Изобретение относится к новым пиридинсодержащим гетероциклическим соединениям, в частности новым 1,3-диоксан-5-иловым производным алкеновых кислот, содержащим пиридиловый остаток, присоединенный к положению 4 1,3-диоксанового кольца. Кислоты предлагаемого изобретения обладают ценными фармацевтическими свойствами.
Известно, что тромбоксан А2 метаболит арахидоновой кислоты (далее обозначаемый как ТХА2) оказывает выраженное вазоконстрикторное действие и является сильнодействующим агрегатором тромбоцитов. Кроме того, ТХА2оказывает сильное констрикторное действие на гладкую мускулатуру бронхов и трахей. Таким образом, ТХА2 может быть использован для лечения различных заболеваний, например ишимической болезни сердца, такой как инфаркт миокарда, ангины, заболеваний, связанных с нарушением мозгового кровообращения, например, временной ишемии мозговых сосудов, приступов мигрени, заболеваний, связанных с нарушением кровообращения периферических сосудов, например атеросклероза, микроангиопатии, гипертонии и тромбозов, вызванных нарушением липидного обмена.
Предполагают, что ТХА2 проявляет свое физиологической действие через тромбоксановый рецептор, при влиянии которого рецепторы других простаноидных веществ, образованных из арахидоновой кислоты, таких как простагландины Н2, простагландин F 2α и простагландин D2, которые обладают сокращательной способностью, могут проявлять контрактурное действие. Существуют два основных способа, с помощью которых можно повысить активность ТХА2. Первый способ заключается в введении лекарственного средства, которое преимущественно воздействует на тромбоксановый рецептор, хотя и не создает констрикторного эффекта, который проявляется после связывания с ТХА2 (или с простагландинами Н2, F 2α и/или D2). Такое средство обладает свойствами антагониста ТХА2. Второй путь заключается в введении лекарственного вещества, которое ингибирует один или несколько ферментов, участвующих в образовании ТХА2, в частности, которые тормозят фермент, известный как тромбоксансинтаза (ТХА2-синтаза). Такое вещество является ингибитором ТХА2-синтазы. Отсюда вытекает предположение, что средства, обладающие свойствами антагониста ТХА2 и ингибирующие ТХА2-синтазу, могут представлять терапевтическую ценность при лечении одного или нескольких вышеуказанных и других заболеваний, в патологическом процессе которых участвует ТХА2. Кроме того, средства, которые обладают свойствами антагониста ТХА2, могут быть использованы при лечении таких заболеваний, в которых принимают участие простагландины Н2, P 2α и/или D2, главным образом, например, при лечении астматических заболеваний и воспалительных процессов. Хотя известно использование 1,3-диоксановых соединений в качестве антагонистов ТХА2 (см. например, Европатент, N 94239В1) [1] которые в некоторой степени являются ингибиторами ТХА2-синтазы (см.например, заявку на Европатент, N 98690А2) [2] нигде не раскрывается получение соединений, которые в требуемой степени проявили бы одновременно оба эти свойства.
Однако в ходе исследований установлено (и этот вывод является основой предлагаемого изобретения), что 1,3-диоксан-5-иловые производные алкеновых кислот формулы (1), приведенной вместе с другими химическими структурами в конце описания, содержащие остаток пиридила, присоединенный к положению четыре 1,3-диоксанового кольца, являются хорошими ингибиторами ТХА2-синтазы, и ко всему проявляют выраженное действие в качестве антагониста ТХА2.
В соответствии с изобретением предлагается 1,3-диоксановое производное алкеновых кислот формулы (1), приведенной вместе с другими химическими формулами далее в описании с обозначением римскими цифрами, где А1 означает С1-С6 алкилен; R1 означает С1-С6-алкил, трифторметил, С3-С6 циклоалкил или С1-С4 алкокси-С1-С4-алкил, или группу формулы R3, А2, где R3 означает пиридил, фенил или фенил, имеющий 1 или 2 заместителя, выбранных из группы гологена, трифторметила, нитро- и цианогруппы, а А2 означает С1-С6-алкилен, окси-С1-С6 алкилен, С2-С6-алкенилен или прямую связь с R3; R2 означает оксигруппу, физиологически приемлемый спиртовый остаток или С1-С4-алкансульфонамидо, Х означает водород, оксигруппу или С1-С4-алкокси, Y означает винилен, а n равно 1 или 2 или его фармацевтически приемлемая соль.
Следует иметь в виду, что соединения формулы (1) содержат асимметрические атомы углерода и могут существовать или их можно выделить в рацемических или оптически активных формах. Предлагаемые соединения представлены как в виде рацематов, так и любой оптически активной форме (или ее смесях), которая способна противодействовать одному или нескольким действиям ТХА2 или ингибировать синтез ТХА2. Известны способы получения отдельных оптических изомеров (например, при синтезе из оптически активных исходных материалов или хроматографическом разделении рацематов) и методы определения активности соединения в качестве антагониста ТХА2 и ингибиторного действия относительно ТХА2-синтазы при использовании одного или нескольких стандартных испытаний, которые приведены далее в описании.
Следует иметь в виду, что группы в положениях 2,4 и 5 1,3-диоксанового остатка формулы (1) имеют относительную цис-стереоизомерию, которую имеют группы, примыкающие к виниловой группе V, (т.е. последние соединения существуют в форме Z изомера). Хотя конкретная конфигурация показана в прилагаемых химических формулах (фиг.1 и 2), она не обязательно соответствует абсолютной конфигурации.
Следует также иметь в виду, что обобщающий термин "алкилен" означает алкиленовые группы как с разветвленными, так и неразветвленными цепями, например, как этилен и этилиден, то же самое можно сказать и о других обобщающих терминах. Однако, если используют уточняющий термин, например "бутил", под этим подразумевают группу с неразветвленной цепью или нормальную бутильную группу, названия изомеров с разветвленными цепями, например трет-бутил обозначены конкретно, когда это необходимо.
Частные значения для R1, когда он означает С1-С6-алкил включают, например, метил, этил, изопропил и трет-бутил, два последних из которых предпочтительны, когда R1 означает С3-С6-циклоалкил, в качестве подходящих групп используют, например, циклопентил или циклогексил, когда R1 означает С1-С4-алкокси С1-С4-алкил к подходящим группам относятся, например, 1,1-диметил-2-метоксиэтил и 1-метил-1-пропоксиэтил.
Частные значения для R2, когда он представляет физиологически приемлемый спиртовый остаток, представляют группы, которые приводят к биологическому распаду сложные эфиры, выбранные, например, из С1-С6-алкила, возможно содержащего окси- или С1-С4-замещающих группы, таких как метил, этил, 2-оксиэтил, 2-метоксиэтил, пропил или 3-оксипропил, фенил и бензил, причем две из последних могут не обязательно содержать 1 или 2 возможных заместителя, выбранных из группы гологена (например. фтор, хлор, бром или йод), С1-С4-алкила (например, метила или этила) и С1-С4-алкокси (например, метокси и этокси).
Когда А1 означает С1-С6-алкилен к подходящим группам относятся, например, метилен, этилен, триметилен, тетраметилен, 1,1-диметилэтилен и 1,1-диметилтриметилен.
Когда А2 означает С1-С6-алкилен к подходящим группам относятся, например, С1-С4 алкилен (например, метилен, этилен, триметилен, изопропилен и 1,1-диметилэтилен) и 3,3-пентилидин, когда А2 означает С2-С6-алкенилен к подходящим группам относятся, например, винилен, 1,3-пропенилен и 1,4-бутен-2-илен, когда А2 означает окси-С1-С6-алкилен к подходящим группам относятся, например, оксиметилен, окситетраметилен (т. е. группа формулы -O˙(СН2)4-), 1-окси-1-метилэтил (т.е. группа формулы -О˙С(СН3)2-) и 2-окси-1,1-диметилэтил (т.е. группа формулы -О˙С(СН3)2-, следует иметь в виду, что окси-связь относится к группе R3, а не 1,3-диоксановому кольцу.
Когда R3 означает пиридил к подходящей группе относится, например, 3-пиридил.
К подходящей необязательной галогензамещающей группе, когда R3означает галоидфенил, относится, например, фтор, хлор или бром.
К подходящей группе Х, когда Х означает С1-С4-алкокси, относится, например, метокси и этокси. Предпочтительно, когда Х означает, например, водород.
В качестве примера, обычно предпочтительное значение для -1, Y означает цис-винилен, а А1-этилен или триметилен.
В группу соединений предлагаемого изобретения, которые представляют особенный интерес, относятся соединения формулы (II), где А3 означает С1-С4-алкилен. R4 означает трифторметил, разветвленный С3-С6 алкил или группу формулы R5.A4 где R5 означает пиридил, фенил или фенил, содержащий 1 или 2 заместителя, выбранных из группы галогена, трифторметила, нитро и циано, и где А4 означает С1-С4-алкилен, окси С1-С4-алкилен или прямую связь с R5; Y означает винилен, О означает 3-пиридильный или 4-пиридильный остаток, а Х, R2 и n имеют любое из вышеуказанных значений, и их фармацевтически приемлемые соли, где R2означает оксигруппу или С1-С2 алкансульфонамидо.
В вышеуказанной группе соединений предпочтительно, чтобы Y означал цис-винилен, R2-гидрокси-, n-1, а Х водород.
К еще одной группе соединений, которые представляют собой интерес, относятся соединения формулы (III), где А3 означает С1-С4-алкилен, R5означает пиридил, фенил или фенил, содержащий 1 или 2 заместителя, выбранных из группы гологена, трифторметила, нитро, и циано, а А4означает С1-С4-алкилен, окси-С1-С4-алкилен или прямую смесь с R5, вместе с их фармацевтически приемлемыми солями.
К подходящим группам для А3 относятся, например, группы, имеющие указанные выше для А1 значения, когда А1 представляет С1-С4-алкилен, например этилен, триметилен и 1,1-диметилэтилен, из которых этилен обычно предпочтителен.
К подходящим значениям для А4 относятся, например, указанные выше для А2 значения, когда А2 означает прямую связь, С1-С4-алкилен или окси-С1-С4-алкилен, например, с прямой связью, изопропилиден, 1,1-диметилэтилен и 1-окси-1-метил (т.е. группу формулы -О˙С(СН3)2-, а к подходящим группам для R5 относятся, например, 3-пиридил, фенил, 4-галоидфенил (например, 4-хлор, или 4-бромфенил-), 2-галоидфенил (например, 2-фтор- или 2-хлорфенил), дигалоидфенил (например, 3,4-дифтор-, 3,4-дихлор- или 2,4-дихлорфенил), нитрофенил (например, 2-нитро-, 3-нитро- или 4-нитрофенил), 2-цианофенил, 4-цианофенил, 2-трифторметилфенил и 4-трифторметилфенил.
В качестве примера подходящих значений для R1 представлены группы трифторметил, изопропил, трет-бутил, циклогексил, фенил, 2-хлорфенил. 3-хлорфенил, 2-цианофенил, 4-цианофенил, 4-нитрофенил, 2-хлор-5-нитрофенил, 3,4-дихлорфенил, 2,4-дихлорфенил, 2-трифторметилфенил, 4-трифторметилфенил, 2-фенокси-1-метилэтил (феноксигруппа может необязательно содержать заместители, выбранные из групп 2-фтор, 2-нитро, 2-трифторметил, 3-фтор, 3-бром, 3-нитро, 4-фтор, 4-бром, 4-циано, 4-нитро, 2,4-дихлор, 3,4-дифтор или 3,4-дихлор), 3-пиридил, 4-пиридил, 1-метил-1-(3-пиридилокси)этил, 1-пропокси-1-метилэтил и 1,1-диметил-2-фенилэтил (фенильная часть может необязательно содержать заместители, выбранные из групп 3-бром, 3-нитро, 4-фтор, 4-нитро, 4-трифторметил, 3,4-дифтор или 3,4-дихлор), стирил или 2-нитростирил.
В вышеуказанных соединениях предлагаемого изобретения, особенно предпочтительное значение для R2, когда R2, например, представляет оксигруппу, а для З, когда Х, например, означает водород.
Конкретные новые соединения предлагаемого изобретения раскрыты в приведенных примерах, при этом в качестве дополнительного признака изобретения они представлены вместе с их фармацевтически приемлемыми солями. К наиболее предпочтительным соединениям относятся соединения, приведенные в примерах 4,8, 11 и 28.
Хотя любое из соединений формулы (1) может образовывать соли с соответствующими кислотами, следует учитывать, что соединения формулы (1) амфотерны, когда R2 означает оксигруппу или алкансульфонамидо и могут образовывать соли как с кислотами, так и с основаниями. К характерным фармацевтически приемлемым солям таких соединений относятся, например, соли щелочных и щелочно-земельных металлов, соли аммония, соли с органическими аминами и четвертичными основаниями, образующими физиологически приемлемые катионы, например соли с метиламино, диметиламином, триэтиламином, этилендиамином, пиперидином, морфолином, пирролидином, пиперазином, этаноламином, триэтаноламином, N-метилглюкамином, тетраметиламмонийгидроксидом и бензилтриметиламмонийгидроксидом, а также соли с кислотами, образующими физиологически приемлемы анионы, такие как соли с минеральными кислотами, например галогениды (хлористый и бромистый водород), серной и фосфорной кислотами и с сильными органическими кислотами, например, с пара-толуолсульфо- и метансульфокислотами.
Соединения формулы (1) можно по- лучить традиционными методами, известными в органической химии для синтеза синтетических аналогов со структурой природных соединений. Такие способы представлены в качестве дополнительного признака предлагаемого изобретения и проиллюстрированы приведенными ниже методами получения предлагаемых соединений, где R1, R2, X, Y, A1 и n имеют любое из указанных выше значений.
(а) Для получения соединений формулы (1), где R2 означает оксигруппу, альдегид формулы (IV) взаимодействует с реактивом Виттига формулы R3P CH˙A1˙CO2=M+, где R означает С1-С6-алкил или арил (фенил особенно предпочтителен), а М+ означает соответствующий катион металла, например катион щелочного металла (катион лития, натрия или калия).
В предлагаемом способе получают требуемые соединения формулы (1), где заместители, прилегающие к виниленовой группе Y, имеют в предпочтительном варианте преимущественно относительную цис-стереоизомерию, т.е. в форме "Z" изомера. Однако в предлагаемом способе также образуется небольшое количество аналогичных соединений, имеющих относительную транс-стереоизомерию (т.е. в форме "Е" изомера), которые можно выделить, используя традиционные методы, например, хроматографию или кристаллизацию.
Предлагаемый способ для удобства можно проводить в соответствующем растворителе или разбавителе, например ароматическом растворителе, таком как бензол, толуол или хлорбензол, простом эфире, например 1,2-диметоксиэтан, простой трет-бутилметиловый эфир, простой дибутиловый эфир или тетрагидрофуран, в диметилсульфоксиде или тетраметиленсульфоне или в смеси, состоящей из одного или нескольких таких растворителей или разбавителей. Предлагаемый способ обычно проводят при температуре в интервале, например, от -80 до 40оС, однако предпочтительно осуществлять указанный способ при комнатной или почти при комнатной температуре, например, в интервале 0-35оС.
(b) Для получения соединений, где Х означает оксигруппу, в соединении формулы V, где R означает защищенную оксигруппу снимают защиту традиционными методами.
К примерам особенно подходящих защищенных оксигрупп относятся С1-С4-алкокси (как например, метокси), бензилокси, аллилокси, тетрагидропиран-2-илокси, С1-С4-алкансульфонилокси (особенно метансульфонилокси) и триалкиосилилокси, содержащего до 10 атомов углерода.
Используемые условия для снятия защиты обязательно зависят от характера защищенных оксигрупп. Снятие конкретных гидроксильных защитных групп подробно описано в обычных учебниках по органической химии и такие традиционные методы, которые известны в данной области, включены в объем предлагаемого способа. Таким образом, конкретные группы могут быть удалены по следующей методике:
(1) аллил или тетрагидропиран-2-ильную группу при обработке сильной кислоты, например, трифторуксусной кислотой, при температуре, например, 10-40оС;
(2) триалкилсилильную группу (например, трет-бутилдиметилсилил, который предпочтителен) при взаимодействии с водным раствором фтористого тетрабутиламмония или для удобства фтористого натрия в подходящем растворителе или разбавителе, например как тетрагидрофуран или простой трет-бутилметиловый эфир, обычно при или близко в температуре окружающей среды, например, в интервале от 10 до 35оС;
(3) алкансульфонильную группу реакцией гидролиза в присутствии основания (например, как NaOH или KOH) в соответствующем водном растворе растворителя (например водный раствор С1-С4-алканода) при температуре, например, 0-60оС;
(4) алкильную группу при обработке тиоалкоксидом или дифинилфосфидом щелочного металла (например, тиоэтоксид натрия в растворителе, таком как N, N-диметилформамид) при температуре, например, 50-60оС или дифенилфосфид лития при температуре, например, 0-60оС;
(5) бензильную группу при катализируемом палладием гидрогенолизе в алканоле, например этаноле, при или близко к температуре давлению окружающей среды или при использовании щелочного металла, например, натрия в жидком аммиаке.
(c) Производное диола формулы (VI), где одно из значений Т1 и Т2представляет водород, а другое водород или группу формулы -CRaRb˙OH (где Ra и Rb имеют одинаковые или разные С1-С2 алвильные группы) взаимодействует с альдегидным производным формулы R1˙CHO или его ацеталем, полуацеталем или гидратом).
Производное альдегида (или его гидрат, либо ацеталь или полуацеталь в С1-С4-алканоле, например, метаноле или этаноле) для удобства можно брать в избытке.
Реакцию обычно осуществляют в присутствии кислоты: соляной, бромистоводородной, серной, фосфорной, метансульфокислоты или паратолуолсульфокислоты предпочтительно в присутствии подходящего растворителя или разбавителя, например дихлорметана, толуола, ксилола или простого эфира, например тетрагидрофуран дибутилового эфира, трет-бутилметилового эфира, или 1,2-диметоксиэтан при температуре в интервале, например, от 0 до 80оС.
Исходные соединения формулы (VI), где Т1 и Т2 оба означают водород, можно получить, например, катализируемым кислотой мягким гидролизом или алкоголизом диоксанового кольца соединения формулы (VII), где одно из значений Ra и Rb представлено водородом или С1-С4-алкилом (например, метилом или этилом), а другое С1-С2-алкилом, полученным по аналогичной методике относительно раскрываемого выше способа (а), например, аналогично описанному в заявке на Европатент N 93439. Гидролиз или алкоголиз обычно проводят при температуре в интервале 10-80оС при использовании водной неорганической кислоты, например, соляной кислоты в алканоле, например этаноле или 2-пропаноле или простом эфире (например, тетрагидрофуране).
Исходные материалы формулы (VI), где одно из значений Т1 и Т2представляет водород, а другое группу формулы -CRaRb˙ОН являются промежуточными соединениями при получении вышеуказанных исходных соединений формулы (VI), где Т1 и Т2 оба означают водород. Однако указанные промежуточные соединения обычно не выделяют.
В соответствии с вышеуказанным, изобретение также предлагает улучшенный метод (d) осуществления способа (с), заключающийся в том, что осуществляют взаимодействие 1,3-диоксана формулы (VII), где одно из значений Ra и Rb представляет водород, метил или этил, а другое метил или этил с избыточным количеством альдегида формулы R1˙CHO (или его гидратом, ацеталем или полуацеталем) в присутствии кислоты (например, одной из указанных выше) при температуре в интервале, например, 10-80оС, возможно в присутствии подходящего растворителя или разбавителя (такого, как один из указанных выше).
В некоторых случаях, необходимо модифицировать методики способа (с) и (d), где альдегид формулы R1˙CHO не имеет достаточной реакционной способности или имеет тенденцию в образовании ациклического полуацетата при взаимодействии с соединением формулы (VI) или (VII), например, когда используют 2,2,2-трифторацетальдегид для получения соединений формулы 1, где R1 означает трифторметил. Таким образом, способ (е) предлагаемого изобретения заключается в том, что осуществляют взаимодействие соединения формулы (VI), где Т2 означает водород, а Т1 означает алкансульфонил (особенно бензол- или толуолсульфонил) с альдегидом формулы R1˙CHO или его гидратом, ацеталем или полуацеталем, например с 2,2,2-трифторацетальдегидом или его гидратом в присутствии подходящей кислоты, обычно при таких же условиях, которые раскрыты выше в методе (с) с последующей катализируемой основанием циклизацией ациклического соединения, полученного, например, при использовании соответствующего основания (например, карбоната калия или гидрата натрия) в соответствующем растворителе или разбавителе (например, простом эфире, указанном выше) и при температуре в интервале, например, от 20 до 50оС. Указанный метод проиллюстрирован в примере 34, приведенном далее, при использовании соединения формулы (VI), где R2 означает метокси, причем эта группа превращается в оксигруппу при гидролизе после образования диоксанового кольца.
Необходимые исходные сложные эфиры алкансульфонила или аренсульфонила формулы (VI), указанные выше, можно в предпочтительном варианте получить из соответствующего диола формулы (VI) (Т1 Т2означает водород) при взаимодействии с одним молекулярным эквивалентом соответствующего алкансульфонил- или аренсульфонилгалогенида (например, метансульфонилхлорида или пара-толуолсульфонил хлорида) в подходящем растворителе или разбавителе (например, простом эфире или дихлорметане) при или близкой к температуре окружающей среды и в присутствии подходящего основания (например, триэтиламина или пиридина).
(f) Разложение сложного эфира формулы (VIII), где R6 означает С1-С6-алкил (особенно метил, этил, пропил или трет-бутил), фенил или бензил, которые содержат 1 или 2 заместителя, выбранных из группы галогена, С1-С4-алкила или С1-С4-алкокси.
Реакцию разложения можно проводить при использовании одного или нескольких традиционных реагентов и в условиях, которые известны в данной области при превращении сложных эфиров в кислоты. Таким образом, разложение можно осуществлять, например, путем катализируемого основанием гидролиза, например, при использовании гидроксида щелочного металла, такого как гидроокись калия, лития или натрия в водяной системе, предпочтительно в присутствии соответствующего растворителя или разбавителя, например тетрагидрофурана, метанола, этанола или трет-бутилметилового эфира при температуре в интервале, например, от 10 до 60оС, предпочтительно при или близко к температуре окружающей среды. В альтернативном варианте, когда R6 означает трет-бутил, реакцию разложения можно проводить тепловым методом при нагревании соединения формулы (VIII) при температуре в интервале, например, 80-150оС без или в присутствии соответствующего разбавителя, например, простого дифенилового эфира или дифенилсульфона.
Требуемые исходные материалы для использования в вышеуказанных способах (а)-(f) можно получить по известным методам, используемым для получения структурно родственных соединений, например, используя аналогичные методы, описанные в Европатенте N 94239В1 и заявке на Европатент N 98690А2 (2).
Альдегиды формулы (IV) можно по- лучить, например, из аллильных соединений формулы (XI), как показано на схемах 1 и 2 (фиг.3 и 4) и проиллюстрировано в примерах, проводимых далее. Следует учитывать, что когда необходимо получить конкретный стереоизомер, может возникнуть потребность регулирования последовательностью селективной очистки, а любые смеси изомеров разделяют при использовании, например, хроматографии. В альтернативном варианте, когда нужен конкретный изомер, его можно получить из конкретного анантиомера 3-[2-(1-гидрокси-1-пиридилметил)пент-4-енил] оксазолидин-2-она формулы (XI), где R7 означает С1-С4-алкил (особенно изопропил), который получают при алдольной конденсации соответствующего 3-(4-пентеноил)оксазолидин-2-она с поликарбоксальдегидом, как показано далее на схеме 3 (фиг.5). [Этот метод особенно для получения чистых энантиомеров соединений формулы 1]
Производные защищенных оксигрупп формулы (V) можно получить, например, путем осуществления процесса (с) или (d) с соответствующим соединением, аналогичным 1,3-диоксану формулы (VII), но где Х предпочтительно означает защищенную оксигруппу. Такое соединение легко получают при использовании стандартных методов, аналогичных описанным выше и представленных в примерах.
Диолы формулы (VI), необходимые для получения диоксанов формулы (1) или (VII), где пиридильная часть содержит Х, а алкеновая кислота в боковой цепи имеет относительную цис-стереоизомерию, можно получить, например, при использовании аналогичного метода, описанного в заявке на Европатент, N 142323 [3] используя в качестве исходного материала соответствующий пиридинсодержащий карбоксальдегид и ангидрид янтарной кислоты, а в качестве подходящего основания такое, которое используют при альдольной конденсации в схеме 3.
Сложные эфиры формулы (VIII) можно получить, например, при осуществлении способа (с), используя соответствующий сложный эфир диола формулы (VI).
Требуемые реактивы Виттига можно получить традиционными методами, например, при обработке галогениза фосфония сильным основанием, например, такой как гидрид натрия, лития, диизопропиламид, трет-бутоксид калия или бутиллития. Обычно их получают in situ до выполнения вышеуказанной реакции конденсации (а).
Следует иметь в виду, что соединения формулы (I), где R2 означает оксигруппу, можно также получить другими известными в данной области методами, например. катализируемым основанием гидролизом соответствующих амидов или нитрилов. Кроме того, соединения формулы (1), где R2 имеет другие значения, кроме оксигруппы, можно получить общепринятыми методами этерификации или сульфонамидирования из соединений, где R2 означает окси- (или ее реакционноспособное производное), и соответствующего спирта, фенола или С1-С4-алкансульфонамида. Такие методы также находятся в объеме предлагаемого изобретения.
В дальнейшем, когда необходимо получить соль соединения формулы (1), ее можно получить при взаимодействии с подходящим основанием кислотой, образующей физиологически приемлемый ион, или любым другим традиционным методом, используемым для получения солей.
Кроме того, когда требуется получить оптически активную форму соединения формулы (1), можно осуществлять один из вышеуказанных реакционных процессов, используя оптически активный исходный материал (например, который раскрыт в схеме 3 и проиллюстрирован в примере 40). В альтернативном варианте, рацемическую форму соединения формулы (1) можно по- лучить при взаимодействии с оптически активной формой подходящего органического основания или кислоты, например, камфарсульфокислоты, эфедрина, N,N,N-триметил(1-фенилэтил)аммония гидроксида или 1-фенилэтиламина с последующим традиционным осуществлением разделения полученных солей в виде диастереизомерной смеси, например, дробной перекристаллизацией из соответствующего растворителя, например С1-С4-алканола, а затем оптически активная форма соединения формулы (1) может быть выделена при обработке кислотой (или основанием) по традиционному методу, например, с помощью водных растворов неорганических кислот, например, таких как разведенная соляная кислота (или водного раствора щелочи, например, таких как водный раствор NaOH).
Обычно предпочтительно получение энантиомерной формы соединения формулы (1), где группы у диоксанового кольца имеют 2S, 4S, 5R-конфигурацию.
Большинство из промежуточных соединений, раскрытых выше, являются новыми, например, соединения формул (IV), (V), (VII), (VIII), (IX) и (XI), представляя дополнительные, отличительные признаки настоящего изобретения. Следует указать, что кроме этого, некоторые соединения формулы (VII) (такие, как соединения, где Ra и Rb означают оба метил или этил) имеют выраженную ингибиторную активность относительно ТХА2-синтазы и могут по существу быть использованы в качестве лекарственных средств как в чистом виде, так и в виде фармацевтических составов, которые также входят в объем предлагаемого изобретения.
Как указывалось ранее, соединение формулы (1) проявляют значительную активность в качестве антагониста ТХА2, а также являются ингибиторами ТХА2-синтазы. Подтверждение антагонистического действия соединений формулы (1) можно продемонстрировать на одном или другом из следующих стандартных тестов:
(а) на модели тонкого слоя аорты крысы, аналогично той, которая известна из работы Nature, вып.223, с.29-35, 1969 г. используя в качестве антагониста ТХА2 миметический агент, известный как V 46619 (описанный R.L.Iones, и др. в "Chemistry, Biochemistry and Pharmacological Activity of Trostanoids, с.211, под изд. I.M.Roberts и F.Scheinmann, Pergamon Press, 1979);
(b) пробе на агрегацию тромбоцитов, выполненной по методике, описанной Born (Nature, вып.194, с.927-929, 1962), заключающейся в том, что
(i) агрегацию осуществляют на подкисленной лимонной кислотой плазме человека с изобильным количеством тромбоцитов путем прибавления миметического агента V 46619 в качестве антагониста ТХА2, в результате чего получают кривую зависимости "доза-реакции";
(II) получение кривой зависимости "доза-реакция" при агрегации стимулированных миметическим агентом V 46619 тромбоцитов осуществляют при введении возрастающих доз испытуемого соединения (обычно в пределах от 10-5 до 10-10 М), и
(III) расчете значения Кв, показывающего активность испытуемого соединения в качестве антагониста ТХА2, усредненную по нескольким концентрациям, от расчетного значения при 50%-ной ответной реакции при агрегации с помощью миметического агента V 46619 в присутствии и отсутствии испытуемого соединения, или
(с) испытание на сужение сосудов бронхов заключается в определении степени ингибирования испытуемым соединением сужения бронхиальных сосудов, индуцированного по Konzett-Kossler на модели наркотизированной морской свинки (по методике, модифицированной Collier и Iames, Brit.I.Pharmacol, вып. 30, с. 283-307, 1967) при внутривенном введении миметического агента V 46619, антагониста ТХА2 и включает
(I) получении кривой зависимости "накопленная доза-реакция" на бронхоконструкцию, индуцированную миметическим агентом V 46619 при внутривенном вливании V 46619 c увеличением его концентрации (0,2-4 мг/кг), разведенного в постоянном объеме физиологического раствора и представлении сужаемости сосудов бронхов как максимум от теоретически рассчитанной без подачи воздуха к испытуемому животному;
(II) получении кривой зависимости "накопленная доза-реакция" на бронхоконстрикцию, индуцированную миметическим агентом V 46619 через 30 минутные промежутки в течение 3 ч после орального введения испытуемого соединения и
(III) расчете величины доза-соотношение для испытуемого соединения (т.е. соотношение концентрации V 46619, необходимой для 50% сужения бронхиальных сосудов в присутствии или отсутствии испытуемого соединения), показывающей активность антагонизма ТХА2.
Методику испытания (b) можно для удобства модифицировать для демонстрации антагонизма эффектов ТХА2 in vivo при оценке влияния испытуемого соединения на агрегацию тромбоцитов после введения испытуемого соединения лабораторному животному, например кролику, крысе, морской свинке или собаке. Однако, когда исследуют агрегацию тромбоцитов собаки, необходимо использовать аденозина дифосфат, агрегатора тромбоцитов при установленной максимально допустимой концентрации (примерно 0,4-1,2х10-6 М) вместе с миметическим агентом V 46619, антагонистом ТХА2.
Антагонизм эффектов от ТХА2 на сосудистую систему можно также продемонстрировать, например, на крысе, используя следующую методику:
(d) крыс-самцов (линии Alderley Park) наркотизируют пентобарбиталом натрия и затем измеряют кровяное давление сонной артерии. Затем через яремную вену вливают агент-антагонист ТХА2 V 46619 в концентрации 5 мг/кг для повышения систолического кровяного давления в интервале 20-30 мм/Hg (2640-3970 паскаль), процесс повторяют дважды для обеспечения адекватности ответной реакции. Затем испытуемое соединение вводят либо внутривенно (через яремную вену), либо перорально (через каннюлю) непосредственно в желудок, и через пять минут после введения испытуемого соединения вводят провокационную пробу, используя V 46619, а затем через каждые 9 мин, пока не установится гипертензивное действие агента V 46619.
Ингибиторное действие испытуемого соединения относительно ТХА2-синтазы можно продемонстрировать, используя стандартную методику для испытаний in vitro [испытание (е)] описанную Howa rth и других (Niochem.Soc.Transactions, вып.10, с,293-240, 1982) с применением препарата микросомальной ТХА2-синтазы с тромбоцитами человека и для оценки превращения (1-14С) арахидоновой кислоты в тромбоксан В2 (ТХВ2) метаболита ТХА2 проводят количественную тонкослойную радиохроматографию.
Ингибиторное действие испытуемого соединения относительно ТХА2-синтазы можно также продемонстировать при использовании стандартного метода [испытание (f)] заключающегося в том, что у лабораторных животных (обычно крыс, но также морских свинок, кроликов или собак) берут пробы крови, после введения испытуемого соединения, как правило, перорально. Пробы крови, обработанные антикоагулянтом, вначале инкубируют при температуре 37оС коллагеном (примерно 100 мкм), затем смешивают с индометацином, ингибитором циклооксигеназы (примерно при 10-3 М), центрифугируют и определяют уровень метаболита ТХА2, ТХВ2стандартным методом радиоиммунологического анализа. Ингибиторную активность относительно ТХА2-синтазы можно оценить путем сравнительного анализа количества ТХВ2, содержащегося в плазме животных, которым введено испытуемое соединение с количеством ТХВ2, присутствующим в плазме контрольной группы животных при введении плацебо.
Как правило, соединения формулы (1), где R1 и R2 означают оксигруппу, демонстрируют следующие результаты в одном или нескольких вышеуказанных испытаниях: испытание (а) рА2 > 5,5; испытание (b): Kв < 1,5х10-6 М; испытание (с): соотношение дозы > 5,1 через 1 ч после введения при дозе 10 мг/кг; испытание (d): значительное ингибирование гипертензии, спровоцированной V 46619 по меньшей мере в течение 1 ч после перорального приема в дозе 50 мг/кг или при меньшей дозировке; испытание (e): ИК50 < 1,0х10-6 М; испытание (f): значительное ингибитование ТХВ2 через 1 ч после введения дозы 100 мг/кг или меньшего количества.
При испытаниях не наблюдалось никакого токсического или другого побочного действия испытуемых соединений формулы (1), проявляющих выраженною действие в испытаниях in vivo (с), (d) или (f) при повторных приемах минимально эффективной дозы.
Соединения формулы (1), где R2 имеет другие значения, нежели оксигруппа, как правило показывает более низкую активность в вышеуказанных испытаниях in vitro, но проявляют аналогичную активность, когда R2 означает оксисоединения формулы (1) в испытаниях in vivo.
В качестве иллюстрации соединение, описанное в примере 2, обладает способностью как антагониста ТХА2, так и ингибитора ТХА2-синтазы, подтверждаемых значением Кв, равным 6,5х10-7 М в испытании (b, ИК50 4,8х10-8М в испытании (е), и обнаруживает почти полное ингибирование образуемого ТХВ2 в течение 5 ч после перорального введения в дозе 25 мг/кг крысам в испытании (f) без каких-либо наблюдаемых признаков токсического действия на испытуемых животных.
Как указывалось ранее, благодаря совместному действию соединений формулы (1) в качестве антагониста ТХА2 и ингибитора ТХА2-синтазы их можно использовать для лечения и профилактики заболеваний или патологических процессов, в которых участвует ТХА2 (или простагландины Н2, D2 и/или F2)2 у теплокровных животных. Как правило, соединение формулы (1) вводят для этой цели перорально, ректально, внутривенно, подкожно, внутримышечно или путем ингаляций таким образом, чтобы доза, например, в интервале 0,01-15 мг/кг массы тела была разделена на 4 приема в день, варьируя дозировку при лечении с учетом метода приема, тяжести болезни, массы и возраста больного.
Соединения формулы (1) обычно применяют в виде фармацевтических композиций, состоящих из соединения формулы (1) или, как указывалось выше, фармацевтически приемлемой его соли в комбинации с фармацевтически приемлемым разбавлением или носителем. Такая композиция представлена как дополнительный признак предлагаемого изобретения и может быть составлена в различных дозированных формах. Например, композиция может быть выполнена в виде таблеток, капсул, растворов или суспензий для перорального приема, в форме свеч для ректального приема, в виде стерильных растворов или суспензий для внутривенных и внутримышечных инъекций, в виде аэрозоля или раствора или суспензии, используемых из распыливателя для ингаляций, в виде порошка, содержащего фармацевтически приемлемый разбавитель, такой как лактоза для инсуффляции.
Фармацевтические композиции можно получить традиционными методами, используя известные в данной области фармацевтически приемлемые разбавители или носители. Таблетки и капсулы для перорального приема можно для удобства покрывать энтеросолюбильной оболочкой, например, из фталата триацетата целлюлозы, для уменьшения контакта активного ингредиента формулы (1) с желудочными кислотами.
Фармацевтические составы предлагаемого изобретения могут также содержать один или несколько известных средств, которые играют огромную роль для лечения заболеваний или патологических состояний например, известный ингибитор агрегации тромбоцитов, гиполипидемный агент, противогипертоническое средство. бронхолитическое средство, бета-адренергический блокатор или вазодилататор могут быть также включены в фармацевтическую композицию предлагаемого изобретения для лечения сердечно-сосудистых заболеваний или состояний. Аналогичным образом, в качестве примера, при лечении легочных заболеваний в композицию предлагаемого изобретения можно также включать антигистамин, стероид (например, дипропионат беклометазона), кромогликат натрия, ингибитор фосфодиэстеразы или бета-адренергический стимулятор. Кроме того, известный антагонист ТХА2, например, как предпочтительное соединение, раскрытое в заявке на Европатент, N публикации 201354 (4), или известный ингибитор ТХА2-синтазы, например, как базоксибен или фурегрелат (V 63557) можно также включать вместе с соединением формулы (1) или ее приемлемой солью в предлагаемую композицию для изменения суммарного равновесия при действии антагониста ТХА2 и ингибитора ТХА2-синтазы для получения требуемого терапевтического эффекта в любой из вышеуказанных болезней или патологических состояний.
Кроме вышеуказанного применения соединений формулы (1) в медицине внутренних болезней у человека их можно с успехом также использовать в ветеринарии для лечения аналогичных состояний у теплокровных животных, представляющих интерес с коммерческой точки зрения, например, собак, кошек, лошадей и крупного рогатого скота. Вообще для такого лечения соединения формулы (1) обычно вводят в аналогичных дозировках или аналогичными способами, которые описаны выше при использовании на человеке. Соединения формулы (1) также представляют ценность в качестве фармакологического инструмента при разработке и стандартизации тестовых систем при оценке влияния ТХА2 на лабораторных животных, например, кошек, собак, кроликов, обезьян, крыс и мышей, как часть постоянного поиска для разработки новых и усовершенствованных лекарственных препаратов. Соединения формулы (1) могут быть также использованы из-за их способности действовать как антагонист ТХА2, так и ингибитор ТХА2-синтазы для поддержания жизнеспособности кровяных сосудов у теплокровных животных (или на отдельных их участках) при искусственном (экстракорпоральном) кровообращении, например, в трансплантантах конечностей или внутренних органов. При использовании соединения формулы (1) или его фармацевтически приемлемого производного для этой цели обычно вводят таким образом, чтобы достигнуть устойчивой концентрации их в крови, например 0,1-10 мг/л.
Изобретение далее проиллюстрировано следующими не ограничивающими объем примерами, в которых примеры 1-17 описывают получение используемых промежуточных соединений и, если не оговорено особо:
(I) концентрацию и упаривание осуществляют роторным выпариванием в вакууме;
(II) операции осуществляют при комнатной температуре, т.е. в интервале 18-26оС;
(III) флэш-хроматографию выполняют на кизельгеле Fluka Kieselgel 60 (N каталоговый 60738), поставляемого фирмой "Fluka Ab. Buchs, Швейцария, СН-9470);
(IV) выходы, когда приводятся, представлены только для справки и не обязательно являются максимально достижимыми при тщательной разработке указанного способа;
(V) ПЯМР-спектры обычно определяют на частоте 90-200 Гц в CDCl3, используя тетраметилсилан (TMS) в качестве внутреннего стандарта и выражают как химические сдвиги (дельта-значение) в частях на млн.относительно ТMS, используя общепринятые сокращения для обозначения главных пиков: С синглет; М мультиплет; т триплет; br шир. d дублет.
(VI) все целевые продукты выделяют в форме рацематов, которые имеют хороший микросостав;
(VII) для удобства рацемические конечные продукты обозначены по номенклатуре "цис" или "транс" для отражения относительной конфигурации заместителей вокруг диоксанового кольца, т.е. в таких рацематах заместители в положениях 4,5 указаны как (4,5-цис) вместо более точного обозначения (4SR, 5RS), причем последнее обозначение используют для указания энантиомерных форм, описанных в примере 40.
П р и м е р 1. Раствор, содержащий 2-[(4,5-цис)-2,2-диметил-4-(3-пиридил)-1,3-диоксан-5-ил] ацетальдегид (1), (0,20 г) в сухом тетрагидрофуране (ТГФ) (7мл) прибавляют в атмосфере аргона к перемешанному и охлажденному льдом раствору илида, полученного при реакции бромистого (3-карбоксипропил)трифенилфосфина (0,91 г) с трет-бутоксидом калия (0,48 г) в сухом ТГФ (30 мл). Полученную смесь перемешивают в течение 2 ч и затем обрабатывают охлажденной льдом водой (50 мл). Раствор концентрируют и прибавляют еще воды (25 мл), рН доводят до 7 путем прибавления нескольких кристаллов щавелевой кислоты с последующим экстрагированием раствора этилацетатом (3х40 мл). Водную фазу подкисляют до рН 4 щавелевой кислотой и экстрагируют этилацетатом (3х50 мл). Указанные соединенные экстракты промывают насыщенным солевым раствором (50 мл), сушат (MgSO4) и упаривают. Остаток очищают флэш-хроматографией, используя в качестве элюента смесь дихлорметан/метанол (95:5 об/об), с выходом 4 (Z)-6-[2,2-диметил-4-(3-пиридил)-1,3-диоксан-цис-5-ил]гексеновой кислоты в виде масла (0,19 г); ЯМР: 1,55 (3Н,с), 1,57 (3Н,с) 1,5-2,6 (7Н,м), 3,85 (1Ндд, I 12 Гц, 1,5 Гц), 4,15 (1Н, дм I 12 Гц), 5,15-5,50 (3Н, м), 7,3-7,4 (1Н,м), 7,7-7,8 (1Н,м) 8,1 (1Н,шир.) и 8,45-8,60 (2Н,м).
Требуемые исходные материалы получают по следующей методике:
(1) Метиловый эфир 2-(никотиноил)уксусной кислоты (17,9 г), полученной по методу E. Weuhert и др. I.Org.Chem. вып.48, с.5006, 1983), прибавляют в атмосфере аргона к раствору, содержащему металлический натрий (2,3 г) в метаноле (200 мл), с последующим перемешиванием полученной смеси при 25оС в течение 30 мин. Затем прибавляют бромистый аллил (12,0 г) и смесь перемешивают в течение ночи. Прибавляют дополнительное количество (примерно 2 г) бромистого аллила с последующим перемешиванием смеси в течение 48 ч, а затем упаривают. Полученное масло разделяют между водой и простым эфиром и водный слой экстрагируют три раза простым эфиром. Объединенные экстракты промывают насыщенным раствором соли, сушат (MgSO4) и упаривают. Остаток промывают флэш-хроматографией и после элюирования смесью из петролейного простого эфира (т.кип. 60-80оС) и этилацетата (1:1, об/об) получают метил 1-2-никотиноил-4-пентеноат (А) в виде масла бледно-желтого цвета (13,8 г); ЯМР: 2,6-2,9 (2Н, м), 8,7 (3Н,с), 4,4 (1Н,м), 4,9-5,2 (2Н, м)/, 5,5-6,0 (1Н,м), 7,2-7,5 (1Н,м), 8,1-8,3 (1Н,м), 8,7-8,8 (1Н,м) и 9,1-9,2 (1Н,м).
(II) Раствор, содержащий А (8,8) в сухом ТГФ (40 мл) прибавляют к суспензии литийалюминийгидрида (1,8 г) в сухом ТГФ (80 мл) в атмосфере аргона со скоростью, при которой температура не превышала бы 10оС. Через 2 ч смесь охлаждают на льду. Для разрушения избытка реагента затем прибавляют этилацетат (20 мл) с последующим введением водного насыщенного раствора хлористого аммония (50 мл). Осадок отфильтровывают и промывают этилацетатом. Водную фазу отделяют и экстрагируют этилацетатом (3х50 мл). Объединенные органические фракции промывают насыщенным солевым раствором, сушат (MgSO4) и концентрируют. Остаток очищают флэш-хроматографией и после элюирования смесью этилацетат/метанол (95:5, об/об) получают 2-аллил-1-(3-пиридил) 1,3-пропандиол (В) (5,3 г) в виде масла (смесь эпимеров); ЯМР: 1,8-2,2 (3Н,м), 3,6-4,1 (4Н, м), 4,7-5,2 (3Н,м), 5,6-5,9 (1Н,м), 7,2-7,4 (1Н,м), 7,65-7,8 (1Н,м) и 8,4-8,6 (2Н,м).
(III) Смесь, состоящую из В (5,2 г) пара-толуолсульфокислоты (5,2 г) и 2,2-диметоксипропан (50 мл), перемешивают в течение ночи при комнатной температуре, рН доводят до 8-10 путем введения триэтиламина, а затем раствор сгущают при пониженном давлении. Остаток очищают флэш-хроматографией и после элюирования смесью из простого петролейного эфира (т.кип. 40-60оС) и этилацетата (60:40, об/об) получают 5-аллил-2,2-диметил-4-(3-пиридил) 1,3-диоксан (С) (смесь 4,5-цис и транс-изомеров) в виде масла (4,6 г); ЯМР: 1,4-1,6 (6Н, м), 1,6-2,5 (3Н,м), 3,65-4,25 (2Н,м), 4,5-5,7 (4Н,м), 7,2-7,4 (1Н,м), 7,6-7,8 (1Н,м), и 8,45-8,65 (2Н,м).
(IV) Озон в кислороде барботируют через раствор, содержащий С (3,4 г) в этилацетате (130 мл) при температуре -70оС, пока не появится голубая окраска. Аргон затем пропускают через раствор для разрядки избыточного озона и прибавляют раствор трифенилфосфина (6 г) в этилацетате (50 мл). Смесь нагревают в течение ночи до комнатной температуры с последующим перемешиванием в течение ночи. Раствор концентрируют и для осаждения трифенилфосфиноксида прибавляют (50 мл) простой эфир. Смесь затем фильтруют, а фильтрат концентрируют. Получают масло, которое очищают флэш-хроматографией. После элюирования смесью (60:40, об/об) этилацетат-простой петролейный эфир (т.кип. 40-60оС) получают первоначально 2-[(4,5-цис]-2,2-диметил-4-(3-пиридил)-1,3-диоксан-5-ил] ацетальдегид (1) в виде масла (0,8 г); ЯМР: 1,5 (3Н, м), 1,55 (3Н, с). 2,0-2,3 (1Н, м), 2,3-2,5 (1Н,м), 2,8-3,0 (1Н,м), 3,8 (1Н, дд, I 12 Гц, 1,5 Гц), 4,3 (1Н, дм, I 12 Гц), 5,25 (1Н, д, I 3 Гц), 7,25-7,35 (1Н, м), 8,45-8,60 (2Н, м) и 9,61 (1Н,с); а затем соответствующий 4,5-транс-изомер; ЯМР: 1,47 (3Н,с), 1,57 (3Н,с), 2,0-2,6 (3Н,м), 3,75-4,05 (2Н,м), 4,68 (1Н, д, I 10 Гц), 7,25-7,40 (1Н,м), 7,70-7,80 (1Н,м), 8,50-8,65 (2Н,м) и 9,5 (1Н, шир,с), в виде масла (0,7).
П р и м е р 2. Смесь, состоящую из 4(Z)-6-[2,2-диметил-4(3-пиридил)-1,3-диоксан-цис-5-ид] гексеновой кислоты [0,458 г] 2-хлорбензальдегида (0,84 мл), и пара-толуолсульфокислоты (0,314 г) перемешивают в течение 60 ч при температуре 25оС. Раствор подщелачивают путем прибавления триэтиламина и всю реакционную смесь затем очищают флэш-хроматографией с элюированием первоначально дихлорметаном для получения непрореагировавшего альдегида, а затем смесью дихлорметан:метанол (95:5, об/об) с выходом 4 (Z)-6-[(2,4,5-цис)-2-(2-хлорфенил)-4-(3-пиридил)-1,3-диоксан-5-ил] -гексеновой кислоты (0,16 г) в виде масла; ЯМР: 1,6-2,7 (7Н, м), 4,1-4,4 (2Н,м), 5,20-5,55 (3Н,м), 6,05 (1Н,с), 7,2-7,5 (5Н, м), 7,65-7,95 (2Н,м) к 8,4-8,6 (2Н,м).
П р и м е р 3. По аналогичной методике, описанной в примере 2, но используя в качестве исходного материала 5(Z)-7-[2,2-диметил-4-(3-пиридил)-1,3-диоксан-цис-5-ул)гептеновую кислоту (Е) и 2-хлорбензальдегид, получают 5(Z) 7-[(2,4,5-цис)-2-(2-хлорфенил)-4-(3-пиридил)-1,3-диоксан-5-ул] гептеновую кислоту в виде масла при 47% выходе; ЯМР: 1,5-2,7 (9Н,м), 4,1-4,4 (2Н, м), 5,2-5,5 (3Н,м), 6,05 (1Н,с), 7,2-7,5 (5Н,м), 7,7-7,9 (2Н,м) и 8,45-8,65 (2Н,м).
Исходную гептеновую кислоту (Е) получают по аналогичной методике, описанной в примере 1, для получения соответствующей гексеновой кислоты, за исключением того, что вместо (3-карбоксипропил) трифенилфосфонийбромида используют (4-карбоксибутил)трифинилфосфонийбромид. Гептеновую кислоту получают в виде масла при 40% выходе, ЯМР: 1,55 (3Н,с), 1,57 (3Н,с), 1,5-2,6 (9Н,м), 3,85 (1Н,дд, I 12 Гц; 1,5 Гц), 4,15 (1Н, дм, I 12Гц); 5,15-5,50 (3Н, м), 6,6 (1Н, шир.с), 7,3-7,4 (1Н,м), 7,7-7,8 (1Н, м) и 8,45-8,60 (2Н,м).
П р и м е р 4. По аналогичной методике, описанной в примере 2, но используя вместо 2-хлорбензальдегида в качестве исходного материала 2-фенокси-2-метилпропаналь, получают 4(Z)-6-[(2,4,5-цис)-2-(1-метил-1-феноксиэтил)-4-(3-пиридил)-1,3- диоксан-5-ил]гексеновую кислоту в виде бесцветного масла (28% выход), которое отверждается при хранении; ЯМР: 1,35 (3Н,с), 1,40 (3Н, с), 1,5-2,6 (7Н, м), 3,9-4,3 (2Н, м), 4,75 (1Н,с), 5,1 (1Н, д, I 2 Гц), 5,15-5,55 (2Н, м), 6,95-7,15 (3Н, м), 7,2-7,4 (3Н,м0, 7,60-7,75 (1Н,м) и 8,5-8,6 (2Н,м).
Исходный альдегид получают по методике, описанной в заявке на Европатент N 201351 А2, пример 6.
П р и м е р 5. 3-Пиридинкарбоксальдегид (0,365 мл) и пара-толуолсульфокислоту (1,08 г) прибавляют к раствору, содержащему 4 (Z)-6-[2,2-диметил-4-(3-пиридил-1,3-диоксан-цис-5-ил] гексеновую кислоту (0,393 г) в ацетонитриле (8 мл) в атмосфере аргона. Полученную реакционную смесь нагревают в обратным холодильником в течение 4 ч, а затем охлаждают. Прибавляют этилацетат (10 мл) с последующим экстрагированием смеси 1М раствора NaOH (50 мл). Объединенные экстракты подкисляют до установления рН 4 уксусной кислотой и экстрагируют в этилацетате (4х20 мл). Соединенные экстракты органического слоя сушат (MgSO4) и концентрируют с получением масла, которое очищают колоночной флэш-хроматографией с использованием в качестве элюента смесь метанол/дихлорметан (1:10-1:5 об/об). Получают 4 (Z)-6-[(2,4,5-цис)-2,4-бис(3-пиридил)-1,3-диоксан-5-ил] гексеновую кислоту, (0,231 г) в виде масла. ЯМР: 1,6-1,9 (2Н,м), 2,3-2,7 (5Н,м), 4,1-4,35 (2Н,м), 5,2-5,55 (3Н, м), 5,8 (1Н,с), 7,3-7,4 (2Н,м), 7,9-8,0 (1Н,м) и 8,5-8,85 (4Н,м).
П р и м е р ы 6-16. Используя аналогичную методику, которая описана в примере 5, но заменяя 3-пиридинкарбоксальдегид на соответствующий альдегид формулы R4.CHO, получают следующие кислоты формулы (III) (A3означает этилен) в виде масла при 14-86% выходах:
R41H ЯМР (ч/млн) 6 (СН3)3СН= 1,0 (9Н,с), 1,5-1,75 (2Н,м), 2,2-2,55 (5Н, м),
3,85-3,95 (1Н,м), 4,1-4,2 (1Н,м), 4,35 (1Н,с),
5,0-5,5 (3Н,м), 7,3-7,4 (1Н,м), 7,7-7,75 (1Н,м),
8,5-8,6 (2Н,м) 7 3-Ру˙О˙С(СН3)2- 1,4(3Н,с), 1,43 (3Н,с), 1,5-1,8 (2Н,м), 2,2-2,6
(5Н,м), 3,95-4,3 (2Н,м), 4,8 (1Н,с), 5,1-5,55 (3Н,м),
7,2-7,7 (4Н,м), 8,3-8,6 (4Н,м) 8 4CN-Ph 1,45-2,6 (7Н,м), 4,05-4,25 (2Н, м), 5,15-5,45 (3Н,м), 5,9 (1Н,с), 7,35-7,45 (1Н,м), 7,7-7,9 (5Н,м),
8,5-8,6 (2Н,м) 9 2CN-Ph 1,5-2,7 (7Н,м), 4,1-4,3 (2Н,м), 5,15-5,5 (3Н,м),
6,0 (1Н,с), 7,35-7,95 (6Н,м), 8,45-8,6 (2Н,м). 10 3Br-PhO˙C(CH3)2- 1,38 (3Н,с, 1,42 (3Н,с), 1,5-1,8 (2Н,м),
2,2-2,6 (5Н,м), 3,95-4,(2Н, м), 4,75
(1Н,с), 5,1-5,55 (3Н,м), 6,9-7,75 (6Н,м),
8,5-8,6 (2Н, м). 11 4Br-PhO˙C(CH3)2- 1,37 (3Н,с), 1,4 (3Н,с), 1,5-1,8 (2Н,м),
3,95-4,25 (2Н,м), 4,75 (1Н,с),
5,05-5,5 (3Н, м), 6,9-7,7 (6Н, м), 8,5-8,6 (2Н,м). 12 4F-PhO˙C(CH3)2- 1,35(3Н,с0, 1,4 (3Н,с), 1,55-1,8 (2Н,м),
2,2-2,6 (5Н,м), 3,95-4,25 (2Н,м), 4,75 (1Н,с),
5,05-5,5 (3Н,м), 6,85-7,05 (4Н,м), 7,3-7,7 (2Н,м),
8,5-8,6 (2Н, м). 13 4F-PhO˙C(CH3)2- 1,38 (3Н,с), 1,42 (3Н,с0, 1,55-1,8 (2Н,м),
2,2-2,55 (5Н, м), 3,95-4,25 (2Н,м), 4,8
(1Н,с), 5,1-5,5 (3Н,м), 6,75-6,85 (3Н,м),
7,15-7,75 (3Н, м), 8,5-8,6 (2Н,м). 14 PhCH2˙C(CH3)2- 1,0 (6Н,с), 1,55-1,75 (2Н,м), 2,25-2,55 (5Н,м),
2,75 (2Н,с), 3,85-4,2 (2Н,м), 4,3 (1Н,с),
5,0-5,5 (3Н, м), 7,1-7,75 (7Н,м), 8,5-8,6 (2Н,м). 15 4CN-PhO˙C(CH3)2- 1,43(3Н,с), 1,46 (3Н,с), 1,55-1,8 (2Н,м),
2,2-2,55 (5Н,м), 3,95-(2Н,м), 4,8 (1Н,с),
5,1-5,5 (3Н, м), 7,1-7,75 (6Н, м), 8,5-8,6 (2Н,м). 16 2NO2-Ph˙CH=CH- 1,6-1,85 (2Н,м), 2,2-2,9 (5Н,м), 4,05-4,3 (2Н,м),
5,15-5,5 (4Н,м), 6,25-6,35 (1Н,м), 7,3-8,0
(7Н,м), 8,5-8,6 (2Н, м).
(Примечание: Ру пиридил и Ph фенил, возможно замещенный, как указано).
Исходный альдегид для примера 7, 2-метил-2-(3-пиридил-окси)-пропиональдегид получают по следующей методике:
(1) Раствор, содержащий 3-гидроксипиридин (4,75 г) в 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидиноне (DMPV) (10 мл) прибавляют по каплям в течение 30 мин к перемешанной, охлажденной на льду суспензии гидрида натрия (50% мас/мас дисперсия в минеральном масле, 2,4 г) в DMPV (40 мл), Смесь нагревают до 50оС с получением прозрачного раствора, который затем охлаждают до 4оС, Затем прибавляют 2-бром-2-метилпропионат (4,38 мл) и иодистый калий (100 мг) с последующим перемешиванием реакционной смеси при температуре окружающей среды в течение 16 ч. Затем смесь выливают в воду (50 мл) и экстрагируют простым эфиром (3х50 мл). Объединенные экстракты промывают последовательно водой (2х25 мл), насыщенным раствором соли (25 мл), сушат (MgSO4) и упаривают. После очистки с использованием флэш-хроматографии с элюированием смесью простой эфир/гексан (1:1, об/об) получают этиловый эфир 2-метил-2-(3-пиридилокси)пропионовой кислоты (А) в виде прозрачного масла (34%), ЯМР: 1,27 (3Н, т, I 7 Гц), 1,61 (6Н,с), 4,25 (2Н,к, I 7 Гц), 7,19 (2Н,м), 8,27 (2Н,м).
(II) 1,5М раствор, содержащий гидрид диизобутилалюминия в толуоле (21 мл) прибавляют по каплям в атмосфере аргона к перемешанному раствору А (2,09 г) в толуоле (75 мл) при -70оС. Перемешивание продолжают в течение 5 мин после завершения прибавления. Затем в реакционную смесь прибавляют 10% (об/об) раствор метанола в толуоле (15 мл). Полученную смесь сливают в воду (300 мл), интенсивно перемешивают в течение 30 мин и отфильтровывают через кизельгур. Органическую фазу отделяют, а водную фазу насыщают хлористым натрием с последующим эктрагированием простым эфиром (2х100 мл). Объединенные органические соли промывают насыщенным соленым раствором (3х100 мл), сушат (MgSO4) и упаривают. После очистки полученного осадка методом жидкостной хроматографии при среднем давлении с элюированием смесью этилацетат/гексан (1:1, об/об) получают 2-метил-2-(3-пиридилокси)-пропиональдегид в виде прозрачного масла (56%); ЯМР: 1,46 (6Н,с), 7,20 (2Н,м), 8,31 (2Н,м), 9,34 (1Н, с).
Исходный альдегид для примера 10 2-(3-бромфенокси)-2-метилпропаналь получают по следующей методике.
(I) Раствор, содержащий метиловый эфир дихлоруксусной кислоты (77,18 г, 0,54 моль) в безводном простом эфире (50 мл), прибавляют к перемешанному раствору метилмагнийиодида, полученному из магниевой стружки (32,8 г, 1,35 моль) и метилиодида (84,1 мл, 1,35 моль) в безводном простом эфире (750 мл) при 0оС в атмосфере аргона с такой скоростью, при которой бы температура смеси не превышала 15оС. Смесь перемешивают при 25oС в течение 30 мин с последующим ее охлаждением до 0оС. Добавляют воду (100 мл), а затем подкисляют до установления рН 4 концентрированной соляной кислотой. Органический слой отделяют, а водный слой промывают простым эфиром (3х100 мл). Объединенные органические слои сушат (MgSO4) и упаривают. После перегонки остаточного масла при пониженном давлении получают 1,1-дихлор-2-гидрокси-2-метилпропан (А) (57,81 г) в виде масла: т.кип. 48-50оМ при 20 ммHg; ЯМР: 1,45 (6Н,с), 2,15 (1Н,шир.с) и 5,65 (1Н,с).
(II) Цетилтриметиламмонийбромид (0,28 г, 0,77 ммоль) прибавляют к раствору м-бромфенола (6,66 г, 38,5 ммоль) в 3,85 М водном растворе NaOH (10 мл) с последующим прибавлением раствора А (1,37 г, 9,6 ммоль) в простом эфире (20 мл). Полученную смесь перемешивают в атмосфере аргона в течение 18 ч с последующим разбавлением простым эфиром (50 мл). Для выделения непрореагировавшего фенола смесь экстрагируют 2М-водным раствором NaOH (4х30 мл). Объединенные водные экстракты обрабатывают простым эфиром (50 мл), а органическую фазу промывают сначала 2М-водным раствором NaOH (20 мл), а затем водой 50 мл. Объединенные органические слои сушат (MgSO4), упаривают. После очистки колоночной флэш-хроматографией с элюированием смесью этилацетат/гексан (1:1, об/об) получают 2-(3-бромфенокси)-2-метилпропаналь (0,89 г) в виде масла: ЯМР: 1,45 (6Н,с), 6,75-7,20 (4Н,м), 9,8 (1Н,с).
Используя аналогичную методику, которая описана при получении 2-(3-бромфенокси)-2-метилпропионового альдегида, за исключением того, что в качестве исходного материала берут замещенный соответствующим образом фенол, получают следующие альдегиды, которые используют в примерах 11,12,13 и 15 2-(4-бромфенокси)-2-метилпропионовый альдегид; ЯМР: 1,4 (6Н,с), 6,7-7,4 (4Н,м), 9,8 (1Н,с).
2-(4-фторфенокси)-2-метилпропионо- вый альдегид;ЯМР: 1,4 (6Н,с), 6,8-7,0 (4Н,м), 9,8 (1Н,с);
2-(3-фторфенокси)-2-метилпропионо- вый альдегид; ЯМР: 1,45 (6Н, с), 6,55-7,3 (4Н,м), 9,8 (1Н,с); и
2-(4-цианофенокси)-2-метилпропионо- вый альдегид; ЯМР: 1,5 (6Н, с), 6,85-7,6 (4Н,м), 9,75 (1Н,с).
Исходный альдегид для примера 14 по- лучают по методике, описанной H.K. Diefl, K.C.Brannock, Tetrahedron Letters, выа.14, с.1273, 1973.
П р и м е р 17. В соответствии с методикой, описанной в примере 1, за исключением того, что в качестве исходного материала используют 2-[(4,5-цис)-2,2-диметил-4-(4-пиридил)-1,3-диоксан-5-ил] ацетальдегид, получают 4(Z)-6-[2,2-диметил-4-(4-пиридил)-1,3-диоксан-цис-5-ил] гексеновую кислоту в виде масла, которое отверждается при хранении, с выходом 7% т.пл. 167-169оС (после перекристаллизации из смеси этилацетат/петролейный эфир); ЯМР: 1,42 (3Н, с), 1,49 (3Н,с), 1,7-2,5 (7Н,м), 3,66 (1Н,д, I12 Гц), 4,12 (1Н,д, I 12 Гц), 5,1-5,42 (3Н,м), 7,30 (2Н, д) и 8,52 (2Н,д).
Вышеуказанный исходный материал получают в виде масла при 50% выходе в соответствии с методикой, описанной в примере 1; ЯМР: 1,5 (3Н,с), 1,55 (3Н, с), 2,0-2,3 (1Н, м), 2,3-2,5 (1Н,м). 2,8-3,0 (1Н,м), 3,8 (1Н, дд, I 12 Гц, 1,5 Гц), 4,3 (1Н, дм, I12 Гц), 5,2 (1Н, д, I 3 Гц), 7,25 (2Н,д), 8,6 (2Н, д) и 9,62 (1Н,с) для его получения используют метиловый эфир 3-(4-пиридил)-3-оксо-пропионовой кислоты, полученной по аналогичной методике, которая описана E.Wenkert и др. I.Org.Chem, вып.48, с.5006, 1983.
Следующие аналоги промежуточных соединений примера 1 получены в виде масел, которые используют без последующей очистки:
(I) метил-2-изоникотиноил-4-пентеноат, выход 65%
(II) 2-аллил-1-(4-пиридил-1,3-пропандиол), выход 77%
(III) 5-аллил-2,2-диметил-4(4-пиридил)-1,3-диоксан (смесь 4,5-цис и транс-изомеров), выход 44%
П р и м е р 18. В соответствии с методикой, описанной в примере 5, но в качестве исходного материала используют 4(Z)-6-[2,2-диметил-4-(4-пиридил)-1,3-диоксан-цис-5-ил] гексеновую кислоту и 2-хлорбензальдегид, получают 4(Z)-6-[2,4,5-цис)-2-(2-хлорфенил)-4-(4-пиридил-1,3-диоксан-5-ил] гексеновую кислоту в виде масла при 21% выходе; ЯМР: 1,6-2,7 (7Н,м), 3,59 (1Н,д, I10,7 Гц), 4,35 (1Н, дд, I 10,7 Гц, 4,8 Гц), 4,6 (1Н, д, I 10,7 Гц), 5,18-5,5 (2Н, м), 5,98 (1Н,с), 7,2-7,8 (6Н,м) и 8,63 (2Н, шир.с).
П р и м е р ы 19-29. В соответствии с методикой. описанной в примере 5, за исключением того, что вместо 3-пиридинкарбоксальдегид используют соответствующий альдегид формулы R4˙СНО, получают следующие кислоты формулы (III) (А3 означает этилен).
R41Н ЯМР (ч/млн) 19 4NO2-Ph 1,5-2,6 (7Н,м), 4,1-4,3 (2Н,м), 5,15-5,5 (3Н,м),
5,95 (1Н, с), 7,35-8,6 (8Н,м). 20 2,4-Cl2-Ph 1,5-2,5 (7Н,м), 4,05-4,25 (2Н,м), 5,15-5,45 (3Н,м),
6,05 (1Н,с), 7,35-7,85 (5Н,м), 8,45-8,55 (2Н,м). 21 3,4-Cl2-Ph 1,6-2,65 (7Н,м), 4,05-4,3 (2Н,м), 5,2-5,5 (3Н,м),
5,7 (1Н, с). 7,3-7,8 (5Н,м), 8,5-8,65 (2Н,м). 22 2-Cl,5-NO2-Ph 1,7-2,7 (7Н,м), 4,15-4,4 (2Н,м), 5,2-5,55 (3Н,м),
6,05 (1Н, с), 7,3-8,7 (7Н,м). 23 3Br-BhCH2C(CH3)2- 1,0 (6Н,с), 1,5-1,75 (2Н,м), 2,3-2,8 (7Н,м),
3,8-4,2 (2Н,м), 4,25 (1Н,с), 4,95-5,5 (3Н,м),
7,05-7,4 (5Н,м), 7,65-7,7 (1Н,м), 8,5-8,6
(2Н,м). 24 3,4-Cl2-PhO˙C(CH3)2- 1,38 (3Н,с), 1,4 (3Н,с), 1,6-1,8 (2Н,м),
2,2-2,6 (5Н,м), 3,95-4,24 (2Н,м), 4,75 (1Н,с),
5,1-5,5 (3Н,м), 6,9-7,75 (5Н,м), 8,5-8,6 (2Н,м). 25 4F-PhCH2C(CH3)2- 1,0 (6Н,с), 1,55-1,75 (2Н,м), 2,2-2,55 (5Н,м),
3,8-4,2 (2Н,м), 4,3 (1Н,с), 4,95-5,5 (3Н,м),
6,9-7,75 (6Н, м), 8,5-8,65 (2Н, м). 26 4NO2PhO˙C(CH3)2- 1,45 (6Н,с), 1,8-2,5 (7Н,м), 3,95-4,1 (2Н,м),
4,9 (1Н,с), 5,05-5,5 (3Н,м), 7,2-7,7 (4Н,м),
8.1-8,2 (2Н, м), 8,45-8,5 (2Н,м). 27 2F-PhO˙C(CH3)2- 1,38 (3Н,с), 1,42 (3Н,с), 1,55-1,8 (2Н,м),
2,2-2,55 (5Н,м) 3,95-4,3 (2Н,м), 4,85 (1Н,с),
5,1-5,5 (3Н,м), 6,95-7,7 (6Н,м), 8,45-8,6 (2Н,м). 28 3,4-F2-PhO˙C(CH3)2- 1,35 (3Н,с), 1,38 (3Н,с), 1,55-1,8 (2Н,м),
2,2-2,55 (5Н,м), 3.95-4,25 (2Н,м), 4,75 (1Н,с),
5,1-5,5 (3Н, м), 6,7-7,75 (5Н,м), 8,5-8,6 (2Н,м). 29 4CF3-Ph 1,6-2,65 (7Н,м), 4,15-4,35 (2Н,м), 5.2-5,5 (3Н,м),
5,78 (1Н,с), 7,3-7,8 (6Н,м), 8,5-8,7 (2Н,м).
В соответствии с методикой, описанной R.Subramanian. Chem. and Ind, с, 731, 1978, которую используют для получения 2,2-диметил-3-фенилпропионового альдегида, за исключением того, что в качестве исходного материала используют соответственно замещенный галоидный бензил, получены следующие альдегиды, которые используют в примерах 23 и 25: 3-(3-бромфенил)-2,2-диметилпропионовый альдегид; ЯМР: 1,05 (6Н,с), 2,75 (2Н,с), 7,0-7,4 (4Н,м), 9,55 (1Н, с); 3-(4-фторфенил)-2,2-диметилпропионовый альдегид; ЯМР: 1,05 (6Н,с), 2,75 (2Н,с), 6.9-7,1 (4Н,м), 9,55 (1Н,с).
В соответствии с аналогичной методикой, описанной для получения 2-(3-бромфенокси)-2-метилпропионового альдегида, но в качестве исходного материала используют соответственно замещенный фенол, получены следующие альдегиды, которые применяют в примерах 24,27 и 28: 3-(3,4-дихлорфенокси)-2-метилпропионо- вый альдегид; ЯМР: 1,45 (6Н,с), 6,7-7,35 (3Н,м), 9,75 (1Н,с); 2(2-фторфенокси)-2-метилпропионовый альдегид; ЯМР: 1,45 (6Н,с), 6,9-7,15 (4Н,м), 9,8 (1Н,с).
Исходный альдегид для примера 26, 2-(4-нитрофенокси)-2-метилпропионовый альдегид, получают по аналогичной методике, описанной для получения 2-метил-2-(3-пиридилокси)-пропионового альдегида, за исключением того, что вместо 3-гидроксипиридина используют 4-нитрофенол. ЯМР: 1,55 (6Н,с), 6,9 (2Н,д, I 7 Гц), 8,15 (2Н,д, I 7 Гц), 9,8 (1Н,с).
П р и м е р 30. Р-пара-толуолсульфокислоту (0,33 г) прибавляют к раствору, содержащему 4(Z)-6-[2,2-диметил-4-(3-пиридил)-1,3-диоксан-цис-5-ил] гексеновую кислоту (0,482 г) в ацетонитриле (15 мл) и смесь перемешивают в течение 30 мин. Затем диэтилацеталь 2-феноксиацетальдегид (1,04 г) прибавляют к реакционной смеси с последующим нагреванием ее до 90оС в течение 15 ч. Затем смесь охлаждают и упаривают. Остаточное масло, состоящее из смеси требуемого кислотного продукта, и сложный этиловый эфир растворяют в метаноле (6 мл). Прибавляют 2М-водный раствор гидроксида натрия (3 мл), а затем полученную смесь перемешивают в течение 1 ч. Прибавляют этилацетат (25 мл) и воду (25 мл), а затем смесь подкисляют уксусной кислотой и экстрагируют этилацетатом (4х25 мл). Объединенные органические экстракты сушат (MgSO4) и упаривают. После очистки полученного масло методом флэш-хроматографии с элюированием сначала в дихлорметане, а затем в смеси метанол/дихлорметан (7:93, об/об) получают 4(Z)-6-[(2,4,5-цис4)-2-феноксиметил-4-(3-пиридил)-1,3-диоксан-5-ил] гексеновую кислоту (0,146 г) в виде масла: ЯМР: 1,65-1,85 (2Н, м), 2,2-2,6 (5Н, м), 4,0-4,25 (4Н,м), 5,1-5,45 (4Н,м), 6,9-7,45 (6Н,м), 7,75-7,8 (1Н,м) и 8,5-8,6 (2H,м).
Исходный материал, диэтилацеталь 2-феноксиацетальдегида получают по следующей методике:
Гидрид натрия (5,83 г, 55% дисперсии в минеральном масле) прибавляют к раствору фенола (12,56 г) в ДМТР (DMPV) (25 мл) при 5оС с последующим перемешиванием полученной смеси в течение 30 мин. Деэтилацеталь бромацетальдегида (10,05 мл) прибавляют к реакционной смеси и затем нагревают до 110оС в течение 5 ч с последующим ее охлаждением. Смесь разделяют этилацетатом (100 мл) и водой (100 мл), органическую фазу отделяют и последовательно промывают 2М-водным раствором NaOH (2х50 мл) и водой (50 мл). Водные фракции объединяют и повторно экстрагируют этилацетатом (100 мл). Объединенные органические экстракты сушат (MgSO4) и упаривают. Полученное масло очищают флэш-хроматографией и, используя в качестве элюента смесь этилацетат/гексан (1:10, об/об), получают диэтилацеталь 2-феноксиацетальдегида (9,43 г) в виде масла; ЯМР: 1,25 (6Н,т, I 7,0 Гц), 3,6-3,85 (4Н,м), 4,05 (2Н,д, I 6,0 Гц), 4,85 (1Н,т, I 6,0 Гц) и 6,9-7,35 (5Н,м).
П р и м е р 31. р-Пара-толуолсульфокислоту (0,358 г) прибавляют к раствору, содержащему 4(Z)-6-[2,2-диметил-4-(3-пиридил -2,3-диоксан-цис-5-ил] гексеновую кислоту (0,522 г) в ацетонитриле (12 мл) с последующим перемешиванием смеси в течение 30 мин. Прибавляют раствор,содержащий 2-(2,4-дифторфенокси)-2-метилпропионовый альдегид (1,02 г) в ацетонитриле, а затем триметилортоформат (0,21 мл) с последующим нагреванием смеси с обратным холодильником в течение 3 ч в атмосфере аргона. Для полного завершения этерификации прибавляют метанол (1 мл), а затем раствор кипятят с обратным холодильником в течение еще 2 ч. Затем реакционную смесь постепенно охлаждают и экстрагируют 1М-водный раствором гидроксида натрия (2 мл) и этилацетатом (25 мл). Органический слой отделяют, сушат (MgSO4) и упаривают. После очистки полученного масла флэш-хроматографией с элюированием в смеси метанол/дихлорметан (1: 100-1: 20, об/об) получают метиловый эфир 4(Z)-6-[(2,4,5-цис)-2-(1-метил-1-(3,4-дифторфенокси)этил-4-(3-пиридил)- 1,3-диоксан-5-ил] гексеновой кислоты (0,333 г) в виде масле; ЯМР: 1,37 (3Н,с), 1,40 (3Н, с), 1,5-1,8 (2Н,м), 2,2-2,6 (5Н,м), 3,65 (3Н,с), 3,9-4,3 (2Н,м), 4,75 (1Н,с). 5,05-5,5 (3Н,м), 6,7-7,7 (5Н,м) и 8,5-8,6 (2Н,м).
П р и м е р 32. Смесь, состоящую из 4(Z)-6-[2,2-диметил-4(3-пиридил)-1,3-диок- сан-цис-5-ил] гексеновой кислоты (0,500 г), 2-метил-2-пропоксипропионового альдегида (2,13 г) и пара-толуолсульфокислоты моногидрата (0,342 г), перемешивают в течение 18 ч, 0,2М раствор гидроокиси натрия (20 мл) прибавляют к реакционной смеси с последующим ее промыванием простым эфиром (2х10 мл) подкислением до рН 5 уксусной кислотой и экстрагированием простым эфиром (3х25 мл). Объединенные эфирные экстракты промывают водой (2х10 мл), насыщенным раствором хлорида натрия (10 мл) и затем сушат над безводным сульфатом магния. Органические экстракты упаривают, давая коричневое масло, которое очищают жидкостной хроматографией при среднем давлении (MLPC), используя в качестве элюента смесь из этилацетата, гексана и уксусной кислоты (80:20:1, об/об), получают прозрачное масло, дающее при порошковании простым эфиром 4(Z)-6-[(2,4,5-цис)-2-(1-метил-2-пропоксиэтил-4-(3-пиридил-1,3-диоксан-5-ил] гексеновую кислоту, 0,25 гидрат (0,053 г) в виде твердого вещества, т. пл. 116-118оС; ЯМР: 200 МГц, d6DMSO): 0,83 (3Н,т, I 7 Гц), 1,18 (3Н, с), 1,20 (3Н, с), 1,42 (3Н,м0, 1,84 (1Н,м), 2,14 (4Н,м), 2,35 (1Н,м), 3,40 (2Н,т, I 6 Гц), 3,94 (2Н,м), 4,65 (1Н,с); 5,15 (1H,д, I 2Гц). 5,18 (1Н, м), 5,34 (1Н,м), 7,38 (1Н,м), 7,68 (1Н,дм, I 7 Гц), 8,49 (2Н,м); элементный анализ:
Найдено: C 65,8; H 8,3; N 3,7
C21H31NO5 0,25H2O;
Вычислено: C 66,0; H 8,3; N 3,7.
Исходный альдегид получают по методике, описанной в заявке на Европатент, N 201351 А2, пример 7.
П р и м е р 33. В соответствии с методикой, описанной в примере 5, за исключением того, что вместо 3-пиридинкарборксальдегида используют циклогексанкарбоксальдегид и реакцию проводят при температуре окружающей среды в присуствии только 1,1 эквивалентов пара-толуолсульфокислоты моногидрата, получают 4(Z)-6-[2,4,5-цис)-2-циклогексил-4-(3-пиридил)-1,3-диоксан-5-ил] гексеновой кислоты гидрат в виде белого твердого продукта (47% выход), т.пл. 121-125оС; ЯМР: (200 МГц, CDCl3); 1,22 (5Н,м), 1,74 (8Н,м), 2.29 (4Н,м), 2,44 (1Н,м), 3,89 (1Н,д, I 11 Гц), 4,12 (1Н,д I 11 Гц), 4,51 (1Н,д,I 4 Гц), 5,00 (1Н, д, I 1,5 Гц), 5,22 (1Н,м), 5,38 (1Н,м), 7,33 (1Н,м), 7,72 (1Н,д, I 7 Гц), 8,53 (2Н,м); элементный анализ:
Найдено: C 67,2; H 8,1; N 3,7%
Вычислено для C21H29NO4, 1H2O: C 66,8; H 8,2: N 3,7.
П р и м е р 34. 1М раствора гидроксида натрия (6,28 мл) прибавляют к перемешанному раствору, содержащему метиловый эфир 4(Z)-6-[(2,4,5-цис)-4-(3-пиридил)-2-трифторметил-1,3-диоксан-5-ил] гексеновой кислоты (А) (563 мл) в метаноле (10 мл). Через 2 ч прибавляют воду (40 мл) и полученную смесь промывают диэтиловым эфиром (2х20 мл), подкисляют до рН 5 уксусной кислотой с последующим экстрагированием этилацетатом (3х30 мл). Объединенные органические экстракты промывают водой (20 мл), насыщенным раствором хлористого натрия (2х20 мл) и затем сушат над безводным сульфатом магния. После упаривания растворителя получают масло, которое очищают жидкостной хроматографией при среднем давлении, и, используя в качестве элюента смесь этилацетат/метанол/уксусная кислота (95:5:1, об/об), получают 4(Z)-6-[(2,4,5-цис)-4-(3-пиридил)-2-трифторметил-1,3-диоксан-5-ил] гексеновой кислоты аддукт моноацетата в виде масла (587 мг); ЯМР: (200 МГц, CDCl3): 1,71 (1Н,м), 1,83 (1Н, м), 2,10 (3Н,с), 2,30 (4Н,м), 2,51 (1Н,м), 4,05 (1Н,дм, I 11 Гц), 4,30 (1Н, д, I 11 Гц), 5,12 (1Н,к, I 3Гц), 5,20 (1Н,д, I 2 Гц), 5,22 (1Н,м), 5,46 (1Н,м); 7,42 (1Н,м), 7.80 (1Н,д, I 7 Гц), 8,59 (2Н, шир.).
Элементный анализ для C16H18NO4F3, ICH3COOH
Найдено: C 53,3; Н 5,6; N 3,4%
Вычислено: C 53,5; H 5,4; N 3,5%
Исходный материал, требуемый для А получают по следующей методике:
(I) 1М раствор соляной кислоты (10 мл) прибавляют к раствору, содержащему 4(Z)-6-[2,2-диметил-4-(3-пиридил)-1,3-диоксан-цис-5-ил] гексеновую кислоту (1,42 г в тетрагидрофуране (15 мл) с последующим перемешиванием смеси в течение 2 ч. Вливают воду и рН доводят до 12 2М-раствором гидроксида натрия. Смесь промывают этилацетатом (2х25 мл), подкисляют до рН 5 уксусной кислотой, а затем насыщают твердым хлоридом натрия. Водную смесь затем экстрагируют этилацетатом (12х50 мл) и объединенные фракции сушат, используя MgSO4. После упаривания растворителя получают 4(Z)-эритро-8-гидрокси-7-гидроксиметил-8-(3-пиридил)-4-октеновую кислоту (В) в виде коричневого масла (1,114 г), используемого в дальнейшем без очистки. Для определения характеристик, образец очищают флэш-хроматографией с элюированием смесью метанол/дихлорметан (1: 5, об/об); ЯМР (200 МГц, CDCl3): 1,91 (3Н,м), 2,23 (5Н, м), 3,59 (2Н, м), 5,02 (1Н,м), 5,35 (3Н,м); 7,30 (1Н,м); 7,76 (1Н,м), 8,46 (1Н,дд, I 4 и 1 Гц). 8,60 (1Н,д, I 2Гц).
(II) пара-толуолсульфокислоты моногидрат (1,06 г) прибавляют к раствору, содержащему В (1,114 г) в метаноле (25 мл) с последующим перемешиванием смеси в течение 3 ч. Прибавляют триэтиламин (0,83 мл), а затем смесь упаривают. После упаривания прибавляют насыщенный раствор хлористого натрия (20 мл) с последующим экстрагированием этилацетатом (4х25 мд). Объединенные органические экстракты промывают насыщенным раствором хлористого натрия (10 мл), сушат, используя MgSO4, и растворитель упаривают. Полученное масло очищают жидкостной хроматографией при среднем давлении, и, используя в качестве элюента смесь метанол/дихлорметан (1:12, об/об), получают метиловый эфир 4(Z)-эритро-8-гидрокси-7-оксиметил-8-(3-пиридил)-4-октеновой кислоты (С) в виде масла (1,044 г): ЯМР: (250 МГц, CDCl3): 1,82 (2Н,м), 2.16 (1Н,м), 2,44 (4Н, м), 4.91 (2Н,шир.), 3,67 (3Н,с), 3.81 (2Н,д, I 3 Гц), 5,20 (1Г, д. I 2 Гц). 5,30 (2Н,м), 7,33 (1Н,м), 7,79 (1H, м), 8,51 (1Н,м), 8,61 (1Н,м).
(III) Раствор метансульфонилхлорида (0,32 мл) в дихлорметане (2,0 мл) прибавляют в течение 10 мин к перемешанному раствору, содержащему соединение С (995 мг) и триэтиламин (0,59 мл) в дихлорметане (20 мл). Смесь перемешивают еще 1 ч, а затем разбавляют этилацетатом (50 мл). Полученную смесь промывают водой (2х15 мл), насыщенным раствором хлористого натрия (15 мл), и сушат, используя (MgSO4). При упаривании растворителя получают масло, которое очищают жидкостной хроматографией при среднем давлении, и, элюируя смесью метанол/дихлорметан (1:32, об/об), получают метиловый эфир 4(Z)-эритро-8-гидрокси-7-(метилсульфонилоксиме- тил)-8-(3-пиридил-4- октеновой кислоты (D) в виде бесцветного масла (886 мг); ЯМР: (250 МГц, CDCl3): 2,24 (8Н,м), 3.01 (3Н, с), 3,68 (3Н,с), 4,10 (1Н,м), 4,31 (1Н,м), 5,02 (1Н,д, I 2 Гц), 5,38 (2Н,м), 7,34 (1Н,м), 7,77 (1Н,д, I 7 Гц), 8,57 (2Н,м).
(IV) Безводный карбонат калия (994 мг) и трифторацетальдегида гидрат (1,13 г) прибавляют к раствору, содержащему соединение D (857 мг) в сухом тетрагидрофуране (10 мл). Смесь перемешивают в течение 15 мин при температуре окружающей среды, а затем при 60оС в течение 5 ч. Затем смесь разводят этилацетатом (75 мл) и промывают водой (25 мл) и насыщенным раствором хлористого натрия (25 мл). Органическую фазу сушат (MgSO4) и растворитель упаривают. После очистки полученного масла жидкостной хроматографией при среднем давлении с элюированием в смеси этилацетат[гексан (7:3, об/об) получают вначале метиловый эфир 4(Z)-6-[2,4-транс, 4,5-цис)-4-(3-пиридил-2-трифторметил-1,3-дио- ксан- 5-ил] гексеновой кислоты в виде бесцветного масла (137 мг). ЯМР: (250 МГц), CDCl3): 1,66 (1Н,м), 2,02 (1Н,м), 2,29 (4Н,м), 2,43 (1Н, м), 3,66 (3Н, с), 3.96 (1Н, дд, I= 11 и 2 Гц), 4,36 (1Н,дм, I 11 Гц), 5,20 (1Н, м), 5,30 (1Н,к, I 6 Гц), 5,42 (1Н,м), 5,48 (1Н,д, I 2 Гц). 7,35 (1Н,м), 7,71 (1Н,м), 8,59 (2Н,м), а затем метиловый эфир 4(Z)-6-[(2,4,5-цис)-4-(3-пиридил-2-трифторметил-1,3-диоксан-5-ил] гексеновой кислоты (А) в виде бесцветного масла (578 мг); ЯМР: (250 МГц, CDCl3): 1,60 (1Н,м), 1,81 (1Н, м), 2,30 (4Н,м), 2,55 (1Н,м), 3.66 (3Н,с), 4,04 (1Н, дм, I 11 Гц), 4,29 (1Г, д, I 11 Гц), 5,12 (1Н,к, I 3 Гц), 5,19 (1Н,д, I 2 Гц), 5,22 (1Н,м), 5,45 (1Н,м), 7,38 (1Н,м), 7,74 (1Н,м), 8,58 (2Н,м).
П р и м е р ы 35-39. В соответствии с аналогичной методикой, описанной в примере 5, за исключением того, что вместо 3-пиридинкарбоксальдегида используют альдегид формулы R4˙CHO, получают следующие кислоты формулы (III) (A3 означает этилен).
R41H ЯМР (ч/млн) 35 3NO2-PhO˙C(CH3)2- 1,45(3Н,с), 1,47 (3Н,с), 1,55-1,8 (2Н,м),
2,2-2,55 (5Н,м), 3,95-4,3
(2Н,м), 4,8 (1Н,с), 5,1-5,55 (3Н,м), 7,3-8,0 (6Н,м),
8,5-8,6 (2Н, м). 36 2NO2-PhO˙C(CH3)3- 1,45 (6Н,с), 1.5-1,8 (2Н,м), 2,15-2,5 (5Н,м),
3,9-4,2 (2Н,м), 4,85 (1Н,с), 5.1%5,5 (3Н,м),
7,1-7,8 (6Н, м), 8,4-8,6 (2Н,м). 37 2,4-Cl2-PhO˙C(CH3)2- 1,46 (3Н,с), 1.48 (3Н,с), 1,55-1,8 (2Н,м),
2,15-2,55 (5Н,м), 3,95-4,25 (2Н,м). 4,95 (1Н,с),
5,1-5,55 (3Н,м), 7,1-7,65 (5Н,м), 8,45-8,65 (2Н,м). 38 4NO2-PhCH2C(CH3)2- 1,0 (3Н,с), 1,02 (3Н,с), 1,5-1,75 (2Н,м),
2,25-2,6 (5Н,м), 2,85 (2Н,с), 3,8-4,2 (2Н,м),
4,3 (1Н,с0, 5.0-5,55 (3Н,м), 7,25-8,1 (6Н,м),
8,5-8,65 (2Н,м) 39 3NO2-PhCH2C(CH3)2- 1,02 (3Н,с), 1,04 (3Н,с), 1,5-1,75 (2Н,м),
2,25-2,6 (5Н,м), 2,85 (2Н,с), 3,85-4,25 (2Н,м),
4,3 (1Н,с), 5,0-5,55 (3Н,м), 7,35-8,1
(6Н,м), 8,5-8,65 (2Н,м).
Согласно методике, описанной для получения 2-метил-2-3-пиридилокси/пропионового альдегида, за исключением того, что в качестве исходного материала используют соответственно замещенный фенол, получают следующие альдегиды, которые используют в примерах 35-37.
2-(3-нитрофенокси)-2-метилпропионо- вый альдегид; ЯМР: 1,5 (6Н, с), 7,15-7,95 (4Н, м), 9,85 (1Н,с) 2-(2-нитрофенокси)-2-метилпропионовый альдегид; ЯМР: 1,5 (6Н,с), 6,9-7,8 (4Н,м), 9,85 (1Н,с). 2-(2,4-дихлорфенокси)-2-метилпропионо- вый альдегид; ЯМР: 1,45 (6Н,с), 6,8-7,4 (3Н,м), 9,85 (1Н, с).
По аналогичной методике, раскрытой R.Subramanian, Chem. ans Ind. с.731, 1978 для получения 2,2-диметил-3-фенил-пропионового альдегида, за исключением того, что в качестве исходного материала используют соответственно замещенный галоидный бензил, получают следующие альдегиды, которые используют в примерах 38 и 39: 3-(4-нитрофенил)-2,2-диметилпропионовый альдегид; ЯМР: 1,1 (6Н, с), 2,9 (2Н,с), 7,3 (2Н,д, I 8 Гц), 8,15 (2Н,д, I 8 Гц), 9,55 (1Н,с). 3-(3-нитрофенил)-2,2-диметилпропионовый альдегид; ЯМР: 1,1 (6Н,с), 2,9 (2Н, с), 7.45-8,15 (4Н,м), 9,6 (1Н,с).
П р и м е р 40. По аналогичной методике, описанной в примере 5, но в качестве исходного материала используя 4(Z)-6-[(4S,5R)-2,2-диметил-4-(3-пиридил)-1,3-диоксан-5- ил] гексеновую кислоту и 2-(4-бромфенокси)-2-метилпропионовый альдегид, получают 4(Z)-6-[(2S, 4S, 5R)-2-[1-(4-бромфенокси)-1-метилэтил] -4-(3-пиридил)-1,3-диоксан-5-ил] гексеновую кислоту в виде масла с 25[α]D -113,3 этанол С 0,465, и данные ЯМР совпадают с полученными по рацемическому соединению, описанному в примере 11.
Исходное 2,2-диметил-1,3-диоксановое производное получено по следующей методике:
(I) 1,53 М раствор, содержащий бутиллитий в гексане (23,9 мл). прибавляют к раствору 4S-(-)-изопропил-2-оксазолидинона (4,68 г) с сухом ТГФ (75 мл), охлажденному до -78оС в атмосфере аргона. Смесь постепенно нагревают до -50оС и затем перемешивают в течение 30 мин. Затем смесь повторно охлаждают до -78оС и прибавляют по каплям раствор 4-пентеноилхлорида (4,33 г) в сухом тетрагидрофуране (10 мл). После того, как закончат введение, смесь перемешивают при -78оС в течение 30 мин с последующим нагреванием до -20оС. Прибавляют насыщенный водный раствор хлористого аммония (20 мл) и смесь экстрагируют этилацетатом (3х100 мл). Объединенные органические слои сушат (MgSO4) и упаривают. После очищения осадка флэш-хроматографией с элюированием смесью этилацетат/гексан (20: 80, об/об) получают (4S)-4-изопропил-3-(4-пентеноил)оксазоли- дин-2-он (А) (6,34 г) в виде масла. ЯМР: 0,85-0,95 (6Н,м), 2.3-2,5 (3Н,м), 2,9-3,2 (2Н,м), 4,15-4,5 (3Н,м), 4,95-5,15 (2Н,м), 5,75-6,0 (1Н,м).
(II) 1М раствор дибутилборотрифталата в дихлорметане (32,7 мл) прибавляют к раствору, содержащему соединение А (6,28 г) в сухом дихлорметане (110 мл), охлажденному до 5оС в атмосфере аргона с последующим добавлением диизопропилэтиламина (6,25 мл). Реакционную смесь перемешивают в течение 30 мин при 5оС, а затем охлаждают до -78оС. По каплям прибавляют 3-пиридинкарбоксальдегид (3,1 мл). Смесь перемешивают в течение 30 мин при -78оС с последующим постепенным нагреванием до -50оС в течение 30 мин. Охлажденную баню удаляют и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Затем смесь охлаждают до 5оС и прибавляют перекись водорода (11,5 мл, 30%-ный водный раствор). Смесь перемешивают в течение 30 мин, а затем выливают в воду (50 мл) и экстрагируют дихлорметаном (3х100 мл). Объединенные экстракты сушат (MgSO4) и упаривают. После очистки остатка флэш-хроматографией на колонке, элюируя смесью этилацетат/гексан (1:1, об/об) с постепенным доведением до 100% этилацетата), получают (4S)-(3-[(2S)-2-[(1S-1-окси-1-(3-пиридил)метил] пент-4-еноил)-4-изопропилоксазолидин-2-он (В) в виде твердого продукта, т. пл. 112-113оС (после перекристаллизации из толуола); 25[α] +136,0 (EtOH, с.0,311); ЯМР: 0,85 (6Н, дд, I 7 Гц), 2,15-2,7 (4Н,м), 4,0-4,2 (2Н,м), 4,3-4,55 (2Н,м), 4.95-5,1 (3Н,м), 5,65-5,9 (1Н,м), 7,25-7,35 (1Н,м), 7,75-7,85 (1Н,м), 8,5-8,65 (2Н,м).
(III) 30 мас. раствор метоксида натрия в метаноле (3,65 мл) прибавляют к раствору, содержащему соединение В (5,76 г) в метаноле (40 мл), охлажденном до 5оС. Смесь перемешивают в течение 15 мин, а затем прибавляют насыщенный водный раствор хлористого аммония (10 мл) и простой эфир (50 мл). Для растворения любого непрореагировашего неорганического реактива прибавляют необходимое количество воды. Затем смесь экстрагируют диэтиловым эфиром (3х50 мл). Объединенные слои сушат (MgSO4) и упаривают. После очистки остатка флэш-хроматографией с элюированием этилацетатом получают метиловый эфир (2S)-2[(1S)-1-окси-1-(3-пиридил)метил]пент-4-еновой кислоты (С) (3,245 г) в виде масла; ЯМР: 2,3-2,6 (2Н,м), 2,8-2,9 (1Н,м), 3,6 (3Н,с), 4,95-5,1 (3Н, м), 5,65-5,85 (1Н,м), 7,25-7,35 (1Н,м), 7,7-7,75 (1Н,м), 8,45-8,6 (2Н,м).
(IV) Раствор, содержащий соединение С (3,88 г) в ТГФ (10 мл), прибавляют по каплям к охлажденной суспензии алюмогидрида лития (767 мг) в ТГФ (50 мл) с такой скоростью, при которой бы температура удерживалась при -10оС. После завершения прибавления смесь перемешивают при 5оС в течение 4 ч. Прибавляют этилацетат (20 мл) с последующим добавлением водного насыщенного раствора хлористого аммония (10 мл и 10 мл). Смесь экстрагируют этилацетатом (3х50 мл). Объединенные экстракты сушат (MgSO4) и упаривают. Остаток очищают флэш-хроматографией, используя в качестве элюента этилацетат, который постепенно разводят метанолом, получая смесь метанол/этилацетат (1:9). Получают (1S,2R)-2-аллил-1-(3-пиридил)-1,3-пропандиол (D) (2,69 г) в виде масла; ЯМР: 1,65-1,8 (1Н,м), 1,95-2,15 (2Н,м), 3,15-3,45 (2Н,м), 4,4-4,5 (1Н,м), 4,75-5,0 (3Н,д, I 7 Гц), 5,6-5,85 (1Н,м), 7,3-7,4 (1Н,м). 7,65-7,7 (1Н,м), 8,4-8,5 (2Н,м).
(V) пара-толуолсульфокислоты моногидрат (2,91 г) прибавляют к раствору, содержащему соединение (D) (2,68 г) в 2,2-диметоксипропане (15 мл), а затем смесь перемешивают в течение 18 ч. Прибавляют триэтиламин (10 мл) с последующим разделением смеси при добавлении простого эфира (50 мл) и воды (20 мл). Органический слой сушат (MgSO4) и упаривают. Остаток очищают флэш-хроматографией и, используя в качестве элюента смесь этилацетат/гексан (1:1, об/об), получают (4S, 5R)-5-аллил-2,2-диметил-4-(3-пиридил)-1,3-диоксан (E) (2,39 г) в виде масла; ЯМР: 1,53 (3Н,с), 1,55 (3Н,с), 1,6-1,75 (1Н,м), 1,9-2,0 (1Н,м), 2,3-2,5 (1Н,м), 3,85-4,2 (2Н,м), 4,9-5,0 (2Н,м), 5.27 (1Н,д, I 3 Гц), 5,45-5,7 (1Н,м), 7,25-7,35 (1Н,м), 7,65-7,7 (1Н, м), 8,5-8,6 (2Н, м).
(VI) Озон пропускают через раствор, содержащий аллильное соединение (E) (530 мг) в метаноле (30 мл), охлажденный до -78оС, пока не появится голубая окраска. Смесь продувают через аргон до того как введут метилсульфид (1,6 мл). Реакционную смесь затем перемешивают при комнатной температуре в течение 18 ч с последующим упариванием в вакууме и разделением ее при использовании диметилового эфира (50 мл) и воды (20 мл). Органический слой сушат (MgSO4) и упаривают. После очистки остатка флэш-хроматографией при элюировании смесью метанол/метиленхлорид (5:95, об/об) получают 2-[4S,5R)-2,2-диметил-4-(3-пиридил)-1,3-диоксан-5-ил]ацеталь- дегид (F) в виде масла; ЯМР: 1,53 (3Н, с), 1,55 (3Н,с), 2,15-2,4 (2Н,м), 2,85-2,95 (1Н,м), 3,8-3,85 (1Н, м), 4,25-4,35 (1Н,м), 5,28 (1Н,д, I3 Гц), 7,25-7,7 (2Н,м), 8,5-8,6 (2Н,м), 9,6 (1Н,с).
Примечание: оптическую чистоту оценивают как 99% методом протонного ЯМР при введении (R)-(-)-2,2,2-трифтор-1-(9-антрил) этанола и наблюдении в области 2,7-2,9 (дельта), где обнаружены 4 дублета, с центром при 2.77, 2.71. 2.82 и 2.85 (1Н. СН-СНО).
(VII) Ацетальдегид (F) затем превращают в 4(Z)-6-[(4S, 5R)-2,2-диметил-4-(3-пиридил)-1,3-диоксан-5-ил]гексеновую кислоту, имеющую 25[α]D (EtOH, с 0,465) и ЯМР данные, совпадающие с полученными по рецепту, описанному в примере 1, используя аналогичную методику, которая раскрыта в первой части примера 1.
П р и м е р 41. Приводимые ниже в качестве иллюстративного примера формы лекарственных дозировок включают составы в виде таблетки, капсулы, состава для вливаний и аэрозоли, которые можно получить традиционными методами, известными в области фармации и предназначенными для лечения и профилактики болезней. (а) Таблетка I мг/таблетка Соединение Z 1,0 Лактоза 93,25 N2-кроскармелоза 4,0 Паста из кукурузного крахмала 0,75 Стеарат магния м/об водная паста 1,0 (b) Таблетка II мг/таблетка Соединение 50 Лактоза 223,75 N2-кроскарлилоза 6,0 Кукурузный крахмал 15,0 Поливинилпирролидон (5% м/об водная паста) 2,25 Стеарат магния 3,0 (d) Капсула мг/капсула Соединение 10 мг Лактоза 488,5 Стеарат магния 1,5 (е) Состав для инъекций (50 мг/мл) Соединение Z (в форме свободной кислоты) 5,0% м/об 1М раствор гидроокиси натрия 15,0% об/об Соляная кислота (для доведения рН до 7,6) Полиэтиленгликоль 400 4,5% м/об Стерильная вода до 100% (f) Состав для инъекций II (10 мг/мл) Соединение Z (в форме свободной кислоты) 1,0% м/об Фосфат натрия ЕР 3,6% м/об 0,1М гидроокись нат- рия разбавитель 15,0% об/об Стерильная вода до 100% (g) Состав для инъекций III (1 мг/мл, за- буференный до рН 6) Соединение Z (в форме свободной кислоты) 0,1% м/об Фосфат натрия ЕР 2,26% м/об Лимонная кислота 0,38% м/об Полиэтиленгликоль 400 3,5% м/об Стерильная вода до 100% (h) Аэрозоль I мг/мл Соединение Z 10,0 Триолеат сорбита 13,5 Трихлорфторметан 910,0 Дихлордифторметан 490,0 (I) Аэрозоль II мг/мл Соединение Z 0,2 Триолеат сорбита 0,27 Трихлорфторметан 70,0 Дихлордифторметан 280,0 Дихлортетрафторметан 1094,0 (j) Аэрозоль III мг/мл Соединение Z* 2,5 Триолеат сорбита 3,38 Трихлорфторметан 67,5 Дихлордифторметан 1086,0 Дихлортетрафторэтан 191,6 (К) Аэрозоль IV Соединение Z* 2,5 Соевый лецитин 2,7 Трихлорфторметан 67,5 Дихлордифторметан 1086,0 Дихлортетрафторэтан 191,6
П р и м е ч а н и е. *Соединение Z представляет соединение формулы (I) или ее соль, например соединение формулы (I), раскрыток в любом из представленных примеров и особенно описано в примерах 4, 8, 11 или 28.
Составы в форме таблеток (а)-(с) могут содержать энтеросолюбильную оболочку, выполненную традиционными методами, например, для покрытия из фталата триацетата целлюлозы. Аэрозольные составы (h)-(k) можно использовать со стандартными распылителями аэрозолей в определенной дозе, а суспендирующие вещества триолеат сорбита и соевый лецитин можно заменить на предпочтительное суспендирующее средство, например монолеат сорбита, по- луторный олеат сорбита, полисорбит 80, полиглицериновый олеат или олеиновая кислота.
Данные микроанализа приведены в табл.1.
В табл.2 приведены данные биологических испытаний, которые демонстируют способность испытуемых соединений действовать в качестве антагонистов ТХА2 и ингибиторов синтазы ТХА2.
Антагонизм ТХА2 демонстрировали с помощью испытания на агрегацию пластинок крови, основанное на испытании, описанном Born (Nature, 1961, 194, 927-929) и включающем:
(i) агрегацию богатой тромбоцитами, citrated плазмы человека с помощью добавления подражательного ТХА2 средства u 46619 таким образом, чтобы получить кривую реакцию на дозу;
(ii) получение кривой реакции на дозу для стимулированной u 46619 агрегации тромбоцитов в присутствии возрастающих количеств испытываемого соединения (обычно в области 10-5-10-10М);
(iii) расчет величины Кв, показывающей возможность антагонизма ТХА2для испытываемого вещества, среднего по нескольким концентрациям из рассчитанной 50% величины агрегации u 46619 в присутствии и отсутствии испытуемого вещества.
В таблице результаты испытаний представлены в виде величины рА2, которую определяют, как отрицательный логарифм величины Кв.
Ингибирование синтазы ТХА2 демонстрировали с использованием стандартного способа испытаний in vitro, описанного Howarth et alia (Biochem. Soc.Transections, 1982, 10, 239-240), используя микросоматический, содержащий ТХА2 синтазу препарат тромбоцитов человека, и используя количественный тонкослойный радиохроматографический метод оценки превращения [I-14C]арахидоновой кислоты в ТХА2 метаболический тромбоксан В2(ТХВ2).
В табл.2 результаты приведены в единицах μ М.
Прилагаются данные биологических испытаний для характерных соединений формулы (I) и известных соединений аналогичной структуры. Известные соединения раскрыты в примерах ЕР-А-201351 (5), 201354 (4) и 202086 (6).
Соединения формулы (I) представляют собой антагонисты ТХА2 и ингибиторы синтазы ТХА2. Эту желаемую биологическую активность нельзя было предсказать ни из одной цитируемой ссылки. В частности, нельзя было предсказать, что при замещении 4-фенильного заместителя на 4-пиридильный заместитель сохранится активность антагониста тромбоксана, и возникнет новая активность ингибирования синтезы тромбоксана. Как подтверждают данные испытаний, известные соединения неактивны в качестве ингибиторов синтазы тромбоксана.
В табл.3 приведены сравнительные данные для соединений примеров из ЕР-А-201351, 201354 и 202086.
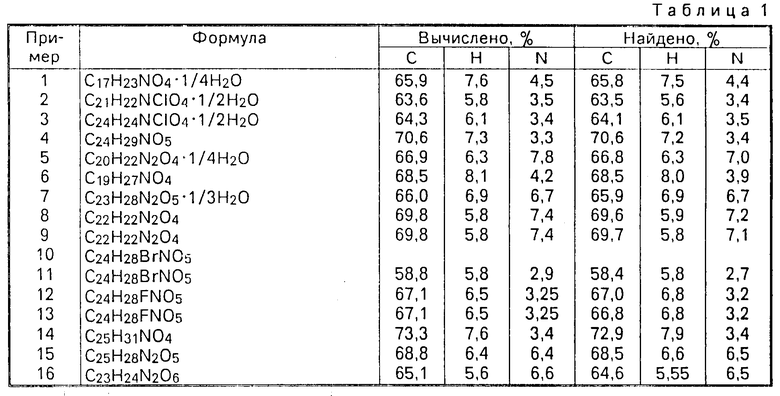
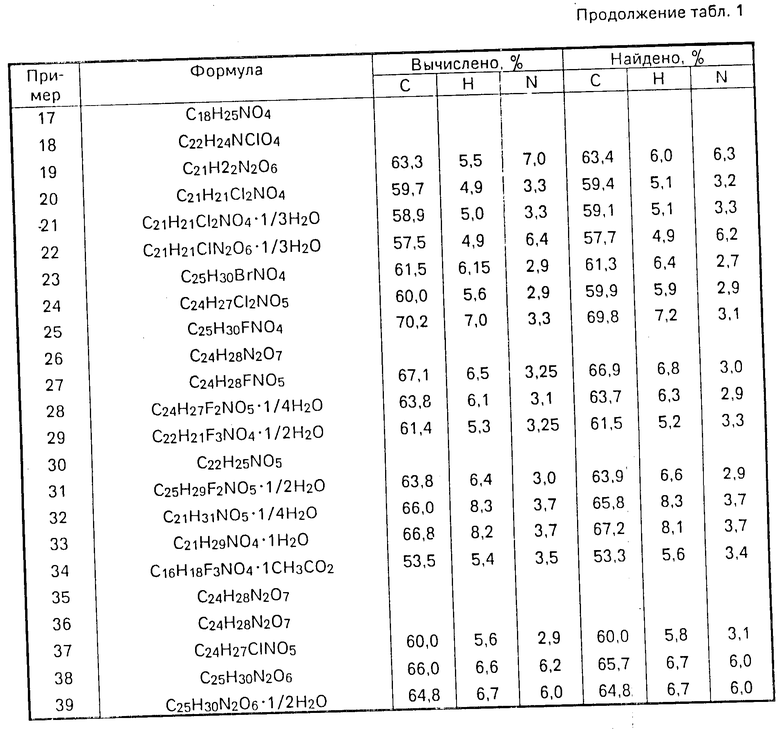
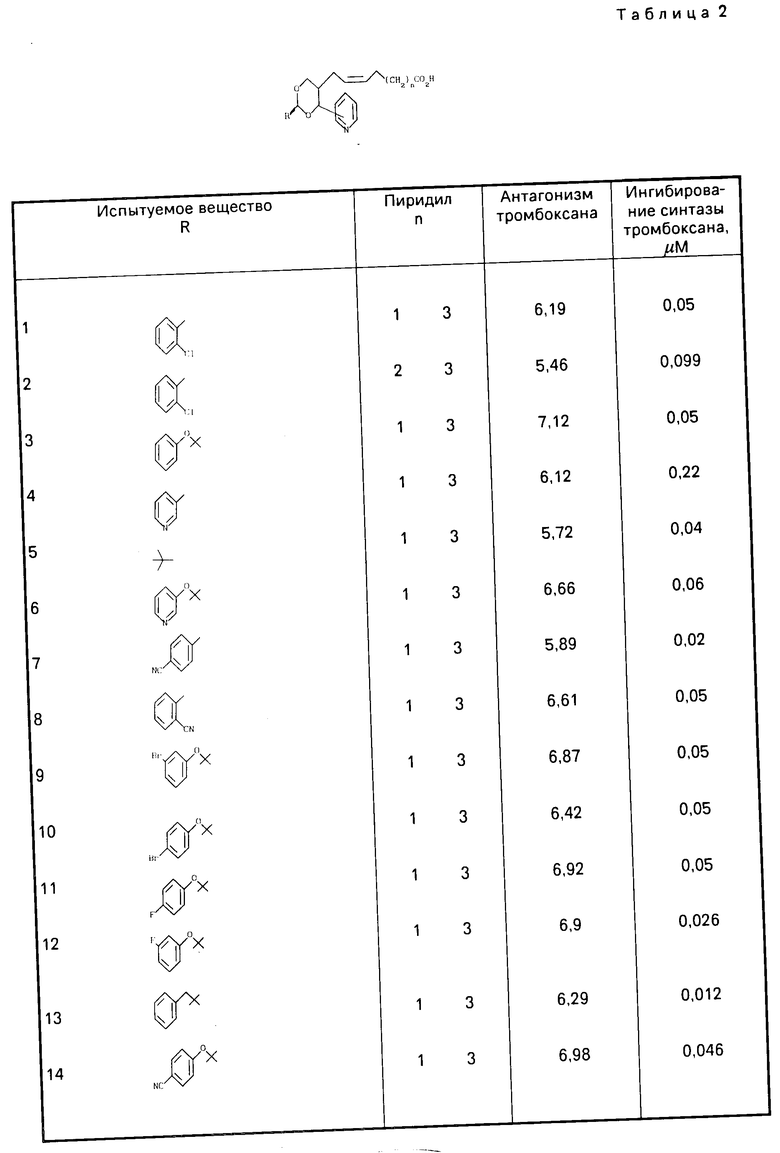
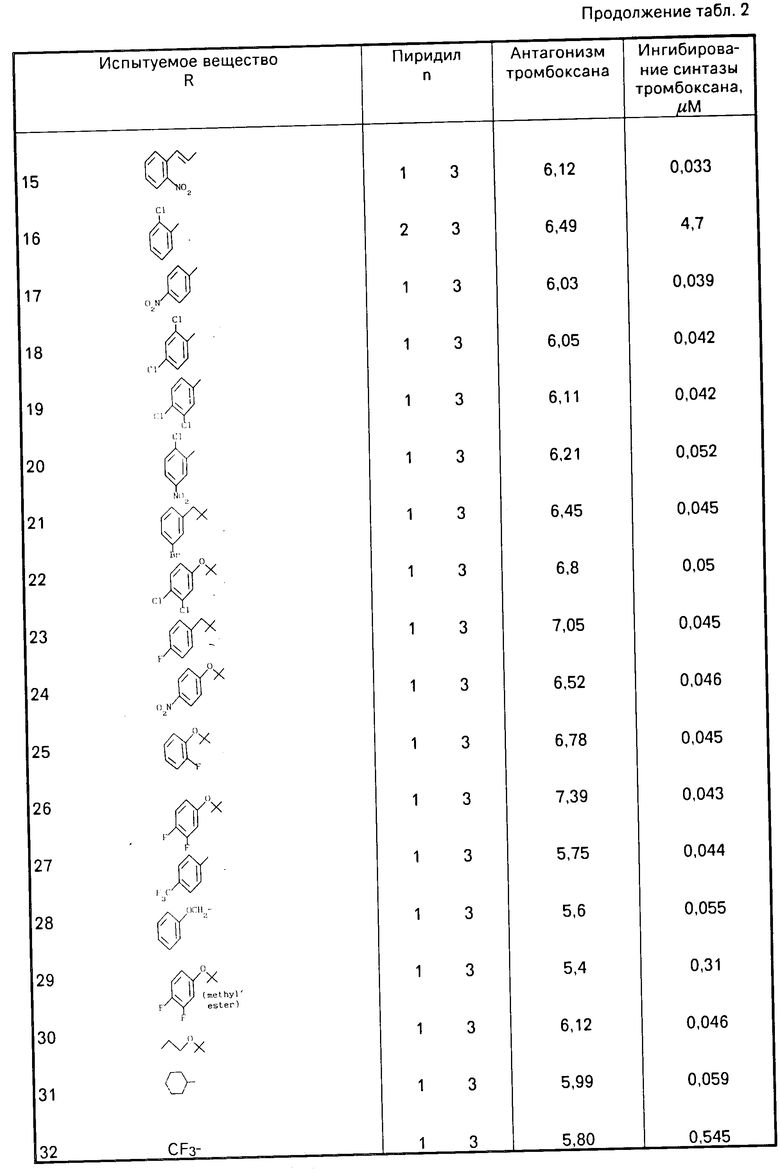
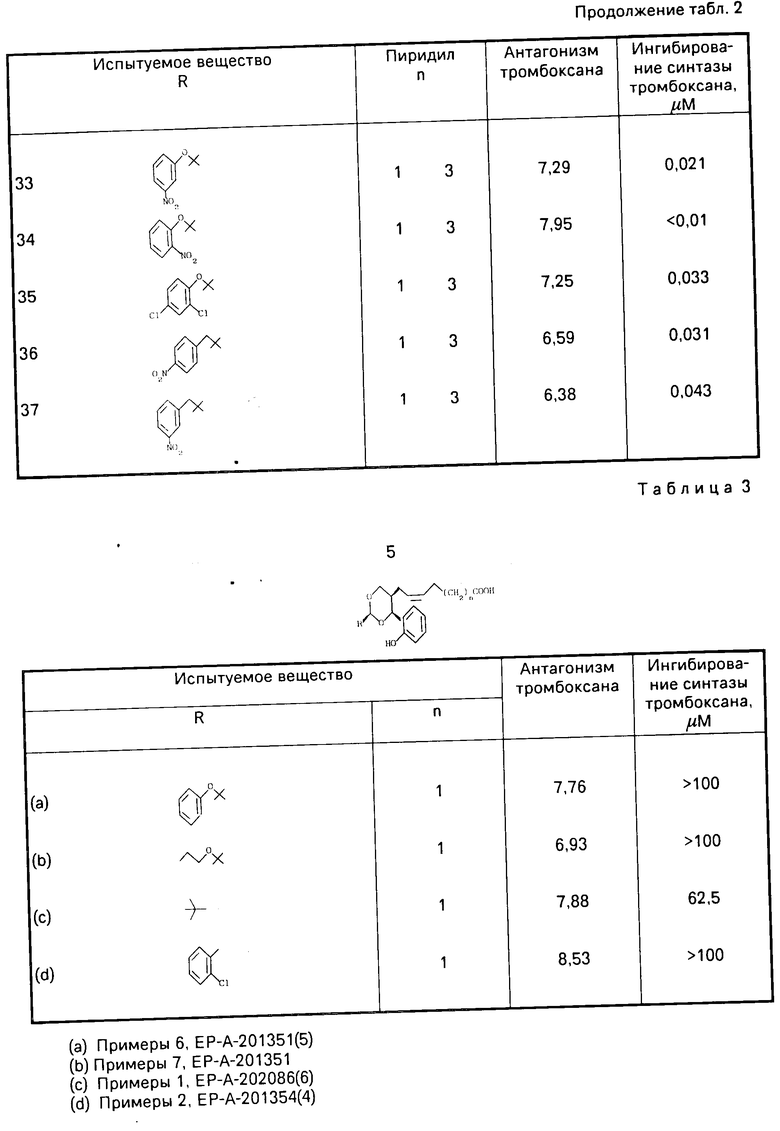
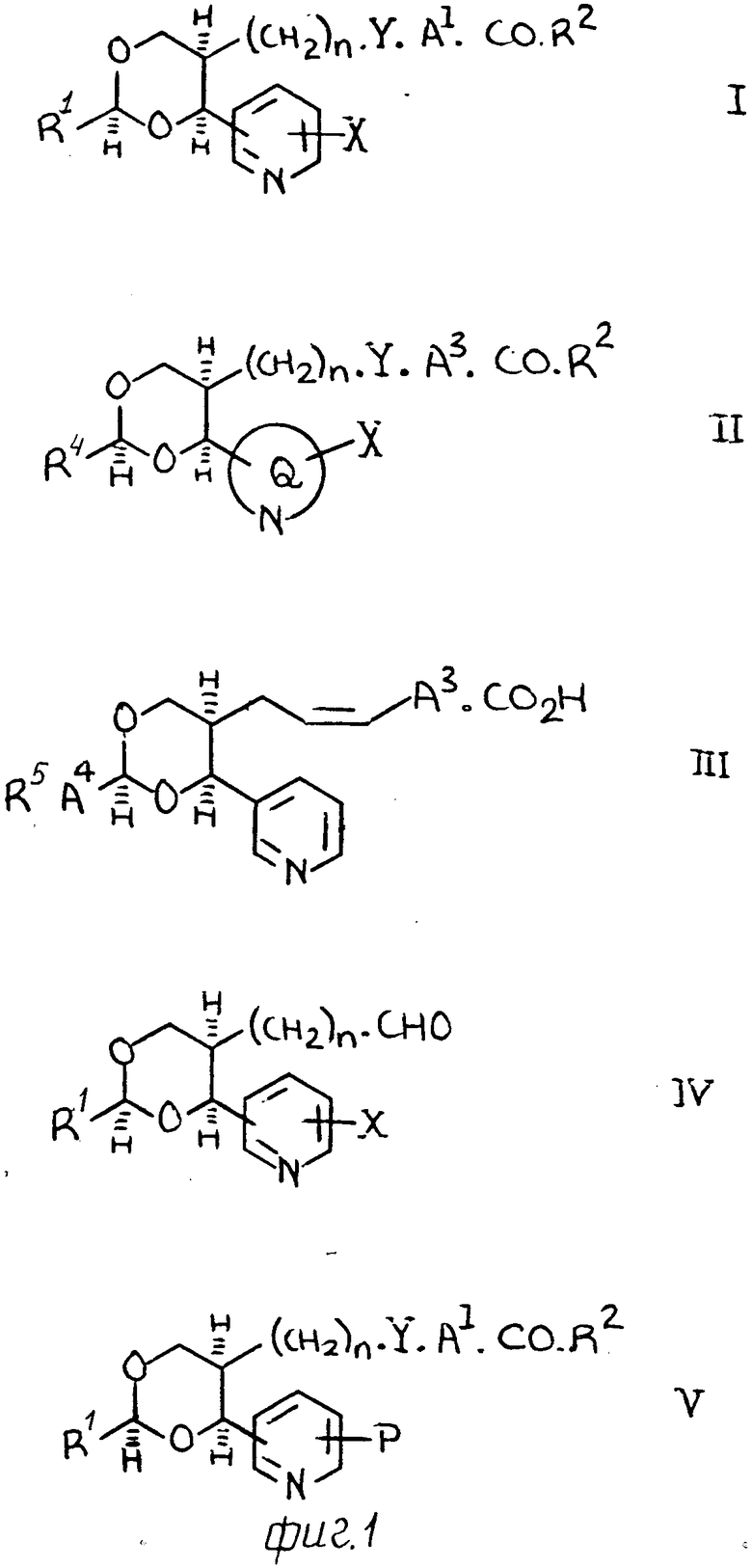
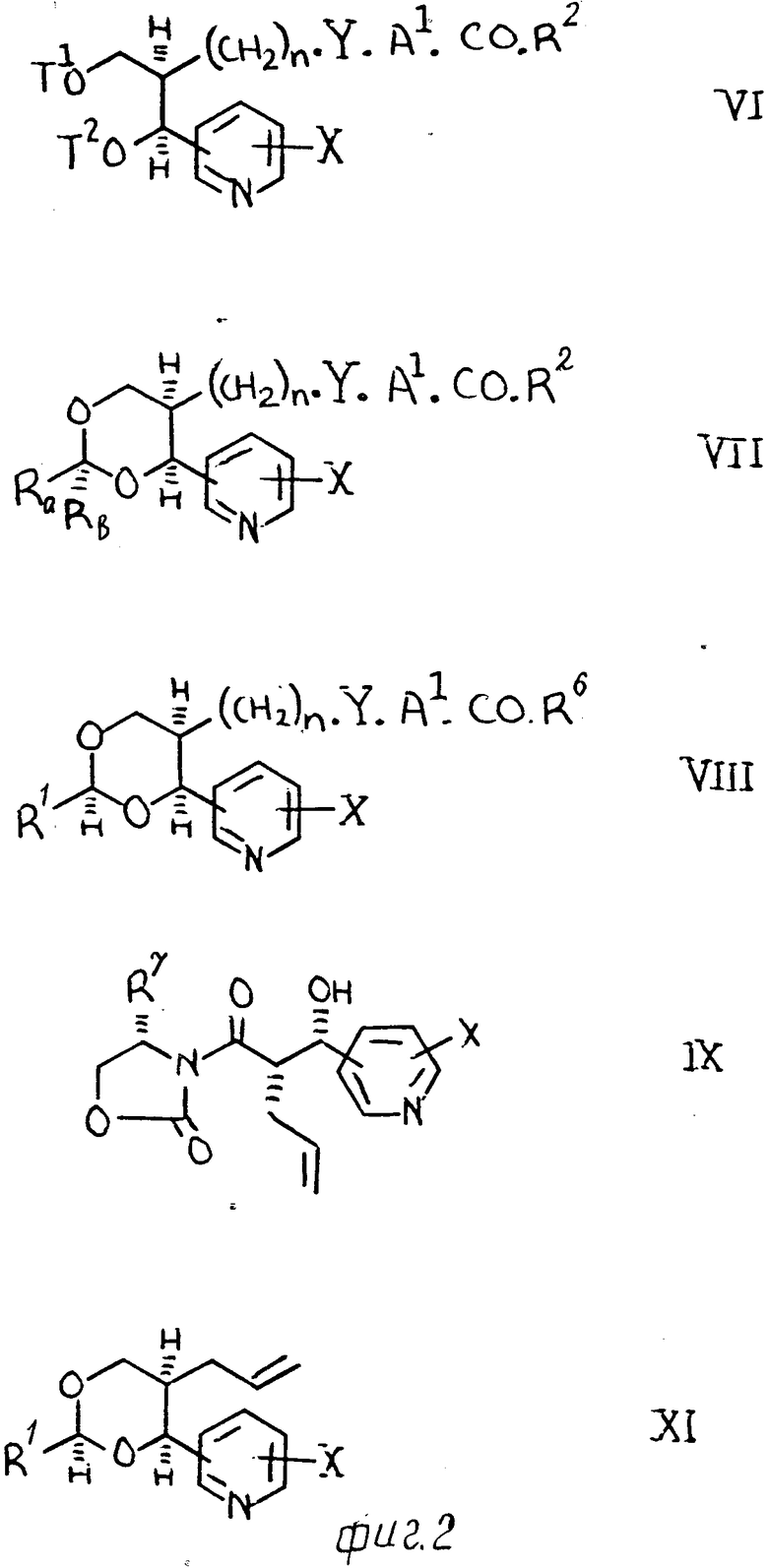
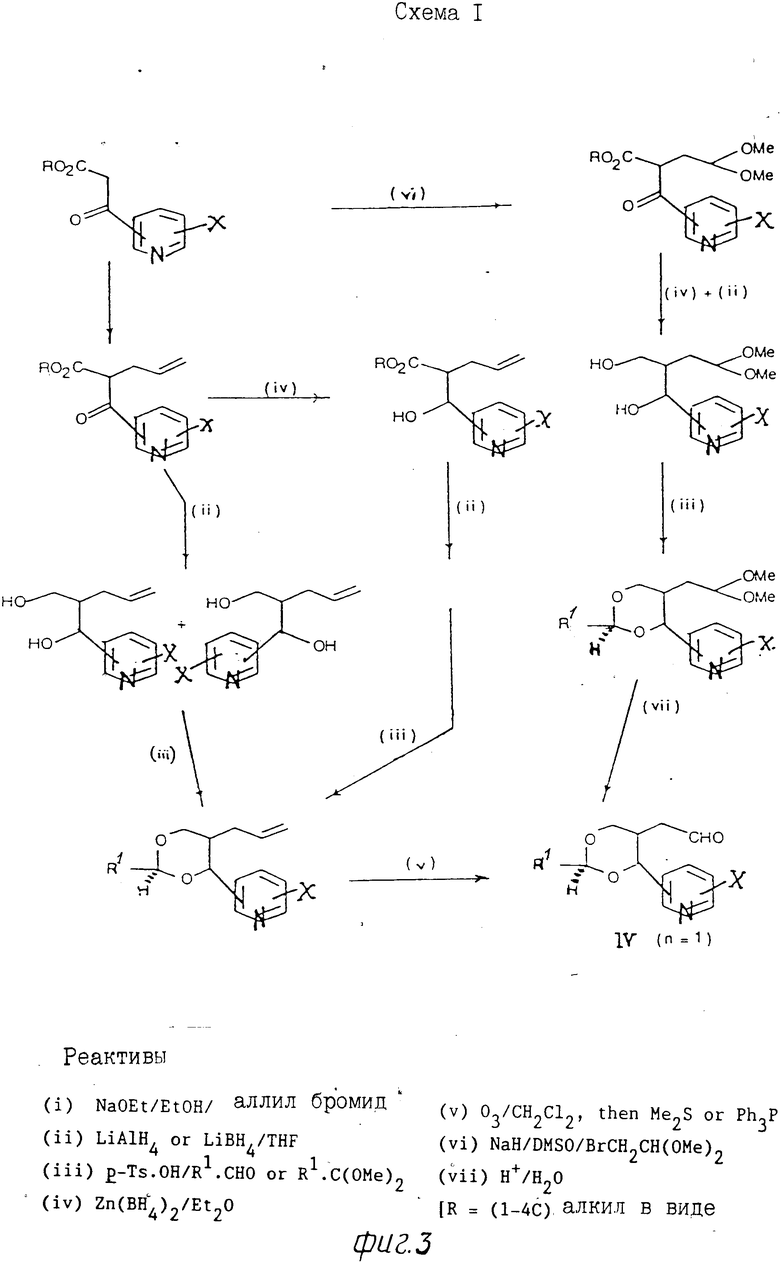
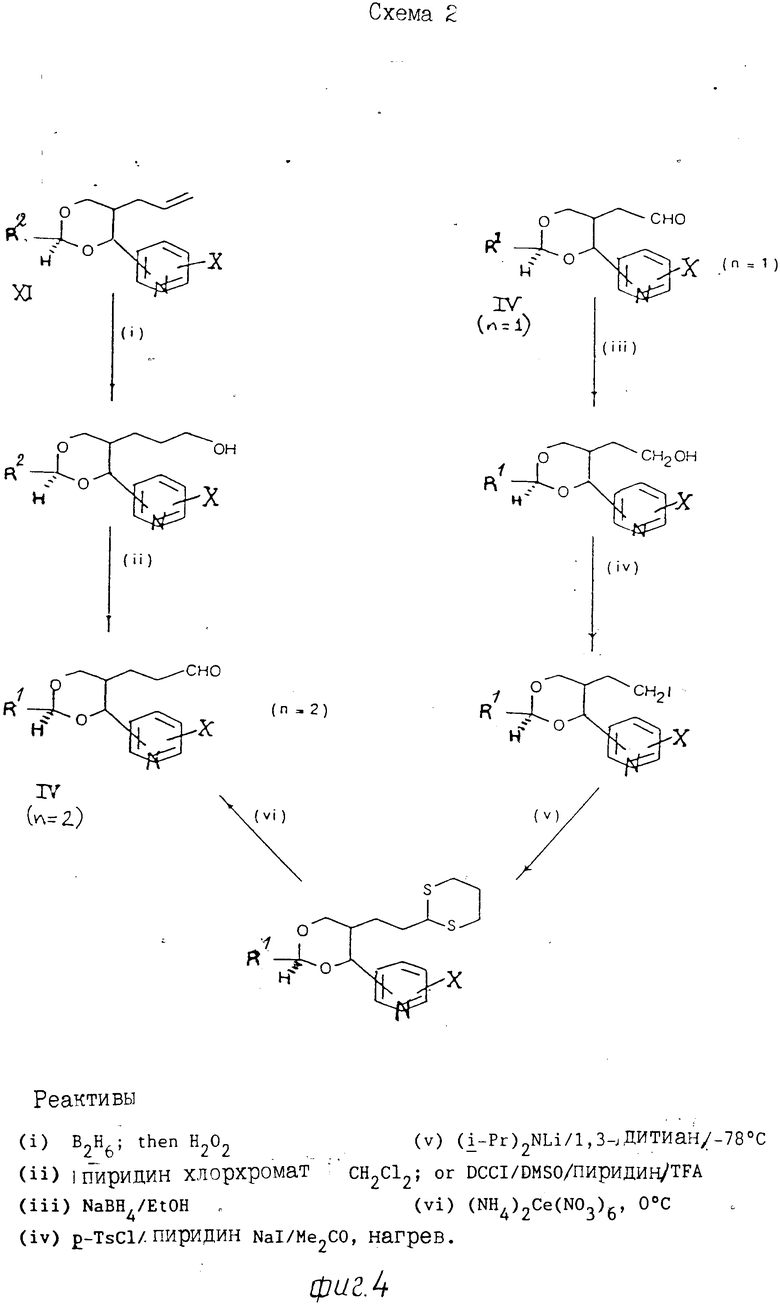
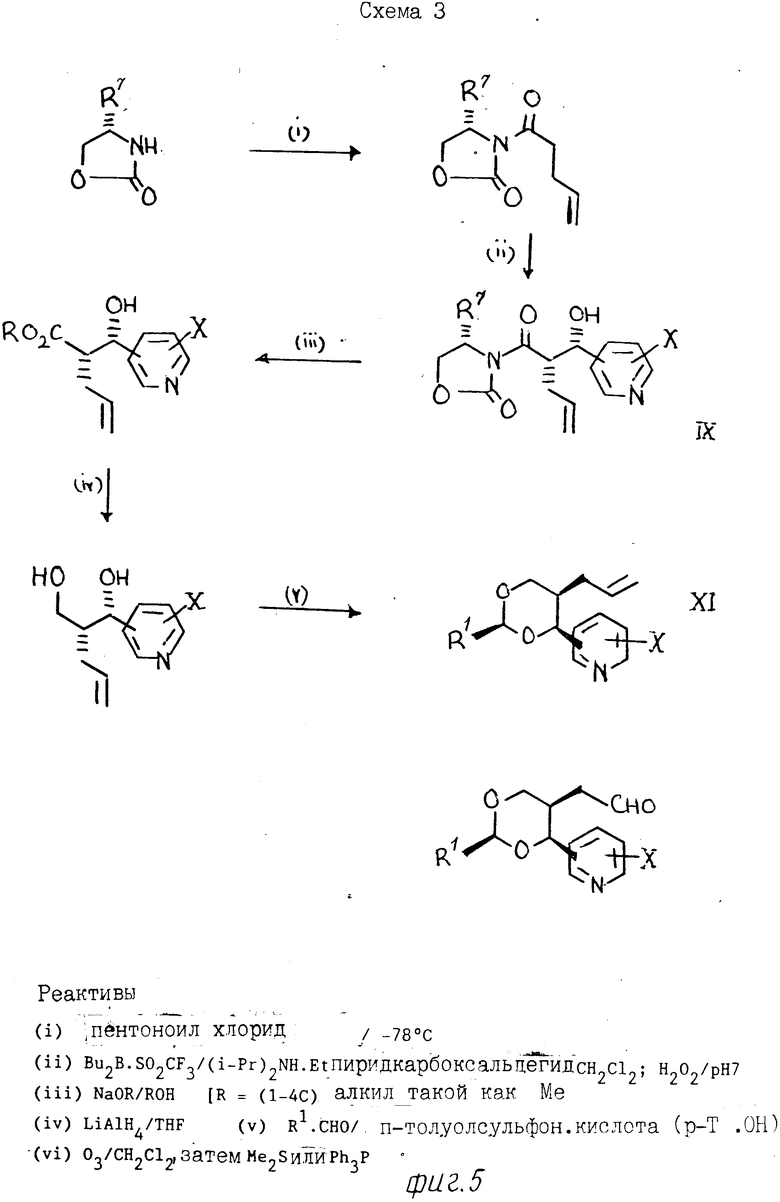
Использование: в медицине для лечения теплокровных животных, например человека, в частности в качестве антогонистов TXA2 и ингибитора синтетазы TXA2 Сущность изобретения: продукт 1,3-диоксановые производные алкеновой кислоты ф-лы 1, где A1-C1-C6 алкилен; R1-C1-C6 -алкил, трифторметил, C3-C6 -циклоалкил, C1-C4 -алкокси- C1-C4 -алкил или группа; R3-A2 при R3 -пиридил, фенил, не- или моно- или дизамещенный водородом, трифторметилом, нитро или цианогруппой, а A2 C1-C6 -алкилен, окси- C1-C6 -алкилен, C2-C6 -алкенилен или прямая связь с R3 R2 -оксигруппа, или физиологически приемлемый спиртовой остаток; X -водород, гидроксил или C1-C4 -алкоксигруппа; Y винилен, n 1 или 2, или их фармацевтически приемлемая соль. У веществ ф-лы 1 величина pA2, показывающая возможность антогонизма TXA2 составляет 5,4 7,95. Ингибирование синтетазы проводят с использованием микросоматического метода и его результаты составляют 0,05 0,31 ммкм. 5 ил.
где А1 C1-C3-алкилен;
R1 C1-C6-алкил, трифторметил, C3-C6-циклоалкил, или C1-C4-алкокси, C1-C4-алкил, или группа общей формулы R3 · А2-,
где R3 пиридил, фенил или фенил, содержащий 1 или 2 заместителя, выбранных из группы: галоген, трифторметил, нитро- или цианогруппа;
А2 C1-C6-алкилен, окси-C1-C6-алкилен, C2-C6-алкенилен или прямая связь с R3;
R2 гидроксигруппа или физиологически приемлемый спиртовый остаток;
Х водород, гидрокси- или C1-C4-алкоксигруппа;
Y винилен;
n 1 или 2,
или их фармацевтически приемлемая соль.
R3 · A2-,
где R3 3-пиридил, фенил или фенил, содержащий 1 или 2 заместителя, выбранных из группы: фтор, хлор, бром, трифторметил, нитро- или цианогруппа;
А2 метилен, этилен, триметилен, изопропилиден, 1,1-диметилэтилен, 3,3-пентилидин, оксиметилен, окситетраметилен, 1-окси-1-метилэтил, 2-окси-1,1-диметилэтил, винилен, 1,3-пропенилен, 1,4-бутен-2-илен или прямая связь с R3;
R2 гидрокси-, C1-C6-алкоксигруппа, возможно содержащая окси- или C1-C4-алкокси заместитель, или R2 - фенокси- или бензилоксигруппа, каждая из которых, возможно, содержит 1 или 2 заместителя, выбранных из группы; галоген, C1-C4-алкил и C1-C4-алкоксигруппа;
Х водород, гидрокси-, метокси- или этоксигруппа.
где A3 C1-C4-алкилен;
R5 пиридил, фенил или фенил, содержащий 1 или 2 заместителя, выбранных из галогена, трифторметила, нитро- или цианогруппа;
А4 C1-C4-алкилен, окси-C1-C4-алкилен или прямая связь с R5,
и его фармацевтически приемлемые соли.
| СПОСОБ ОЧИСТКИ ТРИФТОРИДА КАЛИЯ | 0 |
|
SU202086A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Пневматический водоподъемный аппарат-двигатель | 1917 |
|
SU1986A1 |
