Изобретение относится к новому способу получения известного противоракового средства, более конкретно, к способу получения (S)-(-)- и (R)-(+)-изомерам сложного этилового эфира (5-амино-1,2 - дигидро-2-метил-3-фенилпиридо-[3,4-b] пиразин-7-ил)карбамидной кислоты или их фармацевтически приемлемым солям, а также к новым промежуточным (S)-(-)- и (R)-(+)-изомерам сложного этилового эфира [6-амино-4-[[2-(алкоксиалкил-амино)-1- метил-2-оксоэтил] амино]-5-нитро-2-пиридинил]карбамидной кислоты, которые используют в данном способе.
Известен способ получения (S)-(-)- и (R)-(+)-изомеров сложного этилового эфира (5-амино-1,2-дигидро-2-метил-3- фенилпиридо[3,4-b] пиразин-7-ил)карбамидной кислоты общей формулы (I)
который заключается в том, что сложный этиловый эфир 2- амино-4-хлор-3-нитропиридин-6-карбамидной кислоты формулы (II)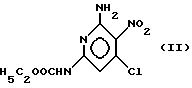
подвергают взаимодействию с норэфедрином в присутствии триэтиламина в среде инертного органического растворителя при кипячении, получаемое при этом соединение формулы (III)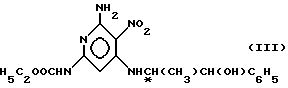
подвергают окислению комплексом трехокиси хрома и пиридина в среде органического растворителя и получаемое при этом соединение формулы (IV)
подвергают восстановительной циклизации в среде органического растворителя в присутствии никеля Ренея с последующим выделением (S)-(-)- или (R)-(+)-изомера целевого продукта в свободном виде или в виде фармацевтически приемлемой соли (патент США N 4866059, МКИ: C 07 D 213/75, 1989).
Недостатком известного способа является применение ядовитой трехокиси хрома для получения окислительного комплекса. Поэтому необходимы меры предосторожности, чем, однако, осложняется технология процесса.
Задачей изобретения является упрощение процесса за счет предотвращения применения ядовитых веществ.
Данная задача решается в способе получения (S)-(-) и (R)-(+)-изомеров сложного этилового эфира (5-амино-1,2-дигидро-2-метил-3-фенилпиридо[3,4-b] пиразин-7-ил)карбамидной кислоты вышеприведенной формулы (I), включающем взаимодействие сложного этилового эфира 2-амино-3-нитро-4-хлорпиридин-6-карбамидной кислоты с агентом аминирования в среде низшего спирта в качестве растворителя в присутствии третичного алкиламина и восстановительную циклизацию сложного этилового эфира (S)- или (R)-[6-амино-4-[(1-метил-2-оксо-2-фенилэтил)амино] -5-нитро-2-пиридинил] карбамидной кислоты в среде инертного растворителя в присутствии никеля Ренея с последующим выделением целевого продукта в свободном виде или в виде фармацевтически приемлемой соли, за счет того, что в качестве агента аминирования используют (R)- или (S)-2-амино-N-алкокси-N-алкилпропанамид общей формулы (V)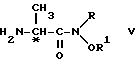
где R и R1 означают низший алкил, или его соль и продукт аминирования формулы (VI),
где R и R1 имеют вышеуказанные значения, подвергают взаимодействию с фенилсодержащим реактивом Гриньяра с получением сложного эфира (S)- или (R)-[6-амино-4-[(1-метил-2-оксо-2-фенилэтил)амино] -5-нитро-2-пиридинил карбамидной кислоты формулы (IV), который подвергают восстановительной циклизации.
R- и S-изомеры соединения формулы (VI) представляют собой новые промежуточные соединения, являющиеся дальнейшим объектом изобретения.
Соединения общей формулы (V) можно получать путем взаимодействия соединения (VII)
где Z защитная группа, такая, как, например, карбобензилокси, бензгидрилоксикарбонил, трет.-бутоксикарбонил и т.п. группы,
с трет.-алкиламином и низшим алкихлорформиатом в среде подходящего растворителя, такого, как, например, тетрагидрофуран, диоксан или диэтиловый эфир, с последующим взаимодействием с ди(низший)алкилгидроксиламином общей формулы RNHOR1, где R и R1 имеют вышеуказанные значения, в среде хлороформа или дихлорметана. Низший алкилхлорформиат имеет неразветвленную или разветвленную алкильную группу с 1-5 атомами углерода. Предпочтительной алкильной группой является метил. Реакцию осуществляют при температуре от -25oC до +25oC, предпочтительно при температуре -10oC. В качестве третичного алкиламина можно применять, например, триэтиламин или же предпочтительно N-метилпиперидин.
Получаемый амид общей формулы (VII)
где R, R1 и Z имеют вышеуказанные значения, подвергают каталитическому дебензилированию, например, водородом в присутствии палладия на активном угле в среде растворителя, такого, как, например, низший спирт, например, метанол или этанол, или простого эфира, такого, как, например, диоксан, тетрагидрофуран или диэтиловый эфир, если защитная группа Z представляет собой карбобензилокси или бензгидрилоксикарбонил, а если защитная группа Z представляет собой трет. -бутоксикарбонил, то ее снимают, например, с применением трифторуксусной кислоты, при этом получают соединение вышеприведенной формулы (V) в виде трифторацетатной соли.
Соединение формулы (V) можно без предварительного выделения подвергать взаимодействию со сложным метиловым эфиром 2-амино-3-нитро-4-хлорпиридин-6-карбамидной кислоты вышеприведенной формулы (II) в среде низшего спирта в качестве растворителя, такого, как, например, метанол или этанол, в присутствии третичного алкиламиамина, такого, как, например, триэтиламин или N-метилпиперидин. Если соединение формулы (V) имеется в виде ацетатной или трифторацетатной соли, то реакцию осуществляют в присутствии дополнительного эквивалента третичного алкиламина.
Реакцию нового промежуточного соединения вышеприведенной формулы (VI) с фенилсодержащим агентом Гриньяра, таким, как, например, бромид фенилмагния, йодид фенилмагния или фениллитий, осуществляют в среде простого эфира в качестве растворителя, такого, как, например, тетрагидрофуран, диэтиловый эфир, или диоксан, при температуре от 0 до 65oC, предпочтительно при 0oC. В результате восстановительной циклизации соединения вышеприведенной формулы (IV) в присутствии уксусной кислоты получают целевой продукт в виде ацетатной соли. Если же проводить эту реакцию в среде этанола, то целевой продукт получают в виде свободного основания.
Наиболее предпочтительный вариант предлагаемого способа представлен на следующей реакционной схеме, представленной в конце описания. На этой схеме Me означает метил, Et этил, Cbz карбобензилокси, Ph фенил, THF - тетрагидрофуран, RaNi никель Ренея, а Pd/C палладий на активном угле.
Если же вместо S-(-)-энантиомера N-карбобензилоксиаланина применяют в вышеприведенной реакционной схеме соответствующий R(+)-энантиомер, то целевой продукт получают в виде R(+)-изомера.
Фармацевтически-приемлемые соли целевого продукта вышеприведенной формулы (I) получают известным образом, например, путем взаимодействия целевого продукта в виде свободного основания с фармацевтически приемлемой кислотой. Наиболее предпочтительной солью является соль, образующаяся с 2-оксиэтансульфокислотой.
Пример 1. Получение сложного этилового эфира (+)-(R)-(5-амино-1,2-дигидро-2-метил-3-фенилпиридо[3,3-b] пиразин-7-ил)карбамидной кислоты в виде соли с 2-оксиэтансульфокислотой.
Стадия 1. Получение сложного этилового эфира (R)-[6-амино-4-[[2-(метоксиметиламино)-1-метил-2-оксоэтил]амино]-5-нитро-2- пиридинил]карбамидной кислоты
К перемешиваемому раствору 102 г (0,45 моль) (R)-2-амино-N-метокси-N-метил-пропанамида в 1,8 л абсолютного этанола добавляют 60,6 г (83,2 мл 0,6 моль) триэтиламина и 126 г ( 0,48 моль) сложного этилового эфира 2-амино-3-нитро-4-хлорпиридин-6-карбамидной кислоты. Получаемую смесь в атмосфере азота нагревают с обратным холодильником в течение 24 ч, упаривают и остаток растворяют в 2 л этилацетата. Получаемый этилацетатный раствор экстрагируют 2 л воды и получаемый водный слой экстрагируют 1 л этилацетата. Объединенные этилацетатные слои последовательно промывают водой (2 х по 500 мл) и 1 л насыщенного хлористого натрия и сушат над сульфатом магния. В результате упаривания получаемой смеси получают 152 г остатка, который подвергают хроматографии на силикагеле с применением в качестве элюента смесей петролейного эфира и этилацетата в соотношениях 70:30oC50:50. Получают 135 г (84%) вышеприведенного соединения с точкой плавления 95oC97oC.
[α
ИК (KBr): 3467, 3344, 1740, 1670, 1653, 1646, 1604-1.
1H ЯМР(CDCl3): 9,56 (д, lH, J 6,8 Гц); 7,77( с, lH); 6,71 (с, 1H); 4,68-4,59 (м, lH); 4,21 (кв, 2H); 3,86 (с, 3H); 1,54 (д, 3H, J 6,7 Гц); 1,29 (т, 3H, J 7,1 Гц).
Масс-спектр (ударная ионизация): М + 1 357,341, 327,311,296,280,268.
C13H20N6O6
Рассчитано, C 43,82 H 5,66 N 23,58.
Найдено, C 43,58; H 5,76; H 23,07.
Стадия 2.Получение сложного этилового эфира (R)- [6-имино-4-[(1-метил-2-оксо-2-фенилэтил)амино]-5-нитро-2- пиридинио]карбамидной кислоты.
1,9-молярный раствор бромида фенилмагния в 3 л диэтилового эфира получают из 46,2 г (1,9 моль) магниевых стружек, 298,3 г (200 мл, 1,9 моль) бромбензола и 2,14 г йода. Получаемый свежий темный раствор держат в атмосфере азота. В другой колбе раствор 130 г (0,365 моль) соединения со стадии 1 в 3 л безводного тетрагидрофурана перемешивают и охлаждают до 0oC в атмосфере азота. При интенсивном перемешивании медленно добавляют свежеприготовленный раствор бромида фенилмагния при помощи до трубки при 0oC. Получаемую темную суспензию перемешивают при комнатной температуре в течение 16 ч, после чего смесь подают в 2 л насыщенного раствора хлористого аммония и органический слой отделяют. Водный слой экстрагируют 2 л этилацетата. Объединенные этилацетатные слои последовательно промывают водой (4 х по 1 л) 1 л насыщенного раствора хлористого натрия и сушат над сульфатом магния. В результате упаривания растворителя получают 180 г остатки, который подвергают хроматографии на силикагеле с применением в качестве элюента смесей петролейного эфира и этилацетата в соотношениях 7:3oC1:1. Получают 62,5 г (46%) вышеприведенного соединения с точкой плавления 95-97oC (размягчается при 90oC. Согласно данным высокоскоростной колоночной жидкостной хроматографии чистота целевого продукта составляет 89%
[α
ИК (KBr): 3477, 3344, 1740, 1717, 1695, 1690, 1685, 1599, 1576, 1563, 1559, 1418, 1300, 1278, 1255, 1211, 1096, 972, 702 см-1.
1H-ЯМР (CDCl3: δ 10,04 (д, 1H, J 6,1 Гц); 8,04 (д, 2H, J 7,2 Гц); 7,68-7,51 (м, 3H); 6,86 (с, 1H) + 5,31-5,19 (м, 1H); 1,61, 4,23 (кв, 2H), (д, 3H, J 7 Гц); 1,32 (т. 3Н, J 7,1 Гц).
Масс-спектр (ударная ионизация): М+1=374, 358, 326, 324, 310, 278, 268.
Стадия 3. Получение сложного этилового эфира (+)-(R)-(5 - амино-1,2-дигидро-2-диетил-3-фенилпиродо[3,4-b] пиразин-7- ил)карбамидной кислоты в виде соли с 2-оксиэтансульфокислотой.
Раствор 52 г (0,139 моль) соединения со стадии 2 в 1 л абсолютного этанола подвергают гидрированию на 10 г никеля Ренея под давлением 3,55 кг/см2 в течение 36 ч. Катализатор удаляют и растворитель упаривают. Получают 48,9 г коричневой пены, которую подвергают хроматографии на силикагеле с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 1: 1. Получают 25,7 г (56%) сложного этилового эфира (+)-(R)-(5-амино-1,2-дигидро-2-метил-3-фенилпиридо[3,4-b] пиразин-7-ил)карбамидной кислоты в качестве желтого твердого вещества с точкой плавления 148-151oC (размягчается при 120oC). Согласно данным высокоскоростной колоночной жидкостной хроматографии чистота составляет 92%
[α
ИК (KBr): 3393, 2976, 1733, 1728, 1719, 1702, 1611, 1579 см-1.
1H-ЯМР (CDCl3: δ 8,21 (шир. м, lH); 7,94-7,9 (м, 2H); 7,46-7,40(м, 3H); 6,72 (с, 1H), 5,24 (шир. с, 2H); 4,82-4,79 (м, 1H); 4,53 (шир. с, 1H); 4,22 (кв, 2H); 1,33-1,25 (м, 6H).
Масс-спектр (ударная ионизация): M + 1 326, 235, 310, 280, 264, 251, 238.
Рассчитано, C 62,76 H 5,89 N 21,52.
Найдено, C 63,07 H 5,95 H 19,59.
К раствору 18,95 г (58,2 нмоль) полученного карбамата в 200 мл метанола, размешивая, добавляют 0,168 нормальный металнольный раствор 2-оксиэтансульфокислоты (329 мл, 55,3 ммоль). По истечении 1 ч растворитель упаривают и получаемую темную пену растворяют в 440 мл ацетонитрила в атмосфере аргона при незначительном нагревании. Получаемый раствор фильтруют и оставляют стоять при комнатной температуре. Получают 15,7 г (56%) тонких желто-коричневых кристаллов со степенью чистоты 96% (согласно данным высокоскоростной колоночной жидкостной хроматографии). Дальнейшую очистку осуществляют путем растворения 13,5 (29,9 ммоль) полученной соли в 700 мл ацетонитрила в присутствии 0,7 г аскарбиновой кислоты и последующего кипячения. Получаемый раствор фильтруют в горячем состоянии и медленно охлаждают с получением 0,7 г тонкого желто-коричневого кристаллического продукта с точкой плавления 164-167oC, который согласно данным высокоскоростной колоночной жидкостной хроматографии имеет степень чистоты 98,5% Изомерная чистота: 100% (по высокодисперсной колоночной жидкостной хроматографии).
[α
ИК (KBr): 3434, 3420, 3295, 3285, 3273, 3192, 3186, 3173, 3162, 3158, 3148, 2980, 1727, 1662, 1635, 1607, 1591, 1572, 1533, 1467, 1448, 1250, 1222, 1215, 1182, 1148, 1124, 1068, 1035, 696 см-1.
1H-ЯМР(d6-ДМСО): δ 11,62 (шир. с, 1H); 11,05 (шир. с, 1H); 8,49 (шир. с, 1H); 8,16-8,14 (м, 2H), 7,71 (шир. с, 2H); 7,49-7,47 (м, 3H); 5,92 (с, 1H); 5,15-5,08 (м, 1H); 4,24 (кв, 2H, J 7 Гц); 3,63 (м, 2H, J 7 Гц); 2,62 (т, 2H, J 6,7 Гц); 1,29 (т, 3H, J 7,1 Гц); 1,15 (д, 3H, 6,5 Гц).
Масс-спектр (бомбардировка быстрыми атомами): M + 1 325 (451 127 M+ HOSO3H); 310, 277, 264, 251, 238, 223, 202, 194, 167, 155, 133, 104, 91, 84, 77, 66, 65, 51, 45.
C17H19N5O2 • C2H6SO4:
Рассчитано, C 50,54; H 5,58; N 15,51; S 7,10.
Найдено, C 50,52; H 5,46; N 15,58; S 6,97.
(R)-2-амино-N-метокси-N-метил-пропанамид получают следующим образом.
Стадия А
Перемешиваемую суспензию 102,4 г (1,05 моль) гидрохлорида N,O-диметилгидроксиламина в 600 мл дихлорметана охлаждают до температуры -10oC и в атмосфере азота добавляют 109,1 г (1,10 моль) N-метилпиперидина с получением прозрачного раствора. В другой колбе 223,2 г (1 моль) N - [(фенилметокси)карбонил] -D-аланина растворяют в 600 мл тетрагидрофурана. К получаемому прозрачному раствору в атмосфере азота добавляют 2 л дихлорметана и 109,1 г (1,1 моль) N-метилпиперидина. Раствор охлаждают до 0oC, интенсивно перемешивают и одной порцией добавляют 99,2 (81,1 мл, 1,05 моль) метилхлороформиата. Реакционную смесь перемешивают при 0 oC -10oC в течение 10 мин, после чего с помощью трубки добавляют холодный раствор N,O-диметилгидроксиламина. Смесь перемешивают при 0oC в течение 1 ч еще в течение 12 ч без наружного охлаждения. Смесь охлаждаются до температуры 0oC, последовательно экстрагируют 1 л 0,5-нормальной холодной соляной кислотой, насыщенным раствором бикарбоната натрия (2 x по 1 л) и 1 л насыщенного раствора хлористого натрия и сушат над сульфатом магния. Раствор упаривают в высоком вакууме (0,2 мм рт. ст. ). Получают 228,3 г (86%) сложного фенилметилового эфира (R)-[2-метоксиметиламино)-1-метил-2-оксоэтил] карбомидной кислоты с точкой плавления 82-84oC.
ИК (KBr): 3288, 1720, 1656, 1616, 1540 см-1.
1H-ЯМР (d6-ДМСО): 7,60 (д, 1H, J 7,6 Гц); 7,36-7,30 (м, 5H); 5,01 (с, 2H); 4,51-4,42 (м, 1H); 3,73 (с, 3H); 3,10 (с, 3H); 1,17 (д, 3H, J= 7,1 Гц).
Масс-спектр (бомбаридровка быстрыми атомами): M + 1 267, 223, 206, 178, 159, 139, 119, 91.
C13H18N2O4:
Рассчитано, C 58,64; H 6,76; N 10,51.
Найдено, C 58,70; H 6,99; N 10,35.
Стадия Б
Раствор 120 г (0,45 моль) соединения со стадии А в 1,2 л абсолютного этанола подвергают гидрированию на 5 г 20%-ного палладия на активном угле под давлением 3,515 кг/см2. Катализатор удаляют и раствор упаривают. Получают 102 г (100%) (R)-2-амино-N-метокси-N -метил-пропанамида в качестве бесцветного вязкого масла, которое хранят в высоком вакууме и применяют без дальнейшей очистки.
Пример 2. Получение сложного этилового эфира (-)-(S)-(5-амино-1,2-дигидро-2-метил-3-фенилпиридо[3,4-b] пиразин-7-ил) карбамидной кислоты в виде соли с 2-оксиэтансульфокислотой.
Стадия 1. Получение сложного этилового эфира (S)-[6 - амино-4-[[2-(метоксиметиламино)-1-метил-2-оксоэтил] амино] -5-нитро-2- пиридинил]карбамидной кислоты.
К перемешиваемому раствору 4,6 г (20 ммоль) (S)-2-амино-N-метокси-N-метил-пропанамида в 100 мл абсолютного этанола добавляют 2,5 г (3,45 мл, 25 ммоль) триэтиламина и 5,5 г (21 ммоль) сложного этилового эфира 2-амино-3-нитро-4-хлорпиридин-6-карбамидной кислоты. Получаемую смесь в атмосфере азота нагревают с обратным холодильником в течение 19 ч, после чего упаривают и остаток растворяют в 100 мл этилацетата. Этилацетатный раствор промывают водой (2 x по 50 мл) и сушат над сульфатом магния. Смесь упаривают с получением 8,5 г остатка, который подвергают хроматографии на силикагеле с применением в качестве элюента смесей петролейного эфира и этилацетата в соотношениях 3:2 oC 2:3. Получают 5 г (70%) указанного соединения с точкой плавления 85-92oC.
[α
ИК (KBr): 3333, 1741, 1669, 1597, 1591 см-1.
1H-ЯМР (CDCl3): δ 9,58 (д, 1H, J 5,9 Гц); 7,65 (шир. с, 1H); 6,71 (с, 1H); 4,68-4,56 (м, 1H); 4,22 (кв, 2H); 3,86 (с, 3H); 3,27 (с, 3H); 1,54 (д, 3H, J 6,7 Гц); 1,30 (т, 3H, J 7,3 Гц).
Масс-спектр (ударная ионизация), M + 1: 357, 356, 341, 323, 311, 280, 269, 268, 252, 223, 222.
C13H20N6O6:
Рассчитано, C 43,82; H 5,66; N 23,58.
Найдено, C 43,62; H 5,49; N 23,02.
Стадия 2. Получение сложного этилового эфира (S)-[6-амино-4- [(1-метил-2-оксо-2-фенилэтил)амино]-5-нитро-2-пиридинил]карбамидной кислоты.
0,74-молярный раствор бромида фенилмагния в диэтиловом эфире свеже подготовляют из 26,7 г (1,1 моль) магниевых стружек и 157 г (105,3 мл, 1 моль) бромбензола в 1 л диэтиловом эфире. Смесь фильтруют в атмосфере азота с тем, чтобы удалить непрореагировавший магний. Аликвот раствора титруют против второбутанола с применением фенил антраценхинолина в качестве индикатора для определения молярности (0,74 моль). В другой колбе раствор 3,8 г (10,7 ммоль) соединения со стадии 1 в 200 мл безводного тетрагидрофурана перемешивают в атмосфере азота при комнатной температуре. Интенсивно перемешивая, 0,74-молярный раствор бромида фенилмагния добавляют 6 порциями по 14,83 мл с соблюдением 15-мин интервалов. Получаемую темную суспензию перемешивают при комнатной температуре в течение 14 ч, после чего подают в 250 мл насыщенного раствора хлористого аммония и органический слой отделяют. Водный слой дважды экстрагируют этилацетатом, взятым в количестве по 200 мл. Объединенные органические слои последовательно промывают водой (2 x по 200 мл) и 100 мл насыщенного хлористого натрия и сушат над сульфатом магния. После упаривания растворителя получают 4,54 г остатка, который подвергают хроматографии на силикагеле с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 1:1. Получают 1,94 г (49%) вышеуказанного соединения с т.пл. 102-106oC.
[α
K (KBr): 3421, 1740, 1736, 1718, 1695, 1685, 1677, 1653, 1653, 1599 cм-1
1H-ЯМР CDCl 3 ): 9,94 (c, 1H); 9,78 (д, 1H, J 6,2 Гц); 8,08 (д, 2H, J 7,2 Гц); 7,76-7,59 (м, 3H), 6,76 (с, 1H); 5,48-5,43 (м, 1H); (м, 1H); 4,12 (кв, 2H); 1,47 (д, 3H, J 6,8 Гц); 1,22 (т, 3H, J 7,2 Гц).
Масс-спектр (бомбардировка быстрыми атомами): M + 1 374,1, 358, 329, 302, 268, 252, 223.
C17H19N5O5:
Рассчитано, C 54,64; Hh 5,09; N 18,75.
Найдено, C 54,78; H 5,11; N 18,71.
Стадия 3. Получение сложного этилового эфира (-)-(S)-(5-амино-1,2-дигидро-2-метил-3-фенилпиридо[3,4-b] пиразин-7-ил)карбамидной кислоты в виде соли с 2-оксиэтансульфокислотой.
Раствор 1,67 г (4,5 ммоль) соединения со стадии 2 в 100 мл абсолютного этанола подвергают гидрированию на 0,5 г никеля Ренея в течение 90 мин под давлением 3,515 кг/см2. После удаления катализатора и упаривания растворителя получают 1,36 г (93%) сложного этилового эфира (-)-(S)-(5-амино -1,2-дигидро-2-метил-3-фенилпиридо[3,4-b] пиразин-7-ил)карбамидной кислоты в качестве желтой пены, имеющей степень чистоты 94% (согласно данным высокоскоростной колоночной жидкостной хроматографии). Он содержит 3,8% R-энантиомера.
ИК (KBr): 3407, 3397, 3391, 3374, 2975, 1733, 1724, 1719, 1701, 1611, 1578, 1558, 1539, 1533, 1497, 1446, 1410, 1220 см-1.
1H-ЯМР (CDCl3): δ 8,05 (шир.с. 1H); 7,94-7,91 (м, 2H); 7,46-7,41 (м, 3H); 6,72 (с, 1H); 5,19 (шир.с, 2H); 4,85-4,77 (м, 1H); 4,50 (с, 1H); 4,22 (кв, 2H); 2,57 (шир.с, 1H); 1,33-1,25 (м, 6H).
Масс-спектр (ударная ионизация): M + 1 326.
К раствору 0,47 г (1,4 ммоль) свободного основания в 20 мл метанола в атмосфере аргона добавляют 0,153-нормальный (9,0 мл, 1,37 ммоль) метанольный раствор 2-оксиэтансульфокислоты и получаемый прозрачный раствор фильтруют. Растворитель упаривают и оставшуюся пену растворяют в 25 мл ацетонитрила при кипячении. Раствор фильтруют в горячем состоянии и медленно охлаждают с получением тонких желто-коричневых кристаллов. После сушки получают 0,46 г (74% ) кристаллического продукта со степенью чистоты 97% (согласно данным высокоскоростной колоночной жидкостной хроматографии), который имеют точку плавления 164-166oC. Согласно данным высокоскоростной жидкостной хроматографии энантиомерная чистота составляет 97,2%
[α
Полученный продукт можно далее очищать путем хроматографии свободного основания на силикагеле с применением в качестве элюента смеси гексана и этилацетата в соотношении 6: 4. При этом степень чистоты составляет 99,5% Свободное основание снова переводят в указанную соль описанным выше образом.
ИК (KBr): 3444, 3435, 3429, 3422, 3306, 3295, 3284, 3270, 3263, 3256, 3243, 3227, 3199, 3187, 3164, 3149, 1727, 1658, 1635, 1607, 1591, 1534, 1448, 1251, 1218, 1212, 1182, 1149, 1123, 1068, 1035, 696 см-1.
1H-ЯМР (ДМСО): δ 11,60 (шир. с, 1H), 11,00 (с, 1H); 8,5 (д, 1H, J 2 Гц); 8,17-8,14 (м, 2H); 7,72 (шир. с, 2H); 7,49-7,47 (м, 3H); 597 (с, 1H); 5,14-5,10 (м, 1H); 4,24 (кв, 2H); 4,14 (шир. с, 1H), 2,67 (т, 2H, J 6,8 Гц); 2,68 (т, 2H, J 6,8 Гц); 1,29 (т, 3H, J 7,1 Гц); 1,20 (д, 3H, J 6,5 Гц).
Масс-спектр (бомбардировка быстрыми атомами): M + 1 325 (451 127), 310, 296, 279, 278, 264, 252, 238, 221, 210, 194, 167.
C17H19N5 O 2•C2H6O4S:
Рассчитано, C 50,54; H 5,58; N 15,51; S 7,10.
Найдено, C 50,71; Hh 5,67; N 15,50; S 6,84.
(S)-2-амино-N-метокси-N-метил-пропанамид получают следующим образом.
Стадия А.
В перемешиваемую суспензию 10,73 г (10 ммоль) N,O- диметилгидроксиламина в виде гидрохлорида в 100 мл дихлорметана охлаждают до 0oC в атмосфере азота, после чего добавляют 12 г (14,7 мл, 121 ммоль) N- метилпиперидина с получением прозрачного раствора. В другой колбе 24,5 г (110 ммоль) N-[(фенилметокси)карбонил] -L-аланина растворяют в 100 мл тетрагидрофурана. К получаемому прозрачному раствору в атмосфере азота добавляют 200 мл дихлорметана и 12 г (14,7 мл, 121 ммоль) N метилпиперидина. Раствор охлаждают до температуры -10 oC -15oC, интенсивно перемешивают и одной порцией добавляют 10,4 г (8,5 мл, 110 ммоль) метилхлорформиата. Реакционную смесь перемешивают при температуре -10 oC -15oC в течение 10 мин, после чего при помощи трубки добавляют при температуре 0oC свышеприготовленный раствор N,O-диметилгидроксиламина. Реакционную смесь перемешивают при 0oC в течение 1 ч и затем в течение 16 ч без охлаждения, после чего ее разбавляют 200 мл дихлорметана, охлаждают до 0oC, последовательно экстрагируют 0,1 н. соляной кислотой (2 х по 200 мл), насыщенным раствором бикарбоната натрия (2 х 100 мл) и 200 мл насыщенного раствора хлористого натрия и сушат над сульфатом магния. Раствор упаривают в высоком вакууме (0,02 мм рт.ст.), в результате чего получают 26,4 г (90%) сложного фенилметилового эфира (S)-[2-(метоксиметиламино)-1-метил-2-оксоэтил] карбамидной кислоты в качестве белого твердого вещества с т.пл. 86-88oC.
[α
ИК (KBr): 3284, 1723, 1719, 1662, 1655 см-1.
1Hh-ЯМР (CDCl3): δ 7,35 (с, 5H); 5,55 (д, 1H, J 8 Гц); 5,13 (д, 1H, J 12,2 Гц); 5,06 (д, 1H, J 12,8 Гц); 4,79 4,56 (м, 1H); 3,77 (с, 3H); 3,21 (с, 3H); 1,34 (д, 3H, J 6,7 Гц).
Масс-спектр (электронная ионизация) M + 1: 267, 223, 207, 206, 178, 151, 134, 107, 92, 91.
C13H18N2O4:
Рассчитано, C 58,64; H 6,81; N 10,52.
Найдено, C 58,58; H 6,83; N 10,42.
Стадия Б
Раствор 5,4 г (20,3 ммоль) соединения состадии А в 75 мл метанола подвергают гидрированию на 0,5 г 20%-ного палладия на активном угле под давлением 3,515 кг/см2 в течение 90 мин. После удаления катализатора и упаривания раствора получают 4,6 г (100%) (S)-2-амино-N-метокси-N-метилпропанамида в качестве бесцветного вязкого масла, которое хранят в высоком вакууме и может и может использоваться без дальнейшей очистки.

Способ получения (S)-(-)- и (R)-(+)-изомеров сложного этилового эфира (5-амино-1,2-дигидро-2-метил-3-фенилпиридо /3,4-b/пиразин-7-ил)карбамидной кислоты или их фармацевтически приемлемых солей и промежуточные (S)-(-)- и (R)-(+)-изомеры сложного этилового эфира [6-амино -4-[/2-(алкоксиалкиламино)-1-метил-2-оксиоэтил]амино]-5-нитро-2-пиридинил]-карбамидной кислоты. Сущность изобретения: усовершенствованный способ получения (S)-(-)- и (R)-(+)-изомеров сложного этилового эфира (5-амино-1,2- дигидро-2-метил-3-фенилпиридино/3,4-b /пиразин-7-ил)карбамидной кислоты формулы
путем взаимодействия сложного этилового эфира 2-амино-3-нитро-4-хлорпиридин-6-карбамидной кислоты с (R)- или (S)-2-амино-N-алкокси-N-алкилпропанамид формулы V ,
,
где R и R' - низший алкил; продукт аминирования VI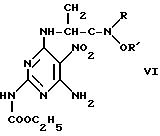
подвергают взаимодействию с фенилсодержащим реактивом Гриньяра с получением сложного эфира (S) -или (R)-/6-амино-4/(1-метил-2-оксо-2-фенилэтил)амино/-5-нитро-2-пиридинил карбамидной кислоты формулы IV:
который подвергают восстановительной циклизации; и промежуточный продукт (S)-(-) и (R)-(+)-изомеры сложного этилового эфира (6-амино-4-[/2-(алкоксиалкиламино)-1-метил-2-оксоэтил/амино/ -5-нитро-2-пиридинил] карбамидной кислоты формулы VI. 2 с. и 4 з.п. ф-лы.
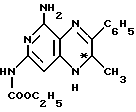
включающий взаимодействие сложного этилового эфира 2-амино-3- нитро-4-хлорпиридин-6-карбамидной кислоты формулы II
с агентом аминирования в среде низшего спирта в качестве растворителя в присутствии третичного алкиламина и восстановительную циклизацию сложного этилового эфира (S)- или (R)-[6-амино-4-[(1-метил-2-оксо-2-фенилэтил)амино] -5-нитро-2-пиридинил]карбамидной кислоты формулы IV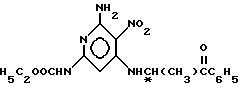
в среде инертного растворителя в присутствии никеля Ренея с последующим выделением целевого продукта в свободном виде или в виде фармацевтически приемлемой соли, отличающийся тем, что в качестве агента аминирования используют (R)- или (S)-2-амино-N-алкокси-N-алкилпропанамид общей формулы V
где R и R' низший алкил,
или его соль и продукт аминирования общей формулы VI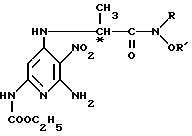
где R и R' имеют указанные значения,
подвергают взаимодействию с фенилсодержащим реактивом Гриньяра с получением сложного этилового эфира (S)- или (R)-[6-амино-4-[(1-метил-2-оксо-2-фенилэтил)амино] -5-нитро-2-пиридинил] - карбоновой кислоты формулы IV, который подвергают восстановительной циклизации.
где R и R' имеют указанные значения,
получают путем взаимодействия соединения общей формулы VII
где Z защитная группа, такая, как, например, карбобензилокси, бензгидрилоксикарбонил или трет-бутоксикарбонил,
с третичным алкиламином и низшим алкилхлорформиатом в среде инертного растворителя с последующим взаимодействием с ди(низший)алкилгидроксиламином общей формулы RNHOR', где R и R' имеют указанные значения, в среде хлороформа или дихлорметана и получаемый при этом амид общей формулы VIII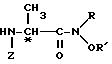
где R, R' и Z имеют указанные значения,
подвергают снятию защитной группы.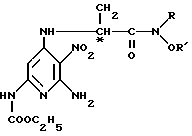
где R и R' низший алкил,
представляющие собой промежуточные соединения для получения (S)-(-)- и (R)-(+)-изомеров сложного этилового эфира (5-амино-1,2-дигидро-2-метил-3-фенилпиридо[3,4-b] пиразин-7-ил)карбамидной кислоты формулы I по п.1 или их фармацевтически приемлемых солей.
| EP, 336345, кл.C 07D 471/04, 1989 | |||
| US, патент, 4950761, кл.C 07D 471/04, 1990. |

