Изобретение относится к области биотехнологии. Настоящее изобретение касается экспрессионной системы Streptomyces и способа экспрессии чужеродных последовательностей ДНК в Streptomyces, а также обеспечения секреции полипептидов и белков, кодируемых чужеродной последовательностью ДНК.
Виды Streptomyces являются хорошо известными продуцентами разнообразных внеклеточных энзимов, включая протеазы, фосфатазы, ксиланазы, целлюлозы, амилазы, липазы и нуклеазы.
В дополнение к этому, члены рода Streptomyces продуцируют большое количество антибиотиков, пигментов и других вторичных метаболитов и имеют свойства, требующие сложной методики дифференциации, приводящей к образованию спор. B экспериментальных сериях культур Streptomyces обычно присутствует совпадение в производстве внеклеточных энзимов, активизации продуцирования антибиотиков, биосинтезе пигмента и споруляции. Все эти процессы подавляются условиями питания, благоприятствующими высокой скорости роста и активируются ограничением источников фосфора, или углерода, или азота. Вероятно, что секреция энзимов, образование вторичных метаболитов и дифференциация полностью независимы, но отвечают на сходные спусковые механизмы.
Ранее клонировали некоторые гены Streptomyces, кодирующие внеклеточные энзимы. Эти энзимы включают агарозу от Streptomyces coelicolor, эндогликозидазу H от Streptomyces plicatus, ксиланазу от Streptomyces Lividans, альфа-амилазу от Streptomyces hydroscopicus, целлюлозу от штамма SpA2 Streptomyces, бета-галактозудазу от strep. Lividans и бета-лактамазы от strep. cocaoi badius и fradial.
Однако, регуляторные механизмы, которые управляют экспрессией этих генов, все еще остаются неизвестными. В дополнение к специфическим регуляторным механизмам, таким как индукция амилазы декстринами или мальтотриозой и углеродно-метаболитное регулирование амилазы или агаразы, вероятно, происходит снятие подавления образования нескольких внеклеточных энзимов, поскольку после снижения питания у streptomyces наблюдалось одновременное продуцирование нескольких энзимов, разлагающих полимерные субстраты. Такие трансактивирующие регуляторные гены были обнаружены в Bacillus subtilis (J. Bacterid 169: 324 - 333, 1987), Bacillus natto (J. Bacteriol. 166: 20 - 28, 1986) и Bacillus Licheniformis.
Позитивные регуляторные гены, воздействующие на синтез энзимо и/или секрецию, могли клонироваться при поиске повышенной секреции внеклеточных энзимов у слабо секретирующих штаммов, как S.Lividans.
Системы для экспрессии чужеродных последовательностей ДНК в streptomyces ранее были описаны, например, в EP 148.552 и WO-88/07079. Эти системы используют эндогенные промоторы внеклеточных энзимов, продуцируемых streptomyces.
Основным объектом изобретения является способ получения внеклеточных энзимов или желаемого полипептида путем культивирования трансформированного организма-хозяина согласно настоящему изобретению и выделения продукта из культурального бульона.
Еще одним аспектом изобретения является промотор saf-гена (т.е. гена, кодирующего фактор активирования вторичного метаболизма).
И еще один аспект изобретения заключается в получении клонирующих носителей (векторов), которые включают промотор saf-гена, оперативно связанного с сигнальной областью секреции эндогенного внеклеточного энзима, и оба они оперативно связаны с чужеродной последовательностью ДНК; этот аспект также касается организмов или клеток-хозяев, трансформированных такими клонирующими носителями, приводящими при этом к экспрессии и секреции чужеродного полипептида, кодированного чужеродной последовательностью ДНК.
Другим аспектом изобретения является интеграция такого клонирующего носителя, несущего чужеродную последовательность ДНК, в хромосомную ДНК streptomyces. В предпочтительном выполнении, вектор экспрессии, содержащий saf-ген также инкорпорируется в такие трансформируемые организмы.
Эти и другие объекты изобретения описаны более подробно в следующем ниже описании.
Краткое описание изобретения.
Фрагмент ДНК от штамма АТСС 10137 streptomyces qriseus, кодирующий ген saf (фактор активации вторичного метаболизма), который включен в обычный механизм управления по меньшей мере пятью энзимами, выделяемыми клетками вида S. Lividans и S. coelicolor. Этот saf-ген присутствует у всех испытывавшихся streptomyces, что позволяет предположить важную роль, которую ген играет в метаболизме. Действительно, фрагмент ДНК, содержащий ген, подобный гену saf, клонировался заявителем из streptomyces oriseus IMPV 3570, штамма, продуцирующего кандицидин. У некоторых streptomyces, например, S. Lividans и S. lactamdurans модель гибридизации предполагает, что saf-ген присутствует в хромосомной ДНК в нескольких копиях.
Ниже следует первый пример гена, вовлеченного в продуцирование внеклеточных энзимов и споруляции у streptomyces.
Ген saf кодирует полипептид из 113 аминокислот (SАF-полипептид); исследование транскрипции-трансляции ин витро у E. coli показало, что образуется белок правильного размера, около 15000 дальтон, SAF-полипептид имеет сильный положительный заряд (18 положительно заряженных аминокислот) и не демонстрирует состава, характерного для лидерного пептида или трансмембранного белка и поэтому представляет маловероятным непосредственное действие на экскрецию протеина. Положительно заряженный белок может легко взаимодействовать с ДНК. Действительно, в SAF-полипептида наблюдается ДНК-связывающая область, типичная для нескольких регуляторных протеинов (Ann. Rew. Biochem. 53: 293 - 321, 1984). Неожиданно было обнаружено, что saf-промотор более эффективен, чем природный промотор внеклеточных энзимов. Например, ген амилазы выдает больше амилазы, когда saf-промотор заменяет природный амилазный промотор. Таким образом, saf-промотор может быть использован для повышения экспрессии любого эндогенного полипептида или белка вместо белкового природного промотора.
SAF-полипептид обладает плейотропным действием, т. е. оказывает воздействие более чем в одном направлении на внеклеточные энзимы, на продуцирование пигмента и на дифференциацию. В соответствии с изобретением достигается повышенное производство эндогенных (внеклеточных) протеинов.
Более широко saf-ген и соответствующий полипептид могут быть использованы для увеличения экспрессии у streptomyces выбранных гетерологичных протеинов путем инсерции гена, кодирующего желаемый протеин в подходящий сайт расщепления гена, кодирующего внеклеточный энзим, экспрессия которого повышается SAF, или его части, трансформируя хозяина streptomyces, содержащего saf-ген с рекомбинированным геном, и культивируя трансформированные бактерии для их выделения выбранного протеина или его части. Предпочтительно, гетерологичная ДНК под контролем saf-промотора инсертируется (интегрируется) в хромосомную ДНК, совместно с ней инсертируется вектор экспрессии, содержащий ДНК, кодирующую SAF-полипептид. Вектор экспрессии не требуется интегрировать в хромосомную ДНК.
Сообщалось о плейотропных генах, воздействующих на биосинтез антибиотиков и дифференциацию у streptomyces. Ген afSB, который позитивно управляет продуцированием A-фактора, актинородина и продигиозина у streptomyces coelicolor и streptomyces lividans, клонировались из s. coelicolor A3-/2-/ (Horinouchi и др., J. Bacteriol. 155: 1238 - 1248, 1983).
Ген saf отличается от afSB и, действительно, они оказывают противоположное действие на производство активнородина. Общим у них является то, что их аминокислотные последовательности типичны для ДНК-связывающих протеинов. Оба гена saf и afSB имеют промоторы без -10 и -35 последовательностей согласования и связаны потенциальными трансляционными регуляторными сигналами (факторами), и сильным стволом, и петлевыми структурами, которые могут действовать в качестве транскрипционных терминаторов (Horinouchi и др., J. Bacteriol. , 168: 257 - 269, 1986). Если транскрипция образует очень стабильную петлю, то связывание рибосом сделается невозможным. Такие "ослабляющие" механизмы контроля генной экспрессии были найдены для управляемого контроля генов резистентности к антибиотикам у streptomyces (Horinouchi и др., Proc. Natl. Acad. Sci. США, 77: 7079 - 7083, 1989). Поскольку ген saf может рассматриваться как "главный" ген, который включает разнообразие генов для экскреции энзимов, его экспрессия, вероятно, строго контролируется в клетке. Все энзимы, стимулируемые saf-геном, участвуют в разложении сложных полимеров и поэтому мы полагаем, что этот ген является частью общей регуляторной системы, вовлеченной в поиск альтернативных источников питания, как предполагается также у Bacillus (Henner и др., J. Bacteriol 170: 296 - 300, 1988).
Фиг. 1 - Карты рестрикции фрагментов хромосомной ДНК от s. grisous АТСС 10137 клонов Bgl II сайте pIJ 702, которые несут генетический детерминант суперпродуцирования щелочной фосфатазы.
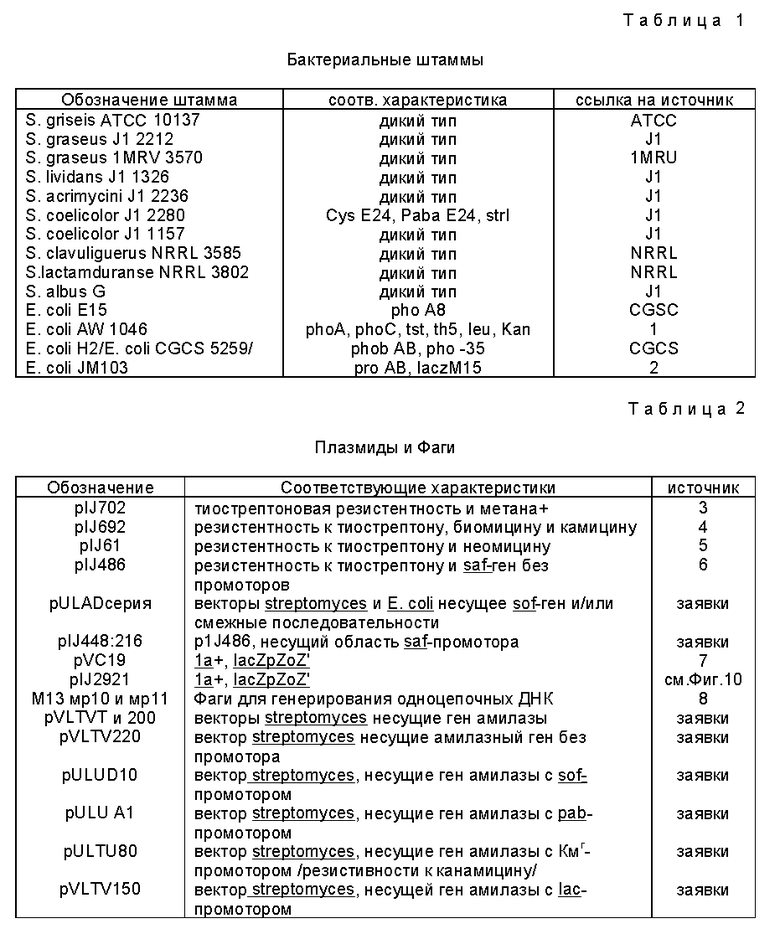
Фиг. 2 - Анализ (саузерн-гибридизацией) гомологии между 1 кб Bgl II фрагментом pIJ AD3 и геномной ДНК других видов streptomyces.
A/ фрагменты ДНК, полученные Ba mHI перевариванием геномных ДНК (полосы 2 - 7).
B/ гибридизация ДНК, показанных в панели A, с фрагментом величиной 1 кб в качестве образца /зонда/.
Полосы: 1. ДНК, переваренная Hind III с получением фрагментов размерами 23, 9.59, 6.68, 4.29, 2.28, 1.94 и 0.58 кб;
2. S. lactamdurans;
3. S. acrimycini;
4. S. coelicolor 1157;
5. S. qriseus 2212;
6. S. qriseus 3570;
7. S. Lividans 1326.
Фиг. 3 Опыты по испытанию протеазной /A/, амилазной /B/ и липазной /C/ активности S. Lividans, трансформированных pIJ 702 /слева/ и трансформированных p VL AD3 /справа/.
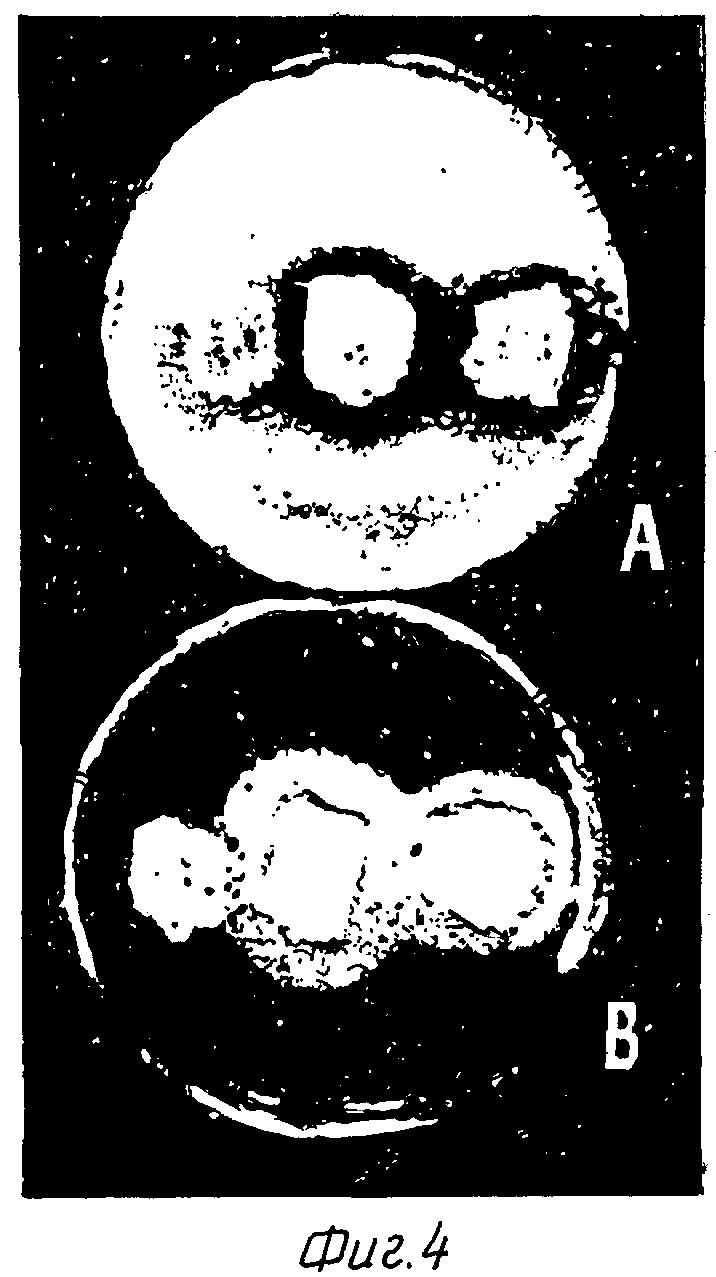
Фиг. 4 Эффект дозировки гена на экспрессию saf-гена. Продуцирование протеазы /A/ и амилазы /B/ S. Lividans, трансформированными pIJ 702 /слева/ S. Lividans, трансформированными p VLAD3 /в середине/ и S. Lividans, трансформированными p VLAD3 /справа/.
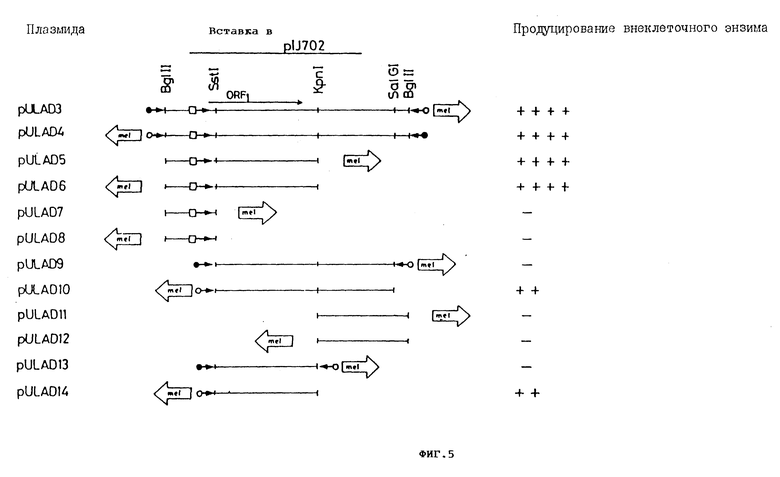
Фиг. 5 Обрезка /"тримминг"/ saf-гена. 1 кб Bgl II. Фрагмент p VLAD3 использовался в качестве исходного материалов. Продуцирование внеклеточного энзима для всех плазмидных конструкций измерялось на чашках с твердой средой, как указано в описании, означает продуцент дикого типа, два, три или четыре креста указывают различные степени суперпродуцирования. Короткие открытые стрелки показывают локализацию и направление транскрипции гена тирозиназы /mel/ в плазмиде pIJ 702. Закрытые кружки показывают промотор mel-гена; открытые /светлые/ кружки представляют активность предполагаемого по часовой стрелке промотора в pIJ 702, а открытые квадратики показывают saf-промотор. Направление транскрипции показано стрелками, ORFI соответствует рамке считывания saf, выведенной из данных по нуклеотидной последовательности (см. фиг. 6A).
Фиг. 6 Нуклеотидная последовательность 1 кб. Bgl II фрагмента p VLAD3 и фиг. 6A - выведенная аминокислотная последовательность продукта saf-гена. Подчеркнут второй кодон предполагаемой АТГ-инициации. Инвертированные комплементарные повторные последовательности показаны сходящимися стрелками. Волнистая линия показывает аминокислотную последовательность, подобную ДНК-связывающим областям известных ДНК-связывающих протеинов. Показаны подходящие сайты рестрикции. Нуклеотиды пронумерованы, начиная от сайта Bgl II /номера справа/.
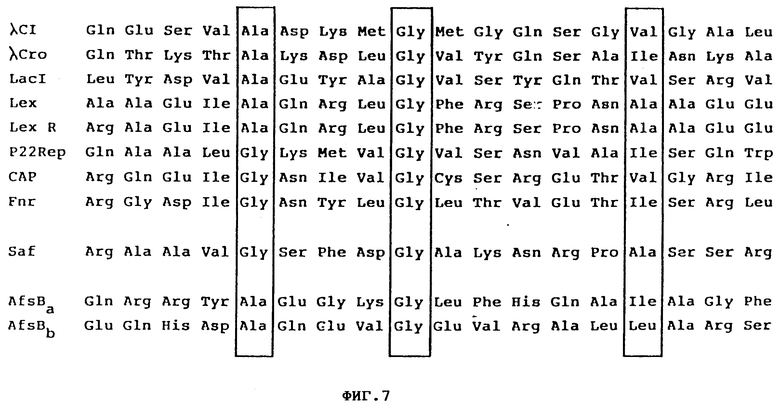
Фиг. 7 Сравнение области из выведенной аминокислотной последовательности saf-генного продукта с ДНК-связывающими областями в известных ДНК-связывающих протеинов. Данные взяты у Pabo и Sauer, Ann. Rev. Biochem. 53: 293 - 321, 1984 и Horinouchi и Beppu, Genetics of Industrial Microorganisms p. 41, изд. Плива, Загреб, Югославия, 1986.
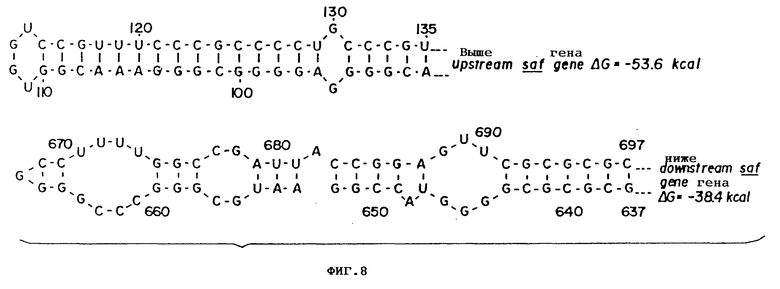
Фиг. 8 Повторные нуклеотидные последовательности "вверх" и "вниз" от saf-гена.
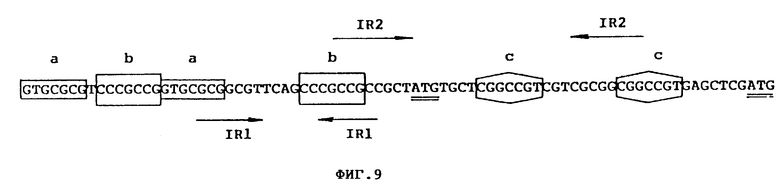
Фиг. 9 Повторные группы нуклеотидов, присутствующие в saf-гене.
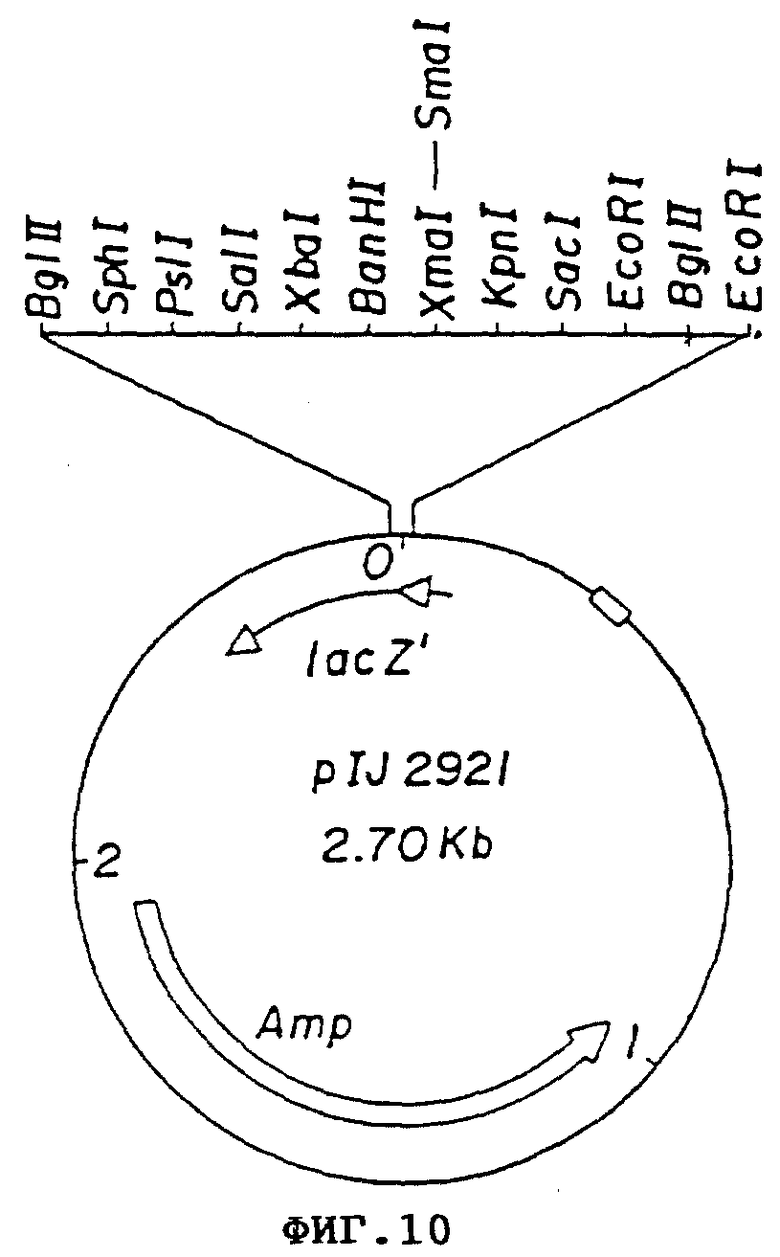
Фиг. 10 Плазмида pIJ 702.
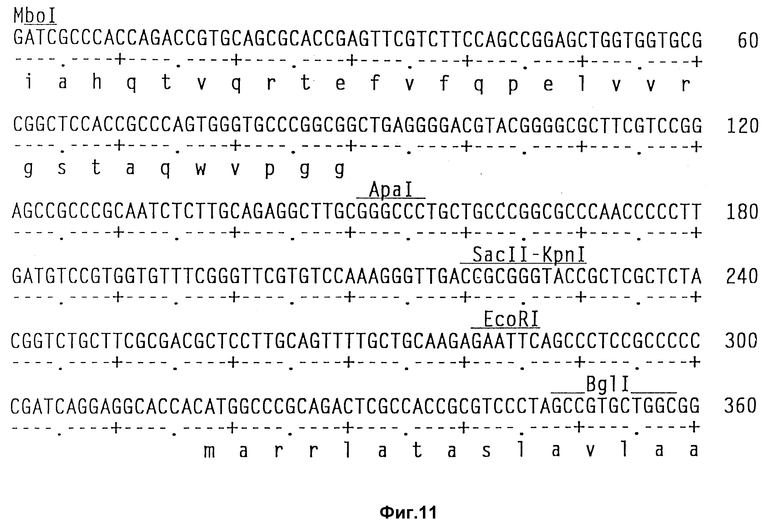
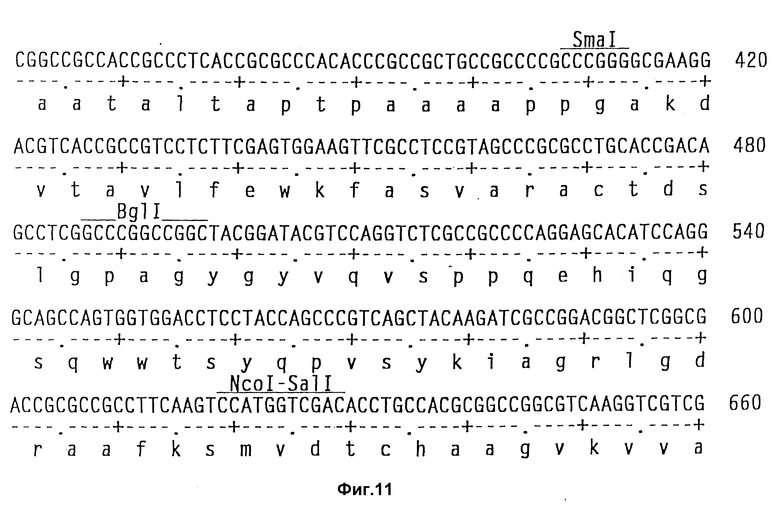
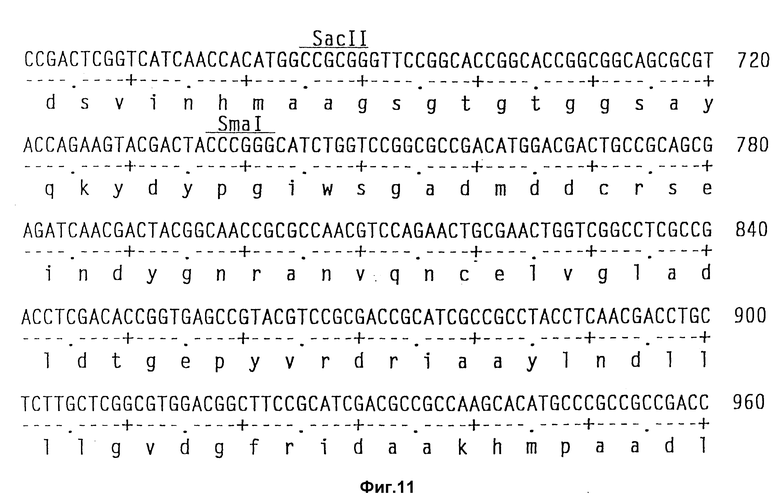
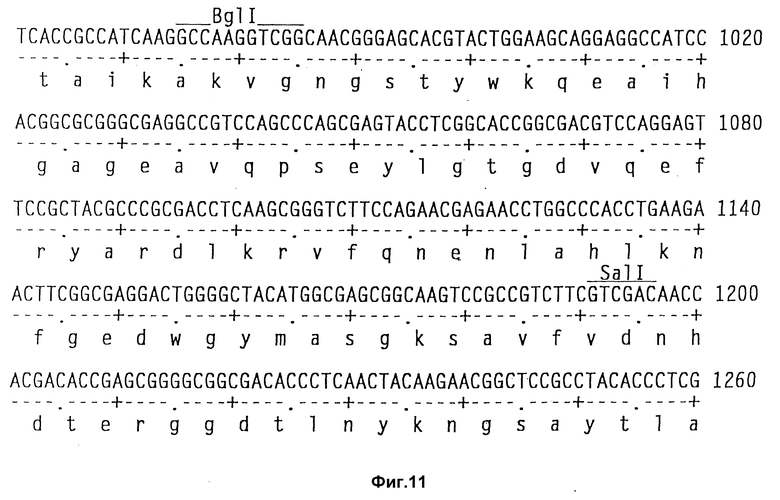
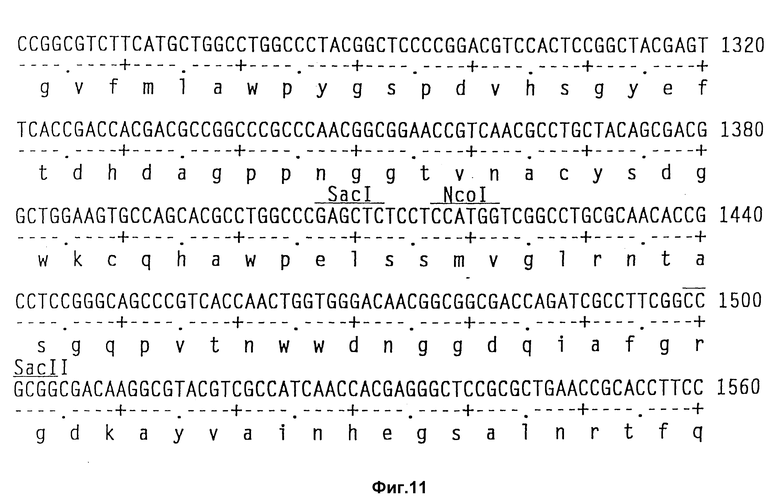
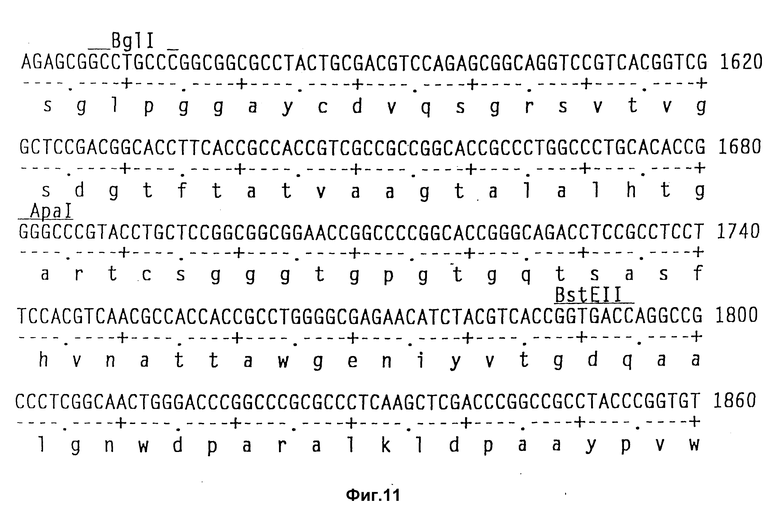
На фиг. 11 показаны полная последовательность нуклеотидов выведенной последовательности аминокислот амилазного гена S. griseus.
Полагают, что разрез Bst EII является оптимальным местом сшивания чужеродного гена.
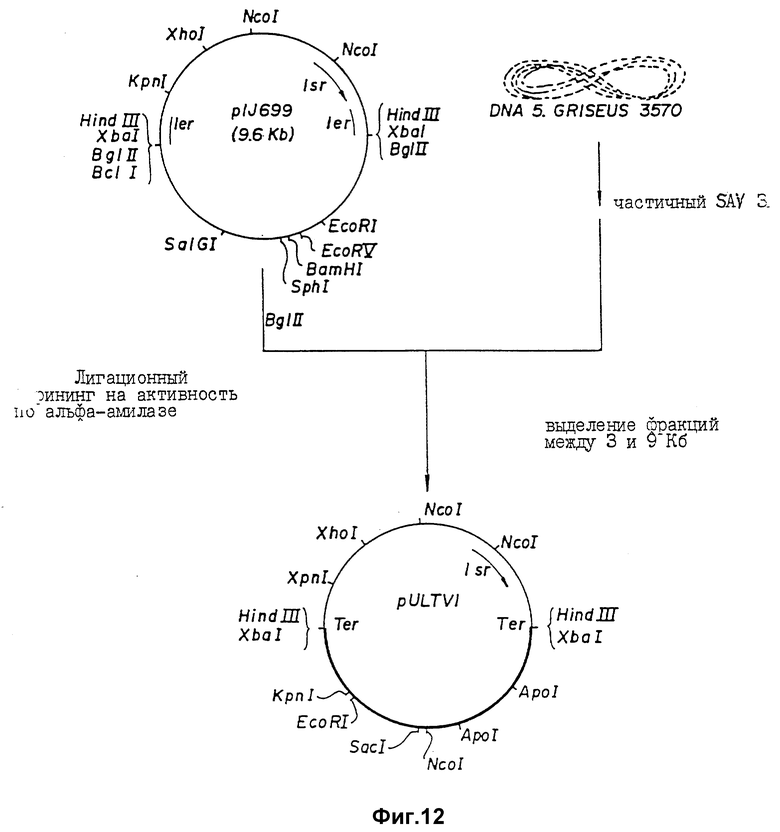
Фиг. 12 Стратегия, используемая при клонировании а-амилазного гена штамма IMRU 3570 S. griseus с применением плазмиды pIJ 699.
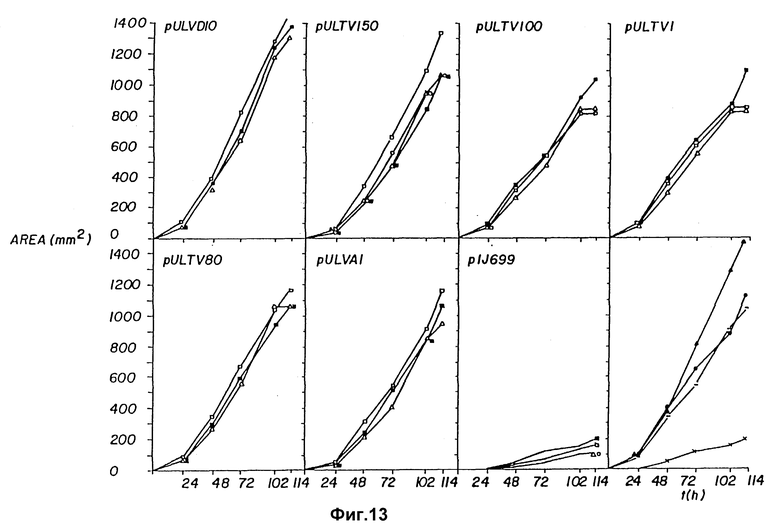
Фиг. 13 Серия графиков, показывающая продуцирование а-амилазы на чашках различными плазмидами в зависимости от времени, с различным составом сред:
(Δ) ММ + крахмал /1%/ + тио
(□) ММ 1 мМР + крахмал /1%/ + тио
(▪) ММ без фосфата + крахмал /1%/ + тио
(•) ММ + крахмал /1%/ + тио + IRTG
Нижняя панель с правой стороны показывает оценки продуцирования плазмиды PUL VD10 в сравнении с pULTVI, S. Lividans и p ULTV 100 в вышеописанных средах в наилучшей продуцентной среде (фиг. 6a) S. Lividans (pIJ 699) в ММ без фосфата + % крахмала + тио:/./ p ULTVI в вышеописанных средах; /-/ p ULTV 100 в вышеописанных средах; и  p ULVD 10 в ММ 1 мМР + 1% крахмала + тио.
p ULVD 10 в ММ 1 мМР + 1% крахмала + тио.
Фиг. 14. Схематически представляет конструкцию рекомбинантной клетки streptomyces, содержащей хромосомный ген, продуцирующий синтез-протеин, включающий молекулу CRF и плазмиды, содержащие ДНК: кодирующую SAF-полипептид.
Описанную здесь работу осуществляли с применением следующих материалов и методов.
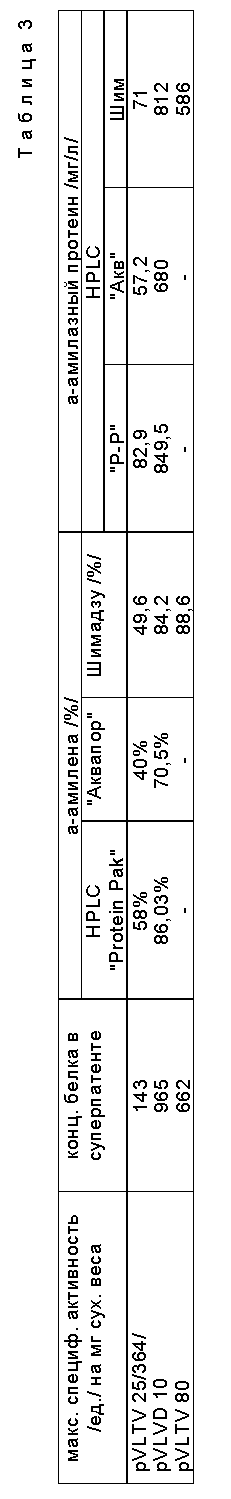
Штаммы бактерий и плазмиды. В этом исследовании использовались штаммы streptomyces и E. coli, которые приведены в табл.1. Плазмиды и фаги показаны в табл. 2.
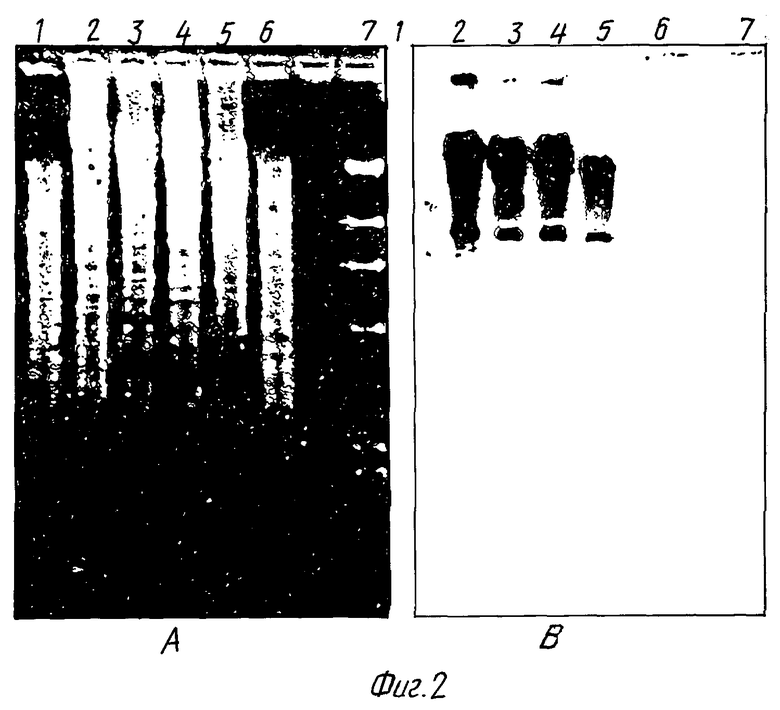
Среды и условия культивирования. Штаммы streptomyces выращивали в Р2УЕ, минимальный среде /ММ/, TSB /Дифко/ или YEME с добавлением 34% сахарозы и 5 мМ MgCl2 /Hopwood и др., Genetic Manipulation of streptomyces, Лабораторный учебник, изд. John Innes Foundation, Норвич, Великобритания, 1985/. Штаммы streptomyces выращивали в тройных перегороженных фляжках при 28oC в ротационном шейкере при его вращении со скоростью 220 об./мин.
Штаммы E. coli выращивали в бульоне Luria /LB/ (см. Миллер и др., EXPERIMENTS in Molecular genetics стр. 433, Cold Spring Harbor Lab. Нью-Йорк 1972) или агаре Luria /LA/ при 37oC.
Чашечные опыты с энзимами. Исследование streptomyces на щелочную фосфатазу проводили в среде РРММ /ММ/ без глюкозы, содержащей пониженный уровень /1 мМ/ фосфатазы с добавлением 30 мкг/мл 5-бром-4-хлор-3-индолилфосфат-р-толуидина /ХР/. Колонии, продуцировавшие щелочную фосфатазу, были синие, а неспособные ее произвести, были белые. Для E. coli использовали среду ВХР, которая содержит /на литр/: 10 г бактопектона, 12 г основания Трисма, 10 г соли, 20 г агара Дифко, с pH 7,5 и 40 мкг/мл ХР/.
Амилазная активность колоний streptomyces испытывалась на ММ /без глюкозы/ с добавлением 1% крахмала. После 3 дней роста чашки обрабатывали парами иода. Зоны просветлений вокруг колоний возникают вследствие разложения крахмала. Липазную активность измеряли путем выращивания streptomyces в ММ, с добавлением 2% /вес./об./ оливкового масла, 0,5 /вес/об./ Твин 80 и 0,5% /вес/об./ Твин 20. После 20 дней роста в чашки выливали по 1 мл 1М CaCl2.
Продуцирование липазы наблюдали в виде преципитата Ca2+-жирная кислота-комплекса /Odera и др., Ferment. Technol. 64: 363 - 371, 1986/.
Протеазуню активность испытывали в ММ /без глюкозы с добавлением 0,5% казеина и 10 мМ CaCl2. Энзимную активность определяли как зоны осветления вокруг колоний.
Активность на агарозу S. coelicolоr испытывали в ММ без глюкозы с заполнением чашек грамм-иодным раствором. Бета-галактизидазную наблюдали в виде синего окрашивания колоний, растущих на ММ без глюкозы с добавлением X-gal /36 мкг/мл/ и IRTG /10 мкг/мл/.
Выделение ДНК. Общая /вся/ ДНК из streptomyces приготавливалась как описано Hopwood и др. , Лабораторный учебник (см. выше). Плазмидную ДНК из streptomyces или E. coli выделяли по методу Кизера /Kieser и др., Plasmid 12: 19 - 36, 1984/.
Процедуры клонирования. 10 мкг хромосомной ДНК штамма АТСС 10137 S. griseus и 0,5 мкг pIJ 702 полностью переваривали Bgl II и лигировали в течение 12 часов при 14oC с использованием T4 ДНК-лигазы. Лигационную смесь использовали непосредственно для трансформирования. Субклонирование фрагментов ДНК проводили путем переваривания 1-2 мкг ДНК плазмид с адекватным рестрикционным энзимом /энзимами/, а продукты реакции разделяли гелевым электрофорезом в агарозе с низкой температурой плавления /LMPA/. Требуемые нити ДНК экстрагировали с использованием методов с СТАВ /Langridge и др., Anal. Biochem. 103: 264 - 272, 1980/.
Методы трансформирования. Штаммы streptomyces трансформировали как описано Хопвудом и др. в Лабораторном учебнике (см. выше). После трансформирования протопласты помещали на среду R2YE и оставляли генерировать в течение 15 - 20 часов при 30oC. Затем 1 мл водного раствора тиострептона /300 мкг/ наливали в чашки, сушили 1 или 2 часа и инкубировали в течение 2 или 3 дней. Трансформанты реплицировали на среду РРММ, содержащую тиострептон /50 мкг/мл/ и ХР /30 мкг/мл/.
Трансформирование E. coli осуществляли согласно Cohen и др. /Proc. NAtl. Acad. Sci США. 69: 2110 - 2114, 1972/.
Трансформанты подвергали селекции на чашках, содержащих ампициллин /100 мкг/мл/. В случае необходимости в LA чашки добавляли X-gal /36 мкг/мл/ и IRTG /10 мкг/мл/.
Исследование гибридизации. Перенос ДНК с агарозного геля на нитроцеллюлозные фильтры и гибридизацию проводили как описано Хопвудом и др. в Лабораторном Учебнике (см. выше). 7,2 кб. Bgl II фрагмент плазмиды p ULADI и 1 кб. фрагмент Bgl II из плазмиды p ULAD3 использовали в качестве зондов. Гибридизации осуществляли при 70oC в течение 24 часов. Фильтры промывали дважды по 30 мин в 2 г SSC, 0,1% SDS и затем еще два раза снова на 30 мин в 0,2 г SSC и 0,1% SDS при 70oC.
Анализ нуклеотидной последовательности. Нуклеотидную последовательность определяли методом терминации цепи по Зангеру /Sanger и др., Prog. NAtl. Acad. Sci. США 74: 5463 - 5467, 1977/. Фрагменты ДНК субклонировали в М13мр10 и М13мр11 для получения инсерта в любой ориентации. Лигационные смеси трансфецировали в компетентные клетки штамма J М103 E. coli и белые /гемолитические/ бляшки скринировали для селекции инсертов. В обеих нитях осуществляли секвенирование с использованием Amersham International pIc/. Великобритания/ и "Секвеназы" /Биохимическая корпорация Соединенных Штатов/ - наборы для анализа. Все фрагменты секвенировали с использованием dGTP, но DITP при необходимости также использовали вместо dGTP. Реакционные смеси разделяли на 6%-ном или 8%-ном полиакриламидных секвенирующих гелях, которые затем переносили на рентгеновскую пленку авторадиографии.
Клонирование промоторов. Фрагменты с активностью по инициации транскрипции отбирали с использованием плазмиды мультикопирования промотора-зонда pIJ 486 -/Ward и др. Mol. Gen. Genet. 203: 468 - 478, 1986/. Трансформанты с резистентностью к канамицину /Км/ выделяли путем реплицирования колоний на ММ, содержащей 15 мкг/мл Км.
Транскрипция-трансляция ин витро. Плазмиды p ULAD 300 и p UC19 транскрибировали и транслировали с использованием набора лабораторных средств для направленной трансляции прокариотической ДНК /из Amersham Int. PIc./ L /35S/-метионин использовали в качестве радиоактивной метки. Для анализа меченных протеинов использовали 12,5% полиакриламидные гели, содержащие додецил-сульфат натрия.
Продуцирование щелочной фосфатазы различными streptomyces.
Испытывалось в нескольких твердых средах продуцирование щелочной фосфатазы десяти разных штаммов streptomyces /список см. в табл. 1/. S. griseus IMRV3570 и S. griseus АТСС 10137 оказались лучшими продуцентами во всех испытывавшихся средах. Лучшей твердой средой для продуцирования щелочной фосфатазы была PRММ /содержащая 30 мкг/мл ХР/. В этой среде S. griseus IMRU 3570 и S. griseus АТСС 10137 демонстрируют темно-синее окрашивание после 48 часов роста S. Lividans 1326 и S. Coelicolor J1 2280 были плохими продуцентами щелочной фосфатазы: через 20 часов роста на PRMM, содержащей ХР, наблюдалось только светло-голубое окрашивание.
Клонирование гена, вовлеченного в продуцирование щелочной фосфатазы.
Общая ДНК от штамма АТСС 10137 S. griseus переваривалась Bgl II, лигировалась к переваренному Bgl II pIJ 702 и лигационную смесь вводили путем трансформации в протопласты S. Lividans 1326. Трансформанты реплицировали на PRMM, содержащую тиострептон /50 мкг/мл и ХР /30 мкг/мл/. Очень темно-синяя колония была обнаружена среди 2.800 меланин-негативных трансформантов S. Lividans. Эта темно-синяя колония содержала производное pIJ 702, несущее 7,2 кб. Bgl II инсерт: названный p ULAD (см фиг. 1).
Плазмида p ULADI - S. Lividans была нестабильной и при трансформировании вызывала белые и синие колонии. Все синие колонии, содержавшие исходный p ULADI, и белые, содержащие дефектную форму /с делецией/ плазмиды p ULADI, далее не исследовали. Поскольку нестабильность могла быть вызвана большим размером инсерта в такой плазмиде как pIJ 702, которая известна своей некоторой нестабильностью, то 7,2 кб инсерт субклонировали в 5 кб Bgl II фрагмент плазмиды pIJ 699 /Kieser и др., Gene 65: 83 - 91, 1988/. Были получены две плазмиды - p ULAD 100 и p ULAD 101 с инсертом, ориентированным в противоположных направлениях. Обе плазмиды были стабильны и ген экспрессировался в обоих ориентациях в S. Lividans.
Локализация SAF-гена.
7,2 кб. Bgl II фрагмент частично переваривали с помощью a 3A1 и лигировали к Bgl II-переваренной pIJ 702. Лигационную смесь трансформировали в протопласты S. Lividans, и трансформанты реплицировали на PRMM, содержащую ХР. Плазмидную ДНК выделяли из нескольких синих колоний. Были изучены подробно четыре небольшие плазмиды: p VLAD2, p VLAD3, p VLAD 16 и p VLAD 18, несущие детерминант продуцирования щелочной фосфатазы; эти плазмиды были стабильны, будучи ретрансформированными в прототпласты S. Lividans и несли инсерты размером 2.4, 1, 2.1 и 1.9 кб /фиг. 1/ соответственно. Плазмида p VLAD3 имела самый маленький инсерт размером 1 кб., который мог быть вырезан обратно с помощью Bgl II. Будучи введенным в S. Lividans, pVLAD3 повышал продуцирование щелочной фосфатазы, как pVLAD1. pVLAD3 депонирован с депозитарным номером 1-859 19.05.89 в Национальной коллекции Культур Микроорганизмов, 28-я ул. Доктора Ру, 175724 Paris, Cedex 15, France, согласно Будапештскому Договору и Правилу 28 EPC.
Гибридизация эромосомной ДНК нескольких streptomyces.
Общую ДНК из штамма 10137 S. Griseus; переваренные BamH1 или Bgl II, гибридизировали к 7,2 кб bGl II фрагменту pVLAD1, меченному "ник-трансляцией с /32 р/d CTP. Гомологичный этому зонду 7,2 кб Bgl II фрагмент присутствовал в ДНК штамма АТСС 10137 S. Griseus, переваренный Bgl II, как ожидалось. Там имелся /в этом фрагменте/ внутренний сайт BamEH1, который приводил к двум четким полосам гибридизации /размером 7,9 кб и 9,4 кб, когда общую ДНК переваривали BamH1.
Для выяснения, тот ли самый ген присутствовал в других streptomyces, общую ДНК от S. acrimycini, s. coelicolor 1157, s. griseus 2212, s. griseus 3570, s. Lividans 1326 и s. lactamdurans NRRL 3802 переваривали BamH1 и гибридизировали с 1 кб Bagl II фрагментом зонда. Гибридизацию с 9,5 кб с обычной полосой наблюдали в первых четырех выше, тогда как у ДНК S. Lividans и S. lactamdurans наблюдали 4 и 5 слабых полос гибридизации соответственно /фиг. 2/. В дополнение, также наблюдали гибридизацию с ДНК S. albus G, S. coelicolor J1 2280, S. clavuligerus NRRL 3585 и S. fradiae АТСС 10475. Дальнейших попыток характеризовать полосы гибридизации не делалось.
Некомплементарность рпо-мутантов E. col.
Была сделана попытка установить комплементарность E. coli phoA-мутантов E15 и A 1046, когда мы клонировали структурный ген щелочной фосфатазы от штамма АТСС 10137 S. griseus 7,2 кб Bgl II фрагмент от pVLAD1 и 1 кб Bgl II фрагмент от p VLAD3 субклонировали раздельно в обеих ориентациях в Bam H1-переваренной pVC 19. Все эти плазмидные конструкции проверялись в клетках E. coli M 103 и затем использовались для трансформирования E. coli E15 /Sartly и др., J. Bacterial, 145: 288 - 292, 1981/ и E. coli A 1046, в В=ХР не было обнаружено никаких синих колоний, позволяющих предположить, что полный структурный ген щелочной фосфатазы не присутствует в 7,2 кб фрагменте S. griseus, или что этот ген не экспрессируется в E. coli. Структурный ген щелочной фосфатазы pho или Bacillus Lichenifotmisi Hullet J. Bacteriol, 158: 978 - 982, 1984/ не гибридизировался с клонированным 1 кб фрагментом /данные не приведены/. В дополнение, phoB /регуляторный/ E. coli мутант H2 /Kreuzer и др. , Genetics 81: 459 - 468, 1975/ не был комплементирован ни с 1 кб Bgl II фрагментом pVLAD3, ни 7,2 Bgl II фрагментом pVLAD1.
Суперпродуцирование других внеклеточных энзимов.
Когда pVLAD3 использовался для ретрансформации протопластов S. Lividans, то наблюдалась ясная отложенная /т.е. с задержкой/ пигментация и споруляция. Эти плейотропные эффекты стимулировали нас к изучению роли этого гена в секреции белка или в контрогенной экспрессии. Как показано на фиг. 3, некоторые внеклеточные энзимы, включающие амилазу, протеазу и липазу, суперпродуцировались S. Lividans, трансформированным с помощью pVLAD3. Продуцирование бета-галактозидазы также увеличивалось, начавшись примерно на 24 - 30 часов ранее, чем у нетрансформированного S. coelicolor. Благодаря плейотропным эффектам клонированного гена на внеклеточные энзимы, продуцирование пигмента и на дифференцирование, этот ген был назван saf/ т.е. фактор активизации вторичного метаболизма/.
Эффект дозы гена.
Количество копий pIJ 702 оценивают порядка 100 - 200 на хромосому. Хотя точное количество копий pVLAD3 не было определено: интенсивность обеих полос pIJ 702 и pVLAD3 не предполагалось наличие сходного количества копий.
С целью изучения эффекта дозировки гена мы субклонировали 1 кб Bgl II фрагмент pVLAD3 в сайт BamH1 плазмиды pIJ 61 с малым количеством копий /от 3 до 4 копий на клетку/ /см. Хопвуд и др., Лабораторный Учебник/. Эта новая плазмида была названа pVLAD30. На фиг. 4 показано, что продуцирование внеклеточных энзимов S. Lividans трансформированными pVLAD30 отчетливо понижается по сравнению с S. Lividans несущими pVLAD3. В дополнение, S. Lividans, несущие pVLAD30, продемонстрировали сходное пигментное продуцирование и модель споруляции как интрансформированные S. Lividans.
Экспрессия в E. coli и тримминг гена.
Хотя pho мутанты E. coli не комплементировались saf-геном, мы наблюдали, что 1 кб Bal II инсерт pVLAD3, будучи субклонирован в одном направлении в pV C19 /плазмида, названная pVLAD300/, экспрессировал в E. coli, вызывая ненормальную морфологию при росте на твердой среде, но не экспрессировал при инсерции его в обратном направлении /плазмида pVLAD302/. Поазмида pVLAD300 содержит ORF /см. ниже/ вниз от 1acZ промотора. Эти результаты предполагают, что saf-ген экспрессирует в E. coli от 1acZ промотора, присутствующего в pV C19.
Исследования транскрипции-трансляции в E. coli /с плазмидой pVLAD300/ проводились также ин витро, и продукты реакции загружали в 12,5%-ный полиакриламидный гель. Полоса мол. весом в 15000 присутствовала на дорожке, соответствующей pVLAD300, и отсутствовала в контрольной дорожке pV C19.
Для точной локации saf-гена было решено продолжать расщепление 1 кб фрагмента плазмиды pVLAD300. В этих попытках использовалось существование уникальных сайтов разреза для энзимов рестрикции SstI/nt216/Hp h/ht 648/ и Sal GI /nt 936/ внутри 1 кб фрагмента. Различные суб-фрагменты из 1 кб инсерта pVLAD300 клонировали на первой стадии в pIJ 2921, производное V C18, содержащее модифицированный полилинкер, фланкировали Bgl II сайтами /фиг. 10/ и затем высвобождали с помощью Bgl II липкие концы и клонировали в Bgl II-переваренной pIJ 702. Продуцирование щелочной фосфатазы, амилазы и протеазы изучали у stertomyces Lividans со всеми плазмидными конструкциями. Результаты этих исследований показаны схематично на фиг. 5, и из интерпретации этих результатов можно вывести следующие заключения:
1. 1 кб фрагмент содержит полный saf-ген, включающий его собственный промотор, поскольку он был экспрессирован на сходном уровне в обеих ориентациях плазмиды pIJ 702 /плазмиды pVLAD3 и pVLAD4/.
2. Этот saf-ген, вероятно, расположен в 648-нуклеотидном Bgl II-Крп1 фрагменте /плазмиды pVLAD5 и pVLAD6/.
3. Фрагмент Sst1-Крn1 /432 нуклеотида/, вероятно, содержит генетический детерминант для суперпродуцирования внеклеточного энзима, но, кажется, теряет свою область промотора, поскольку он экспрессировался только в одной ориентации /плазмида pVLAD14 и pVLAD10/, но не в противоположной /плазмиды pVLAD9 и pVLAD13/.
4. 215-нуклеотидный фрагмент Bgl II-SstI содержит область промотора saf-гена.
5. Все эффекты на внеклеточные энзимы и на дифференциацию сохранялись и терялись вместе, указывая этим, что за эти действия на все энзимы ответственен единственный генетический продукт.
6. Неожиданным оказалось отсутствие экспрессии фрагмента SstI-Kpn1 /saf-ген без промотора/, происходившего от промотора тирозиназы ме1-гена, присутствующего в pIJ 702, который тем не менее экспрессировался в противоположном направлении/ по часовой стрелке/. Это открытие подразумевает, что имеется фрагмент с промоторной активностью, расположенный перед ме1-геном, у Bgl II клонирующего сайта /Gi и Hopwood, GENE, 25: 119 - 132, 1983/. Возможность функционального ORF в обратном направлении /из KpnI-SstI/ была отброшена по нескольким причинам;
A. Такой ORF не был обнаружен при анализе нуклеотидной последовательности.
B. Если бы такой ORF существовал, то ясно, что он нес бы свой собственный промотор, поскольку фрагмент Bgl II-Kpn1 экспрессировался в обоих направлениях /плазмиды pVLAD5 и pVLAD6/. Кроме того, выглядит бессмысленным, чтобы Kpn1-фрагмент, который бы нес промотор в зоне Kpn1, мог экспрессироваться только в одном направлении.
C. Экспрессия в E. coli всегда происходит так, что промотор 1acZ - перед предполагаемым ORF и никогда - в обратном направлении.
Промоторная активность ORF1 фрагмента верхнего участка ДНК.
Чтобы установить существование промотора в Bgl II-SstI фрагменте, из предыдущих экспериментов можно сделать вывод, что этот фрагмент несет ген, который кодирует аминогликозидфосфотрансферазу /neo/ без его промотора. Экспрессия этого гена делает S. Lividans резистентным к канамидину и неомицину. Когда этот фрагмент был субклонирован в pIJ 486 /с SstI концом: ближайшим к neo-гену/, то была создана плазмида pIJ 484 : 216. Наблюдали, что S. Lividans, трансформированная pIJ 486 : 216, растет в ММ с содержанием канамицина более 100 мкг/мл, тогда как S. Lividans, трансформированный pIJ 486, не рос на ММ с добавлением канамицина в количестве 5 мг/мл. Эти результаты показывают, что такой фрагмент имел промоторную активность и поддерживает предполагаемый ORF.
Другие рассуждения, которые выведены из нуклеотидной последовательности вероятного ORF, включают:
1. Ген saf окружен двумя областями с инвертированными и комплементанными повторными последовательностями, которые могут образовывать в мРНК очень стабильные связи G = - 38,4 Ккал в присутствии гена saf /нуклеотид nt 637-697/ /фиг. 8/.
2. Секвенированный фрагмент содержит высокий G + C процент, что естественно у генов streptomyces.
3. Перед геном saf, между ATG / nt 219/ и повторными структурами вверху имеется очень интересная нуклеотидная область: в ней есть три пары повторных нуклеотид: каждый с 7 нт /см. фиг. 9/. Весьма похоже, что эта область, особенно промоторная зона, играет очень важную роль в регулировании экспрессии гена saf.
4. Следует упомянуть о том, что SAF-протеин содержит 18 суммарных положительных зарядов. Исходя из теоретических положений, это предполагает существование значительно большей аффинности к ДНК. Даже в выведенной аминокислотной последовательности можно было наблюдать очень похожую область к областям ДНК-связывающим протеинов /Pobo и Sauer, 1984/ /фиг. 7/.
Нуклеотидная последовательность saf-гена.
Нуклеотидная последовательность всего 1 кб Bgl II инсерта pVLAD2 была определена с использованием плазмиды pVLAD300 в качестве исходного материала. Поскольку М13мР10 и М13мр11 не имеют Kpn1 сайта, фрагменты с Kpn1-концами сначала субклонировались в pV C19 и затем высвобождались в виде EcoR1-Hind 111 фрагментов и вводились в мр10 и мр11, переваренных EcoR1 и Hind 111. Полная нуклеотидная последовательность активной области показывает ORF1 из 229 нуклеотидов, которые кодируют предполагаемый SAF-полипептид /фиг. 6/. Имеется только одна возможная открытая рамка считывания, содержащая Bgl II-Kpn1 фрагмент, начиная от ATG-кодона инициации у нт 183 и заканчиваясь TGA стопкодоном у нт 524 /ORF1 на фиг. 5 и 6, 6A/. Поскольку SstI-Kpn1 инсерт, содержащийся в плазмиде pVLAD14, показал более низкую степень активности, чем плазмиды, также содержавшие верхнюю область SstI сайта /pVLAD3, pVLAD4, pVLAD5 и pVLAD6/, очень вероятно, что ATG /нт 219/ также в рамке с предположительной рамкой считывания saf может действовать как кодон инициации в pVLAD10 и pVLAD14. Никакие другие альтернативные триплеты инициации невозможны, поскольку TGA кодон терминации существует выше первого ATG. Длинная инвертированная повторная область, которая может образовывать очень стабильный стебель /ствол/ и петлевую структуру /G = 53,6 Ккал/ присутствует выше от saf-гена, от нт 90 до нт 135. Другая, с дефисом, инвертированная повторная последовательность наблюдалась вниз по ходу от терминантного триплета ORF1, простираясь от нт 637 до 897 /G = -38,4 Ккал/.
Ген saf имеет высокое G + C содержание /78,3%/ и маркированные гены /Hopwood и др., Regulation of Gene Expression 25 Jearson, издательство Кембриджского Университета, стр. 251 - 276, 1986/.
Промоторная активность фрагмента ДНК вверх от ORF1.
Поскольку плазмида, не имеющая фрагмента Bgl II-SstI /от нт 1 до нт 216/, экспрессировалась только в одной ориентации /плазмиды pVLAD9 и pVLAD13/, кажется вероятным, что ORF1 промотор был расположен в этом фрагменте. Присутствие последовательности инициации транскрипции в этой области было подкреплено путем субклонирования этого фрагмента в промотор-зондовую плазмиду pIJ 486 /SstI и др., Mol. Gen. Genet 203: 468 - 478, 1986/, которая несла беспромоторный ген аминокликозид-фосфотрансферазы /neo/. Экспрессия этого гена придает S. lividans устойчивость к канамицину и неомицину. Bgl II-SstI фрагмент субклонировали в pIJ 486 /SstI конец проксимально к neo-гену/ с получением плазмиды, названной pIJ 486 : 216. S. Lividans, трансформированный pIJ 486 : 216 мог расти на ММ, содержащей более 100 мкг/мл канамицина, тогда как S. Lividans, несущий pIJ 486, не растет на ММ с содержанием 5 мкг/мл канамицина. Этот результат показывает, что фрагмент Bgl II-SstI обладает промоторной активностью. Однако нуклеотидной гомологией с другими промоторами streptomyces не было выявлено типичного "консенсуса" областей - 10 или - 35. /Hopwood и др., Regulation of Gene Expression 25 Jearson, Издательство Кембриджского Университета, стр. 251 - 276, 1986/.
Клонирование гена амилазы.
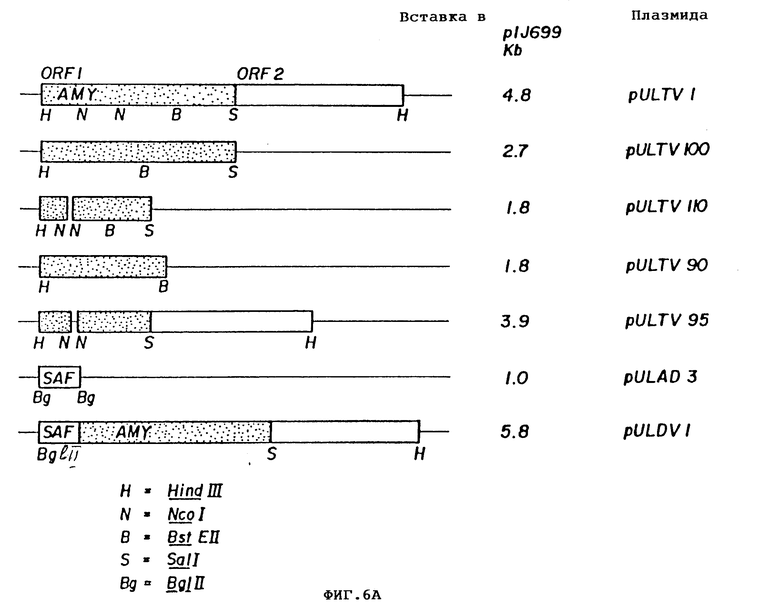
Теперь была объяснена дефинитивная последовательность амилазного гена /аму/ S. Griseus 1MPV 3570. Полная последовательность нуклеотидов и выведенная последовательность аминокислот амилазного гена S. griseus показана на фиг. 11. Протеин с лидерным пептидом имеет 566 аминокислот /от нт 318 до нт 2155, оба включительно/, что соответствует РМ 59, 713Д. Плазмида pVLTV1, содержащая весь а-амилазный ген, была сконструирована путем переваривания ДНК S. griseus 1MPV 3570 с выделением фракций размером от 3 до 9 кб и лигированием к Bgl II-переваренной pIJ 699. После скрининга на а-амилазную активность pVLTV1 плазмиду подвергали селекции на содержание всего а-амилазного гена. Эта конструкция плазмиды pVLTV дана на фиг. 12. Другим путем получения плазмиды с целым геном а-амилаз является конструктирование ее из двух фрагментов по меньшей мере. Среди плазмид, полученных клонированием всей библиотеки ДНК S. griseus, имеются также плазмиды, которые содержат частичный ген, начиная с 5'-конца и такие, у которых частичный ген начинается с 3'-конца. Из названного первым можно получить 1,3 кб фрагмент BamH1-SacI, который включает 5'-конец гена, и из последнего можно выделить 1,2 кб SacI-SalI фрагмент, включающий 3'-конец этого гена. Оба фрагмента затем могут быть соединены с pIJ 2921, обработанной SalI-BamH1, с получением плазмиды с интактным нативным геном а-амилазы /pVLTV200/.
Повышенная эффективность saf-промотора.
С целью испытать экспрессию гена аму под контролем разных промоторов изучали продуцирование амилазы штаммов 1166 S. Lividans. Этот аму-ген помещали под контроль разных промоторов и продуцирование амилазы сравнивали с ее продуцированием с использованием нативного промотора /Раму/. Испытанные промоторы были от гена saf /Psaf/, от гена резистентности к канамицину /Pneo/, от генов синтетазы PABA /Ppab/ и от гена бета-галактозидазы E. coli /PIae/. Плазмида pVLTV220, содержащая ген амилазы без промотора, была получена лигированием фрагмента EcoR1-Bgl II, выделенного из pVLTV200, к плазмиде p C18, переваренной EcoR1-BamH1, EcoR1-Hind 111, фрагмент из pVLTV220 был лигирован непосредственно к промоторам saf, pab или Км /резистентности к канамицину/ генов с получением pVLTV10, pVLTV A1 и pVLTV80 плазмид соответственно, pVLTV150, содержащие промотор 1ac, были получены непосредственно путем Hind 111 переваривания pVLTV220 и лигирования к 5 кб Hind 111 фрагменту pIJ 699.
Исследование продуцирования полученных плазмид проводилось на чашках с ММ + крахмал /1%/, + тиострептон, ММ /1 мМ Р/ + крахмал /1%/ + тиострептон, ММ без фосфата + крахмал + тиострептон и ММ + крахмал /1%/ + тиострептон + 1PTG. В качестве контроля использовали плазмиды pIJ 699, pVLTV100 /фиг. 6a/ и pVLTV1. Относительное измерение продуцирования амилазы осуществляли оценкой площади кольца вокруг каждой колонии при окрашивании ростовой среды йодом /12/.
Графики продуцирования различными конструкциями представлены на фиг. 13. За исключением контроля, наиболее высокое продуцирование было достигнуто в среде ММ /1 мМ Р/ + крахмал + тиострептон. Ясно видно, пик продуцирования был получен pVLTV10 /которая имеет промотор Saf-гена/ - 1482 кв. мм по сравнению с 1335 кв. мм другого наивысшего продуцента pVLTV150, который несет Plac-промотор, при инкубировании в течение 114 часов.
На фиг. 13 /нижняя панель, справа/ оценки продуцирования pVLVD10 представлены в сравнении с контролем в наилучших известных условиях для каждого типа конструкции. Увеличение продуцирования pVLVD10 по сравнению с нативным pVLTV1 является довольно значительным: 1482 кв. мм по сравнению 1105 кв. мм.
Таким образом, промотор saf-гена может быть использован в качестве заменителя других нативных промоторов для улучшения экспрессии различных эндогенных полипептидов и протеинов streptomyces. Подобным образом, если инсертируют чужеродную ДНК, ожидается, что промотор saf-гена обеспечивает подобные улучшенные результаты с амилазой, как было продемонстрировано.
Экспрессия чужеродного полипептида или протеина
Для экспрессии чужеродного полипептида или протеина, чужеродной ДНК предпочтительно инсертируют в подходящий сайт распознавания эндогенный ген, предпочтительно ген, кодирующий внеклеточный энзим, так, чтобы обеспечить секрецию чужеродного полипептида или протеина. Когда этим геном является ген амилазы, то подходящим сайтом распознавания является сайт BstE11 /фиг. 11/. Чужеродную ДНК инсекритируют предпочтительно таким образом, чтобы сохранить секрецию сигналов настолько, насколько возможно, их гена внеклеточного энзима. Поскольку эти сигналы расположены главным образом на лидерной последовательности, в отношении гена амилазы имеется свидетельство, что терминальный карбоксильный конец также важен для секреции. Таким образом, может быть лучше всего создать сшитый белок инсерцией чужеродной ДНК в эндогенную ДНК, чем удалять транскрипционную часть эндогенной ДНК и заменять ее чужеродной ДНК.
Известно, что sag-ген контролирует продуцирование внеклеточных энзимов в хромосомной ДНК. Предположительно, в хромосомной ДНК имеются последовательности распознавания, которые узнают SAF-полипептид и вызывают увеличение производства внеклеточного энзима. Сочетание гена saf с геном амилазы на плазмиде не вызывает повышенного производства амилазы.
Таким образом, предпочтительно следует помещать чужеродный протеин, оперативно связанный с сигнальной последовательностью секреции эндогенного внеклеточного энзима, который может контролироваться SAF и, предпочтительно, далее оперативно связанный с saf-промотором /в отсутствие нативного промотора/, в хромосомную ДНК как в противоположность плазмиде. Этим путем продуцирование чужеродного полипептида a или протеина может быть еще более увеличено нативным SAF. В другом предпочтительном выполнении saf-ген инсертируют посредством плазмиды, в те же организмы, в которых чужеродная ДНК была инсертирована в хромосомную ДНК. Это обеспечивает повышенную секрецию чужеродного полипептида или протеина. Фрагменты ДНК могут клонироваться в хромосомную ДНК streptomyces посредством любого из известных бактериофаговых векторов для streptomyces, такого как ⊘ C31 или R4 /Chater и др., "Gene Cloning in streptomyces" в книге "Gene Cloning in Organisms Other Than E. Coli", изданной в Берлине, изд. Шпрингер, 1982, стр. 87 - 95/.
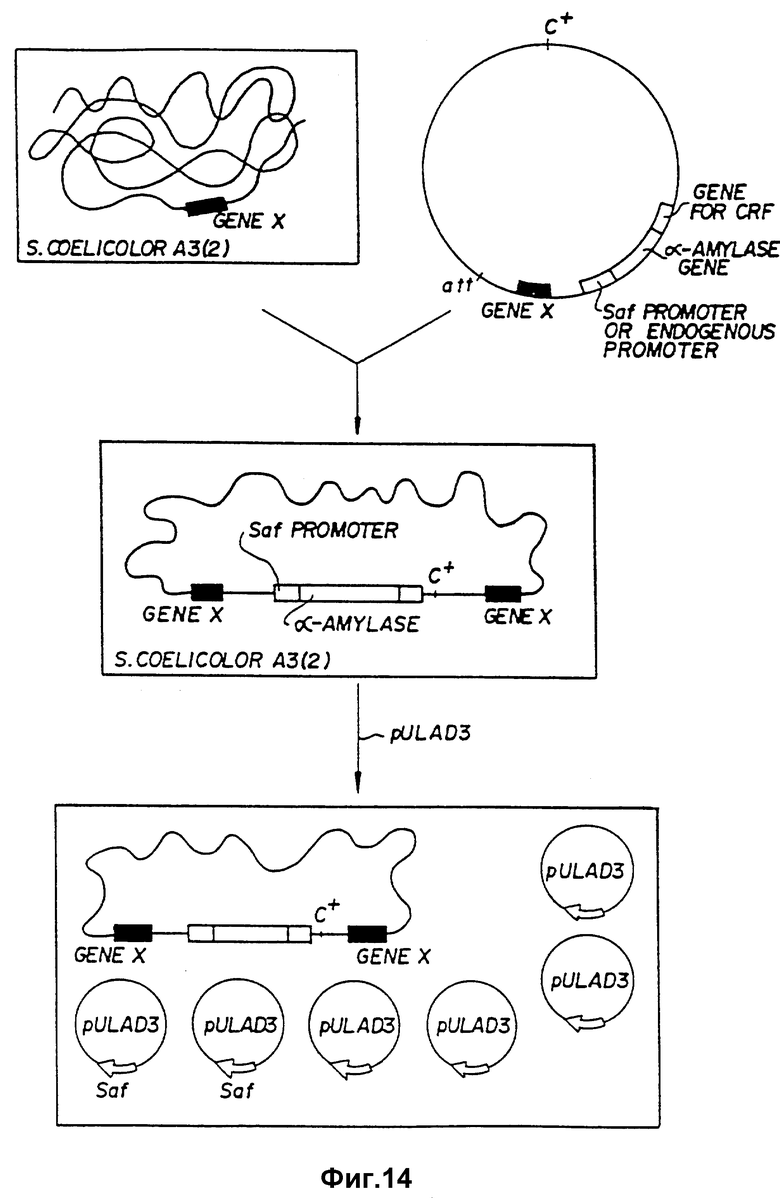
Диаграмма на фиг. 14 показывает предпочтительную технологию для получения секреции чужеродного протеина, в этом случае CRF от штамма KC400 streptomyces - это ⊘ C31 производное, которое не имеет сайта связывания - /att/ и имеет C+ген. См. Chater и др., "Gene" 26: 67 - 78 /1983/, C+ген кодирует репрессор, необходимый для поддержания лизоненного состояния. Сайт att в фаге дикого типа управляет присоединением фага к ДНК клетки-хозяина и управляет освобождением оттуда. Без att-сайта этот фаг не может быть интерирован в хромосому-хозяина, пока в нее не клонирован гомологичный фрагмент ДНК. Для этой цели может быть использован любой эндогенный ген в штамме-хозяина и, таким образом, он просто обозначается как "ген X" на фиг. 14. Фаг ⊘ 31 KC 400 обработан известными технологиями /см. Хопвуд, Лабораторный Учебник/ для инсерции гомологичного фрагмента гена /ген X/ так же, как гена, включающего saf-промотор и ген эндогенной а-амилазы с геном для CRF, оперативно связанного и в рамке считывания с геном а-амилазы. После заражения штамма-хозяина streptomyces, предпочтительно штамма S. coelicolor A3/2 /доступного из коллекции Института John Innes. Norwich, Великобритания/, этим фагом вся фаговая ДНК будет инсертирована в хромосомную ДНК клетки-хозяина, начиная у гомологичного гена X, предпочтительно, с использованием рекомбинации Кэмпбелл-типа. Как указывалось, все конкретные методики для осуществления этих процедур находятся в пределах знаний специалиста в этой области, так что такая инсерция может быть осуществлена без лишнего экспериментирования.
Эти клетки с инсертированным хромосомным геном затем обрабатываются для включения плазмиды pVLAD3. Такие клетки будут экспрессировать SAF-полипептид, который, в свою очередь, увеличит секрецию а-амилазы хромосомным геном. Присутствие saf-промотора на этом гене будет далее увеличивать секрецию а-амилазы. Эта а-амилаза в виде секрета будет содержать присоединенную к ней молекулу CRF, которая может быть легко отделена соответствующим ферментным перевариванием.
В то время, как а-амилазный ген streptomyces был проиллюстрирован конкретными примерами, следует учитывать, что с целью повышения экспрессии посредством применения saf-промотора используемый ген для любого полипептида или белка, продуцируемых видами Streptomyces может быть модифицирован путем удаления нативного промотора и замены его saf-промотором. Сходным образом чужеродный полипептид или протеин может быть инсертирован в любой такой эндонгенный ген. Предпочтительно, однако, чтобы выбранный селекцией эндогенный ген был и геном, который экспрессирует протеин через клеточную стенку и в культуральную среду, в этом случае можно поддержать сигнальную последовательность секреции. Наилучшие результаты будут получены, если выбираемый селекцией ген для инсерции его в чужеродный ген является тем, который контролируется SAF; а инсерция осуществляется в хромосомную ДНК, предпочтительно, с одновременной инсерцией плазмиды, содержащей saf-ген.
В то время, как CRF был упомянут особо, следует понимать, что последовательность чужеродной ДНК может быть любой последовательностью ДНК, производной не от streptomyces, кодирующей протеин или полипептид, в частности, эукариотического или вирусного происхождения. Примерами таких эукариотических и вирусных последовательностей ДНК являются последовательности, кодирующие лейкоцитарный интерферон животных и человека /1FN-a/, фибробластовый интерферон /1FN-бета/ и иммунный интерферон /1FN-гамма/, человеческий инсулин, человеческого и животного ростового и других гормонов, таких как кортикотропин-освобождающий фактор /CRF/, сывороточного альбумина человека и различные факторы крови человека, и плазминогенные активаторы - как тканевой, так урокиназный, антигены сердцевины и поверхности вируса гепатита B, FMD вирусные антигены и другие человеческие, животные и вирусные полипептиды и протеины.
В то время, как предпочтителен saf-промотор, продуцирование чужеродного полипептида или протеина будет далее увеличено путем трансформирования клетки-хозяина экспрессионным вектором, содержащим saf-ген. Этот экспрессионный вектор может находиться в плазмиде ДНК этой клетки. Таким образом, может быть использован эндогенный или любой другой промотор, и при этом будет получена секреция чужеродного полипептида и протеина.
Предыдущее описание конкретных выполнений настолько полно раскрывает общую сущность изобретения, что другие смогут с применением общих знаний в этой области легко модифицировать и/или приспосабливать изобретение для различных его применений в виде конкретных выполнений, не отходящих от общей концепции, и поэтому такие адаптации и модификации включаются в значение и объем раскрытых выполнений. Следует также учесть, что фразеология и терминология данного описания использована не с целью ограничения, а лишь общей ясности.
Количественное определение белка-носителя /амилазы/ в культуральном бульоне /супернатанты/ культур Streptomyces Lividans, трансформированных различными плазмидами.
Мы использовали три разных метода определения количества секретированного протеина:
1. Спектроденситометрия полос белка в гелях SDS-PaGE, окрашенных серебром, или фотонегативов /пленок/ в тех же гелях, используя спектроденситометр Шимадзу.
2. Анализ количеств различных протеинов с использованием HPLC, оснащенной колонкой 08 Аквапор РП-300 /200 • 4,6 нм Лаборатории Браунли США/.
3. Анализ посредством HPLC, оснащенной молекулярным ситов /гель-фильтрацией/ колонны Protein-pek300 sw.
Результаты.
Споры S. Lividans /pVLTV100/, S. Lividans /pVLVD10, и S. Lividans /pVLTV80/ инокулировали в среду Лешевалье + 1% крахмала. Максимальная амилазная активность была получена соответственно при 36, 60, 60 ч ферментации этих спор. Общий протеин во внеклеточной жидкости /супернатанте/ был определен количественно методом Бредфорда. 5μг протеина из каждого бульона переносили в SDS-PAGE и окрашивали серебром.
1. Спектроденситометрия проводилась на полосах в SDS-PAGE /10% акриламида/ супернатантов S. Lividans /pVLTV100/ , 5 μг протеина/, S. Lividans /pVLVD10/, 5μг протеина/ и S. Lividans /pVLTV80/ /5μг протеина/.
2. HPLC в колонне Аквапор РН-300.
Протеины элюировали 50 мМ уксусной кислоты - ацетат аммониевым буфером и градиентом метанола: 0 - 20 мин 0% метанола, 30 - 35 мин 100% метанола, 45 мин 0% метанола. Амилаза элюируется 100%-ным метанолом на 33 - 34 мин. Результаты показаны в табл. 3.
3. HPLC в Protein=Pak300SW.
Супернатанты культуральных бульонов тех же самых конструкций, как указано выше, были предварительно диализованы и концентрированы лиофилизацией. Протеины элюировали уксусно-кислотным аммоний-ацетатным буфером со скоростью потока 0,5 мл/мин. Амилаза элюирует в виде пика с центром на 21 - 22 мин.




Изобретение относится к биотехнологии. Рекомбинантная плазмидная ДНК puLDVI состоит из векторной плазмиды pIj 699 и последовательности ДНК. Последовательность ДНК встроена в BglII-сайт плазмиды pIj 699 и включает SAF-промотор, последовательность, операбельно связанную с 5'-концом полной последовательности amy-гена, находящегося в составе HindIII-фрагмента размером 4,8 кб из геномной ДНК S. Griseus Плазмидная ДНК содержит уникальные сайты: HindIII, NcoI, BstEII, SalI, BglII. Выделяют полную последовательность гена α -амилазы, содержащей промотор, участок, кодирующий сигнальный пептид и последовательность, кодирующую собственно ферментный белок. Указанную последовательность гена операбельно связывают с SAF промотором и встраивают ее в вектор. Проводят трансформацию полученным рекомбинантным вектором штамма streptomyces. Отбирают трансформанты. Культивируют их в условиях, обеспечивающих экспрессию введеннного гена. Изобретение позволяет обеспечить экспрессию чужеродных последовательностей ДНК в streptomyces. 2 с.п. ф-лы, 14 ил., 3 табл.
Приоритет по пунктам:
18.05.90 по п.1;
22.09.89 по п.2.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| СПОСОБ ИЗМЕРЕНИЯ КРИВИЗНЫ ПОВЕРХНОСТИ | 0 |
|
SU196375A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ПАТЕНТНО- t ^ тсхничгси'АЯ ^••БИБЛИОТЕКАВ. А. Черных18 | 0 |
|
SU179449A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Шланговое соединение | 0 |
|
SU88A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Методы генетической инженерии Миниатис Г | |||
| и др | |||
| Молекулярное клонирование | |||
| - М.: Мир, 1984, с | |||
| Способ получения гидроцеллюлозы | 1920 |
|
SU359A1 |

