Настоящее изобретение относится к аналогам нуклеозидов и их применению в медицине. Конкретно, настоящее изобретение касается дидезокси-аналогов нуклеозидов, фармацевтических препаратов, содержащих эти вещества, и применения их при лечении вирусных инфекций.
Единственными соединениями, в настоящее время одобренными и разрешенными для лечения состояний, вызванных ВИЧ, являются D-3'-aзидo-3'-дезoкситимидин (АЗТ, зидовудин, BW 509U) и β-D-2',3'-дидезоксиинозин (ддИ, диданозин), который применяют при лечении пациентов, не чувствительных к АЗТ. Кроме того, β-D-2',3'-дидезоксицитидин (ддЦ) разрешен только в сочетании с АЗТ. Вышеупомянутые соединения, полученные из физиологически значимых нуклеозидов, обладают значительными побочными эффектами и лимитирующей дозу токсичностью.
Помимо этого, наблюдается также резистентность к АЗТ, ддЦ и ддИ ( K.J. Connolly и S.M. Hammer, Antimicrob. Adent. Chemother. 1992; 36, 245 - 254).
Соответственно, существует постоянная нужда в соединениях, эффективных в отношении ВИЧ, но обладающих значительно лучшим терапевтическим индексом (т. е., более избирательного действия).
Все вышеупомянутые соединения используются в форме их естественных энантиомеров (D-сахара). Обнаружено, что соответствующие ненатуральные энантиомеры АЗТ (L-АЗТ) и ддИ (β -L-ддИ) неактивны в отношении ВИЧ (J.Wendel и др. J. Org. Chem. , 1991; 56, 3591 - 3594; и M.M. Mansuri и др., Bio Med. Chem. Lett. , 1991; 1, 65 - 68), в то время как ненатуральный энантиомер ддЦ (β L-ддЦ) был неактивен или слабо активен в отношении ВИЧ (M. Okabe и др., J. Org. Chem., 1988; 54, 4780 - 4786 и M.M. Mansur; и др., Bio Med. Chem. Lett. , 1991; 1, 65-68), без упоминания о селективности. Помимо этого, в литературе не было сообщений об активности β -L-ддЦ в отношении вируса гепатита B (HBV).
К нашему удивлению, недавно мы обнаружили, что β -L-ддЦ, ненатуральный энантиомер (-) ддЦ активен в отношении ВИЧ с неожиданно высокой избирательностью.
Помимо этого, мы также обнаружили, что β -L-ддЦ обладает отличной активностью в отношении вируса гепатита B.
Помимо этого, 5-фтор-аналог ддЦ (5F-ддЦ) был описан и испытан в форме его натурального энантиомера (β -D-5F-ддЦ), и было обнаружено, что он активен в отношении ВИЧ (Kim и др., J. Med. Chem., 1987; 30, 862-866). Однако о его активности в отношении вируса гепатита B не сообщалось.
Мы обнаружили, что натуральный энантиомер 5F-ддЦ (β -D-5F-ддЦ) активен в отношении HBV.
Помимо этого, в литературе нет сообщений об активности его соответствующего ненатурального энантиомера (β -L-5F-ддЦ) в отношении ВИЧ или HBV.
Мы также неожиданно обнаружили, что ненатуральный энантиомер 5F-ддЦ (β -L-5F-ддЦ) обладает активностью в отношении ВИЧ и HBV в концентрациях ниже цитотоксических.
Одним из аспектов настоящего изобретения является разработка применения (-) энантиомера ддД (β -L-ддЦ) и его фармацевтически приемлемых производных для лечения ВИЧ-инфекции.
Другим аспектом настоящего изобретения является разработка применения β -L-ддЦ и его фармацевтически приемлемых производных для лечения HBV-инфекции.
Помимо этого, еще одним аспектом настоящего изобретения является разработка применения β -D-5F-ддЦ и его фармацевтически приемлемых производных для лечения HBV-инфекции.
Кроме того, еще одним аспектом настоящего изобретения является разработка применения β -L-5F-ддЦ и его фармацевтически приемлемых производных для лечения ВИЧ-инфекции.
Еще одним аспектом настоящего изобретения является разработка применения β -L-5F-ддЦ и его фармацевтически приемлемых производных для лечения HBV-инфекции.
Эти соединения представлены формулой (I):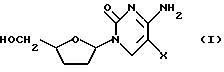
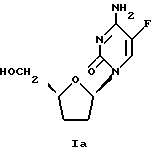
где X представляет водород или фтор. Соединения формулы (I) являются рацемическими смесями двух энантиомеров формул (Ia) и (Ib):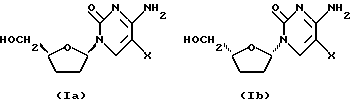
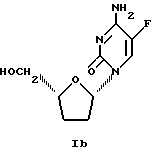
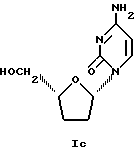
(-) Энантиомер ддЦ имеет абсолютную конфигурацию 1'S на углероде, несущем основание, и 4'R на углероде, несущем группу CH2ОН. Он имеет абсолютную стереохимию соединения формулы (Ib) и химическое название β-L-2', 3'-дидезоксицитидин или (1'S,4'R)-2'3'-дидезоксицитидин (далее упоминается как соединение A).
(+) Энантиомер 5F-ддЦ имеет абсолютную стереохимию соединения формулы (Ia) и химическое название β -D-5-фтор-2',3'-дидезоксицитозин (далее упоминается как соединение B).
(-)-Энантиомер 5F-ддЦ также имеет абсолютную стереохимию соединения формулы (Ib) и химическое название β-L-5-фтор-2',3'-дидезоксицитозин (далее упоминается как соединение C).
Предпочтительно соединения A и C получают не содержащими большого количества соответствующего (+)энантиомера, то есть содержащими не более 5% вес/вес (+)энантиомера, более предпочтительно, не более 2%, в частности, обычно присутствует менее 1% вес/вес.
Предпочтительно соединение B также получают не содержащим большого количества соответствующего (-)энантиомера, то есть содержащим не более 5% вес/вес (-)энантиомера, более предпочтительно, не более 2%, в частности, обычно присутствует менее 1% вес/вес.
Под "фармацевтически приемлемым производным" подразумевается любая фармацевтически приемлемая соль или эфир или соль такого эфира соединения A, B или C или любого другого соединения, которые способны при введении пациенту отщеплять (прямо или опосредованно) соединения A, B или C или метаболит или остаток, обладающий противовирусной активностью.
Специалисты оценят тот факт, что соединения A, B и C могут быть модифицированы по функциональным группам как в основной части, так и в группе гидроксиметила оксатиоланового кольца, для получения их фармацевтически приемлемых производных. Модификация по всем этим функциональным группам также входит в настоящее изобретение. Особый интерес представляют фармацевтически приемлемые производные, полученные путем модификации 2-гидроксиметильной группы при 4'-атоме углерода сахарного кольца.
Предпочтительные сложные эфиры соединений A, B и C включают соединения, в которых водород 2-гидроксиметильной группы замещен ацильной группой R-C(О)-, в которой некарбонильная часть R этого сложного эфира выбирается из водорода, прямого или разветвленного алкила (например, метила, этила, н-пропила, т-бутила, н-бутила), алкоксиалкила (например, метоксиметила), аралкила (например, бензила), арилоксиалкила (например, феноксиметила), арила (например, фенила, необязательно замещенного галогеном, C1-4 алкилом или C1-4 алкокси); сульфонатные сложные эфиры, такие как алкил- или аралкилсульфонил (например, метансульфонил); аминокислотные сложные эфиры (например, L-валил или L-изолейцил) и моно-, ди- или трифосфатные сложные эфиры.
Что касается вышеописанных сложных эфиров, если не указывается иной вариант, любая алкильная часть предпочтительно содержит от 1 до 16 атомов углерода, в частности, от 1 до 4 атомов углерода. Любая арильная часть в этих сложных эфирах предпочтительно включает фенильную группу.
Конкретно, сложные эфиры могут представлять C1-16 алкильный сложный эфир, незамещенный бензиловый сложный эфир или бензиловый эфир, замещенный, по меньшей мере, одним галогеном (бромом, хлором, фтором или йодом), C1-6 алкилом, C1-6 алкокси, нитро- или трифторметильной группами.
Фармацевтически приемлемые соли соединений A, B и C включают соли, полученные с фармацевтически приемлемыми неорганическими или органическими кислотами и основаниями.
Примеры подходящих кислот включают хлористоводородную, бромистоводородную, серную, азотную, перхлорную, фумаровую, малеиновую, фосфорную, гликолевую, молочную, салициловую, янтарную, толуол-п-сульфоновую, винную, уксусную, лимонную, метансульфоновую, муравьиную, бензойную, яблочную, нафталин-2-сульфоновую и бензолсульфоновую кислоты. Другие кислоты, такие как оксалевая, сами по себе будучи фармацевтически неприемлемыми, могут быть полезны в качестве промежуточных реагентов при получении соединений настоящего изобретения и их фармацевтически приемлемых кислых солей.
Соли, полученные с помощью подходящих оснований, включают щелочные металлы (например, натрий), щелочноземельные металлы (например, магний), аммоний и NR+ 4 (где R представляет C1-4 алкил).
В настоящем документе упоминание о соединении настоящего изобретения включает в себя соединения A, B, C и их фармацевтически приемлемые производные.
Соединения настоящего изобретения могут обладать противовирусной активностью сами по себе, либо метаболизироваться до таких соединений. В частности, эти соединения являются эффективными в ингибировании репликации ретровирусов, включая ретровирусы человека, такие как вирус иммунодефицита человека (ВИЧ), являющийся возбудителем СПИДа.
Таким образом, настоящим изобретением предложены соединения A, B, C и их фармацевтически приемлемые производные в качестве активных терапевтических агентов, а именно, противовирусных агентов, например, для лечения инфекций, вызванных ретровирусами, или инфекций, о которых известно, что они вызываются вирусами, обладающими активностью обратной транскриптазы (такими как вирус гепатита B).
Еще одним аспектом настоящего изобретения является создание способа лечения вирусных инфекций, в частности инфекций, вызываемых ретровирусами, такими как ВИЧ, или вирусами, обладающими активностью обратной транскриптазы, такими как HBV, млекопитающих, включая человека, который включает введение эффективного количества соединений A, B или C или их фармацевтически приемлемых производных.
Разработано также применение соединений A, B или C или их фармацевтически приемлемых производных для получения лекарственного средства для лечения вирусных инфекций.
Соединения настоящего изобретения полезны также при лечении HBV или состояний, связанных со СПИДом, таких как комплекс, связанный со СПИДом, прогрессирующая генерализованная лимфаденопатия (ПГЛ), связанные со СПИДом неврологические состояния (такие как деменция или тропический парапарез), ВИЧ-положительные состояния и состояния с положительными анти-ВИЧ антителами, саркома Капоши, тромбоцитопеническая пурпура и ассоциированные условно-патогенные инфекции, например, Pheumocystis carinii.
Соединения настоящего изобретения полезны также для предотвращения прогрессирования до клинического проявления болезни у пациентов, положительных по анти-ВИЧ или анти-HBV антителам или по ВИЧ- или HBV-антигенам, и для профилактики последующего воздействия на них ВИЧ или HBV.
Соединения A, B, C или их фармацевтически приемлемые производные можно применять также в целях предотвращения вирусной контаминации физиологических жидкостей, таких как кровь или сперма in vitro.
Специалисты оценят также тот факт, что терапевтическая широта соединений настоящего изобретения распространяется как на профилактику, так и на лечение установившихся симптомов или инфекций.
Следует также учесть, что количество соединения, которое требуется для лечения, варьирует не только в зависимости от конкретного выбранного соединения, но также и от способа введения, природы состояния, по поводу которого производится лечение, и возраста и состояния пациента и, в конечном итоге, подбирается по усмотрению лечащего врача или ветеринара. Однако обычно доза варьирует приблизительно от 0,1 до 750 мг/кг веса тела в день, предпочтительно, от 0,5 до 60 мг/кг в день, наиболее предпочтительно, от 1 до 20 мг/кг в день.
Желаемая доза может заключаться в разовой дозе или в разделенной дозе, вводимой с подходящими интервалами, например, в виде двух, трех, четырех или более субдоз в день.
Соединения настоящего изобретения удобно вводить в единичных лекарственных формах, содержащих, например, от 10 до 1500 мг, удобнее, от 20 до 1000 мг, наиболее удобно, от 50 до 700 мг активного ингредиента на единицу лекарственной формы.
В идеале следует вводить активный ингредиент таким образом, чтобы пик содержания его в плазме крови составлял от 1 до 75 мкМ, предпочтительно, приблизительно от 2 до 50 мкМ, наиболее предпочтительно, приблизительно от 3 до 30 мкМ. Это достигается, например, путем внутривенной инъекции от 0,1 до 5% раствора активного ингредиента, необязательно, в физиологическом растворе, или путем перорального введения болюса, содержащего приблизительно от 1 до 100 мг активного ингредиента. Желаемые уровни в крови можно поддерживать продолжительной инфузией, составляющей приблизительно от 0,01 до 5 мг/кг в час, или прерывистыми инфузиями, содержащими приблизительно от 0,4 до 15 мг/кг активного ингредиента.
Несмотря на то, что соединение настоящего изобретения можно вводить в виде необработанного химического вещества, предпочтительно представлять активный ингредиент в составе лекарственного препарата.
Таким образом, настоящее изобретение создает фармацевтическую композицию, содержащую соединения A, B или C или их фармацевтически приемлемые производные в сочетании с одним или более фармацевтически приемлемыми носителями и необязательно другими терапевтическими и/или профилактическими ингредиентами. Носитель (носители) должен быть "приемлемым" в том смысле, что он должен быть совместимым с другими ингредиентами композиции и не причинять вреда пациенту.
Фармацевтические композиции включают композиции для перорального, ректального, назального, местного (включая трансбуккальное и сублингвальное), вагинального или парентерального (включая внутримышечное, подкожное и внутривенное) введения или композиции для введения путем ингаляции или инсуффляции. Эти композиции могут изготавливаться в форме дискретных дозовых единиц и любым способом, известным в области фармации. Все эти способы включают смешивание активного ингредиента с жидкими носителями или в конечном счете разделяемыми твердыми носителями или и то и другое, и, если необходимо, придание продукту желаемой формы.
Фармацевтические композиции для перорального приема могут представлять собой дискретные единицы, такие как капсулы, пастилки или таблетки, содержащие определенное количество активного ингредиента; порошки, гранулы, растворы, суспензии или эмульсии. Активный ингредиент может также представлять собой болюс, электуарий или пасту. Таблетки и капсулы для перорального приема могут содержать стандартные вспомогательные вещества, такие как связующие агенты, наполнители, лубриканты, дезинтегрирующие агенты или увлажняющие агенты. Таблетки могут быть покрыты оболочкой согласно стандартным технологиям. Жидкие препараты для перорального приема могут представлять собой водные или масляные суспензии, растворы, эмульсии, сиропы или эликсиры или сухие продукты для последующего растворения в воде или ином подходящем средстве перед употреблением. Такие жидкие препараты могут содержать стандартные добавки, такие как суспендирующие агенты, эмульгирующие агенты, неводные носители (которые могут включать пищевые масла) или консерванты.
Соединения настоящего изобретения могут также изготавливаться в форме композиций для парентерального применения (например, путем инъекции или продолжительной инфузии) и заключаться в ампулы, заранее наполненные шприцы, многодозовые контейнеры или контейнеры для инфузий малыми объемами с добавлением консервантов. Композиции могут принимать форму суспензий, эмульсий в водной или масляной фазе и могут содержать вспомогательные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Активный ингредиент может представлять собой порошок, полученный асептическим выделением стерильного твердого вещества или путем лиофилизации из раствора, для последующего разведения подходящим средством, например стерильной непирогенной водой, перед употреблением.
Для местного применения на коже соединения настоящего изобретения могут помещаться и такие лекарственные формы, как мази, кремы или лосьоны, или представлять собой пластырь. Мази или кремы могут быть приготовлены, например, на водной или масляной основе с добавлением подходящих загустителей и/или желирующих агентов. Лосьоны могут быть приготовлены на водной или масляной основе и содержать один или более эмульгирующих агентов, стабилизирующие агенты, диспергирующие агенты, суспендирующие агенты, загустители или красители.
Композиции для местного применения в полости рта включают таблетки, содержащие активный ингредиент в ароматизированной основе, обычно, сахарозе или акации или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и акация; полоскания, содержащие активный ингредиент в подходящем жидком носителе.
Фармацевтические композиции для ректального применения, в которых носитель является твердым веществом, предпочтительно представляют собой суппозитории. Подходящие носители включают масло какао и другие материалы, хорошо известные специалистам. Суппозитории могут быть приготовлены путем смешивания активного ингредиента с размягченным или расплавленным носителем (носителями), с последующим охлаждением и формованием.
Композиции для вагинального применения могут представлять собой пессарии, тампоны, кремы, гели, пасты, пены или спреи, содержащие помимо активного ингредиента носители, обычно используемые для этих целей.
Для интраназального применения соединения настоящего изобретения можно использовать в виде жидкого спрея или расплавленного порошка или в форме капель.
Капли можно изготавливать на водной или неводной основе, содержащей один или более диспергирующих агентов, солюбилизирующих агентов или суспендирующих агентов. Жидкие спреи удобно помещать в баллончики под давлением.
Для применения путем ингаляции соединения настоящего изобретения удобно помещать в баллончики под давлением, инсуффляторы, ингаляторы и прочие приспособления для аэрозольного распыления. Баллончики под давлением могут содержать подходящий пропеллент, такой как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, двуокись углерода или другой подходящий газ. Дозу можно отмеривать путем установки на баллончике дозирующего клапана.
С другой стороны, для применения путем ингаляции соединения настоящего изобретения могут принимать форму сухого порошка, например, порошковой смеси активного ингредиента и подходящей основы, такой как лактоза или крахмал. Порошок можно выпускать в виде единиц дозы, например, в капсулах или гильзах или, например, в желатиновых или иных упаковках, из которых порошок извлекается с помощью ингалятора или инсуффлятора.
При необходимости можно адаптировать вышеописанные композиции для получения форм с замедленным высвобождением активного ингредиента.
Фармацевтические композиции настоящего изобретения могут содержать также другие активные ингредиенты, такие как антимикробные агенты или консерванты.
Соединения настоящего изобретения можно использовать также в сочетании с другими терапевтическими агентами, например, с другими агентами против инфекции, в частности, соединения настоящего изобретения можно применять вместе с другими противовирусными агентами.
Таким образом, настоящее изобретение предусматривает комбинацию, включающую соединения A, B или C или их фармацевтически приемлемые производные, с другим терапевтически активным агентом, в частности противовирусным агентом.
Подобные комбинации могут быть приготовлены в форме фармацевтических композиций, и, таким образом, такие фармацевтические композиции, включающие вышеописанные комбинации и фармацевтически приемлемый носитель, составляют еще одну особенность настоящего изобретения.
Подходящие терапевтические агенты для использования в таких комбинациях включают ациклические нуклеозиды, такие как ацикловир или ганцикловир, интерфероны, такие как α,β или γ -интерферон, ингибиторы почечной экскреции, такие как пробенецид, ингибиторы транспорта нуклеозидов, такие как дипиридамол, 1,3-оксатиолановые аналоги нуклеозидов, такие как ЗTС (WO, 91/17159), 2', 3' -дидезоксинуклеозиды, такие как АЗТ, 2',3'-дидезоксиаденозин, 2',3'-дидезоксиинозин, 2', 3'-дидезокситимидин, 2',3'-дидезокси-2',3'-дидегидротимидин и 2', 3'-дидезокси-2',3'-дидегидроцитидин, FIAU, иммуномодуляторы, такие как интерлейкин II (ИЛ2) и гранулоцитарно-макрофагальный колониестимулирующий фактор (ГМ-КСФ), эритропоэтин, амплиген, тимомодулин, тимопентин, фоскарнет, рибавирин и ингибиторы связывания ВИЧ с CD4 рецепторами, например, растворимый CD4, фрагменты CD4, CD4 гибридные молекулы, ингибиторы гликозилирования, такие как 2-дезокси-D-глюкоза, кастаноспермин и 1-дезоксинойиримицин.
Отдельные компоненты таких комбинации могут вводиться как последовательно, так и одновременно в составе отдельных или комбинированных фармацевтических композиций.
Когда соединения A, B или C или их фармацевтически приемлемые производные применяются в комбинации с другим терапевтическим агентом, обладающим активностью в отношении этого же вируса, доза каждого соединения может быть той же самой или отличаться от дозы соединения, если оно применяется отдельно. Подходящие дозы без труда определит специалист.
Соединения A, B или C или их фармацевтически приемлемые производные могут быть приготовлены любым способом, известным специалистам для приготовления соединений аналогичной структуры, например, как описано в международной публикации N WO 92/20696, включенной в настоящий документ в качестве ссылки.
Специалистам известно, что в ряде способов желаемую стереохимию соединений A, B или C можно получить, как начиная с оптически чистого исходного материала, так и разделяя рацемическую смесь на подходящей стадии синтеза. Во всех случаях оптически чистый желаемый продукт может быть получен разделением конечного продукта каждой реакции.
Пример 1
Противовирусная активность и цитотоксичность
A) Изучение формазана на МТ-4
Противовирусную активность определяли на клетках МТ-4 путем ингибирования конверсии формазана (Вава и др., (1987) Biochem. Biophys. Res. Commun. , 142, 128 - 134; Mosman (1983) J. Immun. Meth; 65, 55-57).
B) Определение ингибирования образования синцития
Клетки C8166 заражали ВИЧ-1 (штамм RF) в дозе 1 • 10-3 инфекционных единиц на клетку и оставляли при комнатной температуре на 60 минут. После абсорбции клетки отмывали три раза в питательной среде. В каждую лунку 24-луночного планшета добавляли аликвоты по 105 клеток. Лунки содержали серийные разведения тестовых соединений в конечных концентрациях от 50 мкг/мл до 0,05 мкг/мл в среде RPMI® 1640. Необработанные инфицированные клетки и необработанные неинфицированные клетки служили контролем. Планшеты инкубировали при 37oC/5% CO2 в течение 3-4 дней в увлажняемых контейнерах. Клетки ежедневно проверяли на наличие ВИЧ-1-индуцированного образования синцития. Синцитий оценивали количественно в сравнении с необработанным инфицированным контролем и рассчитывали дозу испытуемого соединения, которая требовалась для уменьшения цитопатического эффекта на 50% (ИД50).
C) Цитотоксичность
Цитотоксичность соединений определяли на пяти клеточных линиях CD4: H9, JM, CEM, C8166 и U937.
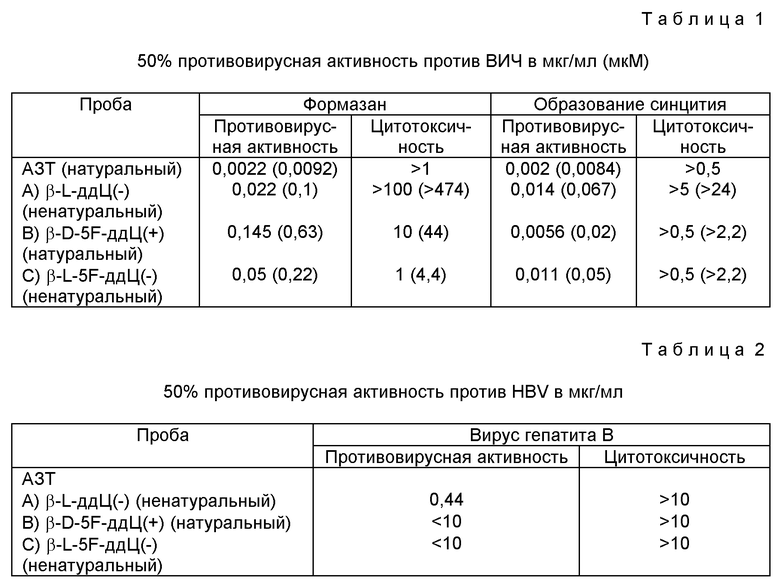
Испытуемые соединения серийно разводили от 100 мкг/мл до 0,3 мкг/мл (конечные концентрации) в 96-луночных микротитрационных планшетах. В каждую лунку планшетов помещали до 3,6 • 104 клеток, включая контроль без лекарства. После инкубации при 37oC в течение 5 дней определяли количество выживших клеток путем забора образца клеточной суспензии и подсчитывали количество клеток, не воспринявших трипановый синий, в гемоцитометре. Результаты представлены в таблице 1.
D) Ингибирование вируса гепатита B человека
Примененная методика подробно описана Korba и др., Antiviral Research 15, 217-228 (1992). Вкратце описывается следующим образом:
Клетки Hep G2, трансфектированные геномной ДНК вируса гепатита B человека (2.2.15 клеток), выращивали на среде RPMI 1640, содержащей 5% сыворотки плода коровы, 2 мМ глутамина и 50 мкМ/мл гентамицина сульфата, и испытывали с помощью стандартной методики на устойчивость к G418. Культуры 2.2.15 клеток выращивали до сливания в 24-луночных планшетах для культур тканей и поддерживали еще 2-3 дня в этом состоянии до начала воздействия лекарствами.
Лекарства растворяли в стерильной воде или стерильном 50% растворе ДМСО в воде до концентраций в 100 раз выше высшей тестовой концентрации. Эти растворы разводили до нужных концентраций культуральной средой.
Культуральную среду до слившихся клеток заменяли за 24 часа до начала воздействия испытуемыми веществами. В течение 10 дней воздействия культуральную среду меняли ежедневно. Спустя 10 дней воздействия культуральную среду собирали и замораживали при -70oC для исследования на ДНК HBV.
Для анализа внеклеточной ДНК HBV образцы по 0,2 мл культуральной среды инкубировали в течение 20 минут при 25oC в 1 М NaOH/IOX SSC (IX SSC составляет 0,15 М NaCl/0,015 цитрат натрия, pH 7,2), а затем помещали на мембраны из нитроцеллюлозы, предварительно увлажненные 20 х SSC. Фильтры затем отполаскивали в 2X SSC и прогревали при 80oC в течение 1 часа под вакуумом.
Фрагмент EcoR1 очищенной ДНК HBV в 3,2 т.п.н. метили [32P] α CTP путем "ник" - трансформации и использовали в качестве зонда для обнаружения ДНК HBV при дот-блоттинге путем ДНК-гибридизации. После отмывания гибридизированный блот высушивали и количественно определяли 32P с помощью бета-сканнера Ambir.
Результаты представлены в таблице 2.
Пример 5
β-L-2',3'-Дидезоксицитидин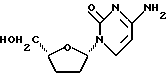
Смесь 1'S-/N-4-ацетилцитозин-1-ил. -4'R-карбоэтокситетрагидрофурана (49 мг, 0,158 ммоль, содержал приблизительно 4% соответствующего 1'R-изомера) и трифторуксусной кислоты (24 мкл, 2 эквивалента) в этаноле (1 мл) нагревали с обратным холодильником в среде аргона в течение 2 часов 40 минут. Полученную смесь, состоящую из 1'S-/цитозин-1-ил/-4'R-карбоэтокситетрагидрофурана и его 1'-эпимера, охлаждали до комнатной температуры и затем разбавляли этанолом (0,5 мл). Затем вводили натрия борогидрид (18 мг, 3 эквивалента), и реакционную смесь перемешивали 1,5 часа. Добавляли еще восстановителя (6 мг) и продолжали перемешивать еще 1 час 20 минут. Реакцию резко прекращали путем добавления 2 капель концентрированного аммонийгидроксида с последующим интенсивным перемешиванием в течение 15 минут. Выпаривали при пониженном давлении растворитель, и полученный неочищенный продукт подвергали колоночной хроматографии (30% метанолэтилацетат), получив в результате 28 мг (84%) указанного в заголовке соединения. 1H ЯМР-спектр этого вещества показал наличие приблизительно 3% соответствующего 1'R-изомера. Этот материал растворяли в минимальном количестве метанола. Добавление к этому раствору диэтилового эфира дало 20 мг (60%) указанного в заголовке соединения в виде кристаллического белого осадка, свободного от 1'R-изомера (1H ЯМР). Указанное в заголовке соединение показало следующие спектральные характеристики: 1H ЯМР (CD3OD): δ 1,60 - 2,00 (м, 3H), 2,25 - 2,43 (м, 1H), 3,59 (дд, 1H, J=4,1 и 12,2 Гц), 3,78 (дд, 1H, J=3,1 и 12,2 Гц), 4,00 - 4,12 (м, 1H), 5,78 (д, 1H, J=7,4 Гц), 5,92 (дд, 1H, J=3,1 и 6,7 Гц), 8,02 (д, 1H, J=7,5 Гц).
Пример 8
β -L-(5-фтор)-2',3'-дидезоксицитидин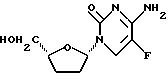
К холодной (0oC) перемешанной суспензии 1'R-(5-фторцитозин-1-ил)-4'R-карбоэтокситетрагидрофурана и 1'S-(5-фторцитозин-1-ил)-4'R-карбоэтокситетрагидрофурана 307 мг, 1,133 ммоль, отношение изомеров в смеси [/1'R,4'R/: /1'S, 4'R/= 4: 1] в 4 мл этанола добавляли борогидрид натрия (86 мг, 2 эквивалента). Полученную смесь перемешивали 5 минут, и ванну охлаждения убирали. Продолжали перемешивание 75 минут при комнатной температуре. Реакцию резко прекращали путем добавления 4 капель концентрированного аммонийгидроксида. После перемешивания смеси в течение 15 минут удаляли растворитель при понижении давления, и полученный неочищенный продукт подвергали колоночной хроматографии (25% метанолэтилацетат), получив в результате 197 мг (76%) ожидаемых 4'-гидроксиметиловых продуктов в виде смеси (4:1). Как было обнаружено (1H ЯМР), одна из уловленных фракций содержала указанное в заголовке соединение с 97%-ной чистотой. Эту фракцию концентрировали и получили 14 мг пены светло-бежевого цвета.
УФ (λмакс): 282,7, 236,4, 206,7 нм (метанол);
[α] D-81o (с 0,7 метанол);
1H ЯМР (CD3OD): δ 1,77 - 1,90 (м, 2H), 1,90 - 2,03 (м, 1H), 2,25 - 2,42 (м, 1H), 3,61 (дд, 1H, J=3,3 и 12,3 Гц), 3,82 (дд, 1H, J=2,8 и 12,3 Гц), 4,06 (м, 1H), 5,87 (м, 1H), 8,32 (д, 1H, J=7,0 Гц).
Пример 9
β-D-(5-фтор)-2',3'-дидезоксицитидин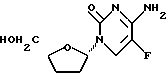
К холодной (0oC) перемешанной суспензии 1'R-(5-фторцитозин-1-ил)-4'S-карбоэтокситетрагидрофурана и 1'S-(5-фторцитозин-1-ил)-4'S-карбоэтокситетрагидрофурана [199 мг, 0,734 ммоль, отношение изомеров в смеси (1'R,4'S): (1'S, 4'S) = 7:1] в 3 мл этанола добавляли борогидрид натрия (56 мг, 2 эквивалента). Полученную смесь перемешивали 5 минут, и ванну охлаждения убирали. Продолжали перемешивание всю ночь (примерно 16 часов) при комнатной температуре. Потом быстро прекращали путем добавления 4 капель концентрированного аммонийгидроксида. После перемешивания смеси в течение 15 минут удаляли растворитель при пониженном давлении, и полученный неочищенный продукт подвергали колоночной хроматографии (20% метанолэтилацетат), что дало 112 мг (67%) ожидаемых 4'-гидроксиметиловых продуктов в виде смеси с отношением (1'R,4'S):(1'S,4'S)=7:1 (1H ЯМР). Было обнаружено (1H ЯМР), что одна из уловленных фракций содержала только указанное в заголовке соединение. Эту фракцию концентрировали в вакууме и получили в результате 27 мг белой пены.
УФ (λмакс): 283,6, 283,2, 202,4 нм (MeOH);
[α] D + 96o (с 0,7 MeOH);
1H ЯМР (CD2OD): δ 1,77-1,90 (м, 2H), 1,90-2,03 (м, 1H), 2,25-2.42 (м, 1H), 3,61 (дд, 1H, J=3,3 и 12,3 Гц), 3,82 (дд, 1H, J=2,8 и 12,3 Гц), 4,06 (м, 1H), 5,87 (м, 1H), 8,32 (д, 1H, J=7,0 Гц).
Изобретение относится к новым способам изготовления лекарств и новым способам лечения инфекций вируса гепатита В. Активными ингредиентами лекарств являются β-L-5-фтор-2',3'-дидезоксицитидин β-L-5F-ддЦ формулы Ib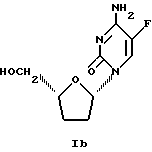
β-L-2',3'-дидезоксицитидин β-L-ддЦ формулы Iс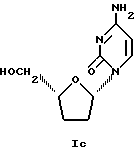
или β-D-5-фтор-2',3'-дидезоксицитидин формулы Iа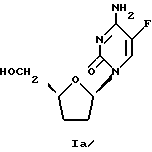
и их фармацевтически приемлемые производные. Изобретение расширяет арсенал средств указанного назначения. 2 с. и 46 з.п. ф-лы, 2 табл.
или их фармацевтически приемлемых солей, сложных эфиров или солей сложных эфиров.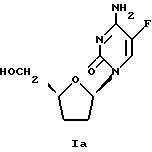
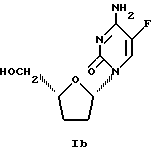
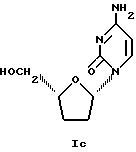
или их фармацевтически приемлемых сложного эфира, их соли или соли их сложного эфира с фармацевтически приемлемым носителем.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| 0 |
|
SU352248A1 | |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Огнетушитель | 0 |
|
SU91A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| ЯХОНТОВ О.И | |||
| и др | |||
| Лечение гепатопротекторами больных хроническими заболеваниями печени | |||
| Врачебное дело, Киев: Здоровья, 1990, N 5, c.18 - 20. | |||

