Изобретение относится к новым производным 2-амино-7-(CH2R2R3)-3H,5H-пирроло[3,2-d] пиримидинам, обладающим свойствами селективного ингибитора Т-лимфоцитов, способам их получения и способу селективного ингибирования пролиферации Т-лимфоцитов млекопитающего и не оказывающего воздействия на B-лимфоциты.
Известно, что пуриннуклеозидфосфорилаза (PNP) катализирует фосфоролиз пуриннуклеозидов в обратимой реакции. У индивидуумов, лишенных PNP, проявляется ослабленное развитие Т-лимфоцитов, приводящее к пониженному межклеточному иммунитету, и нормальное развитие базофильного инсулоцита, приводящее к нормальному гуморальному иммунитету. Соответственно специфические ингибиторы PNP, которые селективно ингибируют развитие Т-лимфоцитов без нарушения гуморального иммунитета, могут быть потенциально эффективны против болезней, в которых активированные Т-лимфоциты являются патогенными.
В журнале (Science, 1981, v. 214) описаны 8-аминогуанозины, обладающие селективным действием на Т-клетки без воздействия на B-клетки.
Новые производные пирролопиримидинонов, в основе которых лежит бициклическая гетероциклическая система, неожиданно проявили свойство селективно ослаблять функции Т-лимфоцитов млекопитающего без ослабления влияния на гуморальный иммунитет и благодаря этим найденным и исследованным свойствам могут найти применение для лечения тяжелых заболеваний таких, как аутоиммунных нарушений, отторжение трансплантированных органов, а также в терапии онкологических заболеваний.
Согласно изобретению предлагаются производные 2-амино-7-(CHR2R3)-3H, 5H-пирроло-[3,2-d]пиримидин-4-она формулы (I)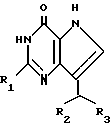
где R1 означает H или NH2;
R2 означает насыщенный пятичленный гетероцикл, содержащий в качестве гетероатома серу или кислород, или шестичленный гетероцикл, содержащий в качестве гетероатома N, которые могут быть необязательно замещены (C1-C4)алкокси, трифторметилом или галогеном; (C6-C10)циклоалкил, незамещенный или галоидзамещенный фенил;
R3 означает H, (CH2)nCN, (CH2)nCOOH, CO2CH3, (CH2)nCONH2, (CH2)nCO2CH3, (CH2)nOH, n 1-4, R2 и R3 вместе с CH-группой, к которой они присоединены, образуют циклогексил или циклогексенильную группу. Предпочтительны соединения, в которых R2 и R3 вместе с CH-группой, к которой они присоединены, образуют циклогексил или циклогексенил.
Предпочтительны соединения, где R1-NH2 группа, и соединения, где R2 и R3 вместе с прилегающей CH-группой образуют циклогексильное или циклогексенильное кольцо, либо R2 фенил, соединения, где R3-CH2CN, CH2COOH, CH2CONH2, либо соединения, где R2 возможно замещенные C1-C4-алкокси, галогеном, трифторметилом, 3-пиперидинил, 2-тетрагидрофуранил, 2-тетрагидротиенил, 3-, 4-пиридинил, циклопентил, циклогексил, циклогептил, 2-адамантил; либо фенил, 3-хлорфенил, 2,3,5-трихлорфенил.
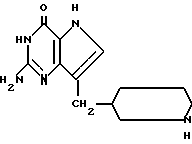
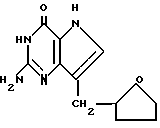
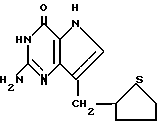
Среди предпочтительных соединений можно назвать 2-амино-7(CH2R)-3H,5H-пиролло[3,2-d] пиримидин-4-оны(I'), где R 2- или 3-тетрагидротиенил, 2-, 3- или 4-пиперидинил, 2- или 3-тетрагидрофуранил, 2-, 3- или 4-тетрагидрофуранил. В частности можно назвать соединения, предлагаемые согласно изобретению, выбранные из группы: 2-амино-7-(2-пиперидинилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IB), 2-амино-7-(3-пиперидинилметил)-3Н,5Н-пирроло[3,2-d] пиримидин-4-он (I'C), 2-амино-7-(4-пипepидинилмeтил)-3H,5H-пиppoлo[3,2-d] пиримидин-4-он (I'D), 2-амино-7-(2-тетрагидрофуранилметил)-3H,5H-пирроло[3,2-d] пиримидин-4- -он (I'E), 2-амино-7-(3-тетрагидрофуранилметил)-3H,5H-пирроло[3,2-d]пиримидин-4-он (I'F), 2-амино-7-(2-тетрагидротиенилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (I'G), 2-амино-7-(3-тетрагидротиенилметил)-3H,5H-пирроло[3,2-d] пиримидин-4-он (I'H).
В указанных соединениях гетероциклический радикал R в положении 7 может быть замещен одним или двумя заместителями, выбранными из группы: галоген, (C1-C4)алкокси, трифторметил.
В качестве галогена предпочтительным является хлор или фтор. В качестве алкокси предпочтительными являются низший алкокси, метокси, этокси, пропокси и бутокси.
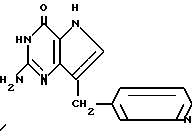
Среди соединений, охватываемых изобретением, можно назвать 2-амино-7-(CH2R)-3H, 5H-пирроло[3,2-d] пиримидин-4-оны (II), в которой R-пиридинил, в частности, соединения II, выбранные из группы, 2-амино-7-(3-пиридинилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IIA), 2-амино-7-(2-пиридинилметил)-3H,5H-пирроло[3,2-d]пиримидин-4-он (IIB), или 2-амино-7-(4-пиридинилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IIC), в которых пиридинил может иметь один или два заместителя, выбранных из группы, содержащей галоген, (C1-C4)алкокси или трифторметил. В качестве галогена предпочтительны хлор или фтор. В качестве алкокси предпочтительны метокси, этокси, пропокси и бутокси.
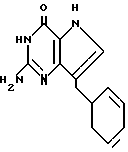
Другой группой предпочтительных соединений являются (III) 2-амино-7-(R)-3H, 5H-пирроло[3,2-d] пиримидин-4-оны, в которых R 1-, 2- или 3- циклогексенил или циклогексил. Среди них можно назвать соединения III, выбранные из группы: 2-амино-7-(1-циклогексенил)-3H, 5H-пирроло[3,2-d]пиримидин-4-он (IIIA), 2-амино-7-(2-циклогексенил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IIIB), 2-амино-7-(3-циклогексенил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IIIC) или 2-амино-7-(циклогексил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IIID).
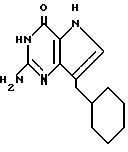
Согласно изобретению предлагаются также 2-амино-7-(CH2R)-3H,5H-пирроло[3,2-d]пиримидин-4-оны (IV), в которых R однокольцевые циклопарафины, как, например, циклопентил, циклогексил и циклогептил; многокольцевые циклопарафины, как, например, 1- и 2-адамантил, 1-норборнанил, 2-экзо-норборнанил, 2-эндо-норборнанил, 1- и 2-бицикло[2.2.2]октанил, 1-, 2-, 3-, 6- и 8-бицикло[3.2.1] октанил, и 1-, 2- и 3-бицикло[3.3.1]нонанил, и циклоолефины, как, например, 1- и 2-норборненил. Примерами этой группы веществ (IV) являются 2-амино-7-(2-адамантилметил)-3H,5H-пирроло[3,2-d]пиримидин-4-он (IVA), 2-амино-7-(1-адамантилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IVB), 2-амино-7-(циклопентилметил)-3H, 5H-пирроло[3,2-d]пиримидин-4-он (IVC), 2-амино-7-(циклогексилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IVD), 2-амино-7-(циклогептилметил)-3H, 5H-пирроло[3,2-d]пиримидин-4-он (IVE), 2-амино-7-(1-норборнанилметил)-3H,5H-пирроло[3,2-d]пиримидин-4-он (IVF), 2-амино-7-(2-экзо-норборнанилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IVG), 2-амино-7-(2-эндо-норборнанилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IVH), 2-амино-7-(1-норборненилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IVI), 2-амино-7-(2-норборненилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IVJ), 2-амино-7-(1-бицикло[2.2.2] октанилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он (IVK), 2-амино-7-(1-бицикло[3.2.1] октанилметил)-3H,5H-пирроло[3,2-d] пиримидин-4-он (IVL) и 2-амино-7-(1-бицикло[3.3.1] нонанилметил)-3H,5H-пирроло[3,2-d] пиримидин-4-он (IVM), и 2-амино-7-(1-норадамантилметил)-3H,5H-пирроло[3,2-d] пиримидин-4-он (IVN).
Среди соединений, полученных согласно изобретению, можно назвать группу производных 2-амино-7-(CHR2R3)-3H, 5H-пирроло[3,2-d]пиримидин-4-она формулы (I), где R1, R2 имеют значения указанные выше, а R3 является группой, выбранной из H, CH2CN, CH2CO2H, CH2CONH2, CO2CH3, CH2CH2OH, CH2CO2CH3.
Другим объектом изобретения является способ селективного ингибирования пролиферации Т-лимфоцитов млекопитающего, неоказывающий воздействия на B-лимфоциты, заключающийся в том, что млекопитающему вводят вышеуказанные соединения формулы (I) в эффективном количестве, при этом указанное соединение ингибирует пуриннуклеозидфосфорилазу и тем самым образование Т-лимфоцитов.
Возможно также введение млекопитающим эффективного количества соединения формулы (I), не оказывающего ослабляющего действия на гуморальный иммунитет и оказывающего селективное ослабляющее действие на Т-лимфоциты, с фармацевтически переносимым разбавителем.
Особенностью предлагаемого способа селективного ослабления функции T-лимфоцитов млекопитающего без ослабления влияния на гуморальный иммунитет, является то, что введение млекопитающему вещества формулы (I) ингибирует пуриннуклеозидфосфорилазу и тем самым образование T-лимфоцитов.
Для энтерального такого, как пероральное или ректальное, трансдермальное или парентеральное, введения млекопитающим, включая человека, которые пригодны для ингибирования активности пуриннуклеозидфосфорилазы и для лечения болезней, восприимчивых к ней, возможно введение эффективного количества фармакологически активного вещества изобретения одного или в комбинации с одним или более фармацевтически переносимыми носителями.
Предпочтительной используемой формой композиций являются таблетки и желатиновые капсулы, включающие активный ингредиент вместе с: а) разбавителями, например, лактозой, глюкозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином; б) смазывающими веществами, например, двуоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток также с: в) связующими веществами, например, магнийалюмосиликатом, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, натрий карбоксиметилцеллюлозой и/или поливинилпирролидоном; при желании: г) дезинтегрантами, например, крахмалами, агар-агаром, альгиновой кислотой или ее натриевой солью, или шипучими смесями; и/или д) абсорбентами, красителями, корригентами и подслащивателями. Вспрыскиваемые композиции являются предпочтительно водными изотоническими растворами или суспензиями, и суппозитории преимущественно приготавливают из жировых эмульсий или суспензий. Указанные композиции могут быть стерилизованными и/или содержать адъюванты такие, как например, предохраняющие, стабилизирующие, смачивающие или эмульгирующие агенты, растворенные промоторы, соли для регулирования осмотического давления и/или буферные растворы. Кроме того, они могут также содержать другие терапевтически ценные вещества. Указанные композиции приготавливают в соответствии со стандартными способами перемешивания, гранулирования или покрытия соответственно и они содержат обычно около 0,1-75,0% предпочтительно около 1-50% активного ингредиента.
Для трансдермального применения используют эффективное количество вещества изобретения с носителем. Полезные носители включают абсорбируемые фармакологически приемлемые растворители для того, чтобы помочь проходить в организм через кожу. Существенным является то, что трансдермальные аппараты могут иметь форму бандажа, включающего поддерживающую деталь, резервуар, содержащий предпочтительное вещество с носителями, перегородку для доставки вещества к коже организма с контролируемой и предварительно определенной скоростью в течение продолжительного периода времени, и средства для прикрепления аппарата к коже.
Предлагаемый способ селективного ингибирования активности пуриннуклеозидфосфорилазы может быть использован при лечении болезней и сред, восприимчивых к ним, например, при аутоиммунных нарушениях, отторжениях при трансплантации, псориазе.
Способ также пригоден для ингибирования фосфоролиза и метаболического расщепления противовирусных или противоопухолевых пуриннуклеозидов у млекопитающих путем введения соединения формулы (I) самостоятельно или с одним или более фармацевтически переносимыми носителями. В частности, это относится к способу ингибирования фосфоролиза и метаболического расщепления известных пуриннуклеозидов, например, 2'-дезоксигуанозина, 2',3'-дидезоксиинозина, 2',3'-дидезоксигуанозина или 2',3'-дидезоксиаденозина.
Таким образом, изобретение относится также к способу ингибирования фосфоролиза или метаболического расщепления противовирусного или противоопухолевого эффекта 2'- или 3'-монодезоксипуриннуклеозидов или 2',3'-дидезоксипуриннуклеозидов у млекопитающих, который включает введение млекопитающим при необходимости, или отдельно, или в комбинации с указанным нуклеозидом эффективного количества вещества изобретения, ингибирующего пуриннуклеозидфосфорилазу, предпочтительно в комбинации с одним или более фармацевтически переносимыми носителями. Более конкретно, это относится к способу активации или усиления влияния 2',3'-дидезоксипуриннуклеозидов, известных в технике, например, 2',3'- дидезоксиинозина, 2',3'-дидезоксигуанозина или 2',3'-дидезоксиаденозина для лечения ретровирусных инфекций, например, ВИЧ-ретровирусных инфекций (приобретенный иммунодефицитный синдром, AIDS). 2',3'-Дидезоксипуриннуклеозиды известны в качестве ингибиторов ВИЧ-ретровирусной инфекции и метаболически расщепляются посредством PNP, например, как описано в Biochemical Pharmacology, 22, 3797 (1987). Они вводятся в фармацевтически переносимой дозе, которая эффективна в ингибировании ВИЧ-ретровирусных инфекций. Предпочтительно используется самая низкая возможная эффективная доза.
Фармацевтически переносимая эффективная дозировка активного вещества изобретения, которую вводят, зависит от вида теплокровных животных (млекопитающих), веса тела, возраста, индивидуального состояния и от формы введения.
Фармацевтическая композиция может быть пероральной, парентеральной, суппозиторием или другой формой, которая доставляет вещество в ток крови млекопитающего, которое необходимо вылечить. Пероральная форма имеет от около 1 до около 150 мг активного вещества для взрослого человека (50-70 кг), которое смешивается вместе с фармацевтически переносимыми разбавителями, например, лактозой. В обычной капсуле 25 мг вещества смешивают вместе с 192 мг лактозы, 80 мг модифицированного крахмала и 3 мг стеарата магния. Возможно также использование форм, пригодных для вспрыскивания.
Использование соединений настоящего изобретения также возможно с другими терапевтическими средствами. Ежедневная дозировка для человека (весом 50-70 кг составляет 1-50 мг/кг) ингибирует метаболическое разрушение определенных противораковых средств, например, бета-2'-дезокси-6-тиогуаназина и противовирусных средств, например, 2',3'-дидезоксиинозина, анти-AIDS средства. Эти типы медикаментов, как известно, неустойчивы и разрушаются, теряя свою эффективность. Вещества изобретения способны уменьшить такую неустойчивость, что увеличивает эффективность других химиотерапевтических средств.
Объектом изобретения являются также способы получения производных 2-амино-7-(CHR2,R3)-3H,5H-пирроло[3,2-d]пиримидин-4-онов общей формулы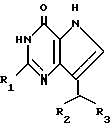
где R1 означает H, NH2;
R2 и R3 означают незамещенный или галоидзамещенный фенил или циклогексил;
R2 означает CH2CN.
Один способ предназначен для получения соединений указанной формулы, где R1 означает NH2, а другой для соединений указанной формулы, где R1 означает H.
Способ получения производных 2-амино-7-(CHR2R3)-3H,5H-пирроло[3,2-d]пиримидин-4-она (способ A) формулы (I')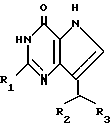
где R1 NH2;
R2 фенил, незамещенный или замещенный галоидом, или циклогексил;
R3 CH2CN, заключается в том, что осуществляют следующие последовательные стадии:
а) реакцию соответствующего R2-замещенного альдегида с циануксусной кислотой в присутствии ацетата аммония с получением 3(R2)-замещенного пентандинитрила;
б) реакцию полученного 3(R2)-пентандинитрила с алкиловым эфиром муравьиной кислоты в присутствии основания, с получением 3(R2)-2-формилпентандинитрила,
в) который взаимодействует с гидрохлоридом метилового эфира глицина в присутствии ацетата натрия или аммония с получением метил-N-[(3(R2)-2,4-дициано)-2-бутенил]глицина;
г) полученный при этом N-[(3(R2)-2,4-дициано)-2-бутенил]глицин взаимодействует с алкиловым эфиром хлоруксусной кислоты в присутствии 1,5-диазабицикло[4.3.0]-нон-5-еном или 1,8-диазабицикло[5.4.0]ундек-7-еном с получением метил-3-амино-4-(2-циано-1(R2)-этил)-1-этил-1H-пирроло-1,2- дикарбоксилата;
д) получаемый метил-3-амино-4-(2-циано-1(R2)-этил)-1-этил-1H-пирроло-1,2- дикарбоксилат обрабатывают основанием с получением метил-3-амино-4-(2-циано-1(R2)-этил)-1H-пирроло-2-карбоксилата;
е) последний подвергают взаимодействию с бензоилизотиоцианатом с получением N-бензоил-N'-[4-(2-циано-1(R2)-этил)-2-метоксикарбонил-1Н-пиррол-3-ил]тиомочевины;
ж) полученную N-бензоил-N'-[4-(2-циано-1(R2)-этил)-2-метоксикарбонил-1H-пиррол-3-ил] тиомочевину алкилируют галоидным алкилом с получением N-бензоил-N'-[4-(2-циано-1(R2)-этил)-2-метоксикарбонил-1H-пиррол-3-ил] -S-метилтиомочевины;
з) затем N-бензоил-N'-[4-(2-циано-1(R2)-этил)-2-метоксикарбонил-1H-пиррол-3-ил] -S-метилтиомочевины обрабатывают метанольным или этанольным раствором аммиака с получением смеси 3(R2)-3-[2-амино-4-оксо-3H,5H-пирроло[3,2-d] пиримидин-7-ил] пропаннитрила и 3(R2)-3-[2-меркапто-4-оксо-3H,5H-пирроло[3,2-d]пиримидин-7-ил]пропаннитрила.
Предпочтительно этот способ использовать для соединений I', где R2 фенил.
Способ получения производных 7(CHR2R3)-3H,5H-пирроло-[3,2-d]пиримидин-4-она формулы (I") (способ B)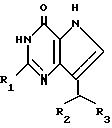
где R1 H;
R2 фенил, незамещенный или замещенный галоидом, или циклогексил;
R3 CH2CN, заключается в том, что осуществляют последовательные стадии:
а) реакцию соответствующего циклического альдегида с циануксусной кислотой в присутствии ацетата аммония с получением 3(R2)-замещенного пентандинитрила;
б) реакцию полученного 3(R2)-пентандинитрила с алкиловым эфиром муравьиной кислоты в присутствии основания с получением 3(R2)-2-формилпентандинитрила;
в) последний подвергают взаимодействию с гидрохлоридом метилового эфира глицина в присутствии ацетата натрия или аммония с получением метил-N-[(3(R2)-2,4-дициано)-2-бутенил]глицина;
г) полученный метил-N-[(3(R2)-2,4-дициано)-2-бутенил]глицин подвергают взаимодействию с алкиловым эфиром хлоруксусной кислоты в присутствии 1,5-диазабицикло[4.3.0] нон-5-ена или 1,8-диазабицикло[5.4.0]ундек-7-ена с получением метил-3-амино-4-(2-циано-1(R2)-этил)-1-этил-1H-пиррол-1,2- дикарбоксилата;
д) метил-3-амино-4-(2-циано-1(R2)-этил)-1-этил-1H-пиррол-1,2-дикарбоксилат обрабатывают основанием с получением метил-3-амино-4-(2-циано-1(R2)-этил)-1H-пирроло-2-карбоксилата;
е) затем метил-3-амино-4-(2-циано-1(R2)-этил)-1H-пирроло-2-карбоксилат подвергают взаимодействию с диметилацеталем диметилформамида с получением метил-4-(2-циано-1(R2)-этил)-3-[N-(диметиламино-метилен)амино] -1H-пирроло-2-карбоксилата;
ж) метил-4-(2-циано-1(R2)-этил)-3-[N-(диметиламинометилен)амино]-1Н-пирроло-2-карбоксилат обрабатывают метанольным раствором аммиака с получением 3(R2)-3-[4-оксо-3H, 5H-пирроло[3,2-d] пиримидин-7-ил] пропаннитрила. Более подробно способ для получения соединений формулы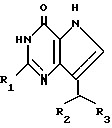
где R1 аминогруппа (способ A), проводится следующим образом.
Первая стадия способа включает реагирование соответствующего R2-замещенного циклического альдегида с циануксусной кислотой при молярном отношении около 1: 1 до 1:5 в присутствии ацетата аммония при температуре около температуры кипения с обратным холодильником в течение около 10 ч до 8 дней с получением 3R2-замещенного пентандинитрила в качестве промежуточного соединения. Во второй стадии 3(R2)-замещенный пентандинитрил реагирует с алкиловым эфиром муравьиной кислоты, например, этиловым эфиром муравьиной кислоты, в присутствии сильного основания, например, металлсодержащего основания, гидрида натрия или алкоксида натрия, в частности метоксида натрия, при молярном отношении около 1-2:3-6:1-3 и при температуре около 20-65oC в течение около 10 ч до 8 дней с получением 3(R2)-2-формилпентандинитрила. Следующая стадия включает взаимодействие 3(R2)-2-формилпентандинитрила с гидрохлоридом метилового эфира глицина и в присутствии ацетата натрия или аммония при молярном отношении около 1-2:1,5-4:1,5-4 и при температуре около 20-60oC в течение около 10-48 ч с получением метил-N-[(3(R2)-2,4-дициано)-2-бутенил] глицина в качестве промежуточного соединения. В последующей стадии метил-N-[(3(R2)-2,4-дициано)-2-бутенил] глицин реагирует с алкиловым эфиром хлормуравьиной кислоты, например, этиловым эфиром хлормуравьиной кислоты, в присутствии 1,5-диазабицикло[4.3.0] нон-5-еном (DBN) или 1,8-диазабицикло[5.4.0]ундек-7-еном (DBU) при молярном соотношении около 1-2:1,5-5:1,5-4 и при температуре около 0-50oC в течение около 10 ч до 10 дней с получением метил-3-амино-4-(2-циано-1(R2)этил)-1-этил-1Н-пиррол-1,2-дикарбоксилата в качестве промежуточного соединения. Следующая стадия включает взаимодействие метил-3-амино-4-(2-циано-1(R2)этил)-1-этил-1Н-пиррол-1,2-дикарбоксилата с основанием, например, карбонатом натрия, при молярном соотношении около 2:1 до 1:5 и при температуре около комнатной в течение около 10-48 ч с получением метил-3-амино-4-(2-циано-1(R2)этил)-1H-пиррол-2-карбоксилата в качестве промежуточного соединения. В следующей стадии метил-3-амино-4-(2-циано-1(R2)-этил)-1Н-пиррол-2-карбоксилат реагирует с бензоилизотиоцианатом при молярном соотношении около 2:1 до 1:2 и при температуре около комнатной в течение около от 30 мин до 3 ч с получением N-бензоил-N-[4-(2-циано-1(R2)этил)-2-метоксикарбонил-1H-пиррол-3-ил] тиомочевины в качестве промежуточного соединения. Следующую стадию взаимодействия N-бензоил-N-[4-(2-циано-1(R2)этил)-2-метоксикарбонил-1H-пиррол-3-ил] тиомочевины с галоидным алкилом, например, йодистым метилом, проводят при молярном отношении около 1:1 до 1:6 и при температуре около 0-30oC в течение от приблизительно 10 мин до 10 ч с получением N-бензоил-N-[4-(2-циано-1(R2)-этил)-2-метоксикарбонил-1Н-пиррол-3-ил]-S-метилтиомочевины в качестве промежуточного соединения. В следующей стадии N-бензоил-N-[4-(2-циано-1(R2)-этил)-2-метоксикарбонил-1Н-пиррол-3-ил] -S-метилтиомочевину (около 1-2 моль) обрабатывают метанольным или этанольным раствором аммиака при молярном соотношении около 1:1 до 1:20 и при температуре около 20-130oC в течение около 16-60 ч с получением смеси 2-аминосоединения изобретения 3(R2)-3-(2-амино-4-оксо-3H,5H-пирроло [3,2-d] пиримидин-7-ил) пропаннитрила и 3(R2)-3-(2-метилмеркапто-4-оксо-3H,5H-пирроло[3,2-d] пиримидин-7-ил)пропаннитрила, которое может быть использовано в качестве промежуточного соединения для получения другого вещества изобретения.
В другом способе получения соединений, где R1 водород (способ B) метил-3-амино-4-(2-циано-1(R2)-этил)-1Н-пиррол-2-карбоксилат, полученный выше в способе A, обрабатывают диметилацеталем диметилформамида при молярном соотношении около 1:1 до 1:4 и при температуре около 25-100oC в течение около 1-10 дней с получением метил-4-(2-циано-1(R2)этил)-3-[N-(диметиламинометилен)амино] -1H-пирроло-2-карбоксилата в качестве промежуточного соединения. Следующая стадия включает обработку метил-4-(2-циано-1(R2)этил)-3-[N-(диметиламинометилен)амино] -1H-пирроло-2-карбоксилата метанольным или этанольным раствором аммиака при молярном соотношении около 1: 1 до 1:20 и при температуре около 20-130oC в течение около 10-68 ч с получением соединения изобретения 3(R2)-3-(4-оксо-3H,5H-пирроло[3,2-d]пиримидин-7-ил)-пропаннитрила.
Итак, для получения соединений формулы (I) используют 3-замещенные пропионитрилы в качестве исходных веществ. Такие исходные вещества могут быть получены с помощью целого ряда способов, которые хорошо известны в литературе. Соединение формулы (I) получается затем из указанного исходного вещества с использованием ряда известных реакций (M.I.Lim, R.S.Klein and J.J. Fox, J. Org. Chem. 44,3826 (1979); M.I.Lim, R.S.Klein and J.J.Fox, Tetrahedron Lett. 21,1013 (1980); M.I.Lim and R.S.Klein, Tetrahedron Lett. 22,25 (1981); M. I. Lim, W. I.Ren, B.A.Otter and R.S.Klein, J. Org. Chem. 48,780 (1983)).
Для получения группы соединений формулы (II), где R пиридинил, используют 3-(пиридинил)пропионитрил в качестве исходного вещества. Соответствующий 3-(пиридинил)пропионитрил может быть получен путем превращения соответствующего 3-(пиридинил)пропионилхлорида в соответствующий амид реакцией амминолиза, например, обработкой аммония, который затем дегидратируется в желаемый нитрил при дистилляции с дегидратирующим агентом, например, POCl3 или SOCl2. Либо исходное вещество получают конденсацией 3-альдегида с цианоуксусной кислотой с последующим декарбоксилированием с получением соответствующего замещенного акрилонитрила, который гидрируют для получения соответствующего 3-(пиридинил)пропионитрила или каталитически, или при растворении металлического магния в метаноле при 0oC (Profitt, J. et al. J. Org. Chem. 40, 127 (1975)). Далее вещества (II) получают из указанного вещества по методике (M.I.Lim, R.S.Klein and J.J.Fox, J. Org. Chem. 44,3826 (1979); M. I. Lim, R.S.Klein and J.J.Fox, Tetrahedron Lett. 21,1013 (1980); M.I.Lim and R. S.Klein, Tetrahedron Lett. 22,25 (1981); M.I.Lim, W.I.Ren, B.A.Otter and R.S.Klein, J. Org. Chem. 48, 780 (1983)).
Возможно использование в качестве исходного вещества циклогексенилацетонитрила для получения соединений группы (III) в результате взаимодействия этого исходного вещества с использованием методики (M.I.Lim, R.S.Klein and J. J.Fox, J. Org. Chem. 44,3826 (1979); M.I.Lim, R.S.Klein and J.J.Fox, Tetrahedron Lett. 21,1013 (1980); M.I.Lim and R.S.Klein, Tetrahedron Lett. 22,25 (1981); M.I.Lim, W.I.Ren, B.A.Otter and R.S.Klein, J. Org. Chem. 48, 780 (1983)). Каталитическое гидрирование одного из (IIIA), (IIIB) или (IIIC) дает вещество (IIID).
Способ получения группы соединений (IV) изобретения использует соответствующие 3-замещенные пропионитрилы в качестве исходных веществ. Такие исходные вещества могут быть получены посредством ряда способов, которые хорошо известны в литературе. Вещество (IV) затем получают из этого исходного вещества посредством использования методик (M.I.Lim, R.S.Klein and J.J.Fox, J. Org. Chem. 44,3826 (1979); M.I.Lim, R.S.Klein and J.J.Fox, Tetrahedron Lett. 21,1013 (1980); M.I.Lim and R.S.Klein, Tetrahedron Lett. 22,25 (1981); M.I. Lim, W.I.Ren, B.A.Otter and R.S.Klein, J. Org. Chem. 48, 780 (1983)).
Как очевидно для специалистов в данной области техники, варианты перечисленных способов являются полезными при получении ряда веществ изобретения без отклонения от существа изобретения. Для реакций, включающих некоторые промежуточные соединения, требующие защиты атомов азота или кислорода в промежуточных соединениях, используют известные способы.
Чтобы более полно описать изобретение, ниже приводятся следующие примеры, не ограничивающие область его применения. В примерах все части и проценты даны в масс. за исключением особо оговоренных случаев. Пропорции смесей растворителей, использованных в качестве хроматографических растворителей для элюирования, даны по объему.
Испытания (как следует из материалов заявки) проводились на млекопитающих. В частности, в примерах, приведенных в описании, опыты проводились на крысах линии Lewis, которым делалось внутрибрюшинное введение полученных по изобретению веществ в виде инъекционных растворов с указанием дозировок, частоты инъекций и сравнительных опытов с контрольными группами животных. Результаты оговорены там же (примеры 22, 53, 65, 70, 81, 89 и др.), а также в приведенной табл. 1.
Пример 1. Способ получения 3-(3-пиридинил)пропионитрила. В трехгорлую колбу, снабженную магнитной мешалкой, термометром, капельной воронкой и обратным холодильником, вводят аргон через холодильник. Свежеизмельченный гидроксид калия (6,6 г, 0,1 моль) и безводный ацетонитрил (150 мл) загружают в колбу и нагревают до кипения с обратным холодильником, добавляют 3-пиридинкарбоксальдегида (10,7 г, 0,1 моль) в безводном ацетонитриле (50 мл) по каплям в течение около 5 мин и кипячение с обратным холодильником продолжают в течение следующих приблизительно 3 мин. Полученную горячую реакционную смесь выливают в смесь лед/вода (100 г) и полученный раствор экстрагируют с CH2Cl2 (3 x 100 мл), высушивают с Na2SO4 и выпаривают, получая сырой 3-(3-пиридинил)акрилонитрил, который очищают посредством колоночной хроматографии над силикагелем, используя CHCl3 в качестве растворителя для элюирования; выход 3,3 г (25,6%).
В атмосфере аргона перемешиваемый раствор акрилонитрила (2,662 г, 0,02 моль) в 99%-ном этаноле (100 мл) обрабатывают каплей 4%-ного водного гидроксида натрия, затем боргидридом натрия (0,378 г, 0,01 моль). Дополнительно боргидрид натрия (0,378 г) добавляют еще дважды с 4-часовыми интервалами. Смесь перемешивают при комнатной температуре в течение ночи, разбавляют водой, экстрагируют с EtOAc и высушивают Na2SO4. Растворитель удаляют при пониженном давлении, полученный сырой продукт хроматографируют через колонку с силикагелем, используя хлороформ: метанол (40:1) в качестве растворителя для элюирования, получая 2,2 г (84,6%) продукта в виде бесцветного масла.
3-(3-Пиридинил)пропионитрил, полученный в примере 1, используют далее для синтеза соединений изобретения.
Пример 2. В атмосфере сухого N2 смесь 3-(3-пиридинил)пропионитрила (0,661 г, 5,0 ммоль), гидрида натрия (0,240 г, 10,0 ммоль) и этилового эфира муравьиной кислоты (1,11 г, 15,0 ммоль) в безводном тетрагидрофуране (20 мл) перемешивают в течение 48 ч с защитой от воздуха и влаги. Летучее вещество выпаривают и раствор твердого остатка в 15 мл холодной воды доводят при 0oC до pH 6 с помощью холодной 6N HCl. Полученную маслянистую смесь экстрагируют CHCl3, экстракт промывают водой, высушивают, используя Na2SO4, и выпаривают, получая темное масло, которое является смесью 2-формил-3-(3-пиридинил)пропионитрила и нитрила исходного вещества. Этот сырой продукт используют в следующей реакции без дальнейшей очистки.
Пример 3. Гидрохлорид метилового эфира глицина (0,942 г, 7,5 ммоль) и безводный ацетат натрия (0,615 г, 7,5 ммоль) добавляют к раствору сырого формильного производного (0,89 г) в MeOH/H2O (4:1,50 мл). Спустя 24 ч MeOH выпаривают в вакууме, смесь воды и масла экстрагируют с CHCl3. Слой CHCl3 высушивают (Na2SO4) и выпаривают, получая янтарное масло, которое направляют в колонну с силикагелем. Элюирование с CHCl3 дает два основных слоя: 3-(3-пиридинил)пропионитрил (использованный в качестве исходного вещества в предыдущей стадии); желаемый енамин.
Пример 4. В атмосфере азота этиловый эфир хлоруксусной кислоты (0,521 г, 4,8 ммоль) добавляют по каплям к раствору енамина (пример 3) (0,513 г, 3,2 ммоль) и 1,5-диазабицикло[4.3.0]нон-5-ена (DBN, 1,37 г, 11,1 ммоль) в сухом CH2Cl2 (15 мл) с внешним охлаждением на ледяной бане. После перемешивания при 0oC в течение 1 ч раствор оставляют при комнатной температуре в течение ночи. После проверки хода реакции с помощью ТСХ добавляют дополнительное количество ClCO2Et (0,1 мл) и DBN (1,0 г) для завершения реакции, раствор выдерживают в течение 24 ч. Летучее вещество выпаривают в вакууме и вязкий остаток очищают в короткой колонке с силикагелем (главная цель которой удаление менее подвижного DBN), получая N-защищенный пиррол, который используют для следующей стадии без дальнейшей очистки.
Пример 5. К раствору N-защищенного пиррола примера 4 (0,635 г, 2,0 ммоль) в MeOH (50 мл) добавляют твердый Na2CO3 (0,212 г, 2,0 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 48 ч с выделением полученного деблокированного пиррола. Смесь упаривают досуха и остаток тщательно растирают в порошок с H2O (25 мл) для растворения неорганических веществ и экстрагируют с CHCl3 (3 x 100 мл). Экстракт высушивают (Na2SO4) и выпаривают до получения вязкой смолы, которая кристаллизуется при высушивании в вакууме для использования в качестве промежуточного вещества без дальнейшей очистки. Более высокая очистка может быть проведена или колоночной хроматографией при использовании силикагель/CHCl3, или перекристаллизацией из смеси толуол/циклогексан (1:3).
Пример 6. Бензоилизотиоцианат (0,232 г, 1,42 ммоль) добавляют по каплям к раствору деблокированного пиррола примера 5 (0,290 г, 1,18 ммоль) в сухом CH2Cl2 (100 мл). После 1 ч при комнатной температуре раствор выпаривают и смолистый остаток перемешивают в смеси Et2O/циклогексан (1:1,20 мл). Полученную суспензию желтого твердого вещества отфильтровывают под давлением азота, и тиоуреидное соединение высушивают в вакууме над P2O5.
Пример 7. Йодистый метил (0,228 г, 1,61 ммоль) добавляют к раствору тиоуреида примера 6 (0,383 г, 0,94 ммоль) и DBN (0,140 г, 1,12 ммоль) в сухом CH2Cl2 (10 мл) при 0oC. Раствор перемешивают при 0oC в течение 15 мин при температуре окружающей среды в течение 1 ч и затем выпаривают в вакууме. Раствор остатка в CHCl3 хроматографируют в колонне с силикагелем со смесью CHCl3/метанол в качестве растворителя для элюирования, получая гомогенные фракции метилтиозамещенного промежуточного соединения.
Пример 8.
Раствор метилтиосоединения примера 7 (0,358 г, 0,85 ммоль) в 100 мл MeOH, который был насыщен NH3 при 0oC, нагревают при 90-95oC в течение 24 ч в футерованной стеклянными плитками бомбе из нержавеющей стали. Содержимое охлажденной бомбы выпаривают в вакууме, получая смесь желаемого 2-аминопроизводного, бензамида и побочного продукта, представляющего собой 2-метилтиопроизводное. Смесь растворяют в метаноле и раствор выпаривают с силикагелем (около 5 г). Смесь затем осторожно наносят на верхнюю часть хроматографической колонки с силикагелем, которую затем элюируют смесью CHCl3/MeOH (9: 1), получая побочное метилтиосоединение и желаемый 2-амино-7-(3-пиридинилметил)-3H, 5H-пирроло[3,2-d]пиримидин-4-он, как промежуточное соединение. Дальнейшую очистку проводят перекристаллизацией из кипящего изопропилацетата в аппарате Сокслета.
Анализ: вычислено для C12H11N5O: C 59,74; H 4,60; N 29,03; найдено: C 59,58; H 4,86; N 28,89.
MS (FAB): 242 (M+H)+; UV: 0,1 N HCl 233 (18,2), 269 (17,4); pH 7 233 (18,2), 269 (17,4); pH 7 233 (22,4); 269 (12,3); 0,1 N NaOH 227 (22,2), 263 (10,5), 290 (sh). IC50(PNP) 25 nM.
Раствор пиридинилметильного промежуточного соединения в 0,1N HCl гидрируют на платиновом катализаторе при давлении H2 60 фунтов/дюйм2 (4,2186 кг/см2). Катализатор генерируют посредством кратковременного гидрирования PtO2 в 0,1N HCl. Когда реакция заканчивается, катализатор удаляют фильтрованием под давлением N2 и фильтрат выпаривают. Остаток в минимальном количестве этанола разбавляют медленно большим количеством EtO2 и получают гидрохлоридную соль в виде белого гигроскопичного твердого вещества (IC). Т.пл. (в oС) при 200o размягчается.
MS (FAB) m/e 248 (M+H)+; UV 0,1 N HCl 234 (15,7), 273 (13,8) pH 7 буффер 232 (19,1), 271 (9,3), 0,1 N NaOH 229 (19,0), 266 (7,1), 288 (sh); 1H-ЯМР (DNSO-d6) δ 1,19, 1,61, 1,77 (m, 4H, H-4's and 5's пиперидина), 1,96 (m, 1H, H-3 пиперидина), 2,53 (d, 2H, -CH2-), 2,69 (m, 2H, H-2's пиперидина), 3,14 (d, 2H, H-6's пиперидина), 7,25 (d, J 2,8, 1H, H-6), 7,76 (s, 2H, NH2), 8,81, 9,18 (уш. 1 каждый, NH+ обменивается с D2О), 12,24 (s, 1H, S-NH).
Анализ: вычислено для C12H17N6O•2,8HCl•0,2 C2H6O•H2O: C, 39,55; H, 6,16; N, 18,60; Cl, 26,36; найдено: C 39,75; H 6,24; N 18,65; Cl 26,08.
IC50 1 мкМ.
Пример 9. Вещество (IC) примера 8 испытывают на ферментативную ингибирующую активность. Проводят ферментативный анализ пуриннуклеозидфосфорилазы (PNP), в котором изучают PNP активность (IC50) для вещества, которую определяют радиохимически посредством измерения образования [14C]-гипоксантина из [14C] -инозина (Biomedicina, 33,39 (1980)), используя селезенку задней части голени в качестве источника энзима.
Примеры 10-14. Следующие вещества изобретения, представляющие собой 2-амино-7-(CH2R)-3H,5H-пирроло[3,2-d]пиримидин-4-оны, в которых R группа означает следующее:
пример 10: R 2-метил-3-пиперидинил;
пример 11: R 2-хлор-3-пиперидинил;
пример 12: R 2-трифторметил-3-пиперидинил;
пример 13: R 2-метокси-3-пиперидинил;
пример 14: R 2-фтор-3-пиперидинил
получают, следуя способам, приведенным в примерах 1-8, используя соответствующие 3-(замещенные 3-пиридинил)-пропионитрилы в качестве исходных веществ.
Пример 15. 3-(2-Пиридинил)пропионитрил получают в этом примере, используя способ (V. Boekelheide, et al. J. Am. Chem. Soc. 75, 3243 (1953)). Раствор цианида калия (83,74 г) в воде (160 мл) добавляют к раствору свежеперегнанного 2-винилпиридина (67,59 г) в ангидриде уксусной кислоты (131,30 г) со скоростью, отрегулированной для поддержания осторожного кипения с обратным холодильником. Когда добавление заканчивают, полученную темно-красную смесь нагревают в течение примерно 17 ч при 105oC в масляной бане с энергичным перемешиванием. Охлажденную реакционную смесь доводят до pH 8 насыщенным раствором Na2CO3. Смесь экстрагируют CHCl3 (4•150 мл), промывают водой, высушивают Na2SO4 и выпаривают до темного вязкого масла. Масло фракционированно перегоняют в вакууме через Vigreaux колонну. После малого предгона, содержащего 2-винилпиридин (0,35 г), желаемый 3-(2-пиридинил)пропионитрил собирают в виде прозрачного, флуоресцирующего желто-зеленого вязкого масла: выход 59,8 г; 70,4% т. кип. 86oC при 1,0 ммHg.
Примеры 16-20. Получают следующие вещества изобретения: 2-амино-7-(CH2R)-3H, 5H-пирроло[3,2-d] пиримидин-4-оны, в которых группа R имеет следующие значения:
пример 16: R 2-пиперидинил;
пример 17: R 4-пиперидинил;
пример 18: R 3-трифторметил-4-пиперидинил;
пример 19: R 3-метокси-2-пиперидинил;
пример 20: R 3-фтор-4-пиперидинил,
следуя способам, приведенным в примерах 1-8 и 15, используя соответствующие 3-(2- или 4-пиридинил)пропионитрилы в качестве исходных веществ.
Пример 21. Получают фармацевтическую композицию для интраперитональной инъекции для испытания вещества (IC). Для этого интраперитональный инъекционный раствор, содержащий вещество примера 8, растворяют в водном носителе, который содержит 10% DMSO.
Пример 22. Вводят интраперитонально в Lewis Rats композицию примера 21, содержащую вещество (IC) с дозой 30 мг вещества (IC) с инъекцией дважды в день. Используют контрольные группы, которые получают только наполнитель. Через определенное время после введения животных усыпляют и получают образцы плазмы. Плазму экстрагируют холодной 0,5N HClO4 и нейтрализуют твердым NH4HCO3. После удаления перхлоратных солей экстракт подвергают HPLC (ЖХВД) жидкостная хроматография высокого давления) на колонне с обратимой фазой (Spherisorb ODSI). Наблюдают значительное увеличение плазменного инозина в плазме, взятой из животных, получающих вещество (IC).
Примеры 23-33. Вещества, полученные в примерах 10-20, использованы для приготовления лекарственных средств в соответствии со способом приготовления примера 21. Полученные вспрыскиваемые растворы испытывают в соответствии со способом примера 22. Наблюдают значительное увеличение плазменного инозина в плазме, взятой из животных, получающих вещества изобретения.
Пример 34. Получают 3-(2-фуранил)пропионитрил. В трехгорлую колбу, снабженную конденсатором и осушительной трубкой, добавляют магниевые стружки (20 г) к раствору 3-(2-фуранил)акрилонитрила (67,2 г) в сухом метаноле (2 л). Дополнительное количество магния добавляют по частям в зависимости от скорости реакции до тех пор, пока не будет добавлено все количество (145 г). Спустя около 5 ч растворитель выпаривают до тех пор, пока содержимое не образует твердую пасту, которую доводят до pH около 6,5 с помощью 6N HCl при охлаждении. Смесь экстрагируют с помощью нескольких порций CHCl3, сушат с помощью Na2SO4 и концентрируют до темного масла. Перегонка в вакууме через короткую Vigreaux колонку дает пропионитрил в виде бесцветного масла; выход 49,5 г (72%); т. кип. 46,0-46,5oC при 0,5 ммHg.
3-(2-Фуранил)пропионитрил из примера 34 далее используют в синтезах изобретения.
Пример 35. В атмосфере сухого N2 пропионитрил (48,7 г), гидрид натрия (10,28 г), этиловый эфир муравьиной кислоты (32,74 г) и безводный тетрагидрофуран (200 мл) перемешивают при комнатной температуре с защитой от влаги в течение 18 ч. Летучее вещество затем выпаривают, полученное желтое твердое вещество растворяют примерно в 20 мл холодной воды с охлаждением на ледяной бане, раствор доводят до pH 6,0 с помощью холодной 6N HCl. Полученный тяжелый маслянистый осадок экстрагируют в CHCl3 и экстракт промывают водой, высушивают с помощью Na2SO4 и выпаривают, получая жидкое масло, которое содержит сырое формильное производное; выход 53,06 г.
Пример 36. Гидрохлорид метилового эфира глицина (67,05 г) и безводный ацетат натрия (43,79 г) добавляют к раствору сырого формильного производного предыдущего примера (53,05 г) в MeOH/H2O (4:1,1500 мл). Через 24 ч MeOH выпаривают в вакууме и смесь воды и масла экстрагируют с помощью CHCl3. Слой CHCl3 высушивают (Na2SO4) и выпаривают, получая янтарное масло (59,1 г).
Пример 37. В атмосфере азота этиловый эфир хлормуравьиной кислоты (43,68 г) добавляют по каплям к раствору янтарного масла примера 36 (59,1 г) и DBN (133 г) в сухом CH2Cl2 (400 мл) с внешним охлаждением на ледяной бане. После перемешивания при 0oC в течение 1 ч раствор оставляют стоять при комнатной температуре в течение ночи. После проверки течения реакции с помощью ТСХ вводят дополнительное количество ClCO2Et и DBN для завершения реакции и раствор оставляют стоять в течение 24 ч. Летучее вещество выпаривают в вакууме, вязкий остаток очищают на колонне с силикагелем (главная цель которой удаление менее мобильного DBN), получая N-защищенный пиррол, соответствующий пиррол без этоксикарбонильной защитной группы на пиррольном азоте и исходный пропионитрил.
Пример 38. К раствору N-защищенного пиррола примера 37 (11,66 г) в MeOH (200 мл) добавляют твердый Na2CO3 (4,23 г) и реакционную смесь перемешивают при комнатной температуре в течение 24 ч с отделением полученного деблокированного пиррола. Смесь выпаривают досуха и остаток тщательно растирают в порошок с H2O (200 мл) для растворения неорганических веществ, затем экстрагируют с помощью CHCl3 (3 x 200 мл). Экстракт сушат (Na2SO4) и выпаривают, получая вязкую смолу, которая кристаллизуется при высушивании в вакууме, для использования в качестве промежуточного соединения без дальнейшей очистки. Более глубокая очистка может быть проведена колоночной хроматографией с использованием силикагель/CHCl3, или перекристаллизацией из смеси толуол/циклогексан (1:3).
Пример 39. Бензоилизотиоцианат (4,58 г) добавляют по каплям к раствору деблокированного пиррола примера 38 (5,15 г) в сухом CH2Cl2 (75 мл). Через 1 ч при комнатной температуре раствор выпаривают, смолистый остаток растворяют в Et2O (100 мл) с почти немедленным выделением кристаллического твердого вещества, которое собирают фильтрованием. Фильтрат Et2O нагревают до кипения и разбавляют с помощью равного объема горячего циклогексана. При медленном охлаждении раствор дает дополнительное количество тиореидопроизводного.
Пример 40. Йодистый метил (8,70 г) добавляют к раствору тиоуреидопроизводного примера 39 (10,68 г) и DBN (4,15 г) в сухом CH2Cl2 (250 мл) при 0oC. Раствор перемешивают при 0oC в течение 15 мин, при температуре окружающей среды в течение 1 ч и затем выпаривают в вакууме. Раствор остатка в CHCl3 хроматографируют в колонне с силикагелем с использованием CHCl3 в качестве растворителя для элюирования, получая гомогенные фракции метилтиопромежуточного соединения.
Пример 41.
Раствор метилтиосоединения из примера 40 (0,80 г, 2,0 ммоль) в 25 мл MeOH, который был насыщен NH3 при 0oC, нагревают при 90-95oC в течение 24 ч в футерованной стеклянными плитками бомбе из нержавеющей стали. Содержимое охлажденной бомбы выпаривают в вакууме, получая смесь 2-аминосоединения, бензамида и побочного продукта, который является 2-метилтиопроизводным. Смесь перемешивают энергично в течение нескольких минут с 30 мл смеси 2:1 Et2O/циклогексан, и нерастворимое белое твердое вещество отфильтровывают и промывают Et2O. Фильтрат содержал большую часть бензамида и 2-метилтиопроизводного. Раствор нерастворимого в смеси Et2O/циклогексан твердого вещества (0,425 г) в MeOH выпаривают с приблизительно 10 г силикагеля. Измельченный в порошок остаток наносится равномерно на верх колонны с силикагелем, которая затем элюируется с помощью смеси CHCl3/MeOH/HOAc (95:5:1), давая 2-метилтиоипроизводное как побочный продукт и желаемое фуранильное промежуточное соединение. Промежуточное соединение перекристаллизовывают экстракцией в кипящем изопропилацетате в аппарате Сокслета. Белые кристаллы собирают в три порции и сушат в вакууме над P2O5 при 110oC в течение 7 ч.
Раствор промежуточного соединения (116 г, 0,5 ммоль) в метаноле (50 мл) гидрируют с помощью 30%-ного палладия на древесном угле (40 мг) при давлении H2 62 фунта/дюйм (4,3592 кг/см) в течение около 36 ч. Катализатор удаляют фильтрованием под давлением N2 и фильтрат выпаривают, затем повторно выпаривают с толуолом. Твердый остаток перекристаллизовывают экстракцией в кипящем изопропилацетате в аппарате Сокслета. 2-Тетрагидрофуранильное соединение (IE) получают в виде белого кристаллического твердого вещества, которое высушивают в вакууме над P2O5 при 110oC в течение примерно 6 ч; выход 73 мг (61,8%); т.пл. 284-286oC (разл.).
MS (FAB) m/e 235 (M+H)+, UV 0,1 N HCl 235 (17,1), 273 (14,7), pH 7 буффер 232 (20,0), 271 (10,5), 0,1 N NaOH 228 (20,2), 264 (7,0), 286 (6,2); 1H-ЯМР (DMSO-d6) δ 1,50, 1,78 (m, 4H, H-3's и 4's тетрагидрофуранила), 2,52, 2,69 (2d, 2H, -CH2R), 3,57, 3,76 (2m, 1H каждый, H-5's тетрагидрофуранила), 5,82 (s, 2H, NH2), 6,97 (d, J=2,8, 1H, H-6), 10,34 (s, 1H, 3-NH), 11,18 (s, 1H, 5-NH).
Анализ: вычислено для C11H14N4O2: C 56,40; H 6,02; N 23,92; найдено: C 56,33; H 6,49; N 23,95.
Пример 42. Продукт примера 41 испытывают на ферментативную ингибирующую активность в соответствии со способом примера 9 и наблюдают PNP активность (IC50 11 мкМ) для вещества.
Пример 43. Приготавливают фармацевтическую композицию для интраперитональной инъекции для испытания вещества (IE). Для этого интраперитональный инъектируемый раствор, содержащий вещество примера 41, растворяют в водном носителе, который содержит 10% DMSO.
Пример 44. Используя способ примера 16, вводят интраперитонально в Lewis Rats композицию примера 43, содержащую вещество (IE) с дозой 30 мг вещества (IE), результаты анализируют по сравнению с контрольными. Наблюдают значительное увеличение плазменного инозина в плазме, взятой из животных, получающих вещество (IE).
Примеры 45-49. Получают следующие вещества изобретения: 2-амино-7-(CH2)-3H,5H-пирроло[3,2-d]пиримидин-4-оны, в которых группа R означает:
пример 45: R 3-тетрагидрофуранил;
пример 46: R 3-хлор-2-тетрагидрофуранил;
пример 47: R 3-трифторметил-2-тетрагидрофуранил;
пример 48: R 3-метокси-3-тетрагидрофуранил;
пример 49: R 3-фтор-2-тетрагидрофуранил.
Вещества получают, следуя способам, приведенным в примерах 34-41, используя соответствующие 3-(фуранил)-акрилонитрилы в качестве исходных веществ.
Пример 50.
В атмосфере азота раствор тетрагидрофуранильного производного (IE) (500 мг), полученного в примере 41, и кристалл фенола в 2N HBr (20 мл) перемешивают в течение 18 ч при 40oC. Растворитель выпаривают в вакууме при низкой температуре, остаток промывают с помощью нескольких мл Et2O, декантацией удаляя любой трибромфенол. Т.пл. 311-313oC.
MS (FAB) m/e 251 (M+H)+; UV 0,1 N HCl 234 (17,4), 274 (15,2), pH 7 буффер 232 (20,9), 272 (10,6), 0,1 N NaOH 228 (21,3), 265 (7,3), 287 (6,5); ИК (KBr) 3211, 3203, 3127, 3108, 2936, 1661, 1609, 1567, 1524, 1369 см-1; 1H-ЯМР (DMSO-d6) δ 1,62, 1,87, 2,01 (комплекс m, 4H, H-3'S и 4'S тетрагидротиенила), 2,72 (m, 4H, -CH2-перекрывающий H-5'S тетрагидротиенила), 3,64 (p, 1H, 6-2 тетрагидротиенила), 5,82 (s, 2H, NH2), 6,97 (d, J=3,0, 1H, H-6), 10,34 (s, 1, 3-NH), 11,17 (s, 1,5-NH);
Анализ: вычислено для C11H14N4OS: C 52,78; H 5,64; N 22,38; найдено: C 52,56; H 5,64; N 22,50.
Из фильтрата получают 2-амино-1,5-дигидро-7-(1,2-дитиан-3-илметил)-4H-пирроло[3,2-d] пиримидин-4-он; т.пл. 311-313oC.
MS (FAB) m/e 283 (M+H)+; IR (KBr) 3139, 3135, 3131, 2926, 1673, 1658, 1609, 1569, 1523, 1370 см-1; 1H-ЯМР (DMSO-d6) d 1,53, 1,67, 2,04 (комплекс m, 4H, H-4's и 5's дитиенила), 2,67 (m, 2H, -CH2R); 2,75 (m, 2H, H-6's дитиенила), 3,17 (m, 1H, H'3 дитиенила), 5,84 (s, 2H, NH2, 7,00 (d, J=3,0, 1H, H-6), 10,34 (s, 1H, 3-NH), ll,24 (s, 1H, 5-NH).
Анализ: вычислено для C11H14N4OS2: C 46,80; H 5,00; N 19,85; найдено: C 46,78; H 4,97; N 19,81.
Суспензию остатка в воде доводят до pH 7 1N NaOH, получая бромоспирт в виде свободного основания. Твердое вещество собирают, промывают холодной водой и высушивают при комнатной температуре.
Раствор бромоспирта в безводном диметилацетамиде охлаждают в ледяной бане и обрабатывают эквимолярной порцией PBr3. Раствор перемешивают при комнатной температуре в течение 1 ч, затем диметилацетамид выпаривают в вакууме при низкой температуре, используя воздушный насос и ловушку с сухим льдом. Суспензию остатка в смеси лед/вода доводят до pH 7, используя 1N NaOH, и получают сырой продукт реакции, содержащий 1,4-дибромсоединение. После фильтрования, промывания холодной водой и высушивания при комнатной температуре полученный продукт кипятят с обратным холодильником с сульфидом натрия в смеси 50% этанол/вода, либо нагревают с Na2S в растворе N,N-диметилацетамида; растворитель удаляют под вакуумом, остаток промывают водой для удаления NaBr и pH доводят до примерно 7 для осаждения 2-тетрагидротиенильного вещества (IG).
Пример 51. Вещество примера 50 испытывают на ферментативную ингибирующую активность, как в примере 9, и наблюдают PNP активность (IC50 11 nM) для вещества. IC50 18 nM для дитиан-3-ильного соединения.
Пример 52. Готовят фармацевтическую композицию для интраперитональной инъекции для испытания вещества (IG). Интраперитональный инъекцируемый раствор, содержащий вещество примера 50, растворяют в водном носителе, который содержит 10% DMSO.
Пример 53. Используя способ примера 16, вводят интраперитонально в Lewis Rats опытную композицию примера 52, содержащую вещество (IG) с дозой 30 мг вещества (IG), результаты анализируют по сравнению с контрольными. Наблюдают значительное увеличение плазменного инозина в плазме, взятой из животных, получающих вещество (IG).
Примеры 54-58. Получают следующие вещества изобретения, 2-амино-7-(CH2R)-3H,5H-пирроло[3,2-d]пиримидин-4-оны, в которых R означает:
пример 54: R 3-тетрагидротиенил;
пример 55: R 3-хлор-2-тетрагидротиенил;
пример 56: R 3-трифторметил-2-тетрагидротиенил;
пример 57: R 3-метокси-2-тетрагидротиенил;
пример 58: R 3-фтор-2-тетрагидротиенил.
Вещества получают, следуя способам, приведенным в примерах 34-41 и 50, используя соответствующие 3-(фуранил)-акрилонитрилы в качестве исходных.
Пример 59. Используя способ примера 1, 3-(2-пирролил)-пропионитрил получают, применяя пиррол-2-карбоксальдегид в качестве исходного вещества. Следуя примерам 2-8, промежуточное вещество 2-амино-7-(2-пирролилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он получают из пропионитрила, из которого 2-пирролидинильное вещество (II) получают восстановлением промежуточного вещества.
Пример 60. Используя способ примера 1, 3-(3-пирролил)-пропионитрил получают, применяя пиррол-3-карбоксальдегид в качестве исходного вещества. Следуя примерам 2-8, промежуточное соединение 2-амино-7-(3-пирролилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он получают из пропионитрила, из которого 3-пирролидинильное вещество (IJ) получают восстановлением промежуточного вещества.
Пример 61. Используя способ примера 1, 3-(2-пиранил)-пропионитрил получают, применяя 2-пиранкарбоксальдегид в качестве исходного вещества. Следуя примерам 2-8, промежуточное вещество 2-амино-7-(2-пиранилметил)-3H,5H-пирроло[3,2-d] пиримидин-4-он получают из пропионитрила, из которого 2-тетрагидропиранильное вещество (IL) получают восстановлением промежуточного вещества.
Пример 62. Используя способ примера 1, 3-(3-пиранил)-пропионитрил получают, применяя 3-пиранкарбоксальдегид в качестве исходного вещества. Следуя примерам 2-8, промежуточное вещество 2-амино-7-(3-пиранилметил)-3H,5H-пирроло[3,2-d] пиримидин-4-он получают из пропионитрила, из которого 3-тетрагидропиранильное вещество (IM) получают восстановлением промежуточного соединения.
Пример 63. Используя способ примера 1, 3-(4-пиранил)-пропионитрил получают, применяя 4-пиранкарбоксальдегид в качестве исходного вещества. Следуя примерам 2-8, промежуточное вещество 2-амино-7-(4-пиранилметил)-3H,5H-пирроло[3,2-d] пиримидин-4-он получают из пропионитрила, из которого 4-тетрагидропиранильное вещество (IN) получают восстановлением промежуточного соединения.
Пример 64. Раствор метилтиопроизводного примера 7 (0,358 г, 0,85 ммоль) в 100 мл MeOH, который был насыщен NH3 при 0o, нагревают при 90-95oC в течение 24 ч в футерованной стеклянными плитками бомбе из нержавеющей стали. Содержимое охлажденной бомбы выпаривают в вакууме, получая смесь желаемого 2-аминопромежуточного вещества, бензамида, и побочного продукта, который является 2-метилтиопроизводным. Смесь растворяют в метаноле и раствор выпаривают с силикагелем (около 5 г). Затем смесь осторожно наносят на верх хроматографической колонны с силикагелем, которую затем элюируют с помощью смеси CHCl3/MeOH (9:1), получая метилтиопобочный продукт и желаемый 2-амино-7-(3-пиридинилметил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он, вещество (IIA). Дальнейшую очистку проводят перекристаллизацией из кипящего изопропилацетата в аппарате Сокслета.
Пример 65. Вещество (IIA) примера 64 испытывают на ферментативную ингибирующую активность. Проводят ферментативный анализ пуриннуклеозидфосфорилазы (PNP), в котором наблюдают PNP активность (IC50) для вещества, определяемую радиохимически посредством измерения образования [14C]-гипоксантина из [14C] -инозина (Biomedicina, 33, 39, 1980), используя селезенку задней части голени в качестве источника энзима.
Примеры 66-67. Получают следующие вещества изобретения, 2-амино-7-(CH2R)-3H,5H-пирроло[3,2-d]пиримидин-4-оны, в которых R означает:
пример 66: R 2-пиридинил;
пример 67: R 3-хлор-2-пиридинил.
Вещества получают, следуя способам, приведенным в примерах 1-7 и 64, используя соответствующие 3-(пиридинил)-пропионитрилы в качестве исходных.
Пример 68. 2-Амино-4-оксо-3H,5H-7-[(4-пиридил)метил]пирроло[3,2-d]пиримидин гидрохлорид гидрат (2:4:1)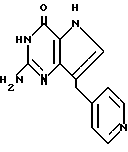
Метил 3-[(N-бензоил-S-метилизотиокарбамоил)амино]-4-[(4- пиридил)метил] -1H-пиррол-2-карбоксилат (BCX-180, 9,5 г, 23,25 ммоль) добавляют в 300-мл выложенный стеклянной футеровкой стеклянный сосуд высокого давления к 90 мл метилата аммония (полученного путем пропускания аммиака в безводный метанол при температуре 0-5oC до насыщения) и реакционный сосуд закупоривают. Этот сосуд нагревают до температуры 85-90oC в течение 12 ч и затем охлаждают до комнатной температуры. Летучие вещества выпаривают с помощью вращающегося испарителя и к полученному остатку добавляют 150 мл 1N HCl и 100 мл хлороформа. Полученную смесь энергично перемешивают и выделяют экстракт HCl. Экстракт HCl нейтрализуют, используя водный раствор карбоната натрия. Выпавшее в осадок твердое вещество выделяют путем фильтрации, высушивают на воздухе и экстрагируют 2 л этанола в аппарате Сокслета в течение 48 ч. Этанол выпаривают и образовавшийся светло-коричневый твердый остаток (3,1 г) размешивают с 20 мл деионизированной (DI) воды. Концентрированную (37%) соляную кислоту добавляют по каплям до тех пор, пока все твердое вещество не растворяется (2,1 мл, 25,7 ммоль). Прозрачный раствор обрабатывают активированным углем (700 мг) при температуре 50oC в течение 5 мин и фильтруют через короткую колонку, заполненную цеолитом. Полученный фильтрат концентрируют на вращающемся испарителе при температуре 50- 55oC с получением белого твердого вещества, высушивают сначало на воздухе, а затем в вакууме при температуре перегонки толуола. Масса полученного белого твердого вещества составляет 3,6 г (48%), т.пл. 340-342oC.
Анализ: вычислено для C12H11N5O•2HCl•0,5H2O: C 44,59; H 4,67; N 21,67; найдено: C 44,59; H 4,57; N 21,47.
Пример 69. Готовят фармацевтическую композицию для интраперитональной инъекции для испытания вещества (IIA). Для этого интраперитональный инъекцируемый раствор, содержащий вещество примера 64, растворяют в водном носителе, который содержит 10% DMSO.
Пример 70. Вводят интраперитонально в Lewis Rats опытную композицию примера 69, содержащее вещество (IIA), в дозе 30 мг вещества (IIA) инъекцией дважды в день. Используют контрольные группы, которые получают только наполнитель. Через определенное время после введения животных усыпляют и получают образцы плазмы. Плазму экстрагируют с помощью холодной 0,5N HClO4 и нейтрализуют с помощью твердого NH4HCO3. После удаления перхлоратных солей экстракт подвергают ЖХВД на колонне с обратимой фазой (Spherisorb ODSI). Наблюдали значительное увеличение плазменного инозина в плазме, взятой из животных, получающих вещество (IIA).
Примеры 71-73. Вещества такие, как в примерах 66-68, использованы для получения фармкомпозиций в соответствии со способом примера 69 и полученные инъекцируемые растворы испытаны в соответствии со способом примера 70. Наблюдают значительное увеличение плазменного инозина в плазме, взятой у животных, получающих вещество изобретения.
Пример 74. 1-Циклогексенилацетонитрил используют в синтезах для получения соединений изобретения. В атмосфере сухого N2 раствор 1-циклогексенилацетонитрила (9,2 г, 75,92 ммоль) в безводном тетрагидрофуране (THF, 10 мл) добавляют к перемешиваемой смеси гидрида натрия (3,18 г, 132,86 ммоль) и этилового эфира муравьиной кислоты (30,14 г, 406,93 ммоль) в 50 мл THF, полученную смесь перемешивают при комнатной температуре в течение около 18 ч. Летучее вещество выпаривают в вакууме при комнатной температуре. Воду (30 мл) добавляют к остатку при 0oC, раствор доводят до pH 6 добавлением по каплям 6N HCl. Полученный осадок в виде тяжелого масла экстрагируют CHCl3. Экстракт промывают водой и высушивают с помощью Na2SO4, полученный органический слой выпаривают, получая сырое формильное производное в виде красно-коричневого масла (9,6 г).
Пример 75. Гидрохлорид метилового эфира глицина (16,68 г, 132,85 ммоль) и безводный ацетат натрия (10,89 г, 132,85 ммоль) добавляют к раствору сырого формильного производного (9,6 г) примера 74 без дальнейшей очистки в смеси MeOH/H2O (4:1,150 мл). Спустя 24 ч MeOH выпаривают в вакууме и смесь воды и масла экстрагируют с помощью CHCl3. Слой CHCl3 высушивают (Na2SO4) и выпаривают, получая янтарное масло, которое подают на колонку с силикагелем. Элюирование с помощью CHCl3 дает желаемый енамин: выход 4,5 г.
Пример 76. В атмосфере азота этиловый эфир хлормуравьиной кислоты (3,04 г, 28,06 ммоль) добавляют по каплям к раствору енамина примера 75 (4,12 г, 18,70 моль) и 1,5-диазабицикло[4.3.0]нон-5-ена (DBN, 6,96 г, 56,11 ммоль) в сухом CH2Cl2 (100 мл) с внешним охлаждением в ледяной бане. После перемешивания при 0oC в течение 1 ч раствору дают постоять при комнатной температуре в течение ночи. После проверки хода реакции с помощью TCX вводят дополнительное количество ClCO2Et (0,5 мл) и DBN (3,0 мл) для завершения реакции, затем раствор оставляют на 24 ч. Летучее вещество выпаривают в вакууме, вязкий остаток очищают на короткой колонке с силикагелем (главная цель которой удаление менее мобильного DBN), получая N-защищенный пиррол, который используют для следующей стадии без дальнейшей очистки.
Пример 77. К раствору N-защищенного пиррола примера 76 (3,0 г, 10,26 ммоль) в MeOH (100 мл) добавляют твердый Na2CO3 (2,71 г, 25,65 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 48 ч с отделением полученного деблокированного пиррола. Смесь выпаривают досуха, остаток тщательно растирают в порошок с водой (50 мл) для растворения неорганических веществ и экстрагируют с помощью CHCl3 (3 x 50 мл). Экстракт сушат (Na2SO4) и выпаривают до получения вязкой смолы, которую очищают на колонне с силикагелем, используя CHCl3 в качестве элюента; выход 2,04 г; т.пл. 125oC.
Пример 78. Бензоилизотиоцианат (0,76 г, 4,65 ммоль) добавляют по каплям к раствору деблокированного пиррола примера 77 (0,91 г, 4,13 ммоль) в сухом CH2Cl2 (30 мл). Спустя 1 ч при комнатной температуре раствор выпаривают и смолистый остаток растирают в порошок с метанолом, получая тиоуреидопроизводное; выход 0,70 г, т.пл. 170oC.
Пример 79. Йодистый метил (0,678 г, 4,78 ммоль) добавляют к раствору тиоуреидопроизводного примера 78 (0,630 г, 1,64 ммоль) и DBN (0,230 г, 1,85 ммоль) в сухом CH2Cl2 (50 мл) при 0oC. Раствор перемешивают при 0oC в течение 15 мин, при температуре окружающей среды в течение 1 ч, а затем выпаривают в вакууме. Раствор остатка в CHCl3 хроматографируют на колонке с силикагелем с помощью CHCl3 в качестве элюента, получая гомогенные фракции метилтиосоединения как промежуточного соединения; выход 0,7 г.
Пример 80.
Раствор метилтиосоединения примера 79 (0,70 г, 1,76 ммоль) в 50 мл MeOH, который был насыщен NH3 при 0oC, нагревают при 90-95oC в течение 24 ч в футерованной стеклянными плитками бомбе из нержавеющей стали. Содержимое охлажденной бомбы выпаривают в вакууме, получая смесь вещества (IIIA), бензамида и побочного продукта, который является 2-метилтиопроизводным, в отличие от 2-аминосоединения (IIIA). Смесь энергично перемешивают в течение нескольких минут с приблизительно 50 мл Et2O, нерастворимое белое твердое вещество отфильтровывают и промывают с помощью Et2O. Фильтрат содержал большую часть бензамида и 2-метилтиопроизводного. Раствор нерастворимого в Et2O твердого вещества (0,342 г) в MeOH выпаривают с приблизительно 10 г силикагеля. Измельченный в порошок остаток наносят равномерным слоем на верхнюю часть колонны с силикагелем, который затем элюируют с помощью смеси CHCl3/MeOH/HOAc (95:5:1), получая 2-метилтиопроизводное как побочный продукт и желаемое 2-аминосоединение (IIIA). Соединение (IIIA) перекристаллизовывают экстракцией в кипящем изопропилацетате в аппарате Сокслета. Белые кристаллы собирают в три массы и высушивают в вакууме над P2O5 при 110oC в течение 7 ч; выход 44% т.пл. 280oC (разл.).
Анализ для C12H14N4O•0,6H2O: вычислено: C 59,78; H 6,35; N 23,23; найдено: C 59,98; H 6,46; N 23,15.
Пример 81. Вещество примера 80 испытывают на ферментативную ингибирующую активность. Проводят ферментативный анализ пуриннуклеозидфосфорилазы (PNP), в котором определяют PNP активность для вещества радиохимическим измерением образования [14C]-гипоксантина из [14C]-инозина (Biomedicina, 33, 39, 1980), используя селезенку задней части голени в качестве источника энзима. При 1 mM фосфата IC50 равно 1,9 мкМ, при 50 mM фосфата IC50 равно 19 мкМ.
Пример 82. Следуя способам, приведенным в примерах 74-80, вещества (IIIB) и (IIIC) получают, используя 2- и 3-циклогексенилацетонитрил соответственно в качестве исходных веществ. Вещества испытывают так, как в примере 81, и наблюдают значительную ферментативно-ингибирующую активность.
Примеры 83-87. Получают следующие вещества изобретения: 2-амино-7-(R)-3H,5H-пирроло[3,2-d]пиримидин-4-оны, в которых R группа следующая:
пример 83: R 3-метил-2-циклогексенил;
пример 84: R 2-хлор-3-циклогексенил;
пример 85: R 3-трифторметил-1-циклогексенил;
пример 86: R 3-метокси-1-циклогексенил;
пример 87: R 2-фтор-3-циклогексенил.
Вещества получают, следуя способам, приведенным в примерах 74-81, используя соответствующие циклогексенилацетонитрилы в качестве исходных.
Пример 88. Готовят фармацевтическую композицию для интраперитонального введения для испытания вещества (IIIA). Интраперитональный инъекцируемый раствор, содержащий вещество (IIIA), растворяют в водном носителе, который содержит 10% DMSO.
Пример 89. Вводят интраперитонально в Lewis Rats опытную композицию, содержащую вещество (IIIA) примера 88, в дозе 30 мг вещества (IIIA) с инъекцией, дважды в день. Используют контрольные группы, которые получают только наполнитель. Через определенное время после введения животных усыпляют и получают образцы плазмы. Плазму экстрагируют с помощью холодной 0,5N HClO4 и нейтрализуют с помощью твердого NH4HCO3. После удаления перхлоратных солей экстракт подвергают ЖХВД на колонке с обратимой фазой (Spherisorb ODSI). Наблюдают значительное увеличение плазменного инозина в плазме, взятой из животных, получающих вещество (IIIA).
Примеры 90-94. Вещества, полученные так, как в примерах 83-87, использованы для приготовления лекарственных средств в соответствии со способом приготовления примера 88 и полученные инъекцируемые растворы испытывают в соответствии со способом примера 89. Наблюдают значительное увеличение плазменного инозина в плазме, взятой у животных, получающих вещества изобретения.
Пример 95.
Вещество (IIID) получают, используя 2-амино-7-(1-циклогексенил)-3H,5H-пирроло[3,2-d] пиримидин-4-он в качестве промежуточного соединения. Раствор промежуточного соединения (0,2 г, 0,86 ммоль) в этаноле (50 мл) гидрируют с помощью 10%-ного Pd-C (50 мг) при давлении 45 фунтов/дюйм2 в течение 16 ч и горячим фильтруют через цеолит. Фильтрат выпаривают досуха, остаток кристаллизуют из горячего этанола, получая вещество (IIID);
выход 157 мг (78%); т.пл. более 300oC (разл.).
Анализ для C12H16N4O•0•EtOH: вычислено: C 61,80; H 7,10; N 23,51; найдено: C 61,95; H 7,43; N 23,56.
Пример 96. Вещество (IIID), полученное в примере 95, испытывают на ферментативно-ингибирующую активность так, как в примере 81. При 1 mM фосфата IC50 равно 1,3 мкМ, при 50 mM фосфата IC50 равно 145 мкМ.
Пример 97. 3-(2-Адамантил)-пропионитрил получают в этом примере, используя методику способа M. Ohno et al. J. Org. Chem. 53, 1285 (1988). Раствор 2-бромадамантана (20 г, 92,96 ммоль), Bu3SnH (32,46 г, 111,5 моль), акрилонитрила (9,86 г, 185,92 ммоль) и AIBN (740 мг) в толуоле (280 мл) перемешивают при температуре кипения с обратным холодильником в течение 3 ч. Реакционную смесь промывают аммиачной водой (0,4 M, 500 мл), органический слой промывают водой, высушивают над MgSO4 и выпаривают. Остаток перегоняют при температуре между 110-118oC и около 0,2 mmHg; фракции соединяют, получая сырой образец загрязненного 3-(2-адамантил)- пропионитрила с комплексами олова, который очищают на колонне с силикагелем с помощью гексанов, за которыми следуют гексан/этилацетат 97:3 и гексан/этилацетат 95:5; выход 9,4 г (53,4%), т.пл. полутвердый.
Пример 98. 3-(2-Адамантил)-пропионитрил примера 97 используют далее для синтеза соединений изобретения. В атмосфере сухого N2 смесь 3-(2-адамантил)-пропионитрила (7,0 г, 36,99 ммоль), гидрида натрия (1,7 г, 73,95 ммоль) и безводного тетрагидрофурана (75 мл) нагревают при 52oC в водяной бане в течение 15 мин, раствор этилового эфира муравьиной кислоты (13,69 г, 184,89 ммоль) в THF (100 мл) добавляют в течение 45 мин. Через 2 ч при 50-55oC вторую порцию NaH (0,8 г) и HCO2Et (7,5 мл) добавляют и реакционную смесь перемешивают в течение примерно 2 дней. Третью порцию HCO2Et (7,5 мл) и NaH (0,8 г) добавляют и оставляют при комнатной температуре в течение около 24 ч (непрореагировавший нитрил инертен в следующей стадии и может быть регенерирован в первой стадии очистки). Крутую пасту перемешивают в течение ночи и дают охладиться до комнатной температуры. Летучее вещество выпаривают при пониженном давлении, оставшуюся бледно-желтую корку растворяют в минимальном объеме холодной воды (приблизительно 150 мл) при 0oC. Раствор доводят до pH 6,0 добавлением 6N HCl и экстрагируют с помощью CHCl3 (3 x 100 мл). Экстракт промывают водой, высушивают над Na2SO4 и выпаривают в вакууме до густого янтарного масла. Этот сырой продукт используют в следующей реакции без дальнейшей очистки.
Пример 99. Гидрохлорид метилового эфира глицина (6,96 г, 55,46 ммоль) и безводный ацетат натрия (4,55 г, 55,46 ммоль) добавляют к раствору сырого формильного производного (8,0 г) в MeOH/H2O (4:1,500 мл). Спустя 24 ч MeOH выпаривают в вакууме и смесь воды и масла экстрагируют CHCl3. Слой CHCl3 высушивают (Na2SO4) и выпаривают, получая янтарное масло, которое подают в колонку с силикагелем. Элюирование с помощью CHCl3 дает два основных слоя: 3-(2-адамантил)-пропионитрил (использованный в качестве исходного вещества в предыдущей стадии) и желаемый енамин; выход 6 г.
Пример 100. В атмосфере азота этиловый эфир хлормуравьиной кислоты (2,82 г, 26,0 ммоль) добавляют по каплям к раствору енамина примера 99 (5,0 г, 17,34 ммоль) и 1,5-диазабицикло[4.3.0]нон-5-ена ("DBN", 6,46 г, 52,0 ммоль) в сухом CH2Cl2 (50 мл) с внешним охлаждением в ледяной бане. После перемешивания при 0oC в течение 1 ч раствору дают отстояться при комнатной температуре в течение ночи. После контролирования хода реакции по TCX вводят дополнительное количество ClCO2Et (1,81 мл) и DBN (3,23 г) для завершения реакции и раствор оставляют стоять в течение 24 ч. Летучее вещество выпаривают в вакууме, вязкий остаток очищают на короткой колонне с силикагелем (главная цель которой удаление менее мобильной DBN), получая N-защищенный пиррол, который используют для следующей стадии без дальнейшей очистки.
Пример 101. К раствору сырого N-защищенного пиррола примера 100 (6,0 г, 16,59 ммоль) в MeOH (100 мл) добавляют твердый Na2CO3 (4,39 г, 41,49 ммоль), реакционную смесь перемешивают при комнатной температуре в течение 48 ч с отделением полученного деблокированного пиррола. Смесь выпаривают досуха, остаток тщательно растирают с водой в порошок (50 мл) для растворения неорганических веществ и экстрагируют CHCl3 (3•100 мл). Экстракт сушат (Na2SO4) и выпаривают, получая вязкую смолу, которую кристаллизуют с помощью эфира; выход 4 г; т.пл. 162-163oC.
Пример 102. Бензоилизотиоцианат (1,22 г, 7,47 ммоль) добавляют по каплям к раствору деблокированного пиррола примера 101 (1,91 г, 6,62 ммоль) в сухом CH2Cl2 (50 мл). Спустя 1 ч при комнатной температуре раствор выпаривают, смолистый остаток растворяют в Et2O (100 мл) с почти немедленным выделением кристаллического твердого вещества, которое отфильтровывают. Маточник Et2O нагревают до кипения и разбавляют равным объемом горячего циклогексана. При медленном охлаждении раствор дает дополнительное количество тиоуреидопроизводного; выход 2,81 г (95%); т.пл. 193-194oC.
Пример 103. Йодистый метил (2,46 г, 17,39 ммоль) добавляют к раствору тиоуреидопроизводного примера 102 (2,7 г, 5,98 ммоль) и DBN (0,82 г, 6,57 ммоль) в сухом CH2Cl2 (50 мл) при 0oC. Раствор перемешивают при 0oC в течение 15 мин, при температуре окружающей среды в течение 1 ч и затем выпаривают в вакууме, получая сырой образец метилтиосоединения как промежуточного вещества; выход 2,78 г (сырой).
Пример 104.
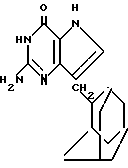
Раствор метилтиосоединения примера 103 (2,78 г, 5,18 ммоль) в 150 мл MeOH, который был насыщен NH3 при 0oС, нагревают при 90-95oC в течение 24 ч в футерованной стеклянными плитками бомбе из нержавеющей стали. Содержимое охлажденной бомбы выпаривают в вакууме, получая смесь вещества (IVA), бензамида и побочного продукта, который является 2-метилтиопроизводным в отличие от 2-аминосоединения (IVA). Смесь энергично перемешивают в течение нескольких минут с приблизительно 75 мл Et2O и нерастворимое белое твердое вещество отфильтровывают с помощью Et2O. Фильтрат содержал большую часть бензамида и 2-метилтиопроизводное. Раствор нерастворимого в Et2O твердого вещества (1,38 г) в MeOH выпаривают с приблизительно 25 г силикагеля. Измельченный в порошок остаток наносится равномерно на верхнюю часть колонки с силикагелем, которую затем элюируют с помощью CHCl3/MeOH/HOAc (95:5:1), получая 2-метилтиопроизводное как побочный продукт и желаемое 2-аминосоединение (IVA). (IVA) продукт перекристаллизовывают при экстракции в кипящем изопропилацетате в аппарате Сокслета. Белые кристаллы собирают в три массы и высушивают в вакууме над P2O5 при 110oC в течение 7 ч; выход 51,5% т.пл. более 350oC (разл.).
Анализ для C17H22N4O•0,21MeOH•0,22H2O: вычислено: C 66,88; H 7,59; N 18,12; найдено: C 66,86; H 7,59; N 18,12.
Пример 105. Вещество примера 104 испытывают на ферментативную ингибирующую активность. Для этого проводят ферментативный анализ пуриннуклеозидфосфорилазы (PNP), в котором определяют PNP активность (IC50) для вещества радиохимическим измерением образования [14C]-гипоксантина из [14C]-инозина (Biomedicine, 33, 39, 1980), используя селезенку теленка в качестве источника энзима. При 1 mM фосфата IC50 равно 0,090 мкМ и при 50 mM фосфата IC50 равно 2,5 мкМ.
Примеры 106-110. Получают следующие вещества изобретения: 2-амино-7-(CH2R)-3H, 5H-пирроло[3,2-d]пиримидин-4-оны, в которых R - замещенная 2-адамантильная группа:
пример 106: R 2-(1-метил)-адамантил;
пример 107: R 2-(1-хлор)-адамантил;
пример 108: R 2-(1-трифторметил)-адамантил;
пример 109: R 2-(1-метокси)-адамантил;
пример 110: R 2-(1-фтор)-адамантил.
Вещества получают, следуя способам, приведенным в примерах 98-104, используя соответствующие 3-(2-адамантил)-пропионитрилы в качестве исходных веществ.
Примеры 111-116. Получают следующие вещества изобретения: 2-амино-7-(CH2R)-3H, 5H-пирроло[3,2-d] пиримидин-4-оны, в которых R - следующая 1-адамантильная группа:
пример 111: R 1-(2-метил)-адамантил;
пример 112: R 1-(2-хлоро)-адамантил;
пример 113: R 1-(2-трифторметил)-адамантил;
пример 114: R 1-(2-метокси)-адамантил;
пример 115: R 1-(2-фтор)-адамантил;
пример 116: R 1-адамантил.
Вещества получают, следуя способам, приведенным в примерах 98-104, используя соответствующие 3-(1-адамантил)-пропионитрилы в качестве исходных веществ.
Пример 117. Готовят фармацевтическую композицию для интраперитонального инъекцирования для испытания вещества (IVA). Интраперитональный инъекцируемый раствор, содержащий вещество (IVA), растворяют в водном носителе, который содержит 10% DMSO.
Пример 118. Вводят интраперитонально в Lewis Rats опытную композицию, содержащую вещество (IVA) примера 117 в дозе 30 мг вещества (IVA) инъекцией дважды в день. Используют контрольные группы, которые получают только наполнитель. Через определенное время после введения животных усыпляют и получают образцы плазмы. Плазму экстрагируют с помощью холодной 0,5N HClO4 и нейтрализуют твердым NH4HCO3. После удаления перхлоратных солей экстракт подвергают ЖХВД на колонке с обратимой фазой (Spherisorb ODSI). Наблюдают значительное увеличение плазменного инозина в плазме, взятой у животных, получающих вещество (IVA).
Примеры 119-129. Вещества, полученные как в примерах 106-116, использованы для приготовления лекарственных средств в соответствии со способом приготовления примера 117, полученные инъекцируемые растворы испытывают в соответствии со способом примера 118. Наблюдают увеличение плазменного инозина в плазме, взятой у животных, получающих вещества изобретения.
Пример 130. В этом примере получают 3-циклопентилпропионитрил. 3-Циклопентилпропионитрил хлорид (57,7 г, 0,36 моль) добавляют по каплям к большому избытку концентрированного гидроксида аммония (400 мл), охлажденного в бане лед/соль. Тяжелую суспензию белого твердого вещества перемешивают в течение ночи, собирают фильтрованием, промывают холодной водой и перекристаллизовывают из примерно 2 л кипящей воды. Блестящие белые пластинки амида сушат в вакууме над P2O5; выход 31,6 г (62,3%), т.пл. 122oC.
С защитой от атмосферной влаги раствор амида (23,5 г, 0,166 моль) в POCl3 (150 мл) нагревают при 120oC в течение 1 ч. Масляную баню охлаждают до около 70oC, избыток POCl3 отгоняют в вакууме, охлажденный остаток наливают на лед (около 300 г). Смесь нейтрализуют путем осторожного добавления твердого Na2CO3 и экстрагируют с помощью нескольких порций Et2O. Высушенный над Na2SO4 экстракт выпаривают, получая прозрачное бледно-желтое масло, которое перегоняют в вакууме, получая желаемый нитрил; выход 16,86 г (82%), т.кип. 88,0-88,5oC/8,7 мм. MS (EI): m/z 122 (M-H)+; ИК (сар. пленка), 2245 см-1 (CN); 1H ЯМР, δ 1,67 (q, 2, CH2CH2CN), 2,36 (t, 2, -CH2CN), комплексные мультиплеты, центрированные около 1,11, 1,63, 1,86 (циклопентильные протоны).
Пример 131. 3-Циклопентилпропионитрил предыдущего примера далее обрабатывают в синтезах изобретения. В атмосфере сухого N2 смесь 3-циклопентилпропионитрила (14,8 г, 0,12 моль), гидрида натрия (5,8 г, 0,24 моль) и безводного тетрагидрофурана (300 мл) нагревают при 52oC на водяной бане в течение 15 мин, раствор этилового эфира муравьиной кислоты (13,3 г, 0,18 моль) в ТГФ (100 мл) добавляют в течение 45 мин. Через 2 ч при 50-55oC добавляют вторую порцию NaH (1,9 г) и HCO2Et (5,0 мл), за которой в течение 30 мин следует третья порция HCO2Et (непрореагировавший нитрил инертен в следующей стадии и может быть регенерирован при первой стадии очистки). Крутую пасту перемешивают в течение ночи и дают возможность охладиться до комнатной температуры. Летучее вещество выпаривают при пониженном давлении, оставшуюся бледно-желтую корку растворяют в минимальном объеме холодной воды (около 150 мл) при 0oC. Раствор доводят до pH 6,0 добавлением 6N НС1 и экстрагируют CHCl3 (3 x 100 мл). Экстракт промывают водой, сушат над Na2SO4 и выпаривают в вакууме до густого янтарного масла. Этот сырой продукт (15,6 г) используют в следующей реакции без дальнейшей очистки.
Пример 132. Гидрохлорид метилового эфира глицина (19,31 г, 0,154 моль) и безводный ацетат натрия (12,61 г, 0,154 моль) добавляют к раствору сырого формильного производного предыдущего примера (15,6 г) в MeOH/H2O (4:1, 500 мл). Через 24 ч MeOH выпаривают в вакууме, смесь воды и масла экстрагируют с помощью CHCl3. Слой CHCl3 высушивают (Na2SO4) и выпаривают, получая янтарное масло, которое подают на колонку с силикагелем. Элюирование с помощью CHCl3 дает два основных слоя: 3-циклопентилпропионитрил (8,22 г, 66,7 ммоль или 55,6% нитрила, использованного в качестве исходного вещества в предыдущей стадии); желаемый енамин (3,45 г, 29,1% от теоретического выхода, с поправкой на количество нитрила, присутствующего в исходном веществе; MS (FAB): 223 (M+H)+).
Пример 133. В атмосфере азота этиловый эфир хлормуравьиной кислоты (2,53 г, 23,3 ммоль) добавляют по каплям к раствору енамина предыдущего примера (3,45 г, 15,5 ммоль) и DBN (5,78 г, 46,6 ммоль) в сухом CH2Cl2 (50 мл) с внешним охлаждением на ледяной бане. После перемешивания при 0o в течение 1 ч раствор выдерживают при комнатной температуре в течение ночи. После проверки хода реакции по TCX вводят дополнительное количество ClCO2Et (0,5 мл) и DBN (3,0 мл) для завершения реакции, раствор выдерживают в течение 24 ч. Летучее вещество испаряют в вакууме, вязкий остаток очищают на короткой колонке с силикагелем (главная цель которой - удаление менее мобильного DBN), получая N-защищенный пиррол (4,50 г, 98%), который используют для следующей стадии без дальнейшей очистки.
Пример 134. К раствору N-защищенного пиррола предыдущего примера (4,50 г, 15,3 ммоль) в MeOH (100 мл) добавляют твердый Na2CO3 (1,62 г, 15,3 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 48 ч с отделением полученного деблокированного пиррола. Смесь выпаривают досуха, остаток тщательно растирают в порошок с H2O (50 мл) для растворения неорганических веществ и экстрагируют CHCl3 (3 x 100 мл). Экстракт сушат (Na2SO4) и выпаривают, получая вязкую смолу, которую кристаллизуют при высушивании в вакууме; выход 2,97 г (87,4%) вещества, пригодного для использования в качестве промежуточного соединения без дальнейшей очистки. Более глубокая очистка может быть осуществлена колоночной хроматографией с применением силикагеля/CHCl3 или перекристаллизацией из смеси толуол/циклогексан (1:3).
Пример 135. Бензоилизотиоцианат (2,62 г, 16,03 ммоль) добавляют по каплям к раствору деблокированного пиррола примера 134 (2,97 г, 13,36 ммоль) в сухом CH2Cl2 (100 мл). Через 1 ч при комнатной температуре раствор выпаривают и смолистый остаток растворяют в Et2O (100 мл) с почти немедленным отделением кристаллического твердого вещества; выход 1,75 г. Фильтрат Et2O нагревают до кипения и разбавляют с помощью равного объема горячего циклогексана. При медленном охлаждении раствор дает дополнительные 1,58 г тиоуреидопроизводного; общий выход 3,33 г (64,6%). Небольшое количество тиоуреидосоединения перекристаллизовывают из горячей смеси Et2O/циклогексан (15 мл каждого); т.пл. 123-125oC. MS (FAB): 386 (M+H)+.
Анализ для C20H23N3O3S•0,45C6H12: вычислено: C 64,40; H 6,76; N 9,93; найдено: C 64,51; H 7,10; N 9,93.
Пример 136. Йодистый метил (2,60 г, 18,32 ммоль) добавляют к раствору тиоуреидосоединения примера 135 (3,21 г, 8,33 ммоль) и DBN (1,24 г, 9,99 ммоль) в сухом CH2Cl2 (80 мл) при 0oC. Раствор перемешивают при 0oC в течение 15 мин при температуре окружающей среды в течение 1 ч, а затем испаряют в вакууме. Раствор остатка в CHCl3 хроматографируют на колонне с силикагелем с помощью CHCl3 в качестве растворителя для элюирования, получая гомогенные фракции метилтиосоединения как промежуточного продукта; выход 2,46 г (74%).
Пример 137.
Раствор метилтиосоединения примера 136 (2,07 г, 5,18 ммоль) в 150 мл MeOH, который был насыщен NH3 при 0oC, нагревают при 90-95oC в течение 24 ч в футерованной стеклянными плитками бомбе из нержавеющей стали. Содержимое охлажденной бомбы выпаривают в вакууме, получая смесь вещества (IVC), бензамида и побочного продукта, который является 2-метилтиопроизводным в отличие от 2-аминосоединения (IVC). Смесь энергично перемешивают в течение нескольких минут с приблизительно 75 мл Et2O и нерастворимое белое вещество отфильтровывают и промывают Et2O. Фильтрат содержал большую часть бензамида и 2-метилтиосоединения. Раствор нерастворимого в Et2O твердого вещества (1,13 г) в MeOH выпаривают с приблизительно 10 г силикагеля. Измельченный в порошок остаток наносят равномерно на верхнюю часть колонки с силикагелем, которую затем элюируют с помощью смеси CHCl3/MeOH/HOAc (95:5:1), получая 2-метилтиопроизводное как побочный продукт (252 мг, MS (FAB): 264 (M+H)+) и желаемое 2-аминосоединение (IVC) (679 мг, 56,4%). (IVC) перекристаллизовывают при экстракции в кипящем изопропилацетате в аппарате Сокслета. Белые кристаллы собирают в три порции и высушивают в вакууме над P2O5 при 110oC в течение 7 ч; выход 540 мг (44,9%); т.пл. 324-326oC (разл.); MS (FAB): 233 (M+H)+.
Анализ для C12H16N4О: вычислено: C 62,05; H 6,94; N 24,12; найдено: C 62,04; H 7,11; N 24,48.
Пример 138. Вещество примера 137 испытывают на ферментативно-ингибирующую активность в соответствии со способом примера 9. При 1 mM фосфата IC50 равно 0,029 мкМ и при 50 mM фосфата IC50 равно 1,8 мкМ.
Примеры 139-142. Получают следующие вещества изобретения: 2-амино-7-(CH2R)-3H, 5H-пирроло[3,2-d]пиримидин-4-оны, в которых R - замещенный циклопентил:
пример 139: R 3-метилциклопентил;
пример 140: R 2-хлороциклопентил;
пример 141: R 3-трифторметилциклопентил;
пример 142: R 3-метоксициклопентил.
Вещества получают, следуя способам, приведенным в примерах 130-137, используя соответствующие 3-(замещенные циклопентил)-пропионитрилы в качестве исходных веществ.
Пример 143. Готовят фармацевтическую композицию для интраперитонального инъекцирования для испытания вещества (IVC). Интраперитональный инъекцируемый раствор, содержащий вещество (IVC), растворяют в водном носителе, который содержит 10% DMSO.
Пример 144. Используя способ примера 112, вводят интраперитонально в Lewis Rats опытную композицию, содержащую вещество (IVC) примера 143 и результаты сравнивают с контрольными. Наблюдают значительное увеличение плазменного инозина в плазме, взятой у животных, получающих вещество (IVC).
Пример 145. Получают 3-циклогексилпропионитрил. Раствор циклогексанпропионовой кислоты (50 г, 0,32 моль) и хлористого тионила (152 г, 1,28 моль) в 100 мл бензола выдерживают в течение ночи и затем выпаривают до маслообразного остатка. Остаток добавляют порциями к 28%-ному водному раствору аммиака (270 мл) при 25oC и смесь перемешивают в течение примерно 2 ч. Полученный продукт собирают фильтрованием, промывают холодной водой и перекристаллизовывают из около 2 л кипящей воды. Блестящие белые пластинки амида высушивают в вакууме над P2O5; выход 45,5 г.
С защитой от атмосферной влаги раствор амида (45,5 г, 0,293 моль) в SOCl2 (200,3 г, 1,68 моль) кипятят с обратным холодильником в течение примерно 6 ч. Масляную баню охлаждают до около 70oC и избыток SOCl2 отгоняют под вакуумом, охлажденный остаток выливают на лед (около 300 г). Смесь нейтрализуют путем осторожного добавления твердого Na2CO3 и экстрагируют с помощью нескольких порций Et2O. Высушенный (Na2SO4) экстракт выпаривают, получая прозрачное, бледно-желтое масло, которое перегоняют в вакууме, получая желаемый нитрил; выход 42,0 г.
Пример 146. 3-Циклогексилпропионитрил примера 145 далее используют в синтезе для получения соединений изобретения. В атмосфере сухого N2 смесь 3-циклогексилпропионитрила (22,3 г, 0,16 моль), гидрида натрия (5,38 г, 0,224 моль) и безводного тетрагидрофурана (120 мл) нагревают при 52oC на водяной бане в течение 15 мин, добавляют раствор этилового эфира муравьиной кислоты (55,4 г, 0,75 моль) в THF (50 мл) в течение 45 мин. Через 2 ч при 50-55oC добавляют вторую порцию NaH (2,0 г) и HCO2Et (5,0 мл) (непрореагировавший нитрил инертен в следующей стадии и может быть регенерирован при первой стадии очистки), реакционную смесь перемешивают в течение примерно 3 дней при 55oC и затем дают охладиться до комнатной температуры. Летучее вещество выпаривают при пониженном давлении, оставшуюся бледно-желтую корку растворяют в минимальном количестве холодной воды (около 75 мл) при 0oC. Раствор доводят до pH 6,0 добавлением 6N HCl и экстрагируют CHCl3 (3 x 100 мл). Экстракт промывают водой, сушат над Na2SO4 и выпаривают в вакууме до густого янтарного масла. Этот сырой продукт используют в следующей реакции без дальнейшей очистки.
Пример 147. Гидрохлорид метилового эфира глицина (30,60 г, 0,24 моль) и безводный ацетат натрия (19,99 г, 0,24 моль) добавляют к раствору сырого формильного производного предыдущего примера (25,22 г) в смеси MeOH/H2O (4: 1,500 мл). Спустя 24 ч MeOH испаряют в вакууме, смесь воды и масла экстрагируют CHCl3. Слой CHCl3 высушивают (Na2SO4) и выпаривают, получая янтарное масло, которое подают на колонку с силикагелем. Элюирование CHCl3 дает два основных слоя: 3-циклогексилпропионитрил (использованный в качестве исходного вещества в предыдущей стадии); желаемый енамин; выход 16 г.
Пример 148. В атмосфере азота этиловый эфир хлормуравьиной кислоты (1,38 г, 12,7 ммоль) добавляют по каплям к раствору енамина примера 147 (2,0 г, 8,46 ммоль) и DBN (2,1 г, 16,9 ммоль) в сухом CH2Cl2 (50 мл) с внешним охлаждением в ледяной бане. После перемешивания при 0oC в течение 1 ч раствор выдерживают при комнатной температуре в течение ночи. После проверки хода реакции по TCX вводят дополнительное количество ClCO2Et (0,5 мл) и DBN (1,5 мл) для завершения реакции, раствору дают постоять в течение 24 ч. Летучее вещество выпаривают в вакууме, вязкий остаток очищают на короткой колонне с силикагелем (цель которой удаление менее мобильного DBN), получая N-защищенный пиррол, который используют для следующей стадии без дальнейшей очистки.
Пример 149. К раствору N-защищенного пиррола примера 148 (2,6 г, 8,43 ммоль) в MeOH (100 мл) добавляют твердый Na2CO3 (2,23 г, 21,07 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 48 ч с отделением полученного деблокированного пиррола. Смесь выпаривают досуха, остаток тщательно растирают в порошок с водой (50 мл) для растворения неорганических веществ и экстрагируют с помощью CHCl3 (3 x 100 мл). Экстракт сушат (Na2SO4) и испаряют, получая вязкую смолу, которую очищают на колонне с силикагелем, используя CHCl3 в качестве растворителя для элюирования; выход 1,67 г (84%), т.пл. 73-74oC.
Пример 150. Бензоилизотиоцианат (0,74 г, 4,02 ммоль) добавляют по каплям к раствору деблокированного пиррола примера 149 (0,95 г) в сухом CH2Cl2 (20 мл). Спустя 1 ч при комнатной температуре раствор выпаривают и смолистый остаток растворяют в Et2O (100 мл) с почти немедленным отделением кристаллического твердого вещества. Маточник Et2O нагревают до кипения и разбавляют равным объемом горячего циклогексана. При медленном охлаждении раствор дает дополнительно тиоуреидопроизводное; выход 1,41 г (88%), т.пл. 156-157oC.
Пример 151. Йодистый метил (1,1 г, 7,6 ммоль) добавляют к раствору тиоуреидопроизводного примера 150 (0,96 г, 2,61 ммоль) и 1,5-диазабицикло[4.3.0] нон-5-ена (0,38 г, 3,0 ммоль) в сухом CH2Cl2 (20 мл) при 0oC. Раствор перемешивают при 0oC в течение 15 мин, при температуре окружающей среды в течение 1 ч и затем выпаривают в вакууме. Раствор остатка в CHCl3 хроматографируют на колонке с силикагелем с CHCl3 в качестве растворителя для элюирования, получая гомогенные фракции метилтиосоединения как промежуточного вещества; выход 0,92 г.
Пример 152.
Раствор метилтиосоединения примера 151 (0,8 г, 1,93 ммоль) в 50 мл MeOH, который был насыщен NH3 при 0oC, нагревают при 90-95oC в течение 24 ч в футерованной стеклянными плитками бомбе из нержавеющей стали. Содержимое охлажденной бомбы выпаривают в вакууме, получая смесь вещества (IVD), бензамида и побочного продукта, который является 2-метилтиопроизводным, в отличие от 2-аминосоединения (IVD). Смесь энергично перемешивают в течение нескольких минут с приблизительно 75 мл Et2, нерастворимое твердое белое вещество отфильтровывают и промывают с помощью Et2O. Фильтрат содержал большую часть бензамида и 2-метилтиосоединения. Раствор нерастворимого в Et2O твердого вещества (0,390 г) в MeOH выпаривают с приблизительно 10 г силикагеля. Измельченный в порошок остаток наносят равномерно на верхнюю часть колонки с силикагелем, которую затем элюируют смесью CHCl3/MeOH/HOAc (95:5: 1), получая 2-метилтиопроизводное как побочный продукт и желаемое 2-аминосоединение (IVD). (IVD) перекристаллизовывают при экстракции в кипящем изопропилацетате в аппарате Сокслета. Белые кристаллы собирают в три порции и высушивают в вакууме над P2O5 при 110oC в течение 7 ч; выход 49% т.пл. более 300oC.
Анализ для C13H18N4O: вычислено: C 63,39; H 7,36; N 22,74; найдено: C 63,50; H 7,74; N 22,67.
Пример 153. Вещество примера 152 испытывают на ферментативную ингибирующую активность как в примере 105. При 1 mM фосфата IC50 равно 0,037 мкМ, при 50 mM фосфата IC50 равно 2,2 мкМ.
Пример 154. Вещество примера 152 испытывают для определения его эффективности по ингибированию токсичности 2'-дезоксигуанозина (d-GuO) (D.A. Schewach et al. Cancer es. 46, 519 (1986) и J.C.Sirear et al. Agents and tions, 21, 253 (1987)). Для этого выращивают CCRF-CEM клетки в RPMI-1640 среде. К суспендированным культурам этих клеток добавляют d-GuO при стационарной концентрации (5,62 мкМ) и вещество при переменной концентрации, определяют число клеток в Coulter счетчике через 24, 48 и 72 ч после этого. Из этих данных рассчитывают IC50, равную 2,0 мкМ, как концентрацию вещества, требуемую для снижения роста числа клеток между 0 и 72 ч на 50% от клеток контрольных культур.
Пример 155.
Получают 2-амино-7-(3-метилциклогексилметил)-3H, 5H-пирроло[3,2-d]пиримидин-4-он. Сначала используя способы, приведенные в примерах 146 152, но с 3-(3-метилбензил)-пропионитрилом в качестве исходного вещества, получают арильное производное 2-амино-7-(3-метилбензил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он. Раствор арильного производного (0,2 г, 0,78 ммоль) в трифторуксусной кислоте (TFA) (20 мл) гидрируют с помощью PtO2 при давлении 60 фунтов/дюйм в течение 24 ч. Катализатор отфильтровывают через цеолитный слой и фильтрат выпаривают. Остаток растирают в порошок с помощью метанола и оставляют в холодильнике на ночь. Полученная кристаллическая трифторацетатная соль осаждается из раствора и собирается фильтрованием. TFA-соль суспендируют в 8 мл воды, доводя до pH 8 конц. NH4OH, измельчают ультразвуком. Чистый продукт собирают, промывают водой и сушат; выход 165 мг (81%), т.пл. 282oC.
Анализ для C14H20N4O: вычислено: C 64,60; H 7,74; N 21,52; найдено: C 64,24; H 7,96; N 21,51%
Пример 156.
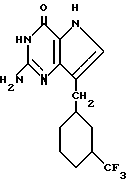
Способ, описанный в примере 155, повторяют для получения 2-амино-7-(3-трифторметилциклогексилметил)-3H, 5H-пирроло [3,2-d] пиримидин-4-она, используя 3-(3-трифторметилбензил)-пропионитрил в качестве исходного вещества; выход 69% т.пл. 165oC.
Анализ для C14H17N4OF3•0,6H2O: вычислено: C 51,72; H 5,64; N 17,23; найдено: C 51,82; H 5,71; N 16,81%
Пример 157. Вещество, полученное в примере 155, испытывают на ферментативно-ингибирующую активность, как в примере 105. При 1 mM фосфата IC50 равно 0,025 мкМ, при 50 mM фосфата IC50 равно 0,820 мкМ.
Пример 158. Вещество, полученное в примере 156, испытывают на ферментативно-ингибирующую активность, как в примере 105. При 1 mM фосфата IC50 равно 0,020 мкМ, при 50 mM фосфата IC50 равно 0,740 мкМ.
Пример 159. В этом примере 3-циклогептилпропионитрил получают в соответствии со способом примера 97, используя раствор 2-бромциклогептана (25,57 г, 144,38 ммоль), Bu3SnH (50,42 г, 173,26 ммоль), акрилонитрила (15,32 г, 288,77 ммоль) и AIBN (1,13 г) в толуоле (300 мл); выход 16 г в виде масла.
Пример 160. 3-Циклогептилпропионитрил примера 159 далее используют в синтезе для получения соединений изобретения. В атмосфере сухого N2 смесь 3-циклогептилпропионитрила (8,5 г, 56,19 ммоль), гидрида натрия (2,6 г, 112,39 ммоль) нагревают на водяной бане в течение 15 мин, раствор этилового эфира муравьиной кислоты (20,81 г, 280,99 ммоль) в THF (100 мл) добавляют в течение 45 мин. Спустя 2 ч при 50-55oC добавляют вторую порцию NaH (1,35 г) и HCO2Et (10,4 г), за которой через 30 мин следует третья порция HCO2Et (непрореагировавший нитрил инертен в следующей стадии и может быть регенерирован на первой стадии очистки). Густую пасту перемешивают в течение ночи и дают охладиться до комнатной температуры. Летучее вещество выпаривают при пониженном давлении, оставшуюся бледно-желтую корку растворяют в минимальном объеме холодной воды (около 150 мл) при 0oC. Раствор доводят до pH 6,0 добавлением 6N HCl и экстрагируют с помощью CHCl3 (3 x 100 мл). Экстракт промывают водой, высушивают над Na2SO4 и выпаривают в вакууме до густого янтарного масла. Этот сырой продукт используют в следующей реакции без дальнейшей очистки.
Пример 161. Гидрохлорид метилового эфира глицина (9,35 г, 74,47 ммоль) и безводный ацетат натрия (6,10 г, 74,47 ммоль) добавляют к сырому формильному производному примера 160 (8,9 г, 49,65 ммоль) в смеси MeOH/H2O (4:1,250 мл). Спустя 24 ч MeOH выпаривают в вакууме, смесь воды и масла экстрагируют с помощью CHCl3. Слой CHCl3 сушат (Na2SO4) и выпаривают, получая янтарное масло, которое подают на колонку с силикагелем. Элюирование CHCl3 дает два основных слоя: 3-циклогептилпропионитрил (использованный в качестве исходного вещества в предыдущей стадии); желаемый енамин, который перекристаллизовывают из смеси CHCl3/Et2O; выход 6,18 г, т.пл. 57-58oC.
Пример 162. В атмосфере азота этиловый эфир хлормуравьиной кислоты (4,01 г, 37,03 ммоль) добавляют по каплям к раствору енамина примера 161 (6,18 г, 24,69 ммоль) и DBN (9,19 г, 74,04 ммоль) в сухом CH2Cl2 (100 мл) с внешним охлаждением на ледяной бане. После перемешивания при 0oC в течение 1 ч раствору дают постоять при комнатной температуре в течение ночи. После проверки хода реакции по TCX добавляют дополнительное количество ClCO2Et (0,5 мл) и DBN (3,0 мл) для завершения реакции, раствору дают постоять в течение 24 ч. Летучее вещество выпаривают в вакууме, вязкий остаток очищают на короткой колонке с силикагелем (главная цель которой удаление менее мобильного DBN), получая N-защищенный пиррол, который используют для следующей стадии без дальнейшей очистки.
Пример 163. К раствору N-защищенного пиррола примера 162 (7,8 г, 24,19 ммоль) в MeOH (100 мл) добавляют твердый Na2CO3 (6,41 г, 60,48 ммоль), реакционную смесь перемешивают при комнатной температуре в течение 48 ч с отделением полученного деблокированного пиррола. Смесь выпаривают досуха, остаток тщательно растирают в порошок с водой (50 мл) для растворения неорганических веществ и экстрагируют с помощью CHCl3 (3 x 100 мл). Экстракт сушат (Na2SO4) и выпаривают до получения вязкой смолы, которую очищают колоночной хроматографией, используя силикагель/CHCl3; выход 4 г, т.пл. 88- 89oC.
Пример 164. Бензоилизотиоцианат (1,5 г, 8,96 ммоль) добавляют по каплям к раствору деблокированного пиррола примера 163 (1,99 г, 7,95 ммоль) в сухом CH2Cl2 (50 мл). Спустя 1 ч при комнатной температуре раствор выпаривают и смолообразный остаток растворяют в Et2O (100 мл) с почти немедленным отделением кристаллического твердого вещества. Маточник Et2O нагревают до кипения и разбавляют равным объемом горячего циклогексана. При медленном охлаждении раствор дает дополнительное тиоуреидосоединение; выход 2,89 г (88% ), т.пл. 158-159oC.
Пример 165. Йодистый метил (1,7 г, 11,96 ммоль) добавляют к раствору тиоуреидосоединения примера 164 (1,7 г, 4,1 ммоль) и DBN (0,56 г, 4,52 ммоль) в сухом CH2Cl2 (80 мл) при 0oC. Раствор перемешивают при 0oC в течение 15 мин, при температуре окружающей среды в течение 1 ч, затем выпаривают в вакууме. Раствор остатка в CHCl3 хроматографируют на колонне с силикагелем с CHCl3 в качестве растворителя для элюирования, получая гомогенные фракции метилтиосоединения как промежуточного вещества.
Пример 166.
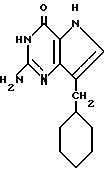
Раствор метилтиосоединения примера 165 (1,72 г, 4,02 ммоль) в 50 мл MeOH, который был насыщен NH3 при 0oC, нагревают при 90-95oC в течение 24 ч в футерованной стеклянными плитками бомбе из нержавеющей стали. Содержимое охлажденной бомбы выпаривают в вакууме, получая смесь вещества (IVE), бензамида и побочного продукта, который является 2-метилтиопроизводным, в отличие от 2-аминосоединения (IVE). Смесь энергично перемешивают с приблизительно 75 мл Et2O, нерастворимое белое твердое вещество отфильтровывают и промывают Et2O. Фильтрат содержал большую часть бензамида и 2-метилтиосоединения. Раствор нерастворимого в Et2O твердого вещества (0,850 г) в MeOH выпаривают с приблизительно 10 г силикагеля. Измельченный в порошок остаток наносят равномерно на верхнюю часть колонны с силикагелем, которую затем элюируют с помощью смеси CHCl3/MeOH/HOAc (95:5:1), получая 2-метилтиопроизводное как побочный продукт и желаемое 2-аминосоединение (IVE). (IVE) продукт перекристаллизовывают при экстракции в кипящем изопропилацетате в аппарате Сокслета. Белые кристаллы собирают в три порции и высушивают в вакууме над P2O5 при 110oC в течение 7 ч; выход 54% т.пл. более 300oC (разл.).
Анализ для C14H20N4O: вычислено: C 64,60; H 7,74; N 21,52; найдено: C 64,78; H 8,01; N 21,61.
Пример 167. Вещество примера 166 испытывают на ферментативную ингибирующую активность как в примере 105. При 1 mM фосфата IC50 равно 0,030 мкМ, при 50 mM фосфата IC50 равно 0,840 мкМ.
Используя способ примера 97, из 1-бромнорборнана получают 3-(1-норборнанил)-пропионитрил, и из 2-бромнорборнана получают 3-(2-норборнанил)-пропионитрил (смесь 2-экз и 2-эндо). Следуя примерам 98-104, пропионитрилы превращают в вещества (IVF), (IVG) и (IVH).
Пример 169. Используя способ примера 97, 3-(1-бицикло[3.2.1]октанил)-пропионитрил, 3-(2-бицикло[3.2.1]октанил)-пропионитрил, 3-(3-бицикло[3.2.1] октанил)-пропионитрил и 3-(8-бицикло[3.2.1] октанил)-пропионитрил получают соответственно из 1-бромбицикло[3.2.1] октана, 2-бромбицикло[3.2.1]октана, 3-бромбицикло[3.2.1] октана и 8-бромбицикло[3.2.1] октана. Следуя примерам 98-104, пропионитрилы превращают в вещество (IVI) и родственные 2-бицикло[3.2.1]октанил-; 3-бицикло[3.2.1]октанил- и 8-бицикло[3.2.1]-октанилпроизводные.
Пример 170. Используя методику способа, описанного в D. Farcasiu, Synthesis, 615 (1972), 6-бицикло[3.2.1]октанкарбоксальдегид получают реакцией бицикло[3.2.1] октан-6-она с триметилсульфоксоний йодидом с образованием промежуточного эпоксида, который затем превращают в альдегид обработкой эфиратом трифторида бора. Следуя способу Netherlands Pat. 6, 610, 204, альдегид конденсируют с циануксусной кислотой при кипячении с обратным холодильником в растворе пиридин/толуол с каталитическим количеством ацетата аммония в течение 48-72 ч, получая соответствующий акрилонитрил. Акрилонитрил затем гидрируют, используя палладий-на-угле как катализатор, в метаноле, как описано Profitt et al. в J. Org. Chem. 40, 127, (1975), для получения 3-(6-бицикло[3.2.1] октанил)-пропионитрила. Следуя примерам 98-104, пропионитрил превращают в 6-бицикло[3.2.1]октанильное производное, аналогичное веществу (IVL).
Пример 171. Используя способ примера 97, 3-(1-бицикло[3.3.1]нонанил)-пропионитрил и 3-(3-бицикло[3.3.1]нонанил)-пропионитрил соответственно получают из 1-бромбицикло[3.3.1] нонана и 3-бромбицикло[3.3.1]нонана. Следуя примерам 98-104, пропионитрилы превращают в вещество (IVM) и родственное 3-бицикло[3.3.1]нонанилпроизводное.
Пример 172. Следуя способу примера 171, бицикло[3.3.1]нонан-9-он реагирует с образованием соответствующего альдегида, из которого получают соответствующий 3-замещенный пропионитрил, который затем превращают в 9-бицикло[3.3.1]нонанильное производное, аналогичное веществу (IVM).
Пример 173. Следуя способу примера 170, 2-бицикло[3.3.1]нонанкарбоксальдегид реагирует с образованием соответствующего 3-замещенного пропионитрила, который затем превращают в 2-бицикло[3.3.1]нонанильное производное, аналогичное веществу (IVM).
Пример 174. Используя способ примера 97, 3-(1-норадамантил) -пропионитрил получают из 1-бромнорадамантана и 3-(2-норадамантил)-пропионитрил получают из 2-бромнорадамантана. Следуя примерам 98-104, пропионитрил превращают в конечное вещество (IVN).
Пример 175. Следуя способу примера 170, 3-норадамантанкарбоксальдегид реагирует с образованием соответствующего 3-замещенного пропионитрила, который затем превращают в 3-норадамантильное производное, аналогичное веществу (IVN).
Пример 176. Следуя способу примера 170, норадамантан-7-он реагирует с образованием соответствующего альдегида, из которого получают соответствующий 3-замещенный пропионитрил, который затем превращают в 7-норадамантильное производное, аналогичное веществу (IVN).
Пример 177. Используя способ примера 97, 3-(1-бицикло[2.2.2]октанил)-пропионитрил получают из 1-бромбицикло[2.2.2]октана и 3-(2-бицикло[2.2.2]октанил)-пропионитрил получают из 2-бромбицикло[2.2.2]октана. Следуя примерам 98-104, пропионитрилы превращают в вещество (IVK) и аналогичное 2-бицикло[2.2.2]октанильное производное.
Пример 178. Используя способ примера 97, 3-(1-норборненил)пропионитрил получают из 1-бромнорборнена. Следуя примерам 98-104, пропионитрил превращают в вещество (IVI).
Пример 179. Следуя способу примера 170, 5-норборнен-2-карбоксальдегид (смесь 2-эндо и 2-экзо) реагирует с образованием соответствующего 3-замещенного пропионитрила, который затем превращают в вещество (IVJ).
Пример 180.
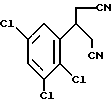
Приведенное выше промежуточное соединение получают в этом примере по методике способа Schiemenz G.P. Engelhard Н. (Chem. Ber. 1962, 95, 195).
Смесь циануксусной кислоты (25,38 г, 298,38 ммоль), 2,3,5-трихлорбензальдегида (25,0 г, 119,35 ммоль), ацетата аммония (500 мг), толуола (120 мл) и пиридина (65 мл) нагревают при температуре кипения с обратным холодильником в течение 16 ч в колбе, снабженной ловушкой Дина-Старка и холодильником. Растворители выпаривают в вакууме, остаток экстрагируют с помощью CHCl3, промывают его водой, высушивают (Na2SO4), выпаривают, получая сырой продукт, который очищают колоночной хроматографией с силикагелем, используя смесь гексан-EtOAc в качестве растворителя для элюирования. Выход 23,69 г (73%), т.пл. 90-91oC.
Пример 181.
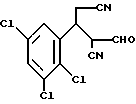
В этом примере получают упомянутое промежуточное соединение. К перемешиваемой смеси NaH (1,56 г, 65,05 ммоль) и этилового эфира муравьиной кислоты (14,78 г, 199,51 ммоль) в THF (100 мл) добавляют замещенный пентандинитрил примера 180 (10,17 г, 37,17 ммоль) при комнатной температуре в атмосфере азота, полученную реакционную смесь перемешивают в течение 24 ч. Летучее вещество выпаривают в вакууме при комнатной температуре. Воду (50 мл) добавляют к остатку при 0-5oC и раствор доводят до pH 5-6 20%-ной концентрации HCl (о/о). Тяжелое масло экстрагируют этилацетатом, промывают водой (1 x 100 мл) и высушивают (MgSO4). Слой этилацетата выпаривают, получая красно-коричневое масло (11,0 г), которое используют в следующей стадии без дальнейшей очистки.
Пример 182.
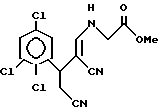
В этом примере получают приведенное промежуточное вещество. Гидрохлорид метилового эфира глицина (8,17 г, 65,06 ммоль) и ацетат натрия (5,33 г, 65,06 ммоль) добавляют к раствору сырого формильного соединения примера 181 (11,0 г) в смеси MeOH (80 мл) и воды (20 мл), полученный раствор перемешивают при комнатной температуре в течение 22 ч. После испарения растворителя при комнатной температуре остаток экстрагируют этилацетатом. Промытый (H2O) и высушенный (MgSO4) органический слой выпаривают, получая масло. Испарительная колоночная хроматография (силикагель), использующая CHCl3 в качестве растворителя для элюирования, дает чистый желаемый енамин в виде смеси цис-транс изомеров, которые перекристаллизовывают из MeOH; выход 10,41 г (75%), т.пл. 142-143oC.
Пример 183.
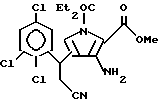
В этом примере получают приведенное промежуточное вещество. Раствор енамина примера 182 (10,0 г, 26,84 ммоль) в сухом CH2Cl2 (100 мл) охлаждают до 0oC и обрабатывают 1,5-диазабицикло[4.3.0] нон-5-еном (10,53 г, 84,79 ммоль) в атмосфере азота, за которым следует этиловый эфир хлормуравьиной кислоты (6,90 г, 63,57 ммоль). Раствор перемешивают при 0oC в течение 1 ч, затем при комнатной температуре в течение 48 ч. Летучие вещества испаряют в вакууме, получая вязкую темную смолу, которую очищают испарительной колоночной хроматографией над силикагелем, используя CHCl3 в качестве растворителя для элюирования. Все фракции, содержащие желаемый N-защищенный пиррол, объединяют и выпаривают, получая пенистое, легкое, бледно-желтое вещество, которое перемешивают в MeOH (100 мл), получая кристаллическое вещество, которое перекристаллизовывают из CHCl3-MeOH; выход 8,92 г (74,7%), т.пл. 160-161oC.
Пример 184.
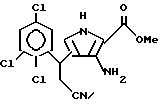
В этом примере получают приведенное промежуточное вещество. Суспензию N-защищенного пиррола примера 183 (8,6 г, 19,34 ммоль) в MeOH (300 мл) обрабатывают Na2CO3 (5,12 г, 48,34 ммоль), реакционную смесь перемешивают при комнатной температуре в течение 17 ч с отделением деблокированного пиррола в течение первого часа. Твердый карбонат натрия удаляют фильтрованием и хорошо промывают MeOH. Фильтрат уменьшают до объема 25 мл, выдерживают в холодильнике в течение 1 ч, получая 5,23 г кристаллического продукта. Дальнейшая концентрация маточного раствора дает дополнительные 0,14 г чистого продукта; общий выход 6,45 г (89,5%), т.пл. 178-181oC.
Пример 185.
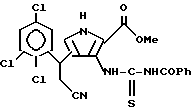
В этом примере получают приведенное промежуточное вещество. К суспензии пиррола примера 184 (5,83 г, 15,64 ммоль) в дихлорметане (100 мл) добавляют бензоилизотиоцианат (2,88 г, 17,64 ммоль) при комнатной температуре под азотом. Реакционную смесь перемешивают в течение 30 мин с отделением желаемого тиоуреидосоединения. Добавляют дополнительное количество бензоилизотиоцианата (0,5 мл) и снова перемешивают в течение 30 мин. Растворитель выпаривают досуха и легкий желтый остаток растирают в порошок с метанолом. Белое кристаллическое вещество выделяют фильтрованием и перекристаллизовывают из смеси хлороформ - эфир, получая требуемое тиоуреидопроизводное в виде аналитически чистого образца; выход 7,71 г (92%), т.пл. 210-211oC.
Пример 186.
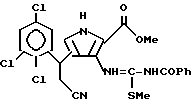
В этом примере получают приведенное промежуточное вещество. Раствор тиоуреидопроизводного примера 185 (6,75 г, 12,6 ммоль) и 1,5-диазабицикло[4.3.0] нон-5-ена (1,76 г, 14,20 ммоль) в сухом CH2Cl2 (200 мл) охлаждают до 0oC и обрабатывают йодистым метилом (5,20 г, 36,65 ммоль). Реакционную смесь перемешивают при 0oC в течение 10 мин и затем в течение 1 ч при комнатной температуре. Растворитель выпаривают при комнатной температуре, остаток экстрагируют CHCl3, промывают водой (2 x 50 мл), сушат (Na2SO4) и выпаривают, получая стекловидную пену (6,95 г), которую используют в следующей стадии без очистки.
Пример 187.
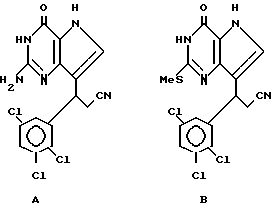
В этом примере получают приведенные вещества A и B. Вещество A вещество настоящего изобретения, вещество B промежуточное соединение. Раствор метилтиопроизводного промежуточного вещества примера 186 (6,90 г, 12,54 ммоль) в MeOH (200 мл) насыщают при 0oC аммиаком и нагревают при 100oC в течение 20 ч в футерованной стеклянными плитками бомбе из нержавеющей стали. Реакционную смесь доводят до комнатной температуры и растворитель выпаривают при комнатной температуре. Очистка сырой смеси испарительной колоночной хроматографией над силикагелем, используя CHCl3 в качестве растворителя для элюирования, дает 8B (1,1 г, 21%), т.пл. 290-291oC, затем CHCl3-MeOH (95:5) дает чистый 8A (2,76 г, 57,5%), т.пл. 284-285oC.
MS (EI) 381 (M+), 346 (381-Cl)+, 341 (381-CH2CN)+; ИК (KBr) 3323, 3180, 3162 2255, 1685, 1630, 1604, 1558, 1525, 1414, 1393; 1H-ЯМР (DMSO-d6) δ 3,27-3,50 (m, 2H, CH2CN), 4,86 (t, 1H, CH-CH2CN), 5,90 (s, 2H, NH2), 7,15 (d, J= 2 Hz, 1H, пиррольное кольцо CH), 7,57 (d, J=1,5 Hz, ароматич. CH), 7,79 (d, J= 1,7 Hz, 1H, ароматич. CH), 10,50 (s, 1H, NH-CO), 11,58 (s, 1H, пиррольное кольцо NH).
Анализ: вычислено для C15H10Cl3N5O: C 47,08; H 2,63; N 18,30; найдено: C 46,92; H 2,94; N 17,05.
Пример 188. Вещество изобретения примера 187 испытывают на ферментативную ингибирующую активность. Проводят ферментативный анализ пуриннуклеозидфосфорилазы (PNP), в котором определяют PNP активность (IC50) для вещества (8A) посредством радиохимического измерения образования [14C]-гипоксантина из [14C]-инозина (Biomedicine, 1980, 33, 39), используя селезенку теленка в качестве источника энзима. При 1 mM фосфата IC50 составляет 0,64 мкМ, при 50 mM фосфата IC50 составляет 10 мкМ.
Пример 189.
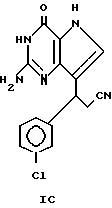
Следуя способу, приведенному в примерах 180-187, 3-(3-хлорфенил)-3-(2-амино-4-оксо-3H, 5H-пирроло[3,2-d] пиримидин-7-ил)пропаннитрил получают, используя 3-(3-хлорфенил)-пентандинитрил в качестве исходного вещества; выход 54,5% т.пл. 157-158oC.
MS(FAB), m/z 314(M+H)+, 273 (313-CH2CN)+; ИК (KBr) 3313, 3166, 3148, 2920, 2240, 1682, 1625, 1596, 1573, 1525; 1H-ЯМР (DMSO-d6) δ 3,24-3,48 (m, 2H, CH2CN), 4,41 (t, 1H, CH-CH2CN), 5,88 (s, 2H, NH2), 7,13 (d, J=3,5 Hz, 1H, пиррольное кольцо CH), 7,20-7,50 (m, 4H, ароматич. CH), 10,44 (s, 1H, NHCO), 11,45 (d, 1H, пиррольное кольцо NH).
Анализ: вычислено для C15H12ClN5O•0,2MeOH: C 57,02; H 4,03; N 21,87; найдено: C 57,03; H 3,95; N 21,88.
Пример 190. Следуя способу, приведенному в примерах 180-187, могут быть получены следующие группы веществ (1-9) общей формулы: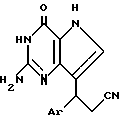
3-Aрил-3-(2-амино-4-оксо-3H,5H-пирроло[3,2-d]-пиримидин-7-ил)пропаннитрил
Где Ar имеет одно из следующих значений: (1) фенил, 2,3-дихлорфенил, 3-метилфенил и 3-метоксифенил; (2) тиенил (2- и 3-); (3) фуранил (2- и 3-); (4) пиридинил (2-, 3- и 4-); (5) пирролил (2- и 3-); (6) тиазолил (2-, 4- и 5-); (7) 2-пиразинил; (8) пиридазинил (3- и 4-); (9) пиразолил.
Пример 191. Следуя способу, приведенному в примерах 180-187, могут быть получены следующие вещества 10-14 и 21, исходя из соответственно замещенного пентандинитрила. Вещества 15-20 и 22 получают из соответствующих ненасыщенных Ar аналогов в примере 190. В этом способе нитрильная группа в ненасыщенном аналоге превращается в амидную группу гидролизом, катализируемым кислотой или основанием, затем ненасыщенную Ar группу переводят в насыщенную R2 группу с помощью каталитического гидрирования, за которым следует вновь превращение амида в нитрил с помощью известных способов дегидратации.
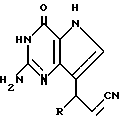
3-(Замещенный)-3-(2-амино-4-оксо-3H, 5H-пирроло[3,2-d] -пиримидин-7-ил)пропаннитрил
где R2 означает: (10) 1-адамантил; (11) 2-адамантил; (12) циклогексил; (13) циклогептил; (14) циклопентил; (15) тетрагидрофуранил; (16) тетрагидротиенил; (17) тетрагидропиранил; (18) пиразолидинил; (19) тиазолидинил; (20) пиперазинил; (21) морфолинил; (22) гексагидропиридазинил.
Пример 192.
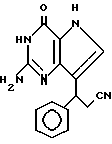
В этом примере получают приведенное вещество 3-(2-амино-4-оксо-3H,5H-пирроло[3,2-d] пиримидин-7-ил)-3-фенилпропаннитрил. Раствор вещества A, полученного в примере 187 (2,0 г, 5,22 ммоль), в горячем этаноле (250 мл) и диметилформамиде (DMF) (150 мл) гидрируют над 30%-ным Pd/C катализатором (1,0 г) в присутствии триэтиламина (2,64 г, 5,0 эквивалентов) при атмосферном давлении. Спустя 5 ч реакция завершается и катализатор отфильтровывают под давлением N2. Твердое вещество, полученное после испарения фильтрата, растирают в порошок, разрушают ультразвуком с H2O и высушивают; выход 1,28 г (88%), т.пл. 168-170oC.
MS (FAB), m/z280 (M+H)+, 239 (280-CH2CN)+; 1H-ЯМР (DMSO-d6) δ 3,20-3,46 (m, 2H, CH2CN), 4,37 (t, 1H, CHCH2CN), 5,86 (s, 2H, NH2), 7,05 (d, J=1,5 Hz, 1H, пиррольное кольцо CH), 7,21 (t, 1H, ароматич. CH), 7,30 (t, 2H, ароматич. CH), 7,39 (d, J=7,5 Hz, 2H, ароматич. CH), 10,41 (s, 1H, NHCO), 11,40 (s, 1H, пиррол NH).
Анализ: вычислено для C15H13N5O: C 64,50; H 4,69; N 25,07; найдено: C 64,68; H 4,57; N 25,19.
Пример 193. Вещество, полученное в примере 192, испытывают на ферментативную ингибирующую активность как в примере 188. При 1 mM фосфата IC50 равно 0,023 мкМ, а при 50 mM фосфата IC50 равно 4,7 мкМ.
Пример 194.
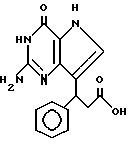
В этом примере получают приведенное вещество 3-(2-амино-4-оксо-3H,5H-пирроло[3,2-d] пиримидин-7-ил)-3- фенилпропановую кислоту. Раствор вещества, полученного в примере 192 (0,200 г, 0,72 ммоль) в 6N HCl (3,0 мл) нагревают при температуре кипения с обратным холодильником в течение 18 ч. Растворитель выпаривают в вакууме, остаток растирают в порошок с H2O (6 мл), доводят до pH 10 концентрации гидроксидом аммония. Нерастворимое вещество собирают фильтрованием, фильтрат повторно доводят до pH 6,8. Белое вещество, которое осаждается, собирают, промывают водой и высушивают; выход 0,19 г (89%), т. пл. 290oC (разл.).
MS (FAB), m/z 299 (M+H)+; ИК (KBr) 3187, 3181, 1688, 1636, 1557, 1524, 1495, 1398, 1376; UV 0,1 N HCl 234 (17,15), 273 (15,05); pH 7 231 (20,05), 272 (10,72); 0,1 N NaOH 227 (22,3), 264 (7,29), 288 (6,66); 1H-ЯМР (DMSO-d6) δ 2,86-3,0 (dd, 1H, -CHH-), 3,02-3,14 (dd, 1H, -CHH-), 4,47 (t, 1H, CH-CH2CN), 5,85 (s, 2H, NH2), 6,95 (d, J=3 Hz, 1H, пиррольное кольцо CH), 7,14 (t, 1H, ароматич. CH), 7,24 (t, 2H, ароматич. CH), 7,33 (d, J 7,0 Hz, 2H, ароматич. CH), 10,38 (плечо, 1H, NHCO), 11,25 (плечо, 1H, пиррольное кольцо NH), 11,1-12,1 (очень широкий, виден в интеграле, 1H, COOH).
Анализ: вычислено для C15H14N4O3•0,3H2O: C 59,38; H 4,85; N 18,45; найдено: C 59,42; H 5,04; N 18,56.
Пример 195. Вещество, полученное в примере 194, испытывают на ферментативную ингибирующую активность как в примере 188. При 1 mM фосфата IC50 равно 0,012 мкМ, а при 50 mM фосфата IC50 равно 0,19 мкМ.
Пример 196.

В этом примере получают приведенное вещество 3-(2-амино-4-оксо-3H,5H-пирроло[3,2-d] пиримидин-7-ил)-3- фенилпропанамид. Раствор вещества, полученного в примере 192 (0,200 г, 0,72 ммоль) в концентрированной H2SO4 (0,5 мл) перемешивают при комнатной температуре в течение 20 ч, затем выливают на дробленый лед (5,0 г) и доводят до pH 6,8 посредством концентрации NH4OH. Осажденное твердое вещество собирают, промывают водой и высушивают; выход 0,180 г, т.пл. 199-201oC (разл.).
MS(FAB), m/z 298 (M+H)+; ИК (KBr) 3324, 3259, 3185, 3178, 1670, 1626, 1561, 1524, 1411; 1H-ЯМР (DMSO-d6) δ 2,60 (dd, 1H, -CHH-), 2,86 (dd, 1H, -CHH-), 4,52 (t, 1H, CH-CH2, 5,98 (плечо, 2H, NH2, 6,67 (s, 2H, CONH2), 6,99 (d, J 2,5 Hz, 1H, пиррольное кольцо CH), 7,06-7,31 (m, 5H, ароматич. CH), 10,60 (очень широкий, 1H, NHCO), 11,37 (плечо, 1H, пиррольное кольцо NH).
Анализ: вычислено для C15H15N5O2•0,8H2O: C 57,80; H 5,36; N 22,46; найдено: C 57,89; H 5,18; N 22,36.
Пример 197. Вещество, полученное в примере 196, испытывают на ферментативную ингибирующую активность как в примере 188. При 1 mM фосфата IC50 равно 0,20 мкМ, при 50 mM фосфата IC50 равно 6,6 мкМ.
Пример 198.
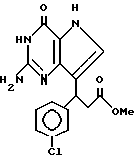
В этом примере получают вышеприведенное вещество 3-(2-амино-4-оксо-3H, 5H-пирроло[3,2-d] пиримидин-7-ил)-3-(3-хлорфенил)пропановую кислоту, метиловый эфир. Хлористый тионил (0,2 г, 0,17 ммоль) добавляют к перемешиваемому метанолу (4,0 мл) при 0oC. Вещество, полученное в примере 194 (0,2 г, 0,67 ммоль), добавляют, смесь перемешивают при комнатной температуре в течение 18 ч. Растворитель удаляют на водном аспираторе (30oC) и вакуумном насосе (лиофилизация), получая полутвердую массу, которую очищают на колонне с силикагелем, используя CHCl3-MeOH в качестве растворителя для элюирования; выход 0,1 г, т.пл. 302-303oC (разл.)
MS (FAB), m/z 347 (M+H), 273 (346-CH2CO2CH3)+; UV 0,1 N HCl 220 (sh), 232 (sh), 273 (15,1), pH 7 буффер 232 (18,1), 270 (7,4), 0,1 N NaOH 262 (6,5), 288 (5,9); 1H-ЯМР (DMSO-d6) δ 3,15 (dd, J, 2H, CH2CO2CH3), 3,48 (s, 3H, CH2CH3), 4,48 (t, 1H, -CHCH2-), 5,83 (s, 2, NH2), 7,06 (d, J, 1H, пиррольное кольцо CH), 7,19, 7,26, 7,39, (комплекс m, 4H, хлорфенил CH), 10,36 (s, 1H, 3-NH), 11,30 (d, J, 1H, 5NH).
Анализ: вычислено для C16H15ClN4O3•0,3CH3OH: C 54,93; H 4,58; N 15,72; найдено: C 54,99; H 4,78; N 15,85.
Пример 199. Вещество, полученное в примере 198, испытывают на ферментативную ингибирующую активность. Обнаружена значительная активность (IC50 55 nM).
Пример 200.
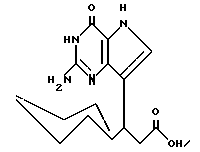
В этом примере получают 3-(2-амино-4-оксо-3H,5H-пирроло[3,2-d]пиримидин-7-ил)-3-циклогексанпропановую кислоту. Раствор вещества, полученного в примере 194 (83 мг, 0,28 ммоль) в трифторуксусной кислоте (TFA) (15 мл) гидрируют с помощью PtO2 (83 мг) при давлении 60 фунтов/дюйм2 в течение 24 ч. Катализатор отфильтровывают через слой цеолита, фильтрат выпаривают при 25oC. Остаток суспендируют в воде (8 мл), доводят до pH 8,5 концентрированной NH4OH и фильтруют через Whatman фильтровальную бумагу для удаления примесей коричневого цвета. Бесцветный фильтрат доводят до pH 6,8, осажденное вещество фильтруют, промывают водой и высушивают; выход 65 мг (77%), т. пл. более 300oC.
MS(FAB), m/z 305 (M+H)+; ИК (KBr) 3330, 3194, 2926, 2852, 1683, 1639, 1557, 1522, 1401, 1377; 1H-ЯМР (DMSO-d6): 0,85, 1,06, 1,60 (m, 11H, циклогексил H), 2,56 (m, 2H, CH2CO2H), 3,05 (m, 1H, CH-CH2), 5,79 (s, 2H, NH2), 6,89 (d, J 2,5 Hz, 1H, пиррольное кольцо CH), 10,31 (плечо, 1H, пиррол NH), 11,16 (s, 1H, NHCO), 11,76 (плечо, 1H, пиррол NH).
Анализ: вычислено для C15H20N4O•1,75H2O: C 53,64; H 7,05; N 16,68; найдено: C 54,03; H 6,71; N 16,30.
Пример 201. Вещество, полученное в примере 200, испытывают на ферментативную ингибирующую активность как в примере 188. При 1 mM фосфата IC50 равно 0,097 мкМ, а при 50 mM фосфата IC50 равно 1,0 мкМ.
Пример 202. Получают вещество изобретения, в котором R2-CH2OPO(OH)2. Нитрильную группу вещества примера 192 превращают в соответствующий амид посредством обработки серной кислотой. Используя реакцию дегидратации по Гофману, амид превращают в соответствующий амин, который затем переводят в соответствующую пиридиниевую соль, используя пиррилиевую соль. Превращение соли в соответствующую соль галоидоводородной кислоты достигают, используя бромид натрия, которую затем превращают в эфир фосфиновой кислоты, используя триэтилфосфит. Гидролиз эфира с использованием триметилсилилбромида дает соответствующую фосфиновую кислоту.
Пример 203. В этом примере получают вещество изобретения посредством изменения числа углеродных атомов. Нитрильную группу вещества примера 192 переводят в соответствующий альдегид, который затем превращают в соответствующий спирт. Используя трехбромистый фосфор, спирт превращают в соответствующий алкилбромид, который затем превращают в ацетонитрил, используя цианид калия.
Пример 204. В этом примере получают вещество изобретения, в котором R2 CH2OP(O)(OH)2. Спирт, полученный в качестве промежуточного соединения в предыдущем примере, превращают в соответствующий диэтилфосфонометиловый эфир, используя диэтилхлорметилфосфонат. Удаление этильной группы эфира выполняют, используя триметилсилилбромид, с получением фосфиновой кислоты.
Пример 205. В этом примере получают вещество изобретения, в котором R2 CH2NHSO2NH2. Нитрильную группу вещества примера 192 восстанавливают до амина, используя стандартное каталитическое гидрирование с помощью палладия в кислой среде (обычно 0,01N до 1N HCl), который затем превращают в сульфамид, используя сульфамоилхлорид.
Пример 206. В этом примере получают вещество изобретения, в котором R2 CH2NHCH2COOH, путем реакции промежуточного метиламиносоединения, полученного в предыдущем примере, с хлоруксусной кислотой.
Пример 207. В этом примере получают вещество изобретения, в котором R2 CH2NHPO(OH)2, реакцией промежуточного производного метиламина, полученного в примере 206, с диэтилхлорметилфосфонатом и обработкой полученного продукта с триметилсилилбромидом.
Пример 208. В этом примере получают соединение изобретения, в котором R3 OSO2NH2, реакцией промежуточного спирта, полученного в примере 203, с сульфамоилхлоридом.
Пример 209. В этом примере получают соединение изобретения, в котором R1 Н, R2 фенил, R3 CH2CN. Используют методику способа, описанного в Mu Ill Lim. et al. J. Org. Chem. vol.44, N 22, 3826 (1979). Вещество примера 184 и диметилацеталь диметилформамида взаимодействуют при комнатной температуре в течение 2 дней и затем выпаривают досуха в вакууме. Остаток кристаллизуется, давая чистое производное N-(диметиламино)метилена, которое циклизуют в присутствии насыщенного метанольного раствора аммиака, получая желаемый конечный продукт.
Пример 210. В этом примере получают вещество изобретения, в котором R1 OCH3, R2 фенил, R3 CH2CN. Используя вещество B примера 187, S-метильную группу окисляют до метилсульфона, который затем превращают в конечное метоксисоединение обработкой метоксидом натрия в метаноле.
Пример 211. В этом примере получают соединение изобретения, в котором R2 тетразол. Вещество примера 192 обрабатывают азидом лития в присутствии хлорида аммония в качестве катализатора в диметилформамиде (DMF) при 100oC, получая желаемый тетразол.
Пример 212. В этом примере получают соединение изобретения, в котором R2 триазол. Вещество примера 198 обрабатывают гидратом гидразина, получая соответствующий гидразид, который затем обрабатывают иминоэфиром, получая желаемый триазол.
Пример 213. Вещество, полученное в примере 189, испытывают на ферментативную ингибирующую активность как в примере 188. При 1 mM фосфата IC50 равно 0,012 мкМ, а при 50 mM фосфата IC50 равно 2,0 мкМ.
Пример 214. В этом примере получают вещество изобретения, в котором R3 в приведенной общей формуле CNH(NH2). Вещество A из примера 187 взаимодействует с метоксидом натрия в метаноле при комнатной температуре в течение примерно 2 дней, образуя промежуточное вещество метил-имидат. Промежуточное вещество затем реагирует с аммиаком в метаноле, образуя соответствующий имидин.
Пример 215. 2-амино-7-(3-хлорФенил-3-гидроксипропил)-3H, 5H-пирроло[3,2-d] пиримидин-4-он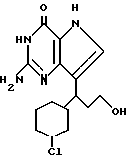
Размешанную суспензию метилового сложного эфира примера 198 (6,00 г, 17,3 ммоль) и сухого (NH4)2SO4 (100 мг) в гексаметилдисилазане (400 мл) кипятят с обратным холодильником в течение 8 ч, одновременно обеспечивая защиту от атмосферной влаги. Прозрачный раствор уменьшают в объеме до приблизительно половины его первоначального объема путем дистиллирования при атмосферном давлении, а оставшиеся гексаметилдисалазаны выпаривают в вакууме, получая триметилсилилпроизводное в виде вязкой смолы, которую затем высушивают над P2O5. Сырой продукт затем используют в последующей реакции без дальнейшей очистки: MS (FAB), m/z 491 (M+H)+.
Раствор LiAlH4 в тетрагидрофуране (26 мл 1М, 26 ммолm) добавляют по каплям в раствор триметилсилилированного сложного эфира (предположительно 17,3 ммоль) в сухом тетрагидрофуране (200 мл). После размешивания в течение 1 ч избыток гидрида разрушают путем добавления EtOAc (100 мл) и все растворители выпаривают в вакууме. В размешанную суспензию остатка в холодной H2O (200 мл) добавляют разведенную HCl до тех пор, пока pH не достигнет 1 и по истечении 15 мин pH доводят до 7 с помощью раствора NaOH. Твердое вещество собирают фильтрованием, промывают холодной H2O, слегка высушивают в вакууме, затем порошкуют путем размешивания со смесью 1:1 (100 мл) циклогексана и Et2O. Твердое вещество отфильтровывают и частично высушивают в воронке под потоком N2. Раствор твердого вещества в горячем MeOH выпаривают досуха с силикагелем (отверстия 230-400 меш, 50 г), смесь осторожно наносят на верх колонны с силикагелем, затем элюируют с помощью смеси CHCl3/MeOH в соотношении 17: 3 и 4:1, получая желаемый спирт (4,65 г, 84%). С помощью двух перекристаллизаций из H2O/EtOH в соотношении 2:1 получают белые кристаллы, которые затем высушивают в вакууме над P2O5 в течение 30 ч при 110oC; выход 3,36 г (61%).
MS(FAB), m/z319 (M+H)+; 321 (Cl изотопный пик 319)+, 301 (318-OH)+, 151 (B + 2H)+; UV 0,1 NHCl 233 (17,9), 274 (15,7), pH 7 буффер 230 (22,3), 272 (10,6), 0,1 N NaOH 266 (7,3), 288 (6,7); 1H-ЯМР (DMSO-d6) δ 2,07, 2,21 (2 секстета 1H каждый, -CHCH2CH2OH), 3,29 (m, 1H, -CH2OH), 4,17 (t, 1H, -CHCH2CH2OH), 4,45 (t, 1H, -CH2OH), 5,79 (s, 2H, NH2), 7,09 (d, J, 1H, пиррольное кольцо CH), 7,18, 7,27, 7,34 (комплекс m, 4H, хлорфенил CH), 10,36 (s, 1H, 3-NH), 11,29 (d,J, 1H, 5-NH).
Анализ: вычислено для C15H15ClN4O2: C 56,52; H 4,74; N 17,58; найдено: C 56,51; H 4,84; N 17,49.
Пример 216. 2-амино-7-(1-циклогексил-3-гидроксипропил)-3H, 5H-пирроло[3,2-d]пиримидин-4-он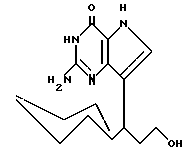
Раствор спирта из примера 215 (100 мг, 0,314 ммоль) в CF3COOH (20 мл) гидрогенизируют путем размешивания с платиновым катализатором (100 мг PtO2) в аппарате Парра при первоначальном давлении в 60 фунтов/дюйм. Катализатор удаляют путем фильтрации через тонкий слой цеолита под N2. Фильтрат выпаривают, остаток дважды растворяют в MeOH (5,0 мл), а раствор выпаривают. Мутному раствору остатка в H2O/конц. NH4OH (20 мл, 1:1) дают постоять в течение 4 ч при комнатной температуре, прежде чем подвергнуть его фильтрованию и выпариванию досуха. Раствор остатка в H2O (30 мл), содержащий 2 капли 1N NaOH нейтрализуют 1N HCl и охлаждают до 5oC. Осадившееся белое твердое вещество собирают фильтрацией, промывают холодной водой и высушивают в вакууме над P2O5 при температуре 110oC в течение 6 ч; выход 63 мг (67%), т.пл. 181-186oC.
MS(FAB), m/z 291 (M+H)+; UV 0,1 N HCl 236 (16,2), 275 (14,9), pH 7 буффер 231 (18,8), 274 (12,1), 0,1 N NaOH 230 (19,6), 268 (7,8), 288 (плечо); 1H-ЯМР (DMSO-d6) δ 0,85, 1,08, 1,59 (комплексные мультиплеты, 11H, циклогексил CH), 1,78 (m, 2Н, -CHCH2CH2OH), 2,62 (m, 1H, -CHCH2CH2OH), 3,14, 3,17, 3,24 (комплексные мультиплеты, 2Н, -CH2OH), 4,25 (уш. 1H, -CH2OH), 5,82 (s, 2Н, -NH2, 6,86 (d, J, 1H, H-6), 10,35 (уш. 1H, 3-NH), 11,17 (d, J, 1H, 5-NH).
Анализ: вычислено для C15H22N4O2•0,5H2O: C 60,18; H 7,74; N 18,71; найдено: C 60,09; H 7,68; N 18,73.
Пример 217. 2-Амино-7-[2-карбокси-1-(3-хлорфенил)этил] -3H, 5H-пирроло[3,2-d] пиримидин-4-он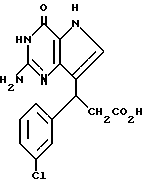
2-Амино-7-[2-циано-1-(3-хлорфенил)этил] -3H, 5H-пирроло[3,2-d] пиримидин-4-он (20 г, 63,75 ммоль) гидролизуют до получения соответствующей кислоты с помощью способа, описанного в примере 194. Выход: 18 г (84,9%), т.пл. 295-296oC.
MS(FAB), m/z 333 (M+H)+; ИК(KBr) 3182, 3170, 1686, 1639, 1596, 1572, 1525, 1477, 1402, 1377; UV 0,1 N HCl 235 (17,2), 273 (15,42); pH 7 233 (20,2), 272 (1058), 0,1 N NaOH 231 (20,9), 263 (7,2), 288 (6,67); 1H-ЯМР (DMSO-d6) δ 2,96 (m, 2H, CH2CN), 4,48 (t, 1H, CH-CH2CN), 6,06 (уш. 2H, NH2), 7,11 (d, J 2,6 Hz, 1H, пиррольное кольцо CH), 7,21 (d, J 8 Hz, 1H, ароматич. CH), 7,25-7,46 (m, 3H, ароматич. CH), 10,3-11,0 (очень широкий, виден в интеграле, 1H, NH или COOH), 10,42 (уш. 1H, пиррольное кольцо NH), 11,7-12,3 (очень широкий, виден в интеграле, 1H, NH или COOH).
Анализ: вычислено для C15H14N4O3•0,3H2O: C 59,38; H 4,85; найдено: C 59,42; H 5,04; N 18,56.
Пример 218. 7-[2-циано-1-(3-хлорфенил)этил]-3H,5H-пирроло[3,2-d]пиримидин-4-он
К раствору 3-(2-метилтио-4-оксо-3H,5H-пирроло[3,2-d]пиримидин-7-ил)-3-(3-хлорфенил)пропаннитрила (1,0 г) в этаноле (100 мл) добавляют 30% Pd/C и образовавшуюся суспензию кипятят с обратным холодильником в течение нескольких минут. Затем при постоянном помешивании добавляют гидрат гидразина (300 мл), реакционную смесь кипятят с обратным холодильником в течение 2 дней. Затем добавляют дополнительные порции гидрата гидразина (300 мл) и Pd/C (0,5 г) и реакционную смесь кипятят с обратным холодильником еще 4 дня. Катализатор удаляют фильтрацией, фильтрат уменьшают в объеме до 75 мл, фильтруют и выпаривают, получая желаемый продукт в виде белого твердого вещества; выход 0,82 г (94,7%); т.пл. 230oC.
MS(FAB), 299 (M+1)+; ИК (KBr) 3166, 3155, 3150, 3142, 3133, 3093, 2248, 1673, 1595, 1414; UV (0,1N HCl), 237 (27,8); pH 7; 262(9,9), 233(31,l); 0,1N NaOH, 268(8,9); 1H-ЯМР (DMSO-d6) δ 3,3-3,52 (m, CH2CN), 4,6 (t, 1H, CHCH2-CN), 7,24-7,56 (m, ароматич. CH и H-6), 7,84 (s, H-2), 11,95 (s, NHCO), 12,11 (s, пиррол NH).
Анализ: вычислено для C15H11ClN4O: C 60,31; H 3,71; N 18,75; найдено: C 60,19; H 3,92; N 18,41.
IC50 для PNP 10 nM.
В таблице приведены данные по ингибированию пролиферации Т-лимфоцитов a.
aCCRF-CEM лейкемические Т-лимфоциты.
bКонцентрация, необходимая для ингибирования пролиферации 50% клеток по сравнению с необработанными клетками контрольных опытов.
Вышеуказанные соединения не были способны ингибировать пролиферацию B-лимфоцитов в концентрации 100 мкМ, тем самым демонстрируя присущую им селективность по отношению к Т-лимфоцитам.
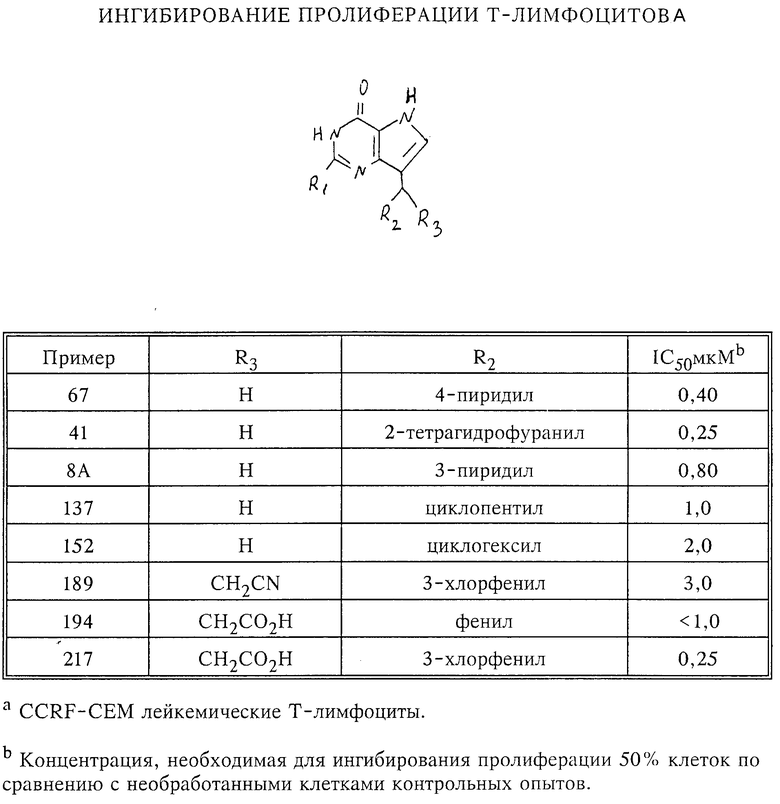
Предложены производные 2-амино-7-(CHR2R3)-3H,5H-пирроло[3,2-d]пиримидин-4-она формулы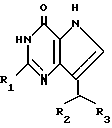
где R1 - H, NH2; R2 - насыщенный пятичленный гетероцикл, содержащий в качестве гетероатома S или O, или шестичленный N-содержащий гетероцикл, которые могут быть замещены; R3 - H, CH2CN, CH2CO2H, CH2CONH2, CO2CH3, CO2H или CH2CH2OH, или R2 и R3 вместе с CH образуют циклогексил- или циклогексенильную группу, которые являются ингибиторами пуриннуклеозидфосфорилазы. Предложены также способы их получения и способ селективного ингибирования пролиферации Т-лимфоцитов млекопитающего и не оказывающий воздействия на B-лимфоциты. 4 с. и 6 з.п. ф-лы, 1 табл.
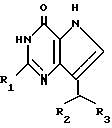
где R1 Н или NH2;
R2 насыщенный пятичленный гетероцикл, содержащий в качестве гетероатома серу или кислород, или шестичленный гетероцикл, содержащий в качестве гетероатома N, и которые могут быть необязательно замещены С1 С4-алкокси, трифторметилом или галогеном, С6 - С1 0-циклоалкил, незамещенный или галоидзамещенный фенил;
R3 H, (CH2)nCN, (CH2)nCO2H, (CH2)nCONH2, CO2CH3, (CH2)nCO2CH3 или (CH2)nOH, где n 1-4, или R2 и R3 вместе с CH-группой, к которой они присоединены, образуют циклогексил- или циклогексенильную группу.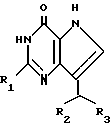
где R1 NH2;
R2 фенил, незамещенный или замещенный галогеном, или циклогексил;
R3 CH2CN,
отличающийся тем, что осуществляют следующие последовательные стадии: а) реакцию соответствующего R2-замещенного циклического альдегида с циануксусной кислотой в присутствии ацетата аммония с получением 3(R2)-замещенного пентандинитрила, б) реакцию полученного 3(R2)-замещенного пентандинитрила с алкиловым эфиром муравьиной кислоты в присутствии отнования с получением 3(R2)-2-формилпентандинитрила, в) последний взаимодействует с гидрохлоридом метилового эфира глицина в присутствии ацетата натрия или аммония с получением метил-N-[(3(R2)-2,4 -дициано)-2-бутенил]-глицина, г) полученный при этом метил N -[(3(R2)-2,4-дициано)-2-бутенил]-глицин взаимодействует с алкиловым эфиром хлоруксусной кислоты в присутствии 1,5-диазабицикло [4.3.0]нон-5-ена или 1,8-диазабицикло[5.4.0]ундек-7-ена с получением метил-3-амино-4-(2-циано-1(R2)-этил)- 1-этил-1Н-пиррол-1,2-дикарбоксилата, д) полученный метил-3-амино-4-(2-циано-1(R2)этил)-1-этил-1Н-пиррол-1,2 -дикарбоксилат обрабатывают основанием с получением метил-3-амино-4- (2-циано-1(R2)этил)-1Н-пирроло-2-карбоксилата, е) последний подвергают взаимодействию с бензоилизотиоцианатом с получением N-бензоил-N'-[4-(2-циано-1(R2)-этил) -2-метоксикарбонил-1Н-пиррол-3-ил] тиомочевины, ж) полученную N-бензоил-N'-[4-(2-циано-1(R2)-этил)-2-метоксикарбонил -1Н-пиррол-3-ил] тиомочевину алкилируют галоидным алкилом с получением N-бензоил-N'-[4-(2-циано-1(R2)-этил)-2-метоксикарбонил-1Н -пиррол-3-ил]-S-метилтиомочевины, з) затем N-бензоил-N' -[4-(2-циано-1(R2)-этил)-2-метоксикарбонил-1Н-пиррол-3-ил] -S -метилтиомочевину обрабатывают метанольным или этанольным раствором аммиака с получением смеси 3(R2)-3-[2-амино-4-оксо-3Н, 5Н-пирроло[3,2-d]пиримидин-7-ил]пропаннитрила и 3(R2)-3 -[2-метилмеркапто-4-оксо-3Н,5Н-пиролло[3,2-d]пиримидин-7-ил] -пропаннитрила.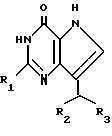
где R1 H;
R2 фенил, незамещенный или замещенный галоидом, или циклогексил;
R3 CH2CN,
отличающийся тем, что осуществляют следующие последовательные стадии: а) реакцию соответствующего циклического альдегида с циануксусной кислотой в присутствии ацетата аммония с получением 3(R2)-замещенного пентандинитрила, б) реакцию полученного 3(R2)-пентандинитрила с алкиловым эфиром муравьиной кислоты в присутствии основания с получением 3(R2)- 2-формилпентандинитрила, в) последний подвергают взаимодействию с гидрохлоридом метилового эфира глицина в присутствии ацетата натрия или аммония с получением метил-N-[(3(R2)-2,4-дициано) -2-бутенил] глицина, г) полученный при этом метил -N-[(3(R2)2,4-дициано)-2-бутенил] глицин подвергают взаимодействию с алкиловым эфиром хлоруксусной кислоты в присутствии 1,5-диазабицикло[4.3.0]нон-5-ена или 1,8-диазабицикло[5.4.0] ундек-7-ена с получением метил-3-амино-4-(2-циано-1(R2) -этил)-1-этил-1Н-пиррол-1,2-дикарбоксилата, д) метил-3-амино-4-(2-циано-1(R2)-этил)-1-этил-1Н-пиррол -1,2-дикарбоксилат обрабатывают основанием с получением метил-3-амино-4-(2-циано-1(R2) -этил)-1Н-пиррол-2-карбоксилата, е) затем метил-3-амино-4- (2-цикло-1(R2)этил)-1Н-пирроло-2-карбоксилат подвергают взамиодействию с диметилацеталем диметилформамида с получением метил-4-(2-циано-1(R2)-этил)-3-[N-(диметиламинометилен)амино] - 1Н-пирроло-2-карбоксилата, и ж) метил-4-(2-циано-1(R2) этил)-3-[N-(диметиламинометилен)амино] -1Н-пирроло-2-карбоксилат обрабатывают метанольным раствором аммиака с получением 3(R2)3- [4-оксо-3Н,5Н-пирроло[3,2-d] пиримидин-7-ил]пропаннитрила.
Приоритет по признакам:
31.10.89 N 429,097 соединения по п.1, где R2 необязательно замещенная гетероалициклическая группа, N 429,098 соединения по п. 1, где R2 необязательно замещенный циклогексенил или циклогексил, N 429,100 соединения по п. 1, где R2 необязательно замещенный пиридинил, N 429,099 соединения по п. 1, где R2 необязательно замещенная алициклическая группа;
29.11.89 соединения по п. 1, где R2 необязательно замещенная циклическая группа, необязательно содержащая гетероатом, R3 водород, CO2H, CH2CONH2, CO2CH3, CH2CN, CH2CO2H.
| Irenc S | |||
| Kuzmers et al | |||
| Science, 1981, v | |||
| Устройство для вытяжки и скручивания ровницы | 1923 |
|
SU214A1 |
| ЦЕПНОЙ ВЕТРЯНОЙ ДВИГАТЕЛЬ | 1924 |
|
SU1137A1 |
