Изобретение относится к новому способу получения производных мутилина формулы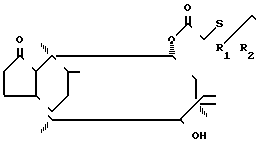

 (I)
(I)
где R1 и R2 одинаковые или различные и каждый представляет собой алкил с 1-4 атомами углерода.
Эти соединения являются известными производными мутилина и находят применение в ветеринарии в качестве необходимых составляющих, в частности, для химиотерапевтического лечения свиней.
Данные соединения могут быть использованы, например, в форме свободного основания или в форме кислотного присоединения с образованием соли.
По известному двухстадийному способу [1] соединения формулы (I) получают обработкой соответствующего производного мутилина аминомеркатаном с последующим ацилированием полученного амина соответствующей карбоновой кислотой.
К недостатком известного способа относятся его двухстадийность и использование для защиты аминогрупп дорогих, токсичных и/или агрессивных эфиров.
Целью изобретения является упрощение процесса за счет использования другой защиты и проведения реакции в одном реакторе.
Данное изобретение описывает способ получения соединения формулы I, который включает стадию образования неустойчивого соединения, имеющего формулу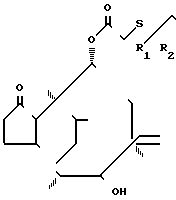
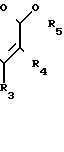 (III)
(III)
где R1, R2 имеют вышеуказанные значения;
R3 и R5 алкил с 1-4 атомами углерода;
R4 водород или алкил с 1-4 атомами углерода. Реакция может быть проведена в традиционной форме гидролиза енаминовой группы. Например, реакция может проводиться в кислой среде, т.е. с соляной кислотой. Реакцию можно проводить приблизительно от 10 до 50оС, предпочтительно при 20оС.
Соединение формулы III можно легко получить ацилированием соединения, имеющего формулу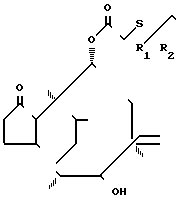
 (II) где R1 и R2 описаны выше. Ацилирование можно проводить традиционным методом. Легко ацилирование производится с D-валинзащищенным соединением как енамин, например, в форме соли Дейна. Это может быть осуществлено взаимодействием D-валина с β-кетосложным эфиром формулы R3 CO-CH (R4) COOR5, где R3, R4 и R5- как определено выше, например, ацетоуксусный кислый метиловый эфир.
(II) где R1 и R2 описаны выше. Ацилирование можно проводить традиционным методом. Легко ацилирование производится с D-валинзащищенным соединением как енамин, например, в форме соли Дейна. Это может быть осуществлено взаимодействием D-валина с β-кетосложным эфиром формулы R3 CO-CH (R4) COOR5, где R3, R4 и R5- как определено выше, например, ацетоуксусный кислый метиловый эфир.
Предпочтительно, когда D-валин переходит в активную форму ангидрида угольной кислоты, например, получается из хлороформовой кислоты. Реакция ацилирования достаточно легко проводится, например, при температуре от -20 до 0оС. Растворителем для реакции ацилирования служит, например, третбутилметиловый эфир.
Для ацилирования соединения формулы II применяется защищенный активированный D-валин. До сих пор традиционные защищающие группы соответствующие сложные эфиры, которые применяли для защиты, дороги и также иногда токсичны и/или являются едкими. По сравнению с этим настоящий процесс предлагает осуществлять защиту аминовой функциональной группы реакцией D-валина с кетосложным эфиром, например, в форме D-валиновой соли Дейна. До настоящего времени активацию проводили с дициклогексилкарбодиимидом и нитрофенолом с образованием активного сложного эфира. В настоящем процессе образуется смешанный ангидрид. Это имеет такое преимущество, что не образуется никаких отходов (дициклогексилмочевина) и реакционная технология может быть упрощена (отсутствие фильтрации, выпаривания, разделения). Ацилирование плевромутилинцистеамин производного формулы II с использованием смешанного ангидрида угольной кислоты приводит к образованию только СО2 и этанола как побочных продуктов, применение 4-нитрофенола в данном случае не сложно.
Удаление защитной группы может быть осуществлено, например, гидролизом, к примеру, с соляной кислотой и значительно легче, чем удаление ранее использованных защитных групп.
Форма свободного основания любого из соединений может быть переведена в формы солей присоединений кислоты и наоборот.
П р и м е р 1. 2-Изопропил-5,5-диметилтиазомедин.
78,6 г 2-изопропил-5,5-диметилтиазолина суспендируют в 80 мл дистиллированной воды в однолитровой четырехгорлой колбе с мешалкой, термометром, рН-электродом и двумя капельными воронками. рН суспензии устанавливают на рН 2 с помощью 2 н. НСl (около 20 мл). Суспензию энергично перемешивают и охлаждают льдом и одновременно обрабатывают соляной кислотой, добавляемой по каплям (в начале 80 мл 2 н. HCl, затем около 80 мл 6 н. HCl) и NaBH4-раствором (10 г в 10 мл Н2О) таким образом, что внутренняя температура не превышает 20оС и рН 2. По завершении реакции (жидкостная хроматография контроль) реакционную смесь дважды подвергают экстракции к третбутилметиловым эфиром (1х20 мл, 1х50 мл). Фазы эфира отделяют. Бесцветный водный раствор обрабатывают 250 мл третбутилметилового эфира и устанавливают рН 5,8 с помощью 10н. NaOH. Фазы разделяют. Водная фаза экстрагируется с 50 мл третбутилметилового эфира (где значение рН не должно упасть ниже 5,5). Эфирную фазу отделяют и отбрасывают. Водный раствор 2-изопропил-5,5-диметилтиазолидин гидрохлорида непосредственно используется в следующей стадии.
Указанное соединение также может быть выделено выпариванием из эфирной фазы вторичной экстракции и дистилляцией в вакууме, посредством чего образуется бесцветная жидкость, точка кипения 76-78оС.
Восстановление и проведение процесса контролируется с помощью жидкостной хроматографии (TLC). Часть реакционной смеси обрабатывают 10 ч. метанола и 1 ч. 2н. раствора гидроокиси натрия, непосредственно переносят в тарелку, этилацетат используют в качестве элюента. Обнаружение осуществляется с ультрафиолетовым светом или разбрызгиванием с молибдатфосфорной кислотой и нагреванием до 150оС.
Водную фазу, полученную в примере 1 в однолитровой четырехгорлой колбе, снабженной термометром, рН-электродом и обратным холодильником, покрывают слоем 300 мл толуола и обрабатывают в атмосфере инертного газа 50 мл фенилгидразина. Смесь энергично перемешивают в атмосфере инертного газа при орошении в течение 1,5-2 ч, до тех пор, пока не будет обнаружен исходный реагент (ТLC) (контроль). Смесь охлаждают до 20-25оС, обрабатывают 20 мл ацетона и перемешивают в течение 10 мин. Фазы отделяют и водную фазу подвергают экстракции 4 раза с 50 мл толуола каждый раз. Воду отгоняют под слабым вакуумом при 90оС.
Твердый осадок хорошо высушивают в реакционном сосуде (температура бани 60оС) и затем растворяют в 100 мл горячего абсолютного этанола, раствор охлаждают до комнатной температуры при перемешивании и медленно обрабатывают 300мл третбутилметилового эфира, добавляя его по каплям. Смесь оставляют на ночь в холодильнике для кристаллизации. Бесцветные кристаллы отфильтровывают, промывают смесью 20 мл этанола и 100 мл третбутилметилового эфира и сушат при 50оС в вакуумной полочной сушилке, что дает 49,2 г (70%) указанного соединения, точка плавления 238оС (сублимации).
Расщепление кольца и проведение процесса контролируется ТLС. Фазы толуола непосредственно помещают на ТLС-тарелки, водные фазы вначале разбавляют метанолом, (1:10) и переводят в щелочную формулу 1-й частью 2 н.раствора гидроокиси натрия.
Элюент: CH2Cl2 (CH3OH)NH4OH 90 (10).
Обнаружение: разбрызгивание с молибдатфосфорной кислотой и нагревание до 150оС.
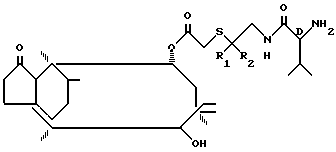
П р и м е р 2. 14-0-1[(D-2-Амино-3-метилбутириламино)-2-метилпропан-2-ил-тио- ацетил]-мутилин. Гидрохлорид.
а) N-(3-Метокси-1-метил-3-оксо-1-пропенил)-D-валин, калиевая соль (D-валин соль Дейна).
36,6 г твердого КОН растворяют в 1250 мл изопропанола при слабом нагревании. Добавляют 65 г D-валина, с последующим добавлением 65,9 мл метилацетоацетата. Смесь перемешивают в течение 10 мин до образования светло-желтого раствора, который подвергают дефлегмации в течение 2 ч. Обратный холодильник заменяют на холодильник Кляйзена и короткую колонку (около 10 см), воду, образующуюся в процессе реакции конденсации (2 моль ˙ экв) удаляют отгонкой с около 1100 мл изопропанола. После того как добавили 500 мл изопропанола, вновь отгоняют 500 мл изопропанола. Все еще теплый раствор (который отвердевает на холоде) либо выливают в 3 л третбутилметилового эфира, либо разбавляют таким же количеством третбутилметилового эфира, либо перемешивают при охлаждении льдом в течение около 3 ч. Полученную суспензию оставляют на ночь при 4оС при отсутствии влаги (продукт гигроскопичен), отфильтровывают, промывают 50 мл третбутилметилового эфира и сушат при 40-50оС в течение ночи в вакуумной полочной сушилке, при этом получают указанное соединение, точка плавления 212-218оС.
б) 14-0-[(1-Амино-2-метилпропан-2-ил)тиоацетил]-мутилин.
75,7 плевромутилина, 42 г паратолуолсульфонил хлорида в 200 мл третбутилметилового эфира, и 40 мл воды медленно обрабатывают в 0,5 л реактора Шмизо с мешалкой, внутренним термометром и обратным холодильником с 50 мл 10 н. раствора гидроокиси натрия. Реакционную смесь нагревают до температуры около 30оС. Смесь энергично перемешивают и подвергают орошению в течение 1 ч, после чего реакция завершается с образованием соответствующего тозилата. Смесь охлаждают до 25оС и обрабатывают при перемешивании 31,2 г диметилцистеамин-гидрохлорида, 3,2 г бензилтрибутиламмоний хлорида и 50 мл 10 н. раствором гидроокиси натрия. Внутренняя температура поднимается до 30оС. Реакционную смесь энергично перемешивают и нагревают до 40-45оС. Спустя около 1 ч реакция завершается. Смесь разбавляют 500 мл воды, охлаждают до 0оС и перемешивают при 0оС 15 мин. Затем смесь отфильтровывают. Продукт промывают водой и холодным третбутилметиловым эфиром (дважды, каждый раз со 100 мл) и сушат ночь в вакуумной сушилке при 55оС, при этом получают указанное соединение, точка плавления 153-155оС.
Если это желательно, можно не проводить повторного добавления 50 мл 10 н. гидроокиси натрия, и вместо этого 100 мл 10 н. гидроокиси натрия добавляют вместо первичного добавления 50 мл 10 н.гидроокиси натрия (к плевромутилину).
в) 14-0-1-[(2-Амино-3-метилбутириламино)-2-метилпропан-2-ил-тиоацетил] мутилин- гидрохлорид.
14 г D-валин соли Дейна суспендировали в 200 мл третбутилметилового эфира в 0,5 л реактора Шмизо с двойной рубашкой, снабженного термометром и мешалкой. Добавляют 6,6 мл N-метилморфолина и суспензию добавляют до -10оС. К охлажденной и перемешанной суспензии в течение 5 мин добавляют 5 мл этилового эфира хлороформовой кислоты. Смесь перемешивают 30 мин при -10оС, 23,3 г твердого 14-0(1-амино-2-метилпропан-2-ил)тиоацетил мутилина добавляют с последующим прибавлением 50 мл третбутилметилового эфира. Смесь либо перемешивают в течение 60 мин при 0оС 30 мин при 20оС, либо 30 мин при -10оС и 60 мин при 0оС. Затем смесь экстрагируют со 150 мл воды.
Получают раствор соединения формулы III в третбутилметиловом эфире. Органическую фазу обрабатывают 200 мл воды, и рН 1,0-1,2 устанавливается с помощью около 14 мл 6 н. соляной кислоты при энергичном перемешивании. Смесь тщательно перемешивают при комнатной температуре от 1 до 2 ч и значение рН поддерживают постоянным с помощью добавления 2н. соляной кислоты. После завершения гидролиза енаминовой защищающей группы используют высокоэффективную жидкостную хроматографию (НPLС). Органическую фазу отделяют и водную фазу трижды экстрагируют третбутилметиловым эфиром, 125 мл каждый раз, чтобы удалить эфир ацетоуксусной кислоты. К перемешанной водной фазе добавляют 150 мл третбутилметилового эфира и поддерживают рН около 7 при помощи 10 н. раствора гидроокиси натрия и затем устанавливают рН 8,0-9,0 добавлением 2 н. раствора гидроокиси натрия. Фазы разделяют, органическую фазу дважды экстрагируют водой, каждый раз по 100 мл. Эфирную фазу обрабатывают 150 мл воды, и устанавливают рН 2,5-3,0 при помощи 2 н. соляной кислоты при энергичном перемешивании. Смесь перемешивают в течение 5 мин и фазы отделяют. Третбутилметиловый эфир удаляют в ротационном испарителе под вакуумом при 30оС. Указанное соединение получают из водного раствора сушкой с разбрызгиванием или сушкой с вымораживанием.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПЛЕКС ПРОИЗВОДНОГО ПЛЕВРОМУТИЛИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ЕГО СОДЕРЖАЩАЯ | 1992 |
|
RU2099324C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЛЕВРОМУТИЛИНОВ | 2007 |
|
RU2512591C2 |
| ГЕРБИЦИДНОЕ СРЕДСТВО И СПОСОБ БОРЬБЫ С СОРНЯКАМИ | 1991 |
|
RU2054871C1 |
| ПРОИЗВОДНЫЕ ТИЕНИЛОКСИАЛКИЛАМИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ СОВМЕСТИМЫЕ КИСЛЫЕ АДДИТИВНЫЕ СОЛИ | 1990 |
|
RU2027716C1 |
| АНТИБАКТЕРИАЛЬНЫЕ ЗАМЕЩЕННЫЕ 7-АЦИЛАМИНО-3-(МЕТИЛГИДРАЗОНО)МЕТИЛЦЕФАЛОСПОРИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ МИКРООРГАНИЗМАМИ | 1998 |
|
RU2201933C2 |
| ПОЛУЧЕНИЕ ЦЕФОТАКСИМА И НАТРИЕВАЯ СОЛЬ ЦЕФОТАКСИМА | 1995 |
|
RU2169150C2 |
| ПРОМЕЖУТОЧНОЕ ПРОИЗВОДНОЕ ЦЕФАЛОСПОРИНА И СПОСОБ ПОЛУЧЕНИЯ ЦЕФАЛОСПОРИНА | 1995 |
|
RU2150471C1 |
| АНТИБАКТЕРИАЛЬНЫЕ ЦЕФАЛОСПОРИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2183212C2 |
| Способ получения плевромутилинов | 1972 |
|
SU523632A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕНИЦИЛЛИНА | 1969 |
|
SU240565A1 |
Использование: в химии, в частности в способе получения производных мутилина. Сущность изобретения: продукт - производные мутилина. Реагент 1: аминомутилин. Реагент 2: D-валин, предварительно защищенный енамином и активированный смешанным ангидридом карбоновой кислоты. Условия реакции: в среде третбутилметилового эфира при температуре от - 20 до 0°С. Реагент 4: соляная кислота. Условия реакции: температура 10 - 50°С. Обе стадии проводят в одном реакторе.
где R1 и R2 одинаковые или различные, C1 - C4-алкил,
или их солей с кислотами взаимодействием соединения общей формулы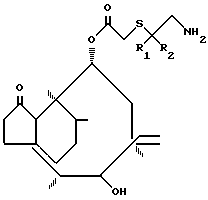
где R1 и R2 имеют указанные значения,
с ацилирующим агентом с последующим снятием защитной группы у полученного промежуточного соединения путем гидролиза и выделением целевого продукта в свободном виде или в виде соли с кислотой, отличающийся тем, что в качестве ацилирующего агента используют D-валин, защищенный в виде енамина и активированный в виде смешанного ангидрида карбоновой кислоты, а снятие защитной группы проводят у промежуточного соединения общей формулы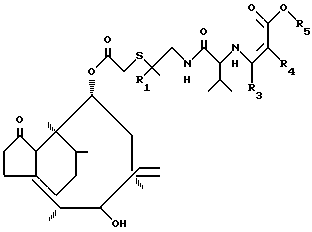
где R1 и R2 имеют указанные значения;
R3 и R5 одинаковые или различные, C1 - C4-алкил;
R4 водород или C1 C4-алкил.
| Патент США N 4675330, кл | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
