Изобретение относится к получению биологически активных полисахаридов, которые могут быть использованы в медицине.
Под термином глюкозаминргликан понимают вещества, образованные последовательностями уроновых кислот (D-глюкуро- новая кислота или L-идуроновая кислота) и аминосахаров, причем эти последние представляют собой глкжрзэмины или галакта- замины.
Глюкозаминогликаны природного происхождения образованы более или менее однородными смесями последовательно соединенных дисахаридных единиц: образованных уроновой субъединицей (глюкоро- новая кислота или идуроновая кмслота) и озаминной субъединицей (глюкозамин или галактозамин), объединенных связью 1 или 1 .
В глюкозаминдиканах гидроксильные группы по разному замещены функциональ00
W
00
VJ
Сл
ными группами, в особенности сульфатными группами, причем амино-группы озаминов замещены сульфатными и/или ацетильными группами.
Глюкозаминогликаны, рассматриваемые здесь, включают 6 типов продукта: гепарин, гепарин-сульфат, хондроитинсуль- фат А, хондроитин-сульфат С, хонДроитин- сульфат В, более точно называемый дерматан-сульфат, и гиалуроновая кислота. Они характеризуются двумя следующими основными дисахаридными структурами (А) и (В);.
ovy
NHR JH
в которой R обозначает группу 50з и/или ацетильную группу и знак|указывает, что карбоксильная группа может быть ниже плоскости цикла (идуроновое звено) или выше плоскости цикла (глюкуроновое звено), и
(В)
i3Jn
Глюкозаминликаны, обладающие основной структурой (А), представленной выше, называют глюкоэаминогликанами (или GAGS) гепаринового типа и включают гепа- рины и гепарин-сульфаты, а глюкозами- ногликаны, имеющие структуру основания (В), представленную выше, называют GAGS типа хондроитин-сульфата и включают хон- дроитин-сульфаты А и С и дерматан-сульфат.
Гиалуроновая кислота имеет основную структуру (В), в которой галактоэамин заменен на глюкозамин.
Как указано выше, как в GAGS гепаринового типа, так и в GAGS типах хондроитин-сульфата, более или менее высокое процентное содержание гидроксильных групп в п диса- харидных единицах находится в форме сложных эфиров серной кислоты.
Так, гепарин может быть представлен основной структурой (А), указанной выше, в которой п может изменяться от 1 до 80..R в большинстве п единиц обозначает группу 50з. и, в остальных случаях, ацетильную группу; группы ОН в положении 6 глюкоза- мина и в положении 2 уроновой кислоты в большинстве случаев сульфатированы, тогда как группа ОН в положении 3 глюкозами
10
15
на сульфатирована только в меньшинстве случаев; ОН в положении 3 уроновой кислоты практически не сульфатирована, причем вышеуказанная уроновая кислота в большинстве случаев представляет собой идуро- новую кислоту.
Продукты, имеющие основную структуру (А), включают также М-десульфатирован- ный N-ацетилированный гепарин. Этот гепарин представлен формулой (А), в которой R обозначает ацетильную группу.
Гепарин также содержит формы, имеющие структуру (А), с п изменяющимся от 3 до 15, и тот же самый химический профиль. Вышеуказанные формы могут быть выделены фракционно согласно известным методам и называются гепаринами низкого молекулярного веса (н.м.в.).
Гепарины н.м.в,, имеющие молекулярное распределение и обладающие биологическими свойствами, по существу идентичны таковым гепаринов н.м.в., получаемыми путем фракционирования, могут быть получены фрагментацией гепарина согласно следующим методам:
-метод азотистокислой деполимеризации и восстановления (2, 3), который разрезает молекулу, атакуя субъединицы глюкозамин-М-сульфата, чтобы дать концевой 2,5-ангидромэннитол:
СН2СИ
- о -о-(он(Ь}
Ун
СН7ОН
в известных случаях сульфатированных по первичной гидроксильной группе в положении 6.
-метод ферментативной деполимери- 40 зации (4,5) или щелочной деполимеризации
(6), который разрезает молекулу, атакуя уро- новые субъединицы, чтобы дать окончание в виде а,/3-ненасыщенной уроновой кислоты:С00
О
О- (А)
онв известных случаях 2-сульфатированной;
50 - метод окислительной деполимеризации (7,8), который разрезает молекулу путем окисления, сохраняя природные окончания.
Другие гепарины н.м.в., которые имеют
55 молекулярное распределение, отличающееся от такового природного гепарина, обладают особыми биологическими свойствами, могут быть получены йодным окислением природного гепарина, с последующей депо20
25
30
35
45
лимеризацией, путем / -элимирования или кислотного гидролиза. Йодное окисление разрезает несульфатированные звенья уро- новой кислоты между углеродами в положении 2 и 3. Известно, что такие звенья присутствуют в участке связи с антитромбином HI (AT III), плазматическом ингибиторе коагуляции различных серин-протеаз, и активность которых сильно увеличивается за счет гепарина, действующего как кофактор, Йодное окисление, следовательно, позволяет получать продукты, лишенные участка связи с AT IH, и, следовательно, по существу лишенные антикоагулирующей активности.
Гепарины н.м.в. этого типа могут быть получены в особенности при осуществлении способа, включающего следующие стадии:
-осторожное окисление гепарина, реализуемое путем введения в реакцию гепарина, в виде водного раствора с конечной концентрацией 0,5-5% (вес/обьем), с солью йодной кислоты при конечной концентрации 0,5-1% (вес/обьем), при рН. равном 4,5-6,5, предпочтительно при рН 5, при температуре 0-10°С, в течение 15-48-часов, в отсутствие света;
-деполимеризация полученных гепариновых цепей путем присоединения сильного основания, такого как гидроксид натрия при рН выше примерно 11, а именно от 11 до 12, предпочтительно при 11,2-11,6, предпочтительно вблизи 11,5;
-восстановление фрагментов деполяризации с помощью восстановителя и, после удаления, в желательном случае, восстановителя, который не прореагировал;
-рекуперация восстановленных фрагментов гепарина путем осаждения с помощью растворителя, в котором они нерастворимы;
-выделение искомых фрагментов путем фракционирования с помощью спирта, в присутствии неорганической соли, полученного водного раствора, снова растворяя в воде, выделенный ранее осадок, и рекуперируя образовавшийся осадок.
Эти гепарины н.м.в. отвечают формуле
(С):
соо чон
о
СН7ОН -Q
он
Оч
(С)
OSOjNHR,Jn
в которой п может изменяться от 6 до 15; R по крайней мере в 90% п единиц представляет собой группу 50з, и, в остальных случаях, ацетильную группу, причем группы ОН в положении 3 глюкозамина могут быть сульфатированы, а группы ОН в положении
б глюкозамина сульфатированы на 70%. Более того, по 1 звену на 2 цепи по крайней мере этого гепарина н.м.в. имеют замену идуроновой кислоты на звено уроновой кислоты (D-глюкуроновой или L-идуроновой), не сульфатированной, раскрытой между углеродами в положениях 2-й 3, следующей формулы:
СОО
-О
:снгон снгон
Такие гепарины н.м.в. могут быть еще фракционированы гель-фильтрацией согласно известным способам, чтобы получить смеси фар фрагментов, однородных с точки зрения их молекулярной массы, лишенные участка связи с AT III.
Другие глюкозаминогликаны гепаринового типа образованы GAGS формулы (А) или (С), в которых карбоксильная функция уроновой кислоты этерифицирована, например, путем конденсации со спиртом в
присутствии агента конденсации карбоди- мидного типа (9) или путем алкилирования карбоксила с помощью алкилгалогенида в присутствии слабого основания,
Точно также, глюкозаминогликаны типа
хондроитин-сульфата образованы глюкоза- мингликанами формулы (В), в которых карбоксильные функции уроновых кислот этерифицированы, например, путем алкилирования карбоксила с помощью алкилгалоген ид а в присутствии слабого основания.
Такие сложные эфиры для гиалуроновой кислоты получают путем обработки гиалуроновой кислоты в форме соли четвертичного аммония спиртом в присутствии катализатора или этерифицирующего (превращающего в простой эфир) агента (10).
Гепар н-сульфат представлен вышеприведенной формулой (А), в которой п может изменяться от 1 до 80, R обозначает в большинстве видов ацетильную группу и в меньшинстве видов группу S )з. Кроме того, в преобладающем большинстве форм, группы ОН в положении 6 глюкозамина сульфатированы, причем уроновая кислота в
большинстве форм представляет собой глю- куроновую кислоту.
Хондроитин-сульфаты А и С представлены вышеприведенной формулой (В), в которой п изменяется от 1 до 80, соответственно, группы 4-ОН и б-ОН глюкозамина сульфатированы, причем уроновая кислота в большинстве форм представляет собой глюкуроновую кислоты.
Дерматан-сульфат имеет структуру, по существу идентичную таковой хондроитинсульфата А, но субъединица уроновой кислоты представляет собой идуроновую кислоту в большинстве случаев.
Фрагменты дерматан-сульфата, отвечающие вышеприведённой формуле (В), в которой п может изменяться от 1 до 20, могут быть получены йодным окислением с последующим восстановлением брргидридом натрия и кислотным гидролизом, как описано у FRAN.
Глюкозаминогликаны обладают многочисленными биологическими активностями, среди которых можно назвать их активности по отношению к факторам коагуляции, которые могут осуществляться через посредство различных плазматических протеинов. В том, что касается гепарина, также из литературы известно, что гепарин или некоторые из его производных, обладающие или нет антикоагулирующей активностью, могут иметь регуляционную активность пролиферации гладких мышечных клеток сосудистой стенки или еще ингиби- рующую активность гепараназы, фермента, принимающего участие в механизмах мета- стазного распространения. Кроме того, глю- козамингликаны составляют часть более широкого семейства сульфатированных полисахаридов, причем некоторые в более или менее значительной степени обладают противовирусной активностью и в особенности анти-УМ-Ьактивностью (вирус человеческого иммунодефицита) В ABA и др.
В нижеследующем тексте используется термин GAGS для обозначения глюкозами- ногликана. который либо имеет природную структуру, такую, которая получается экстракцией, полусинтетическую или синтетическую, либо имеет химически модифицированную структуру на функциональных карбоксильных или аминогруппах, сначала по реакции ацилирования, дающей GAGS, селективно 0-ацилированные,
Несмотря на большой интерес к их фармакологическим активностям, природные GAGS обладают тем недостатком, что имеют относительно короткий период полураспада, что приводит к необходимости повторных введений. Этот недостаток отчасти временно сглаживается за счет использования, для предохранения и лечения венозных тромбозов, производных гепарина незначительной молекулярной массы, подкожно, уменьшая таким образом частоту введения инъекций в день.
Тем не менее, большой интерес представляет возможность иметь в распоряжении производные, обладающие пролонгирующим действием, что позволяет еще снижать
частоту введения этих продуктов, увеличивая продолжительность их действия.
Также может представлять большой интерес располагать пролонгированными, неантикоагулирующими производными, такими, как производные N-десульфатиро- ванного N-ацетилированного гепарина, активность которых как ингибитора гепараназы, фермента, вовлекаемого в метастаз0 ные явления распределения, оценивается очень высоко.
Известны многочисленные производные модифицированных глюкозаминогли- канов, чтобы улучшить их фармакокинетику,
5 а именно сложные эфиры с карбоксильной функцией уроновых кислот или с гидро- ксильными функциями,
В особенности используют частичную или полную этерификэцию карбоксильных
0 функций гепарина, обрабатываемых в форме соли четвертичного аммония, в инертном растворителе, спиртом или галогенирован- ным производным, в присутствии, в известных случаях, агента конденсации .
5 Производные гиалуровой кислоты, полученные этерификацией с помощью спирта в присутствии катализатора или путем реакции с этерифицирующим агентом до получения простого эфира, также описаны.
0 Кроме того, известны различные способы для этерификации GAGS по первичным и вторичным гидроксильным функциям.
Известен способ получения неполных гидролизуемых эфиров гепарина и неток5 сичной органической кислоты, например, 4- хлорфеноксиизомасляной, 4-хлорфенокси- уксусной, холиновой, никотиновой, пириди- луксусной и др.
При этом указанные соединения получа0 ют реакцией четвертичной соли гепарина с кислотой, активированной карбодиимидом. Эта реакция дает не только о-ацилирован- ное производное, но и вторичный стабильный продукт в виде сложноэфирного
5 о-ацилиэомочевинного производного. Кроме того условия реакции благоприятны для образования пангидридов и межмолекулярных реакций между кислотными и гидро- ксильными функциями гепарина,
0 Наиболее близким к предлагаемому является способ этерификации гепарина или низкомолекулярного гепарина воздействием хлорангидрида кислоты в формамиде в присутствии пиридина при температуре
5 40°С, в течение 3-4 ч. Получаемые продукты обладают повышенной трансмембранной проницаемостью.
По окончании реакции добавляется вода и осуществляется диализ против NaCI и воды, и последующая миофилиэация.
Недостатком этого способа является то, что получаемые продукты претерпевают ча стичную десульфатацию. Кроме того, они 0- и N-ацилированы. В связи с этим соотношение групп сульфат/карбоксил в получаемом глюкозаминогликане изменено.
По указанным причинам получить продукты, обладающие нужными и воспроизво- димыми биологическими свойствами, известным способом нельзя.
Цель изобретения - обеспечение селективности 0-ацилирования и получение продуктов с воспроизводимыми биологическими свойствами.
Указанная цель достигается тем, что в способе получения 0-ацилированных глю- козаминогликанов, включающем обработку глюкозаминогликанов ацилирующими агентами в присутствии акцептора кислоты, в среде полярного органического растворите- ля, с последующей очисткой целевых продуктов, глюкозаминогликаны используют в виде три- или тетрабутиламмониевых солей, в качестве ацилирующих агентов применяют ангидриды карбоновых кислот, и обработку осуществляют при комнатной температуре.
При этом в качестве акцептора кислоты используют одно или несколько соединений, выбранных из группы, включающей пиридин, диметиламинопиридин, триэтила- мин и трибутиламин.
После ацилирования продукты могут быть осаждены с помощью раствора ацетата натрия в этаноле, осадок растворен в воде, а полученный раствор диализован против воды.
Диализ может быть осуществлен в присутствии слабого основания, В частности, в качестве слабого основания может быть использован бикарбонат натрия.
Введением во взаимодействие растворимой в органической среде соли GAGS, такой, как соль третичного или четвертичного аммония, с ангидридом карбоновой кислоты в апротонном полярном органическом растворителе, получают селективное ацили- рование свободных гидроксидов, без изменения функциональных карбоксильных или аминных групп используемого GAGS. Использование GAGS в форме растворимой в органической среде соли позволяет предпочтительно контролировать реакцию ацилирования с большой точностью. Более того, степень ацилирования легко модулируется и может быть увеличена, не вызывая при этом изменения остальной части молекулы. Особенно можно достигать степени ацилирования 0,1-3-ацмльных групп на ди- сахаридную единицу, в особенности 0,5-2- ацильные группы.
Найдено, что при этом не образуется низкое N-ацилированное производное и что, когда ангидриды образуются на уровне карбоксильных функций, использование слабого основания позволяет легко их снова превратить в свободную кислоту.
Предпочтительно, селективное ацили- рование GAGS согласно способу изобретения позволяет осуществлять модулирование их биологических активностей, которые в некоторых случаях сильно увеличиваются.
Наконец, найдено,что 0-ацилирован- ные GAGS, полученные таким образом, обладают более пролонгированной фармакологической активностью.
В предпочтительном способе изобретения, выбранные глюкозаминогликаны представляют собой .глюкозаминогликаны гепаринового типа, а именно гепарин, производные гепарина, полученные либо путем фракционирования, либо полусинтетическим или синтетическим путем, гепарин- сульфат и его производные, полученные путем фракционирования, полусинтетическим или синтетическим путем.
Для получения смесэй фрагментов гепарина, имеющих молекулярную массу ниже 10000 дальтонов, также можно использовать метод ферментативной деполимеризации.
В другом предпочтительном аспекте изобретения, можно использовать смесь фрагментов гепарина, однородных по их молекулярной массе; фрагмент гепарина, получаемый путем синтеза, однородный в том, что касается его молекулярной массы, а также в том, что касается его функционализа- ции.
В другом интересном аспекте изобретения, в качестве глюкозаминогликана можно выбирать производные гепарина, лишенные участков связи с антитромбином III (AT HI), либо когда гепариновые цепи фракционированы, чтобы удалить олигоса- харидные цепи, включающие участок связи с AT III, прибегая, например, к афинной хроматографии на смоле Сефзроза-АТ III или к ионообменным хроматографиям, либо когда эти участки разрушены, например, деполимеризацией периодатом с последующим / -элиминированием или кислотным гидролизом.
Особенно предпочтительные соединения могут быть получены из соединений, приготовленных по способу, использующему йодного окисление с последующим щелочным / -элиминированием, восстановлением и фракционированием.
Другими предпочтительными соединениям, отвечающими формуле VI, являются
смеси соединений, гомогенных по их молекулярной массе, получаемые путем гель- фильтрации, в которых п обозначает целое число 2-12,
Из соединений с гепариновой структурой, лишенных участка фиксации с AT III, и следовательно, лишенных антикоагулмрующей активности, семейство предпочтительных соединений соответствует N-десульфатиро- ванным М-ацетилированнымселективно 0-. ацилированным производным гепарина.
В другим предпочтительном аспекте изобретения, выбираются глюкозамингли- каны типа хондроитин-сульфата, а именно хлондроитин-4 и 6-сульфаты, дерматан- сульфат, и их фрагменты.
Способ настоящего изобретения отличается тем, что гяюкозаминогликаны превращают в растворимую в органической среде соль и эту соль обрабатывают ацили- рующим агентом, способным селективно ацилировать первичные и вторичные гидро- ксилъные группы этого глюкозаминоглика- на, не затрагивая функциональные группы NHRa, или COOR, присутствующие в этом глюкозаминогликане до реакции ацилйро- вания, причем реакция осуществляется в. апротрнном полярном растворителе в присутствии катализатора и в присутствии основания, способного улавливать выделяющуюся во время ацилирования кислоту, при комнатной температуре.
Продукт затем осаждают с помощью смешивающегося с водой растворителя, такого как этанол, к которому можно добавлять добавку, благоприятствующую осаждению, такую как минеральную соль, 0-Ацилированный глюкозаминогликан выделяют путем растворения в воде осадка и предпочтительно диализирования в присутствии слабого основания.
Предпочтительно, способ изобретения осуществляется согласно следующим стади- ям:. . : . --.. - ;
(1)Глюкозаминогликан предотвращают в соль вышеуказанного глюкозаминоглика- на, растворимую в апротонном полярном органическом растворителе;
(2)эту соль обрабатывают ангидридом формулы: ацил-О-ацил, в вышеуказанном апротонном полярном органическом растворителе, в присутствии каталитических количеств пиридина, диметиламинопири- дина, или триэтиламина или трибутилами- на.. . - ;
(3)таким образом полученный продукт осаждают воздействием раствора ацетата натрия в этанола; и
(4)селективно-0-ацилированный глюкозаминогликан выделяют путем растворения
в воде таким образом полученного осадка и путем диализа в присутствии слабого основания, и в известных случаях натриевую соль таким образом полученного селективно 0-ацилированного глюкозаминогликака превращают в другую фармацевтическую совместимую соль, :
В предпочтительном аспекте способа изобретения, используемый в стадии (1)
0 глюкозаминогликан выбирается в группе, образованной гепарином, смесью фрагментов гепарина, имеющих молекулярную массу ниже 10000 дальтонов, смесью фрагментов гепарина, имеющих среднюю моле5 кулярную массу 2000-7000 дальтонов, Смесью фрагментов гепарина, имеющих среднюю молекулярную массу около 4500 дальтонов, смесью фрагментов гепарина, имеющих среднюю молекулярную массу
0 около 2500 дальтонов, смесью фрагментов гепарина, гомогенных по своей молекулярной массе: фрагментом гепарина, получен- иым путем синтеза, гомогенным в том, что касается его молекулярной массы, и в том,
5 что касается.его функционализации.
В другом предпочтительном аспекте способа изобретения, используемым в стадии (1) глюкоэаминогликаном является гепарин, фракция или фрагмент гепарина.
0 лишенные участка связи с антитромбином III.
Глюкозаминогликан, используемый в стадии (1) способа изобретения, предпочтительно может быть еще выбран в группе,
5 образованной дерматансульфатом и его фрагментами, или 4-и 6-сульфат-хондроити- нами и их фрагментами.
Предпочтительно, соль глюкозаминог- ликана, используемая в стадии (1), способа
0 изобретения, представляет собой соль третичного амина соль трибутиламмония, или соль четвертичного аммония соль тетрабу- тиламмония,
В другом предпочтительном аспекте
5 способа изобретения, используемым в стадии (2) ангидридом является ангидрид алка- новой кислоты, содержащей 2-ЮС-атомов, предпочтительно 4-10, особенно 4 или 6 С-атомов.
0 Апротонный полярный растворитель, в котором осуществляется стадия (2) способа изобретения, предпочтительно может быть выбран из группы, включающей диметил5 формамид, гексаметилфрсфортриамид, пиридин, или еще смесь этих растворителей друг с другом или с дихлорметаном, и катализатор выбирается в группе, образованной аминами, такими, как пиридин, и диметила- минопиридин.
Предназначенным для нейтрализации кислотности основанием может быть пиридин (который может служить одновременно растворителем, катализатором и основанием), триэтиламин или трибутиламин.
Предпочтительно, диализ, осуществляемый в стадии (4), проводится в присутствии слабого основания, такого, как бикарбонат натрия, чтобы удалить возможные вторичные продукты, такие, как ангидриды.
Продолжительность реакции может меняться в зависимости от желательной степени ацилирования, например, 1-24 ч.
Соединения изобретения, селективно 0-ацилированные,обладают ин виво пролонгированной активностью. Например, когда эти соединения представляют собой соединения гепаринового типа, обладающие участком связи с А Т III, и следовательно, способны проявлять энтитромботическую активность, то эта активность четко пролонгирована во времени по сравнению с таким же соединением, не подвергнутым селективному 0-ацилированию.
Селективно 0-ацилированные гепариновые производные, согласно изобретению, имеющие или нет участок связывания с AT III, примем эти последние практически лишены антикоагулирующей активности, обладают также различными биологическими активностями, а именно:
-ингибирование пролиферации гладких мышечных клеток, которое представляет большой интерес для избежания рецидивов стеноза при вмешательствах, таких, как ангиопластика, обходы и сшивка вен и артерий, трансплантация органов, в особенности трансплантации сердца:
-ингибирование гепариназы и гепари- тиназы, ферментов, принимающих участие в метастазных диссеминировэниях;
-противовирусные свойства, в особенности по отношению к ретро-вирусу, а именно по отношению к различным НУ (вирус человеческого иммунодефицита), что представляет большой интерес при лечении СПИДа.
-свойства ингибирования лейкоцитарной эластазы. повышения имеющегося процента ингибиторов эластазы, также как селективного ингибирования синтеза коллагена типа III и фибронектина, что представляет большой интерес для лечения заболеваний, в которых наблюдается нарушение равновесия системы эластазанти- эластаза, таких, как эмфизема, также для лечения дегенеративных заболеваний инъ- юнктивной ткани, таких, как артериосклероз и диабет.
Селективно-0-ацилированные глюкоз- миногликаны изобретения, следовательно,
могут образовывать действующее начало лекарств большого интереса в многочисленных показаниях.
Изобретение также позволяет получить
5 биологические объекты (селективно 0-аци- лированные глкжозаминогликаны), которые могут быть использованы в качестве стандартов или этанолов в сравнительных изучениях для исследований отношений
0 структура/активность в различных физиологических системах, в которых могут содержаться глюкозаминогликзны.
Изобретение будет лучше понятно с помощью нижеследующих примеров.
5 Пример 1. Получение О-ацетилиро- ванного гепарина (1C 1938) из тетрабути- ламмониевой соли гепарина.
а)Получение тетрабутиламмониевой соли гепарина
0 Натриевую соль гепарина (10 г) растворенную в воде (500 мл), перколируют через колонну с катйонообменной смолой (Dowex 50 W х 4, в форме Н4), Полученный раствор нейтрализуют гидроксидом тетрабути5 ламмония. После лиофилизации получают тетрабутиламмониевую соль гепарина
(19,55 г).
б)Ацитилирование:
1,05 г Тетрабутиламмонийгепарината
0 растворяют в безводном диметилформа- миде (5 мл), После охлаждения до 0°С прикапывают уксусный ангидрид (1 мл: 10:46 ммоль), затем триэтиламин (1,45 мл, 10:46 ммоль) и диметиламинопиридин
5 (64 мг, 0,5 ммоль), Смесь выдерживают 24 часа при комнатной температуре. После добавления воды (5 мл), раствор диализуют в течение 72 ч против дистиллированной воды. Тетрабутиламмониевую соль превраща0 ют в натриевую соль пропусканием через смолу Dowex 50, Н+, при 0°С, с последующей нейтрализацией 1н.раствором гидроксида натрия. После лиофилизации получают 0,49 г гепарината натрия, селективно 0-ацетили5 рованного и имеющего следующие характеристики:
соотношение сульфат/карбоксил: 2,28 мэкв/г (исходный продукт 2,20 мэкв/г}. Титр АРТТ: 9.1 nl/мг
0 13С-ЯМР-спектр (метанол, 51,6 м.д., внутренний стандарт):
сигнал при 23,4 м.;д.: СНз от СНзСОО; сигнал при 24,5 м.д.: (слабый): СНз от СНз- CO-NH- (идентичен исходному продукту).
5 ЯМР-сп:ектр указывает на наличие около двух ацетильных групп на дисахаридную единицу.
Пример 2. Получение Оацетилиро- ванного гепарина (1C 1938) из трибутилам- мониевой соли гепарина.
а)Получение трибутиламмониевой соли гепарина:
Натриевую соль гепарина (10 г) раство-1 ряют в воде (500 мл), затем перколируют через колонну с катионообменной смолой (Dowex 50 W х 4, в Н -формё). Раствор нейтрализуют добавлением 10%-ного раствора трибутиламина в этаноле. После промывки и лиофилизации, получают тетрабутиламмо- ниевую соль гепарина (14,77 г),
б)Ацетилирование
К охлажденному до 0°С раствору вышеуказанной соли(4 г) в безводном диме- тилформамиде (50 мл), добавляют диметиламинолиридин (250 мг), уксусный ангидрид (3,9 мл) и трибутиламин (9,7 мл). После выдерживания 24 ч при комнатной температуре, добавляют воду (1,5 мл). Смесь затем выливают в насыщенный раствор в этаноле ацетата натрия. После промывки этанолом, осадок растворяют в воде, после чего диализуют против 5%-но.го бикарбоната в воде, затем против воды. После лиофилизации получают гепаринат натрия, который 0-ацетилирован (1,81 г),
ЙК-спектр имеет сильную полосу, относимую к сложному эфиру, при 1730см -,
После омыления, продукт имеет:
-соотношение сульфат/карбоксил 2,38 мэкв/г (исходный продукт: 2,40 мэкв/г),
-титр АРТТ: 85 nl/мг,
Пример 3. Получение О-ацетилиро- ванного гепарина (1C 1938), используя катализ пиридином,
Тетрабутиламмониевую соль гепарина (0,58 г) растворяют в смеси (1/1 от объема) пиридина и диметилформамида (10 мл). Добавляют уксусный ангидрид (0,5 мл), затем смесь выдерживают при комнатной температуре. Спустя 24 часа добавляют водный раствор ацетата натрия (1М, 15 мл), затем смесь выливают в этанол (80 мл) охлажденный до 0°С. После центрифугирования осадок растворяют в воде (10 мл), затем добавляют этанол (160 мл), а после него- водный ацетат натрия (Ш, 10 мл). Осадок затем обрабатывают водной и лиофмлизу- ют, получая 0-ацетилированный гепарин (0,22 г).
П р и мер 4. Получение 6-пропионили- рованного гепарина (1C 1939) с различными степенями пролионилирования.
К охлажденному до 0°С раствору гепа- рината тетрабутияаммония (1,85 г), полученному как описано в примере 1, в диметилформамиде (10 мл) прикапывают пропионовый ангидрид (2,7 мл), затем триэтиламин (2,9 мл) и диметиламинопири- дин (128 мг). Во время 1 ч, 2 ч, 4ч, 8 ч и 24 ч отбирают часть реакционной смеси, разбавляют равным объемом воды и диализуют в течение 24 ч в дистиллированной воде. После пропускания через смолу Dowex 50, Н. и нейтрализации раствором гидроксида натрия, полученные продукты лиофилизуют и они имеют характеристики, представленные в табл.1.
ПМР-спектр продуктов зарегистрирован в воде с 3,3,3-триметилсилилпропио- натрм (ТСП) в качестве внутреннего стандарта. Он имеет сигналы при: 1,1 м.д. и 2,4 м.д., характерные относительно остатков СНз и СН2, СНз-СНг-СО-О, и сигналы 5-5 м.д., характерные для протонов озидие- вого скелета.
Сравнение интенсивности сигналов лрипи- олила и скелета между обработанным .в течение 1 часа продуктом и таковым, подвергнутым в течение 24-х часов реакции, иллюстрирует влияние продолжительности реакции, в самом деле, наблюдают уменьшение сигналов протонов скелета, что указывает на увеличение степени замещения.
С-ЯМР-спектр (метанол, 51,6 м.д., внутренний стандарт), имеет сигналы при 10,8 и 30 м.д.( характерные для радикалов СНз и СН2 пропионатов.
П р и м е р 5. Получение 0-бути л и рованного гепарина (1C 1940)
А) Использование тетрабутиламмоние- вой соли гепарина:
К раствору соли тетрабутиламмония гепарина (0,55 г), полученной как описано в примере 1, в диметилформамиде (5 мл) добавляют при 0°С масляный ангидрид и ди- метиламинопиридин. После выдерживания 24 ч при комнатной температуре, добавляют воду (5 мл), затем реакционную смесь диализуют а течение 72 ч против дистиллированной воды. После пропускания через ионообмен ник Dowex 50, Н, нейтрализации раствором гидроксида натрия и лиофилизации получают Обутилированный гепарин в форме соли натрия (0,32 г).
Соотношение сульфат/карбоксил: 2,15 мэкв/г (исходный продукт: 2.20 мэкв/г),
Титр АРТТ: 59 nl/мг.
ПМР-спектр (ГСП в качестве внутреннего стандарта) показывает наличие сигналов при 0,9 м.д., 1,6 м.д. и 2,4 м.д., характерные для групп СНз-СНг из СНз-СНа-СНа-СО-О-, и сигналы между 3 и 6 м.д., характерные для озидиевого скелета.
Анализ ЯМР-спектра указывает на наличие около одной бутирильной цепи на диса- харидную единицу.
Б) Использование трибутиламмоние- вой соли гепарина:
К охлажденному до 0°С раствору трибу- тиламингепарината (4 г), полученного как описано в примере 2, в димётилформамиде (50 мл), добавляют диметиламинопиридин (0,25 г), масляный ангидрид (6,7 мл) и трибутиламин (9,7 мл). Оставляют реакционную смесь на 24 часа при комнатной температуре. Добавляют воду (1,5 мл), затем, спустя 30 минут, насыщенный раствор ацетата натрия в этаноле (250 мл). Осадок затем промывают три раза этанолом, затем диализуют против 5%-ного раствора бикарбоната, затем против воды. Получают, после лиофилизации, натриевую соль 0-бутилиро- ванного гепарина (2,1 г).
Титр АРТТ: 29 ni/мг
После омыления сложных эфиров 0,5 М раствором гидроксида натрия в течение 2 часов при 0РС, полученный продукт имеет соотношение сульфат/карбоксил 2,36 мэкв/г (2,40 мэкв/г в исходном продукте).
Пример 6. Получение 0-гексаноили- рованного гепарина 0С 1941).
А) Использование тетрабутиламмоние- вой соли гепарина:
К раствору тетрабутиламмрниевой соли гепарина (0,55 г), полученной как описано в примере 1, в диметилформамиде (5 мл),:До- бавляют, при 0°С, капроновый ангидрид (1 мл, 6 ммоль), триэтиламин (0,84, мл, 6 ммоль) и диметиламинопиридин. Спустя 24 часа стояния при комнатной температуре добавляют воду (5 мл), затем реакционную смесь диализуют в течение 72 ч против 5%-ного раствора бикарбоната, затем против дистиллированной воды. После ионноОбмена на смоле Sowex 50, Н+, нейтрализации раствором гидроксида натрия и лиофилизации, получают 0-гексаноилированный гепарин в форме натриевой соли (0,30 г).
Титр АРТТ: 25 nl/мг
ПМР-спектр (ГСП, внутренний стандарт), показывает сигналы при 0,8, 1,2, 1,5 и 2,3 м.д. характерные для СНз-СН2 из СНз(СН2)8-СО-0-.
Б) Использование трибутияаммониевой соли гепарина:
К охлажденному до 0°С раствору гепа- рината трибутиламмония (4 г), полученного как описано в примере 2, в диметилформамиде (50 мл), добавляют диметиламинопиридин (0,25 г), трибутиламин (9,7 мл) и ангидрид гексановой кислоты (10,6 мл). Спустя 24 часа стояния при 20°С, добавляют воду (1,5 мл), затем насыщенный раствор ацетата натрия в этаноле. После промывки этанолом, диализа и лиофилизации, получают 0-гексаноили- зированный гепарин (2,5 г).
Пример 7. Получение 0-октаноили- рованного гепарина (1C 1942).
К охлажденному до 0°С раствору гепа- рйната трибутиламмония (4 г), полученного как описано в примере 2, в диметилформамиде (50 мл), добавляют диметиламинопи5 ридин (0,25 г), ангидрид октановой кислоты .(12,1 мл) и трибутиламин (9,7 г). Спустя 24 часа стояния при комнатной температуре, добавляют воду, затем, спустя 30 минут, насыщенный раствор ацетата натрия в этано0 ле. После диализа против 20%-ного раствора ацетата натрия в этаноле, затем против дистиллированной воды, и ультрафильтрации, продукт подвергают катирнно- му обмену путем пропускания через смолу
5 Dowex 50, Н , с последующей нейтрализацией гидроксидом натрия. После лиофилизации получают 0-актаноилированный гепарин (2,5 г).
Пример 8. Получение 0-деканоили0 рованного гепарина (1C 1943).
А) Использование тетрабутиламмоние- вой соли гепарина.
К раствору тетрабутиламмониевой соли гепарина (0,55 г), полученной как описано в
5 примере 1, в диметилформамиде (5 мл), при ОС добавляют капроновый ангидрид (Т мл, 6 ммоль), триэтиламин (0,84 мл, 6 ммоль) и диметиламинопиридин. После стояния 24 часа:при комнатной температуре добавляют
0 воду (5 мл), затем реакционную смесь диализуют в течение часов против 5%-ного раствора бикарбоната, затем против дистиллированной воды. После ионообмена на смоле Dowex 50, Н , нейтрализации раство5 ром гидроксидэ натрия и лиофилизации, получают 0-деканоилированный гепарин в форме натриевой соли (0,30 г).
ЯМР-слектр (ГСП в качестве внутреннего стандарта) имеет два сигнала при 0,8,1,2,
0 1.5 и 2,4 м.д, характерные для СНз и СН2 от СНз-(СН2)8-СО-0-.
Б).Использование трибутиламмониевой соли гепарина.
К охлажденному до 0°С раствору трибу5 тиламмонийгепарината {4 г), полученного как описано в примере 2, в диметилформамиде (50 мл), добавляют диметиламинопиридин (0,25 г), ангидрид декановой кислоты (13,3 г), растворенный в 20 мл диметилфор0 мамида) и трибутиламин (9,7 мл). После стояния 24 часа при комнатной температуре, добавляют воду (5 мл), затем насыщенный раствор ацетата натрия в этаноле. Осадок растворяют в диметилсульфоксиде, затем
5 диализуют против воды, бикарбоната натрия и снова воды. После лиофилизации, получают 0-деканойлированный гепарин (2,38 г).
При мер 9. Получение 0-олеоилиро- ванного гепарина (1C 2013)
Трибутиламмониевую соль гепарина (3 г) и N.N-AHMeTMnaMHHonnpHflMHa (244 г) растворяют в безводном диметилформамиде (50 мл). После охлаждения до 0°С, прикапывают олеиновый ангидрид (21 г) в виде рас- твора в дихлорметане (20 мл), затем трибутиламин (9,5 мл). Спустя 24 часа реакции vi охлаждения при 0°С, вводят 5%-ный бикарбонат натрия (10 мл), затем, спустя час, насыщенный спиртовой раствор ацета- та натрия. После промывки абсолютным этанолом, растворения в смеси диметил- сульфоксида с водой (4/1, по объему, 750 мл), диализуют против водного 10%-ного этанола, в течение 2-х дней, затем против воды в течение 3-х дней. Натриевую соль олеоилированного гепарина выделяют, после лиофилизации и осаждения в диэтиловом эфире лиофилизата, снова растворенного в ДМФ (2,77 г). Содержание олеиновой кисло- ты: 1,44 мкмоль/мг.
Прим ер 10. Получение СНюнзоили- рованного гепарина (1C 1944).
Тетрабутиламмониевую соль гепарина, полученную как описано в примере 2, бен- зоилируют с помощью бензойного ангидрида в условиях, описанных выше для получения 0-декзноилироаанного гепарина (пример 8) ЯМР-13С-спектр полученного продукта имеет сигналы при 131, 132 и 136 м.д., характерные для бензоильных групп.
Продукт лишен антикоагулирующей активности ин витро.
П р и м е р 11. Получение 0-3-циклопен- тилпропионилированного гепарина (1C 2014).
Ангидрид 3-циклопентилпропионовой кислоты получают после присоединения кислоты (23 мл, 150 ммоль), в виде раствора в дихлорметане (400 мл), к перемешиваемой и охлаждаемой до -10°С смеси, содержащей тетрабутиламмонийбромид (15 ммоль), 20%-ный раствор гидроксида натрия (60 мл) и дихлорметан (80 мл), и после декантации, промывок 5%-ным бикарбонатом натрия и водой, и концентрирования в виде масла (96%). Характеристическая частота в ПК-спектре: 1800, 1745, 1040 трибути- ламмониевую соль гепарина (4 г) и М,М-ди- метиламинопиридин (253 мг) растворяют в безводном диметилформамиде (40 мд). После охлаждения до 0°С прикапывают 3-цик- лопентилпропионовый ангидрид (11 г), затем трибутиламин (9,8 мл). После 24 ч реакции при комнатной температуре, а затем охлаждения до 0°С добавляют воду (1 мл), и затем, час спустя, насыщенный спиртовой раствор ацетата натрия. После промывки абсолютным этанолом, осадок диализируют 5%-ным бикарбонатом натрия в течение 24
часов, затем водой, в течение 3 дней. После лиофилизации получают 0-3-циклопентил- пропионил-содержащий гепарин в форме его соли натрия (2.64 г).
ЯМ Р-13 С-спектр в РзО (метанол 51,6 м.д., внутренний стандарт), показывает характеристические сигналы при 27,4 м.д.; 31,1 м.д.,34,6м.д.,35,9м.д. и41,7м.д, группы 0-циклопентилпропионил.
Соотношение сульфат/карбоксил: 2,2.
Пример 12. Получение 0-ацетилиро- ванного гепарина с небольшой молекулярной массой (средняя мол.масса 4500 дальтрнов, интервал мол.массы 1800-8000 дальтоновХЮ 1945).
Этот гепарин с небольшой молекулярной массой получают путем частичной деполимеризации с помощью азотистой кислоты и спиртового фракционирования как описано в европейском патенте 181252 и называют ниже как СУ 216.
А) Использование тетрабутиламмоние- вой соли СУ 216.
Натриевую соль СУ 216(1 г) превращают в тетрабутиламмониевую соль путем пропускания через колонну со смолой Dowex 50, Н+, с последующей нейтрализацией гидро- ксидом тетрабути я аммония.
Таким образом полученную соль (1:7 г) сушат под вакуумом в течение трех часов при 50°С. затем растворяют в безводном диметилформамиде (10 мл). После охлаждения до 0°С, добавляют уксусный ангидрид (1,7 мл) по каплям, затем триэтиламин (2,4 мл) и диметиламинопиридин (102 мг). Спустя 20 ч протекания реакции, продукт хроматографируют на колонне с Сефадек- сом-ЧЗ-25, элюируя водой. Получают, после конверсии в натриевую соль и лиофилизации, СУ 216 О-ацетилированный (0,89 г).
ЯМР-ТЗ С-спектр (метанол, 51,6 м.д., как внутренний стандарт) имеет сигнал при 23 м.д., характерный для ацетатов.
Сигнал СНз от СНз-CO-NH-при 24,5 м.д. идентичен таковому исходного продукта.
Соотношение сульфат/карбоксил составляет 2,09 мэкв/r (исходный продукт: 2,05 мзкв/г)..
-ТитрАРТТ: 18п|/мг
- Титр анти-Ха: 205 n/мг (анализ по Jin и др., J.Lab Clin. Meo) 1973. 81, 298-310).
Б) Использование трибутиламмониевой солиСУ216.
Натриевую соль СУ 216 превышают в Трибутиламмониевую соль путем пропускания через колонну со смолой Dowex 50, Н, затем нейтрализации трибутиламином, как описано для гепарина. Трибутиламмониевую соль СУ 216 получают после промывки
эфиром, лиофилизации и высушивания в сушильном шкафу под вакуумом.
Вышеуказанную соль (4 г) и М,М-димети- ламинопиридин (288 г) растворяют в диметилформамиде. После охлаждения до 0°С, прикапывают уксусный ангидрид (4.4 мл) и затем трибутиламин (11.2 мл). После 24 ч при комнатной температуре и при охлаждении до 0°С добавляют воду (1,7 мл), затем, спустя 1 час, насыщенный спиртовой рас- твор ацетата натрия. Осадок промывают этанолом, растворяют в воде, диализуют в течение 36 часов против 5%-ного бикарбоната, затем против воды в течение 3 дней. Получают после лиофилизации а.цетили- рованный СУ 216 в форме натриевой соли
(1,4 Г).:
С-ЯМР-спектр в Д2 О (метанол при 51,6 м.д. в качестве внутреннего стандарта) имеет при 23 м.д. сигнал, характерный для ацетилированной группы. Соотношение сульфат/карбоксил: 2,07.
П р и м е р 13. Получение Обутирили- рованного гепарина незначительной молекулярной массы (средняя мол.масса 4500 дальтонов, интервал мол.масс 1800- 8000 дальтонов (1C 1957).
Тетрабутиламмониевую соль СУ 216 (4 г), полученную как описано в примере 12, и М,М-диметиламинопиридин (288 мг) рас- тврряют в безводном диметилформамиде (40 мл): после охлаждения до 0°С добавляют масляный ангидрид (7,68 мл), по каплям, затем добавляют трибутиламин (11,2 мл). После протекания реакции в течение 24-х часов при комнатной температуре, затем охлаждения до 0°С, добавляют воду (1,7 мл), затем, спустя час, насыщенный спиртовой раствор ацетата натрия. После промывки этанолом, растворения в апирогенной воде. диализуют против 5%-ного бикарбоната на- трия в течение 36 часов, затем против воды в течение 3-х дней: 0-бутирилированный СУ 216 выделяют в форме натриевой соли после лиофилизации (2,14 г).
13С-ЯМр-спектр в ДаО (метанол при 51,6 м.д. в качестве внутреннего стандарта) имеет при 15..6 м.д., 20,5 м;д. и 39.4 м.д. сигналы, характерные для 6-бутирилиро- ванной группы. Соотношение сульфат/кар- боксил: 2,08.
Пример 14. Получение 0-гексаноили- рованного гепарина незначительной молекул ирной массы (средняя мол. масса 4500 дальтонов, интервал мол.масс. 1800-80рО дапьтонов) (С 1958).
Трибутиламмониевую соль СУ 216 (4 г), полученную как описано в примере 12, и N.N-диметиламинопиридин (288 мг) растворяют в диметилформамиде (40 мл). Посдв
охлаждения до 0°С прикапывают капроновый ангидрид (10,8 мл) и трибутиламин .(11.2. мл). После протекания реакции в течение 24-х часов при комнатной температуре, затем охлаждения до 0°С, добавляют воду (1,7 мл), затем спустя 1 ч насыщенный спиртовой раствор ацетата натрия, После промывки абсолютным этанолом, растворения в апирогенной воде, диализуют против 5%-ного бикарбоната натрия в течение 36 часов, затем против воды в течение 3-х Дней, 0-Капроилированный СУ 216 выделяют в форме натриевой соли после лиофилизации (2,5 г}.
13С-ЯМР-спектр в ДаО (метанол с 51,6 м.д. в качестве внутреннего стандарта) имеет при 15,9 м.д., 24,2 м.д.. 26,4 м.д:, 33,1 м.д. и 36,4 м.д. сигналы, характерные для капроильной группы.
Соотношение сульфат/карбоксил: 2,08.
Прим ер 15. Получение 0-октаноили- рованного гепарина небольшой молекулярной массы (средняя мол.масса 4500 дальтонов, интервал мол.массы 1800-8000 дальтонов) (1C 1959).
Трибутиламмониевую соль СУ 216 (4 г), полученную как описано в примере 12, и М,М-диметиламинопиридин (288 мг) растворяют .в диметилформамиде (40 мл). После охлаждения до 0°С прикапывают ангидрид каприловой кислоты (14 мл), затем трибутиламин (1 1,2 мл). После протекания реакции в течение 24 ч при комнатной температуре и охлаждения до 0°С добавляют воду (1,7 мл), затем, спустя час, насыщенный спиртовой раствор ацетата натрия. После промывки абсолютным этанолом, растворения в апирогенной воде; диализуют 5%-ным бикарбонатом натрия в течение 36 часов, затем водой в течение 3 дн., получают после лиофилизации, 0-октаноилированный СУ 216 в форме натриевой соли (.1,76 г). С-ЯМР- спектр. в ДМСО-de (метанол при 5:1,6 м.д. в качестве внутреннего стандарта) имеет сигналы при 16,8 м.д,, 25,0 м.д., 27,3 м.д., 30,9 м.Д., 31,4, 34,1 и 36,4 м.д., характерные для каприлоильной группы. Соотношение сульфат/карбоксил: 2,07.
Пример 16. Получение 0-деканои- лированного гепарина небольшой молекулярной массы (средняя мол.масс 4500 дальтонов, интервал мол.масс 1800-8000 дальтонов) (1C 1960),
Трибутиламмониевую соль СУ 216 (4 г), полученную как описано в примере 12, и N.N-диметиламинопиридин (288 мг) растворяют в диметилформамиде (40 мл). После охлаждения до 0°С, прикапывают ангидрид декановой кислоты (15,3 мл) в виде раствора в безводном диметилформамиде
(10 мл), ззтем трибутиламин (11,2 мл). После протекания реакции в течение 24 ч при комнатной температуре, и-охлаждения до 0°С. добавляют воду (1,7 мл), затем, спустя час, насыщенный спиртовой раствор ацетата натрия.
После промывки абсолютным этанолом и суспендирования в апирогенной воде, ди- ализуют 5%-ным бикарбонатом натрия в течение 2-х дней, 10%-ным NaCI в течение 2-х дней, затем водой в течение 5 дней. 0-Деканоилированный СУ 216 выделяют в форме натриевой соли после лиофилизации (2,76 г).
13С-ЯМР-спектр в Д20 (метанол при 51,6 м.д. в качестве внутреннего стандарта) имеет сигналы при 16,4 м.д., 22,3 м.д., 25,1 м.д., 29,2 м.д., 31,9, 34,4 и 36,5 м.д., характерные для деканоильной группы.
Соотношение сульфат/карбоксил: 2,13. П р и м е р 17. Получение 0-ацетилиро- ванного гепарина незначительной молекулярной массы (средняя мол.масс 2500 дальтонов, интервал мол.масс 1500-8000 дальтоновХ С 1946).
Этот гепарин небольшой молекулярной массы получают путем частичной деполимеризации с помощью азотистой кислоты согласно способу, описанному в европейском патенте 37319, и называют далее СУ 222.
Натриевую соль СУ 222 превращают в трибутиламмониевую соль путем пропускания через колонну со смолой Dowex 50, Н , затем нейтрализации трибутиламином.
Полученную после лиофилизации соль (1,5 г) растворяют в диметилформамиде (5 мл), затем, после охлаждения до 0°С, добавляют по каплям уксусный ангидрид (1,35 мл), затем триэтиламин (2 мл) и диме- тиламинопиридин (85 мг). После протекания реакции в течение 18ч, добавляют воду (20 мл), затем смесь диализуют в течение 3 дн против дистиллированной воды. После конверсии в натриевую соль и лиофилиэа- ции получают СУ 222, 0-ацетилированный (0,86 г).
Продукт имеет соотношение сульфат/карбоксил 1,98 мэкв/г (исходный продукт: 1,97 мэкв/г).
ЯМР13 С-спектр (метанол 51,6 м.д. в качестве внутреннего стандарта) продукта содержит сигнал при 23 м.д., характерный для 0-ацетила.
Сравнение интенсивностей сигналов N-эцетила (при 24,5 м.д.) между исходным продуктом и полученным продуктом показывает, что ацетилирование селективное.
-Титр АРТТ: 8ni/Hi- Титр анти-Ха: 19.1 и/мг.
Пример 18. Получение фрагмента гепарина, лишенного сродства к антитромбину III (1C 1772).
1)Разрезание гепариновых цепей с по- мощью йодной кислоты:
10 г гепарина, инъекцируемого в mucus de pore, в форме натриевой соли, с титром 157 nl/мг в дозировке Codex и 155 и/мг в дозировке анти-фактора Ха по Jin и др.,
растворяют в 250 мл деминерализованной воды при 4°С; рН раствора доводят до 5,0 с помощью концентрированной соляной кислоты. При умеренном перемешивании добавляют 10 г метапериодата натрия (Na 164,
5 мол.масса: 213,89) в виде раствора в 250 мл диминерализованной воды при 4°С, рН совокупности доводят до 5,0 с помощью концентрированной соляной кислоты. Раствор выдерживают 24 часа, в темноте, в холод- 0 ном помещении при+4°С. .
2)Элиминирование остаточного перио- дата:
Затем реакционный раствор размещают в три диализных рукава (пористость 3- 5 4.000 Da) и подвергают его диализу в течение 15 ч против проточной деминерализованной воды.
3)Деполимеризация в основной среде: К 780 мл полученного после диализа
0 раствора добавляют 16 мл 10 н. раствора гидроксида натрия, и всю совокупность подвергают перемешиванию в течение 3-х часов при комнатной температуре (порядка ,18-21°С).
5 4) Восстановление:
500 мг Боргидрида натрия NaBH/j, (мол.масса: 37.83) добавляют после этого и раствор перемешивают снова 4 ч при комнатной температуре. Его рН затем доводят
0 до 4 с помощью концентрированной соляной кислоты. После перемешивания в течение 15 мин рН доводят до 7 с помощью концентрированного раствора гидроксида натрия.
5 к 820 мл таким образом полученного раствора добавляют 16,4 г NaCI, затем 1270 мл этанола. Всю совокупность оставляют стоять в течение 3 ч, затем центрифугируют при 2500 об/мин, в течение 20 мин. Осадок
0 собирают, суспендируют снова его в 200 мл чистого этанола, размельчают и окончательно рекупирируют фильтрацией на воронке Бюхнер§. После этого высушивают под вакуумом при 40°С в течение 5 ч. Получают
5 таким образом 8,9 г продукта.
5) Спиртовое фракционирование:
Эти 8,9 г растворяют примерно в 120 мл
деминерализованной воды при комнатной
температуре. Добавляют 1.78 г NaCI и рН
раствора снижают до 3.5 с помощью соляной кислоты. Объем раствора доводят до 178 мл с помощью деминерализованной воды. 151 мл чистого этанола добавляют при перемешивании. Перемешивание продолжают в течение 15 минут после окончания добавления, затем всю совокупность оставляют стоять в течение 10 ч при комнатной температуре.
Образовавшийся осадок получают путем центрифугирования в течение 20 минут при 2500 об/мин. Снова суспендируют в 150 мл чистого этанола, размельчают, рекуперируют путем фильтрации на воронке Бюхнера, промывают 300 мл чистого этанола, и, наконец, высушивают под вакуумом при 40°С в течение 24 ч.
Таким образом, выделяют в форме порошка белого цвета 5 г продукта 1C 1772, обладающего следующими характеристиками:.
-50з: 3.55 мэкв/г; 1,54 мэкв/г; S + С: 5,09 мэкв/г; S/C: 2,31 мэкв/г.
Он практически лишен N-ацетил-глюко- замина (отсутствие сигнала при 24,5 м.д. на спектр ЯМР-13 С).
Титр Codex: 11 nl/мг.
-Титрр АРТТ: 9 nl/иг
-Титр анти-Ха: 12 и/.мг.
Б) Получение 0-ацетилированного фрагмента гепарина, лишенного сродства к антитромбину III ОС 1924).
Полученный в стадии А) продукт превращают в тетрабутиламмониевую соль путем пропускания через смолу Dowex 50. Н , затем нейтрализации с помощью гидрокси- да тетрабутиламмония. Из 9,5 г натриевой соли получают 18 г соли тетрабутиламмония.
К раствору 6 г таким образом полученной соли, в диметилформамиде (55 мл), до- бавляют, после охлаждения до 0°С, уксусный ангидрид (6,2 мл, 5,6 ммоль), затем триэтиламин (9 мл, 65,6 ммоль) и диметила- минопиридин (403 мг, 3,3 ммоль). Спустя 24 ч добавляют насыщенный раствор ацетата натрия в этаноле (250 мл). После центрифугирования и промывки осадко. этанолом, твердое вещество обессоливают на Сефа- декса-С-25, затем подвергают ионообмену путем пропускания на Dowex 50, Н+, с последующей нейтрализацией гидроксидом натрия. После лиофилизации получают продукт 1C 1924(3 г).
Этот продукт имеет следующие характеристики:
-. соотношение сульфат/карбоксил 2,28 мэкв/г
С-ЯМР-спектр (метанол. 51,6 м.д., как внутренний стандарт) ясно показывает, что продукт 0-ацилирован. Сигнал, характерный для С2 N-ацетйл-глюкозамина, около 56 м.д., такой же,-как в исходном продукте, отсутствует в спектре,
Пример 19. Получение 0-бутирили- 5 рованного фрагмента гепарина, лишенного сродства к антитромбину III (IC 1925).
6 г соли тетрабутилэммония 1C 1772, полученной как описано в примере 18. бути- ри лиру ют с помощью масляного ангидрида 0 в условиях, описанных для ацетилирования. Получают продукт 1C 1925 (2,96 г), который имеет следующие характеристики:
-соотношение сульфат/карбоксил: 2,31 мэкв/г
5 Спектр 1 С-ЯМР (метанол. 51,6 м.д. в качестве внутреннего стандарта) содержит
пы при 15,6, 20,4 и 38,4 м.д.
Пример 20. Получение 0-г1й63ййв1ли0 рованного фрагмента гепарина, лишенного сродства к антитромбину III (1C 1926).
6 г соли тетрабутиламмония 1C 1772, полученной как описано в примере 12, обрабатывают ангидридом гексановой кис5 лоты таким же образом, как для ацетилирования. Получают продукт 1C 1326 (3 г), который имеет следующие характеристики: - соотношение сульфат/карбоксил: 2,20 мэкв/г
013С-ЯМР-спектр (метанол, 51,6 м.д,, в
качестве внутреннего стандарта) имеет сигналы, характерные для СНз и СНа гек- сильной группы при 15,5, 23,9, 26,2, 32,7 и 36,1 м.д.
5ПМР-спектр указывает на наличие примерно одной гексильной группы на дисаха- ридную единицу.
П р и м е р 21. Получение смеси фрагментов, лишенных сродства к антитромбину
0 III. гомогенных с точки зрения их молекулярной массы и 0-бутирилированных.
Смесь фрагментов, лишенных антикоа- гулирующей активности, описанных в примере 12,. фракционируют на ее различные
5 составляющие путем гель-фильтрации. Таким образом получают roi-.огенные по молекулярной массе фракции, массы которых составляют 7700, 6500, 5800, 5300, 4980, 4400, 3900, 3400, 2600, 1860 и 1210.
0 Пример этерификации фракции:
Фракцию с массой 2600 (0,20 г) превращают в соль трибутиламмония, затем лио- филизируют и высушивают (0,34 г). Этот продукт затем растворяют а ДМФ (2 мл),
5 затем последовательно добавляют диэтила- минопиридин (18 мг), масляный ангидрид (0,49 мл) и трибутиламин (0,7 мл), после охлаждения до 0°С. После выдерживания 24 часа при комнатной температуре, добавля- ют бикарбонат натрия (1 мл 5%-ного раствоpa), затем через 2 ч, смесь хроматографиру- ют на колонне с Сефадексом С 25 (1 л), элюируя 0,2 М раствором хлорида натрия. Продукт лиофилизмруют после обессолива- ния, получая порошок светло-бежевого цвета (0,24 г).
Другие фракции обрабатывают таким же образом, получая .соответствующие продукты,ИК-анализ которых обнаруживает наличие сложноэфирной полосы при 1734 . Степень сульфатации продуктов (отношение сульфата к карбоксилу, не изменена после ацилирования.
Определение степени ацилирования: . Ее определяют путем хроматографии в газовой фазе, после бутанолиза продуктов бутано-сернокислотной смесью., затем путем экстракции хлороформом сложных бутиловых эфиров и удаления избыточного метанола промывкой водой.
Пример 22. Получение 0-ацетилиро- еэнного дерматан-сульфата (1C 1947).
К охлажденному до 0°С раствору тетра- бутиламмониевой соли дерматансульфата (0.9Т г), полученной в тех же условиях, что и таковые, описанные для получения тетрабу- тиламмонийгеларината в примере 1, вдиме- тилформзмиде (20 мл), прикапывают уксусный ангидрид (1,35 мл, 14,2 ммоль), затем триэтиламин (1,97 мл, 14,2 ммоль) и диметиламинопиридин (0,7 ммопь). Спустя 24 часа выдерживания при комнатной температуре, добавляют воду (40 мл), затем ди- ализуют в течение 72 ч, Натриевую соль получают путем пропускания через колонну С Dowex 50., Н+, при ОеС, е последующей нейтрализацией гидроксидом натрия. Получают, после лиофилизацим. порошок бежевого цвета (0,62 г).
0-Ацеталированный демантан-сульфат имеет следующие характеристики:
-соотношение сульфат/карбоксил: 0,99 мэкв/r (исходный продукт: 1,05 мэкв/г)
-13С-ЯМР-спектр (метанола 51,6 м.д. в качестве внутреннего стандарта), сигнал при 25,2 м.д. СНзот СНз-COH-NH-(идентичный исходному продукту).
Сигнал при 23,0 м.д. СНз от СНз-СО-0, Пример 23. Получение 0-сукцинили- рованного дерматан-сульфата (1C 2020).
Тетрэбутиламмониевую соль дерматан- сульфата (1 г) и N.N-диметилзминопмрйдин (110 мг) растворяют в безводном диметил- формамиде. После добавления янтарного ангидрида (396 мг) и трибутиламина (0,94 мл), среду инкубируют а безводных условиях в течение 2 ч при 60°С, После охлаждения и добавления воды (2 мл) осуществляют осаждение в охлажденном льдом спиртовом растворе, насыщенном ацетатом
натрия. Осадок промывают этанолом, растворяют в пирргенной воде, затем диализу- ют против 5%-ного бикарбоната натрия в . течение 36 часов, затем против воды в течение 3 дн. Получают, после лиофилизации, 0-сукцинилированный дермант-сульфат в форме натриевой соли (0,83 мг).
ИК-спектр (KB) имеет при 1730 и 1420 частоты, характерные (волновое
0 число) для карбоксильной группы,
1 С-ЯМР-спектр в ДгО (метанол 51,6 м.д. в качестве внутреннего стандарта) имеет сигналы при 33, 34,5 и Ф83.6 м.д.. характерные для СуКЦИНИЛЬ:НОЙ ГруППЫ, И При
5 .25,3 м.д. сигнал, характерный для метила ацетамидной группы, идентичный исходному продукту.
, Соотношение сульфат/карбоксил: 0,51. П р и м е р 24. Получение О-бутирили0 рованного гепарина, предварительно N-де- .сульфатированного и 4-aцeтилиpoвaннoгo (ГС 1948).
Гепарин N-десульфатируют, затем N- ацетилируют.
5 Трибутиламмониевую соль (1 г) получают путем пропускания через смолу Dowex 50, Н, затем нейтрализации с помощью раствора трибутиламина в этаноле,
После лиофилизации и высушивания,
0 эту соль растворяют в диметилформамйде (10 мл); затем, после охлаждения до 0°С, добавляют масляный ангидрид (2 мл), трибу- тиламин (2 мл) и диметиламинопиридин (65 мг). Спустя 24 ч добавляют воду (5 мл),
5 затем смесь диализуют против 5%-ного раствора бикарбоната натрия, после чего -против воды. После пропускания через Dowex 50, Н4 с последующей нейтрализацией гидроксидом натрия, получают натриевую соль
0 0-бутирилированного N-ацетилированного гепарина.
Спектр ЯМР-23 С (метанол 51.6 м.д. в качестве внутреннего стандарта) имеет сигнал при 24,6 м. д., характерный для М-ацетилз,
5 и.сигналы при 16, 20 и 38 м.д..характерные для сложных масляных эфиров.
Эксперимент может быть повторен с частично N-десульфатированным М-ацетилиро- ванным гепарином и проводить к частично
0 N-десульфатированному N-ацетилированно- му и 0-бутирилированному продукту.
Пример 25. Получение парацетили- рованного дерматан-сульфата (1C 1950). Тетрабутиламмониевую соль дерма5 тан-сульфата (0,8 г), растворенную в диме- тилформамиде (20 мл), ацетилируют воздействием диметиламинопиридмна (76 мг), уксусного ангидрида (1,2 мл) и триэтилами- на (1,7 мл). Смесь нагревают при 80°С в течение 1ч.
После возвращения к комнатной температуре, добавляют воду (0,45 мл), затем 0,3 М раствор ацетата натрия в этаноле (100 мл). После центрифугирования, осадок растворяют в воде, затем диализуют против дис- тиллированной воды. Натриевую соль получают путем обмена на колонке с Dowex 50, Н+, затем путем нейтрализации гидро- ксидом натрия. Получают, после лиофилиза- ции, перацетилированный дерматан- сульфат (0,51 г).
Этот продукт имеет соотношение сульфат/карбоксил 1,07 мэкв/г (исходный продукт: 1,05 мэкв/г) и содержит около трех ацетильных групп на дисахаридную едини-
цу.
При м е р 26. Получение О-ацетилиро- ванного гепарин-бензилового сложного эфира (1C 1949).
К раствору тетрабутиламмонийгепари- ната (1 г) в диметилформамиде (10 мл) добавляют бензилбромид (0,17 мл). Спустя 24 часа выдерживания при комнатной температуре, добавляют тетрабутиламмонийаце- тат (220 мг). Спустя 24 ч, осуществляют ацетилирование образовавшегося сложного бензилового эфира. Для этого, вводят диметиламинопиридин (57 мг), затем триэтиламин (1,3 мл) и уксусный ангидрид (0,9мл).
После возвращения к комнатной температуре реакционную смесь перемешивают в течение 24 ч. Тогда добавляют воду, затем продукт осаждают насыщенным раствором ацетата натрия в этаноле. После диализа против дистиллированной воды, пропускание через Dowex 50, Н. нейтрализации гидроксидом натрия и лиофилизации, получают натриевую соль 0-ацетилирован- ного сложного бензилового эфира гепарина (0,57 г).
Продукт имеет соотношение сульфат/карбоксил 3,6 мэкв/г (исходный продукт, не бензилированный: 2.20 мэкв/г),
С-ЯМР-спектр(метанол,51,6м.д.,вка- честве внутреннего стандарта) имеет сигналы при 23,3 м.д. (0-ацетил) и при 131,6 м.д. (бензил).
Сигнал СНз от СНз-CO-NH при 24,5 м.д. идентичен таковому исходного продукта.
П р и м е р 27. Получение 0-ацетилиро- ванного бемзилового эфира дерматансуль- фэта(1С 1953).
Тетрабутиламмониевую соль дерматан- сульфата (1 г) растворяют в безводном диметилформамиде (15 мл). К этому раствору, охлажденному до 0°С, добавляют бензил- бромид (0,25 мл), затем оставляют стоять в течение 24 ч при комнатной температуре.
Тетрабутиламмонийацетат(0,32 г) затем добавляют, после чего, спустя 24 часа при комнатной температуре, осуществляют аце- тилировэние.
Вводят уксусный ангидрид (1,5 мл), затем триэтиламин (2,2 мл) и диметиламинопиридин (96 мг). Спустя 24 ч добавляют воду (0,6 мл), затем продукт осаждают добавлением насыщенного этанольного раствора ацетата натрия.
Продукт диализуют против 10%-ного хлорида натрия, затем против волы.
Получают, после лиофилизации, сложный бензиловый эфир 0-ацетилированного дерматан-сульфата (0,63 г).
П р и м е р 28. Получение 0-бутирили- рованного дерманат-сульфата (1C 2018).
Трибутиламмониевую соль дерматан- сульфата (2 г), полученную в тех же условиях, что и таковые, описанные для получения трибутиламмонийгепарината в примере 2, и N.N-диметиламинопиридин (220 мг) растворяют в безводном диметилформамиде (25 мл). После охлаждения до 0°С прикапывают масляный ангидрид (5,9 мл), затем три- бутиламин (8,6 мл), После инкубации в течение 24-х часов, и охлаждения до 0°С добавляют воду (1 мл); осаждение проводят в спиртовом, охлажденном льдом, растворе, насыщенном ацетатом натрия. Осадок промывают этанолом, растворяют в апирогенной воде и диализуют против 5%- ного бикарбоната натрия в течение 36 часов, затем против воды в течение 3-х дней. Получают, после лиофилизации, 0-бутири- лированный дерматан-сульфат в виде натриевой соли (1,3 г).
3С-ЯМР-спектр в Д20 (метанол при 51,6 м.д. в качестве внутреннего стандарта) имеет при 15,6; 20,5 и 38,4 м.д. сигналы, характерные для бутирильной группы, и при 25,2 м,д. сигнал, характерный для метила ацетамидо-группы идентичный исходному продукту.
Соотношение сульфат/карбоксил: 1,05.
Пример 29. Получе. ие 0-гексаноили- рованного дерматан-сульфата (1C 2019).
Трибутиламмониевую соль дерматан- сульфата (2 г) и М,1М-диметиламинопиридин (220 мг) растворяют в безводном диметилформамиде (25 мл). После охлаждения до 0°С, прикапывают ангидрид гексановой кислоты (9,4 мл), затем трибутиламин (8,6 мл). После выдерживания 24 ч при комнатной температуре и охлаждения до 0°С, добавления воды (1 мл), осуществляют осаждение в ледяном спиртовом растворе, насыщенном ацетатом натрия. Осадок промывают абсолютным этанолом, растворяют в апирогенной воде и диализуют против 5%ного бикарбоната натрия в течение 36 часов, затем против воды в течение 3 дн. Получают после лиофилизации 0-гексаноилирован- ный дерматан-сульфат в виде натриевой соли (1 г).
13С-ЯМР-спектр в ДаО (метанола при 51,6 м.д, в качестве внутреннего стандарта) имеет при 15,9, 24,2, 26,5, 33,1 и 36,4 м.д., сигналы, характерные для 0-гексаноилиро- вэнной группы, а при 25,2 м.д. сигнал, харак- терный для метила ацетамйдной группы, идентичный исходному продукту.
Соотношение сульфат/карбоксил: 1,02. . При м е р 30. Осуществление селективного 0-ацетилирования по способу иэобре- тения и сравнения с другими способами ацетилирования.
Способ настоящего изобретения сравнивается со способами патента Франции № 2100735 и европейского патента 256880. В особенности, характеристики сложного уксусного эфира гепарина, полученного по способу изобретения 1C 1938, пример 1, сравниваются с таковыми сложного уксусного эфира гепарина, полученного при при- менении условий работы, описанных в патенте Франции 2100735 (продукт А), и сложными уксусными эфирами гепарина, полученными по способам работы примеров 3 и 4 европейского патент 256880 (про- дукты В и С).
(а)Получение продукта А
В раствор тетрабутиламмонийгепари- ната (1 г), растворенного в безводном диме- тилформамиде (10 мл), вводят дициклогекси- лкарбодиимид(4,2 г), растворенный в диме- тилформамиде (15 мл); затем уксусную кислоту (1,16 мл), растворенную в диметил- формамиде (25 мл) прикапывают в течение 45 мин при+4°С.
После выдерживания 24 часа при комнатной температуре, реакционную смесь фильтруют и концентрируют под вакуумом. Остаток снова суспендируют в эфире. После фильтрации и промывки, осадок диалиэуют против дистиллированной воды. Натриевую соль получают путем пропускания через колонну со смолой Dowex Н. затем нейтрализации гидроксидом натрия. Получают 0,493 г продукта А.
Это приготовление также реализуют в течение 48 ч при +4°С.
(б)Получение продуктов В и С. Продукты В и С получают путем ацети-
лирования гепарина в смеси формамида с пиридином с помощью ацетилхлорида, используя 2 мл ацетилхлорида для продукта В и 40 мл для продукта С в условиях, описанных в примерах 3 и 4 европейского патента
256880, Уксусный эфир затем растворяют в воде и диализуют против хлорида натрия.
(в) Результаты
Характеристики продуктов даны в табл.2. .Характеристика исходного гепарина, используемого в каждом опыте, дана в качестве ссылки.
Результаты показывают, что, хотя титры АРТТ и JW ниже для всех продуктов, чем таковые исходного гепарина, и это более сильно выражено для продуктов А, В и С, только один продукт 1C 1938 сохраняет соотношение сульфат/карбоксил практически идентичным таковому исходного гепарина. Это указывает на то, что способ изобретения позволяет селективно ацилировать гид- роксильные функции без затрагивания функциональных групп гепарина, что подтверждается химическим анализом продуктов.
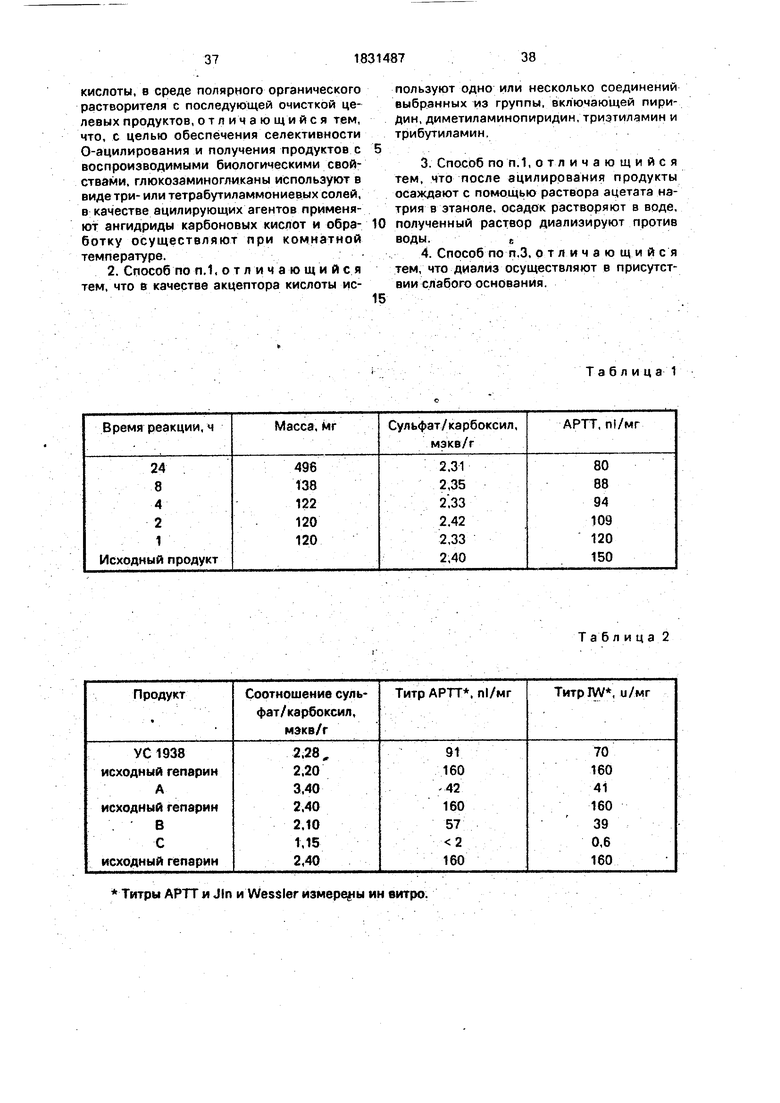
Продукты t C 1938, А, В и С проанализированы с помощью С-ЯМР (метанола с 51,6 м.д. в качестве внутреннего стандарта). Спектры этих продуктов, также, как таковой исходного гепарина, даны в следующем виде: ...- ; /
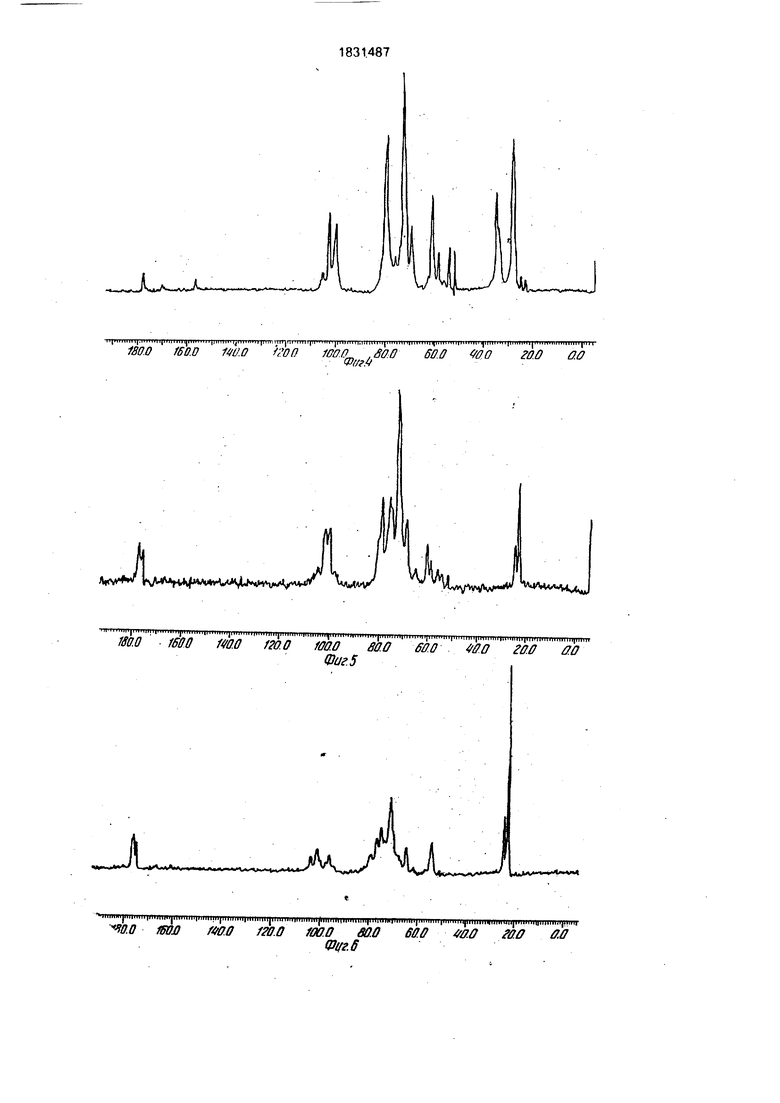
Фиг. 1: исходный гепарин
Фиг. 2: 1C 1938
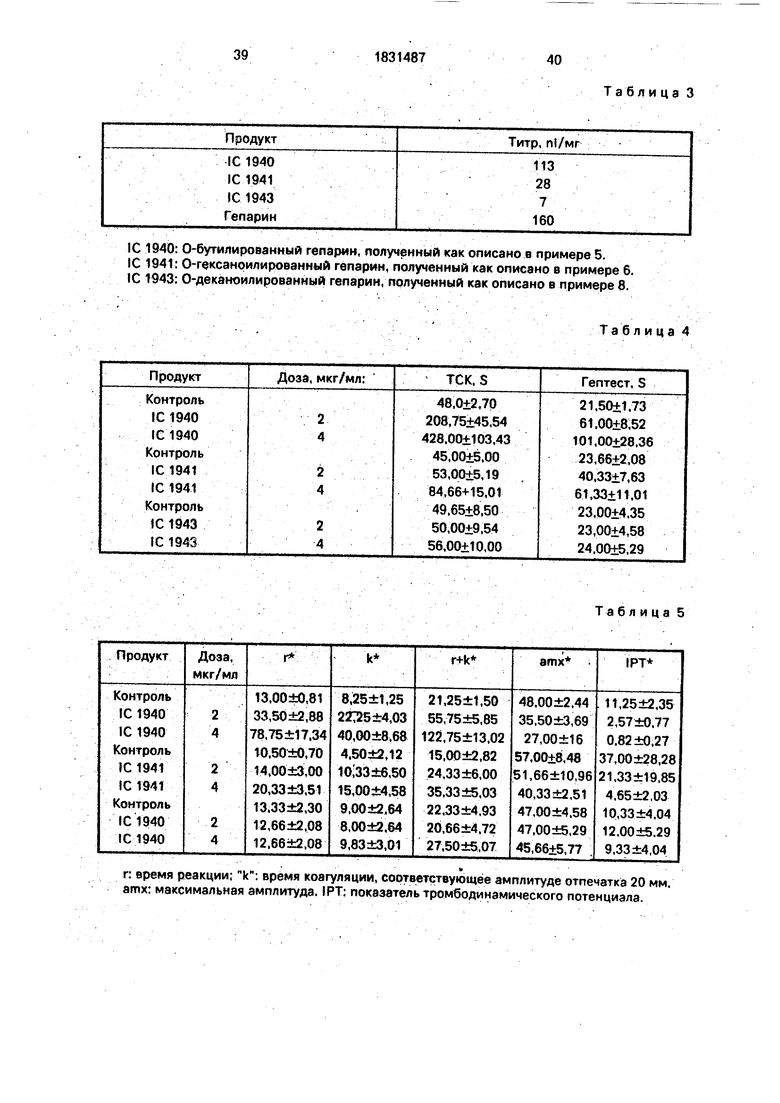
Фиг. 3: продукт А после реакции в течение 24 ч
Фиг. 4: продукт А после реакции в течение 48 ч
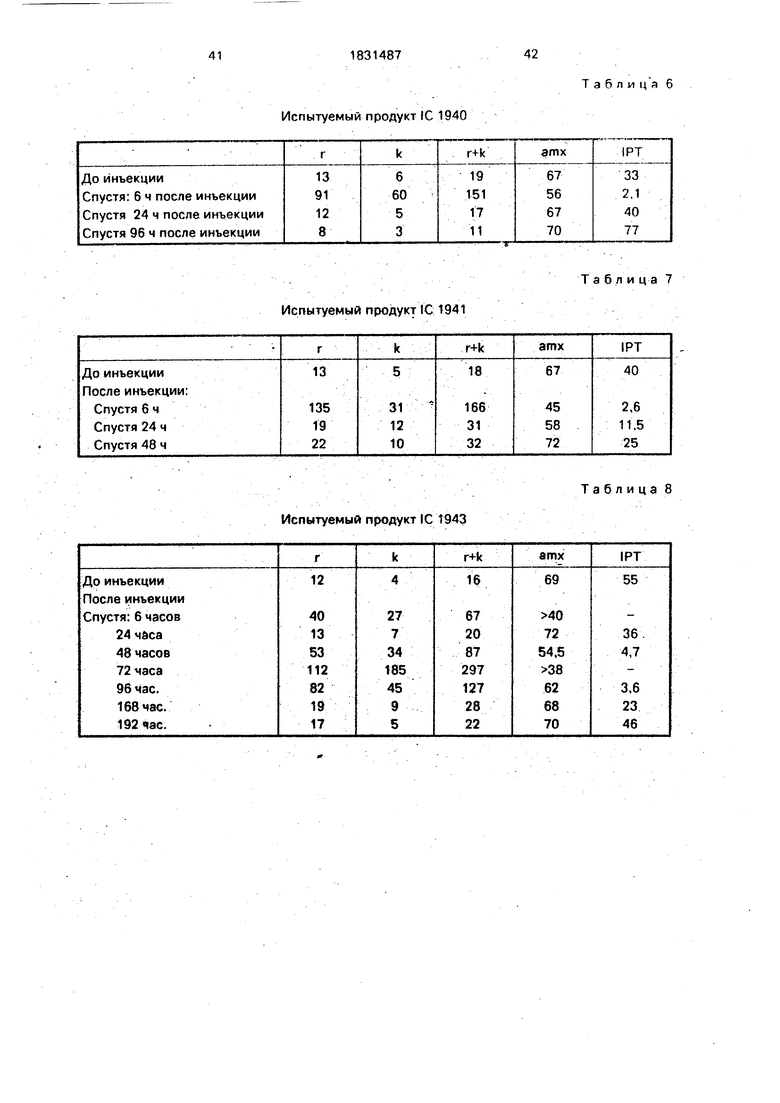
Фиг. 5: продукт В
Фиг. 6: продукт С.
Результаты следующие:
1,Продукт 1C 1938 имеет в спектре углерода (фиг.2): сигнал при 23,4 м.д., соответствующий СНз из СНз СО-0 сигнал при 24,4 м.д., соответствующий СНз из СНз-СО-МН, идентичный сигналу, имеющемуся в исходном гепарина..
Эти сигналы показывают, что аминные и карбоксильные группы не затрагиваются и что происходит селективное ацетилирова- ние на уровне гидроксильных групп.
2.Анализ продукта А 13С-ЯМР-спектро- скопией показывает, что полученный в большинстве продукт, также хорошо спустя 14 часа, как и после 48 ч(фиг.З и 4) представляет собой изомочевинное производное гепарина, карбоксильные функции которого замещены группой:
O-NH-C N-Q
вследствие использования дициклогексил- карбодиимида, В самом деле, наблюдают значительные сигналы, соответствующие атомам углерода вышеуказанной группы jnpH 28, 34, 54 и 156 м.д., тогда как сигнал,
соответствующий СНз из СНз-СО-0 при 23 м.д, очень слабый.
Следовательно, способ не позволяет получать селективное 0-ацилирование карбоксильных функций, что отражает увеличение соотношения сульфат/карбоксил.
3.Продукт В имеет в углеродном спектре (фиг.З):
-сигнал при 23,1 м.д., соответствующий СНз из СНз-СО-0
-сигнал при 24,8 м.д., соответствующий СНз из СНз-CO-NH, четко более интенсивные чем таковые исходного гепарина и 1C 1938.
Эти сигналы показывают, что наблюдают не только 0-ацетилирование, но и также сильное N-ацетилирование. Используемый способ ацилирования, который не селективный, вызывает частичную П-десульфа- тацию, с последующим ацетилированием аминов, что также вызывает уменьшение соотношения сульфат/карбоксил, что также вызывает уменьшение соотношения сульфат/карбоксил.
4.Продукт С имеет в углеродном спектре:.
-сигнал при 22 м.д., соответствующий СНз из СНз-СО-0
-сигнал при 24 м.д., соответствующий СНз из СНз-CO-NH, четко более интенсивные, чем таковые исходного гепарина и 1C 1938.
Как и для продукта В, наблюдают сразу О- и N-ацетилирование, N-десульфатация, более значительная; чем для продукта В, вызывает сильное уменьшение соотношения сульфат/карбоксил.
Фармакологическая активность продуктов изобретения
А) Антикоагулирующая активность ин витро:
1.Оценка титра ин витро по отношению к gamme-эталону:
Это измерение осуществляют с помощью теста время цефалина, сенсибилизированного каолином (Франция), на человеческой плазме. Результаты даны в табл.3.
Полученные результаты показывают, что селективно 0-ацилировэнные продукты изобретения имеют титр ин витро ниже такого гепарина, их антикоагулирующая ак: тивность уменьшается, когда возрастает длина ацилирующей цепи.
2.Измерение антикоагулирующей активности инвитров продуктов изобретения на человеческой крови:
Опыты осуществляются с двумя дозами продуктов изобретения: 2 мкт/мл и 4 мкг/мл, на 5 мл человеческой крови, Контрольный
опыт также осуществляется для каждого продукта, заменяя испытуемый продукт изотоническим раствором NaCI. После инкубации 30 мин при комнатной температуре, 5 кровь центрифугируют в течение 20 мин при 3000 об/мин. Л пшенную тромбоцитов плазму декан-тиру ют, чтобы реализовать следующие тесты:
- ТСК (время Цефалин-Каолина)
0 - Гегттестк
Результаты даны в табл.4. Каждый результаты соответствует среднему из трех опытов.
Результаты показывают в обоих мето5 дахудлинение времени коагуляции для продуктов 1C 1940 и 1C 1941. Это удлинение пропорционально дозе.
Взамен, в этих методах ин витро, продукт 1C 1943 показывает только очень не0 значительное воздействие на удлинение времени коагуляции.
3... Измерение антикоагулирующей активности ин витро продуктов изобретения путем тромбоэластографии на человеческой
5 крови:
Продукты изобретения, при дозах 2 мкг/мл и 4 мкг/мл, испытывают на 5 мл человеческой крови, как описано в предыдущем испытании, причем контроль реализуют
0 для каждого продукта, заменяя испытуемый продукт на изотонический раствор NaCI.
После инкубации 30 мин при комнатной температуре, тромбоэластографический отпечаток реализуют с помощью тромбоэла5 стогрэфии Helllge на 0,25 мл крови, рекаль- цифированной с помощью 0,1 мл 0,058 М CaCl2. Результаты даны в табл.5.
Результаты показывают, что 1C 1940 и 1C 1941 вызывают увеличение 1с.
0 уменьшение amx и IPT, что соответствует увеличению гипокоагуляции. Эта гипокоагу- ляция возрастает сразу в зависимости от используемой дозы и длины ацилирующей цепи.
5 1C 1943, в этом тесте, не вызывает заметной модификации параметров.
Б) .Антикоагулирующая активность ин виво:
0 1) Измерение антикоагулирующей активности ин вивов продуктов изобретения у кролика (внутривенно):
Опыты осуществляются на самцах новозеландских кроликов. Отбор крови осущест- 5 ел я ют на уровне медиальной артерии уха перед инъекцией продукта.
Затем, в краевую вену уха вводят путем инъекции раствор испытуемого продукта в количестве 25 мг продукта в 5 мл изотонического раствора NaCI.
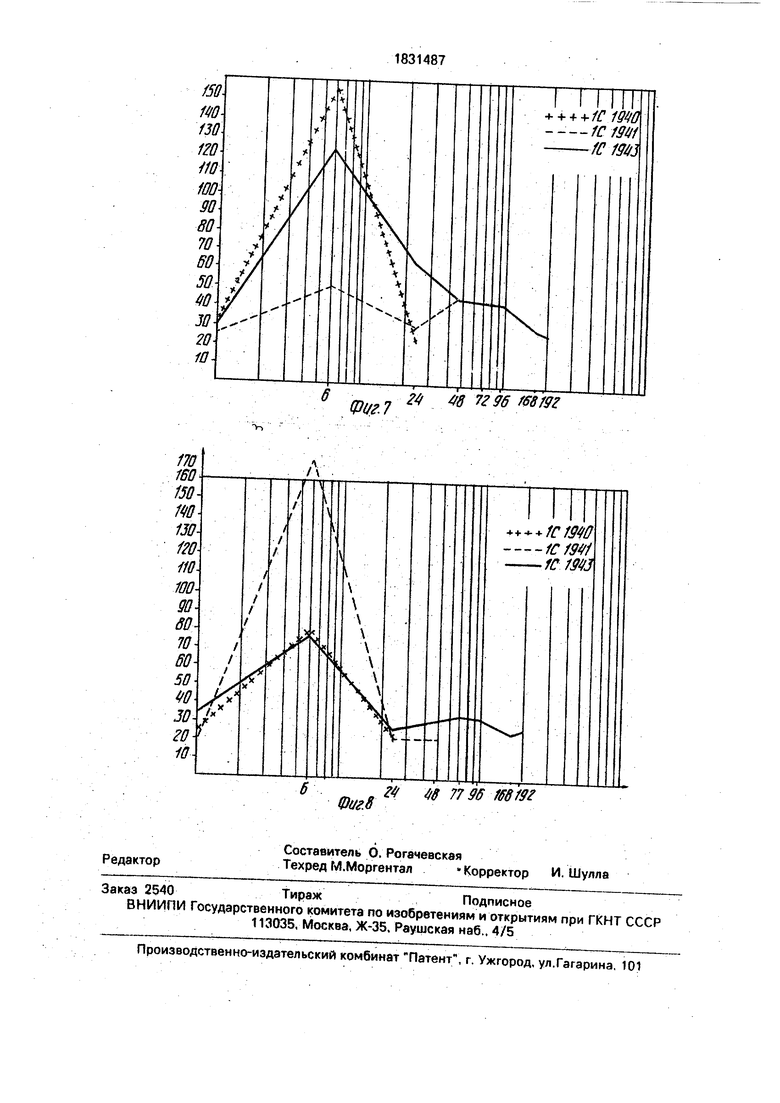
Тест ТСК и HEPTEST реализуются на рови, отобранной перед инъекцией проукта, и отборах крови, осуществленных пустя 6, 24, 48 и 96 ч после инъекции.
Результаты даны на фиг.7 и 8. Они покаывают чистое удлинение во времени анти- оагулирующей активности, причем это длинение увеличивается с длиной ацили- ующейцепи. В самом деле, антикоагулирующая активность продукта 1C 1940 еще тчетливая плюс 6ч после инъекции, тогда как она еще отчетливая спустя 24 ч после инъекции для продукта 1C 1941 и она проонгируется вплоть До 96 ч для продукта 1C 1943.
2.Измерение а нти коагулирующей акивности ин виво продуктов изобретения путем тромбоэластографии:
Испытуемые продукты вводят путем инъекции внутривенно как описано в предыдущем опыте (25 мг в 5 мл изотонического раствора NaCI). Тромбоэластографический отпечаток реализуют с помощью тромбоэла- стографа Helllge на 0,25 мл образцов крови, отобранных перед инъекцией, и спустя 6,24, 48 и 96 ч после инъекции, причем дополнительные отборы осуществляются, когда ан- тикоагулирующая активность длится сверх 96 ч. Результаты даны в табл. 6,7 и 8.
Полученные результаты показывают, что для всех продуктов отмечается сильное увеличение г + k и уменьшение IPT на б-ом часу, что указывает на удлинение гипо- коагуляемости, Она пролонгируется вплоть до по крайней мере 24 ч после инъекции для продукта 1C 1941 и еще измерима 96 ч спустя после инъекции для продукта 1C 1943.
Следовательно, констатируют, что все продукты имеют сильную антикоагулирую- щую активность ин виво, что эта активность пролонгирована во времени и что 1C 1943, который только мало или вообще неактивен в опытах ин витро, оказывается очень активным и сильно пролонгирован в опытах ин виво.
3.Антикоагулирующая активность продуктов 1C 1945, 1C 1957 и 1C 1958. полученных соответственно примерам 12, 13 и 14, испытывается йн виво у кролика.
Результаты показывают удлинение фар- макокинетики при введении продуктов внутривенно и подкожно, в отношении продуктов 1C 1957 и 1C 1958.
В) Активность на модели ин витро инги- бирования вирусов HIV-1 и-2:
Продукты, полученные согласно изобретению, испытываются на модели инги- бирования вирусов HIV-1 и HIV-2 ин витро.
Все испытуемые продукты становятся активными при дозах, полностью лишенных
цитотоксичности. В особенности, констатируют, что продукты 1C 1925 и 1C 1926 более активны, чем исходный продукт.
Г) Активность на модели ин витро инги- бирования образования Syncltla
Syncltla представляет собой гигантские клетки, с многими ядрами, образуемые путем слияния здоровых лимфоцитов Т 4 и инфицированными лимфоцитами Т4. 0 Продукты, полученные согласно изобретению, испытываются на модели ко- культуры клетки MOLT 4 (здоровые лимфоциты Т4) и клеток HUT-78/HT V III В (-инфицированные лимфоциты Т 4 челове- 5 ческой линии).
Все испытуемые продукты оказываются активными в этой модели, при дозах, полностью лишенных цитотоксичности. В частности, продукты 1C 1924, 1C 1925 и 1C 1926 0 найдены более активными, чем исходный продукт.
Д) Активность в модели ин витро инги- бирования различных покровных вирусов
Активность продуктов, полученных со5 гласно изобретению, по отношению к ингибированию различных покровных вирусов с
ADN или ARN, в особенности следующих
вирусов:
-HSY 1 и 2 (Herpes Simp Lex virus 1 и 2, 0 вирус к ADN)
-YSY (Vaslcular Stomatitis Virus) вирус с ARN .
...- Slndbis -вирус
Все испытуемые продукты оказались ак- 5 тивными при ингибировании этих вирусов, при дозах, лишенных цитотоксичности. В особенности, продукты 1C 1924,1C 1925 и 1C 1926 обладают сильно увеличенной активностью по отношению к YSY и Slndbis виру- 0 су, вирус с ARN по сравнению с исходным продуктом.
Е) Активность в модели ин виво ингиби- рования пролиферации гладких мышечных клеток,
5 Продукты, полученные согласно изобретению, испытываются на модели инги- бирования пролиферации гладких мышечных клеток ин виво у крысы (модель катетер- ного баллона).
0 Испытуемые продукты оказались активными. В особенности, продукты 1C 1924, 1C 1925 и 1C 1926 обладают возросшей активностью по сравнению с исходным продуктом. 5
Формула изобретения 1. Способ получения 0-ацилированных глюкозаминогликанов, включающий обработку глюкозаминогликанов ацилирующи- ми агентами в присутствии акцептора
кислоты, в среде полярного органического растворителя с последующей очисткой целевых продуктов, отличаю щи и с я тем, что, с целью обеспечения селективности О-ацилирования и получения продуктов с воспроизводимыми биологическими свойствами, глюкозаминогликаны используют в виде три- или тетрабутиламмониевых солей, в качестве ацилирующих агентов применяют ангидриды карбоновых кислот и обработку осуществляют при комнатной температуре.
2. Способ по п.1, отличающийся тем, что в качестве акцептора кислоты ис
пользуют одно или несколько соединений выбранных из группы, включающей пиридин, диметиламинопиридин, триэтияамин и трибутиламин,
3.Способ по п. 1,отличающийся тем, что после ацилиррвания продукты осаждают с помощью раствора ацетата натрия в этаноле, осадок растворяют в воде, полученный раствор диализируют против воды.е
4.Способ по п.З, отличающийся тем, что диализ осуществляют в присутствии слабого основания.

















Использование: в медицине. Сущность изобретения: -о-ацилированные глюкоза- миногликаны получают ацилированием глюкозаминогликанов в виде три- или тет- рабутиламмониевых солей ангидридами карбоновых кислот при комнатной температуре, в полярном органическом растворителе в присутствии акцептора кислоты с последующей очисткой целевых продуктов. Акцептор кислоты - пиридин, диметилами- нопиридин, триэтиламин и трибутиламин. После ацилирования продукты осаждают с помощью раствора ацетата натрия в этаноле, осадок растворяют в воде, и раствор диализуют против воды. Диализ осуществляют в присутствии слабого основания. 3 з.п,ф-лы, 8 табл., 8 ил.
Титры АРТТ и Лп и Wessler измерены ин витро.
Т а б л и ц а 1
Т а б л и ц а 2
1C 1940: 0-бутилированный гепарин, полученный как описано в примере 5. 1C 1941: 0-гексанрилированныЙ гепарин, полученный как описано в примере 6. 1C 1943: О-деканоилированный гепарин, полученный как описано в примере 8.
г: время реакции; время коагуляции, соответствующее амплитуде отпечатка 20 мм. атх: максимальная амплитуда. IPT: показатель тромбодинэмического потенциала.
Таблица 3
Т а б л и ц а 4
Таблица 5
Испытуемый продукт 1C 1940
Испытуемый продукт 1C 1941
Испытуемый продукт 1C 1943
Таблица 6
Т а б л и ц а 7
Таблица 8
я
tAM «Ои
1Ц|. Н|||| |11|||Ц|Ц||111111|||1111Ч11|Ч111И1Ь1 1|||11111|11||1111111Г||||.111И|1111|1|11||1|И|11|| И ЦЙ1|1 Ч|ii|miiiiii Jl l liil|tilil illvVJ| 1 1
Фиг.1
т.о то та ко.о 100.0 то
®иг.2.
то 12й.о юо.о BOM бо.о 4о.о то ао
фцг.З
.ышМ
I
.iVt/Vvtr d
д-гЛф 0у ояг 0ff# оаэ 009 о oot оог вам оя& ош.
.....,..1.|И..|..ЧiJ......II....lllll.ll4l.lllll.mllllll4l.l.irilllllllll.llll.lll.M.I,.nil.III..IlLuiH.lll.II,,II,,I,UU.M,S Wd
ffЈругqw -Q ffff-.ffffff owt оог ош 0т от
VWwVytv,
,№&.,.
ov оог oto 009 и off ош оог ami а ош 0ш
„I,..,..„....„L,,.....,.........ti,.,,.|Iniimirimiiiiil IIni .....HIbin.null1.1.и.-Мнит..i
.
r
,№&.,.
ош
.
Фиг.7
24 48 7296 f$8f9Z
