Изобретение относится к новым производным имидазола, в частности производным имидазола нижеследующей формулы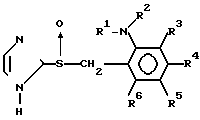
(1) где R1 представляет водород или алкил с 1-6 атомами углерода, R2представляет собой алкил с 2-6 атомами углерода, замещенный алкоксигруппой с 1-4 атомами углерода, каждый из R3, R4, R5 и R6независимо представляет водород, галоид, алкил с 1-6 атомами углерода, алкоксигруппу с 1-6 атомами углерода, фторзамещенный алкил с 1-6 атомами углерода или фторзамещенную алкоксигруппу с 1-6 атомами углерода.
Изобретение также относится к способу получения производных имидазола формулы (I) и противоязвенному средству, содержащему в качестве активного компонента производное имидазола формулы (I).
Известны производные бензимидазола формулы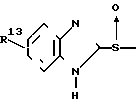 C
C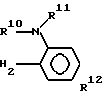
(A) где каждый из R10 и R11 представляет водород или низший алкил, один из R12 и R13 представлен галоидом, трифторметилом, низшим алкилом, низшей алкоксигруппой, низшим алкоксикарбонилом или аминогруппой, являются эффективными противоязвенными средствами, обладающими ингибирующим действием по отношению к H+ +K+ АТфазе.
Известны также сульфоксиды формулы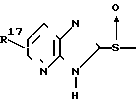 C
C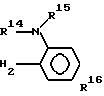
(B) где каждый из R10 и R11 представляет водород или низший алкил, и каждый из R16 и R17 представляет водород, низшую алкоксигруппу или низший алкил, являются эффективными противоязвенными средствами, обладающими ингибирующей активностью по отношению к H+ +K+ АТфазе.
Цель настоящего изобретения заключается в создании нового соединения с повышенным противоязвенным действием и одновременно улучшенной безопасностью.
Создателями настоящего изобретения найдено, что новые производные имидазола формулы (I) обладают прекрасным ингибирующим действием на выделение желудочного сока.
В отличие от вышеуказанных известных производных бензимидазола и производных имидазопиридина, в которых имидазольный цикл сконденсирован с ароматическими циклами, производные имидазола по изобретению не имеют ароматического цикла, сконденсированного с имидазольным циклом, т.е. такие производные имидазола по своему строению резко отличаются от вышеупомянутых известных соединений.
Предлагаемые производные имидазола являются эффективными противоязвенными средствами, обладают высокой безопасностью и улучшенной стабильностью, в частности прекрасной стабильностью в растворе.
Настоящим изобретением даются новые производные имидазола формулы (I), являющиеся эффективными противояз- венными средствами.
Изобретением также дается новый способ получения производных имидазола формулы (I).
Кроме того, изобретением дается противоязвенное средство, содержащее производное имидазола формулы (I).
Производное имидазола формулы (I) может быть получено, например, способом, включающим стадии:
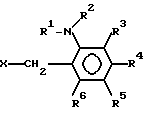
реакции соединения нижеследующей формулы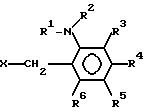
(II) где R1 представляет водород или алкил с 1-6 атомами углерода, R2представляет алкил с 2-6 атомами углерода, в котором один водород замещен алкоксигруппой с 1-4 атомами углерода, каждый из R3, R4, R5 и R6независимо представляет водород, галоид, алкил с 1-6 атомами углерода, в котором водород может быть замещен фтором или алкоксигруппой с 1-6 атомами углерода, в которой водород может быть замещен фтором, и Х представляет отходящую группу, такую как галоид, тозилоксигруппа или мезилоксигруппа,
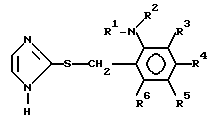
с 2-меркаптоимидазолом с получением производного имидазола формулы (III) и окисления производного имидазола формулы (III):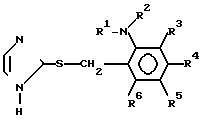
(III) где каждый из R1, R2, R3, R4, R5 и R6 принимают значения, указанные для формулы (II).
Вышеприведенная реакция между соединениями формулы (II) и 2-меркаптоимидазолом может быть осуществлена в температурном интервале от комнатной температуры до температуры кипения инертного растворителя, такого как бензол, этанол или ацетон. Реакция может быть осуществлена в присутствии щелочного средства, такого как: NaOH, KOH, K2CO3, NaHCO3 и триэтиламин для связывания образующейся в реакции кислоты.
Методика окисления соединения формулы (III) с получением производного сульфинильного типа может быть самой обычной. К примеру, соединение формулы (III) может быть окислено с помощью окислителя, такого как водная перекись водорода в присутствии иона металла (например, ванадия, молибдена или вольфрама), органическая перекись (например, м-хлорнадбензойная кислота или трет-бу- тилгидроперекись) или гипохлорит натрия. Реакция может быть осуществлена в инертном растворителе, такой как хлороформ, дихлорметан, метанол или этилацетат в температурном интервале от -30 до 50оС, предпочтительно от -15 до 5оС.
В описании примеры алкилов с 1-6 атомами углерода включают: метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, изопентил неопентил, н-гексил и изогексил. Примеры алкоксигрупп с 1-4 атомами углерода включают: метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, изобутокси-, и трет-бутоксигруппу и циклопропокси. Примеры алкилов с 2-6 атомами углерода включают: этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил и изогексил. Что касается R2 (т.е. алкила с 2-6 атомами углерода, замещенного алкоксигруппой с 1-4 атомами углерода) в формулах (I), (II) и (III), то алкоксигруппа с 1-4 атомами углерода предпочтительно присоединена к концевому атому углерода (который не связан с атомом азота) алкила с 2-6 атомами углерода. Примеры алкоксигрупп с 1-6 атомами углерода включают: метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, изобутокси-, трет-бутокси-, пентилокси- и гексилоксигруппу. Что касается фторзамещенных алкилов (каждый из R3, R4, R5 и R6), то в их числе: фторзамещенный метил, фторзамещенный изопропил, фторзамещенный н-бутил, фторзамещенный изобутил, фторзамещенный трет-бутил, фторзамещенный н-пентил, фторзамещенный изопентил, фторзамещенный неопентил, фторзамещенный н-гексил или фторзамещенный изогексил, и примеры фторзамещенной алкоксигруппы включают: фторзамещенную алкоксигруппу, фторзамещенную этоксигруппу, фторзамещенную н-пропоксигруппу, фторзамещенную изопропоксигруппу, фторзамещенную трет-бутоксигруппу, фторзамещенную пен- тилоксигруппу или фторзамещенную гексилоксигруппу. Атом галоида - это фтор, хлор, бром или йод.
Примеры производных имидазола изобретения включают нижеследующие соединения.
Представительные примеры производных имидазола формулы (I) включают соединения, для которых значения R1, R2, R3, R4, R5 и R6 приведены в табл. 1.
Фармакологическое действие испытано по отношению к представительному соединению формулы (I) изобретения. Ниже приведены результаты испытаний.
(1) Ингибирующее действие на H+ +К+ АТфазу (испытание 1).
По известной методике из слизистой желудка кролика выделяют клетки. Пузырек, содержащий H+ +K+ АТфазу, получают центрифугированием клеток в Фиколле с непрерывным градиентом плотности. После инкубирования фермента при комнатной температуре в течение 15 мин в 0,5 мл раствора, содержащего 5 ммоль имидазольного буфера (рН 6) и 2 х 10-4 М 2-[2-(2-метоксиэтиламино)бензилсульфинил] имидазола (соединение примера 1 - испытуемое соединение), смесь нагревают до 37оС. После этого смесь оставляют еще на 5 мин. К смеси добавляют 0,5 мл раствора, содержащего 4 ммоль хлорида магния, 80 ммоль имидазольного буфера (рН 7,4), 20 ммоль хлорида калия и 4 ммоль АТФ. Полученную смесь оставляют реагировать на 15 мин при 37оС, после чего для прекращения реакции добавляют 1 мл 24%-ной трихлоруксусной кислоты. Выделившийся неорганический фосфор определяют количественно.
Вышеприведенная методика повторена, но без добавления хлорида калия с целью определения активности АТФ в отсутствие хлорида калия. Целевую К+-зависимую активность АТфазы подсчитывают вычитанием значения активности АТфазы, определенную в отсутствии KCl, из значения активности АТфазы, определенной в присутствии KCl.
В результате таких измерений найдено, что значение ингибирующего действия испытуемого соединения (соединение примера 1) на H+ +K+ АТфазу равно 73,1%.
(2) Ингибирующее действие на H+ +K+ АТфазу (испытание 2).
Фермент получают по методике, приведенной выше в испытании 1. После инкубирования фермента при комнатной температуре в течение 25 мин в 0,5 моль раствора, содержащего 5 ммоль имидазольного буфера (рН 6 или 7,4) и переменные концентрации испытуемого соединения, смесь нагревают до 37оС. После этого смесь оставляют еще на 5 мин. Значение ингибирующего действия на H+ +K+ АТфазу каждого испытуемого соединения определяют так, как описано выше в испытании 1.
Полученные результаты приведены в табл. 2.
(3) Ингибирующее действие на секрецию желудочной кислоты (испытание I).
Самцов крыс линии Danryu массой 200-250 г связывают (но со свободным доступом к воде) на 24 ч по известной методике. Под эфирной анестезией перевязывают привратник желудка и внутрь двенадцатиперстной кишки вводят 2-[2-(2-метокси- этиламино)бензилсульфинил]имидазол (соединение примера 1 - испытуемое соединение). Спустя 4 ч крыс умерщвляют и в удаленном желудке собирают желудочный сок. Ингибирующее действие определяют путем сравнения выхода кислоты, определяемого титрованием до рН 7 гидроксидом натрия (0,1 н) с помощью автоматического титратора, с соответствующим значением для контрольной крысы, обработанной аналогичным образом, но с введением ей только носителя.
В результате подобных измерений установлено, что ингибирующее действие испытуемого соединения (соединение примера 1) равно 75,9 и 94,5% при дозах соответственно 10 и 30 мг/кг.
(4) Ингибирующее действие на секрецию желудочной кислоты (испытание II).
У самцов связанных беспородных собак создают дивертикул Хайденхейна. Затем собакам непрерывно вводят внутривенно в дозировке 160 мкг/кг/ч хлоргидрат гистамина (средство, индуцирующее секрецию желудочного сока). Желудочный сок отбирают с интервалом в 15 мин с измерением его количества и выхода кислоты с целью определения количества секретируемой кислоты (мЭкв/15 мин).
Испытуемое соединение вводят собакам внутривенно через час после начала введения гистамина.
Ингибирующее действие определяют путем сравнения количества секретируемой кислоты между предлекарственным значением (до применения лекарства) и послелекарственным значением (спустя 30 мин после применения лекарства).
Результаты при дозировке 3 мг/кг приведены ниже. Испытуемое Подавляющее соединение действие, % по примеру 1 89 2 97 3 85 4 89 5 98 6 92
Согласно другому эксперименту испытуемые соединения назначали орально за 2 ч до инициирования с применением гистамина и ингибирующее действие определяли путем сравнения количества секретируемой кислоты с соответствующим количеством для контрольной собаки, обработанной таким же образом, за исключением того, что вводят один лишь носитель.
Результаты при дозе 10 мг/кг приведены ниже. Испытуемое Подавляющее соединение действие, % по примеру 3 100 8 100
Испытание на острую токсичность.
Самцам крыс вида SD с массой около 190 г орально вводили соединение по примеру 8. За крысами наблюдали затем в течение 2 дн. MLD составляет 1000 мг/кг или более.
Соединения формулы (I) настоящего изобретения могут быть введены либо перорально, либо парентерально. Препараты для перорального введения могут иметь вид, например: таблеток, капсул, порошков, гранул, сиропа и т.п. Препараты для парентерального введения могут иметь вид инъектируемых препаратов и т. п. В состав таких препаратов могут быть добавлены наполнители, диспергирующие добавки, связующие добавки, смазки, красители, разбавители и аналогичные обычно применяемые добавки. Наполнители могут включать: декстрозу, лактозу и т. п. В качестве диспергирующих добавок могут быть использованы крахмал, карбоксиметилцеллюлоза кальция и т.п. В качестве смазок могут быть использованы тальк, стеарат магния и т.п. Связующей добавкой может служить гидроксипропилцеллюлоза, желатин, поливинилпирролидон и т.п.
Применяемая доза может составлять 1-50 мг/день в случае инъектируемого препарата и 10-500 мг/день - при пероральном введении (в обоих случаях для взрослого человека). Доза может быть увеличена или уменьшена в зависимости от возраста пациента и других условий.
Ниже приведены примеры настоящего изобретения.
П р и м е р 1. 2-[2-(2-Метоксиэтиламино)бензилсульфинил]имидазол.
(I) Метиловый эфир 2-метоксиацетамидобензойной кислоты.
К раствору 3,02 г (20 ммоль) метилового эфира антраниловой кислоты в 30 мл хлористого метилена добавляют 2,22 г (22 ммоль) триэтиламина и к полученному раствору при охлаждении льдом по каплям прибавляют раствор 2,39 г (22 моль) метоксиацетилхлорида. Полученную смесь перемешивают 1 ч. После добавления льда перемешиваемую соль подщелачивают добавлением 1 н. водного раствора гидроксида натрия и затем экстрагируют хлороформом. Органическую часть отделяют и последовательно промывают 3 н. HCl, 6 н. HCl, водой и насыщенным водным раствором хлорида натрия. Промытую хлороформенную часть сушат над безводным сульфатом натрия. Растворитель удаляют при пониженном давлении и получают 3,6 г целевого соединения.
1Н-ЯМР (CDCl3), δ: 3,56 (с, 3Н), 3,93 (с, 3Н), 4,05 (с, 2Н), 6,9-8,8 (м, 4Н).
(II) 2-(2-Метоксиэтиламино)бензиловый спирт.
К суспензии 912 мг (24 ммоль) литийалюминийгидрида в 40 мл сухого тетрагидрофурана (ТГФ) при перемешивании и охлаждении льдом в течение 10 мин по каплям прибавляют 3,6 г (16,1 ммоль) метилового эфира 2-метоксиацетамидобензойной кислоты (получение см. стадию (I), после чего смесь перемешивают 1 ч. Затем к реакционной смеси добавляют насыщенный водный раствор сульфата натрия. Органическую часть отделяют и растворитель отгоняют при пониженном давлении. Очисткой остатка колоночной хроматографией на силикагеле получают 1,2 г целевого соединения в виде бесцветного масла.
1Н-ЯМР (CDCl3), δ: 3,38 (с, 3Н), 3,2-3,4 (м, 2Н), 3,5-3,7 (м, 2Н), 4,63 (с, 2Н), 6,5-7,3 (м, 4Н).
(III) 2-[2-(2-Метоксиэтиламино)бензилтио]имидазол.
К раствору 1,2 г (6,6 ммоль) 2-(2-метоксиэтиламино)бензилового спирта (получение см. стадию (II) в 10 мл хлористого метилена при перемешивании и охлаждении льдом в течение 10 мин по каплям прибавляют 0,62 мл (8,6 ммоль) хлористого тионила в 2 мл хлористого метилена. После перемешивания смеси в течение 15 мин при комнатной температуре растворитель отгоняют при пониженном давлении. К полученному остатку добавляют 1,32 г (13,2 ммоль) 2-меркаптоимидазола и 10 мл этанола, после чего полученную смесь перемешивают 1,5 ч при комнатной температуре. Растворитель отгоняют при пониженном давлении. После добавления льда остаток подщелачивают добавлением 1н. водного раствора гидроксида натрия и экстрагируют хлороформом. Органическую часть отделяют и промывают водой и насыщенным водным раствором хлорида натрия. Промытую хлороформенную часть сушат над безводным сульфатом натрия. Затем растворитель удаляют отгонкой при пониженном давлении. Очисткой остатка колоночной хроматографией на силикагеле получают 1,52 г целевого соединения в виде бесцветного масла.
1Н-ЯМР (CDCl3), δ: 3,44 (с, 3Н), 3,2-3,5 (м, 2Н), 3,6-3,8 (м, 2Н), 4,13 (с, 2Н), 6,4-7,2 (м, 4Н), 7 (с, 2Н).
(IV) 2-[2-(2-Метоксиэтиламино)бензилсульфонил]имидазол.
В смеси 13 мл хлористого метилена, 13 мл метанола и 1,3 мл уксусной кислоты растворяют 1,45 г (5,5 ммоль) 2-[2-(2-метоксиэтиламино)бензилтио] имидазола (получение, см. стадию (III) и к полученному раствору при охлаждении льдом прибавляют 2,6 мл 35%-ной водной перекиси водорода и 37 мг метаванадата аммония. Полученную смесь перемешивают при той же температуре 2,5 ч. После завершения реакции к перемешиваемой смеси добавляют насыщенный водный раствор гидрокарбоната натрия и полученную смесь экстрагируют хлороформом. Органическую часть отделяют и подвергают дальнейшему экстрагированию 1н. раствором гидроксида натрия. В водной части добавлением 20%-ного водного раствора хлористого аммония создают щелочно-аммонийную среду. Осажденный маслянистый продукт экстрагируют хлороформом. Хлороформенную часть промывают насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и перекристаллизацией остатка из эфира получают 760 мг целевого соединения в виде белого кристаллического порошка.
ИК (KBr), ν: 3390, 1605, 1585, 1525, 1475, 1310, 1105, 1000, 885, 745, 505 см-1.
1Н-ЯМР (CDCl3-CD3OD, 1: 1 об/об), δ: 3,04-3,38 (м, 2Н), 3,34 (с, 3Н), 3,44-3,7 (м, 2Н), 4,26 и 4,56 (каждый д., 2HI=14 Гц), 5,08 (м, 1Н), 6,4-7,3 (м, 6Н).
Т.пл. 126-128оС (с разл.).
П р и м е р 2. 2-[2-(2-Изопропоксиэтиламино)бензилсульфинил]имидазол.
(I) Изопропоксиуксусная кислота.
В 160 мл изопропилового спирта при нагревании растворяют 8,4 г металлического натрия и к полученному раствору примерно при 50оС в течение 10 мин прибавляют раствор 20,9 г бромуксусной кислоты в 20 мл изопропилового спирта. Полученную смесь затем кипятят 1 ч. После добавления конц. соляной кислоты растворитель отгоняют при пониженном давлении. К остатку добавляют эфир и осадившуюся неорганическую соль отфильтровывают. Полученный фильтрат сушат над безводным сульфатом натрия, эфир отгоняют и перегонкой остатка при пониженном давлении получают 5,37 г целевого соединения в виде бледно-желтого масла (выход 30,3%).
1Н-ЯМР (CDCl3), δ: 1,23 (д, 6Н, I=6 Гц), 3,73 (м, 1Н), 4,12 (с, 2Н), 8,79 (уш. с. 1Н).
Т.кип. 83-87оС /5-6 мм рт.ст.
(II) Изопропоксиацетилхлорид.
Смесь 5,37 г (46 ммоль) изопропоксиуксусной кислоты и 3,7 мл хлористого тионила перемешивают около суток при комнатной температуре и затем нагревают и перемешивают еще 1,5 ч при 55оС. Разгонкой полученного раствора при пониженном давлении получают 4,43 г целевого соединения в виде бесцветного масла (выход 71,3%).
1Н-ЯМР (CDCl3), δ: 1,21 (д. 6Н, I=6 Гц), 3,72 (м, 1Н), 4,4 (с, 2Н).
Т.кип. 70оC/77 мм рт.ст.
(III) Метиловый эфир (2-изопропоксиацетамидо)бензойной кислоты.
К раствору 4,9 г (33 ммоль) метилового эфира антраниловой кислоты и 6,1 мл триэтиламина в 50 мл дихлорметана при охлаждении льдом по каплям в течение 15 мин прибавляют раствор 4,43 г (33 ммоль) изопропоксиацетилхлорида в 20 мл дихлорметана и полученную смесь перемешивают около суток при комнатной температуре. Реакционную смесь последовательно промывают 5%-ным водным раствором карбоната натрия, 3н. соляной кислотой и 5%-ным водным раствором карбоната натрия и затем сушат над безводным сульфатом натрия. Отгонкой растворителя получают 5,59 г целевого соединения в виде остаточного бледно-коричневого масла (выход 68,6%).
1Н-ЯМР (CDCl3), δ: 1,2 (д, 6Н, I=6 Гц), 3,74 (м, 2Н), 3,92 (с, 3Н), 4,09 (с, 2Н), 6,9-8,8 (м, 4Н), 11,74 (уш. с. 1Н).
(IV) 2-(2-Изопропоксиэтиламино)бензиловый спирт.
К суспензии 2,12 г литийалюминийгидрида в 50 мл ТГФ при охлаждении льдом в течение 15 мин по каплям прибавляют раствор 5,59 г метилового эфира (2-изопропоксиацетамидо)бензойной кислоты в 20 мл ТГФ, после чего смесь кипятят 1 ч. Для разложения избытка литийалюминийгидрида к полученной смеси добавляют насыщенный водный раствор сульфата натрия, органическую часть отделяют и растворитель удаляют. Остаток растворяют в хлороформе, полученный раствор промывают водой и сушат над безводным сульфатом натрия. После отгонки хлороформа получают 5,19 г целевого соединения (чистота 89,7%) в виде бледно-коричневого масла.
1Н-ЯМР (CDCl3), δ: 1,17 (д, 6Н, I=6 Гц), 3,28 (т, 2Н, I=5 Гц), 3,3-3,8 (м, 3Н), 4,63 (с, 2Н), 6,4-7,3 (м, 4Н).
(V) 2-[2-(2-Изопропоксиэтиламино)бензилтио]имидазол.
К раствору 5,19 г (22 ммоль) 2-(2-изопропоксиэтиламино)-бензилового спирта, полученного на стадии (IV), в 50 мл дихлорметана при охлаждении льдом в течение 10 мин по каплям прибавляют раствор 2,4 мл хлористого тионила в 10 мл дихлорметана и полученную смесь перемешивают 30 мин. Отгонкой растворителя получают бледно-желтый кристаллический продукт, который добавляют к раствору 2,66 г 2-меркаптоимидазола в 27 мл этанола в течение 15 мин, после чего смесь перемешивают 30 мин при комнатной температуре. Этанол отгоняют, к остатку добавляют хлороформ и 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и сушат над безводным сульфатом натрия. Хлороформ отгоняют, остаток очищают колоночной хроматографией на силикагеле и кристаллизацией из смеси эфир-гексан получают 2,4 г целевого соединения в виде белого кристаллического порошка (выход 37%).
1Н-ЯМР (CDCl3), δ: 1,2 (т, 6Н, I=6 Гц), 3,34 (т, 2Н, I=5 Гц), 3,4-3,9 (м, 3Н), 4,12 (с, 2Н), 6,3-7,3 (м, 4Н), 6,98 (с, 2Н).
(VI) 2-[2-(2-Изопропоксиэтиламино)бензилсульфинил]имидазол.
В смеси 15 мл дихлорметана, 15 мл метанола и 1,5 мл уксусной кислоты растворяют 1,5 г (5,2 ммоль) 2-[2-(2-изопропокси- этиламино)бензилтио]имидазола и к полученному раствору при охлаждении льдом прибавляют 2,3 мл 35% -ной перекиси водорода в воде и 48 мг метаванадата аммония, после смесь перемешивают 2 ч с охлаждением льдом. После завершения реакции к перемешиваемой смеси добавляют 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и экстрагируют 20 мл 0,2н. водным раствором гидроксида натрия. К отделенной водной части постепенно прибавляют 60 мл 1н. водного раствора хлорида аммония. Осадившийся маслянистый продукт экстрагируют хлороформом, хлороформенный экстракт сушат над безводным сульфатом натрия, после чего хлороформ отгоняют. Кристаллизацией остатка из эфира получают 1,2 г целевого соединения в виде белого кристаллического порошка (выход 75,8%).
ИК (KBr), ν: 3360, 2970, 2950, 2850, 1600, 1580, 1520, 1460, 1320, 1310, 1265, 1150, 1130, 1100, 1080, 1035, 750, 500 см-1.
1Н-ЯМР (CDCl3-CD3OD, 1:1 об/об), δ : 1,21 (д, 6Н, I=6 Гц), 3,26 (т, 2Н, I= 5 Гц), 3,5-3,8 (м, 3Н), 4,31 (д, 1Н, I=14 Гц), 4,54 (д, 1Н, I=14 Гц), 6,4-7,3 (м, 4Н), 7,21 (с, 2Н). Т.пл. 140-142оС (с разл.).
П р и м е р 3. 2-[2-(2-Метоксиэтиламино)-5-метилбензилсульфинил]имидазол.
(I) Метиловый эфир (2-метоксиацетамидо-5-метил)бензойной кислоты.
К раствору 4 г (24 ммоль) метилового эфира 5-метилантраниловой кислоты и 5,2 мл триэтиламина в 40 мл дихлорметана при охлаждении льдом в течение 15 мин по каплям прибавляют раствор 3,42 г (32 ммоль) метоксиацетилхлорида в 10 мл дихлорметана и полученную смесь перемешивают около суток при комнатной температуре. Затем реакционную смесь последовательно промывают 5%-ным водным раствором карбоната натрия, 3н. соляной кислотой и 5%-ным раствором карбоната натрия и сушат над безводным сульфатом натрия. После отгонки растворителя получают 5,5 г целевого соединения в виде бледно-коричневых кристаллов (выход 95,7%).
1Н-ЯМР (CDCl3), δ: 2,33 (с, 3Н), 3,56 (с, 3Н), 3,93 (с, 3Н), 4,05 (с, 2Н), 7,1-8,7 (м, 3Н).
(II) 2-(2-Метоксиэтиламино)-5-метилбензиловый спирт.
К суспензии 1,76 г литийалюминийгидрида в 55 мл ТГФ при охлаждении льдом в течение 15 мин по каплям прибавляют 5,5 г (23 ммоль) метилового эфира (2-метоксиацетамидо-5-метил) бензойной кислоты и полученную смесь кипятят 1 ч. Затем для разложения избытка литийалюминийгидрида к реакционной смеси при охлаждении льдом прибавляют насыщенный водный раствор сульфата натрия. Органическую часть отделяют и растворитель удаляют. Остаток растворяют в хлороформе, полученный раствор промывают водой и сушат над безводным сульфатом натрия. Отгонкой хлороформа получают 4,88 г целевого соединения (чистота 92,7%) в виде остаточного бледно-коричневого масла.
1Н-ЯМР (CDCl3), δ: 2,23 (с, 3Н), 3,1-3,7 (м, 4Н), 3,37 (с, 3Н), 4,6 (с, 2Н), 6,4-7,1 (м, 3Н).
(III) 2-[2-(2-Метоксиэтиламино)-5-метилбензилтио]имидазол.
К раствору 4,88 г (23 ммоль) 2-(2-метоксиэтиламино)-5-метилбензилового спирта, полученного на стадии (II), в 50 мл дихлорметана при охлаждении льдом в течение 15 мин по каплям прибавляют 2,5 мл хлористого тионила в 10 мл дихлорметана и полученную смесь перемешивают 30 мин при комнатной температуре. Отгонкой растворителя в остатке получают маслянистый продукт, который растворяют в 15 мл дихлорметана. Дихлорметановый раствор прибавляют 15 мин к раствору 2,78 г 2-меркатоимидазола в 30 мл этанола и смесь перемешивают 30 мин при комнатной температуре. Этанол отгоняют, к остатку добавляют хлороформ и 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и сушат над безводным сульфатом натрия. Хлороформ отделяют, остаток очищают колоночной хроматографией на силикагеле и кристаллизацией из смеси эфиргексан с получением 4,91 г целевого соединения в виде бледно-коричневого кристаллического порошка (выход 76,4%).
1Н-ЯМР (CDCl3), δ: 2,15 (с, 3Н), 3,35 (т, 2Н, I=5 Гц), 3,43 (с, 3Н), 3,72 (т, 2Н, I=5 Гц), 4,11 (с, 2Н), 6,4-7 (м, 3Н), 7,01 (с, 2Н).
(IV) 2-[2-(2-Метоксиэтиламино)-5-метилбензилсульфинил]имидазол.
В смеси 15 мл дихлорметана, 15 мл метанола и 1,5 мл уксусной кислоты растворяют 1,5 г (5,4 ммоль) 2-[2-(2-метокси- этиламино)-5-метилбензилтио] имидазола. К полученной смеси добавляют при охлаждении льдом 2,3 мл 35%-ной водной перекиси водорода и 46 мг метаванадата аммония, после чего перемешивают 2 ч с охлаждением льдом. После завершения реакции к перемешиваемой смеси добавляют 5%-ный водный раствор карбоната натрия. Органическую часть отделяют, промывают 20 мл 0,05н. водного гидроксида натрия и затем экстрагируют 10 мл 1н. водного раствора гидроксида натрия. К собранной водной части понемногу добавляют 40 мл 1н. водного раствора хлорида аммония. Выпавший в осадок кристаллический продукт отфильтровывают и после промывания достаточным количеством эфира и воды получают 1,04 г целевого соединения в виде бледно-коричневого кристаллического порошка (выход 65,5%).
ИК (KBr), ν: 3320, 2990, 2910, 2850, 2810, 1600, 1525, 1475, 1440, 1420, 1310, 1125, 1110, 1020, 970, 795, 785 см-1.
1Н-ЯМР (CDCl3-CD3OD): 1: 1, об/об), δ: 2,14 (с, 3Н), 3,24 (т, 2Н, I=5 Гц), 3,42 (с, 3Н), 3,64 (т, 2Н, I=5 Гц), 4,3 (д, 1Н, I=13 Гц), 4,5 (д, 1Н, I=13 Гц), 6,4-7,1 (м, 3Н), 7,23 (с, 2Н).
Т.пл. 147-149оС (с разл.).
П р и м е р 4. 2-[2-(2-Метоксиэтиламино)-6-метилбензилсульфинил]имидазол.
(I) Метиловый эфир (2-метоксиацетамидо-6-метил)бензойной кислоты.
К раствору 5 г (30 ммоль) метилового эфира (2-амино-6-метил)бензойной кислоты и 5 г триэтиламина в 50 мл дихлорметана при охлаждении льдом в течение 15 мин по каплям прибавляют раствор 4,67 г (43 ммоль) метоксиацетилхлорида и полученную смесь перемешивают 30 мин при комнатной температуре. Перемешиваемую смесь последовательно промывают водой, 3н. соляной кислотой и водой, после чего сушат над безводным сульфатом натрия. После отгонки хлороформа получают 7,67 г целевого соединения в виде остаточного бледно-коричневого масла (чистота 93,6%).
1Н-ЯМР (CDCl3), δ: 2,44 (с, 3Н), 3,52 (с, 3Н), 3,94 (с, 3Н), 4 (с, 2Н), 6,8-7,3 (м, 3Н).
(II) 2-(2-Метоксиэтиламино)-6-метилбензиловый спирт.
К суспензии 2,28 г литийалюминийгидрида в 45 мл сухого эфира при охлаждении льдом в течение 20 мин прибавляют по каплям раствор 7,76 г (30 ммоль) метилового эфира (2-метоксиацетамидо-6-метил)-бензойной кислоты, полученного на стадии (I), в 20 мл сухого эфира и смесь кипятят 1 ч. Для разложения избытка литийалюминийгидрида к смеси при охлаждении льдом прибавляют насыщенный раствор сульфата натрия. Органическую часть отделяют и сушат над безводным сульфатом натрия. Отгонкой растворителя получают 5,42 г целевого соединения в виде остаточных бледно-коричневых кристаллов (выход 91,7% ).
1Н-ЯМР (CDCl3), δ: 2,31 (с, 3Н), 3,27 (т, 2Н, I=5 Нц), 3,36 (с, 3Н), 3,6 (т, 2Н, I=5 Гц), 4,69 (с, 2Н), 6,3-7,2 (м, 3Н).
(III) 2-[2-(2-Метоксиэтиламино-6-метил)бензилтио]имидазол.
К раствору 5,42 г (28 ммоль) 2-(2-метоксиэтиламино)-6-метилбензилового спирта в 55 мл дихлорметана при охлаждении льдом в течение 10 мин по каплям прибавляют раствор 2,7 мл хлористого тионила в 10 мл дихлорметана и полученную смесь перемешивают 30 мин с охлаждением льдом. После отгонки растворителя получают бледно-желтый кристаллический продукт. Кристаллический продукт прибавляют в течение 15 мин к раствору 3,36 г 2-меркаптоимидазола в 30 мл этанола и полученную смесь перемешивают 30 мин при комнатной температуре. Этанол отгоняют, к остатку добавляют хлороформ и 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и сушат над сульфатом натрия. Хлороформ отгоняют, остаток очищают колоночной хроматографией на силикагеле и кристаллизацией из смеси эфир-гексан с получением 5,27 г целевого соединения в виде белых кристаллов (выход 68,4%).
1Н-ЯМР (CDCl3), δ: 2,21 (с, 3Н), 3,32 (т, 3Н, I=5 Гц), 3,42 (с, 3Н), 3,71 (т, 2Н, I=5 Гц), 4,2 (с, 2Н), 6,3-7,1 (м, 3Н), 7,03 (с, 2Н).
(IV) 2-[2-(2-Метоксиэтиламино)-6-метилбензилсульфинил]имидазол.
В смеси 15 мл хлороформа, 15 мл метанола и 1,5 мл уксусной кислоты растворяют 1,5 г (5,4 ммоль) 2-[2-(2-метоксиэтиламино)-6-метилбензилтио]имидазола и к полученному раствору при охлаждении льдом прибавляют 2,3 мл 35% -ной водной перекиси водорода и 48 мг метаванадата аммония, после чего полученную смесь перемешивают 1,5 ч с охлаждением льдом. После завершения реакции к перемешиваемой смеси добавляют 5%-ный водный раствор карбоната натрия. Органическую часть отделяют, промывают 20 мл 0,05н. водным раствором гидрооксида натрия и затем экстрагируют 20 мл 0,5н. водным раствором гидроксида натрия. К собранной водной части постепенно добавляют 15 мл 1н. водного раствора хлорида аммония. Осажденный маслянистый продукт экстрагируют хлороформом. Хлороформенный экстракт сушат над безводным сульфатом натрия, после чего хлороформ отгоняют. Кристаллизацией остатка из эфира получают 1,09 г целевого соединения в виде белого кристаллического порошка (выход 68,7%).
ИК (KBr), ν: 3340, 2870, 1585, 1520, 1470, 1440, 1305, 1105, 1020, 960, 770 см-1.
1Н-ЯМР (CDCl3-CD3OD, 2:1 об/об), δ: 2,22 (с, 3Н), 3,1-3,8 (м, 4Н), 3,42 (с, 3Н), 4,39 (д, 1Н, I=14 Гц), 4,62 (д, 1Н, I=14 Гц), 6,4-7,2 (м, 3Н), 7,27 (с, 2Н).
Т.пл. 129-132оС (с разл.).
П р и м е р 5.
2-[2-(2-Этоксиэтиламино)бензилсуль- финил]имидазол.
(I) Метиловый эфир (2-этоксиацетамидо)бензойной кислоты.
К раствору 4,53 г (30 ммоль) метилового эфира антраниловой кислоты и 6,1 мл триэтиламина в 50 мл дихлорметана при охлаждении льдом в течение 15 мин по каплям прибавляют раствор 4,41 г (36 ммоль) этоксиацетилхлорида в 10 мл дихлорметана и полученную смесь перемешивают 1 ч при комнатной температуре. Перемешиваемую смесь последовательно промывают водой, 4н. соляной кислотой и 5%-ным водным раствором карбоната натрия, после чего сушат над безводным сульфатом натрия. После отгонки хлороформа получают 7,66 г целевого соединения (чистота 92,8%) в виде остаточного бледно-коричневого масла.
1Н-ЯМР (CDCl3), δ: 1,37 (т, 3Н, I=7 Гц), 3,7 (к, 2Н, I=7 Гц), 3,93 (с, 3Н), 4,1 (с, 2Н), 6,9-8,8 (м, 4Н).
(II) 2-(2-Этоксиэтиламино)бензиловый спирт.
К суспензии 3,42 г литийалюминийгидрида в 90 мл ТГФ при охлаждении льдом в течение 10 мин по каплям прибавляют раствор 7,66 г (30 ммоль) метилового эфира (2-этоксиацетамидо)бензойной кислоты, полученного на стадии (I), в 15 мл ТГФ, после чего смесь кипятят 1 ч при 70оС. Для разложения избытка литийалюминийгидрида к полученной смеси при охлаждении прибавляют насыщенный водный раствор сульфата натрия. Органическую часть отделяют и растворитель удаляют. Остаток растворяют в хлороформе, полученный раствор промывают водой и сушат над безводным сульфатом натрия. После отгонки хлороформа получают 5,27 г целевого соединения в виде бесцветного масла (выход 90,1%).
1Н-ЯМР (CDCl3), δ: 1,2 (т, 3Н, I=7 Гц), 3,3 (т, 2Н, I=5 Гц), 3,53 (к, 2Н, I=7 Гц), 3,66 (т, 2Н, I=5 Гц), 4,62 (с, 2Н), 6,4-7,4 (м, 4Н).
(III) 2-[2-(2-Этоксиэтиламино)бензилтио]имидазол.
К раствору 5,27 г (27 ммоль) 2-(2-этоксиэтиламино)бензилового спирта в 50 мл дихлорметана при охлаждении льдом в течение 10 мин прибавляют по каплям раствор 3 мл хлористого тионила в 10 мл дихлорметана и полученную смесь перемешивают 30 мин с охлаждением льдом. После отгонки растворителя получают бледно-желтый кристаллический продукт, который добавляют в течение 15 мин к раствору 3,24 г 2-меркаптоимидазола в 35 мл этанола, после чего смесь перемешивают 30 мин при комнатной температуре. Этанол отгоняют, к остатку добавляют хлороформ и 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и сушат над безводным сульфатом натрия. Хлороформ отгоняют, остаток очищают колоночной хроматографией на силикагале и кристаллизацией из смеси эфир-гексан с получением 3,25 г целевого соединения в виде бледно-желтых кристаллов (выход 43,4%).
1Н-ЯМР (CDCl3), δ: 1,22 (т, 3Н, I=7 Гц), 3,37 (т, 2Н, I=5 Гц), 3,61 (к, 2Н, I=7 Гц), 3,78 (т, 2Н, I=5 Гц), 4,13 (с, 2Н), 6,4-7,3 (м, 4Н), 6,99 (с, 2Н).
(IV) 2-[2-(2-Этоксиэтиламино)]бензилсульфенил]имидазол.
В смеси 15 мл хлороформа, 15 мл метанола и 1,5 мл уксусной кислоты растворяют 1,5 г (5,4 ммоль) 2-[2-(2-этоксиэтиламино)бензилтио]имидазола, к полученному раствору при охлаждении льдом добавляют 2,3 мл 35%-ной водной перекиси водорода и 48 мг метаванадата аммония и полученную смесь перемешивают 3 ч с охлаждением льдом. После завершения реакции к перемешиваемой смеси добавляют 5%-ный водный раствор карбоната натрия. Органическую часть отделяют, промывают 0,1н. водным раствором гидроксида и экстрагируют 20 мл 1н. водного раствора гидроксида натрия. К отделенной водной части понемногу прибавляют 30 мл водного раствора хлорида аммония (1н). Осажденный маслянистый продукт экстрагируют хлороформом, хлороформенный экстракт сушат над безводным сульфатом натрия и хлороформ затем отгоняют. Кристаллизацией остатка из эфира получают 0,84 г целевого соединения в виде белого кристаллического порошка (выход 52,9%).
ИК (KBr) ν: 3370, 2870, 2840, 1605, 1585, 1525, 1310, 1130, 1115, 1110, 1040, 750, 500 см-1.
1Н-ЯМР (CDCl3-CD3OD, 1:1 об/об) δ: 1,24 (т, 3Н, I=7 Гц), 3,28 (т, 2Н, I=5 Гц), 3,59 (к, 2Н, I=7 Гц), 3,69 (т, 2Н, I=5 Гц), 4,31 (д, 1Н, I=13 Гц), 4,53 (л, 1Н, I=13 Гц), 6,4-7,3 (м, 4Н), 7,21 (с, 2Н).
Т.пл. 119-121оС (с разл.)
П р и м е р 6. 2-[2-(3-Метоксипропиламино)бензилсульфинил]имидазол.
(I) Натриевая соль 3-метоксипропионовой кислоты
К раствору 25 г (212 ммоль) метилового эфира 3-метоксипропионовой кислоты в 250 мл метанола при охлаждении льдом по каплям прибавляют раствор 8,9 г (212 ммоль) гидроксида натрия в 25 мл воды, смесь перемешивают 30 мин при той же температуре и затем помещают на ночь в холодильник. Растворитель отгоняют при пониженном давлении и сушкой остатка при пониженном давлении получают 26,2 г целевого соединения (выход 98,1%).
1Н-ЯМР (CD3OD) δ: 2,41 (т, 2Н, I=7 Гц), 3,33 (с, 3Н), 3,63 (к, 2Н, I=7 Гц).
(II) Метиловый эфир N-(3-метоксипропаноил)антраниловой кислоты.
К суспензии 12,6 г (100 ммоль) полученной на стадии (I) натриевой соли в 100 мл бензола при комнатной температуре и перемешивании в течение 5 мин по каплям прибавляют 8 мл (110 ммоль) хлористого тионила и полученную смесь кипятят 30 мин. После охлаждения льдом нерастворимые вещества отфильтровывают, к фильтрату добавляют 15,1 г (100 ммоль) метилового эфира антраниловой кислоты и 13,8 г (100 ммоль) карбоната калия, после чего полученную смесь кипятят 4 ч. После добавления ледяной воды реакционную смесь последовательно промывают 6 н. соляной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия и сушат над безводным сульфатом натрия. Отгонкой растворителя получают 8,8 г маслянистого продукта, кристаллизацией которого из гексана получают 8,3 г целевого соединения в виде бледно-желтых кристаллов.
1Н-ЯМР (CDCl3) δ: 2,69 (т, 2Н, I=6 Гц), 3,4 (с, 3Н), 3,76 (т, 2Н, I=6 Гц), 3,91 (с, 3Н), 7,05 (дв. т., 1Н, I=1 и 8 Гц), 7,52 (дв. т, 1Н, I=2 и 8 Гц), 8 (дв.т., 1Н, I=2 и 8 Гц), 8,73 (дв.т., 1Н, I=1 и 8 Гц).
(III) 2-(3-Метоксипропил)аминобензиловый спирт.
К суспензии 0,96 г (25,3 ммоль) литийалюминийгидрида в 40 мл сухого ТГФ при охлаждении льдом в течение 15 мин по каплям прибавляют раствор 4 г (16,9 ммоль) метилового эфира N-(3-метоксипропаноил)антраниловой кислоты в 10 мл сухого ТГФ и полученную смесь кипятят 1 ч. Для разложения избытка литийалюминийгидрида к реакционной смеси при охлаждении льдом прибавляют насыщенный водный раствор сульфата натрия. После добавления 40 мл эфира нерастворимые вещества удаляют фильтрованием через целит. Органическую часть собирают и сушат над безводным сульфатом натрия. После отгонки растворителя при пониженном давлении получают 3,1 г целевого соединения в виде бледно-желтого масла (выход 94%).
1Н-ЯМР (CDCl3) δ: 1,92 (М, 2Н), 3,25 (т, 2Н, I=6 Гц), 3,34 (с, 3Н), 3,51 (т, 2Н, I=6 Гц), 4,62 (с, 2Н), 6,4-7,5 (м, 4Н).
(IV) 2-[2-(3-Метоксипропиламино)бензилтио]имидазол.
К раствору 2,9 г (14,8 ммоль) 2-(3-метоксипропил)аминобензилового спирта в 29 мл дихлорметана при охлаждении льдом в течение 5 мин по каплям прибавляют раствор 1,3 мл (17,8 ммоль) хлористого трионила в 3 мл дихлорметана и полученную смесь перемешивают 15 мин при комнатной температуре. Растворитель отгоняют при комнатной температуре и пониженном давлении, а к остатку добавляют 10 мл дихлорметана. Полученный дихлорметановый раствор постепенно прибавляют к раствору 2,22 г (22,2 ммоль) 2-меркаптоимидазола в 22 мл этанола и затем смесь перемешивают 15 мин при комнатной температуре. Этанол отгоняют при пониженном давлении, к остатку добавляют лед и насыщенный раствор гидрокарбоната натрия, после чего полученную смесь экстрагируют эфиром. Органическую часть отделяют и последовательно промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и после очистки остатка колоночной хроматографией на силикагеле получают 2,7 г целевого соединения в виде бледно-желтого масла (выход 65,8%).
1Н-ЯМР (CDCl3) δ; 1,92 (м, 2Н), 3,23 (т, 2Н, I=6 Гц), 3,32 (с, 3Н), 3,54 (т, 2Н, I=6 Гц), 4,14 (с, 2Н), 6,3-7,2 (м, 4Н), 7,02 (с, 2Н).
(V) 2-[2-(3-Метоксипропиламино)бензилсульфинил]имидазол.
К раствору 2,7 г (9,74 ммоль) 2-[2-(3-метоксипропиламино)-бензилтио/имидазола в 27 мл хлороформа при охлаждении льдом в течение примерно 10 мин прибавляют 2,1 г (9,74 ммоль) м-хлорнадбензойной кислоты (чистота 80%) и после завершения реакции добавляют насыщенный водный раствор гидрокарбоната натрия. Образовавшуюся смесь экстрагируют 30 мл хлороформа. Хлороформенную часть отделяют и последовательно промывают насыщенным водным раствором гидрокарбоната натрия и 6 мл 0,1н. водного раствора гидроксида натрия. Промытый раствор обрабатывают с целью экстракции 6 мл 1н. водного раствора гидроокиси натрия (6 ммоль). В щелочной части добавлением насыщенного раствора хлорида аммония создают щелочно-аммонийную среду, после чего экстрагируют хлороформом. Органическую часть отделяют и промывают насыщенным водным раствором хлорида натрия, затем сушат над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении и после кристаллизации остатка из смеси ацетонитрил-эфир получают 890 мг целевого соединения в виде желтых кристаллов (выход 31,2%).
ИК (KBr) ν: 3390, 2870, 1600, 1580, 1510, 1310, 1115, 1100, 1035, 1000, 890, 745 см-1.
1Н-ЯМР (CDCl3) δ: 1,86 (м, 2Н), 2,9-3,2 (м, 2Н), 3,32 (с, 3Н), 3,49 (т, 2Н, I= 6 Гц), 4,23, 4,52 (каждый, д, 2Н, I=13 Гц), 4,95 (уш. полоса, 1Н)= 6,4-7,3 (м, 6Н).
Т.пл. 100-107оС (с разл.)
П р и м е р 7. 2-[2-(2-Этоксиэтиламино)-5-метилбензилсульфинил]имидазол.
(I) Метиловый эфир (2-этоксиацетамидо-5-метил)бензойной кислоты
К раствору 4,95 г (30 ммоль) метилового эфира 5-метилантраниловой кислоты и 6,5 мл триэтиламина в 50 мл дихлорметана при охлаждении льдом в течение 15 мин по каплям прибавляют раствор 4,78 г (39 ммоль) этоксиацетилхлорида в 15 мл дихлорметана и полученную смесь перемешивают около суток при комнатной температуре. Затем реакционную смесь последовательно промывают 5% -ным водным раствором карбоната натрия, 3н. соляной кислотой 50-ным водным раствором карбоната натрия, после чего сушат над безводным сульфатом натрия. Отгонкой дихлорметана получают 7,6 г целевого соединения (чистота 99,1% ) в виде остаточных бледно-коричневых кристаллов (выход теоретический).
1Н-ЯМР (CDCl3) δ: 1,37 (т, 3Н, I=7 Гц), 2,33 (с, 3Н), 3,69 (к, 2Н, I=7 Гц), 3,92 (с, 3Н), 4,09 (с, 2Н), 7,1-8,7 (м, 3Н).
(II) 2-(2-Этоксиэтиламино)-5-метилбензиловый спирт.
К суспензии 2,28 г литийалюминийгидрида в 70 мл ТГФ при охлаждении льдом в течение 15 мин по каплям прибавляют раствор 7,6 г (30 ммоль) метилового эфира (2-этоксиацетамидо-5-метил)-бензойной кислоты в 20 мл ТГФ и полученную смесь кипятят 1 ч. Для разложения избытка литийалюминийгидрида к образовавшейся смеси при охлаждении льдом добавляют насыщенный водный раствор сульфата натрия. Органическую часть отделяют декантированием и растворитель удаляют. Остаток растворяют в хлороформе, полученный раствор промывают водой и сушат над безводным сульфатом натрия. Отгонкой хлороформа получают 7,19 г целевого соединения (чистота 87,2%) в виде остаточного коричневого масла (выход теоретический).
1Н-ЯМР (CDCl3) δ: 1,2 (т, 3Н, I=7 Гц), 2,23 (с, 3Н), 3,28 (т, 2Н, I=6 Гц), 3,52 (к, 2Н, I=7 Гц), 3,64 (т, 2Н, I=6 Гц) 4,6 (с, 2Н), 6,4-7,1 (м, 3Н).
(III) 2-[2-(2-Этоксиэтиламино)-5-метилбензилтио]имидазол
К раствору 7,19 г (30 ммоль) 2-(2-этоксиэтиламино)-5-метилбензилового спирта, полученного на стадии (II), в 60 мл дихлор-фметана при охлаждении льдом в течение 15 мин по каплям прибавляют раствор 2,8 мл хлористого тионила в 10 мл дихлорметана и полученную смесь перемешивают 30 мин при комнатной температуре. После отгонки растворителя в остатке получают маслянистый коричневый продукт, который растворяют в дихлорметане. Дихлорметановый раствор добавляют к раствору 3,6 г 2-меркаптоимидазола в 36 мл этанола и полученную смесь перемешивают 30 мин при комнатной температуре. Этанол отгоняют, к остатку добавляют хлороформ и 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и сушат над безводным сульфатом натрия. Хлороформ отгоняют и кристаллизацией остатка из смеси эфир-гексан получают 6,82 г целевого соединения в виде бледно-коричневого кристаллического порошка (выход 78,1%).
1Н-ЯМР (CDCl3) δ: 1,23 (т, 3Н, I=7 Гц), 2,15 (с, 3Н), 3,35 (т, 2Н, I=5 Гц), 3,62 (к, 2Н, I=7 Гц), 3,78 (т, 2Н, I=5 Гц), 4,1 (с, 2Н), 6,4-7,1 (м, 3Н), 6,99 (с, 2Н).
(IV) 2-[2-(2-Этоксиэтиламино)-5-метилбензилсульфинил]имидазол.
В смеси 60 мл дихлорметана, 60 мл метанола и 6 мл уксусной кислоты растворяют 6 г (21 ммоль) 2-[2-(2-этоксиэтиламино)-5-метилбензилтио]имидазола и к полученному раствору при охлаждении льдом прибавляют 9 мл 35%-ной водной перекиси водорода и 120 мг ванадата аммония, после чего смесь перемешивают 2,5 ч с охлаждением льдом. После завершения реакции к перемешиваемой смеси добавляют 5%-ный водный раствор карбоната натрия. Органическую смесь отделяют и дважды промывают 40 мл 0,1н. водного раствора гидроксида натрия и затем дважды экстрагируют 40 мл 1н. водного раствора гидроксида натрия. Водную часть собирают и нерастворимые вещества отфильтровывают. К фильтрату постепенно прибавляют 100 мл 1н. водного раствора хлорида аммония, осажденный кристаллический продукт отфильтровывают и промывают достаточным количеством воды. Сушкой полученного продукта при пониженном давлении и комнатной температуре получают 3,61 г целевого соединения в виде бледно-коричневого кристаллического порошка (выход 57,4%).
ИК (KBr) ν: 3320, 2960, 2900, 2860, 1615, 1520, 1470, 1435, 1420, 1335, 1310, 1105, 1020, 795, 780 см-1.
1Н-ЯМР (CDCl3-CD3OD, 1:1 об/об) δ: 1,23 (т, 3Н, I=7 Гц), 2,13 (с, 3Н), 3,24 (т, 2Н, I=6 Гц), 3,58 (к, 2Н, I=7 Гц), 3,69 (т, 2Н, I=6 Гц), 4,29 (д, 1Н, I=13 Гц), 4,42 (д, 1Н, I=13 Гц) 6,4-7 (м, 3Н), 7,22 (с, 2Н).
Т.пл. 145-146оС (с разл.).
П р и м е р 8. 2-[4,6-Диметил-5-метокси-2-(2-метоксиэтиламино)бензилсульфинил]имидазол.
(I) N-(3,5-диметил-4-метоксифенил)-2-метоксиацетамид.
К раствору 5 г (33 ммоль) 3,5-диметил-4-метоксианилина и 7,1 мл триэтиламина в 50 мл дихлорметана при охлаждении льдом в течение 30 мин по каплям прибавляют раствор 4,31 (40 ммоль) метоксиацетилхлорида в 20 мл дихлорметана и полученную смесь перемешивают 30 мин при комнатной температуре. Затем реакционную смесь последовательно промывают 5%-ным водным раствором карбоната натрия и 3н. соляной кислотой, и сушат над безводным сульфатом натрия. Дихлорметан отгоняют и кристаллизацией остатка из смеси эфир-гексан получают 6,7 г целевого соединения в виде белых кристаллов (выход 90,7%).
1Н-ЯМР (CDCl3) : 2,27 (с, 6Н), 3,48 (с, 3Н), 3,69 (с, 3Н), 3,98 (с, 2Н), 7,21 (с, 2Н), 8,06 (уш. с., 1Н).
(II) 3,5-Диметил-4-метокси-N-(2-метоксиэтил)анилин.
К суспензии 2,5 г литийалюминийгидрида в 70 мл ТГФ при охлаждении льдом в течение 10 мин добавляют 6,7 г N-(3,5-диметил-4-метоксифенил)-2-метоксиацетамида и полученную смесь кипятят 1 ч. Для разложения избытка литийалюминийгидрида к полученной смеси добавляют насыщенный водный раствор сульфата натрия с охлаждением льдом. Органическую часть отделяют декантированием и растворитель отгоняют. К остатку добавляют дихлорметан и воду. Дихлорметановую часть отделяют и сушат над безводным сульфатом натрия. Отгонкой растворителя получают 6,5 г целевого соединения (чистота 96,6%) в качестве остаточного бледно-желтого масла (выход теоретический).
1Н-ЯМР (CDCl3) : 2,22 (с, 6Н), 3,22 (т, 2Н, I=5 Гц), 3,37 (с, 3Н), 3,58 (т, 2Н, I=5 Гц), 3,65 (с, 3Н), 6,29 (с, 2Н).
(III) [3,5-Диметил-4-метокси-N-(2-метоксиэтил)-2-метилтиометил]анилин.
К раствору 6,5 т (30 ммоль) 3,5-диметил-4-метокси-N-(2-метоксиэтил)анилина, полученного на стадии (II), и 3,3 мл диметилсульфида в 100 мл дихлорметана при охлаждении льдом в течение 30 мин прибавляют 5,98 г N-хлорсукцинимида (НХС) и полученную смесь перемешивают 10 мин с охлаждением льдом. После добавления 6,3 мл триэтиламина смесь кипятят с перемешиванием 1 ч. После завершения реакции добавляют 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и сушат над безводным сульфатом натрия. Дихлорметан отгоняют, остаток очищают колоночной хроматографией на силикагеле. Отгонкой растворителя получают 5,62 г целевого соединения в виде бледно-желтого масла (выход 69,6%).
1Н-ЯМР (CDCl3) δ: 2,06 (с, 3Н), 2,25 (с, 3Н), 2,27 (с, 3Н), 3,27 (т, 2Н), 3,4 (с, 3Н), 3,63 (с, 3Н), 3,65 (т, 2Н), 3,75 (с, 2Н), 6,37 (с, 1Н).
(IV) 3,5-Диметил-4-метокси-N-(2-метоксиэтил)-2-(метилсульфинилметил)анилин.
В смеси 50 мл хлороформа и 5 мл метанола растворяют 5,62 г (21 ммоль) [3,5-диметил-4-метокси-N-(2-метоксиэтил)-2-метил- тиометил]анилин и к полученному раствору при охлаждении льдом в течение 15 мин прибавляют 3,61 г 85% -ной м-хлорнадбензойной кислоты. После завершения реакции добавляют хлороформ и 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и сушат над безводным сульфатом натрия. Хлороформ отгоняют, остаток очищают колоночной хроматографией на силикагеле. После отгонки растворителя получают 3,74 г целевого соединения в виде бледно-коричневых кристаллов (выход 62,8%).
1Н-ЯМР (CDCl3) δ: 2,22 (с, 3Н), 2,26 (с, 3Н), 2,59 (с, 3Н), 3,22 (т, I= 5 Гц, 2Н), 3,39 (с, 3Н), 3,61 (т, I=5 Гц, 2Н), 3,63 (с, 3Н), 3,96 (д, I=14 Гц, 1Н), 4,28 (д, I=14 Гц, 1Н), 6,43 (с, 1Н).
(V) 2-[4,6-Диметил-5-метокси-2-(2-метоксиэтиламино)бензилтио]-имидазол.
Через раствор 3,74 г (13 ммоль) [3,5-диметил-4-метокси-N-(2-метоксиэтил)2-(ме- тилсульфинилметил)] анилина при комнатной температуре в 40 мл дихлорметана 10 мин продувают газообразный хлористый водород, затем смесь перемешивают 10 мин. После отгонки растворителя остаток растворяют 20 мл дихлорметана. Полученный раствор в течение 10 мин прибавляют к раствору 3,9 г 2-меркаптоимидазола в 40 мл этанола, после чего перемешивают 30 мин при комнатной температуре. Этанол отгоняют, к остатку добавляют дихлорметан и 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и сушат над безводным сульфатом натрия. Дихлорметан отгоняют, остаток очищают колоночной хроматографией на силикагеле. Растворитель отгоняют и кристаллизацией остатка из смеси эфир-гексан получают 3,15 г целевого соединения в виде белого кристаллического порошка (выход 75%).
1Н-ЯМР (СDCl3) δ: 2,16 (с, 3Н), 2,24 (с, 3Н), 3,3 (т, 2Н, I=5Гц) 3,42 (с, 3Н), 3,6 (с, 3Н), 3,67 (т, 2Н, I=5 Гц), 4,18 (с, 2Н), 6,42 (с, 1Н), 7,04 (с, 2Н).
(VI) 2-[4,6-Диметил-5-метокси-2-(2-метоксиэтиламино)бензилсульфинил]имид- азол.
В 20 мл хлороформа и 2 мл метанола растворяют 2 г (6,2 ммоль) 2-[4,6-диметил-5-метокси-2-(2-метоксиэтиламино)бензил- тио]имидазола и к полученному раствору при охлаждении смесью NaCl-лед в течение 20 мин прибавляют 1,27 г 85% -ной м-хлорнадбензойной кислоты. После завершения реакции добавляют хлороформ и 5%-ный водный раствор карбоната натрия. Органическую часть отделяют и дважды промывают 20 мл 0,05н. водным раствором гидроксида натрия, после чего полученную смесь экстрагируют 30 мл 1н. водным раствором гидроксида натрия. Водную часть отделяют и промывают хлороформом. К промытому раствору добавляют 45 мл 1н. водного раствора хлорида аммония. Осадившийся маслянистый продукт экстрагируют дихлорметаном. Дихлорметановую часть отделяют и сушат над безводным сульфатом натрия. Дихлорметан отгоняют и кристаллизацией остатка из смеси эфир-гексан получают 0,78 г целевого соединения в виде бледно-коричневого кристаллического порошка (выход 37%).
ИК (KBr) ν: 3200, 2900, 2880, 1580, 1500, 1460, 1410, 1330, 1240, 1210, 1120, 1090, 1050, 1040, 1020, 1000, 860, 750 см-1.
1Н-ЯМР (CDCl3-CD3OD, 1:1 об/об) δ: 2,15 (с, 3Н), 2,26 (с, 3Н), 3,23 (т, 2Н, I=5 Гц), 3,42 (с, 3Н), 3,63 (с, 3Н), 3,67 (т, 2Н, I=5 Гц), 4,39 (д, 1Н, I=14 Гц), 4,61 (д, 1Н, I=14 Гц), 6,46 (с, 1Н), 7,27 (с, 2Н).
Т.пл. 110-111оС (с разл.).
Препаративный пример 1.
Пример препарата (таблетки)
Каждая таблетка (220 мг) содержит следующие компоненты, мг: Активный компонент 50 Лактоза 103 Крахмал 50 Стеарат магния 2 Гидроксипропилцеллюлоза 15
Препаративный пример 2.
Пример препарата (капсулы)
Каждая твердая желатиновая капсула (350 мг) содержит следующие компоненты, мг: Активный компонент 40 Лактоза 200 Крахмал 70 Полицинилпирролидон 5 Кристаллическая целлюлоза 35
Препаративный пример 3.
Пример препарата (гранулы)
Каждая гранула (1 г) содержит следующие компоненты, мг: Активный компонент 200 Лактоза 450 Кукурузный крахмал 300 Гидроксипропилцеллюлоза 50
Сравнительные эксперименты осуществлялись с использованием известных соединений, которые довольно близки к соединениям, отвечающим данному изобретению. Испытываемые для сравнения соединения представляют собой следующие:
Соединение А (описано в английском патенте N 2163747):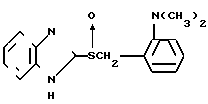
Соединение В (также описано в английском патенте N 216747):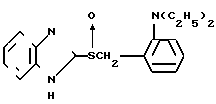
Соединение С (описано в Европейском патенте N 234690А):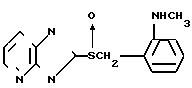
Соединение D (также описано в Европейском патенте N 234690А):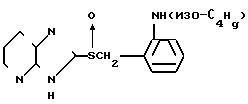
Результаты испытания при той же дозе, составляющей 3 мг/кг, приведены ниже. Испытываемое Подавляющее соединение действие, % Соединение А 22 Соединение В нет Соединение С 20 Соединение Д 35
Таким образом, новые соединения, полученные предлагаемым способом, имеют преимущество перед аналоговыми соединениями, уже известными ранее, в отношении их ингибирующего действия против секреции желудочной кислоты.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 1993 |
|
RU2069212C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА | 1995 |
|
RU2137770C1 |
| АЛКИЛЕНДИАМИНОВЫЕ ПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1993 |
|
RU2069661C1 |
| АКТИВИРУЮЩИЙ АГЕНТ ДЛЯ РЕЦЕПТОРА, АКТИВИРУЕМОГО СТИМУЛИРУЮЩИМИ РОСТ ПЕРОКСИСОМ АГЕНТАМИ | 2009 |
|
RU2501794C2 |
| ПРОИЗВОДНЫЕ БУТЕНОВОЙ ИЛИ ПРОПЕНОВОЙ КИСЛОТЫ | 1992 |
|
RU2041871C1 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛА, ОПТИЧЕСКИ ИНЕРТНЫЕ ИЛИ ИМЕЮЩИЕ R- ИЛИ S-КОНФИГУРАЦИЮ C-2 И C-3 АСИММЕТРИЧНЫХ ЦЕНТРОВ, ИЛИ ИХ СОЛИ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2039050C1 |
| Способ получения пиперазиновых производных или их фармацевтически приемлемых солей (его варианты) | 1982 |
|
SU1318161A3 |
| ПРОИЗВОДНЫЕ ДИФЕНИЛМЕТИЛИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1990 |
|
RU2024521C1 |
| ПРОИЗВОДНЫЕ ПИРИДИНА | 1992 |
|
RU2035461C1 |
| Способ получения производных хинальдинамида или их кислотно-аддитивных солей | 1986 |
|
SU1450741A3 |
Использование: в медицине, в частности в качестве соединений с повышенной противоязвенной активностью и меньшей токсичностью. Сущность изобретения: продукт - соединение ф-лы I, где R1 = H, C1-C6 -алкил, R2 -алкил, замещенный C1-C4 - алкоксигруппой; каждый из R3-R6 - независимы - Н, галоид, C1-C6 -алкил, C1-C6 -алкоксигруппа, фторзамещенный C1-C6 -алкил или фторзамещенная C1-C6 -алкоксигруппа. Реагент 1: соединение ф-лы II, где X - отщепляемая группа. Реагент 3: 2-меркаптоимидазол. Реагент 4: соединение ф-лы III. Условие реакции: окисление указанных реагентов. Структура соединений ф-лы 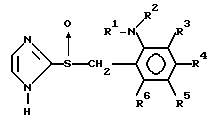
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ИМИДАЗОЛА общей формулы I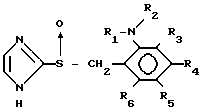
где R1 - водород или С1 - С6-алкил;
R2 - С2 - С6-алкил, замещенный С1 - С4-алкоксигруппой;
R3 - R6 каждый независимо - водород, галоид, С1 - С6-алкил, С1 - С6-алкоксигруппа, фторзамещенный С1 - С6-алкил, фторзамещенный С1 - С6-алкил, фторзамещенная С1 - С6-алкоксигруппа,
отличающийся тем, что осуществляют реакцию соединения общей формулы II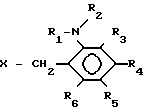
где R1 - R6 имеют указанные значения;
Х - отщепляемая группа,
с 2-меркаптоимидазолом с получением производного имидазола общей формулы III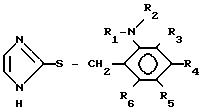
где R1 - R6 имеют указанные значения,
и окисляют полученное производное имидазола общей формулы III.
| Вейганд-Хильгетаг, Методы эксперимента в органической химии, М.: Химия, 1968, с.592. |
