Изобретение относится к новым производным алкеновой кислоты, содержащим пиридин, точнее к новым 1,3-диоксан-5-илалкеновым кислотам, содержащим остаток пиридина в положении 4 1,3-диоксанового кольца. Алконовые кислоты по изобретению обладают ценными фармацевтическими свойствами и изобретение включает фармацевтические композиции, содержащие новые кислоты, и способы получения и медицинского использования указанных новых кислот.
Известно, что метаболит арахидоновой кислоты тромбоксан А2 (далее обозначается "ТХА2") является мощным вазоконстриктером и потенциальным агрегатором тромбоцитов. ТХА2 представляет собой также эффективный сократитель гладкой мускулатуры бронхов и трахеи. Поэтому ТХА2 может участвовать в различных болезнях, например в ишемической болезни сердца, в частности в инфаркте миокарда, ангине, в цереброваскулярных заболеваниях, таких как перемежающаяся церебральная ишемия, миграина и удар, в заболеваниях периферических сосудов, в частности в атеросклерозе, микроангиопатии, гипертонии и в дефектах свертывания крови, обусловленных липидным дисбалансом.
Считается, что ТХА2 оказывает физиологическое действие через тромбоксановый рецептор, и через этот рецептор другие простаноидные сократительные соединения, такие как простагладины Н2, F2-альфа и простагландин D2, являющиеся производными арахидоновой кислоты, могут оказывать сокращающее действие. Существуют два принципиальных способа предотвращения действия ТХА2. Первый состоит во введении фармакологического агента, преимущественно занимающего тромбоксановый рецептор, но не вызывающего сокращения, которое следует за связыванием ТХА2 или простагландинов Н2, F2 альфа и/или D2). Действие такого агента называют ТХА антагонистическим. Второй подход состоит во введении фармакологического агента, ингибирующего один или больше фермент, участвующий в образовании ТХА2, в частности агента, ингибирующего фермент, известный как тромбоксаносинтаза (ТХА2-синтаза). Такой агент называют ингибитором ТХА синтазы. В соответствии с этим агенты, обладающие ТХА2 антагонистическими и ингибирующими ТХА2 синтазу свойствами могут рассматриваться как терапевтически эффективные при лечении одного или больше из вышеуказанных заболеваний или других заболеваний, в которых участвует ТХА2. Кроме того, агенты, являющиеся ТХА2 антагонистами, могут оказаться эффективными и при лечении заболеваний, в которых участвуют простагландины Н2, F2 альфа и/или D2, в частности при лечении астматических и воспалительных заболеваний. Хотя 1,3-диоксановые ТХА2-антагонисты известны [1] как и некоторые ингибиторы ТХА2-синтазы [2] однако соединения, обладающие обоими свойствами в приемлемой степени до сих пор не получены.
Тем не менее, что является основой изобретения, некоторые 1,3-диоксан-5-илалкеновые кислоты формулы (1), содержащие пиридильный остаток в положении 4 1,3-диоксанового кольца, являются хорошими ингибиторами ТХА2-синтазы, обладают также существенными ТХА2 антагонистическими свойствами и являются пригодными фармацевтическими агентами.
В изобретении предлагаются производные 1,3-диоксаналкеновой кислоты формулы (1) (вынесена вместе с другими химическими формулами), где n является целым числом, равным 1 или 2; Х представляет собой водород, окси-группу, (1-4С) алкокси-группу ((1-4С) алкокси-группа группа, содержащая 1-4 атома углерода); Y является метиленокси-группой, виниленом или этиленом; А1 представляет собой (1-6С) алкилен;
а) R2 является водородом; R1 нафтилом или фенилтио(1-6С)-алкилом, возможно содержащим 1 или 2 заместителя, выбранных из галогена, цианогруппы, нитрогруппы (1-4С)-алкила, (1-4С) алкокси-группы и трифторметила, либо R1 представляет собой группу формулы R3, А2-, где R3является фенилом, замещенным (1-4С)-алкилом, (1-4С)-алкокси-группой, окси-группой, (2-5С), алкенилом (1-4С), алкилтио-группой, (1-4С) алкилсульфинилом, (1-4С) алкилсульфонилом, (2-5С)-алканоилом, карбокси-группой, (1-4С) алкокси (карбонилом) (N-/-1-4С) алкил/карбамоилом, (1-5С) алканоиламиногруппой и (1-4С)-алкилом, замещенным (1-4С) алкокси-группой, цианогруппой, карбоксигруппой или (1-4С) алкокси) карбонилом и фенилом, возможно содержащим второй заместитель, выбранный из (1-4С)-алкила, (1-4С)-алкокси-группы, галогена, трифторметила, нитрогруппы и цианогруппы; или R3 представляет собой тиенил или фурил, возможно замещенный 1-2 заместителями, независимо выбранными из галогена, (1-4С) алкила, нитрогруппы и циагруппы; и А2 представляет собой (1-6С)-алкилен, окси (1-6С)-алкилен или (2-6С)-алкилен или (2-6С)-алкилен, где до трех любых атомов углерода может быть частично или полностью фторировано, либо А2 является прямой связью с R3, либо R1 является группой формулы Q2.А3.Q1, где Q1 и Q2 ароматические остатки, один из которых представляет собой остаток бензола, а другой является бензольными, пиридиновым или нафталиновым остатком, любой из которых возможно замещен галогеном, цианогруппой, нитрогруппой (1-4С) алкилом, (1-4С) алкокси-группой или трифторметилом, а А3 представляет собой окси-группу, тиогруппу, сульфинил, сульфонил, карбонил, карбамоил, иминокарбонил, уреидогруппу, (1-6С) алкилен окси (1-6С) алкилен, (2-6С) алкенилен или прямую связь между и
(b) R1 представляет собой пентафторэтил и R2 является водородом, либо как R1, так и R2 являются трифторметиламин или
(с) R1 и R2 независимо представляют собой алкилы или вместе образуют алкилен, где R1 и R2 вместе содержат 5-9 атомов углерода; R4представляет собой окси-группу, остаток физиологически приемлемого спирта или (1-4С) алкансульфонамидогруппу: или их фармацевтически приемлемые соли.
Следует отметить, что соединения формулы (1) имеют асимметрические атомы углерода и могут существовать и выделяться в рацемической и оптически активной формах. Изобретение включает как рацемические, так и оптически активные формы (или их смеси), способные выступать антагонистами по отношению к одному или больше действию ТХА2 и ингибировать синтез ТХА2, при этом в данной области хорошо известно как получать индивидуальные оптические изомеры (например, синтезом из оптически активных исходных соединений или разделением рацемической формы) и как определять ТХА2 антагонистические свойства и свойство ингибировать ТХА2 синтазу с использованием одного или больше стандартного испытания, которые будут описаны ниже.
Понятно, что группы в положениях 4 и 5 (и в положении 2, если
R2 является водородом) 1,3-диоксанового остатка в формуле (1) имеют цис-относительную стереохимию, также как и группы, присоединенные к Y, если это винилен (т.е. последние соединения существуют в виде "Z"-изомера). Кроме того, хотя в прилагаемых химических формулах указаны конкретные конфигурации, однако в действительности это необязательно соответствует абсолютной конфигурации.
Следует понимать, что общий термин "алкилен" включает как разветвленные, так и неразветвленные алкиленовые группы, такие как этилен и этилиден и другие общие термины имеют тот же смысл. Однако если используется конкретный термин, такой как "бутил", то это соответствует неразветвленной или "нормальной" бутильной группе, а изомеры с разветвленной цепью, такие как "трет-бутил" указываются при необходимости конкретно.
Конкретные значения для R1 и R2, если они являются алкиламин, включают, например, метил, этил, пропил, изопропил, бутил и пентил; а если они вместе образуют алкенил, то он включает, например, тетраметилен, пентаметилен и гексаметилен, каждый из которых может содержать 1 или 2 метильных заместителя.
Конкретными значениями R1, если он представляет собой фенилтио (1-6С)-алкил, являются, например, 1-метил-1-(фенилтио)-этил или фенилтиометил, возможно замещенный так, как указано выше.
Конкретные значения заместителей, которые могут присутствовать в R1, если он является нафтилом или фенилтио (1-6С)-алкилом или в указанных выше ароматических остатках Q1 и Q2 включают, например, для (1-4С) алкила; метил и этил; для (1-4С) алкокси-группы: метокси-группу и этоксигруппу; и для галогена; фтор, хлор и бром. Конкретными значениями Х, если он представляет собой алкокси-группу, являются, например, метокси-группа или этокси-группа.
Конкретные значения заместителей, которые могут присутствовать, если вышеописанный R3 является фенилом, тиенилом или фурилом, включают, например, для (1-4С) алкила метил и этил; для (1-4С) алкокси-группы метокси-группу и этокси-группу; для (1-4С) алкокси-группы метокси-группу и этокси-группу; для галогена фтор, хлор, бром; для (2-5С) алкенила винил, 2-пропенил и 3,3-диметилпропенил; для (1-4С) алкилтиогруппы метилтиогруппу и этил тиогруппу; для (1-4) алкилсульфинила метилсульфинил и этилсульфинил; для (1-4С) алкилсульфонила метилсульфонил и этилсульфонил; для (2-5С) алканоила ацетил, пропионил, бутирил и 2-оксопропил; для (1-4С) алкокси) карбонила метоксикарбонил, этоксикарбонил и т-бутоксикарбонил; для (N-(1-4С) алкил) карбамоила N-метилкарбамоил, N-этилкарбамоил и N-пропилкарбамоил; для (1-5С) алканоиламиногруппы формамидогруппу, ацетамидогруппу и пропионамидогруппу; и для замещенного (1-4С) алкила метил, 1-этил, 2-этил или 1-, 2- или 3-пропил, содержащий в качестве заместителя (1-4С) алкокси-группу (такую как метокси- или этокси-группа), циано-группу, карбоксил или ((1-4С)алкокси)карбонил (такой, как метоксикарбонил или этоксикарбонил).
Конкретные значения для R4, если это остаток физиологически приемлемого спирта, придающий сложному эфиру биодеградируемость, выбирают, например, из (1-6С) алкила, возможно замещенного окси-группой или (1-4С) алкокси-группой, такого как метил, этил, 2-оксиэтил, 2-метоксиэтил, пропил или 3-оксипропил; фенил; бензил; последние две группы могут содержать 1 или 2 заместителя, выбранных из галогена (например, фтора, хлора, брома или иода), (1-4С) алкила (например, метила или этила) и (1-4С) алкокси-группы (такой как метокси- или этокси-группа).
Конкретными значениями для R4, если он представляет собой (1-4С) алкансульфонамидогруппу, являются, например, метансульфонамидогруппа, этансульфонами- догруппа и бутансульфонамидогруппа.
Конкретные значения А1, когда он представляет собой (1-6С) алкилен, включают например, метилен, этилен, триметилен, тетраметилен, 1,1-диметилэтилен и 1,1-диметилтриметилен, среди которых этилен и триметилен в целом предпочтительны, причем этилен особенно предпочтителен.
Если А2 является (1-6С) алкиленом, то его конкретные значения включают в себя, например, (1-4С) алкилен (такой, как метилен, этилен, триметилен, изопропилиден и 1,1-диметилэтилен) и 3,3-пентилидин; если он является (2-6С) алкениленом, то эти значения включают в себя, например, винилен, 1,3-пропенилен, и 1,4-бутен-2-илен; и если он является окси (1-6С) алкиленом, то эти значения включают в себя, например, оксиметиленокситетраметилен (т.е. группу формулы -0, (СН2)4(1-окси-1-метилэтил) (т. е. группу формулы -0,С(СН3)2 и 2-окси-1,1-диметилэтил (т. е. группу формулы -0, СН2.С(СН3)2 и понятно, что окси-связь присоединена к группе R3, а не к 1,3-диоксановому кольцу.
Конкретными значениями А2, если он содержит фторзаместители, являются, например, дифторметилен или 2,2,2-трифтор-1-окси-1-трифторметилэтил (т.е. группа формулы -0, С(СF3)2).
Конкретными значениями R3, если он представляет собой тиенил или фурил являются, например, 2-тиенил, 3-тиенил или 2-фурил, возможно с 1 или 2 заместителями, независимо выбранными из метила, этила, хлора, брома, нитрогруппы и цианогруппы.
Q1 Предпочтительно представляет собой бензольный остаток, а Q2обычно является остатком бензола, пиридина или нафталина, возможно замещенным так, как указано выше.
Конкретные значения А3, если он является (1-6С) алкиленом, включают в себя, например, (1-4С) алкилен (такой как метилен, этилен, триметилен, изопропилиден и 1,1-диметилэтилен) и пентаметилен; если он является (2-6С) алкениленом, то это, например, винилен, 1,3-пропенилен и 1,4-бутен-2-илен, если он является окси (1-6С) алкиленом, то это, например, оксиметилен, оксиэтилен и окситетраметилен (т.е. группа формулы -О,(СН2)4-) и понятно, что окси-связь может быть соединена с Q1или Q2.
Как правило, предпочтительным значением для n является 1, для Х водород, для Y-цис-винилен и для А1 этилен и для R4 окси-группа.
Конкретными значениями R1 и R2 являются, например, следующие:
а) R1 им R2 оба являются трифторметиламин;
b) R1 является тиенилом или фурилом, возможно замещенным галогеном, цианогруппой или нитрогруппой, а R2 представляет собой водород;
с) R1 является фенокси (1-4С) алкилом (в частности, 1-метил-1-феноксиэтилом), фенильный остаток которого содержит один заместитель, выбранный из (1-4С) алкила и (1-4С) алкокси-группы, и возможно второй заместитель, выбранный из (1-4С) алкила, (1-4С) алкокси-группы, галогена, нитрогруппы, трифторметила и цианогруппы, а R2представляет собой водород;
d) R1 является фенилтио (1-4С)-алкилом (в частности, 1-метил-1-фенилтиоэтилом), фенильный остаток которого возможно содержит 1 или 2 заместителя, независимо выбранных из (1-4С)алкила, (1-4С)-алкокси-группы, галогена нитрогруппы, трифторметила и цианогруппы, а R2 является водородом;
е) R1 является нафтилом, возможно замещенным 1 или 2 заместителями, выбранными из галогена (1-4С) алкила и нитрогруппы, а представляет собой водород;
f) R1 является бензилфенилом, бензилоксифенилом, (пиридилметокси)фенилом, (нафтилметокси)фенилом, феноксифенилом и (феноксиметил)фенилом.
Группа соединений по изобретению, представляющих особый интерес, включает соединения формулы (2), где R1 такой, как определено выше; А4представляет собой (1-4С) алкилен; Х1 является водородом или окси-группой; и R5 является нафтилом или тиенилом, возможно замещенным цианогруппой, нитрогруппой, галогеном, (1-4С) алкилом, либо R5 является группой формулы R6.А5-, где R6 является фенилом, замещенным первым заместителем, выбранным из (1-4С) алкила, (1-4С) алкокси-группы, окси-группы (2-5С) алкенила, (1-4С) алкилтиогруппы, (1-4С) алкилсульфинила), (1-4С) алкилсульфонила, (2-5С) алканоила, карбоксила, //1-4С/ алкокси/карбонила, /N/1-4С(алкил)карбамоила, (1-5С)алканоила- миногруппы и /1-4С/-алкила, где последний содержит /124С/алкокси-группу, цианогруппу, карбоксил или (1-4С/алкокси/ карбонил, где указанный фенил может, иметь второй заместитель, выбранный из (1-4С) алкила, (1-4С) алкокси-группы, галогена, трифторметила, нитрогруппы и цианогруппы, а А5представляет собой (1-4С) алкилен, окси (1-4С) алкилен или прямую связь с R5, а также их фармацевтически приемлемые соли.
Конкретные значения заместителей, которые могут присутствовать как часть R5 или R6 включают в себя, например, те, которые указаны выше для R1. Конкретные значения для R6 включают в себя, например, фенил, имеющий первый заместитель, выбранный из (1-4С) алкила (например метила, этила), (1-4С) алкокси-группы (например, метокси-группы) или окси-группы, возможно со вторым заместителем, выбранным из нитрогруппы, галогена (например, фтора, хлора и брома) или трифторметила.
Если А4 представляет собой, например, (1-4С) алкилен, то его конкретные значения включают например, этилен, триметилен и 1,1-диметилэтилен, среди которых, как правило, предпочтительны этилен и триметилен.
Конкретные значения для А5 включают в себя, например, те, которые указывались выше для А2, если тот представляет собой прямую связь, (1-4С) алкилен или окси (1-4С) алкилен, например прямую связь изопропилен, 1,1-диметилэтилен, 1-окси-1-метилэтил (т.е. группу формулы -О.С(СН3)2-).
Конкретные значения R5 включают в себя, например, 1-нафтил, 2-нафтил, 2-хлор-1-нафтил, 2-тиенил, 3-тиенил, 5-циано-2- тиенил, 5-бром-2-тиенил, 4-бром-2-тиенил, 4-хлор-2-тиенил, 5-хлор-2-тиенил, 2-фурил, 5-бром-2-фурил, 1-(4-метоксифенокси)-1-метилэтил, 1-(4-т-бутилфенокси)-1- метилэтил,
1-(2-метоксифенокси)-1-метилэтил,
1-(2-метилтиофенокси)-1-метилэтил,
1-(4-метилтиофенокси)-1-метилэтил,
1-(2-метоксифенокси)-1-метилэтил, 1-(2-метилтиофенокси)-1-метилэтил,
1-(4-метилтиофенокси)-1-метиэтил,
1-(2-метилсульфонилфенокси)-1-метилэтил,
1-(4-метилсульфонилфенокси)-1-метилэтил, 1-метил-1-(2-метилфенокси)этил,
2-фенилтиофенил,
2-фенилсульфонилфенил, 2-дифенилил, 2-бензоилфенил, альфа, альфа-дифторбензил, 1-метил-1-/4-метокси-2-нитрофенокси/этил, 1-метил-1-/4-метил-2-нитро- фенокси/этил, 1-метил-1-/2-циано-4-метилфенокси/этил,
1-метил-1-/4-хлор-2-цианофенокси/этил,
1-метил-1-(2-циано-4-метоксифенокси/этил, 1-метил-1-(2-циано-5-метилфенокси)-этил,
1-метил-1-(2-нитрофенокси)этил,
1-(2-оксифенокси)-1-метилэтил или (Е)-2-метоксистирил.
Другая группа соединений по изобретению, представляющая особый интерес, включают соединения формулы (3), где А4 является (1-4С) алкиленом; Х1 является водородом или окси-группой; А6 является окси-группой, тиогруппой, сульфонилом, карбонилом, карбамоилом, иминокарбонилом, (1-6) алкиленом, окси (1-6С) алкиленом или прямой связью с Q3; Q3 представляет собой бензол, пиридин или нафталин; R7 и R8независимо представляют собой водород, галоген, цианогруппу, нитрогруппу, (1-4С) алкил, (1-4С) алкокси-группу и трифторметил. R4имеет указанное выше значение; также их фармацевтически приемлемые соли.
Конкретные значения А4 в соединениях формулы (3) являются, например, такими, как в указанных выше соединениях формулы (2).
Конкретные значения А6 включают, например, определенные выше значения для А3, когда он представляет собой (1-6С) алкилен или окси (1-6С) алкилен, например, метилен, этилен, изопропилиден и оксиметилен, а также оксигруппу, сульфонил, карбонил, карбамоил, иминокарбонил и прямую связь с Q3.
Примеры конкретных значений R7 или R8 включают в себя фтор, хлор и бром для галогена; метил и этил для алкила; метокси-группу и этокси-группу для алкокси-группы, а также водород, цианогруппу, нитрогруппу и трифторметил. Q3 обычно является фенилом или пиридилом.
Конкретные значения для группы формулы R8.Q3.А6 в соединениях формулы (3) включают, например, феноксигруппу, фенилсульфонил, фенил, бензоил, бензил, бензилоксигруппу, 4-цианобензилокси-группу, 2-пиридилметоксигруппу, 3-пиридилметокси-группу, 4-пиридилметокси-группу, фенокси-метил, 2-нафтил- метокси-группу, 2,5-диметоксибензилокси-группу, 4-нитро- бензилокси-группу и 3-цианобензилокси-группу.
Особенно предпочтительным значением для R4 является окси-группа, для Х1-водород и для А4-этилен.
Конкретные новые соединения по изобретению, описанные ниже в примерах, представляют вместе с их фармацевтически приемлемыми солями, дополнительный предмет изобретения. Среди них особый интерес представляют соединения, описанные в примерах 18,19,20,46 и 48, а также их фармацевтически приемлемые соли, физиологически приемлемые биодеградируемые сложные эфиры и (1-4С) алкансульфонамиды как дополнительные объекты изобретения.
Следует отметить, что соединения формулы (1) являются амфотерными, если R4 представляет собой окси-группу или алкансульфонамидогруппу и могут образовывать соли как с кислотами, так и с основаниями. Поэтому конкретные фармацевтически приемлемые соли включают, например, соли щелочных и щелочноземельных металлов, аммония, соли с органическими аминами и четвертичными основаниями, образующими физиологически приемлемые катионы, например соли с метиламином, диметиламином, триметиламином, этилендиамином, пиперидином, морфолином, N-пирролидином, пиперазином, этаноламином, триэтаноламином, -метилглюкамином, гидроксидом тетраметиламмония и бензилтриметиламмония, а также соли с кислотами, дающими физиологически приемлемые анионы, такие как соли с неорганическими кислотами, например с водородгалогенидами (например, с хлористым и бромистым водородом), с серной и фосфорной кислотами и с сильными органическими кислотами, например с n-толуолсульфокислотой и метансульфокислотой.
Соединения формулы (1) можно получать обычными способами органической химии, известными в области для получения структурно аналогичных соединений. Такие методики являются дополнительным предметом изобретения и иллюстрируются представленными ниже методиками, где R1,R2,R4,Х,Y,А1 и n такие, как определено выше.
(а) Производные диола формулы (4), где один из Т1 и Т2 является водородом, а другой представляет собой водород или группу формулы СRа Rb.ОН (где Rа и Rb являются одинаковыми или различными (1-4С) алкилами) вводят в реакцию с альдегидным производным формулы R1.СНО или с его ацеталем, полуацеталем или гидратом.
Указанный альдегид (или его гидрат, ацеталь или полуацеталь с (1-4С) алканолом (таким как метанол или этанол)) обычно может присутствовать в избытке.
Реакцию, как правило, проводят в присутствии такой кислоты, как хлористый или бромистый водород, серная кислота, фосфорная кислота метаносульфокислота или n-толуолсульфокислота, обычно в присутствии проходящего растворителя или разбавителя, такого как дихлорметан, толуол, ксилол или простой эфир, например, в тетрагидрофуране, дибутиловом эфире, метил-т-бутиловом эфире или 1,2-диметоксиэтане, в температурном интервале, например, 0-80оС.
Исходные соединения формулы (4), где оба Т1 и Т2 являются водородами можно получать, например, мягким катализируемым кислотой гидролизом или алкоголизом диоксанового кольца соединения формулы (5), где один из Rа и Rb является водородом или (1-4С) алкилом (таким как метил или этил), а другой (1-4С) алкилом, получаемым способом, аналогичным описанному ниже способу (d), например как в [1] Гидролиз или алкоголиз обычно проводят при температуре в интервале 10-80оС с использованием водной неорганической кислоты, такой как соляная кислота, в таком спирте, как этанол или 2-пропанол или эфире (таком как тетрагидрофуран) как растворителе.
Исходные соединения формулы (4), в которых один из Т1 и Т2 является водородом, а другой группой формулы -СRа Rb. ОН являются промежуточными соединениями при вышеуказанном образовании исходных соединений формулы (4), в которых оба Т1 и Т2 являются водородами. Однако указанные промежуточные соединения обычно не выделяют и не охарактеризовывают.
Таким образом, в изобретении предлагается также предпочтительная модифицированная методика (b) способа (а), включающая взаимодействие 1,3-диоксана формулы (5), где один из Rа и Rb являются водородом, метилом или этилом, а другой представляет собой метил или этил, с избытком альдегида формулы R1.СНО (или его гидратом, ацеталем или полуацеталем) в присутствии кислоты (например, одной из указанных выше), обычно в температурном интервале, например, 10-80оС и возможно в присутствии подходящего разбавителя или растворителя (такого, как указано выше).
В некоторых случаях необходимо модифицировать методики (а) и (b), если альдегид формулы R1.СНО не очень реакционноспособен или имеет тенденцию образовывать ациклические полуацетали при взаимодействии с соединениям формулы (4) или (5), например, если пентафторпропиональдегид используют для получения соединений формулы (1), в которых R1 является пентафторметильной группой. Таким образом, дополнительная методика (с) по изобретению включает взаимодействие соединения формулы (4), где один из Т1 и Т2 является водородом, а другой алкансульфонилом (например, метансульфонилом) или аронсульфонилом) например, бензол- или толуолсульфонилом) с альдегидом формулы R1.СНО) или его гидратом, ацеталем или пол ацеталем) в присутствии кислотного катализатора в тех же общих условиях, как описано для методики (а), с последующей катализируемой основанием циклизацией ациклического получаемого промежуточного соединения, например, с использованием карбоната щелочного металла или гидрида щелочного металла, такого как карбонат калия, гидрид натрия, в подходящем растворителе или разбавителе (таком как описанный выше простой эфир) и при температуре, например, 20-50оС.
Необходимые исходные алкансульфонильные или аренсульфонильные сложные эфиры формулы (4), указанные выше, удобно получать из соответствующего диола формулы (4) (Т1-Т2-водород) реакцией с одним молекулярным эквивалентом соответствующего алкансульфонил- или арилсульфонилгалогенида (такого как метансульфонилхлорид или п-толуолсульфонилхлорид) в подходящем растворителе или разбавителе (таком как простой эфир или дихлорметан) при температуре, близкой к комнатной, и в присутствии подходящего основания (такого как триэтиламин или пиридин).
(d) Для тех соединений формулы (1), в которых Y является виниленом, а R4 представляет собой окси-группу, альдегид формулы (4) вводят в реакцию с реагентом Виттига формулы R3Р=СН.А1.СО2-М+, где R является (1-6С) алкилом или арилом (преимущественно фенилом, который предпочтителен), а М+ является катионом, например, такого щелочного металла, как литий, натрий или калий.
Этим способом получают, как правило, требуемые соединения формулы (1), где заместители присоединены к двойной связи, имеющей в основном предпочтительную цис-конфигурацию, т.е. как "Z"-изомер. Однако в способе получают, как правило, также небольшие количества аналогичных соединений, имеющих транс-стереохимию (т.е. "Е" изомер), которые можно удалить обычными способами, например хроматографированием или кристаллизацией.
Способ удобно осуществлять в подходящем растворителе или разбавителе, например в ароматическом растворителе, таком как бензол, толуол или хлорбензол, в таком простом эфире, как 1,2-диметоксиэтан, т-бутилметиловый эфир, дибутиловый эфир или тетрагидрофуран, и диметилсульфоксиде или тетраметиленсульфоне или в смеси одного или больше таких растворителей или разбавителей. Способ, как правило, осуществляют при температуре от -80 до 40оС, но его удобно проводить при температуре, близкой к комнатной, например в интервале 0-35оС.
(е) Для тех соединений, где Х является окси-группой, соединений формулы (7), где Р является защищенной окси-группой (включая (1-4С) алкокси-группу, снимают защиту обычными способами.
Примеры особенно пригодных защищенных окси-групп включают в себя, например, (1-4С) алкокси-группу (такую как метокси-группы), бензилокси-группу, аллилокси-группу, тетрагидропиран-2-илокси-группу, (1-4С) алкансульфонилокси-группу (преимущественно метансульфонилокси-группу) и триалкилсилилокси-группу, имеющую до 10 атомов углерода.
Условия снятия защиты зависят от природы защищенной оксигруппы. Удаление конкретных групп, защищающих оксигруппу, хорошо освещено в обычных книгах по химии и эти известные обычные методики входят в способ по изобретению. Так, например, конкретные группы можно удалить следующими образом.
(1) Аллил или тетрагидропиран-2-ил: обрабатывают сильной кислотой, такой как трифторуксусная кислота, при температуре, например, 10-40оС: (2) триалкилсилильная группа (такая, как т-бутилдиметилсилильная группа, которая предпочтительна): реакция с водным раствором тетрабутил- аммоний фторида или фторида натрия, обычно в подходящем разбавителе или растворителе, таком как тетрагидрофуран, т-бутилметиловый эфир и, как правило, при температуре, близкой к комнатной, например, при 10-35оС; (3) алкансульфонил: гидролизом в присутствии основания (такого, как гидроксид натрия или калия) в подходящем водном растворителе (таком, как водный (1-4С) алканол) при температуре, например, 0-60оС; (4) алкил: обработка тиоалкоголятом или дифенилфосфидом щелочного металла (например, тиоэтилатом натрия в таком растворителе, как N, N-диметилформамид, например, при 50-160оС или дифенилфосфидом лития в метил-т-бутиловом эфире или тетрагидрофуране, например, при 0-60оС; или (5) бензил; катализируемым палладием гидрогенолизом в таком спирте, как этанол, при температуре, близкой к комнатной, и повышенном давлении или с использованием щелочного металла, такого как натрий в жидком аммиаке.
Изобретение включает также методику (f), аналогичную
(е) где окси-группу, нужную как заместитель в R1 вводят удалением подходящей защитной группы (такой как (1-4С) алкил, преимущественно метил) на последней стадии, например, в условиях, указанных выше для получения оксипиридильной группы.
(g) Разложение сложного эфира формулы (8), где R9 представляет собой (1-6С) алкил (в частности, метил, этил, пропил или т-бутил), фенил или бензил, причем последние два могут содержать 1-2 заместителя, выбранных из галогена, (1-4С) алкила или (1-4С) алкокси-группы.
Разложение можно проводить с использованием одного или больше обычных реагентов и условий, хорошо известных в данной области для превращения сложных эфиров в кислоты. Так, например, разложение удобно проводить катализируемым основанием гидролизом, например, с использованием гидроксида щелочного металла, такого как гидроксид лития, калия или натрия в водной системе, преимущественно в присутствии подходящего растворителя или разбавителя, такого как тетрагидрофуран, метанол, этанол, т-бутилметиловый эфир и температуре 10-60оС, обычно при температуре, близкой к комнатной. Кроме того, если R9 является т-бутилом, то разложение можно проводить термически, нагревая соединение формулы (8) при температуре, например, 80-150оС либо индивидуально, либо в присутствии подходящего разбавителя, такого как дифениловый эфир или дифенилсульфон.
(h) Для соединений формулы (1), где Y является этиленом, гидрирование соединения формулы (9), где Y2 является виниленом или этиниленом.
Гидрирование предпочтительно проводить в присутствии подходящего катализатора, например, на основе благородного металла, такого как металлические палладий или платина, преимущественно на инертной подложке, такой как углерод, сульфат бария, карбонат бария или кальция, с использованием водорода под давлением около 1-2 атм. Способ осуществляют, как правило, в подходящем разбавителе или растворителе, например в (1-4С) алканоле (таком, как метанол, этанол или пропанол) при температуре, например, 15-35оС.
Изобретение включает также модификацию вышеуказанной методики, адаптированной для получения тех соединений формулы (1), где Y является виниленом, включающую частичное гидрирование соединения формулы (9), где Y2 является этиниленом. В этой модификации используют подходящий отравленный катализатор, например катализатор Линдлара (такой, как палладий на карбонате кальция, отравленный свинцом) с подходящим растворителем при температурах способа (h).
(i) Для соединений, где Y является метиленоксигруппой, спирт формулы (10) вводят в реакцию с производным алкановой кислоты формулы (11), где L является отщепляемой группой, например галогеном (таким, как хлор, бром или иод), алкансульфонилокси-группой (такой, как метансульфонилоксигруппа) или аренсульфонилокси-группой (такой, как бензол- или толуолсульфонилокси-группа).
Реакцию предпочтительно проводить в присутствии подходящего основания, например алкоголята щелочного металла (например, метилата или этилата натрия), гидрида (например, гидрида натрия) или алканового производного (такого, как бутиллитий) в подходящем растворителе или разбавителе, например в (1-4С) алканоле, если используют алкоголят щелочного металла, в N,N-диметилформамиде или простом эфире, таком, как тетрагидрофуран или т-бутилметиловый эфир, если используют гидрид щелочного металла, либо в простом эфире, если используют алкановое производное. Реакцию проводят, как правило, при 0-50оС. Во многих случаях предпочтительно сначала получать соль спирта формулы (10) реакцией с соответствующим основанием и затем вводить эту соль в реакцию с производным алкановой кислоты формулы (11) в подходящем растворителе или разбавителе, таком как один из указанных выше. Ясно, что если заместитель (10) является окси-группой, то, как правило, необходимо проводить реакцию (i) и затем удалять защитную группу в условиях, аналогичных условиям описанного выше способа (е).
Необходимые исходные соединения для вышеописанных способов (а)-(i) можно получать по общим методикам, хорошо известным для получения структурно аналогичных соединений, например способами, аналогичными способами, описанным в [1] Альдегиды формулы [6] можно получать, например, как показано ниже на реакционных схемах 1 и 2 и проиллюстрировано в примерах. Кроме того, если нужен конкретный энантиомер, то его можно получать, исходя из конкретного энантиомера 3-/2-/1-окси-1-пиридилметил/пент-4-енил/оксазолидин-2-она формулы (14), где R10 представляет собой (1-4С) алкил (в частности, изопропил), который в свою очередь получают альдольной конденсацией соответствующего 3/4-пентеноил/оксазолидин-2-она с пиридилкарбоксальдегидом, как показано ниже на реакционной схеме 3.
Защищенное окси-производное формулы (7) можно получать, например, проводя описанные выше реакции (а) или (b) с подходящими аналогами 1,3-диоксана формулы (5), но где Х является подходящей защищенной окси-группой, и это соединение получается легко по стандартным методикам, аналогичным описанным выше и проиллюстрированным ниже в примерах.
Соответствующие диоды формулы (4) для получения диоксанов формулы (1) или (5), где пиридильный остаток, содержащий Х, и боковая цепь алкеновой кислоты имеют цис-конфигурацию, можно получать исходя из соответствующего пиридинкарбоксиальдегида и сукцинангидрида и подходящего основания, например такого, как используют для альдольной конденсации части (ii) реакционной схемы 3.
Сложные эфиры формулы (8) можно получать, например, способом
(а) с использованием соответствующего сложного эфира диола, соответствующего формуле (4). Соединения формулы (9), где Y2 является этиниленом, можно получать так, как, например, показано на реакционной схеме 4. Спирты формулы (10) можно получать из соответствующих аллильных соединений формулы (12) (получая соединения Х с n=2) обычным гидроборированием (гидрид бора с последующей обработкой перекисью водорода) или восстановлением соответствующих альдегидов (например, боргидридом натрия) формулы (6), например так, как показано на реакционной схеме 11 (получая соединения Х с n=1).
Альдегиды или кетоны формул R1. ОНО или R1.ОО.R2, являющиеся новыми, можно получать обычными методиками, хорошо известными в данной области, проиллюстрированными ниже в примерах. Нужные реагенты Виттига можно получать по обычным методикам, например, обработкой соответствующих фосфоний галогенидов сильным основанием, таким как гидрид натрия, диизопропиламид лития, т-бутиллат калия или бутиллития. Их как правило получают in situ непосредственно перед проведением вышеописанной конденсации (d).
Понятно, что соединения формулы (1), где R4 является окси-группой, можно также получать другими обычными способами, известными в данной области, например катализируемым основанием гидролизом соответствующих амидов или нитрилов. Кроме того, те соединения формулы (1), где R4отличается от окси-группы, можно получать обычной этерификацией или сульфонамидированием из соединений, где R4 является оксигруппой (или их реакционноспособных производных) и соответствующих спиртов, фенола или (1-4С) алкансульфонамида. Эти методики также входят в данное изобретение.
Если нужна соль соединения формулы (1), то ее моно получать реакцией с соответствующим основанием или кислотой, дающими физиологически приемлемый ион, или другими любыми обычными способами получения солей.
Кроме того, если нужна оптически активная форма соединения формулы (1), то один из вышеописанных способов можно осуществлять с использованием оптически активного исходного соединения (например, как описано в примерах 46 и 47). Кроме того, рацемическую форму соединения формулы (1) можно ввести в реакцию с оптически активной формой подходящего органического основания или кислоты, например камфорсульфокислоты, эфедрина, N,N,N-триметил(1-фенил-этил)аммоний гидроксида или 1-фенилэтиламина с последующим обычным разделением диастереомерной смеси, полученной таким образом, в частности дробной кристаллизацией из подходящего растворителя, такого как (1-4С) алканол и затем оптически активную форму указанного соединения формулы (1) можно выделить обработкой кислотой (или основанием) обычным образом, например водным раствором неорганической кислоты, такой как разбавленная соляная кислота (или водным раствором щелочи, например гидроксида натрия).
Многие указываемые здесь промежуточные соединения являются новыми, например соединения формулы (5) (Rа-Rb-этил), (6-10) и являются отдельным предметом данного изобретения.
Как указывалось выше, соединения формулы (1) обладают существенными ТХА2-антагонистическими свойствами и ингибируют ТХА2-синтезу, ТХА2-антагонизм можно показать одним из следующих стандартных испытаний.
(а) Модель аортальной полоски крыс, аналогичная описанной Пипером и Ваном Nature, 1969, 223, 29-35) с использованием в качестве антагониста ТХА2-миметического агента, известного как U46619 (описанного Р.Л.Джонсом и др. в "Сhemistry, Вiochemistry and Рharmacological Аctivity of Рrostanoids " ed. S.М. Roberts, F.Scheinmаnn, р.211, Рergamon Рress, 1979).
(b) Испытание на агрегацию тромбоцитов, основанное на испытании, описанном Борном Nature 1962, 194, 927-929) и включающее:
(i) агрегацию человеческой цитратной обогащенной тромбоцитами плазмы при прибавлении ТХА2-миметического агента U46619 с получением кривой зависимости реакции от дозы;
(ii) получение кривой зависимости реакции от дозы для U46619-стимулированной агрегации тромбоцитов в присутствии нарастающих количеств испытуемого соединения (как правило, в интервале 10-5-10-10М); и
(iii) расчет КВ, отражающей эффективность испытуемого соединения как антагониста ТХА2, усредненного для нескольких концентраций из рассчитанного значения 50% реакции для U46619-агрегации в присутствии и отсутствии испытуемого соединения.
(с) Бронхоспазматическое испытание, включающее измерение ингибирования испытуемым соединением спазма бронхов, вызываемого модельном эксперименте Концетт-Росслера с анестезированной морской свинкой (в модификации Коллиера и Джеймса, Вril. J. Рharmacol. 1967, 30, 283-307) при внутривенном введении ТХА2-миметического агента, U46619, включающее:
(i) получение кумулятивной зависимости реакции от дозы для U46619-индуцированного спазма бронхов при внутривенном введении постоянных объемов с повышающимися концентрациями Г46619 (0,2-4 мкг/кг) в физиологическом солевом растворе, выражая спазм бронхов через теоретический максимум, где воздух вообще не проходит в испытуемое животное;
(ii) получение кумулятивной зависимости реакции от дозы для U46619-индуцированного спазма бронхов через 30 мин, интервалы в течение 3 ч после орального введения испытуемого соединения;
(iii) расчет зависимости от дозы для испытуемого соединения (выражаемой в концентрации U46619, нужной для вызывания 50% спазма бронхов в присутствии и отсутствии испытуемого соединения), указывающей на эффективность ТХА2-антагонизма.
Испытание (b) удобно модифицировать, чтобы показать эффективность ТХА2-антагонизма in vivo, оценивая действие испытуемого соединения на агрегацию тромбоцитов после введения испытуемого соединения лабораторному животному, такому как кролик, крыса, морская свинка или собака. Однако при изучении агрегации тромбоцитов собак необходимо использовать заданную пороговую концентрацию агента агрегации тромбоцитов аденозиндифосфата (около 0,4-1,2х10-6 М) вместе с ТХА2-миметическим агентом, U46619.
Антагонизм действию ТХА2 на мускулатуру сосудов можно показать, например, на крысах по следующей методике.
(d) Крыс-самцов (линия Аlderley Раrk) анестезируют пентабарбиталом натрия и кровяное давление регистрируют на сонной артерии, ТХА2-миметический агент U46619 вводят внутривенно в дозе 5 мкг/кг через яремную вену с достижением повышения систолического кровяного давления на 20-30 мм рт.ст. (2640-3970 Па). Операцию повторяют дважды для оценки адекватности ответа. Затем испытуемое соединение вводят либо внутривенно (через яремную вену) либо орально (через канюлю) непосредственно в желудок и животному вводят U46619 через 5 мин после введения испытуемого соединения и затем через каждые 10 мин до тех пор, пока гипертонический эффект U46619 не будет более блокироваться.
Способность испытуемого соединения ингибировать ТХА2-синтазу можно показать стандартным испытанием in vitro (испытание (е)) описанным Ховарсем и др. (Вiochem.Soc. Тransactions 1982, 10, 239-240) с использованием препарата человеческой тромбоцитарной микросомальной ТХА2-синтезы и количественного тонкослойного радиохроматографического анализа для оценки превращения (1-14С) арахидоновой кислоты в ТХА2-метаболит тромбоксан В (ТХВ2).
Способность испытуемого соединения ингибировать ТХА2-синтазу можно также показать стандартным испытанием (испытанием (f)), включающим получение образцов крови лабораторных животных (как правило, крыс, но также и морских свинок, кроликов или собак), которым ввели дозу испытуемого соединения, как правило перорально. Образцы обрабатывают антикоагулянтом и сначала инкубируют при 37оС с коллагеном (приблизительно при 100 мкм), а затем смешивают с циклооксигеназным ингибитором индометацином (приблизительно 10-3 М), центрифугируют и определяют концентрацию ТХА2-метаболита, ТХВ2 стандартным радиоиммуноанализом. Из сравнения количества ТХВ2, присутствующего в плазме животных с веденным испытуемым соединением, с количеством в плазме контрольной группы с введением плацебо можно оценить способность ингибировать ТXА2-синтазу.
Как правило, большинство соединений формулы (1), где R1 и R4являются окси-группами, в вышеописанных испытаниях обнаруживают эффективность в следующих пределах:
испытание (а): рА2 более 5,5;
испытание (b) КВ менее 1,5х10-6 М;
испытание (с): зависимость дозы более 5 при измерении через 1 ч дозы 10 мг/кг;
испытание (d); значительное ингибирование U46619 индуцированной гипертонии в течение по крайней мере 1 ч после орального введения 50 мг/кг или меньше;
испытание (е): ИК50 менее 1,0х10-6 М;
испытание (f): значительное ингибирование ТХВ2 образования через 1 ч после введения дозы 100 мг/кг или меньшей.
Никакого токсического или другого нежелательного действия не наблюдалось для характерных соединений формулы (1), обнаруживающих эффект in vivo в испытаниях (с), (d) или (f) при дозах, в несколько раз превышающих минимальную эффективную дозу.
Как правило, соединения формулы (1), где R4 отличается от окси-группы, обнаруживают более низкую активность вышеописанных испытаниях in vitro, но обладают активностью, аналогичной активности соединений формулы (1), в которых R4 является окси-группой, в испытаниях in vivo.
Соединение, описанное в примере 1, обладает как ТХА2-антагонистическими свойствами, так и свойством ингибировать ТХА2-синтазу, что обнаруживается по КВ=3,0 и 10-7М в испытании (b) и ИК50=4,0х10-8 М в испытании (е).
Как отмечалось выше, в силу того, что соединения формулы (1) обладают ТХА2-антагонистической активностью и одновременно способны ингибировать и ТХА-синтазу, то их можно использовать при терапии и предупреждении тех заболеваний и нежелательных состояний теплокровных животных, которые протекают с участием ТХА2 (либо простагландинов Н2, D2и/или F2-альфа). Как правило, соединение формулы (1) можно вводить перорально, ректально, внутривенно, подкожно, внутримышечно или ингаляцией в результате чего дозу, например, 0,01-15 мг/кг веса тела можно вводить до 4-х раз в день, варьируя в зависимости от способа введения, тяжести состояния, веса и возраста пациента.
Соединение формулы (1) можно, как правило, использовать в форме фармацевтической композиции, содержащей соединение формулы (1) или его фармацевтически приемлемую соль, как указывалось выше, вместе с фармацевтически приемлемым разбавителем или носителем. Эта композиция является дополнительным предметом изобретения и может представлять собой различные дозированные формы. Например, она может иметь форму таблеток, капсул, растворов или суспензий для орального введения, форму суппозиториев для ректального введения, форму стерильного раствора или суспензии для внутривенной или внутримышечной инъекции, форму аэрозоля или пульверизируемых раствора или суспензии для ингаляции и форму порошка вместе с фармацевтически приемлемыми инертными твердыми разбавителями, такими как лактоза для введения вдуванием.
Фармацевтические композиции можно получать обычными методами с использованием известных фармацевтически приемлемых разбавителей и носителей. Таблетки и капсулы для орального введения удобно получать с желудочным покрытием, например, содержащим целлюлозацетатфталат для сведения к минимуму контакта активного ингредиента формулы (1) с кислотами желудка.
Фармацевтические композиции по изобретению могут также содержать один или более агент, известный как полезный при лечении заболеваний и состояний. Например, в фармацевтические композиции по изобретению можно успешно вводить известный ингибитор агрегации тромбоцитов, гиполипедимический агент, противогипертонический агент, тромболитический агент (такой, как стрептокиназа), бета-адренергический блокатор или вазодилатор при лечении сердечных или сосудистых заболеваний или состояний. Аналогично при лечении пульмонологических заболеваний и состояний в фармацевтических композициях по изобретению могут присутствовать также, например, антигистамин, стероид (такой, как беклометазондипропионат), хромогликат натрия, фосфодиэстеразный ингибитор или бета-адренергический стимулянт. Кроме того, известный ТХА2-антагонист или известный ингибитор ТХА2-синтазы, как дезоксибен или фурегрелат (U63557), могут присутствовать кроме соединения формулы (1) или его фармацевтически приемлемой соли в композиции по изобретению для изменения общего баланса ТХА2-антагониста и эффекта ингибирования ТХА2-синтазы и достижения нужного терапевтического эффекта при лечении любого из указанных выше заболеваний или болезненных состояний.
Кроме терапии, соединения формулы (1) пригодны также как фармакологические инструменты при разработке и стандартизации испытательных систем оценки действия ТХА2 на таких лабораторных животных, как кошки, собаки, кролики, обезъяны, крысы и мыши, как часть для исследования новых терапевтических агентов. Поскольку соединения формулы (1) обладают ТХА2 антагонистическими и синтез-ингибирующими свойствами, то их можно использовать как вспомогательные агенты при поддержании жизнеспособности крови и кровеносных сосудов теплокровных животных (или их частей) в ходе искусственной экстракорпоральной циркуляции, например, при трансплантации конечностей или органов. При использовании для этой цели соединение формулы (1) или его фармацевтически приемлемую соль вводят, как правило, таким образом, чтобы в крови поддерживалась постоянная концентрация, равная, например, 0,1-10 мг/л.
В примере 14 описано получение исходного соединения формулы (5), где за исключением специально указанных случаев:
(i) концентрирование и выпаривание проводят на роторном испарителе под вакуумом;
(ii) операции проводят при комнатной температуре в интервале 18-26оС;
(iii) колоночное хроматографирование проводят на Fluka kuselgel (кат. N 60738) производства Fluka А.G. Вuchs, Switzerland СН-9470;
(w) выходы указаны лишь для иллюстрации и необязательно являются максимальными, которые можно достичь при развитии способа;
(y) протонные ЯМР-спектры обычно записывают при 90 или 200 МГц в СDCl3 с использованием тетраметилсилана (ТМS) в качестве внутреннего стандарта и выражают в виде химсдвигов (дельта-значения) в частях на миллион относительно ТМS с использованием стандартных сокращений для обозначения основных пиков: с синглет; м мультиплет; т триплет; ш- широкий; д дублет;
(vi) все конечные продукты выделяют в виде рацематов и их состав удовлетворительно подтверждается микроанализом.
П р и м е р 1. п-Толуолсульфокислоту (0,325 г) прибавляют к раствору 4(z)-6-(2,2-диметил-4-(3-пиридил)-1,3-диоксан-цис-5-ил)гексановой кислоты (А) (0,469 г) в ацетонитриле (7 мл) и смесь перемешивают 0,5 ч. Раствор 2-(4-метоксифенокси)-2-метилпропаналя (0,894 г) в ацетонитриле (5 мл) прибавляют и смесь кипятят 18 ч с обратным холодильником в атмосфере аргона. Затем смесь оставляют охлаждаться. Раствор подщелачивают 2М водным гидроксидом натрия и обрабатывают смесью воды и этилацетата. Водную фазу подкисляют уксусной кислотой и трижды экстрагируют этилацетатом. Соединенные органические экстракты сушат над сульфатом магния и концентрируют до масла, которео очищают колоночной хроматографией, сначала элюируя дихлорметаном, а затем смесью метанол/дихлорметан (1: 10 объем/объем/ с получением 4(z)-6-//2,4,5-цис/-2-/1-(4-метоксифенокси/-1-метилэтил/-4-/3-пиридил/ -1,3-диоксан-5-ил/-гексановой кислоты (0,345 г) в виде масла, отверждающегося при стоянии;
ЯМР: 1,35 (3Н, с), 1,38 (3Н, с), 1,55-1,8 (2Н, м), 2,25-2,55 (5Н, м), 3,75 (3Н, с), 3,95-4,25 (2Н, м), 4,75 (1Н, с), 5,1-5,5 (3Н, м), 6,75-7,0 (4Н, м), 7,35-7,75 (2Н, м), 8,5-8,6 (2Н, м).
Исходное соединение А получают следующим образом.
(i) Метил-2-(никотиноил)ацетат (17,9 г) получен способом, описанным в Е. Wenbеrt et al. J.Оrg. Chem. 1983, 48, 5006) в атмосфере аргона прибавляют к раствору металлического натрия (2,3 г) в метаноле 200 мл) и результирующую смесь перемешивают 30 мин при 25оС. Затем прибавляют аллилбромид (12,0 г) и перемешивают ночь. Прибавляют дополнительное количество аллилбромида (около 2 г), смесь перемешивают 48 ч и затем концентрируют. Результирующее масло обрабатывают смесью воды и эфира и водный слой трижды экстрагируют эфиром. Соединенные экстракты промывают насыщенным водным раствором соли, сушат над сульфатом магния и концентрируют. Остаток очищают колоночной хроматографией. Элюируя смесью петролейного эфира (т. кип. 60-80оС) и этилацетата (1:1, объем/объем) и получают метил-2-никотиноил-4-пентеноат (В) в виде бледно-желтого масла (13,8 г);
ЯМР: 2,6-2,9 (2Н, м); 3,7 (3Н, с), 4,4 (1Н, м), 4,9-5,2 (2Н, м), 5,5-6,0 (1Н, м), 7,2-7,5 (1Н, м), 8,1-8,3 (1Н, м), 8,7-8,8 (1Н, м) и 9,1-9,2 (1Н, м).
(ii) Раствор В (8,8 г) в сухом тетрагидрофуране (40 мл) прибавляют к суспензии литийалюмогидрата (1,8 г) в сухом тетрагидрофуране (80 мл) в атмосфере аргона с такой скоростью, что температура не превышает 10оС. Через 2 ч смесь охлаждают на льду. Затем для разрушения избытка реагента прибавляют этилацетат (20 мл) и затем насыщенный водный раствор хлорида (50 мл). Осадок удаляют фильтрованием и промывают этилацетатом. Водную фазу отделяют и экстрагируют этилацетатом (3х50 мл). Соединенные органические фракции промывают насыщенным раствором соли, сушат над сульфатом магния и концентрируют. Остаток очищают колоночной хроматографией, элюируя смесью этилацетата и метанола ( 95: 5 объем/объем) и получают 2-аллил-1-/3-пиридил/-1,3-пропандиол (С) (5,3 г) в виде масла (смесь диастереомеров);
ЯМР: 1,8-2,2 (3Н, м), 3,6-4,1 (4Н, м), 4,7-5,2 (3Н, м). 5,6-5,9 (1Н, м), 7,2-7,4 (1Н, м), 7,65-7,8 (1Н, м), 8,4-8,6 (2Н, м).
(iii) Смесь С (5,2 г), п-толуолсульфокислоты (5,2 г) и 2,2-диметоксипропана (50 мл) перемешивают ночь при комнатной температуре. рН доводят до 8-10 добавлением триэтиламина и раствор концентрируют при пониженном давлении. Остаток очищают колоночным хроматографированием, элюируя смесью петролейного эфира (т.кип. 40-60оС) и этилацетата (60:40, объем/объем с получением 5-аллил-2-, 2-диметил-4-/3-пиридил/-1,3-диоксана (D) (смесь 4,5-цис- и транс-изомеров) в виде масла (4,6 г);
ЯМР: 1,4-1,6 (6Н, м); 1,6-2,5 (3Н, м); 3,65-4,25 (2Н, м); 4,5-5,7 (4Н, м), 7,2-7,4 (1Н, м), 7,6-7,8 (1Н, м), 8,45-8,65 (2Н, м).
(iv) Озон в кислороде продувают через раствор D (3,4 г) в этилацетате (130 мл) при (-70оС) до исчезновения голубой окраски. Затем через раствор продувают аргон до удаления избытка озона и прибавляют раствор трифенилфосфина (6,0 г) в этилацетате (50 мл). Смесь оставляют нагреваться до комнатной температуры и затем перемешивают ночь. Раствор концентрируют и прибавляют эфир (50 мл) для осаждения трифенилфосфиноксида. Смесь фильтруют, фильтрат концентрируют и получают масло, которые очищают колоночной хроматографией, элюируя смесью (60:40 объем/объем) этилацетата и петролейного эфира (т.кип. 40-60оС) с получением первоначально 2,2-диметил-4-/3-пиридил/-1,3-диоксан-цис-5-илацетальдегида (Е) в виде масла (0,8 г).
ЯМР: 1,5 (3Н, м), 1,55 (3Н, с), 2,0-2,3 (1Н, м), 2,3-2,5 (1Н, м), 2,8-3,0 (1Н, м), 3,8 (1Н, двойной д, J=12 Гц, 1,5 Гц), 4,3 (1Н, дм, J=12 Гц), 5,25 (1Н, д, J=3 Гц), 7,25-7,35 (1Н, м), 8,45-8,60 (2Н, м), 9,6 (1Н, с); и затем соответствующий 4,5-транс-изомер.
ЯМР: 1,47 (3Н, с), 1,57 (3Н, с), 2,0-2,6 (3Н, м), 3,75-4,05 (2Н, м), 4,68 (1Н, д, J=10 Гц), 7,25-7,40 (1Н, м), 7,70-7,80 (1Н, м), 8,50-8,65 (2Н, м)= 9,5 (1Н, шс) в виде масла (0,7 г).
(Y) Раствор Е (0,20 г) в сухом тетрагидрофуране (ТНF) (7 мл) в атмосфере аргона прибавляют к перемешиваемому охлаждаемому льдом раствору илида, полученного из (3-карбоксипропил) трифенилфосфоний бромида (0,91 г) и т-бутилата калия (0,48 г) в сухом ТНF (30 мл). Смесь перемешивают 2 ч и затем обрабатывают водой, охлажденной льдом (50 мл). Раствор концентрируют и прибавляют еще воду (25 мл). рН доводят до 7 прибавлением нескольких кристаллов щавелевой кислоты и раствор экстрагируют этилацетатом (3х40 мл). Водную фазу подкисляют затем до рН= 4 щавелевой кислотой и экстрагируют этилацетатом (3х50 мл). Эти объединенные экстракты промывают затем насыщенным раствором соли (50 мл), сушат над сульфатом магния и концентрируют. Остаток очищают колоночной хроматографией, элюируя смесью дихлорметан (метанол) 95:5, объем/объем/ и получают 4(z)-6-(2,2-диметил-4-(3-пиридил)-1,3-диок-сан-цис-5-ил/гексеновую кислоту (А) в виде масла (0,19 г).
ЯМР: 1,55 (3Н, с), 1,57 (3Н, с), 1,5-2,6 (7Н, м), 3,85 (1Н, двойной д, J= 12 Гц, 1,51 Гц), 4,15 (1Н, дм, J=12 Гц), 5,15-5,50 (3Н, м), 7,3-7,4 (1Н, м), 7,7-7,8 (1Н, м), 8,1 (1Н, шс) и 8,45-8,60 (2Н, м).
2-/4-метоксифенокси/-2-метилпропа-нель получают следующим образом.
(vi) Перемешиваемый раствор метилиодида магния (полученного из магниевых стружек (32,8 г, 1,35 М), и метилиодида (84,1 мл, 1: 35 М)) в безводном эфире (750 мл) обрабатывают при 0оС в атмосфере аргона раствором метилдихлорцетата (77,18 г, 0,54 М) в безводном эфире (50 мл) с такой скоростью, что температура не поднимается выше 15оС. Смесь перемешивают 30 мин при 25оС и затем охлаждают до 0оС. Прибавляют воду (100 мл) и смесь подкисляют до рН=4 концентрированной хлористоводородной кислотой. Слои разделяют и водную фазу экстрагируют эфиром (3х100 мл). Объединенные экстракты сушат над сульфатом магния и концентрируют. Остаточное масло перегоняют под вакуумом и получают 1,1-дихлор-2-окси-2-метилпропан (57,81 г) в виде масла; т.кип. 48-50оС при 20 мм рт.ст.
ЯМР; 1,45 (6Н, с), 2,15 (1Н, шс) и 5,65 (1Н, с).
(vii) Раствор 4-метоксифенола (21,72 г, 0,175 моля) в водном растворе гидроксида натрия (5,8 М, 30 мл) обрабатывают диметилтриметиламмоний бромидом (0,255 г, 0,7 ммоля), а затем раствором 1,1-дихлор-2-окси-2-метилпропана (5,01 г, 35 ммоль) в эфире (70 мл). Смесь перемешивают в атмосфере аргона 18 ч и затем разбавляют эфиром (100 мл) и экстрагируют водным раствором гидроксида натрия (2М, 4х50 мл) для удаления непрореагировавшего фенола. Соединенные водные экстракты экстрагируют эфиром (100 мл), органическую фазу отмывают водным раствором гидроксида натрия (2М, 250 мл) и водой (100 мл). Соединенные органические экстракты сушат над сульфатом магния, концентрируют, очищают колоночной хроматографией, элюируя смесью этилацетат (гексан (10% объем/объем и получают 2-/4-метокси-фенокси/-2-метилпропаналь (3,61 г) в виде масла.
ЯМР: 1,36 (6Н, с), 3,76 (3Н, с), 6,7-6,9 (4Н, м), и 9,85 (1Н, с).
Вышеуказанное исходное соединение можно также получить по известной методике для получения 2-фенокси-2-метилпропаналя.
П р и м е р 2. Аналогично примеру 1, но исходя из 2-(4-т-бутилфенокси)-2-метилпропаналя получают 4(Z)-6-(//2,4,5-цис/-2-(1-/4-т-бутил фенокси)-1-метилэтил)-1-4-3- пиридил/-1,3-диоксан-5-ил/гексеновую кислоту в виде масла, отверждающегося при стоянии с выходом 24%
ЯМР: 1,3 (9Н, с), 1,35 (3Н, с), 1,4 (3Н, с), 1,55-1,8 (2Н, м), 2,25-2,55 (5Н, м), 3,95-4,25 (2Н, м), 5,1-5,5 (3Н, м), 6,9-7,75 (6Н, м) и 3,5-8,6 (2Н, м).
Исходный альдегид получают в виде масла аналогично примеру 1 из 4-т-бутилфенола и 1,1-дихлор-2-окси-2-метилпропана, имеющего следующий спектр ЯМР: 1,26 (9Н, с), 1,41 (6Н, с), 6,7-7,3 (4Н, м) и 9,85 (1Н, с).
П р и м е р 3. 5-цианотиофен-2-карбоксальдегид (500 мг) и п-толуолсульфокислоту (220 мг) прибавляют к перемешиваемой суспензии 4(Z)-6-(2,2-диметил-/3-пиридил)-4-/3-пиридил)-1,3-диоксан-цис-5-ил (гексановой кислоты (305 мг) и ацетонитриле (10 мл). Смесь перемешивают 18 ч. Прибавляют воду (20 мл) и 1М раствор гидроксида натрия (30 мл) и смесь промывают этилацетатом (3х20 мл). Водную фазу подкисляют уксусной кислотой до рН=4-5 и экстрагируют этилацетатом (3х30 мл). Экстракты промывают насыщенным раствором соли, сушат над сульфатом магния и выпаривают. Остаточную смолу перекристаллизовывают из смеси гексан/этилацетат и получают 4(Z)-6-((2,4,5-цис/2-5-циано-2-тиенил/-4-(3-пи-ридил)-1,3-диоксан-5-ил)-гекс ановкислоту (148 мг) в виде бесцветного твердого соединения с т.пл. 129-132оС.
ЯМР: 1,68 (1Н, м), 1,84 (1Н, м), 2,32 (4Н, м), 2,54 (1Н, м), 4,20 (2Н, м), 5,05-5,95 (4Н, м), 5,96 (1Н, с), 7,17 (1Н, д), 7,36 (1Н, м), 7,53 (1Н, д), 7,72 (1Н, д) и 8,57 (2Н, шс); m/е 383 (М-Н).
Исходный 5-цианотиофен-2-карбоксальдегид получают окислением 5-циано-2-метилтиофена триоксидом хрома (Оrg. Synthesis, Collected volunu II, 441, 1943) в виде бесцветного твердого соединения (60% выход), т.пл. 91-93оС.
ИК: 2210 (CN) и 1670 (СНО) см-1 m/е=137 137 (М+).
П р и м е р ы 4-11. Аналогично примеру 3, но исходя из соответствующего замещенного гетероциклического альдегида формулы R1.СНО, получают следующие кислоты формулы (13) с выходами 24-42% (см.табл.1).
П р и м е р 12. Аналогично примеру 1, но используя в качестве альдегида 2-метил-2-/4-метилсульфонилфенокси/пропанель, получают 4-/Z/-6-//2, 4,5-цис/2-1-метил-1-/4-метилсульфонилфенокси/этил/-4-/3-пиридил-1,3-диоксан- 5-илкислоту в виде масла с выходом 20%
ЯМР: 1,45 (6Н, с), 1,8-2,45 (7Н, м), 3,2 (3Н, с), 3,95-4,1 (2Н, м), 4,9 (1Н, с), 5,15-5,45 (3Н, м), 7,2-7,85 (6Н, м), 8,45-8,5 (2Н, м).
Нужный альдегид получают аналогично примеру 19, исходя из 4/метилтио/фенола, который превращают в этил-2-метил-2-/4-/метилтио-фенокси/пропионат/ масло, выход 20%
ЯМР: 1,25 (3Н, т, J=7 Гц), 1,6 (6Н, с)= 2,45 (3Н, с), 4,25 (2Н, к, J=7 Гц), 6,75-7,2 (4Н, м).
Этот сложный эфир окисляют м-хлорпербензойной кислотой в дихлорметане при комнатной температуре и после обычной обработки получают этил-2(метил-2-/4-метилсульфонилфенокси) пропионат (масло, медленно отвердевающее, выход 92%).
ЯМР: 1,25 (3Н, к, J=7 Гц), 1,65 (6Н, с), 3,0 (3Н, с), 4,25 (2Н, к, J=7 Гц), 6,9-6,95 (2Н, м), 7,8-7,85 (2Н,м)), который затем восстанавливают с помощью DIВАL и получают 2-метил-2-/4-метилсульфонилфенокси) пропаналь в виде твердого соединения (выход 66%).
ЯМР: 1,5 (6Н, с), 3,05 (3Н, с), 6,9-7,0 (2Н, м), 7,8-7,9 (2Н, м), 9,8 (1Н, с).
П р и м е р ы 13-15 проводят аналогично примеру 1, но с использованием соответствующих альдегидов получают следующее.
П р и м е р 13. 4(Z)-6-//2,4,5-цис/-2-/1-/2-метоксифенокси/-1-метилэтил/-4-/3-пи- ридил/1,3- диоксан-5-ил/гексеновая кислота в виде масла, отверждающегося при хранении, дающее твердое соединение с выходом 40%
ЯМР: 1,35 (3Н, с), 1,40 (3Н, с), 1,55-1,80 (2Н, м), 2,20-2,55 (5Нм), 3,78 (3Н, с), 3,95-4,25 (2Н, м), 4,80 (1Н, с), 5,10-5,50 (4Н, м), 6,80-7,10 (4Н м), 7,40-7,75 (2Н, м), 8,45-8,60 (2Н, м), исходя из 2-метил-2-/2-метоксифенокси/пропаналя.
П р и м е р 14. 4(Z)-6-[(2,4,5-цис)-2-(1-[2-метилфенокси]-1-метилэтил)-4-(3-пиридил)- -1,3- диоксан-5-ил]гексановая кислота в виде масла, которое отверждается при хранении, давая твердое вещество с 15% выходом.
ЯМР; 1,40 (3Н, с), 1,45 (3Н, с), 1,50-1,80 (2Н, м), 2,25 (3Н, с), 2,15-2,60 (5Н, м), 3,95-4,25 (2Н, м), 4,85 (1Н, с), 5,10-3,50 (4Н, м), 6,90-7,15 (4Н, м), 7,30-7,75 (2Н, м), 8,50-8,60 (2Н, м), исходя из 2-метил-2-(2-метилфенокси)пропаналя.
П р и м е р 15. 4(Z)-6-[(2,4,5-цис)-2-(1-[2-нитро-4-метилфенокси)]-1-метилэтил)-4-(3-пиридил )-1, кислота в виде масла, которое отверждается при хранении, давая продукт с 25% выходом.
ЯМР; 1,45 (6Н, с), 1,50-1,80 (2Н, м), 2,35 (3Н, с), 2,15-2,50 (5Н, м), 3,90-4,20 (2Н, м), 4,80 (1Н, с), 5,05-5,50 (3Нм), 7,10-7,70 (5Н, м), и 8,45-8,60 (2Н, м), исходя из 2-метил-2-(2-нитро-4-метилфенокси)пропаналя.
Необходимые исходные альдегиды получали следующим образом.
(13) 2-метил-2-(2-метоксифенокси)пропанель получали в виде масла;
ЯМР: 1,35 (6Н, с), 3,75 (3Н, с) и 6,80-7,25 (4Н, м), используя методику, аналогичную описанной в примере 1, но исходя из 2-метоксименола и 1,1-дихлор-2-окси-2-метилпропана.
(14) 2-метил-2-(2-метилфенокси)пропаналь получали в виде масла.
ЯМР: 1,45 (6Н, с), 2,25 (3Н, с) и 6,60-7,20 (4Н, м), используя методику, аналогичную описанной в примере 1, но исходя из 2-метилфенола и 1,1-дихлор-2-окси-2-метилпропана.
(15) 2-метил-2-(2-нитро-4-метилфенокси)пропаналь получали в две стадии.
(I) Раствор 2-нитро-4-метилфенола (11,48 г) в 1,3-диметил/3, 4,5,6-тетрагидро-2 (1Н)-пиримидиноне (DМРИ) (50 мл) охлаждали до 5оС и обрабатывали гидридом натрия (55 мас. дисперсия в минеральном масле, 3,25 г). Смесь перемешивали при комнатной температуре 2 ч, затем охлаждали до 5оС и обрабатывали этил-2-бром-2-метилпропионатом (13,15 г). Смесь нагревали при температуре около 100оС в течение 18 ч, затем охлаждали до комнатной температуры и вливали в смесь водного раствора гидроокиси натрия (1М) и этилацетата. Органический раствор отделяли и промывали дважды 1М раствором гидроокиси натрия. Затем сушили (над МgSО4) и упаривали. Полученное в результате масло очищали на хроматографической колонке, элюируя смесью этилацетата и гексана (увеличивая от 5:95 об/об. до 10:90 об./об.), что дало этиловый эфир 2-метил-2-(2-нитро-4-метилфенокси)пропи- оновой кислоты в виде масла.
ЯМР: 1,25 (3Н, т), 1,60 (6Н, с), 2,35 (3Н, с), 4,25 (2Н, к), и 6,8-7,6 (3Н, м).
(II) Раствор этилового эфира 2-метил-2-(2-нитро-4-метилфенокси)пропионовой кислоты (4,47 г) в толуоле (50 мл) охлаждали до (-78оС) и обрабатывали гидридом диизобутилалюминия (DIВАL 11,3 мл, 1,5 М раствор в толуоле), добавляя последний по каплям. Смесь перемешивали 2 ч и затем добавляли дополнительное количество DIВАL до полного завершения реакции (около 4,4 мл), определяя этот момент методом тонкослойной хроматографии (t1е). Реакцию прекращали добавлением водного раствора хлористого аммония и диэтилового эфира. Полученную в результате смесь осветляли фильтрацией через кизельгур. Органическую фазу отделяли, сушили (над МgSО4) и упаривали, получая масло. Продукт очищали на хроматографической колонке, элюируя смесью этилацетата и гексана (увеличивая их соотношение от 10:90 об./об до 15:85 об./об.), что дало 2-метил-2-(2-нитро-4-метил-фенокси)пропаналь в виде масла (1,89 г).
ЯМР: 1,45 (6Н, с), 2,35 (3Н, с), 6,8-7,6 (3Н, м) и 9,85 (1Н, с).
П р и м е р 16. Используя методику, аналогичную описанной в примере 1, но применяя в качестве альдегидного компонента 2-метоксикоричный альдегид в качестве альдегидного компонента, была получена 4(Z)-6-[(2,4,5-цис)-2-(2Е-[метоксифенил] этенил)-4-(3-пиридил)-1,3-диоксан-5- ил]гексеновая кислота в виде масла, которое отверждается при хранении, давая твердое вещество с 18% выходом.
ЯМР: 1,60-1,80 (2Н, м), 2,15-2,65 (5Н, м), 3,85 (3Н, с), 4,00-4,30 (2Н, м), 5,10-5,50 (4Н, м), 6,30-6,40 (1Н, м), 6,85-7,85 (7Н, м) и 8,50-8,65 (2Н, м).
П р и м е р 17. 2-нефтальдегид (0,468 г) и пара-толуолсульфокислоту (0,22 г) добавляли к раствору 4(Z)-6-[2,2-диметил-4-(3-пиридил)-1,3-диоксан-цис-5-ил гексеновой кислоты (А, как описано в примере 1) (0,305 г) в ацетонитриле (10 мл) под током аргона. Смесь кипятили в сосуде с обратным холодильником 18 ч и затем охлаждали. Добавляли этил-ацетат (10 мл) и смесь экстрагировали 2М водным раствором гидроокиси натрия (60 мл). Основной экстракт подкисляли уксусной кислотой до рН 4 и экстрагировали этилацетатом (90 мл). Объединенные органические экстракты сушили (над МgSО4) и упаривали, получая масло, которое очищали на хроматографической колонке, элюируя смесью метанол/дихлор- метан (от 1: 10 до 1:5 об./об.). В результате получали 4(Z)-6-[2,4,5-цис)-2-(2-нафтил)-4-(3-пиридил)-1,3-диоксан-цис-5-ил]гексенову ю кислоту в виде твердого вещества (0,224 г) с температурой плавления 139-142оС.
ЯМР: 1,74 (2Н, м), 2,28 (4Н, м), 2,64 (1Н, м), 4,18-4,34 (2Н, м), 5,27 (3Н, м), 5,43 (1Н, м), 5,88 (1Н, с), 7,33-7,68 (4Н, м), 7,85-8,02 (5Н, м) и 8,52-8,63 (2Н, м) м/е 404 (М+Н)+;
данные микроанализа:
Найдено% С 73,5, Н 6,3, N 3,3.
С25Н25NО4 0,25 Н2О.
Вычислено% С 73,6, Н 6,3, N 3,4.
П р и м е р 18. Используя методику, аналогичную описанной в примере 14, но исходя из 3-бензилоксибензальдегида, была получена 4(Z)-6[(2,4,5-цис)-2-(3-бензилоксифенил)-4-(3-пиридил)-1,3-диоксан- цис-5-ил]гексеновая кислота в виде бесцветного твердого вещества с температурой плавления 125-128оС и 43%-ным выходом.
ЯМР: 1,75 (2Н, м), 2,28 (4Н, м), 2,55 (1Н, м), 4,12-4,28 (2Н, м), 4,85 (1Н, м), 5,12 (2Н, с), 5,27 (2Н, м), 6,98-7,75 (11Н,м), и 8,58 (2Н, м); m/е 459 (М+Н)+; данные микроанализа:
Найдено: С 72,8; С 6,4; N 2,9%
С28Н 29NО5
Вычислено: С 73,2; Н 6,4; N 3,0%
П р и м е р 19-39. Используя методику, аналогичную описанной в примере 1, но исходя из соответствующего альдегида формулы R1.СНО и диоксангексеновой кислоты А, были получены следующие соединения формулы (8).
П р и м е р 19. R1-3-(4-цианобензилокси)фенил; выделенный в виде твердого вещества с температурой плавления 149-150оС; неполный спектр ЯМР: 5,70 (1Н, с), 6,95 (1Н, м), 7,28 (4Н, м), 7,55 (2Н, м), 7,65 (2Н, д), 7,73 (1Н, д), 8,53 (1Н, м), 8,62 (1Н, с).
П р и м е р 20. R1-1-нафтил; выделенный в виде твердого вещества с температурой плавления 171-172оС; неполный спектр ЯМР: 6,3 (1Н, с), 7,32 (1Н, м), 7,52 (3Н, м), 7,85 (4Н, м), 8,22 (1Н, с), 8,52 (11, д), 8,63 (1Н, с).
П р и м е р 21. R1-4(4-цианобензилокси)фенил; выделенный в виде твердого вещества с температурой плавления 162-164оС; неполный спектр ЯМР: 5,69 (1Н, с), 6,97 (2Н, д), 7,36 (1Н, м), 7,52 (4Н, м), 7,70 (3Н, м), 8,53 (2Н, м).
П р и м е р 22. R1-2-бензилоксифенил; выделенный в виде твердого вещества с температурой плавления 142-144оС; неполный спектр ЯМР: 6,15 (1Н, с), 6,93 (1Н, д), 7,05 (1Н, т), 7,31 (7Н, м), 7,78 (2Н, с), 8,51 (1Н, д), 8,58 (1Н, с).
П р и м е р 23. R1-4-бензилоксифенил; выделенный в виде твердого вещества с температурой плавления 200-204оС; неполный спектр ЯМР: 5,72 (1Н, с); 7,02 (2Н, д), 7,18 (1Н, м), 7,40 (7Н, м), 7,72 (1Н, с), 8,48 (1Н,д), 8,53 (1Н, с).
П р и м е р 24. R1-4-(3-пиридилметокси)фенил; выделенный в виде твердого вещества с температурой плавления 174-177оС; неполный спектр ЯМР: 5,62 (1Н, с), 6,93 (2Н, д), 7,25 (2Н, д), 7,43 (2Н, д), 7,69 (2Н, с), 8,50 (4Н, м).
П р и м е р 25. R1-4/феноксифенил; выделенный в виде твердого вещества с температурой плавления 142-144оС; неполный спектр ЯМР: 5,72 (1Н, с); 7,07 (5Н, с), 7,35 (3Н,с), 7,55 (2Н, с), 7,78 (1Н, м), 8,57 (2Н, м).
П р и м е р 26. R1-3-феноксифенил; выделенный в виде твердого вещества с температурой плавления 130-132оС; неполный спектр ЯМР: 5,72 (1Н, с), 7,07 (4Н, м), 7,35 (6Н, с), 7,82 (1Н, м), 8,55 (2Н, м).
П р и м е р 27. R1-3-(3-пиридилметокси)-фенил; выделенный в виде твердого вещества с температурой плавления 104-105оС.
ЯМР: 5,70 (1Н, с), 6,98 (1Н, м), 7,18 (1Н, м), 7,33 (4Н, м), 7,71 (1Н, м), 7,88 (1Н, м), 8,55 (3Н, м), 8,82 (1Н, м).
П р и м е р 28. R1-2(4-цианобензилокси)фенил; выделенный в виде твердого вещества с температурой плавления 159-162оС; неполный спектр ЯМР: 6,15 (1Н, с), 6,88 (1Н, д), 7,09 (1Н, т), 7,37 (2Н, м), 7,47 (2Н, д), 7,58 (2Н, д), 7,77 (2Н, м), 8,55 (2Н, м).
П р и м е р 29. R1-2(3-пиридилметокси)фенил; выделенный в виде твердого вещества с температурой плавления 131-135оС; неполный спектр ЯМР: 6,15 (1Н, с), 6,92 (1Н, д), 7,08 (1Н, т), 7,30 (3Н, м), 7,72 (3Н, м), 8,60 (4Н, м).
П р и м е р 30. R1-4-бензилокси-3-нитрофенил; выделенный в виде твердого вещества с температурой плавления 150-152оС; неполный спектр ЯМР: 5,70 (1Н, с), 7,12 (1Н, д), 7,38 (6Н, с), 7,73 (2Н, м), 8,04 (1Н, д), 8,57 (2Н, д).
П р и м е р 31. R1-3-(-нафтилметокси)фенил; выделенный в виде твердого вещества с температурой плавления 115-117оС; неполный спектр ЯМР: 5,70 (1Н, с), 6,52 (1Н, м), 7,03 (1Н, м), 7,18 (1Н, д), 7,33 (3Н, м), 7,47 (3Н, м), 7,60 (1Н, д), 7,81 (3Н, м), 8,06 (1Н, м), 8,55 (2Н, м);
П р и м е р 32. R1-3(2,5-диметоксибензилокси)фенил; выделенный в виде твердого вещества с температурой плавления 53-54оС; неполный спектр ЯМР: 5,72 (1Н, с), 6,82 (2Н, м), 7,00 (1Н, м) 7,15 (2Н, м), 7,31 (4Н, м), 7,82 (1Н, м), 8,57 (2Н, м).
П р и м е р 33. R1-2-(4-пиридилметокси)фенил; выделенный в виде твердого вещества с температурой плавления 115-117оС, неполный спектр ЯМР: 6,14 (1Н, с); 6,87 (1Н, д), 7,0 (1Н,т), 7,12 (1Н, м), 7,60 (7Н, м), 8,6 (2Н, м).
П р и м е р 34. R1-2-хлор-1-нафтил; выделенный в виде твердого вещества с температурой плавления 188-190оС; неполный спектр ЯМР: 6,73 (1Н, с), 7,2 (1Н, с), 7,46 (2Н, м), 7,62 (1Н, м), 7,75 (3Н, м), 8,53 (2Н, м), 9,02 (1Н, д).
П р и м е р 35. R1-2-(2-пиридилметокси)фенил; выделенный в виде твердого вещества с температурой плавления 86-88оС; неполный спектр ЯМР: 6,18 (1Н, с), 6,92 (1Н, д), 7,05 (1Н, т), 7,18 (1Н, т), 7,29 (2Н, м), 7,45 (1Н, д), 7,57 (1Н, д), 7,76 (2Н, дд), 8,55 (3Н, м).
П р и м е р 36. R1-2(4-нитробензилокси)фенил; выделенный в виде твердого вещества с температурой плавления 166-168оС; неполный спектр ЯМР: 6,14 (1Н, с), 6,88 (1Н, д), 7,11(1Н, и), 7,32 (2Н, м), 7,50 (2Н, д), 7,76 (2Н, м), 8,16 (2Н, м), 8,59 (2Н, м).
П р и м е р 37. R1-3-бензилокси-4-метоксифенил, выделенный в виде твердого вещества с температурой плавления 128-130оС; неполный спектр ЯМР: 5,62 (Н, с), 6,90 (1Н, д), 7,12 (2Н, м), 7,44 (6Н, м), 7,68 (1Н, м), 8,55 (2Н, м).
П р и м е р 38. R1-3-(3-цианобензилокси)-4-метоксифенил; выделенный в виде твердого вещества с температурой плавления 148-149оС.
Неполный спектр ЯМР: 5,63 (1Н, с), 6,92 (1Н, д), 7,13 (2Н, м), 7,32 (1Н, м), 7,45 (1Н, м), 7,58 (1Н, м), 7,68 (2Н, м), 7,80 (1Н, с), 8,55 (2Н, м).
П р и м е р 39. R1-4-бензилокси-3-цианофенил; выделенный в виде твердого вещества с температурой плавления 164-165оС; неполный спектр ЯМР: 7,41 (7Н, м), 7,78 (3Н, м), 8,48 (1Н, дд), 8,55 (1Н, д).
Новые исходные бензальдегиды формулы R1.СНО были получены по одной и той же общей методике, исходя из соответствующих оксибензальдегида бензилбромида или (бромметил) пиридина. Эти исходные вещества совместно кипятили в присутствии избытка безводного карбоната калия в этилметилкетоне в сосуде с обратным холодильником 2-18 ч до того, как реакция не завершилась, о чем судили, используя метод тонкослойной хроматографии (ТLС) на силикагеле. Затем продукт выделяли упариванием всплывшей наверх реакционной смеси и оставшееся вещество очищали на хроматографической колонке, используя в качестве элюента систему гексан/этилацетат (вплоть до 30 об./об.). Полученные в результате бензальдегиды формулы R1.СНО использовали по возможности сразу же. Данные вещества имели следующие характеристики.
(1) 3(4-цианобензилокси)бензальдегид; температура плавления 96-97оС; неполный спектр ЯМР: 5,18 (2Н, с), 7,24 (1Н, м), 7,50 (5Н, м), 7,68 (2Н, д), 9,98 (1Н, с).
(2) 4-(4-цианобензилокси)бензальдегид; температура плавления 105-106оС; неполный спектр ЯМР: 5,23 (2Н, с), 7,06 (2Н, д), 7,54 (2Н,д), 7,69 (2Н,д), 7,86 (2Н, д), 9,90 (1Н, с).
(3) 2-бензилоксибензальдегид; температура плавления 42-44оС, неполный спектр ЯМР: 5620 (2Н, с), 7,03 (2Н, с), 7,44 (6Н, м), 7,86 (1Н, м), 10,58 (1Н, с).
(4) 4-(3-пиридилметокси)бензальдегид; температура плавления 75-77оС; неполный спектр ЯМР: 5,16 (2Н, с), 7,02 (2Н, м), 7,30 (1Н, м), 7,76 (3Н, м), 8,63 (2Н, м), 9,95 (1Н, с).
(5) 3-(3-пиридилметокси)бензальдегид; температура плавления 48-50оС; неполный спектр ЯМР: 5,09 (2Н, с), 7,47 (5Н, м), 7,70 (1Н, м), 8,47 (2Н, м), 9,90 (1Н, с).
(6) 2-(4-цианобензилокси)бензальдегид; температура плавления 102-103оС; неполный спектр ЯМР: 5,28 (2Н, с), 7,01 (2Н, м), 7,55 (3Н, м), 7,70 (2Н, м), 7,88 (1Н, м), 10,55 (1Н, с).
(7) 2-(3-пиридилметокси)бензальдегид; температура плавления 58-59оС.
(8) 4-бензилокси-3-нитробензальдегид; температура плавления 86-88оС; неполный спектр ЯМР: 5,34 (2Н, с), 7,24 (2Н, м), 7,42 (4Н, м), 8,03 (1Н, дд), 8,37 (1Н, д), 99,93 (1Н, с).
(9) 3-(1-нафтилметокси)бензальдегид; температура плавления 63-64оС; неполный спектр ЯМР: 5,55 (2Н, с), 7,27 (7Н, м), 7,52 (2Н, м), 7,87 (2Н, м), 8,03 (1Н, м), 8,98 (1Н, с).
(10) 3-(2,5-диметоксибензилокси)бензальдегид; получен в виде масла: неполный спектр ЯМР: 5,28 (2Н, с), 6,83 (2Н, д), 7,05 (1Н, м), 9,97 (1Н, с).
(11) 2-(4-пиридилметокси)бензальдегид; температура плавления 137-140оС; неполный спектр ЯМР: 5,33 (2Н, с), 7,20 (2Н, м), 7,52 (2Н, д), 7,71 (2Н, м), 8,58 (2Н, д), 10,48 (1Н, с).
(12) 2-(2-пиридилметокси)бензальдегид; температура плавления 67-68оС; неполный спектр ЯМР: 5,32 (2Н, с), 7,06 (2Н, м), 7,25 (1Н, м), 7,53 (2Н, м), 7,75 (1Н, м), 7,84 (1Н, дд), 8,61 (1Н, д), 10,62 (1Н, с).
(13) 2-(4-нитробензилокси)бензальдегид; температура плавления 110-111оС; неполный спектр ЯМР: 5,31 (2Н, с), 7,0 (1Н,д), 7,1 (1Н, т); 7,59 (3Н, м), 7,88 (1Н, дд), 8,28 (2Н, м), 10,36 (1Н, с).
(14) 3-бензилокси-4-метоксибензальдегид; получен в виде масла; неполный спектр ЯМР: 5,19 (2Н, с), 6,99 (1Н, м), 7,38 (7Н, м), 9,82 (1Н, с).
(15) 3-(3-цианобензилокси)-4-метоксидбензальдегид; температура плавления 113-114оС; неполный спектр ЯМР: 5,20 (2Н, с) 7,02 (1Н, д), 7,42 (1Н, д), 7,49 (2Н, м), 7,65 (2Н, м), 7,80 (1Н, м), 9,83 (1Н, с).
(16) 4-бензоилокси-3-цианобензальдегид; температура плавления 117-118оС; неполный спектр ЯМР: 5,32 (2Н, с), 7,14 (1Н, д), 7,42 (5Н, м), 8,03 (1Н, дд), 8,11 (1Н, д), (1Н, с).
П р и м е р 40-41. Используя методику, аналогичную описанной в примере 1, но исходя из 4(Z)-6-[(4,S,5R-2,2-диметил-4-(3- пиридил)-1,3-диоксан-5-ил] гексеновой кислоты и 2-метил-2-(2-нитро-4-метилфенокси)пропеналя и 2-(4-метоксифенокси)-2-метилпропаналя, соответственно были получены следующие вещества.
П р и м е р 40. 4(Z)-6-[(2S, 4S, 5 R)-2-[1-метил-1-(2-нитро-4-метилфенокси)этил-4-(3- пиридил)-1,3-диоксан- 5-ил] гексеновая кислота в виде твердого вещества с выходом 25% с 25[альфа]D -117,6o (ЕtОН, с 0,635).
ЯМР: 1,45 (6Н, с), 1,5-1,75 (2Н, м), 2,2-2,4 (8Н, м), 3,9-4,2 (2Н, м), 4,8 (1Н, с), 5,05-5,5 (3Н, м), 7,15-7,0 (5Н, м), 8,4-8,55 (2Н, м).
П р и м е р 41. 4(Z)-6-[(2S, 4S, 5R)-2-1(4-метоксифенокси)-1-метил-этил-4-(3-пири- дил)-1,3-диоксан-5-ил] гексеновая кислота в виде твердого вещества с выходом 28% с 25[альфа]D-122,9о (ЕtОН, с, 0,59).
ЯМР: 1,35 (3Н, с), 1,4 (3Н, с), 1,55-1,8 (2Н, м), 2,2-2,6 (5Н, м), 3,75 (3Н, с), 3,9-4,2 (2Н, м), 4,75 (1Н, с), 5,05-5,5 (3н, м), 6,75-7,7 (6Н, м), 8,5-8,6 (2Н, м).
Исходное оптическое активное производное 2,2-диметил-1,3-диоксангексеновой кислоты получали следующим образом.
I) К 1,53 М раствор бутиллития и гексана (23,9 мл) добавляли раствор 4S-(-)-изопропил-2-оксазолидинона (4,68 г) в сухом ТНF (75 мл), охлаждали до (-78)оС в токе аргона. Смеси давали возможность нагреться до (-50)оС и затем перемешивали 30 мин. Затем смесь вновь охлаждали до (-78оС) и по каплям добавляли раствор 4-пентеноилхлорида (4,33 г) в сухом ТНF (10 мл). После добавления смесь перемешивали при (-78оС) в течение 30 мин и затем давали возможность нагреться до (-20)оС. Добавляли насыщенный водный раствор хлористого аммония (20 мл) и смесь экстрагировали этилацетатом (3х100 мл). Объединенные органические фазы сушили (над МgSО4 и упаривали. Остаток очищали на хроматографической колонке, элюируя системой этилацетат/гексан (20:80 об. /об. ), получая (4S)-4-изопропил-3-(4-пентеноил) оксазолидин-2-он (А) (6,34 г) в виде масла.
ЯМР: 0,85-0,95 (6Н, м), 2,2,3-2,5 (3Н, м), 2,9-3,2 (2Н, м), 4,15-4,5 (3Н, м), 4,95-5,15 (2Н, м), 5,75-6,0 (1Н, м).
II) 1М раствор трифлата дибутилбора в дихлорометане (32,7 мл) добавляли к раствору соединения А (6,28 г) в сухом дихлорметане (110 мл), охлаждали до 5оС в токе аргона и затем добавляли диизопропилэтиламин (6,25 мл). Реакционную смесь перемешивали 30 мин при 5оС и затем охлаждали до (-78)оС. По каплям добавляли 3-пиридинкетбоксиальдегид (3,1 мл). Смесь перемешивали 30 мин при (-78)оС и затем давали нагреться до (-50)оС в течение 30 мин.
Охлаждающую ванну удаляли и реакционную смесь перемешивали при комнатной температуре 2 ч. Затем смесь охлаждали до 5оС и добавляли перекись водорода (11,5 мл, водный раствор, 30 мас./об.). Смесь перемешивали 30 мин, затем вливали в воду (50 мл) и экстрагировали дихлорметаном (3х100 мл). Объединенные экстракты сушили над МgSО4 и упаривали. Остаток очищали на хроматографической колонке, элюируя системой этилацетат/гексан (1:1 об./об. постепенно переход к 100% этилацетату), получая (4S)-3-(3-[(2S)-2-[(1S-1-окси-1-(3-пиридил/метил] пент-4-оксил)-4-изопро- пилоксазолидин-2-он (В) в виде твердого вещества с температурой плавления 112-113оС (после перекристаллизации из толуола);
25[альфа]D+136,0 (ЕtОН, с, 0,311);
ЯМР: 0,85 (6Н, дд, J=7 Гц), 2,15-2,7 (4Н, м), 4,0-4,2 (2Н, м), 4,3-4,55 (2Н, м), 4,95-5,1 (3Н, м), 5,65-5,9 (1Н, м), 7,25-7,35 (1Н, м), 7,75-7,85 (1Н, м), 8,5-8,65 (2Н, м).
III) Раствор метоксида натрия (30 мас.) в метаноле (3,65 мл) добавляли к раствору вещества В (5,76 г) в метаноле (40 мл), охлаждали до 5оС. Смесь перемешивали 15 мин и затем добавляли насыщенный водный раствор хлористого аммония (10 мл) и диэтиловый эфир (50 мл). Добавляли воду в количестве, достаточном для растворения любых выпавших в осадок неорганических веществ, и затем смесь экстрагировали диэтиловым эфиром (3х50 мл). Объединенные экстракты сушили (над МgSО4) и упаривали.
Остаток очищали на хроматографической колонке, элюируя этилацетатом, получая метиловый эфир (2S)-2-[(1S)-1-окси- 1-(3-пиридили)метил]-4-еновой кислоты (С) (3,245 г) в виде масла;
ЯМР: 2,3-2,6 (2Н, м), 2,8-2,9 (1Н, м), 3,6 (3Н, с), 4,95-5,1 (3Н, м), 5,65-5,85 (1Н, м), 7,25-7,35 (1Н, м), 7,7-7,75 (1Н, м), 8,45-8,6 (2Н, м).
IV) Раствор вещества С (3,88 г) в тетрагидрофуране (10 мл) добавляли по каплям к охлажденной суспензии литийалюминий гидрида (767 мг) в тетрагидрофуране (50 мл) с такой скоростью, чтобы температура сме- си не превышала 10оС. После завершения этой операции смесь перемешивали при 5оС в течение 4 ч. Добавляли этилацетат (20 мл), затем насыщенный водный раствор хлористого аммония (10 мл) и воду (10 мл). Смесь экстрагировали этилацетатом (3х50 мл). Объединенные экстракты сушили над МgSО4 и упаривали. Остаток очищали на хроматографической колонке, элюируя этилацетатом, постепенно переходя к смеси метанол/этилацетат (1: 9 об./об.), получая (1S, 2R)-2-аллил-1-(3-пиридил)-1,3-пропандиол (D) (2,69 г) в виде масла;
ЯМР: 1,65-1,8 (1Н, м), 1,95-2,15 (2Н, м), 3,15-3,45 (2Н, м), 4,4-4,5 (1Н, м), 44,75-5,0 (3Н, м), 5,25 (1Н, д, J=7 Гц), 5,6-5,86 (1Н, м), 7,3-7,4 (1Н, м), 7,65-7,7 (1Н, м), 8,4-8,5 (2Н, м).
V) Моногидрат пара-толуолсульфокислоты (2,91 г) добавляли к раствору вещества D (2,68 г) в 2,2-диметоксипропане (15 мл) и смесь перемешивали 18 ч. Добавляли триэтиламин (10 мл) и смесь распределяли в системе диэтиловый эфир (50 мл) вода (20 мл). Органический слой сушили (над МgSО4) и упаривали. Остаток очищали на хроматографической колонке, элюируя смесью этилацетат/гексан (1: 1) об./об.), получая (4S, 5R)-5-аллил-2,2-диметил-4-(3-пиридил)-1,3-диоксан.
(Е) (2,39 г) в виде масла;
ЯМР: 1,53 (3Н, с), 1,55 (3Н, с), 1,6-1,75 (1Н, м), 1,9-2,0 (1Н, м), 2,3-2,5 (1Н, м), 3,85-4,2 (2Н, м), 43,9-5,0 (2Н, м), 5,27 (1Н, д, J=3 Гц), 5,45-5,7 (1Н, м), 7,25-7,35 (1Н, м), 7,65-7,7 (1Н, м), 8,5-8,6 (2Н, м).
VI) Через охлажденный до (-78)оС раствор аллилового соединения (Е) (530 мг) в метаноле (30 мл) пропускали озон до тех пор, пока система не окрасилась в синий цвет. Смесь продували аргоном и затем добавляли метилсульфид (1,6 мл). Далее смесь перемешивали при комнатной температуре 18 ч, упаривали под вакуумом и распределяли в системе диэтиловый эфир (50 мл) вода (20 мл). Органический слой сушили (над МgSО4) и упаривали. Остаток очищали на хроматографической колонке, элюируя смесью метанола и хлористого метилена (5:95 об. /об.), получая 2-[4S,5R-2,2-диметил-4-(3-пиридил)-1,3-диоксан-5- ил]ацетальдегид (F) в виде масла;
ЯМР: 1,53 (3Н, с), 1,55 (3Н, с), 2,15-2,4 (2Н, м), 2,85-32,95 (1Н, м), 3,8-3,85 (1Н, м), 4,25-4,35 (1Н, м), 5,28 (1Н, д, J=3 Гц), 7,25-7,7 (2Н, м), 8,5-8,6 (2Н, м), 9,6 (1Н, с).
Оптическую чистоту оценили, как превышающую 99% с помощью протонного ЯМР путем добавления (R)-(-)-2,2,2-трифтор-1-(9-нитрил)этанола и исследования области 2,7-2,9 (дельта), где были обнаружены 4 дублета, локализованных на 2,77, 2,71, 2,82 и 2,85 (1Н, СН-СНО).
VII) Ацетальдегид (F) затем превращали в 4 (Z)-6-[(4S,5R)2,2-диметил-4-(3-пиридил)-1,3-диоксан-5-ил] гексеновую кислоту, имеющую 25[альфа]D-113,3 (ЕtОН, с 0,465) и спектр ЯМР, по существу идентичный спектру рацемического соединения, описанного в примере 1, используя методику, аналогичную описанной в разделе (V) примера 1.
П р и м е р 42. Моногидрат пара-толуолсульфокислоты (409 г) добавляли к перемешиваемому раствору 4(Z)-6-[2,2-диметил-4- (3-пиридил)-1,3-диоксан-цис-5-ил] гексено-вой кислоты (570 мг) и 2-(2-циано-4-метилфенокси)-2-метилпропаналя (568 мг) в ацетонитриле (5 мл) и смесь перемешивали при 80оС в течение 18 ч. Добавляли воду (40 мл) и 2М раствор гидроокиси натрия (4 мл) и смесь промывали диэтиловым эфиром (2х20 мл), водную фазу подкисляли уксусной кислотой и экстрагировали этилацетатом (3х25 мл). Экстракты промывали насыщенным раствором соли (2х15 мл), сушили (над МgSО4) и упаривали. Оставшееся смолистое вещество очищали методом МРLС, элюируя смесью этилацетат (гексан) уксусная кислота (70:30:1) об./об.), получая 4(Z)-6-[(2,4,5-цис)-2-(1-(2-циано-4-метилфенокси)-1-метил- этил)-4-(3-пиридил)-1,3-диоксан-5-ил]гексе- новой кислоты полугидрат (249 мг) в виде белой пены;
ЯМР: 1,48 (3Н, с), 1,50 (3Н, с), 1,57 (1Н, м), 1,72 (1Н, м), 2,09 (0,75Н, с), 2,27 (4Н, м), 2,30 (3Н, с), 2,43 (1Н, м), 4,02 (1Н, дм, J=11 Гц), 4,19 (1Н, дд, J=11, 1,5 Гц), 4,98 (1Н, с), 5,18 (1Н, д, J=2 Гц), 5,25 (1H, м), 5,41 (1Н, м), 5,77 (1Н, ш), 7,09 (1Н, д, J=9 Гц), 7,28 (3Н, м), 7,57 (1Н, дм, J=8 Гц), 8,45 (1Н, шс), 8,51 (1Н, шд. J=4 Гц); данные микроанализа:
Найдено: С 68,0, Н 6,5, N 6,0%
С26Н30N2О5.1/2 Н2О.
Вычислено: С 68,0, Н 6,7, N 6,1%
m/е 451 (М+NН)+
Необходимый исходный альдегид получали следующим образом.
I) Безводный карбонат калия (14,0 г) добавляли к перемешиваемому раствору 2-бром-4-метилфенола (19,0 г) и этилового эфира 2-бром-2-метилпропионов и кислоты (27,7 г) и 2-бутенона (80 мл), смесь кипятили в сосуде с обратным холодильником 3 ч, затем охлаждали до окружающей температуры и добавляли воду (400 мл). Эту смесь экстрагировали диэтиловым эфиром (1х300 мл, 2х150 мл) и экстракты, собранные вместе, промывали 1М раствором гидроокиси натрия (2х100 мл), водой (2х100 мл) и насыщенным раствором соли (1х100 мл), затем ушили (над МgSО4) и испарили растворитель. Оставшееся в результате масло перегоняли при пониженном давлении, получая этиловый эфир 2-(2-бром-4-метилфенокси)-2-метилпропионовой кислоты (А) (18,7 г) в виде масла с температурой кипения 116-118оС при 0,5 мм Нg;
ЯМР: 1,28 (3Н, т, J=7 Гц), 1,60 (6Н, с), 2,26 (3Н, с), 4,26 (2Н, к, J=7 Гц), 6,78 (1Н, д, J=8 Гц), 6,96 (1Н, дд, J=8, 1,5 Гц), 7,35 (1Н, д, J=1,5 Гц), m/е 318 (М+NН4)+.
II) Цианид меди (I) (3,76 г) добавляли к перемешиваемому раствору соединения А (10,54 г) в диметилформамиде (20 мл) и затем нагревали в сосуде с обратным холодильником (температура бани 180оС) в течение 4,5 ч. Получившуюся смесь после охлаждения добавляли к перемешиваемому раствору состава: хлорид железа (III)/12М хлористоводородная кислота/вода (14 г:3,5 мл:55 мл) и перемешивали 30 мин. Этот раствор экстрагировали дихлорметаном (1х100 мл, 2х50 мл), затем сушили (над МgSО4) и растворитель удaляли в вакууме. Получившийся продукт очищали методом МРLС, элюируя 10 об. этилацетатом в гексане. В результате был получен этиловый эфир 2-(2-циено-4-метилфенокси)-2-метил-прописной кислоты (В) (7,01 г) в виде бесцветного масла, которое медленно кристаллизовалось при хранении при 4оС; температура плавления соединения 54-56оС;
ЯМР: 1,24 (3Н, т, J=7 Гц), 1,65 (6Н, с), 2,91 (3Н, с), 4,23 (2Н, к, J=7 Гц), 6,74 (1Н, д, J= 9 Гц), 7,21 (1Н, м), 7,35 (1Н, д, J=2Гц), m/е 265 (М+NН4)+.
III) К перемешиваемому охлажденному до (-70)оС раствору вещества В (2,47 г) в сухом толуоле под током аргона по каплям добавляли 1,5 М раствор диизобутилалюминийгидрида в толуоле (7,3 мл). Перемешивание продолжали еще в течение 30 мин при (-70)оС, когда добавляли 10 об. раствор метанола в толуоле (2 мл) и смесь доводили до окружающей температуры. Раствор добавляли к энергично перемешиваемой смеси воды со льдом (50 мл), перемешивали 2 ч и затем фильтровали через кизельгур. Органическую фазу отделяли, а водную фазу экстрагировали диэтиловым эфиром (2х75 мл). Органические фазы, собранные вместе, промывали насыщенным раствором соли (2х40 мл), сушили (над МgSО4) и упаривали. Полученный в результате продукт очищали методом МРLС, элюируя 10 об. этилацетатом в гексане, получая масло, которое медленно кристаллизовалось. Растирание вещества в порошок в гексане и последующая фильтрация привели к получению 2-(2-циано-4-метилфенокси)-2-метилпропаналя (600 мг) с температурой плавления 60-64о;
ЯМР: 1,50 (6Н, с), 2,31 (3Н, с), 6,74 (1Н, д, J=9 Гц), 7,26 (1Н, м), 7,39 (1Н, д, J=2 Гц), 9,83 (1Н, с); m/е 221 (М+NН4)+.
П р и м е р 43. Способом, аналогичным указанному в примере 42 но, исходя из 2-(2-циано-5-метилфенокси)-2-метилпропаналя вместо 2-(2-циано-4-метилфенокси)-2-метилпропаналя и нагревая реакционную смесь в течение 48 ч вместо 18 ч, был получен полугидрат 4(Z)-6[(2,4,5-цис)-2-(1-(2-циано-5-метилфенокси)-1-метилэтил)-4-(3- пиридил)-1,3-диоксан-5-ил]гексановой кислоты (17%) в виде белой пены;
ЯМР: 1,49 (3Н, с), 1,52 (3Н, с), 1,59 (1Н, м), 1,73 (1Н, м), 2,28 (4Н, м), 2,36 (3Н, с), 2,43 (1Н, м), 4,03 (1Н, дм, J=11 Гц), 4,20 (1Н, дд, J=11,1 Гц), 4,97 (1Н, с), 5,17 (1Н, д, J=2 Гц), 5,23 (1Н, м), 5,41 (1Н, м), 6,92 (1Н, дд, J=7,1 Гц), 7,02 (1Н, с), 7,30 (1Н, м), 7,36 (1Н, д, J=7 Гц), 7,57 (1Н, м), 8,49 (2Н, м); данные микроанализа:
Найдено: С 68,1, Н 6,5, N 5,9%
С26Н30N2О5 1/2 Н2О
Вычислено: С 68,0, Н 6,7, N 6,1%
m/е 451 (М+NН)+
Необходимый альдегид получали следующим образом.
I) Используя тот же способ, что и описанный в примере 48 (I), но, исходя из 2-хлор-5-метилфенола вместо 2-бром-4-ме- тилфенола и кипятя реакционную смесь в сосуде с обратным холодильником 18 ч вместо 3 ч, был получен этиловый эфир 2-(2-хлор-5-метилфенокси)-2-метилпропионовой кислоты (72%) в виде бесцветного масла с температурой кипения 109-110оС при 0,5 мм Нg;
ЯМР: 1,28 (3Н, т, J=7 Гц), 1,60 (6Н, с), 2,26 (3Н, с), 4,26 (3Н, с), 4,26 (2Н, к, J=7 Гц), 6,71 (2Н, м), 7,21 (1Н, д, J=7 гц); m/е 274 (М+NН4)+
II) Способом, аналогичным описанному в примере 42 (II), но исходя из этилового эфира (2-(2-хлор-5-метилфенокси)-2-метил-фенокси)-2-метилпропионовой кислоты и используя DМРU в качестве растворителя вместо диметилформамида, а также нагревая реакционную смесь 18 ч при 200оС, после испарения дихлорметанового экстракта была получена жидкость. Ее растворяли в диэтиловом эфире (200 мл), раствор промывали водой (3х50 мл), чтобы удалить DМРU, затем сушили (над МgSО4) и упаривали. Остаток очищали методом МРLМ, элюируя 10 об. этилацетатом в гексане, получая этиловый эфир 2-(2-циано-5-метилфенокси)-2-метилпропионовой кислоты (38%) в виде бесцветного масла;
ЯМР (250 МГц, СDCl3): 1,24 (3Н, т, J=7 Гц), 1,67 (6Н, с), 2,33 (3Н, с), 4,24 (2Н, к, J=7 Гц), 6,60 (1Н, с), 6,84 (1Н, дм, J=8 Гц), 7,44 (1Н, д, J=8 Гц);
m/е 265 (М+NН4)+.
III) Способом, аналогичным описанному в примере 42 (III), но исходя из этилового эфира 2-(2-циано-5-метилфенокси)-2-метил-пропионовой кислоты вместо этилового эфира 2-(2-циано-4-метилфенокси)-2-метилпропионовой кислоты, был получен 2-(2-циано-5-метилфенокси)-2-метилпропаналь (28%) в виде твердого кристаллического вещества с температурой плавления 57-59оС;
ЯМР: (250 МГц, СDCl3); 1,53 (6Н, с), 2,34 (3Н, с), 6,62 (1Н, с), 6,91 (1Н, д, J=8 Гц), 7,47 (1Н, д, J=8 Гц), 9,34 (1Н, с), m/е 221 (М+NН4)+.
П р и м е р 44. Используя методику, аналогичную описанной в пример 43, но, исходя из 2-(2-циано-4-метоксифенокси)2-метилпропаналя вместо 2-(2-циано-4-метилфенокси)-2-метилпропаналя, была получена 4-(Z)-6[(2,4,5-цис)-2-(1-(2-циано-4-метокси-фенокси)-1-метилэтил)-4-(3-пирид ил)-оксан-5ил] гексеновая кислота с выходом в 10% температура плавления 130-140оС.
ЯМР: 1,46 (3Н, с), 1,48 (3Н, с), 1,58 (1Н, м), 1,72 (1Н, м), 2,26 (4Н, м), 2,42 (1, м), 3,76 (3Н,м), 4,03 (1Н, дм, J=11 Гц), 4,20 (1Н, дд, J=11, 1,5 Гц), 4,97 (1Н, с), 5,17 (1Н, д, J=2 Гц), 5,24 (1Н, м), 5,40 (1Н, м), 6,93 (1Н, д, J=2,5 Гц), 7,02 (1Н, дд, J=8 Гц, 2,5), 7,12 (1Н, д, J=8 Гц), 7,30 (1Н, м), 7,57 (1Н, м), 8,50 (2Н, м).
Данные микроанализа:
Найдено: С 66,5, Н 6,2, N 5,8%
С26Н30N2О6
Вычислено: С 66,9, Н 6,5 N 6,0%
m/е 467 (М+NН)+.
Исходный альдегид получали следующим образом.
I) 2М раствор гидроокиси натрия медленно добавляли к перемешиваемому раствору 2-окси-5-метоксибензальдегида (9,4 г) и хлористоводородного гидроксиамина (5,37 г) в метаноле (100 мл) до тех пор, пока не достигли рН 7. Перемешивание продолжали еще 20 мин и затем смесь выливали в воду (600 мл). После выдерживания смеси при температуре около 4оС в течение 2 ч отфильтровывали белое твердое вещество, промывали его водой и сушили, получая 2-окси-5-метоксибензальдекси (А) (9,84 г) в виде белых кристаллов с температурой плавления 119-120оС;
ЯМР: 3,77 (3Н, с), 6,70 (1Н, д, J=2 Гц), 6,90 (2Н, м)= 7,47 (1Н, с), 8,19 (1Н, с), 9,39 (1Н, m)
m/е 168 (М+NН)+.
II) Раствор соединения А (9,84 г) в уксусном ангидриде (50 мл) кипятили 6 ч в сосуде с обратным холодильником. Смесь оставляли на ночь при комнатной температуре и затем вливали в энергично перемешиваемую смесь воды со льдом (400 мл). Спустя 1 ч образовавшееся в результате твердое вещество отделяли фильтрованием. К раствору этого твердого вещества в метаноле (50 мл) при перемешивании добавляли 2М раствор гидроокиси натрия (150 мл). Перемешивание продолжали еще 1 ч. Полученный прозрачный раствор был промыт диэтиловым эфиром (50 мл), подкислен 2М раствором соляной кислоты до рН 4 и экстрагирован этилацетатом (3х50 мл). Экстракты, собранные вместе, промывали водой (20 мл), затем насыщенным раствором соли (20 мл), сушили над МgSО4 и растворитель упаривали, получая 2-циено-4-метоксифенол (В) (783 г) в виде белого твердого вещества с температурой плавления 128-138оС;
ЯМР (250 МГц, СDCl3 (DМSО.d6);
3,66 (3Н, с), 6,86 (3Н, м); m/е 167
(М+NН4)+.
III) Безводный карбонат калия (1,38 г) добавляли к перемешиваемому раствору соединения В (745 мг) и 2-бром-2-метилпропаналя (755 мг) в тетрагидрофуране (20 мл). Смесь кипятили в сосуде с обратным холодильником в 1 ч и затем составляли на ночь при комнатной температуре. Добавляли воду (20 мл), смесь экстрагировали диэтиловым эфиром (2х20 мл). Экстракты, собранные вместе, промывали водой (2х10 мл), затем насыщенным раствором соли (10 мл) и сушили (над МgSО4). Испарение растворителя и очистка оставшегося масла методом хроматографии среднего давления (МРLС) с элюированием 25 об. этилацетатом в гексане привели к получению 2-(2-циано-4-метоксифенокси)-2-метилпропаналя (677 мг) в виде бесцветного масла;
ЯМР: 1,47 (6Н, с), 3,80 (3Н, с), 6,83 (1Н, д, J=9 Гц), 7,03 (2Н, м), 9,86 (1Н, с); m/е 237 (М+NН4)+.
П р и м е р 45. Используя методику, аналогичную описанной в примере 1, но, исходя из 2-метил-2-(2-метилсульфонилфенокси)пропаналя в качестве альдегида, была получена 4 (Z)-6-[(2,4,5-цис)-2-(1-метил-1-(4-метилсульфонилфенокси)этил)-4-метилсуль- фoнилфенокси)этил)-4-(3-пиридил)-1,3-дио- ксан-5-ил]гексеновая кислота в виде масла с 29% выходом;
ЯМР: 1,6 (6Н, с), 1,7-2,6 (7Н, м), 3,2 (3Н, с), 4,0-4,25 (2Н, м), 5,1 (1Н, с), 5,2-5,5 (3Н, м), 7,1-8,0 (6Н, м), 8,5-8,6 (2Н, м).
Необходимый альдегид был получен, исходя из 2-(метилтио)фенола по методике, аналогичной описанной для 2-метил-2-(4-метилсульфонилфенокси) пропаналя в примере 12 2-метил-2-(2-метилсульфонилфенокси) пропаналь был получен в виде твердого вещества с 51% выходом (от карбоксилата);
ЯМР: 1,55 (6Н, с)= 3,25 (3Н, с), 6,85-7,55 (3Н, м), 8,0-8,5 (1Н, м), 9,85 (1Н, с) с выделением следующим промежуточных продуктов:
а) этилового эфира 2-метил-2-(2-(метилтио)фенокси) пропионовой кислоты в виде масла (38% выход по фенолу),
ЯМР: 1,25 (3Н, т, J=7 Гц), 1,6 (6Н, с), 2,4 (3Н, с), 4,25 (2Н, к, J=7 Гц), 6,7-7,2 (4Н, м) и
б) этилового эфира 2-метил-2-(2-метилсульфонилфенокси) пропионовой кислоты в виде масла (89% выход по соединению (а)
ЯМР: 1,25 (3Н, т, J=7 Гц), 1,7 (6Н, с), 3,25 (3Н, с), 4,3 (2Н, к, J= 7 Гц), 6,9-7,55 (3Н, м), 7,95-8,05 (1Н, м). примере 12 2-метил-2-(2-метилсульфонилфенокси) пропаналь был получен в виде твердого вещества с 51% выходом (от карбоксилата);
ЯМР: 1,55 (6Н, с), 3,25 (3Н, с), 6,85-7,55 (3Н, м), 8,0-8,5 (1Н, м), 9,85 (1Н, с) с выделением следующих промежуточных продуктов:
а) этилового эфира 2-метил-2-(2-(метилтио)фенокси) пропионовой кислоты в виде масла (38% выход по фенолу),
ЯМР: 1,25 (3Н, т, J=7 Гц), 1,6 (6Н, с), 2,4 (3Н, с), 4,25 (2Н, к, J=7 Гц), 6,7-7,2 (4Н, м) и
б) этилового эфира 2-метил-2-(2-метилсульфонилфенокси) пропионовой кислоты в виде масла (89% выход по соединению (а)
ЯМР: 1,25 (3Н, т, J=7 Гц), 1,7 (6Н, с), 3,25 (3Н, с), 4,3 (2Н, к, J= 7 Гц), 6,9-7,55 (3Н, м), 7,95-8,05 (1Н, м).
П р и м е р ы 46-47. Используя методику, аналогичную описанной в примере 1, но исходя из 2-метил-2-(метилтиофенокси)пропаналя и 2-метил-2-(4-метилтиофенокси)про- паналя, соответственно были получены:
П р и м е р 46. 4(Z)-6-(2,4,5)-2-(1-метил-1-(2-метилтиофенокси)этил)-4-(3-пиридил)1,3- диоксан/5-ил гексановая кислота в виде твердого вещества с 45% выходом,
ЯМР: 1,45 (3Н, с), 1,5 (2Н, м), 1,55 (2Н, м), 2,2-2,5 (8Н, м), 4,0-4,25 2Н (2Н, м), 5,0-(1Н, с), 5,1-5,5 (3Н, м), 7,0-7,7 (6Н, м), 80,5-8,6 (2Н, м).
П р и м е р 47. 4(Z)-6-[(2,4,5-цис)-2-(1-метил-1-(4-метилтиофенокси)этил)-4-4-(3-пи-ридил)-1 3- диоксан-5-ил]гексеновая кислота в виде твердого вещества с 26% выходом,
ЯМР: 1,35 (3Н, с), 1,4 (3Н, с), 1,6-1,8 (2Н, м), 2,2-2,5 (8Н, м), 3,95-4,25 (2Н, м), 4,75 (1Н, с), 5,1-5,5 (3Н, м), 6,0-7,75 (6Н, м), 8,5-8,6 (2Н, м).
Исходные альдегиды были получены способом, аналогичным описанному для 2-метил-2-(4-метилсульфонилфенокси)пропа- наля в примере 12:
а) 2-метил-2-(2-метилтиофенокси)пропаналь, выделенный в виде масла;
ЯМР: 1,45 (6Н, с), 2,4 (3Н, с), 6,7-7,2 (4Н, м), 9,9 (1Н, с); выход 53% путем восстановления этилового эфира 2-метил-2-(2-метилтиофенокси)пропионовой кислоты: и
б) 2-метил-2-(4-метилтиофенокси)пропаналь, выделенный в виде масла; ЯМР: 1,4 (6Н, с), 2,45 (3Н, с), 6,75-7,25 (4Н, м), 9,85 (1Н, с).
П р и м е р 48. Используя методику, аналогичную описанной в примере 1, но исходя из 2-тиофеноксибензальдегида в качестве альдегида, была получена 4(Z)-6-[2,4,5-цис)-4-(3-пиридил)-2-(2-тио-фе- ноксифенил)-1,3-диоксан-5-ил] гексеновая кислота в виде твердого вещества с 26% выходом;
ЯМР: 1,7-1,9 (2Н, м), 2,2-2,5 (5Н, м), 4,1-4,3 (2Н, м), 5,2 5,5 (3H, м), 6,15 (1H, м), 7,1-7,9 (11Н, м), 8,45-8,6 (2Н, м).
2-тиофеноксибензальдегид был получен известным способом из 2-фторбензальдегида и тиофенола.
П р и м е р ы 49-50. Используя методику, аналогичную описанной в примере 1, но, исходя из 2-метил-2-(2-нитро-4-метоксифенокси)пропаналя и 2-метил-2-(2-метил-6-нитрофенокси)пропаналя, соответственно были получены:
П р и м е р 49. 4(Z)-6-[(2,4,5-цис)-2-(1-метил)-1-(2-нитро-4-метоксифенокси)этил)-4- (3-пиридил)-1,3-диоксан-5-ил] гексеновая кислота в виде твердого вещества с 10%-ным выходом;
ЯМР: 1,35 (6Н, с), 1,45-1,7 (2Н, м), 2,1-2,4 (5Н, м), 3,75 (3Н, с), 3,85-4,1 (2Н, м), 4,75 (1Н, с), 5,0-5,4 (3Н, м), 6,9-7,55 (5Н, м), 8,4-8,5 (2Н, м); и
П р и м е р 50. 4(Z)-6-[(2,4,5-цис)-2-(1-метил-1-(2-метил-6-нитрофенокси)этил)-4-(3-пиридил) 1,3-диоксан-5-ил] гексеновая кислота в виде твердого вещества с 7% выходом;
ЯМР: 1,25 (3Н, с); 1,3 (3Н, с); 1,45-1,65 (2Н, м), 2,05-2,4 (8Н, м), 3,8-4,05 (2Н, м), 4,7 (1Н, с), 5,0-5,5 (3Н, м), 6,95-7,5 (5Н, м), 8,35-8,5 (2Н, м).
Исходные альдегиды были получены способом, аналогичным описанному в примере 12 для 2-метил-2-(4-метилсульфонилфенокси)пропаналя:
а) 2-метил-2-(2-нитро-4-метоксифенокси)пропаналь, выделяемый в виде масла оранжевого цвета;
ЯМР: 1,45 (6Н, с), 3,8 (3Н, с), 6,9-7,3 (3Н, м), 9,85 (1Н, с); с выходом 41% путем восстановления этилового эфира 2-метил-2-(2-нитро-4-метоксифенокси)пропионовой кисло- ты, в свою очередь получаемого в виде масла;
ЯМР: 1,3 (3Н, т, J=7 Гц), 1,6 (6Н, с), 3,8 (3Н, с), 4,25 (2Н, к, J=7 Гц), 7,0-7,3 (3Н, м);
с выходом 8% путем алкилирования 4-метокси-2-нитрофеноксола; и
б) 2-метил-2-(2-метил-6-нитрофенокси)пропаналь, выделяемый в виде масла желтого цвета;
ЯМР: 1,35 (6Н, с), 2,3 (3Н, с), 7,1-7,6 (3Н, м), 9,85 (1Н, с); с выходом 65% путем восстановления этилового эфира 2-метил-2-метил-2-(2-метил-6-нитрофенокси)пропионовой кислоты, в свою очередь получаемого в виде масла;
ЯМР: 1,35 (3Н, т, J=7 Гц), 1,5 (6Н, с), 2,3 (3Н, с), 4,25 (2Н, к, J= 7 Гц), 7,05-7,55 (3Н, м);
с выходом 8% путем алкилирования 2-метил-6-нитрофенола.
П р и м е р 51. Используя методику, аналогичную описанной в примере 1, но исходя из 2-бензоилбензальдегида в качестве альдегида, была получена 4(Z)-6-[(2,4,5-цис(2-(2-бензоилфенил)-4-(3-пиридил)2,3- диоксан-5-ил] гексеновая кислота в виде твердого вещества с выходом 64%
ЯМР: 1,15-1,45 (2Н, м), 1,85-2,25 (5Н, м), 3,6-3,9 (2Н, м), 4,7-5,3 (3Н, м), 5,55 (1Н, м), 6,8-7,65 (11Н, м), 8,05-8,2 (2Н, м).
2-бензоилбензальдегид получали в виде твердого вещества с выходом 82%
[ЯМР: 7,4-8,1 (9Н, м), 10,05 (1Н, с)] путем окисления 2-(альфа-оксибензил)бензилового спирта в системе оксалилхлорид/диметилсульфоксид при изменении температуры от (-50)оС до комнатной; 2-(альфа-оксибензил)бензиловый спирт в свою очередь получали в виде твердого вещества с выходом 26%
ЯМР: 2,95 (1Н, шс), 4,45-4,65 (2Н, м), 6,05 (1Н, с), 7,2-7,4 (9Н, м)
путем восстановления 2-бензоилбензойной кислоты литийалюминий гидридом.
П р и м е р ы 52-53. Используя методику, аналогичную описанной в примере 17, но, исходя из соответствующего бензальдегида формулы R1.СНО, были получены следующие соединения:
П р и м е р 52. 4(Z)-6-[(2,4,5-цис)-2-(1-метил-1-(фенилтио)этил)-4-(3-пиридил)-1,3-ди- оксан -5-ил]гексановая кислота в виде бесцветного твердого вещества с температурой плавления 118-120оС
[ЯМР: 1,32 (6Н, с), 1,65 (2Н, м), 2,31 (5Н, м), 3,90 (1Н, д), 4,18 (1н, д), 4,55 (1Н, с), 5,00 (1Н, д), 5,22 (1Н, м), 5,40 (1Н, м), 5,70 (1Н, м), и 7,28 (4Н, м), 7,53 (2Н, м), 7,62 (1Н, д), 8,50 (2Н, м); m/е 428 (М+Н)+]
с выходом 63% исходя из 2-метил-2-(фенилтио)пропаналя; и
П р и м е р 53. 4(Z)-6-[(2,4,5-цис)-2-(1-метил-1-(4-фторфенилтио)этил)-4-(3-пиридил)-1,3-дио ксанкислота в виде бесцветного твердого вещества с температурой плавления 109-111оС
[ЯМР: 1,33 (6Н, с), 1,64 (2Н, м), 2,34 (5Н, м), 3,90 (1Н, м), 4,18 (1Н, м), 4,56 (1Н, с), 5,02 (1Н, д), 5,31 (2Н, м), 6,94 (2Н, м), 7,35 (1Н, м), 7,55 (3Н, м), 8,52 (2Н, м);
m/е 444 (М-Н+)] с входом 61% исходя из 2-метил-2-(4-фторфенилтио)пропаналя.
Исходные бензальдегиды были получены по методике, аналогичной описанной в разделе (VII) примера 1, и имели следующие свойства.
а) 2-метил-2-(фенилтио)пропаналь получали с 40% выходом в виде масла
[ЯМР: 1,25 (6Н, с), 7,25 (5Н, м), 9,28 (1Н, с);
m/е 194 (М+NН4)+] исходя из фенилтиола и 1,1-дихлор-2-окси-2-метилпропана.
б) 2-метил-2-(4-фторфенилтио)пропаналь получали с 14% выходом в виде масла
[ЯМР: 1,31 (6Н, с), 6,99 (2Н, м), 7,36 (2Н, м), 9,28 (1Н, с); m/е 216 (М+NН4)+]
исходя из 4-фторфенилтиоола и 1,1-дихлор-2-окси-2-метилпропана.
П р и м е р 54. Катализируемые кислотой реакции обмена в системе альдегид/катон, описанные в любом из предшествующих примеров 17-50, могут быть также проведены с использованием 4(Z)-6-[2,2-диэтил-4-(3-пиридил)-1,3-диоксан-цис-5-ил]гексановой кислоты (описанной в примере 14) вместо 2,2-диметилпроизводного [соединение (А), описанное в разделе (V) примера 1] При этом могут быть получены соединения формулы I с, как правило, такими же выходами.
Используя процедуру, подобную описанной в примере 1, но исходя из соответствующего альдегида, и, если требуется оптически активный продукт, 4(Z)-6-/(4S, 5R)-2,2-диметил-4-(3-пиридил)-1,3-диоксан- 5-ил/гексиновая кислота (получаемая как описано в примере 35), могут быть получены следующие соединения:
П р и м е р 55. 4(Z)-6-/(2S,4S,5R)-2-(1-циано-4-этилфенокси)-1-метилэтил-4-(3-пири- дил)1,3- диоксан-5-ил/гексеновая кислота, т.пл. 108-110оС (простой эфир); ЯМР: 1,21 (3Н, m, J=2 Гц), 1,48 (3Н, с), 1,50 (3Н, с), 1,59 (1Н, м), 1,74 (1Н, м), 2,26 (4Н, м), 2,42 (1Н, м), 2,60 (2Н, кв, J=7 Гц), 4,03 (1Н, дм, J=11 Гц), 4,20 (1Н, д, J=11 Гц), 4,57 (1Н, ш), 4,97 (1Н, с), 5,18 (1Н, д, J=2 Гц), 5,22 (1Н, м), 5,41 (1H, м), 7,11 (1Н, м), 7,31 (3Н, м), 7,58 (1Н, дм, J=8 Гц), 8,50 (2Н, м).
Соответствующий альдегид может быть получен следующим образом.
I) Раствор 4-этилфенола (2,44 г) в дихлорметане (10 мл) добавляли к перемешиваемому, охлаждаемому льдом раствору трихлорида бора 1М дихлорметане (24 мл) в атмосфере аргона, за эти следовал метилтиоцианат (1,64 мл), а после этого безводный хлорид алюминия (2,66 г). Смесь перемешивали 1 ч при 4oС, затем нагревали с обратным холодильником (температура ванны 60оС) 4 ч и наконец перемешивали 16 ч при окружающей температуре. Смесь добавляли осторожно к перемешиваемому, охлаждаемому льдом раствору гидроокиси натрия 4М, и полученную смесь нагревали при 70оС в течение 30 мин, после чего охлаждали. Водную фазу отделяли, промывали дихлорметаном (2х25 мл), затем подкисляли до рН 2 с помощью соляной кислоты 6М и экстрагировали этилацетатом (3х75 мл). Объединенные органические экстракты промывали насыщенным соляным раствором (3х50 мл), сушили (МgSО4) и растворитель удаляли под вакуумом до получения твердого коричневого вещества. Очистка хроматографией вспышки, элюирование 20% этилацетата в гексане с последующей перекристаллизацией из гексана давали 2-циано-4-этилфенол (А) (2,56 г), т.пл. 95-98оС; ЯМР: 1,21 (3Н, m, J= 7 Гц), 2,59 (2Н, кв, J=7 гц), 5,70 (1Н, ш), 6,90 (1Н, м), 7,29 (2Н, м); миллиэквивалент 165 (М+NН4)+.
II) Твердый А (1,47 г) добавляли порциями к перемешиваемой, охлаждаемой льдом суспензии гидрида натрия (60 мас.-ная дисперсия в минеральном масле, 420 мг) в DMPU(10 мл) в атмосфере аргона. Перемешивание продолжали 1 ч при окружающей температуре, смесь охлаждали до 4оС и добавляли 2-бром-2-метил-пропанал (2,26 г) в DМРU (2 мл). Перемешивание продолжали 18 ч при окружающей температуре. Воду (50 мл) добавляли и полученную смесь экстрагировали простым эфиром (3х50 мл). Объединенные экстракты промывали водой (2х30 мл) и насыщенным соляным раствором (2х30 мл), затем сушили (МgSО4). Далее выпаривание растворителя и очистка остаточного масла хроматографией вспышки, элюирование 20 об.-ным этилацетатом в гексане давали 2-(2-циано-4-этилфенокси)-2-метилпаропанал (В) (2,04 г) в виде бесцветного масла;
ЯМР: 1,21 (3Н, m, J=7 Гц), 1,51 (6Н, с), 2,61 (2Н, кв, J=7 Гц), 6,76 (1Н, g, J= 7 Гц), 7,28 (1Н, gg, J=7,2 Гц), 7,41 (1Н, d, J=2 Гц), 9,85 (1Н, S); миллиэквивалент 235 (М+NН4)+.
П р и м е р 56. 4(Z)-6-/(2,4,5-цис)-2-(2-циано-4-этилфенокси)-1-метилэтил)-4-(3-пири- дил)- 1,3-диоксан-5-ил/гексановая кислота, т.пл. 118-119оС (из эфира/гексана); ЯМР: подобно энантиомеру, описанному в примере 55.
П р и м е р 57. 4(Z)-6-/(2S,4S,5R)-2-(1-(4-циано-2-метокси-фенокси)-1-метилэтил)-4-(3- пиридил)-1,3-диоксан-5-ил/гексеновая кислота; т.пл. 133-134оС (из этилацетата/эфира); ЯМР: 1,40 (3Н, с), 1,43 (3Н, с), 1,61 (1Н, м), 1,73 (1Н, м), 2,28 (4Н, м), 2,41 (1Н, м), 3,81 (3Н, с), 4,00 (1Н, дм, J=11 Гц), 4,21 (1Н, g, J=11 Гц), 4,88 (1Н, с), 5,12 (1Н, g, J=2 Гц), 5,22 (1Н, м), 5,40 (1Н,м), 7,05 (1Н, g, J=2 Гц), 7,16 (2Н, м), 7,34 (1Н, м), 7,59 (1Н, дм, J=8 Гц), 8,54 (2Н, ш).
П р и м е р 58. 4(Z)-6-/(2,4,5-цис)-2-(2,4-диметилфенокси)-1-метилэтил)-4-(3-пири- дил)-1,3- диоксан-5-ил/гексановая кислота; т.пл. 122-124оС (из эфира/гексана); ЯМР: 1,36 (3Н, с), 1,41 (3Н, с), 1,69 (2Н, м), 2,21 (3Н, с), 2,25 (3Н, с), 2,28 (4Н, м); 2,49 (1Н, м), 4,02 (1Н, дм, J=11 Гц), 4,24 (1Н, ш, J= 11 Гц), 4,85 (1Н, с), 5,13 (1Н, g, J=2 Гц), 5,24 (1Н, м), 5,42 (1Н, м), 6,89 (3Н, м), 7,35 (1Н, м), 7,71 (1Н, д.м. J=8 Гц), 8,53 (1Н, gg, J=4 Гц), 8,57 (1Н, g, J=1 Гц).
П р и м е р 59. 4(Z)-6-/(2,4,5-цис)-2-(1-(2-бром-4-метилфенокси)-1-метилэтил)-4-(3- пиридил)-1,3-диоксан-5-ил/гексеновая кислота; т.пл. 145-147оС (из эфира/гексана); ЯМР: 1,43 (3Н, с), 1,48 (3Н, с), 1,67 (2Н, м), 2,26 (3Н, с), 2,28 (4Н, м); 2,45 (1Н, м), 4,03 (1Н, д.м, J=11 Гц), 4,23 (1Н, g, J=11 Гц), 4,98 (1Н, с), 5,15 (1Н, g, J=2 Гц), 5,24 (1Н, м), 5,41 (1Н, м), 7,00 (2Н, м), 7,33 (2Н, м), 7,65 (1Н, д.м. J=8 Гц), 8,51 (2Н, м).
П р и м е р 60. 4(Z)-6-/(2S,4S,5R)-2-(1-(2-нитро-4-тиометилфенокси)1-метилэтил)-4- (3-пиридил)-1,3-диоксан-5-ил/гексеновая кислота; т.пл. 99-102оС (из простого эфира); ЯМР: 1,45 (6Н, с), 1,50-1,80 (2Н, м), 2,15-2,45 (5Н, м), 2,48 (3Н, с), 3,90-4,20 (2Н, м), 4,80 (1Н, с), 5,05-5,5 (3Н, м), 7,18-7,60 (5Н, м), 8,40-8,60 (2Н, м).
П р и м е р 61. 4(Z)-6-/(2S,4S,5R)-2-(1-(4-ацетил-2-нитрофенокси)-1-метилэтил)-4-(3-пиридил) -1,3кислота; т.пл; 151-155оС (из простого эфира); ЯМР 1,55 (6Н, с), 1,55-1,75 (2Н, м), 2,15-2,45 (5Н, м), 2,55 (3Н, с), 3,90-4,20 (2Н, м), 4,82 (1Н, с), 5,05-5,50 (3Н, м), 7,25-7,60 (3Н, м), 8,0-8,20 (2Н, м), 8,40-8,55 (2Н, м).
Исходные альдегиды, необходимые для получения соединений из примеров 57-60, были получены с использованием процедуры, аналогичной процедуре, описанной в примере 55, следующим образом:
2-(4-циано-2-метоксифенокси)-2-метилпропанал для использования в примере 57 может быть получен после хроматографии вспышки элюированием 20%-ным по объему этилацетатом в гексане с 71%-ным выходом в виде бесцветного масла; ЯМР: 1,38 (6Н,с), 3,81 (3Н, с); 6,95 (1Н, g, J=8 Гц), 7,12 (1Н, g, J=2 Гц), 7,21 (1Н, gg, J=8 Гц, 2 Гц), 9,77 (1Н, с); миллиэквивалент 237 (М+NН+);
2-(2,4-диметилфенокси)-2-метилпропа- нал для использования в примере 58 может быть получен после элюирования с хроматографией вспышки с применением 2% -ного по объему этилацетата в гексане, с 73%-ным выходом в виде бесцветного масла; ЯМР: 1,41 (6Н, с), 2,22 (3Н, с), 2,25 (3Н, с), 6,55 (1Н, g, J=8 Гц), 6,85 (1Н, gg, J=8 Гц), 6,97 (1Н, ш.с), 9,86 (1Н, с); миллиэквивалент 210 (М+NН4)+;
2-(2-бром-4-метилфенокси)-2-метилпро- панал для использования в примере 59 может быть получен после элюирования при хроматографии вспышки с помощью 1,5 об. этилацетата в гексане с выходом 82% в виде бесцветного масла; ЯМР: 1,44 (6Н, с), 2,28 (3Н, с), 6,74 (1Н, g, J=8 Гц), 6,99 (1Н, gg, J=8 Гц), 7,39 (1Н, g, J=2 Гц), 9,90 (1Н, с); миллиэквивалент 274 (М+NН4)+;
2-(2-нитро-4-тиометилфенокси)-2-ме- тилпропанал для использования в примере 60 может быть получен после элюирования при хроматографии вспышки с помощью 15 об. этилацетата в гексане, с 45%-ным выходом в виде масла; ЯМР: 1,48 (6Н, с), 2,49 (3Н, с), 6,99 (1Н, g, J=4 Гц), 7,33 (1Н, gg, J=4 Гц= 1ГЦ), 7,63 (1Н, g, J=1 Гц); миллиэквивалент 273 (М+NН4)+;
2-(4-ацетил-2-нитрофенокси)-2-метил-пропанал для использования в пример 61 может быть получен после элюирования при хроматографии вспышки с помощью 30 об. этилацетата в гексане, с 49%-ным выходом в виде масла; ЯМР: 1,58 (6Н, с), 2,60 (3Н, с), 6,96 (1Н, g, J=9 Гц), 8,05 (1Н, gg, J=9 Гц, 2 Гц), 8,36 (1Н, g, J=2 Гц), миллиэквивалент 269 (М+NН4)+.
2-Нитро-4-тиометилфенол, используемый для получения исходного альдегида для примера 60, может быть получен следующим образом:
раствор 4-(метилмеркапто)фенола (1,4 г) в смеси простого эфира (30 мл) и метанола (5 мл) добавляли к раствору нитрата натрия (0,85 г) и нитрата лантана (III) (0,044 г) в концентрированной соляной кислоте (8 мл), разбавленной водой (8 мл). Спустя 48 ч, когда реакция дала оранжевый цвет, смесь экстрагировали хлороформом (2х30 мл). Собранные вместе экстракты сушили МgSО4 и выпаривали до получения оранжевого масла, которое очищали элюированием при хроматографии вспышки с помощью 20 об. этилацетата в гексане для получения 2-нитро-4-тиометилфенола (0,82 г) в виде оранжевого твердого вещества; ЯМР: 2,50 (3Н, с), 7,10 (1Н, g, 9 Гц), 7,53 (1Н, gg, J=9 Гц, 2 Гц), 7,96 (1Н, g, J=2 Гц), 10,48 (1Н, с); миллиэквивалент 185 (М)+.
П р и м е р 62. Поясняющие формы фармацевтического дозирования соединений, пригодные для использования в терапевтических или профилактических целях, включают следующие рецептуры в виде таблеток, капсул, инъекций и аэрозолей, которые могут быть получены с помощью обычных, хорошо известных и в современной фармации методов и пригодны для терапевтического и практического использования в организме человека. а) Таблетка I мг/таблетка Соединение Z* 1,0 Соединение Рh Eur 93,25 Кроскармеллоза натрия 4,0 Паста кукурузного крахмала (5 мас. /об. водная паста) 0,75 Стеарат магния 1,0 b) Таблетка II мг/таблетка Соединение Z* 50 Лактоза Рh Eur 223,75 Кроскармеллоза натрия 6,0 Кукурузный крахмал 15,0 Поливинилпирролидон (5 мас./об. водная паста) 2,25 Стеарат магния 3,0 с) Таблетка III мг/таблетка Соединение Z* 100 Лактоза Рh Eur 182,75 Кроскармеллоза натрия 12,0 Паста кукурузного крахмала (5 мас./об. водная паста) 2,25 Стеарат магния 3,0 d) Капсула мг/капсула Соединение Z* 10 Лактоза Рh Eur 488,5 Стеарат магния 1,5 е) Инъекция I (50 мг/мл) Соединение Z* (в форме свободной кислоты) 5,0 мас./об. IМ Раствор гидроокиси натрия 15,0 об./об. 0,1М Раствор соляной кислоты (для дове- дения до рН 7) Полиэтиленгли- коль 400 4,5 мас./об. Вода для инъекции до 100% f) Инъекция II (10 мг/мл) Соединение Z* (в форме свободной кислоты) 1,0 мас./об. Фосфат натрия ЕР 3,6 мас./об. 0,1М Раствор гидро- окиси натрия 15,0 об./об. Вода для инъекции до 100% g) Инъекции III (1 мг/мл, стабилизирована буфером с рН 6) Соединение Z* (в форме свобод- ной кислоты) 0,1 мас./об. Фосфат натрия ВР 2,26 мас./об. Лимонная кислота 0,38 мас./об. Полиэтиленгликоль 400 3,5 мас. /об. Вода для инъекции до 100% h) Аэрозоль I мг/мл Соединение Z* 10,0 Сорбит триолеат 13,5 Трихлорфторметан 910,0 Дихлордифторметан 490,0 i) Аэрозоль II мг/мл Соединение Z* 0,2 Сорбита триолеат 0,27 Трихлорфторметан 70,0 Дихлордифторметан 280,0 Дихлортетрафторэтан 1094,0 j) Аэрозоль III мг/мл Соединение Z* 2,5 Сорбита триолеат 3,38 Трихлорфторметан 67,5 Дихлордифторметан 1086,0 Дихлортетрафторэтан 191,6 k) Аэрозоль IV мг/мл Соединение Z* 2,5 Соевой лецитин 2,7 Трихлорфторметан 67,5 Дихлордифторметан 1086,0 Дихлортетрафторэтан 191,6
Активным ингредиентом соединения является соединение формулы (1) или его соль, например соединение формулы (1), описанное в любом из предшествующих примеров, и, особенно в примерах 18, 19, 20, 46 или 48. Составы таблеток (а)-(с) могут быть покрыты обычными способами, например фталатом ацетата целлюлозы. Аэрозольные композиции h-k могут быть использованы в сочетании со стандартными измеренно-дозирующими аэрозольными устройствами, а стабилизаторы суспензии триолеат сорбита и соевый лецитин могут быть заменены другими стабилизаторами такими, как моноолеат сорбита, полуторный олеат сорбита, полисорбит 80, олеат полиглицерина или олеиновая кислота.
Прилагающие данные демонстрируют способность значительного числа испытуемых соединений действовать в качестве нейтрализаторов ТХА2 и ингибиторов синтазы ТХА2. Испытания проводили по методике испытания (в) и (е). Таблица также содержит сравнительные данные для четырех соединений [2] демонстрирующие, что эти соединения не имеют активности в качестве ингибиторов синтазы ТХА2.
Данные биологических испытаний
В табл. 2 приводятся данные биологических испытаний, которые демонстрируют способность испытываемых соединений действовать как нейтрализаторы ТХА2 и ингибиторы синтазы ТХА2.
Способность к нейтрализации ТХА2 была продемонстрирована испытанием слипания тромбоцитов крови на основе описанного Воrn (Nature, 1962, 194, 927-929) и включающего
агрегирование богатой тромбоцитами плазмы крови человека с добавлением цитратом путем добавления миметического агента ТХА2-U46619, в результате чего получают кривую реакции на дозу;
получение кривой реакции на дозу для агрегирования тромбоцитов, стимулированного U46619, в присутствии возрастающих количеств испытуемого соединения (обычно в диапазоне 10-5-10-10 М) и
подсчет значения КВ, указывающего силу способности к нейтрализации ТХА2 для испытываемого соединения, усредненную на основе нескольких концентраций, от расчетного 50%-ного значения реагирования для агрегирования U46619 в присутствии и отсутствии испытываемого соединения.
В этой таблице результаты испытаний даны в виде величины рА2, которая определена как отрицательны логарифм от значения КВ.
Ингибирование синтазы ТХА2 было продемонстpировано с использованием стандартной процедуры ин витро, описанной в Нowarth et alia (Вiochem.Soc.Тransactions 1982, 10, 239-240) с использованием препарата синтазы ТХА2 микросомы тромбоцита человека и с применением количественного тонкослойного рентегно-хроматографического метода для оценки превращения (1-14С) арахидоновой кислоты для метаболита ТХА2-тромбоксана В2 (ТХВ2). Таблица биологических испытаний прилагается. Результаты даны в единицах мкМ.
Далее приведены формулы поясняющие текст описания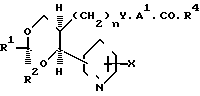 (1)
(1)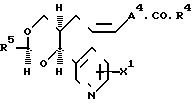 (2)
(2)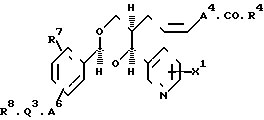 (3)
(3)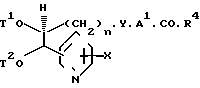 (4)
(4)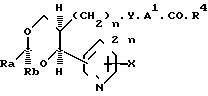 (5)
(5)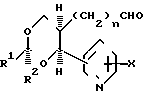 (6)
(6)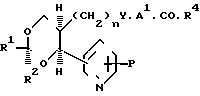 (7)
(7)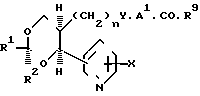 (8)
(8)
R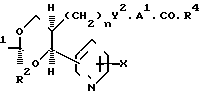 (9)
(9)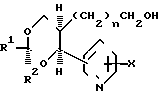 (10)
(10)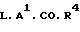 (11)
(11)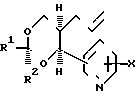 (12)
(12)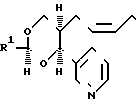
 (13)
(13)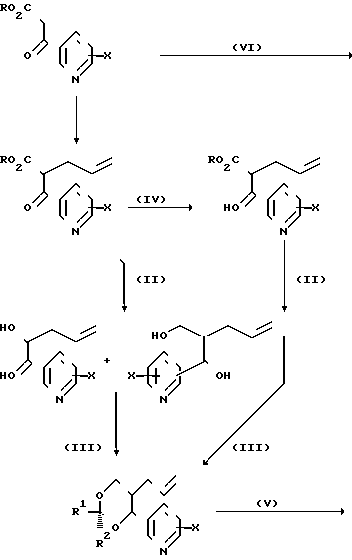
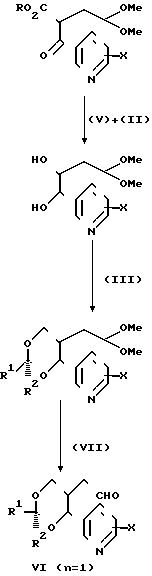
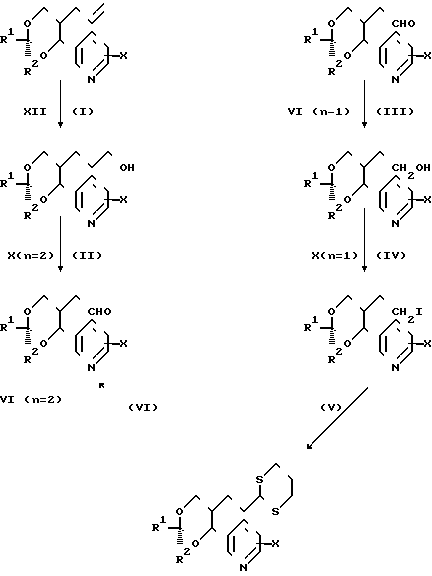
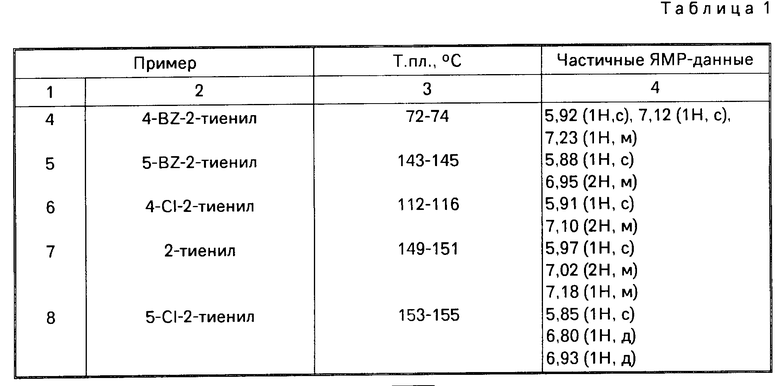



Использование: в медицине, в частности в качестве ингибитора ТХА-синтазы, а также ТХА-антагонистов при лечении ишемической болезни сердца, цереброваскулярного заболевания системмы и/или воспаления. Условия реакции: процесс ведут в присутствии кислоты, а если необходимо получить оптически активную форму целевого продукта, то используют соединение ф-лы, указанной в тексте описания, в оптически активной форме. 2 табл.
ПРОИЗВОДНЫЕ 1,3-ДИОКСАНАЛКЕНОВОЙ КИСЛОТЫ общей формулы I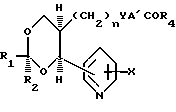
где n 1 или 2;
X водород, гидрокси- или С1 С4-алкоксигруппа;
Y метиленоксигруппа, винилен или этилен;
A′ С1 С6-алкилен;
R2 водород;
R1 нафтил или фенилтио(С1 С6)алкил, необязательно содержащий 1 или 2 заместителя, выбранных из группы, включающей в себя галоген, цианогруппу, нитрогруппу, С1 С4-алкил, С1 С4-алкоксигруппу или трифторметил, или R1 группа общей формулы R3A2, где R3 фенил, содержащий заместитель, который выбирают из группы, включающей в себя С1 - С4-алкил, С1 С4-алкоксигруппу, оксигруппу, С2 С5-алкенил, С1 - С4-алкилтиогруппу, С1 С4-алкилсульфинил, С1
С4-алкилсульфонил, С2 С5-алканоил или фенил, необязательно содержащий второй заместитель, выбранный из С1 С4-алкила, С1 С4-алкоксигруппы, галогена, нитро- или цианогруппы, или R3 тиенил или фурил, необязательно содержащим 1 или 2 заместителя, независимо выбранных из группы, включающей в себя галоген или цианогруппу;
А2 С1 С6-алкилен, окси(С1 С6)алкилен, где в любой из этих групп вплоть до трех атомов углерода могут быть полностью или частично фторированы или А2 непосредственно связан с R3, или R1 является группой общей формулы Q2A3Q1, где Q1 и Q2 ароматические заместители, один из которых является бензольным заместителем, а другой бензольным, придиновым или нафталиновым заместителем, любые из которых необязательно содержат заместитель, выбранный из группы, включающей в себя цианогруппу, нитрогруппу, С1 С4 алкоксигруппу или трифторметил, а А3 простой эфирный мостик, тиогруппа, карбонил, окси(С1 С6)алкилен, или фрагменты Q1 и Q2 связаны непосредственно, или R1 пентафторметил, а R2 водород, или R1 и R2 оба трифторметил или оба независимые С1 С6-алкилы так, что R1 и R2 вместе содержат 5 9 атомов углерода, а R4 оксигруппа или физиологически приемлемый остаток спирта.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЕР N 228887, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
