Изобретение относится к области получения прокариотических клеток со стабильной амплификацией гена путем рассеянного нетандемного встраивания в хромосому по крайней мере двух копий определенной последовательности ДНК.
Бациллы широко используются для производства промышленно важных ферментов, таких как альфа-амилаза, нейтральная протеаза и щелочная или сериновая протеаза (сравни Debabov, "The Industrial Use of Bacilli" in "The Molecular Biology of Bacilli", Acad. Press, New York, 1982).
Усовершенствование производства ферментов Bacillus может быть достигнуто как классическими генетическими способами, такими как мутации и последующая селекция, так и современными молекулярно-биологическими способами. В последнем случае описаны несколько путем достижения высоких уровней экспрессии гомологичных и гетерологичных генов в определенных прокариотических и эукариотических микроорганизмах при помощи генетической инженерии.
Один из подходов к достижению высокого уровня экспрессии гена состоит в обеспечении гена эффективными регуляторными последовательностями. Другой подход, часто использующийся в комбинации с первым, состоит в увеличении количества копий нужного гена. Амплификация обычно достигается путем вставки гена в многокопийную внехромосомную молекулу ДНК, такую как плазмида. Однако, значительным препятствием для использования плазмид в качестве векторов для экспрессии и амплификации генетической информации является их нестабильность. Для широкомасштабного использования стабильность амплифицированного гена является предпосылкой для поддержания высокого уровня продукции продукта экспрессии, кодируемого амплифицированным геном, так как должно произойти большое количество клеточных делений прежде, чем сформируется достаточная биомасса для достижения значительного образования продукта.
Существуют две формы нестабильности: сегрегационная нестабильность, где во время культивирования происходит утрата плазмиды, и структурная нестабильность, при которой утрачивается участок плазмиды.
Способ, используемый для предотвращения сегрегационной нестабильности, заключается в отборе клеток, содержащих многокопийные плазмиды, которые несут гены, дающие преимущество плазмидосодержащей клетке, например, сообщают устойчивость к антибиотику, на фоне добавления соответствующего антибиотика к среде ферментации. Однако в широкомасштабных промышленных производственных процессах введение антибиотиков является нежелательным средством селекции.
Другой способ, используемый для уменьшения вероятности потери плазмиды, связанной с сегрегационной нестабильностью, заключается во встраивании в векторную молекулу гена, который функционально важен для хозяйской клетки (Ferrari et al. Biotechnology, 3 (1985) 1003-1007). Однако этот метод не гарантирует структурную стабильность вектора.
Способы, используемые для решения проблемы структурной плазмидной нестабильности, включают попытку исключить экспрессию гена в фазе экспотенциального роста, например, путем использования регуляторных последовательностей, таких как температурочувствительные регуляторные последовательности, также встраивание экзогенной ДНК в хромосому хозяйской клетки. Другие известные способы включают отказ от использования автономно реплицирующихся векторных молекул и использование взамен этого приемов, благоприятствующих встраиванию введенной ДНК в хромосому хозяйской клетки.
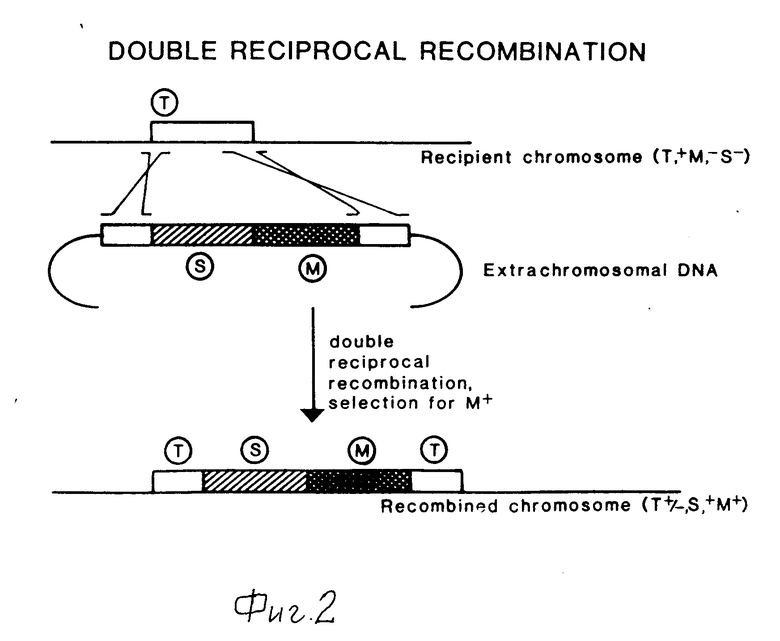
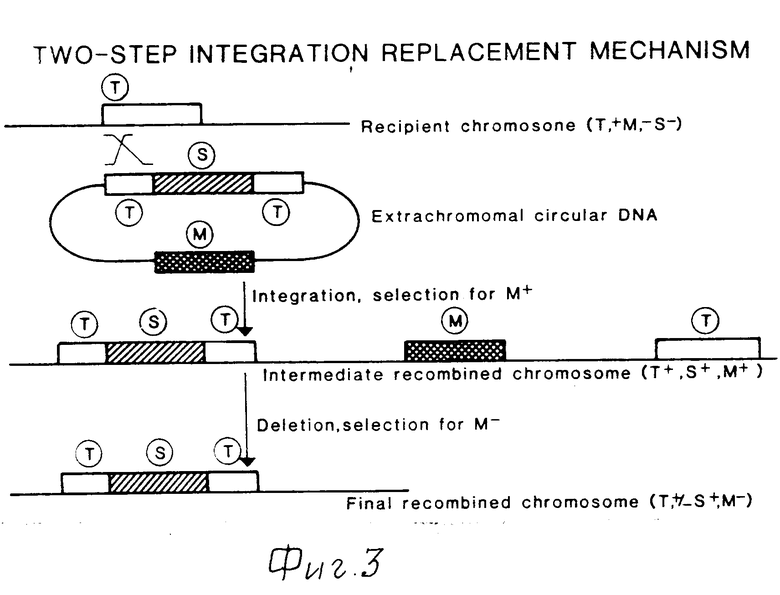
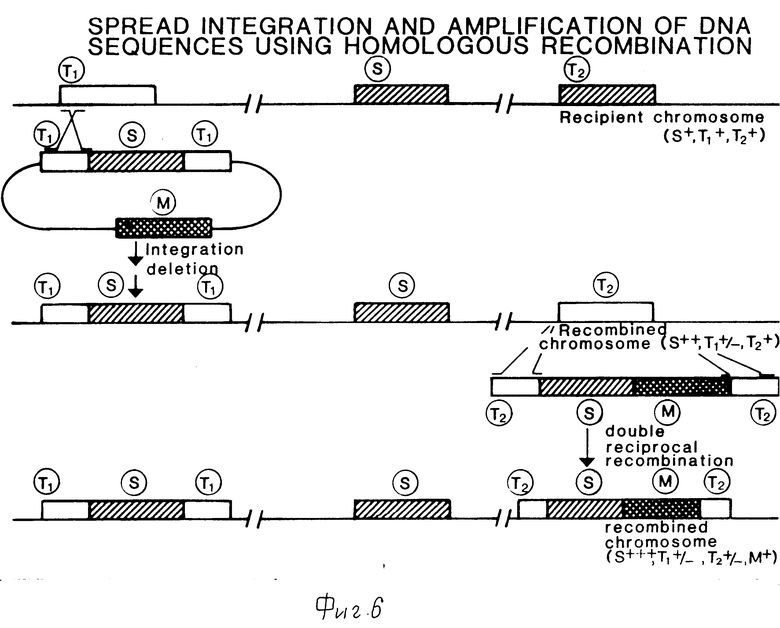
Способы встраивания чужеродной ДНК в хромосому хозяйской клетки включают гомологичную рекомбинацию и незаконную рекомбинацию. Существуют два способа встраивания последовательности ДНК в определенные места на хромосоме путем гомологичной рекомбинации: гомологичная рекомбинация Campbell-типа и двойная реципрокная рекомбинация, показанные, соответственно, на фиг.1 и 2. Третий путь введения последовательностей ДНК в хромосому это способ, использующий механизм двухстадийного замещения, показанный на фиг.3. В принципе, используется та же рекомбинация Campbell -типа, но конечным результатом является организация хромосомы, которая не содержит дуплицированных, последовательностей, и, следовательно, амплифицированной единицы, в рекомбинантной части хромосомы. Таким образом, он схож с двойной реципрокной рекомбинацией.
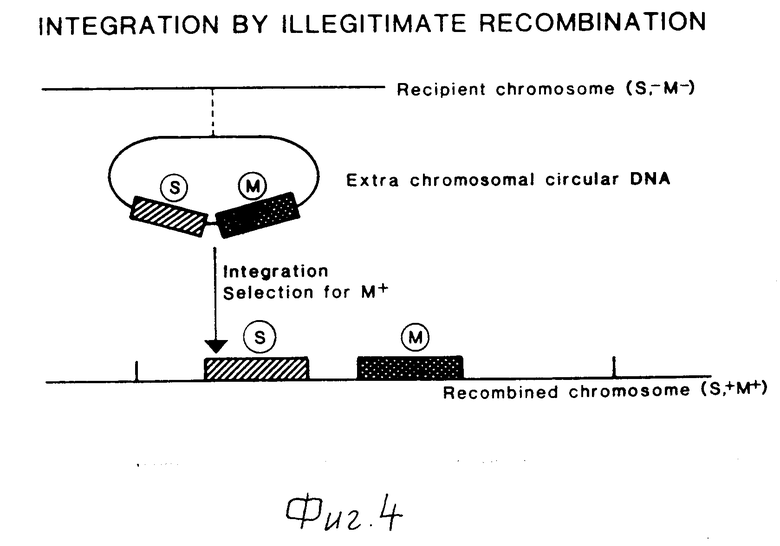
Кроме использования гомологичной рекомбинации для встраивания чужеродной ДНК в хромосому, также можно использовать незаконную рекомбинацию. Интегрированные векторные молекулы могут быть отобраны в условиях, которые ингибируют автономную репликацию неинтегрированных векторных молекул. Использование незаконной рекомбинации для встраивания изображено на фиг.4. Отсутствие тандемных дупликаций в полученной организации хромосомной последовательности делает способы, показанные на фиг.2-4, предпочтительными для стабильного введения последовательностей ДНК в геном. Гены, встроенные в хромосому, включают как гомологичные, так и гетерологичные гены, при этом амплификация встроенной ДНК осуществляется в тандемном порядке. Сообщают, что эти хромосомно амплифицированные гены нестабильны, хотя в ряде случаев сообщают и о стабильности. Таким образом, желательно разработать методы, посредством которых ДНК, встроенная в хромосому, поддерживается стабильно.
Встраивание экзогенной ДНК в хромосому Bacillus subtilis путем гомологичной рекомбинации описано Duncan et al. Proc. Nati. Acad. Sci. USA 75 (1978) 3564-3668, а для Anacystis nidulans Williams and Sgalay, Iene 24 (1983) 37-51 и в Международной патентной заявке WO 84/00381. Встраивание путем гомологичной рекомбинации гетерологичного гена, который не может стабильно поддерживаться в составе плазмидного вектора, в хромосому микроорганизма, описано в ЕР-А-0127328.
Описана амплификация встроенных в хромосому генов, как гемологичных, так и гетерологичных. Смотри, например, Saito et al. Материалы четвертого Междунаролного Симпозиума по генетике промышленных микроорганизмов, Киото, Япония, 1982, с. 125-130; Young, Y. Gen. Microbiol. 130 (1984) 1613-1621; Janniere et al. Yene 40 (1985) 47-55; Sargent and Bennett, j. Bacteriol. 161 (1985) 589-595; Jutterson and Koshland, Proc. Natl. Acad. Sci. USA, 80 (1983) 4894-4898; Hashiguchi et al. Agric. Biol. Chem. 49 (1985) 545-550; Wilson and Morgan, J. Bacteriol, 163 (1985) 445-453; Патентная заявка Франции N 8406701; и EP-A-0134048).
О спонтанной амплификации в прокариотических клетках также сообщалось ранее. Смотри, например, обзор Anderson and Roth, Ann. Rev. Microbiol. 31 (1977) 473-505.
Во всех случаях, относящихся к упомянутым выше, амплификация встроенной в хромосому ДНК имела тандемный порядок. Сообщают, что этот тип хромосомной амплификации последовательности нестабилен, хотя в отдельных случаях была показана достаточная стабильность, как описано Ganniere et al. Iene 40 1985, 47-55.
Сообщают о стабилизации встречающихся в природе амплифицированных прокариотических генов благодаря присутствию других важных генов между этими амплифицированными последовательностями. Например, для наборов из 9-10 копий гена рибосомальной РНК, встречаемых в хромосоме B. subtilis, два тандемно расположенных набора разделены кластером генов тРНК (Wawrousek and Hansen, G. Biol. Chem. 258 (1983) 291-298). В других случаях встречающиеся в природе тандемно повторяющиеся опероны рибосомальной РНК были делетированы (как в E. coli, так и в B. subtilis) без заметного влияния на фенотипические свойства организма: Elwood and Mamura, G. Bacteriol. 143 (1980) 1077-1080 и Loughney et al. G. Bacteriol. 154 (1983) 525-532, соответственно.
Встраивание плазмид в хромосому B. Subtilis путем незаконной рекомбинации с использованием вектора pE 194 описано Hofemeister et al. Mol. Ien. Ienet. 189 (1983) 58-68 и Prorozov et al. Iene 34 (1985) 39-46.
Успешно клонированы несколько генов внеклеточных ферментов бацилл, таких как гены альфа-амилазы B. amyloligue-faciens (Palva et al. Iene 15 (1981) 43-51). B. licheniformis (Ortlepp, Iene 23 (1983) 267), B. stearothermophilus (Mieleng et al. Proc. Natl. Acad. Sci. USA 80 (1983) 5975-5979; EP-A-0057976) и B. subtilis (Yang et al. Nucleic Acids Res. 11 (1983) 237); ген левансахарозы, B. subtilis (Iay ey al. G. Bacteriol. 153 (1983) 1424); гены, кодирующие нейтральную протеазу B. stearothermophilus (Fuji et al. G. Bacteriol. 156 (1983) 831), B. amyloliquefaciens (Honjo et al. G. Biotech. 1 (1984) 165) и B. subtilis (Yang et al. G. Bacteriol. 160 (1984) 115; гены, кодирующие сериновую или щелочную протеазу B. subtilis (Wong et al. Proc. Natl. Acad. Sci. USA 81 (1984) 1184, B. licheniformis (Jacobs et al. Nucleic Acids Res. 13 (1985) 8913 и B. amyloliquefaciens (Wells et al. Nucleic Acids Res. 11 (1983) 7911).
Сообщают о трансформации протопласта для некоторых видов грамположительных микроорганизмов (EP A 0124374, кл. C 12 N 15/00, 1983). Для B. subtilis Chand and Cohen (Mol. Ien. Ienet. 168 (1979) 111-115) был описан способ трансформации протопласта, который широко используется. Похожий успешный способ описан для трансформации протопластов B. Megaterium (Vorobjeva et al. FEMS Microbiol. Letters 7 (1980) 261-263, B. amyloliquefaciens (Smith et al. Appl. and Env. Microbiol. 51 (1986) 634); B. thuiringiensis (Fisher et al. Arch. Microbiol. 139 (1981) 213-217); B. sphaericus (McDonald, G. Ien. Microbiol. 130 (1984) 203) и B. larval (Bakhiet et al. Appl. and Env. Microbiol. 49 (1985) 577; в той же публикации сообщалось о неудачных результатах с B. popillae. Этот способ был успешен для B. polymixa, B. licheniformis, B. macerans и B. laterosporus, но не для B. coagulans, B. cereus и B. pumilus, хотя и наблюдалось хорошее образование протопластов (Mann et al. Current Microbiol. 13 (1986) 132-135).
Другие способы введения ДНК в протопласты включают слияние с липосомами, содержащими ДНК (Holibova, Folia Microbiol. 30 (1985) 97), или слияние протопласта с использованием легко трансформируемого организма в качестве промежуточной хозяйской клетки (EP-A-0134048).
Краткое изложение сущности изобретения.
Предлагается способ получения прокариотических хозяйских клеток, который включает в себя по крайней мере две стабильно встроенные в хромосому хозяйской клетки копии последовательности ДНК, кодирующей интересующий полипептид, в частности фермент гидролазу. Стабильное поддержание последовательности чужеродной ДНК получено путем встраивания двух или более копий последовательности в хромосому хозяйской клетки таким образом, что эти копии разделены последовательностями эндогенной хромосомной ДНК, жизненно важными для хозяйской клетки.
Фиг. 1-4 схематически представляют четыре способа встраивания последовательностей внехромосомной ДНК в хромосому микроорганизмов.
T обозначает последовательность-мишень, то есть последовательности ДНК, представленные на хромосоме и плазмиде, между которыми может происходить гемологичная рекомбинация.
S обозначает последовательность ДНК, которая должна встраиваться в хромосому.
M обозначает последовательность маркерного гена, используемого для отбора рекомбинантного штамма.
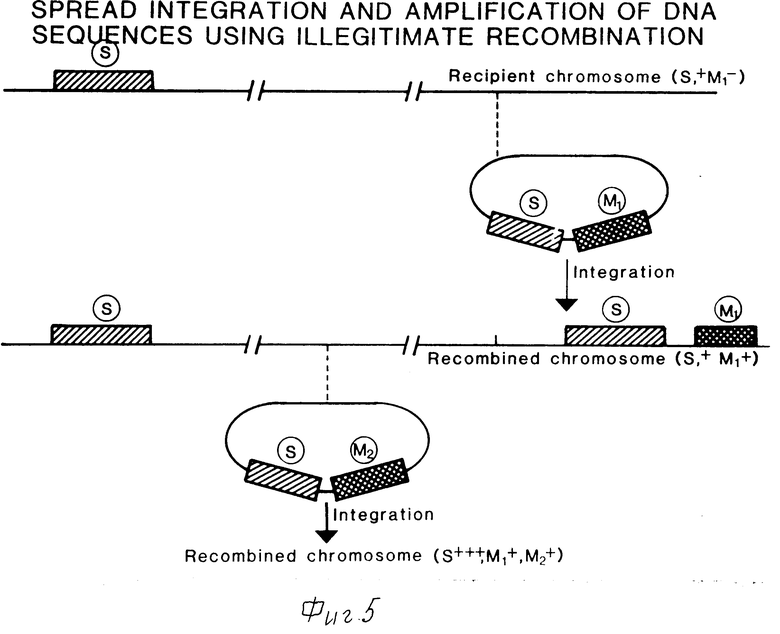
Фиг. 5 и 6 схематично представляют два способа получения стабильной амплификации гена в прокариотической хромосоме.
Фиг.7 показывает результаты гистидин (MOPS гельэлектрофореза, проведенного с супернатантом из культур B. subtilis DB 104, содержащих pUB 110 и pM 58, соответственно, в сравнении с несколькими субтилизинами:
1 дорожка: субтилизин Carlsberg
2 дорожка: протеаза Bacillus PB 92
3 дорожка: субтилизин Bacillus subtilis
4 дорожка: Bacillus subtilis DB 104 (pM 58)
5 дорожка: Bacillus subtilis DB 104 (pB 110).
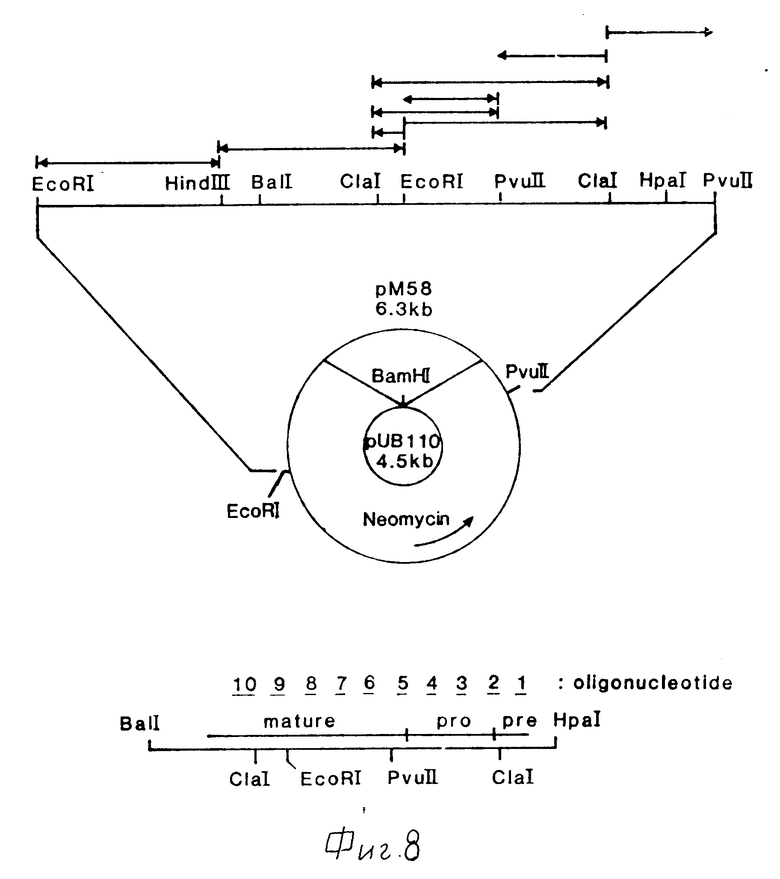
Фиг. 8 показывает рестрикционную карту плазмиды pM 58. В верхней части рисунка показана стратегия секвенирования. Сплошные линии со стрелками представляют фрагменты, клонированные в векторах mp 10, mp 11 mp 18 фага M 13. Нижняя часть рисунка показывает стратегию секвенирования с использованием десяти олигонуклеотидов, локализованных через равные промежутки на гене протеазы.
Фиг.9-11 показывают нуклеотидную последовательность кодирующей ДНК, соотнесенную с аминокислотной последовательностью сериновой протеазы Bacillus PB 92. Также показаны промоторы (P1,P2), сайт связывания рибосом (rbs) и районы терминации (term) последовательности ДНК. Пронумерованные сплошные линии представляют место размещения десяти олигонуклеотидов, используемых для секвенирования.
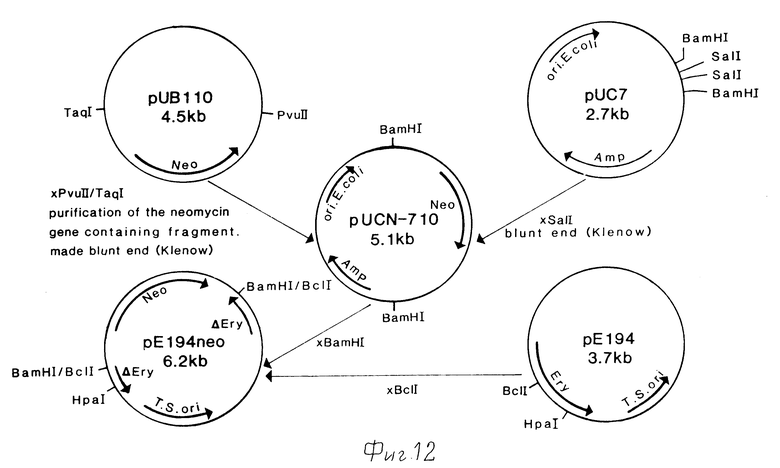
Фиг.12 показывает конструирование плазмиды pE 194 neo.
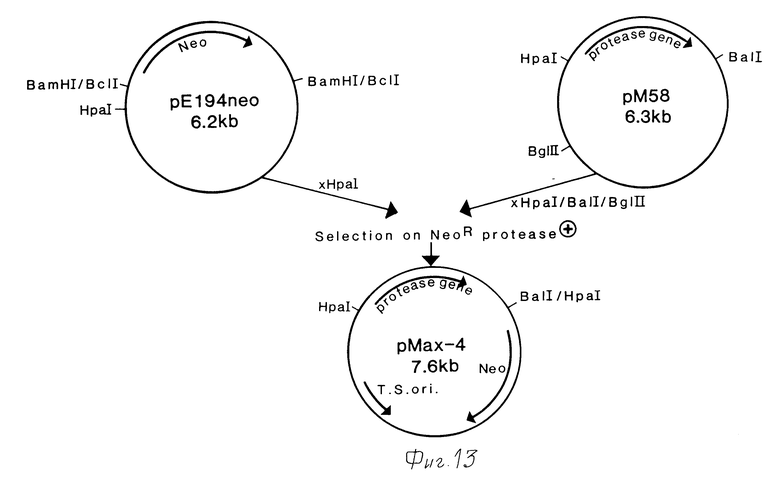
Фиг.13 показывает конструирование плазмиды pMAX 4.
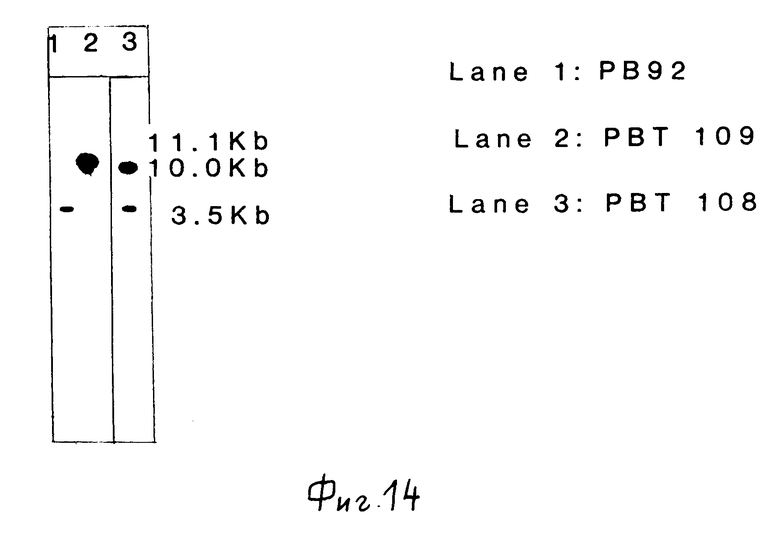
Фиг.14 продукты расщепления хромосомной ДНК штаммов PB 92, PBT 109 и PBT 108 Hind 111 подвергали электрофорезу в 0,5%-ном агарозном геле, перенесли на нитроцеллюлозу, как описано Southern, и гибридизовали с меченой 32P никтранслированной ДНК pM 58. Рисунок показывает радиоавтограф.
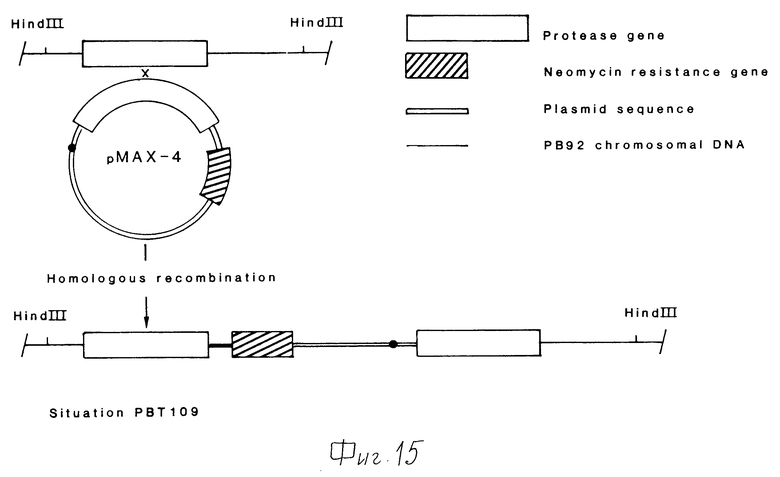
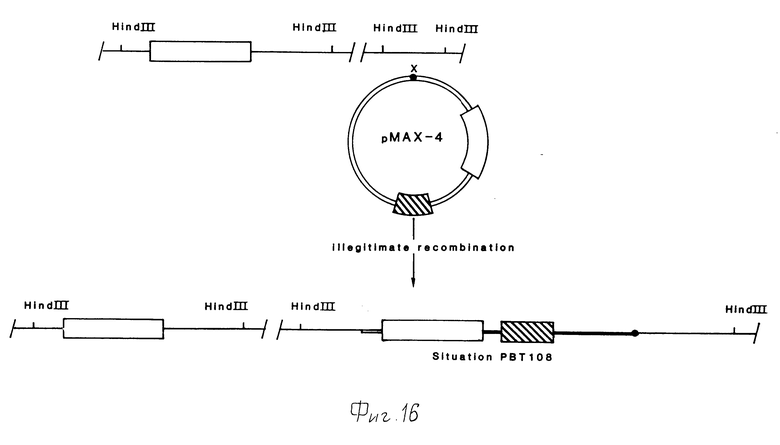
Фиг. 15 и 16 иллюстрируют интеграционные события, происходящие в случае гомологичной (B) рекомбинации и незаконной (C) рекомбинации между pMAX 4 и хромосомой Bacillus PB 92.
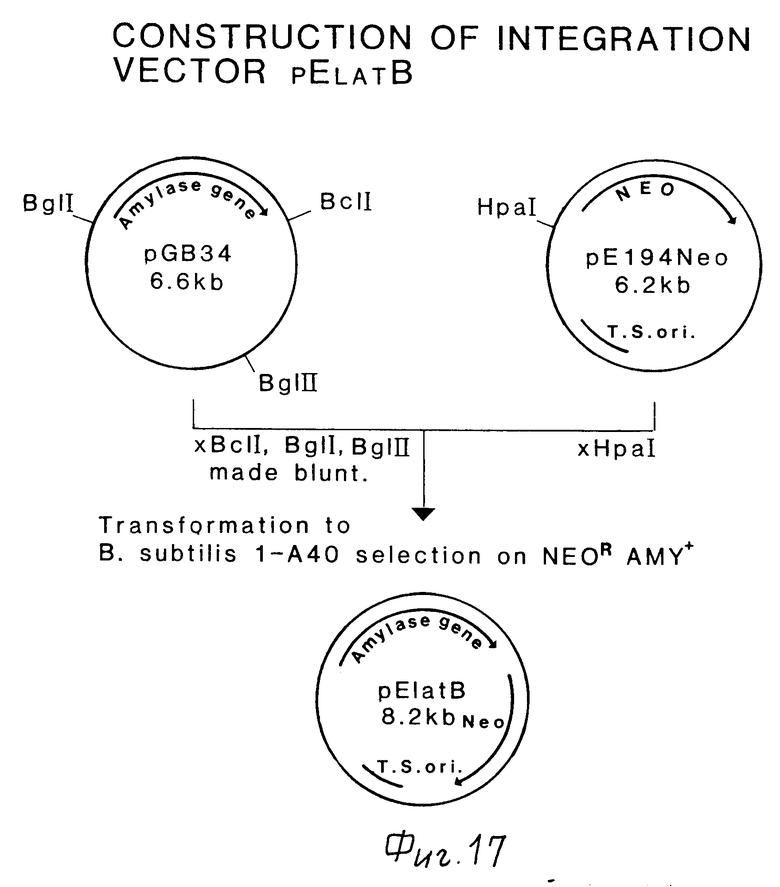
Фиг.17 показывает конструирование интеграционного вектора pElatB.
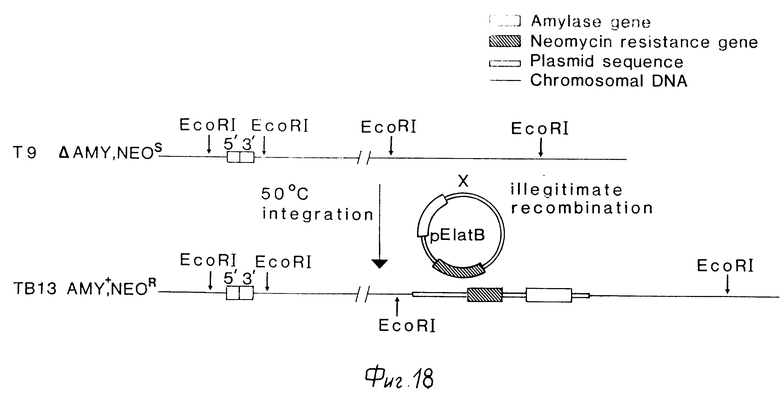
Фиг. 18 иллюстрирует встраивание плазмиды pElatB в хромосому штамма T 9 B. licheniformis, приводящее к получению штамма TB 13 B. licheniformis.
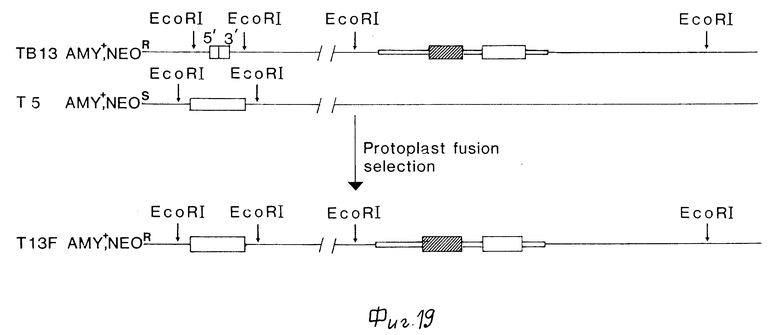
Фиг.19 иллюстрирует хромосомную рекомбинацию штаммов TB 13 и T 5 B. licheniformis при слиянии протопластов этих штаммов, приводящую к получению штамма T 13F B. licheniformis.
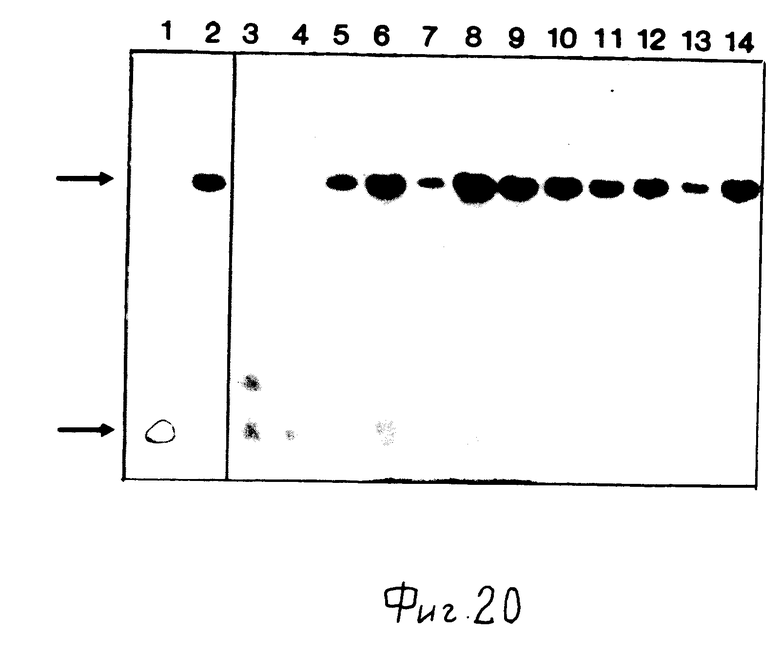
Фиг. 20 показывает хромосомный анализ девяти разных колоний, полученных после ферментации штамма T 13, как описано в примере 11. Выделенная хромосомная ДНК была переварена EcoRI, разделена на 0,8% агарозном геле и перенесена на нитроцеллюлозу. Гибридизация велась с меченой 32P никтранслированной ДНК pElatB. Рисунок показывает радиоавтограф. Верхняя стрелка указывает на положение, в которое мигрирует EcoRI фрагмент ДНК длиной около 15000 пар оснований, содержащий полную последовательность pElatB, которая была встроена в хромосому в участок, не примыкающий к исходному гену альфа-амилазы, как изображено для штамма ТВ13 на фиг.18. Нижняя стрелка указывает на положение, в котром мигрирует EcoRI фрагмент ДНК длиной около 33000 пар оснований, который содержит полный ген альфа-амилазы, первоначально присутствующий в штамме Т5 B. licheniformis (смотри также фиг.19). Анализировали следующие образцы ДНК:
1 дорожка ДНК Bacillus licheniformis T5
2 дорожка ДНК Bacillus licheniformis TB13
3 дорожка ДНК Bacillus licheniformis T390
4 дорожка ДНК из чувствительного к неомицину производного Bacillus licheniformis T390, выделенного после ферментации, как описано в примере 12.
5 дорожка ДНК Bacillus licheniformis T13F
6-14 дорожки ДНК из 9 разных колоний, выделенных после ферментации штамма T13F, как описано в примере 12.
Согласно данному изобретению, предлагаются методы приготовления прокариотических клеток, в которых две или более копий последовательности интересующей ДНК стабильно встроены в хромосому. Хозяйская клетка, включающая последовательность ДНК, кодирующую интересующий полипептид, трансформируется рекомбинантной ДНК, включающей такую же последовательность ДНК. Затем отбираются трансформированные клетки, в которых встроенные последовательности ДНК разделены эндогенными хромосомными последовательностями из гена, подлежащего амплификации.
Эндогенные промежуточные последовательности должны быть существенными для хозяйской клетки. Потери амплифицированных последовательностей из-за гомологичной рекомбинации будут летальными для хозяйской летки. Таким образом, будет существовать селективное давление на клетки, несущие амплифицированные последовательности без необходимости использования антибиотинов и подобных средств селекции. Встраивание может быть достигнуто путем либо гомологичной рекомбинации, либо незаконной рекомбинации. Способы, которые могут быть использованы для получения желаемых клеток, показаны соответственно, на фиг. 5 и 6.
При использовании гомологичной рекомбинации в векторных молекулах могут присутствовать некоторые участки, которые гомологичны хромосоме хозяйской клетки, особенно, когда одна или более копий амплифицируемого гена уже встроены в хозяйскую клетку. Векторная молекула, таким образом, может включать интересующую последовательность ДНК; последовательность мишень и маркерную последовательность ДНК.
Надо обратить внимание на то, что отбираются только желаемые рекомбинантные хромосомные структуры. Это может быть достигнуто путем использования для рекомбинации линейных молекул ДНК. Кольцевая векторная молекула, которую надо встроить, расщепляется рекстрикционным ферментом в участке гомологии с последовательностью-мишенью. В этом случае рекомбинация и встраивание будут предпочтительно происходить в этом специфическом месте. Кроме того, что интересующая последовательность ДНК представлена в векторной молекуле, она также может быть представлена в хромосоме хозяйской клетки. Последовательность ДНК может быть последовательностью ДНК, кодирующей любой структурный ген, который желательно амплифицировать. Последовательность ДНК может быть эндогенной для хозяйского организма или может быть вставлена в хозяйскую хромосому на предварительной стадии трансформации.
Последовательности-мишени для описываемой нетандемной амплификации предпочтительно выбираются среди несущественных генов, например, в случае Bacilli в качестве хозяйских организмов как последовательности-мишени могут быть использованы гены, кодирующие внеклеточные ферменты, или гены, включенные в споруляцию. Встраивание последовательностей ДНК в эти гены будет, как правило, инактивировать ген. Потеря экспрессии гена затем может быть проверена и использована для отбора желаемых рекомбинантных штаммов.
При использовании незаконной рекомбинации для амплификации хромосомного гена, как показано на фиг.4 и 5, предпочтительны такие условия встраивания и отбора, в которых гомологичная рекомбинация не превышает незаконную рекомбинацию. Средства для предотвращения гомологичной рекомбинации заключаются в трансформации первой и второй хозяйских клеток, лишенных интересующего структурного гена, вектором, включающим последовательность ДНК, кодирующую интересующий полипептид, и маркерный ген. Затем первая и вторая хозяйские клетки, в которых последовательность ДНК представлена в разных местах, могут быть отобраны и объединены в условиях слияния для получения трансформированной клетки по крайней мере с двумя копиями последовательности ДНК, кодирующей интересующий структурный ген, расположенными в несмежных участках второго хозяйского генома. Для облегчения селекции первый хозяин должен быть убит перед слиянием.
Интересующим геном (генами) может быть прокариотический или эукариотический ген. Структурные гены могут быть получены множеством путей, включая синтез, выделение из геномной ДНК, приготовление из кДНК или их комбинацией. Различные способы манипуляции с генами хорошо известны и включают рестрикцию, переваривание, вырезание, лигирование, мутагинез in vitro, первичную репарацию, использование линкеров и адаптеров и др. Таким образом, последовательности ДНК, полученные от хозяина, могут быть обработаны множеством методов, в зависимости от требований конструкции ДНК. Смотри Maniatis et al. Molecular Cloning, Cold Sprind Harbor Laboratory, Cold Sprind Harbor, NY, 1982.
Интересующие гены могут быть предназначены для экспрессии различных полипептидов и белков, таких как ферменты, гормоны, лимфокины, поверхностные белки, белки крови, структурные белки, иммуноглобулины и т.д. Особенный интерес представляют ферменты, в частности, различные гидролазы, и особенно протеазы и амилазы. Примером таких ферментов служат сериновые протеазы, включая высокощелочные сериновые протеазы, несериновые протеазы, альфа- и бетаамилазы. Предпочтительным источником сериновой протеазы является Bacillus novo species pB 92, а для альфаамилазы штамм T5 B. licheniformis, а также мутанты и разновидности этих штаммов.
Способ получения гена обычно включает приготовление геномной библиотеки из подходящего организма. Геномную библиотеку удобно приготовить, например, лигируя фрагменты ДНК донорного штамма в подходящий вектор.
В векторе для экспрессии структурный ген должен быть соединен в нужной ориентации с контролирующими участками, такими как промоторная последовательность, последовательность, формирующая участок связывания рибосом, и последовательности, контролирующие терминацию трансляции и транскрипции структурного гена, функциональными в хозяйской клетке. Там, где хозяйская клетка обладает слишком низкой частотой трансформации и интеграции, для применения прямой селекции на встраивание без промежуточного выделения клеток, содержащих плазмиды, вектор может дополнительно включать origin репликации, способный автономно реплицироваться в хозяйской клетке.
Там, где ген получен из донорной клетки, которая имеет регуляторные сигналы инициации и терминации транскрипции и трансляции, которые распознаются хозяйским штаммом, обычно удобно поддерживать исходные регуляторные последовательности структурного гена. Кроме того, участок регуляции транскрипции может обеспечивать конститутивную или индукцибельную экспрессию, так что в соответствующих ситуациях хозяин может выращиваться до высокой плотности прежде, чем получаются высокие уровни экспрессии интересующих структурных генов.
Там, где структурные гены получены из источника, чьи регуляторные сигналы не распознаются хозяйской клеткой, будет необходимо получить регуляторные сигналы, распознаваемые хозяйской клеткой, и встроить структурный ген между регуляторными сигналами инициации и терминации. В некоторых случаях экзогенный структурный ген со своим собственным стопкодоном (кодонами) может быть вставлен в рамке считывания после N-концевых кодонов эндогенного структурного гена, который сохраняет свои природные регуляторные сигналы.
Желательно, чтобы продукт экспрессии секретировался. Там, где продукт экспрессии секретируется естественным образом и лидерные сигналы и сигнал (сигналы) процессинга распознаются хозяйской клеткой, это не повлечет за собой трудностей. Однако там, где продукт не секретируется из-за того, что хозяйская клетка не распознает секреторные лидерные сигналы и/или сигнал (сигналы) процессинга, или эти сигналы не функционируют в удовлетворительной степени в хозяйской клетке, может быть необходимо выделить или синтезировать последовательности ДНК, кодирующие секреторные лидерные сигналы и сигнал (сигналы) процессинга полипептида хозяйской клетки и присоединить их в правильной рамке считывания к 5'-концу структурного гена.
Вектор может дополнительно включать маркерный ген, придающий устойчивость к антибиотику, к которому хозяйская клетка исходно чувствительна. При использовании в хромосомной интеграции вектора маркерный ген должен выполнять требование, чтобы отбор на выживание был возможным, даже если только одна или несколько копий маркерного гена представлены в хозяйском штамме. Под маркером подразумевается структурный ген, способный к экспрессии в хозяине, который обеспечивает отбор на выживание. Под отбором на выживание подразумевается придание фототрофности ауксотрофному хозяину, устойчивому к биоцидам или вирусам. Что касается фототрофности, могут быть использованы различные гены, такие как leu, his, trp и т.д. Что касается биоцидной устойчивости, то она может включать устойчивость к антибиотикам, например, neo, cam, tet, tun, kan и т. д. Другие маркеры включают устойчивость к тяжелым металлам, иммунитет и подобное.
Различные последовательности ДНК могут быть получены из разных источников и соединены вместе для получения вектора, который включает один (или более), удобный, желательно уникальный рестрикционный сайт, что позволяет вставлять или замещать структурные гены в эти сайты или на место утерянных фрагментов для получения плазмидной конструкции.
Отбору на хромосомное встраивание может способствовать использование плазмиды с origin репликации, имеющим мутацию, делающую его функционирование в хозяйской клетке температурозависимым. Смотри, например, Ehrlich, Proc. Natl. Acad. Sci. USA 75 (1978) 1433.
Когда плазмидная конструкция приготовлена, она сейчас же может быть клонирована в подходящем хозяине клонирования. Может быть использован любой подходящий хозяин, который легко трансформируется, позволяет репликацию плазмидной конструкции и перенос ее в хозяйскую клетку. Доступно большое число штаммов, имеющих высокую эффективность трансформации; они обычно относятся к числу ауксотрофных и/или чувствительных к антибиотику. Когда хозяйской клеткой является промышленный штамм Bacillus, использование этого организма в качестве хозяйской клетки для клонирования плазмидной конструкции имеет много приемуществ, в частности заключающееся в том, что он позволяет использование одной репликационной системы, а также одного или того же маркера для отбора на выживание как в хозяине клонирования, так и в системе экспрессии. Смотри, например, Европейскую заявку ЕР-А-134048, которая включена здесь путем ссылки.
Плазмидная конструкция может быть введена в хозяине клонирования в согласии с удобными способами, такими как трансформация, использование кальциевой преципитации ДНК, конъюгация или другим подходящим способом. Хозяин клонирования затем может быть выращен на подходящей питательной среде в селективных условиях для отбора хозяина, содержащего плазмидную конструкцию. Для автотрофных хозяев питательная среда недостаточна по необходимому питательному веществу, в то время как для биоцидной устойчивости, например, устойчивости к антибиотику, применяются цитотоксические количества биоцида (биоцидов) в питательной среде.
Могут быть применены различные хозяйские клетки, включая штаммы E. coli, Bacillus, особенно Bacillus subtilis, Pseudomonas и Streptomyces. При выборе хозяйской клетки во внимание принимаются различные факторы, включая факторы, которые могут влиять на экспрессию амплифицируемого гена и производство желаемого продукта. С учетом этого желательно использовать хозяйскую клетку, в которой распознаются регуляторные сигналы, легко идет секреция, снижена деградация желаемого продукта и так далее. Предпочтительная хозяйская клетка уже продуцирует интересующий полипептид и может быть либо организмом дикого типа, либо мутантным организмом. Хозяйская клетка также может быть мутантом организма, продуцирующего интересующий полипептид, который сам, однако, не является продуцентом. Когда интересующим полипептидом является протеаза или амилаза, предпочтительные штаммы включают Bacillus novo species PB 92 и T5 Bacillus licheniformis, соответственно, и мутанты и разновидности этих штампов.
Кроме того, могут использоваться промышленные штаммы, используемые для продукции ферментов, такие как B. licheniformis, B. amyloliguefaciens и щелочефильные бациллы. Промышленные штаммы являются очень сильными и стабильными. Более того, упомянутые штаммы устойчивы к фаговой инфекции и к генетическому обмену, каким является введение ДНК с помощью пригодных способов трансформации. Подходящие промышленные штаммы фототрофны для того, чтобы избежать добавления дорогих аминокислот к питательной среде. Другими характеристиками промышленных штаммов являются их высокая продуктивность до конца ферментации, которая может длиться до недели, стабильная клеточная концентрация до истощения бульона и высокая продуктивность, обычно по крайней мере 5 г/л (0,5 мас. объем) специфического секретируемого белка.
Могут быть получены трансформанты, имеющие гены либо в тандемной организации, либо рассеянные по хромосоме. Обычно, возможно отобрать траснсформанты, содержашие рассеянные гены, из смеси двух упомянутых типов трансформантов путем выделения хромосомной ДНК каждого отдельного трансформанта, последовательным анализом упомянутой ДНК на предмет относительной локализации упомянутых генов, например, способом Southern, J. Mol. Biol. 98 (1975) 508-517 или способами, известными специалистам в данной области.
Способы получения трансформантов с тандемно встроенными генами с помощью двойной реципрокной рекомбинации (фиг.3 и 6), включают использование линеаризованных конструкций ДНК, которая должна быть амплифицирована, маркерный ген и последовательность-мишень для рекомбинации.
Более того, специфические способы получения трансформантов с рассеянными генами включают использование незаконной рекомбинации (фиг.5), при котором можно избежать выделения тандемных трансформантов путем отбора на основе дифференциальной экспрессии маркерного гена, например гена, кодирующего устойчивость к антибиотику, где чувствительность к антибиотику в штаммах с тандемным встраиванием гена и нетандемным встраиванием различна. Обычно длина встроенных эндогенных последовательностей ДНК будет меньше 10000 пар оснований.
Кроме того, способы получения трансформантов с рассеянными генами, предотвращающие тандемную дупликацию, включают использование убитых протопластов гомологичного донорного штамма, несущего конструкцию ДНК, включающую структурный ген и маркерный ген, причем структурный ген встроен в хромосому в другом месте относительно акцепторного штамма.
Трансформация хозяйских клеток предпочтительно включает использование протопластов, приготовленных из хозяйского штамма. Обычно протопласты готовят из клеток в соответствии с общепринятыми способами, например, обработкой лизоцимом или зимолиазой, и протопласты аккуратно суспендируют в подходящей среде, обладающей подходящей осмотичностью для поддержания целостности протопласта. Для промышленных штаммов Bacillus способы приготовления протопластов описаны в EP-A-0134048, приводимой здесь в виде ссылки. Когда хозяйским штаммом является щелочефильный штамм Bacillus, протопласты могут быть приготовлены при щелочном pH, предпочтительно около 8,0. Этот способ раскрыт в Европейской заявке N EP-A-87200358.7, которая приводится здесь в виде ссылки.
Хозяйская клетка может быть трансформирована объединением плазмидной конструкции или протопласта, содержащего плазмиду хозяина с протопластом хозяйской клетки, в присутствии подходящего реагента для слияния. Может быть применен любой реагент, обеспечивающий желаемую степень эффективности; обнаружено, что полиэтиленгликоль в основном обеспечивает весьма высокую эффективность слияния. Через короткий промежуток времени реакционная смесь заменяется подходящей питательной средой, и клетки регенерируют в селективной среде обычно путем высева на чашку с агаром.
Трансформанты, полученные соединением хозяйской клетки с подходящей конструкцией ДНК, могут содержать упомянутую конструкцию ДНК или ее часть либо в виде вставки в хромосому, либо как свободные векторные молекулы, когда конструкции ДНК содержат origin репликации, функционирующий в упомянутой хозяйской клетке.
Способ селекции трансформантов, в которых ДНК-конструкция встроена в хромосому, заключается в использовании плазмиды, содержащей температурочувствительный origin репликации. Трансформанты выращиваются на селективной среде при пермиссивной температуре, затем температура сдвигается до непермиссивного уровня. Колонны, экспрессирующие маркерный ген при непермиссивной температуре, выделяются и культивируются на селективной среде при пермиссивной температуре. Отсутствие плазмиды может быть подтверждено либо выделением суммарной ДНК из колоний и электрофорезом в агарозном геле, либо демонстрацией отсутствия способности трансформантов трансформировать компетентные клетки. Определение пути, по которому произошло встраивание в хромосому, может быть проведено анализом хромосомной ДНК, например по способу Southern, G. Mol. Biol. 98(1975) 503-517 или другими способами, известными специалистам в данной области.
Когда имеется дифференциальная чувствительность к селективному агенту между трансформантами, содержащими добавочные копии маркерного гена в тандемной организации, и трансформантами, в которых маркерный ген встроен в различные участки хозяйского генома, трансформанты можно вырастить на среде, содержащей подходящую концентрацию селективного агента, с отбором трансформантов с нетандемным встраиванием.
Другим способом получения трансформантов с рассеянным встраиванием копий интересующей последовательности ДНК является использование протопласта, приготовленного из гомологичной донорной клетки, содержащей по крайней мере одну копию интересующей последовательности ДНК в участке ее хромосомы, отличном от участка встраивания этой последовательности в реципиентной хозяйской клетке.
Гомологичная донорная клетка может быть приготовлена, например, трансформацией клетки, не содержащей данного структурного гена, вектором, включающим этот структурный ген. Встраивание последовательности ДНК в хромосому донорной клетки может быть облегчено путем использования плазмиды, содержащей температурочувствительный origin репликации и выращивания трансформантов в селективных условиях сначала при допустимой, а затем при недопустимой температуре, как описано выше, с последующей изоляцией колоний, экспрессирующих маркерный ген.
После подтверждения отсутствия плазмидной ДНК хромосомная ДНК может быть изолирована и проанализирована по способу Southern (см. выше), путем гибридизации с зондом, меченым, например, 32P или биотинилированными нуклеотидами. Зондом может быть как кДНК, кодирующая интересующий полипептид, а также ее фрагменты, так и конструкции ДНК или ее фрагменты, включающие интересующую последовательность ДНК, например, вектор. Трансформанты, содержащие интересующий ген в другом месте по сравнению с местоположением гена в донорном штамме, могут затем использоваться в качестве гомологичной донорной клетки. Реципиентным хозяйским штаммом предпочтительно является тот же самый штамм, который используется в качестве источника интересующей последовательности ДНК, или штамм, в котором интересующая последовательность ДНК находится в ином районе хромосомы, чем в трансформированной донорной клетке.
Для облегчения селекции донорная клетка предпочтительно отрабатывается цитотоксическим агентом перед или во время образования протопласта. Могут быть применены различные агенты для "убийства" донорной клетки, включая антибиотики, но обнаружено, что удобным, эффективным, не влияющим на последующее слияние является иодоацетамид. При использовании "убитых" протопластов клона отношение их к акцепторному хозяйскому штамму будет в основном не менее 1:1, но может быть использован и избыток "убитых" протопластов.
После слияния протопласта мертвой донорной клетки и протопласта реципиентной хозяйской клетки трансформанты могут быть отобраны с помощью маркерного гена. Затем ДНК может быть выделена и проанализирована, как описано выше, для идентификации трансформантов, в которых более одной копии интересующего гена было встроено в геном и разделено эндогенными хромосомными последовательностями.
Трансформанты, содержащие два разделенных гена, затем скринируются подходящими способами для обнаружения увеличенной экспрессии интересующего полипептида. Могут использоваться различные методы, в частности, те, в которых используются ферменты, имеющие хорошо разработанные методы обнаружения. С другой стороны, там, где не привлекаются ферменты или нет пригодной системы обнаружения скрининга клонов с целью определения наличия плазмидной конструкции и экспрессии интересующего структурного гена, могут быть применены биоанализ, антитела, ДНК или РНК гибридизация.
Хозяйская клетка, содержащая встроенные в хромосому плазмидные конструкции или их фрагменты, затем выращивается на питательной среде в условиях, пригодных для ферментации. Ферментация может продолжаться, пока не истощится бульон. Там, где секретируется продукт, он может быть выделен из бульона удобным способом, например, экстракцией, хроматографией, электрофорезом и т. п. Там, где продукт остается в цитоплазме, клетки можно собрать центрифугированием, фильтрацией и так далее, лизировать механической обработкой, детергентом, лизоцимом или другими способами и выделить продукт, как описано ранее. При использовании способа, являющегося предметом изобретения, как способа генной амплификации, можно достигнуть стабильного встраивания по крайней мере двух копий последовательности ДНК.
Следующие примеры предлагаются как иллюстрация, но не как ограничение.
Экспериментальная часть
Пример 1
Приготовление библиотеки геномной ДНК из щелочефильного Bacillus novo sp. PB92 и выделение гена сериновой протеазы
Хромосомная ДНК, выделенная из Bacillus novo sp. PB92 (хранится под N OR-60 в лаборатории микробиологии, Технический университет Дельфта, Нидерланды, патент США N Re. 30602) по способу, описанному Saito Miuva, Biochim, Biophys. acta 72 (1963) 619-632, была частично переварена рестрикционным ферментом Sau 3A и лигирована в Bam H1 сайт плазмиды puB110 (Yryczan et al. J. Bacteriol. 134 (1978) 318-329). Плазмидная ДНК puB110 была приготовлена, как описано Birnboim and Doly (nucl. acids Res. 7 (1979) 1513-1523).
Лигированной смесью трансформировали B. subtiles 1A40 (Генетический центр хранения бацилл) по способу Spizizen et al. J. Bacteriol. 81 (1961) 741-746, используя 0,6-1 мкг ДНК на мл компетентных клеток. Клетки из трансформационной смеси высевали на минимальные чашки, содержащие 2,8% K2HPO4; 1,2% KH2PO4; 0,4% (NH4)2SO4; 0,2% три-Na-цитрат•2H2O; 0,04% MgSO4•7H2O; 0,00005% MnSO4•4H2O; 0,4% L-глутаминовую кислоту; 0,5% глюкозу; 0,02% казаминовых кислот; 50 мкг/мл метионина; 20 мкг/мл лизина; 20 мкг/мл неомицина; 0,4% казеина и 1,5% агара. После инкубации чашек в течение ночи при 37oC одна из 50000 колоний, устойчивых к неомицину, показала возросшую продукцию протеазы, что определили по увеличению преципитации ореола продуктов расщепления казеина вокруг колонии на пластинке агара. Плазмидная ДНК была выделена из этой колонии по способу, описанному Bimboim and Doly (nucleic acids Res. 7 (1979) 1513-1523) и обозначена pM58.
Пример 2
Экспрессия гена сериновой протеазы PB92
Bacillus subtilis 1A40, содержащая pM58, была выращена на минимальной среде (spizizen et al. Prjc. Natl. acad. sci. USA 44 (1958) 1072-1078), к которой было добавлено 0,02% казаминовых кислот; 50 мкг/мл триптофана; 20 мкг/мл метионина; 20 мкг/мл лизина и 20 мкг/мл неомицина. Через 24 ч культуру центрифугировали и супернатант проверяли на протеазную активность, используя в качестве субстрата диметилказеин (Zin et al. j. Biol. Chem. 244 (1969) 789-793). Культура B. subtilis 1A40, содержащая плазмиду pUB110, использованная в качестве контроля, показала меньше чем 1/60 протеазной активности, показанной культурой, трансформированной pM58. Протеазная активность полностью ингибировалась обработкой 1 мМ фенилсульфанилфлуоридом (PMSF), но не 20 мМ ЭДТА.
Аликвоты супернатантов, описанных выше, анализировались с помощью белкового геля по способу Zaemmli, Nature 227 (1970) 680. Пробы для анализа на этих гелях были приготовлены обработкой супернатантов 5% трихлоруксусной кислотой (ТХУ). После центрифугирования образца осадок преципитированного белка промывался дважды ацетоном, затем растворялся в 40 мкл буфера для образцов 0,5 М Трис/HCl pH 7,5; 10 об. 2-меркаптоэтанола; 50 об. глицерина и 0,05% бромфенолового синего при кипячении в течение 10 мин. После электрофореза гели окрашивали, используя Кумасси Бриллиантовый Синий. Затем пробы супернатанта культур анализировались электрофорезом. Использовались три разных штамма B. subtilis 1A40: штамм, содержащий puB110, штамм с pM58 и штамм без плазмиды, и протеаза Bacillus PB92 в качестве контроля. После электрофореза гели окрашивали, используя Кумасси Бриллиантовый Синий, и отмывали. Образец из B. subtilis штамма 1A40, содержащего pM58, содержал белок весом 31 кДа, который мигрирует совместно с протеазой Bacillus PB92. Этот белок не обнаруживался в контрольной дорожке штамма B. subtilis 1A40, содержащего puB110.
Все сериновые протеазы имеют одинаковые молекулярные веса. Поэтому клонированная сериновая протеаза Bacillus PB92 дифференцировалась от известных сериновых протеаз (субтилизина B. subtilis, субтилизина Carlsbery) с помощью трансформации pM58 и puB110 в беспротеазный штамм B. subtilis DB104 (R. Doi, j. Bacteriol. 160 (1984) 442-444) и анализа продуцируемой внеклеточной протеазы. Полученные трансформанты выращивались на минимальной среде (Spizizen et al. выше), содержащей 0,02% казаминовых кислот, 50 мкг/мл гистидина и 20 мкг/мл неомицина. Взятые через 24 ч образцы были отцентрифугованы и без дополнительной обработки проанализированы с помощью гистидин/MOPS гелей, содержащих 75 мМ KOH; 40 мМ гистидина; 100 мМ MOPS (3-(N-морфолино)-пропансульфоновая кислота), pH 7,5 и 5% полиакриламид. Электрофоретический буфер содержал 40 мМ гистидина, 100 мМ MOPS, pH 6,6. Образцы двигались по направлению к катоду. Полосы протеазы обнаруживались с помощью профессиональных пленок Agfa Pan 100 (zuidweg et al. Biotechnol and Bioengin, 14 (1972) 685-714). Эти результаты приведены на фиг.7. Как видно, pM58 несет ген, кодирующий протеазу Bacillus PB92.
Пример 3
Секвенирование гена сериновой протеазы
Bacillus PB92
Полная последовательность Bal I Hpal фрагмента pM58 была определана способом Sanger, Proc. Natl. Acad. Sci. USA 77 (1977) 6463. Рестрикционные фрагменты pM58 (фиг.8) были клонированы в векторах mp10, mp11 и mp18 фага M13 (Messing et al. Nucleic Acids Res. 9 (1981) 309-321). Встраивание фрагментов pM58 определяли с помощью гибридизации бляшек. После секвенирования были синтезированы десять олигонуклеотидов, локализованных через равные промежутки на гене, которые использовали для повторного секвенирования. Оно подтвердило последовательность, показанную на фиг.9-11.
Пример 4
Конструирование плазмиды pMAX-4, содержащей сериновую протеазу
Для конструирования плазмиды pUCN 710 (фиг.12) puB110 была переварена Tag I и Pvu II. Фрагмент, содержащий ген, придающий устойчивость к неомицину, был очищен с помощью низкоплавкой агарозы и затуплен фрагментом полимеразы Кленова и НТФ (Maniatis, Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, 1982). Плазмида pUC7 (Vieira et al. Jene 19 (1982) 259-268) была линеаризована Sal I и затуплена, как описано выше. Оба фрагмента были лигированы T4 лигазой (Maniatis) и трасформированы в E. coli JMl 03. Отбор вели на 2xTY-чашках (1,6% вес/объем Бакто-триптона; 1% вес/объем дрожжевого экстракта; 0,5% NaCl, содержащих 50 мкг/мл ампициллина и 10 мкг/мл неомицина). Полученная плазмида, названная pUCN 710, была переварена Bam H1. Плазмида pEl94 (Jordanescu, Plasmid 1 (1978) 468-479) была переварена Bcl 1. Фоагменты из обоих расщеплений были лигированы T4 лигазой и трансформированы в B. subtilis 1A40. Отбор вели на минимальных чашках, содержащих 20 мкг/мл неомицина (пример 1). Полученная плазмида pEl 94-neo (фиг.12) содержала ген неомицина и температурочувствительный origin репликации.
Субклонирование гена протеазы в вектор pEl94-neo вели следующим образом: pM58 (пример 1) переваривали Hpal и Bal 1 и Bgl II. Плазмиду pEl94-neo переваривали Hpal. Эти фрагменты лигировали T4-лигазой и трансформировали ими B. subtilis 1A40. Трансформанты отбирали на основе устойчивости к неомицину и увеличения продукции протеазы, о чем судили по преципитации продуктов расщепления казеина (образованию ореола, пример 1). Была получена плазмида pMAX-4, структура которой подтверждена анализом рестрикционными ферментами (фиг.13).
Пример 5
Трансформация протопласта штамма
Bacillus PB92 pMAX-4
Штамм Bacillus PB92 растили в течение ночи в 100 мл NBSG-X среды (Thorne et al. J. Bacteriol 91 (1966) 1012-1020). Культуру центрифугировали в течение 10 мин при 4500 об/мин в роторе Sorvall модель GSA. Протопласты были приготовлены инкубированием бацилл в течение одного часа при 37oC в 10 мл щелочной среды (AHM), содержащей 0,5 М сахарозу; 0,02 М MgCl2 и 0,02 М Трис/малеат, pH 8,0, в стерильной воде, к которой было добавлено 0,4 мг/мл лизоцима. Протопласты осаждались 10 мин при 4500 об/мин, затем были ресуспендированы в 5 мл буферной смеси AHM+, pH 8,0 (AHM буфер, к которому добавлено 3,5 мас%/объем Бакто Penassay бульона и 0,04 мас%/объем Merieux альбумина), перемешаны и переосаждены, как указано выше. После ресуспендирования в 5,0 мл AHM 0,5 мл этой суспензии протопластов было смешано с 5 мкг диминерализованной воды, содержащей 1 мкг плазмидной ДНК; смесь инкубировали в течение 2 мин в присутствии 30 мас%/объем полиэтиленгликоля 8000, pH 8,0. После трехкратного разбавления средой AHM+, pH 8,0 и центрифугирования осадок ресуспендировали в малом объеме (1 мл) AHM+ и инкубировали в течение 2-3 ч. Аликвоты по 100 мкл были высеяны на свежеприготовленные регенерационные чашки, содержащие 0,5 М сукцинат (HCl, pH 8,0, 1,5 (мас%/объем) агар; 0,5 (мас%/объем) казаминовые кислоты; 0,5 (мас%/объем) дрожжевой экстракт; 0,031 М фосфатный буфер; pH 8,0, 0,5 (мас%/объем) глюкозу, 0,02 М MgCl2 и 0,02 (мас%/объем) Merieux альбумин. Эти чашки также содержали 1000 мкг/мл неомицина для селекции. После инкубации при 37oC в течение по крайней мере 72 ч колонии были перенесены на чашки с агаром, содержащим сердечный экстракт и 20 мкг/мл неомицина.
Пример 6
Встраивание pMAX-4 в хромосому штамма
Bacillus PB92
Трансформант Bacillus PB92, содержащий плазмиду pMAX-4, инкубировали в Триптон-соевом бульоне (TSB), содержащем либо 1 мкг/мл, либо 20 мкг/мл неомицина в течение 24 ч при 37oC.
Порции суспензии клеток по 2 мл были затем разведены в 100 мл TSB, содержащего 1 мкг/мл или 20 мкг/мл неомицина, соответственно, и инкубированы в течение 24 ч при 50oC. Спустя 24 ч образцы (по 5 мл) обоих культур вновь были разведены, как описано выше, и вновь инкубированы в течение 24 ч при 50oC в присутствии 1 мкг/мл или 20 мкг/мл соответственно. Последняя процедура была посторена еще раз. Затем клеточные суспензии были разведены в 100 раз и высеяны на агаровые чашки с экстрактом сердечной мышцы (HI-агар), содержащим 1 мкг/мл неомицина для образцов из флаконов, содержащих 1 мкг/мл неомицина, и 20 мкг/мл неомицина для образцов из флаконов, содержащих 20 мкг/мл неомицина. Чашки инкубировали в течение 16 ч при 50oC. Колонии, резистентные к неомицину, изолировали и культивировали в 10 мл TSB среды, содержащей 1 мкг/мл неомицина, в течение 16 ч при 37oC. Из этих колоний была выделена тотальная ДНК (Holmes et al. Anal. Biochem 114 (1981) 193-197). Отсутствие плазмиды проверяли путем электрофореза ДНК в агарозном геле. Отсутствие плазмидной ДНК в образцах, в которых плазмидная ДНК не была обнаружена, было подтверждено трансформацией тотальной ДНК в B. subtilis 1A40. Образцы, не проявлявшие способности трансформировать B. subtilis 1A40, оценивались как свободные от плазмиды.
Для исследования факта и способа встраивания pMAX-4 в хромосому хромосомную ДНК выделяли, передавали Hind 111, разгоняли на 0,5% ДНК-агарозных гелях, переносили на нитроцеллюлозу (Southern, J. Mol. Biol. 98 (1975) 503-517) и гибридизировали с меченой 32P ник-транслированной pM58 (Maniatis, 1982). Результат этого анализа показан на фиг.14.
Селекция на фоне 1 мкг/мл неомицина приводила в результате к получению протеазных генов, тандемно локализованных и разделенных в хромосоме плазмидными последовательностями (штамм PBT109) в результате гомологичной рекомбинации Campbell-типа. При селекции на 1 мкг/мл неомицина было накоплено 30 независимо выделенных интегрантов. Был выделен также один интегрант, который содержал плазмиду pMAX-4 на случайном участке в хромосоме в результате незаконной рекомбинации (штамм PBT122). К получению копии плазмиды pMAX-4 на случайном месте в хромосоме в результате незаконного типа рекомбинации приводила и селекция при 20 мкг/мл неомицина. Последний штамм был обозначен pBT108. Генетическая организация штаммов PBT109 и 108 изображена на фиг.15 и 16 соответственно. Анализ хромосом показал, что встраивание в PBT122 и PBT108 происходит в различные места.
Пример 7
Стабильность дуплицированных протеазных генов в штаммах PBT108 и PBT109
В 100 мл производственной среды (содержащей 1% крахмала, 4% лактозы, 0,8% K2HPO4, 0,5% дрожжевого экстракта, 0,5% (NH4)2HPO4, 0,2% три-Na-цитрата • 2H2O, 0,05% MgSO4•7H2O, 0,07% CaCl2, 0,068% FeSO4 •7H2O и 1 мл/л противовспенивателя) без неомицина, помещенные в 500 мл качалочные колбы, инокулировали 0,2 мл штаммов PBT108 или PBT109, выращенных в течение ночи при 37oC. После инкубации в течение 44 ч при 37oC и постоянной аэрации культура была испытана на колонии, устойчивые к неомицину, и протеазную активность.
Штамм PBT108 и штамм PBT109 были также испытаны в ферментерах Eschweiler, содержащих вышеупомянутую производственную среду, для выяснения эффекта увеличения объема до 1 л. Результаты экспериментов по ферментации суммированы в табл.1.
Анализы колиний, полученных при Eschweiler ферментации PBT109 после 2 дн культивирования, показали, что продукция 3-25% этих колоний была на уровне штамма, содержащего только один ген протеазы. Было обнаружено, что вышеупомянутые колонии являются неомицин-чувствительными благодаря вырезанию последовательности pMAX-4 путем гомологичной рекомбинации. Однако, анализы колоний, полученных в эксперименте по ферментации штамма PBT108, показали, что все эти клетки были неомицин-устойчивыми. Сто из этих неомицин-устойчивых колоний были взяты для случайного и индивидуального тестирования на предмет продуцирования протеазы, для определения один или два продуктивных протеазных гена они содержат. Все 100 индивидуально тестированных колоний давали продукцию на уровне, отвечающем содержанию двух генов, показывая, что два случайно встроенных протеазных гена в PBT108 стабильно поддерживаются в использованных условиях ферментации.
Пример 8
Конструирование интегральнного вектора pElaTB
Плазмида pGB34, описанная в EPA 0134048, была переварена рестрикционными ферментами BCl1, Bgl1 и Bgl II. Концы полученных фрагментов рестрикции затупляли полимеразой Кленова и затем лигировали в Hpa1 сайт pE194-neo (пример 6). ДНК плазмиды pE194-neo использовали как описано Birnboim and Doly (Nucl. Acids Res. 7 (1979) 1513-1523).
Лигазную смесь трансформировали в B. subtilis 1A40 по способу Spizizen et al. (J. Bacteriol. 81 (1961) 741-746), используя 0,5-1 мкг ДНК на мл компетентных клеток. Клетки из трансформационной смеси высевали на минимальные чашки, содержащие 2,8% K2HPO4; 1,2% KH2PO4; 0,4% (NH4)2SO4; 0,2% три-Na-цитрат•2H2O; 0,04% MgSO4•7H2O, 0,00005% MnSO4•4H2O; 0,4% глутамициновую кислоту, 0,5% глюкозу, 0,02% казаминовые кислоты, 50 мкг/мл триптофана, 20 мкг/мл метионина, 20 мкг/мл лизина, 20 мкг/мл неомицина, 0,4% казеин, 0,5% крахмал и 1,5% агар.
Из колоний, продуцирующих альфа-амилазу, изолировали ДНК, как описано Birnboim и Doly, и проверяли ее рестрикционными ферментами. Из одного из этих трансформаторов была выделена плазмида pElaTB (фиг.17).
Пример 9
Трансформация негативного по альфа-анализу штамма
Bacillus licheniformis T9 при помощи pElatB
Трансформацию штамма Bacillus licheniformis T9 проводили, как описано в EP-A-0253455, за исключением того, что всю процедуру проводили при 30oC вместо 37oC. Отбор трансформантов проводили на минимальных чашках, содержащих 20 мкг/мл неомицина. Все трансформанты продуцировали амилазу. Рестрикционный анализ, проведенный на ДНК, приготовленной, как описано Birnboim и Doly, показал, что все трансформанты содержат pElatB.
Пример 10
Встраивание pElatB в хромосому B. licheniformis
Штамм T9 Bacillus licheniformis, содержащий плазмиду pElat B, инокулировали в Триптон-соевый бульон (TSB), содержащий 20 мкг/мл неомицина, и инкубировали в течение 16 ч при 30oC. Порции клеточной суспензии (по 5 мл) разводили в 100 мл вышеназначенной среды и инкубировали при 50oC в течение 24 ч.
Этот процесс повторяли еще раз. Клеточную суспензию затем разводили в 100 раз и высевали на чашки с агаром, содержащим экстракт сердечной мышцы, и 10 мкг/мл неомицина. После 40 ч инкубации при 50oC колонии, устойчивые к неомицину, выделяли и культивировали в 10 мл TSB-среды, соедржащей 10 мкг/мл неомицина, в течение 16 ч при 30oC. Из этих культур выделяли тотальную ДНК (Holmes et al Anal. Biochem. 114 (1981) 193-197). Отсутствие плазмид в этих клетках устанавливали путем электрофореза ДНК в агарозном геле. Образцы, в которых ДНК с низким молекулярным весом, фактически отсутствовали, были перепроверены на присутствие плазмидной ДНК путем трансформации ДНК в B. Subtilis 1-A40 (Spizizen et al. 1961). Образцы с отсутствием способности к трансформации B. Subtilis 1-A40 в устойчивый к неомицину считали плазмидоотрицательными.
Для проверки того, происходит ли встраивание pElatB и как это происходит, выделяли хромосомную ДНК из трансформантов (Saito-Minwa, Biochem. Biophys. Acta 72 (1963) 619-632), переваривали ее EcoRI, фракционировали на 0,5% агарозных гелях, переносили на нитроцеллюлозу (Southern, J. Mol. Biol. 98 (1975) 503-517) и гибридизовали с 32P меченой никтраслянцией pGB33 (EP-A-0134048). Результаты этих анализов показаны на фиг.18. Эти данные показывают, что имеет место незаконная рекомбинация pElatB, в результате чего штамм содержит отдельный амилазный ген в другом месте генома по сравнению с исходным амилазным штаммом Bacillus licheniformis T5. Полученный штамм, содержащий pElatB, был обощначен TB13.
Пример 11
Конструирование штамма Tl3F, содержащего два амилазных гена, разделенных эндогенными хромосомными последовательностями
Для того, чтобы сконструировать штамм, содержащий два амилазных гена, разделенных занчительными для хозяйской клетки эндогенными хромосомными последовательностями, был проведен эксперимент по слиянию между штаммом Bacillus licheniformis T5 (амилазный штамм, содержащий исходный ген амилазы, EP-A-0134048) и штаммом TBl3 (штамм, содержащий случайно встроенный ген амилазы). Слияние протопластов проводили, как описано в EP-A-0134048, включенной здесь путем ссылки. Штамм TBl3 перед формированием протопластов был убит при помощи иода-цетамида. Штамм T5 (чувствительный к неомицину) был сохранен жизнеспособным. Отбор слившихся клеток проводили на регенерационных чашках, содержащих 10 мкг/мл неомицина.
Для проверки и идентификации потенциальных слившихся клеток выделяли хромосомную ДНК, переваривали ее EcoR1, фракционировали на 0,5% агарозных гелях, переносили на нитроцеллюлозные фильтры (Southern, J. Mol. Biol. 98 (1975) 503-517) и гибридизовали с 32P меченой ник-трансляцией pGB33 (EP-A-0134048). Результат этого анализа показан на фиг.19. Один из полученных клонов слившихся клеток (Tl3F) содержал два амилазных гена, разделенных эндогенными хромосомными последовательностями.
Пример 12
Стабильность дуплицированных амилазных генов в штаммах T390 и Tl3F. Стабильность штамма TL3F, содержащего два хромосомных амилазных гена, разделенных существенными хромосомными последовательностями, сравнивали со стабильностью штамма T 390 с двумя хромосомными амилазными генами, локализованными в тандемном порядке. Приготовление штамма T 390 раскрыто в EP-A-0134048 (табл.1), где это относится к B. Licheniformis T 5 (pGB 33). Штаммы T13 F и T 390 испытывали в условиях ферментации, 0,2 мл TSB культуры, выращенной в течение ночи при 37oC, инокулировали в 500 мл качалочные колбы, содержащие 100 мл производственной среды (см. пример 7, после стерилизации pH доводили до 6,9 при помощи NaOH) без неомицина. После инкубации в течение 6 дн при 40oC при постоянной аэрации культура была испытана на устойчивость к неомицину и амилазную активность. Результаты экспериментов по ферментации суммированы в табл.2.
Для исключения возможности вырезания одного амилазного гена без сопутствующей потери гена неомицина в штамме T13 F, были проанализированы 20 колоний, полученных после ферментации T13 F. Хромосомная ДНК из 20 случайно выбранных колоний была выделена и охарактеризована путем экспериментов по гибридизации, как описано выше. Результаты 9 таких анализов показаны на фиг. 20. Все проанализированные штаммы содержали два амилазных гена, что было доказано присутствием двух EcoR I фрагментов альфа-амилазных генов в составе хромосомных ДНК.
В противоположность генетической стабильности штамма T13 F было обнаружено, что штамм T 390 является нестабильным во время ферментации, что приводит к появлению 12% колоний, чувствительных к неомицину. Одну из этих колоний проанализировали и обнаружили содержание только одного альфа-амилазного гена (фиг.20, дорожка 4). Это показывает, что случайно встроенные амилазные гены более стабильны в условиях ферментации, чем тандемно встроенные гены.
Из приведенных выше результатов очевидно, что могут быть получены прокариотические клетки, в которых стабильная амплификация гена достигнута отбором трансформированных клеток, в которых нетандемно встроено по крайней мере две копии структурного гена. Встраивание может происходить путем гомологичной рекомбинации или незаконной рекомбинации.
Так как изобретение теперь полностью описано, то возможность многих изменений и модификации в нем без отхода от содержания и объема, заявленных приложенной формулой изобретения, будет, очевидно, лишь обычным делом техники.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СЕРИНОВОЙ ПРОТЕАЗЫ, ШТАММ ЩЕЛОЧЕФИЛЬНЫХ BACILLUS-ПРОДУЦЕНТ СЕРИНОВОЙ ПРОТЕАЗЫ | 1988 |
|
RU2023723C1 |
| ДНК, КОДИРУЮЩАЯ ФИТАЗУ ASPERGILLUS NIGER, РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК ДЛЯ ЭКСПРЕССИИ ФИТАЗЫ (ВАРИАНТЫ), ШТАММЫ-ПРОДУЦЕНТЫ ФИТАЗЫ (ВАРИАНТЫ), СПОСОБ ПОЛУЧЕНИЯ ФИТАЗЫ И РЕКОМБИНАНТНАЯ ФИТАЗА ASPERGILLUS NIGER | 1990 |
|
RU2113468C1 |
| СПОСОБ ПОЛУЧЕНИЯ МУТАНТНОЙ ПРОТЕАЗЫ | 1989 |
|
RU2069695C1 |
| СПОСОБ КАТАЛИЗА РЕАКЦИИ ФЕРМЕНТАЦИИ СУБСТРАТА, СПОСОБ УЛУЧШЕНИЯ ПОТРЕБЛЕНИЯ ПИЩЕВОГО РАЦИОНА ЖИВОТНЫМ, ПЛАЗМИДА pMOG413, ПЛАЗМИДА pMOG429 И ПЛАЗМИДА pMOG227 | 1991 |
|
RU2129609C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИАЛУРОНАНА В РЕКОМБИНАНТНОЙ КЛЕТКЕ-ХОЗЯИНЕ | 2002 |
|
RU2346049C2 |
| МУТАНТНАЯ ГЛЮКОЗОИЗОМЕРАЗА С ИЗМЕНЕННОЙ СУБСТРАТНОЙ СПЕЦИФИЧНОСТЬЮ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1991 |
|
RU2096457C1 |
| СПОСОБ ЭФФЕКТИВНОГО ПРОИЗВОДСТВА 7-АДЦК ЧЕРЕЗ 2-(КАРБОКСИЭТИЛТИО) АЦЕТИЛ-7-АДЦК И 3-(КАРБОКСИМЕТИЛТИО)ПРОПИОНИЛ-7-АДЦК | 1994 |
|
RU2139349C1 |
| СПОСОБ ЭФФЕКТИВНОГО ПРОИЗВОДСТВА 7-АДЦК ЧЕРЕЗ 3-(КАРБОКСИЭТИЛТИО)ПРОПИОНИЛ-7-АДЦК | 1994 |
|
RU2139350C1 |
| СПОСОБ ПОЛУЧЕНИЯ 7-АМИНОЦЕФАЛОСПОРАНОВОЙ КИСЛОТЫ, ВЕКТОР ЭКСПРЕССИИ, РЕКОМБИНАНТНЫЙ ШТАММ | 1992 |
|
RU2202616C2 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК, СПОСОБ ЭКСПРЕССИИ В КЛЕТКАХ STREPTOMYCES ГЕНА α -АМИЛАЗЫ | 1990 |
|
RU2124559C1 |






Использование: биотехнология. Сущность изобретения: штамм Bacillus - продуцент гидролитического фермента (в частности, Bac. novo sp. PB 92 - продуцент высокощелочной протеазы или Bac. licheniformis ____→ T5 - продуцент α- амилазы), в геном которого интегрирована последовательность ДНК, кодирующая целевой продукт, обрабатывают либо рекомбинантной плазмидой, включающей вторую копию названной последовательности ДНК, маркерный ген и температурочувствительную область начала репликации, либо геномной ДНК штамма-реципиента с интегрированной в хромосому последовательностью, кодирующей целевой фермент; выращивают полученные клоны на среде с антибиотиком и отбирают формы, содержащие две копии последовательности ДНК, кодирующие гидролитический фермент, разделенные последовательностью, жизненно важной для хозяйской клетки. 2 з.п. ф-лы, 20 ил., 2 табл.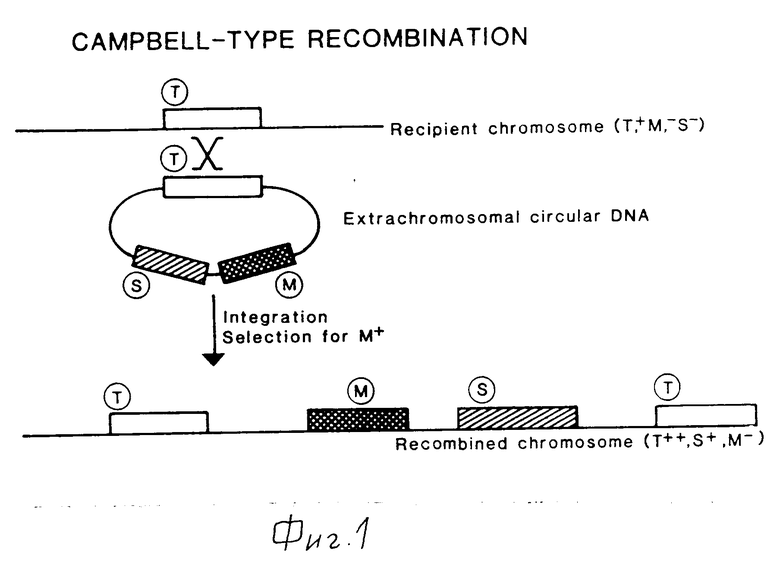
| Способ предупреждения беременности | 1959 |
|
SU124374A1 |
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
