Изобретение относится к оптически активным производным карбоксамидов, имеющим сильную аналгазирующую активность и низкую токсичность, или их фармакологически пригодным солям.
Прояснено присутствие в центральной нервной системе трех видов рецепторов, μ -, k- и d- рецепторов, участвующих в аналгезирующем действии. Сообщается, что агонист, который среди этих рецепторов имеет сродство к k-рецептору, проявляет сильное аналгезирующее действие и не обладает такими побочными действиями, как появление привыкания, толерантности, респираторное подавление, ингибирование движения гладких мышц и т.д. которые наблюдаются в случае морфина, являющегося агонистом k-рецептора. В соответствии с этим было желательно разработать агонист k-рецептора, обладающий сильным аналгезирующим действием и меньшим побочным действием.
Авторы настоящего изобретения синтезировали ряд гетероциклических соединений и исследовали их фармакологическую активность в течение многих лет. В результате они нашли производные карбоксамидов или их фармакологически пригодные соли, обладающие сильной аналгезирующей активностью, и подали заявку на Японский патент [JP Kakoai Hei 3-163068 (Hei 3.Juli 15)]
Авторы настоящего изобретения нашли, что оптически активные производные карбоксамидов, имеющие определенную стереохимическую конфигурацию соединений, включенных в указанную выше заявку, обладают очень сильной аналгезирующей активностью в качестве агониста k-рецептора по сравнению с рацематами и другими стереоизомерами и меньшими побочными действиями, полезны в качестве аналгезирующих средств и выполнены настоящим изобретением.
Оптически активные производные карбоксамидов настоящего изобретения имеют общую формулу
В формуле R1 представляет собой атом водорода или галогена, R2 представляет собой атом галогена; R3 представляет собой пирролидино- или пиперидиногруппу, Y представляет собой метиленовую или карбонильную группу и n является целым числом 1 или 2.
Аналгезирующее средство или средство, являющееся агонистом k-рецептора, настоящего изобретения содержит указанные выше соединения или их фармакологически пригодные соли в качестве активного компонента.
Атомы галогена, представленные R1 и R2, включают например атомы фтора, хлора, брома и йода; предпочтительно атом фтора или хлора, в частности предпочтительно атом хлора.
Соединения общей формулы (I), описанные выше, можно если нужно, превратить в фармакологически пригодные соли их обычными способами.
Примеры таких солей включают например соли минеральных кислот, таких как соляная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота или фосфорная кислота, и соли органических кислот, таких как метансульфокислота, этансульфокислота, бензолсульфокислота, п-толуолсульфокислота, щавелевая кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, миндальная кислота, лимонная кислота или винная кислота, предпочтительно соли соляной кислоты, метансульфокислоты, фумаровой кислоты или янтарной кислоты
Предпочтительными соединениями общей формулы (I) являются соединения, у которых:
(1) R1 является атомом водорода, фтора или хлора;
(2) R2 является атомом фтора или хлора;
(3) R3 является пирролидиногруппой;
(4) Y является метиленовой группой и
(5) n является 1;
наиболее предпочтительными соединениями общей формулы (I) являются те соединения, у которых:
(6) R1 является атомом водорода или хлора и
(7) R2 является атомом хлора,
в частности предпочтительными соединениями общей формулы (I) являются те соединения, у которых
(8) R1 является атомом хлора.
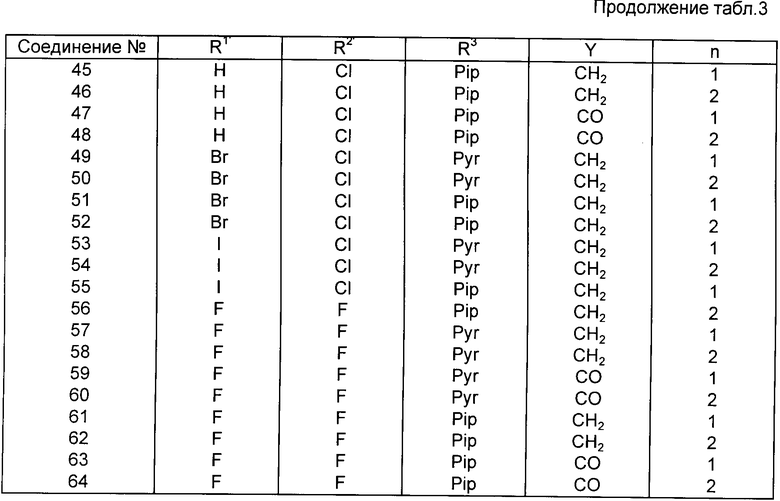
Предпочтительными конкретными соединениями являются соединения N 1, 2, 3, 4, 5, 6, 7, 8, 9, 17, 19, 33, 41, 42, 43, 44, 57 и 59, причем номер этого соединения такой же, как в приведенной ниже таблице 3.
Наиболее предпочтительными конкретными соединениями являются:
Соединение N 1: (3R)-3-(1-пирролидинилметил)-4-[(1S)- -5,6-дихлор-1-инданкарбонил]тиоморфолин;
Соединение N 3: (3R)-3-(1-пирролидинилметил)-4-[(1S)- -3-оксо-5,6-дихлор-1-инданкарбонил]тиоморфолин;
Соединение N 4: (3R)-3-(1-пирролидинилметил)-4-[(1S)- -6,7-дихлор-4(3H)-оксо-1,2-дигидро-1-нафтоил]тиоморфолин;
Соединение N 6: (3R)-3-(1-пиперидинилметил)-4-[(1S)- -6,7-дихлор-1,2,3,4-тетрагидро-1-нафтоил]тиоморфолин;
Соединение N 8: (3R)-3-(1-пиперидинилметил)-4-[(1S)- -6,7-дихлор-4(3H)-оксо-1,2-дигидро-1-нафтоил]тиоморфолин и
Соединение N 43: (3R)-3-(1-пирролидинилметил)-4-[(1S)- -3-оксо-5-хлор-1-инданкарбонил]тиоморфолин.
Соединения общей формулы (I) настоящего изобретения можно легко получить следующим способом:
Способ A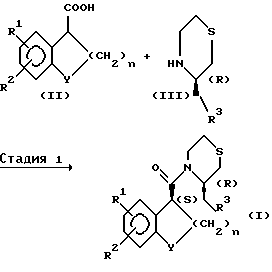
В указанных выше формулах R1, R2, R3, Y и n имеют указанные выше значения.
Способ A включает способ получения соединения формулы (I)
Стадия A1 является стадией получения соединения общей формулы (I), ее проводят реакцией соединения общей формулы (II) или его реакционноспособного производного с соединением общей формулы (III) в инертном растворителе и затем разделением полученных смесей изомеров.
Реакцию соединения формулы (II) или его реакционноспособного производного с соединением общей формулы (III) проводят, например, способом с галогенангидридом кислоты, способом со смешанным ангидридом кислоты, способом с активированным сложным эфиром или способом конденсации.
Способ с галогенангидридом кислоты проводят реакцией соединения формулы (II) с галогенирующим агентом (например хлористым тионилом, пятихлористым фосфором или подобным реагентом) для получения галогенангидрида кислоты и затем реакцией его с соединением формулы (III) в инертном растворителе в присутствии или отсутствии основания.
Пригодные амины включают например органические амины, такие как триэтиламин, N-метилморфолин, пиридин, 4-диметиламинопиридин и 1,8-диазабицикло [5.4.0] ундец-7-ен (DBU); бикарбонаты щелочных металлов, такие как бикарбонат натрия или бикарбонат калия, карбонаты щелочных металлов, например карбонат натрия или карбонат калия, гидроксиды щелочных металлов, таких как гидроксид натрия или гидроксид калия, предпочтительно органические амины.
Не имеется особого ограничения для применения инертного растворителя на основании его природы, если он не оказывает обратного действия на реакцию. Такие растворители включают например углеводороды, такие как гексан, циклогексан, бензол, толуол или ксилол, галогенированные углеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод или 1,2-дихлорэтан, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран или диоксан, кетоны, такие как ацетон, амиды, такие как N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон или гексаметилфосфорамид, сульфоксиды, такие как диметилсульфоксид, предпочтительно галогенированные углеводороды или простые эфиры.
Температура реакции зависит от природы исходных соединений формул (II) и (III), а также природы растворителя, но температура для обеих реакций соединения формулы (II) с галогенирующим агентом и галогенангидрида с соединением формулы (III) обычно находится в пределе от -20 до 150oC, предпочтительно около комнатной для реакции соединения формулы (II) c галогенирующим агентом и 0-100oC для реакции галогенангидрида кислоты с соединением формулы (III).
Время реакции изменяется в зависимости от температуры реакции и других условий, но обычно оно находится в пределе от 30 мин до 24 ч (предпочтительно 1-10 ч).
Способ со смешанным ангидридом проводят реакцией соединения формулы (II) с C1-C4-алкилгалогенкарбонатом, например этилхлорформиатом или изобутилхлорформиатом, для получения смешанного ангидрида кислоты и затем реакцией его с соединением формулы (III).
Реакцию получения смешанного ангидрида кислоты проводят реакцией соединения формулы (II) с C1-C4 -алкилгалогенкарбонатом, предпочтительно в инертном растворителе в присутствии основания.
Применяемые основания и инертный растворитель аналогичны тем, которые применяют в способе с галогенангидридом кислоты.
Температура реакции зависит от природы исходного соединения формулы (II) и природы применяемого растворителя, она обычно находится в пределе от -20 до 50oC (предпочтительно 0-30oC). Время реакции изменяется в зависимости от температуры реакции и других условий, но обычно оно составляет от 30 мин до 24 ч (предпочтительно 1-10 ч).
Реакцию соединения формулы (III) со смешанным ангидридом кислоты предпочтительно проводят в инертном растворителе в присутствии или отсутствии основания. Применяемые основания и инертный растворитель аналогичны тем, которые применяют в способе с галогенангидридом кислоты.
Температура реакции зависит от природы исходного соединения формулы (III), а также природы применяемого растворителя, но температура обычно находится в пределе от -20 до 100oC (предпочтительно от 0oC до комнатной температуры). Время реакции изменяется в зависимости от температуры реакции и других условий, но обычно оно составляет от 30 мин до 24 ч (предпочтительно 1-10 ч).
Этот способ получения предпочтительно проводят в присутствии соединения формулы (II), соединения формулы (III) и C1-C4 -алкилгалогенкарбоната без выделения смешанного ангидрида кислоты.
Способ с активированным эфиром проводят реакцией соединения формулы (II) с активным этерифицирующим агентом (например, N-гидроксисоединением, таким как N-гидроксисукцинимид, N-гидроксибензотриазол или подобное) в присутствии конденсирующего средства (например дициклогексилкарбодиимида, карбонилдиимидазола и подобного соединения) или реакцией соединения формулы (II) с активным этерифицирующим агентом (таким как Ди (C1-C4-алкил) цианофосфонаты, например, диэтилцианофосфонатом, диарилфосфорилазиды, например дифенилфосфорилазидом или динафтилфосфорилазидом или подобные соединения) для получения активированного эфира и затем реакцией его с соединением формулы (III).
Реакцию получения активированного эфира предпочтительно проводят в присутствии инертного растворителя. Применяемый для этой реакции инертный растворитель аналогичен тем, которые применяют в способе с галогенангидридом кислоты.
Температура реакции зависит от природы исходных соединений формул (II) и (III), а также природы применяемого растворителя, но температура для реакции с активным этерифицирующим средством обычно находится в пределе от -20 до 50oC (предпочтительно от -10 до 30oC). Температура реакции активированного эфира с соединением формулы (III) от -20 до 50oC (предпочтительно 0-30oC). Время для обеих реакций изменяется в зависимости от температуры реакции и других условий, но оно для обеих реакций составляет от 30 мин до 24 ч (предпочтительно 1-10 ч).
Этот способ предпочтительно проводят в присутствии соединения формулы (II), соединения формулы (III) и активного этерифицирующего агента без выделения активированного эфира.
Способ конденсации проводят реакцией непосредственно соединения формулы (II) с соединением формулы (III) в присутствии конденсирующего агента (например дициклогексилкарбодиимида, карбонилдиимидазола, гидрохлорида 1-(N, N-диметиламинопропил)-3-этилкарбодиимида, трифенилфосфин-2,2'-дипиридинилдисульфида или подобного соединения). Эту реакцию проводят подобно проведению реакции по способу с активированным эфиром.
После окончания реакции целевые соединения в каждой стадии можно выделять из реакционной смеси обычным образом. Например, осажденные кристаллы отделяют фильтрованием или в реакционную смесь добавляют воду и образованную смесь экстрагируют несмешиваемым с водой органическим растворителем, например этилацетатом, экстракт сушат и растворитель экстракта удаляют перегонкой. Продукт, если нужно, можно далее очищать обычным образом, например перекристаллизацией, колоночной хроматографией или подобным образом.
Разделение образованных смесей изомеров можно проводить обычными способами разделения оптических изомеров. Например оптические изомеры можно разделить колоночной хроматографией на силикагеле с использованием в качестве подвижной фазы смеси этилацетата и триэтиламина с соотношением их (50-100):1.
Целевое соединение формулы (I) можно также получить реакцией оптически активного соединения общей формулы (IIa), которое получают разделением соединения (II) на оптические антиподы, с соединением формулы (III).

Разделение соединения формулы (II) на оптические антиподы проводят обычными способами. Например разделение проводят с применением бруцина в качестве средства для разделения изомеров и перекристаллизацией из смеси простого эфира, например этилового эфира или изопропилового эфира (предпочтительно изопропилового эфира) и кетона (предпочтительно ацетона).
Реакцию соединения формулы (IIa) с соединением формулы (III) проводят аналогично указанной выше реакции соединения формулы (II) с соединением формулы (III), предпочтительно способом со смешанным ангидридом кислоты, способом с активированным эфиром или способом конденсации.
Исходное соединение формулы (II), применяемое в способе A, является или известным соединением, или его легко получить известными способами (например, Японского патента Kokai N Hei 2-149560).
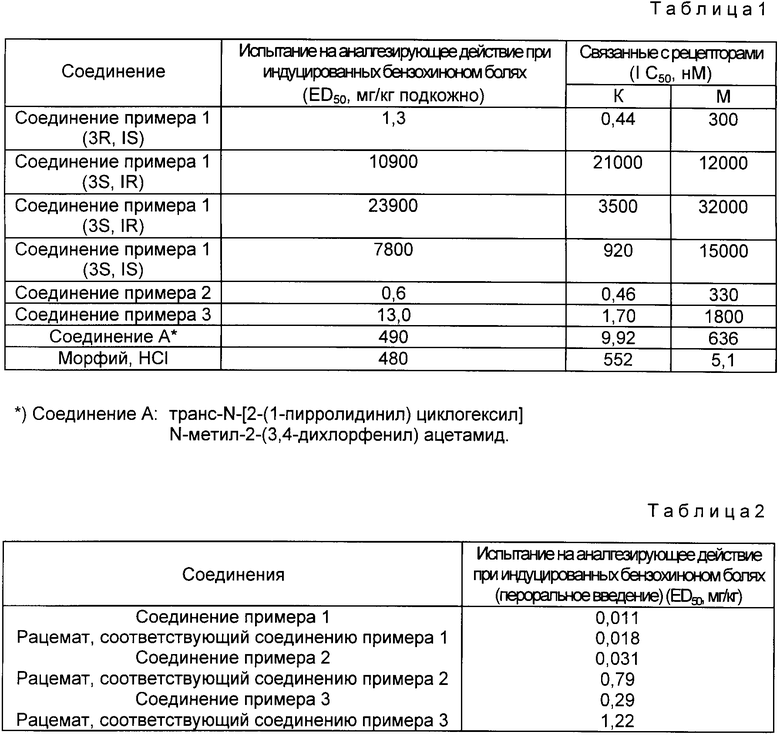
Указанные выше соединения, имеющие общую формулу (I), обладают сильной аналгезирующей активностью, как показано ниже, и не обладают такими побочными действиями, как появление привыкания, толерантности, респираторное подавление и ингибирование движения гладких мышц. Поэтому эти соединения пригодны в качестве аналгезирующего средства.
(Пример 1 на испытание)
Испытание на индуцированную п-бензохиноном боль у мышей
(гиподермальное введение)
Заглавное испытание проводили по методике Siegmund et al. [Proc. Soc. Exptl. Biol. Med. 95, 729 (1957)]
Самцы мышей ddy (Japan SLC), каждый из которых весил около 20 г, голодали в течение около 16 ч со дня перед проведением испытания. Их разделили на группы, каждая из которых включала 5-10 мышей.
Испытываемое соединение растворили в физиологическом солевом растворе и ввели подкожной инъекцией. Через 15 мин внутрибрюшинной инъекцией ввели 0,1 мл/мышь 0,03%-ного раствора п-бензохинона. В течение 5-10 мин после инъекции подсчитывали число болевых реакций (восприимчивость к боли), наблюдаемых у каждой мыши. Тех мышей, у которых обнаружили 1/2 или менее от средней величины числа болевых реакций, которые наблюдали у контрольных мышей, инъецированных только физиологическим солевым раствором, рассматривали как эффективно аналгезированных мышей. Соотношение числа эффективно аналгезированных животных и числа применяемых животных получили для каждой дозы и ED50 (50% эффективной дозы) подсчитали в соответствии с методом вероятности.
(Пример 2 на испытание)
Испытание на сродство к рецептору
(1) Получение сырого церебро-мембранного препарата (препарат мембран мозга)
Подготовительную операцию проводили по методике Pasuternak et al. [Mol. Pharmacol. 11, 340 (1975)]
Целые мозги (удален только мозжечок) самцов морских свинок Hertley (Japan SLC), каждый из которых весил 400-700 г, гомогенизировали в 30 ч охлажденного льдом 50 мМ трис-буфера (pH 7,4) с применением политрона и затем центрифугировали в течение 15 мин при 49000 х.г. Осажденный осадок суспендировали снова в том же виде буфера, как указано выше. Суспензию инкубировали при 37oC в течение 30 мин и затем центрифугировали в указанных выше условиях. Осадок суспендировали в 30 ч буфера и сохраняли при -80oC. Этот сырой препарат "расплавляли" перед применением, гомогенизировали при помощи гомогенизатора Даунса и затем разбавляли до конечной концентрации белка 0,5 мг/мл.
(2) Связывание с k-рецептором
Испытание на связывание проводили по методике Magnan et al. [Arch. Pharmacol. 319, 197 (1982)] В качестве лиганда применяли 0,6 нМ меченого тритием этилкетоциклазоцина. Его связывание с церебро-мембранным препаратом испытывали в условиях, при которых μ и d- рецепторы насыщены 10 нМ DAGO (D-Ala2, MePhe4, Gly-oL5-энкефалин) и 10 нМ DADLE [(D-Ala2, D-Len5)-энкефалин]
Церебро-мембранный препарат, меченый и немеченый лиганды и испытываемое соединение инкубировали в 1 мл трис-буферного раствора, предварительно охлажденного льдом, смесь фильтровали при пониженном давлении через фильтровальную бумагу Watmann GF/B и два раза промыли. В меченый лиганд, соединенный с фильтровальной бумагой, добавили эмульгированный сцинтиллятор (ACS-11), оставили на ночь и затем измерили счетчиком жидкостной сцинтилляции. Сродство испытываемого соединения к рецептору обозначали концентрацией, которая подавляется связыванием меченого лиганда на 50% (IC50), нМ).
(3) Связывание с μ- рецептором
Испытание на связывание проводили по указанной выше методике Magnan et al. С применением 1 нМ меченого тритием DAGO в качестве лиганда испытание проводили аналогично процедуре испытания связывания с k-рецептором. Сродство испытываемого соединения к этому рецептору обозначали указанным выше образом.
В табл. 1 приведены результаты примеров испытаний 1 и 2.
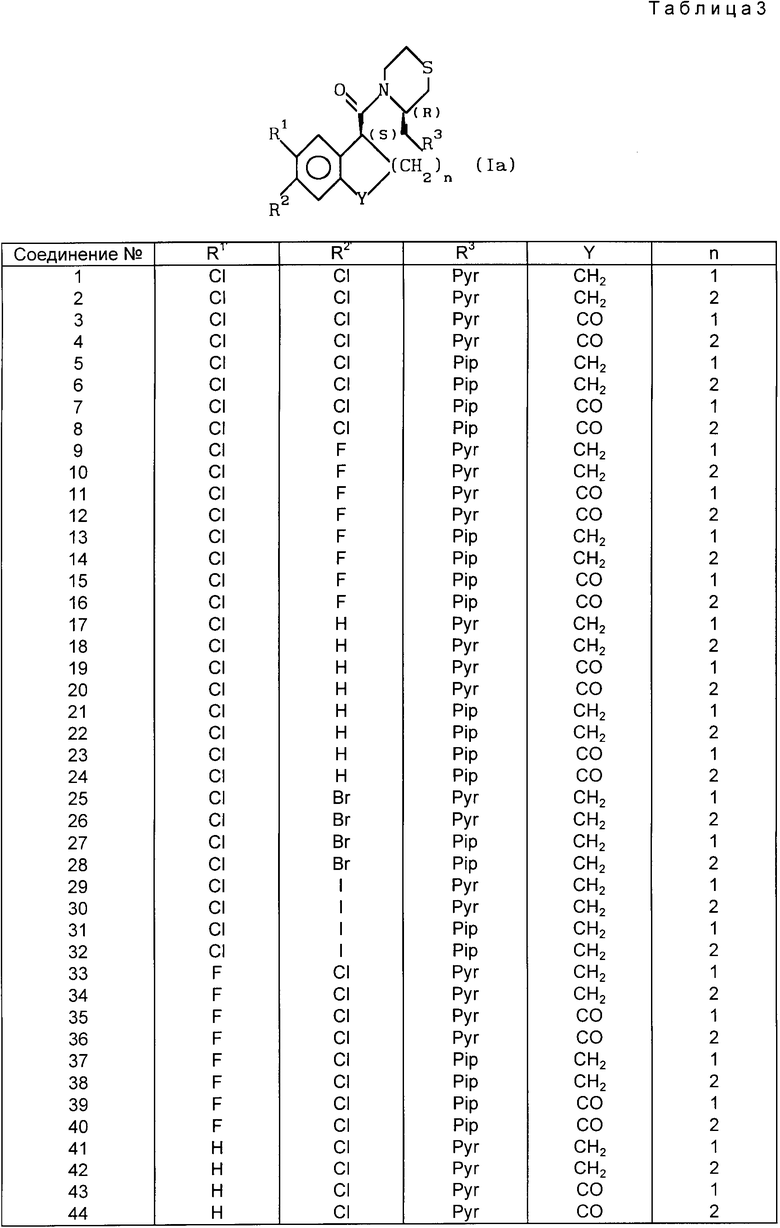
(Пример 3 на испытание)
Испытание на индуцированную п-бензохиноном боль у мышей
(пероральное введение)
Заглавное испытание проводили по методике Siegmund et al. [Proc. Soc. Exptl. Biol. Med. 95, 729 (1957)] Самцы мышей ddy (Japan SLC), каждый из которых весил около 20 г голодали около 16 ч. со дня перед проведением испытания. Мышей разделили на группы, каждая из которых включала 5-10 мышей.
Испытываемое соединение растворили в 0,5% трагаканте и ввели мышам перорально. Через 15 мин каждой мыши инъецировали внутрибрюшинно 0,1 мл/мышь 0,03% раствора п-фенилхинона. В течение 5-10 мин после инъекции подсчитывали число болевых реакций (восприимчивость к боли), наблюдаемых у каждой мыши. Тех мышей, которые обнаружили 1/2 или менее от средней величины числа болевых реакций, наблюдаемых у контрольных мышей, инъецированных только физиологическим соляным раствором, рассматривали как эффективно аналгезированных мышей. Соотношение числа эффективно аналгезированных мышей и числа применяемых мышей было получено для каждой дозы и ED50 (50% эффективной дозы) подсчитывали способом вероятности.
В табл. 2 приведены результаты примера испытания 3
В случае, когда соединения формулы (1) применяют в качестве аналгезирующего соединения, эти соединения или смеси их с фармакологически пригодными носителями, наполнителями, разбавителями и подобными компонентами можно удобно вводить перорально или неперорально (внутривенно, внутримышечно, внутрибрюшинно и т. д. ) в виде фармацевтических препаратов, например, порошка, гранул, таблеток, капсул, сиропа, инъекций, суппозитория, мази, крема, пасты и подобных форм. Доза изменяется в зависимости от симптомов пациента и метода введения, но эти соединения обычно вводят как суточную долю около 0,001-100 мг, в частности около, 0,01-10 мг в случае перорального введения или как суточную дозу около 0,001-10 мг, в частности, около 0,002-5 мг в случае внутривенного или внутримышечного введения в виде однократной дозы или разделенных доз.
Следующие примеры и ссылочные примеры приведены для того, чтобы настоящее изобретение можно было понять более полно. Такие примеры не должны быть истолкованы как ограничение изобретения.
(Пример 1).
Гидрохлорид (3R)-3-(1-пирролидинилметил)-4-[(1S)-(5,6-дихлор-1- инданкарбонил)] тиоморфолина
Раствор 2,74 г. хлорангидрида 1-(5,6-дихлориндан)карбоновой кислоты в 30 мл дихлорметана добавили по каплям в раствор 1,96 г. (R)-3-(1-пирролидинил) метилтиоморфолина и 1,67 мл триэтиламина в 30 мл дихлорметана при -10oC в струе азота. Полученную смесь перемешивали при той же температуре 20 мин. и затем при комнатной температуре 1,5 ч. Реакционную смесь вылили в насыщенный водный раствор бикарбоната натрия и экстрагировали дихлорметаном. Экстракт промыли насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом натрия. После отгонки растворителя остаток очищали колоночной хроматографией на силикагеле с применением в качестве подвижной фазы смеси (50: 1) этилацетата и триэтиламина, получая 1,52 г свободного основания заглавного соединения из менее полярных фракций. Продукт добавили в раствор избытка хлористого водорода в диоксане и раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель отогнали и получили 1,55 г, заглавного соединения.
T.пл. 245-248oC.
[α]
Аналитические данные. Рассчитано для C19H25Cl3N2OS: C, 52,36% H, 5,78% Cl, 24,40% N, 6,43% S, 7,36%
Найдено: C, 52,40% H, 5,79% Cl, 24,33% N, 6,39% S, 7,18%
Из более полярной фракции в указанной выше колоночной хроматографии получили 1,09 г. свободного основания (R,R)-оптического изомера заглавного соединения. Этот продукт обработали хлористым водородом аналогичным образом, получая 1,12 г. оптического изомера заглавного соединения.
T.пл. 230-232oC
[α]
Аналитические данные. Рассчитано для C19H25Cl3N2OS: C, 52,36% H, 5,78% Cl, 24,40% N, 6,43% S, 7,36%
Найдено: C, 52,39% H, 5,73% Cl, 24,18% N, 6,42% S, 7,54%
(Пример 2).
Гидрохлорид (3R)-3-(1-пирролидинилметил)-4-[(1S)-3-оксо-5,6- дихлор-1-инданкарбонил]тиоморфолина
Реакцию, аналогичную реакции примера 1, проводили с применением 1,0 г (3R)-3-(1-пирролидинил)метилтиоморфолина, 2,0 г триэтиламина и 3,0 г хлорангидрида 3-оксо-5,6-дихлор-1-инданкарбоновой кислоты. Неочищенный продукт очищали колоночной хроматографией на силикагеле с применением смеси (100:1) этилацетата и триэтиламина в качестве подвижной фазы, получая 0,82 г свободного основания заглавного соединения из менее полярных фракций. Продукт обработали хлористым водородом аналогичным образом последней части примера 1, получая 0,85 г заглавного соединения.
Т.пл. 257 261oC
[α]
Аналитические данные. Рассчитано для C19H23Cl3N2O2S C, 50,73% H, 5,15% Cl, 23,64% N, 6,23% S, 7,13%
Найдено C, 50,48% H, 5,20% Cl, 23,49% N, 6,16% S, 7,09%
Из более полярных фракций в указанной выше колоночной хроматографии получили 0,99 г свободного основания (R, R) оптического изомера заглавного соединения. Этот продукт обработали хлористым водородом способом, аналогичным последней части примера 1, и получили 1,00 г оптического изомера заглавного соединения.
Т.пл. 247 249oC.
[α]
Аналитические данные. Рассчитано для C19H23Cl3N2O2S C, 50,73% H, 5,15% Cl, 23,64% N, 6,23% S, 7,13%
Найдено C, 50,69% H, 5,33% Cl, 23,54% N, 6,17% S, 7,24%
(Пример 3).
Гидрохлорид (3R)-3-(1-пиперидинилметил)-4-[(1S)- 6,7-дихлор 4 (3H)-оксо-1,2-дигидро-1-нафтоил] тиоморфолина.
Реакцией, аналогичной реакции примера 1, с применением 2,0 г (R)-3-(1-пиперидинилметил) тиоморфолина, 1,7 мл триэтиламина и 3,1 г хлорангидрида 1-(4-оксо-6,7-дихлортетранол) карбоновой кислоты получили неочищенный продукт, который очищали колоночной хроматографией, применяя смесь (100:1) этилацетата и триэтиламина в качестве подвижной фазы. Получили 1,6 г свободного основания заглавного соединения из менее полярных фракций. Этот продукт обработали хлористым водородом способом, аналогичным последней части примера 1, получая 1,65 г заглавного соединения.
Т.пл. 255 258oC.
[α]
Аналитические данные. Рассчитано для C21H27Cl3N2O2S C, 52,78% H, 5,70% Cl, 22,26% N, 5,86% S, 6,71%
Найдено C, 52,58% H, 5,76% Cl, 22,15% N, 5,76% S, 6,94%
Из более полярных фракций указанной выше колоночной хроматографии получили 1,1 г свободного основания (R, R) оптического изомера заглавного соединения. Продукт обработали хлористым водородом способом, аналогичным последней части примера 1, получая 1,05 г оптического изомера заглавного соединения.
Т.пл. 245 248oC.
[α]
Аналитические данные. Рассчитано для C22H27Cl3N2O2S C, 52,78% H, 5,70% Cl, 22,26% N, 5,86% S, 6,71%
Найдено C, 52,49% H, 5,88% Cl, 22,11% N, 5,65% S, 6,67%
(Пример 4).
(3R) 3 (1 пирролидинилметил) 4 [(IS) 5,6 -дихлор 1 - инданкарбонил] тиоморфилин.
9,0 мл триэтиламина добавили по каплям в раствор 14,7 г (S) 5,6 - дихлоринданкарбоновой кислоты в 200 мл тетрагидрофурана при температуре от -25 до -20oC в потоке азота и затем при той же температуре также по каплям добавили раствор 10,9 г (R) 3 (1 пирролидинилметил) тиоморфолина в 50 мг тетрагидрофурана. После перемешивания раствора в течение 10 мин в указанный выше раствор добавили по каплям раствор 10,5 г диэтил цианофосфоната в 50 мл тетрагидрофурана и перемешивали при этой же температуре еще 30 мин и затем при 0-5oC 1 ч. После отгонки тетрагидрофурана к остатку добавили водный раствор бикарбоната натрия и экстрагировали этилацетатом. Экстракт промыли водой и сушили над безводным сульфатом натрия. После отгонки органического растворителя остаток перекристаллизовали из смеси этилацетата и гексана, получая 13,1 г заглавного соединения.
Т.пл. 123 124oC.
Аналитические данные. Рассчитано для C19H24Cl2N2OS C, 57,14% H, 6,06% N, 7,01% Cl, 17,75% S, 8,03%
Найдено C, 57,28% H, 6,09% N, 7,04% Cl, 17,78% S, 8,30%
Аналогично способам примеров 1-4 можно получить предпочтительные соединения, приведенные в следующей табл. 3.
В табл. 3 применяемая аббревиатура имеет следующие значения:
Pyr: пирролидиногруппа
Pip: пиперидиногруппа
(Ссылочный пример 1)
(1S)-5,6-дихлор-1-инданкарбоновая кислота
138,4 г 5,6-дихлор-1-инданкарбоновой кислоты (0,509 моля) и 257,9 г дигидрата бруцина (0,599 моля) растворили в 500 мл ацетона при нагревании и в раствор добавили 500 мл изопропилового простого эфира и затем выдерживали в течение ночи. Осажденные кристаллы отделили фильтрованием при пониженном давлении и получили 198,0 г соли (1S)-5,6-дихлор-1-инданкарбоновой кислоты с бруцином.
Т. пл. 78-80oC
[α]
Аналитические данные. Рассчитано для C33H34Cl2N2O6•3H2O: C, 58,32% H, 5,76% N, 4,12% Cl, 10,43%
Найдено: C, 58,03% H, 5,76% N, 4,18% Cl, 10,36%
Полученную, как описано выше, соль (1S)-5,6-дихлор-1-инданкарбоновой кислоты и бруцина растворили в 250 мл воды и pH раствора установили до 1 при помощи 60 мл 6N соляной кислоты. После энергичного перемешивания образовавшееся желтое масло экстрагировали этилацетатом. Экстракт промыли водой и сушили над безводным сульфатом магния. После отгонки растворителя получили 68,7 г заглавного соединения.
Т. пл. 135-136oC.
[α]
Аналитические данные. Рассчитано для C10H8Cl2O2: C, 51,98% H, 3,49% Cl, 30,68%
Найдено: C, 51,98% H, 3,59% Cl, 30,64%
(Ссылочный пример 2).
(R)-3-(1-пирролидинил) тиоморфолин
32,6 г ди-трет-бутилдикарбоната и 94,2 мл триэтиламина добавили в раствор 0,20 г тиоморфолин-(R)-3-карбоновой кислоты в 800 мл водного диоксана (1: 1) и смесь перемешивали при комнатной температуре в течение 3 ч. После отгонки растворителя к остатку добавили 400 мл этилацетата и смесь промыли 70 мл насыщенного водного раствора лимонной кислоты и сушили над безводным сульфатом натрия. После отгонки растворителя остаток перекристаллизовали из смеси этилацетата и гексана, получая 27,2 г (N-трет-бутоксикарбонил) тиоморфолин (R)-3-карбоновой кислоты.
Т. пл. 136-137oC.
В раствор 12,37 г полученной выше карбоновой кислоты, 7,64 мл триэтиламина и 5,01 мл пирролидина в 150 мл тетрагидрофурана добавили 10,14 мл диэтилцианофосфоната. Смесь перемешивали при комнатной температуре в течение 3 ч. После отгонки растворителя остаток растворили в этилацетате. Раствор промыли насыщенным водным раствором бикарбоната натрия и сушили над безводным сульфатом натрия. После отгонки растворителя неочищенный кристаллический остаток перекристаллизовали из смеси этилацетата и гексана, получая 13,73 г (N-трет-бутоксикарбонил) тиоморфолин-(R)-3-карбокси(1-пирролидинил) амида.
Т. пл. 121-123oC.
12,02 г полученного выше производного амида растворили в 25 мл 4N раствора хлористого водорода в диоксане и раствор перемешивали при комнатной температуре 0,5 ч. Растворитель отогнали, получая 6,42 г тиоморфолин-(R)-3-карбокси(1-пирролидинил).
Т. пл. 79-80oC.
6,00 г полученного выше производного амида растворили в 150 мл тетрагидрофурана, в раствор добавили 3,41 г алюмогидрида лития и смесь перемешивали при комнатной температуре в течение 1,0 ч. После окончания реакции медленно в реакционную смесь добавили 30 г декагидрата сульфата натрия для остановки реакции. Смесь фильтровали и фильтрат концентрировали, получая 5,12 г заглавного соединения.
Т. кип. 124-126oC/12 мм рт.ст.
[α]
Аналитические данные. Рассчитано для C9H18N2S: C, 58,02% H, 9,74% N, 15,04% S, 17,21%
Найдено: C, 57,94% H, 9,68% N, 14,78% S, 17,14%
Приготовление таблеток
Соединение примера 1 0,1 мг
Лактоза 70,0 мг
Целлюлоза 23,0 мг
Натрий-карбоксиметилкрахмал 4,9 мг
Ангидрид кремниевой кислоты 0,5 мг
Стеарат магния 1,5 мг
100 мг
Порошок, содержащий вышеуказанные компоненты, перемешивают и формируют в таблетки с помощью таблеточной машины, получая 100 мг искомого препарата в виде таблеток. При необходимости, на таблетки может быть нанесено сахарное покрытие.
Использование: в химии гетероциклических соединений, обладающих анальгезирующей активностью. Сущность изобретения: оптически активные производные карбоксамидов формулы I ,
,
где
R1 представляет собой атом водорода или галогена,
R2 представляет собой атом галогена, R3 представляет собой пирролидино- или пиперидиногруппу, Y представляет собой метиленовую или карбонильную группу и n является целым числом 1 или 2, или их фармакологически пригодные соли, а также фармацевтическая композиция на их основе. 2 с и 91 з.п. ф-лы, 3 табл.
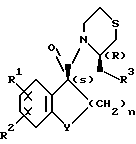
где R1 водород или галоген;
R2 галоген;
R3 пирролидино- или пиперидиногруппа;
Y метиленовая или карбонильная группа;
n 1 или 2,
или их фармакологически пригодные соли за исключением (3R)-3-(1-пирролидинилметил)-4-[(1s)-3-оксо-5,6-дихлор-1-инданкарбонил]тиоморфолина.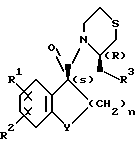
где R1 водород или галоген;
R2 галоген;
R3 пирролидино- или пиперидиногруппа;
Y метиленовая или карбонильная группа;
n 1 или 2,
или его фармацевтически пригодные соли, за исключением (3R)-3-(1-пирролидинилметил)-4-[(1S)-3-оксо-5,6-дихлор-1- инданкарбонил]тиоморфолина.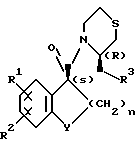
где R1 водород или галоген;
R2 галоген;
R3 пирролидино- или пиперидиногруппа;
Y метиленовая или карбонильная группа;
n 1 или 2,
или их фармакологически пригодные соли в качестве активного компонента, за исключением (3R)-3-(1-пирролидинилметил)-4-[(1S)-3-оксо-5,6-дихлор-1- инданкарбонил]тиоморфолина.
| EP, 356247, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
