Настоящая заявка относится к способам получения 1,3-оксатиолановых нуклеозидов и имеет приоритет в соответствии с предварительными заявками на патент США №60/096214 от 12 августа 1998 г. и 60/122841 от 3 марта 1999 г.
Предпосылки создания изобретения
Способность различных синтетических нуклеозидов, таких как AZT, D4T, DDI и DDC, ингибировать репликацию ВИЧ in vitro и in vivo привело исследователей в конце 1980-х годов к разработке и испытанию нуклеозидов, в которых атом углерода в 3’-положении замещен гетероатомом. Норбек с соавт. (Norbeck et аl.) раскрыли, что (±)-1-[цис-(2,4)-2-(гидроксиметил)-4-диоксоланил]тимин (обозначаемый как (±)-диоксолан-Т) проявляет умеренную активность против ВИЧ (со значением ЭК50 на АТН8 клетках в 20 мкМ) и является нетоксичным для неинфицированных контрольных клеток в концентрации 200 мкМ (Tetrahedron Letters 30 (46), 6246, (1998)). В публикации по заявке на Европейский патент №337713 и в патенте США №5041449, принадлежащем БиоХемфарма Инк. (BioChemPharma, Inc.), раскрываются рацемические 2-замещенные-4-замещенные-1,3-диоксоланы, которые проявляют противовирусную активность. В опубликованных РСТ заявках US 91/09124 и US 93/08044 раскрываются выделенные β-D-1,3-диоксоланиловые нуклеозиды, применяемые для лечения ВИЧ-инфекции. В документе WO 94/09793 раскрывается использование выделенных β-D-1,3-диоксоланиловых нуклеозидов, применяемых для лечения HBV (вирус гепатита В) инфекции.
Публикация РСТ US 95/11464 раскрывает применимость (-)-(2S,4S)-1-(2-гидроксиметил-1,3-диоксолан-4-ил) цитозина в лечении опухолей и других процессов аномальной клеточной пролиферации.
В патенте США №5047407 и в публикации по заявке на Европейский патент №0382526, также принадлежащей БиоХемфарма, Инк (BioChemPharma, Inc.), показано, что множество рацемических 2-замещенных-5-замещенных-1,3-оксатиоксолановых нуклеозидов обладают противовирусной активностью и конкретно указывается, что рацемическая смесь 2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиоксолана (обозначаемая ниже как ВСН-189) обладает примерно той же активностью против ВИЧ, что и AZT, но характеризуется меньшей токсичностью. (-)-Энантиомер ВСН-189 (патент США №5539116, принадлежащий Лиотта с соавт.. (Liotta et al) ), известный как 3ТС, в настоящее время коммерчески доступен и применяется для лечения ВИЧ у человека в Соединенных Штатах. См. также ЕР 513200 В1.
Было также раскрыто, что цис-2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-оксотиолан (FTC) проявляет мощную активность в отношении ВИЧ. (См. Schinazi et al., "Selective Inhibition of Human Immunodeficiency Viruses by Racemates and Enantiomers of cis-5-Fluoro-1-[2-(Hydroxymethyl)-1,3-Oxathiolane-5-yl]Cytosine", Antimicrobial Agents and Chemotherapy, November 1992, page 2423-2431. А также патенты США №5814639, 5914331, 5210085 и патент США 5204466, а также WO 91/11186 и WO 92/14743.)
В связи с коммерческой важностью 1,3-оксатиолановых нуклеозидов в патентах и научной литературе описано множество способов их получения. При разработке способа их получения должны приниматься во внимание три ключевых аспекта их синтеза. Во-первых, реакционная схема должна предусматривать эффективный путь создания кольцевой структуры 1,3-оксатиолана, предпочтительно с наличием замещающих групп в соответствующих положениях для использования в последующих реакциях. Во-вторых, схема реакции должна предусматривать эффективные возможности конденсации 1,3-оксатиоланового кольца с защищенным соответствующим образом основанием, которое в случае 3ТС представляет собой цитозин, а в случае FTC представляет собой 5-фторцитозин. В-третьих, такая реакция должна быть стереоселективной, то есть обеспечивать образование нужного энантиомера. Заместители на хиральных углеродах (конкретном пуриновом или пиримидиновом основании) (обозначаемые как С5 заместитель)) и на СН2ОН (обозначаемые как С2 заместитель) в 1,3-оксатиолановых нуклеозидах могут находиться либо в цис-положении (на той же самой стороне), либо в транс-положении (на противоположных сторонах) по отношению к плоскости оксатиолановой кольцевой системы. При этом и цис-, и транс-рацематы включают два оптических изомера. Исходя из этого, каждое соединение имеет четыре индивидуальных оптических изомера. Четыре оптических изомера изображаются следующими конфигурациями (при ориентации оксатиолановой группы в горизонтальной плоскости, так что -S-СН2-группа находится сзади): (1) цис-конфигурация (обозначаемая также как β-конфигурация) - в том случае, когда обе группы находятся “сверху”, и этот вариант представляет собой встречающуюся в природе L-конфигурацию; (2) цис-конфигурация, когда обе группы находятся “снизу”, и это представляет собой не встречающуюся в природе β-цис-конфигурацию; (3) транс-конфигурация (также обозначаемая как α-конфигурация), когда С2 заместитель находится “сверху” и С5 заместитель “снизу”; и (4) трансконфигурация, когда С2 заместитель находится “снизу” и С5 заместитель находится “сверху”. Два цис-энантиомера вместе обозначаются как рацемическая смесь β-энантиомера, а два транс-энантиомера вместе обозначаются как рацемическая смесь α-энантиомеров. В целом, разделение пары рацемических цис-оптических изомеров от пары рацемических транс-оптических изомеров является хорошо разработанной стандартной процедурой. Значительно труднее разделить или каким-то образом выделить индивидуальные энантиомеры в цис-конфигурации. Для 3ТС и FTC желательная стереохимическая конфигурация представляет собой β-L-изомер.
Пути получения 1,3-оксатиоланового кольца
Ниже дана пронумерованная схема 1,3-оксатиоланового кольца:

Краус с соавт. (Kraus et al., "Synthesis of New 2,5-Disubstituted 1,3-Oxathiolanes. Intermediates in Nucleoside Chemistry", Synthesis, pages 1046-1048 (1991)) описывают проблемы, возникающие при проведении реакции альдегида глиоксилата или гликолевой кислоты с меркаптоуксусной кислотой в толуоле в присутствии п-толуолсульфоновой кислоты. Краус отмечает, что для успешности указанной реакции необходимо, чтобы гликолевое производное, находящееся в форме гидрата, было превращено в свободный альдегид при азеотропном удалении воды с толуолом перед проведением циклоконденсации. После этого для восстановления лактоновой группы и группы карбоновой кислоты используют различные каталитические восстановители. При этом восстановление с использованием боргидрида натрия не привело к желаемым результатам, тогда как применение боранметилсульфидного комплекса (БМС) приводит к восстановлению только фрагмента карбоновой кислоты. При повышении температуры или при использовании большого избытка БМС происходящее раскрытие кольца ведет к получению полимерного материала. Восстановление 2-карбокси-1,3-оксатиолан-5-она с помощью гидрида бис(2-метоксиэтокси) алюминия-натрия в толуоле дает смесь продуктов. Гидрид трибутилолова не приводит к нужному восстановлению. И, наконец, при проведении восстановления на защищенных лактонах также не удалось выделить целевое соединение, независимо от условий восстановительного катализа.
В связи с наличием указанных трудностей Краус с соавт. предложили реакцию, которая включает проведение циклоконденсации безводных глиоксилатов с диэтилацеталем 2-меркаптоацетальдегида при температуре кипения с обратным холодильником в толуоле с получением производных 5-этокси-1,3-оксатиолана, которые далее могут быть восстановлены с помощью БМС до соответствующего 2-гидроксиметил-1,3-оксатиолана с 50% выходом, и который после бензоилирования дает смесь цис- и транс-2-бензоилоксиметил-5-этокси-1,3-оксатиолана. Указанный способ описан также в патенте США №5047407.
В патенте США №5248776 раскрывается способ получения энантиомерно чистых β-L-1,3-оксатиолановых нуклеозидов из 1,6-тиоангидро-L-гулозы.
В патенте США №5204466 раскрывается способ получения 1,3-оксатиоланового кольца реакцией меркаптоуксусной кислоты (тиогликолевой кислоты) с гликолевым альдегидом с образованием 2-(R-окси)-метил-5-оксо-1,3-оксатиолана.
Патент США №5466806 описывает способ получения 2-гидроксиметил-5-гидрокси-1,3-оксатиолана посредством реакции димера меркаптоацетальдегида с соединением формулы RwOCH2CHO в нейтральных или щелочных условиях, где Rw обозначает защитную группу гидроксила. (См. также Mclntosh et al., "2-Mercaptoaldehyde dimers and 2,5-dihydrothiophenes from 1,2-oxathiolan-5-ones,"Can. J. Chem. Vol.61, 1872-1875 (1983).)
Белло с соавт. (Belleau et al.) раскрывают способ получения 1,3-диоксоланового нуклеозида посредством окислительного разложения L-аскорбиновой кислоты (Belleau et al., "Oxidative Degradation of L-ascorbic Acid Acetals to 2’, 3’-Dideoxy-3’-Oxaribofuranosides. Synthesis of Enantiomerically Pure 2’,3’-Dideoxy-3’-Oxacytidine Stereoisomers as Potential Antiviral Agents", Tetrahedron Letters, vol.33, №46, 6949-6952 (1992)).
В патенте США №5204466 раскрывается получение 1,3-оксатиоланового кольца посредством озонолиза аллилового простого или сложного эфира формулы CH2=CHCH2OR, где R обозначает защитную группу, с образованием гликолевого альдегида формулы OHCCH2OR, и при добавлении тиогликолевой кислоты к гликолевому альдегиду с образованием лактона формулы 2-(R-окси)-метил-5-оксо-1,3-оксатиолан.
Пути конденсации 1,3-оксатиолана с защищенным основанием
В патенте США №5204466 раскрывается способ конденсации 1,3-оксатиолана с защищенным пиримидиновым основанием при использовании хлорида олова в качестве кислоты Льюиса, причем этот способ обеспечивает практически полную β-стереоселективность (см. также Choi et al., "In Situ Complexation Directs the Stereochemistry of N-Glycosylation in the Synthesis of Oxathiolanyl and Dioxolanyl Nucleoside Analogues," J. Am Chem. Soc. 1991, 213, 9377-9379). Использование хлорида олова приводит к получению в ходе реакции нежелательных остатков и побочных продуктов, которые трудно удалить.
Множество патентов США раскрывают способ получения 1,3-оксатиолановых нуклеозидов через реакцию конденсации 1,3-оксатиоланового интермедиата, который включает хиральный эфир во 2-м положении кольца с защищенным основанием в присутствии кремнийсодержащей кислоты Льюиса. Сложный эфир, находящийся во 2-м положении, далее восстанавливают до соответствующей гидроксиметильной группы, что дает готовый продукт. См. патенты США №5663320; 5864164: 5693787; 5696254; 5744596 и 5756706.
В патенте США №5763606 раскрывается способ получения преимущественно цис-2-карбоновой или тиокарбоновой кислоты 1,3-оксатиолановых нуклеозидов, который включает конденсацию целевого предварительно силилированного пуринового или пиримидинового основания с бициклическим интермедиатом в присутствии кислоты Льюиса.
Патент США №5272151 описывает способ получения 1,3-диоксолановых нуклеозидов, которые включают взаимодействие 2-О-защищенного-5-О-ацилированного-1,3-диоксолана с защищенным по кислороду или азоту пуриновым или пиримидиновым основанием в присутствии титанового катализатора.
Чои с соавт. (Choi et al., "In Situ Complexation Directs the Stereochemistry of N-Glycosylation in the Synthesis of Oxathiolanyl and Dioxolanyl Nucleoside Analogues," J. Am Chem. Soc. 1991, 213, 9377-9379) показали, что в случае использования HgCl2, Et2AlCl или TiCl2(O-изопропил)2 не происходит связывания 1,3-оксатиолана с защищенным пиримидиновым основанием (см. примечание 2). Чои также указал, что взаимодействие между аномерными ацетатами 1,3-оксатиолана с силилированным цитозином и практически любой обычно применяемой кислотой Льюиса, отличной от хлорида олова, приводит к образованию неразделяемых смесей N-гликозилированных аномеров.
В патенте США №5922867 раскрывается способ получения диоксоланового нуклеозида, который включает гликозилирование пуринового или пиримидинового основания с использованием 2-защищенного-оксиметил-4-галоген-1,3-диоксолана.
Пути получения 1,3-оксатиоланового нуклеозида в желательной стереоконфигурации
В патенте США №5728575 заявляется способ получения 3ТС и FTC энзиматической реакцией 5’-ацил-защищенного рацемического нуклеозида с использованием эстеразы из печени свиньи, липазы из поджелудочной железы свиньи или субтилизина. В патенте США №5539116 заявляется продукт ЗТС, получаемый в результате разделения продукта по способу патента ‘575.
В патенте США №5827727 (Liotta) заявляется способ получения 3ТС и FTC реакцией стереоселективного дезаминирования с использованием цитидиндезаминазы.
В патенте США №5892025 (Liotta et al.) заявляется способ разделения конденсации энантиомеров цис-FTC при пропускании цис-FTC через колонку с хиральным ацетилированным β-циклодекстрином.
В патенте США №5663320 заявляется способ получения хирального 1,3-оксатиоланового интермедиата, который включает разделение рацемического интермедиата на основе методики, основанной на использовании хиральных свойств.
В связи с важностью 1,3-оксатиолановых нуклеозидов для лечения заболеваний, вызванных вирусом иммунодефицита человека и вирусом гепатита В, настоящее изобретение ставит своей целью разработку способов получения 1,3-оксатиолановых нуклеозидов, которые могут использоваться в производственном масштабе.
Краткое описание сущности изобретения
Предлагаются способы получения 1,3-оксатиолановых нуклеозидов, которые включают эффективные методы образования 1,3-оксатиоланового кольца с последующей конденсацией 1,3-оксатиолана с пиримидиновым или пуриновым основанием. С использованием приведенных в описании способов указанное соединение может быть получено в виде изолированного энантиомера.
Было показано, что 2-[R1С(О)OCH2]-1,3-оксатиоланил-5-он может быть получен с высоким выходом при непосредственном взаимодействии ацеталя формулы (R1O)2CHR, где R обозначает -(СН2-О-С(О)R1 и R1 обозначает алкил, арил, гетероарил, гетероциклический радикал, алкарил, алкилгетероарил или алкилгетероциклический радикал, или аралкил, с меркаптоуксусной кислотой в органическом растворителе, например в ацетонитриле, в присутствии кислоты Льюиса или протонной кислоты в органическом растворителе с минимальным количеством воды. В альтернативном варианте может использоваться предшественник альдегида (ОН)2СНR или (R1O) (OH)CHR. Ацеталь может также использоваться в виде смеси полуацеталя, мономера ацеталя или его продуктов с более высокой степенью конденсации. При взаимодействии меркаптоуксусной кислоты непосредственно с ацеталем количество побочных продуктов снижается, что в свою очередь повышает чистоту продукта и выход указанного исходного материала. Ацеталь получают обычным способом, например при взаимодействии диэфира спирта с н-бутирилхлоридом.
(R1O)2CHR может быть получен любым подходящим способом, в частности, например, либо (i) реакцией соединения формулы ОН-СН2-С=С-СН2-ОН с RC(O)Cl с образованием RC(O)OCH2C(H)=C(H)OC(О)R, который озонируют или каким-либо другим способом расщепляют с получением целевого соединения, либо (ii) при восстановлении (R1O)2CHC(О)Н с образованием (R1O)2CHCH2OH, который далее взаимодействует с ClC(O)R с образованием целевого соединения.
Альтернативно НС(О)СН2ОС(О)R1 взаимодействует с меркаптоуксусной кислотой с образованием целевого 1,3-оксатиоланового кольца. НС(О)СН2OС(О)R1 может быть получен с помощью любого приемлемого способа, например, способов А и В, приведенных на фиг.2.
5-(O-защищенная группа)-2-защищенный гидроксиметил-1,3-оксатиолан или его 5-ацетилокси производное могут быть конденсированы с соответствующим силилированным пиримидиновым или пуриновым основанием, включая цитозин или 5- фторцитозин, с использованием кислоты Льюиса, такой как хлорид олова, (Сl)3Тi. (изопропоксид), триметилсилилтрифлат, триметилсилилиодид или другая кислота Льюиса, которая известна своей способностью катализировать конденсацию, включая те кислоты Льюиса, которые были описаны в патентах США №5663320; 5864164; 5693787; 5696254; 5744596 и 5756706, с получением соответствующего нуклеозида с высокой β-селективностью. Было обнаружено, что (Сl)3Тi(изопропоксид) может применяться в качестве катализатора для конденсации 1,3-оксатиолана с защищенным основанием, тогда как ранее сообщалось, что в случае защищенного пиримидинового основания и 1,3-оксатиолана не происходит конденсации при использовании HgCl2, Et2AlCl или ТiСl2(O-изопропил)2.
В альтернативном варианте осуществления изобретения гликолевая кислота используется вместо меркаптоуксусной кислоты в присутствии кислоты Льюиса с образованием соответствующего 1,3-диоксолана, который может быть конденсирован с пуриновым или пиримидиновым основанием с образованием 1,3-диоксоланового нуклеозида. Предпочтительно проводить циклоконденсацию ацеталя (или альдегида) с гликолевой кислотой в присутствии кислоты Льюиса, такой как диэтилэтерат трифторида бора, а не протонной кислоты, такой как п-толуолсульфоновая кислота.
Было также показано, что 1,3-оксатиолановый нуклеозид может быть получен посредством: (i) образования 5-галоген-2-защищенного-оксиметил-1,3-оксатиолана, и (ii) взаимодействия 5-галоген-2-защищенного-оксиметил-1,3-оксатиолана с защищенным пуриновым или пиримидиновым основанием при низкой температуре, предпочтительно ниже 25 градусов Цельсия и более предпочтительно ниже 10 градусов Цельсия. Было обнаружено, что реакция конденсации может быть эффективно проведена и без использования кислоты Льюиса. В предпочтительном варианте реализации изобретения галогеновый заместитель в 5 положении оксатиолана представляет собой хлор. В типичном варианте реакция приводит к образованию смеси β- и α-аномеров, которые должны быть разделены. В типичном случае β-аномер образуется в избытке относительно α-аномера. Разделение β- и α-аномеров может быть осуществлено любым известным способом, включая фракционную кристаллизацию, хроматографию (ахиральную или хиральную) или получение и разделение диастереомерных производных. В одном варианте реализации изобретения рацемический 5-ацилированный-2-защищенный-оксиметил-1,3-оксатиолан хлорируют при низкой температуре (например, при температуре 0 градусов Цельсия), затем конденсируют с защищенным основанием, таким как 5-фторцитозин или цитозин, с получением смеси диастереомеров (в типичном случае при значительном избытке β-соединения). В другом варианте реализации изобретения хиральный 5-ацилированный-2-защищенный-оксиметил-1,3-оксатиолан хлорируют и затем используют в реакции взаимодействия с защищенным основанием. Может использоваться любой 5-ацилированный-2-защищенный-оксиметил-1,3-оксатиолан, который обеспечивает получение желательного продукта. Неограничивающие примеры соответствующих ацильных групп включают, не ограничиваясь ими, ацетат, пропионат, бутират, бензоат, п-метоксибензоат и п-(т-бутил)-бензоат. Реакция галогенирования может быть проведена в любом приемлемом органическом растворителе, включая толуол, хлороформ, уксусную кислоту, тетрагидрофуран, диэтиловый эфир, бензол и др. На коэффициент соотношения аномеров α к β, достигаемый в реакции конденсации, может оказывать влияние растворитель, выбранный для использования в данной реакции. При этом любой специалист может путем подбора выбрать тот органический растворитель, который обеспечивает оптимальный выход желательного продукта.
Краткое описание фигур
Фиг.1 представляет собой иллюстрацию одного из способов получения 1,3-оксатиоланового нуклеозида согласно изобретению, который включает получение 2-[R1C(О)ОСН2]-1,3-оксатиоланил-5-она посредством взаимодействия ацеталя формулы (R1O)2CHR, где R обозначает -(СН2-О-С(О)R1), с меркаптоуксусной кислотой.
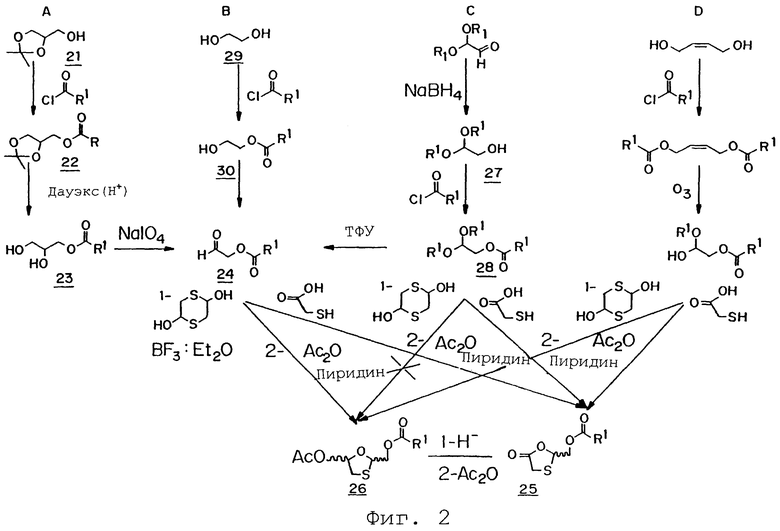
Фиг.2 представляет собой схематическую иллюстрацию четырех альтернативных способов (A-D) образования 1,3-оксатиоланового кольца согласно изобретению.
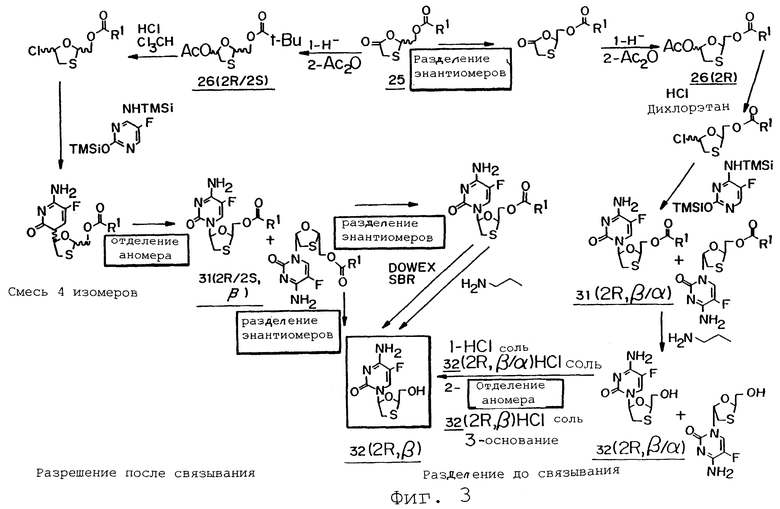
Фиг.3 представляет собой схематическую иллюстрацию получения энантиомеров 1,3-оксатиоланового нуклеозида с использованием процедуры разделения до и после связывания.
Подробное описание изобретения
Предлагается способ получения 1,3-оксатиолановых нуклеозидов, которые включают эффективные способы образования 1,3-оксатиоланового кольца и последующую реакцию конденсации 1,3-оксатиолана с пиримидиновым или пуриновым основанием.
Было показано, что 2-[R1C(О)OCH2O]-1,3-оксатиоланил-5-он может быть получен с высоким выходом при непосредственном взаимодействии ацеталя формулы (алкилО)2CHR, где R обозначает -(СН2-О-С(О)R1), и R1 обозначает алкил, арил, гетероарил, алкарил, алкгетероарил или аралкил с меркаптоуксусной кислотой в присутствии кислоты Льюиса или протонной кислоты в органическом растворителе с минимальным количеством воды. Ацеталь может использоваться в виде смеси полуацеталя, мономера ацеталя или его продукта конденсации. При непосредственном взаимодействии меркаптоуксусной кислоты с ацеталем количество побочных продуктов снижается, что, в свою очередь, повышает чистоту продукта и выход исходного материала.
5-(O-защищающая группа)-2-защищенный гидроксиметил-1,3-оксатиолан или его 5-ацетилокси производное могут быть подвергнуты конденсации с защищенным силилсодержащим пиримидиновым или пуриновым основанием, включая цитозин или 5-фторцитозин, с использованием кислоты Льюиса, такой как хлорид олова, (Сl)3Тi(изопропоксид), триметилсилилтрифлат, триметилсилилиодид или другая кислота Льюиса, катализирующая реакцию конденсации, включая те кислоты Льюиса, которые были описаны в патентах США №5663320; 5864164; 5693787; 5696254; 5744596 и 5756706, с получением соответствующего нуклеозида с высокой β-селективностью. Было обнаружено, что (Сl)3Ti(изопропоксид) может применяться в качестве катализатора для конденсации 1,3-оксатиолана с защищенным основанием, несмотря на имевшиеся сообщения о том, что 1,3-оксатиолан с защищенным пиримидиновым основанием не конденсируется в присутствии HgCl2, Et2AlCl или TiCl2(O-изопропил)2.
В альтернативном варианте реализации изобретения меркаптоуксусную кислоту используют вместо гликолевой кислоты с образованием соответствующего 1,3-диоксолана, который может быть конденсирован с пуриновым или пиримидиновым основанием с образованием 1,3-диоксоланового нуклеозида. Предпочтительно реакцию циклоконденсации ацеталя (или альдегида) с гликолевой кислотой проводят в присутствии кислоты Льюиса, такой как диэтилэтерат трифторида бора, а не протонной кислоты, такой как п-толуолсульфоновая кислота.
Было также показано, что 1,3-оксатиолановый нуклеозид может быть получен посредством: (i) образования 5-ацилированного-2-защищенного-оксиметил-1,3-оксатиолана и (ii) взаимодействия 5-галоген-2-защищенного-оксиметил-1,3-оксатиолана с защищенным пуриновым или пиримидиновым основанием при низкой температуре, предпочтительно ниже 25 градусов Цельсия и более предпочтительно ниже 10 градусов Цельсия. Было обнаружено, что реакция конденсации может быть эффективно проведена без использования кислоты Льюиса. В предпочтительном варианте реализации изобретения галогеновый заместитель в 5-м положении оксатиолана представляет собой хлор. В типичном случае реакция приводит к образованию смеси β- и α-аномеров, которые должны быть разделены. В типичном случае β-аномер образуется в избытке относительно α-аномера. Разделение β- и α-аномеров может быть осуществлено любым известным способом, включая фракционную кристаллизацию, хроматографию (ахиральную или хиральную) или получение и разделение диастереомерных производных. В одном варианте реализации изобретения рацемический 5-ацилированный-2-защищенный-оксиметил-1,3-оксатиолан хлорируют при низкой температуре (например, при температуре 0 градусов Цельсия) и затем конденсируют с защищенным основанием, таким как 5-фторцитозин или цитозин, с получением смеси диастереомеров (в типичном случае при значительном избытке β-соединения). В другом варианте реализации изобретения хиральный 5-ацилированный-2-защищенный-оксиметил-1,3-оксатиолан хлорируют и затем используют в реакции взаимодействия с защищенным основанием. Может использоваться любой 5-ацилированный-2-защищенный-оксиметил-1,3-оксатиолан, который обеспечивает получение целевого продукта. Неограничивающие примеры соответствующих ацильных групп включают, не ограничиваясь ими, ацетат, пропионат, бутират, бензоат, п-метоксибензоат и п-(т-бутил)-бензоат. Реакция галогенирования может быть проведена в любом приемлемом органическом растворителе, включая толуол, хлороформ, уксусную кислоту, тетрагидрофуран, диэтиловый эфир, бензол и др. На коэффициент соотношения аномеров α к β, достигаемый в реакции конденсации, может оказывать влияние вид растворителя, выбранного для использования в данной реакции. При этом любой специалист в данной области может методом подбора выбрать тот органический растворитель, который обеспечивает оптимальный выход желательного продукта.
Выбранный 5-ацилированный-2-защищенный-оксиметил-1,3-оксатиолан может быть галогенирован, например, до получения 5-хлор, 5-бром или 5-йод производного с использованием известных методов.
Хиральные стационарные фазы для проведения хиральной хроматографии описаны во множестве работ, включая, например, работу Стради с соавт. (Stradi et al. Analytical Enantioseparations, Polysaccharides and their Derivatives as Chiral Stationary Phases. Perkin Elmer, 1992).
Вместо 5-ацильной группы может использоваться любая другая удаляемая группа, которая может быть замещена и заменена галогеном и предпочтительно хлоридом. Такие примеры включают алкокси, алкоксикарбонил, амидо, азидо и изоцианато.
I. Определения
В контексте настоящего описания термин “выделенный энантиомер” относится к нуклеозидной композиции, которая включает по меньшей мере примерно от 95% до 100% или более предпочтительно свыше 97% чистого энантиомера указанного нуклеозида.
Термин “пуриновое или пиримидиновое основание” включает, не ограничиваясь приведенным списком, 6-алкилпурин и N6-алкилпурины, N6-ацилпурины, N6-бензилпурин, 6-галогенпурин, N6-ацетиленовый пурин, N6-ацилпурин, N6-гидроксиалкилпурин, 6-тиоалкилпурин, N2-алкилпурины, N4-алкилпиримидины, N4-ацилпиримидины, 4-галогенпиримидины, N4-ацетиленовые пиримидины, 4-амино- и N4-ацилпиримидины, 4-гидроксиалкилпиримидины, 4-тиоалкилпиримидины, тимин, цитозин, 6-азапиримидин, включая 6-азацитозин, 2- и/или 4-меркаптопиримидин, урацил, C5-aлкилпиpимидины, С5-бензилпиримидины, C5-гaлoгeнпиpимидины, С5-винилпиримидин, С5-ацетиленовый пиримидин, C5-aцилпиpимидин, С5-гидроксиалкилпурин, C5-aмидoпиpимидин, C5-циaнoпиpимидин, С5-нитропиримидин, С5-аминопиримидин, N2-aлкилпypины, N2-aлкил-6-тиопурины, 5-азацитидинил, 5-азаурацилил, триазолопиридинил, имидазолопиридинил, пирролопиримидинил и пиразолопиримидинил. Функциональные кислородные и азотные группы на основании могут быть защищены, если это необходимо или желательно. Приемлемые защитные группы хорошо известны специалистам в данной области и включают триметилсилильные, диметилгексилсилильные, т-бутилдиметилсилильные и т-бутилдифенилсилильные, тритильные, алкильные группы, а также ацильные группы, такие как ацетил и пропионил, метансульфонил и п-толуолсульфонил. Предпочтительные основания включают цитозин, 5-фторцитозин, урацил, тимин, аденин, гуанин, ксантин, 2,6-диаминопурин, 6-аминопурин, 6-хлорпурин и 2,6-дихлорпурин.
Термин “алкил” в контексте настоящего описания, если не указано иное, относится к насыщенному линейному, разветвленному или циклическому, первичному, вторичному или третичному углеводороду, в типичном случае от C1 до C18 и конкретно включает метил, этил, пропил, изопропил, бутил, изобутил, т-бутил, пентил, циклопентил, изопентил, неопентил, гексил, изогексил, циклогексил, циклогексилметил, 3-метилпентил, 2,2-диметилбутил и 2,3-диметилбутил. Алкильная группа может быть необязательно замещена одной или большим числом групп, выбранных из группы, состоящей из гидроксила, групп карбоновой кислоты или сложного эфира, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, либо незащищенных, либо защищенных, если это необходимо, в соответствии с известной специалистам в данной области методикой, например, приведенной в руководстве Грина с соавт. (Green et al., "Protective Groups in Organic Synthesis", John Wiley and Sons, Second Edition, 1991), включенном в настоящее описание в качестве ссылки.
Термин “защищенный” в контексте настоящего описания, если особо не оговорено иное, относится к группе, которая добавляется к атому кислорода, азота или фосфора для предупреждения его дальнейшего взаимодействия или для других целей. Специалистам в области органического синтеза известен широкий перечень защищающих групп для кислорода и азота. Приемлемые защищающие группы описаны, например, в руководстве Грина с соавт. (Green et al., "Protective Groups in Organic Synthesis", John Wiley and Sons, Second Edition, 1991), включенном в настоящее описание в качестве ссылки.
Термин "арил" в контексте настоящего изобретения, если особо не оговорено иное, относится к фенилу, бифенилу или нафтилу и обозначает предпочтительно фенил. Арильная группа может быть необязательно замещена одной или большим числом групп, выбранных из группы, состоящей из гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, которые могут быть либо незащищенные, либо защищенные, если это необходимо, с использованием методик, известных специалистам в данной области техники и приведенной, например, в руководстве Грина с соавт. (Green et al., "Protective Groups in Organic Synthesis", John Wiley and Sons, Second Edition, 1991), включенном в настоящее описание в качестве ссылки.
Термин “алкарил”, или “алкиларил” относится к алкильной группе с арильным заместителем.
Термин “аралкил”, или “арилалкил” относится к арильной группе с алкильным заместителем.
Термин “галоген” в контексте настоящего описания включает хлор, бром, йод и фтор.
Термин “ацил” относится к группе формулы -C(O)R’, где R’ обозначает алкил, арил, алкарил, аралкил, гетероароматическую, гетероциклическую группы, алкоксиалкил, включающий метоксиметил; арилалкил, включающий бензил; арилоксиалкил, такой как феноксиметил; арил, включающий фенил, необязательно замещенный галогеном, C1-C4алкилом или C1-C4алкокси или аминокислотным остатком.
В контексте настоящего описания удаленная группа относится к функциональной группе, которую в соответствующих условиях отщепляют от молекулы, к которой она была присоединена.
Термин “гетероарил”, или “гетероциклический радикал” в контексте настоящего описания относится к циклической группе, которая включает в кольце по меньшей мере один атом серы, кислорода или азота. Неограничивающие примеры указанной группы включают фурил, пиридил, пиримидил, тиенил, изотиазолил, имидазолил, тетразолил, пиразинил, бензофуранил, бензотиофенил, хинолил, изохинолил, бензотиенил, изобензофурил, пиразолил, индолил, изоиндолил, бензимидазолил, пуринил, карбазолил, оксазолил, тиазолил, изотиазолил, 1,2,4-тиадиазолил, изоксазолил, пирролил, хиназолинил, пиридазинил, пиразинил, циннолинил, фталазинил, хиноксалинил, ксантинил, гипоксантинил и птеридинил. Функциональные кислородные и азотные группы на гетероциклическом основании могут быть защищены, если это необходимо или желательно. Подходящие защищающие группы хорошо известны специалистам в данной области и включают триметилсилил, диметилгексилсилил. Алкильная группа может быть необязательно замещена одним или большим числом групп, выбранных из группы, состоящей из гидроксила, группы карбоновой кислоты или сложного эфира, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, которые могут быть либо незащищены, либо защищены, если это необходимо, в соответствии с известной специалистам данной области методикой и приведенной, например, в руководстве Грина с соавт. (Green et al., "Protective Groups in Organic Synthesis", John Wiley and Sons, Second Edition, 1991), которое включено в настоящее описание в качестве ссылки.
Термин “алкилгетероарил” относится к алкильной группе, замещенной гетероарильным заместителем.
II. Получение лактонового кольца 1,3-оксатиолана
На фиг.1 показан один путь осуществления описанного процесса. 2-бутен-1,4-диол взаимодействует с хлорангидридом карбоновой кислоты или другим эфирным предшественником с образованием 2-бутен-1,4-диолового диэфира. Выбор хлорангидрида карбоновой кислоты или другого предшественника сложного эфира определяется группой, которую желательно иметь во 2-м положении образующегося 1,3-оксатиоланового кольца. Так, например, если бутирилхлорид взаимодействует с 2-бутен-1,4-диолом, то в получаемом 2-[R1C(О)ОСН2О]-1,3-оксатиоланил-5-оне R1 будет пропилом. В другом варианте реализации изобретения хлорангидрид карбоновой кислоты или другой эфирный предшественник выбирают так, чтобы R1 был алкилом, арилом, гетероарилом, алкарилом, алкилгетероарилом или аралкилом.
На второй стадии реакции 2-бутен-1,4-диэфир расщепляют предпочтительно путем озонолиза с получением ацеталя формулы (алкилО)2CHR, в которой R обозначает (СН2-О-С(О)R1), где R1 обозначает алкил, арил, гетероарил, алкарил, алкилгетероарил или аралкил. Реакцию озонолиза в типичном случае проводят при очень низких температурах, обычно при температуре -70°С или ниже. В случае проведения реакции при более высокой температуре, например при -10°С, необходимы специальные низкотемпературные реакторы. Реакция, приводящая к образованию ацеталей, может быть проведена с использованием различных спиртовых растворителей при наличии или в отсутствие дополнительных растворителей, таких как дихлорметан. Предпочтительным спиртовым растворителем является метанол. Реакцию озонолиза чаще всего гасят диметилсульфидом, однако было показано, что применение тиомочевины позволяет получать целевой продукт с более высокой степенью чистоты.
В альтернативном варианте ацеталь формулы (алкилО)2CHR, где R обозначает (CH2OC(O)R’) и R7 обозначает алкил, арил, гетероарил, алкилгетероарил или аралкил, может быть получен при ацилировании (алкилО)2CHCH2OH галогенидом или ангидридом соответствующей кислоты в присутствии основания, такого как триэтиламин.
На ключевой стадии процесса ацеталь непосредственно взаимодействует с меркаптоуксусной кислотой в присутствии кислоты Льюиса или протонной кислоты в органическом растворителе с минимальным количеством воды. Ацеталь может быть использован в виде смеси полуацеталя, мономера ацеталя или продуктов его конденсации. При этом для использования в рамках настоящего способа приемлема любая протонная кислота или кислота Льюиса, которая приводит к получению целевого продукта. Было показано, что реакция циклоконденсации ацеталя с меркаптоуксусной кислотой приводит к эффективному образованию 1,3-оксатиолана. И наоборот, циклоконденсация альдегида с меркаптоуксусной кислотой зачастую сопряжена с рядом проблем, что сказывается на более низких выходах целевого продукта 1,3-оксатиолана, загрязненного непрореагировавшим альдегидом, а также ведет к образованию альдегидных побочных продуктов.
На следующей стадии 2-защищенный гидроксиметил-5-оксо-1,3-оксатиолан разделяют с использованием большого числа приемлемых методов, известных в технике. Заместитель во 2-м положении выбирают с учетом легкости его разделения на этой стадии. Таким заместителем может быть, например, группа, которая стереоселективно расщепляется ферментом. В патенте США №5204466 (Liotta et al.) описывается способ разделения оксатиолана путем стереоселективного ферментного гидролиза с использованием липазы из поджелудочной железы свиньи, субтилизина или эстеразы из печени свиньи. В патенте США №5663320 заявлен способ получения хирального интермедиата 1,3-оксатиолана, который включает разделение рацемического интермедиата с использованием хиральной техники. В WO 91/17159 раскрывается использование хиральных колонок с триацетатом целлюлозы или с β-циклодекстрином для разделения энантиомеров 1,3-оксатиолановых нуклеозидов.
Выделенный целевой (2R)-энантиомер 2-защищенного гидроксиметил-5-оксо-1,3-оксатиолана, который в случае 3ТС и FTC дает β-L-энантиомер, восстанавливают до соответствующего 5-O-защищенного соединения, например до 5-ацетата, с использованием восстановителя, предпочтительно три-трет-бутоксиалюмогидрида лития.
На фиг.2 показаны четыре дополнительных варианта реализации настоящего изобретения (способы A-D), применяемые для получения 1,3-оксатиоланового кольца. В качестве неограничивающего примера, поясняющего способ А, показанный на фиг.2, можно привести процедуру получения (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата в ходе четырехстадийного процесса, в рамках которого не требуется очистка от промежуточных продуктов. На первой стадии получают (2,2-диметил-1,3-диоксолан-4-ил)метилбутаноат из солкеталя и н-бутирилхлорида в т-бутилметиловом эфире, ДМАП и триэтиламине. Затем (2,2-диметил-1,3-диоксолан-4-ил)метилбутаноат помещают в раствор со смолой Дауэкс 50W Х8-100 H+ (Dowex SOW X8-100 Н+) в метаноле с получением 2,3-дигидроксипропилбутаноата. Полученный диол затем вступает в реакцию с раствором периодата натрия в дистиллированной воде с образованием 2-оксоэтилбутаноата. С использованием 2-оксоэтилбутаноата получают (5-оксо-1,3-оксатиолан-2-ил)метилбутаноат в ходе его реакции с меркаптоуксусной кислотой, такой как п-TsOH·H2O в ацетонитриле. Далее из (5-оксо-1,3-оксатиолан-2-ил) метилбутаноата получают 5-ацетилокси производное в реакции с три-т-бутоксиалюмогидридом лития в ТГФ.
Одним из неограничивающих примеров реализации изобретения по способу В, показанному на фиг.2, является получение (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата или его 5-ацетилокси производного в ходе реакции взаимодействия 1,2-дигидроксиэтана с н-бутирилхлоридом в триэтиламине. Эта реакция дает 2-гидроксиэтилбутаноат, который впоследствии взаимодействует с P2O5 в сухом ДХМ и затем с ДМСО и триэтиламином с образованием 2-оксиэтилбутаноата. Полученный 2-оксиэтилбутаноат может быть далее превращен в 5-ацетилокси производное (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата по указанному выше способу или может быть превращен в (5-оксо-1,3-оксатиолан-2-ил) метилбутаноат взаимодействием с меркаптоуксусной кислотой и CSA в сухом ДХМ.
Неограничивающим примером получения (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата или его 5-ацетилокси производного согласно изобретению с использованием способа С, показанного на фиг.2, является способ, который включает взаимодействие 2,2-диэтоксиэтилбутаноата в ДХМ с последующей стадией обработки ТФУ и водой. Указанная реакция дает 2-оксоэтилбутаноат, который может дальше вступать в реакцию с меркаптоуксусной кислотой в CSA и ДХМ с образованием целевого (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата или с 1,4-дитиан-2,5-диолом в ТГФ с образованием 5-ацетилокси производного.
Способ D на фиг.2 аналогичен описанному выше способу на фиг.1.
Указанные стадии поясняются ссылкой на приведенные ниже примеры, которые не ограничивают область настоящего изобретения.
Пример 1
В реактор на 200 галлонов (909,2 л), снабженный эффективной охлаждающей системой, вводят метил трет-бутиловый эфир (МтБЭ, 278 л), DMAP (391 г, 3,2 моль), триэтиламин (102,3 л, 74,4 кг, 736,2 моль) и 2-бутен-1,4-диол (26,4 л, 28,2 кг, 320 моль). Начинают перемешивание и снижают температуру реакции примерно до 4°С. Затем к реакционной смеси добавляют бутирилхлорид (69,6 л, 71,5 кг, 672 моль) с такой скоростью, чтобы поддерживать температуру реакции ниже 20°С. При добавлении выпадает осадок гидрохлорида триэтиламина, и реакционная смесь становится густой, но перемешиваемой взвесью. Анализ реакционной среды методом тонкослойной хроматографии (пластинки с силикагелем; Analtech №02521, элюация смесью гексан/EtOAc, 9:1, визуализация (проявка) с помощью красителя РМА) указывает на то, что через час дополнительного перемешивания после проведенного добавления реакция завершилась. В реактор добавляют воду (120 л) и полученную смесь перемешивают до полного растворения твердого вещества. Разделяют фазы. В нижнем (водном) слое с помощью ТСХ определяют отсутствие продукта (если продукт имеется, такой слой сохраняют для дальнейшего восстановления продукта).
Верхний органический слой промывают водой (72 л), насыщенным водным раствором бикарбоната калия (72 л), убеждаются в том, что отобранный водный слой имеет щелочную реакцию, и выпаривают при пониженном давлении с получением 69,4 кг 2-бутен-1,4-дибутирата (95% выход) в виде бледно-золотистого масла. Спектр ЯМР соответствует спектру стандартного продукта.
Пример 2: Озонолиз полуацеталя метил 2-оксоэтилбутирата
В 12-литровую трехгорлую кругодонную колбу, снабженную механической мешалкой, термометром (погруженным), выходом газового барботера, заполненным маслом, и трубкой для ввода озона, вводят 2-бутен-1,4-дибутират (1005,0 г, 4,4 моль) и метанол (5 л). Включают генератор озона Ozonia CFS-2, 1200 Вт, 1 атмосфера кислорода, со скоростью потока 1 м3/час и начинают перемешивание при охлаждении смеси до -20°С в бане лед/метанол. Раствор барботируют озоном. В ходе процесса добавления озона температуру смеси поднимают до -10°С. Через два часа анализ реакционной смеси методом тонкослойной хроматографии (пластинки с силикагелем; Analtech №02521, проводят элюацию смесью гексан/EtOAc, 9:1 и визуализацию с использованием красителя РМА) указывает на полное потребление исходного материала. Через перемешанную реакционную смесь пропускают азот в течение 15 минут и ее вновь охлаждают до -20°С. Порциями по 17 г в течение 1,5 часов добавляют тиомочевину (170 г, 2,23 моль, Johnson Matthey I0B16). Температуру реакционной смеси повышают до 0°С. Через час после завершения добавления тиомочевины проводят анализ методами тонкослойной хроматографии и 1H ЯМР, которые указывают на полное отсутствие озонида. Смесь вновь охлаждают до температуры -20°С и фильтруют. Фильтрат выпаривают с получением 1,5 кг полуацеталя метил-2-оксоэтилбутирата (97% выход) в виде бледно-желтого масла. Спектр ЯМР соответствует спектру стандартного вещества.
Пример 3. Получение 2-бутирилоксиметил-1,3-оксатиолан-5-она
В 72-литровую круглодонную колбу, снабженную механической мешалкой, термометром (погруженным), входом для азота, капельной воронкой с уравновешенным давлением и дистилляционной насадкой, вводят толуол (31 л, Fisher) и полуацеталь метил-2-оксоэтилбутирата (10 кг, фактически 9,3 кг с учетом остаточного МеОН). Исходный материал представляет собой фактически смесь ацеталя, полуацеталя, димера и тримера. Начинают перемешивание и в течение двух часов добавляют по каплям через капельную воронку меркаптоуксусную кислоту (4,5 л, 64,7 моль). Температуру реакционной смеси повышают до 28°С в ходе процесса добавления. Проводят анализ реакционной смеси методом тонкослойной хроматографии (пластинки с силикагелем; Analtech №02521, проводят элюацию смесью гексан/EtOAc, 7:3 и визуализацию с использованием красителя РМА), который показывает полное потребление исходного материала после завершения добавления. Далее смесь нагревают до 85°С (внутренняя температура). Дистиллят (5 л смеси толуола и водного метанола) собирают при 75°С (температура насадки). Анализ реакционной смеси методом тонкослойной хроматографии (пластинки с силикагелем; Analtech №02521, проводят элюацию смесью гексан/EtOAc, 7:3 и визуализацию с использованием красителя РМА) показывает, что реакция закончилась после 8 часов нагревания. Далее реакционной смеси дают охладиться до комнатной температуры и медленно нагнетают ее в 100-литровый реактор, содержащий 16 л перемешанного насыщенного водного раствора бикарбоната калия. Смесь перемешивают в течение 20 минут, после чего прекращают перемешивание и позволяют слоям разделиться. Органический слой промывают 6 л насыщенного водного раствора хлорида натрия и выпаривают досуха. Неочищенный продукт пропускают через 2-дюймовый пленочный дистиллятор (Pope Scientific) (температура колонки 90°С, разрежение воздуха 0,5 мм, скорость примерно 0,5 кг в час). Низкокипящая примесь остается в колбе с дистиллятом, тогда как продукт собирают в нижней колбе. Выход составляет 5,8 кг (53,8%). Полученный материал характеризуется 92% чистотой по данным ГХ анализа (колонка НР-1 с метилсиликоновой смолой (Methyl Silicone Gum Column), в качестве газа-носителя используют азот со скоростью течения 50 мл/мин, пламенно-ионизационный детектор работает в следующем режиме: 280°С, выдерживают в течение 1 минуты 65°С, затем температуру поднимают со скоростью 12,5°С/мин до 250°С и выдерживают эту температуру в течение 1 минуты, инъецируемый объем составляет 1-2 мкл раствора EtOAc). Спектр ЯМР соответствует спектру стандартного вещества.
Пример 4: Получение 5-ацетокси-2-бутирилоксиметил-1,3-оксатиолана
В 50-литровую четырехгорлую круглодонную колбу, снабженную внешней механической мешалкой, двумя N2-барботерами и термопарой/карманом для термопары, вводят безводный ТГФ (4,1 л, Aldrich). К указанной смеси порциями по 100 г медленно добавляют шарики гидрида алюминия-лития (334 г, 8,8 моль; Aldrich lot # 04414KR). Полученную взвесь дальше разбавляют дополнительным количеством ТГФ (4,1 л) и перемешивают в течение 15 часов. После добавления температуру сначала поднимают до 37°С и затем постепенно охлаждают до 22°С. Полученную серую смесь охлаждают до -5°С с использованием бани лед/МеОН. Пробку заменяют 5-литровой капельной воронкой с уравновешенным давлением и вводят смесь трет-бутанола (2,0 кг, 2,6 л, 27,6 моль) и ТГФ (600 мл). Указанную смесь медленно добавляют к реакционной смеси в течение 2,5 часов. Температуру реакции повышают до 15,9°С в течение всего периода добавления. Удаляют охлаждающую баню и заменяют ее теплой водяной баней, повышая температуру реакции до 33°С. Указанную температуру поддерживают в течение 1,5 часов до прекращения выделения газа. Реакционную смесь охлаждают до -6°С с использованием бани лед/МеОН. Через капельную воронку вводят смесь 2-бутирилоксиметил-1,3-оксатиолан-5-она [1410,6 г, 6,9 моль и ТГФ (350 мл)]. Указанную смесь медленно добавляют к реакционной смеси, поддерживая внутреннюю температуру ниже 5°С. Реакционную смесь перемешивают в течение 1,5 часов, после чего аликвоту реакционной смеси (5 капель) гасят уксусным ангидридом/4-диметиламинопиридином и разбавляют этилацетатом (примерно 1 мл). Проводят ГХ анализ аликвоты из смеси (колонка НР-1 с метилсиликоновой смолой (Methyl Silicone Gum), в качестве газа-носителя используют азот со скоростью тока 50 мл/мин, пламенно-ионизационный детектор работает в следующем режиме: 280°С, 65°С выдерживают в течение 1 минуты и затем повышают температуру со скоростью 12,5°С/мин до 250°С и выдерживают эту температуру в течение 1 минуты, инъецируемый объем составляет 1 мкл гашеной реакционной смеси), который показывает отсутствие исходного лактона (RT=7,4 мин). В охлаждающую баню лед/МеОН вводят свежую смесь и охлаждают все до -9,0°С. К полученной реакционной смеси зеленоватого цвета добавляют в виде одной порции 4-диметиламинопиридин (42 г, 0,35 моль). Через капельную воронку вводят порциями уксусный ангидрид (7065,5 г, 6,5 л, 69,0 моль). Указанную смесь медленно добавляют к реакционной смеси в течение 1,5 часов, поддерживая температуру ниже 0°С. Полученную реакционную смесь зеленоватого цвета оставляют перемешиваться в течение 13 часов, постепенно нагревая ее до 19°С. Проводят анализ методом ГХ (НР-1 колонка с метилсиликоновой смолой (Methyl Silicone Gum), скорость поступления газа-носителя азота составляет 50 мл/мин, пламенно-ионизационный детектор работает в следующем режиме: 280°С, 65°С выдерживают в течение 1 минуты, затем температуру поднимают со скоростью 12,5°С/мин до 250°С и выдерживают эту температуру в течение 1 минуты, инъецируемый объем составляет 1-2 мкл реакционной смеси), который показывает завершение реакции (образование 2-х новых пиков со значениями RT=8,4 мин и 8,6 мин).
Коричнево-оранжевую реакционную смесь разбавляют этилацетатом (13 л). Половину объема реакционной смеси фильтруют через слой целита (7,5 см толщиной, с использованием 18-дюймовой (45,72 см) воронки с верхней пластиной). Фильтрование идет чрезвычайно медленно. Ко второй половине реакционной смеси добавляют целит (1,5 кг). Указанную смесь перемешивают в течение четырех часов и затем фильтруют через слой целита в рамках указанной выше процедуры. Фильтрование проходит равномерно. Объединенные фильтраты переносят в колбу с каплевидным дном на 72 л, снабженную внешней механической мешалкой. В нее добавляют насыщенный водный раствор бикарбоната натрия (20 л). Полученную двухфазную смесь перемешивают в течение одного часа, после чего слои разделяют и органический слой промывают дополнительным количеством насыщенного водного раствора бикарбоната натрия (10 л) и затем насыщенным водным раствором хлорида натрия (20 л). Слои разделяют и органический слой высушивают над безводным сульфатом магния (3,0 кг) с использованием легкой мешалки для возбуждения суспензии. Фильтрованием в вакууме удаляют сульфат магния и полученный фильтрат выпаривают в вакууме (водяная баня с температурой 35°С), что приводит к образованию жидкости красного цвета. Полученный материал далее концентрируют с использованием насоса под высоким вакуумом (23 ммНg; 40°С) в течение 1,5 часов с выходом неочищенного 5-ацетокси-бутирилоксиметил-1,3-оксатиолана в виде красного масла (1483,0 г, 87% выход).
Порцию в 10 г неочищенного 5-ацетокси-бутирилоксиметил-1,3-оксатиолана растворяют в гексане (100 мл, 10 объемов) и энергично перемешивают до тех пор, пока на дне колбы останется небольшая часть красного масла. К указанной перемешанной смеси добавляют силикагель (2 г) и смесь перемешивают еще в течение 10 минут. Образовавшуюся взвесь фильтруют через слой целита с получением бледно-желтого фильтрата. Выпаривание растворителя в вакууме дает 5-ацетокси-бутирилоксиметил-1,3-оксатиолан в виде желтого масла (7,7 г, 77% восстановление). Поскольку имевшиеся основные примеси при ТСХ были удалены, ГХ анализ не выявил изменений.
Пример 5: Конденсация 5-ацетокси-бутирилоксиметил-1,3-оксатиолана с 5-фторцитозином с использованием иодтриметилсилана в качестве кислоты Льюиса
В 3-литровую трехгорлую круглодонную колбу, снабженную механической мешалкой, пробкой и обратным холодильником с водным охлаждением, соединенным с устройством для барботирования азотом, добавляют 5-фторцитозин (51,6 г, 0,40 моль), гексаметилдисилазан (665 мл, 3,10 моль) и сульфат аммония (2,0 г). Полученную взвесь нагревают при температуре кипения с обратным холодильником в течение 2,5 часов, после чего на внутренней стенке холодильника образуется белое твердое вещество. Полученный желтый раствор оставляют охлаждаться до комнатной температуры, после чего в реакционном растворе образуется белое твердое вещество. Избыток гексаметилдисилазана удаляют при пониженном давлении при сохранении инертной атмосферы. К указанному белому твердому веществу добавляют метиленхлорид (890 мл), что приводит к образованию прозрачного желтого раствора. Реакционный сосуд снабжен термопарой/карманом для термопары, колба Кляйзена снабжена капельной воронкой, уравновешивающей давление, и барботером азота. Реакционный раствор охлаждают до температуры -5°С в бане лед-метанол, после чего раствор ацетата оксатиолана (175,6 г (62% чистоты по ГХ), 0,41 моль) в метиленхлориде (300 мл) переносят порциями в капельную воронку и по каплям в течение 45 минут добавляют к реакционной смеси. Температуру реакционного раствора поддерживают от -5°С до 0°С. После добавления капельную воронку промывают 100 мл метиленхлорида и промывную жидкость добавляют к реакционной смеси. Раствор иодтриметилсилана (89,0 мл, 0,62 моль) в метиленхлориде (150 мл) переносят в капельную воронку и затем добавляют к реакционной смеси в течение 45 минут, поддерживая внутреннюю температуру смеси в интервале от -5°С до 0°С. В начале добавления отмечается некоторое образование белой дымки, которая к концу добавления быстро рассеивается. Полученную реакционную смесь оставляют нагреваться до комнатной температуры с перемешиванием в течение ночи. Далее реакционную смесь осторожно гасят добавлением насыщенного водного раствора бикарбоната натрия и расслаивают ее. Органический слой промывают солевым раствором и концентрируют при пониженном давлении с получением 228 г желто-коричневого полутвердого вещества. Анализ методом ВЭЖХ показывает, что смесь состоит из α- и β-аномеров в соотношении примерно 1:1. Часть указанного материала повторно кристаллизуют из толуола с получением чистых разделенных α- и β-аномеров.
Пример 6: Удаление бутиратной защищающей группы
Образец 8,0 г (25 ммоль) бутиратного эфира (SA.494.89.1) растворяют в 160 мл метанола, начинают энергичное перемешивание и раствор погружают в баню лед/вода. Через 10 минут указанный раствор обрабатывают 6,4 г сильно щелочной анионообменной (ОН-) смолы Дауэкс SBR (DOWEX SBR) (Sigma cat#I-9880, p.1803). После перемешивания в течение 3 часов удаляют баню и перемешивание продолжают до тех пор, пока анализ методом ТСХ не покажет полное потребление исходного материала. Затем смесь разбавляют 100 мл метанола и фильтруют. Смолу промывают 100 мл метанола, и объединенный раствор концентрируют с получением бледно-желтого твердого вещества. Твердое вещество растирают с 20 мл холодного этилацетата и полученное твердое вещество высушивают с образованием 5,0 г (81%) 9/152-15 в виде желтовато-белого твердого вещества.
Следует отметить, что смола перед использованием должна быть тщательно промыта метанолом и затем высушена. Хорошей системой ТСХ для такой реакционной смеси является 15% метанол/85% хлороформ.
Альтернативно бутиратный эфир может быть удален обработкой эфира первичным или вторичным амином в спиртовом растворе. Предпочтительными аминами являются аммиак и бутиламин, а предпочтительным растворителем является метанол.
Пример 7: Синтез (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата (25) и (5-ацетилокси-1,3-оксатиолан-2-ил)метилбутаноата (26) из (2,2-диметил-1,3-диоксолан-4-ил)метанола
Синтез (2,2-диметил-1,3-диоксолан-4-ил)метил бутаноата (22)

К хорошо перемешанному раствору солкеталя (21, 62,6 мл, 500 ммоль), Et3N (83,6 мл, 600 ммоль) и ДМАП (5 г, 40,9 ммоль) в трет-бутилметиловом эфире (1 л) при 0°С добавляют по каплям в течение 75 минут н-бутирилхлорид (52,4 мл, 500 ммоль). Смесь перемешивают еще в течение часа при 0°С и затем при комнатной температуре в течение 5 часов. Далее смесь разбавляют AcOEt (1 л), промывают водой (1 л), высушивают (MgSO4), фильтруют и выпаривают с получением 22 (104,6 г, 500 ммоль, 100%) в виде масла. Материал используют на следующей стадии без дополнительной очистки.
Синтез 2,3-дигидроксипропилбутаноата (23)
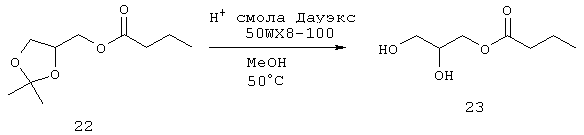
Раствор 22 (50,6 г, 250 ммоль) и смолу Дауэкс 50W Х8-100 Н+ (Dowex 50W Х8-100 Н+) (76,5 г) в МеОН (500 мл) нагревают при 50°С в течение 2 часов, охлаждают до комнатной температуры, фильтруют и смолу промывают МеОН (1×200 мл). Метанольные фракции объединяют и концентрируют в вакууме. Неочищенный продукт пропускают через силикагелевый слой с использованием в качестве элюента смеси этилацетат/гексаны (1:1). Фракции, содержащие продукт, объединяют и концентрируют в вакууме с получением 23 (32,8 г, 200 ммоль, 81%) в виде масла. Полученный материал используют на следующей стадии без дальнейшей очистки.
Синтез 2-оксоэтилбутаноата (24)
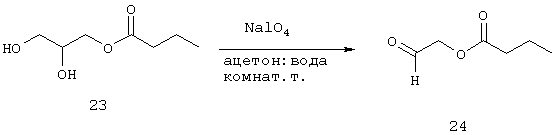
Готовят раствор периодата натрия (89,4 г, 418 ммоль) в дистиллированной воде (450 мл) при нагревании смеси при 45°С в течение примерно 20 минут. Указанный раствор добавляют по каплям в течение 60 минут к раствору диола 23 (30,8 г, 190 ммоль) в ацетоне (225 мл). По завершении добавления смесь перемешивают еще в течение 2 часов при комнатной температуре. Ацетон удаляют с помощью роторного испарителя (температура бани не должна превышать 35°С). Далее реакционную смесь разбавляют водой (250 мл) и водный слой экстрагируют AcOEt (3×250 мл). Органические фракции объединяют, промывают водой (250 мл), высушивают (МgSO4), фильтруют и выпаривают (температура бани не должна превышать 35°С) с получением 24 (20,5 г, 157 ммоль, 83%) в виде масла. Полученный материал используют на следующей стадии без дальнейшей очистки.
Синтез (5-оксо-1,3-оксатиолан-2-ил) метилбутаноата (25)
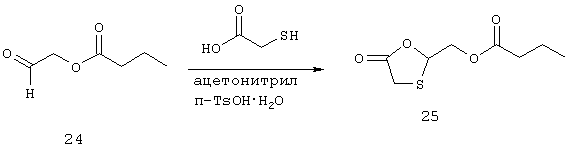
Раствор 24 (3,90 г, 0,030 моль), меркаптоуксусной кислоты (3,32 г, 0,036 моль) и п-TsOH·Н2О (0,28 г, 1,5 ммоль) в ацетонитриле (600 мл) нагревают при температуре кипения с обратным холодильником в течение 3,5 часов. Во время кипения с обратным холодильником с помощью ловушки Дина-Старка отбирают 4 порции по 25 мл каждая (для удаления водно-ацетонитрильного азеотропа). Анализ реакционного раствора методом ТСХ (гексан : AcOEt, 6:1) показывает наличие одного основного нового компонента и отсутствие непрореагировавшего альдегида (визуализацию проводят с помощью красителей РМА и 2,4-ДНФ). Реакционный раствор оставляют перемешиваться при комнатной температуре в течение 16 часов и затем выпаривают досуха. Остаток распределяют между концентрированной NaHCO3 (50 мл) и AcOEt (75 мл); водную часть экстрагируют дополнительным количеством AcOEt (2×75 мл). Органические фракции объединяют, высушивают (МgSO4), фильтруют и концентрируют в вакууме. Неочищенный материал (6 г) очищают флэш-хроматографией (125 г силикагеля с использованием 20% этилацетата в гексане). Получают соединение 25 (3,27 г, 16 ммоль, 53%) в виде масла. Анализ методом ТСХ (3:1, гексан : AcOEt) выявляет одно пятно с Rf=0,41; Анализ методом 1H-ЯМР (CDCl3) показывает, что спектр продукта соответствует структуре; масс-спектральный анализ (FAB) - m/z=205,1 (М+1).
Синтез (5-ацетилокси-1,3-оксатиолан-2-ил) метилбутаноата (26)
 К раствору 25 (0,50 г, 2,5 ммоль) в безводном ТГФ (15 мл) при температуре от -5°С до -10°С с помощью шприц-насоса в течение 2 часов добавляют раствор 1,0 М три-т-бутоксигидрида алюминия-лития в ТГФ (2,7 мл), поддерживая температуру от -5°С до - 10°С. После завершения добавления раствор выдерживают при 3°С в течение 18 часов и затем нагревают до комнатной температуры. Добавляют ДМАП (1,7 ммоль, 0,20 г) и уксусный ангидрид (25,0 ммоль, 2,4 мл) и полученный оранжевый раствор перемешивают при температуре окружающей среды в течение 3 часов, после чего добавляют концентрированную NaHCO3 (25 мл). После перемешивания в течение 1 часа разделяют фазы и водную фазу экстрагируют дополнительными порциями AcOEt. Органические фракции объединяют, высушивают (MgSO4), фильтруют и выпаривают с получением неочищенного продукта (0,77 г). После флэш-хроматографии (20 г силикагеля с использованием 20% этилацетата в гексане) выделяют в виде масла соединение 26 (0,50 г, 2,0 ммоль, 80%): ТСХ (25% этилацетат: гексан) показывает наличие одного пятна с Rf=0,51; 1H-ЯMP (CDCl3) подтверждает соответствие стандарту по структуре.
К раствору 25 (0,50 г, 2,5 ммоль) в безводном ТГФ (15 мл) при температуре от -5°С до -10°С с помощью шприц-насоса в течение 2 часов добавляют раствор 1,0 М три-т-бутоксигидрида алюминия-лития в ТГФ (2,7 мл), поддерживая температуру от -5°С до - 10°С. После завершения добавления раствор выдерживают при 3°С в течение 18 часов и затем нагревают до комнатной температуры. Добавляют ДМАП (1,7 ммоль, 0,20 г) и уксусный ангидрид (25,0 ммоль, 2,4 мл) и полученный оранжевый раствор перемешивают при температуре окружающей среды в течение 3 часов, после чего добавляют концентрированную NaHCO3 (25 мл). После перемешивания в течение 1 часа разделяют фазы и водную фазу экстрагируют дополнительными порциями AcOEt. Органические фракции объединяют, высушивают (MgSO4), фильтруют и выпаривают с получением неочищенного продукта (0,77 г). После флэш-хроматографии (20 г силикагеля с использованием 20% этилацетата в гексане) выделяют в виде масла соединение 26 (0,50 г, 2,0 ммоль, 80%): ТСХ (25% этилацетат: гексан) показывает наличие одного пятна с Rf=0,51; 1H-ЯMP (CDCl3) подтверждает соответствие стандарту по структуре.
Пример 8: Синтез (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата (25) из (2,2-диэтоксиэтанола) (27)
Синтез (2,2-диэтоксиэтилбутаноата) (28)
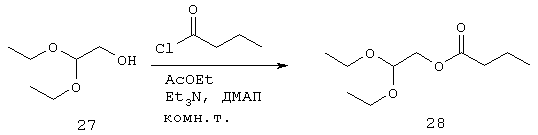
К хорошо перемешанному раствору соединения 27 (Lancaster 6282, 13,4 г, 100 ммоль), ДМАП (61 мг, 0,5 ммоль) и EtaN (16 мл, 11,64 г, 115 ммоль) в EtOAc (50 мл) при температуре 0°С медленно добавляют н-бутирилхлорид (10,90 мл, 11,19 г, 105 ммоль). После перемешивания в течение 1 часа при комнатной температуре реакционную смесь разбавляют большим количеством EtOAc (50 мл) и последовательно промывают концентрированным NаНСО3 (2×100 мл) и солевым раствором (2×100 мл), высушивают, фильтруют и выпаривают с получением 28 (21,5 г, 100 ммоль, 100%) в виде желтой жидкости, которую используют на следующей стадии без дальнейшей очистки.
Синтез (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата (25)

Хорошо перемешанный раствор 28 (6,13 г, 30 ммоль), меркаптоуксусной кислоты (4,14 г, 3,13 мл, 45 ммоль) и п-TsOH·Н2О (60 мг, 0,31 ммоль) в сухом толуоле нагревают при температуре кипения с обратным холодильником в течение 2 часов. Растворитель удаляют с помощью ловушки Дина-Старка и добавляют свежий сухой толуол. После охлаждения до комнатной температуры реакционную смесь разбавляют AcOEt (50 мл) и последовательно промывают концентрированным NaHCO3 (2×100 мл) и солевым раствором (2×100 мл), высушивают, фильтруют и выпаривают с получением 25 (5,2 г, 25,5 ммоль, 85%) в виде желтой жидкости, которую используют на следующей стадии без дальнейшей очистки.
Пример 9: Синтез (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата (25) и (5-ацетилокси-1,3-оксатиолан-2-ил)метилбутаноата (26) из (2,2-диэтоксиэтанола) (27) через 2,2-диэтоксиэтилбутаноат (28) и 2-оксоэтилбутаноат (24)
Синтез 2-оксоэтилбутаноата (24)
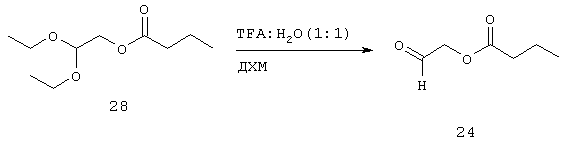
Хорошо перемешанный раствор 28 (8,16 г, 40 ммоль) в ДХМ (200 мл) обрабатывают при комнатной температуре ТФУ (44,4 г, 30 мл, 390 ммоль) и водой (7,2 г, 7,2 мл, 400 ммоль). После перемешивания при комнатной температуре в течение 2 часов раствор выпаривают при 35°С. Затем его перегоняют несколько раз вместе с гексаном для удаления следов ТФУ. Соединение 24 (5,2 г, 40 ммоль, 100%) получают в виде бесцветной жидкости, которую используют на следующей стадии без дальнейшей очистки.
Синтез (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата (25)
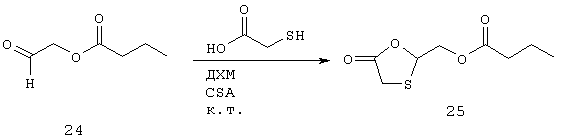
К хорошо перемешанной суспензии 24 (1,3 г, 10 ммоль) и CSA (116 мг, 0,50 ммоль) в сухом ДХМ (10 мл) медленно добавляют раствор меркаптоуксусной кислоты (2,76 г, 2,08 мл, 30 ммоль) в сухом ДХМ (5 мл). Реакционную смесь оставляют при комнатной температуре на 16 часов при перемешивании. Затем реакционную смесь разбавляют ДХМ (20 мл) и последовательно промывают концентрированным NаНСО3 (3×30 мл) и солевым раствором (2×30 мл), высушивают, фильтруют и выпаривают с получением 25 (0,9 г, 4,4 ммоль, 44%) в виде неокрашенного сиропа.
Синтез (5-ацетилокси-1,3-оксатиолан-2-ил)метилбутаноата (26)
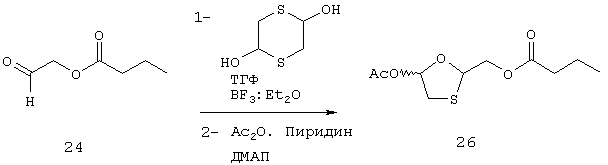
К хорошо перемешанному раствору 24 (2,6 г, 20 ммоль) и 1,4-дитиан-2,5-диола (1,68 г, 11 ммоль) в сухом ТГФ (10 мл) добавляют BF3:Et2O (312 мг, 278 мл, 2,2 ммоль). Смесь перемешивают в течение 16 часов при комнатной температуре. Твердый материал удаляют фильтрованием и к оставшемуся раствору добавляют сухой пиридин (2,3 г, 2,4 мл, 29 ммоль), ДМАП (18 мг, 0,15 ммоль) и затем Ас2O (30 г, 2,77 мл, 29 ммоль). Раствор перемешивают в течение 16 часов при комнатной температуре. Далее реакцию гасят добавлением 8% НСl и экстрагируют AcOEt. Отделяют органическую фазу и промывают ее последовательно 8% НСl, солевым раствором, концентрированным NaHCO3 и солевым раствором, высушивают, фильтруют и выпаривают с получением 26 (3,5 г, 14 ммоль, 70%, 60% чистоты) в виде желтоватого сиропа.
Пример 10: Синтез (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата (25) и (5-ацетилокси-1,3-оксатиолан-2-ил)метилбутаноата (26) из 1,2-диэтанола (29)
Синтез 2-гидроксиэтилбутаноата (30)
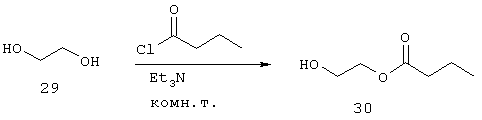
К хорошо перемешанному раствору 29 (834 г, 750 мл, 13,5 моль) и Et3N (116 г, 160 мл, 1,15 моль) медленно добавляют при 0°С н-бутирилхлорид (122 г, 120 мл, 1,15 моль). Реакционную смесь перемешивают в течение 16 часов при комнатной температуре.
Далее раствор разбавляют солевым раствором (1,5 л) и перемешивают еще в течение часа. Затем его экстрагируют гептаном (3×700 мл) для удаления диэфира. Водный слой экстрагируют EtOAc (3×600 мл). Объединенную органическую фазу промывают водой для удаления остаточных количеств этиленгликоля (29), высушивают, фильтруют и выпаривают с получением соединения 30 (39,7 г, 0,3 моль, 26%).
Синтез 2-оксоэтилбутаноата (24)
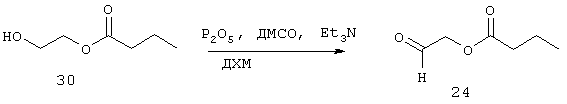
К хорошо перемешанной суспензии P2O5 (42,53 г, 150 ммоль) в сухом ДХМ (100 мл) при температуре 0°С медленно добавляют 30 (11,0 г, 83 ммоль) и затем ДМСО (13 г, 11,8 мл, 166 ммоль). После перемешивания в течение 1 часа при температуре 0°С ледяную баню убирают и смесь далее перемешивают при комнатной температуре в течение 1,5 часов. Затем ее охлаждают до 0°С и медленно добавляют Et3N (42 г, 58 мл, 416 ммоль).
Реакционную смесь оставляют при перемешивании на 6 часов при комнатной температуре. После этого реакцию гасят добавлением при 0°С 1,0 М НСl (60 мл) и оставляют все перемешиваться на 30 минут при 0°С. Органический слой промывают водой (2×250 мл), высушивают, фильтруют и выпаривают с получением 24 (6,60 г, 51 ммоль, 61%) в виде желтой жидкости, которую используют на следующей стадии без дальнейшей очистки.
Синтез (5-оксо-1,3-оксатиолан-2-ил)метилбутаноата (25)
К хорошо перемешанной суспензии 24 (1,3 г, 10 ммоль) и CSA (116 мг, 0,50 ммоль) в сухом ДХМ (10 мл) медленно добавляют раствор меркаптоуксусной кислоты (2,76 г, 2,08 мл, 30 ммоль) в сухом ДХМ (5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов.
Далее реакционную смесь разбавляют ДХМ (20 мл) и последовательно промывают концентрированным NaHCO3 (3×30 мл) и солевым раствором (2×30 мл), высушивают, фильтруют и выпаривают с получением 25 (1,4 г, 6,8 ммоль, 68%) в виде желтого сиропа.
Синтез (5-ацетилокси-1,3-оксатиолан-2-ил) метилбутаноата (26)

К хорошо перемешанному раствору 24 (2,6 г, 20 ммоль) и 1,4-дитиан-2,5-диола (1,68 г, 11 ммоль) в сухом ТГФ (10 мл) добавляют BF3:Et2O (312 мг, 278 мкл, 2,2 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Твердый материал удаляют фильтрованием и к оставшемуся раствору добавляют сухой пиридин (2,3 г, 2,4 мл, 29 ммоль), ДМАП (18 мг, 0,15 ммоль) и затем АС2O (30 г, 2,77 мл, 29 ммоль). Раствор перемешивают при комнатной температуре в течение ночи. Далее реакцию гасят добавлением 8% НСl, экстрагируют AcOEt. Отделяют органическую фазу и последовательно промывают 8% НСl, солевым раствором, концентрированным NаНСО3 и солевым раствором, высушивают, фильтруют и выпаривают с получением 26 (4,75 г, 19 ммоль, 95%, 95% чистоты) в виде желтоватого сиропа.
III. Связывание 1,3-оксатиолана при наличии защищенного основания
Пример 11: Связывание 1,3-оксатиолана при наличии защищенного основания с ТiСl3(OiРr)
Защищенный ацетат (150 мг, 0,604 ммоль, 1 экв.) растворяют в 1,5 мл безводного дихлорметана в атмосфере аргона. В другом контейнере в атмосфере аргона растворяют в 1,5 мл безводного дихлорметана бис-силилированный цитозин (154 мг, 0,604 ммоль, 1 экв.) и все перемешивают с одним эквивалентом свежеприготовленного ТiСl3(OiРr) (полученного из 0,75 экв. TiCl4 в виде 1М раствора в дихлорметане и 0,25 экв. чистого Ti(OiPr), и оба получены от компании Олдрич (Aldrich)). Раствор комплекса основания и ТiСl3(OiРr) добавляют по каплям к ацетату и полученный слегка желтый прозрачный раствор оставляют перемешиваться при комнатной температуре в течение примерно 20 минут, после чего медленно добавляют 0,6 мл TiCl4 (1 М раствор в дихлорметане, от компании Олдрич (Aldrich)). Полученный красный раствор оставляют перемешиваться при комнатной температуре в течение примерно 2 часов, после чего добавляют 1 мл гидроксида аммония. Через 30 минут смесь фильтруют через силикагель с использованием в качестве элюентов смеси гексана и этилацетата в соотношении 4:1 и смеси этилацетата и этанола в соотношении 9:1 с получением белой пены, которая после, как показал анализ методом ядерно-магнитного резонанса, соответствует в основном защищенному нуклеозидному аналогу 3ТС. В альтернативном варианте на стадии связывания могут использоваться другие кислоты Льюиса, такие как триметилсилилтрифлат и иодтриметилсилан или их смесь.
Пример 12: Синтез [5-(4-амино-5-фтор-2-оксо-1(2Н)-пиримидинил)-1,3-оксатиолан-2-ил]метилбутаноата (2R/2S, β) [31 (2R/2S, β]

Хлорирование рацемического ацетата: в течение 75 минут раствор 26 (2R/2S) (49,6 г, 0,2 моль) в Сl3СН (0,5 л) при температуре 0°С барботируют газообразным НСl. Гомогенный темно-желтый раствор оставляют перемешиваться в течение 30 минут, после чего добавляют толуол (100 мл) и полученный раствор концентрируют досуха при пониженном давлении при 48°С. Добавление толуола повторяют дважды. Полученное неочищенное масло разбавляют Сl3СН (100 мл) и указанный раствор используют для связывания (см. ниже).
Силилирование 5-фторцитозина: Суспензию 5-фторцитозина (30,96 г, 0,24 моль), сульфата аммония (1 г) и 1,1,1,3,3,3-гексаметилдисилазана (100 мл, 0,48 моль) в Сl3СН (0,5 л) нагревают при температуре кипения с обратным холодильником в течение 4 часов, получая гомогенный раствор. Затем раствор охлаждают до комнатной температуры.
Связывание силилированного 5-фторцитозина с рацемическим хлоридом: К раствору силилированного 5-фторцитозина добавляют раствор рацемического хлорида. Полученный раствор нагревают при температуре кипения с обратным холодильником в течение 3 часов и охлаждают до комнатной температуры. Затем раствор разбавляют EtOAc (300 мл) и добавляют концентрированный NаНСО3 (300 мл). Смесь перемешивают в течение 1 часа при комнатной температуре и разделяют слои. Водный слой экстрагируют один раз ДХМ (100 мл) и объединенные органические слои высушивают (Na2SO4), фильтруют и выпаривают досуха при пониженном давлении. Неочищенный материал хроматографируют на силикагеле с получением желательного материала 31 (2R/2S) (48,8 г, 77%) в виде смеси β:α аномеров в соотношении 3,5:1 (AUC).
Выделение β-аномера: Смесь аномеров в соотношении 3,5:1 (48,8 г) добавляют к EtOAc (290 мл). Суспензию нагревают при температуре кипения с обратным холодильником в течение 10 минут, получая при этом гомогенный раствор. Удаляют масляную баню и в раствор вносят β-аномер (10 мг). Смесь оставляют выстаиваться при комнатной температуре в течение 2 часов. Полученные белые кристаллы собирают фильтрованием, что дает соединение 31 (2R/2S) (25,4 г, 52% восстановление после рекристаллизации) в виде смеси β:α аномеров в соотношении 97:3 (AUC) по данным ВЭЖХ.
Оксоацетаты, отличные от бутирата, такие как бензоат, п-метоксибензоат и п-(т-бутил)-бензоат, связывают с силилированным 5-фторцитозином с использованием описанной выше процедуры с получением соответствующих продуктов в виде смесей β:α аномеров (AUC) в соотношении 2,2:1, 2,2:1 и 2:1 соответственно.
В реакции хлорирования может использоваться любой подходящий органический растворитель, включая толуол, хлороформ, уксусную кислоту, ТГФ, эфиры, бензол и другие обычно применяемые растворители. При этом не наблюдается заметного влияния растворителей на процесс хлорирования и стереоселективность готовых продуктов.
Однако на стереоселективность реакции связывания оксоацетата с силилированным 5-фторцитозином существенное влияние оказывают растворители. Так, коэффициент соотношения β:α аномеров (AUC) при проведении реакции связывания в хлороформе составляет 3,0-5,0:1, тогда как при проведении реакции в толуоле он составляет 2,8:1.
Пример 13: Синтез [5-(4-амино-5-фтор-2-оксо-1(2Н)-пиримидинил)-1,3-оксатиолан-2-ил]метилбутаноата (2R, β/α,) [31 (2R, β/α)]
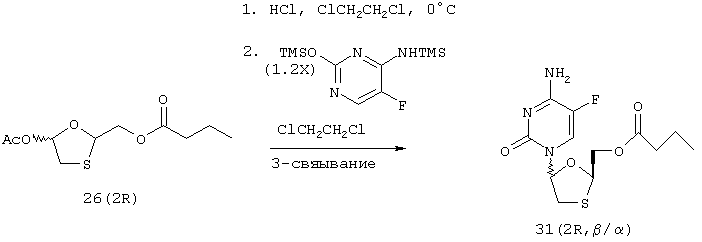
Хлорирование хирального ацетата: К раствору, содержащему хиральный ацетат 26 (2,7 г, 8,0 ммоль) [74% AUC по данным ГХ] в 1,2-дихлоэтане (40 мл), при 0°С добавляют раствор HCl (16 ммоль) в 1,2-дихлорэтане (26 мл). После перемешивания в течение 0,5 часа добавляют еще HCl (8 ммоль) в 1,2 дихлорэтане (13 мл). Указанный раствор перемешивают в течение 1 часа и затем обрабатывают HCl (16 ммоль) в 1,2 дихлорэтане (26 мл) и перемешивают в течение 1 часа. После потребления ацетата раствор энергично дегазируют азотом в течение 0,25 часа и хранят в азоте при 0°С до применения.
Силилирование 5-фторцитозина: Суспензию, включающую 5-фторцитозин (1,55 г, 12,0 ммоль), сульфат аммония (155 мг) и 1,1,1,3,3,3-гексаметилдисилазан (7,6 мл, 36 ммоль) в 1,2-дихлорэтане (80 мл) нагревают при температуре кипения с обратным холодильником в течение 2 часов. (Примерно через 1 час смесь становится бледно-желтым гомогенным раствором). После завершения реакции раствор охлаждают до 0°С и хранят в атмосфере азота до применения.
Связывание силилированного 5-фторцитозина с хиральным хлоридом: Полученный выше раствор хлорида осторожно добавляют в атмосфере азота к силилированному основанию. Полученную мутную смесь нагревают до температуры кипения с обратным холодильником и поддерживают эту температуру в течение 2 часов. Гомогенный бледно-желтый раствор охлаждают до комнатной температуры и гасят добавлением 1/2 объема концентрированного NаНСО3. После расслаивания высушивают органический слой (Na2SO4), фильтруют его и концентрируют при пониженном давлении с получением 2,5 граммов вязкого коричневого масла. Указанное масло очищают с помощью хроматографии на силикагеле, используя 5% EtOH:DCM с получением 31 (2R) (1,9 г, 76%) в виде смеси β:α аномеров в соотношении 60:40. Попытки разделить аномеры дробной кристаллизацией были безуспешны.
Пример 14
Синтез 4-амино-5-фтор-1-(2-гидроксиметил-1,3-оксатиолан-5-ил)-2(1Н)-пиримидинона (2R, β/α) [32 (2R, β/α)]
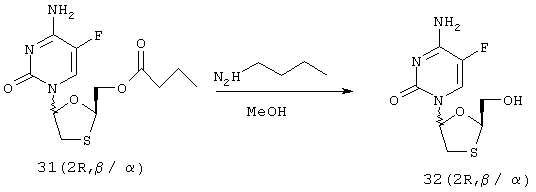
Раствор 31 (2R, β/α) (29,61 г, 93,3 ммоль) и н-бутиламина (30 мл, 304 ммоль) в МеОН (400 мл) перемешивают при комнатной температуре в течение 16 часов. Затем реакционную смесь концентрируют в вакууме. Добавляют EtOAc (3×400 мл) и удаляют его в вакууме. Затем добавляют МеОН (250 мл) и удаляют его в вакууме. Неочищенный продукт растирают с ДХМ (250 мл), фильтруют и промывают дополнительным количеством ДХМ (2×100 мл). Продукт в виде рыжевато-коричневого твердого вещества высушивают в вакуумной печи при 45°С в течение 1 часа с получением 32 (2R) (18 г, 72 ммоль, 77%) в виде смеси β:α аномеров в соотношении 60:40. Полученный материал используется на следующей стадии без дальнейшей очистки. Попытки разделить аномеры дробной кристаллизацией были безуспешны.
Образование α:β (-)-FTC HCl соли [32 (2R, β/α) соли]
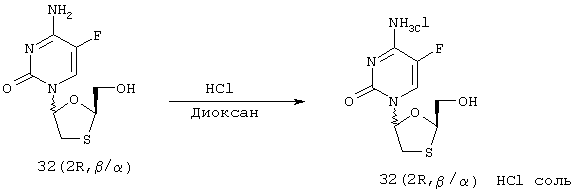
Смесь (-)-FTC [32 (2R, β/α)] (смесь аномеров β:α в соотношении 60:40, 3,0 г) растворяют в метаноле (30 мл), охлаждают до 0°С и обрабатывают 4,0 М раствором HCl в 1,4-диоксане (3,3 мл [1,1х]). Раствор перемешивают в течение 20 минут и затем концентрируют досуха с получением желтовато-белого твердого вещества.
Пример 15: Рекристаллизация α:β (-)-FTC HCl соли [32 (2R, β/α)HCl соли]

Неочищенную (-)-FTC HCl соль (32 (2R, β/α) НСl соль) [смесь β:α аномеров в соотношении 60:40, 3,0 г) растворяют в горячем EtOH (20 мл). Полученный гомогенный раствор оставляют на ночь при комнатной температуре. Собирают образующиеся кристаллы. Получают 0,9 грамм образца чистого β-материала. Маточный раствор концентрируют и указанную смесь повторно кристаллизуют из этанола с получением 0,5 г чистого α-изомера. Объединенные маточные растворы концентрируют и указанный материал повторно кристаллизуют из этанола с получением 0,5 г β-изомера. Объединенный показатель восстановления 1,4 г β-аномера соответствует 78% выходу (теоретический выход желательного β-изомера составлял 1,8 г). Результаты анализа методом хиральной ВЭЖХ показывают, что при образовании соли не происходит рацемизации.
Пример 16
Синтез эмтрицитабина ((-)-FTC или 32 (2R, β))
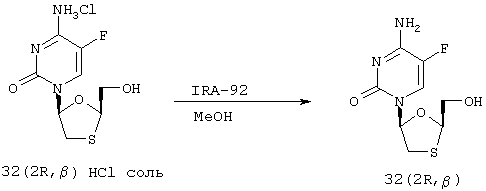
Для восстановления свободного основания хлористоводородную соль (32 (2R, β) HCl соль) отбирают десятью объемами метанола и обрабатывают 3,0 эквивалентами смолы IRA 92. Смесь перемешивают в течение 16 часов и отфильтровывают смолу. Растворитель удаляют под вакуумом с получением свободного основания (32 (2R, β) с 90% выходом. При дальнейшей очистке может быть получена взвесь либо в AcOEt, либо в ТГФ.
Указанное изобретение описано со ссылкой на предпочтительные варианты его реализации. Специалистам в данной области очевидны изменения и усовершенствования, которые могут быть внесены в приведенное выше детальное описание изобретения. При этом все такие вариации и усовершенствования включены в область настоящего изобретения.
Описывается способ получения 1,3-оксатиолановых нуклеозидов или способ получения производных 1,3-оксатиоланил-5-она, которые включают эффективные методы образования 1,3-оксатиоланового кольца с последующей конденсацией 1,3-оксатиолана с пиримидиновым или пуриновым основанием. С использованием приведенных способов рассматриваемые соединения могут быть получены в виде отдельных энантиомеров с высокой селективностью. 2 с. и 25 з.п.ф-лы, 3 ил.

Приоритеты по пунктам и признаками:
| US 5041449 А, 20.08.1991 | |||
| US 5539116 А, 03.07.1996 | |||
| Кулачковый механизм | 1975 |
|
SU513200A2 |
| Schinazi et al | |||
| Antimicrobial Agents and Chemotherapy, november, 1992, p.2423-2431. | |||

