Настоящее изобретение предлагает некоторые хиназолиновые соединения, которые проявляют антипролиферативную активность, такую как противоопухолевая активность, способы получения этих соединений, фармацевтические композиции, содержащие эти соединения, и применение этих соединений для ингибирования роста и пролиферации клеток высших организмов и микроорганизмов, таких как бактерии, дрожжи и грибы. Предпочтительные соединения настоящего изобретения способны ингибировать ферментативную тимидилатсинтазу. Эффект от ингибирования ферментативной тимидилатсинтазы включает действие, рассмотренное выше.
Большой класс антипролиферативных агентов включает соединения антиметаболитов. Особым подклассом антиметаболитов, известных как антифолаты или антифолы, являются антагонистами витамина, фолиевой кислоты. Обычно антифолаты очень напоминают структуру фолиевой кислоты и включают характерную часть фолиевой кислоты, п-бензоилглутамат. Глутаматная часть фолиевой кислоты несет при физиологических pH двойной отрицательный заряд. Поэтому это соединение и его аналоги имеют активную энергетическую транспортную систему для прохождения клеточной мембраны и проявления метаболического эффекта. С другой стороны, соединение без глутаматной группы может пассивно диффундировать в клетку.
Действительной мишенью для антифолата является ферментативная тимидилатсинтаза. Тимидилатсинтаза катализирует C-метилирование 2'-деоксиуридилата ("dUMP") до получения 2'-деокситимидилата ("dTMP"). Эта одно-углеродная реакция переноса является решающей для деления клетки. Таким образом, синтезирован ряд фолатных налогов и изучена их способность ингибировать ферментативную тимидилатсинтазу. Прототипный, специфичный, трудно-связывающий ингибитор тимидилатсинтазы, 10-пропаргил-5,8-дидеазафолиевая кислота (T.R. Jones et al., "a Potent antitumor Quinazoline Inhibitor of Thymidylate Synthetase: Synthesis Biological Properties and Therapeutic Results in Mice", Eur. J. Cancer 17 : 11 (1981)), показывает эффективность против рака яичников, печени и грудных желез, однако обладает тревожной токсичностью для печени и почек (A.H. Calvert et al., "a Phase I Evaluation N 10 - Propargyl - 5,8-Dideazofolic Acid CB 3717", J. Clin. Oncol. 4 : 1245 (1986)). С помощью выявления двух свойств в этом классе молекул (растворимость и способность к внутриклеточному образованию полиглутаматов) выделен лучший агналог второй генерации (ICI D1694).
Недавно разработаны несколько липофильных ингибироторов тимидилатсинтазы. (Смотри, например, E.M. Berman et al., Substituted Quinazolinones as Anticancer Agents". пат. США N 4857530, T.R. Jones et al., "Antiproliferative Cyclic Compounds", Copending заявка США N 07/432338, которая является продолжением заявки 07/251765 от сентября 30, 1988, M.D. Varney et al. , "Antiproliferative Substituted Naphthalene Compounds", заявка США N 07/583970 от 17 сентября 1990; S.H. Reich et al., "Antiproliferative Substituted Fricyclic Compounds", заявка США N 07/587666 от 25 сентября 1990; Z.R. Hughes et al., "Anti-fumour Agents", заявка европейского патента N 373891 от 12 сентября 1989. T.R. Jones et al., "Antifolate Quinozolines", заявка США N 07/812274 от 20 декабря 1991).
Настоящее изобретение предлагает новые [иназолиновые соединения, проявляющие антипролиферативную активность, такую как противоопухолевая. Эти соединения эффективно ингибируют рост и пролиферацию клеток высших организмов и микроорганизмов, таких как бактерии, дрожжи и грибы. Изобретение предлагает способы получения этих соединений, фармацевтические составы, содержащие эти соединения и применение этих соединений. Предпочтительные хиназолиновые соединения согласно настоящему изобретению пригодны для ингибирования ферментативной тимидилатсинтазы. Эффект от ингибирования ферментативной тимидилатсинтазы включает действие, рассмотренное выше.
Настоящее изобретение касается хиназолиновых соединений формулы I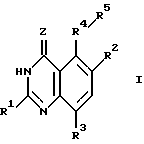
где R1 означает водород, галоген, алкил, -OH, -O-алкил, -O- (арил или гетероарил), -S-алкил, -S- (арил или гетероарил), -NH2, -NH-алкил, -N-(алкил)2, -NHCHO, -NHOH, -NHO-алкил, -NHNH2, замещенный -NHNH2, -NHC(= NH)NH2, -NHC(= NH)алкил, фторалкил, циклоалкил, алкенил, алкинил, арил или гетероцикл,
R2 и R3, которые могут быть одинаковыми или различными, представляют собой водород, галоген, алкил, циклоалкил, -OH, -O-алкил, -S-алкил, -NH2, -NH-алкил, -N-(алкил)2, -NHCHO, -NO2, -NHOH, -NHO-алкил, -NHNH2, замещенный -NHNH2, -CH, -CO2H, -CO2-алкил, CONH2, -CONH-алкил, -CON (алкил)2, -CSNH2, -CSNH-алкил, -CSN(алкил)2, -C(=NH)NH2, -NHC(=NH)NH2, -NHC(=NH)алкил, -SO-алкил, -SO2-алкил, фторалкил, -O-фторалкил, -S-фторалкил, -NHCO(алкил), -NHCO(фторалкил), -SO-фторалкил, -SO2-фторалкил, -SH, -SO3H, -SO2NH2, -SO2NH(алкил), -SO2N(алкил)2, алкенил, алкинил, арил или гетероцикл.
Z означает O или S,
R4 представляет собой O, S, SO, SO2, NH, N-алкил, CH2, CH-алкил, CH-(арил или гетероарил), CHOH, CHO-алкил, CHO-(арил или гетероарил), C(алкил)2, C(арил или гетероарил)2, C(алкил) (арил или гетероарил), CHS-алкил, CHS- (арил или гетероарил), C(OH)(алкил), C(OH) (арил или гетероарил), C(OH)(циклоалкил), N(OH), N-циклоалкил, N(арил или гетероарил), C(циклоалкил)2, С(арил или гетероарил)(циклоалкил), С(алкил)(алкенил), С(алкил)(алкинил), С(алкенил)2, С(алкинил)2, С(алкинил)(арил или гетероарил), С(алкинил)(алкенил), С(алкенил)(арил или гетероарил), С(циклоалкил)(алкенил), С(циклоалкил)(алкинил), C(алкил) (арил или гетероарил), CH(циклоалкил), CH(алкенил), CH(алкинил), С(алкил)(циклоалкил), С(алкил)(O-алкил), С(алкенил)(O-алкил), С(алкинил)(O-алкил), С(алкил)(O-циклоалкил), С(алкенил)(O-циклоалкил), С(алкинил)(O-циклоалкил), C(арил или гетероарил)(O-алкил), C(арил или гетероарил)(O-циклоалкил), C(алкинил)(S-алкил), C(алкинил)(S-циклоалкил), C(алкенил)(S-алкил), C(алкенил)(S-циклоалкил), C(алкил)(S-алкил), C(алкил)(S-циклоалкил), C(арил или гетероарил)(S-алкил), C(арил или гетероарил)(S-циклоалкил), N(NH2), N[NH(алкил)], N[N(алкил)2], N[NH(циклоалкил)], N[N((алкил)циклоалкил)], CH(NH2), CH[NH(алкил)] , CH[NH(циклоалкил)], CH[N(алкил)2], CH[N(алкил)(циклоалкил)], CH[N(циклоалкил)2], C(алкил)(NH2), C(алкил)[NH(алкил)], C(алкил)[N(циклоалкил)2] , C(алкил)[NH(циклоалкил)] , C(алкил)[N(алкил)2], C(алкил)/[N(алкил)(циалоалкил)], C(арил или гетероарил)(NH2), C(арил или гетероарил)[NH(алкил)], C(арил или гетероарил)[NH(циклоалкил)], C(арил или гетероарил)[N(алкил)2], C(арил или гетероарил)[N(циклоалкил)2], или C(арил или гетероарил)[N(алкил)(циклоалкил)], и
R5 является замещенной или незамещенной арил или гетероарил группой.
Используемый здесь термин "способное ингибировать ферментативную тимидилатсинтазу" или другие подобные означает соединение, имеющее константу ингибирования тимидилатсинтазы ("TSki") менее или равной приблизительно 10-4 М. Предпочтительные соединения согласно настоящему изобретению имеют TSki величину в ряду менее приблизительно 10-5 М, более предпочтительно менее приблизительно 10-6 М, особенно предпочтительно менее приблизительно 10-7 М.
Тимидилатсинтаза является просто образцом активности хиназолиновых соединений настоящего изобретения. Действительно, некоторые соединения могут показывать антифолатную активность, кроме или даже в дополнение к ингибированию тимидилатсинтазы. Кроме того, некоторые соединения могут показывать антипролиферативную активность, обусловленную полностью другим местом действия, чем ингибирование метаболического пути фолиевой кислоты.
Некоторые хиназолиновые соединения согласно настоящему изобретению могут обладать одним или более асимметрическими атомами углерода и поэтому могут существовать в рацемической или оптически активной формах. Таким образом настоящее изобретение имеет в виду включение рацемических форм хиназолиновых соединения этого изобретения, так же как и любых оптически активных форм их, которые обладают антиопухолевой активностью.
Используемый здесь термин "алкил" включает прямые или разветвленные алкильные группы. Аналогичное соглашение применяют и к другим основным терминам, таким как "алкенил", "алкинил" и другие подобные. Кроме того, используемые здесь термины "алкил", "алкенил", "алкинил" и другие подобные включают замещенные и незамещенные группы.
Термин "алкил" означает группу, содержащую от одного до восьми, предпочтительно от одного до шести, атомов углерода. Например "алкил" может означать метил, этил, н-пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, трет-пентил, гексил, изогексил и другие подобные. Подходящие замещенные алкилы включают, но не ограничиваются этим, фторметил, дифторметил, трифторметил, 2-фторэтил, 3-фторпропил, гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил и другие подобные.
Термин "алкенил" означает группы, содержащие от двух до восьми, предпочтительно от двух до шести, атомов углерода. Например, "алкенил" может означать проп-2-енил, бут-2-енил, бут-3-енил, 2-метилпроп-2-енил, гекс-2-енил, гекс-5-енил, 2,3-диметилбут-2-енил и другие подобные. Термин "алкинил", который также означает группы, содержащие от двух до восьми, предпочтительно от двух до шести, атомов углерода, включает, но не ограничивается этим, проп-2-инил, бут-2-инил, бут-3-инил, пент-2-инил, 3-метилпент-4-инил, гекс-2-инил, гекс-5-инил и другие подобные.
Используемый здесь термин "циклоалкил" означает группы, содержащие от трех до семи, предпочтительно от трех до шести, атомов углерода. Пригодные циклоалкилы включают, но не ограничиваются этим, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и другие подобные. Термин "гетероцикл", который означает группы, содержащие один или более гетероатомов и предпочтительно от трех до семи атомов всего, включает, но не ограничивается этим, оксетан, тетрагидрофуранил, тетрагидропиранил, азиридин, азетидин, пирролидин, пиперидин, морфолин, пиперазин и другие подобные.
Заместитель "галоген" согласно настоящему изобретению может быть фтором, хлором, бромом или иодом.
Термин "арил" и "гетероарил", используемый здесь, означает и моноциклические, и полициклические группы, которые могут быть или замещенные, или незамещенные. Например, употребительные арил группы включают фенил, 1,2,3,4-тетрагидро-нафтил, нафтил, фенантрил, антрил, фенантро и другие подобные. Примерами обычных гетероарил колец являются 5-членные моноциклические группы, такие как тиенил, пирролил, имидазолил, пиразолил, фурил, изотиазолил, фуразанил, изоксазолил, тиазолил и другие подобные, 6-членные моноциклические группы, такие как пиридил, пиразинил, пиримидинил, пиридазинил, триазинил и другие подобные, и полициклические группы, такие как бензо[в]тиенил, нафто[2,3-b]тиенил, тиантренил, изобензофуранил, хроменил, ксантенил, феноксатиенил, индолизинил, изоиндолил, индолил, индазолил, пуринил, изохинолил, хинолил, фталазинил, нафтиридинил, хиноксалинил, хиназолинил, бензотиазол, бензимидазол, тетрагидрохинолин, циннолинил, птеридинил, карбазолил, бета-карболинил, фенантридинил, акридинил, перимидинил, фенантролинил, феназинил, изотиазолил, фенотиазинил, феноксазинил и другие подобные.
Как обсуждено выше, заместитель R1 формулы I может означать водород, галоген, алкил, -OH, -O-алкил, -O-(арил или гетороарил), -S-алкил, -S-(арил или гетероарил), -NH2, -NH-алкил, -N-(алкил)2, -NHCHO, -NHOH, -NHO-алкил, -NHNH2, замещенный -NHNH2, -NHC(=NH)NH2, -NHC(=NH)алкил, фторалкил, циклоалкил, алкенил, алкинил, арил или гетероцикл. Заместитель R1 является предпочтительно метилом или амино группой.
Заместители R2 и R3 формулы I согласно настоящему изобретению, которые могут быть одинаковыми или различными, могут представлять собой водород, галоген, алкил, циклоалкил, -OH, -O-алкил, -S-алкил, -NH2, -NH-алкил, -N-(алкил)2, -NHCHO, -NO2, -NHOH, -NHO-алкил, -NHNH2, замещенный -NHNH2, -CN, -CO2H, -CO2-алкил, -CONH2, -CONH(алкил), - CON(алкил)2, -CSNH2, -CSNH-алкил, -CSN(алкил)2, -C(= NH)NH2, -NHC(= NH)NH2, -NHC(= NH)алкил, -SO-алкил, -SO2-алкил, фторалкил, -O-фторалкил, -S-фторалкил, -NHCO(алкил), -NHCO(фторалкил), -SO-фторалкил, -SO2-фторалкил, -SH, -SO3H, -SO2NH2, -SO2NH(алкил), -SO2N(алкил)2, алкенил, алкинил, арил или гетероцикл.
Заместитель R2 предпочтительно является водородом или метилом, этилом, гидрокси, метокси, хлором или трифторметилом.
Более предпочтительно R2 означает водород или метил, хлор или трифторметил группу. Заместитель R3 предпочтительно является водородом.
Заместитель Z формулы I согласно изобретению является или кислородом, или серой. В предпочтительном воплощении заместитель означает кислород.
Заместитель R4 формулы I согласно настоящему изобретению может означать кислород, серу, SO, SO2, NH, N-алкил, CH2, CH-алкил, CH-(арил или гетероарил), CHOH, CHO-алкил, CHO-(арил или гетероарил), C(алкил)2, C(арил или гетероарил)2, C(алкил) (арил или гетероарил), CHS-алкил, CHS-арил, C(OH)(алкил), C(OH) (арил или гетероарил), C(OH)(циклоалкил), N(OH), N-циклоалкил, N(циклоалкил)SO2 , N(арил или гетероарил), C(циклоалкил)2, C(арил или гетероарил)(циклоарил), C(алкил)(алкенил), C(алкил)(алкинил), C(алкенил)2, C(алкинил)(арил или гетероарил), C(алкинил)(алкенил), C(алкенил)(арил или гетероарил), C(циклоалкил)(алкенил), C(циклоалкил)(алкинил), C(алкил)(арил или гетероарил), CH(циклоалкил), CH(алкенил), CH(алкинил), C(алкил)(циклоалкил), C(алкил)(O-алкил), C(алкенил)(O-алкил), C(алкинил)(O-алкил), C(алкил)(O-циклоалкил), C(алкенил)(O-циклоалкил), C(алкинил)(O-циклоалкил), C(арил или гетероарил)(O-алкил), C(арил или гетероарил)(O-циклоалкил), C(алкинил)(S-алкил), C(алкинил)(S-циклоалкил), C(алкенил)(S-алкил), C(алкенил)(S-циклоалкил), C(алкил)(S-алкил), C(алкил)(S-циклоалкил), C(арил или гетероарил)(S-алкил), C(арил или гетероарил)(S-циклоалкил), N(NH2), N([NH(алкил)/, N/N(алкил)2], N[NH(циклоалкил)] , N[N(алкил)(циклоалкил)] , CH(NH2), CH[NH(алкил)] , CH[NH(циклоалкил)], CH[N(алкил)2], CH[N(алкил)(циклоалкил)], CH[N(циклоалкил)2], C(алкил)(NH2), C(алкил)[NH(алкил)], C(алкил)[N(циклоалкил)2] , C(алкил)NH(циклоалкил)] , C(алкил)[N(алкил)2], C(алкил)[N(алкил)(циклоалкил)] , C(арил или гетероарил)(NH2), C(арил или гетероарил)(NH(алкил), C(арил или гетероарил)[NH(циклоалкил)], C(арил или гетероарил)[N(алкил2] , C(арил или гетероарил)[N(циклоалкил)2], или C(арил или гетероарил)[N(алкил)(циклоалкил)].
Заместитель R4 является предпочтительно кислородом, серой или метиленом, C= O, NH, NCH3, CH(OH) или C(OH)(фенил) группой. Более предпочтительно заместитель R4 означает серу.
Заместитель R5 формулы I может быть любым из большого ряда арильных или гетероарильных соединений, включая, но не ограничиваясь этим, арильные или гетероарильные кольца, обсуждаемые ранее. Заместитель R5 может быть незамещенным или замещенным. Подходящие заместители R5 включают широкое разнообразие электронодонорных и электроноакцепторных заместителей. Используемый здесь термин "электроноакцепторный" включает, но не ограничивается этим, группы, такие как -NO2, -CF3, -CN, карбокси, галоген, -SO2R6, где R6 означает алкил, арил или гетероарил группу, определенные выше, или R6 является -NR7R8 группой, где R7 и R8 представляют собой алкил группы, и другие подобные. Термин "электронодонорный" включает, но не ограничивается этим, группы, такие как -NH2, -NH-(алкил), -NHOH, -NHNH2, -O-(алкил), -S-(алкил), -NR7R8, где R7 и R8 означает алкил группы, и другие подобные.
Типичные заместителями R5 являются галоген, гидрокси, алкокси, алкил, гидроксиалкил, фторалкил, амино, -NH-(алкил), -N-(алкил)2, -CO-аминокислота, -CH, -NO2, -CF3, карбалкокси, карбамил, карбонил, карбокси, карбониламинокислоты, -SO2NHCO, SO2-аминокислота, сульфониламинокислоты, сульфамил, сульфанилил, сульфгидрил, сульфино, сульфинил, сульфо, сульфонамидо, сульфонил, (алкил)тио, замещенный или незамещенный фенилсульфонил, фенилмеркапто, фосфазо, фосфинико, фосфино, фосфо, фосфоно, фосфоро, фосфорозо, меркаптоарил и другие подобные.
Особенно предпочтительные структуры для R5 включают:
Предпочтительный класс соединений согласно настоящему изобретению включает соединения формулы I, где R3 является водородом. Особенно предпочтительными соединениями этого класса являются соединения, где Z означает кислород.
Другой предпочтительный класс соединений настоящего изобретения включает соединения формулы I, в которых R3 является водородом и R1 является или метилом, или амино группой. Особенно предпочтительными соединениями этого класса являются такие соединения, где Z является кислородом.
Другим предпочтительным классом настоящего изобретения являются такие соединения формулы I, где R3 означает водород и R2 означает водород или метил, этил, гидрокси или метокси группу. Более предпочтительно, R2 является водородом или метил группой. Особенно предпочтительными соединениями этого класса являются соединения, где Z является кислородом.
Другим предпочтительным классом соединений настоящего изобретения являются такие соединения формулы I, где R3 означает водород и R4 означает кислород, серу или метилен, C= O, CH(OH) или C(OH) (фенил) группу. Более предпочтительно, когда R4 является серой. Особенно предпочтительными соединениями этого класса являются соединения, где Z является кислородом.
Другим предпочтительным классом соединений настоящего изобретения являются соединения формулы I, где R3 является водородом и R5 является одной из следующих структур:
Особенно предпочтительны соединения этого класса, у которых Z является кислородом.
Другим предпочтительным классом соединений настоящего изобретения являются соединения формулы I, где R3 означает водород, R1 означает или метил, или амино группу, R2 является водородом или метилом, этилом, гидрокси или метокси группой, R4 означает кислород, серу или метилен, C=O, CH(OH) или C(OH) (фенил) группу и R5 является одной из следующих структур:
Особенно предпочтительными соединениями этого класса являются соединения, где Z является кислородом.
Согласно предпочтительному воплощению настоящего изобретения R3 означает водород, R1 является или метилом, или амино группой, R2 означает водород или метил группу, R4 представляет серу и R5 является одним из колец, рассмотренных в предшествующих пунктах. Особенно предпочтительными соединениями этого класса являются соединения, где Z означает кислород.
Особенно предпочтительные соединения настоящего изобретения проиллюстрированы в табл. 1.
Особенно предпочтительны соединения 14A, 24A и 25A.
Другой аспект настоящего изобретения касается способов получения хиназолиновых соединений формулы I с антипролиферативной активностью.
Один из способов настоящего изобретения для получения хиназолиновых соединений формулы I включает реакцию замещения для соединения формулы
где Z и R1 - R3 имеют те же значения, что определены предварительно, и L является удаляемой группой, с соответствующим соединением для замены удаляемой группы на требуемый -R4-R5-заместитель для случая (i) или на соответствующий R5-заместитель в случае (ii). Способ проводят при широко варьирующихся условиях, но обычно этот процесс выполняют в присутствии соответствующего основания, растворителя и катализатора при температуре, изменяющейся от приблизительно 70oC до приблизительно 165oC, предпочтительно от приблизительно 80oC до приблизительно 140oC, особенно предпочтительно при от приблизительно 90oC до приблизительно 100oC.
Удаляемые группы, пригодные для использования в этом способе, так же как и для использования в других способах настоящего изобретения, включают атомы галогена, такие как Br, Cl, F и I.
Предпочтительный способ получения хиназолиновых соединений формулы I, обладающих антипролиферативной активностью, где Z и R1 - R5 имеют ранее определенные значения, включает стадии:
(1) реакция соединения формулы
где L является удаляемой группой, например атомом галогена, таким как Br, Cl, F и I и R2 имеет ранее определенные значения, с гидрохлоридом гидроксиламина и хлоральгидратом с образованием изонитрозоацетанилида формулы
(2) обработка изонитрозоацетанилида стадии (1) серной кислотой, затем льдом и очистка этанолом с получением изатинового соединения формулы
(3) реакция изатинового соединения, полученного на стадии (2), с водной щелочной перекисью, такой как водный раствор NaOH и H2O2 до образования соединения о-аминобензойной кислоты формулы
(4) реакция соединения O-аминобензойной кислоты, полученного на стадии (3), с уксусным ангидридом до образования ацетилантранильного соединения формулы
(5) реакция ацетилантранильного соединения, полученного на стадии (4), с безводным аммиаком, с последующей обработкой NaOH, затем соляной кислотой до образования хиназолина формулы
(6) реакция замещения для хиназолинового соединения, полученного на стадии (5), обеспечивающая замену удаляемой группы L одним из требуемых R4-R5-заместителей, описанных предварительно, и таким образом получение соединения формулы I.
Стадию (1) могут выполнять при широко изменяющихся условиях, но обычно она проводится в присутствии воды, хлоральгидрата, соляной кислоты, сульфата натрия и гидрохлорида гидроксиламина при температуре, изменяющейся от приблизительно 0oC до приблизительно 100oC, предпочтительно от приблизительно 20oC до приблизительно 100oC и более предпочтительно при приблизительно 100oC.
Стадия (2) может выполняться при широко изменяющихся условиях, но обычно она проводится в присутствии концентрированной H2SO4 при температуре, изменяющейся от приблизительно 50oC до приблизительно 100oC, предпочтительно от приблизительно 65oC до приблизительно 100oC и более предпочтительно при приблизительно 80oC.
Стадия (3) может проводиться при широко изменяющихся условиях, но обычно она выполняется в присутствии воды, гидроокиси натрия и перекиси водорода при температуре от приблизительно 0oC до приблизительно 80oC, предпочтительно от приблизительно 20oC до приблизительно 80oC и более предпочтительно при приблизительно 80oC.
Стадия (4) может проводиться при широко изменяющихся условиях, но обычно она выполняется в присутствии уксусного ангидрида при температуре, изменяющейся от приблизительно 70oC до приблизительно 140oC, предпочтительно от приблизительно 100oC до приблизительно 140oC и более предпочтительно при приблизительно 140oC.
Стадия (5) может проводиться при широко изменяющихся условиях, но обычно она выполняется в присутствии аммиака при температуре, изменяющейся от приблизительно -33oC до приблизительно 20oC, предпочтительно при приблизительно 20oC.
Стадия (6) может выполняться при широко изменяющихся условиях, но обычно проводится она в присутствии соответствующего основания, растворителя и катализатора при температуре, изменяющейся от приблизительно 70oC до приблизительно 165oC, предпочтительно от приблизительно 80oC до приблизительно 140oC и более предпочтительно при от приблизительно 90oC до приблизительно 100oC.
Модификация шести стадий процесса, обсужденного выше, включает альтернативные стадии:
(5a) обработка ацетилантранилового соединения, полученного на стадии (4), MeOH с последующей обработкой соляной кислотой до получения соединения формулы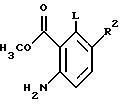
(5a') обработка о-аминобензойной кислоты, полученной на стадии (3), фосгеном или трифосгеном до образования соединения формулы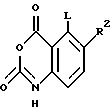
которое далее обрабатывают метанолом,
(5b) реакция продукта стадии (5a) или (5a') с гидрохлоридом хлорформамидина до получения хиназолинового соединения формулы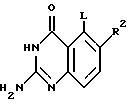
затем образовавшееся хиназолиновое соединение по реакции замещения, приведенной в методике стадии (6), описанной ранее, превращают в соединение формулы I.
Стадию (5a) могут выполнять при широко изменяющихся условиях, но обычно проводят (i) в присутствии метанола при температуре, изменяющейся от приблизительно 0oC до приблизительно 100oC, предпочтительно от приблизительно 20oC до приблизительно 70oC и наиболее предпочтительно при приблизительно 70oC и затем (ii) в присутствии концентрированной соляной кислоты при температуре, изменяющейся от приблизительно 70oC до приблизительно 100oC, более предпочтительно при приблизительно 100oC.
Стадию (5a') могут проводить при широко изменяющихся условиях, но обычно выполняют (i) в присутствии трифосгена при температуре от приблизительно 0oC до приблизительно 20oC, и затем (ii) в присутствии метанола при температуре от приблизительно 0oC до 70oC, более предпочтительно при температуре от приблизительно 0oC до приблизительно 20oC и наиболее предпочтительно при приблизительно 20oC.
Стадия (5b) может проводиться при широко изменяющихся условиях, но обычно ее выполняют в присутствии диглима и гидрохлорида хлорформамидина при температуре от приблизительно 100oC до приблизительно 175oC, предпочтительно от приблизительно 160oC до 175oC и особенно предпочтительно при приблизительно 170oC.
В особенно предпочтительном воплощении шестистадийного способа, приведенного выше, стадию (6) выполняют реакцией продукта или стадии (5), или стадии (5b) с анионом 4-тиопиридина в присутствии гидрида натрия, бромида меди (2) и окиси меди (1). Предпочтительный способ получения анионов 4-тиопиридинов для использования в этом изобретении включает реакцию 4-меркаптопиридина с NaH в безводном N,N-диметилацетамиде. Процесс получения 4-тиопиридинов могут проводить при широко варьирующихся условиях, но обычно выполняют в присутствии гидрида натрия и диметилформамида при температуре от приблизительно -20oC до приблизительно 20oC, предпочтительно от приблизительно 0oC до приблизительно 20oC, более предпочтительно при приблизительно 20oC.
Другой особенно предпочтительный способ настоящего изобретения для получения хиназолиновых соединений формулы I включает стадии:
(1) реакции соединения формулы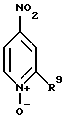
где R9 - заместитель является водородом, -CH3, -OCH3, -СF3, N(CH3)2 и другими подобными,
с бензилмеркаптаном до образования соединения формулы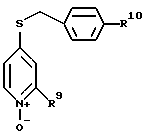
где заместитель R10 означает водород или -OCH3,
(2) восстановление продукта стадии (1),
(3) снятие защиты у продукта стадии (2), и
(4) реакция продукта стадии (3) с соединением формулы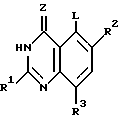
где Z и R1 - R3 имеют определенные ранее значения, L - удаляемая группа до получения соединения формулы
Стадию (1) этого способа могут проводить при широко изменяющихся условиях, но обычно выполняют в присутствии соответствующего основания и растворителя при температуре от приблизительно 0oC до приблизительно 80oC, предпочтительно от приблизительно 0oC до 20oC.
Стадию (2), стадию восстановления, могут выполнять при широко изменяющихся условиях, но обычно проводят в присутствии PCl3 и CHCl3 при температуре от приблизительно 0oC до приблизительно 80oC, предпочтительно от приблизительно 20oC до приблизительно 80oC и более предпочтительно при приблизительно 20oC.
Стадию (3), стадию снятия защиты, могут проводить при широко изменяющихся условиях, но обычно выполняют в присутствии соответствующего растворителя и металла или соли металла при температуре от приблизительно -78oC до приблизительно 20oC, предпочтительно от приблизительно -78oC до приблизительно 0oC и более предпочтительно от приблизительно -33oC до приблизительно 0oC.
Стадию (4) могут проводить при широко изменяющихся условиях, но обычно выполняют в присутствии диметилацетамида, гидрида натрия, бромида меди (I) и окиси меди (I) при температуре от приблизительно 70oC до приблизительно 165oC, предпочтительно от приблизительно 90oC до приблизительно 100oC и особенно предпочтительно при приблизительно 90oC.
Другой способ в сравнении с четырехстадийным способом, приведенным выше, включает стадии:
(1) восстановление соединения формулы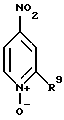
с образованием соединения формулы
(2) реакция продукта стадии (1) с ксантогенатом до получения соединения формулы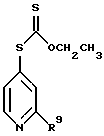
(3) гидролиз продукта стадии (2) и последующая реакция с соединением формулы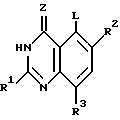
где Z и R1-R3 имеют определенные ранее значения и L является удаляемой группой, в присутствии N,N-диметилацетамида, бромида меди (I) и окиси меди (I) до получения соединения формулы
Стадию (1), стадию восстановления, могут проводить при широко изменяющихся условиях, но обычно выполняют в присутствии водорода (газа), соответствующего растворителя и каталитического количества палладия, предпочтительно при комнатной температуре приблизительно 20oC. Конечно, в некоторых случаях могут быть использованы повышенные температуры для ускорения реакции.
Стадию (2) могут проводить при широко изменяющихся условиях, но обычно выполняют в присутствии водной кислоты и Na2NO2 с последующей обработкой ксантогенатом калия при температуре от приблизительно -40oC до приблизительно 20oC, предпочтительно от приблизительно 0oC до приблизительно 5oC и более предпочтительно при приблизительно 0oC.
Гидролиз стадии (3) могут выполнять при широко изменяющихся условиях, но предпочтительно проводят с NaOH/CH3OH при температуре от приблизительно 0oC до приблизительно 20oC. Следующую после гидролиза реакцию стадии (3) также проводят при широко изменяющихся условиях, но обычно выполняют в присутствии соответствующего основания, растворителя и катализатора при температуре от приблизительно 70oC до приблизительно 165oC, предпочтительно от приблизительно 90oC до приблизительно 100oC и более предпочтительно при приблизительно 90oC.
Другой предпочтительный способ получения хиназолинового соединения формулы I, где Z и R1 - R5 имеют определенные ранее значения, включает стадии:
(1) реакция соединения формулы
где R1 - R3 имеют ранее определенные значения, с соединением, пригодным для введения защитной группы P, до образования соединения формулы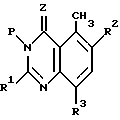
(2) превращение продукта стадии (1) до соединения формулы
где L является удаляемой группой,
(3) реакция замещения хиназолинового соединения стадии (2) с образованием соединения формулы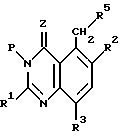
где R5 имеет значения, определенные ранее, и
(4) снятие защиты у продукта стадии (3).
Стадию (1) описанного ранее процесса могут проводить при широко изменяющихся условиях, но обычно выполняют в присутствии соответствующих алкил или ацил галогенида, основания и растворителя при температуре от приблизительно 0oC до приблизительно 20oC, предпочтительно при приблизительно 20oC.
Хотя для описанного выше процесса может быть использовано в качестве защитной группы P разнообразие заместителей, защитная группа P предпочтительно означает CH2OCH2CH2Si(CH3)3, CH2OCH3, CH2OC(O) tBu или CO tBu группы.
Согласно предпочтительному воплощению P является CH2OCH2CH2 Si(CH3)3.
Стадия (2), стадия превращения, может проводиться при широко изменяющихся условиях для того, чтобы обеспечить широкое разнообразие удаляемых групп, но предпочтительно выполняется в присутствии N-бромсукцинимида, брома, N-хлорсукцинимида или N-иодсукцинимида при температуре от приблизительно 20oC до приблизительно 100oC, предпочтительно от приблизительно 50oC до приблизительно 100oC и более предпочтительно при приблизительно 80oC. В предпочтительном воплощении процесс выполняют в присутствии N-бромсукцинимида, CCl4 и света.
Стадию (3) могут проводить при широко изменяющихся условиях, но обычно выполняют в присутствии соответствующих нуклеофила, основания и растворителя при температуре от приблизительно 0oC до приблизительно 50oC, предпочтительно от приблизительно 20oC до приблизительно 100oC и более предпочтительно при приблизительно 20oC.
А предпочтительном воплощении этого процесса стадию (3) выполняют реакцией продукта стадии (2) с NaOEt (этилат натрия) и 2-нитропропаном с последующей обработкой фенилмагнием до образования соединения формулы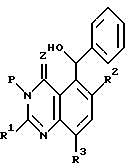
По другому воплощению стадию (3) проводят реакцией продукта стадии (2) с 5-хлориндолом.
Стадию (4) которая может выполняться при широко изменяющихся условиях, обычно проводят в присутствии соответствующего кислого или щелочного фторида при температуре от приблизительно 0oC до приблизительно 100oC, предпочтительно от приблизительно 20oC до приблизительно 100oC и более предпочтительно при приблизительно 20oC.
Материалы и условия на стадии (4) по снятию защитной группы зависят от разнообразных факторов. Конечно, одним из факторов является заместитель, используемый в качестве защитной группы P. Например, если P является CH2OCH2CH2 Si (CH3)3 группой, то стадия (4) предпочтительно выполняется реакцией продукта стадии (3) с тетрабутиламмонийфторидом.
В модификации способа, описанного выше, до стадии (4) снятия защитной группы продукт стадии (3) окисляют до образования соединения формулы
затем вслед за стадией (4) снятия защитной группы продукт стадии (4) реагирует с фениллитием до образования соединения формулы
Как показано выше может быть необходимо использовать защитные группы или до, или после, или в течение процесса получения соединения настоящего изобретения.
Подходящей защитной группой азота кольца, например входящего в гетероциклический остаток, является, например, пивалоилоксиметил группа, которая может удаляться гидролизом с основанием, таким как гидроокись натрия, трет-бутилоксикарбонил группа, которая может удаляться гидролизом с кислотой, такой как соляная или трифторуксусная кислота, или с основанием, таким как тетра-н-бутиламмонийфторид (TBAF") или гидроокись лития; метоксметил группа, которая может удаляться соляной кислотой и п-толуолсульфокислотой, или 2-(триметилсилил) этоксиметил группа, которая может удаляться TBAF или кислотой, такой как соляная кислота.
Подходящей защитной группой для гидроксил группы является, например, этерифицирующая группа, такая как ацетил или бензоил группа, которая может быть удалена гидролизом с основанием, таким как гидроокись натрия. Альтернативно, если присутствуют в исходном материале другие группы, не содержащие алкенильной или алкинильной группы, то защитная группа может быть, например альфа-арилалкильной группой, такой как бензильная группа, которая может удаляться гидрированием в присутствии катализатора, такого как палладий на углероде или никель Ренея. Дополнительной защитной группой для гидроксильной группы является группа, такая как трет-бутилдифенилсилил (-Si -трет- Bu - Ph2), которая может удаляться обработкой TBAF.
Подходящей защитной группой для меркаптогруппы является, например, этерифицирующая группа, такая как ацетил группа, которая удаляется гидролизом с основанием, таким как гидроокись натрия.
Пригодной защитной группой для амино группы может быть, например, алкилкарбонил группа, такая как ацетил группа (CH3CO-), которая может быть удалена обработкой водной неорганической кислотой, такой как азотная, серная или соляная кислота. Другой защитной группой для амино группы является алкоксикарбонил группа, такая как метоксикарбонил или трет-бутилоксикарбонил группа. Эти группы могут быть удалены при обработке органической кислотой, такой как трифторуксусная кислота.
Подходящей защитной группой для первичной амино группы является, например, ацетил группа, которая может быть удалена обработкой водной неорганической кислотой, такой как азотная, серная или соляная кислота, или фталоил группа, которая может быть удалена обработкой алкиламином, таким как диметиламинопропиламином, или гидразином.
Пригодной защитной группой для карбокси группы может быть эфирная группа, например, метил или этил группа, которая может быть удалена гидролизом с основанием, таким как гидроокись натрия. Другой защитной пригодной группой является трет-бутил группа, которая может быть удалена обработкой органической кислотой, такой как трифторуксусной кислотой.
В то время как предпочтительные способы получения антипролиферативных соединений настоящего изобретения подробно описаны, по предшествующим работам, видно, что разнообразные другие способы, так же как и изменения, и модификации представленных способов могут быть использованы для получения соединений настоящего изобретения.
Антипролиферативные хиназолиновые соединения настоящего изобретения, которые могут быть использованы в фармкомпозициях настоящего изобретения, включают все эти описанные выше соединения, так же как и фармацевтически приемлемые соли этих соединений. Фармацевтически приемлемые кислые соли соединений изобретения, содержащих основную группу, образуются с сильными или умеренно сильными органическими или неорганическими кислотами в присутствии основного амина с помощью известных методов. Примерами кислых солей, которые включены в это изобретение, являются: (1) соли органических кислот, такие как малеат, фумарат, лактат, оксалат, метансульфонат, этансульфонат, бензолсульфонат, тартрат, глюкуронат, цитрат и ацетат, и (2) соли неорганических кислот, такие как гидробромид, гидрохлорид, гидросульфат, фосфат и нитрат. Фармацевтически приемлемые основные соли соединений изобретения, содержащих кислотные группы, получают известными методами из органических и неорганических оснований, которые включают нетоксичные основания щелочных и щелочноземельных металлов, например гидроокиси кальция, натрия и калия, гидроокиси аммония, и нетоксичные органические основания, такие как триэтиламин, бутиламин, пиперазин и три(гидроксиметил)метиламин.
Как указано выше, соединения изобретения обладают антипролиферативной активностью, свойство, которое выражается в форме противоопухолевой активности. Соединение изобретения может действовать само по себе или может применяться как пропрепарат, который превращается in vivo в действующее соединение. Предпочтительные соединения изобретения действуют как ингибиторы ферментативной тимидилатсинтазы. Особенно предпочтительны соединения, которые действуют как ингибиторы роста клеток линии 11210, линии лейкемических клеток мышей, которые могут быть выращены в культурах тканей. Такие соединения изобретения также ингибируют рост бактерий, таких как грам-отрицательных бактерий Escherichia coli, которые могут быть выращены в культуре. Соединения изобретения могут также ингибировать рост бактерий.
Антипролиферативные соединения настоящего изобретения, так же как и их фармацевтически приемлемые соли, могут быть включены в удобные препаративные формы, такие как капсулы, таблетки или препараты для инъекций. Могут быть использованы твердые или жидкие фармацевтически приемлемые носители. Твердые носители включают крахмал, лактозу, дигидрат сульфата кальция, сахарозу, тальк, желатину, агар, пектин, аравийскую камедь, стеарат магния и стеариновую кислоту. Жидкие носители включают сироп, арахисовое масло, оливковое масло, солевой раствор и воду.
Аналогично носитель или разбавитель могут включать материал, обеспечивающий пролонгированное высвобождение, такой как глицерин моностеарат или глицерил дистеарат, один или в смеси с парафином. При использовании жидкого носителя препаративная форма может быть в виде сиропа, эликсира, эмульсии, мягкой желатиновой капсулы, стерильной жидкости для инъекций (например, раствора), как например ампулы, или в виде водной или неводной жидкой суспензии.
Фармацевтическое препаративные формы получают по обычным методикам с использованием таких стадий как смешение, гранулирование или прессование, если необходимо получить таблетки, или смешение, наполнение и растворение ингредиентов, если уместно, для того, чтобы получить требуемые продукты для орального, парентерального, местного, внутривагинального, внутриносового, внутрибронхиального, внутриглазного, внутриушного и ректального введения.
Кроме того, фармкомпозиции изобретения могут включать одно или более соединений, которые являются антиопухолевыми агентами, такие как ингибиторы деления клеток (например, винбластин), алкилирующие агенты (например, цис-платин, карбоплатин и циклофосфамидин) ингибиторы дигидрофолатредуктазы (например, метотрексат, пиритрексим и триметрексат), другие ингибиторы тимидилатсинтазы, антиметаболиты (например 5-фторурацил и цитозинарабинозид), интеркаляционные антибиотики (например, адриамицин и блеомицин), ферменты (например, аспарагиназа), ингибиторы топоизомеразы (например, этопозид) или модификаторы биологической ответной реакции (например, интерферон).
Фармсоставы изобретения могут также включать одно или более соединений, являющихся антифунгальными, антипаразитическими, антивирусными, антипсориазными и антикокковыми агентами. Примерами антибактериальных агентов являются, например, сульфонамиды, такие как сульфаметоксозал, сульфадиазин, сульфаметер или сульфадоксин, ингибиторы дигидрофолатредуктазы, такие как триметоприм, бромдиаприм, или триметрексат, пенициллины, цефалоспорины, аминогликозиды, бактериостатические ингибиторы синтеза протеинов, хиналинкарбоновые кислоты и их конденсированные изотиазолиновые аналоги.
Другим аспектом изобретения является терапевтический способ ингибирования роста и пролиферации клеток высших организмов и микроорганизмов, такой способ включает введение реципиенту позвоночному животному (например, млекопитающим или птицам) эффективного количества соединения настоящего изобретения. Особенно предпочтителен терапевтический способ, включающий введение реципиенту эффективного количества соединения настоящего изобретения для ингибирования ферментативной тимидилатсинтазы. Соединения изобретения особенно полезны при лечении млекопитающих, таких как человек, и для лечения птиц.
Любые описанные выше антипролиферативные соединения или их фармацевтически приемлемые соли могут быть использованы для терапевтического способа изобретения. Соединения изобретения могут вводиться в виде фармацевтически приемлемых композиций, включающих разбавитель или носитель, такие как описаны ранее. Дозы соединений предпочтительно включают единицы фармацевтических доз, содержащих эффективное количество активного соединения. Под эффективным количеством понимают количество, достаточное для ингибирования фолатного метаболического пути и получения удовлетворительного эффекта от введения одной или более единиц фармацевтических доз. Примерная единица суточной дозы для позвоночного включает вплоть до приблизительно 1 г активного соединения на килограмм веса, предпочтительно 0,5 г, более предпочтительно 100 мг и особенно предпочтительно приблизительно 50 мг на килограмм веса позвоночного.
Выбранная доза может вводиться теплокровным животным или млекопитающим, например человеку, при необходимости лечения посредством ингибирования метаболического пути фолиевой кислоты любым известным способом введения, например местным (например, мазь или крем), оральным, ректально (например, как суппозитории), парентерально, введением инъекций или непрерывным вливанием, внутривагинально, внутриносовым, внутрибронхиальным, внутриушным или внутриглазным.
Соединения настоящего изобретения могут характеризоваться как продуцирующие любой один или более антипролиферативный, антибактериальный, антипаразитический, антивирусный, антипсориазный, антипротозойный, антикокковый или антифунгальный эффект. Соединения особенно полезны для противоопухолевого действия на позвоночных, имеющих опухоли.
Примеры
Как установлено предварительно, таблица 1 включает ряд предпочтительных соединений настоящего изобретения. Примеры способа, использованного для получения некоторого количества этих предпочтительных соединений, представлены ниже.
Структура всех соединений настоящего изобретения подтверждена данными спектроскопии протонного магнитного резонанса, ИК спектроскопии, элементного микроанализа и/или масс-спектрометрии. Спектры поглощения в ИК-области сняты на спектрофотометре Midac FT или Perkin Elmer Model 457. Спектры снимают как в таблетках KBr, так и в тонком слое, величину пика сообщают в см-1.
Спектры протонного магнитного резонанса снимают с использованием спектрометра General Electric QE-300 при напряженности магнитного поля 300 МГц. Химический сдвиг приводят в частях на миллион ( δ ) с указанием такой ссылки, как в CДCl3 CHCl3 пик присутствует при 7.26 ppm и в ДMCO-d6, пик ДMCO присутствует при 2.49 ppm. Стандарт и мультиплетность пика обозначаются следующим образом: с, синглет, д, дублет, дд, дублет дублетов, т, триплет, шир.с., широкий синглет, шир. д, широкий дублет, шир, широкий сигнал, м, мультиплет.
Масс-спектры снимают при помощи масс-спектрометра высокого разрешения VG 7070E-HF при использовании метода прямой вставки, ионизирующего потенциала 70 эВ и температуры источника ионов 200oC. Элементный анализ предлагает данные по элементам обычно в пределах ± 0.4% от теоретической величины.
Основные методики
N, N-Диметилформамид (ДМФА) сушат над активированными 3- молекулярными ситами, N, N-диметилацетамид (ДМА) (Aldrich Gold Label) сушат аналогично. Тетрагидрофуран (ТГФ) отгоняют от бензофенона натрия в атмосфере азота. Термин "эфир" означает диэтиловый эфир.
молекулярными ситами, N, N-диметилацетамид (ДМА) (Aldrich Gold Label) сушат аналогично. Тетрагидрофуран (ТГФ) отгоняют от бензофенона натрия в атмосфере азота. Термин "эфир" означает диэтиловый эфир.
Хроматография выполняется при использовании силикагеля 60 (Merck Art 9385). Если сырой твердый продукт нерастворим в выбранном элюенте, то его растворяют в более полярном растворителе и добавляют двуокись кремния Merck Art 7734. Суспензию упаривают досуха на роторном испарителе с использованием стеклянного фритта для предупреждения распыления двуокиси кремния. Двуокись кремния с нанесенным веществом помещают в колонку. Тонкослойную хроматографию (ТСХ) проводят на пластинках с нанесенной двуокисью кремния 60F254 (Merck Art 5719). Экстракты сушат над безводными Na2SO4 или MgSO4. Температура плавления определяется на аппарате MeI-Temp и не корректируется.
Пример 1: Получение соединений 8A и 14A
Соединения 8A и 14A получают по следующей схеме реакций: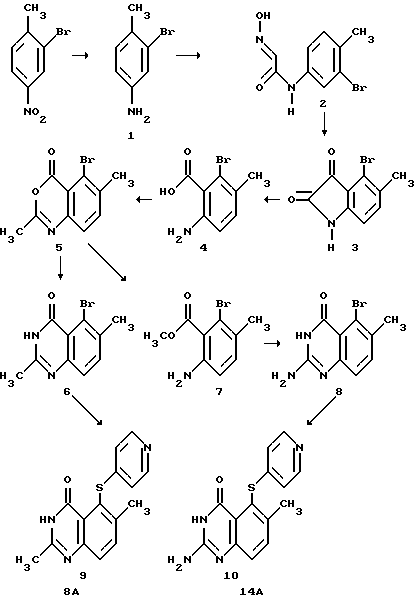
Получение промежуточного соединения (1) 3-бром-4-метиланилина
В аппарат гидрирования Парра помещают раствор 50.0 г (0.23 молей) 2-бром-4-нитротолуола в 500 мл метанола, добавляют 5.0 г никеля Ренея. Смесь гидрируют при 30 фунт/дюйм2 H2 в этом аппарате в течение 3 часов при перемешивании. Аппарат соединяют с атмосферой, реакционную смесь фильтруют через кизельгур (целит), фильтрат упаривают и получают 41.0 г (95%) желтого масла. ИК (чистый) 3329, 3144, 2604, 1609, 1288, 1030, 312 см-1, 1H-ЯМР (ДМСО-d6) δ 2.13 (с, 3H), 5.60 (шир. с, 2H), 6.46 (дд, 1H, J = 8.1 Гц, 2.3 Гц), 6.79 (д, 1H, J = 2.3 Гц), 6.94 (д, 1H, J = 8.2 Гц), HRMS для C7H8BrN: вычислено 184.9843, найдено 184.9840.
Получение промежуточного соединения (2) 3-бром-4-метил α -изонитрозоацетанилида
Смесь 45.0 г гидрата хлораля (0.27 молей), 65.0 г сульфата натрия (0.46 молей), 40.0 г 3-бром-4-метиланилина (1) (0.21 молей) 20 мл концентрированной HCl, 55.0 г гидрохлорида гидроксиламина (0.79 молей) и 1.5 л H2O нагревают 1 час при 100oC, охлаждают до 0oC, выпавший осадок отфильтровывают, промывают его водой, сушат и получают 41.0 г (76%) желтовато-коричневого вещества: т. пл. 195-197oC. ИК (KBr) 3439, 3310, 3110, 2998, 2876, 2749, 1636, 1591, 1466, 1256, 905, 691 см-1;
1H-ЯМР (ДМСО-d6) δ 2.28 (с, 3H), 3.50 (шир. с. 1H), 7.28 (д, 1H, J = 8.3 Гц), 7.53 (дд, 1H, J = 8.2 Гц, 2.1 Гц), 8.02 (д, 1H, J = 2.0 Гц), 10.26 (с, 1H), 12.21 (с, 1H).
Элементный анализ для C9H9BrN2O2:
Вычислено: C 42.04 H 3.53 Br 31.08 N 10.90
Найдено: C 42.71 H 3.57 Br 31.44 N 11.09
Получение промежуточного соединения (3) 4-бром-5-метилизатина
К 160 мл концентрированной серной кислоты при 80oC прибавляют 40 г (0.156 молей) (2), перемешивают 1 час, охлаждают до комнатной температуры, выливают в 2 л измельченного льда. Выпавший осадок отфильтровывают, промывают водой, затем бензолом. Красное твердое вещество прибавляют к 800 мл кипящего этанола. Раствор охлаждают до комнатной температуры, собирают, промывают холодным этанолом. 6-Бром-5-метилизатин, так же как и некоторый требуемый продукт, остается в жидком маточнике, он может быть отделен на хроматографической колонке с силикагелем. Оставшийся на фильтре материал сушат и получают 19 г (50.7%) твердого вещества красного цвета:
т.пл. 245-248oC. ИК (KBr) 3302, 1750, 1609, 1466, 1273, 675 см-1.
1H-ЯМР (CДCl3) δ 2.26 (c, 3H), 6.8 (д, 1H, J = 7.9 Гц), 7.5 (д, 1H, J = 8.3 Гц), 11.06 (c, 1H).
Элементный анализ для C9H6BrNO2:
Вычислено: C 45.02, H 2.52, Br 33.28, N 5.86
Найдено: C 45.10, H 2.54, Br 33.19, N 5.84
Получение промежуточного соединения (4) 5-метил-6-бром-о-аминобензойной кислоты
Смесь 80 мл 3 N NaOH и 19 г изатина (3) (0.08 молей) нагревают при 80oC, прибавляют 18 мл 30% H2O2, перемешивают 1 час, охлаждают до 5oC, подкисляют до pH 5 концентрированной соляной кислотой. Раствор упаривают досуха, вносят 300 мл метанола, фильтруют, фильтрат упаривают и получают 18 г желтовато-коричневых кристаллов (97.8%), т.пл. (гидрохлорид) 290-294oC.
ИК (KBr) 3619, 3229, 1578, 1478, 1412, 1381, 1084, 1010, 820, 706 см-1,
1H-ЯМР (ДМСО-d6) δ 2.13 (с, 3H), 4.9 (c, 2H), 6.4 (д, 1H, J = 7,9 Гц), 6.74 (д, 1H, J = 7.8 Гц).
Получение промежуточного соединения (5) 5-бром-6-метил ацетилантранила
(5-бром-2,6-диметил-4H-3,1-бензоксазин-4-она)
Смесь 18 г о-аминобензойной кислоты (4) (0.078 молей) в 300 мл уксусного ангидрида кипятят 3 часа, охлаждают до 0oC, фильтруют. Остаток на фильтре промывают ацетоном и получают 16 г (81% теоретически) в виде твердого белого вещества, которое используется без дальнейшей очистки.
ИК (KBr) 3460, 1750, 1660, 1574, 1416, 1260, 1070, 841 см-1,
1H-ЯМР (CДCl3) δ 2.45 (с, 3H), 2.55 (c, 3H), 7.40 (д, 1H, J = 8.2 Гц), 7.64 (д, 1H, J = 8.0 Гц).
HRMS для C10H8BrNO2: вычислено 252.9738, найдено 252.9743.
Получение промежуточного соединения (6) 5-бром-3,4-дигидро-2,6- диметилхиназолин-4-она
Безводный аммиак (50 мл) конденсируют в колбе, содержащей 8,5 г (34 ммолей) антранила (5), перемешивают 3 часа, упаривают растворитель, остаток обрабатывают 75 мл 1 N NaOH, кипятят 1 час, охлаждают до 0oC, подкисляют до pH 4 концентрированной соляной кислотой, фильтруют. Остаток на фильтре промывают водой, сушат и получают 7.1 г (82.5% от теоретического) соединения 6 в виде желтовато-коричневого твердого вещества, которое используют без дальнейшей очистки.
ИК (KBr) 2910, 2620, 1680, 1630, 1460, 1377, 1298, 1128, 872 см-1,
1H-ЯМР (ДMCO-d6) δ 2.33 (с, 3H), 2.43 (c, 3H), 7.49 (д, 1H, J = 8.3 Гц), 7.70 (д, 1H, J = 8.3 Гц), 12.20 (шир. с, 1H), HRMS для C10H9BrN2O: вычислено 251.9898, найдено 251.9908.
Получение соединения (7)
Метилового эфира 2-амино-6-бром-5-метилбензойной кислоты
Смесь 10 г (0.039 молей) антранила (5) в 75 мл метанола кипятят 2 часа, прибавляют 10 мл концентрированной соляной кислоты, нагревают дополнительно 2 часа, упаривают досуха. Остаток растворяют в 20 мл воды, нейтрализуют до pH 7 триэтиламином. Водный раствор экстрагируют хлористым метиленом. Слои разделяют, органический слой сушат над сульфатом магния, фильтруют, упаривают досуха и получают 6,0 г соединения (7) в виде оранжевого масла (63% от теоретического).
ИК (чистый) 3483, 3410, 3220, 3000, 2950, 2851, 1720, 1620, 1560, 1430, 1288, 1120, 1015, 816 см-1,
1H-ЯМР (CДCl3) δ 2.31 (с, 3H), 3.95 (c, 3H), 4.10 (шир.с., 2H), 6.60 (д, 1H, J = 8.2 Гц), 7.05 (д, 1H, J = 8.1 Гц),
HRMS для C9H10BrNO2: вычислено 242.9890, найдено 242.9895.
Получение промежуточного соединения (8)
2-амино-5-бром-3,4-дигидро-6-метилхиназолин-4-она
К раствору метилового эфира (7) (6 г, 24 ммолей) в 50 мл диглима прибавляют 3 г (24 ммолей) гидрохлорида хлорформамидина. Смесь кипятят 1 час, охлаждают до 0oC, фильтруют, промывают эфиром, сушат и получают 6.25 г (88% от теоретического) желтовато-коричневого твердого вещества; т.пл. (гидрохлорид) > 390oC. Продукт используется без дальнейшей очистки.
ИК (KBr) 3140, 2950, 1670, 1620, 1471, 1402, 816, 600 см-1
1H-ЯМР (ДМСО-d6) δ 2.28 (с, 3H), 6.75 (шир.с., 2H), 7.0 (д, 1H, J = 8.3 Гц), 7.40 (д, 1H, J = 8.0 Гц), 11.8 (шир.с., 1H).
HRMS для C9H8BrN3O: вычислено 253.9927, найдено 253.9929.
Получение соединения (9) (соединения 8A)
3,4-дигидро-2,6-диметил-4-оксо-5-(4-пиридилтио)-хиназолина
К раствору 3.2 г 4-меркаптопиридина (28.8 ммолей) в 50 мл безводного N, N-диметилацетамида при 0oC прибавляют 1.24 г (28.8 ммолей) ПаH (60% дисперсия в минеральном масле), перемешивают 1 час, прибавляют 3.1 г бромхиназолина (6) (0.012 молей), 1.4 г бромида меди (1) и 0.70 г окиси меди (1), нагревают 4 часа при 90oC, упаривают досуха. Остаток обрабатывают 50 мл раствора H2S/метанола (10 г/л), перемешивают 1 час, фильтруют, фильтрат упаривают досуха. Твердый продукт очищают хроматографически на силикагеле при использовании MeOH/CH2Cl2 (5:95) и получают 1.7 г (48% от теоретического) желтовато-коричневого твердого вещества: т.пл. 235-238oC, ИК (KBr) 3430, 1670, 1633, 1575, 1460, 1408, 1300, 841, 820, 714 см-1,
1H ЯМР (ДМСО-d6) δ 2.28 (с, 3H), 2.40 (с, 3H) 6.80 (д, 2H), J = 5.9 Гц), 7.60 (д, 1H, J = 8.3 Гц), 7.80 (д, 1H, J = 8.5 Гц), 8.24 (д, 2H, J = 6.5 Гц), 12.10 (шир с. 1H).
Элементный анализ для C15H13N3OS • H2O
Вычислено: C 59.80, H 4.98 N 13.95 S 10.63
Найдено: C 59.58 H 4.90 N 13.89 S 10.62
HRMS для C15H13N3OS : вычислено 283.0773, найдено 283.0779.
Получение соединения (10) (соединения 14A) 2-амино-3,4-дигидро-6-метил-4-оксо-5-(4-пиридилтио)хиназолина
К раствору 17.2 г 4-меркаптопиридина (15.5 молей) в 250 мл безводного N, N-диметилацетамида при 0oC прибавляют 6.2 г (15.5 молей) NaH (60% дисперсия в минеральном масле), перемешивают 1 час, вносят 15 г аминохиназолина • HCl (8) (51.3 ммолей), 4.5 г бромида меди (I) и 4.5 г окиси меди (I). Смесь нагревают 4 часа при 90oC, упаривают в вакууме. Остаток обрабатывают 150 мл H2S/MeOH (20 г/л), темную смесь перемешивают, выпаривают, выпавшую CuS отфильтровывают, фильтрат упаривают. Остаток промывают хлористым метиленом, этиловым эфиром и кипящим изопропанолом и получают 7.5 г (50 % от теоретического) соединения (10) в виде желтовато-коричневого твердого вещества: т. пл. 301 - 302oC,
ИК (KBr) 3320, 3150, 2750, 1670, 1575, 1466, 1305, 1220, 804, 710, 482 см-1,
1H ЯМР (ДМСО-d6) δ 2.30 (с, 3H), 6.35 (шир с, 2H), 6.80 (д. 2H, J = 5.9 Гц), 7.26 (д, 1H, J = 8.4 Гц), 7.58 (д, 1H, J = 8.5 Гц), 8.25 (шир с, 2H), 10.85 (шир с, 1H).
Элементный анализ для C14H12N4OS • 1,5 H2O:
Вычислено: C 54.00 H 4.86 N 18.00 S 10.30
Найдено: C 53.81 H 4.25 N 17.71 S 10.28
HRMS для C14H12N4OS: вычислено 284.0734, найдено 284.0732
Пример 2: Получение соединений 13A и 15A
Соединения 13A и 15A получают по следующей схеме реакций:
Получение промежуточного соединения (11) 3-бром-4-метоксианилина
К раствору 38.0 г 1-бром-4-нитроанизола (0.164 молей) в 300 мл метанола/ТГФ (1: 1) прибавляют 5 мл безводного гидразина и 4.0 г активированного никеля Ренея, суспендированного в этаноле. Смесь перемешивают, нагревают до мягкого кипения. Смесь начинает выделять газ. В течение 3 часов в реакцию дополнительно вносят 7 мл гидразина и 4 г никеля Ренея. Теплую реакционную смесь отфильтровывают в вакууме через прокладку силикагеля для удаления катализатора, промывают этилацетатом. Фильтрат упаривают, темно-коричневое масло выдерживают в высоком вакууме для удаления следов растворителя. Продукт используют сразу после получения, так как он быстро разлагается.
1H ЯМР (CДCl3) δ 3.46 (с, 2), 6.60 (дд, 1H, J = 8.6, 2.7 Гц), 6.73 (д, 1H, J = 8.6 Гц), 6.92 (д, 1H, J = 2.7 Гц).
Получение промежуточного продукта (12) 3-бром-4-метокси- α -изонитрозоацетанилида
В 250 мл 3-х горловую колбу помещают 6.3 г (37.8 ммолей) хлоральгидрата, прибавляют 84 мл воды, снабжают механической мешалкой, обратным холодильником, при постоянном перемешивании в течение 1 минуты прибавляют 90 г порошка безводного сульфата натрия, затем добавляют раствор 6.3 г (31.2 ммолей) анилина (11) в 3.0 мл концентрированной HCl и 21 мл воды, затем раствор 7.7 г (112 ммолей) H2NOH•HCl в 35 мл воды. Смесь медленно нагревают до кипения при постоянном перемешивании, кипятят 2 минуты до образования кристаллов коричневого цвета. Смесь охлаждают, твердый продукт отфильтровывают, промывают водой, сушат в вакууме до постоянного веса и получают 5.65 г (66% от теоретического) твердого продукта, пригодного для следующей стадии без дальнейшей очистки. Аналитически чистый образец получают при перекристаллизации.
Т. пл. 202-203oC (гексан, EtOAc). ИК (KBr) 3409, 2875, 2056, 2023, 1643, 1634, 1543, 1502, 1295, 1270, 1047, 799 см-1,
1H ЯМР (CДCl3 + одна капля ДМСО-d6) δ 3.88 (с, 3H), 6.87 (д, 1H, J = 8.9 Гц), 7.53 (м, 2H), 7.83 (д, 1H, J = 2.5 Гц), 8.49 (с, 1H), 11.60 (с, 1H, ПH),
Элементный анализ для C9H9BrN2O3• 0,11 EtOAc:
Вычислено: C 40.09 H 3.52 Br 28.26 N 9.91
Найдено: C 40.45 H 3.44 Br 27.86 N 10.34
Получение промежуточного соединения (13) 4-бром-5-метоксиизатина
К 8 мл концентрированной H2SO4 при 50oC при перемешивании прибавляют медленно высушенный в вакууме α -изонитрозоацетанилид (12) (3.0 г 11 ммолей). Реакционная смесь вначале становится желтой, затем темнеет, температура в течение 10 минут повышается до 65oC, реакцию контролируют ТСХ (EtOAc/гексан, 40:60). Нагревание смеси при 65-70oC проводят до тех пор, пока по ТСХ присутствует исходный материал. После окончания реакционную смесь охлаждают и прибавляют к 80 г измельченного льда при перемешивании. Образовавшийся твердый продукт темно-красного цвета отфильтровывают, промывают от кислоты водой, сушат в вакууме. Остаток хроматографируют на колонке с двуокисью кремния при использовании системы EtOAc/гексан с градиентом 40:60, 50: 50, 60:40, 70:30, 80:20. Нежелательный изомер, 6-бром-5-метоксиизатин, элюируют первым, затем элюируют требуемый изомер (13), который выделяют в виде твердого вещества красного цвета (0.71 г, 25% выход). Т.пл. 250-251oC.
ИК (KBr) 2064, 1758, 1750, 1634, 1278 см-1,
1H ЯМР (CДCl3 + одна капля ДМСО-d6) δ 3.91 (с, 3H), 6.84 (д, 1H, J = 8.8 Гц), 7.09 (д, 1H, J = 8.8 Гц), 10.88 (с, 1H).
Элементный анализ для C9H6BrNO3:
Вычислено: C 42.19 H 2.34 Br 31.25 N 5.47
Найдено: C 42.57 H 2.37 Br 31.30 N 5.42
Получение промежуточного соединения (15)
5-бром-6-метоксиацетилантранила (5-бром-2,6-диметил-4H-3,1-бензоксазин-4-она)
К раствору 2.28 г (8.9 ммолей) изатина (13) в 13.4 мл 2 N водного NaOH (26.7 ммолей) при охлаждении до 0oC при перемешивании магнитной мешалкой прибавляют постепенно 0.90 мл 30% H2O2 (8.9 ммолей), поддерживая температуру ниже 20oC. Течение реакции контролируют ТСХ (EtOAc/гексан, 40:60). Дополнительно вносят 0.20 мл 30% H2O2, . перемешивают 20 минут при комнатной температуре и наблюдают по ТСХ исчезновение исходного материала. Смесь подкисляют ледяной уксусной кислотой до pH 4 и концентрируют через криогенную ловушку при -78oC, удаляя сырой продукт 6-бром-5-метоксиантраниловую кислоту (14) в виде серого полукристаллического продукта, который обрабатывают 28 мл уксусного ангидрида, кипятят 40 минут, упаривают. Остаток обрабатывают избытком этилацетата:гексана (2:1), смесь нагревают, отфильтровывают через силикагель, удаляя нерастворившиеся частицы. Раствор частично упаривают, охлаждают и выделяют закристаллизовавшийся продукт (1.71 г) (71% по исходному изатину (13)). Т.пл. 228 - 229oC (разложение).
ИК (KBr) 3397, 2039, 1717, 1651, 1625, 1543, 1295, 1055, 881, 617 см-1, 1H-ЯМР (CДCl3) δ 2.42 (с, 3H), 3.98 (с, 3H), 7.34 (д, 1H, J = 8.9 Гц), 7.51 (д, 1H, J = 8.9 Гц).
Элементный анализ для C10H8BrNO3:
Вычислено: C 44.44 H 2.96 Br 29.62 N 5.19
Найдено: C 44.32 H 3.04 Br 29.53 N 5.09
Получение промежуточного соединения (16)
5-бром-3,4-дигидро-6-метокси-2-метил-хиназолин-4-она
К 1.25 г (4.6 ммолей) антранила (15), помещенного в сухую круглодонную колбу, снабженную конденсатором с сухим льдом, конденсируют приблизительно 50 мл безводного NH3. Смесь перемешивают магнитной мешалкой в течение 40 минут, затем конденсатор удаляют, NH3 позволяют испариться. После испарения добавляют 15 мл воды, 1.5 мл 2 N NaOH, кипятят 1 час, охлаждают до комнатной температуры, доводят pH до приблизительно 9 добавлением 1 N HCl, выпавший хиназолин белого цвета отфильтровывают, промывают водой, сушат и получают 0,71 г (57%). Т. пл. 273 - 274oC. ИК (KBr) 3189, 3074, 2990, 2974, 2899, 2362, 1676, 1643, 1552, 1461, 1303, 1286, 1063, 872, 832 см-1,
1H-ЯМР (CДCl3) δ 2.39 (с, 3H), 3.98 (с, 3H), 7.39 (д, 1H, J = 9.0 Гц), 7.59 (д, 1H, J = 9.0 Гц), 11,60 (с, 1H).
Элементный анализ для C10H9BrN2O2:
Вычислено: C 44.61 H 3.35 Br 29.74 N 10.41
Найдено: C 44.56 H 3.40 Br 29.63 N 10.36
Получение промежуточного соединения (17) (соединение 13A)
3,4-дигидро-6-метокси-2-метил-4-оксо-5-(4-пиридилтио) хиназолина
К 78 мг (0.7 ммолей) 4-меркаптопиридина прибавляют 34 мг (0.5 ммолей) твердого NaOH в 1 мл сухого ДМА, вносят 134 мг (0.5 ммолей) хиназолинона (16), растворенного в 2 мл сухого ДМА. Смесь выдерживают в атмосфере N2, вносят каталитическую смесь 44 мг CuBr и 22 мг Cu2O, перемешивают магнитной мешалкой при 135oC до окончания реакции (по ТСХ: безводный NH3/MeOH/CHCl3 - 0,5 : 4,5 : 9,5). Растворитель упаривают в высоком вакууме через криогенную ловушку при охлаждении до -78oC. Требуемый продукт выделяют хроматографией (безводный NH3/MeOH/CHCl3 - 0,5 : 4,5 : 9,5) на двуокиси кремния и получают 130 мг (89%) соединения (17) в виде порошка белого цвета. Т.пл. 248 - 249oC (разложение). ИК (KBr) 3358, 3073, 2933, 1682, 1634, 1574, 1475, 1462, 1318, 1277, 1059, 835, 710 см-1,
1H-ЯМР (CДCl3) δ 2.36 (с, 3H), 3.84 (с, 3H), 6.90 (д, 2H, J = 5.1 Гц), 7.48 (д, 1H, J = 9.1 Гц), 7.79 (д, 1H, J = 9.1 Гц), 8.28 (д, 2H, J = 5.1 Гц), 10.86 (с, 1H).
Элементный анализ для C15H13N3O2S:
Вычислено: C 60.18 H 4.38 N 14.04 S 10.71
Найдено: C 60.28 H 4.43 N 14.07 S 10.63
HRMS для C15H13N3O2S: вычислено 299.0730, найдено 299.0718.
Получение промежуточного соединения (18) (соединение 15А)
3,4-дигидро-6-гидрокси-2-метил-4-оксо-5-(4-пиридилтио) хиназолина
Для расщепления метилового эфира хиназолина (17) (100 мг, 0.30 ммолей) мягко в течение 8 часов кипятят с 2 мл смеси (1 : 1) 48% водной HBr и ледяной AcOH, затем упаривают растворитель в высоком вакууме с криогенной ловушкой при -78oC. Полученный остаток растворяют в 10% безводном NH3 в MeOH, хроматографируют на колонке с двуокисью кремния (безводный NH3/MeOH/CHCl3 0,5 : 4,5 : 9,5) и получают 62 мг соединения (18) в виде порошка белого цвета (65%), т. пл. 246 - 247oC (с разложением). ИК (KBr) 3450, 3240, 3073, 1667, 1634, 1580, 1464, 629 см-1, 1H ЯМР (CДCl3) δ 2.40 (с, 3H), 6.83 (д, 2H, J = 6.0 Гц), 7.40 (д, 1H, J = 9.0 Гц), 7.59 (д, 1H, J = 9.0 Гц), 8.20 (д, 2H, J = 6.0 Гц), 8.51 (c, 1H), 11.51 (c, 1H).
Элементный анализ для C14H11N3O2S:
Вычислено: C 58.94 H 3.13 N 11.97 S 11.22
Найдено: C 58.98 H 3.16 N 12.00 S 11.61
HRMS для C14H11N3O2S: вычислено 285.05733, найдено 285.05720.
Пример 3: Получение соединения 12А
Соединение 12А получают согласно реакционной схеме: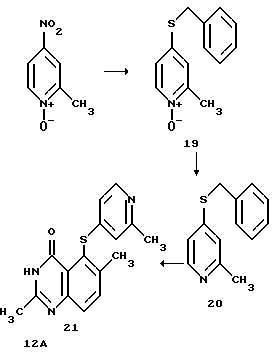
Получение соединения (19)
4-бензилтио-2-пиколин-N-оксида
Минеральное масло удаляют из гидрида калия (0.11 М, 35 вес.% дисперсия в минеральном масле) с помощью нескольких промываний с петролейным эфиром (5 x 50 мл). Оставшийся петролейный эфир удаляют в вакууме, к остатку прибавляют 350 мл безводного ТГФ, перемешивают, охлаждают до 0oC, прибавляют по каплям 14.1 мл (0.12 молей) бензилмеркаптана в течение 30 минут. Результирующую смесь молочно-белого цвета нагревают до комнатной температуры, дополнительно перемешивают 30 минут, охлаждают до - 30oC и вносят порциями 15.41 г (0.1 молей) 4-нитро-2-пиколин-N-окиси. Смесь окрашивается в темный оранжево-коричневый цвет, нагревают до комнатной температуры, кипятят 1 час, охлаждают до 0oC, вносят 50 мл воды, смесь подкисляют до pH приблизительно 6 2 М HCl, экстрагируют дихлорметаном (3 x 300 мл). Объединенный органический слой сушат (безводный Na2SO4), растворитель упаривают при уменьшенном давлении. Сырой остаток хроматографируют на силикагеле с MeOH/CH2Cl2 (градиент 3:97, 4: 96, 5:95). Выделяют чистый продукт (6.94 г, 30% выход) в виде желтовато-коричневого твердого вещества, т.пл. 98 - 99oC,
ИК (KBr) 3063, 3028, 1612, 1466, 1236, 831, 715, 675 см-1, 1H ЯМР (CДCl3) δ 2.45 (с, 3H), 4.16 (с, 2H), 6.97 (дд, 1H, J = 6.8, 2.7 Гц), 7.07 (д, 1H, J = 2.7 Гц), 7.32 (м, 5H), 8.09 (д, 1H, J = 6.8 Гц).
Элементный анализ для C13H13NOS:
Вычислено: C 67.50 H 5.66 N 6.05 S 13.86
Найдено: C 67.51 H 5.69 N 6.08 S 13.77
Получение промежуточного соединения (20)
4-бензилтио-2-пиколина
Соединение (19) (1.97 г, 8.5 ммолей) растворяют в 50 мл хлороформа. Раствор перемешивают, охлаждают до 0oC, вносят по каплям 1.75 мл (17.4 ммолей) треххлористого фосфора. После окончания добавления, реакционную смесь доводят до комнатной температуры, осторожно нагревают при температуре кипения (приблизительно 55oC) до тех пор, пока на ТСХ не исчезнет пятно П-окиси (MeOH/CH2Cl2 5:95). Раствор повторно охлаждают до 0oC, прибавляют при энергичном перемешивании 10 г льда, подщелачивают (до pH 8) осторожным добавлением 1 М NaOH. Органический слой отделяют. Водный слой экстрагируют дихлорметаном (3 x 50 мл). Органические слои объединяют, сушат (безводный Na2SO4), упаривают растворитель при уменьшенном давлении и получают масло, которое хроматографируют на короткой колонке с двуокисью кремния при использовании в качестве элюента MeOH/CH2Cl2, 3 : 97. Продукт выделяют в виде белого твердого вещества (1.54 г, 84% выход). Т.пл. 69 - 70oC, ИК (KBr) 3028, 3003, 2920, 1583, 1454, 864, 815, 719, 702 см-1,
1H ЯМР (CДCl3) δ 2.55 (с, 3H), 4.22 (с, 2H), 7.03 (м, 2H), 7.35 (м, 5H), 8.28 (д, 1H, J = 5.5 Гц),
Элементный анализ для C13H13NS:
Вычислено: C 72.52 H 6.08 N 6.50 S 14.90
Найдено: C 72.46 H 6.11 N 6.50 S 14.80
Получение соединения (21) (соединения 12А)
3,4-дигидро-2,6-диметил-4-оксо-5-[4-(2-пиколинилтио)]хиназолина
К раствору 5 мл NH3, сконденсированного в 5 мл ТГФ при -78oC, прибавляют 115 мг металлического натрия (5.0 ммолей). Раствор глубокого голубого цвета перемешивают 15 минут, прибавляют 1.0 г (4.65 ммолей) 4-бензилтио-2-пиколина (20), перемешивают 1 - 1/2 часа при 0oC. Растворитель упаривают в вакууме, к остатку прибавляют 10 мл безводного N,N-диметилацетамида, 0.5 г хиназолина (6) (2.0 ммолей) и 0.25 г бромида меди (I). Смесь нагревают 4 часа при 90oC, растворитель упаривают в вакууме, остаток обрабатывают 10 мл H2S/MeOH раствором (20 г/л). Нерастворимый CuS отфильтровывают, фильтрат упаривают досуха. Остаток хроматографируют на силикагеле с MeOH/CH2Cl (5 : 95) и получают 400 мг (84% от теоретического) желтовато-коричневого твердого вещества, т.пл. 225 - 227oC, ИК (KBr) 3480, 3160, 3053, 2960, 1670, 1630, 1590, 1460, 1298, 831 см-1,
1H ЯМР (ДМСО-d6) δ 2.28 (с, 6H), 2.36 (с, 3H), 6.60 (шир с, 1H), 6.80 (с, 1H), 7.60 (д, 1H, J = 8.4 Гц), 7.80 (д, 1H, J = 8.4 Гц).
Элементный анализ для C16H15N3OS • 0,5 H2O:
Вычислено: C 62.73 H 5.22 N 13.72 S 10.46
Найдено: C 63.08 H 5.20 N 13.73 S 10.50
HRMS для C16H15N3OS: вычислено 297.0936, найдено 297.0936.
Пример 4: Получение соединения 16А
Соединение 16А получают по следующей схеме реакций: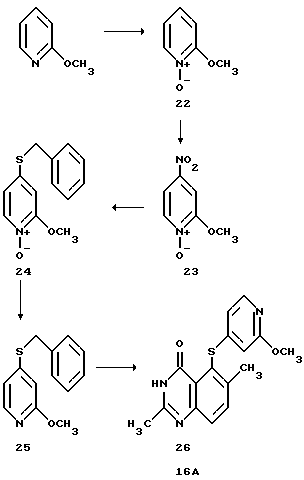
Получение промежуточного соединения (22) 2-Метоксипиридин-N-оксида
Это соединение, впервые полученное H.J.Den Hertog и M.Van Ammers, Rec. Trav. Chem. 1955, 74, 1160, синтезируют с помощью различных методик. К раствору 21.83.2-метоксипиридина (0.2 молей) в ледяной уксусной кислоте (80 мл) осторожно прибавляют 30% перекись водорода (20 мл), нагревают при перемешивании 3 часа при 80oC, охлаждают до комнатной температуры, дополнительно вносят 20 мл 30% H2O2, прозрачный раствор нагревают 12 часов при 80oC. Раствор упаривают до половины объема, прибавляют 100 мл воды, повторно упаривают, процесс повторяют 2 раза (2 х 100 мл H2O). Для удаления остатков воды и уксусной кислоты сироп выдерживают в вакууме и получают с количественным выходом твердый продукт белого цвета, который используют далее без дальнейшей очистки т.пл. 128-130oC, ИК (KBr) 3447, 1613, 1570, 1508, 1447, 1316, 1214, 1015, 764 см-1,
1H ЯМР (CДCl3) δ 4.05 (с, 3H), 6.91 (д, 1H, J = 8.0 Гц), 6.92 (м, 1H), 7.33 (дт, 2H, J = 8.0, 1.6 Гц), 8.3 (дд, 1H, J = 6.3, 1.6 Гц), HRMS для C6H7NO2: вычислено 125.0477, найдено 125.0474.
Получение промежуточного продукта (23)
2-метокси-4-нитропиридин-N-окиси
Нитрование проводится по методу Den Hertog и Van Ammers3. Результаты, полученные по этому эксперименту отличают от сообщенных. К концентрированной H2SO4(35 мл), охлажденной до 0oC, 15.3 г N-оксида (22) (0.12 мл) порциями осторожно прибавляют, затем при перемешивании при 0oC добавляют по каплям нитрующую смесь (концентрированная H2SO4, 35 мл: дымящая HNO3, 60 мл), баню со льдом удаляют, смесь нагревают 90 минут при 75oC. Смесь охлаждают до 0oC, осторожно выливают в 150 г льда, затем при энергичном перемешивании порциями прибавляют твердый K2CO3 до pH 7. Жидкую часть экстрагируют несколько раз CH2Cl2 (3 х 200 мл). Водный слой непрерывно экстрагируют CHCl3. Органические слои объединяют, сушат над безводным Na2SO4, упаривают и получают твердое вещество желтого цвета. Твердый остаток хроматографируют на колонке с двуокисью кремния при использовании системы MeOH/CH2Cl2 с градиентом 2:98, 3: 97, 4:96, 5:95. Смесь 2-метокси-4-нитропиридина и 2-метокси-5-нитропиридина (2.9 г) элюируется первой, затем следует 2-метокси-4-нитропиридин-N-окись (6.4 г) и затем 2-метокси-5-нитропиридин-N-окись (2.9 г). Соединение (23) получают в виже желтого твердого вещества (30%): т.пл. 176-178oC (с разложением), (по литературным данным: 154.5 - 158.5oC, (с разложением) 2, ИК (KBr) 3106, 3082, 1601, 1528, 1346, 1296, 1231, 1088, 1011, 660 см-1.
1H ЯМР (СДСl3) δ 4.18 (с, 3H), 7.73 (д, 1H, J = 2.9 Гц), 7.78 (дд, 1H, J = 7.1, 2.9 Гц), 8.35 (д, 1H, J = 7.1 Гц).
Элементный анализ для C6H6N2O4:
Вычислено: C 42.36 H 3.56 N 16.47
Найдено: C 42.42 H 3.57 N 16.41
Получение промежуточного продукта (24)
4-бензилтио-2-метоксипиридин-N-окиси
N-окись пиридина (24) получают по методике, аналогичной методике для соединения (19) со следующими изменениями. Как только прибавляют 4-нитро-2-метоксипиридин-N-окись, реакционной смеси позволяют нагреваться до комнатной температуры, непрерывно перемешивают 12 часов, выпавший твердый осадок отфильтровывают, промывают холодным ТГФ. Твердый продукт сушат в вакууме, он показывает одно пятно на ТСХ (MeOH/CH2Cl2, 10:90). Фильтрат упаривают, хроматографируют на двуокиси кремния MeOH/CH2Cl2 (градиент 4:96, 5:95, 6:94). Выделяют аналитически чистый желтовато-коричневый твердый продукт. Общий выход составляет 70%. Т. пл. 131-133oC, ИК (KBr) 3105, 3038, 3005, 1670, 1610, 1543, 1483, 1290, 1211, 1132, 1016, 802 см-1, 1H ЯМР (CДCl3) δ 3.95 (с, 3H), 4.19 (c, 2H), 6.64 (д, 1H, J = 2.4 Гц), 6.78 (дд, 1H, J = 6.9, 2.4 Гц), 7.33 (м, 5H), 8.09 (д, 1H, J = 6.9 Гц).
Элементный анализ для C13H13NO2S:
Вычислено: C 63.13 H 5.30 N 5.66 S 12.96
Найдено: C 62.88 H 5.28 N 5.62 S 12.89
Получение промежуточного соединения (25)
4-бензилтио-2-метоксипиридина
Исходную N-окись пиридина (24) (1.85 г) восстанавливают по методу, приведенному для соединения (20), за исключением того, что отсутствует необходимость нагревать смесь. Время реакции - приблизительно 90 минут. После хроматографии на двуокиси кремния при элюировании эфиром/петролейным эфиром 5: 95 получают 1.57 г (90%) соединения (25) в виде желтовато-коричневого твердого продукта. Т.пл. 35-36oC,
ИК (KBr) 3028, 2943, 1589, 1543, 1385, 1307, 1037, 715 см-1,
1H ЯМР (CДCl3) δ 3.98 (c, 3H), 4.23 (с, 2H), 6.64 (д, 1H, J= 1.6 Гц), 6.84 (дд, 1H, J = 5.9 и 1.6 Гц), 7.35 (м, 5H), 7.98 (д, 1H, J = 5.9 Гц).
Элементный анализ для C13H13NOS:
Вычислено: C 67.50 H 5.66 N 6.05 S 13.86
Найдено: C 67.60 H 5.70 N 6.10 S 13.80
Получение соединения (26) (соединения 16A)
3,4-дигидро-2,6-диметил-4-оксо-5-[4-(6-метоксипиридилтио)]хинозолина
Это соединение с выходом 6-7% получают по методике, приведенной для соединения (21) (соединение 12A). Получено желтовато-коричневое твердое вещество, т.пл. 223-226oC,
ИК (KBr) 3445, 1684, 1675, 1669, 1452, 1394, 1320, 1038 см-1,
1H ЯМР (ДMCO-d6) δ 2.28 (с, 3H), 2.35 (c, 3H), 3.70 (c, 3H), 6.05 (c, 1H), 6.49 (дд, 1H, J = 4.1, 2.9 Гц), 7.60 (д, 1H, J = 8.5 Гц), 7.78 (д., 1H, J = 8.4 Гц), 7.85 (д, 1H, J = 5.4 Гц), 12.10 (с, 1H). HRMS для C16H15N3O2S: вычислено 313.0885, найдено 313.0882.
Пример 5: Получение соединений 17A и 18A
Соединения 17A и 18A получают по следующей схеме реакций: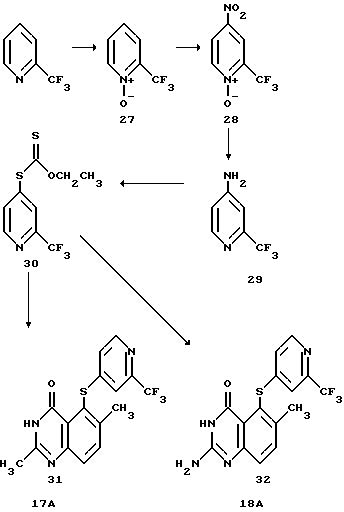
Получение промежуточного соединения (27)
2-трифторметилпиридин-N-окиси
По методике получения промежуточного продукта (22) синтезируют П-окись 2-трифторметилпиридина с 72% выходом из исходного 2-трифторметилпиридина. Соединение представляет собой желтое масло, ИК (чистый) 3125, 3085 1721, 1615, 1439, 1329, 1269, 1115, 1071, 1044, 852, 771, 662 см-1,
1H ЯМР (CДCl3) δ 7.38 (т, 1H, J= 7.9 Гц), 7.48 (дт, 1H, J = 7.0, 2.1 Гц), 7.71 (дд, 1H, J = 7.9, 2.1 Гц), 8.35 (д, 1H, J = 6.5 Гц).
Элементный анализ для C6H4F3NO•0,5 H2O
Вычислено: C 41.8 H 2.93 F 33.12 N 8.14
Найдено: C 41.84 H 2.81 F 33.19 N 8.26
Получение промежуточного соединения (28)
4-нитро-6-трифторметилпиридин-N-окиси
Нитрование пиридин-N-окиси (27) проводят по методике получения соединения (23) со следующими изменениями. Реакционную смесь нагревают при 125-130oC в течение 3-1/2 часов, при проведении процесса нет необходимости в постоянном экстрагировании водного слоя. Сырой твердый продукт очищают хроматографически на колонке с двуокисью кремния при элюировании этилацетатом/гексаном, 20:80. Продукт выделяют в виде желтого твердого вещества (т. пл. 112-114oC) с 38% выходом.
ИК (KBr) 3416, 3125, 1620, 1591, 1537, 1449, 1354, 1306, 1281, 1165, 1130, 916, 693 см-1.
1H ЯМР (CДCl3) δ 8.28 (дд, 1H, J = 7.2, 3.1 Гц), 8.36 (д, 1H, J = 7.2 Гц), 8.52 (д, 1H, J = 3.1 Гц).
Элементный анализ для C6H3F3N2O3:
Вычислено: C 34.63 H 1.45 F 27.39 N 13.46
Найдено: C 34.86 H 1.35 F 27.16 N 13.66
Получение промежуточного соединения (29) 4-амино-6-трифторметилпиридина
В аппарат гидрирования Парра помещают 8.32 г нитропиридин N-окиси (28) (0.04 молей) в 275 мл 95% этанола, аппарат наполняют аргоном, вносят 0.83 г 10% палладия на активированном углероде. Аппарат встряхивают при 35 фунт/дюйм2 в течение 45 минут на гидрогенизаторе Парра, затем катализатор отфильтровывают через прокладку целита. Фильтрат упаривают в вакууме, оставшееся масло растворяют в 50 мл дихлорметана. Раствор фильтруют через небольшую прокладку силикагеля для удаления следов катализатора и углерода. Фильтрат упаривают, следы растворителя удаляют в вакууме. Масло немедленно кристаллизуется и получают 5.77 г (89% выход) аналитически чистого светло-оранжевого твердого продукта. Т.пл. 56 - 58oC, ИК (KBr) 3501, 3335, 3175, 1657, 1611, 1472, 1373, 1300, 1169, 1117, 993, 850 см-1, 1H ЯМР δ 4.40 (шир с, 2H), 6.64 (дд, 1H, J = 5.6, 2.3 Гц), 6.89 (д, 1H, J = 2.3 Гц), 8.30 (д, 1H, J = 5.6 Гц).
Элементный анализ для C6H5F3N2:
Вычислено: 44.45 H 3.11 F 35.16 N 17.28
Найдено: 44.56 H 2.95 F 35.14 N 17.28
HRMS для C6H5F3N2: вычислено 162.0405, найдено 162.0402.
Получение промежуточного соединения (30)
этил-4-(6-трифторметилпиридил)ксантогената
Раствор 4.86 г амина (29) (0.03 молей) в 30 мл концентрированной H2SO4 охлаждают до 0oC и прибавляют по каплям в течение 15 минут охлажденный до 0oC водный раствор (30 мл воды) 2.69 г NaNO2 (39 ммолей). Смесь коричневого цвета непрерывно перемешивают 5 минут при этой температуре, вносят по каплям охлажденный льдом раствор 8.17 г этилоксантогената калия (51.0 ммолей) в 30 мл воды, температуру реакции при этом поддерживают 0 - 5oC. Смесь нагревают до комнатной температуры, прибавляют дихлорметан (125 мл). Водный слой нейтрализуют до pH 7 твердым NaCO3. Органический слой отделяют, водный слой экстрагируют этилацетатом (3 х 50 мл). Органические слои объединяют, сушат над безводным Na2SO4, упаривают. Остаток хроматографируют при использовании колонки с силикагелем при элюировании системой растворителей этилацетат/гексан с градиентом (2:98, 2,5: 97,5, 3:97). Соединение (30) выделяют в виде желтого масла с 36% выходом и используют без дальнейшей очистки. ИК (в частом виде) 3061, 2988, 2901, 1738, 1584, 1555, 1406, 1323, 1252, 1184, 1146, 1038, 845, 720 см-1,
1H ЯМР (CДCl3) δ 1.38 (т, 3H, J = 7.1 Гц), 4.66 (кв. 2H, J = 7.1 Гц), 7.60 (дд, 1H, J = 5.0, 1.3 Гц), 7.83 (д, 1H, J = 1.0 Гц), 8.77 (д, 1H, J = 5.00 Гц),
HRMS для C9H8F3NOS2 (М+1): вычислено 268,0077, найдено: 268,0065.
Получение соединения 31 (соединение 17A)
3,4-дигидро-2,6-диметил-4-оксо-5[4(6-трифторметилпиридилтио)]-хиназолина
К раствору 0.67 г ксантогената (30) (2.5 ммолей) в 3 мл MeOH прибавляют 2.5 мл 1 N КОН в метаноле, смесь перемешивают 1-1/2 часа, упаривают досуха, к остатку прибавляют 10 мл безводного N,N-диетилацетата, 0.25 г хиназолина (6) (10,0 ммолей), 0.1 г бромида меди (1) и 0,1 г окиси меди (1). Смесь нагревают 6 часов при 90oC, упаривают растворитель. Твердый продукт обрабатывают 50 мл H2S/MeOH раствором (20 г/л) в течение 1 часа. Смесь фильтруют, фильтрат упаривают досуха. Остаток очищают хроматографически на двуокиси кремния при элюировании MeOH/CH2Cl2 (5:95) и получают 65 мг (18,5% от теоретического) твердого вещества желтого цвета: т.пл. 240-245oC, ИК (KBr) 3440, 3190, 3057, 2950, 1675, 1630, 1595, 1321, 1140, 720 см-1
1H ЯМР (ДМСО-d6) δ 2.28 (с, 3H), 2.42 (с, 3H), 6.97 (д, 1H, J = 5.2 Гц), 7.46 (д, 1H, J = 1.1 Гц), 7.67 (д, 1H, J = 8.4 Гц), 7.84 (д, 1H, J = 8.4 Гц), 8.37 (д, 1H, J = 5.2 Гц), 12.05 (шир с, 1H).
HRMS для C16H12F3N3O:
вычислено 351.0656, найдено 351.0653.
Получение соединения 32 (соединения 18А)
2-амино-3,4-дигидро-6-метил-4-оксо-5-[4-(6-трифторметилпиридилтио)] -хиназолина
Соединение получают с 22% выходом по методике, приведенной выше. Желтовато-коричневое твердое вещество, т. пл. 247 - 249oC, ИК (KBr) 3421, 2056, 1650, 1625, 1485, 1419, 1328, 1146, 815, 724 см-1, 1H ЯМР (ДМСО-d6) δ 2,30 (с, 3H), 6.50 (шир с, 2H), 6.7 (дд, 1H, J = 4.1, 1.2 Гц), 7.30 (д, 1H, J = 8.4 Гц), 7.39 (д, 1H, J = 1.0 Гц), 7.62 (д, 1H, J = 8.6 Гц), 8.36 (д, 1H, J = 5.2 Гц), 12.10 (шир с, 1H). HRMS для C15H11F3N4OS (M+1): вычислено 353.0677, найдено 353.0684.
Пример 6: Получение соединения 26A
Соединение 26A получают по следующей схеме реакций: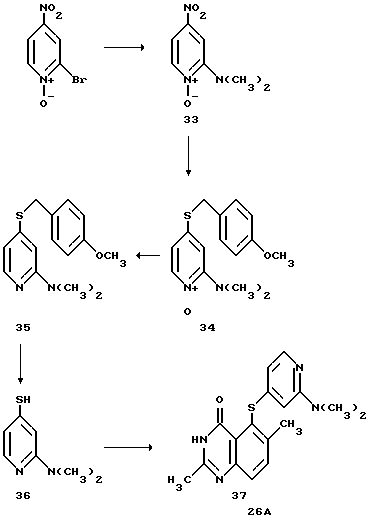
Получение промежуточного соединения (33)
6-диметиламино-4-нитропиридин-N-окиси
К раствору 5.0 г (23 ммолей) 2-бром-4-нитропиридин-N -окиси4 в 75 мл тетрагидрофурана прибавляют 1.1 г (24 ммолей) диметиламина, перемешивают 3 часа, отфильтровывают гидробромид диметиламина. Фильтрат упаривают, хроматографируют на колонке с двуокисью кремния при элюировании метанолом/дихлорметаном 4:96. Продукт выделяют в виде твердого вещества оранжевого цвета (т.пл. 128-130oC), с 83% выходом.
1ЯМР (CДCl3) δ 3.14 (с, 6H), 7.67 (с, 2H), 8.24 (д, 1H, J = 7. Гц). Элементный анализ для C7H9N3O3:
Вычислено: C 45.60 H 4.95 N 22.94
Найдено: C 46.00 H 5.00 N 22.96
Получение промежуточного соединения (34)
6-диметиламино-4-(4-метоксибензилтио)-пиридин-N-окиси
К раствору 1.1 г (7.1 ммолей) 4-метокси- α-толуолтиола в 75 мл безводного ДМФА прибавляют 0.28 г (7,0 ммолей, 60 вес.% дисперсия в минеральном масле), перемешивают 1 час, добавляют по каплям 1.2 г (6.55 ммолей) пиридин-N-окиси 33 в 25 мл безводного ДМФА, перемешивают 2 часа, выливают в 200 мл воды. Водный раствор экстрагируют 500 мл диэтилового эфира, разделяют слои. Эфирный слой сушат над безводным MgSO4, эфир упаривают и получают соединение 34 в виде желтовато-коричневого твердого вещества с выходом 63%.
1H ЯМР (CДCl3) δ 3.06 (с, 6H), 3.83 с, 3H), 4.16 (с, 2H), 6.60 (д, 1H, J = 2.5 Гц), 6.70 (д, 1H, J = 7.0 Гц), 6.90 (д, 2H, J = 8.7 Гц), 7.30 (д, 2H, J = 8.7 Гц), 8.0 (д, 1H, J = 7.0 Гц).
Получение промежуточного продукта (35)
6-диметиламино-4-(4-метоксибензилтио)пиридина
Исходную N-окись пиридина 34 (0.60 г, 2.07 ммолей) восстанавливают по методу получения соединения 25. После окончания реакции смесь выливают в 200 мл воды, pH доводят до 7. Водный раствор экстрагируют этилацетатом (500 мл), органический слой сушат над безводным Na2SO4, отфильтровывают и упаривают. Выделяют без хроматографической очистки продукт 35 в виде твердого вещества желтого цвета с 88% выходом.
1H ЯМР (CДCl3) δ 3.08 (с, 3H), 3.82 (с, 3H), 4.17 (с, 2H), 6.34 (д, 2H, J = 1.3 Гц), 6.48 (д, 1H, J = 5.5 Гц), 6.87 (д, 2H, J = 8.7 Гц), 7.33 (д, 2H, J = 8.7 Гц), 8.0 (д, 1H, J = 5.5 Гц).
Получение промежуточного соединения (36)
6-диметиламино-4-меркаптопиридина
К раствору пиридина (35) (0.40 г, 1.46 ммолей) в муравьиной кислоте (10 мл) при 0oC прибавляют 1.2 г Hg(OAc)2 в 3 мл воды, удаляют баню со льдом, перемешивают 12 часов, значение pH регулируют добавлением водного аммиака. Образовавшийся серый осадок отфильтровывают, промывают избыток воды, сушат на воздухе, обрабатывают насыщенным раствором H2S /метанолом. Образовавшийся черный твердый осадок (HgS) отфильтровывают, фильтрат упаривают досуха и получают твердое вещество желтого цвета (89%), которое используют далее без дальнейшей очистки.
1H ЯМР (CДCl3) δ 3.12 (с, 3H), 3.5 (шир с, 1H), 6.5 (д, 1H, J = 5.3 Гц), 6.57 (д, 1H, J = 3.6 Гц), 7.69 (д, 1H, J = 5.5 Гц).
Получение соединения 37 (соединение 26A)
3,4-дигидро-2,6-диметил-4-оксо-5-[4-(6-диметиламинопиридилтио)] -хиназолина
Это соединение получают из промежуточных продуктов 6 и 36 при использовании методики получения соединения 9 (8A). Сырой продукт хроматографируют на колонке с силикагелем при использовании MeOH/CH2Cl2 (8:91) и получают желтовато-коричневый продукт с 21% выходом.
1H ЯМР (ДМСО-d6) δ 1.97 (с, 3H), 2.08 (с, 3H), 2.56 (с, 3H), 5.55 (д, 1H, J = 5.4 Гц), 5.82 (д, 1H, J = 1.2 Гц), 7.27 (д, 1H, J = 8.4 Гц), 7.44 (д, 1H, J = 5.2 Гц), 7.45 (д, 2H, J = 8.4 Гц), 12.75 (шир с, 1H).
Элементный анализ для C17H18N4OS • 0,5 H2O:
Вычислено: C 60.82 H 5.66 N 16.69 S 9.54
Найдено: C 61.01 H 5.63 N 16.55 S 9.42
Пример 7: Получение соединений 27A и 28A
Соединение 27A и 28A получают по схеме реакций:
Получение промежуточного соединения (38)
Метил-4-[3,4-дигидро-2,6-диметил-4-оксо-5-хиназолинилил)тио]бензоат
Это соединение получают из промежуточного продукта 6 и метил-4-меркаптобезоата [(P. R.Marsham et al., J.Med. Chem., 34, 2209 (1991), и E. Campaigne, et al., J. Org. Chem., 27. 2835 (1962)/ по методике, предложенной для синтеза соединения 9 (8A). После нагревания смеси при 90oC в течение 16 часов, упаривают в вакууме ДМФА, твердый остаток суспендируют в метаноле. Через суспензию при перемешивании медленно пропускают газообразный H2S в течение приблизительно 5 минут. Темный осадок (CuS), который образовывается при этом отфильтровывают. Фильтрат упаривают, остаток очищают хроматографически на колонке с двуокисью кремния при элюировании метанол/дихлорметаном, 5:95 и получают с 85% выходом желтовато-коричневое твердое вещество.
1H ЯМР (ДМСО-d6) δ 2.26 (c, 3H), 2.43 (с, 3H), 3.75 (с, 3H), 6.94 (д, 2H, J=8.4 Гц), 7.22 (д, 1H, J=8.5 Гц), 7.52 (д, 1H, J=8.5 Гц), 7.71 (д, 2H, J= 8.4 Гц), 11.7 (шир с, 1H). HRMS для C18H16N2O3S: вычислено 340.0898, найдено 340.0882.
Получение соединения (39) (соединение 28A)
4-[(3,4-дигидро-2,6-диметил-4-оксо-5-хиназолинил)тио]бензойной кислоты
Этанольный раствор (5 мл), содержащий 0.186 г (0.55 ммолей) метилового эфира 38 и 0.5 мл водного 1 N NaOH нагревают 4 часа при 50oC, упаривают досуха, полученную натриевую соль растворяют в 3 мл воды. Раствор осторожно подкисляют до pH 4 концентрированной HCl. Свободную кислоту отфильтровывают, промывают 5 мл холодной воды. Твердый продукт сушат в эксикаторе над CaSO4 и получают 0.15 г (84%) кислоты 39 (28A) в виде твердого вещества бежевого цвета.
1H ЯМР (ДМСО-d6) δ 2.29 (с, 3H), 2.45 (с, 3H), 7.0 (д, 2H, J=8.5 Гц), 7.44 (д, 1H, J=8.6 Гц), 7.71 (д, 2H, J=8.4 Гц), 7.74 (д, 1H, J=8.3 Гц), HRMS для C17H14N2O3S: вычислено 326.0742, найдено 326.0725.
Получение промежуточного соединения (40)
диэтил-N-[4-((3,4-дигидро-2,6-диметил-4-оксо-5-хиназолинил)тио) бензоил] -L-глутамата
К раствору бензойной кислоты 39 (60.0 мг, 18.4 ммолей) и гидрохлорида диэтилового эфира глутаминовой кислоты (0.144 г, 0.6 ммолей) в 5 мл безводного ДМФА при 0oC при перемешивании прибавляют дифенилфосфорилазид (0.15 мл, 0.7 ммолей), выдерживают 15 минут, прибавляют 0.2 мл (1.4 ммолей) триэтиламина, перемешивают 12 часов при комнатной температуре, растворитель упаривают в вакууме. Твердый остаток обрабатывают 5 мл воды, значение pH осторожным добавлением концентрированной HCl доводят до 6, водный раствор экстрагируют CHCl3 (3 х 10 мл). Органические слои объединяют, сушат над MgSO4. фильтруют, упаривают досуха. Продукт очищают хроматографически на двуокиси кремния при элюировании метанол/дихлорметаном, 10:90. Выделяют твердый продукт желтовато-коричневого цвета (78.0 мг, 82%).
1H ЯМР (ДМСО-d6) δ 1.11 (м, 6H), 1.61 (м, 2H), 1.79 (м, 2H) 2.26 (с, 3H), 2.37 (с, 3H), 3.26 (м, 1H), 4.05 (м, 4H), 6.96 (д, 2H, J=8.4 Гц), 7.55 (д, 1H, J=8.5 Гц), 7.64 (д, 2H, J=8.4 Гц), 7.71 (д, 1H, J=8.5 Гц), 8.60 (д, 1H, J=5.3 Гц), 12.10 (шир с, 1H), HRMS для C26H29N3O6S (M+1):
вычислено 512.1843, найдено 512.1855.
Получение соединения (41) соединение 27A)
N-[4-((3,4-дигидро-2,6-диметил-4-оксо-5-хиназолинил)тио)бензоил] -L-глутаминовой кислоты
К диэтиловому эфиру 40 (78.0 мг, 0.15 ммолей) в 5 мл этанола прибавляют 0.5 мл водного 1 N NaOH, перемешивают 3 часа при 50oC до исчезновения на ТСХ исходного соединения, затем раствор упаривают досуха. Динатриевую соль растворяют в 2 мл воды, подкисляют до pH 4 концентрированной HCl, отфильтровывают образовавшийся осадок, промывают его 5 мл холодной воды, сушат над CaSO4 и получают 50 мг (72%) не совсем белого твердого вещества. 1H ЯМР (ДМСО-d6) δ 1.95 (м, 2H), 2.05 (м, 2H), 2.26 (с, 3H), 2.46 (с, 3H), 4.40 (м, 1H), 6,93 (д, 2H, J=8.4 Гц), 7.55 (д, 1H, J=8.4 Гц), 7.65 (д, 2H, J=8.5 Гц), 7.71 (д, 1H, J=8.40 Гц), 8.40 (шир д, 1H, J=5.4 Гц), 12.00 (шир с, 1H).
Элементный анализ для C22H21N3O6S • 2HCl
Вычислено: C 50.05 H 4.36 N 7.96 S 6.06
Найдено: C 50.38 H 4.69 N 7.60 S 5.77
Пример 8: Получение соединений 3A и 5A
Соединения 3A и 5A получают по схеме реакций:
Получение промежуточного соединения (42)
3,4-дигидро-2,5-диметил-4-оксохиназолинона
Это соединение получают через соответствующий бензоксазинон из 6-метилантраниловой кислоты при использовании методики получения хиназолинона (6). Твердый продукт перекристаллизовывают из этанола (т.пл. 258-259oC).
1H ЯМР (CДCl3) δ 2.53 (с, 3H), 2.89 (с, 3H), 7.20 (д, 1H, J=7.3 Гц), 7.50 (д, 1H, J=8.0 Гц), 7.59 (дд, 1H, J=8.1, 7.3 Гц), 11.52 (шир с, 2H).
Элементный анализ для C10H10N2OS:
Вычислено: C 68.95 H 5.79 N 16.08
Найдено: C 69.03 H 5.82 N 16.03
Получение промежуточного соединения (43)
2,5-диметил-3-[2'-(триметилсилил)этоксиметил]-хиназолин-4-она
К 70 мл сухого ДМФА прибавляют 2.175 г (12.5 ммолей) хиназолинона (42), охлаждают до 0oC, вносят порциями при перемешивании 0.55 н NaH (13.75 ммолей, 60% дисперсия в масле), смесь окрашивается в зеленый цвет, затем позволяют смеси нагреться до комнатной температуры, перемешивают до окончания выделения газа (H2). Раствор повторно охлаждают до 0oC, прибавляют по каплям 2-(триметилсилил)этоксиметилхлорид (SEM-CI) (2.45 мл, 13.75 ммолей) и наблюдают образование мути (NaCl). После добавления всего SEM-Cl баню со льдом удаляют, реакционную смесь перемешивают 12 часов при комнатной температуре, выливают в воду (300 мл), экстрагируют гексаном (3х150 мл). Органические слои объединяют, сушат над безводным MgSO4, фильтруют и частично упаривают. Образующийся белый осадок отфильтровывают и с помощью ТСХ и 1H ЯМР показывают, что это исходное вещество (42). Фильтрат упаривают и получают масло светло-желтого цвета, которое пропускают через хроматографическую колонку с силикагелем при элюировании смесью диэтиловый эфир/петролейный эфир, 1:1, и получают 3.0 г (79%) продукта (43) в виде масла. ИК (в частом виде) 2980, 1675, 1600, 1572, 1460, 1380, 1287, 1248, 1075, 858, 835 см-1.
1H ЯМР (CДCl3) δ 0.00 (с, 9H), 0.95 (дд, 2H, J=8.2, 7.2 Гц), 2.66 (с, 3H), 2.84 (с, 3H), 3.68 (дд, 2H, J=8.2, 7.1 Гц), 5.52 (с, 2H), 7.18 (д, 1H, J=6.8 Гц), 7.43 (дд, 1H, J=8.0, 7.3 Гц), 7.55 (дд, 1H, J=8.0, 7.5 Гц).
Получение промежуточного соединения (44)
5-бромметил-2-метил-3-[2'-(триметилсилил)этоксиметил] хиназолин-4-она
К раствору защищенного SEM хиназолинона (43) (2.28 г, 7.5 ммолей) в 30 мл CCl4 прибавляют 1.47 г (8.23 ммолей) N-бромсукцинимида, светло-желтый раствор мягко нагревают до кипения получения гомогенной массы, затем начинают под действием лампы 200 Вт реакцию бензилбромирования, наблюдают окрашивание в темно-оранжевый цвет, выдерживают приблизительно 15 минут, цвет постепенно исчезает и выпадает сукцинимид. Реакцию охлаждают, фильтруют, промывают 25 мл CCl4. Фильтрат промывают минимальным количеством воды (приблизительно 5 мл), разделяют, сушат над MgSO4, повторно фильтруют и упаривают с удалением образующийся в процессе упаривания твердый остаток, который далее очищают на хроматографической колонке с двуокисью кремния при элюировании системой диэтиловый эфир/петролейный эфир с градиентом 15/85, 20/80, 25/75, 30/70, 35/65. Чистый бромид (1.25 г) выделяют в виде белого твердого продукта с выходом 43% (54% по превращению (43): т.пл. 78-80oC, ИК (KBr) 3085, 2980, 1675, 1608, 1382, 1340, 1293, 1248, 1075, 860, 830, 710 см-1.
1H ЯМР (CДCl3) δ 0.00 (с, 9H), 0.93 (дд, 2H, J=8.3, 7.1 Гц), 2.66 (с, 3H), 3.68 (дд, 2H, J=8.4, 7.1 Гц), 5.24 (с, 2H), 5.55 (с, 2H), 7.38 (дд, 1H, J=7.2, 1.5 Гц), 7.55 (дд, 1H, J=7.5, 1.5 Гц), 7.61 (дд, 1H, J=7.5, 7.2 Гц).
Элементный анализ для C16H23BrN2O2Si:
Вычислено: C 50.12 H 6.04 Br 20.84 N 7.30
Найдено: C 50.35 H 6.06 Br 21.01 N 7.32
Получение промежуточного соединения (45)
5-хлор-N-[2-метил-3'-(2''-триметилсилил)этоксиметил)-4'- оксо-5-хиназолил)метил]индола
К 0.417 г (2.75 ммолей) 5-хлориндола в 6.5 мл безводного ДМФА при 0oC при перемешивании прибавляют порциями 0.11 г (2.75 ммолей, 60% дисперсия в масле) NaH. Как только образуется анион (приблизительно 30 минут), вносят 0.958 г (2.5 ммолей) бромметилхиназолина (44), растворенного в 0.5 мл безводного ДМФА. Окончание реакции фиксируют по исчезновению пятна исходного материала на ТСХ (40% эфир/петролейный эфир). Добавляют лед для того, чтобы остановить образование избытка аниона, затем вносят 20 мл воды, экстрагируют диэтиловым эфиром ( 3 х 50 мл). Органические слои объединяют сушат, над MgSO4, фильтруют, упаривают, остаток очищают на хроматографической колонке с двуокисью кремния при элюировании смесью диэтиловый эфир/петролейный эфир, 40: 60. Кристаллический твердый продукт белого цвета выделяют (0.927 г, 82%, т. пл, 98-99oC). ИК (KBr) 3095, 2980, 1715, 1595, 1565, 1440, 1345, 1280, 1245, 1175, 1060, 932, 834, 795, 755, 720, 612 см-1,
1H ЯМР (CДCl3) δ 0.03 (с, 9H), 1.00 (м, 2H), 2.71 (с, 3H), 3.74 (м, 2H), 5.55 (с, 2H), 6.04 (с, 2H), 6.25 (дд, 1H, J=7.4, 1.3 Гц), 6.54 (дд, 1H, J= 3.1, 0.3 Гц), 7.08 (м, 2H), 7.18 (д, 1H, J=3.1 Гц), 7.45 (м, 2H), 7.63 (дд, 1H, J=1.6, 1.0 Гц). Элементный анализ для C24H28ClN3O2Si:
Вычислено: C 63.48 H 6.21 Cl 7.80 N 9.25
Найдено: C 63.41 H 6.13 Cl 7.91 N 9.19
Получение соединения (46) (соединение 3A)
5-хлор-N-[3,4-дигидро-2-метил-4-оксо-5-хиназолил)метил]индола
К защищенному SEM хиназолину (45) (0.75 г, 1.65 ммолей) в 1.5 мл ТГФ прибавляют 6.0 мл 1.0 М ТГФ раствора тетрабутиламмоний фторида, нагревают при перемешивании 7 часов при 50oC, охлаждают до комнатной температуры, прибавляют 20 мл воды, экстрагируют большим избытком (200 мл) этилацетата. Органический слой отделяют, сушат над MgSO4, фильтруют, упаривают, удаляют образующийся в процессе упаривания остаток, который перекристаллизовывают из этилацетата и получают продукт (46) (соединение 3A) с 46% выходом. Т.пл. 251-252oC.
1H ЯМР (ДМСО-d6) δ 2.34 (с, 3H), 6.08 (с, 2H), 6.14 (дд, 1H, J=7.3, 1.0 Гц), 6.53 (дд, 1H, J=3.0, 0.5 Гц), 7.05 (дд, 1H, J=8.7, 2.1 Гц), 7.34 (д, 1H, J=8.8 Гц), 7.46 (м, 2H), 7.56 (д, 1H, J = 3.1 Гц), 7.64 (д, 1H, J = 2.1 Гц), 12.30 (шир с, 1H).
Элементный анализ для C18H14ClN3O • 0,1 EtOAc:
Вычислено: C 66.45 H 4.49 Cl 10.66 N 12.63
Найдено: C 66.68 H 4.62 Cl 10.95 N 12.27
HRMS для C18H14ClN3O: вычислено 323.0825, найдено 323.0813.
Получение промежуточного соединения (47)
5-формил-2-метил-3-/[2'-(триметилсилил)этоксиметил]хиназолин- 4-она
К раствору NaOEt в этаноле, полученному при растворении 34.5 мг (1.5 ммолей) натрия в 1.5 мл абсолютного этанола, прибавляют 0.14 мл (1.56 ммолей) 2-нитропропана, прибавляют 0.575 г (1.5 ммолей) бромметилхиназолина (44), нагревают при перемешивании 12 часов при 40oC, добавляют 10 мл воды, смесь экстрагируют диэтиловым эфиром (250 мл). Органические слои отделяют, сушат над безводным Na2SO4, фильтруют, упаривают. Остаток очищают хроматографически при использовании смеси 70% эфира/петролейный эфир и получают 0.346 г (73%) альдегида (47) в виде белого твердого вещества.
1H ЯМР (CДCl3) δ 0.00 (с, 9H), 0.96 (м, 2H), 2.73 (с, 3H), 3.75 (м, 2H), 5.58 (с, 2H), 7.82 (м, 2H), 7.87 (м, 1H), 11.7 (с, 1H).
Получение промежуточного соединения (48)
5-( α -гидрокситолил)-2-метил-3[2'-(триметилсилил)- этоксиметил]хиназолин-4-она
К раствору альдегида (47) (0.72 г, 2.26 ммолей) в 9.0 мл безводного ТГФ в атмосфере аргона при перемешивании при -78oC прибавляют по каплям фенилмагний бромид (0.33 мл 3.0 М в диэтиловом эфире), оставляют до нагревания смеси до комнатной температуры, перемешивают 1 час, для остановки реакции вносят 10 мл насыщенного водного NH4Cl. Смесь экстрагируют диэтиловым эфиром (3 x 50 мл), разделяют, объединяют органические слои, сушат над безводным MgSO4, фильтруют и упаривают, очищают хроматографически на колонке с двуокисью кремния при элюировании смесью эфир/петролейный эфир, 60:40 и получают 0.621 г бензилового спирта (48) в виде бесцветного масла с 74% выходом. ИК (в чистом виде) 3380, 3070, 3035, 2960, 2900, 1660, 1600, 1540, 1445, 1245, 1135, 1075, 915, 830, 695 см-1,
1H ЯМР (CДCl3) δ 0.00 (с, 9H), 0.88 (м, 2H), 2.68 (с, 3H), 3.54 (м, 2H), 5.50 (с, 2H), 5.77 (д, 1H, J = 8.3 Гц), 6.36 (д, 1H, J = 8.0 Гц), 7.25 (м, 4H), 7.30 (м, 2H), 7.60 (дд, 1H, J = 8.2, 1.5 Гц), 6.67 (дд, 1H, J = 7.7, 7.2 Гц).
Элементный анализ для C22H28N2O3Si:
Вычислено: C 66.63 H 7.11 N 7.06
Найдено: C 66.66 H 6.97 N 7.00
Получение соединения (49) (соединение 5A)
3,4-дигидро-2-метил-4-оксо-5-(α -гидрокситолил)-хиназолина
Используют тот же процесс, что и при получении соединения (46) (соединения 3A), у SEM-хиназолина (48) снимают защитную группу при использовании 3,0 эквивалентов тетрабутиламмоний фторида, реакцию с которым проводят 4 часа при 50oC. Хиназолин (49) (соединение 5A) выделяют в виде белого твердого вещества с выходом 38% после очистки хроматографией на двуокиси кремния при элюировании метанол/дихлорметаном, 5:95. 1H ЯМР (ДМСО-d6) δ 2.29 (с, 3H), 5.93 (д, 1H, J = 5.1 Гц), 7.15 (м, 1H), 7.21 (м, 3H), 7.27 (м, 2H), 7.45 (дд, 1H, J = 5.6, 3.9 Гц), 7.73 (м, 2H), 12.06 (шир с, 1H).
Пример 9: Получение соединений 4A и 6A
Соединения 4A и 6A получают по схеме реакций:
Получение промежуточного соединения (50)
5-бензоил-2-метил-3-[2'-(триметилсилил)этоксиметил]хиназолин- 4-она
Бензиловый спирт (48) (0,569 г, 1,43 ммолей) в 18 мл сухого CH2Cl2 перемешивают в инертной атмосфере, вносят активированный MnO2 (1,43 г), за течением реакции наблюдают ТСХ (эфир/петролейный эфир, 70/30). После исчезновения исходного материала, черную смесь фильтруют через прокладку целита, который затем промывают тщательно CH2Cl2 (100 мл). Фильтрат сушат над безводным Na2SO4, фильтруют, упаривают и получают 0,48 г (85%) белого твердого вещества как аналитически чистого. Т.пл. 124-125oC, ИК (KBr) 3010, 2955, 2900, 1660, 1560, 1440, 1345, 1292, 1245, 1178, 1060, 920, 825, 685 см-1,
1H ЯМР (CДCl3) δ -0.10 (с, 9H), 0.82 (м, 2H), 2.70 (с, 3H), 3.48 (м, 2H), 5.42 (с, 2H), 7.31 (дд, 1H, J = 7.1, 1.3 Гц), 7.39 (м, 2H), 7.51 (м, 1H), 7.72 (м, 1H), 7.74 (м, 2H), 7.80 (дд, 1H, J = 8.2, 7.1 Гц). Элементный анализ для C22H26N2O3Si:
Вычислено: C 66.97 H 6.64 N 7.10
Найдено: C 66.76 H 6.52 N 6.95
Получение соединения (51) (соединения 4A)
5-бензоил-3,4-дигидро-2-метил-4-оксо-хиназолина
К смеси (7 мл) 1: 1 ТГФ: 2 N HCl прибавляют защищенный хиназолин (5) (0.255 г, 0.64 ммолей), кипятят сначала до гомогенности, затем нагревают в течение 3 часов до образования белого осадка. Смесь охлаждают до комнатной температуры, вносят 10 мл холодной воды, вносят при энергичном перемешивании избыток насыщенного водного NaHCO3. Твердый продукт отфильтровывают, промывают тщательно холодной водой (2 x 10 мл), сушат в вакууме над активированным силикагелевым осушителем и получают аналитически чистый твердый продукт белого цвета (0,14 г) с 83% выходом, т.пл. 288-289oC,
ИК (KBr) 3175, 3030, 2880, 1660, 1635, 1325, 1265, 880, 825, 780, 725 см-1, 1H ЯМР (ДМСО-d6) δ 2.35 (с, 3H), 7.30 (дд, 1H, J = 7.3, 1.0 Гц), 7.44 (м, 2H), 7.58 (м, 3H), 7.71 (дд, 1H, J = 8.3, 1.0 Гц), 7.85 (дд, 1H, J = 8.1, 7.3 Гц), 12.22 (шир с, 1H). Элементный анализ для C16H12N2O2:
Вычислено: C 72.71 H 4.57 N 10.60
Найдено: C 72.61 H 4.70 N 10.39
Получение соединения (52) (соединение 6A)
3,4-дигидро-5-(α -дифенил-гидроксиметил)-2-метил-4-оксо- хиназолина
79.3 мг (0.3 ммолей) кетона (51) (соединения 4A) суспендируют в атмосфере аргона в 5.0 мл безводного ТГФ, затем добавляют по каплям при перемешивании при 0oC 0.375 мл фениллития (2.0 М в смеси 70:30, циклогексан:эфир). После добавления первого эквивалента реагента исходный субстрат солюбилизируют, после полного добавления баню со льдом удаляют, перемешивают 1 час, добавляют к раствору смесь лед-вода (приблизительно 2 мл) и выливают в 50 мл CH2Cl2. Водный слой экстрагируют 2 раза CH2Cl2 (50 мл). Органические слои объединяют, сушат над безводным MgSO4, фильтруют, фильтрат упаривают, остаток очищают хроматографически на двуокиси кремния при элюировании CH3OH: CH2Cl2/4: 96. Выделяют продукт в виде белого твердого вещества (57 мг, 58%). 1H ЯМР (ДМСО-d6) δ 2.33 (с, 3H), 6.55 (дд, 1H, J = 5.4, 3.6 Гц), 7.07 (м, 4H), 7.23 (м, 6H), 7.59 (м, 2H), 8.73 (с, 1H), 12.46 (шир с, 1H). HRMS для C22H18N2O2 : вычислено 342.1368, найдено 342.1366.
Биохимическая и биологическая оценка
Определение константы ингибирования ферментативной тимидилатсинтазы относительно 5,10-метилентетрагидрофолата
Активность тимидилатсинтазы измеряют с помощью модифицированного метода высвобождения трения Lomax u Greenberg [M.I.S. Lomax u G.R.Greenberg, J. Biol. Chem., 242 109 (1967). Константы ингибирования, ki наклон и Ki, отрезок на оси [W.W. Cleland, Biochim. Biophys. Acta 67 173 (1963)] определяют относительно кофактора (6R,6S)-5,10-метилен-тетрагидрофолата, который образуется с помощью реакции тетрагидрофолата с формальдегидом [R.G.Kallen u W. P. Jencks, J. Biol. Chem., 241 5851 (1966)]. Кофактор присутствует как изменяющийся субстрат при условии насыщенного радиомеченного 2'-деоксиуредин-5'-монофосфата (dUMP). Образцы для анализа при общем объеме 0,1 мл содержат 50 мМ Tris при pH 7.6, 10 мМ ДТТ (дитиотреитол), 1мМ ЕДТА (этилендиаминотетрауксусная кислота) 25 мМ MgCl2, 15 мл формальдегида, ± 1% ДМСО (в зависимости от растворимости соединения), 25 мкМ [5-3H] dUMP (специфическая активность 2 х 108 число/мин/мкмолей), тетрагидрофолат (восемь концентраций, изменяющихся в ряду от 5 мкМ до 300 мкМ) и фермент (= 30 нг для E. coli TS u = 60 нг для TS человека. Образцы для анализа TS человека также содержат 1-5 мкг/мл альбумина бычьей сыворотки для стабилизации протеина. Реакции инициируются добавлением фермента, выполняются в течение 5 минут при 24oC и затем гасятся добавлением активированного угля (15 мг в 0,1 мл воды). Эти образцы центрифугируют при 10000 об/мин в течение 12-15 минут при 40oC для удаления непрореагировавшего dUMP, который связывается с углем, 0,1 мл супернатанта обсчитывают с помощью сцинтилляции жидкости в присутствии 5 мл объема для определения высвобождения трития по 5 положению dUMP. Стандартная кривая строится в отсутствии ингибитора, определяются три дополнительные кривые, содержащие ингибитор при приблизительно 1/2 - 2Ki. Экспериментальные результаты анализируют EZ-FIT, нелинейным регрессионным анализом программы (Perrella Scientific, Springfield PA), которая используется для всех точек данных одновременно для схемы смешанного неконкурентного ингибирования. Полученные результаты приведены в таблице. Первая графа для каждого соединения представляет Ki, наклон и графа ниже является Ki, отрезок на оси.
Определение ингибирования роста опухолевых клеток при тестировании in vitro
Рост клеток в присутствии обсуждаемых соединений оценивают при использовании трех линий клеток: LI210 лейкемии мышей (ATCC CCL 219), CCFP-CEM, лимфобластическая лейкемическая линия T-клеток человека (ATCC CCL 119) и аденокарциномы тимидинкиназы-дефицитной ободочной кишки человека, GC3/M TK- (приведены Drs. P.J. u J.A.Houghton, St. Jude Children's Research Hospital, Memphis, TN).
Линии клеток сохраняют в RPMI 1640 среде, содержащей 5% (LI210, CCRF-CEM) или 10% (GC3/M TK-) теплотой-неактивированной фетальной бычьей сыворотки без антибиотиков.
IC50 определяется в 150 мкл микрокультур, каждая из которых содержит 150 (LI210) или 10000 (CCRF-CEM, GC3/M TK-) клеток, при оценке на пластинке с 96 отверстиями в питательной среде, к которой добавляют 50 ед/мл пенициллина и 50 мкг/мл стрептомицина. Рост измеряют в течение 3 дней (LI210) или 5 дней (CCRF-CEM, GC3/M TK-) непрерывного воздействия для изменения концентраций каждого тестируемого соединения, выдерживают 4 часа после первоначального посева клеток, с помощью анализа восстановления MTT-тетразоля T.J.Mosmann [J. Immund. Meth. 65 55 (1983)] модифицированного Alley et al. [Cancer Res. 48 589 (1988)]. Нерастворимые в воде производные растворяют в ДМСО, разбавляют до конечной концентрации 0,5% растворителя в культуре клетки.
Результаты, полученные по этой методике, приведены в табл. 2. (Хотя табл. 2 показывает, что некоторые соединения не проявляют особенно хорошее TS ингибирование, эти соединения представляют потенциальный интерес, потому что они могут проявить другую противоопухолевую активность, такую как токсичность к LI210 клеткам в культуре ткани.
В то время как изобретение описано в деталях и со ссылкой на специфические воплощения его, в него согласно предшествующим работам могут быть внесены различные изменения и модификации без отклонения от характера и области изобретения. Таким образом, полагается, что настоящее изобретение охватывает модификации и отклонения, при условии, что они включены в область прилагаемой формулы изобретения и ее эквивалентов.
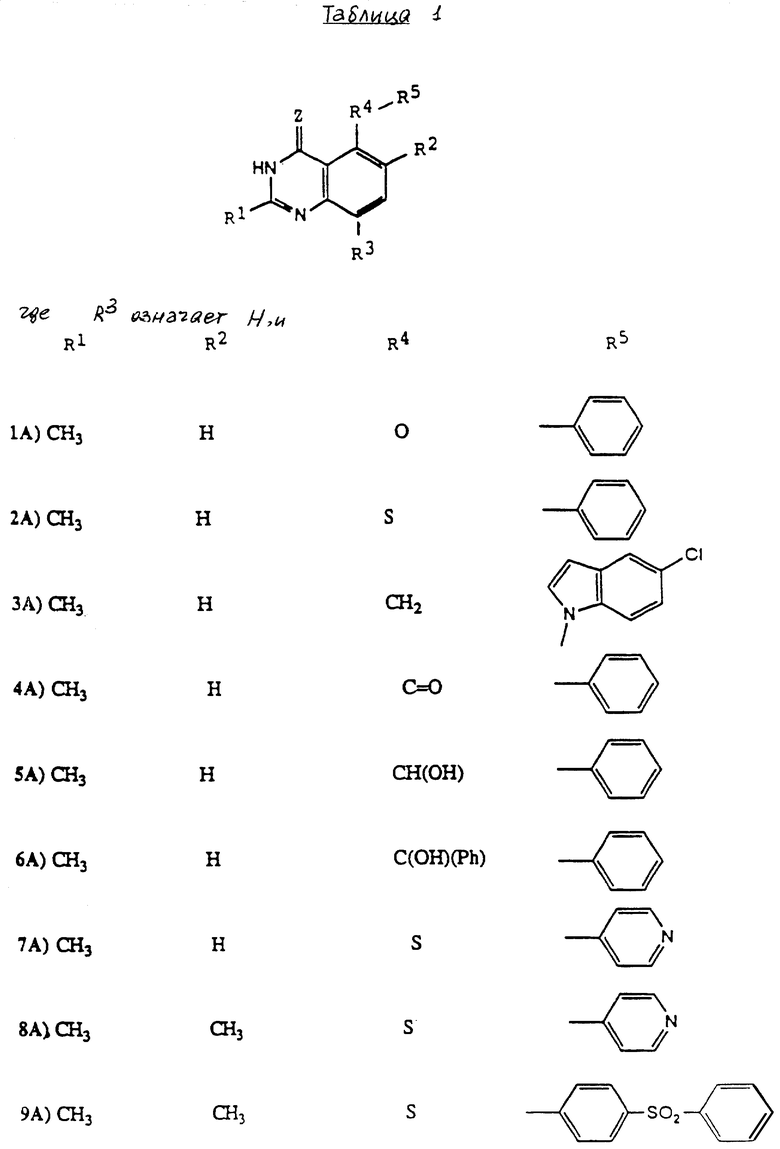
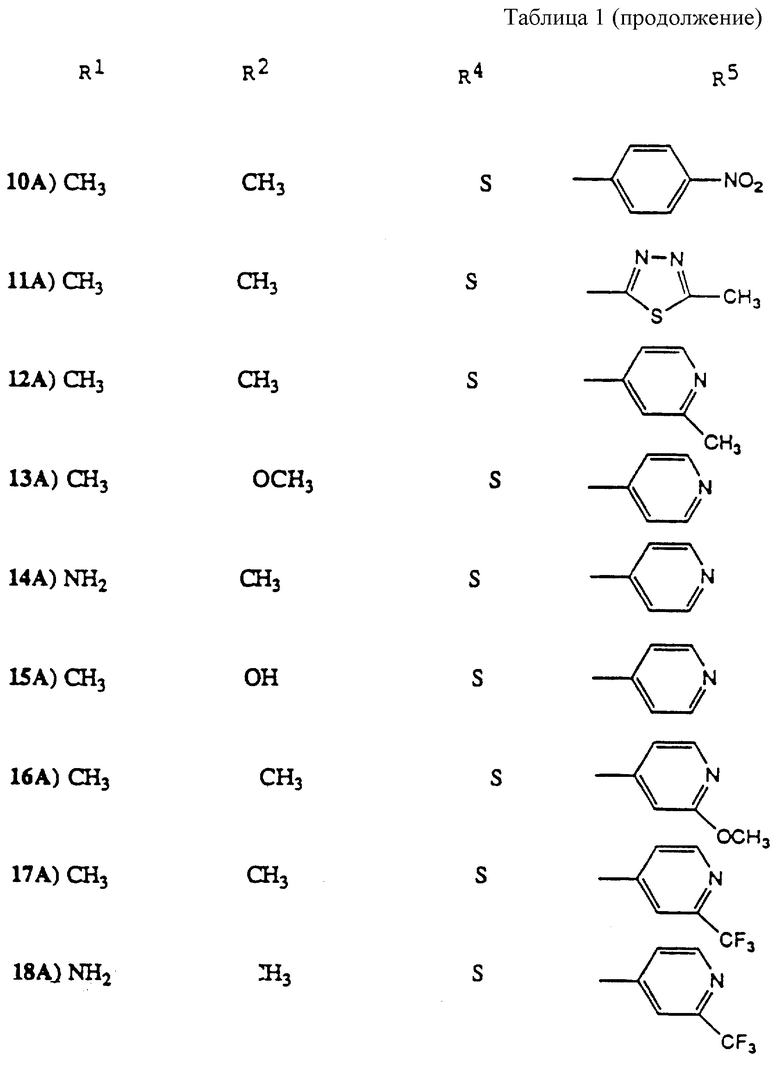
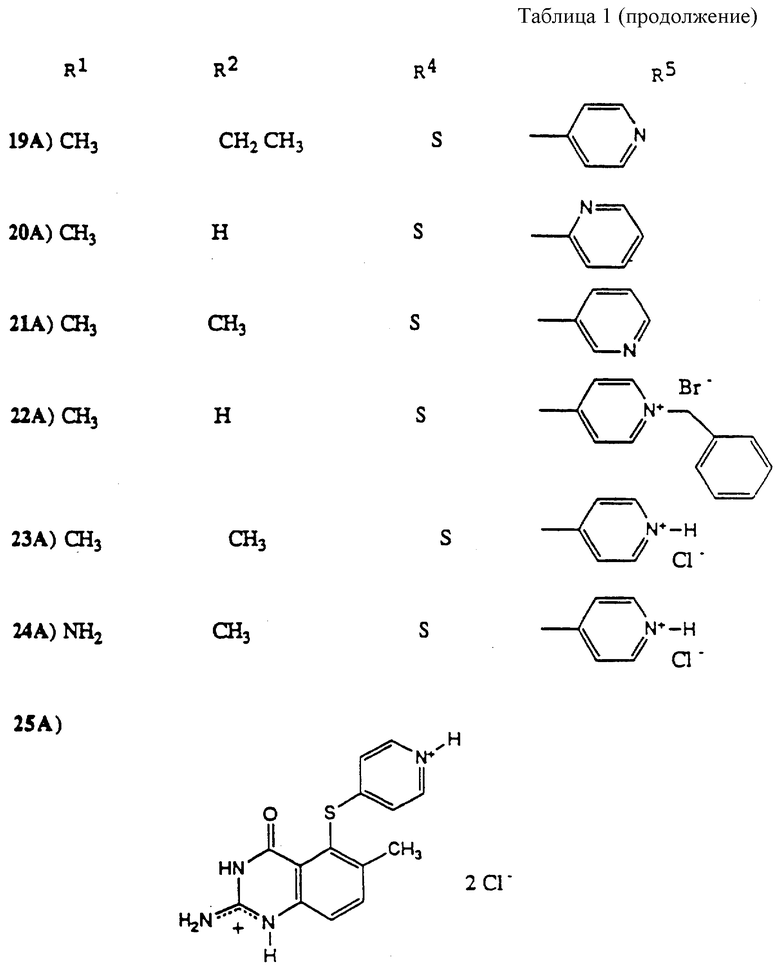


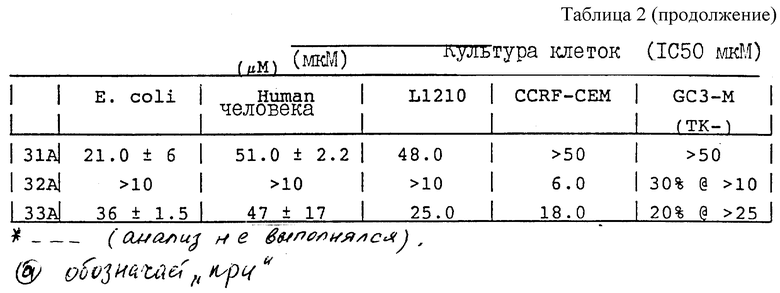
Хиназолиновые соединения формулы I, где R1 - низший алкил или аминогруппа; R2 - водород, низший алкил, гидрокси, низший алкокси; R3 - водород, Z - кислород; R4 - О, S, СН2,С=С, СНОН, ОН-, C(Ph)-; R5 - остаток формулы, указанный в п.1 формулы изобретения, или их фармацевтически приемлемые соли, проявляют антипролиферативную активность, такую как противоопухолевую активность. 2 с. и 17 з. п. ф-лы, 2 табл.
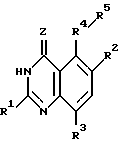
где R1 представляет собой низший алкил или аминогруппу;
R2 представляет собой водород, низший алкил, гидрокси, низшую алкоксигруппу;
R3 представляет собой водород;
Z представляет собой кислород;
R4 представляет собой O, S, CH2, C=O, CHOH, группу OH-C(Ph)-;
R5 представляет собой группу, выбранную из ряда, включающего


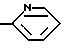
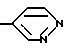
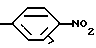


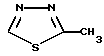


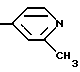
или их фармацевтически приемлемые соли.


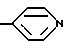
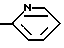





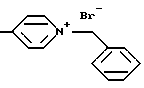

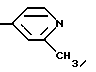

9. Соединения по п. 8 или их фармацевтически приемлемые соли, где R2 представляет собой водород или метильную группу и R4 представляет собой серу.
где R3 представляет собой водород;
Z представляет собой кислород
или соединений формулы
11. Соединения по п.10 или их фармацевтически приемлемые соли, где вышеуказанные соединения выбирают из соединений общей формулы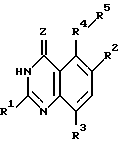
где R3 представляет собой водород;
Z представляет собой кислород
или соединения формулы
12. Соединения по п.11 или их фармацевтически приемлемые соли, где вышеуказанные соединения имеют следующую структуру:
13. Соединения по п.1 или их фармацевтически приемлемые соли, где вышеуказанные соединения или их соли имеют константу ингибирования тимидилатсинтазы менее или равную приблизительно 10-4 М.
17. Фармацевтическая композиция по п.15, отличающаяся тем, что композиция имеет форму, выбранную из группы, включающей формы, пригодные для перорального, парентерального, местного, внутривагинального, внутриносового, внутрибронхиального, внутриглазного, внутриушного или ректального введения.
| Датчик скорости вращения вала | 1972 |
|
SU459730A1 |
| УСТРОЙСТВО для ОБРАБОТКИ РАДИОИМПУЛЬСНОГО | 0 |
|
SU373891A1 |
| ОКОНЦЕВАТЕЛЬ ГЛУБОКОВОДНОГО КАБЕЛЯ1Известны оконцевателн гл}'боководного кабеля, соединение контактных нроводннков которых с жилой кабеля заключено в люнолнт- ный корнус из электроизоляцнонного материала. Недостатком такнх оконцевателеГ! является сложность и трудоемкость их изготовления. Кроме того, герметичиость оконцевателей может быть достаточно надежно проконтролнро- 1зана практически лишь после монтажа кабельной цепп и погруження, что приводит к воз- можностн монтажа дефектных оконцевателей и значнтель]1ым потерям времени на ликвидацию дефектов.В предлагаемом оконцевателе выводы контактных проводников нз корпуса выполнены в внде петель и изолированы тем же материалом, из которого выполнен корпус. | 0 |
|
SU365763A1 |
| СПОСОБ ОЧИСТКИ сточных вод ПРОИЗВОДСТВА ТИОКОЛА | 0 |
|
SU316657A1 |
| Способ получения производных хиназолинона | 1983 |
|
SU1299510A3 |

