Изобретение относится к новым замещенным производным сульфонилмочевины общей формулы (I)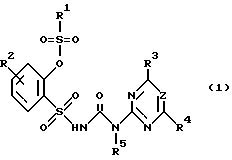
где R1 C1-C4-алкильная группа, необязательно замещенная атомом галогена или C1-C2-алкоксигруппой; C1-C3-алкиламиногруппа или ди-(C1-C4-алкил)аминогруппа;
R2 водород, галоген, метил;
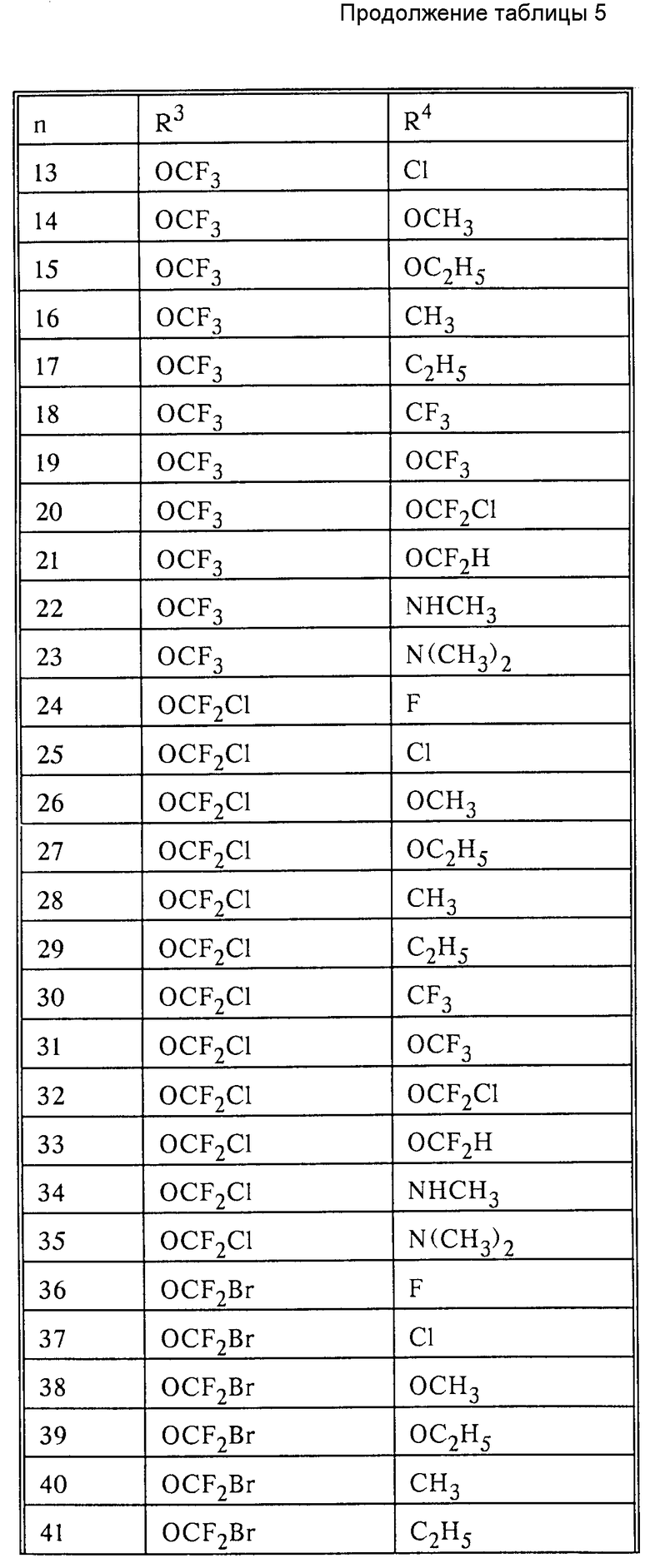
R3 дифторметокси, трифторметокси, бромдифторметокси, хлордифторметокси или фтор;
R4 галоген, при определенных условиях галогензамещенные метил, этил, C1-C2-алкоксигруппа; метокси- или этоксигруппа, метил или диметиламиногруппа;
R5 водород;
Z CH или N, при условии, что
а) когда R3 означает дифторметоксигруппу, R1 не может быть ди(алкил)аминогруппой, а R4 метилом или метоксигруппой,
б) когда R3 означает атом фтора, а Z N, R4 не может быть алкиламиногруппой;
или их применимым в сельском хозяйстве солям, обладающим гербицидной активностью, и гербицидному средству.
В описаниях изобретений (ЕП, NN 30433, 44212, 125205, 135332, 136061, 158600 и в описании изобретения к патентам США NN 4534789 и 4127405) представлены различные замещенные эфиры сложных алкил- или арилсульфокислот, в ЕП N 125205 и в описаниях изобретения к патентам США NN 4576633 и 4515624 описываются замещенные сульфонилмочевины, имеющие аминосульфонатные остатки.
Однако они не удовлетворяют требованиям к гербицидам по активности и селективности.
Соединения формулы (I), их соли и гербицидное средство на их основе могут быть использованы для эффективной борьбы с сорняками для таких культур, как, например, рис, пшеница и кукуруза, при низкой норме расхода и при этом не повреждают культурные растения.
Предпочтительны производные сульфонилмочевин формулы (I), где R1 означает C1-C4-алкильную группу, которая может быть замещена одно- или трехкратно атомом галогена или C1-C2-алкоксигруппу, а R2-R5 и Z имеют значения, указанные выше.
Среди предпочтительных соединений можно назвать соединения формулы (I), где R1 означает C1-C3-алкиламино- или ди-(C1-C4-алкил)аминогруппу, а R2-R5 и Z имеют значения, указанные выше; либо замещенные производные сульфонилмочевины формулы (I), в которой R1 означает C1-C4-алкильную группу, которая может иметь от 1 до 3 атомов галогена; метиламино- или диметиламиногруппу; R3 означает дифторметокси- или трифторметоксигруппу или фтор, а R2, R4, R5 и Z имеют значения, указанные выше; или замещенные производные сульфонилмочевины формулы (I), в которой R1 означает C1-C4-алкильную группу, которая может иметь от 1 до 3 атомов галогена; метиламино- или диметиламиногруппу; R3 означает дифторметокси- или трифторметокси-, или хлордифторметоксигруппу или фтор; R4 означает метоксигруппу, а R2, R5 и Z имеют значения, указанные выше.
C1-C4-алкильные остатки обозначают, например, метил, этил, н-пропил или изопропил, н-бутил, втор.-бутил или трет.-бутил, предпочтительно метил или этил;
C1-C3-галогеналкильные остатки обозначают, например, фторметил, хлорметил, бромметил, 2-фторэтил, 2-хлорэтил, дифторметил, дихлорметил, трифторметил, трихлорметил, 2,2,2-трифторэтил или 2,2,2-трихлорэтил, предпочтительно 2,2,2-трифторэтил;
C2-C5-алкоксиалкильные остатки означают, например, метоксиметил, этоксиметил, 2-метоксиэтил, 2-этоксиэтил, 2-метоксипропил, 3-метоксипропил, 2-этоксипропил или 3-этоксипропил, предпочтительно метоксиметил, этоксиметил или 2-метоксиэтил;
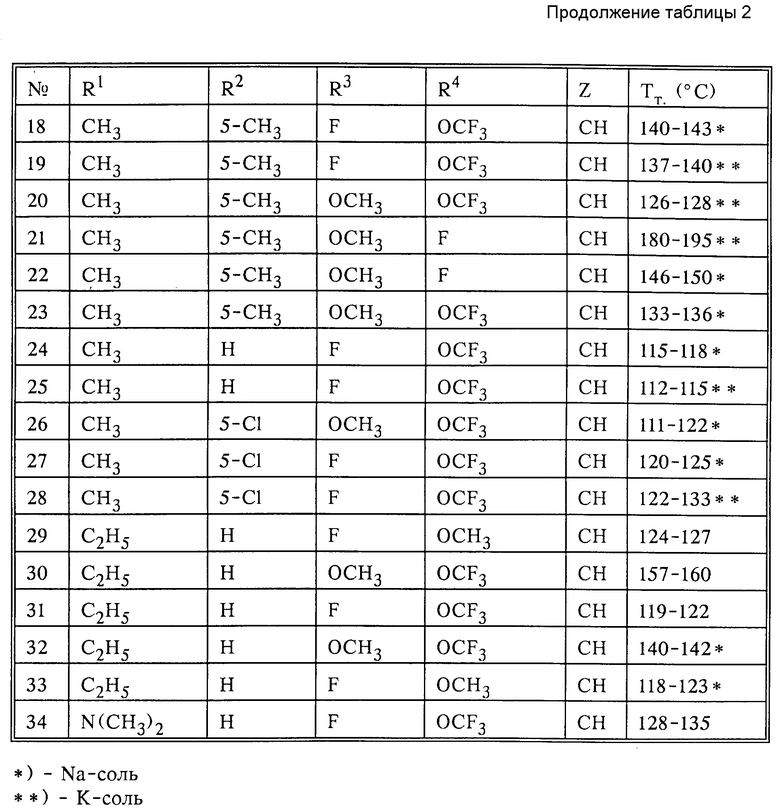
C1-C3-алкиламинорадикалы означают, например, метил-, этил-, н-пропил- или изопропиламино, предпочтительно метиламино; ди(C1-C4-алкил)- радикалы, как, например, диметил-, диэтил-, ди(н-пропил)-, ди(изопропил)-, ди(трет.-бутил)-, метилэтил- или метил(изопропил)амино, предпочтительно диметиламино;
Галоген обозначает фтор, хлор, бром, иод.
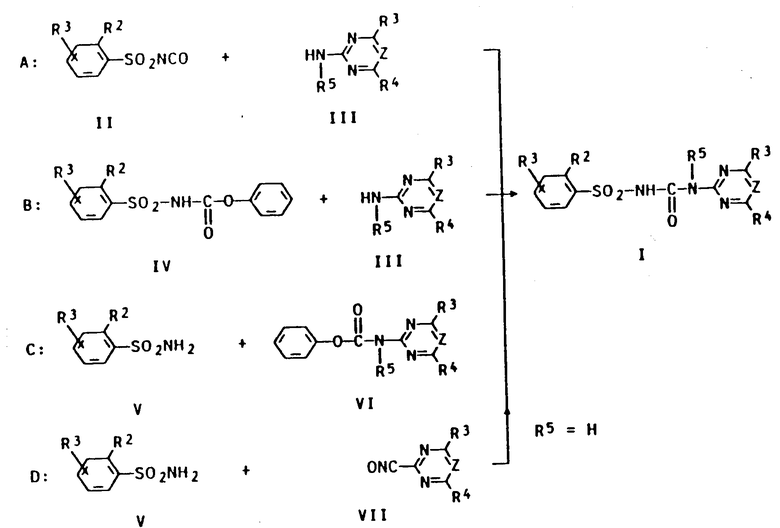
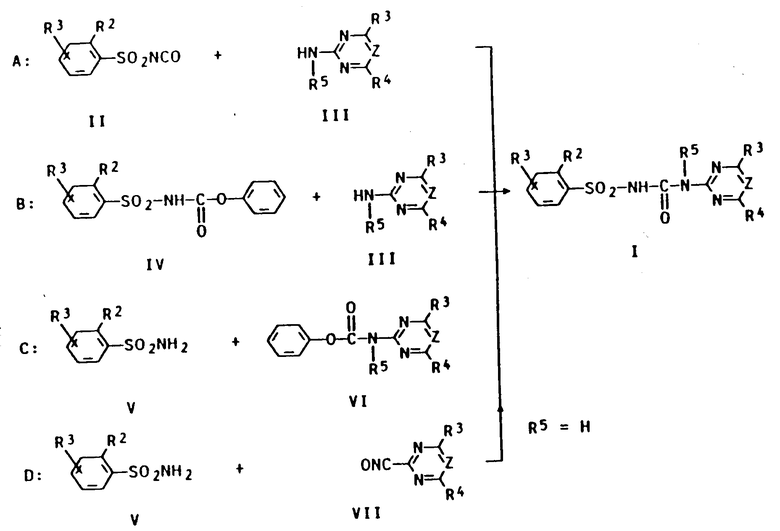
Сульфонилмочевины формулы (I) могут быть получены различными способами, описанными в литературе. Особенно предпочтительными являются способы (A-D), более подробно поясненные в конце текста.
Способ A:
Сульфонилизоцианат II преобразуют известным способом (ЕП 162723 или ЕП 44212) с помощью примерно стехиометрического количества производного 2-амино-1,3,5-триазина или пиримидина III при температуре от 0 до 120oC, предпочтительно от 10 до 100oC. Реакция может осуществляться при нормальном давлении или низком давлении (до 50 бар), предпочтительно при 1-5 бар, непрерывно или периодически.
Для реакции целесообразно использовать инертные растворители и разбавители при соответствующих условиях реакции. В качестве растворителей используются, например, галогенуглеводороды, в частности, хлорпроизводные углеводорода, например, тетрахлорэтилен, 1,1,2,2- или 1,1,1,2-тетрахлорэтан, дихлорпропан, метиленхлорид, дихлорбутан, хлороформ, хлорнафталин, дихлорнафталин, тетрахлорметан, 1,1,1- или 1,1,2-трихлорэтан, трихлорэтилен, пентахлорэтан, о-, м-, п-дифторбензол, 1,2-дихлорэтан, 1,1-дихлорэтан, 1,2-цис-дихлорэтилен, хлорбензол, фторбензол, бромбензол, иодбензол, о-, м-, п-дихлорбензол, о-, п-, м-дибромбензол, о-, м-, п-хлортолуол, 1,2,4- трихлорбензол; простой эфир, например, простой этилпропиловый эфир, простой метил-трет.-бутиловый эфир, простой н-бутилэтиловый эфир, простой ди-н-бутиловый эфир, простой диизобутиловый эфир, простой диизоамиловый эфир, простой диизопропиловый эфир, анизол, фенетол, простой циклогексил-метиловый эфир, простой диэтиловый эфир, простой этиленгликольдиметиловый эфир, тетрагидрофуран, диоксан, тиоанизол, простой β,β′-дихлордиэтиловый эфир; нитроуглеводороды, как, например, нитрометан, нитроэтан, нитробензол, о-, м-, п-хлорнитробензол, о-нитротолуол; нитрилы, как, например, ацетонитрил, бутиронитрил, изобутиронитрил, бензонитрил, м-хлорбензонитрил; алифатические или циклоалифатические углеводороды, например, гептан, пинан, нонан, о-, м-, п-цимол, бензиновые фракции в пределах температуры кипения от 70 до 190oC, циклогексан, метилциклогексан, декалин, петролейный эфир, гексан, лигроин, 2,2,4-триметилпентан, 2,2,3-триметилпентан, 2,3,3-триметилпентан, октан; сложный эфир, например, этилацетат, сложный ацетоуксусный эфир, изобутилацетат; амиды, например, формамид, метилформамид, диметилформамид; кетоны, например, ацетон, метилэтилкетон и соответствующие смеси. Растворитель целесообразно применять в количестве о 100 до 2000 мас. предпочтительно от 200 до 700 мас. относительно исходного вещества II.
Соединение II, необходимое для преобразования, применяется обычно примерно в эквимолярных количествах (с избытком или недостатком, например, от 0 до 20% из расчета на соответствующее исходное вещество III). Исходное вещество III можно добавить предварительно в ранее указанный разбавитель, а затем добавить исходное вещество II.
Однако способ получения новых соединений целесообразно проводить таким образом, чтобы исходное вещество II помещалось соответственно в один из ранее указанных разбавителей и затем добавлялось исходное вещество III. Для окончания реакции после добавления компонентов смесь перемешивают еще от 20 мин до 24 ч при температуре от 0 до 120oC, предпочтительно при температуре от 10 до 100oC, в частности, от 10 до 100oC.
Для ускорения реакции можно добавлять предпочтительно третичный амин, например, пиридин, α,β,γ-пиколин, 2,4-, 2,6-лутидин, 2,4,6-коллидин, п-диметиламинопиридин, триметиламин, триэтиламин, три(н-пропил)амин, 1,4-диаза[2,2,2] бициклооктан (ДАБЦО) или 1,8-диазабицикло[5,4,0]ундец-7-ен в количестве от 0,01 до 1 моль на моль исходного вещества II.
Продукт I выделяется из реакционной смеси обычным путем, например, путем отгонки растворителей или непосредственно путем фильтрования. Оставшийся остаток можно промыть водой или разбавленной кислотой для удаления основных примесей. Кроме того, можно также растворить остаток в растворителе, не смешиваемом с водой, и промыть, как описано выше. Целевые соединения выпадают при этом в чистом виде и, при необходимости, они могут быть очищены путем перекристаллизации, перемешивания в органическом растворителе, поглощающем примеси, или хроматографическим путем.
Это взаимодействие проводят предпочтительно в ацетонитриле, простом метил-трет. -бутиловом эфире, толуоле или метиленхлориде в присутствии от 0 до 100 молярных эквивалентов, предпочтительно от 0 до 50 молярных эквивалентов третичного амина, как, например, 1,4-диазабицикло[2,2,2]октана или триэтиламина.
Способ B.
Соответствующий сульфонилкарбамат формулы IV преобразуют известным способом (ЕП 120814, ЕП 101407) в инертном органическом растворителе при температуре в пределах от 0 до 120oC, предпочтительно от 10 до 100oC, с помощью производного 2-амино-1,3,5-триазина или пиримидина III. При этом могут быть добавлены основания, как, например, третичные амины, что вызывает ускорение реакции и улучшает качество продукта.
Подходящими основаниями для этого являются, например, третичные амины, как указано в способе A, в частности, триэтиламин или 1,4-диазабицикло[2,2,2] октан в количестве от 0,01 до 1 моль на исходное вещество IV. В качестве растворителей целесообразно применять растворители, указанные в способе A.
Растворитель применяют в количестве от 0 до 2000 мас. предпочтительно от 200 до 700 мас. относительно исходного вещества IV.
Соединение IV, необходимое для преобразования, применяется обычно примерно в эквимолярных количествах (с избытком или недостатком, например, от 0 до 20% из расчета на исходное вещество III). Исходное вещество IV можно добавить в один из указанных ранее разбавителей и затем добавить исходное вещество III.
В обоих случаях в качестве катализатора до или во время реакции может быть добавлено основание.
Конечный продукт I может быть выделен из реакционной смеси обычным способом, как указано в способе А.
Способ С.
Сульфонамид формулы V преобразуют известным способом (ЕП 141777 и ЕП 101670) в инертном органическом растворителе с помощью примерно стехиометрического количества фенилкарбамата VI при температуре от 0 до 120oC, предпочтительно при температуре 20-100oC. Реакция может осуществляться при нормальном давлении или при давлении до 50 бар, предпочтительно при 1-5 бар, непрерывно или периодически.
При этом могут быть добавлены основания, как, например, третичные амины, ускоряющие реакцию и улучшающие качество продукта. Подходящими основаниями для этого являются основания, указанные в способе А, в частности, триэтиламин, 2,4,6-коллидин, 1,4-диазабицикло[2,2,2]октан (ДАБЦО) или 1,8-диазабицикло[5,4,0] ундец-7-ен (ДБУ) в количестве от 0,01 до 1 моль на каждый моль исходного вещества V.
В качестве растворителей или разбавителей целесообразно применять указанные в способе А растворители и разбавители.
Растворитель применяют в количестве от 100 до 2000 мас. предпочтительно от 200 до 700 мас. относительно исходного вещества V.
Соединение V, используемое в реакции, применяют обычно примерно в эквимолярных количествах (с избытком или недостатком, например, от 0 до 20% из расчета на исходное вещество VI). Можно исходное вещество VI добавить в один из указанных ранее разбавителей и затем добавить исходное вещество V.
Кроме того, можно также исходное вещество V добавить в указанный растворитель и затем добавить к карбамату VI. В обоих случаях до или во время реакции в качестве катализатора можно добавить одно из указанных оснований.
Для окончания реакции после добавления компонентов смесь перемешивают еще от 20 мин до 24 ч при температуре от 0 до 120oC, предпочтительно от 10 до 100oC, в частности, от 20 до 80oC.
Сульфонилмочевины формулы (I) выделяют из реакционной смеси обычными методами, как описано в способе А.
Способ D.
Сульфонамид формулы V превращают известным способом (ЕП 234352) в инертном органическом растворителе с помощью примерно стехиометрического количества изоцианата VII при температуре от 0 до 150oC, предпочтительно от 10 до 100oC. Реакция может осуществляться при нормальном давлении (до 50 бар), предпочтительно при 1-5 бар, непрерывно или периодически.
При этом во время или до реакции может быть добавлено основание, как, например, третичные амины, ускоряющие реакцию и улучшающие качество продукта. Подходящими основаниями для этого являются основания, указанные в способе A, в частности, триэтиламин или 2,4,6-коллидин в количестве от 0,01 до 1 моль на каждый моль исходного вещества V.
В качестве растворителей целесообразно применять растворители, указанные в способе А, в количестве от 100 до 2000 мас. предпочтительно от 200 до 700 мас. относительно исходного вещества V.
Соединение V применяется обычно примерно в эквимолярных количествах (с избытком или недостатком, примерно от 0 до 20% из расчета на исходное вещество VII). Исходное вещество VII можно добавить в один из указанных разбавителей и затем добавить исходное вещество V. Однако можно также сульфонамид добавить в разбавитель и затем добавить изоцианат VII.
Для окончания реакции после добавления компонентов смесь перемешивают еще от 20 мин до 24 ч при температуре от 0 до 120oC, предпочтительно от 10 до 100oC, в частности, от 20 до 80oC. Целевой продукт I можно выделить из реакционной смеси обычным способом, как описывается в способе A.
Сульфонилизоцианаты формулы II, используемые в качестве исходных веществ, можно получить известным способом из соответствующих сульфонамидов путем фосгенирования [ЕП 44212, Hoybev-Weyl 11/2 (1985) 1106, патент США 4379769) или путем обработки сульфонамидов хлорсульфонилизоцианатами (выложенная заявка ФРГ 3132944).
Синтез гетероциклических аминов общей формулы III и дальнейшее превращение полученных при этом промежуточных продуктов можно осуществлять общими методами, описанными в специальной гетероциклической литературе (пиримидины: D.J. Brown в "The Chemistry of Heterocyclic Compounds", A. Weissberger. E.C. Teylor (Hrsg.), Wiley, New York, 1985, т.16; D.J. Brown в "Comprehensive Heterocyclic Chemistry", A. R. Katritzki (Hrsg.), Pergamon Press, New York, 1984, т. 3, 57 ff. триазины: E.M. Smolin и L. Rapoport в "The Chemistry of Heterocyclic Compounds", A. Weissberger, Interscience Publ. New York, 1959, т. 13; J.E. Quirke в "Comprehensive Heterocyclic Chemistry", A.R. Katritzki (Hrsg.), Pergamon Press. New York, 1984, т. 3, 457 ff.).
2-Аминопиримидины и 2-амино-1,3,5-триазины, которые в 4 или 6 положении имеют трифторметокси- или хлордифторметоксильный заместитель, могут быть получены, в частности, по методикам, описанным в заявках DE 4007316.5, 4007317.3, 4007683.0, 4024761.9, 4024755.4 и 4024754.6.
Таким образом можно получить также производные формулы IIIa, в которой R4 обозначает метиламино, диметиламино, метокси, этокси или C2-галоалкоксигруппу (см. схему 2).
Схема 2.
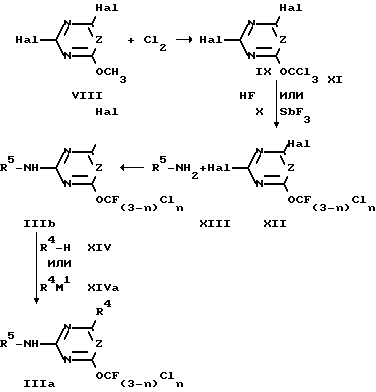
n 0,1; R4 NHCH3, N(CH3)2, OCH3, OC2H5, C1-C2-галоалкокси; Z CH, N.
Соответствующим образом получают производные 2-амино-6-трифторметил-1,3,5-триазина или 2-амино-6-трифторметилпиримидина IIIc, из 2,4-дигалоген-6-трихлорметил-1,3,5-триазина или 2,4-дигалоген-6-трифторметилпиримидина формулы XV согласно схеме 3.
Схема 3.
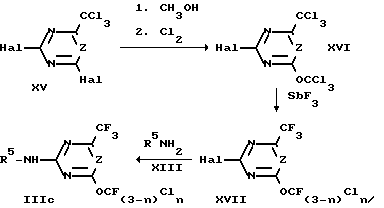
Исходя из промежуточных продуктов XII в схеме 2, замещая атом галогена в 4 положении, аналогично способу, описанному в схеме 3 (1. CH3OH, 2. Cl2, 3. SbF3), с последующей обработкой R5NH2 получают промежуточные продукты IIId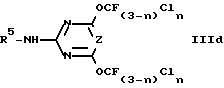
Аналогично получают 4-алкил-2-амино-1,3,5-триазины или 4-алкил-2-аминопиримидины IIIe, если используют 2-алкокси-4-алкил-6-галогентриазины или 4-алкокси-6-алкил-2-галогенпиримидины (см. схему 4).
Схема 4.
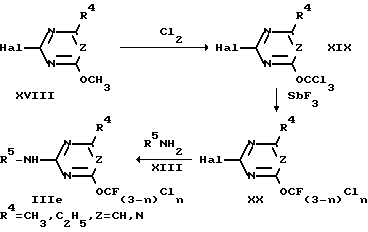
Используемые в качестве исходных веществ 2-алкокси-4-алкил-6-галоген-1,3,5-триазины и 4-алкокси-6-алкил-2-галогенпиримидины известны в литературе (например, 2-хлор-4-метокси-6-метилпиримидин в Bull. Soc. Chim Belg. 68 (1959) 30; 2-хлор-4-метокси-6-метил-1,3,5-триазин в Monatsh. Chem. 101 (1970) 724) или могут быть получены аналогичным путем.
Хлорирование 2-метокси-1,3,5-триазинов или 2-метоксипиримидинов VIII, XV или XVIII хлором в производные трихлорметокси IX, XVI или XIX осуществляется, например, при температуре от 100 до 180oC.
В качестве хлорирующих средств подходят элементарный хлор или вещества, отдающие хлор, как, например, сульфурилхлорид или фосфорпентахлорид.
Преобразование может осуществляться в присутствии инертного растворителя, например, хлорированного углеводорода, как, например, хлорбензола, 1,2-, 1,3- или 1,4-дихлорбензола, нитросоединения, как, например, нитробензола, карбоновой кислоты, как, например, уксусной кислоты, пропионовой кислоты, ангидрида кислоты, как, например, хлорацетилхлорида, хлорида α-хлорпропионовой кислоты, хлорида a,αдихлорпропионовой кислоты, неорганического галогенида кислоты, как, например, фосфортрихлорида или фосфороксихлорида, или предпочтительно без растворителя в расплаве исходного вещества VIII, XV или XVIII.
Соответственно, можно ускорить реакцию путем ускорения образования радикалов. Для этого подходит облучение светом, предпочтительно ультрафиолетовым излучением или добавление a,α′-азоизобутиронитрила в количестве от 0,2 до 7 мол. по отношению к исходным веществам XIII, XV или XVIII. Реакцию можно ускорить также путем добавления катализатора; в качестве катализатора подходит фосфорпентахлорид, предпочтительно в количестве от 0,5 до 7 мол. от исходных веществ VIII, XV или XVIII. В этом случае исходное вещество добавляют вместе с катализатором и начинают хлорирование. Вместо фосфорпентахлорида можно добавить также исходный компонент, образующий его в условиях реакции, например, фосфортрихлорид или желтый фосфор, и затем начать хлорирование.
Исходные вещества VIII, XV или XVIII могут преобразовываться хлором, примерно в стехиометрическом количестве или предпочтительно в избытке, эквивалент метоксигруппы в исходных веществах XIII, XV или XVIII. Преобразование может осуществляться при температуре от 100 до 180oC, предпочтительно от 120 до 150oC, без давления или при давлении, непрерывно или периодически.
Хлорируют при давлении 1 бар, при этом применяют предпочтительно от 3,3 до 5 моль газообразного хлора на один эквивалент метоксильной группы в исходных веществах VIII, XV или XVIII, что приводит к превращению хлора от 91 до 60% Путем применения соответствующих условий, например, умеренного избыточного давления, предпочтительно от 1 до 10 бар, или барботажной колонны можно повысить процент превращения хлора. Как правило, газообразный хлор перемешивают длительное время с органической фазой или пропускают через густой слой органической фазы.
Длительность реакции достигает обычно примерно от 0,5 до 12 ч.
В предпочтительном варианте выполнения способа поступают таким образом, что в течение от 0,5 до 12 ч, предпочтительно от 1 до 10 ч, требуемое количество газообразного хлора при интенсивном перемешивании вводят в жидкие исходные вещества VIII, XV или XVIII, причем сначала начинают при температуре от 120 до 130oC и, с учетом экзотермичности реакции, непрерывно повышают температуру так, что в конце реакцию проводят при температуре от 135 до 150oC. В некоторых случаях, учитывая экзотермичность реакции, применяют внешнее охлаждение или регулируют дозировку количества хлора; в процессе протекания реакции удаляют ванну с охлаждающей смесью и затем нагревают.
Переработка и выделение получаемых веществ может осуществляться обычным способом. Например, из горячей органической фазы с помощью инертного газа можно извлечь остатки хлороводорода, хлора или катализатора; при этом остается уже довольно чистый сырой продукт с высоким выходом. Сырой продукт можно подвергнуть дальнейшей очистке посредством дистилляции или хроматографии или непосредственно применять для дальнейших преобразований.
Преобразование производных трихлорметокси IX, XVI или XIX с помощью галогенообменных средств осуществляют, например, при температуре от 0 до 180oC.
В качестве галогенообменных средств подходят трифторид сурьмы в присутствии или отсутствии каталитических количеств соли сурьмы (V) или фтороводорода.
Целесообразно применять избыток от 1 до 200, предпочтительно от 5 до 25 мол. трифторида сурьмы на эквивалент трихлорметила. Каталитическое количество соли сурьмы (V) достигает от 1 до 20, предпочтительно от 5 до 18 мол. трихлорметила. Исходные вещества IX, XVI или XIX дозируют предпочтительно при температуре от 90 до 130oC для смешивания с галогенообменными средствами и затем нагревают еще в течение от 10 до примерно 240 мин до температуры в пределах от 110 до 180oC. Затем отделяют перегонкой.
Реакцию можно осуществлять также непрерывно, исходные вещества IX, XVI или XIX добавляют при температуре от 110 до 180oC в течение от 10 до примерно 240 мин и одновременно при пониженном давлении отгоняют образовавшиеся конечные вещества XII, XVII или XX. Следы оставшихся солей сурьмы удаляют путем извлечения с помощью концентрированной соляной кислоты.
Если реакцию ведут без добавления соли сурьмы (V) или применяют лишь незначительное количество, например, от 0,5 до 5 мол. и уменьшают количество трифторида сурьмы от 60 до 90 мол. на эквивалент трихлорметила, то реакция идет с образованием хлордифторметокси.
Вместо трифторида сурьмы галогенообмен может осуществляться также с помощью фтороводорода при температуре от 0 до 150oC, предпочтительно при температуре от 40 до 120oC. Для этого смешивают в автоклаве исходные вещества IX, XVI или XIX с избытком от 300 до 700, предпочтительно от 350 до 400 мол. фтороводорода на эквивалент трихлорметила, и перемешивают от 10 мин до 10 ч. Соответственно можно ускорить реакцию тем же способом, который описывается в случае применения трифторида сурьмы, путем добавления катализатора, как, например, пентахлорида сурьмы. После прекращения реакции и удаления летучих компонентов процесс переработки продолжается, как описано выше.
Реакция производных фторметокси XII, XVII или XX осуществляется с аминами XIII, например, при температуре от -80oC до 40oC.
2-Галоген-1,3,5-триазины или -пиримидины XII, XVII или XX могут реагировать в апротонном полярном растворителе с аминами XIII при температуре от -80oC до 40oC; применяют амин XIII либо в избытке, либо используют органическое вспомогательное основание.
Для реакции триазинов или -пиримидинов XII, XVII или XX с амином XIII подходят следующие растворители: простые эфиры, как, например, простой метил-трет. -бутиловый эфир, простой диэтиловый эфир, простой этилпропиловый эфир, простой н-бутилэтиловый эфир, простой ди-н-бутиловый эфир, простой диизобутиловый эфир, простой диизоамиловый эфир, простой диизопропиловый эфир, простой циклогексилметиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан, простой диметиловый эфир диэтиленгликоля и анизол; сложные эфиры, как, например, этилацетат, н-бутилацетат и изобутилацетат; а также хлорированные углеводороды, как, например, метиленхлорид, 1,1,2,2-тетрахлорэтан, 1,1-дихлорэтилен, 1,2-дихлорэтан, хлорбензол, 1,2-дихлорбензол и 1-хлорнафталин и смеси этих растворителей.
Целесообразно применять растворитель в количестве от 100 до 2000 мас. предпочтительно от 400 до 1200 мас. относительно исходных веществ XII, XVII или XX.
К смеси исходных веществ XII, XVII или XX в одном из указанных ранее растворителей при температуре -80oC до 40oC, предпочтительно от -70oC до 25oC, добавляют предпочтительно от 1,8 до 2,5, в частности, от 1,95 до 2,2 моль-эквивалента амина XIII относительно исходных веществ XII, XVII или XX в течение 0,5-2 ч, перемешивают в течение 3 ч и затем нагревают для переработки до 25oC.
Если применяют примерно стехиометрические количества амина XIII, то целесообразно использовать от 0,9 до 1,1 эквивалентов органического основания от исходных веществ XII, XVII или XX. В качестве основания подходят органические основания, как, например, трифторметиламин, триэтиламин, N-этилдиизопропиламин, триизопропиламин, N,N-диметиланилин, N,N-диметилциклогексиламин, N-метилпирролидин, пиридин, хинолин, α-,β-,γ--пиколин, 2,4- и 2,6-лутидин и триэтилендиамин.
Реакция может осуществляться при отсутствии давления или при давлении, непрерывно или периодически.
Для разделения реакционную смесь экстрагируют водой для удаления солей, высушивают и очищают органическую фазу, например, хроматографией способом. Однако можно также непосредственно концентрировать органическую фазу и остаток смешать с растворителем.
2-Амино-4-фторалкокси-1,3,5-триазины или 2-амино-4-фторалкоксипиримидины формулы IIIa получают предпочтительно способом, при котором 2-амино-4-фторалкокси-6-галоген-1,3,5-триазины или -пиримидины формулы IIIb
в которой Hal обозначает фтор, хлор или бром, R5, n и Z имеют указанное ранее значение,
обрабатывают нуклеофилом формулы XIV
R4-H XIV
в которой R4 обозначает метиламино, диметиламино, метокси-, этокси- или C1-C2-галоалкоксигруппу,
или его солью XIVa.
Обработку 2-амино-4-фторалкокси-1,3,5-триазинов или 2-амино-4-фторалкоксипиримидинов IIIb нуклеофилом XIV или его солью XIVa осуществляют, например, при температуре от -80oC до 80oC, предпочтительно от -30oC до 20oC, причем либо нуклеофилы применяются в избытке, либо добавляется органическое основание.
Для обработки 4-галогенпроизводных IIIb подходят следующие растворители: простые эфиры, как, например, простой метил-трет.-бутиловый эфир, простой диэтиловый эфир, простой этилпропиловый эфир, простой н-бутилэтиловый эфир, простой ди-н-бутиловый эфир, простой диизобутиловый эфир, простой диизоамиловый эфир, простой диизопропиловый эфир, простой циклогексилметиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан, простой диметиловый эфир диэтиленгликоля и анизол; сложные эфиры, как, например, этилацетат, н-бутилацетат и изобутилацетат, а также хлорированные углеводороды, как, например, метиленхлорид, 1,1,2,2-тетрахлорэтан, 1,1-дихлорэтилен, 1,2-дихлорэтан, хлорбензол, 1,2-дихлорбензол и 1- хлорнафталин, и смеси этих растворителей.
Целесообразно использовать растворитель в количестве от 100 до 2000 мас. предпочтительно от 400 до 1200 мас. относительно исходного вещества IIIb.
К смеси исходного вещества IIIb в указанном ранее растворителе при температуре от -80oC до 80oC, предпочтительно от -80oC до 25oC, добавляют предпочтительно от 1,8 до 2,5, в частности, от 1,95 до 2,2, моль-эквивалентов нуклеофила XIVa от исходного вещества IIIb в течение 0,5-2 ч, перемешивают до полного завершения реакции (до 3 ч) и затем нагревают до 25oC.
Если используют лишь примерно стехиометрические количества нуклеофила XIV или XIVa, то целесообразно добавить от 0,9 до 1,1 эквивалентов органического основания в расчете на исходное вещество IIIb. В качестве органических оснований подходят органические основания, как, например, триметиламин, триэтиламин, N-этилдиизопропиламин, триизопропиламин, N,N- диметиланилин, N, N-диметилциклогексиламин, N-метилпирролидин, пиридин, хинолин, α-,β-,γ--пиколин, 2,4- и 2,6-лутидин и триэтилендиамин.
Реакция может осуществляться при отсутствии давления или при давлении, непрерывно или периодически.
Для разделения полученную смесь экстрагируют водой для удаления солей, высушивают и органическую фазу очищают, например, хроматографией. Продукты реакции в большинстве случаев достаточно чистые, так что необходимо только отфильтровать выпавшую соль и провести концентрирование органической фазы.
2-Аминопиримидины, имеющие в 4 или 6 положении атом фтора, можно получить по аналогии со способами, описанными в Европейском патенте 378092 или в Yakugaku Zasshi 87 (1967) 1315. Соответствующие 1,3,5-триазины могут быть получены аналогичным способом. Соответствующие исходные вещества, как, например, 2,4-дифтор-6-метокси-1,3,5-триазин известны в литературе (заявка на патент Франции 1561876 (CA 72, 90530), выложенная заявка ФРГ 2901498 (CA91, 194627) или Chem. Ber. 102 (1969) 2330).
2-Аминопиримидины или 1,3,5-триазины формулы III, которые в 4 или 6 положении имеют бромдифторметоксигруппу, описаны в Европейском патенте 169815.
2-Аминопиримидины, которые в 4 или 6 положении имеют дифторметоксигруппу, можно получить по методам, описанным в Европейском патенте 84020.
Сульфонилкарбаматы формулы IV были получены по аналогии с известными реакциями (например, Европейский патент 120814). Однако можно также перевести сульфонилизоцианаты формулы I в обычных условиях в инертном растворителе, как, например, простом эфире или дихлорметане, обработкой фенолом в карбаматы формулы IV.
Карбаматы формулы VI можно получить по аналогии с известными способами (например, ЕП 101670), однако они могут быть получены также из соответствующих изоцианатов VII путем обработки фенолом.
Изоцианаты формулы VII получают из аминов формулы III путем обработки оксалилхлоридом или фосгеном (по аналогии с Angew. Chem. 83 (1971), 407, ЕП 388873).
Сульфонамиды формулы V можно получить путем реакции соответствующих хлоридов сульфокислоты с аммиаком (Houben Weyl, Methoden der organischen Chemie, т. 9, (1955) 605). Хлориды сульфокислоты получают либо путем реакции Меервейна (диазотирование соответствующих аминов и сульфохлорирование, катализированное солью меди), или путем хлорсульфонирования соответствующих ароматических углеводородов (F. Muth в "Methoden der organischen Chemie", Houben Weyl, Thieme Verlag, Stuttgart (1955) 557 ff). Сульфонамиды формулы V можно получить также из соответственно замещенных 2-гидроксибензолсульфонамидов путем обработки соответствующими замещенными сульфонилгалогенидами в присутствии вспомогательного основания.
Типичные примеры получения промежуточных соединений приведены в экспериментальной части.
Соли соединений I можно получить известным способом (ЕП 304282, патент США 4599412). Их получают путем депротонирования соответствующих сульфонилмочевин I в воде или инертном органическом растворителе при температурах от -80oC до 120oC, предпочтительно от 0 до 60oC, в присутствии основания.
Соответствующими основаниями являются, например, гидроксиды, оксиды или алкоголяты щелочных или щелочно-земельных металлов, как, например, гидроксид натрия, калия и лития, метанолят, этанолят и трет.-бутанолят натрия, гидрид натрия и кальция и оксид кальция.
В качестве растворителей, наряду с водой, рассматриваются также, например, спирты, как, например, метанол, этанол и трет.-бутанол; простые эфиры, как, например, тетрагидрофуран и диоксан, ацетонитрил, диметилформамид; кетоны, как, например, ацетон и метилэтилкетон; а также галогенированные углеводороды.
Депротонирование может осуществляться при нормальном давлении или при давлении до 50 бар, предпочтительно при нормальном давлении до 5 бар избыточного давления.
Соединения I или гербицидные средства, содержащие эти соединения, а также их соли с щелочными или щелочно-земельными металлами, приемлемые для окружающей среды, могут эффективно применяться для борьбы с сорняками в таких культурах, как, например, пшеница, рис и кукуруза, не повреждая культурные растения; эффект достигается также при низкой норме расхода.
Поэтому объектом настоящего изобретения также является гербицидное средство, включающее активное вещество производное сульфонилмочевины и целевые добавки, приемлемые в сельском хозяйстве, содержащее в качестве производного сульфонилмочевины соединение общей формулы I, где R1 - C1-C4-алкил, ди-(C1-C4)-алкиламино, R2 - водород, R3 трифторметокси, фтор, R4 метокси, R5 - водород, Z CH, в эффективном количестве.
Средство может быть использовано в виде непосредственно распыляемых растворов, порошков, суспензий, а также высокопроцентных водных, масляных и других суспензий или дисперсий, эмульсий, масляных дисперсий, паст, пылевидных препаратов, разбрасываемых препаратов или гранулятов путем распыления, опыливания, опрыскивания или разбрасывания. Формы используемых составов зависят от поставленной задачи; в каждом случае они должны обеспечивать, по возможности, тончайшее распределение предложенных в изобретении активных веществ.
Соединения I подходят, главным образом, для получения непосредственно распыляемых растворов, эмульсий, паст или масляных дисперсий. В качестве инертных добавок используются в основном минеральные масляные фракции с температурой кипения от средней до высокой, как, например, керосин или дизельное масло, кроме того, масла каменноугольной смолы, а также масло растительного или животного происхождения, алифатические, циклические и ароматические углеводороды, например, толуол, ксилол, парафин, тетрагидронафталин, алкилированные нафталины или их производные, метанол, этанол, пропанол, бутанол, циклогексенон, циклогексенон, хлорбензол, изофорон или сильно полярные растворители, как, например, N,N-диметилформамид, диметилсульфоксид, N-метилпирролидон или вода.
Водные формы могут быть приготовлены из эмульсионных концентратов, дисперсий, паст, смачиваемых порошков или вододиспергируемых гранулятов путем добавления воды. Для получения эмульсий, паст или масляных дисперсий субстраты как таковые или растворенные в масле или растворителе гомогенизируются в воде с помощью смачивающих, адгезионных, диспергирующих или эмульгирующих средств. Могут быть изготовлены концентраты, состоящие из активного вещества, смачивающего, адгезивного, диспергирующего или эмульгирующего средства и соответственно растворителя или масла, которые разбавляются водой.
В качестве поверхностно-активных веществ рассматриваются соли щелочных, щелочно-земельных металлов, аммония, ароматических сульфокислот, например, лигнин-, фенол-, нафталин- и дибутилнафталинсульфокислоты, а также кислоты жирного ряда, алкил- и алкиларилсульфонаты, алкил-, лауриловые простые эфиры и сульфаты спирта жирного ряда, а также соли гекса-, гепта- и октадеканолов, а также простой гликолевый эфир спирта жирного ряда, продукты конденсации сульфированного нафталина и его производных с формальдегидом, продукты конденсации нафталина или нафталинсульфокислоты с фенолом и формальдегидом, простой полиоксиэтиленоктилфеноловый эфир, этоксилированный простой изооктил-, октил- или нонилфенол-, алкилфенол-, трибутилфенилполигликолевый эфир, алкиларилполиэфиры спиртов, изотридециловый спирт, конденсаты окиси этилена и спирта жирного ряда, этоксилированное касторовое масло, простой оксиэтиленалкиловый эфир или полиоксипропилен, ацетат простого полигликолевого эфира лаурилового спирта, сложный сорбитовый эфир, лигнин-отработанный сульфитный щелок или метилцеллюлоза.
Порошковые, распыляемые и разбрызгиваемые препараты могут изготавливаться путем перемешивания или размалывания активных веществ с твердым наполнителем.
Грануляты, например, грануляты в оболочке, пропиточные грануляты и гомогенные грануляты могут быть изготовлены путем присоединения активных веществ к твердым наполнителям. Твердыми наполнителями являются минеральные почвы, как, например, кремневые кислоты, силикагели, силикаты, тальк, каолин, известняк, известь, мел, болюс, лесс, глина, доломит, диатомовая земля, сульфат кальция и магния, окись магния, размолотые синтетические материалы, удобрения, как, например, сульфат аммония, фосфат аммония, нитрат аммония, мочевины и растительные продукты, как, например, зерновая мука, мука из древесной коры, древесины и ореховых скорлупок, целлюлозный порошок или другие твердые наполнители.
Составы содержат обычно от 0,1 до 95 мас. предпочтительно от 0,5 до 90 мас. активного вещества.
Примеры составов:
I. 90 мас.ч. соединения N 1 смешивают с 10 мас.ч. N-метил-α-пирролидина и получают раствор, пригодный для применения в виде мельчайших капель.
II. 20 мас.ч. соединения N 5 растворяют в смеси, состоящей из 80 мас.ч. ксилола, 10 мас.ч. продукта присоединения 8-10 моль окиси этилена к 1 моль N-моноэтаноламида масляной кислоты, 5 мас.ч. соли кальция додецилбензолсульфокислоты и 5 мас.ч. продукта присоединения 40 моль окиси этилена к 1 моль касторового масла. Путем смешивания и тонкого распределения раствора в 100000 мас.ч. воды получают водную дисперсию, содержащую 0,02 мас. активного вещества.
III. 20 мас.ч. соединения N 1 растворяют в смеси, состоящей из 40 мас.ч. циклогексанона, 30 мас. ч. изобутанола, 20 мас.ч. продукта присоединения 7 моль окиси этилена к 1 моль изооктилфенола и 10 мас.ч. продукта присоединения 40 моль окиси этилена к 1 моль касторового масла. Путем смешивания и тонкого распределения раствора в 100000 мас.ч. воды получают водную дисперсию, состоящую из 0,02 мас. активного вещества.
IV. 20 мас.ч. активного вещества N 5 растворяют в смеси, состоящей из 25 мас.ч. циклогексанона, 65 мас.ч. минеральной масляной фракции с температурой кипения от 210 до 280oC и 10 мас.ч. продукта присоединения 40 моль окиси этилена к 1 моль касторового масла. Путем смешивания и тонкого распределения раствора в 100000 мас. ч. воды получают водную дисперсию, содержащую 0,02 мас. активного вещества.
V. 20 мас.ч. активного вещества N 1 хорошо перемешивают с 3 мас.ч. частями соли натрия диизобутилнафталин-a-сульфокислоты, 17 мас.ч. соли натрия лигнинсульфокислоты из отработанного сульфитного щелока и 60 мас.ч. порошкообразного геля кремневой кислоты и размалывают в молотковой мельнице. Путем тонкого распределения смеси в 20000 мас.ч. воды получают раствор для опрыскивания, содержащий 0,1 мас. активного вещества.
VI. 3 мас. ч. активного вещества N 14 смешивают с 97 мас.ч. высокодисперсного каолина. Таким образом получают пылевидный препарат, содержащий 3 мас. активного вещества.
VII. 30 мас.ч. активного вещества N 14 хорошо перемешивают со смесью из 92 мас. ч. порошкообразного геля кремневой кислоты и 8 мас.ч. парафинового масла, которое распылено на поверхность этого геля кремневой кислоты. Таким способом получают подготовленное активное вещество с хорошей адгезионной способностью.
VIII. 20 мас. ч. активного вещества N 5 хорошо перемешивают с 2 мас.ч. соли кальция додецилбензолсульфокислоты, 8 мас.ч. простого полигликолевого эфира спирта жирного ряда, 2 мас.ч. соли натрия конденсата фенол-мочевина-формальдегида и 68 мас.ч. парафинового минерального масла. Получают стабильную масляную дисперсию.
Применение может осуществляться по способу до или после появления всходов. Если активные вещества плохо переносятся некоторыми культурными растениями, то может применяться специальная техника, благодаря которой гербицидные средства с помощью распыляющих приборов наносятся таким образом, что, по возможности, не происходит соприкосновения с листьями чувствительных растений, в то время как активные вещества попадают на листья растущих среди них нежелательных растений или попадают на открытую поверхность.
Нормы расхода активного вещества в зависимости от цели применения, времени года, растений и стадии роста достигают от 0,001 до 1,0 кг/га, предпочтительно от 0,01 до 0,5 кг/га активного вещества.
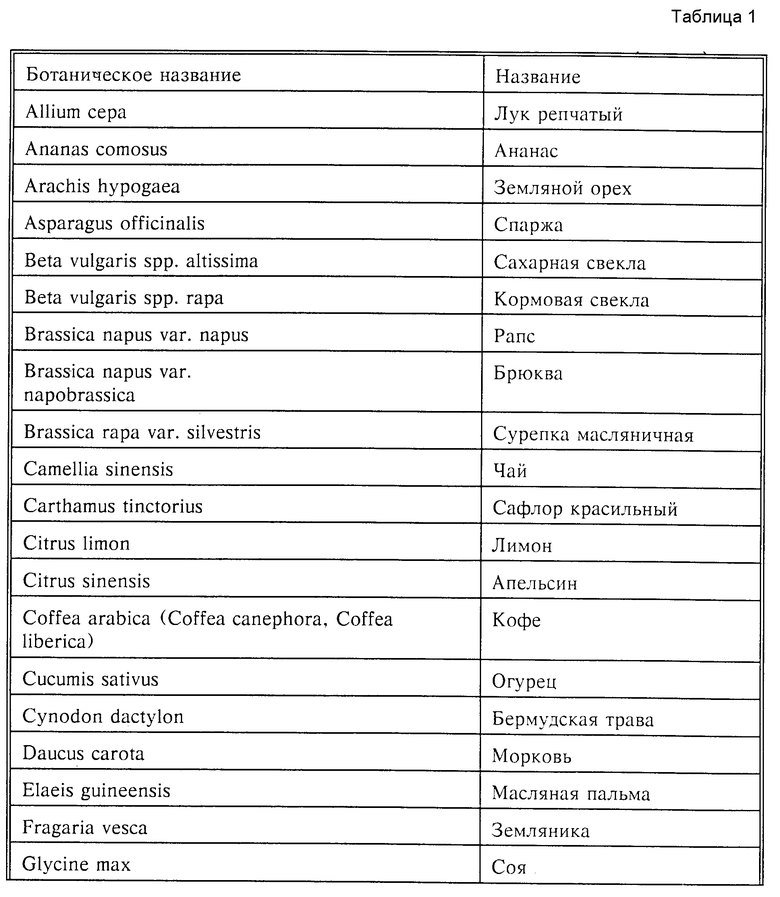
Учитывая разнообразие методов применения, предложенные в изобретении соединения или средства, содержащие эти соединения, могут применяться также для большого количества культурных растений для удаления сорняков. Рассматриваются, например, следующие культуры (табл. 1).
Для расширения спектра действия и достижения синергетического эффекта производные сульфонилмочевины формулы (I) могут смешиваться с многочисленными представителями других гербицидных и регулирующих рост растений групп активных веществ и применяться вместе с ними. В качестве компонентов для смешивания рассматриваются, например, диазин, 4Н-3,1-бензоксазин-производные, бензотиадиазиноны, 2,6-динитроанилины, N-фенилкарбаматы, тиокарбаматы, галогенкарбоновые кислоты, триазины, амиды, мочевины, простой дифениловый эфир, триазиноны, урацилы, бензофуранпроизводные, циклогексан-1,3-дион-производные, производные хинолинкарбоновой кислоты, фенилокси- или гетероарилоксифенилпропионовые кислоты, а также их соли, сложные эфиры и амиды и другие.
Кроме того, полезно соединения формулы I применять не только по отдельности, но и в сочетании с другими гербицидами, также смешанными с другими средствами для защиты растений, например, со средствами для борьбы с сорняками или фитопатогенными грибами или бактериями. Кроме того, представляют интерес смеси с растворами минеральных солей, которые применяются для устранения недостатка питательных веществ и микроэлементов. Могут добавляться также нефитотоксичные масла и масляные концентраты.
Ниже приводятся примеры синтеза соединений I.
Изготовление промежуточных продуктов:
Сложный 2-амино-4-хлорфениловый эфир метансульфокислоты
174,5 г метансульфонилхлорида (1,52 моль) при температуре 10-15oC по каплям добавляют к раствору из 218,7 г 2-амино-4-хлорфенола (1,52 моль) и 153,8 г триэтиламина (1,52 моль) в 400 мл метиленхлорида. Перемешивают в течение 18 ч при температуре 25oC, смесь вводят в ледяную воду и отделяют органическую фазу. Органическую фазу дважды промывают водой и высушивают Na2SO4. После отгонки растворителя остается постепенно кристаллизующийся остаток продукта, достаточно чистого для дальнейшего преобразования (329,5 г, 98% теор. ). Продукт можно перекристаллизовать из метанола/воды (т. пл. 70-71oC).
1Н-ЯМР спектр (250 МГц, CDCl3, int. TMS): 7,10 (d, 1Н), 6,75 (dd, 1Н), 6,67 (dd, 1Н), 4,10 (уш. 2Н), 3,16 (s, 3Н).
Сложный 2-хлорсульфонилфениловый эфир метансульфокислоты
Раствор соли диазония, полученного путем одновременного введения раствора из 39,5 г нитрата натрия (0,57 моль) в 60 мл воды и 105 г сложного 2-аминофенилового эфира метансульфокислоты (0,57 моль) в 200 мл концентрированной соляной кислоты при температуре 0-5oC и перемешивания в течение 1 ч при температуре 0oC, прикапывается в насыщенный диоксидом серы раствор из 1,7 г CuCl2 и 4,5 г хлорида бензилтриэтиламмония в 200 мл 1,2-дихлорэтана и 10 мл воды при температуре 0-10oC. Удаляют охлаждение и перемешивают при температуре 25oC в течение 30 мин, затем при медленном повышении температуры реакционной смеси еще в течение 1 ч при температуре 50oC. Органическую фазу удаляют, промывают ледяной водой и высушивают над Na2SO4. После удаления растворителя остается маслянистый остаток, который можно кристаллизовать путем растирания небольшим количеством этанола. Таким образом получают 139 г соединения, указанного в заголовке (90% теор.) с т. пл. 94-95oC.
1Н-ЯМР (300 МГц, CDCl3, int. TMS): 8,10 (d, 1Н), 7,68-7,85 (m, 2Н), 7,51 (1Н), 3,42 (s, 3Н).
Сложный (2-аминосульфонил)фениловый эфир метансульфокислоты
Аммиак при температуре -30oC вводят в раствор из 130 г сложного (2-хлорсульфонил)фенилового эфира метансульфокислоты в 1 л тетрагидрофурана и контролируют превращения посредством тонкослойной хроматографии. После осуществления реакции тетрагидрофуран отгоняют в вакууме, получаемом с помощью водоструйного насоса, и остаток растирают с водой и простым диэтиловым эфиром. Таким образом получают 110,2 г соединения, указанного в заголовке (91%) с т. пл. 131-132oC.
1Н-ЯМР (300 МГц, CD3SOCD3, int. TMS): 7,90 (d, 1Н), 7,61 (уш, 2Н), 7,45-7,74 (m, 3Н), 3,50 (s, Н).
2-(диметиламиносульфонилокси)бензолсульфонамид
Раствор из 5,0 г 2-оксибензолсульфонамида (29 ммоль) в 200 мл ацетонитрила смешивают при температуре 25oC с 4,0 г карбоната калия (29 ммоль). Через 10 мин перемешивания по каплям добавляют 4,1 г диметиламиносульфонилхлорида (29 ммоль) при температуре 25oC и перемешивают еще 16 ч при этой температуре. Летучие компоненты удаляют в вакууме, получаемом с помощью водоструйного насоса, остаток поглощают сложным этиловым эфиром уксусной кислоты, раствор высушивают над Na2SO4 и растворитель удаляют в вакууме, получаемом с помощью водоструйного насоса. Остаток сильно перемешивают в течение 1 ч со 100 мл простого диэтилового эфира. Кристаллический продукт отсасывают и высушивают при температуре 40oC в вакууме. Таким образом получают 5,7 г соединения, указанного в заголовке.
1Н-ЯМР спектр (250 МГц, CD3SOCD3, int. TMS) d: 7,91 (d, 1Н), 7,57-7,74 (m, 2Н), 7,49 (уш, 2Н), 7,36-7,52 (m, 1Н), 3,02 (s, 6Н).
Получение промежуточных продуктов III
2,4-дифтор-6-трихлорметокси-1,3,5-триазин
В смесь из 300 г (2,041 моль) 2,4-дифтор-6-метокси-1,3,5-триазина и 0,3 г a,α′-азоизобутиронитрила при температуре 130oC и при УФ-излучении вводят поток газообразного хлора таким образом, что в течение 2 ч устанавливается температура от 140 до 145oC. После ЯМР-спектроскопического контроля протекания реакции при внешнем обогреве вводят газообразный хлор еще в течение 3 ч при 135-140oC. После отсасывания выпавшего осадка и отгонки фильтрата в вакууме получают 444 г (87% теор.) указанного соединения с температурой кипения 40-46oC/0,3 мбар.
2,4-дифтор-6-трифторметокси-1,3,5-триазин
К раствору из 187,4 г (1,048 моль) трифторида сурьмы и 35,2 г (0,117 моль) пентахлорида сурьмы добавляют половину от 210 г (0,838 моль) 2,4-дифтор-6-трихлорметокси-1,3,5-триазина сначала при температуре 110oC при помешивании таким образом, что сначала устанавливается температура 125oC; при установлении устойчивого кипения добавление ведут при дополнительном нагревании. В течение 1 ч перемешивают при температуре 125-130oC и кипящую при температуре 100-105oC фракцию отгоняют через 25-сантиметровую насадочную колонну. После затухания реакции в течение 30 мин по каплям добавляют оставшуюся половину трихлорметоксипроизводного и фракцию при температуре 100-105oC непрерывно отгоняют. Общая продолжительность реакции достигает 3 ч. Получают 134,4 г (79,8% теор.) соединения, указанного в заголовке, c n
6-хлордифторметокси-2,4-дифтор-1,3,5-триазин
210 г (0,838 моль) 2,4-дифтор-6-трихлорметокси-1,3,5-триазина в течение 10 мин при помешивании при температуре 110oC добавляют к 110 г (0,614 моль) трифторида сурьмы. После подачи 3/4 от 9,38 г (0,0313 моль) пентахлорида сурьмы нагревают до 145oC и перемешивают в течение 1 ч. Добавляют оставшийся катализатор и еще раз перемешивают в течение 2 ч, причем в виде низкокипящей фракции через 30-сантиметровую насадочную колонну в пределах от 95 до 105oC получают 20 г (11,8% теор. ) 2,4-дифтор-6-трифторметокси-1,3,5-триазина. Остаток перегонки перегоняют без колонны и получают 94,8 г (52% теор.) соединения, указанного в заголовке, с температурой кипения 125-130oC; n
2,4-дихлор-6-трифторметокси-1,3,5-триазин
К смеси из 40,9 г (0,229 моль) фторида сурьмы и 7,03 г (0,0234 моль) пентахлорида сурьмы добавляют при помешивании 52 г (0,183 моль) 2,4-дихлор-6-трихлорметокси-1,3,5-триазина в течение 5 мин при температуре 90oC, причем температуру повышают до 180oC. Перемешивают еще в течение 20 мин при температуре от 170 до 180oC и затем отгоняют сырой продукт при температуре 90-103oC/70 мбар. С помощью повторной перегонки получают 32,3 г (75,5% теор. ) указанного соединения с температурой кипения 165-173oC.
2-амино-4-фтор-6-трифторметокси-1,3,5-триазин
4,4 г (0,295 моль) газообразного аммиака в течение 45 мин при температуре от -70 до -65oC при перемешивании вводят в смесь из 26,0 г (0,1293 моль) 2,4-дифтор-6-трифторметокси-1,3,5-триазина в 100 мл тетрагидрофурана. Смесь перемешивают с течение 2 ч при температуре -70oC и в течение ночи при нагревании до 22oC. После концентрирования в вакууме остаток смешивают с водой, отсасывают и промывают. После высушивания получают 22 г (85,9% теор.) указанного соединения с т. пл. от 138 до 139oC.
2,4-бисметиламино-6-трифторметокси-1,3,5-триазин и
2-метиламино-4-фтор-6-трифторметокси-1,3,5-триазин
5,9 г (0,189 моль) метиламина при -70oC в течение 30 мин вводят при помешивании в смесь из 19,0 г (0,0945 моль) 2,4-дифтор-6-трифторметокси-1,3,5-триазина в 100 мл простого диэтилового спирта. Смесь перемешивают в течение 2 ч при температуре -70oC и в течение ночи при нагревании до 22oC. Реакционную смесь концентрируют в вакууме, поглощают метиленхлоридом и промывают водой. После высушивания разделяют на фракции хроматографическим способом через силикагелевые колонки, причем в первых обеих фракциях получают 5,0 г (25% теор.) 2-метиламина-4-фтор-6-трифторметокси-1,3,5-триазина с т. пл. 68-72oC. В следующих фракциях 4-7 выделяют 10,7 г (51% теор.) плохо растворимого 2,4-бисметиламино-6-трифторметокси-1,3,5-триазина с т. пл. 150-152oC.
2-амино-4-хлордифторметокси-6-фтор-1,3,5-триазин и
2,4-диамино-6-диамино-6-дифторметокси-1,3,5-триазин
7,8 г (0,46 моль) аммиака вводят в течение 45 мин при помешивании при температуре -70oC в смесь из 50,0 г (0,23 моль) 2,4-дифтор-6-хлордифторметокси-1,3,5-триазина в 150 мл тетрагидрофурана. В течение 2 ч при температуре -70oC и в течение ночи при нагревании до 22oC смесь перемешивают. Реакционную смесь концентрируют в вакууме, промывают водой и высушивают. Затем реакционный продукт взмучивают с помощью метиленхлорида на силикагелевой колонне и элюируют подобным растворителем. Во фракциях 1-8 получают 21,5 г (43,6% теор. ) 2-амино-4-фтор-6-хлордифторметокси-1,3,5-триазина с т. пл. 131-133oC. Путем последующей промывки простым уксусным эфиром выделяют из фракций 9-14 плохо растворимый 2,4-диамино-6-хлордифторметокси-1,3,5-триазин (11,2 г, 23% теор.) с т. пл. 114oC.
2-хлордифторметокси-4-фтор-6-метиламино-1,3,5-триазин и
2,4-бисметиламино-6-хлордифторметокси-1,3,5-триазин
5,2 г (0,166 моль) метиламина вводят в течение 20 мин при помешивании при температуре -70oC в смесь из 18,1 г (0,083 моль) 4-дифторхлорметокси-2,6-дифтор-1,3,5-триазина. Смесь перемешивают в течение 2 ч при температуре -70oC и в течение ночи при нагревании до 22oC. Реакционную смесь концентрируют в вакууме, поглощают метиленхлоридом, промывают водой и высушивают. После хроматографического разделения на силикагеле получают в первых фракциях 5,5 г (29% теор.) 2-хлордифторметокси-4-фтор-6-метиламино-1,3,5-триазина с т. пл. 62-64oC. В последнем погоне выделяют 8,7 г (44% теор.) 2,4-бисметиламино-6-хлордифторметокси-1,3,5-триазина с т. пл. 118-120oC.
2-амино-4-метокси-6-трифторметокси-1,3,5-триазин
9,1 г (0,05 моль) 30-ного метилата натрия добавляют при 0oC в течение 15 мин при помешивании в смесь из 10 г (0,05 моль) 2-амино-4-фтор-6-трифторметокси-1,3,5-триазина в 100 мл метанола. После перемешивания в течение 1 ч при температуре 0oC смесь концентрируют в вакууме, поглощают метиленхлоридом и экстрагируют водой. После высушивания и концентрирования получают 10,5 г (99% теор.) соединения, указанного в заголовке, с т. пл. 96-101oC.
2-амино-4-хлордифторметокси-6-метокси-1,3,5-триазин
8,4 г (0,047 моль) 30%-ного метилата натрия добавляют при температуре 0oC в течение 15 мин при помешивании в смесь из 10 г (0,047 моль) 2-амино-4-хлордифторметокси-6-фтор-1,3,5-триазина в 100 мл метанола. После перемешивания в течение 1 ч при 0oC смесь концентрируют в вакууме, поглощают метиленхлоридом и экстрагируют водой. После высушивания и концентрирования получают 10,4 г (98,5% теор. ) соединения, указанного в заголовке, с т. пл. 109-110oC.
2-амино-4-метокси-6-трифторметокси-1,3,5-триазин
9,1 г (0,05 моль) 30%-ного метилата натрия добавляют при 0oC в течение 15 мин при помешивании в смесь из 10 г (0,05 моль) 2-амино-4-фтор-6-трифторметокси-1,3,5-триазина в 100 мл метанола. После перемешивания в течение 1 ч при 0oC смесь концентрируют в вакууме, поглощают метиленхлоридом и экстрагируют водой. После высушивания и концентрирования получают 10,5 г (99% теор.) соединения, указанного в заголовке, с т. пл. 96-101oC.
2-амино-4-хлордифторметокси-6-метокси-1,3,5-триазин
8,4 г (0,047 моль) 30%-ного метилата натрия добавляют при 0oC в течение 15 мин при помешивании в смесь из 10 г (0,047 моль) 2-амино-4-хлордифторметокси-6-фтор-1,3,5-триазина в 100 мл метанола. После перемешивания в течение 1 ч при 0oC смесь концентрируют в вакууме, поглощают метиленхлоридом и экстрагируют водой. После высушивания и концентрирования получают 10,4 г (98,5% теор.) соединения, указанного в заголовке, с т. пл. 109-110oC.
2-амино-4-этокси-6-трифторметокси-1,3,5-триазин
2,3 г (0,093 моль) 97%-ного гидрида натрия порциями при температуре от 20 до 35oC добавляют к 300 мл этанола и перемешивают в течение 15 мин до получения раствора. При температуре 0oC при помешивании добавляют 18,5 г (0,093 моль) 2-амино-4-фтор-6-трифторметокси-1,3,5-триазина в течение 10 мин, перемешивают 1 ч при 0oC и в течение ночи при температуре 22oC. После концентрирования в вакууме остаток поглощают метиленхлоридом, экстрагируют водой и высушивают. После концентрирования получают 17,9 г (85,9% теор.) указанного соединения с т. пл. от 96 до 91oC.
2-амино-4-хлордифторметокси-6-этокси-1,3,5-триазин
1,2 г (0,047 моль) 97%-ного гидрида натрия добавляют при температуре от 20 до 35oC порциями к 150 мл этанола и перемешивают в течение 15 мин до образования раствора. Затем при 0oC при помешивании добавляют 10 г (0,047 моль) 2-амино-4-хлордифторметокси-6-фтор-1,3,5-триазина, перемешивают в течение 1 ч при температуре 0oC и в течение ночи при температуре 22oC. После концентрирования в вакууме остаток поглощают метиленхлоридом, экстрагируют водой и высушивают. После концентрирования получают 10,6 г (94,6% теор.) указанного соединения с т. пл. от 63 до 65oC.
2-амино-4-метиламино-6-трифторметокси-1,3,5-триазин
3,5 г (0,111 моль) метиламина вводят при 0oC в течение 20 мин при помешивании в раствор из 11 г (0,055 моль) 2-амино-4-фтор-6-трифторметокси-1,3,5-триазина в 150 мл тетрагидрофурана. Перемешивают в течение 1 ч при 0oC и в течение ночи при температуре 22oC. Реакционную смесь концентрируют в вакууме, смешивают с водой и высушивают. Получают 10,8 г (93,1% теор.) указанного соединения с т. пл. от 155 до 157oC (разложение).
2-амино-4-хлордифторметокси-6-метиламино-1,3,5-триазин
2,9 г (0,093 моль) метиламина вводят в течение 20 мин при помешивании при 0oC в раствор из 10 г (0,047 моль) 2-амино-4-хлордифторметокси-6-фтор-1,3,5-триазина в 150 мл простого диэтилового эфира. Перемешивают 1 ч при 0oC и в течение ночи при 22oC. После промывания водой, высушивания и концентрирования получают 9,4 г (89,5% теор.) указанного соединения с т. пл. 143oC (разложение).
2-амино-4-диметиламино-6-трифторметокси-1,3,5-триазин
5,0 г (0,111 моль) диметиламина вводят в течение 20 мин при помешивании при температуре 0oC в раствор из 11 г (0,055 моль) 2-амино-4-фтор-6-трифторметокси-1,3,5-триазина в 150 мл тетрагидрофурана. Перемешивают в течение 1 ч при температуре 0oC и в течение ночи при температуре 22oC. После концентрирования, промывания водой и высушивания получают 9,9 г (80,7% теор.) указанного соединения с т. пл. 114-118oC (разложение).
2-амино-4-хлордифторметокси-6-диметиламино-1,3,5-триазин
4,2 г (0,093 моль) диметиламина вводят в течение 20 мин при помешивании при температуре 0oC в раствор из 10 г (0,047 моль) 2-амино-4-хлордифторметокси-6-фтор-1,3,5-триазина в 150 мл простого диэтилового эфира. Перемешивают 1 ч при температуре 0oC и в течение ночи при температуре 22oC. После промывания водой, высушивания и концентрирования получают 9,8 г (87,8% теор. ) соединения, указанного в заголовке, с т. пл. от 130 до 133oC (разложение).
2-хлор-4-трихлорметокси-6-трихлорметилпиримидин
а) 2-хлор-4-метокси-6-трихлорметилпиримидин
293,1 г (1,692 моль) 30%-ного раствора метилата натрия добавляют в течение 1,5 ч при температуре от 0 до 5oC к раствору из 434 г (1,692 моль) 2,6-дихлор-4-трихлорметилпиримидина в 1 л 1,2-дихлорэтана. Перемешивают в течение 1 ч при температуре от 0 до 5oC и 12 ч при температуре 25oC. Реакционную смесь четырехкратно экстрагируют водой и трехкратно насыщенным раствором поваренной соли. После высушивания над сульфатом магния и концентрирования получают 423 г (95% теор.) соединения, указанного в заголовке, в виде почти бесцветного масла.
1Н-ЯМР (CDCl3) (частей на миллион) OCH3 (s/3Н) 4,1; CH (s/1Н) 7,25.
б) 2-хлор-4-трихлорметокси-6-трихлорметилпиримидин
В раствор из 210 г (0,802 моль) 2-хлор-4-метокси-6-трихлорметилпиримидина и 260 мг (0,0016 моль) α,α′-азоизобутиронитрила вводят хлор при УФ-излучении и контроле за ходом протекания реакции с помощью газовой хроматографии при температуре сначала 110oC, причем также после удаления нагревательной бани устанавливается температура реакции 140oC. После затухания реакции в течение 5,5 ч при 20oC вводят всего 341 г (4,8 моль) хлора. К охлаждающейся реакционной смеси от 40oC примешивают н-пентан для образования осадка. Осадок отсасывают, промывают петролейным эфиром и высушивают, причем получают 163 г (55% теор. ) соединения, указанного в заголовке, с т. пл. 67-69oC.
Фильтрат (113,8 г) после газовой хроматограммы состоял из 83% соединения, указанного в заголовке, 4% 2-хлор-4-дихлорметокси-6-трихлорметилпиримидина и 9% 2,4-дихлор-6-трихлорметилпиримидина. Общий выход соединения, указанного в заголовке, составил 87,6% теории.
2,4-дифтор-6-трихлорметоксипиримидин
210 г (2,96 моль) хлора при УФ-излучении и контроле за ходом протекания реакции посредством газовой хроматографии в течение 2,5 ч при помешивании при 130oC вводят в 123 г (0,843 моль) 2,2-дифтор-6-метоксипиримидина, полученного по способу, описанному в Европейском патенте 378089. Реакционную смесь перегоняют через 10-сантиметровую Vigreux-колонну в вакууме, причем получают 190,2 г (90,5% теор.) соединения, указанного в заголовке, с температурой кипения 40 -43oC/0,2 мбар.
2,4-дихлор-6-трихлорметоксипиримидин
303 г (4,27 моль) хлора при УФ-излучении и контроле за ходом реакции с помощью газовой хроматографии, при помешивании вводят в течение 0,5 ч при 80oC, 1 ч при температуре 100oC, 3 ч при температуре 120oC и 3 ч при 150oC в смесь из 209 г (1,168 моль) 2,6-дихлор-4-метоксипиримидин и 2 г (0,012 моль) α,α′-азоизобутиронитрила. Затем реакционную смесь перегоняют в вакууме. Получают 241,3 г (73% теор.) соединения, указанного в заголовке, с температурой кипения 87-88oC/0,4 мбар; температура плавления 55-56oC.
2,4-дифтор-6-трифторметоксипиримидин
49,9 г (0,2 моль) 2,4-дифтор-6-трихлорметоксипиримидина добавляют при температуре 100oC в течение 15 мин при помешивании к смеси из 39,3 г (0,22 моль) трифторида сурьмы и 9,38 г (0,031 моль) пентахлорида сурьмы. В течение 25 мин повышают температуру ванны от 100 до 150oC и еще перемешивают в течение 30 мин, причем при температуре в пределах от 120 до 125oC устанавливается рефлюкс. Путем последующей перегонки получают 37,1 г (92,7% теор.) соединения, указанного в заголовке, с температурой кипения 125-127oC.
6-хлордифторметокси-2,4-дифторпиримидин
93 г (0,373 моль) 2,4-дифтор-6-трихлорметоксипиримидина добавляют при 100oC в течение 10 мин при помешивании к смеси из 44,5 г (0,249 моль) трифторида сурьмы и 0,94 г (0,0031 моль) пентахлорида сурьмы. В течение 25 мин повышают температуру ванны от 100 до 175oC, причем при температуре 145oC наступает рефлюкс. После перемешивания в течение 1,5 ч реакционный продукт отгоняют при 146-150oC. Дистиллят растворяют в метиленхлориде, экстрагируют 6 н. соляной кислотой и высушивают над сульфатом магния. После концентрирования в вакууме получают в виде остатка соединение, указанное в заголовке, с выходом 63,7 г (78,8% теор.).
2-фтор-4-трифторметокси-6-трифторметилпиримидин
80 г (0,219 моль) 2-хлор-4-трихлорметил-6-трихлорметоксипиримидина добавляют в течение 5 мин при помешивании при температуре 100oC к смеси из 93,9 г (0,525 моль) трифторида сурьмы и 18,7 г (0,0627 моль) пентахлорида сурьмы. В течение 10 мин температуру ванны повышают до 140oC и перемешивают еще в течение 1 ч, причем наступает более сильный рефлюкс. Продукт реакции перегоняют при температуре 135-140oC, к концу при 95oC/50 мбар. Дистиллят забирают метиленхлоридом, экстрагируют 6 н. соляной кислотой и высушивают над сульфатом магния. После концентрирования в вакууме получают соединение, указанное в заголовке, с выходом 35,9 г (65,5% теор.).
2,4-дихлор-6-трифторметоксипиримидин
115 г (0,407 моль) 2,4-дихлор-6-трихлорметоксипиримидина добавляют в течение 5 мин при помешивании при 100oC к смеси из 80 г (0,447 моль) трифторида сурьмы и 18,77 г (0,0627 моль) пентахлорида сурьмы, причем температуру реакции повышают до 140oC. Перемешивают еще в течение 45 мин при 150oC. Для дистилляции устанавливается давление 210 мбар, причем соединение, указанное в заголовке, переходит при температуре 128oC; последние летучие компоненты перегоняют при температуре 110oC/22 мбар. Дистиллят растворяют в метиленхлориде, экстрагируют с помощью 6 н. соляной кислоты и высушивают над сульфатом магния. После концентрирования в вакууме получают соединение, указанное в заголовке, с выходом 80 г (84,4% теор.) в виде бесцветного масла, n
2-амино-4-хлордифторметокси-6-фторпиримидин
9,8 г (0,578 моль) газообразного аммиака вводят в течение 1 ч при температуре от -75 до -70oC при помешивании в смесь из 62,5 г (0,289 моль) 2,4-дифтор-6-хлордифторметоксипиримидина в 300 мл тетрагидрофурана. Перемешивают в течение 1 ч при -70oC и затем нагревают до комнатной температуры. Выпавший осадок отсасывают, распределяют между уксусным эфиром и водой и органическую фазу высушивают над сульфатом магния. Реакционный фильтрат концентрируют, растворяют в вышеуказанной фазе уксусного эфира, хроматографируют через силикагель с помощью петролейного эфира и простого эфира в соотношении 5:1 и концентрируют. Получают 46,5 г (75,3% теор.) соединения, указанного в заголовке, в виде бесцветных кристаллов с температурой плавления 77 -80oC.
2-амино-4-фтор-6-трифторметоксипиримидин
8,7 г (0,51 моль) газообразного аммиака вводят в течение 1 ч при температуре от -75 до -70oC при помешивании в смесь из 51 г (0,255 моль) 2,4-дифтор-6-трифторметоксипиримидина в 200 мл простого диэтилового эфира. Перемешивают еще 1,5 ч при -70oC и 1 ч при комнатной температуре. Реакционную смесь концентрируют в вакууме, поглощают метиленхлоридом и экстрагируют с помощью воды. После высушивания органической фазы, концентрирования и хроматографического разделения через силикагель с помощью петролейного эфира и простого эфира в соотношении 8:1 получают 38,1 г (75,6% теор.) соединения, указанного в заголовке, в виде бесцветных кристаллов с температурой плавления 86-89oC.
2-амино-4-хлор-6-трифторметоксипиримидин
4,3 г (0,25 моль) газообразного аммиака вводят в течение 45 мин при помешивании при температуре от -50 до -45oC в смесь из 23,3 г (0,1 моль) 2,4-дихлор-6-трифторметоксипиримидина в 150 мл метил-трет.-бутилового эфира. Перемешивают в течение 30 мин при -50oC, в течение 1 ч при -30oC и в течение 1 ч при температуре 25oC. Выпавший остаток отсасывают, промывают водой и высушивают, причем получают 5,4 г (33,1% теор.) 4-амино-2,4-дихлорпиримидина с температурой плавления 270-272oC в качестве побочного продукта. Фильтрат промывают водой, высушивают, частично концентрируют в вакууме и разделяют на фракции хроматографическим способом с помощью петролейного эфира и простого эфира в соотношении 5:1, причем в первых фракциях получают 3 г (12,8% теор.) исходного материала в виде бесцветного масла и в последнем погоне 9 г (42% теор. ) соединения, указанного в заголовке, в виде бесцветных кристаллов с температурой плавления 55 -56oC. Конверсия составляет 48,3%
4-хлордифторметокси-6-фтор-2-метиламинопиримидин
20,3 г (0,0938 моль) 4-хлордифторметокси-2,6-дифторпиримидина помещают в 150 мл тетрагидрофурана и при температуре от -70 до -60oC смешивают в течение 30 мин с 5,8 г (0,188 моль) газообразного метиламина. Перемешивают соответственно 1 ч при температурах -70oC, 0 и 25oC. После концентрирования реакционной смеси в вакууме остаток смешивают с водой, дважды экстрагируют уксусным эфиром и экстракт высушивают над сульфатом магния. Частично концентрируют в вакууме и затем разделяют на фракции хроматографическим способом через силикагель с помощью простого эфира и петролейного эфира в соотношении 1:5. Первые фракции содержат 12,5 г (58,5%) соединения, указанного в заголовке, с температурой плавления 57-61oC.
2-амино-4-трифторметокси-6-трифторметилпиримидин
4,7 г (0,278 моль) газообразного аммиака вводят при помешивании при температуре от -75 до -70oC в течение 1 ч в смесь из 38,0 г (0,147 моль) 2-фтор(хлор)-4-трифторметокси-6-трифторметилпиримидина в 150 мл простого диэтилового эфира. Перемешивают соответственно в течение 2 ч при температуре -75oC и после нагревания при 25oC. После отсасывания из выпавшего остатка органическую фазу экстрагируют водой, высушивают и частично концентрируют. После хроматографического разделения с помощью метил-трет.-бутилового эфира через силикагель получают 20,4 г (56,1% теор.) соединения, указанного в заголовке, с температурой плавления 47-49oC.
2-амино-4-метокси-6-трифторметоксипиримидин
2,7 г (0,015 моль) 30%-ного метилата натрия в течение 15 мин при помешивании при температуре от -5oC до 0oC добавляют к 2,95 г (0,015 моль) 2-амино-4-фтор-6-трифторметоксипиримидина в 50 мл метанола. После перемешивания в течение 1 ч при 0oC и нагревании до 25oC реакционную смесь концентрируют в вакууме, смешивают с водой и экстрагируют дважды метиленхлоридом. После высушивания и концентрирования в вакууме получают 3,1 г (98% теор.) соединения, указанного в заголовке, c n
2-амино-4-хлордифторметокси-6-фторпиримидин
26,1 г (0,145 моль) 30%-ного метилата натрия добавляют в течение 15 мин при помешивании при температуре от -10oC до 0oC к 31,0 г (0,145 моль) 2-амино-4-хлордифторметокси-6-фторпиримидина в 300 мл метанола. Перемешивают в течение 30 мин при температуре 0oC и в течение 1 ч при температуре 25oC. Реакционную смесь концентрируют в вакууме, перерабатывают, как указывается выше. Получают 31,6 г (96,6% теор.) соединения, указанного в заголовке, в виде бесцветного масла с n
4-хлордифторметокси-2-метиламино-6-метоксипиримидин
4,7 г (0,026 моль) 30%-ного натрия добавляют в течение 10 мин при помешивании при 0oC к 6,0 г (0,0263 моль) 4-хлордифторметокси-6-фтор-2-метиламинопиримидина в 100 мл метанола. Соответственно перемешивают в течение 1 ч при температуре 0oC и при 25oC. После обычной обработки получают 6,3 г (100% теор.) соединения, указанного в заголовке, с температурой плавления 49-53oC.
4-хлордифторметокси-6-диметиламино-2-метиламинопиримидин
1,9 г (0,0417 моль) газообразного диметиламина вводят в течение 10 мин при помешивании при 0oC в смесь из 8,9 г (0,0417 моль) 2-амино-4-хлордифторметокси-6-фторпиримидина в 100 мл тетрагидрофурана. Перемешивают в течение 1 ч при 0oC и 2 ч при температуре 25oC. После обычной обработки получают 9,7 г (97,5% теор. ) соединения, указанного в заголовке, с температурой плавления 127-130oC.
Получение конечных продуктов I.
1. Сложный [2-[[(4-метокси-6-трифторметоксипиримидин-2-ил)амино- карбонил]аминосульфонил]фениловый эфир]метансульфокислоты
Раствор из 3 г 2-амино-4-метокси-6-трифторметоксипиримидина (14 ммоль) в 10 г 1,2-дихлорэтана смешивают при 25oC с 4 г сложного 2-изоцианатосульфонилфенилового эфира метансульфокислоты (14 ммоль). Перемешивают в течение 10 мин, удаляют растворитель в простом эфире/пентане (1:1, объем:объем). Кристаллический продукт отсасывают и высушивают в вакууме, получаемом с помощью водоструйного насоса, при температуре 40oC. Таким образом получают 5 г соединения, указанного в заголовке (73% теор. ), с температурой плавления 146-149oC.
2. Сложный [2-[[(4-фтор-6-метоксипиримидин-2-ил)аминокарбонил]аминосульфонил]фениловый эфир метансульфокислоты
Суспензию из 2 г 2-амино-4-метокси-6-трифторметоксипиримидина (14 ммоль) в 10 г 1,2-дихлорэтана смешивают при температуре 25oC с 4 г сложного (2-изоцианатосульфонилфенилового) эфира метансульфокислоты (14 ммоль). Образуется гомогенный раствор, из которого через примерно 30 мин выделяется объемистый белый осадок. Продукт отсасывают, промывают небольшим количеством 1,2-дихлорэтана и высушивают в вакууме, получаемом с помощью водоструйного насоса, при 40oC. Получают 2 г соединения, указанного в заголовке (34% теор.), с температурой плавления 168 169oC.
3. [2-[[(4-Метокси-6-трифторметокси-1,3,5-триазин-2-ил)аминокарбонил] аминосульфонил]фениловый сложный эфир натриевой соли метансульфокислоты
Раствор из 3 г сложного [2-[[(4-метокси-6-трифторметокси-1,3,5-триазин-2-ил)аминокарбонил] аминосульфонил] фенилового эфира метансульфокислоты (6,2 ммоль) в 30 мл метанола смешивают при температуре 25oC с 1,1 г (6,2 ммоль) раствора метилата натрия (30 мас.) в метаноле. Перемешивают в течение 2 мин при 25oC и удаляют растворитель при температуре 80oC в вакууме, получаемом с помощью водоструйного насоса. Получают соединение, указанное в заголовке, с количественным выходом с температурой разложения 130-135oC.
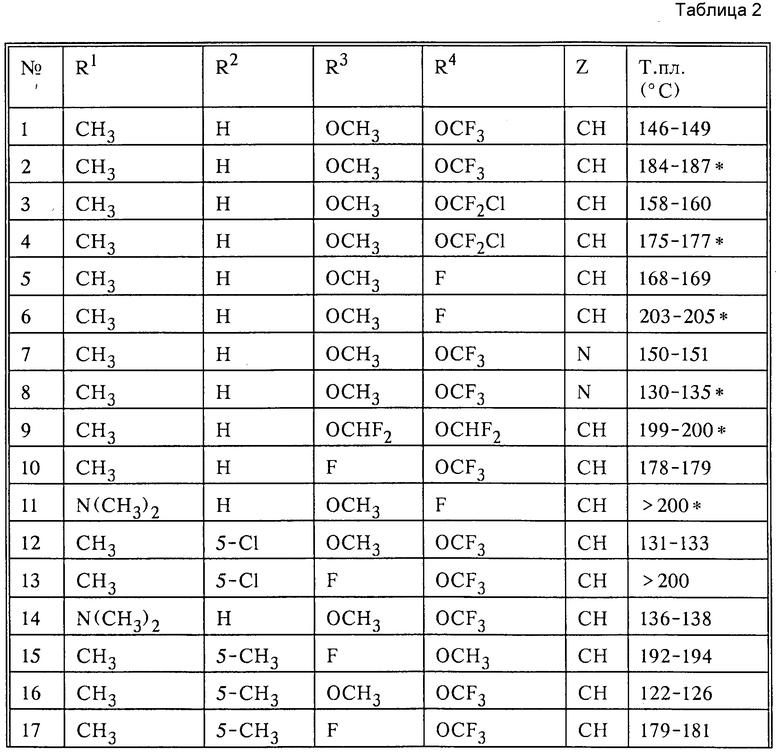
Указанные в нижеследующей табл. 2 активные вещества получают аналогичным способом.
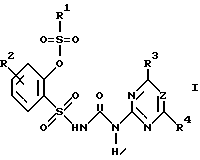
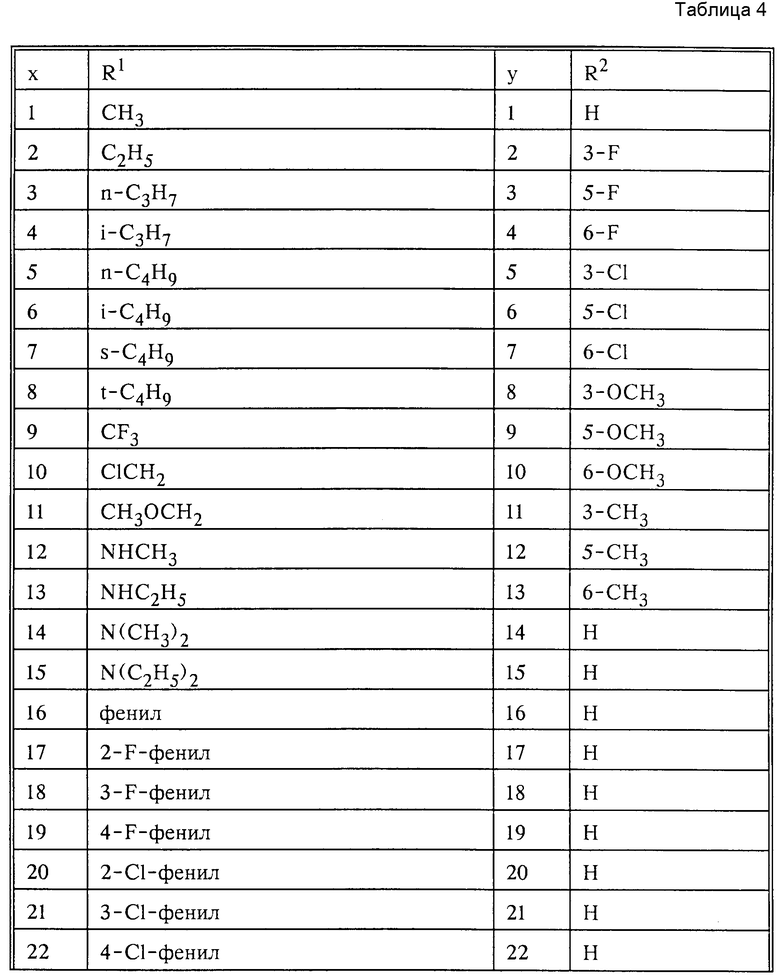
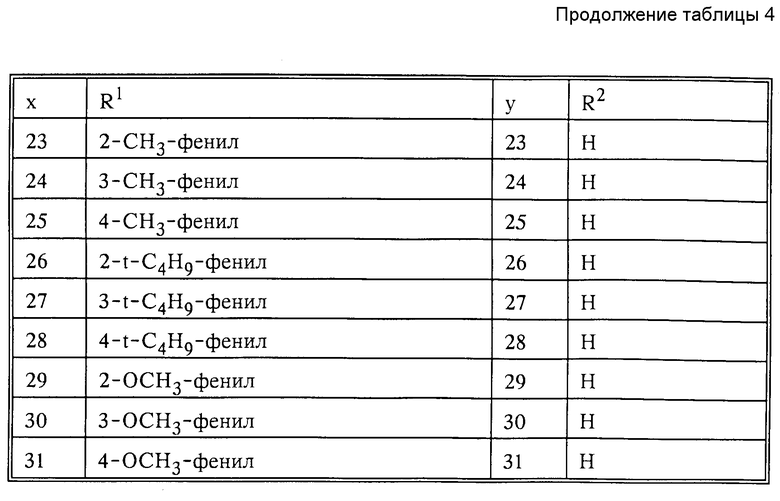
Примеры других гербицидно активных производных сульфонилмочевины I, получаемых аналогичным способом, приведены ниже в табл. 3. Для этого за основу принимают упрощенные формулы (I') и (I'')
Ax-y-SO2-NH-CO-NH-Tn (I')
Ax-y-SO2-NH-CO-NH-Pn (I")
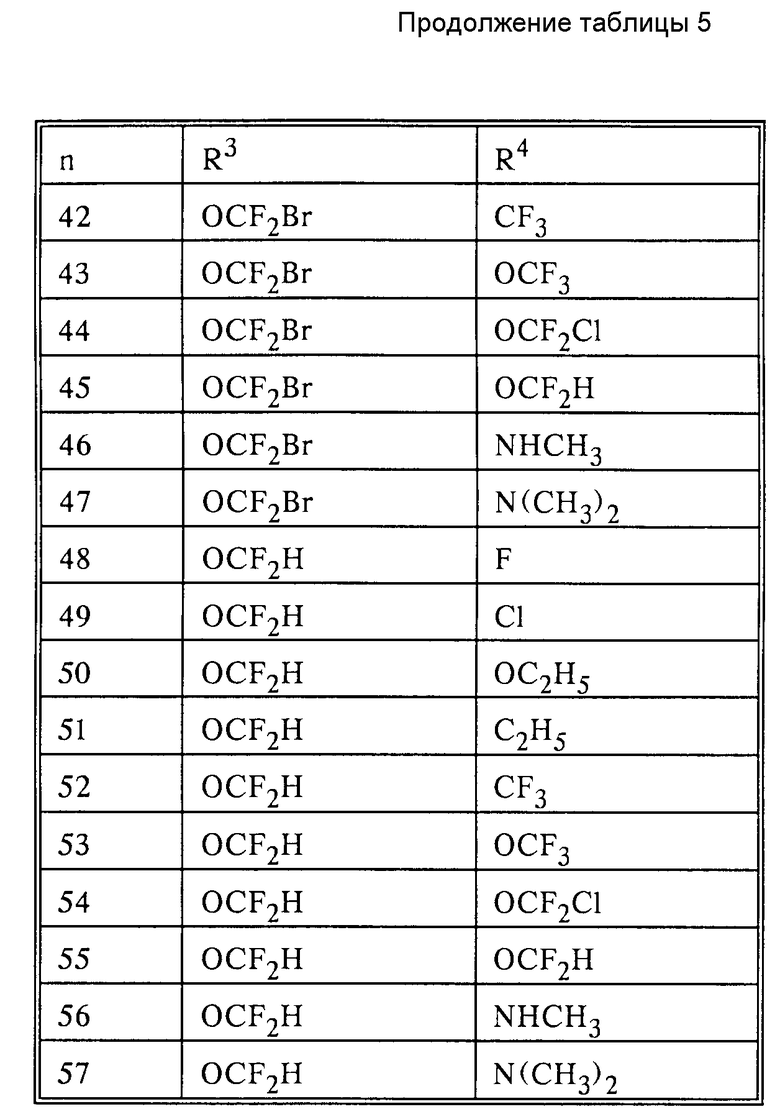
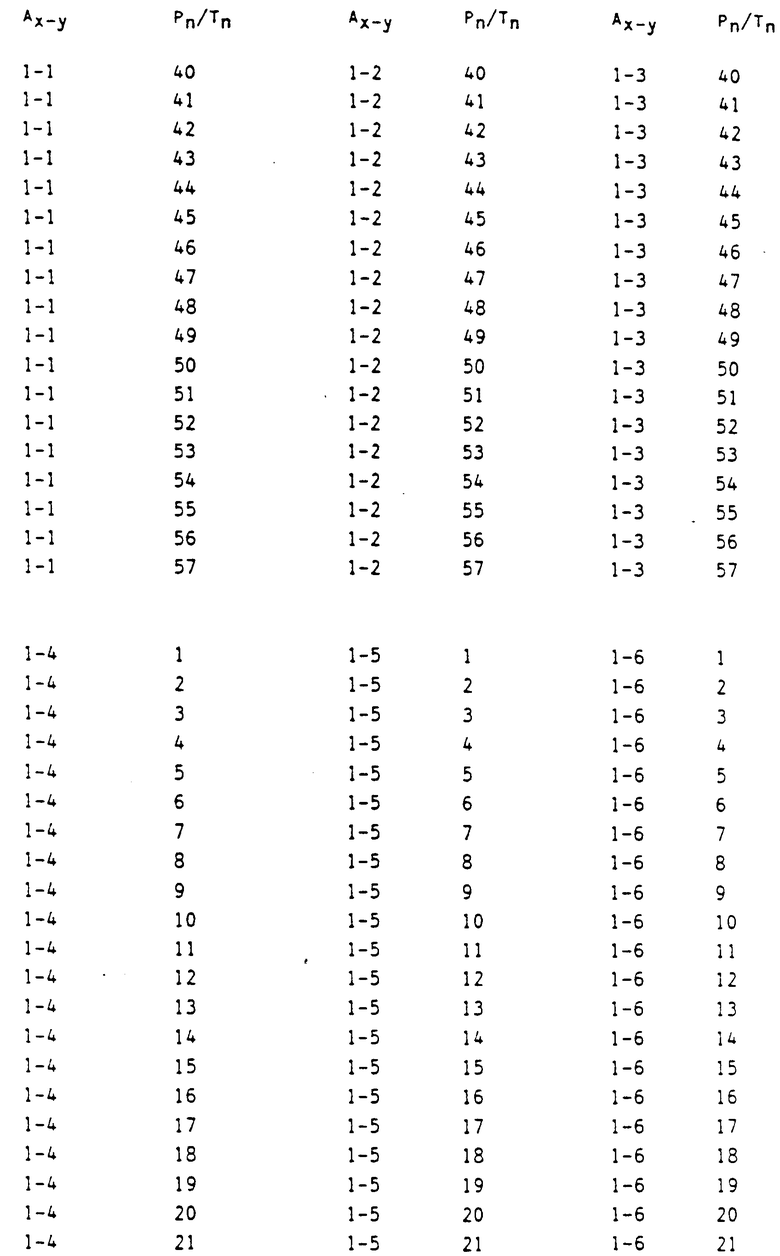
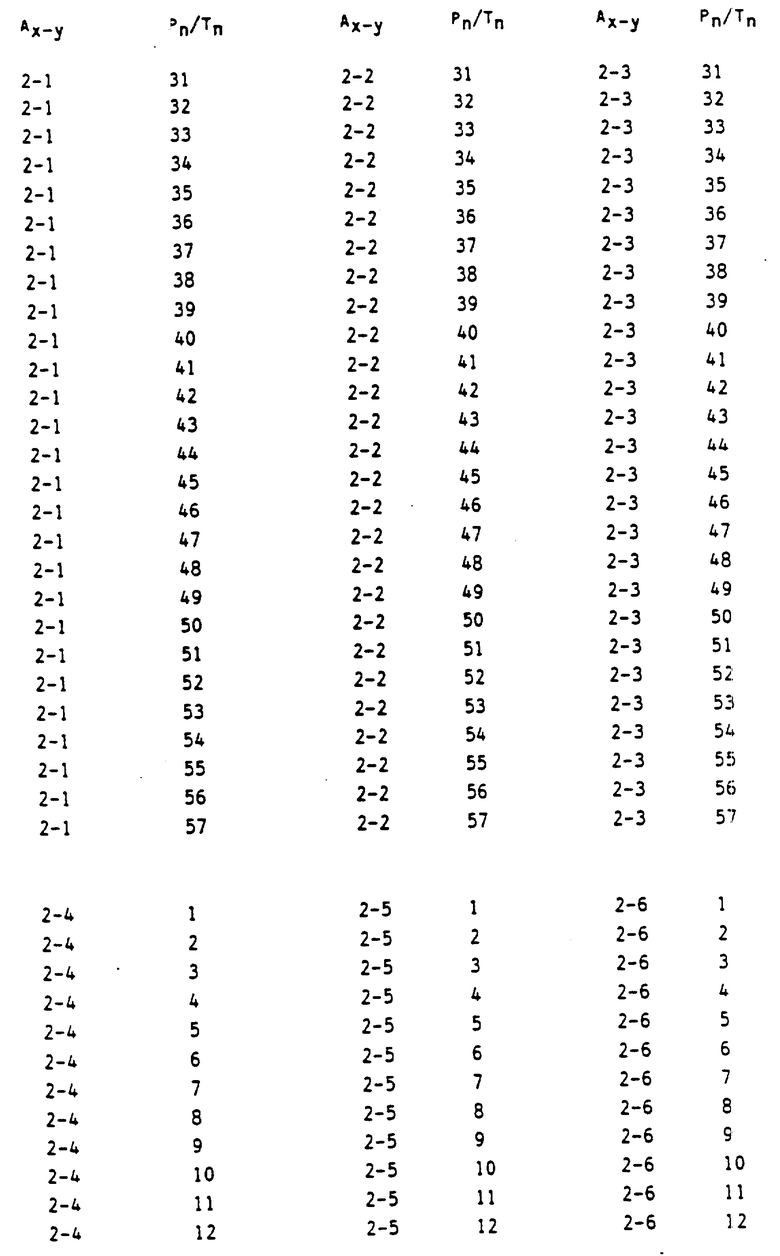
в которых Ax-y обозначает ароматический радикал формулы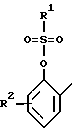
значение x обозначает радикал R1 и значение y радикал R2 (см. табл.4).
Обозначают:
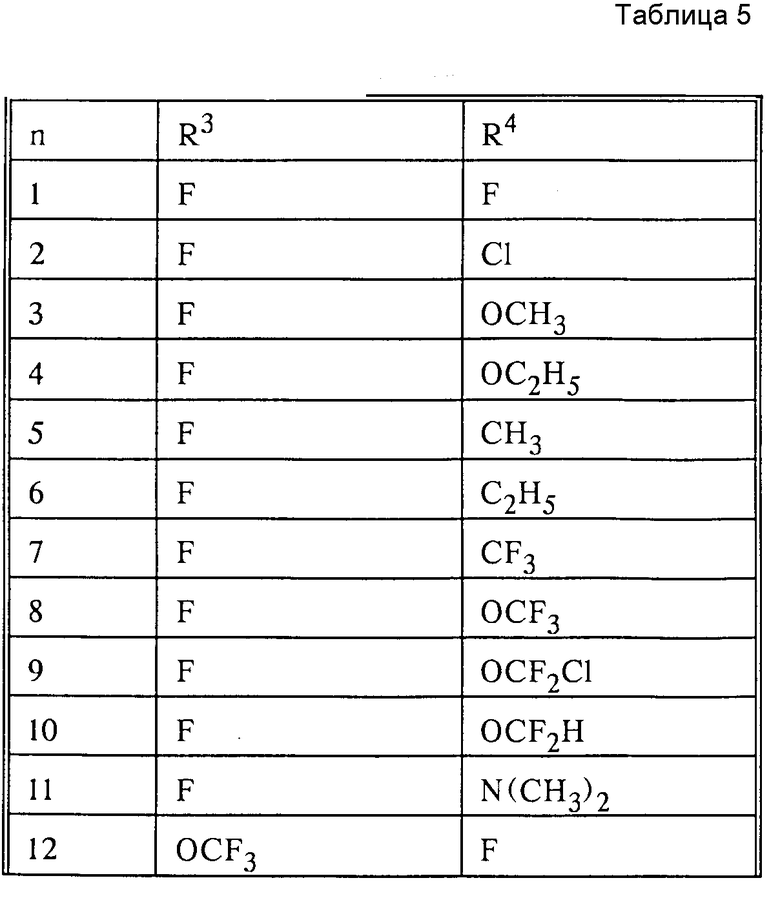
Pn и Tn, соответственно, обозначают пиримидин- или 1,3,5-триазинрадикалы, при этом обозначает: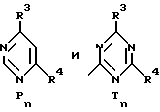
(см. табл. 5).
Сочетание Ax-y 1-1 и Pn/Tn 1 обозначает сульфонилмочевины и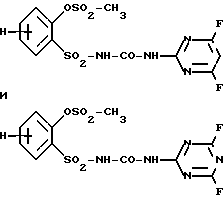
Соответственно остальные числовые сочетания можно отнести к соответствующим производным сульфонилмочевин.
Примеры применения
Гербицидное действие производных сульфонилмочевин формулы I можно проследить на опытах, проведенных в теплице:
В качестве емкости для культур служат пластиковые цветочные горшки с песчаной землей, содержащей примерно 3,0% гумуса в качестве субстрата. Семена испытуемых растений высевают отдельно по типам семян.
При обработке до появления всходов суспендированные или эмульгированные в воде активные вещества непосредственно после посева с помощью тонко распределяющего сопла наносятся на поверхность. Сосуды слегка опрыскивают для ускорения прорастания и роста и затем накрывают прозрачной пластиковой крышкой, пока не прорастут растения. Эта крышка способствует равномерному прорастанию испытуемых растений, поскольку активные вещества не оказывают на них вредного воздействия.
В целях послевсходовой обработки испытуемые растения обрабатываются суспендированными или эмульгированными в воде активными веществами лишь при высоте роста растений от 3 до 15 см. Для этого испытуемые растения либо непосредственно высеваются и проращиваются в одинаковых сосудах, либо проращиваются отдельно и за несколько дней до обработки проростки пересаживают в сосуды для проведения опыта. Норма расхода для послевсходовой обработки составляет 0,015 кг/га активного вещества или 0,5 кг/га активного вещества.
Растения содержатся в зависимости от типа при температурах от 10 до 25oC или от 20 до 35oC. Продолжительность опыта от 2 до 4 нед. В течение этого времени за растениями обеспечивается уход и определяется их реакция на отдельные виды обработки.
Оценка осуществляется по шкале от 0 до 100. При этом 100 обозначает отсутствие всходов растений или полное разрушение по меньшей мере надземных частей и 0 отсутствие повреждения или процесс роста.
Растения, использованные для опытов в теплице, составлены из следующих типов (см. табл.6)
Применение 0,015 кг/га активного вещества при послевсходовом способе оказало очень хорошее воздействие на широколистные нежелательные растения (соединения примеров 1 и 5) при одновременной переносимости культур пшеницы и кукурузы.
Соединение примера 14 при норме расхода 0,5 кг/га при послевсходовом способе показывает очень хорошее гербицидное воздействие против нежелательных растений Amaranthus retroflexus, Galium aparine и Ceantaurea cyanus.
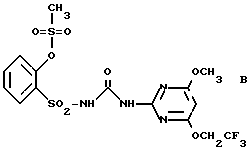
В нижеследующих сравнительных примерах примеру N 1 противопоставляются соединение A, известное из Европейского патента 44212, и соединение В, подпадающее в указанную там общую формулу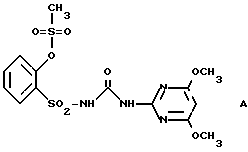
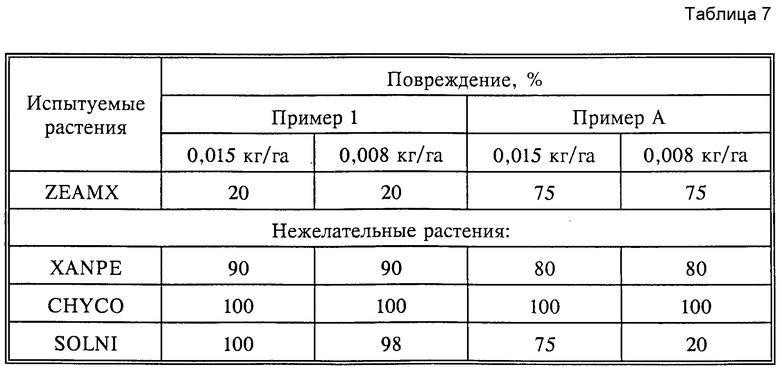
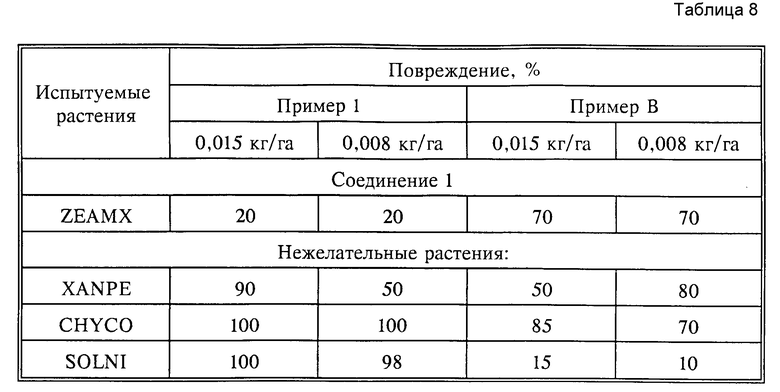
Результаты опытов, указанные в табл. 3 и 7, показывают неожиданно высокую селективность использованного соединения по сравнению со сравнительными веществами при одновременно хорошей гербицидной активности.
Табл. 7 показывает результаты испытаний по борьбе с нежелательными широколистными растениями и переносимость культур, приведенных в качестве примеров, при послевсходовом применении с нормой расхода 0,015, 0,008 кг активного вещества на гектар в теплице.
Табл. 8 показывает результаты испытаний по борьбе с нежелательными широколистными растениями и переносимость культур, приведенных в качестве примеров, при послевсходовом применении с нормой расхода 0,015, 0,008 кг активного вещества на гектар в теплице.
0,5 кг/га
Соединение 14 Повреждение,
AMARE 98
GALAP 98
CENCY 100.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АСИММЕТРИЧНО ЗАМЕЩЕННЫХ ТРИАЗИНОВ | 1994 |
|
RU2125995C1 |
| N-[(1,3,5-ТРИАЗИН-2-ИЛ)АМИНОКАРБОНИЛ]БЕНЗОЛСУЛЬФОНАМИДЫ ИЛИ ИХ СОЛИ, ПРИМЕНИМЫЕ В СЕЛЬСКОМ ХОЗЯЙСТВЕ, ГЕРБИЦИДНОЕ СРЕДСТВО И СПОСОБ БОРЬБЫ С НЕЖЕЛАТЕЛЬНЫМ РОСТОМ РАСТЕНИЙ | 1991 |
|
RU2102388C1 |
| АМИДЫ КАРБАМОИЛКАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СРЕДСТВО И СПОСОБЫ ДЛЯ БОРЬБЫ С ВРЕДОНОСНЫМИ ГРИБАМИ | 1995 |
|
RU2145956C1 |
| ПРОСТЫЕ ЦИКЛОГЕКСЕНОНОКСИМОВЫЕ ЭФИРЫ И ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2110513C1 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУЧЕНИЯ МЕТИЛАМИДОВ α-МЕТОКСИИМИНОКАРБОНОВЫХ КИСЛОТ | 1995 |
|
RU2146247C1 |
| 2-[(ДИГИДРО)ПИРАЗОЛИЛ-3'-ОКСИМЕТИЛЕН]АНИЛИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СРЕДСТВО БОРЬБЫ С СЕЛЬСКОХОЗЯЙСТВЕННЫМИ ВРЕДИТЕЛЯМИ И ВРЕДОНОСНЫМИ ГРИБАМИ И СПОСОБЫ БОРЬБЫ | 1995 |
|
RU2151142C1 |
| ПРОИЗВОДНЫЕ 2-ИМИНООКСИФЕНИЛУКСУСНОЙ КИСЛОТЫ И СРЕДСТВО, СОДЕРЖАЩЕЕ ЭТИ СОЕДИНЕНИЯ | 1996 |
|
RU2170229C2 |
| ПРОИЗВОДНЫЕ 2-[(2-АЛКОКСИ-6-ТРИФТОРМЕТИЛПИРИМИДИН-4-ИЛ)ОКСИМЕТИЛЕН]ФЕНИЛУКСУСНОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СРЕДСТВО ДЛЯ БОРЬБЫ С ВРЕДИТЕЛЯМИ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 1995 |
|
RU2166500C2 |
| Z-1,2,-ДИАРИЛАЛЛИЛХЛОРИДЫ | 1992 |
|
RU2096401C1 |
| ПРОИЗВОДНЫЕ САХАРИНА И ГЕРБИЦИДНЫЙ ПРЕПАРАТ | 1995 |
|
RU2156244C2 |
































Производные сульфонилмочевины формулы (I)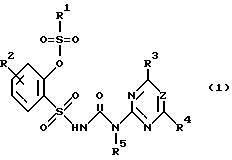
в которой R1 обозначает соответственно замещенный алкил или фенил; алкенил или пропаргил; алкиламино или диалкиламино; R2 обозначает водород, галоген, соответственно галогензамещенный метил, метокси или этокси, алкилсульфонилгруппу, нитро или циано; R3 обозначает дифторметокси, трифторметокси, бромдифторметокси, хлордифторметокси или фтор; R4 обозначает галоген, соответственно галогензамещенный метил, этил, метокси или этокси, или метил- или диметиламиногруппу; R5 обозначает водород, алкил, алкенил или алкинил; Z обозначает CH или N с указанием, что а) если R3 обозначает дифторметоксигруппу, то R1 не обозначает ди(алкил)аминогруппу, R2 не обозначает алкилсульфонилгруппу, R4 не обозначает метил- или метоксигруппу, б) если R3 обозначает атом фтора и Z обозначает N, то R4 не обозначает алкиламиногруппу, а также их соли. Производные сульфонилмочевины применяются в качестве гербицидов. 2 с. и 5 з.п.ф-лы, 8 табл.
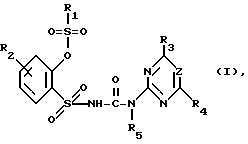
где R1 C1 C4-алкильная группа, необязательно замещенная атомом галогена или С1 С2-алкоксигруппой, С1 С3-алкиламиногруппа или ди-(С1 С4-алкил)-аминогруппа;
R2 водород, галоген, метил;
R3 дифторметокси, трифторметокси, бромдифторметокси, хлордифторметокси или фтор;
R4 галоген, при определенных условия галогензамещенные метил, этил, С1 С2-алкоксигруппа, метокси- или этоксигруппа, метил или диметиламиногруппа;
R5 водород;
Z СН или N, при условии, что а) когда R3 дифторметоксигруппа, R1 не может быть ди(алкил)аминогруппой, а R4 метилом или метоксигруппой; б) когда R3 атом фтора, а Z N, R4 не может быть алкиламиногруппой,
или их применимые в сельском хозяйстве соли.
| ЕР, патент, 0044212, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, патент, 4515624, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
