Настоящее изобретение относится к новому способу получения 14-O-[(N-(3-метил-2-аминобутирил-пиперидинил)сульфанил)ацетил]-мутилинов.
Плевромутилин, соединение формулы:
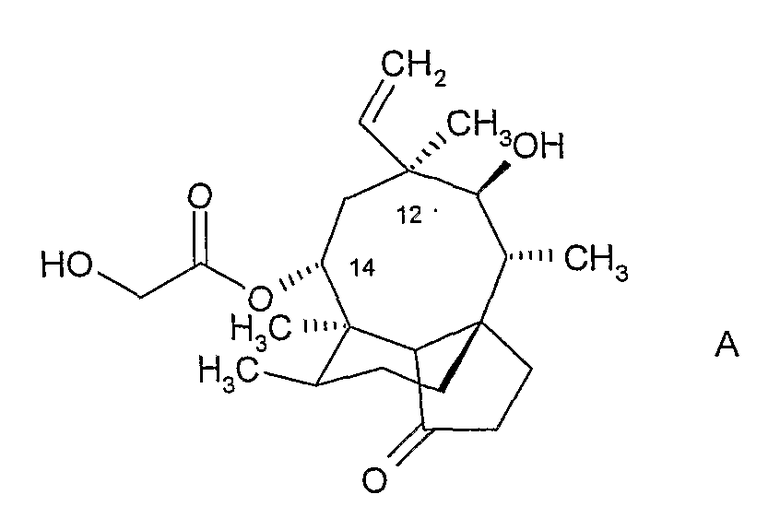
представляет собой природный антибиотик и продуцируется, в частности, базидиомицетами Pleurotus mutilus и P. Passeckerianus, см., например, The Merck Index, 12-е издание, пункт 7694.
Был открыт ряд новых плевромутилинов, имеющих базовую кольцевую структуру плевромутилина и замещенных в положении гидроксильной группы, например, в качестве антимикробных средств. Группа производных плевромутилина, валил-замещенных пиперидинил-сульфанил-ацетилмутилинов вызывает особенно большой интерес в связи с их ярко выраженной антимикробной активностью, как описано в публикации WO 02/22580. Для получения по существу чистых изомеров соединений этой группы существует потребность в разработке способа, который был бы подходящим для использования в промышленном масштабе, и не требовал бы применения дорогостоящих исходных материалов, вредных для окружающей среды реагентов и растворителей или стадий, требующих больших затрат времени и трудоемких стадий очистки.
В одном аспекте настоящее изобретение представляет способ получения соединения формулы:
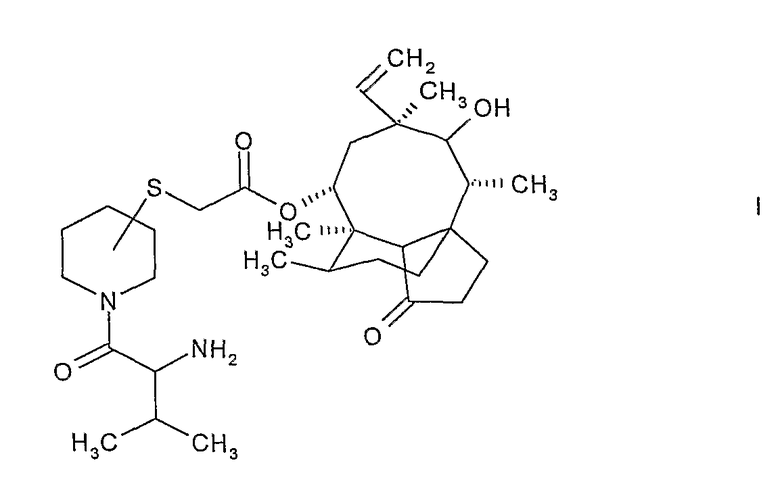
где атом углерода пиперидинового кольца, связанный с атомом серы, находится либо в (S)-конфигурации, либо в (R)-конфигурации и 2-амино-3-метилбутирильная группа, связанная с пиперидиновым кольцом, находится либо в (S)-конфигурации, либо в (R)-конфигурации, включающий следующие этапы:
а) снятие защиты с N-защищенного пиперидинил-сульфанил-ацетилмутилина формулы:
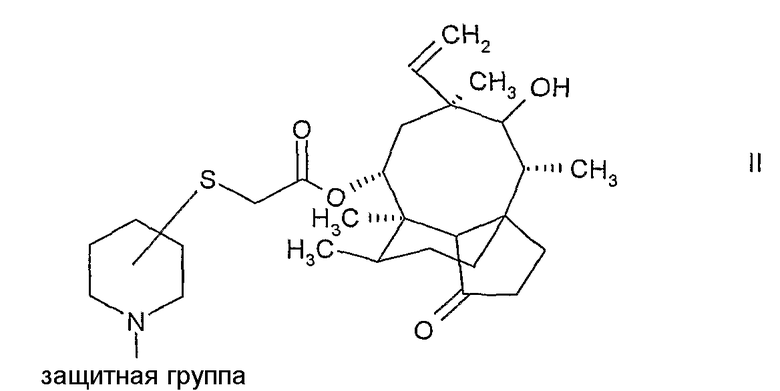
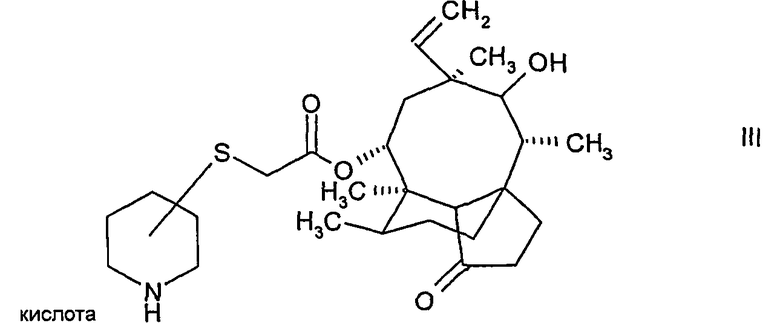
и выделение соединения формулы:
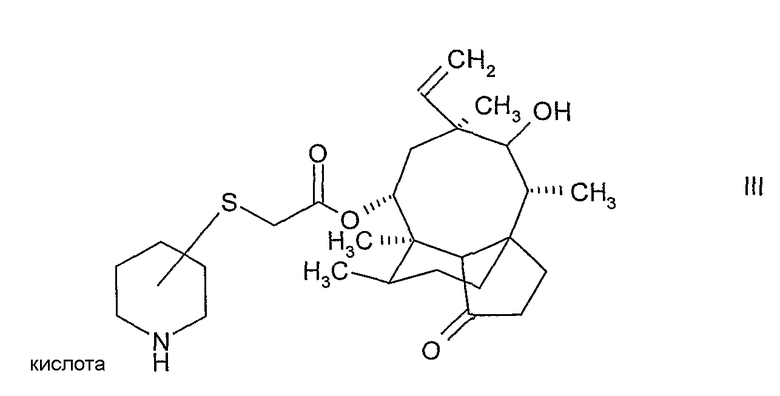
где атом углерода пипиридинового кольца, связанный с атомом серы, находится либо в (S)-конфигурации либо в (R)-конфигурации, в свободной форме или в форме соли присоединения кислоты,
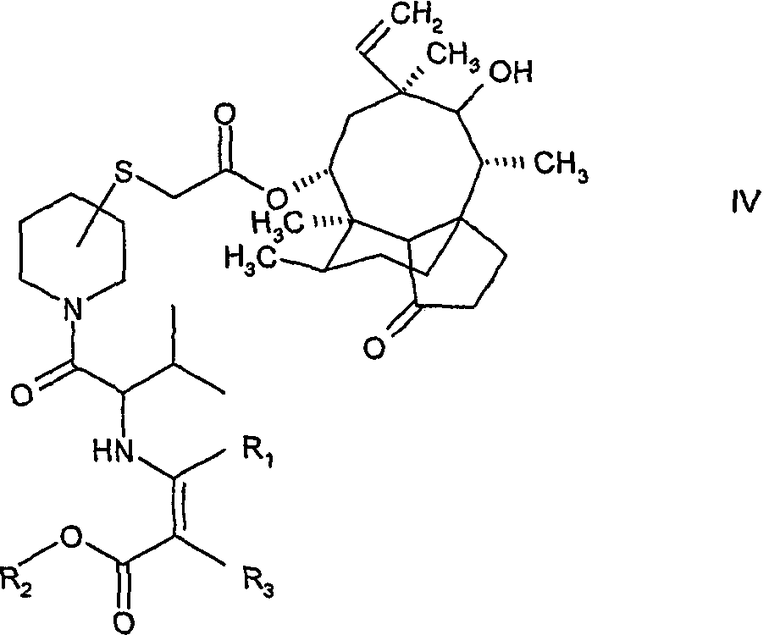
б) ацилирование указанного соединения формулы III либо (R)- либо (S)-валином, защищенным как енамин и активированным как смешанный ангидрид карбоновой кислоты, с получением соединения формулы:
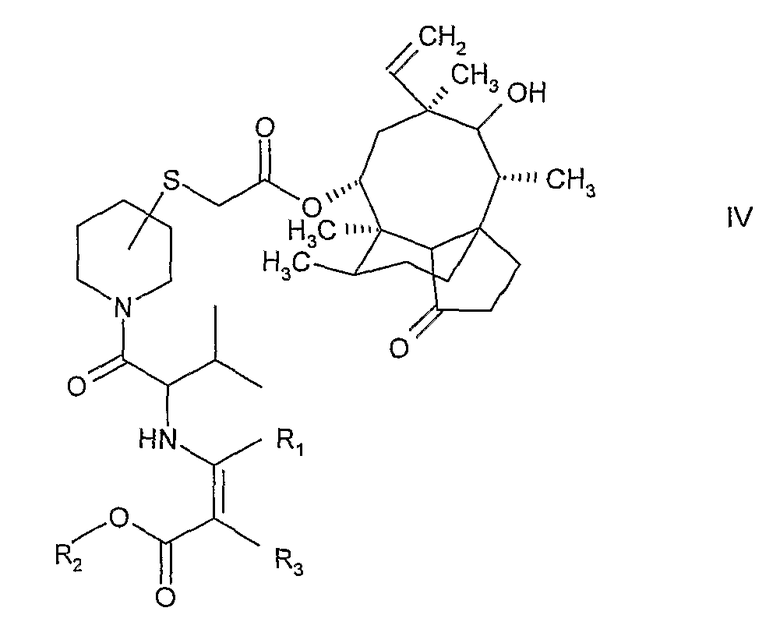
где R1 и R2 означают С1-4алкил, а R3 означает водород или С1-4алкил,
с) снятие защиты с соединения формулы IV и выделение соединения формулы I.
В предпочтительном варианте осуществления соединение формулы I выделяют в форме фармацевтически приемлемой соли. В еще более предпочтительном варианте осуществления соединение формулы I выделяют в форме гидрохлорида.
В соединении формулы I атом углерода пиперидинового кольца связан с атомом серы. Эта связь может находиться в любом положении пиперидинового кольца, например в α-, β- или γ-положении, предпочтительно в β-положении, по отношению к атому азота пиперидинового кольца.
Снятие защиты с N-защищенного соединения формулы II осуществляется путем кислотного гидролиза защитной группы с образованием соли присоединения кислоты в виде смеси изомеров по отношению к конфигурации атома углерода пиперидинового кольца, связанного с атомом серы в соединении формулы II. Предпочтительно, конфигурация мутилинового кольца в соединении формулы II является такой же, как и конфигурация природного мутилина. Смесь изомеров легко разделяется с помощью кристаллизации, в результате чего получаются по существу чистые изомеры, в которых атом углерода пиперидинового кольца, связанный с атомом серы, находится либо в (S)-конфигурации, либо в (R)-конфигурации. Стадия кристаллизации обеспечивает высокую степень чистоты, поэтому соединение формулы III в кристаллической форме очень пригодно в качестве промежуточного соединения, в особенности в промышленных масштабах, в способе получения пиперидинил-сульфанил-ацетилмутилинов. Дополнительным преимуществом применения соединения формулы III в кристаллической форме является то, что другие стадии очистки, такие как хроматография, можно не проводить, поскольку чистота соединения формулы III в кристаллической форме вполне достаточна для производства валил-замещенных пиперидинил-сульфанил-ацетилмутилинов.
Для снятия защиты с соединения формулы II можно использовать неорганическую или органическую кислоты. В предпочтительном варианте осуществления для получения соответствующего мезилата в виде соли присоединения кислоты используют метансульфоновую кислоту. В качестве защитных групп, называемых также «Prot», в соединениях формулы II могут быть использованы традиционные N-защитные группы. Предпочтительно, когда используют для этих целей трет-бутоксикарбонильную группу.
Установлено, что путем подбора подходящего агента для снятия защиты растворимость (S)-конфигурации или (R)-конфигурации может быть селективно улучшена в том смысле, что изомерная (S)-конфигурация может быть отделена от (R)-конфигурации. Например, при использовании в качестве агента для снятия защиты метансульфоновой кислоты выкристаллизовавшийся продукт представляет собой чистый (S)-изомер, в то время как (R)-изомер остается в растворе (см. Пример 1F).
На следующем этапе осуществляется введение валильной группы в практически чистый изомер соединения формулы III путем ацилирования указанного соединения с помощью (R)- или (S)-валина, защищенного как енамин, для получения соединения формулы IV. Защищенный валин получают путем реакции (R)- или (S)-валина со сложным β-кетоэфиром формулы R1-CO-CH(R3)-COOR2, где R1, R2 и R3 такие, как определено выше. Предпочтительно, используется метиловый эфир ацетоуксусной кислоты. Предпочтительно, когда валин активирован по методу смешанных ангидридов карбоновых кислот. Смешанный ангидрид получают in situ, например, при добавлении хлорида триметилуксусной кислоты. После добавления соединения Формулы III, полученного на этапе а), образуется соединение Формулы IV. Эти соединения обладают превосходными кристаллизационными свойствами в сравнении с соединениями, содержащими другие защитные группы, например трет-бутоксикарбонил. Соединения, обладающие такими превосходными кристаллизационными свойствами, удобны в обращении в процессе получения и выделения и имеют преимущества при дополнительной очистке, например, в сравнении с 3-замещенными пиперидинил-сульфанил-ацетилмутилинами формулы I, енамин-защищенное соединение формулы IV выделяют и очищают при помощи кристаллизации, в то время как соответствующее трет-бутоксикарбонил-защищенное производное необхдимо очищать с использованием трудоёмких и длительных стадий хроматографической очистки.
Защитную группу у соединений формулы IV удаляют путем кислотного гидролиза. После удаления сложного эфира ацетоуксусной кислоты и экстракции соединение формулы I может быть выделено в свободной форме или в случае добавления кислоты, которая дает фармацевтически приемлемые соли и после лиофилизации соответствующей водной фазы, в аморфной форме фармацевтически приемлемой соли, например, в форме гидрохлорида.
В предпочтительном варианте осуществления атом углерода пиперидинового кольца, связанный с атомом серы, находится в β-положении относительно атома азота пиперидинового кольца, что означает 3-замещенный пиперидинил-сульфанил-ацетилмутилин формулы I. Более предпочтительно, настоящее изобретение относится к 14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)сульфанил-ацетил]мутилина гидрохлориду.
Настоящее изобретение также относится к новой кристаллической форме 14-O-[((N-3-метил-2-(R)-аминобутирил-пиперидин-3(S)-ил)сульфанил)ацетил]мутилина гидрохлорида. Лиофилизированное аморфное соединение 14-O-[((N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)сульфанил)ацетил]-мутилина гидрохлорида, полученное с помощью последовательности реакций, описанных выше, можно перевести в кристаллическую форму с использованием процесса кристаллизации в водной среде. Процесс можно улучшить и ускорить посредством использования затравки кристаллов. С помощью перекристаллизации кристаллический 14-O-[(N-3-метил-2-(R)-аминобутирил-пиперидин-3(S)-ил)сульфанил-ацетил]мутилина гидрохлорид может быть приведен в форму, имеющую желаемую консистенцию, а также химическую и оптическую чистоту. Получают по существу чистые изомеры, характеризующиеся диастереомерным избытком ≥97% по отношению к 3-(S)-положению.
Кристаллический 14-O-[(N-3-метил-2-(R)-аминобутирил-пиперидин-3-(S)-ил)сульфанил-ацетил]мутилина гидрохлорид характеризуется порошковой рентгенограммой с пиками при значениях углов 2-тета, равных 6,2±0,2; 10,9±0,2; 12,3±0,2; 13,4±0,2; 14,1±0,2; 20,8±0,2 градусам (2-тета, CuK α). Он также может быть охарактеризован при помощи инфракрасного спектра, имеющего характеристические полосы при приблизительно 2927, 1721, 1645, 1462, 1403, 1142 см-1.
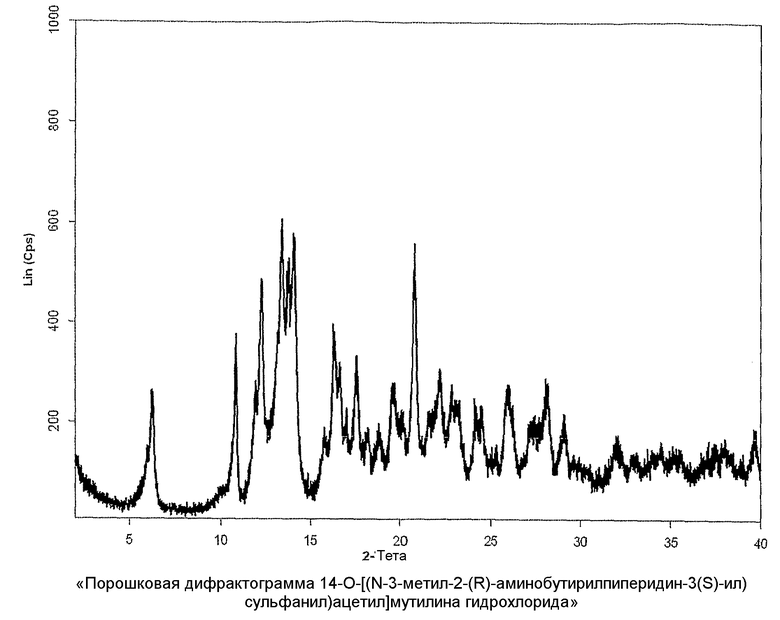
Кристаллический 14-O-[(N-3-метил-2-(R)-аминобутирил-пиперидин-3(S)-ил)сульфанил-ацетил]мутилина гидрохлорид характеризуется высокой чистотой и лучшей стабильностью, чем аморфная лиофилизированная форма, которая более предпочтительна при изготовлении фармацевтических композиций, содержащих кристаллический 14-O-[((N-3-метил-2-(R)-аминобутирил-пиперидин-3(S)-ил)сульфанил)ацетил]мутилина гидрохлорид в качестве активного ингредиента.
N-защищенные пиперидинил-сульфанила-цетилмутилины формулы II могут быть получены путем реакции 14-O-меркаптоацетил-мутилина с N-защищенным гидроксипиперидином, имеющим уходящую группу в α, β или γ-положении по отношению к атому азота пиперидинового кольца, например, N-BOC-3(R)-метилсульфонил-оксипиперидин, как описано в WO 02/22580.
Более предпочтительно и в качестве другого аспекта настоящего изобретения N-защищенные пиперидинил-сульфанил-мутилины могут быть получены путем реакции плевромутилин-22-0-сульфоната (например, мезилата, безилата или тозилата) с N-защищенным пиперидин-тиолом. Защитные группы включают подходящие защитные группы, например традиционно используемые защитные группы.
Предпочтительно, в качестве N-защитной группы используют трет-бутоксикарбонильную группу, а в другом предпочтительном варианте осуществления используют N-защищенный пиперидин, имеющий тиольную группу в положении 3 пиперидинового кольца. Может быть использован рацемически или энантиомерно чистый N-защищенный пиперидин-тиол, имеющий либо (R)-, либо (S)-конфигурацию при атоме углерода, несущем тиольную группу.
Предпочтительно, для того чтобы избежать использования дорогих хиральных исходных материалов, используют N-защищенный пиперидин-тиол. N-защищенный пиперидин-тиол можно получить начиная с подходящего гидроксипиперидина путем добавления N-защитной группы (например, трет-бутоксикарбонила) и реакцией с хлоридом или ангидридом сульфоновой кислоты (например, метансульфонилхлоридом). Тиольная группа вводится путем реакции с серосодержащим нуклеофильным реагентом, например тиоацетатом, и основного гидролиза соответствующего сложного тиоэфира (например, N-BOC-3-(R,S)-ацетилтиопиперидина).
В случае когда гидроксипиперидин используют в форме отдельного энантиомера (например, 3-(R)-гидроксипиперидина), то вышеописанная последовательность реакций, включающая нуклеофильное замещение с использованием тиоцетата, осуществляется управляемым способом (вальденовское обращение) для получения соответствующего N-защищенного пиперидин-тиола, где атом углерода пиперидинового кольца, связанный с атомом серы, находится либо в (S)-конфигурации, либо в (R)-конфигурации (например, 3-(S)-пиперидин-тиол).
Последующая реакция с плевромутилин-22-O-сульфонатом приводит к образованию N-защищенных пиперидинил-сульфанил-ацетилмутилинов формулы II, где атом углерода пиперидинового кольца, связанный с атомом серы, находится либо в (S)-конфигурации, либо в (R)-конфигурации.
В приведенных ниже примерах, которые иллюстрируют настоящее изобретение, ссылки на температуру приведены в градусах Цельсия.
В тексте используются следующие аббревиатуры:
N-BOC = N-бутоксикарбонил,
RT = комнатная температура,
MTBE = метил-трет-бутиловый эфир.
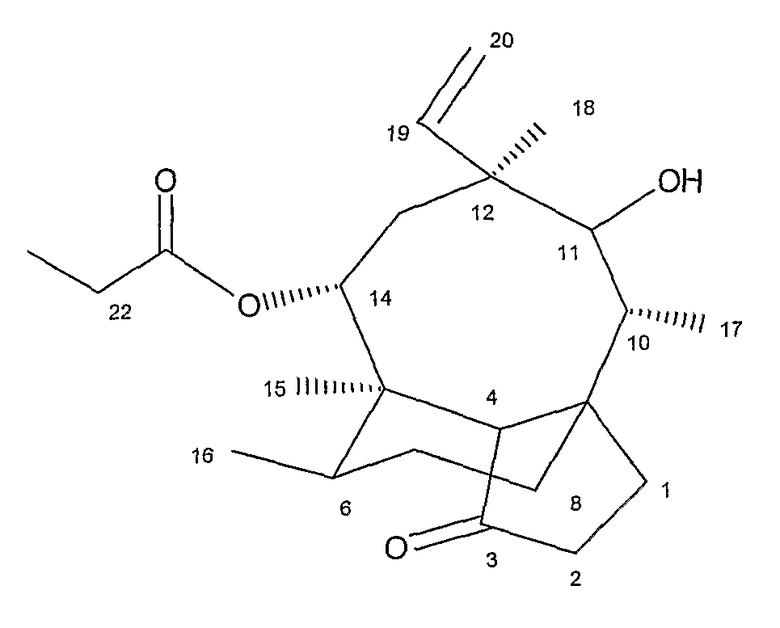
Нумерация мутилиновых циклов, на которые даются ссылки в примерах, приведена в следующей ниже формуле:
Примеры
Пример 1
14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)
сульфанил)ацетил]мутилина гидрохлорид
А. N-BOC-3-(R,S)-гидроксипиперидин
202,4 г 3-(R,S)-гидроксипиперидина растворили в 4,5 л деионизированной воды в 10-литровом реакторе. Добавили 336 г гидрокарбоната натрия, растворенных в 1,1 л воды. При энергичном перемешивании к раствору при комнатной температуре добавили 534 г ди-трет-бутилдикарбоната. После перемешивания в течение ночи смесь экстрагировали CH2Cl2(3×). Смешанные экстракты отмыли деионизированной водой, а растворитель удалили дистилляцией. Полученный осадок снова растворили в CH2Cl2, а раствор выпарили досуха. В результате было получено 423 г N-BOC-3-(R,S)-гидроксипиперидина, который мог быть использован в последующих этапах без дополнительной очистки.
В. N-BOC-3-(R,S)-метил-сульфонил-оксипиперидин
К раствору 216 г N-BOC-3(R,S)-гидроксипиперидина в 5 л CH2Cl2 добавили 222 мл триэтиламина при температуре 0-5°С. Удерживая температуру на уровне 0-5°С в течение 45 минут, по каплям добавляли 137 г метансульфохлорида, растворенных в 300 мл CH2Cl2. После дополнительного перемешивания в течение 70 минут добавили 2 л деионизированной воды и pH смеси довели до 5,9 путем добавления приблизительно 90 мл 2 Н раствора HCl. Водную фазу отделили, а органическую фазу отмыли водой. Раствор выпарили досуха и получили 270-280 г маслянистого осадка. После обработки 1 л н-гептана произошла кристаллизация. Кристаллы выделили и высушили под вакуумом. В результате было получено 250 г N-BOC-3-(R,S)-метил-сульфонил-оксипиперидина.
Т.Пл. 69°С.
1Н ЯМР (CDCl3): 4,71 (м, 1Н, CHOSO2CH3), 3,2-3,6 (м, 4Н, CHN), 3,05 (с, 3Н, CH3SO2), 1,94 (м, 2Н, Н4), 1,83, 1,54 (2×м, 2Н, Н5), 1,46 (м, 9Н, трет-бутил).
С. N-BOC-3-(R,S)-ацетилтиопиперидин
2,7 л диметилформамида поместили в реактор с инертной атмосферой. При нагревании добавили 251,4 г N-BOC-3-(R,S)-метил-сульфонил-оксипиперидина. При достижении внутренней температуры 50°С добавили одной порцией 256,8 г тиоацетата калия. После перемешивания в течение 90 минут при температуре 95°С реакционную смесь перенесли в реактор, содержащий 4 л воды. Добавили 4,2 л петролейного эфира. После энергичного перемешивания в течение 5 минут водную фазу удалили. После повторного добавления воды pH довели до значения >8 путем добавления гидроксида натрия. Органическую фазу отделили, отмыли водой и обработали активным углем. Раствор выпарили досуха. После проведения флэш-хроматографии на диоксиде кремния с использованием петролейного эфира, толуола и этилацетата и концентрирования раствора, содержащего продукт, при охлаждении произошла кристаллизация, в результате было получено 110 г N-BOC-3-(R,S)-ацетилтиопиперидина.
Т. пл. 46-48°С (н-гептан).
1Н ЯМР (CDCl3): 3,79 (дд, 1Н, 2Н), 3,5-3,6 (м, 2Н, Н3, H6), 3,17-3,27 (м, 2Н, Н2, Н6), 2,32 (с, 3Н, CH3SO2), 1,99 (м, 1Н, Н4), 1,55-1,72 (м, Н4, Н5), 1,47 (с, 9Н, трет-бутил).
D. N-BOC-пиперидин-3-(R,S)-тиол
200 г N-BOC-3-(R,S)-ацетилтиопиперидина поместили в 10-литровый реактор, содержащий 3,4 л метанола в инертной атмосфере. К этому раствору добавляли 42 г метилата натрия в метаноле в течение периода времени 15 минут. После дополнительного перемешивания добавили 170 мл 5 Н HCl, в результате чего значение pH изменилось до 2,6-3. Раствор сконцентрировали с использованием испарителя. Полученная двухфазная смесь содержала 1,7 л метил-трет-бутилового эфира (МТБЭ) и 1,7 л воды. После перемешивания, отделения водной фазы и отмывки фазу МТБЭ выделили и выпарили, выход масла составил 170 г.
1Н ЯМР (ДMСO-d6): 3,92 (уш., 1Н, Н6), 3,69 (д, 1Н, H2), 2,7-2,9 (м, 3Н, Н2, Н3, Н6), 2,61 (д, 1Н, SН), 2,0 (м, 1Н, Н4), 1,64 (м, 1Н, Н5), 1,45-1,31 (м, 2Н, Н4, Н5), 1,39 (с, 9Н, трет-бутил).
Е. 14-O-[(N-BOC-пиперидин-3-(R,S)-ил)сульфанилацетил]мутилин
359,7 г 22-O-плевромутилинтозилата суспендировали в 3,2 л метил-трет-бутилового эфира (МТБЭ). Добавили 1350 мл 1 Н раствора гидроксида натрия и 21,1 г хлорида бензил-трибутиламмония. Смесь охладили до 15°С и добавили по каплям раствор, содержащий 161,6 г N-BOC-пиперидин-3-(R,S)-тиола в 800 мл МТБЭ. Двухфазную смесь перемешали в течение одного часа при температуре 20°С. После завершения реакции фазы были разделены. Органическую фазу высушили и выпарили с получением на выходе 521,5 г 14-O-[(N-BOC-пиперидин-3-(R,S)-ил)сульфанил-ацетил]мутилина в виде маслянистой жидкости, которую использовали на следующем этапе без дополнительной очистки.
1Н ЯМР (CDCl3): 6,48 (дд, 1Н, Н19, J=17,4 Гц, J=11,2 Гц), 5,77 (д, 1Н, H14, J=8,4 Гц), 5,34, 5,20 (2×дд, 2Н, Н20, J=11,2 Гц, J=1,3 Гц; J=17,4 Гц, J=1,3 Гц), 4,0 3,75, 2,96, 2,01 (уш., 6Н, пиперидин), 3,37 (дд, 1Н, Н11, J=10,5 Гц, J=10,5 Гц, J=6,6 Гц), 3,19 (м, 2Н, SCH2), 2,85 (м, 1Н, СНS), 2,36 (дкв, 1Н, Н10, J=6,6 Гц, J=6,6 Гц, J=6,5 Гц), 2,11 (уш., 1Н, Н4) 1,47 (с, 12Н, (CH3)3(CH3)15, 1,18 (с, 3Н, (CH3)18), 0,89 (д, 3Н, (CH3)17, J=6,9 Гц), 0,75 (д, 3Н, (CH3)16, J=6,5 Гц).
F. 14-O-[(пиперидин-3-(S)-тиоацетил]мутилин метансульфонат
521 г 14-O-[(N-BOC-пиперидин-3-(R,S)-ил)сульфанилацетил]-мутилина растворили в 4 л 2-пропанола и нагрели до 55°С. После добавления 165 мл метансульфоновой кислоты раствор перемешивали в течение 5 часов в инертной атмосфере при той же температуре. После завершения реакции гидролиза BOC-группы реакционную смесь охладили до 0°С и перемешивали еще в течение двух часов. Выкристаллизовавшийся продукт отфильтровали, промыли 2-пропанолом и высушили под вакуумом. В результате было получено 159,8 г 14-O-[(пиперидин-3-(S)-тиоацетил]мутилин метансульфоната.
1H ЯМР (ДМСО-d6): 8,58 (м, 2H, NH2 +), 6,15 (дд, 1H, H19, J=17,2 Гц, J=11,5 Гц), 5,57 (д, 1H, H14, J=8,2 Гц), 5,07 (м, 2H, H20), 4,5 (уш., 1H, OH), 3,41 (с, 2H, SCH2), 2,4 (уш., 1H, H4), 2,32 (с, 3H, CH3SО3), 1,37 (с, 3H, (CH3)15), 1,07 (с, 3H, (CH3)18), 0,83 (д, 3H, (CH3)17, J=7,0 Гц), 0,64 (д, 3H, (CH3)16, J=6,6 Гц).
G. Калиевая соль N-(3-метокси-1-метил-3-оксо-1-пропенил)-R-валина (соль Дэйн R-валина)
36,6 г твердого KOH растворили в 1250 мл 2-пропанола при небольшом нагревании. Затем к раствору добавили 65 г R-валина и 65,9 мл метилового эфира ацетоуксусной кислоты. Смесь перемешивали и нагревали с обратным холодильником в течение 2 часов. Обратный холодильник заменили на конденсатор Клайзена и короткую колонну, а воду, образующуюся в ходе реакции конденсации, удаляли отгонкой с использованием примерно 1 л 2-пропанола. После этого добавили 500 мл 2-пропанола и снова отогнали 500 мл. Нагретый раствор перелили в 3 л МТБЭ и перемешивали при охлаждении льдом в течение примерно 3 часов. Полученную суспензию оставили на ночь при 4°С (без доступа влаги), затем ее отфильтровали, промыли 500 мл МТБЭ и высушили, получив в результате калиевую соль N-(3-метокси-1-метил-3-оксо-1-пропенил)-R-валина.
H. 14-O-[((N-(3-метил-2-(R)-N-(3-метокси-1-метил-3-оксо-1-пропениламино)бутирил)пиперидин-3-(S)-ил)сульфанилацетил]мутилин (соль Дэйн)
К суспензии 88,2 г соли Дэйн R-валина в 2175 мл МТБЭ добавили 14,5 мл воды и смесь перемешивали в течение 10 минут при комнатной температуре. Затем смесь охладили до 0°С, добавили к ней 3,5 мл 4-метилморфолина и 41 мл хлорангидрида триметилуксусной кислоты и перемешивали смесь в течение 1 часа. Затем к полученной смеси добавили 2175 мл предварительно охлажденной воды (0-4°С) и 166,4 г метансульфоната 14-O-[(пиперидин-3(S)-тиоацетил)]мутилина. pH смеси поддерживали при значении 7 путем добавления приблизительно 210 мл 2 н. раствора NaOH. Охлажденную смесь (0°С) перемешивали в течение 30 минут, что привело к образованию кристаллов соединения Дэйн. Для завершения реакции суспензию нагрели до 30°С и перемешивали в течение 1 часа. После этого установили значение pH на уровне 9,5 путем добавления приблизительно 85 мл 2 н. раствора NaOH. После охлаждения до 0°С и перемешивания в течение еще 2 часов кристаллы отфильтровали, промыли охлажденной водой и МТБЭ, а затем высушили под вакуумом. В результате было получено 195,4 г датского соединения в виде МТБЭ-сольвата.
Т. пл. 136-142°С.
1H ЯМР (ДМСО-d6, 1:1 смесь из двух стабильных ротамеров): 8,87, 8,83 (2×д, 1H, NH, J=9,3 Гц), 6,15 (м, 1H, H19), 5,60, 5,56 (2×д, 1H, H14, J=8,3 Гц), 5,04 (м, 2H, H20), 4,50 (м, 1H, α-H-валин), 4,37 (с, 1H, CH-енамин), 3,50 (с, 3H, OCH3), 2,42, 2,40 (2×уш., 1H, H4), 1,87, 1,85 (2×с, 3H, CH3-енамин), 1,36 (2×с, 3H, (CH3)15), 1,07 (с, 3H, (CH3)18), 0,96-0,77 (м, 9H, (CH3)17, (CH3)2валин), 0,64 (м, 3H, (CH3)16).
I. 14-O-[((N-3-метил-2-(R)-аминобутирил-пиперидин-3-(S)-ил) сульфанил)ацетил]мутилина гидрохлорид
145 г соединения Дэйн суспендировали в смеси 1,8 л МТБЭ и 1,8 л воды. Полученную смесь нагрели до 50°С и энергично перемешали. Значение pH смеси удерживали на уровне 1 путем добавления по каплям 2 Н раствора HCl. После гидролиза защитной группы енамина провели высокоэффективную жидкостную хроматографию. Органическую фазу отделили, а водную фазу дважды экстрагировали МТБЭ (каждый раз по 1,6 л) для удаления метилового эфира ацетоуксусной кислоты. К водной фазе при перемешивании добавили 1,6 л МТБЭ и значение pH удерживали на уровне 10 путем добавления 10 н. раствора NaOH. Фазы разделили, после чего фазу МТБЭ дважды экстрагировали водой порциями по 1,6 л. После добавления 1,5 л воды pH смеси довели до значения 3,2 путем добавления приблизительно 100 мл 2 н. раствора HCl. Водную фазу сконцентрировали в роторном испарителе. После лиофилизации оставшегося раствора получили 117,4 г заявленного соединения.
1H ЯМР (DMSO-d6, ~1:1 смесь из двух стабильных ротамеров): 7,95 (уш., 3H, NH3 +), 6,15 (дд, 1H, H19, J=17,6 Гц, J=11,2 Гц), 5,6 (д, 1H, H14, J=8,2 Гц), 5,05 (м, 2H, H20), 4,53 (м, 1H, OH), 4,24, 4,30 (2×д, 1H, α-H-валин, J=4,8 Гц), 4,08 (дд, 0,5H, H2-пиреридин, J=13,7 Гц, J=3,3 Гц), 3,08 (дд, 0,5 Гц, H2-пиперидин, J=13,7 Гц, J=9,8 Гц), 3,89 (дд, 0,5H, H2-пиперидин, J=13,1 Гц, J=3,2 Гц), 3,41 (м, 0,5H, H2-пиперидин).
АВ-Система: 3,33(2H, SCH2, J=14,9 Гц), 3,42 (м, 1H, H11), 2,83, 2,96 (2×м, 1H, CHS), 2,4 (уш., 1H, H4), 1,34 (с, 3H, (CH3)15), 1,05 (с, 1H, (CH3)18), 0,9, 1,0 (2×м, 6H, (CH3)2валин), 0,81 (д, 3H, (CH3)17, J=6,9 Гц), 0,63 (м, 3H, (CH3)16).
Пример 2
Кристаллическая форма гидрохлорида 14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)сульфанилацетил]мутилина
3,0 л воды поместили в реактор и нагрели до 40°С. Затем добавили 1 кг лиофилизированного 14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)сульфанилацетил]мутилина гидрохлорида, а дополнительные принадлежности ополоснули 1,0 л воды. После истечения примерно 10 минут получили раствор бледно-желтого цвета. В раствор внесли затравку кристаллов и медленно перемешивали его при 40°С в течение 6 часов. Затем прекратили нагревание и суспензию кристаллов перемешивали при температуре окружающей среды в течение еще 64 часов. Полученный продукт отфильтровали, промыли 1,5 л холодной воды и провели повторную кристаллизацию без высушивания или сырой продукт высушили при 40°С под вакуумом, в результате получили заявленное соединение в кристаллической форме.
Повторная кристаллизация
1,5 л воды поместили в реактор и нагрели до 50°С. Добавили сырой продукт, полученный в результате первой кристаллизации, а дополнительные принадлежности ополоснули 1,0 л воды. Суспензию нагревали до 70-75°С до тех пор, пока продукт не растворился. Затем раствор охладили, внесли в него затравку кристаллов и медленно перемешивали при 40°С в течение 6 часов. После этого прекратили нагревание и суспензию кристаллов перемешивали при температуре окружающей среды еще в течение 24 часов. Полученный продукт отфильтровали, промыли 1,3 л холодной воды и высушили при 40°С под вакуумом, в результате получили 765 г заявленного соединения в кристаллической форме.
Т. пл. 150-155°С.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ МУТИЛИНОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2000 |
|
RU2276135C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОКИСЛОТНЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2643146C2 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2006 |
|
RU2443688C2 |
| ПЕПТИДНЫЕ МАКРОЦИКЛЫ ПРОТИВ ACINETOBACTER BAUMANNII | 2016 |
|
RU2729609C2 |
| МЕТА-АЗАЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ АМИНОБЕНЗОЙНОЙ КИСЛОТЫ В КАЧЕСТВЕ АНТАГОНИСТОВ ПАН-ИНТЕГРИНА | 2016 |
|
RU2729518C2 |
| ЛИГАНДЫ МЕЛАНОКОРТИНОВЫХ РЕЦЕПТОРОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2277535C2 |
| НОВЫЕ N-СУЛЬФАМОИЛПИПЕРИДИНАМИДЫ, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ОЖИРЕНИЯ И РОДСТВЕННЫХ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2006 |
|
RU2442773C2 |
| ПРОИЗВОДНЫЕ АМИДИНОФЕНИЛАЛАНИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОИЗВОДНЫЕ АМИДИНОФЕНИЛАЛАНИНА, ОБЛАДАЮЩИЕ АНТИКОАГУЛИРУЮЩЕЙ АКТИВНОСТЬЮ | 1992 |
|
RU2088591C1 |
| АМИДЫ 3-ЗАМЕЩЕННОЙ 5- И 6-АМИНОАЛКИЛИНДОЛ-2-КАРБОНОВОЙ КИСЛОТЫ И РОДСТВЕННЫЕ АНАЛОГИ КАК ИНГИБИТОРЫ КАЗЕИНКИНАЗЫ IΕ | 2005 |
|
RU2369599C2 |
| ИНДАЗОЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2005 |
|
RU2404179C2 |
Настоящее изобретение относится к способу получения 14-O-[(N-(3-метил-2-аминобутирилпиперидинил)сульфанил)ацетил]мутилинов формулы (I), заключающемуся в снятии защиты с N-защищенного пиперидинил-сульфанил-ацетил-мутилина формулы (II), выделением полученного соединения формулы (III) и его ацилированием с получением соединения формулы (IV) и последующим снятием защиты и выделением соединения формулы (I). Также изобретение относится к соединению формулы (IV), метансульфонатной соли соединения формулы (III), кристаллическому гидрохлориду 14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)сульфанил-ацетил]мутилина, способу его получения и фармацевтической композиции на его основе. Технический результат: разработан новый способ получения соединения формулы (I), отличающийся пониженными затратами времени, ресурсов и труда. 6 н. и 12 з.п. ф-лы, 2 пр.
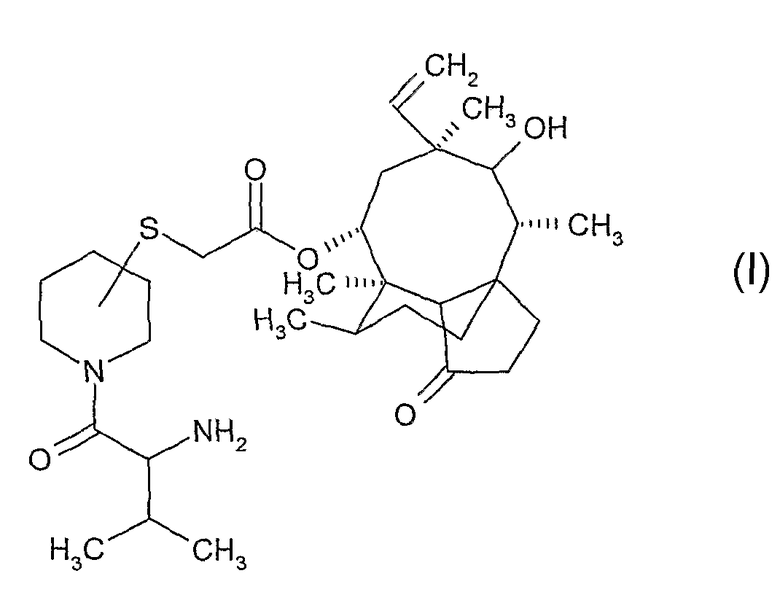
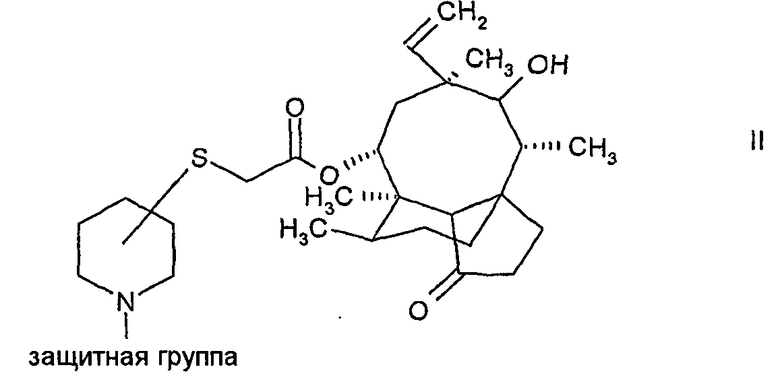
1. Способ получения пиперидинил-сульфанил-ацетилмутилинов формулы:
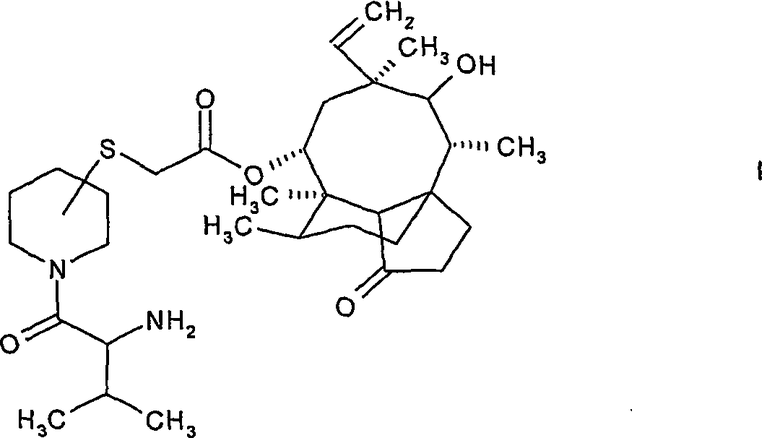
где атом углерода пиперидинового кольца, связанный с атомом серы, находится либо в (S)-конфигурации, либо в (R)-конфигурации, а 2-амино-3-метилбутирильная группа, связанная с пиперидиновым кольцом, находится либо в (S)-конфигурации, либо в (R)-конфигурации, включающий следующие этапы:
a) снятие защиты с N-защищенного пиперидинил-сульфанил-ацетил-мутилина формулы:
и выделение соединения формулы:
где атом углерода пиперидинового кольца, связанный с атомом серы, находится либо в (S)-конфигурации, либо в (R)-конфигурации, в свободной форме или в форме соли присоединения кислоты, в кристаллической форме,
б) ацилирование указанного соединения формулы III (R)- или (S)-валином, защищенным как енамин и активированным как смешанный ангидрид карбоновой кислоты, с образованием соединения формулы:
где R1 и R2 означают C1-4алкил, а R3 означает водород или C1-4алкил,
в) снятие защиты с соединения формулы IV и выделение соединения формулы I.
2. Способ по п.1, в котором соединение формулы I выделяют в форме фармацевтически приемлемой соли.
3. Способ по пп.1-2, в котором соединение формулы I выделяют в форме гидрохлорида.
4. Способ по п.1, в котором указанное соединение формулы III представляет собой соль присоединения метансульфоновой кислоты.
5. Способ по п.1, в котором N-защитная группа представляет собой трет-бутоксикарбонильную группу.
6. Способ по п.1, в котором указанный N-защищенный пиперидинил-сульфанил-ацетилмутилин получают путем реакции плевромутилин-22-О-сульфоната (например, мезилата, безилата или тозилата) с N-защищенным пиперидин-тиолом.
7. Способ по п.6, в котором N-защищенный пиперидинил-сульфанил-ацетилмутилин получают путем реакции плевромутилин-22-O-сульфоната с N-защищенным пиперидин-тиолом, в котором атом углерода пиперидинового кольца, связанный с атомом серы, находится либо в (S)-конфигурации, либо в (R)-конфигурации.
8. Способ по п.1, в котором выделенное соединение формулы I представляет собой 3-замещенный пиперидинил-сульфанил-ацетилмутилин.
9. Способ по п.8, в котором указанный 3-замещенный пиперидинил-сульфанил-ацетилмутилин представляет собой 14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)сульфанилацетил]-мутилина гидрохлорид.
10. Соединение формулы

в котором R1 и R2 означают C1-4алкил, а R3 означает водород или C1-4алкил.
11. Соединение по п.10, в котором R1 и R2 означают метил, а R3 означает водород.
12. Соединение по п.11 в кристаллической форме.
13. Метансульфонатная соль соединения формулы (III)
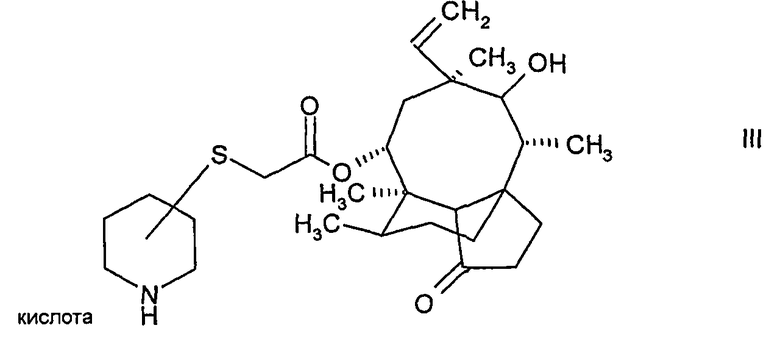
в кристаллической форме.
14. Кристаллический гидрохлорид 14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)сульфанил-ацетил]мутилина.
15. Кристаллический гидрохлорид 14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)сульфанил-ацетил]мутилина по п.14, характеризующийся 3-(S)-диастереомерным избытком ≥97%.
16. Кристаллический гидрохлорид 14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(3)-ил)сульфанил-ацетил]мутилина по п.14, характеризующийся рентгенографическими пиками при следующих значениях угла 2-тета: 6,2±0,2; 10,9±0,2; 12,3±0,2; 13,4±0,2; 14,1±0,2; 20,8±0,2.
17. Способ получения кристаллического соединения по п.14, включающий следующие этапы:
- растворение и нагревание гидрохлорида 14-O-[(N-3-метил-2-(R)-аминобутирилпиперидин-3(S)-ил)сульфанилацетил]мутилина в водной среде;
- необязательное затравливание раствора кристаллами и перемешивание при повышенной температуре;
- охлаждение полученной суспензии и перемешивание при температуре окружающей среды;
- выделение кристаллического продукта и необязательное повторение процедуры.
18. Фармацевтическая композиция, обладающая антимикробной активностью, содержащая кристаллический 14-O-[(N-3-метил-2-(R) -аминобутирилпиперидин-3(S)-ил)сульфанил-ацетил]мутилина гидрохлорид.
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| УСТРОЙСТВО ДЛЯ РАЗРУШЕНИЯ НЕГАБАРИТНЫХ КУСКОВРУДЫ | 1972 |
|
SU421364A1 |
| Способ получения производных плевромутилина или их гидрохлоридов | 1985 |
|
SU1582985A3 |
| RU 2005105576 A, 10.10.2005 | |||

