Настоящее изобретение относится к производным 9-деазагуанинов, которые являются сильными ингибиторами пуриннуклеозидфосфорилазы (ПНФ), к фармацевтическим композициям, содержащим указанные соединения и обладающим способностью ингибировать пуриннуклезид-фосфорилазы, а также лечения заболеваний млекопитающих, восприимчивых к ингибированию пуриннуклеозид-фосфорилазы, с использованием указанных соединений или фармацевтических композиций, содержащих соединения настоящего изобретения.
Соединения настоящего изобретения могут быть использованы в качестве ингибиторов пуриннуклеозид-фосфорилазы /ПНФ/, в частности, в качестве селективных ингибиторов Т-клеток, и в целях подавления клеточного иммунитета. Указанные соединения могут быть использованы для лечения аутоиммунных заболеваний для предотвращения отторжения трансплантата, для лечения псориаза или подагры у млекопитающих. Они могут быть также использованы для усиления противовирусной и противоопухолевой активности противовирусных и противоопухолевых пуриннуклеозидов.
В заявке Европатент N 178178 и, в основном, аналогичном ей патенте США N 4772606 описываются 9-арилметил-замещенные пурины /включая и гуанины/ в качестве ПНФ-ингибиторов.
Данные ПНФ-ингибирования, приведенные в Drugs of the Future 13, 654 /1988/ и Agents and Actions 21 253, /1987/, показывают, что производные 9-арилметилзамещенного гуанина формулы: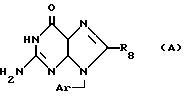
где R9 представляет собой водород, являются значительно менее сильным ПНФ ингибиторами, чем соответствующие соединения, где R9 представляет собой амино /8-аминогуанины/. Настоящее изобретение относится к соединениям формулы: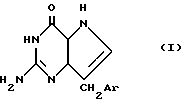
где /а/ CH2Ar представляет собой: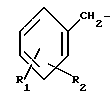
где R1 является водородом, галогеном, С1-С3 - алкилом, С1-C3 алкокси, бензилокси, гидрокси или трифторметилом, и R2 является водородом, галогеном, С1-С3 алкилом, С1-С3 алкокси, бензилокси, гидрокси или трифторметилом, при условии, что R2 является водородом, или С1-С3 алкилом, если R1 является трифторметилом, или R1 является водородом или С1-С3-алкилом, если R2 является трифторметилом, или /b/ CH2Ar представляет собой: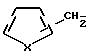
где X является серой или кислородом, и связь с тиофеновым или фурановым кольцом имеет место в 2- или 3-положении; и их таутомерам.
Один из параметров настоящего изобретения относится к соединениям формулы II: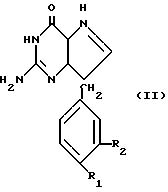
где один из R1 и R2 является водородом, а с другой является водородом, хлором, фтором, С1-С3-алкилом, С1-С3-алкокси, бензилокси, гидрокси или трифторметил или R1 и R2 являются хлором или фтором; или их таутомерам.
Предпочтительными соединениями формулы II являются соединения, в которых один из R1 и R2 является водородом, а другой водородом, хлором, метилом, Метокси, бензилокси, гидрокси или трифторметилом; и их туатомеры.
Более предпочтительный вариант настоящего изобретения относится к соединениям формулы II, где R1 является водородом; а R2 является водородом, хлором, гидрокси, бензилокси или трифторметилом, и таутомерам.
Особенно предпочтительными соединениями форму II являются соединения, где:
/а/ R1 является хлором, а R2 является водородом;
/b/ R1 является водородом, а R2 является хлором;
(с) R1 является водородом, а R2 является трифторметилом,
(d) R1 и R2 является водородом, и их таутомеры.
Другой вариант настоящего изобретения относится к соединениям формулы III: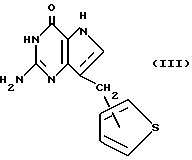
где связь с тиофеновым кольцом имеет место либо в положении 2, либо в положении 3, и их таутомерам.
Еще один вариант настоящего изобретения относится к соединениям формулы IV: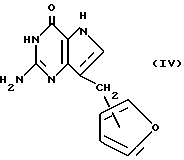
где связь с фурановым кольцом имеет место либо в положении 2, либо в положении 3, и их таутомером.
Предпочтительные варианты настоящего изобретения относятся к конкретным соединениям, раскрытым в примерах.
9-замещенные-9-деазагуанины настоящего изобретения, например, соединения формул I-IV, могут быть также названы 7-замещенными 2-амино-3,5-дигидро-4Н-пирроло[3,2-d] пиримидин-4-онами. Кроме того, указанные соединения могут существовать в таутомерных формах, которые, например, могут быть представлены структурой IA: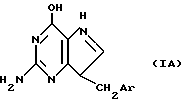
и т. п. при этом все таутомерные формы входят в объем настоящего изобретения.
Основные определения, входящие в объем настоящего изобретения, имеют следующие значения:
С1-С3алкил представляет собой метил, этил или пропил, а предпочтительно метил;
С1-С3-алкокси представляет собой метокси, этокси, пропокси, а предпочтительно метокси;
галоген представляет собой фтор, хлор, бром или йод, предпочтительно фтор или хлор, а более предпочтительно хлор.
Соединения настоящего изобретения являются особенно эффективными при их использовании для селективного подавления опосредованного Т-клетками иммунитета у млекопитающих, а также для лечения млекопитающих, страдающих аутоиммунными заболеваниями, псориазом или подагрой, или для предотвращения отторжения трансплантата. К аутоиммунным заболеваниям относятся такие заболевания, как ревматоидный артрит, системная красная волчанка, миестения, рассеянный склероз, и диабеты типа I.
Соединения настоящего изобретения могут быть также использованы в целях ингибирования метаболического разложения in vivo пуриннуклеозидов посредством фосфорилиза и, таким образом, в целях усиления противовирусной и противоопухолевой активности 2'- и/или 3'-моно или дидезокси-пуриннуклеозидов, например, указанные соединения могут быть использованы для потенцирования, например, 2',3-дидезоксиаденозина, 2',3'-дидезоксигуанозина или 2', 3'-дидезоксинозина в целях лечения ретровирусных инфекций, например, синдрома приобретенного иммунодефицита (СПИДа). Указанные соединения могут быть также использованы для усиления противоопухолевого/цитотоксичного действия, например, 2'-дезоксигуанозина, в организме млекопитающих.
Описанные выше свойства соединений настоящего изобретения были проиллюстрированы испытаниями in vitro и in vivo проведенными с использованием млекопитающих, например крыс, мышей, собак, телят, и клеток, полученных от этих млекопитающих. Указанные соединения могут быть использованы in vitro в виде растворов, например, водных растворов, и in vivo путем перорального или парентерального введения, предпочтительно, перорально и внутривенно In vitro доза варьируется в пределах молярных концентраций от около 10-5 до около 10-8. In vivo доза зависит от способа введения и составляет около 0,01 30 мг/кг.
ПНФ-ингибирование определяли радиохимическим способом путем измерения образования [14-C]-гипоксантина из [14-C]-инозина с использованием 50 мМ фосфата и телячьей селезенки в качестве источника фермента. Полученные результаты выражали в величинах IC50, которые соответствуют концентрации соединения, требуемой для 50%-ного снижения образования гипоксантина.
Усиление соединениями настоящего изобретения активности 2'-дезоксигуанозина (d-GuO) к ингибированию клеточного роста (цитотоксичности) определяли следующим образом: CCRF-CEM клетки выращивали в среде RPMI-1640. К суспензионным культурам этих клеток добавляли d-GuO при фиксированной концентрации (5,62 мкМ) и испытуемый ПНФ-ингибитор при различных концентрациях и подсчитывали число клеток через 24, 28 и 72 часа с помощью счетчика Коултера. На основании этих данных рассчитывали IC50 как концентрацию ПНФ-ингибитора, требуемую для уменьшения роста числе клеток в интервале 0 72 часа до 50% по сравнению с контрольной культурой. Этот способ аналогичен способу, который был использован для определения эффективности ПНФ-ингибиторов в усилении токсичности d-GuO /3/.
ПНФ-ингибирование может быть также определено in vivo способом, описанным в [3] путем измерения увеличения уровней инозина в плазме крыс, вызванного соединениями настоящего изобретения.
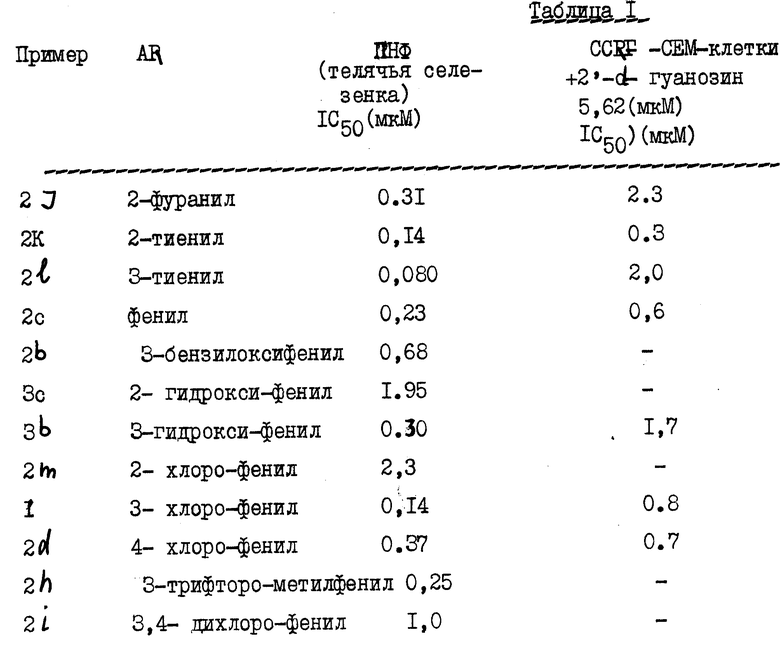
Для иллюстрации настоящего изобретения ниже приводятся результаты, полученные в in vitro испытаниях ПНФ-ингибирования и in vivo испытаниях потенцирование 2'-дезоксигуанозина проведенных для производных 8-незамещенных гуанинов (см. табл. 1).
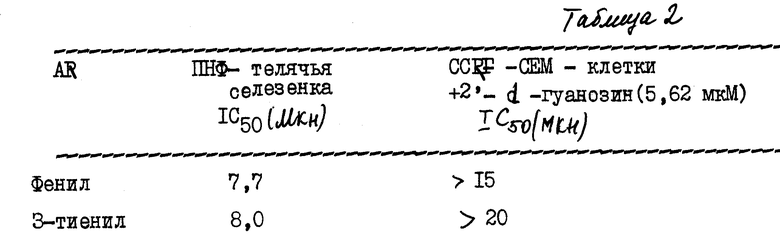
Ниже приводятся данные, полученные в in vitro испытаниях ПНФ-ингибирования и испытаниях потенцирования 2'-дезоксигуанозина, проведенные для соответствующих 8-аминозамещенных соединений, раскрытых в заявке на Европатент N 260 491 (см. табл. 2).
Сравнительные результаты ясно проиллюстрировали неожиданно более высокую ингибиторную активность 8-незамещенных соединений настоящего изобретения.
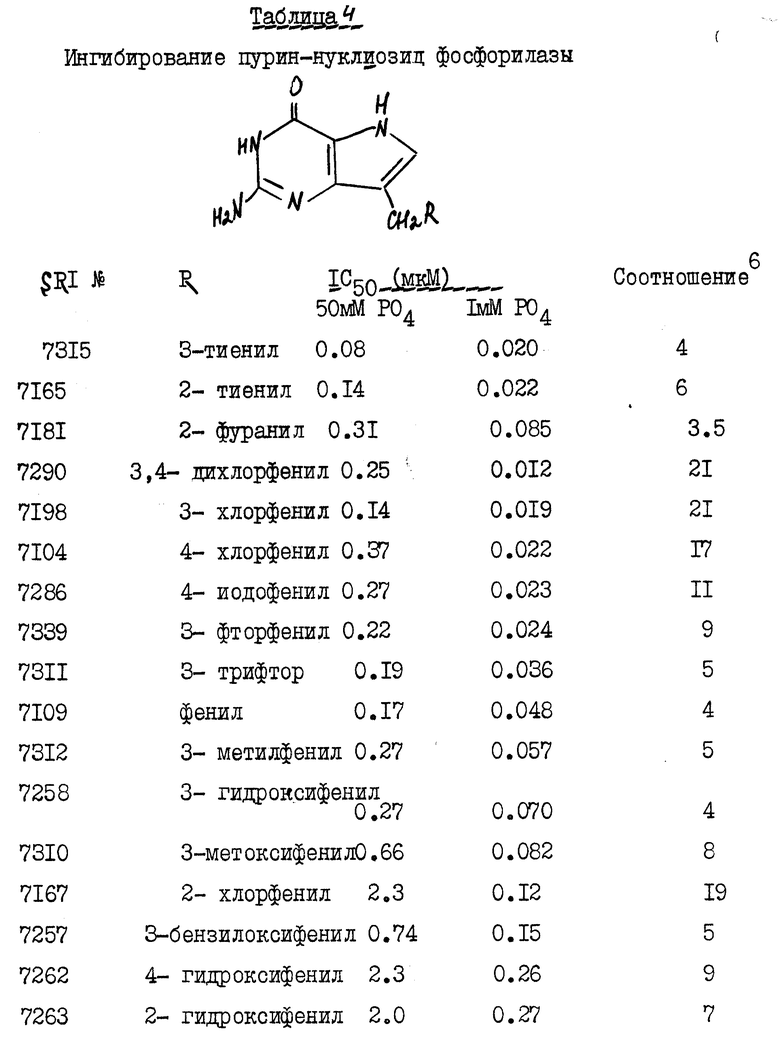
Приведенные ниже таблицы 3 и 4 иллюстрируют токсичность и сравнительные данные величин IC50 со значениями ПНФ (ингибирование) для заявленных и прошедших испытание соединений.
1d-GuO и ПНФ ингибитор добавляют одновременно к клеткам, вырастающим во взвешенных культурах, и количество клеток определяют 72 часа спустя. Значения указываются приблизительно вследствие того, что они являются результатами однократных испытаний.
а телячья селезенка ПНФ
6IC50 мМ PO4/IC50 1 мМ PO4
Соединения настоящего изобретения могут быть получены путем адаптации известной методики синтеза (см. например, 48, 780 (1983), описанной ниже и проиллюстрированной примерами.
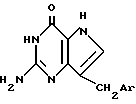
Соединения настоящего изобретения получают преимущественно путем обработки соединения формулы: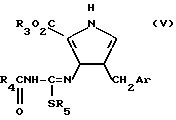
где Ar имеет значения, определенные выше, R3 является низшим алкилом, R4 является карбоциклическим арилом, а R5 является низшим алкилом, c безводным аммиаком, и если требуется, с последующим превращением полученного соединения формулы I в другое соединение настоящего изобретения.
Низший алкил, представленный R3 и R5, является С1-С7 алкилом, предпочтительно метилом или этилом.
Карбоциклический арил, представленный R4, является предпочтительно фенилом.
Конденсацию промежуточного соединения формулы V с аммиаком и циклизацию с получением соединения настоящего изобретения, например, соединения формулы I, II, III и IV, предпочтительно осуществляют в полярном инертном безводном растворителе, таком как низший алифатический спирт, предпочтительно метанол, и предпочтительно при повышенной температуре, например 80 100oC, под давлением в закрытом сосуде.
Полученные бензилокси-замещенные соединения настоящего изобретения, например соединения формулы I(а), где R1 и/или R2 являются бензилоксигруппой, могут быть подвергнуты дебензилированию с получением соответствующих соединений, где R1 и/или R2 являются гидроксигруппой, путем каталитической гидрогенизации при стандартных условиях.
Исходные соединения формулы V получают преимущественно путем обработки пирролового производного формулы VI: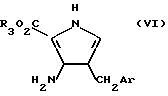
где Ar и R3 определены выше, карбоциклическим ароилизотиоцианатом, предпочтительно бензоилизотиоцианатом в инертном растворителе, таком как дихлорметан, с получением соединения формулы VII: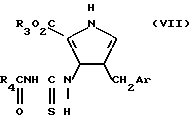
где Ar, R3 и R4 определены выше.
После чего, в результате конденсации промежуточных соединений формулы VII с реактивным производным низшего алкилкарбинола, например низшего алкилгалида, преимущественно метилиодида, в инертном растворителе, таком как метиленхлорид, и в присутствии органического или неорганического основания, например амина, такого как 1,5-ди-азабицикло[4.4.O]-нон-5-ен (ДБН), получают промежуточные соединения формулы V.
Пирроловые исходные соединения формулы VI могут быть получены способом, аналогичным способу, используемому для синтеза 3-амино-4-замещенных 2-пирролокарбоновых кислот и их сложных эфиров.
Указанные пирроловые соединения получают предпочтительно способом, представленным ниже для соединений формулы VI, где R3 является метилом.
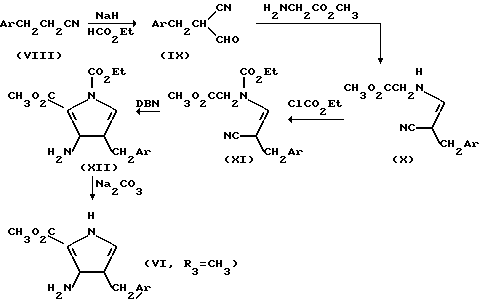
Ar в вышеуказанных соединениях имеет значения, определенные выше.
Вкратце, согласно вышеприведенной схеме, 3-арилпропионитрил (VIII) конденсируют с этилформатом в присутствии, например, гидрида натрия, в безводном тетрагидрофуране, в результате чего получают 2-формил-3-арилпропионитрил (IX), который, в свою очередь, конденсируют со сложным метиловым эфиром глицина в присутствии, например, ацетата натрия, в результате чего получают анамин формулы (Х). Указанный энамин формулы X в свою очередь, подвергают N-блокированию с этилхлороформатом, и полученное N-этоксикарбониловое производное X циклизуют в присутствии основания, например DBN в результате чего получают N-защищенный пиррол формулы XII. Промежуточное соединение формулы XI получают in situ и обычно не выделяют. После разблокирования путем обработки, например, карбонатом натрия в метаноле получают исходный материал формулы VI.
Арилпропионитрилы формулы VII являются известными соединениями, либо они могут быть получены стандартными способами. Указанные способы обычно заключаются в том, что соответствующий ариловый альдегид конденсируют с цианоуксусной кислотой с последующим декарбоксилированием и получают 3-арилакрилонитрил, в целевые 3-арил-пропионитрилы восстанавливают одним из двух способов: каталитической гидрогенизации или с использованием металлического магния в метаноле при 0oC. Последний способ является предпочтительней, если арильная часть содержит чувствительную группу, подобную бензилокси-группе, которая могла бы быть быстро разблокирована путем гидрогенолиза.
Как указывалось выше, упомянутые способы, если это необходимо, могут осуществляться при временном блокировании любой реакционной группы, с последующим высвобождением соединения настоящего изобретения.
В исходных и промежуточных соединениях, превращаемых с помощью вышеуказанных способов в соединения настоящего изобретения, присутствующие функциональные группы, такие как гидрокси-группы, являются не обязательно защищенными стандартными защитными группами, хорошо известными специалистам по органической химии.
Стандартные защитные группы, их введение и удаление описаны, например, в работе J.F.W. Mc Omie "Protective Groups in Organic Chemistry". Пленум-пресс, Лондон, Нью-Йорк, 1973, T. W. Greene "Protective Groups in Organic Synthesis", Wiley, Нью-Йорк, 1984 г. Например, гидроксильная группа является предпочтительно защищенной в виде простого бензилового эфира, который может быть отщеплен путем каталитической гидрогенизации с получением гидроксизамещенного продукта.
Вышеупомянутые реакции осуществляют в соответствии со стандартными способами, в отсутствие или в присутствии разбавителя, предпочтительно инертного а реагентам, их растворителям, катализаторам конденсирования, или другим агентам, и/или инертных атмосферах, при пониженных температурах, комнатной температуре или при повышенных температурах, предпочтительно при (или около) точки кипения используемых растворителей, и при атмосферном или выше атмосферного давления. Предпочтительные растворители, катализаторы и реакционные условия проиллюстрированы в прилагаемых ниже примерах.
Предпочтительными исходными материалами, используемыми в указанных реакциях, являются соединения, с помощью которых можно получить указанные выше предпочтительные соединения настоящего изобретения.
Настоящее изобретение также относится ко всем новых исходным соединениям и способам их получения.
Любые смеси конечных продуктов или промежуточных соединений могут быть разделены стандартными способами, исходя из физико-химического различия их составляющих, с получением чистых конечных продуктов или промежуточных соединений, например с помощью хроматографии, дистилляции, фракционной кристаллизации, или путем образования солей, если это необходимо или возможно в данной конкретной ситуации.
Соединения настоящего изобретения или промежуточные соединения могут быть также получены в виде их гидратов, или могут включать другие растворители, используемые для их кристаллизации.
Кроме того, настоящее изобретение относится к фармацевтическим композициям, приемлемым для введения млекопитающему или человеку, например, перорально или ректально, чрезкожно или парентерально, в целях ингибирования активности пуриннуклеотид-фосфорилазы и лечения связанных с ней заболеваний, при этом указанные композиции включают в себя эффективное количество фармакологически активного соединения настоящего изобретения, взятого отдельно или в сочетании с одним или несколькими фармацевтически приемлемыми носителями.
Предпочтительными формами фармацевтических композиций являются таблетки и желатиновые капсулы, содержащие активный ингредиент в сочетании с: a) разбавителями, такими как лактоза, декстроза, сахароза, маннит, сорбит, целлюлоза и/или глицин, b) замасливателями, такими как двуокись кремния, тальк, стеариновая кислота, ее магниевая или кальциевая соль и/или полиэтиленгликоль, для таблеток также с: c) связующими, такими как алюмомагниевый силикат, крахмальная паста, желатин, трагакант, метилцеллюлоза, натриевая карбоксиметилцеллюлоза и/или поливинилпирролидон, по желаю, с: d) дезинтеграторами, такими как крахмалы, агар, альгиновая кислота или ее натриевая соль, или пенистые смеси, и/или с: e) абсорбентами, окрашивающими агентами, ароматизаторами и подслащивающими агентами. Инъецируемые композиции являются предпочтительно водными изотоническими растворами или суспензиями, а суппозитории получают предпочтительно из жирных эмульсий или суспензий. Указанные композиции могут быть стеарилизованы и/или содержать адъюванты, такие как консерванты, стабилизаторы, смачивающие или эмульгирующие агенты, усилители растворения, соли для регулирования осмотического давления и/или буферы. Кроме того, композиции настоящего изобретения могут также содержать и другие терапевтически ценные вещества. Указанные композиции получают в соответствии со стандартными способами смешивания, гранулирования или покрытия, и эти композиции содержат около 0,1 75% а предпочтительно около 1 50% активного ингредиента.
Соответствующие композиции для чрезкожного введения содержат эффективное количество соединения настоящего изобретения в сочетании с носителем. Предпочтительные носители включают в себя абсорбируемые фармакологически приемлемые растворители, облегчающие прохождение лекарственного средства через кожу. Обычно для чрезкожного введения используют бандаж, состоящий из каркаса, резервуара, содержащего соединение не обязательно в сочетании с носителем, не обязательно скорость регулирующего барьера для доставки соединения через кожу хозяина с регулируемой и заранее определенной скоростью в течение длительного периода времени, и средства для укрепления бандажа на коже.
Настоящее изобретение также относится к способу ингибирования активности пуриннуклеозид- фосфоридазы в организме млекопитающих и лечения связанных с ней заболеваний, например аутоиммунных заболеваний, псориаза или отторжения трансплантата, причем указанный способ заключается во введении млекопитающему эффективного количества соединения настоящего изобретения или фармацевтической композиции, содержащей указанное соединение в сочетании с одним или несколькими фармацевтическими приемлемыми носителями.
Один из вариантов настоящего изобретения относится к способу селективного подавления функции Т-клеток и клеточного иммунитета, заключающемуся во введении млекопитающему соответствующего эффективного ингибирующего количества соединения настоящего изобретения или указанного соединения в комбинации с одним или несколькими фармацевтически приемлемыми носителями.
Другой вариант настоящего изобретения относится к способу ингибирования фосфорилазы и метаболического распада противовирусного или противоопухолевого пуриннуклеозидов в организме млекопитающих, заключающемуся во введении млекопитающим либо отдельно, либо в комбинации с ними эффективного пурриннуклеозид-фосфорилаза-ингибирующего количества соединения настоящего соединения или указанного соединения в сочетании с одним или несколькими фармацевтически приемлемыми носителями. В частности, в указанном варианте настоящее изобретение относится к способу ингибирования фосфорилазы и метаболического распада известных пуриннуклеозидов, таких как 2'-дезоксигуанозин, 2',3'-дидезоксинозин, 2'3'-дидезоксигуанозин или 2'3'-дидезоксиаденозин.
Кроме того, настоящее изобретение относится к способу потенцирования противовирусного или противоопухолевого действия 2'- или 3'-монодезоксипуриннуклеозидов или 2',3'-дидезоксипуриннуклеозидов в организме млекопитающих, заключающемуся во введении млекопитающему либо отдельно, либо в комбинации с указанным нуклеозидом эффективного пуриннуклеозидфосфорилаза - ингибирующего количества соединения настоящего изобретения, предпочтительно в сочетании с одним или несколькими фармацевтически приемлемыми носителями. В частности, в указанном варианте своего осуществления настоящее изобретение относится к способу усиления или потенцирования действия известных 2',3'-дидезоксипуриннуклеозидов, таких как 2',3'-дидезоксиинозин, 2',3'-диедезоксигуанозин или 2', 3'-дидезоксиаденозин, в целях лечения ретровирусных инфекций, таких как инфекции, вызываемые ретровирусом ВИЧ (например, СПИД), 2',3'-дидезоксипуриннуклеозиды известны специалистам как ингибиторы ВИЧ-ретровирусной инфектиности, которые метаболически разлагаются ПНФ'ой. Указанные соединения в фармацевтически приемлемой дозе эффективны для ингибирования ВИЧ-ретровирусной инфекции.
Предпочтительно использовать по возможности наиболее низкую дозу.
Указанная фармацевтически приемлемая эффективная доза активного соединения настоящего изобретения зависит от вида теплокровного млекопитающего, веса его тела, возраста, состояния и способа введения.
Разовая доза для млекопитающего весом 50 70 кг может содержать около 1 150 мг активного ингредиента.
Соединение настоящего изобретения может быть также использовано вместе с другими терапевтическими средствами. Суточная доза 1 50 мг/кг для человека весом 50 70 кг способна ингибировать метаболическую декструкцию некоторых противораковых средств, таких как бета 2'-дезокси-6-тиогуанозин, и противовирусных средств, таких как 2'-3'-дидезоксинозин (средство против СПИДа). Указанные средства, как известно, являются восприимчивыми к расщеплению. После расщепления эти лекарственные средства теряют свою эффективность. Соединения настоящего изобретения обладают способностью к снижению такого расщепления. Поэтому соединения настоящего изобретения способствуют повышению эффективности других терапевтических средств.
Представленные ниже примеры приведены с целью иллюстрации настоящего изобретения и не должны рассматриваться как некие ограничения его возможных вариантов. Все температуры в указанных примерах даны в градусах Цельсия. Если это не оговорено особо, выпаривания проводили при пониженном давлении, предпочтительно в пределах 15 100 ммрт.ст. Структура конечных продуктов, промежуточных продуктов, исходных материалов подтверждалась с помощью стандартных аналитических методов, например, микроанализа и спектральных анализов (таких, как МС, ИК, ЯМР и УФ-спектроскопии).
Пример 1.
А) Гидрид натрия (2,43 г, 101 мМ) суспендировали в безводном ТГФ (80 мл) в атмосфере безводного азота, и к этой суспензии, размешивая, добавляли этилформат (24,69 г, 330 мМ) и 3-(3-хлорфенил)-пропионитрил (12,0 г, 72,46 мМ). Реакционную смесь размешивали при комнатной температуре в течение 24 ч. Летучие вещества выпаривали в вакууме при комнатной температуре. К остатку при 0oC добавляли воду (50 мл) и раствор подкисляли до pH 5 с помощью 10%-ной водной HCl при охлаждении льдом. Тяжелое масло экстрагировали этилацетатом (1•100 мл), экстракт промывали водой (2•40 мл) и осушали сульфатом натрия. Органический слой выпаривали и получали соединение формулы: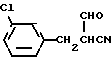
в виде маслянистого вещества бурого цвета, которое использовали в следующей стадии без очистки.
b) Гидрохлорид сложного метилового эфира глицина (13,46 г, 108,6 мМ) и ацетат натрия (8,9 г, 108,6 мМ) добавляли к 134,20 г неочищенного продукта, полученного в стадии (а) в смеси с метанолом (137 мл) и водой (34 мл), и полученный раствор размешивали при комнатной температуре в течение 24 ч. После выпаривания метанола при комнатной температуре остаток экстрагировали этилацетатом. Органический слой промывали водой (2•40 мл), осушали сульфатом натрия и получали маслянистый продукт янтарного цвета, который очищали с помощью колоночной флеш-хроматографии на силикагеле, используя в качестве элюента хлороформ, в результате чего получали соединение формулы: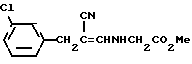
в виде масла, представляющего собой смесь цис-транс-изомеров.
c) К раствору продукта, полученного в стадии (7,35 г, 27,8 мМ) в безводном дихлорметане (80 мл) добавляли DBN (10,35 г, 83,3 мМ) и этилхлороформат (4,52 г, 41,65 мМ) в присутствии азота при комнатной температуре. Полученный раствор размешивали при комнатной температуре в течение 24 ч. Летучие вещества выпаривали в вакууме и получали смолистый остаток, который очищали с помощью флеш-хроматографии на силикагеле, элюируя CHCl3, в результате чего получали продукт формулы: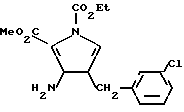
в виде неочищенного масла, который использовали в следующей стадии без очистки.
d) К раствору неочищенного продукта, полученного в стадии (с) (8,19 г, 24,3 мМ) в MeOH (130 мл) добавляли Na2CO3 (6,44 г, 60,84 мМ). Реакционную смесь размешивали при комнатной температуре в течение 24 ч. Нерастворимые соли удаляли путем фильтрации и промывали MeOH. Раствор метанола снижали до 15 мл и выдерживали в холодильнике в течение 2 ч для получения кристаллического продукта. В результате концентрирования маточного раствора получали дополнительный кристаллический продукт формулы: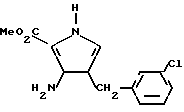
имеющего точку плавления 105 106oC.
а) К раствору соединения, полученного в стадии (d) (1,0 г, 4,26 мМ) в безводном дихлорметане (20 мл), добавляли бензоилизотиоцианат (0,69 г, 4,26 мМ) в атмосфере азота при комнатной температуре. Реакционную смесь размешивали в течение 1 ч, выпаривали досуха и полученный остаток бледно-желтого цвета перетирали с метанолом. Белое кристаллическое вещество выделяли путем фильтрации и после перекристаллизации из смеси CHCl3 и эфира получали тиоруеидо-соединение, имеющее структуру: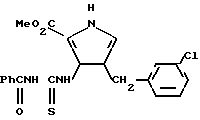
и точку плавления 160 161oC.
(f) К охлажденному льдом раствору соединения, полученного в стадии (e) (0,71 г, 1,66 мМ) в сухом CH2Cl2 (50 мл) добавляли DBN (0,24 г, 1,9 мМ) и метилиодил (0,68 г, 4,8 мМ). Реакционную смесь размешивали при 0oC в течение 1 ч. Растворитель выпаривали, остаток экстрагировали CHCl3, промывали водой (2•30 мл), высушивали сульфатом натрия и после выпаривания получали стеклообразный густой маслянистый продукт, который очищали с помощью флеш-хроматографии на силикагеле с использованием CHCl3 в качестве элюента, в результате чего получали соединение со структурой: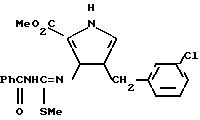
в виде стеклообразной пены, которое кристаллизовали из метанола, т.пл. 121oC.
(g) Раствор промежуточного метилтио-соединения, полученного в стадии (f) (0,6 г, 1,35 мМ) в MeOH, насыщенном аммиаком (40 мл), нагревали при 110oC в течение 20 ч в эмалированном автоклаве. После чего реакционную смесь охлаждали до комнатной температуры и выпаривали досуха. В результате очистки полученной смеси с помощью колоночной флеш-хроматографии на силикагеле, используя CHCl3 в качестве элюента, удаляли 2-метилтио-3,5-дигидро-7-(3-хлорфенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он. После дополнительного элюирования смесью CHCl3-MeOH (95: 5) получали 2-амино-3,5-дигидро-7-(3- хлорфенилметил)-4Н-пирроло-[3,2-d]пиримидин-4-он-формулы: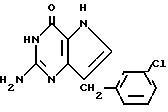
с точной плавления 258oC (разл).
Пример 2.
(а) 2-амино-3,5-дигидро-7-(4-бензилоксифенилметил)-4Н-пирроло[3,2-d] пиримидин-4-он, т.пл. при 250 351oC.
Исходный материал получали следующим образом.
В раствор 3-(4-бензилоксифенил)-акрилонитрила (17,0 г, 72,4 мМ) в безводном MeOH (800 мл) добавляли магниевую стружку (28 г). Как только начиналось выделение H2, полбу погружали в активно охлажденную льдом /водой баню до тех пор, пока начальная бурная реакция не начнет ослабевать, приблизительно через 45 мин. Затем добавляли еще 10 г магния, реакционную смесь перемешивали 4 ч, перемежая размешивание с охлаждением в те периоды времени, когда происходило наибольшее потребление металлического Mg. Смесь выпаривали до получения густой пасты, которую затем охлаждали до 0oC и медленно обрабатывали достаточно холодной 6 н HCl для растворения магниевых солей. Мутный раствор экстрагировали CHCl3, органический слой промывали 0,1 н NaOH и H2O, осушали сульфатом натрия и выпаривали, в результате чего получали белый воскообразный твердый остаток и после его перекристаллизации из смеси (2:1) EtOH/H2O получали 3-(4-бензилоксифенил)-пропионитрил.
b) 2-амино-3,5-дигидро-7-(3-бензилоксифенилметил)-4Н-мирроло [3,2-d]пиримидин-4-он, т.пл. 228 230oC.
с) 2-амино-3,5-дигидро-7-(фенилметил)-4Н-пирроло-[3,2-d] пиримидин-4-он, т.пл. 269 270oC (разл)
d) 2-амино-3,5-дигидро-7-(4-хлорофенилметил)-4Н-пирроло-[3,2-d] пиримидин-4-он, т.пл. 207 208oC.
е) 2-амино-3,5-дигидро-7-(3-фторфенилметил)-4H-пирроло [3,2-d] пиримидин-4-он, т.пл. 297 298oC.
f) 2-амино-3,5-дигидро-7-(3-метилфенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он, т.пл. 253oC.
g) 2-амино-3,5-дигидро-7-(3-метоксифенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он(дигидрат), т.пл. 235oC.
h) 2-амино-3,5-дигидро-7-(3-трифторметилфенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он, т.пл. 239 240oC.
i) 2-амино-3,5-дигидро-7-(3,4-дихлорфенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он, т.пл. 278 280oC (разл).
j) 2-амино-3,5-дигидро-7-(2-фуранилметил)-4Н-пирроло [3,2-d] пиримидин-4-он, т.пл. 244 245oC (разл).
k) 2-амино-3,5-дигидро-7-(2-тиенилметил-4Н-пирроло [3,2-d]пиримидин-4-он, т.пл. 250oC (разл).
l) 2-амино-3,5-дигидро-7-(3-тиенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он, т.пл. 270 271oC (разл).
m) 2-амино-3,5-дигидро-7-(2-хлорофенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он, т.пл. 279 280oC.
h) 2-амино-3,5-дигидро-7-(2-бензилоксифенилметил)-4Н-мирроло [3,2-d] пиримидин-3-он.
o) 2-амино-3,5-дигидро-7-(4-йодофенилметил)-4Н-пирроло [3,2-d] пирмидин-4-он, т.пл. 320 322oC.
Исходный материал получали следующим образом.
Смесь цианоуксусной кислоты (12,76 г, 150,0 мМ) 4-нитробензальдегида (24,60 г, 162,8 мМ), ацетата аммония (500 г), толуола (140 мл) нагревали в течение 64 ч в колбе с обратным холодильником, снабженной ловушкой Дина-Старка. После выпаривания растворителей раствор остатка с CHCl3 фильтровали и промывали водой. Осушенный сульфатом натрия органический слой выпаривали, и образовавшееся желто-оранжевое твердое вещество перекристаллизовывали из бензола. Твердый желтый 3-(4-нитрофенил)-ацетонитрил, полученный в виде смеси цис-транс-изомеров, использовали в качестве промежуточного соединения без дополнительной очистки.
Частичный раствор 3-(4-нитрбензил)-акрилонитрила (24,1 г, 138 мМ) в EtOH (800 мл) гидрировали при атмосферном давлении с 5%-ным палладированным углем (катализатором). Начальная реакция является экзотермической и во избежание перегрева необходимо проводить охлаждение в бане со льдом. Как только восстановление нитрогруппы завершится и образуется почти бесцветный раствор, внешнее охлаждение прекращают. Через 9 ч расход водорода прекращается. Затем катализатор отфильтровывали, растворитель выпаривали, а раствор остатка в виде оранжевого масла в CHCl2/MeOH (99:1) хроматографировали на колонке с силикагелем. Фракции, содержащие 3-(4-аминофенил)-пропионитрил с чистотой, достаточной для использования в качестве промежуточного соединения, собирали и выпаривали досуха.
Смесь 3-(4-аминофенил)-пропионитрила (12,82 г) 87,7 мМ и конц. H2SO4 (18,9 г, 193 мМ) в H2O (85 мл) охлаждали до -4oC в бане со льдом и солью, и полученную суспензию медленно обрабатывали раствором NaNO2 (6,35 г, 98,1 мМ) в воде (30 мл) с такой скоростью, чтобы внутренняя температура оставалась ниже -2oC. Через 10 мин после последнего добавления в целях устранения избытка азотистой кислоты добавляли твердую мочевину (0,53 г, 8,8 мМ). Затем быстро добавляли холодный раствор Kl (20,38 г, 122,8 мМ) в H2O (25 мл), темную реакционную смесь размешивали в течение 4 ч без внешнего охлаждения. Смесь тяжелого масла и воды декантировали из темно-коричневой смолы и экстрагировали EtO2. После выпаривания осушенного (Na2SO4) экстракта маслянистый темный остаток перегоняли в вакууме и получали 3-(4-йодофенил)-пропионитрил.
Пример 3.
а) Частичный раствор 2-амино-3,5-дигидро-7-(4-бензилоксифенилметил)-4Н-пирроло[3,2-d] пиримидин-4-она (249 мг) в EtOH (200 мл) гидрировали в присутствии 10%-ного палладированного угля (75 мг) (катализатора) при атмосферном давлении и в водяной бане, имеющей температуру 55oC. Через 4,5 ч реакция завершалась, а катализатор отфильтровывали в присутствии азота под давлением. После выпаривания фильтрата полученное твердое вещество перекристаллизовывали из EtOH и получали 2-амино-3,5-дигидро-7-(4-гиброксифенилметил)-4Н-пирроло[3.2-d] пирмидин-4-он с т. пл. выше 350oC (разл).
Гидрогенизация, проводимая в вибрационном аппарате Парра при комнатной температуре и начальном давлении водорода 3 атм, протекает быстрее и дает одинаково удовлетворительные результаты.
b) Аналогичным образом получали 2-амино-3,5-дигидро-7-(3-гидроксифенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он, т.пл. 278 280oC.
с) Аналогичным образом получали 2-амино-3,5-дигидро-7(2-гидротксифенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он, т.пл. 360 361oC (разл).
Пример 4.
Раствор 2-амино-3,5-дигидро-7-(2-хлорофенилметил)-4Н-пирроло [3,2-d]пиримидин-4-она (0,5 г, 1,8 мМ) в горячем этаноле (80 мл) обрабатывали 30% Pd-C (1,0 г) в присутствии азота, а затем в течение 10 мин по каплям добавляли моногидрат гидразина (1,5 мл). Реакционную смесь выдерживали при температуре перегонки в течение 16 ч, а затем фильтровали в горячем состоянии через целит. Фильтрат выпаривали досуха, а остаток перетирали с водой (3 мл) и обрабатывали ультразвуком. Продукт собирали, промывали водой и осушали, в результате чего получали 2-амино, 3,5-дигидро-7-(фенилметил)-4Н-пирроло [3,2-d] пиримидин-4-он, т.пл. 269 270oC (разл).
Пример 5.
Получали 1000 капсул, каждая из которых содержала 25 кг активного ингредиента, имели следующий состав (в целом):
2-амино-3,5-дигидро-7-(3-хлорфенил-метил)-4Н-пирроло[3,2-d] пиримидин-4-он 25,00 г
Лактоза 192,00 г
Модифицированный крахмал 80,00 г
Стеарат магния 3,00 г
Процедура. Весь порошок пропускали через сито с отверстиями 0,6 мм. Затем лекарственное вещество помещали в смеситель и смешивали сначала со стеаратом магния, а затем с лактозой и крахмалом до получения гомогенной смеси. Затем с помощью специальной машины для наполнения капсул жесткие желатиновые капсулы N 2 наполняли 300 мг (каждую) указанной смеси. Аналогичные капсулы, содержащие около 1 50 мг других соединений, были получены аналогичным образом.
Пример 6.
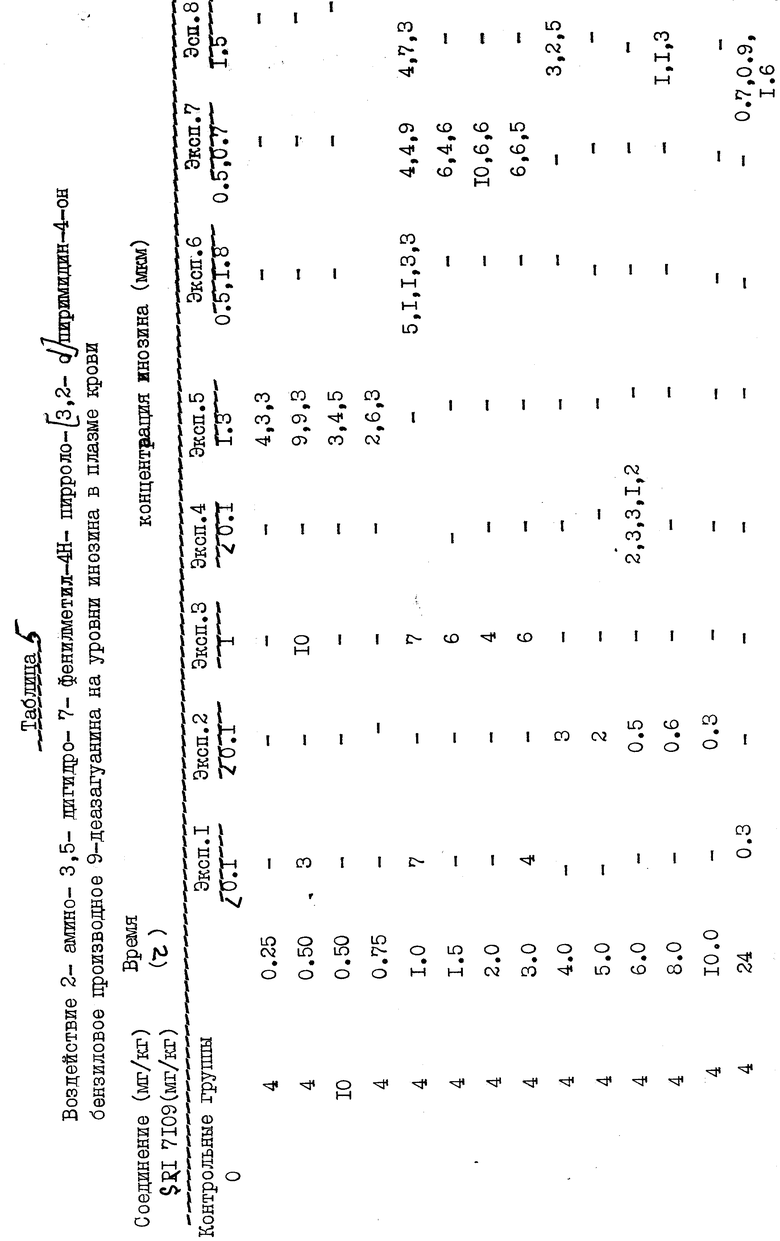
Эффективный метод оцени in vivo действия ПНФ-ингибиторов заключается в оценке уровня нуклеозидов в плазме с последующим введением ингибитора. В частности, было показано, что некоторые ПНФ-ингибиторы, повышающие уровни инозина в плазме крови, способствуют повышению срока жизни кожного алоотрансплантата. В испытаниях настоящего изобретения измеряли уровни инозина в крови лабораторных крыс, в результате чего было продемонстрировано, что соединения настоящего изобретения обладают in vivo активностью.
Крысам Lewis (150-200 г) вводили внутрибрюшинно 2-амино-3,5-дигидро-7(фенилметил)-4Н-пирроло[3,2-d]пиримидин-4-он (бензиловое производное 9-деазагуанина), используя 10% ДМСО в качестве наполнителя. Контрольные группы получали только один наполнитель. Через определенные интервалы времени после введения указанного средства животных забивали и брали образцы плазмы. Плазму экстрагировали холодной 0,5 н HClO4 и нейтрализовали твердым NH4HCO3. После удаления перхлоратных солей экстракт подвергали ВЭЖХ на обращенно-фазовой колонке (Spherisorb ODSI). Результаты эксперимента представлены в табл. 5. Эти результаты показывают значительное повышение уровня инозила в плазме.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 2-АМИНО-7-(CHRR)-3Н,5Н-ПИРРОЛО[3,2-D]-ПИРИМИДИН-4-ОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ ПРОЛИФЕРАЦИИ Т-ЛИМФОЦИТОВ МЛЕКОПИТАЮЩЕГО И НЕ ОКАЗЫВАЮЩИЙ ВОЗДЕЙСТВИЯ НА B-ЛИМФОЦИТЫ | 1992 |
|
RU2097384C1 |
| 2-ГЕТЕРОАРИЛ-5,11-ДИГИДРО-6Н-ДИПИРИДО[3,2-B:2',3'-E][1,4] ДИАЗЕПИН-6-ОНЫ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИЧ-1 | 1995 |
|
RU2142464C1 |
| ИНГИБИТОРЫ Axl ДЛЯ ПРИМЕНЕНИЯ В КОМБИНИРОВАННОЙ ТЕРАПИИ ДЛЯ ПРЕДОТВРАЩЕНИЯ, УСТРАНЕНИЯ ИЛИ ЛЕЧЕНИЯ МЕТАСТAЗИРУЮЩЕГО РАКА | 2010 |
|
RU2555326C2 |
| ЗАМЕЩЕННЫЕ АМИДЫ ТИЕНОПИРРОЛКАРБОНОВОЙ КИСЛОТЫ, АМИДЫ ПИРРОЛОТИАЗОЛКАРБОНОВОЙ КИСЛОТЫ И РОДСТВЕННЫЕ АНАЛОГИ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЗЕИНКИНАЗЫ Iε | 2005 |
|
RU2403257C2 |
| ПРОЛЕКАРСТВЕННЫЕ ПРОИЗВОДНЫЕ 1,3-ДИАМИНО-2-ГИДРОКСИПРОПАНА | 2003 |
|
RU2357962C2 |
| БИЦИКЛИЧЕСКИЕ ПИРИДИНЫ И АНАЛОГИ В КАЧЕСТВЕ МОДУЛЯТОРОВ СИРТУИНА | 2010 |
|
RU2550821C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ АНТАГОНИСТА СRTH2 (ВАРИАНТЫ) | 2004 |
|
RU2361865C2 |
| ХИНАЗОЛИНОВЫЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2135481C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛПИРИМИДИНОНА, СПОСОБ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2263676C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2'-ДЕЗОКСИ-2'-ФТОРРИБОНУКЛЕОЗИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2043361C1 |

Производные 9-деазагуанины или их таутомеры и фармацевтическая композиция, обладающая способностью ингибировать активность пуриннуклюзид фосфорилазы у млекопитающих Назначение: в медицине и биохимии Сущность изобретения: Производные 9-деазагуанина формулы I, приведенной в описании, где R1 - водород, галоген, алкил /С1-С3/, алкокси /С1-С3/, бензилокси, гидрокси, трифторметил, R2 - водород, галоген, /С1-С3/ алкокси, бензилокси, гидрокси, трифторметил при условии: если R2 - водород или алкил /С1-С3/, R1 - трифторметил или R1 = водород, /С1-С3/ алкил, если R2 - трифторметил или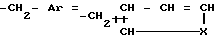
где x - сера, кислород, а связь с тиофеновым или фурановым кольцом имеет место во 2-м или 3-м положении или их таутомеры. Соединения 1 получены путем взаимодействия реагента 1 приведенного в описании, где R3 и R5 - низший алкил, R4 - карбоциклический арил реагента 2: безводный аммиак с последующей циклизацией и, в случае необходимости, отщеплением защитных групп. Фармацевтическая композиция, обладающая способностью ингибировать активность пуриннуклюзид фосфорилазы у млекопитающих, содержащая производное 9-деазагуанина формулы I или его таутомер в качестве активного начала в количестве от 0,1 до 75%, предпочтительно от 1 до 50%, и фармацевтически приемлемый носитель. 5 табл.
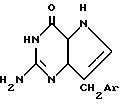
где CH2Ar -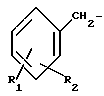
R1 водород, галоген, C1 C3-алкил C1 - C3-алкокси, бензилокси, гидрокси или трифторметил;
R2 водород, галоген, C1 C3-алкил, C1 - C3-алкокси, бензилокси, гидрокси или трифторметил, при условии если R2 водород или C1 C3-алкил, R1 трифторметил, или R1 водород, C1 C3-алкил, если R2 - трифторметил или CH2Ar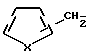
где X сера или кислород, а связь с теофеновым или фурановым кольцом имеет место во 2- или 3-м положении,
или их таутомеры.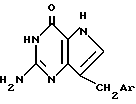
где CH2Ar -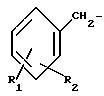
R1 водород, галоген, C1 C3-алкил, C1 - C3-алкокси, бензилокси, гидрокси или трифторметил;
R2 водород, галоген, C1 C3-алкил, C1 - C3-алкокси, бензилокси, гидрокси, трифторметил при условии, если R2 водород или C1 C3-алкил, R1 трифторметил или R1 водород или C1 C3-алкил, если R2 - трифторметил или CH2Ar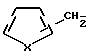
где X сера или кислород, а связь с тиофеновым или фурановым кольцом имеет место во 2- или 3-м положении,
или его таутомер в количестве 0,1 75,0% предпочтительно 1 50%
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ЗАПОМИНАЮЩЕЕ УСТРОЙСТВО | 0 |
|
SU178178A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Патент США N 4772606, кл.C 07D 473/02, 1988 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| H | |||
| Stenzel, B | |||
| Hummer и др | |||
| Деревянное стыковое устройство | 1920 |
|
SU163A1 |
