Техническая область изобретения
Данное изобретение относится к кристаллическим формам производных 1-метилкарбапенема или его фармацевтически приемлемых солей, которые проявляют превосходную антибиотическую активность против различных бактериальных штаммов и являются достаточного стабильными для сохранения в течение продолжительного времени.
Данное изобретение относится к композициям для профилактики (предотвращения) или лечения бактериальных инфекционных заболеваний, содержащим кристаллическую форму настоящего изобретения в качестве активного ингредиента.
Данное изобретение относится к применению кристаллической формы настоящего изобретения для получения лекарственных средств для профилактики или лечения бактериальных инфекционных заболеваний.
Данное изобретение относится к способам профилактики или лечения бактериальных инфекционных заболеваний, которые включают введение теплокровному животному, нуждающемуся в такой профилактике или лечении, эффективного количества кристаллической формы настоящего изобретения.
Данное изобретение далее относится к способам получения кристаллических форм настоящего изобретения.
Предпосылки изобретения
Производное 1-метилкарбапенема формулы (I) описано в публикациях заявок на патент Японии Hei-10-204086 и Hei-11-071277. Данное соединение (I) проявляет превосходную антибиотическую активность не только против штаммов грамположительных бактерий, но также и против штаммов грамотрицательных бактерий, и можно отследить, что оно станет полезным антибиотическим агентом.
Однако соединение (I), полученное по примеру публикации заявки на патент Японии Hei-11-071277, получено лиофилизацией в виде некристаллического порошка. Данный порошок является нестабильным и представляет собой материал, который трудно сохранить в течение продолжительного времени. Имеется много проблем при практическом использовании порошка в качестве лекарственного средства, особенно в качестве антибиотического агента.
Авторы данного изобретения приложили значительные усилия для решения данных проблем и обнаружили, что некоторые кристаллические формы соединения (I) являются чрезвычайно стабильными по сравнению с некристаллическим порошком соединения (I) и являются полезными лекарственными средствами, особенно практически полезными антибиотическими агентами. Стабильные кристаллические формы данного изобретения включают кристаллическую форму 1/2 карбоната соединения (I), содержащего 1/2 этанол (I-1), кристаллическую форму соединения (I), содержащего 1/2 этанол (I-2), кристаллическую форму соединения (I) и кристаллическую форму соединения (I), содержащего 1/4 этанола и 3/2 воды (I-3).
Описание изобретения
Данное изобретение относится к:
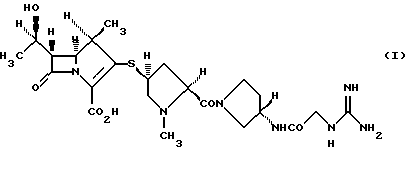
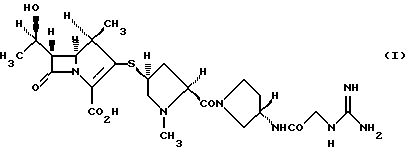
1) производному 1-метилкарбапенема формулы (I) или его фармацевтически приемлемой соли в кристаллической форме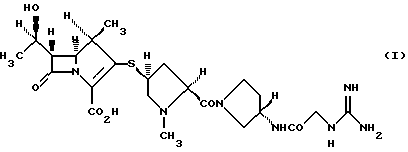
2) производному 1-метилкарбапенема формулы (I-1) в кристаллической форме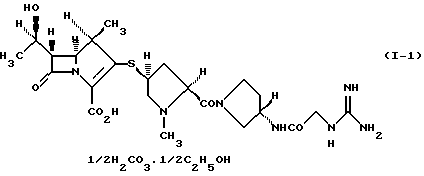
3) производному 1-метилкарбапенема формулы (I-2) в кристаллической форме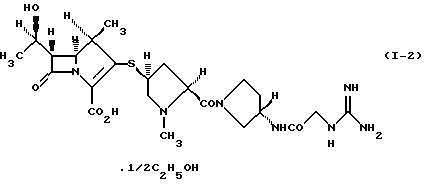
4) производному 1-метилкарбапенема формулы (I) в кристаллической форме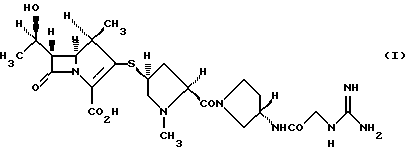
5) производному 1-метилкарбапенема формулы (I-3) в кристаллической форме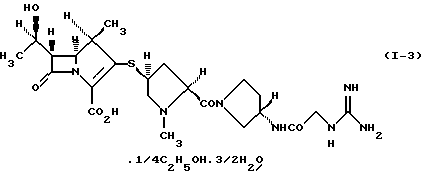
6) фармацевтическим композициям для профилактики или лечения бактериальных инфекционных заболеваний, содержащим кристаллическую форму производного 1-метилкарбапенема по любому одному из п.п.1-5 в качестве активного ингредиента;
7) применению кристаллической формы производного 1-метилкарбапенема по любому из п.п.1-5 для получения лекарственного средства для профилактики или лечения бактериальных инфекционных заболеваний и
8) способам профилактики или лечения бактериальных инфекционных заболеваний, которые включают введение теплокровному животному, нуждающемуся в такой профилактике или лечении, эффективного количества кристаллической формы производного 1-метилкарбапенема по любому из п.п.1-5.
Производные карбапенема формулы (I) описаны в публикациях заявок на патент Японии Hei-10-204086 и Hei-11-071277 и проявляют сильную активность против грамположительных и грамотрицательных бактериальных штаммов.
Производные карбапенема формулы (I) могут существовать в виде фармацевтически приемлемых солей. Имеется в виду, что термин "фармацевтически приемлемая соль", используемый здесь и в формуле изобретения, включает соли, которые обычно можно использовать в качестве лекарственных средств.
Соединение формулы (I) имеет основные группы, такие как третичная аминогруппа и гуанидиногруппа, и может превращаться в фармацевтически приемлемую кислотно-аддитивную соль обработкой подходящей кислотой с применением общепринятых приемов. Такие кислотно-аддитивные соли включают соли неорганических кислот, такие как гидрохлориды, гидробромиды, сульфаты и фосфаты, соли органических кислот, такие как карбонаты, ацетаты, бензоаты, оксалаты, малеаты, фумараты, тартраты и цитраты, и сульфонаты, такие как метансульфонаты, бензолсульфонаты и п-толуолсульфонаты.
Соединение формулы (I) имеет кислотную группу, такую как карбоксильная группа, и может превращаться в фармацевтически приемлемую аддитивную соль основания с помощью обработки подходящим основанием с применением общепринятых приемов. Такие аддитивные соли оснований включают соли щелочных металлов, такие как натриевые соли, калиевые соли и литиевые соли; соли щелочноземельных металлов, такие как соли кальция и магния; соли металлов, такие как соли алюминия, соли железа, соли цинка, соли меди, соли никеля и соли кобальта, и соли четвертичного аммония, такие как аммониевые соли.
Когда их оставляют стоять на воздухе, некоторые формы соединения (I) и его фармацевтически приемлемых солей абсорбируют или адсорбируют воду и могут образовывать гидраты. В некоторых случаях формы соединения (I) и его фармацевтически приемлемых солей абсорбируют некоторые растворители и могут образовывать сольваты. Соединение (I) данного изобретения и его фармацевтически приемлемые соли включают такие гидраты и сольваты. Такими солями, гидратами и сольватами являются, предпочтительно, натриевые соли, гидрохлориды, сульфаты, карбонаты, гидраты или сольваты этанола; наиболее предпочтительно карбонаты, гидраты или сольваты этанола.
Соединение формулы (I-1) представляет 1/2 сольват этанола 1/2 карбонатной соли производного 1-метилкарбапенема формулы (I). Соединение формулы (I-2) представляет 1/2 сольват этанола производного 1-метилкарбапенема формулы (I). Соединение формулы (I-3) представляет 3/2 гидрат и 1/4 сольват этанола производного 1-метилкарбапенема формулы (I).
Кристаллические формы настоящего изобретения являются твердыми веществами, которые имеют регулярное расположение атомов (группы атомов) в трехмерной структуре и повторяют такие расположения. Кристаллы отличаются от аморфного твердого вещества, которое не имеет такого регулярного расположения атомов в трехмерной структуре.
Обычно некоторые соединения образуют множество кристаллических форм (полиморфных кристаллов) в соответствии с условием кристаллизации, кристаллы которых различаются в отношении их трехмерного расположения атомов и физико-химических свойств. Данное изобретение может включать каждую из таких кристаллических форм и смеси не менее чем двух их форм.
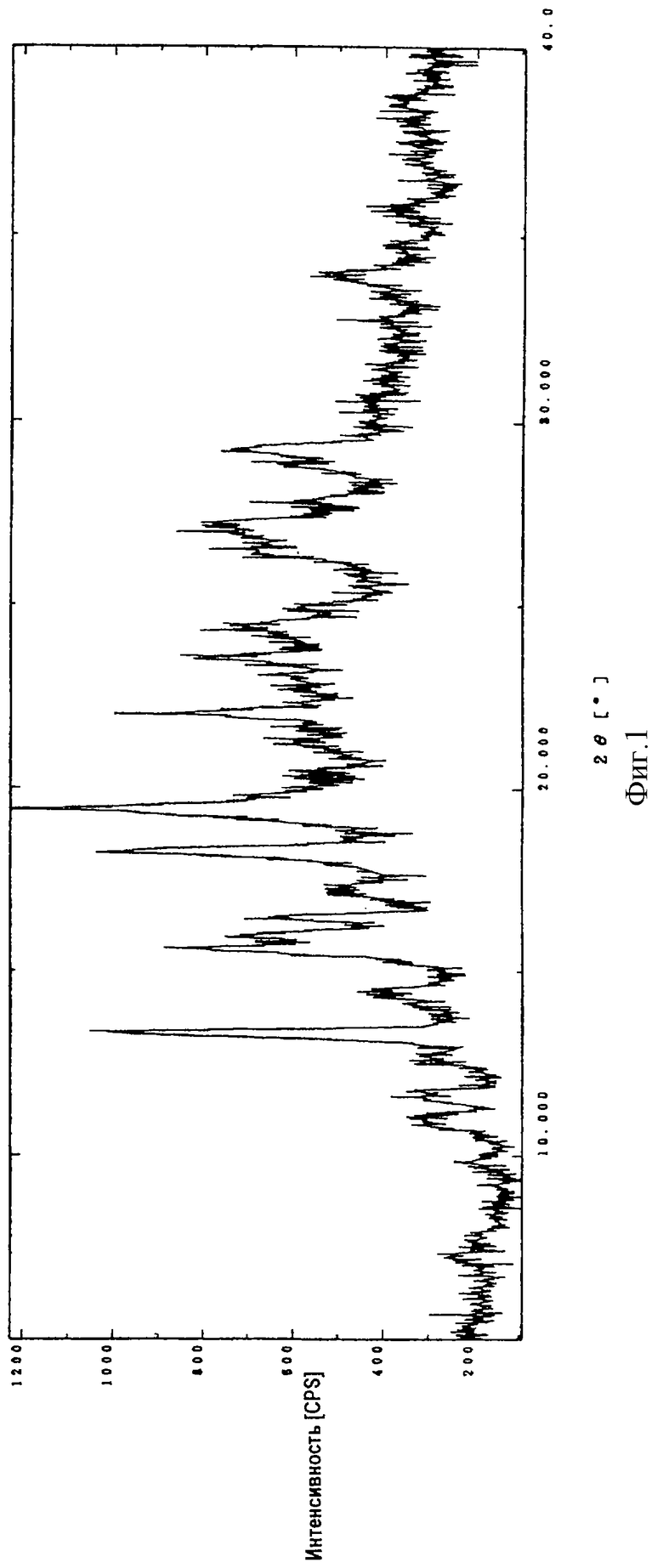
Кристаллическая форма производного 1-метилкарбапенема формулы (I-1) обнаруживает основные пики при межплоскостных расстояниях d=6,65, 5,68, 4,86, 4,57 и 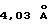 на порошковой рентгенограмме, полученной с Сu Kα облучением
на порошковой рентгенограмме, полученной с Сu Kα облучением  Основные пики имеют интенсивность не меньше, чем 74, которая является относительной интенсивностью, когда интенсивность пика при
Основные пики имеют интенсивность не меньше, чем 74, которая является относительной интенсивностью, когда интенсивность пика при 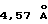 оценивают как 100.
оценивают как 100.
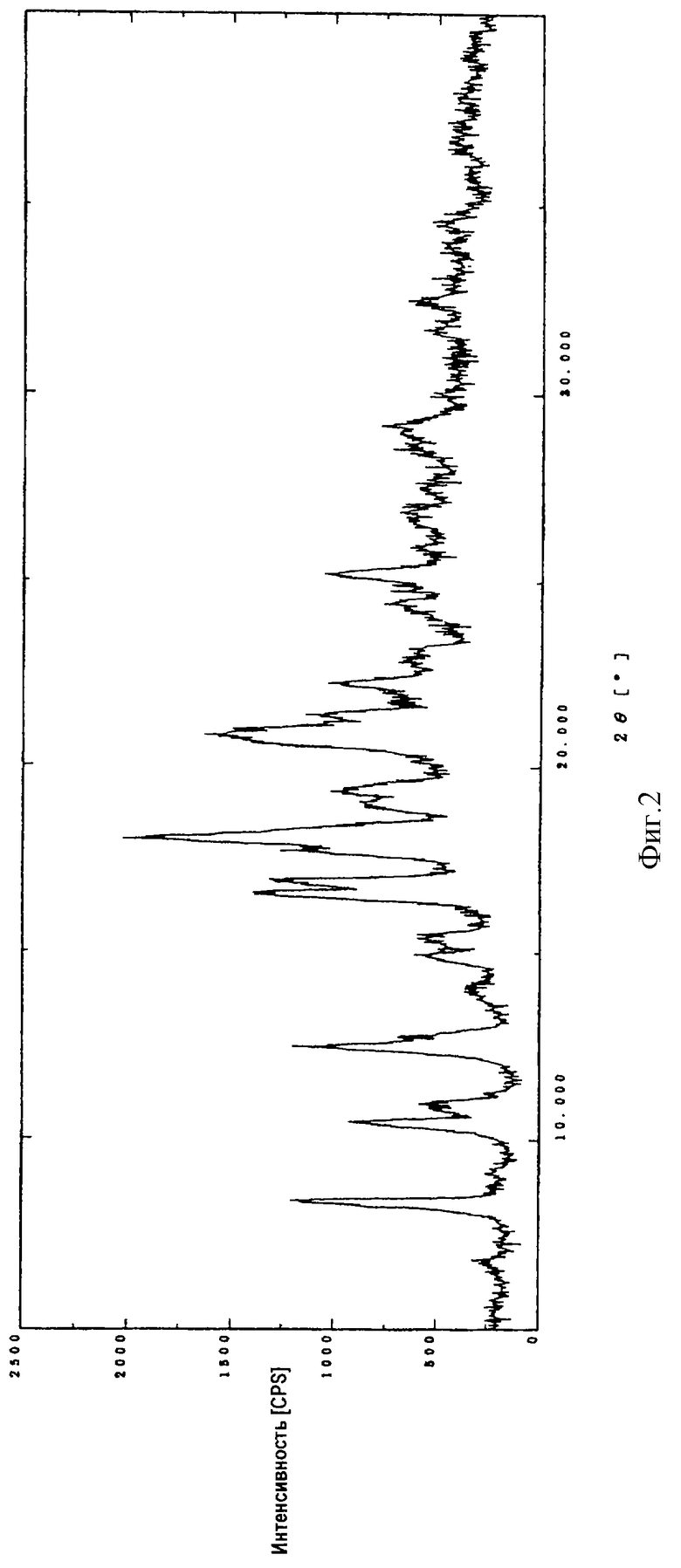
Кристаллическая форма производного 1-метилкарбапенема формулы (I-2) обнаруживает основные пики при межплоскостных расстояниях d=10,57, 7,12, 5,34, 5,23, 4,91 и 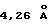 на порошковой рентгенограмме, полученной с Сu Kα облучением
на порошковой рентгенограмме, полученной с Сu Kα облучением 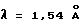 . Основные пики имеют интенсивность не меньше, чем 56, которая является относительной интенсивностью, когда интенсивность пика при
. Основные пики имеют интенсивность не меньше, чем 56, которая является относительной интенсивностью, когда интенсивность пика при 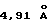 оценивают как 100.
оценивают как 100.
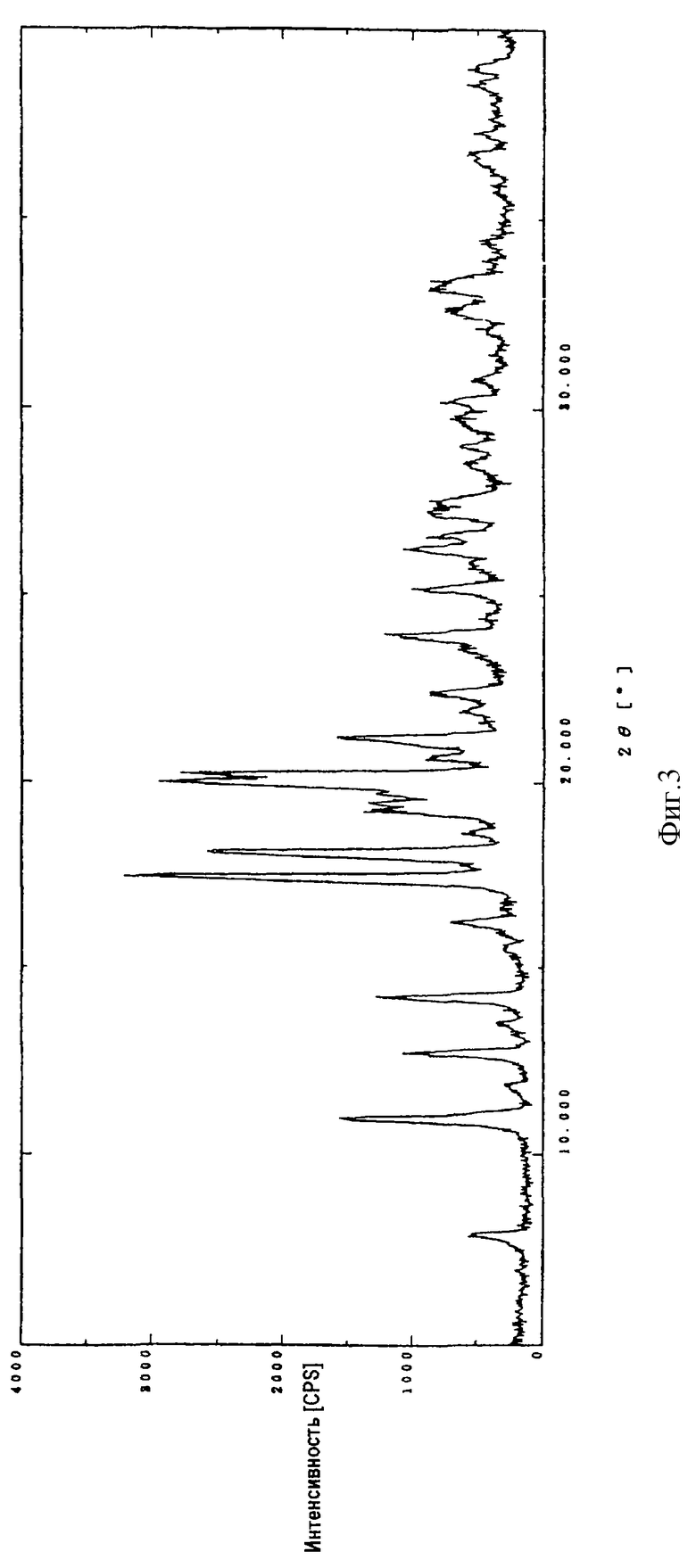
Кристаллическая форма производного 1-метилкарбапенема формулы (I) обнаруживает основные пики при межплоскостных расстояниях d=8,07, 5,08, 4,89, 4,44, 4,39 и 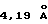 на порошковой рентгенограмме, полученной с Сu Kα облучением
на порошковой рентгенограмме, полученной с Сu Kα облучением 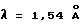 . Основные пики имеют интенсивность не меньше, чем 48, которая является относительной интенсивностью, когда интенсивность пика при
. Основные пики имеют интенсивность не меньше, чем 48, которая является относительной интенсивностью, когда интенсивность пика при 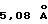 оценивают как 100.
оценивают как 100.
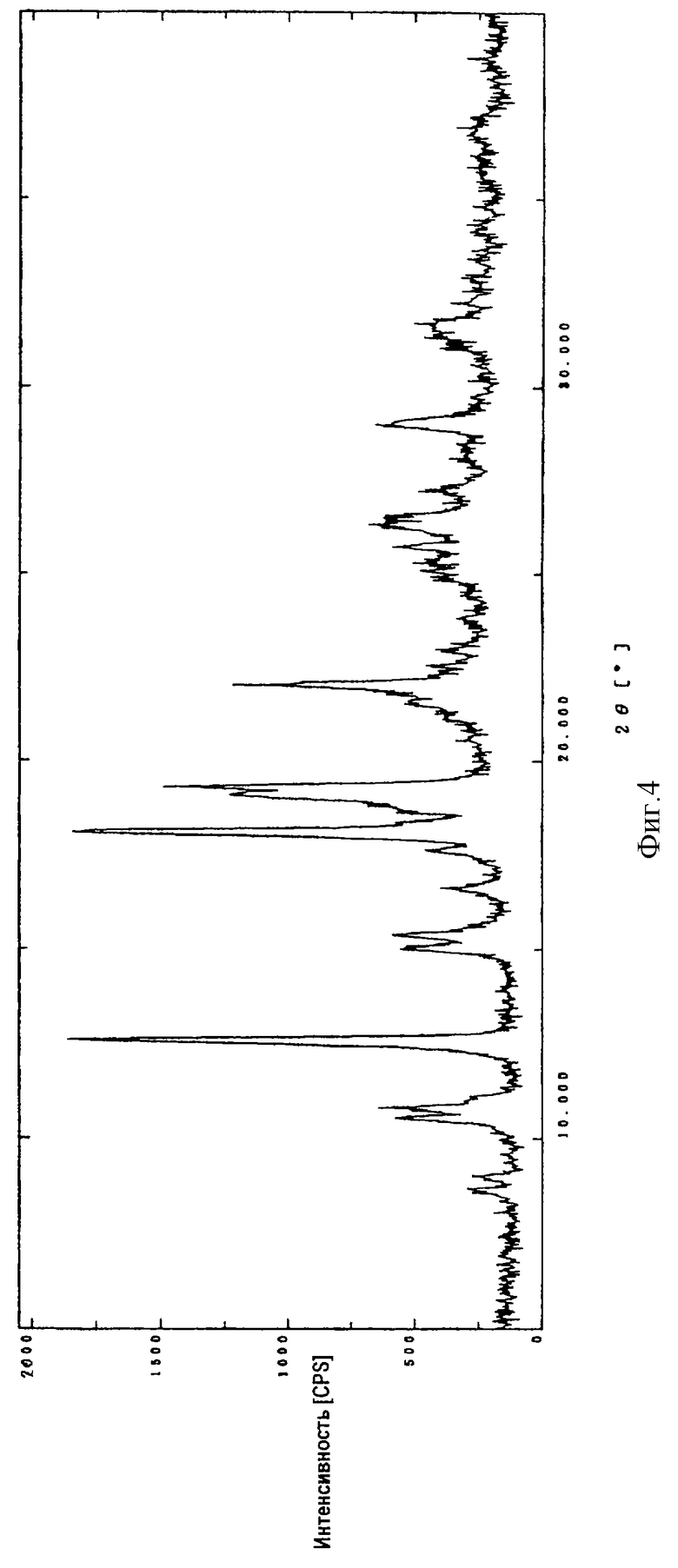
Кристаллическая форма производного 1-метилкарбапенема формулы (I-3) обнаруживает основные пики при межплоскостных расстояниях d=7,02, 4,90, 4,64, 4,59 и 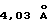 на порошковой рентгенограмме, полученной с Сu Kα облучением
на порошковой рентгенограмме, полученной с Сu Kα облучением 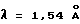 . Основные пики имеют интенсивность не меньше, чем 65, которая является относительной интенсивностью, когда интенсивность пика при
. Основные пики имеют интенсивность не меньше, чем 65, которая является относительной интенсивностью, когда интенсивность пика при 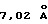 оценивают как 100.
оценивают как 100.
Соединение формулы (I) может быть получено таким же способом, как описано, или с помощью процедуры, аналогичной процедуре, описанной в публикациях заявок на патент Японии Hei-10-204086 и Hei-11-071277.
Кристаллические формы данного изобретения были получены, например,
1) растворением соединения (I) или его фармацевтически приемлемой соли в подходящем растворителе, который может его легко растворить,
2) если необходимо, концентрированием раствора, добавлением к раствору подходящего растворителя, который может слегка растворять соединение (I) или его фармацевтически приемлемую соль, или охлаждением раствора, чтобы привести к перенасыщенному раствору и, следовательно, к кристаллизации, и
3) выделением кристаллов и затем сушкой кристаллов.
Осаждение кристаллов начинается самопроизвольно в сосуде или осаждение можно также начать или ускорить добавлением затравочных кристаллов или механической стимуляцией, такой как облучение ультразвуковой волной и поскребыванием по поверхности сосуда.
Фармацевтически приемлемыми солями соединения (I) являются, предпочтительно, гидрохлориды, сульфаты и карбонаты; наиболее предпочтительно карбонаты. Фармацевтически приемлемые соли могут быть получены добавлением необходимого количества требуемой кислоты или основания к раствору соединения (I).
Когда обрабатывают растворы соединения (I) или его фармацевтически приемлемых солей, растворы этих соединений обычно обрабатывают при температуре между 0 и 60oС во избежание разложения этих соединений.
Предпочтительная температура кристаллизации этих соединений составляет между 0 и 10oС.
Способами концентрирования растворов соединения формулы (I) или его фармацевтически приемлемых солей являются способ упаривания с использованием роторного испарителя при пониженном или нормальном давлении при нагревании и способ концентрирования с использованием обратноосмотической мембраны. Обратноосмотическую мембрану, используемую для концентрирования водного раствора, можно выбрать из полиакрилонитрильных мембран, поливинилспиртовых мембран, полиамидных мембран и целлюлозоацетатных мембран.
Примерами растворителей, которые могут легко растворять соединение (I) или его фармацевтически приемлемые соли, являются вода, диметилсульфоксид, диметилформамид и метанол, предпочтительно вода.
Примерами растворителей, которые могут слегка растворять соединение (I) или его фармацевтически приемлемые соли, являются С2-С4-спирты, такие как этанол, пропанол и бутанол; кетоны, такие как ацетон и метилэтилкетон; простые эфиры, такие как диэтиловый эфир и тетрагидрофуран; и сложные эфиры, такие как метилацетат и этилацетат; предпочтительно этанол и ацетон; наиболее предпочтительно этанол.
Можно использовать исходное соединение (I), которое выделяют в виде лиофилизованного порошка. Можно также использовать неочищенный реакционный раствор, содержащий соединение (I), поскольку его можно очистить кристаллизацией.
Перенасыщение может достигаться концентрированном водного раствора соединения (I) при температуре между 30 и 60oС до получения насыщенного водного раствора с последующим постепенным охлаждением до температуры между 0 и 10oС или оно может достигаться постепенным добавлением подходящего растворителя, который может слегка растворять соединение (I) или его фармацевтически приемлемую соль, такого как этанол или ацетон, к насыщенному водному раствору, если необходимо, с последующим охлаждением.
Кристаллические формы данного изобретения, предпочтительно, осаждаются, когда концентрируют водные растворы соединения (I) или фармацевтически приемлемых солей, если необходимо, с последующим добавлением растворителя, который может слегка растворять данные соединения с последующим охлаждением. Наиболее предпочтительно, кристаллы данного изобретения осаждаются, когда концентрируют водные растворы соединения (I) или его фармацевтически приемлемых солей с последующим добавлением этанола или ацетона и затем охлаждением.
Наиболее предпочтительно, предпочтительная кристаллическая форма соединения (I-1) осаждается, когда концентрируют водный раствор соединения (I) с последующим насыщением диоксидом углерода, добавлением этанола и охлаждением; предпочтительная кристаллическая форма соединения (I-2) осаждается, когда концентрируют водный раствор соединения (I) с последующим добавлением этанола и охлаждением (предпочтительно, облучением ультразвуковыми волнами); предпочтительная кристаллическая форма соединения (I) осаждается, когда концентрируют водный раствор соединения (I) с последующим охлаждением; предпочтительная кристаллическая форма соединения (I-3) осаждается, когда концентрируют водный раствор соединения (I) с последующим добавлением этанола и охлаждением.
Осажденные кристаллы выделяют, например, фильтрованием, центрифугированием или декантацией. Если необходимо, выделенные кристаллы можно промыть подходящим растворителем. Кристаллы, предпочтительно, промывают сначала растворителем, который используют при кристаллизации, затем промывают растворителем, таким как этанол, ацетон и простой эфир.
Выделенные кристаллы сушат при температуре между 10 и 50oС, предпочтительно, между 20 и 30oС, до тех пор, пока вес кристаллов не станет постоянным. Если необходимо, их можно сушить в присутствии осушающих агентов, таких как силикагель и хлорид кальция, при пониженном давлении.
Кристаллические формы соединения (I-1), (I-2), (I) и (I-3) являются удобными для практического лечения в качестве лекарственного средства и крайне стабильными по сравнению с лиофилизованным порошком соединения (I), который описан в публикации заявки на патент Японии Hei-11-071277.
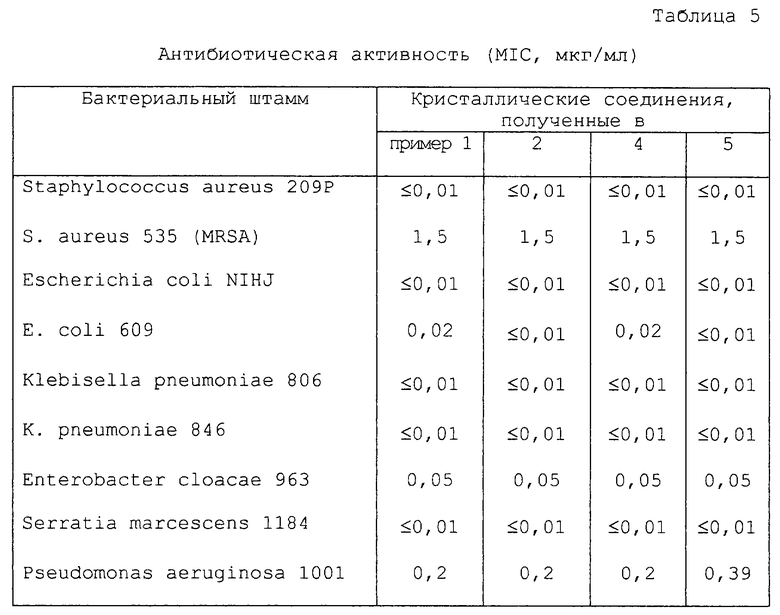
Кристаллические формы данного изобретения проявляют широкий спектр антибиотической активности и сильную антибактериальную активность против грамположительных и грамотрицательных штаммов и анаэробных бактерий, а также бактерий, продуцирующих цефалоспориназу. Когда антибактериальную активность кристаллов данного изобретения определяют методом разведения на пластинах агара, они проявляют сильную антибактериальную активность против различных бактерий, например грамположительных штаммов, таких как Staphylococcus aureus, метициллин-устойчивых Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus и аналогичных; грамотрицательных штаммов, таких как Escherichia coli, Bacillus dysenteriae, Klebsiella pneumoniae, Proteus vulgaris, Serratia, Enterobacteriaceae, Pseudomonas aeruginosa и т.п.; и анаэробных бактерий, таких как Bacteroides fragilis. Кристаллические формы данного изобретения проявляют сильную антибактериальную активность против Helicobacter pylori, которые часто обнаруживают у пациентов с хроническим гастритом и пептическими язвами.
Когда подходящие растворы кристаллических форм данного изобретения вводили мышам, они проявляли продолжительные полупериоды концентрации в крови и хорошее выделение с мочой по сравнению с показателями аналогичных соединений, известных специалистом в данной области.
Когда кристаллические формы данного изобретения вводили подкожно мышам, инфицированным системно Staphylococcus aureus, Streptococcus pneumoniae, Escherichia coli или Pseudomonas aeruginosa, они проявляли превосходный эффект лечения. Кристаллические формы данного изобретения, следовательно, являются полезными лекарственными средствами (особенно антибактериальными агентами).
Когда кристаллические формы данного изобретения используют в качестве лекарственных средств (особенно в качестве антибактериальных агентов), их можно вводить как таковые или в виде смеси указанных кристаллических форм данного изобретения и фармацевтически приемлемого эксципиента(ов) и разбавителя(ей); их можно вводить в различных дозированных лекарственных формах, таких как таблетки, капсулы, гранулы, порошки или сиропы для перорального введения, таких как инъекции для парентерального введения или таких как мази для местного применения.
Такие дозированные формы получают способами, известными специалистам в данной области, с использованием добавок, таких как эксципиенты, связующие, дезинтеграторы, смазывающие вещества, стабилизаторы, корригенты, суспендирующие агенты, разбавители, растворители для получения готовой препаративной формы, вспомогательные агенты для растворения и местные анестезирующие агенты.
Примеры эксципиентов включают производные сахаров, такие как лактоза, сахароза, глюкоза, маннит или сорбит; производные крахмала, такие как кукурузный крахмал, картофельный крахмал, α-крахмал, декстрин и карбоксиметилкрахмал; производные целлюлозы, такие как кристаллическая целлюлоза, низкозамещенная гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза и сшитая внутренними связями натрийкарбоксиметилцеллюлоза; аравийская камедь; декстран; пулулан; силикатные производные, такие как легкий ангидрид кремниевой кислоты, синтетический силикат алюминия и алюминатметасиликат магния; фосфатные производные, такие как фосфат кальция; карбонатные производные, такие как карбонат кальция и сульфатные производные, такие как сульфат кальция.
Примеры связующих включают эксципиенты, описанные выше; желатин; поливинилпирролидон и макрогол.
Примеры дезинтеграторов включают эксципиенты, описанные выше, и химически модифицированные производные крахмала и целлюлозы, такие как натрийкросскармелоза, натрийкарбоксиметилкрахмал и сшитый поливинилпирролидон.
Примеры смазывающих веществ включают тальк; стеариновую кислоту; стеаратные производные металлов, такие как стеарат кальция и стеарат магния; коллоидный диоксид кремния; пчелиную камедь; воски, такие как пчелиный воск и спермацет; борную кислоту; глицерин; производные карбоновых кислот, такие как фумаровая кислота и адипиновая кислота; карбоксилатые производные натрия, такие как бензоат натрия; сульфатные производные, такие как сульфат натрия; лейцин; лаурилсульфатные производные, такие как лаурилсульфат натрия и лаурилсульфат магния; производные кремниевой кислоты, такие как ангидрид кремниевой кислоты и гидрат кремниевой кислоты, и производные крахмала, описанные для эксципиентов.
Примеры стабилизаторов включают эфиры пара-оксибензойной кислоты, такие как метилпарабен и пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт и фенетиловый спирт; хлорид бензалкония; производные фенола, такие как фенол и крезол; тимерозал; уксусный ангидрид и сорбиновая кислота.
Примеры корригентов включают подслащивающие, подкисляющие и ароматизирующие агенты, все из которых используются обычно.
Примеры растворителей для изготовления препаратов включают воду, этанол и глицерин.
Примеры вспомогательных агентов для растворения включают неионные поверхностно-активные вещества и анионные поверхностно-активные вещества.
Примеры местных анестезирующих агентов включают гидрохлорид лидокаина и гидрохлорид мепивакаина.
Дозированные формы для перорального введения включают, например, твердые дозированные формы, такие как таблетки, покрытые таблетки, капсулы, пастилки, порошки, мелкие гранулы и сухие сиропы, и жидкие дозированные формы, такие как сиропы. Формы для парентерального введения включают, например, инъекции, капельные инфузии и суппозитории. Лекарственные формы для местного применения включают, например, мази, настойки, кремы и гели.
Предпочтительными дозированными формами кристаллических производных 1-метилкарбапенема данного изобретения являются инъекции и капельные инфузии. Подходящие уровни доз для кристаллических форм зависят от возраста, массы тела и симптомов пациента и обычно составляют от 10 мг (предпочтительно, 50 мг) до 6000 мг (предпочтительно, 4000 мг) для взрослого человека в день, такую дозу можно вводить в виде одной дозы или разделенной на несколько доз для введения на протяжении дня.
Лучший способ осуществления изобретения
Данное изобретение далее иллюстрируется следующими примерами, ссылочными примерами, примерами испытаний и примерами готовых препаративных форм.
Все спектры ЯМР в примерах и ссылочных примерах определяли в дейтерированной воде с использованием тетраметилсилана или в другом растворителе с использованием 3-(триметилсилил)пропионата натрия в качестве внутренних стандартов, соответственно.
Для всех химических структур используют следующие аббревиатуры со следующими значениями:
PNB: 4-нитробензил
PNZ: 4-нитробензилоксикарбонил.
Пример 1
(1R, 5S, 6S)-2-[(2S,4S)-2-[(3S)-3-(2-Гуанидиноацетиламино)пирролидин-1-илкарбонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил]-1-метил-1-карбапен-2-ем-3-карбоновая кислота•1/2 карбонат•1/2 этанол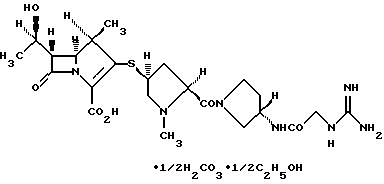
К раствору 4-нитробензил (1R,5S,6S)-2-[(2S,4S)-2-[(3S)-3-[2-[2,3-бис-(4-нитробензилоксикарбонил)гуанидино] ацетиламино]пирролидин-1-илкарбонил]-1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоксилата (9,4 г) в смеси тетрагидрофурана (235 мл) и воды (140 мл) добавляют 7,5% палладий на угле (9,4 г, который содержит воду (53,1%)) и полученную смесь перемешивают в атмосфере водорода при 35oС в течение 2 часов. В конце данного периода времени катализатор удаляют фильтрованием и фильтрат промывают простым эфиром. Эфир и тетрагидрофуран выпаривают в вакууме и полученный остаток хроматографируют на колонке с обращенной фазой (Cosmosil 75C18PREP (товарный знак), производимой фирмой Nacalai Tesque Inc. ) с использованием смеси ацетонитрила и воды в качестве элюента. Фракции, содержащие требуемый продукт, объединяют и концентрируют приблизительно до 50 мл в вакууме. К концентрату добавляют этанол (100 мл) и сухой лед и полученный раствор выдерживают на ледяной бане. Полученный осадок отфильтровывают и промывают последовательно смесью этанола и воды (2:1), этанолом и эфиром с получением указанного в заголовке соединения в виде бесцветных кристаллов (3,15 г).
Точка плавления: 228-233oС (разлож.).
ИК спектр (KBr) ν max cm-1: 3331, 2968, 2875, 2791, 1755, 1669, 1637, 1453, 1386, 1339, 1312, 1283, 1254.
ЯМР спектр (400 МГц, D2O) δ м.д.: 1,13-1,24 (4,5Н, м), 1,30 (3Н, д, J= 6/4 Гц), 1,57-1,72 (1Н, м), 1,93-2,10 (1Н, м), 2,15-2,35 (1Н, м), 2,27, 2,29 (3Н, с х 2), 2,68-2,88 (2Н, м), 3,09 (1Н, д, J=10,6 Гц), 3,29-3,73 (7Н, м), 3,75-3,93 (2Н, м), 4,01 (2Н, с), 4,12-4,30 (2Н, м), 4,38-4,50 (1Н, м).
Элементный анализ: Вычислено для C23H35N7O6S•1/2Н2СО3•1/2C2H6O.
Вычислено: С 49,73%; Н 6,64%; N 16,57%; S 5,42%.
Найдено: С 49,57%; Н 6,86%; N 16,68%; S 5,47%.
Порошковую рентгенограмму кристаллического продукта, показанную на фиг. 1, получали с Cu Kα излучением  . Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунду (CPS). Горизонтальная ось указывает угол диффракции в виде величины 2θ. Межплоскостное пространство d можно вычислить с использованием уравнения 2dsinθ = nλ, в котором n равно 1.
. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунду (CPS). Горизонтальная ось указывает угол диффракции в виде величины 2θ. Межплоскостное пространство d можно вычислить с использованием уравнения 2dsinθ = nλ, в котором n равно 1.
Пример 2
(1R, 5S, 6S)-2-[(2S,4S)-2-[(3S)-3-(2-Гуанидиноацетиламино)пирролидин-1-илкарбонил] -1-метилпирролидин-4-илтио] -6[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоновая кислота•1/2 этанол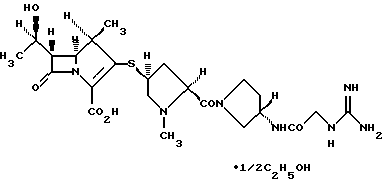
К раствору 4-нитробензил(1R, 5S,6S)-2-[(2S,4S)-2-[(3S)-3-[2-[2,3-бис-(4-нитробензилоксикарбонил)гуанидино] ацетиламино] пирролидин-1-илкарбонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоксилата (10,00 г) в смеси тетрагидрофурана (250 мл) и воды (150 мл) добавляют 7,5% палладий на угле (10,00 г, который содержит воду (53,1%)), и полученную смесь перемешивают в атмосфере водорода при 35oС в течение 2 часов. В конце данного периода времени катализатор удаляют фильтрованием, фильтрат промывают эфиром и фильтруют через мембранный фильтр. Полученный фильтрат концентрируют приблизительно до 50 мл в вакууме. К концентрату добавляют этанол (100 мл) и полученную смесь облучают ультразвуковыми волнами для осаждения кристаллов и затем оставляют стоять на ледяной бане. Осажденные кристаллы отфильтровывают и промывают последовательно смесью этанола и воды (2:1), этанолом и эфиром и затем сушат с получением указанного в заголовке соединения в виде бесцветных кристаллов (3,30 г).
Точка плавления: 235-250oС (разложение).
ИК спектр (KBr) ν max cm-1: 3405, 3344, 3273, 3207, 2969, 2883, 2795, 1760, 1673, 1644, 1591, 1553, 1452, 1415, 1381, 1370, 1341, 1311, 1283, 1255.
ЯМР спектр (400 МГц, D2O) δ м.д.: 1,15-1,25 (4,5Н, м), 1,30 (3Н, д, J= 6,4 Гц), 1,57-1,72 (1Н, м), 1,93-2,13 (1Н, м), 2,15-2,35 (1Н, м), 2,27, 2,29 (3Н, с х 2), 2,68-2,88 (2Н, м), 3,08 (1Н, д, J=10,7 Гц), 3,29-3,73 (7Н, м), 3,75-3,93 (2Н, м), 4,01 (2Н, с), 4,16-4,31 (2Н, м), 4,37-4,49 (1Н, м).
Элементный анализ: Вычислено для С23Н35N7О6S•1/2С2Н6О.
Вычислено: С 51,41%; Н 6,83%; N 17,49%; S 5,72%.
Найдено: С 51,13%; Н 6,96%; N 17,17%; S 5,72%.
Порошковую рентгенограмму кристаллического продукта, показанную на фиг. 1, получали с Cu Kα излучением  . Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунду (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ. Межплоскостное пространство d можно вычислить с использованием уравнения 2dsinθ = nλ, в котором n равно 1.
. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунду (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ. Межплоскостное пространство d можно вычислить с использованием уравнения 2dsinθ = nλ, в котором n равно 1.
Пример 3
(1R, 5S, 6S)-2-[(2S,4S)-2-[(3S)-3-(2-Гуанидиноацетиламино)пирролидин-1-илкарбонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил]-1-метил-1-карбапен-2-ем-3-карбоновая кислота•1/2 этанол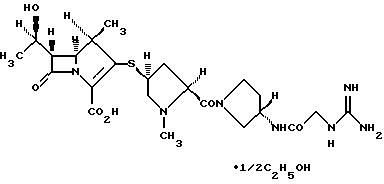
К раствору 4-нитробензил (1R,5S,6S)-6-[(1R)-1-гидрокси-этил]-1-метил-2-[(2S, 4S)-1-метил-2-[(3S)-3-[2-[3-(4-нитробензил-оксикарбонил)гуанидино] ацетиламино] пирролидин-1-ил-карбонил] пирролидин-4-илтио]-1-карбапен-2-ем-3-карбоксилата (112 мг) в смеси тетрагидрофурана (2,2 мл) и воды (2,2 мл) добавляют 7,5% палладий на угле (112 мг, который содержит воду (53,1%)) и полученную смесь перемешивают в атмосфере водорода при 35oС в течение 2 часов. В конце данного периода времени катализатор удаляют фильтрованием и фильтрат промывают эфиром и фильтруют через мембранный фильтр. Фильтрат концентрируют приблизительно до 1 мл в вакууме. К полученному концентрату добавляют этанол (2 мл), и смесь облучают ультразвуковыми волнами для осаждения кристаллов и затем оставляют стоять на ледяной бане. Осажденные кристаллы отфильтровывают и промывают последовательно смесью этанола и воды (2:1), этанолом и эфиром и затем сушат с получением указанного в заголовке соединения (45 мг) в виде бесцветного порошка. Данные точки плавления, инфракрасного спектра, ЯМР спектра, элементного анализа и порошковой рентгенограммы данного продукта были идентичными данным соединения, полученного в примере 2.
Пример 4
(1R, 5S, 6S)-2-[(2S,4S)-2-[(3S)-3-(2-Гуанидиноацетиламино)пирролидин-1-илкарбонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил]-1-метил-1-карбапен-2-ем-3-карбоновая кислота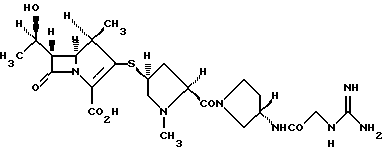
(1) (1R, 5S, 6S)-2-[(2S, 4S)-2-[(3S)-3-(2-Гуанидиноацетиламино]пирролидин-1-илкарбонил]-1-метилпирролидин-4-илтио]-6-[(1R)-1-гидроксиэтил]-1-метил-1-карбапен-2-ем-3-карбоновую кислоту•1/2 этанол (680 мг) растворяют в воде (35 мл). Смесь фильтруют через мембранный фильтр. Фильтрат концентрируют приблизительно до 3 мл в вакууме. Полученный концентрат оставляют при 0oС на ночь. Осажденные кристаллы фильтруют и промывают небольшим количеством воды и затем сушат с получением указанного в заголовке соединения в виде бесцветных кристаллов (294 мг).
Точка плавления: 235-250oC (разложение).
ИК спектр (KBr) ν max cm-1: 3327, 3177, 3068, 2970, 2904, 2880, 2820, 1751, 1681, 1654, 1629, 1594, 1572, 1536, 1481, 1440, 1423, 1382, 1336, 1314, 1286, 1264.
ЯМР спектр (400 МГц, D2O) δ м.д.: 1,20 (3Н, дд, J=7,2, 2,2 Гц), 1,30 (3Н, д, J=6,4 Гц), 1,57-1,72 (1Н, м), 1,93-2,13 (1H, м), 2,15-2,35 (1H, м), 2,27, 2,29 (3Н, с х 2), 2,68-2,88 (2Н, м), 3,09 (1H, д, J=10,5 Гц), 3,29-3,73 (6Н, м), 3,75-3,93 (2Н, м), 4,01 (2Н, с), 4,16-4,31 (2Н, м), 4,37-4,49 (1H, м).
Элементный анализ: Вычислено для С23Н35N7O6S.
Вычислено: С 51,38%; Н 6,56%; N 18,24%; S 5,96%.
Найдено: С 51,14%; Н 6,85%; N 18,26%; S 6,04%.
Порошковую рентгенограмму кристаллического продукта, показанную на фиг. 3, получали с Cu Kα излучением 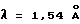 . Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунду (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ. Межплоскостное пространство d можно вычислить с использованием уравнения 2dsinθ = nλ, в котором n равно 1.
. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунду (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ. Межплоскостное пространство d можно вычислить с использованием уравнения 2dsinθ = nλ, в котором n равно 1.
(2) Процедуру, аналогичную процедуре, описанной выше, проводили в том же масштабе, что и в примере 3. К полученному концентрату (приблизительно 1 мл) добавляют небольшое количество бесцветных кристаллов, полученных, как описано в примере 4(1), и полученную смесь оставляют на ночь при 0oС. Осажденные кристаллы собирают фильтрованием и промывают небольшим количеством воды с получением указанного в заголовке соединения (20 мг).
Пример 5
(1R, 5S, 6S)-2-[(2S,4S)-2-[(3S)-3-(2-Гуанидиноацетиламино)пирролидин-1-илкарбонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил]-1-метил-1-карбапен-2-ем-3-карбоксилат•1/4этанол•3/2 гидрат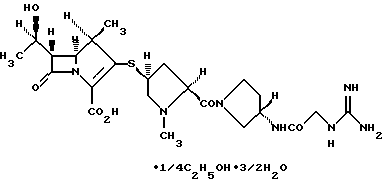
К раствору 4-нитробензил (1R,5S,6S)-6-[(1R)-1-гидроксиэтил]-1-метил-2[(2S, 4S)-1-метил-2[(3S)-3[2-[3-(4-нитробензилоксикарбонил)гуанидино]ацетиламино] пирролидин-1-ил-карбонил] пирролидин-4-илтио]-1-карбапен-2-ем-3-карбоксилата (220 мг) в смеси тетрагидрофурана (2200 мл) и воды (2200 мл) добавляют 7,5% палладий на угле (220 г, который содержит воду (53,1%)) и полученную смесь перемешивают в атмосфере водорода при 30oС в течение 2 часов. В конце данного периода времени катализатор удаляют фильтрованием и фильтрат промывают этилацетатом и фильтруют через мембранный фильтр. Фильтрат концентрируют приблизительно до 600 мл. К концентрату добавляют этанол (1800 мл) и полученную смесь перемешивают до тех пор, пока не осадятся кристаллы, и затем все оставляют стоять на ледяной бане. Осажденные кристаллы фильтруют и промывают последовательно смесью этанола и воды (3:1) и этанолом и затем сушат с получением указанного в заголовке соединения (40 г) в виде бесцветных кристаллов.
Точка плавления: 226-245oС (разложение).
ИК спектр (KBr) ν max cm-1: 3409, 3345, 3275, 3185, 2967, 2884, 1761, 1674, 1644, 1586, 1551, 1452, 1415, 1380, 1369, 1340, 1282, 1254.
ЯМР спектр (400 МГц, D2O) δ м.д.: 1,17-1,21 (4,7Н, м), 1,30 (3Н, д, J= 6,4 Гц), 1,57-1,70 (1H, м), 1,95-2,08 (1Н, м), 2,18-2,31 (1H, м), 2,27, 2,29 (3Н, с х 2), 2,70-2,87 (2Н, м), 3,08 (1H, д, J=10,8 Гц), 3,31-3,72 (7Н, м), 3,76-3,92 (2Н, м), 4,00 (2Н, с), 4,18-4,28 (2Н, м), 4,39-4,48 (1H, м).
Элементный анализ: Вычислено для С23Н35N7O6S•1/4С2Н6О•3/2Н2O.
Вычислено: С 48,99%; Н 6,91%; N 17,02%; S 5,56%.
Найдено: С 48,35%; Н 6,47%; N 17,23%; S 5,67%.
Порошковую рентгенограмму кристаллического продукта, показанную на фиг. 3, получали с Cu Kα излучением 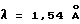 . Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунду (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ. Межплоскостное пространство d можно вычислить с использованием уравнения 2dsinθ = nλ, в котором n равно 1.
. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунду (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ. Межплоскостное пространство d можно вычислить с использованием уравнения 2dsinθ = nλ, в котором n равно 1.
Ссылочный пример 1
4-нитробензил (1R, 5S, 6S)-2-[(2S,4S)-2-[(3S)-3-[2-[2,3-бис(4-нитробензилоксикарбонил)гуанидино] ацетиламино] пирролидин-1-ил-карбонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоксилат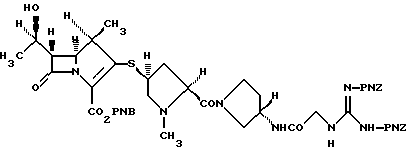
Ацетат гидразина (652 мг) добавляют к раствору (2S,4S)-4-ацетилтио-2-[(3S)-3-[2-[2,3-бис(4-нитробензилоксикарбонил)гуанидино] ацетиламино]пирролидин-1-илкарбонил] -1-метилпирролидина (4,3 г) в N,N-диметилформамиде (86 мл) и перемешивают при комнатной температуре в течение 4 часов. К полученной смеси добавляют 4-нитробензил (1R,5R,6S)-6-[(1R)-1-гидроксиэтил]-1-метил-2-дифенилфосфорилокси-1-карбапен-2-ем-3-карбоксилат (3,53 г) и N,N-диизопропилэтиламин (1,34 мл) и оставляют реагировать при -30oС в течение 3 дней. В конце данного периода времени к реакционной смеси добавляют 1% водный раствор гидрокарбоната натрия и полученный осадок отфильтровывают, промывают водой и растворяют в смеси тетрагидрофурана и этилацетата (3:7). Полученный раствор промывают последовательно насыщенным водным раствором гидрокарбоната натрия, водой и насыщенным водным раствором хлорида натрия, сушат над безводным сульфатом натрия и концентрируют в вакууме. Образовавшийся остаток хроматографируют на колонке с силикагелем с использованием 10% смеси метанол/этилацетат и 20% смеси метанол/этилацетат в качестве элюента с получением сырого требуемого продукта. Продукт растворяют в тетрагидрофуране и осаждают смесью этилацетата и простого эфира (1:1) с получением требуемого соединения (4,02 г) в виде бледно-желтого порошка.
ИК спектр (KBr) ν max cm-1: 3336, 1772, 1741, 1688, 1643, 1610, 1522, 1447, 1378, 1347.
ЯМР спектр (270 МГц, CDCl3) δ м.д.: 1,17-1,40 (6Н, м), 1,64-2,40 (4Н, м), 2,33 (3Н, с), 2,47-2,80 (2Н, м), 3,00-3,38 (3Н, м), 3,46-3,83 (5Н, м), 3,93-4,60 (5Н, м), 5,12-5,54 (6Н, м), 7,21 (1Н, д, J=6,5 Гц), 7,46-7,70 (6Н, м), 8,10-8,28 (6Н, м), 8,80-9,10 (1Н, ушир.), 11,60 (1Н, ушир.).
Ссылочный пример 2
4-Нитробензил-(1R, 5S,6S)-6-[(1R)-1-гидроксиэтил]-1-метил-2-[(2S,4S)-2-[(3S)-3-[2-[3-(4-нитробензилоксикарбонил)гуанидино] ацетиламино] пирролидин-1-илкарбонил]-1-метилпирролидин-4-илтио]-1-карбапен-2-ем-3-карбоксилат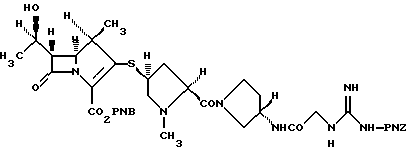
(1) Раствор 28% метилата натрия в метаноле (0,5 мл) добавляют к раствору (2S, 4S)-4-ацетилтио-1-метил-2-[(3S)-3-[2-[3-(4-нитробензилоксикарбонил)гуанидино] ацетиламино] пирролидин-1-ил-карбонил] пирролидин (1,5 г) в метаноле (30 мл) и перемешивают при комнатной температуре в течение 1 часа. В конце данного периода времени к полученной смеси добавляют 1 н хлористовородную кислоту (2,73 мл) и концентрируют в вакууме. Полученный остаток хроматографируют на колонке с обращенной фазой (Cosmosil 75C18REP (товарный знак), производимой фирмой Nacalai Tesque Inc.) с использованием смеси ацетонитрила и воды в качестве элюента. Фракции, содержащие требуемый продукт, объединяют и концентрируют в вакууме. Остаток растирают в порошок в смеси этилацетата и изопропилового эфира. Порошкообразный (2S,4S)-4-меркапто-1-метил-2-[(3S)-3-[2-[3-(4-нитробензилоксикарбонил)-гуанидино] -ацетиламино] пирролидин-1-илкарбонил]пирролидин (806 мг) получают фильтрованием.
ИК спектр (KBr) ν max cm-1: 3391, 3307, 3112, 3078, 2949, 2877, 2786, 1732, 1639, 1548, 1522, 1448, 1380, 1347, 1291, 1211, 1154, 1109.
ЯМР спектр (400 МГц, CDCl3) δ м.д.: 1,64-2,15 (3Н, м), 2,20-2,86 (5Н, м), 2,93-3,93 (9Н, м), 4,16-4,35 (1Н, м), 5,12 (2Н, с), 6,70-6,90 (1Н, ушир. ), 7,00-7,85 (1Н, ушир.), 7,59 (2Н, Д, J=8,5 Гц), 8,23 (2Н, д, J=8,5 Гц), 8,18-8,40 (1H, ушир.).
(2) N, N-Диизопропилэтиламин (0,17 мл) и 4-нитробензил(1R,5R,6S)-6-[(1R)-1-гидроксиэтил] -1-метил-2-дифенилфосфорилокси-1-карбапен-2-ем-3-карбоксилат (585 мг) добавляют к раствору соединения (500 мг), полученного в ссылочном примере 2(1), в N,N-диметилформамиде (5 мл) на ледяной бане и смеси дают взаимодействовать при 0oС в течение ночи. В конце данного периода времени к реакционной смеси добавляют этилацетат и тетрагидрофуран, полученную смесь промывают 10% водным раствором хлорида натрия и концентрируют в вакууме. Полученный остаток хроматографируют на колонке с обращенной фазой (Cosmosil 75C18REP (товарный знак), производимой Nacalai Tesque Inc.) с использованием смеси ацетонитрила и воды в качестве элюента. Фракции, содержащие требуемый продукт, объединяют и концентрируют в вакууме. Полученный остаток растирают в порошок в изопропиловом эфире и фильтруют с получением требуемого соединения (524 мг) в виде бледно-желтого порошка.
ИК спектр (KBr) ν max cm-1: 3384, 3113, 3080, 2970, 2875, 2789, 1770, 1643, 1609, 1522, 1450, 1379, 1346, 1322, 1287, 1209, 1181, 1136, 1109.
ЯМР спектр (400 МГц, CDCl3) δ м.д.: 1,08-2,22 (6Н, м), 1,75-2,26 (6Н, м), 2,44-2,76 (2Н, м), 2,89-3,00 (1Н, м), 3,03-3,15 (1Н, м), 3,18-3,65 (6Н, м), 3,68-3,90 (3Н, м), 3,93-4,06 (1Н, м), 4,13-4,35 (2Н, м), 5/05-5,15 (2Н, м), 5,30, 5,45 (каждый 1Н, д, J=14,1 Гц), 7,58 (2Н, дд, J=8,8, 2,7 Гц), 7,74 (2Н, д, J=8,7 Гц), 8,18-8,33 (4Н, м).
Ссылочный пример 3
4-Нитробензил (1R, 5S,6S)-6-[(1R)-1-гидроксиэтил]-1-метил-2-[(2S,4S)2-[(3S)-3-[2-[3-(4-нитробензилоксикарбонил)гуанидино]-ацетиламино]-пирролидин-1-илкарбонил]-1-метилпирролидин-4-илтио]-1-карбапен-2-ем-3-карбоксилат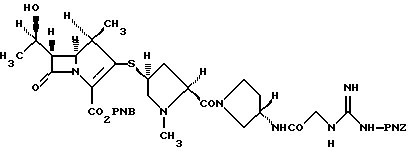
К раствору (2S, 4S)-4-ацетилтио-1-метил-2-[(3S)-3-[2-[3-(4-нитробензилоксикарбонил)гуанидино]ацетиламино]пирролидин-1-илкарбонил]пирролидина (500 мг) в этаноле (5 мл) добавляют 4 н раствор хлористый водород/этилацетат (2,7 мл) и смесь перемешивают при 50oС в течение 3 часов. В конце данного периода времени к реакционной смеси добавляют эфир. Полученный осадок отделяют декантацией и сушат в вакууме. К раствору осадка в N,N-диметилформамиде (10 мл) добавляют N,N-диизопропилэтиламин (0,63 мл) и 4-нитробензил(1R,5R,5S)-6-[(1R)-1-гидроксиэтил] -1-метил-2-дифенилфосфорилокси-1-карбапен-2-ем-3-карбоксилат (541 мг) на ледяной бане и оставляют для реакции на ночь. В конце данного периода времени к реакционной смеси добавляют 1% водный раствор гидрокарбоната натрия. Полученный осадок отфильтровывают, промывают водой и сушат. Сырой порошок хроматограифруют на колонке с силикагелем с использованием раствора 30% метанол/этилацетат и раствора 50% метанол/этилацетат в качестве элюента с получением требуемого соединения (446 мг). Инфракрасные спектры и ЯМР спектры данного соединения идентичны спектрам соединения, полученного в ссылочной примере 2(2).
Ссылочный пример 4
4-Нитробензил(1R, 5S, 6S)-6-[(1R)-1-гидроксиэтил]-1-метил-2-[(2S,4S)-2-[(3S)-3-[2-[3-(4-нитробензилоксикарбонил)-гуанидино] -ацетиламино] пирролидин-1-илкарбонил]-1-метилпирролидин-4-илтио]-1-карбапен-2-ем-3-карбоксилат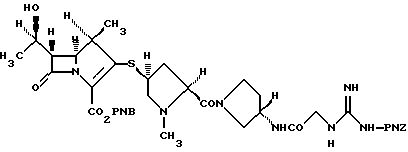
К раствору (2S, 4S)-4-ацетилтио-1-метил-2-[(3S)-3-[2-[3-(4-нитробензилоксикарбонил)гуанидино] ацетиламино] пирролидин-1-илкарбонил] пирролидина (1,00 г) в метаноле (20 мл) добавляют метилат натрия (98,3 мг) при 0oС и смесь перемешивают в течение 1 часа. В конце данного периода времени к реакционной смеси добавляют 4 н раствор хлористый водород/этилацетат (0,46 мл) и концентрируют в вакууме. На ледяной бане к раствору полученного остатка в N, N-диметилформамиде (10 мл) добавляют раствор N,N-диизопропилэтиламина (0,32 мл) и 4-нитробензил(1R,5R,6S)-6-[(1R)-1-гидроксиэтил]-1-метил-2-дифенилфосфорилокси-1-карбапен-2-ем-3-карбоксилат (1,08 г) в N,N-диметилформамиде и оставляют стоять при 0oС в течение ночи. В конце данного периода времени к реакционной смеси добавляют 1% водный раствор гидрокарбоната натрия. Полученный осадок отфильтровывают, промывают водой и сушат. Сырой порошок хроматографируют на колонке с силикагелем с использованием смеси метанол/этилацетат= 1/3 и метанол/этилацетат=1/2 в качестве элюента с получением требуемого соединения (975 мг). Инфракрасные спектры и ЯМР спектры данного соединения идентичны спектрам соединения, полученного в ссылочном примере 2(2).
Пример 1 испытания
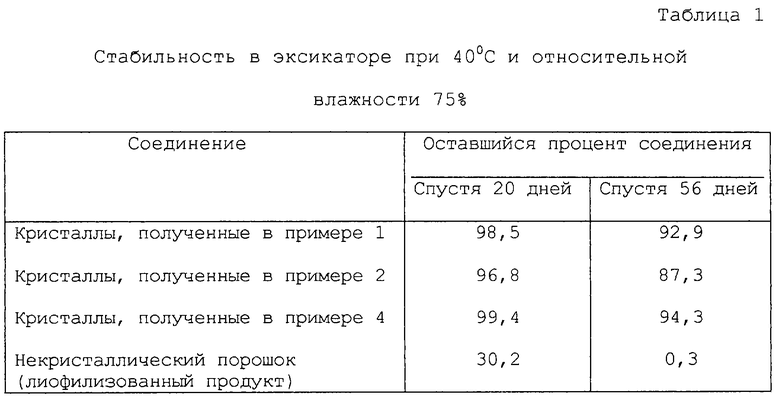
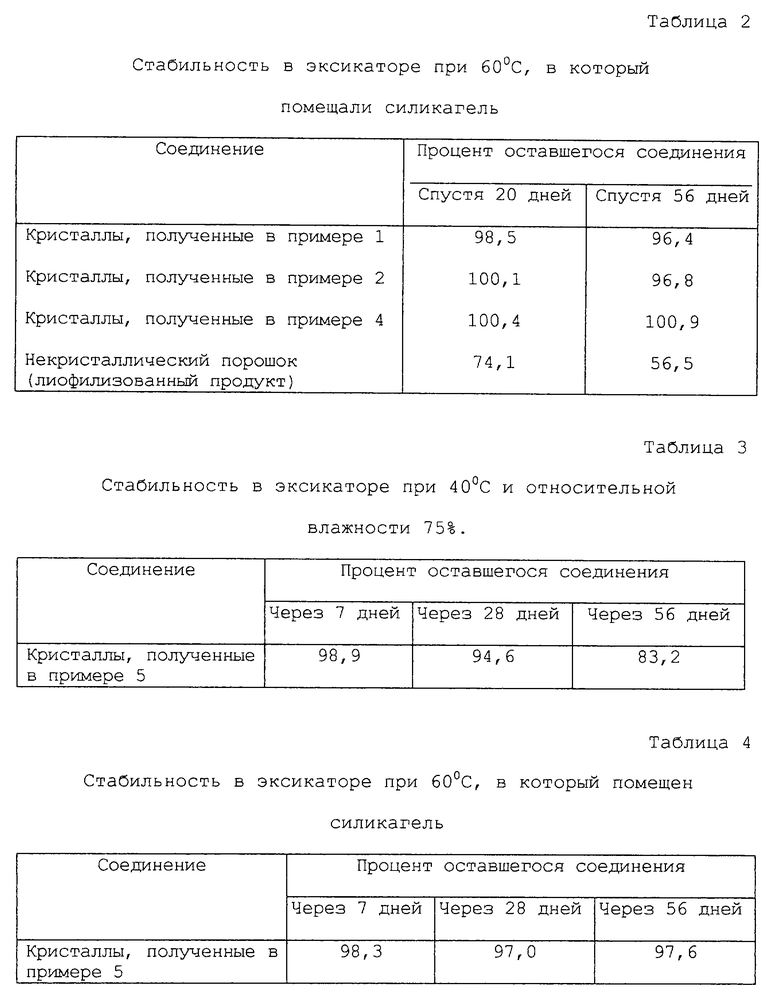
Кристаллические соединения, полученные в примере 1, 2, 4 и 5, хранили в течение примерно 2 месяцев в эксикаторе при 40oС и относительной влажности 75% и в эксикаторе при 60oС, в который помещали силикагель, соответственно. Некристаллическое порошкообразное соединение (лиофилизованный продукт), полученный по процедуре, описанной в публикации заявки на патент Японии Hei-11-071277, которое является ссылочным образцом, хранили в тех же условиях, что описаны выше. Остаточное количество кристаллических соединений и некристаллических порошкообразных соединений анализировали спустя 7, 20, 21, 28 и 56 дней жидкостной хроматографией при высоком давлении на L-колонке ODS (4,6 ф х 150 мм (товарный знак), производимой Kagakuhinn kennsa kyoukai) при элюировании смесью 20 мМ КН2РО4(рН 7,0):СН3СN=96:4 при скорости 1,0 мл/минуту и при 60oС с использованием ультрафиолетовой длины волны 300 нм. Оставшийся процент данных соединений вычисляли по оставшемуся количеству их и он показан в табл.1-4.
Из табл. 1 и 3 ясно, что некристаллический порошок соединения (I) (лиофилизованный продукт) является очень нестабильным при 40oС и относительной влажности 75%, т.е. спустя 20 дней процент оставшегося количества соединения составляет 30,2%, спустя 56 дней он составляет лишь 0,3%, а процент оставшегося количества кристаллических соединений выше, чем 83%, соответственно, в тех же условиях.
Из табл. 2 и 4 ясно, что некристаллический порошок соединения (I) (лиофилизованный продукт) является также нестабильным при 60oС в сухих условиях, т.е. спустя 21 день процент оставшегося количества соединения составляет 71,4%, спустя 56 дней он составляет только 56,5%, а процент оставшегося количества кристаллических соединений данного изобретения выше, чем 96%, соответственно, в тех же условиях.
Данные результаты показывают, что кристаллические соединения данного изобретения являются очень стабильными по сравнению с некристаллическим порошком {лиофилизованный продукт), относящийся к предыдущему изобретению.
Пример испытания 2
Испытание на антибиотическую активность
MIC (мкг/мл), самую низкую концентрацию антибиотика, которая ингибирует рост испытуемого бактериального штамма, определяли по методу разведения на агаровой пластинке. Кристаллические соединения, полученные в примере 1, 2, 4 и 5 данного изобретения, оценивали на действие против различных бактериальных штаммов определением MIC каждого соединения в отношении каждого штамма. В табл.5 приведены результаты экспериментов.
Пример 1 готовой препаративной формы
Инъекции
Кристаллическое соединение, полученное в примере 1 (250 мг), используют для заполнения пузырька и закрывают пробкой в стерильных условиях. Если необходимо, в пузырек можно добавить фармацевтические добавки, известные специалистам в данной области, такие как местный анестезирующий агент, например гидрохлорид лидокаина. Стерильные твердые композиции можно растворять в инъецируемой среде, такой как вода для инъекции, непосредственно перед использованием.
Краткое описание чертежей
На фиг.1 показана порошковая рентгенограмма кристаллического соединения (1R, 5S, 6S)-2-[(2S, 4S)-2-[(3S)-3-(2-гуанидиноацетиламино)пирролидин-1-илкарбонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоновой кислоты•1/2 карбонат•1/2 этанол (I-1).
Диффракционную картину получали с Cu Kα излучением 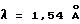 на кристаллах. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунда (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ.
на кристаллах. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунда (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ.
На фиг.2 показана порошковая рентгенограмма кристаллического соединения (1R, 5S, 6S)-2-[(2S,4S)-2-[(3S)-3-(2-гуанидиноацетиламино)пирролидин-1-илкароонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоновой кислоты•1/2 этанол (I-2).
Диффракционную картину получали с Cu Kα излучением 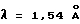 на кристаллах. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунда (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ.
на кристаллах. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунда (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ.
На фиг. 3 показана порошковая рентгенограмма кристаллической (1R,5S, 6S)-2-[(2S, 4S)-2-[(3S)-3-(2-гуанидино-ацетиламино)пирролидин-1-илкарбонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоновой кислоты (I).
Диффракционную картину получали с Cu Kα излучением 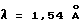 на кристаллах. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунда (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ.
на кристаллах. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунда (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ.
На фиг.4 показана порошковая рентгенограмма кристаллического соединения (1R, 5S, 6S)-2-[(2S, 4S)-2-[(3S)-3-(2-гуанидиноацетиламино)пирролидин-1-илкарбонил] -1-метилпирролидин-4-илтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоновой кислоты•1/4 этанол•3/2 гидрат (I-3).
Диффракционную картину получали с Си Kα излучением 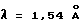 на кристаллах. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунда (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ.
на кристаллах. Вертикальная ось порошковой рентгенограммы указывает интенсивность диффракции в единицах число импульсов/секунда (CPS). Горизонтальная ось указывает угол диффракции как величину 2θ.
Изобретение относится к новым кристаллическим формам производного 1-метилкарбапенема формулы I или его фармацевтически приемлемым солям, которые проявляют антибиотоническую активность против различных бактериальных штаммов, обладая при этом достаточной стабильностью для практического использования, а также к фармацевтической композиции на их основе и способу профилактики и лечению бактериальных инфекционных заболеваний.
Технический результат - повышение стабильности при хранении. 8 с.п.ф-лы, 4 ил., 5 табл.
2. Производное 1-метилкарбапенема формулы (I-1) в кристаллической форме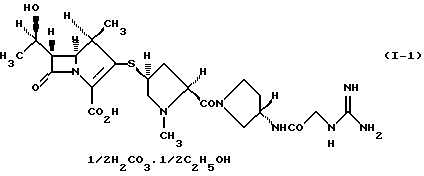
3. Производное 1-метилкарбапенема формулы (I-2) в кристаллической форме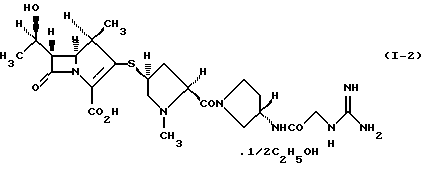
4. Производное 1-метилкарбапенема формулы (I) в кристаллической форме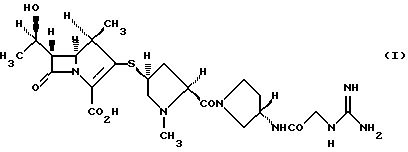
5. Производное 1-метилкарбапенема формулы (I-3) в кристаллической форме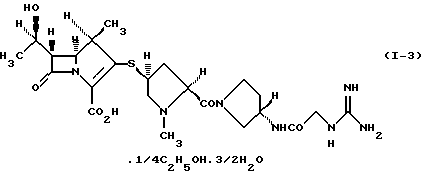
6. Фармацевтическая композиция для профилактики или лечения бактериальных инфекционных заболеваний, содержащая кристаллическую форму производного 1-метилкарбапенема или его фармацевтически приемлемой соли по любому из пп.1-5 в качестве активного ингредиента.

