (3,H, синглет), 3,68 (3H, синглет),
4,11 (2H, триплет, J=7 ),
4,33 /1H, дублет дублетов, J=7 и 9 Гц),
6,73 (2H, дублет, J=9 Гц),
Получение 2 (J A 2 a).
5-[4-(3,5,6-триметил-1,4-бензолинон-2-илокси)бензил]тиазолидин-2,4-дион.
Раствор 2,1 г цериевого нитрита аммония в смеси 2 мл воды и 2 мл ацетонитрила добавлялся по каплям при 0oC к раствору 0,4 г 5-[4-(2,4,5-триметил-3,6-диметоксифенокси)бензил] тиазолидин-2,4-диона (полученного, как описано в примере 1) в 3 мл ацетонитрила, и получающаяся смесь перемешивалась при той же температуре в течение 1 ч. В конце данного периода времени реакционная смесь выливалась в воду, после чего она экстрагировалась этилацетатом. Экстракт промывался насыщенным водным раствором хлорида натрия, а затем сушился над безводным сульфатом натрия. Растворитель затем удалялся из экстракта с помощью перегонки при пониженном давлении, и остаток, полученный таким образом, очищался с помощью хроматографии на колонке из силикагеля с использованием 4:1 по объему смеси бензола и этилацетата в качестве элюента, давая 260 мг целевого соединения, плавящегося при 153-156oC (с разложением).
Получение 3 (JA 2-b).
5-{ 4-[2-(3,5,6-триметил-1,4-бензохинон-2-ил)этокси]бензил} тиазолидин-2,4-дион.
Следуя процедуре, аналогичной описанной в получении 2, но при использовании 5-{ 4-[2-(2,4,5-триметил-3,6-диметоксифенил)-этокси] бензил} тиазолидин-2,4-дион (полученного, как описано в примере 2), получали целевое соединение, плавящееся при 157-158oC.
Получение 4 (JA 2-c).
5-{ 4-[3-(3,5,6-Триметил-1,4-бензолхинон-2-ил)пропокси] бензил} тиазолидин-2,4-дион.
Следуя процедуре, аналогичной процедуре, описанной в получении 2, но используя 5-{4-[3-(2,4,5-триметил-3,6-диметоксифенил)-пропокси]-бензил} тиазелидин-2,4-дион (полученный, как описано в примере 10), получали целевое соединение, плавящееся при 118-120oC (с разложением).
Получение 5 (JA 2-d).
5-{ 4-[4-(3,5,6-Триметил-1,4-бензохинон-2-ил)бутокси]бензил}тиазолидин-2,4-дион.
Следуя процедуре, аналогичной процедуре, описанной в получении 2, но используя 5-{4-[4-(2,4,5-триметил-3,6-диметоксифенил)-бутокси]бензил} тиазолидин-2,4-дион (полученный, как описано в примере II), получали целевое соединение в виде желтого пенистого порошка.
Спектр ЯМР (CDCl3) d млн. дол.
1,63 (2H, мультиплет), 1,83 (2H, мультиплет),
2,01 (6H, синглет), 2,0,3 (3Н, синглет),
2,55 (2H, триплет, J=7 Гц),
3,10 (1H, дублет дублетов, J=9 и 14 Гц),
3,45 (1H, дублет дублетов, J=4 и 14 Гц),
3,96 (2H, триплет, J=6 Гц),
4,50 (1H, дублет, дублетов, J=4 и 9 Гц),
6,83 (2H, дублет, J=9 Гц), 7,13 (2H, дублет, J=9 Гц),
8,24 (1H, широкий сигнлет).
Получение 6 (J A 3).
3-Хлор-2-(4-нитрофенокси)-1,4-нафтохинон.
10 г 2,3-дихлор-1,4-нафтохинона добавлялось к раствору 7 г натриевой соли п-нитрофенола в 100 мл диметилформамида, и получающаяся смесь перемешивалась при комнатной температуре в течение 5 ч. В конце данного периода времени реакционная смесь выливалась в воду, после его она экстрагировалась бензолом. Экстракт промывался водой и сушился над безводным сульфатом натрия. Растворитель затем удалялся из экстракта с помощью перегонки при пониженном давлении, и остаток, полученный таким образом, очищался с помощью хроматографии на колонке из силикагеля с использованием 1:4 по объему смеси гексана и бензола в качестве элюента, давая 10 г целевого соединения, плавящегося при 179-182oC.
Получение 7 (J A 4).
Бутил 2-бром-3-[4-(1,4-диацетокси-3-хлор-2-нафтилокси)фенил]-пропионат.
7(a) 3-Хлор-1,4-дигидро-2-(4-нитрофенокси)нафталин.
1 г боргидрида натрия добавлялся при охлаждении льдом к раствору 11 г 3-хлор-2-/4-нитрофенокси)-1,4-нафтохинона (полученного как описано в получении 6) в 150 мл метанола, и смесь перемешивалась при охлаждении льдом в течение 30 мин. Смесь затем выливалась в смесь льда и 15 мл 2 н. водной соляной кислоты, давая осадок, который собирался фильтрованием, промывался водой и сушился при пониженном давлении в присутствии пятиокиси фосфора, давая 9 г 3-хлор-1,4-дигидрокси-2-/4-нитрофенокси/нафталина.
7 (b) 1,4-диацетокси-3-хлор-2-/4-нитрофенокси/нафталин.
Смесь всех 9 г данного 3-хлор-1,4-дигидрокси-2-/4-нитрофенокси/нафталина (полученного, как описано на стадии (a) (6,6 г уксусного ангидрида, 7 г пиридина и 150 мл бензола затем перемешивалась при комнатной температуре в течение 20 ч. В конце данного периода времени реакционная смесь выливалась в смесь льда и 15 мл 2 н. водной соляной кислоты и экстрагировалась бензолом. Экстракт промывался водой и сушился над безводным сульфатом натрия. Растворитель затем удалялся с помощью перегонки при пониженном давлении, давая 7,8 г 1,4-диацетокси-3-хлор-2-/4-нитрофенокси/нафталина.
Тонкослойная хроматография:
Величина Rf 0,40
Адсорбент: силикагельная пластинка N 5715 (Мерк),
Проявляющийся растворитель: бензол.
7 (c) 1,4-Диацетокси-2-/4-аминофенокси/-3-хлор-нафталин.
Следуя процедуре, аналогичной описанной в получении 1 (c), 8,5 г 1,4-диацетокси-3-хлор-2-/4-нитрофенокси/нафталина [полученного, как описано выше, на стадии [b)] гидрировались в атмосфере водорода и в присутствии 1,7 г 10% палладия на угле в 200 мл тетрагидрофурана при комнатной температуре в течение 5 ч, давая 8,3 г 1,4-диацетокси-2-/4-аминофенокси-3-хлорнафталина в виде маслянистого вещества.
Тонкослойная хроматография:
Величина Rf 0,10
Адсорбент: силикагельная пластина N 5715 (Мерк);
Проявляющий растворитель: 10:0,3 по объему смесь бензола и этилацетата.
7 (d) Бутил 2-бром-3-[4-(1,4-диацетокси-3-хлор-2-нафтилокси)фенил]пропионат.
Следуя процедуре, аналогичной процедуре, описанной в получении 1(d), 8,3 г 1,41-диацетокси-2-/4-аминофенокси/-3-хлор-нафталина (полученного, как описано выше, на стадии (c)) арилировались с использованием 15 г 47 мас./об. водного раствора соляной кислоты, 1,9 г нитрата натрия, 27 г бутилакрилата и 0,5 г бромида меди, давая 5,8 г целевого соединения в виде бледно-желтого масла.
Спектр ядерно-магнитного резонанса (CDCl3, частичный) d млн. дол.
0,91 (3H, триплет, J=7 Гц),
3,19 (1H, дублет, дублетов, J=14 и 7 Гц),
3,41 (1H, дублет, дублетов, J=14 и 8 Гц),
4,34 (1H, дублет, дублетов, J=8 и 7 Гц).
Получение 8 (JA 5).
5-[4-(3-Хлор-1,4-нафтохинон-2-илокси)бензил]тиазолидин-2,4-дион.
Смесь 5,8 г бутил-2-бром-3-[4-(1,4-диацетокси-3-хлор-2-нафтилокси) фенил] пропионата (полученного, как описано в получении 7), 1 г тиомочевины и 10 мл сульфолана нагревалась при 120oC в течение 5 ч в атмосфере азота. В конце данного периода времени 20 мл монометилового эфира этиленгликоля и 10 мл 2 н. водной соляной кислоты добавлялись к смеси в присутствии атмосферного кислорода, и получающаяся смесь нагревалась при 100oC в течение 6 ч. Реакционная смесь затем выливалась в воду, после чего она экстрагировалась бензолом. Экстракт промывался водой и сушился над безводным сульфатом магния. Растворитель затем удалялся из экстракта с помощью перегонки при пониженном давлении и получающийся остаток очищался с помощью хроматографии на колонке из силикагеля с использованием 4:1 по объему смеси бензола и этилацетата в качестве элюента. С помощью перекристаллизации из смеси тетрагидрофурана и бензола получалось около 2,4 г целевого соединения в виде кристаллов, плавящихся при 250-252oC.
Спектр ядерно-магнитного резонанса гаксадейтерированный диметилсульфоксид) d млн. дол.
3,09 (1H, дублет дублетов, J=14 и 9 Гц),
3,37 (1H, дублет дублетов, J=14 и 4 Гц),
4,91 (1H, дублет дублетов, J=9 и 4 Гц),
7,13 (2H, дублет, J=8 Гц), 7,22 (2H, дублет, J=8 Гц),
7,85-7,96 (2H, мультиплет), 7,98-8,01 (1H, мультиплет),
8,11 (1H, дублет, J=7 Гц), 12,04 (1H, широкий синглет, исчезал при добавлении окиси дейтерия).
Получение 9 (JA 6).
5-(4-[3-(3,5,6-Триметил-1,4-бензохинон -2- ил)пропокси] бензилиден/тиазолидин-2,4-дион.
Следуя процедуре, аналогичной процедуре, описанной в получении 2, но используя 15,8 г 5-{4-[3-(2,5-диметокси-3,4,5- триметилфенил)пропокси]бензил}тиазолидин-2,4-диона (полученного, как описано в примере 3), 78,1 г цериевого нитрата аммония и 350 мл ацетонитрила, получали 1,7 г целевого соединения, плавящегося при 230-232oC.
Спектр ядерно-магнитного резонанса (гексадейтерированный диметилсульфоксида d млн. дол.
1,80-1,87 (2H, мультиплет), 1,92 (3H, синглет),
1,94 (6H, синглет), 2,60 (2H, триплет, J=7 Гц),
4,04 (2H, триплет, J=6 Гц), 7,04 (2H, дублет, J=9 Гц),
7,53 (2H, дублет, J=9 Гц, 7,77 (1H, синглет),
12,49 (1H, широкий синглет).
Получение 10 (JA 7).
2-/2,3,4,5-Тетраметокси-6-метилфенил/этанол.
10(а) 1-Аллил-2,3,4,5-тетраметокси-6-метилбензол.
Каталитическое количество иода добавлялось к суспензии 975 мг магния в 20 мл тетрагидрофурана, и получающаяся смесь нагревалась примерно до 45oC, давая толчок образованию белой мути. Затем к смеси добавлялся раствор 10,61 г 2,3,4-тетраметокси-6-метил-бромбензола в 30 мл тетрагидрофурана, после чего смесь нагревалась примерно при 45oC в течение нескольких минут. Смесь затем перемешивалась при комнатной температуре в течение 30 мин, после чего к смеси по каплям добавлялось 3,47 мл аллилбромида; смесь затем перемешивалась при комнатной температуре в течение 2 ч. В конце данного периода времени реакционная смесь смешивалась с насыщенным водным раствором хлористого аммония, а затем экстрагировалась этилацетатом. Растворитель удалялся из экстракта с помощью перегонки при пониженном давлении, и остаток, полученный таким образом, очищался с помощью колоночной хроматографии на силикагеле с использованием 10: 1 по объему смеси гексана и этилацетата в качестве элюента, давая 7,98 г целевого соединения в виде масла.
Спектр ядерно-магнитного резонанса (CDCl3) d млн. дол. сообщаются только сигналы вследствие аллильной группы) примерно 3,4 (2H, мультиплет), 4,85-5,05 (2H, мультиплет), 5,8-6,0 (1H, мультиплет).
10 (b) 2-/2,3,4,5-тетраметокси-6-метилфенил/ацитальдегид.
109 мг четырехокиси осмия добавлялось к раствору 7,98 г 1-аллил-2,3,4,5-трераметокси-6-метилбензола [полученного, как описано в стадии (a) выше] в смеси 300 мл диоксана и 100 мл воды, и получающаяся в результате смесь перемешивалась при комнатной температуре в течение 10 мин. Затем по каплям добавлялся водный раствор 35,6 г периодата натрия, и смесь перемешивалась при комнатной температуре в течение 2 ч. В конце данного периода времени реакционная смесь освобождалась от диоксана с помощью выпаривания при пониженном давлении, и получающийся в результате концентрат вливался в насыщенный водный раствор хлористого натрия, после чего он экстрагировался диизопропиловым эфиром. Растворитель затем удалялся из экстракта с помощью перегонки при пониженном давлении, и получающийся в результате остаток очищался с помощью хроматографии на колонке через силикагель с использованием метода градиентного элюирования смесями гексана и этилацетата, варьирующими от 8: 1 до 5:1 по объему в качестве элюента, давая 4,64 г целевого соединения.
Спектр ядерно-магнитного резонанса (CDCl3) (частичный) d млн. дол.
3,71 (2H, дублет, J=2 Гц); 9,68 (1H, триплет, J=2 Гц),
10 (c) 2-/2,3,4,5-Тетраметокси-6-метилфенил/этанол.
5,38 г 2-/2,3,4,5-Тетраметокси-6-метилфенил/ацетальдегида [полученного, как описано в стадии (b)] выше растворялось в 60 мл этанола и восстанавливалось с использованием 400 мг боргидрида натрия при 0oC. Затем к реакционной смеси добавлялось 150 мл насыщенного водного раствора хлористого натрия, и смесь экстрагировалась этилацетатом. Экстракт сушился над безводным сульфатом магния и концентрировался досуха и с помощью выпаривания при пониженном давлении, давая неочищенный продукт. Данный неочищенный продукт очищался затем с помощью хроматографии на колонке через силикагель с использованием метода градиентного элюирования смесями гексана и этилацетата, варьирующими от 5:1 до 2:1 по объему в качестве элюента, давая 5,27 г целевого соединения в виде бесцветного масла.
Спектр ядерно-магнитного резонанса (CDCl3) d млн. дол.
2,19 (3H, синглет); 2,90 (2H, триплет, J=7 Гц); 3,75 (2Н, триплет, J=7 Гц), 3,78 (3H, синглет), 3,85 (3H, синглет), 3,90 (3H, синглет), 3,81 (3H, синглет).
Получение 11 (JA 8).
1,4-Диметокси-2-нафтилметинол.
11(a) Метил 1,4-диметокси-2-нифтоат.
20,7 г безводного карбоната калия добавлялось к раствору 5,1 г 1,4-дигидрокси-2-нафтойной кислоты в 50 мл диметилформамида и к получающейся в результате смеси по каплям добавлялось 28,4 г йодистого метила, после чего они перемешивались в течение 19 ч. В конце данного периода времени реакционная смесь вливалась в воду, и водная смесь нейтрализовалась 3н. водной соляной кислотой и экстрагировалась этилацетатом. Экстракт сушился над безводным сульфатом натрия, и растворитель удалялся с помощью перегонки при пониженном давлении. Получающийся в результате остаток очищался с помощью хроматографии на колонке через силикагель с использованием 10:1 по объему смеси гексана и этилацетата в качестве элюента, давая 5,45 г целевого соединения в виде желтого масла.
Тонкослойная хроматография:
Величина Rf 0,24;
Адсорбент: силикагельная пластина N 5715 (мерк);
Проявляющий растворитель: 10:1 по объему смесь гексана в этилацетате.
11(b) 1,4-Диметокси-2-нафтилметанол.
Раствор 5,32 метил 1,4-диметокси-2-нафтоата (полученного как описано в стадии (A) выше) в 15 мл тетрагидрофурана добавляется по каплям к суспензии 0,98 г литий алюминий гидрида в 15 мл тетрагидрофурана с одновременным охлаждением льдом. Получающаяся в результате смесь затем перемешивалась при комнатной температуре в течение 1 ч, после чего добавлялось 20 мл насыщенного водного раствора хлористого аммония. Образующийся осадок отфильтровывался, и затем продукт экстрагировался этилацетатом. Экстракт сушился над безводным сульфатом натрия и затем концентрировался с помощью выпаривания при пониженном давлении, давая 3,97 г целевого соединения в виде желтого твердого вещества, плавящегося при 63-66oC.
Спектр ядерно-магнитного резонанса (CDCl3) d млн. дол.
3,92 (3H, синглет), 4,0 (3H, синглет), 4,89 (2H, синглнет), 6,82 (1H, синглет), 7,45 7,6 (2H, мультиплет) 8,04 (1H, дублет, J=8 Гц); 8,23 (1H, дублет, J=9 Гц).
Получение 12 (JA 9).
2-(1,4-Диметокси-2-нафтил)этанол.
12 (a) 1,4-Диметокси-2-нафтилметилтрифенилфсофоний хлорид.
Раствор 4,73 г 1,4-диметокси-2-нафтилметил хлорида (полученного, как описано в получении 20) и 6,29 г трифторфенилфосфина в 50 мл сухого ацитонитрила нагревался с обратным холодильником в течение 2 ч, в конце данного периода времени реакционная смесь освобождалась от растворителя с помощью перегонки при пониженном давлении, и получающийся в результате кристаллический остаток промывался диэтиловым эфиром и сушился воздухом, давая 7,36 г целевого соединения в виде белого порошка, плавящегося при 244-246oC (с разложением).
12 (b) 1,4-Диметокси-2-винилнафтален.
50 мл 10% водного раствора гидроокиси натрия добавлялось по каплям с перемешиванием к смеси 7,36 г 1,4-диметокси-2-нафтилметилтрифенилфосфония хлорида (полученного, как описано в стадии (a) выше) и 75 мл 30 об./об. водного раствора формальдегида, и получающаяся в результате смесь перемешивалась в течение 1 ч. В конце данного периода времени реакционная смесь нейтрализовалась 3 н. водной соляной кислотой, после чего они экстрагировалась этилацетатом. Экстракт сушился над безводным сульфатом натрия, и растворитель удалялся при помощи перегонки при пониженном давлении. Получающийся в результате остаток очищался с помощью хроматографии на колонке через силикагель с использованием 24:1 по объему смеси гексана и этилацетата в качестве элюента, давая 2,45 г целевого соединения в виде бледно-желтого масла.
Тонкослойная хроматография:
Величина Rf 0,53;
Адсорбент: силикагельная пластинка N 5715 (Мерк);
Проявляющий растворитель: 24 1 по объему смесь гексана и этилацетата.
12(c) 2-(1,4-Диметокси-2-нафтил)этанол.
1,61 г четыреххлористого титана добавлялось к смеси 0,65 г боргидрида натрия и 20 мл сухого диметилового эфира этиленгликоля, и получающаяся в результате смесь перемешивалась при комнатной температуре в течение 1 ч. Затем к получающейся в результате смеси добавлялся раствор 1,63 г 1,4-диметокси-2-винилнафталена (полученного, как описано в стадии (b) выше) в 40 мл сухого диметилового эфира этиленгликоля, и смесь перемешивалась в течение 21 ч. В конце данного периода времени реакционная смесь вливалась в воду, после чего она экстрагировалась этилацетатом. Экстракт сушился над безводным сульфатом натрия, и растворитель удалялся при помощи перегонки при пониженном давлении. Получающийся в результате остаток очищался с помощью хроматографии на колонке через силикагель с использованием 1:2 по объему смеси гексана и этилацетата в качестве элюента, давая 0,40 г целевого соединения в виде бесцветного масла.
Спектр ядерно-магнитного резонанса (CDCl3) d млн.дол.
3,07 (2Н, триплет, J=7 Гц), 3,91 (H, синглет),
3,93 (2Н, триплет, J=7 Гц), 3,98 (H, синглет),
6,63 (1Н, синглет), 7,4 7,6 (2Н, мультиплет),
8,02 (1Н, дублет, J=8 Гц), 8,22 (1Н, дублет, J=8 Гц).
Получение 13 (JA 10).
3-(1,4-Диметокси-2-нафтил)пропанол.
13(a) 1,4-Диметокси-2-формилнафтален.
4,18 г двуокиси магнезии добавлялось к раствору 0,87 г 1,4-диметокси-2-нафтилметанола (полученного, как описано в получении 11) и 10 мл метиленхлорида и получающаяся в результате смесь перемешивалась при комнатной температуре в течение 6,5 ч. В конце данного периода времени реакционная смесь фильтровалась для удаления неорганических веществ, и фильтрат сушился над безводным сульфатом натрия, после чего растворитель удалялся с помощью перегонки при пониженном давлении. Получающийся в результате кристаллический остаток промывался гексаном и сушился на воздухе, давая 0,57 г целевого соединения в виде бледно-желтых игловидных кристаллов, плавящихся при 120-123oC.
Тонкослойная хроматография:
Величина Rf: 0,44;
Адсорбент: силикагельная пластинка N 5715 (Мерк),
Проявляющий растворитель: 4:1 по объему смесь гексана и этилацетата.
13(b) Метил транс-3-(1,4-диметокси-2-нафтил)акрилат.
0,40 г триметил фосфонацетата добавлялось к суспензии 0,10 г гидрида натрия (в виде 55% мас./мас. дисперсии в минеральном масле, которое предварительно было промыто сухим гексаном) в 6 мл диметилсульфоксида, и получающаяся в результате смесь перемешивалась в течение 20 мин. Затем к смеси добавлялось 0,43 г 1,4-диметокси-2-формилнафталена (полученного, как описано в стадии (a) выше), при одновременном охлаждении льдом, и смесь перемешивалась в течение 1 ч. В конце данного периода времени реакционная смесь вливалась в воду, после чего она экстрагировалась этилацетатом. Экстракт сушился над безводным сульфатом натрия, и растворитель удалялся с помощью перегонки при пониженном давлении. Остаток очищался с помощью хроматографии на колонке через силикагель, с использованием 4:1 по объему смеси гексана и этилацетата в качестве элюента, давая 0,47 г целевого соединения в виде бледно-желтого масла.
Тонкослойная хроматография:
Величина Rf:0,42;
Адсорбент: силикагельная пластинка N 5715 (Мерк);
Проявляющий растворитель: 4:1 по объему смесь гексана и этилацетата.
13(c) Метил 3-(1,4-диметокси-2-нафтил/пропионат.
0,47 г метил транс-3-/1,4-диметокси-2-нафтил/акрилата (полученного, как описано в стадии (b) выше) растворялось в 20 мл метанола и гидрировалось в атмосфере водорода и в присутствии 0,20 г 10 мас./мас. палладия на угле, давая 0,41 г целевого соединения в виде бесцветного масла.
Тонкослойная хроматография:
Величина Rf:0,66,
Адсорбент: силикагельная пластинка N 5715 (Мерк).
Проявляющий растворитель: 3 2 по объему смесь гексана и этилацетата.
13(d) 3-/1,4-Диметокси-2-нафтил/пропанол
Следуя процедуре, аналогичной той, что описана в получении 11(b), но и с использованием 0,41 г метил 3-/1,4-диметокси-2-нафтил)пропионата [полученного, как описано в стадии (c) выше] 68 мг литий алюминий гидрида и 6 мл тетрагидрофурана, было получено 0,34 г целевого соединения в виде бесцветного масла.
Спектр ядерно-магнитного резонанса (CDCl3) d млн.дол.
1,85 2,0 (2Н, мультиплет), 2,91 (2Н, триплет, J 7 Гц),
3,58 (2Н, триплет, J 6 Гц), 3,91 (3Н, синглет),
3,98 (3Н, синглет), 6,60 (1Н, синглет),
7,4 7,6 (2Н, мультиплет), 8,01 (1Н, дублет, J 8 Гц),
8,21 (1Н, дублет, J 8 Гц).
Получение 14 (JA 11).
4-(1,4-диметокси-2-нафтил/бутанол.
14(a) 4-/1,4-Диметокси-2-нафтил/бутиронитрил.
Раствор 5,08 г 3-/1,4-диметокси-2-нафтил/пропилйодида (полученного, как описано в получении 21) и 0,70 г цианида натрия в 60 мл сухого диметилсульфоксида перемешивался при 60oC (внешняя температура) в течение 80 мин. В конце данного периода времени реакционная смесь охлаждалась и вливалась в воду, после чего экстрагировалась этилацетатом. Экстракт сушился над безводным сульфатом натрия и растворитель удалялся с помощью перегонки при пониженном давлении. Получающийся в результате остаток очищался с помощью хроматографии на колонке через силикагель, с использованием 4:1 по объему смеси гексана и этилацетата в качестве элюента, давая 3,36 г целевого соединения в виде бесцветного масла.
Тонкослойная хроматография:
Величина Rf:0,19,
Адсорбент, силикагельная пластинка N 5715 (Мерк).
Проявляющий растворитель: 7 1 по объему смесь гексана и этилацетата.
14(b) 4-/1,4-Диметокси-2-нафтил/бутиральдегид.
20 мл 1,0 М гексанового раствора диизобутилалюминийгидрида добавлялось при -70oC к раствору 3,36 г 4-/1,4-диметокси-2-нафтил/-бутиронитрила [полученного, как описано в стадии (a) выше] в 100 мл сухого метиленхлорида, и получающаяся в результате смесь перемешивалась в течение 2 ч. В конце данного периода времени вода добавлялась к реакционной смеси и нерастворимые вещества отфильтровывались с помощью Целита (торговая марка) фильтра. Метиленхлоридный слой, который отделялся, сушился над безводным сульфатом натрия, и растворитель удалялся с помощью перегонки при пониженном давлении, давая 2,96 г целевого соединения в виде бесцветного масла.
Тонкослойная хроматография:
Величина
Rf:0,19.
Адсорбент: силикагельная пластинка N 5715 (Мерк).
Проявляющий растворитель: 7:1 по объему смесь гексана и этилацетата.
14 (c) 4-(1,4-Диметокси-2-нафтил)бутанол.
Следуя процедуре аналогичной той, что описана в получении 13(c), но с использованием 2,96 г 4-/1,4-диметокси-2-нафтил/ бутиральдегида (полученного, как описано в стадии (b) выше), 0,87 г боргидрида натрия и 80 мл этанола, было получено 2,84 г целевого соединения в виде бесцветного масла.
Спектр ядерно-магнитного резонанса (CDCl3) d млн.дол.
1,6-1,95 (4Н, мультиплет), 2,83 (2Н, триплет, J= 8 Гц), 3,71 (2Н, триплет, J= 7 Гц), 3,87 (3H, синглет), 3,97 (3H, синглет, 6,61 (1H синглет), 7,4 7,6 (2H, мультиплет), 8,01 (1H, дублет, J=8 Гц), 8,20 (1H, дублет, J=8 Гц).
Получение 15 (JA 12).
3-(2,5-диметокси-3,4,6-триметилфенил)пропил йодид 2,13 мл метансульфонилхлорида добавлялись по каплям при 0oC к смеси 5,47 г 3-(2,5-диметокси-3,4,6-триметилфенил/пропанола, 4,8 мл триэтиламина и 50 мл метиленхлорида, и получающаяся смесь перемешивалась в течение 30 мин. В конце данного периода времени реакционная смесь смешивалась со смесью 50 мл ледяной воды и 50 мл 10 мас. /об. водной соляной кислоты. Органический слой, который отделялся, промывался насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлористого натрия в указанном порядке, после чего он сушился над безводным сульфатом магния. Растворитель затем удалялся с помощью перегонки при пониженном давлении, остаток растворялся в 100 мл ацетона, и к получающейся смеси добавлялось 6,88 г иодистого натрия. Реакционная смесь затем перемешивалась при 50oC в течение 2 ч, после чего растворитель удалялся с помощью перегонки при пониженном давлении. Остаток смешивался со 100 мл насыщенного водного раствора тиосульфата натрия, после чего он экстрагировался этилацетатом. Экстракт освобождался от растворителя с помощью колоночной хроматографии через силикагель с использованием 10:1 по объему смеси гексана и этилацетата в качестве элюента, давая 7,7 г целевого соединения в виде масла.
Спектр ядерно-магнитного резонанса (CDCl3) d млн.дол.
2,00 (2H, квинтет, J=7 Гц), 2,17 (6H, синглет),
2,23 (3H, синглет) 2,71 (2H, дублет дублетов, J=7 Гц),
3,27 (2H, триплет, J=7 Гц), 3,64 (3H, синглет),
3,67 (3H, синглет).
Получение 16-22.
Следуя процедуре, аналогичной описанной в получении 15 выше, следующие соединения формулы (1-7):
(в которой R1, R2, R3, N и HaI имеют значения, определенные в табл.7) получались из соответствующих гидрокси соединением с помощью замещения гидрокси группы гидрокси соединения атомом галогена, показанным в табл.7. Сокращения расшифровываются, как определено в отношении табл. 4. В получениях 20, 21, и 22, R2 и R3 вместе представляют группу, показанную в их столбцах.
Спектр ядерно-магнитного резонанса соединения получения 16, δ млн.дол. CDCl3 (частично благодаря W): 4,66 (2H, синглет).
Спектр ядерно-магнитного резонанса соединения получения 17 d мол. дол. CDCl3 (частично вследствие W):
1,50-1,70 (2H, мультиплет), 1,85 2,00 (2H, мультиплет),
2,63 (2H, дублет, дублетов, J=8 Гц),
3,24 (2H, триплет, J=7 Гц).
Спектр ядерно-магнитного резонанса соединения получения 18, d млн.дол. CDCl3 (частично вследствие W): 4,61 (2 H, синглет).
Спектр ядерно-магнитного резонанса соединения получения 19 d млн. дол. CDCl3 (частично вследствие W)):
1,90-2,10 (2H, мультиплет),
2,67 (2H, дублет дублетов, J=8 Гц),
3,26 (2H, триплет, J=7 Гц).
Спектр ядерно-магнитного резонанса соединения получения 20, d, млн.дол. CDCl3 (частично вследствие W): 4,85 (2H, мультиплет).
Спектр ядерно-магнитного резонанса соединения получения 21, d млн.дол. CDCl3 (частично вследствие W):
2,22 (2H, квинтет, J=7 Гц), 2,90 (2H, триплет, J=7 Гц),
3,26 (2H, триплет, J=7 Гц).
Спектр ядерно-магнитного резонанса соединения получения 22, d млн.дол. CDCl3 (частично вследствие W):
4,92 (2H, синглет).
Получение 23.
5-/4-Гидроксибензил/-3-трифенилметил-тиазолидин-2,4-дион 23(a) -5-/4-ацетоксибензилидене/тиазолидин-2,4-дион.
Смесь, включающая 200 г п-гидроксибензальдегида, 229 г тиазолидин-2,4-диона, 280 мг ацетата натрия и 660 м диметилацетамида, перемешивалась при 150oC в течение 1 ч. Затем она охлаждалась и к реакционной смеси добавлялись 370 мл уксусного ангидрида и 540 мл диметилацетамида. Получающаяся смесь затем перемешивалась при 50oC в течение 1,5 ч, после чего она выливалась в воду. Твердое вещество, которое осаждалось, собиралось с помощью фильтрования, промывалось водой и сушилось в вакууме, давая 390 г названного в заголовке соединения.
23 (b) 5-/4-Ацетоксибензил/тиазолидин-2,4-дион.
2,0 г 5-/4-ацетоксибензилиден (тиазолидин-2,4-диона [полученного, как описано выше, на стадии (a)] растворялось в 80 мл уксусной кислоты и гидрировалось в атмосфере водорода при атмосферном давлении при 90oC в течение 5 ч в присутствии 2,0 г 10 мас./мас. палладия на угле. В конце данного периода времени катализатор отфильтровывался, и фильтрат разбавлялся толуолом. Растворитель уксусная кислота, затем удалялся с помощью перегонки в виде азеотропа с толуолом. Кристаллы, которые отделялись при добавлении к концентрату толуола и гексана, собирались путем фильтрования и сушились, давая 1,8 г названного в заголовке соединения.
23 (c) 5-/4-Ацетоксибензил/-3-трифенилметил-тиазолидин-2,4-дион 3,43 г триэтиламина добавлялись к раствору 9,0 г 5-/4-ацетоксибензил /тиазолидин-2,4-диона [полученного на стадии (b), как описано выше] в 70 мл метиленхлорида и к получающейся смеси по каплям добавлялся раствор 9,45 г трифенилметилхлорида в 30 мл метиленхлорида. Смесь затем перемешивалась при комнатной температуре в течение 1 ч, после чего она оставлялась стоять на протяжении ночи при той же температуре. В конце данного периода реакционная смесь смешивалась с водой и этилацетатом, и органический слой отделялся, промывался насыщенным водным раствором хлористого натрия и сушился над безводным сульфатом натрия. Кристаллы, которые отделялись при отгонке растворителя при пониженном давлении, промывались смесью гексана и этилацетата и сушились, давая 7,86 г целевого соединения.
23 (d) 5-(4-Гидрооксибензил/-3-трифенилметил-тиазолидин-2,4-дион.
Раствор 2,99 г 28 мас./об. метанольного раствора метилата натрия в 10 мл метанола добавлялся по каплям при охлаждении льдом к раствору 7,86 г 5-/4-ацетоксибензил-3-трифенилметил-тиазолидин-2,4-диона (полученного на стадии (c), как описано выше) в 70 мл толуола, и получающаяся смесь перемешивалась при комнатной температуре в течение 1 ч, после чего она оставлялась стоять на протяжении ночи при той же температуре. Величина pH реакционной смеси затем доводилась до значения 4 путем добавления 1н. водяной соляной кислоты, и смесь экстрагировалась этилацетатом. Экстракт промывался водой и сушился над безводным сульфатом натрия. Растворитель затем удалялся с помощью перегонки при пониженном давлении, и кристаллы, которые появлялись в остатке, собирались, промывались гексаном и сушились, давая 6,0 г целевого соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТИАЗОЛИДИН-2,4-ДИОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2103265C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| ПРОИЗВОДНЫЕ ОКСИМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2122998C1 |
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2038354C1 |
| ПРОИЗВОДНЫЕ 13-(ЗАМЕЩЕННОГО ТИО)АЦЕТОКСИМИЛБЕМИЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИНСЕКТИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ ЗАЩИТЫ РАСТЕНИЙ | 1992 |
|
RU2086552C1 |
| ПРОИЗВОДНЫЕ БИФЕНИЛА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2109736C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СЛОЖНЫЕ ЭФИРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2086541C1 |
| АЗАСТЕРОИДНЫЕ СОЕДИНЕНИЯ | 1991 |
|
RU2070204C1 |
| 13-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МИЛБЕМИЦИНА, АКАРИЦИДНАЯ И ИНСЕКТИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ ЗАЩИТЫ РАСТЕНИЙ ОТ ПОВРЕЖДЕНИЯ ПАРАЗИТАМИ | 1995 |
|
RU2109744C1 |
| КРЕМНИЙСОДЕРЖАЩИЕ СОЕДИНЕНИЯ ИЛИ ИХ СОЛИ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2069213C1 |














Использование: в химии гетероциклических соединений, проявляющих гипогликемическую активность. Сущность изобретения: тиазолидиновые соединения общей формулы I: ,
,
где
R1-C1-C5-алкил, R2 и R3-одинаковые или различные и представляют C1-C5-алкил, C1-C5-алкокси, или R2 и R3 вместе образуют бензольное кольцо, в этом случае R1 представляет атом водорода, галогена или C1-C5-алкил, R4 и R5-атом водорода, Y1 и Y2-одинаковые или различные и представляют атом водорода, C1-C5-алкил, алифатическую карбоксильную ацильную группу, имеющую 1-7 атомов углерода, или пиридинкарбонильную группу, W-одинарная связь или C1-C5-алкилен, Z-атом или 1/X эквивалента катиона, где X - заряд у катиона. Предложены 2 способа получения соединений формулы I и способ снижения сахара в крови у млекопитающих с использованием этих соединений. 4 с. и 17 з.п. ф-лы, 7 ил.,7 табл.

в которой
R1 алкильная группа, имеющая 1 5 атомов углерода;
R2 и R3 одинаковые или различные и каждый представляет алкильную группу, имеющую 1 5 атомов углерода, или алкокси группу, имеющую 1 5 атомов углерода, или R2 и R3 вместе образуют бензольное кольцо и когда R2 и R3 вместе образуют указанное бензольное кольцо, R1 представляет атом водорода, галогена, или алкильную группу, имеющую 1 5 атомов углерода;
R4 и R5 атомы водорода;
Y1 и Y2 одинаковые или различные и каждый представляет атом водорода, алкильную группу, имеющую 1 5 атомов углерода, алифатическую карбоксильную ацильную группу, имеющую 1 7 атомов углерода, или пиридинкарбонильную группу;
W одинарная связь или алкиленовая группа, имеющая 1 5 атомов углерода;
Z атом водорода или 1/х эквивалента катиона, где x представляет заряд у катиона.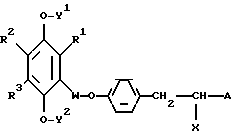
в которой R1, R2, R3, Y1, Y2 и W имеют значения, определенные в п.1, А карбоксильная, алкоксикарбонильная или карбамоильная группа или группа формулы -СООМ, где М атом металла, Х атом галогена, с тиомочевиной с получением промежуточного соединения формулы III
в которой R1, R2, R3, Y1, Y2 и W имеют значения, определенные выше, и затем гидролиз соединения формулы III, необязательно ацилируют полученный продукт, где Y1 и/или Y2 - атом водорода, с получением соединения формулы I, в которой Y1 и/или Y2 ацильная группа, и/или необязательно превращают полученный продукт в соль.
в которой R1, R2, R3, Y1, Y2 и W имеют значения, определенные в п.1, или его активного сложного эфира или галоидированного производного с соединением формулы V
в которой R6 атом водорода или защитная группа,
с получением соединения формулы VI
в которой R1, R2, R3, R6, Y1, Y2 и W имеют значения, определенные выше,
и, если необходимо, удаляют защитную группу; необязательно ацилируют полученный продукт, где Y1 и/или Y2 атом водорода, с получением соединения формулы I, в которой Y1 и/или Y2 представляет ацильную группу; и/или необязательно превращают полученный продукт в соль.
| Коммутатор | 1972 |
|
SU441605A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
