Изобретение относится к стереоселективному процессу приготовления аналогов и производных нуклеозидов. В частности, изобретение относится к процессу приготовления аналогов и производных нуклеозидов, которые в основном находятся в их цис-изомерной конфигурации.
Аналоги и производные нуклеозидов являются важным классом лекарственных средств: так, например, ряд аналогов нуклеозидов проявил антивирусное действие против таких ретровирусов, как вирус иммунодефицита человека (ВИЧ), вирус гепатита B и T-лимфотропный вирус человека (изд. "РСТ" WO 89/04662 и Евр. патент, изд. 0349242 A2). Среди аналогов нуклеозидов, у которых выявлено противовирусное действие, - азидодезокситимидин (АЗТ), n 2',3'-дидезоксицитидин и 2'-дезокси 3'-тиацитидин [(-)-2-гидpoкcимeтил-5-(цитoзин-1'-y1)- 1,3-оксатиолан] (Евр. патент, изд. 0382526 A2).
Большинство аналогов и производных нуклеозидов содержат по крайней мере два хиральных центра (обозначенных знаком "*" в формуле A), а каждый изомер может существовать в двух парах оптических изомеров (энантиомеров) (т.е. два в цис-конфигурации и два - в транс-конфигурации). Однако биологически полезное действие обычно обнаруживают цис-изомеры.
Пуриновое или пиримидиновое основание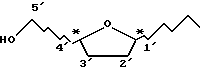
Многие из известных процессов производства аналогов и производных нуклеозидов для добавления сахара к пуриновому или пиримидиновому основанию полагаются на общепринятую методику гликозилирования. Такая методика неизменно дает диастереомерные смеси цис- и транс-изомеров, которые требуют кропотливого разделения и приводят к более низкому выходу искомых биологически активных цис-нуклеозидных аналогов. Усовершенствованная методика, направленная на выход лишь цис-нуклеозидов, требует добавления в сахар, желательно в положении 2'-, арила или ацил-заместителя. Поскольку заместитель 2'-полезен лишь для цис-нуклеозидного синтеза в одной конфигурации (когда заместитель 2'-расположен в положении цис- по отношению к заместителю 4'-), этот заместитель приходится вводить в правильную конфигурацию через целый ряд стадий. После гликозилирования заместитель 2'-должен быть удален, что требует прохождения дополнительных стадий. [L Wilson and D. Liotta. "A general method for controlling stereochemistry in the synthesis of 2'-dcoxyribose nucleoside", Tetrahedron Lett.31, pp, 1815-1818 (1990).]
Поэтому приобретает важность задача нахождения общего и перспективного с экономической точки зрения стереоселективного синтеза биологически активных цис-нуклеозидных аналогов.
Преимуществом описанного в настоящем изобретении процесса является то, что он позволяет осуществлять приготовление аналогов и производных цис-нуклеозидов через меньшее число стадий, используя недорогие и доступные исходные материалы и устраняя необходимость в кропотливых стадиях придания и лишения защитных свойств. Более того, описанный в настоящем изобретении процесс позволяет получать хороший выход искомых аналогов и производных цис-нуклеозидов.
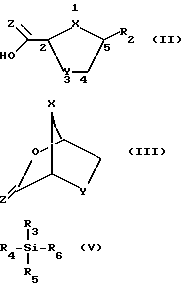
В настоящем изобретении делается попытка найти усовершенствованный процесс получения преимущественно цис-нуклеозидных аналогов и производных ф-лы I, а также их солей и ложных эфиров, пригодных для использования в технологии лекарственных форм: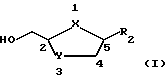 ,
,
где X-S или О; Y-S, CH2 или CH(R), где R - азидо или галоген: и R2 - пуриновое или пиримидиновое основание, или его аналог, или производное.
Предлагаемый в настоящем изобретении процесс охватывает следующие стадии:
стадия 1:
вступление в реакцию соединения ф-лы IV: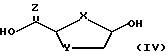
с мягким осушителем;
стадия 2:
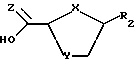
взаимодействие желательного предварительно подвергнутого силилированию (или силилированию на месте) пуринового или пиримидинового основания (R2) или его аналога или производного с новым бициклическим промежуточным продуктом ф-лы III: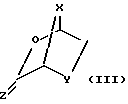 ,
,
где X и Y определены выше; Z - S или О;
причем взаимодействие осуществляется путем применения соответствующей льюисовой кислоты в пригодном растворителе a; для выхода нуклеозидного промежуточного продукта 2- карбоновой или тиокарбоновой кислот ф-лы II: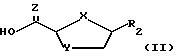
стадия 3:
восстановление промежуточного продукта ф-лы III в соединение ф-лы I путем применения пригодного восстановителя в пригодном растворителе b.
На схеме 1 изображен предпочтительный процесс в том виде, в котором он применим к любому нуклеозидному аналогу в общем, и особенно - к 1,3-оксатиолану, 1,3-диоксолану, 1,3-дитиолану, азидодезокси - или 2',3'-дидезоксинуклеозидным аналогам.
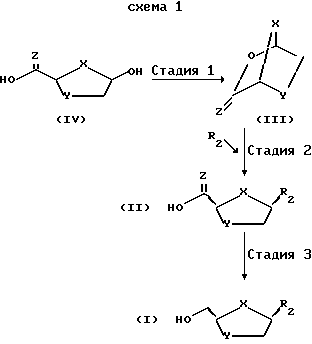
где X - S или O; Y-S, CH2, O или CH(R), где R - азидо или прогон; Z - О или S; и R2 - пуриновое или пиримидиновое основание или его аналог или производное.
Описанный в настоящем изобретении новый процесс осуществляется преимущественно при использовании соединения ф-лы II, где X - O, Y - S и Z - O.
Показанные на схеме 1 различные стадии могут быть вкратце описаны следующим образом:
Стадия 1. 2-карбоновая или тиокарбоновая кислоты сахарных производных ф-лы IV могут быть приготовлены любым известным на настоящем уровне развития технологии способом (напр., изд. "РСТ" WO 92/20669, которое инкорпорируется в настоящем документе путем отсылки). Бициклический промежуточный продукт ф-лы III получается посредством вступления в реакцию сахарного производного ф-лы IV в присутствии пригодного мягкого осушителя, среди которых предпочтительным мягким осушителем является триметилортоформат.
Стадия 2. Предварительно подвергнутый силилированию (или силилированию на месте) пуриновое или пиримидиновое основание или его аналог или производное затем взаимодействует с новым бициклическим промежуточным продуктом ф-лы III в присутствии той или иной льюисовой кислоты, напр. иодотриметилсилана, либо трифторметансульфоната триметилсилила, - для получения 2-кapбoнoвoй или тиокарбоновой кислот нуклеозидного аналога ф-лы II, преимущественно в цис-конфигурации.
В предпочтительном варианте осуществления настоящего изобретения R2 - предпочтительно пуриновое или пиримидиновое основание или его аналог или производное.
В более предпочтительном варианте осуществления настоящего изобретения R2 - предпочтительно пуриновое или пиримидиновое основание или его аналог или производное - выбирается из группы, состоящей из фторцитозина, цитозина и урацила.
Среди предпочтительных льюисовых кислот, применяемых при взаимодействии пуринового или пиримидинового основания или его аналога или производного, - иодотриметилсилан, трифторметансульфонат t-бутил-диметилсилила или триметилсилила.
Предпочтительные льюисовые кислоты, применяемые при взаимодействии пиримидиновых оснований с бициклическим промежуточным продуктом ф-лы II, - трифторметансульфонат t-бутил-диметилсилила и триметилсилила.
Среди предпочтительных растворителей, пригодных к применению при взаимодействии пуринового или пиримидинового основания или его аналога или производного, - по крайней мере один галогенированный органический растворитель. Более предпочтительным среди предпочтительных растворителей является дихлорметан.
В более предпочтительном варианте осуществления настоящего изобретения основание R2 предварительно подвергается силилированию при помощи соответствующего силилирующего вещества, выбранного из группы, в которую входят гексаметилдисилазан и трифторметансульфонат триметилсилила, либо силилированию на месте при помощи силилирующего вещества, выбранного из группы, в которую входят трифторметансульфонат t-бутил-димстилсилнла и триметилсилила.
Стадия 3. Цис-2-карбоновая или тиокарбоновая кислоты нуклеозидного аналога ф-лы II могут быть восстановлены при помощи соответствующего восстановителя в пригодном растворителе для получения конечного соединения ф-лы I. Факультативно, выход на этой последней стадии восстановления может быть улучшен, если соединение ф-лы II первоначально превратить в сложный эфир, напр. этиловый эфир, любым известным на настоящем уровне развития технологии методом, вслед за чем последовало бы восстановление при помощи пригодного реагента - согласно описанному выше.
Среди предпочтительных восстановителей - боргидрид натрия, триэтилборгидрид лития, алюмогидрид лития, боран, а также смесь боранметилового сульфида и триметилбората.
Среди предпочтительных растворителей - по крайней мере один растворитель, который был независимо выбран в группе, состоящей из метанола, этанола, изопропанола, тетрагидрофурана, простого эфира и дихлорметана.
На схеме 1a показано применение процесса в схеме 1 при синтезе рацемической смеси цис-2-гидроксиметил-5-(5'-фторцитозин-1'-y1)- 1,3-оксатиолана. Несмотря на то, что при иллюстрировании этого процесса применяются конкретные реагенты и исходные материалы, специалисту в этой области понятно, что для приготовления аналогичных соединений могут использоваться пригодные аналогичные реагенты и исходные материалы.
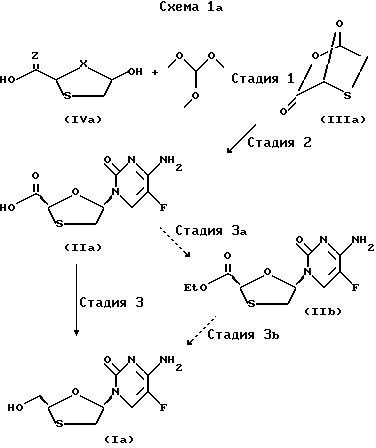
Показанные на схеме 1a различные стадии могут быть вкратце описаны следующим образом:
Стадия 1. Транс-5-гидрокси-1,3-оксатиолан-2-карбоновую кислоту ф-лы IVa можно получить любым известным на настоящем уровне развития технологии методом. Транс-5-гидрокси-1,3-оксатиолан-2- карбоновая кислота ф-лы IVa при кипячении с обратным холодильником реагирует с триметилортоформатом, в результате чего получается новый бициклический промежуточный продукт ф-лы IIIa - 2,7-диокса-3-оксо-5- тиабицикло[2.2.1]гептан.
Стадия 2. Новый бициклический промежуточный продукт ф-лы IIIa - 2,7-диокса-3-оксо-5-тиабицикло[2.2.1]гептан - реагирует с 5-фторцитозином, предварительно подвергнутым силилированию при помощи такой льюисовой кислоты, как гексаметилдисилазан, либо силилированию на месте при помощи такой льюисовой кислоты, как трифторметансульфонат триметилсилила, в таком пригодном растворителе, как дихлорметан, содержащий 2,6 лутидин. Затем добавляется льюисовая кислота, предпочтительно иодотриметилсилан или трифторметансульфонат триметилсилила, для получения нуклеозидного аналога ф-лы IIa - цис-5-(5'-фторцитозин-1'-y1)-1,3-оксатиолан-2-карбоновой кислоты - высокодиастереоселективным образом и с высоким коэффициентом цис.транс.
Стадия 3a. Цис-нуклеозидный аналог ф-лы IIa - цис-5-(5'- фторцитозин-1'-y1)-1,3-оксатнолан-2-карбоновая кислота - затем обрабатывается соответствующим конвертером, таким как смесь CsF и иодоэтана в таком пригодном растворителе, как N,N-диметилформамид, для получения сложного эфира ф-лы IIb - цис-этил-5-(5'-фторцитозин-1'-y1)- 1,3-оксатиолан-2-карбоксила.
Предпочтительным конвертером является смесь CsF и иодоэтана.
Предпочтительным растворителем является-диметилформамид.
Стадия 3b. Этиловый эфир цис-нуклеозидного аналога ф-лы IIIb - цис-этил-5-(5'-фторцитозин-1'-y1)-1,3-оксатиолан-2-карбоксил - затем восстанавливается при помощи соответствующего восстановителя, как, напр., боргидрид натрия, в соответствующем растворителе, как, напр., этанол, с целью получения конечного соединения ф-лы Ia - цис-2-гидроксиметил-5-(5'-фторцитозин-1'-y1)-1,3-оксатиолана.
Аналоги нуклеозида ф-лы I, синтезированные с применением описанного в изобретении процесса, предпочтительно включают 1,3-оксатиолан, 1,3-диоксоланы, 1,3-дитиоланы и 2',3'-дидезокси- аналоги, которые были модифицированы любым из следующих путей (или их комбинаций): изменение основания, напр. добавление заместителя (такого, как 5'-фторцитозин) или замещение одной группы изостерной группой (напр., 7-деазааденин): изменение сахара, напр. замещение гидроксильных групп C-2' и C-З' любым заместителем, в том числе галогеном, азидо или водородом (напр., 2',3'-дидезоксинуклеозидами); изменение сайта связывания сахара к сайту N-1 может, напр., быть прикреплен к сайту N-3 или C-6. а обычно прикрепленные к сайту N-9 пурины, могут, напр., быть прикреплены к сайту N-7; изменение конфигурации связи основания сахара (напр., цис- и транс-конфигурации).
Термин "пуриновое" или "пиримидиновое основание" означает основание, которое можно найти в нуклеозидах, встречающихся в естественном виде. Аналог основания - основание, которое имитирует встречающиеся в природе основания в том смысле, что их структуры (виды атомов и их расположение) являются схожими с основаниями, встречающимися в природе, однако обладать дополнительными функциональными свойствами встречающихся в природе оснований (или, наоборот, в них определенные функциональные свойства могут отсутствовать). Такие аналоги включают полученные замещением составляющей CH атомом азота (напр., такие 5-азапиримидины, как 5-азацитозин), либо замещением атома азота составляющей CH (напр., такие 7-деазапурины, как 7-деазааденин или 7-деазагуанин), либо и то и другое (напр., 7-деаза, 8-азапурины). Под производными таких оснований или аналогов понимаются такие основания, у которых заместители в цикле включаются в обычные, известные на настоящем уровне развития технологии заместители, - напр., галоген, гидроксил, амино, C1-6 алкил, - отщепляются или модифицируются ими. Такие пуриновые или пиримидиновые основания, аналоги и производные - хорошо известны специалистам в этой области, как это встречается у M.J. Robins, "Chemistry of naturally occurring pyrimidine nucleoside and analogues" Nucleosides Analogue, (R.T. Walker et al., Eds.) Plenum Press, pp 165-192 (1979) и у Nasr et al., Antiviral Reg., 14 pp 125-148 (1990).
Льюисовая кислота, польза которой заключается в способствовании взаимодействию промежуточного продукта ф-лы III с предварительно подвергнутым силилированию (или силилированию на месте) пуриновым или пиримидиновым основанием или его аналогом, обладает общей ф-лой V: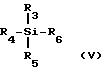
где R3, R4 и R5 - независимо выбраны в группе, состоящей из водорода, C1-6 алкила (напр. , мстила, этила, t-бутила), с факультативным замещением галогенами (F, Cl, Br, I) C1-20 алкокси (напр., метокси) или C6-20 арилокси (напр. , фенокси); C7-20 аралкил (напр., бензил) с факультативным замещением галогеном C1-20 алкилом или C1-20 алкокси (напр. p-метоксибензил); C6-20 арил (напр. , фенил) с факультативным замещением галогенами C1-20 алкилом или C1-20 алкокси; триалкилсилилом; и галогенами (F, Cl, Br, I); а
R6 - выбран в группе, состоящей из галогена (F, Cl, Br, I); C1-20 сульфонатных эфиров с факультативным замещением галогенами (напр., трифторметансульфонат); C1-20 алкиловых эфиров с факультативным замещением галогеном (напр. , трифторацетат); поливалентных галогенидов (напр., трииодид); трижды замещенных силиловых групп общей ф-лы: (R3) (R4) (R5) Si (где R3, R4 и R5 - определены выше); насыщенных или ненасыщенных селенинилов C6-20 арил; замещенного или незамещенного C6-20 арилсульфенила; замещенного или незамещенного C1-20 алкоксиалкила; а также триалкилсилокси.
Предпочтительные группы R3, R4 и R5 - независимо метил или йод. Наиболее предпочтительной группой R3, R4 и R5 является метил. Предпочтительными группами R6 являются йод, хлор, бром или сульфонатные эфиры. Наиболее предпочтительными группами R6 являются иод и трифторметансульфонат.
В наиболее предпочтительном варианте льюисова кислота выбирается из группы, состоящей из иодотриметилсилана; трифторметансульфоната t-бутил-диметилсилила и трифторметансульфоната триметилсилила.
Под солями и сложными эфирами, пригодными для использования в технологии лекарственных форм, подразумеваются любая соль, сложный эфир или соль такого сложного эфира, пригодные для использования в технологии лекарственных форм, соединения ф-лы I. Такая соль, сложный эфир или соль такого сложного эфира, пригодные для использования в технологии лекарственных форм, также включают любое иное соединение, которое, при введении его реципиенту, способно обеспечить (непосредственно или опосредствованно) соединение ф-лы I, либо обладающий противовирусным действие метаболит или его остаток.
Специалистам в этой области будет понятно, что соединение ф-лы I может быть изменено для получения его производных, пригодных для использования в технологии лекарственных форм, в функциональных группах в обоих составляющих основания R2, а также в гидроксиметиле C-2 сахарного кольца. Изменения во всех таких функциональных группах входят в охватываемые настоящим изобретением процессы. Тем не менее, особый интерес представляют приемлемые производные (напр. , сложные эфиры), полученные путем изменения группы 2-гидроксиметила сахарного кольца.
Предпочтительные сложные эфиры ф-лы I, произведенные путем процесса настоящего изобретения включают соединение, в котором ОН замещается функцией карбоксила R1(CO)O-, в которой R1 выбирается из водорода; алкил с прямой или разветвленной цепью (напр., метил, этил, n-пропил, t-бутил, n-бутил); алкоксиалкил (напр., феноксиметил); арил (напр., фенил, факультативно замещенный галогеном, C1-4 алкил или C1-4 алкокси); замещенный дигидропиридинил (напр., N-метилдигидропиридинил). R1(CO)O- тоже может быть заменен такими сульфонатными эфирами, как алкил- или аралкилсульфонилом (напр., метансульфонил); сульфонатные эфиры, сложные эфиры аминокислот (напр., L-валил или L-изолейцинил); и моно- ди- или трифосфатные сложные эфиры. Также включаются в диапазон таких сложных эфиров сложные эфиры, полученные из таких многофункциональных кислот, как фосфорные кислоты или карбоновые кислоты, содержащие более одной группы карбоксила, напр. , дикарбоновые кислоты ф-лы HOOC(CH2)qCOOH, где q - целое число от 0 до 10 (напр., янтарная кислота).
Пригодные для использования в технологии лекарственных форм соли соединений ф-лы I включают соли, полученные из пригодных для использования в технологии лекарственных форм неорганических и органических кислот и оснований. Примеры пригодных кислот включают хлористоводородную, бромистоводородную, серную, азотную, перхлорную, фумаровую, малеиновую, фосфорную, гликолевую, молочную, салициловую, янтарную, p-толуолсульфокислоту, винную, уксусную, лимонную, метансульфоновую, муравьиную, бензойную, малоновую, нафталин-2- сульфоновую, бензолсульфоновую кислоты и p-толуолсульфокислоту. Иные кислоты, такие как щавелевая, хотя сами по себе и не пригодны для использования в технологии лекарственных форм, могут быть полезны в приготовлений солей, полезных в качестве промежуточных продуктов при получении описанных в настоящем изобретении соединений и их с добавлением кислоты солей, пригодных для использования в технологии лекарственных форм.
Соли, полученные из приемлемых оснований, включают щелочной металл (напр. , натрий), щелочно-земельные металлы (напр., магний), аммоний и соли N(R')4 (где R' - C1-4 алкил).
Следующие примеры иллюстрируют настоящее изобретение таким образом, которым оно может применяться на практике, однако сами по себе, не должны истолковываться как ограничения, накладываемые на общую сферу охвата настоящего изобретения.
Пример 1.
2,7-диокса-3-оксо-5-тиабицикло[2.2.1]гептан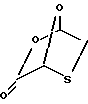
Раствор транс-5-гидрокси-1,3-оксатиолан-2-карбоновой кислоты (200 мг, 1,33 ммоль) и триметилортоформата (15 мл) нагревался в течение 2 часов в графитной ванне при 120oC. После удаления растворителя неочищенная реакционная смесь была очищена путем силикагель-хроматографией, элюированной этилацетатом:гексанами (1:4) для получения 64 мг (35%) желательного продукта; 1H ЯМР (ДМСО): δ 3,33 (dd, 1H, J=11,2 Гц), 3,42 (d, 1H, J=11 Гц), 6,53 (s, 1H), 6,83 (d, 1H, J=2 Гц); 13C ЯМР (ДМСО): δ 38,0, 75,4, 101,9, 167,1.
Пример 2.
Цис-5-(5'фторцитозин-1'-y1)-1,3-оксатиолан-2-карбоновая кислота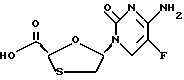
Трифторметансульфонат триметилсилила (0,164 мл, 0,844 ммоль) и 2,6-лутидин (0,098 мл, 0,844 ммоль) были добавлены в 5-фторцитозин (54,4 мг, 0,422 ммоль) в дихлорметане (1 мл) при комнатной температуре в аргоновой атмосфере. Смесь немедленно стала светлой. Был добавлен раствор 2,7-диокса-3-оксо-5-тиабицикло[2.2.1] гептана (пример 1) (56 мг, 0,422 ммоль) в дихлорметане (1 мл), затем - иодотриметилсилан (0,06 мл, 0,422 ммоль). Раствор желтого цвета перемешивался при комнатной температуре в течение 16 часов. Было добавлено дополнительное количество 2,6-лутидина (0,05 мл, 0,422 ммоль) затем - метанола (0,034 мл, 0,844 ммоль). После 5-минутного перемешивания смесь была концентрирована, а остаток был растирен простым эфиром/дихлорметаном для достижения смеси цис- и транс-продуктов взаимодействия в соотношении 10: 1 (99,7 мг, выход 90,6%). Эта смесь была далее растирена метанолом при комнатной температуре для получения почти чистого цис-продукта (78 мг, выход 72,7%). 1H ЯМР (ДМСО-d6): δ 3,20 (1H, dd, J=2,9, 9,3), 3,53 (1H, dd, J=2,5, 9,3); 5,61 (1H, s): 6,25 (м); 7,69 (1H, bs); 7,90 (1H, bs); 8,28 (1H, d, 7,21). 13C ЯМР (ДМСО-d6): δ 36,07, 78,38, 89,46, 125,76 (d, J=32,8), 136,29 (d, J=284,9), 153,28, 157,93 (d, J=18,0), 171,29.
Пример 3ю
Цис-этил-5-(5'-фторцитозин-1'-y1)-1,3-оксатиолан-2- карбоксилат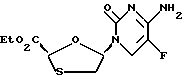
циc-5-(5'-Фтopцитoзин-1'-y1)-1,3-oкcaтиoлaн-2-кapбoнoвaя кислота (пример 2) (10 мг, 0,0383 ммоль) в N,N-диметилформамиде (0,5 мл) была обработана CsF (8,7 мг, 0,057 ммоль) и иодоэтаном (5 μл, 0,57 ммоль). Раствор перемешивался при комнатной температуре всю ночь и N,N-диметилформамид был удален. Остаток был обработан этилацетатом/дихлорметаном (1: 1,8 мл) и профильтрован. Фильтрат был концентрирован и остаток промывался несколько раз простым эфиром для получения продукта в виде белого твердого вещества (8 мг, выход 72%). 1H ЯМР (CD3OD): δ 1,13 (3H, t), 3,01 (1H, dd), 3,36 (1H, dd), 5,43 (1H, s), 6,16 (1H, m), 8,30 (1H, d).
Пример 4.
Цис-2-гидроксиметил-5-(5'-фторцитозин-1'-y1)-1,3- оксатиолан (ВСН-330)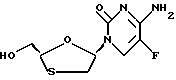
Цис-этил-5-(5'-фторцитозин-1'-y1)-1,3-оксатиолан-2-карбоксилат (пример 3) (5,5 мг, 0,019 ммоль) в этаноле (0,5 мл) был обработан борогидридом натрия (2 мг, 0,057 ммоль) при 0oC. Исходный материал не был полностью растворен. После того, как он перемешивался при комнатной температуре в течение 2 часов, метанол (0,2 мл) был добавлен и перемешивание продолжалось в течение 1,5 часа. Растворители были удалены и смесь была очищена путем силикагель-хроматографией с метанолом/этилацетатом в качестве элюентов для достижения чистого продукта в виде белого твердого вещества (4,2 мг, выход 89%). 1H ЯМР (CD3OD): δ 2,97 (1H, dd), 3,32 (1H, dd), 3,66 (1H, dd), 3,79 (1H, dd), 5,07 (1H, t), 6,03 (1H, m), 8,15 (1H, dd).
Пример 5.
Цис-5-(цитозин-1'-y1)-1,3-оксатиолан-2-карбоновая кислота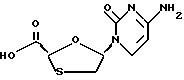
Трифторметансульфонат t-бутил-диметилсилила (0,32 мл, 1,4 м моль) был добавлен в суспензию цитозина (70,3 мг, 0,63 ммоль) и 2,6-лутидин (0,162 мл, 1,4 ммоль) в безводном дихлорметане (1 мл). Смесь перемешивалась при комнатной температуре в течение 10 минут и суспензия стала светлой во время перемешивания. Был добавлен раствор 2,7'диокса-3-оксо-5-тиа6ицикло[2.2.1] гептана (пример 1) (74 мг, 0,56 ммоль) в дихлорметане (1 мл) в раствор цитозина - затем иодотриметилсилан (0,086 мл, 0,61 ммоль). Полученный светлый раствор желтого цвета перемешивался при комнатной температуре в течение 18 часов и был резко охлажден метанолом. Большинство растворов было удалено под вакуумом. Липкий материал был растирен этилацетатом и дихлорметаном для получения белого твердого вещества, которое было тщательно промыто этилацетатом и дихлорметаном для достижения 114 мг продукта (выход 83,2%) (коэффициент цис. транс; 27: 1) 1H ЯМР (ДМСО-D6): δ 3,12 (dd, 1H, J=6 и 12 Гц), 3,51 (dd, 1H, J= 5 и 12 Гц), 5,58 (s, 1H); 5,79 (d, 1H, J=7,5 Гц), 6,27-6,31 (m, 1H), 7,27-7,41 (bd, 2H), 8,02 (d, 1H, J=7,5 Гц). 13C ЯМР (ДМСО-d6): δ 36,1, 78,3, 89,2, 94,5, 141,6, 154,6, 165,7, 171,1.
Пример 6.
Цис-этил-5-(цитозин-1'-y1)-1,3-оксатиолан-2-карбоксилат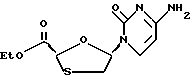
Иодоэтан (0,02 мл, 0,25 ммоль) был добавлен в суспензию цис-5- (цитозин-1'-y1)-1,3-оксатиолан-2-карбоновой кислоты (пример 5) (38 мг, 0,16 ммоль) и безводной CsF (36 мг, 0,24 ммоль) в N,N-диметилформамиде (1 мл) при комнатной температуре. Полученный светлый раствор перемешивался в течение 18 часов. Диметилформамид был удален под вакуумом для получения белого твердого вещества, которое было подвергнуто колоночной хроматографией (этилацетат/гексаны/метанол/2: 2:1) для получения 31 мг (выход 72%) продукта в виде белых гранул. 1H ЯМР (ДМСО-d6): δ 1,3 (t, 3H, J=7,1 Гц), 3,12 (dd, 1H, J= 6,7 и 12 Гц), 3,52 (dd, 1H, J=5,1 и 12 Гц), 4,21 (q, 2H, J=7,1 Гц), 5,7 (s, 1H), 5,79 (d, 1H, J=7,5 Гц), 6,34 (dd, 1H, J=5,1 и 12 Гц), 7,28-7,32 (bd, 1H), 7,95 (d, IH, J=7,5 Гц).
Пример 7.
Цис-2-гидроксиметил-5-(цитозин-1'-y1)-1,3-оксатиолан (ВСН-189)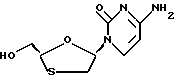
Борогидрид натрия (6 мг, 0,16 ммоль) был добавлен в суспензию цис-этил-5-(цитoзин-1'-y1)-1,3-oкcaтиoлaн-2-кap6oкcилaт (пример 6) (15 мг, 0,055 ммоль) в смеси метанола (1 мл) и дихлорметана (1 мл) при комнатной температуре. Полученный раствор перемешивался в течение 2 часов и растворители были удалены под вакуумом для получения белого твердого вещества, которое было пропущено через силика-колонку короткого пути (этилацетат/гексаны/метанол), для получения выхода 12,5 мг (выход 100%) продукта. 1H ЯМР (ДМСО-d6): δ 2,99 (dd, 1H), 3,40 (dd, 1H), 3,71 (m, 2H), 5,14 (t, 1H), 5,70 (d, 1H), 6,18 (t, 1H), 7,20 (d, 2H), 7,80 (d, 1H). 13C ЯМР (ДМСО-d6): δ 36,22, 62,79, 85,75, 86,47 93,86, 140,91, 154,63, 165,59.
Пример 8.
Цис-5-(урацил-1'-y1)-1,3-оксатиолан-2-карбоновая кислота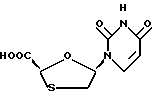
Иодотриметилсилан (65 μл, 0,454 ммоль) был добавлен в раствор 2,7-диокса-3-оксо-5-тиабицикло[2.2.1] гептана (пример 1) (60 мг, 0,454 ммоль) и бис-триметилсилилурацила (140 мг, 0,545 ммоль) в безводном дихлорметане при комнатной температуре в аргоновой атмосфере. Полученный раствор перемешивался в течение 20 часов. Реакция была резко охлаждена путем добавления смеси 1: 1 насыщенного раствора тиосульфата натрия-бикарбоната натрия, затем разбавления дихлорметаном. Смесь перемешивалась в течение 10 минут для получения белой суспензии. Белое твердое вещество было собрано путем фильтрации, затем высушено под вакуумом для получения 21 мг белого порошка. Анализ 1H ЯМР проявил смесь 6:1 желательного продукта и урацила. Водная часть фильтрата была подкислена 1 M HCl по pH 4, затем насыщена хлоридом натрия. Этот раствор была экстрагирован добавлением тетрагидрофурана. Совмещенные экстракты были высушены над безводным сульфатом магния и растворитель был испарен под пониженным давлением для достижения 73 мг белого твердого вещества. Анализ 1H ЯМР показал смесь 5:2 желательного продукта и урацила, на основании анализа 1H ЯМР общий выход был 64% а изомерную чистоту оценили как ≥ 95% цис-изомера. 1H ЯМР (ДМСО-d6): δ 2,26 (dd, 1H, J=4,9, 12,3 Гц), 3,49 (dd, 1H, J=5,2, 12,4 Гц), 5,57 (9, 1H), 5,71 (dd, 1H, J=2,2, 8,0 Гц); [этот сигнал разрушился в дублет при обработке D2O (J=8,2 Гц)], 6,29 (t, 1H, J=5,2 Гц), 8,07 (d, 1H, J=8,2 Гц), 11,41 (ba, 1H, обменено D2O).
Пример 9.
Цис-2-гидроксиметил-5-(урацил-1'-y1)-1,3-оксатиолан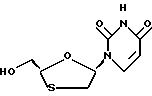
Боранметиловый сульфид добавляется в цис-5-(урацил-1'-y1)-1,3- оксатиолан-2-карбоновую кислоту и триметилборат в тетрагидрофуране. Восстановление производится при комнатной температуре. Окончательный продукт был изолирован по J.L. Kraus и G. Attardo. Synthesis. 1991, 1046.
Описываются способы получения цис-нуклеозидных аналогов общей формулы II, где Х = S или O; Y = S и R2 - остаток пуринового или пиримидинового основания или его аналога или его производного, включающий взаимодействие желаемого предварительно силилированного или силилированного in situ пуринового или пиримидинового основания или его аналога или его производного с соединением формулы III, где Х и Y определены выше; Z = S или 0; причем это взаимодействие осуществляют в растворе с использованием кислоты Льюиса формулы V, где R3, R4 и R5 независимо выбирают из группы, включающей водород, С1-20алкил (например, метил, этил, t-бутил), необязательно замещенный галогенами (F, Сl, Вr, I), С1-20>алкокси (например, метокси) или С6-20арилокси (например, фенокси); С7-20аралкил (например, бензил), необязательно замещенный галогеном С1-20алкилом или С1-20алкокси (например, р - метоксибензил); С6-20арил (например, фенил), необязательно замещенный галогенами; С1-20алкилом или С1-20алкокси, триалкилсилилом; галогенами (F, Сl, Вr, I); R6 выбирают из группы, состоящей из галогена (F, Сl, Вr, I); С1-20сульфонатных эфиров, необязательно замещенных галогенами (например, трифторметансульфонат); С1-20алкиловых сложных эфиров, необязательно замещенных галогеном (например, трифторацетат); поливалентных галогенидов (например, трииодид); трижды замещенных силильных групп общей формулы: (R)(R4)(R5), где R3, R4 и R5 указаны выше: насыщенанных или ненасыщенных соединений С6-20арилов, замещенного или незамещенного С6-20арилсульфенила, замещенного или незамещенного С1-20алкоксиалкила, а также триалкилсилокси. Целевые продукты обладают антивирусной активностью. Технический результат - упрощение процесса получения вышеуказанных соединений с высоким выходом. 3 с. и 16 з.п. ф-лы.
где X = S или O;
Y = S, O;
R2 - остаток пуринового или пиримидинового основания,
включающий взаимодействие желаемого предварительно силилированного или силилированного in situ пуринового или пиримидинового основания, или его аналога, или его производного с соединением формулы III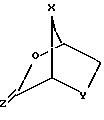
где X и Y определены выше; Z = S или O,
причем это взаимодействие осуществляют в растворе с использованием кислоты Льюиса формулы V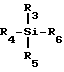
где R3, R4 и R5 независимо выбирают из группы, включающей водород, C1-20 алкил (например, метил, этил, t-бутил), необязательно замещенный галогенами (F, Cl, Br, J), C1-20 алкокси (например, метокси) или C6-20 арилокси (например, фенокси); C7-20 аралкил (например, бензил), необязательно замещенный галогеном C1-20 алкилом или C1-20 алкокси (например, р-метоксибензил); C6-20 арил (например, фенил), необязательно замещенный галогенами, C1-20 алкилом или C1-20 алкокси; триалкилсилил или галоген (F, Cl, Br, J);
R6 выбирают из группы, состоящей из галогена (F, Cl, Br, J); C1-20 сульфонатных эфиров, необязательно замещенных галогенами (например, трифторметансульфонат); C1-20 алкиловых сложных эфиров, необязательно замещенных галогенами (например, трифторацетат); поливалентных галогенидов (например, трииодид); трижды замещенных силильных групп общей формулы: (R3) (R4) (R5) Si (где R3, R4 и R5 - определены выше); насыщенных или ненасыщенных селенинил C6-20 арилов; замещенного или незамещенного C6-20 арилсульфенила; замещенного или незамещенного C1-20 алкоксиалкила, а также триалкилсилокси.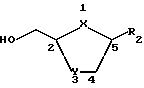
где X, Y и R2 имеют значения, указанные в п.1,
включающий восстановление полученного соединения формулы II.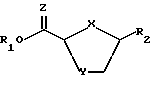
где X, Y, Z и R2 определены по любому из пп.1 - 3 и R1 = C1-6 алкил,
и восстановление соединения формулы IIb с получением соединения формулы I в подходящем растворителе.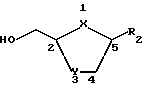
где X, Y и R2 определены выше по любому из пп.1 - 4,
включающий взаимодействие желаемого предварительно силилированного или силилированного in situ пуринового или пиримидинового основания, или его аналога, или его производного с соединением формулы III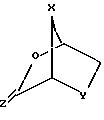
где X, Y и Z определены выше по п.1 или 4,
причем это взаимодействие осуществляют по методике, определенной по любому из пп. 1 и 5 - 8, с тем, чтобы получить соединение формулы II, с последующим восстановлением соединения формулы II с получением соединения формулы I по методике, определяемой по любому из пп.9 - 16.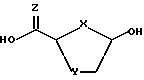
где X, Y и Z имеют вышеуказанные значения,
с мягким дегидратирующим агентом.
| Способ получения производных тиоформамида | 1982 |
|
SU1209029A3 |
| WO 9111186 А, 1991 | |||
| WO 911759 А, 1991 | |||
| WO 9303027 А, 1993 | |||
| КЛАПАН ЦЕНТРАЛИЗОВАННОЙ СИСТЕМЫ ПОДКАЧКИ ШИН ТРАНСПОРТНОГО СРЕДСТВА | 0 |
|
SU382526A1 |
| Пъезокерамический элемент памяти | 1974 |
|
SU515157A1 |
| EP 526253 А, 1992. | |||

