Изобретение относится к технологии производства производных пирролидина, более конкретно к новому способу получения известных производных пирролидина, новым промежуточным продуктам для их синтеза и промежуточному продукту для синтеза антибактериальных агентов на основе хинолона.
Известен способ получения (S)-3-амино-1-фенилпирролидина, который заключается в том, что аминогруппу L-аспарагиновой кислоты защищают трет.-бутилоксикарбонилом, получаемую N-защищенную L-аспарагиновую кислоту подвергают взаимодействию с подходящим реагентом для получения (S)-3-(трет.-бутоксикарбониламино)-янтарного ангидрида с последующим добавлением фениламина и замыканием кольца при нагревании в присутствии уксусного ангидрида, получаемый при этом (S)-3-(трет.-бутоксикарбониламино)-1-фенил-пирролидин-2,5-дион растворяют в смеси хлороформа и бензола с последующим взаимодействием с газообразной хлористоводородной кислотой и получаемый при этом гидрохлорид сукцинимида обрабатывают раствором бикарбоната натрия с последующим восстановлением алюмогидридом лития и выделением целевого (S)-3-амино-1-фенилпирролидина (см. например, Д.Т.Витиак и др. J. Med. Chem. том 14, N 1, стр. 24-29, 1971).
Если вместо фениламина применять бензиламин, то получают соответствующее 1-бензиловое производное, которое известно из: Х.К. Браун и др. J. Org. Chem. 51, стр. 4296, 1986 и Д. Роузен и др. J. Med. Chem. 31, стр. 1586, 1988).
Недостатком известного способа является низкий общий выход.
Задачей изобретения является повышение общего выхода (S)-3-амино-1-замещенных пирролидинов из L-аспарагиновой кислоты.
Данная задача решается в способе получения производных (S)-3-амино-1-замещенного пирролидина, включающем снабжение аминогруппы L-аспарагиновой кислоты защитной группой, переработку N-защищенной L-аспарагиновой кислоты с использованием первичного амина для получения производных (S)-3-амино-1-замещенного пирролидина, снятие используемой защитной группы и выделение целевого продукта за счет того, что снабжение аминогруппы L-аспарагиновой кислоты защитной группой осуществляют путем взаимодействия с алкилсульфонил- или арилсульфонилгалоидом, а переработку N-защищенной L-аспарагиновой кислоты осуществляют путем восстановления последней или ее сложного низшего диалкилового эфира восстанавливающим гидридом, получаемый при этом N-защищенный-1,4-бутандиол подвергают взаимодействию примерно с двумя эквивалентами тионилгалоида или алкилсульфонилокcи- или арилсульфонилоксигалоида в присутствии примерно трех эквивалентов основания, получаемое при этом соединение формулы: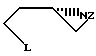
где Z означает алкилсульфонил или арилсульфонил, а L галоид, алкилсульфонилокси или арилсульфонилокси, подвергают взаимодействию с первичным амином формулы R-NH2, где R означает бензил, незамещенный или замещенный низшим алкилом или низшим алкоксилом, или бензгидрил, в присутствии третичного амина для получения 1-R-3-(защищенное амино)-пирролидина, в котором R имеет вышеуказанное значение, а снятие защитной группы осуществляют путем гидролиза или восстановления.
Основание, используемое в качестве вспомогательного вещества при осуществлении взаимодействия N-защищенного-1,4-бутандиола примерно с двумя эквивалентами тионилгалоида или алкилсульфонилоксигалоида или арилсульфонилоксигалоида, может представлять собой органическое или неорганическое основание. В качестве органического основания используют амин, предпочтительно трет. -амин, такой, как, например, триэтиламин, пиридин, хинуклидин и т.п. амины. В качестве неорганического основания используют гидроокись или карбонат щелочного или щелочноземельного металла, как, например, гидроокись или карбонат натрия или калия.
Предлагаемый способ поясняется общей реакционной схемой.
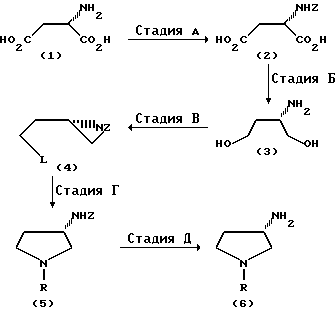
На стадии А осуществляют превращение L-аспарагиновой кислоты до производного, у которого аминогруппа защищена n-толуолсульфонилом или же другим арилсульфонилом, таким, как, например, n-бромбензолсульфонил, или же алкансульфонилом, таким, как, например, метансульфонил. Такие защищенные производные L-аспарагиновой кислоты можно получать с высоким выходом известными из литературы способами. Так, например, N-тозил-L-аспарагиновой кислоты можно получать по способу К. Фройденберга и А. Ное (см. Chem. Ber. 58, стр. 2399, 1925) или же по способу Е.В. Мек-Чесни и В.К. Свон, Дж. (см. J. Am. Chem. Soc. 58, стр. 1116, (1937).
Стадия Б сводится к восстановлению производного N-защищенной L-аспарагиновой кислоты до соответствующего оптически-активного 2-(защищенное амино)-1,4-бутандиола либо путем непосредственного восстановления обеих карбоксильных функций до двух спиртовых групп, либо путем превращения до соответствующего сложного диэфира с последующим восстановлением обеих сложноэфирных функций до двух спиртовых групп. Для восстановления карбоксильных функций производных L-аспарагиновой кислоты можно применять различные гидридные восстанавливающие агенты, такие, как, например, боран (B2H6), алюмогидрид лития (LiAlH4), смесь боргидрида натрия (NaBH4) и йода (I2). Для восстановления сложноэфирных функций соответствующих сложных диэфиров аспарагиновой кислоты можно применять боргидрид натрия (NaBH4) в присутствии хлористого лития, бромистого лития, хлористого кальция, хлористого магния, хлористого цинка или же кислоты Льюиса, а также алюмогидрид лития и нитрид [NaAlH2(OCH2CH2-OCH3)2] Подходящими растворителями при осуществлении реакции восстановления являются апротонные растворители, такие, как, например, толуол, третрагидрофуран, 162-диметоксиэтан и соответствующие эфирные соединения. Если в качестве восстанавливающего агента применять боргидрид, то целесообразно применение в качестве растворителя спирта, такого, как, например, метанол или этанол.
Стадия В сводится к превращению оптически-активного 2-(защищенное амино)-1,4-бутандиола до оптически-активного производного 2-(2'-оксиэтил)-1-(защищенного)азиридина, в котором спиртовая функция заменена на удаляемую группу (L), такую, как, например, хлор или алкансульфонилоксигруппа, как, например, метансульфонилокси, или арилсульфонилоксигруппа, как, например, n-толуолсульфонилокси. Производные азиридина, имеющие сульфонатную удаляемую группу, можно получать путем взаимодействия 2-(защищенное амино)-1,4-бутандиола примерно с двумя эквивалентами алкансульфонилхлорида или арилсульфонилхлорида в присутствии примерно трех эквивалентов основания, такого, как, например, трет. -амин, как, например, триэтиламин, или карбонат, как, например, карбонат калия, в среде апротонного растворителя, такого, как, например, хлористый метилен, тетрагидрофуран или толуол, при температурах от -10 до +35oC в течение 32-240 мин. Производные азиридина, имеющие в качестве удаляемой группы хлор, можно получать путем взаимодействия 2-(защищенное амино)-1,4-бутандиола с тионилхлоридом в присутствии подходящего основания, такого, как, например, триэтиламин или пиридин, в среде апротонного растворителя, такого, как, например, хлористый метилен, тетрагидрофуран или толуол.
Стадия Г сводится к превращению оптически-активного производного 2-(2'-оксиэтил)-1-(защищенного)азиридина, в котором спиртовая группа заменена на удаляемую группу, до 1-замещенного-3-(защищенное амино)-пирролидина путем взаимодействия производного азиридина с первичным амином, таким, как, например, бензиламин или n-метоксибензиламин, в присутствии подходящего основания, такого, как, например, третичный амин, например, триэтиламин, или карбонат, например, карбонат калия, в среде апротонного растворителя, такого, как, например, диметилсульфоксид, диметилформамид, ацетонитрил, толуол, тетрагидрофуран, при температурах от 10oC до 110oC в течение 1 18 ч. Избыточный первичный амин можно отделять от получаемого продукта путем обработки раствора реакционной смеси двуокиси углерода с последующей экстракцией первичного амина в виде производного карбаминовой кислоты разбавленным водным раствором гидроокиси натрия. Вариант отделения избыточного первичного амина заключается в том, что раствор реакционной смеси обрабатывают метилформиатом и получаемое при этом формамидное производное первичного амина подвергают экстракции водной кислотой. Дальнейший вариант отделения избыточного первичного амина заключается в том, что реакционную смесь обрабатывают фталевым ангидридом с последующим отделением получаемого бензиламинового аддукта, моноамида фталевой кислоты, путем экстракции водным основанием.
Стадия Д сводится к снятию защитной группы с 3-аминофункции путем восстановления сульфонамидной группы. 1-замещенный-3-(защищенное амино)пирролидин можно обрабатывать бромистым водородом в среде подходящего растворителя, такого, как, например, вода или уксусная кислота, в присутствии подходящего акцептора кислоты, такого, как, например, фенол, двуокись серы, бисульфит натрия, при температуре 50-130oC в течение 1-10 ч. В результате переработки реакционной смеси получают желаемый 3-амино-1-замещенный пирролидин. Но сульфонаминовую группу можно также снимать путем восстановления металлическим агентом, таким, как, например, амальгама натрия, калий в присутствии катализатора на основе так называемого краун эфира, нафталин натрия.
Целевой (S)-3-амино-1-R-пирролидин формулы (6) можно использовать для получения антибактериальных агентов (патент США N 4916141). Так, например, аминогруппу снабжают подходящей защитной группой на основе карбамата, такой, как, например, трет.-бутоксикарбонил или бензилоксикарбонил, после чего группу R удаляют путем восстановления водородом в присутствии катализатора. Получаемый при этом (S)-3-защищенный аминопирролидин подвергают взаимодействию с подходящим 7-галоген-хинолоном или 7-галоген-нафтирилоном с получением желаемого производного хинолона или нафтиридона. Кроме того, (S)-3-амино-1-R-пирролидин формулы (6) можно также использовать для переведения в промежуточное соединение с двумя асимметричными атомами углерода (см. пример 2), который подвергают взаимодействию с подходящим 7-галоген-хинолоном или 7-галоген-нафтиридоном с тем, чтобы получить антибактериальные агенты в соответствии с патентом США N 4851418.
Пример 1.
Стадия А: (N-(n-толуолсульфонил)-L-аспарагиновая кислота
239,4 г L-аспарагиновой кислоты растворяют в 1152 мл 3н. гидроокиси натрия и получаемый раствор охлаждают до температуры 0oC. Добавляют 9 г бисульфата тетрабутиламмония и 240 мл тетрагидрофурана и pH среды доводят до 12,8 путем добавления 160 мл 3н. гидроокиси натрия. Отдельно и одновременно добавляют раствор 342 г n-толуолсульфонилхлорида в 675 мл тетрагидрофурана и 900 мл 3н. гидроокиси натрия в течение 6 ч при температуре в пределах 0-5oC и pH среды 11,7-12,8. Реакционную смесь размешивают в течение ночи и на следующий день подкисляют до pH 2,6 путем добавления 400 мл 36%-ной соляной кислоты. Реакционную смесь два раза экстрагируют этилацетатом, взятым в количестве по 1350 мл, и объединенные этилацетатные экстракты сгущают с получением масла, которое два раза обрабатывают толуолом, взятым в количестве по 180 мл, получаемую смесь сгущают в вакууме и остаток сушат. Получают 487,7 г (84%) N-(n-толуолсульфонил)-L-аспарагиновой кислоты в виде беловатого твердого вещества, которое без дальнейшей очистки используют на следующей стадии.
Стадия Б: (S)-2-(n-толуолсульфониламино)-1,4-бутандиол
110 г полученной на стадии А N-(n-толуолсульфонил)-L-аспарагиновой кислоты растворяют в 300 мл тетрагидрофурана и получаемый раствор охлаждают до температуры -5oC в атмосфере азота. 1,0м. раствор борана в тетрагидрофуране прикапывают в течение одного часа при температуре 0-5oC и получаемый раствор размешивают при температуре 20-25oC в течение ночи. Раствор снова охлаждают до температуры 0oC, после чего прикапывают 250 мл метанола в течение 30 мин (выделяется газ). Раствор сгущают в вакууме, получаемое масло растворяют в 1 мл метанола и раствор нагревают с обратным холодильником в течение одного часа. Затем раствор сгущают в вакууме, получаемый остаток растворяют в 1 л метанола и получаемый раствор нагревают с обратным холодильником в течение двух часов, после чего раствор сгущают, получаемый остаток сушат в вакууме при температуре 50oC. При этом получают 97 г (98%) (S)-2-(n-толуолсульфониламино)-1,4-бутандиола в виде беловатого твердого вещества, которое без дальнейшей очистки используют на следующей стадии. Часть целевого продукта перекристаллизовывают из изопропилового спирта и гексанола. Получают продукт с точкой плавления 90-92oC.
Стадия В: (S)-2-(2'-метансульфонилоксиэтил)-1-(n-толуолсульфонил) азиридин
46 г полученного на стадии Б (S)-2-(n-толуолсульфониламино)-1,4-бутандиола растворяют в 450 мл хлористого метилена, после чего добавляют 60 г триэтиламина, получаемый раствор охлаждают до температуры -5oC в атмосфере азота и прикапывают раствор 42,0 г метансульфонилхлорида в 110 мл хлористого метилена в течение 3 ч. Получаемую смесь размешивают в течение 30 мин при температуре 0-5oC и осторожно добавляют 150 мл воды и 350 мл хлористого метилена. Слои разделяют и органический слой экстрагируют 100 мл 0,1н. соляной кислоты и 100 мл деминерализованной воды с последующим вакуумным сгущением. Получают 57,2 г (S)-2-(2-метансульфонилоксиэтил)-1-(n-толуолсульфонил)азиридина в качестве светло-желтого твердого вещества, которое без дальнейшей очистки можно использовать на следующей стадии. Часть продукта перекристаллизовывают из этилацетата и гексанов. При этом получают продукт с точкой плавления 78-80oC.
Стадия Г: (S)-1-бензил-3-(n-толуолсульфониламино)-пирролидин и гидрохлорид (S)-1-бензил-3-(n-толуолсульфониламино)пирролидина
54,5 г (S)-2-(2'-метансульфонилоксиэтил)-1-(n-толуолсуьфонил)азиридина растворяют в 50 мл теплого тетрагидрофурана и получаемый раствор прикапывают к раствору 40 г бензиламина и 40 г триэтиламина в 250 мл диметилсульфоксида при температуре 75-85oC в течение 20 мин. Получаемый раствор нагревают при температуре 75-85oC в течение 150 мин, после чего сгущают в вакууме для удаления тетрагидрофурана и диметилсульфоксида, получаемый остаток растворяют в 300 мл толуола и осторожно последовательно обрабатывают 10 г сухого льда и 150 мл 1,0н. гидроокиси натрия. Слои разделяют и толуольный слой осторожно последовательно обрабатывают 3 г сухого льда и 20 мл 1,0н. гидроокиси натрия. Эту экстракцию двуокисью углерода и гидроокисью натрия повторяют еще три раза и получаемый толуольный раствор сгущают в вакууме. Получают (S)-1-бензил-3-(n-толуолсульфониламино)пирролидин в качестве масла янтарного цвета, которую растворяют в 15 мл теплого толуола (60-70oC), после чего добавляют раствор 30 мл изопропилового спирта, насыщенного хлористоводородным газом. Раствор размешивают, охлаждают до температуры -5oC и поддерживают при этой температуре в течение трех часов. Получаемый твердый продукт собирают, последовательно промывают изопропиловым спиртом и диэтиловым эфиром и сушат в вакууме. Получают 46,5 г (74% в пересчете на стадии В и Г) гидрохлорида (S)-1-бензил-3-(n-толуолсульфониламино)пирролидина в виде беловатого твердого вещества. Часть этого продукта перекристаллизовывают из изопропилового спирта. При этом получают продукт с точкой плавления 179-181oC.
Стадия Д: (S)-3-амино-1-бензилпирролидин.
В колбу подают 40,0 г гидрохлорида (S)-1-бензил-3-(n-толуолсульфониламино)пирролидина, 12,0 г фенола и 200 мл 30%-ного бромистого водорода в уксусной кислоте, колбу закрывают и реакционную смесь нагревают при температуре 105-125oC в течение 55 мин. Получаемый красный раствор сгущают в вакууме для удаления избыточной уксусной кислоты и избыточного бромистого водорода и получаемое густое масло растворяют в 500 мл воды и 150 мл толуола. Слои разделяют и водный слой последовательно экстрагируют 50 мл толуола и 30 мл диэтилового эфира, после чего pH среды доводят до 8,5 путем добавления 11 мл 28%-ной гидроокиси аммония. Получаемый раствор экстрагируют 25 мл толуола, органические экстракты удаляют и оставшийся водный слой обрабатывают 145 мл 50% -ной гидроокиси натрия, после чего три раза экстрагируют толуолом, взятым в количестве по 50 мл. Объединенные толуольные экстракты сгущают, в результате чего получают 19,2 г масла, которое перегоняют в вакууме (т. кип. 104-106oC при 2,5 мм рт.ст.). Получают 18,2 г (94%) (S)-3-амино-1-бензилпирролидина в виде бесцветного масла. [α]25 +11,2o.
Следующий пример иллюстрирует получение нового 1-бензил-3-(S)-[(трет. бутокси-карбониламино)пропиониламино] -пирролидина, который представляет собой промежуточный продукт для синтеза антибактериальных агентов на основе производных хинолина.
Пример 2.
1-бензил-3-(S)-[2-(S)-(трет. -бутокси-карбониламино)пропиониламино] пирролидин
12,8 г N-(трет.-бутоксикарбонил)-L-аланина растворяют в 50 мл хлористого метилена, получаемый раствор охлаждают до температуры -5oC и обрабатывают 6,7 г N-метилморфолина. Получаемую смесь размешивают, охлаждают до температуры от -5 до -10oC и при указанной температуре медленно добавляют холодный раствор (-5oC) 8,5 г изобутилхлорформиата в 100 мл хлористого метилена, после чего смесь размешивают в течение 30 мин при температуре от -5 до -10oC. Затем добавляют имеющий температуру 0oC раствор 10,0 г (S)-3-амино-1-бензилпирролидина в 100 мл хлористого метилена при температуре от 0 до -10oC, после чего реакционную смесь размешивают при температуре от 0 до -5oC в течение одного часа и затем при температуре 15-25oC в течение двух часов. Реакционную смесь обрабатывают 300 мл деминерализованной воды и получаемые слои разделяют. Органическую фазу последовательно экстрагируют раствором 18,5 г бикарбоната натрия в 200 мл деминерализованной воды и 300 мл деминерализованной воды. Органическую фазу сушат над сульфатом натрия, взятым в количестве 50 г, и раствор сгущают в вакууме. Получают 21,3 г 1-бензил-3-(S)-[2-(S)-(трет. -бутоксикарбониламино)-пропиониламино] пирролидина в виде беловатого твердого вещества с точкой плавления 102-105oC. Бензильную группу можно удалять следующим путем.
3-(S)-[2-(S)-(трет.бутоксикарбониламино)-пропиониламино]пирролидин
21,3 г 1-бензил-3-(S)-[2-(S)-(трет.-ботоксикарбониламино)пропиониламино] пирролидина растворяют в 250 мл метанола и подвергают гидрированию до окончания поглощения водорода на взятом в количестве 5,0 г катализаторе, представляющем собой 20%-ную гидроокись палладия на активном угле, увлажненную на 50% водой, при температуре 25-40oC и давлении 3,5155 кг/см2. Затем реакционную смесь фильтруют на торговом силикагеле "целите" с тем, чтобы удалить катализатор, и остаток обрабатывают 100 мл метанола. Фильтраты объединяют и сгущают в вакууме. Получают 14,6 г 3-(S)-[2-(S-(трет.-бутоксикарбониламино)пропиониламино] -пирролидина в виде бесцветного масла, которое кристаллизует при стоянии. Согласно данным высокопроизводительной жидкостной хроматографии, чистота S,S-изомера составляет 99% Часть продукта перекристаллизовывают из метил-трет.-бутилового эфира. При этом получают продукт с точкой плавления 137-138oC.
Получение промежуточного бутандиола иллюстрируется следующим примером.
Пример 3.
(S)-2-(n-толуолсульфониламино)-1,4-бутандиол
2,0 г сложного диметилового эфира N-(n-толуолссульфонил)-L-аспарагиновой кислоты (см. И. М. Теобальд, М.В. Виллиемс, Г.Т. Янг, J. Chem. Soc. 1927, 1963) растворяют в 20 мл тетрагидрофурана, после чего добавляют 1,5 г борана натрия и 1,5 г хлористого лития и реакционную смесь размешивают при температуре 0oC в течение 64 ч. Медленно добавляют 10 мл 10%-ной хлористоводородной кислоты (выделяется газ) и реакционную смесь три раза экстрагируют этилацетатом, взятым в количестве по 20 мл. Объединенные органические экстракты сгущают и остаток сушат в вакууме. Получают 1,95 г (S)-2-(n-толуолсульфониламино)-1,4-бутандиола в виде беловатого твердого вещества. Согласно данным высокопроизводительной жидкостной хроматографии, его чистота составляет 97,7%
Сущность изобретения: объектом изобретения является способ получения производных (S)-3-амино-1-замещенного пирролидина, включающий защиту аминогруппы L-аспарагиновой кислоты, обработку N-защищенной L-аспарагиновой кислоты с использованием первичного амина для получения производных (S)-3-амино-1-замещенного пирролидина, снятие используемой защитной группы и выделение целевого продукта, при этом защиту аминогруппы L-аспарагиновой кислоты осуществляют путем взаимодействия с алкилсульфонил- или арилсульфонилгалоидом, а обработку N-защищенной L-аспарагиновой кислоты осуществляют путем восстановления последней или ее сложного низшего диалкилового эфира восстанавливающим гидридом, получаемый при этом N-защищенный-1,4-бутандиол подвергают взаимодействию примерно с двумя эквивалентами тионилгалоида или алкилсульфонилокси- или арилсульфонилоксигалоида в присутствии примерно трех эквивалентов основания, получаемое при этом соединение формулы: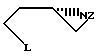
где Z означает алкилсульфонил или арилсульфонил, а L - галоид, алкилсульфонилокси- или арилсульфонилокси, подвергают взаимодействию с первичным амином формулы R-NH2, где R означает бензил, незамещенный или замещенный низшим алкилом или низшим алкоксилом, или бензгидрил, в присутствии третичного амина для получения 1-R-3-(защищенное амино)-пирролидина, в котором R имеет вышеуказанное значение, а снятие защитной группы осуществляют путем гидролиза или восстановления. Другим объектом изобретения являются производные (S)-азиридина указанной формулы, где L означает галоид или алкилсульфонилокси или арилсульфонилокси, а Z - алкилсульфонил или арилсульфонил, представляющие собой промежуточные продукты для синтеза производных (S)-3-амино-1-замещенного пирролидина. Еще одним объектом изобретения является 1-бензил-3-(S)-/(третбутоксикарбониламино)пропиониламино/-пирролидин, представляющий собой промежуточный продукт для синтеза антибактериальных агентов на основе производных хинолона. 3 с. и 2 з.п. ф-лы.
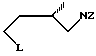
где Z алкил- или арилсульфонильная группа;
L алкил- или арилсульфонилоксигруппа,
подвергают взаимодействию с первичным амином общей формулы
R-NH2,
где R бензил,
в присутствии третичного амина с получением 1-R-3-(аминозащищенного) пирролидина, в котором R имеет указанное значение, и последующим удалением защитной группы путем гидролиза или восстановления.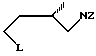
где L алкилсульфонилоксигруппа;
Z п-толуолсульфонильная группа,
в качестве промежуточных соединений для синтеза (S)-3-амино-1-защищенного пирролидина по п.1.
| EP, патент, 304087, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 331960, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, патент, 4916141, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

