Настоящее изобретение относится к ингибиторам тромбина, которые используются как антикоагулянты у млекопитающих. В частности, оно относится к пептидным производным, обладающим высокой антитромботической активностью, антикоагулянтной активностью и пероральной биодоступностью.
Процесс коагуляции крови, тромбоз, вызывается комплексным протеолитическим каскадом, приводящим к образованию тромбина. Тромбин протеолитически уничтожает активацию пептидов из Aα-цепей и Bβ-цепей фибриногена, который растворим в плазме крови, инициируя образование нерастворимого фибриногена.
Антикоагуляции обычно достигают путем введения гепаринов и кумаринов.
Парентеральный фармакологический контроль коагуляции и тромбоза основан на ингибировании тромбина за счет использования гепаринов. Гепарины косвенно воздействуют на тромбин путем ускорения ингибиторных воздействий эндогенного антитромбина-III (главный физиологический ингибитор тромбина). Поскольку уровни антитромбина-III в плазме изменяются и ввиду того, что граничная поверхность тромбина, по-видимому, резистентна по отношению к этому косвенному механизму, гепарины могут быть неэффективными. Так как анализы по коагуляции считаются эффективными и безопасными, уровни гепарина должны контролироваться с помощью анализов по коагуляции (особенно определение активированного парциального тромбопластинового времени /APTT/). Кумарины препятствуют генерации тромбина за счет блокирования посттрансляционного гамма-карбоксилирования в синтезе протромбина и других протеинов этого типа. Вследствие своего механизма, воздействие кумаринов может проявляться только медленно, спустя 6-24 ч после введения. Более того, они являются неселективными антикоагулянтами. Следовательно, кумарины требуют контролирования с помощью анализов по коагуляции (особенно анализ протромбинового времени /PT/).
В настоящее время вырос интерес к небольшим синтетическим пептидам, которые распознаются протеолитическими ферментами таким же образом, что и природные субстраты. Трипептид-альдегиды, такие, как D-Phe-Pro-Arg-H, Boc-D-Phe-Pro-Arg-H, D-MePhe-Pro-Arg-H, Bajuszetal. , J.Med.Chem., 33, 1729-1735 (1990), показывают сильное прямое ингибирование тромбина. Недавние клинические исследования, которые показали, что D-MePhe-Pro-Arg-H сульфат является антикоагулянтом у людей, описаны Simoons и др., Circulation, 90, 1-231, Abstr. 1241 (1994). Многочисленные исследователи синтезировали аналоги в попытке получить фармацевтические агенты, как, например, Shuman и др., J.Med. Chem., 36, 314-319 (1993). В патенте США N 4346078 приводится ряд антикоагулянтных пептидов, содержащих агматиновую (1-амино-4-гуанидинбутановую) группу. Агматиновые производные и родственные соединения описаны как в PCT-заявке с номером международной публикации ВОИС 93/11152, так и в европейской заявке на патент, публикация N 601459 от 15 июня 1994 г. Такие соединения отличаются от предшествующего ряда, в котором агматиновые соединения не содержат карбонильной группы, имеющейся в подобных соединениях с Arg-группой.
Хотя гепарины и кумарины являются эффективными антикоагулянтами, все еще не появилось лекарственное средство на основе известных трипептид-альдегидов, несмотря на сохраняющуюся перспективу для этого класса соединений, поэтому существует необходимость в антикоагулянтах, которые селективно воздействуют на тромбин и, независимо от антитромбина-III, оказывают ингибирующее воздействие вскоре после введения, предпочтительно оральным путем, и не интерферируют с лизисом сгустков крови, необходимого для поддержания гемостаза.
Настоящее изобретение касается обнаружения того, что соединения по настоящему изобретению, такие, как указанные ниже, являются сильными ингибиторами тромбина, могут иметь высокую биодоступность при пероральном введении. Кроме того, некоторые соединения по настоящему изобретению также могут вызывать ингибирование фактора Xa, который вовлекается в коагулянтный каскад.
Следовательно, первым объектом настоящего изобретения являются новые пептидные производные, которые являются сильными ингибиторами тромбина, пригодными в качестве антикоагулянтов.
Другие объекты, признаки и преимущества будут ясны специалистам из нижеследующих описаний и формулы изобретения.
Настоящее изобретение относится к ингибирующему тромбин соединению, имеющему формулу I,
X-Y-NH-(CH2)r-G, (I)
где X обозначает пролинил; гомопролинил; Rm-(CH2)g-NH-CH2-C(O)-,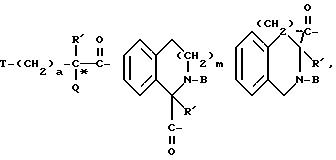
где Rd - карбокси или метилсульфонил;
Re - NHRc, NHCORc или NHCOORc, где Rc - C1-C10алкил, C3-C8циклоалкил или (C3-C8)циклоалкил(C1-C6)алкил с 4-10 атомами углерода;
T - C3-C8циклоалкил, C1-C8алкил,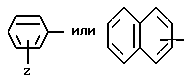
a = 0, 1 или 2 и
Q обозначает -OH, C1-C4алкокси или NH-A;
A - водород; C1-C4алкил, R''SO2-, R''OC(O)-, R''C(O)-, RnC(O)- или -(CH2)g-Rm;
g = 1, 2 или 3;
B - водород или (C1-C4)-алкил;
R' - водород или (C1-C4)-алкил;
R'' - C1-C4алкил, C1-C4перфторалкил, -(CH2)d-Rm, или незамещенный или замешенный арил, где арил обозначает фенил, нафтил, 5- или 6-членное незамещенное или замещенное ароматическое гетероциклическое кольцо, содержащее один или два гетероатома, которые являются одинаковыми или разными и которые выбраны из серы, кислорода и азота, или 9- или 10-членную, незамещенную или замещенную конденсированную бициклическую ароматическую гетероциклическую группу с одним или двумя гетероатомами, которые являются одинаковыми или разными и которые выбраны из серы, кислорода или азота;
Rm - -COORb, SO2(C1-C4)-алкил, -SO3H, -P(O)(ORb)2 или тетразол-5-ил;
Rn - -COORb или тетразол-5-ил;
каждый Rb независимо друг от друга - водород или C1-C4алкил;
d = 1, 2 или 3;
m = 0, 1 или 2;
n = 1 или 2; и
Z - водород, C1-C4алкил, C1-C4алкокси, гидрокси, галоген или RaSO2NH, где Ra - C1-C4алкил;
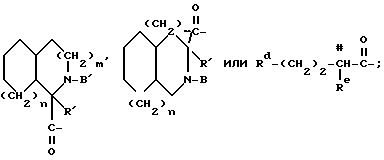
Y обозначает: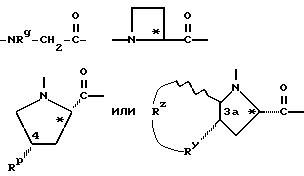
где Rg - C1-C6алкил, C1-C8циклоалкил или - (CH2)p-L-(CH2)q-T';
Rp - водород, C1-C6алкил, C3-C8циклоалкил или -(CH2)p-L-(CH2)q-T';
где p = 0, 1, 2, 3 или 4;
L - связь, -O-, -S- или -NH-;
q = 0, 1, 2 или 3;
T' - водород, C1-C4алкил, C3-C8циклоалкил, -COOH, -CONH2 или Ar, где Ar обозначает незамещенный или замещенный арил, где арилом является фенил, нафтил; 5- или 6-членное незамещенное или замещенное ароматическое гетероциклическое кольцо с одним или двумя гетероатомами, которые являются одинаковыми или разными и выбраны из серы, кислорода и азота; или 9- или 10-членную незамещенную или замещенную конденсированную бициклическую ароматическую гетероциклическую группу с одним или двумя гетероатомами, которые являются одинаковыми или разными и выбраны из серы, кислорода и азота;
RY - -CH2-, -O-, -S- или -NH-;
RZ - связь или вместе с RY и тремя соседними атомами углерода образует насыщенное карбоциклическое кольцо из 5-8 атомов, причем одним атомом может быть -O-, -S- или -NH-;
r = 1, 2 или 3 и
G - -(CH2)s-R, где s = 0-5; -CH=CH-(CH2)t-R, где t = 0-3; или G обозначает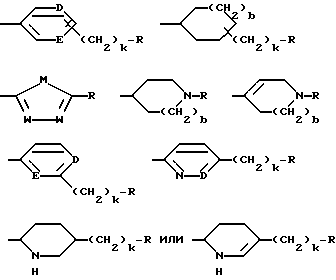
где D и E каждый независимо друг от друга - N или CH;
k = 0 или 1;
b = 0 или 1;
M обозначает S, кислород или NH;
каждый W независимо обозначает N или CH; и
R обозначает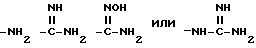
G обозначает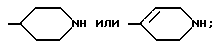
и где от одного до всех незамещенных атомов углерода ароматического или гетероциклического кольца: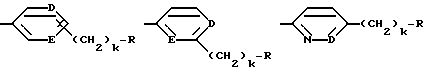
могут содержать фтор в качестве заместителя;
или его фармацевтически приемлемой соли; или к фармацевтически приемлемому сольвату указанного соединения или его соли;
при условии, что A не обозначает водород или трет-бутилоксикарбонил, когда G обозначает -(CH2)s-NH-C(NH)NH2, Y обозначает незамещенный пролинил (Rp является водородом) и T обозначает: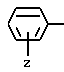
и при условии еще, что R не является амино или гуанидино,
когда r = 1 и s = 0;
и при условии, далее, что A не обозначает водород, C1-C4алкил, метилсульфонил или -(CH2)g-Rm и когда G обозначает (CH2)s-R, где R обозначает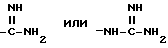
Y обозначает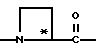
незамещенный пролинил (Rp = водород) или 4-гидроксипролинил (Rp=OH), R' обозначает водород;
T обозначает циклогексил и Q обозначает - NH-A;
и при условии еще, что R''SO2 не обозначает арилсульфонил, когда G обозначает -(CH2)s-R, где R обозначает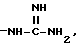
Y обозначает незамещенный пролинил (Rp является водородом) или 4-метилтиопролинил (Rp обозначает - SCH3) и Q обозначает - NH-A;
и при условии еще, что A не обозначает R''SO2-, когда G обозначает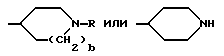
T обозначает (C1-C8)-алкил,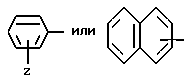
и Q обозначает - NH-A.
Конкретная группа вышеуказанных соединений формулы I включает такие соединения формулы I, где
X обозначает пролинил, гомопролинил;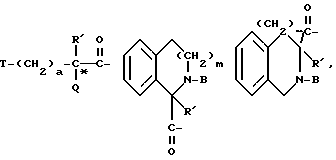
T обозначает C3-C8циклоалкил, C1-C8алкил,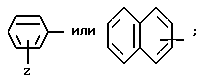
a - 0 или 1;
Q - -OH, C1-C4алкокси или - NH-A;
A - водород, C1-C4алкил, R''SO2-, R''OC(O)-, R''C(O)- или -(CH2)g -COOH;
g = 1, 2 или 3;
B - водород или C1-C4алкил;
R' - водород или C1-C4алкил;
R'' - C1-C4алкил, C1-C4перфторалкил, -(CH2)d-COOH или незамещенный или замещенный арил, где арил обозначает фенил, нафтил; 5- или 6-членное незамещенное или замещенное ароматическое гетероциклическое кольцо с одним или двумя гетероатомами, которые являются одинаковыми или разными и выбраны из серы, кислорода и азота; или 9- или 10-членная, незамещенная или замещенная конденсированная бициклическая ароматическая гетероциклическая группа с одним или двумя гетероатомами, которые являются одинаковыми или разными и выбираются среди серы, кислорода и азота;
d = 1, 2 или 3;
m = 0, 1 или 2;
n = 0, 1 или 2; и
Z - водород, C1-C4алкил, C1-C4алкокси, гидрокси, галоген или RaSO2NH, где Ra - C1-C4алкил; Y обозначает: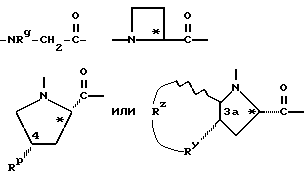
где Rg - C1-C6алкил, C3-C8циклоалкил или - (CH2)p-L-(CH2)q-T';
Rp - водород, C1-C6алкил, C3-C8циклоалкил или -(CH2)p-L-(CH2)q-T';
где p = 0, 1, 2, 3 или 4;
L - связь, -O-, -S- или -NH-;
q = 0, 1, 2 или 3; и T' - водород, C1-C4алкил, C3-C8циклоалкил, -COOH, -CONH2 или Ar, где Ar - незамещенный или замещенный арил, где арил обозначает фенил, нафтил; 5- или 6-членное незамещенное или замещенное ароматическое гетероциклическое кольцо с одним или двумя гетероатомами, которые являются одинаковыми или разными и которые выбраны из серы, кислорода и азота; или 9- или 10-членная незамещенная или замещенная конденсированная бициклическая ароматическая гетероциклическая группа с одним или двумя гетероатомами, которые являются одинаковыми или разными и выбраны из серы, кислорода и азота;
RY - -CH2-, -O-, -S- или -NH-; и
RZ - связь или вместе с RY и тремя соседними атомами углерода образует насыщенное карбоциклическое кольцо с 5-8 атомами, один атом из которых может представлять собой -O-, -S- или -NH-;
r = 1 или 2; и
G - -(CH2)s-R, где s=0-5; -CH=CH-(CH2)t-R,
где t = 0 - 3;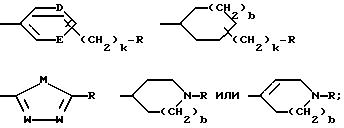
где D и E каждый независимо - N или CH;
k = 0 или 1;
b = 0 или 1;
M - S, O или NH;
каждый W независимо - N или CH; и
R обозначает -NH2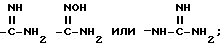
или их фармацевтически приемлемые соли; или фармацевтически приемлемые сольваты вышеуказанных соединений или их соли;
при условии, что A не обозначает водород или трет- бутилоксикарбонил, когда G обозначает -(CH2)s-NH-C(NH)NH2, Y - незамещенный пролинил (Rp - водород) и T обозначает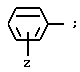
и при условии еще, что R не обозначает амино или гуанидино, когда r = 1 и s = 0;
и при условии еще, что A не обозначает водород, C1-C4-алкил, метилсульфонил или -(CH2)g-COOH, когда G обозначает -(CH2)s-R, где R обозначает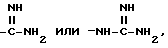
Y обозначает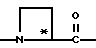
незамещенный пролинил (Rp - водород) или 4-гидроксипролинил (Rp - OH);
R' - водород;
T - циклогексил и Q - -NH-A;
и при условии еще, что R''SO2 не обозначает арилсульфонил, когда G обозначает -(CH2)s-R, где R -
Y - незамещенный пролинил (Rp - водород) или 4-метил-тиопролинил (Rp - -SCH3), и Q = -NH-A;
и при условии еще, что A не обозначает R''SO2-, когда G обозначает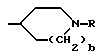
T - C1-C8алкил,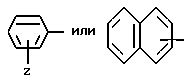
и Q = -NH-A.
В дополнение к соединениям формулы (I), настоящее изобретение относится к фармацевтическим композициям, содержащим соединение формулы (I) в сочетании с фармацевтически приемлемым носителем, разбавителем или наполнителем.
Настоящее изобретение также относится к способу ингибирования тромбоза у млекопитающих, который включает введение млекопитающему, нуждающемуся в лечении, антитромботической дозы соединения формулы I.
Настоящее изобретение, далее, относится к способу ингибирования тромбина, который включает введение млекопитающему, нуждающемуся в лечении, ингибирующей тромбин дозы соединения формулы I.
Изобретение относится к новым ингибиторам тромбина, фармацевтическим композициям, содержащим соединения в качестве активных ингредиентов, и к использованию соединений в качестве антикоагулянтов для профилактики и лечения тромбоэмболических заболеваний, таких как венозный тромбоз, легочная эмболия, артериальный тромбоз, в особенности, миокардиальная ишемия, инфаркт миокарда и церебральный тромбоз; состояний общей и локальной гиперкоагуляции, таких, которые наступают вследствие ангиопластики и операций с коронарным шунтированием; и для генерализации тканевого повреждения, так как оно относится к воспалительному процессу.
Термин "алкил" сам по себе или в качестве части другого заместителя обозначает алкильный радикал с линейной или разветвленной цепью, содержащий определенное число атомов углерода, такой, как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил и втор-бутил. Термин "перфторалкил" сам по себе или в качестве части другого заместителя обозначает алкильный радикал с линейной или разветвленной цепью, содержащий определенное число атомов углерода, где каждый атом водорода замещен атомом фтора, такой как трифторметил, перфторэтил, пер-фтор-н-пропил, перфторизопропил, перфтор-н-бутил, перфтор-трет-бутил, перфторизобутил и перфтор-втор-бутил.
Термин "C3-C8циклоалкил" относится к насыщенным алициклическим кольцам с 3-8 атомами углерода, таким как циклопропил, метилциклопропил, циклобутил, циклопентил, циклогексил, 4-метилциклогексил, циклооктил и т.п.
Термин "алкокси" обозначает алкильный радикал с линейной или разветвленной цепью, имеющий определенное число атомов углерода, связываемый с другой частью молекулы атомом кислорода. Термин "галоген" обозначает хлор, фтор, бром или иод. Термин "ацетил" обозначает CH3-C(O). Термин "трет-бутилоксикарбонил" обозначает (CH3)3 C-O-C(O)- и обычно сокращается как "Boc". Термин "бензилоксикарбонил" обозначает C6H5CH2-O-C(O)- и в сокращении представляет собой "Cbz".
Термин "5- или 6-членное гетероциклическое кольцо" обозначает любое 5- или 6-членное кольцо, которое имеет стабильную структуру и содержит один или два атома азота; один атом серы; один атом кислорода, один атом азота и один атом серы; или один атом азота и один атом кислорода, 5-членное кольцо содержит одну или две двойные связи, а 6-членное кольцо содержит две или три двойные связи. Такие гетероциклические системы включают фурил, тиенил, пирролил, пиразолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиранил, пиридинил, пиримидинил, пиразинил, оксазинил и тиазинил.
Термин "9- или 10-членное гетероциклическое кольцо" обозначает любую бициклическую группу, в которой любое из вышеуказанных 5- или 6-членных колец конденсировано с бензольным кольцом или другим 6-членным гетероциклом, как указано выше, имеющую стабильную структуру. Эти гетероциклические системы включают индолил, бензотиенил, бензофурил, бензоксазолил, бензоизоксазолил, бензопиразолил, хинолинил, изохинолинил, бензимидазолил и бензотиазолил.
Следует принять во внимание, что многие из вышеуказанных гетероциклов могут существовать в таутомерной форме. Все такие формы входят в рамки настоящего изобретения.
Все перечисленные для определения Ar или R'' ароматические или гетероциклические группы, независимо, незамещены или замещены одним или двумя заместителями, приводящими к стабильной структуре независимо выбранными из галогена, гидроксила, C1-C4алкила, C1-C4алкоксила, амино (-NH2), моно-(C1-C4алкил)амино, -(CH2)j-COOH, меркапто, -S(O)h (C1-C4-алкила), -NHS(O)h (C1-C4алкила), -NHC(О) (C1-C4алкила), -S(O)hNH2, -S(O)hNH(C1-C4алкила) или -S(O)hN(C1-C4алкил)2, где
h = 0, 1 или 2 и
j = 0, 1, 2, 3 или 4.
Особенно предпочтительным значение для заместителя R''(C)О - является 1-метилиндол-2-оил.
В представленной формуле I карбонильный радикал X связан с амином радикала Y. Карбонильный радикал Y затем присоединен к аминогруппе, показанной в формуле I.
Группа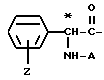
где Z и A оба - водород, относится здесь к фенилглицилу и обозначается Phg. Соединения, где A, например, метил, относятся к Nα- метилфенилглицильнoй группе и обозначаются MePhg. Замещенные соединения, где Z другой, чем водород, относятся по типу и положению заместителя, к замещенной группе, например, 3'-хлорфенилглицил или Phg (3-Cl).
Группа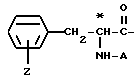
где Z и A оба - водород, относится к фенилаланилу и обозначается Phe. Соединения, где A, например, метил, относятся к Nα- метилфенилаланильной группе, обозначаются MePhe. Замещенные соединения, где Z другой, чем водород, относятся по типу и положению заместителя, к замещенной группе, например, 3'-хлорфенилаланил или Phe(3-Cl).
Группы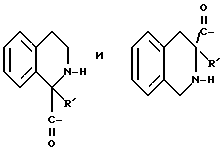
где R' - водород, относится к 1- и 3-тетрагидро-изохинолин-карбонилу, соответственно, которые имеют соответственно аббревиатуры 1-Tiq и 3-Tiq.
Группы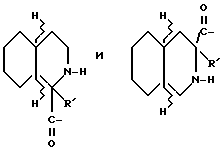
где R' - водород, относятся к 1- и 3-пергидроизохинолинкарбонилу, соответственно, и имеют, соответственно, аббревиатуры 1-Piq и 3-Piq. Как указано волнистыми линиями, существуют различные, образующие при конденсации циклов, изомеры этих заместителей; настоящее изобретение включает каждый индивидуальный изомер и их комбинации.
Группы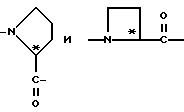
относятся к таким, как пролинил и азетидин-2-карбонил, соответственно, и имеют, соответственно, аббревиатуру Pro и Azt.
Группа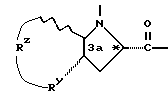
обозначает насыщенную бициклическую систему 4,5; 5,5; 6,5; 7,5 или 8,5 - типа. Стереохимия в положении 3a представляет собой цис- по отношению к карбонилу; другая предмостиковая связь может быть любой цис- или транс-связью, за исключением 4,5- и 5,5-систем, где может быть цис-предмостиковой связью. Определения RY и RZ предусматривают, что изменяемое кольцо, которое включает упомянутые три атома углерода, представляет собой насыщенную карбоциклическую систему с 4-8 атомами. Все атомы кольца могут представлять собой углерод, или одним из атомов кольца может быть гетероатом, выбираемый из -O-, -S- и -NH-. Это определение включает предпочтительную составляющую, происходящую от октагидроиндол-2-карбоновой кислоты, в аббревиатуре "Ohi", как представлено формулой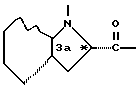
Разные цис- и транс-формы этой структурной единицы входят в рамки настоящего изобретения.
Знак в виде звездочки в радикале Y обозначает хиральный центр, который является (L). Звездочка в радикале X обозначает хиральный центр, который представляет собой (D) или (DL); знак # в радикале X обозначает хиральный центр, который представляет собой (L).
Кроме того, в зависимости от разветвления алкильных заместителей могут существовать диастереомеры. Соединения настоящего изобретения включают как смеси двух или более диастереомеров, так и каждый индивидуальный изомер.
Предпочтительные соединения настоящего изобретения включают такие соединения формулы I, где
гомопролинил, 1- или 3-Tig, или 1-, или 3-Pig, и
Y - пролинил; и их фармацевтически приемлемые соли и сольваты. В особенности, предпочтительны все соединения, где Q - NHA и A - водород или сульфонамид (например, A= R''SO2-), R' - водород, Z - водород и B - водород. Также предпочтительны те соединения, где R - гуанидиногруппа, особенно амидиногруппа.
Одной особенно предпочтительной комбинацией заместителей является такая, где G - R-замещенный фенил (т.е. D=E=CH, k = 0); в особенности, предпочтительны соединения, где G - 4-амидинофенильная группа.
Предпочтительной группой таких соединений, где от одного до всех по-другому незамещенных атомов углерода ароматического или гетероароматического кольца: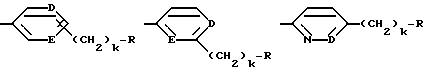
содержат в качестве заместителя фтор, является группа, в которой фтор-заместителя нет в α- или γ-положении к D или E, когда D или E - азот.
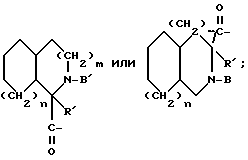
Другая группа предпочтительных соединений по настоящему изобретению включает такие соединения формулы I, как определено выше, где X обозначает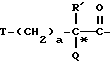
где T - циклогексил, a = 1, R' - водород и Q - -NH-A. Одной особенной подгруппой является такая, в которой A обозначает водород. Второй особенной подгруппой является такая, в которой A - R''SO2-, в особенности, когда R'' - этил. Третьей особенной подгруппой является такая, в которой A - -(CH2)g-COOH; предпочтительно g = 1.
Особыми значениями Y для соединения формулы I, в которой X, r и G имеют вышеуказанные значения, являются (L)-пролинил (Pro), (S)-цис-октагидро-1H-индол-2- карбонил (Ohi) и N-(2-фенилэтил)глицил [NPhCH2CH2Gly].
Для соединения формулы I, в которой R обозначает -NH2, предпочтительно, чтобы значения X и Y выбирались из вышеопределенных и значения r и G выбирались из следующих:
а) r = 1 и G -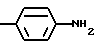
где анилиновое кольцо может содержать один или два атома фтора в качестве заместителей;
б) r = 1 и G обозначает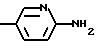
в) r = 1 или 2 и G обозначает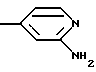
Одной предпочтительной группой соединений формулы I является такая, в которой Y обозначает (L)-пролинил, r = 1 и G обозначает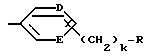
где каждый из D и E обозначает CH, k = 0 и R - амидино, и которая может быть представлена формулой (Ia):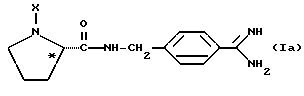
где бензамидиновое кольцо незамещено или может содержать один или два атома фтора в качестве заместителей, предпочтительно, в мета-положении к амидиновому радикалу, и X имеет любое из вышеуказанных значений.
Наиболее предпочтительным значением для соединения формулы Ia является такое, в котором бензамидиновое кольцо незамещено.
Другой особенно предпочтительной группой соединений формулы (I) является такая, в которой Y - (L)-пролинил, r = 1 и G обозначает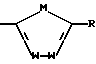
где M - сера, каждый W - CH и R - амидино, и которая может быть представлена формулой (Ib)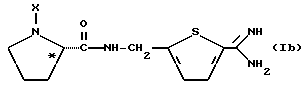
где X имеет любое из вышеуказанных значений.
Дополнительной предпочтительной группой соединений формулы I является такая, в которой Y - (L)-пролинил, r = 1 и G обозначает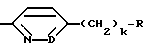
где D - азот или CH, k = 0 и R - амидино, и которая может быть представлена формулой (Ic)
где X имеет любой из вышеуказанных значений и D - азот или CH.
Предпочтительным значением для X в соединении формул Ia, Ib или Ic является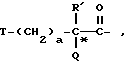
где R' - водород, a = 1, T - циклогексил или фенил и Q обозначает -NH-A. Более предпочтительно, A - водород, этилсульфонил или карбоксиметил. Одним особенно предпочтительным значением для X является N-карбоксиметил-D-циклогексилаланил. Другим предпочтительным значением для X является N-карбоксиметил-D-фенилаланил.
Специфические соединения формулы I изобретения описаны в примерах. Предпочтительный тип соединения, который может быть использован в качестве фармацевтически приемлемой соли или сольвата, может быть выбран из соединений, полученных в примерах 15, 18, 23, 44, 45, 46, 48, 49, 51, 52, 56, 65, 66, 68-72, 80, 86, 87, 88 и 92. Более предпочтительные соединения могут быть выбраны среди соединений, описанных в примерах 45, 46, 48, 51, 65, 70-72. Один из наиболее предпочтительных типов, на основании их неожиданно превосходных свойств, представлен в примере 48. Другим очень предпочтительным типом соединений является тип по примеру 65.
Как указано выше, изобретение включает фармацевтически приемлемые соли соединений доставленной выше формулы I. Конкретное соединение по настоящему изобретению может содержать одну или более достаточно основных функциональных групп и в соответствии с этим может реагировать с любой из числа неорганических и органических кислот с образованием фармацевтически приемлемой соли. Обычно используемыми кислотами для получения солей присоединения являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, фосфорная кислота и т. п. , и органические кислоты, такие как п-толуолсульфокислота, метансульфокислота, щавелевая кислота, п-бромфенилсульфокислота, карбоновая кислота, янтарная кислота, лимонная кислота, бензойная кислота, уксусная кислота и т. п. Примерами таких фармацевтически приемлемых солей являются такие соли, как сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, пропионат, деканоат, каприлат, акрилат, метоксибензоат, формиат, изобутират, капроат, гептаноат, пропионат, оксалат, малонат, сукцинат, суберат, себацинат, фумарат, малеат, бутин-1,4- диоат, гексин-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, фталат, сульфонат, ксилол-сульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, гамма-гидроксибутират, гликолят, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и т.п. Предпочтительными фармацевтически приемлемыми солями присоединения кислот являются образованные неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота и серная кислота.
Соединения по настоящему изобретению могут образовывать гидраты и сольваты с соответствующими растворителями. Предпочтительными растворителями для получения сольватных форм являются вода, спирты, тетрагидрофуран, ДМФ и ДМСО. Предпочтительными спиртами являются метанол и этанол. В зависимости от размера молекулы растворителя могут быть выбраны другие соответствующие растворители. Маленькие молекулы растворителя предпочтительны для облегчения соответствующего образования сольвата. Сольват или гидрат обычно образуются в процессе перекристаллизации или в процессе образования соли. Подходящей ссылкой в отношении сольватов является Sykes, Peter, A Guidebook to mechanism in Organic Chemistry, 6-е изд., (1986, John Villey and Sons, Нью-Йорк). В качестве используемого в настоящем изобретении термин "сольват" включает гидратные формы, такие как моногидраты и дигидраты.
Соединения формулы I получают известными методами связывания пептидов. Согласно одному такому методу, кислоту формулы P-X'-COOH, где -X'-C(O)- обозначает -X-, имеющий указанное для формулы I значение, и P обозначает аминозащитную группу, если необходимо, вводят в реакцию присоединения с карбоксилзащищенным Y-соединением с получением дипептида (a). Карбоксизащитную сложноэфирную группу Y-радикала затем удаляют (снятие защиты или деэтерификация) и свободнокислотную форму дипептида (b) связывают с защищающим реагентом (d). Вышеуказанная реакционная последовательность иллюстрируется следующей схемой 1: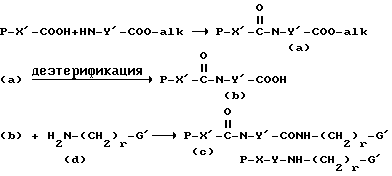
где G' имеет такое же значение, что и G, за исключением R обозначает -CN, - NHP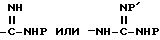
каждый P представляет аминозащитную группу, если необходимо, P' - H или P; alk - низший алкил или подобная защитная карбоксикислотная группа; и -Y'- имеет такое же значение, что и Y, представленный с амино- и карбоксильной функциональными группами, т. е. -Y- имеет такое же значение, как - N-Y'-C(О)-.
Если она имеется, циано-группу в G' переводят в значение R; и защитную группу в (c) затем удаляют известными специалистам способами, такими как гидрирование в присутствии металлического катализатора с получением соединений формулы I.
Связывание P-X'-COOH производного с HN-Y'-COO-alk проводят, сначала защитив аминогруппу аминокислоты, если она есть. Используют обычные аминозащитные группы, обычно применяемые для временной защиты или блокирования аминогруппы.
Аминозащитная группа относится к заместителям аминогруппы, обычно используемым для блокирования или защиты аминогруппы в процессе взаимодействия других функциональных групп соединения. Примерами таких аминозащитных групп являются формильная группа, тритильная группа, фталимидогруппа, трихлорацетильная группа, хлорацетильная, бромацетильная и иодацетильная группы; блокирующие группы уретанового типа, такие, как бензилоксикарбонил, трет- бутоксикарбонил, 4-фенилбензилоксикарбонил, 2-метилбензилоксикарбонил, 4-метоксибензилоксикарбонил, 4-фторбензилоксикарбонил, 4-хлорбензилоксикарбонил, 3-хлорбензилоксикарбонил, 2-хлорбензилоксикарбонил, 2,4-дихлорбензилоксикарбонил, 4-бромбензилоксикарбонил, 3-бромбензилоксикарбонил, 4-нитробензилоксикарбонил, 4-цианобензилоксикарбонил, 2-(4-ксенил)изопропоксикарбонил, 1,1-дифенилэт-1-илоксикарбонил, 1,1-дифенилпроп-1-илоксикарбонил, 2-фенилпроп-2- илоксикарбонил, 2-(п-толуил)-проп-2-илоксикарбонил, циклопентанилоксикарбонил, 1-метилциклопентанилоксикарбонил, циклогексанилоксикарбонил, 1-метилциклогексанилоксикарбонил, 2-метилциклогексанилоксикарбонил, 2-(4-толуилсульфонил)этоксикарбонил, 2-(метилсульфонил)этоксикарбонил, 2-(трифенилфосфино)этоксикарбонил, 9-фтоенилметоксикарбонил ("FMOC"), 2-(триметилсилил)этоксикарбонил, аллилоксикарбонил, 1-(триметилсилилметил)-проп-1-енилоксикарбонил, 5-бензизоксазолилметоксикарбонил, 4-ацетоксибензилоксикарбонил, 2,2,2-трихлорэтоксикарбонил, 2-этинил-2-пропоксикарбонил, циклопропилметоксикарбонил, 4-(децилокси)бензилоксикарбонил, изоборнилоксикарбонил, 1-пиперидилоксикарбонил, и т.п.; бензоилметилсульфонильная группа, 2-(нитро)фенилсульфенильная группа, дифенилфосфиноксидная группа и тому подобные аминозащитные группы. Тип используемой аминозащитной группы не является ограничивающим, только эта замещенная аминогруппа должна быть стабильной в условиях последующего (их) взаимодействия (ий) по другим положениям молекулы и может быть удалена в соответствующий момент без затрагивания остальной части молекулы. Предпочтительными аминозащитными группами являются бензилоксикарбонил, аллилоксикарбонил, трет-бутоксикарбонил и тритил. Подобные аминозащитные группы, используемые в цефалоспорине, пенициллине и пептидах, следовательно, охватываются вышеуказанными терминами. Дальнейшие примеры групп, относящихся к вышеуказанным терминам, описаны J.W.Barton. "Protective Groups in Organic Chemistry", J.G.WcOmie, Ed., Plenum Press, New York, N. Y., 1973, Chapter 2, and T.W.Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, N.Y., 1981, глава 7. Родственный термин "защищенная амино-группа" определяет амино-группу, замещенную защитной для аминной функции группой, обсужден выше.
При осуществлении реакции связывания, для HN-Y'-COOH используют сложноэфирную защитную группу, которая удаляется в условиях, при которых аминозащитная группа остается незатронутой. Аминозащитная группа ацилирующей кислоты P-X'-COOH таким образом сохраняется для защиты амино-группы в течение последующего сочетания с амином (d) при образовании (c).
Карбоксизащитная сложноэфирная группа, как используемая в описании, относится к одному из сложноэфирных производных карбоновой кислоты, обычно используемых для связывания или защиты карбоксильной группы при проведении реакций на других функциональных группах соединения. Примеры таких, защитных для карбоновой кислоты групп включают C1-C4алкил, бензил, 4-нитробензил, 4-метоксибензил, 3,4-диметоксибензил, 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, пентаметилбензил, 3,4-метилендиоксибензил, бензгидрил, 4,4'-диметоксибензил, 2,2', 4,4'-тетраметоксибензгидрил, трет-бутил, трет-амил, тритил, 4-метокситритил, 4,4'-диметокситритил, 4,4', 4''-триметокситритил, 2-фенилпроп-2-ил, триметилсилил, трет-бутил-диметилсилил, фенацил, 2,2,2-трихлорэтил, β- (триметилсилил)этил, β- (ди(н-бутил)метилсилил)этил, п-толуолсульфонилэтил, 4-нитробензилсульфонилэтил, аллил, циннамил, 1-(триметилсилилметил)-проп-1-ен-3-ил, и т.п. группы. Тип используемой для карбоксизамещенной группы не является определяющим, поскольку преобразованная карбоновая кислота стабильна в условиях последовательной (ых) реакции (реакций) по другим положениям молекулы, и защитная группа может быть удалена в соответствующий момент без затрагивания остальной части молекулы. В частности,
важно не подвергать молекулу с защищенной карбоксильной группой воздействию сильных нуклеофильных оснований или восстановительных условий с использованием катализаторов на основе высокоактивированного металла, такого, как никель Ренея. (Таких жестких условий для удаления защитной группы следует также избегать при удалении аминозащитных групп, как это обсуждается далее). Кроме того, примеры этих групп имеются y E.Haslam, "Protecrive Groups in Organic Chemistry", J.G.W.McOmie, Ed., Plenum Press, New York, N. Y., 1973. Chapter 5, and T.W.Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, N.Y., 1981, Chapter 5.
Соединения формулы I также могут быть получены путем синтеза сначала амидного предшественника HN-Y'-CONH(CH2)r-G' и затем реакции с защищенной X-группой. Согласно одному такому способу,
(d) получают и связывают с PN-Y'-COOH (g), как показано ниже, с получением амида (h)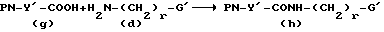
где P обозначает аминозащитную группу, такую, как бензилоксикарбонил (Cbz), трет-бутоксикарбонил (Boc), п-толуолсульфонил и т.п. Предпочтительно, используемая аминозащитная группа удаляется путем гидрирования или обработки слабой кислотой (например, трифторуксусной кислотой) или сильной кислотой (например, HCl). Примеры других подходящих аминозащитных групп приводятся в "Protective Groups in Organic Synthesis", второе издание, T.W.Greene and Peter G.M. Wuts, глава 7, страницы 309-405 (1991), John Wiley and Sons, Inc. Группу Воc или другую подходящую защитную группу удаляют с аминного азота Y-остатка, который затем ацилируют с помощью желаемой аминокислотной ацильной группы с получением дипептида, как показано далее: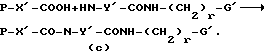
Циано-группу, если она имеется в G', преобразуют и защитные группы на (c) удаляют так, как описано выше.
Связывание соединения P-X'-COOH осуществляют путем сначала защиты амино-группы аминокислоты, если она есть. Применяют стандартные аминозащитные группы, обычно используемые для временной защиты или блокирования амино-группы. Примеры таких защитных групп описаны выше.
Вышеописанные реакции связывания проводят при охлаждении, предпочтительно при температуре между около -20oC и около 15oC. Реакции связывания проводят в инертном органическом растворителе, таком, как диметилформамид, диметилацетамид, тетрагидрофуран, метиленхлорид, хлороформ и т.п., и как в обычных растворителях, так и в смесях таких растворителей. Обычно, когда, в реакциях связывания используют активный сложный эфир ацилирующей кислоты, применяют безводные условия.
Промежуточные продукты (d) и (g) получают стандартными методами органической химии, как представлено на следующих схемах: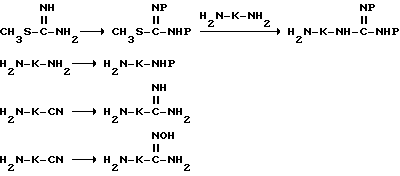
где -K-R обозначает -(CH2)r-G.
Согласно вышеприведенным последовательностям, защищенные гуанидины могут быть получены путем двойной защиты S-метилизотиомочевины. Предпочтительной защитной группой является группа трет-бутилоксикарбонил (Boc), которая может быть введена путем взаимодействия S-метилизотиомочевины в присутствии ди-трет- бутилдикарбоната. Часто используют образующуюся соль кислоты с S-метилизотиомочевиной, которая может быть переведена в свободное основание in situ путем растворения соли в воде и обработки водным основанием. Ди-трет-бутилдикарбонат затем вводят в реакцию в смешивающемся с водой растворителе, таком, как трет-бутанол, с получением дважды защищенной S-метилизотиомочевиной. Желаемый дважды защищенный гуанидин затем получают путем обработки с помощью соответствующего диамина H2N-K-NH2 в инертном растворителе или комбинации растворителей. Обычно эффективно использовать смешивающиеся с водой растворители, такие, как диметилформамид или воду, или их смеси. Такая реакция обычно заканчивается полностью по истечении 3-72 ч. Полученный в результате защищенный гуанидин затем может быть связан, как описано выше, с получением защищенных промежуточных продуктов соединений формулы I, где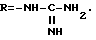
Для соединений формулы I, где R=-NH2, промежуточным соединением является однозащищенный диамин. В большинстве случаев этот промежуточный продукт может быть получен просто взаимодействием незащищенного диамина с одним моль-эквивалентом защитного реагента. Другие методы получения целевого амина (R= -NH2) общеизвестны химикам-органикам. Например, амин может быть получен из любого другого предшественника функциональной группы, например, нитро- или циано-группы. В случае нитро-группы преобразования в амино-группу обычно осуществляют на таких веществах, где нитро-группа непосредственно присоединена к ароматическому кольцу, в частности, к фенильной группе. В таких случаях нитрофенильную группу восстанавливают до соответствующего анилина с помощью какого-либо из многих известных специалисту методов. Одним особенно эффективным методом является обработка нитросоединения гидросульфитом натрия в нереакционноспособном растворителе, таком как этанол, вода, или их смеси. Когда нитросоединение кипятят с обратным холодильником в смеси воды с этанолом в присутствии гидросульфита натрия, то восстановление полностью заканчивается за несколько часов. Циано-группа может быть восстановлена, при желании, в присутствии восстановителя, такого как литийалюминийгидрид, боран, в растворителе, таком, как тетрагидрофуран, или путем восстановления боргидридом натрия, промотированным металлом.
Амидины по настоящему изобретению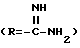
также могут быть получены из нитрильного предшественника. Специалисту известно множество способов осуществления этого превращения. В частности, использование сероводорода в смеси пиридина и триэтиламина с последующей обработкой ацетоном и метилиодидом и, наконец, ацетатом аммония в метаноле представляет собой предпочтительный и эффективный способ осуществления этого преобразования. Альтернативно, для осуществления этого превращения также можно использовать нагревание нитрила с гидрохлоридом гидроксилами основанием, таким, как N, N-диизопропилэтиламин, в содержащем гидроксильную группу растворителе, таком как этанол, с последующим каталитическим гидрированием (например, гидрирование на палладии-на-угле). Этим способом получают гидроксиамидин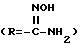
в виде промежуточного продукта, который может быть выделен, если желательно.
Другие соединения, используемые в качестве исходных материалов в синтезе соединений по настоящему изобретению, хорошо известны и, при отсутствии их в продаже, легко синтезируются обычными способами, обычно используемыми для этой цели специалистом.
4-замещенные пролины (Rp= C1-C6алкил, C3-C8циклоалкил или -(CH2)p-L-(CH2)q-T'), которые используют для получения соединений по настоящему изобретению, все имеют цис-конфигурацию заместителя в положении 4 по отношению к карбонильной группе. Промежуточные соединения для введения этой функциональной группы в соединения формулы I получают обычными способами.
Например, 4-замещенные пролиновые производные, где Rp- группа содержит метиленовую группу в точке присоединения пролинового кольца, могут быть получены следующим образом: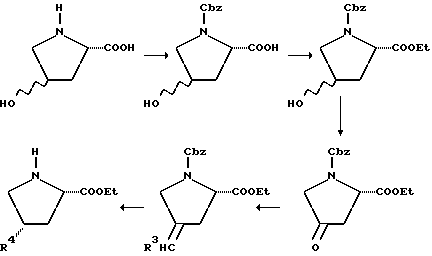
где R4= R3CH2= Rp-группа, содержащая метиленовую группу в месте присоединения к пролиновому кольцу.
4-Гидроксипролин (как цис-, так и транс-формы являются коммерчески доступными) сначала защищают аминозащитной группой, причем в этом случае особенно пригодна Cbz-группа. Полученное в результате промежуточное соединение затем этерифицируют до сложного эфира (особенно пригодны метиловый и особенно этиловый сложные эфиры) и затем окисляют с получением соответствующего кетона. Это окисление осуществляют в условиях любого из множества способов окисления, таких как окисление по Jones или с помощью пиридинийхлорформиата, особенно пригодным для этого превращения является использование пиридинийхлорформиата в сухом нереакционноспособном растворителе, таком, как дихлорметан. Когда реакцию проводят в течение 8-16 часов, то при комнатной температуре взаимодействие обычно полностью заканчивается. Это многофункциональное кетоновое промежуточное соединение затем вводят во взаимодействие с соответствующим реагентом Виттига для получения желаемого олефина. Обычно соответствующий, Rp-замещенный трифенилфосфонийгалогенид добавляют к сухому инертному растворителю (например, тетрагидрофуран), который содержит сильное основание (например, трет-бутоксид калия). Вводя кетон спустя примерно три часа при комнатной температуре, может быть выделен желаемый промежуточный олефин. С целью получения хороших выходов олефина, предпочтительно использовать 0,4-0,6-мольный избыток реагента Виттига по отношению к кетону. Олефин затем восстанавливают до желаемого Rp-замещенного пролина с помощью стандартных методов восстановления. Каталитическое гидрирование является наиболее легким способом осуществления этого превращения в лаборатории. Гидрирование олефина в присутствии катализатора (например, 5%-ный палладий-на-угле) в инертном растворителе, таком как этанол, эффективно при атмосферном давлении. В случае таких промежуточных продуктов, в которых аминозащитной группой является Cbz, гидрирование также удаляет защитную группу, что приводит к соединению, которое может быть использовано для связывания с P-X'-COOH. Как следует учесть специалисту, этот способ не может быть эффективным для получения соединений, где Rp-группа присоединена к пролиновому кольцу через гетероатом или являются ароматическим кольцом. Таким образом, в вышеприведенной схеме R3 должен означать алкил, аралкил (например, бензил), (циклоалкил)алкил и т.д.
Соответствующий способ получения этих промежуточных продуктов представлен в следующей схеме: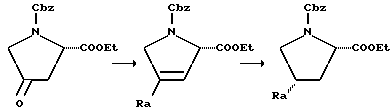
Вышеприведенная реакционная схема альтернативна вышеописанной реакции Виттига и пригодна для получения соединений, которые не могут быть получены с помощью реагентов Виттига. Так, для получения промежуточных продуктов, где Ra обозначает алкил, фенил и т.п., промежуточный пирролидинон вводят во взаимодействие с соответствующим реактивом Гриньяра. Обычно используют незначительный молярный избыток реактива Гриньяра, работая обычно при низкой температуре (например, от -80 до -60oC), в низкозамерзающем инертном растворителе, таком, как тетрагидрофуран. После добавления реагентов, реакционной смеси можно дать нагреться до комнатной температуры, после чего реакция обычно полностью заканчивается за несколько часов. Полученное промежуточное соединение дегидратируют, например, обработкой трифторуксусной кислотой. Промежуточное 3,4-дегидросоединение затем восстанавливают до желаемого промежуточного цис-соединения в тех же условиях восстановления, что описаны выше для восстановления олефинового промежуточного продукта.
Промежуточные соединения, где гетеро-L-группа является кислородом и она непосредственно присоединена к пролиновому кольцу (т.е. p=0), можно получать по реакции Mitsunobu (Mitsunobu, Synthesis, 1 (1981)):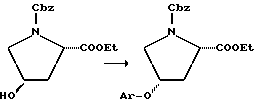
По этой реакции сложный эфир транс-гидроксипирролидинкарбоновой кислоты обрабатывают трифенилфосфином в растворителе, таком как тетрагидрофуран, в присутствии Ar-O-H. Смесь охлаждают примерно до 0oC и добавляют диэтилазодикарбоксилат. После нагревания до комнатной температуры реакционную смесь обрабатывают с получением желаемого промежуточного цис-продукта. Тогда как вышеприведенная схема представляет реакцию для соединения, где L = -O-, p = q = 0 и T=Ar, эта реакционная последовательность пригодна для получения других соединений, где p=0 и L= -O-.
Промежуточные соединения, где L обозначает серу и присоединено непосредственно к кольцу, могут быть получены сначала преобразованием гидрокси-группы в тозилатную или другую подобную удаляемую группу и последующим замещением тиолят-анионом (см. , например, Kropcho и др., J. Med. Chem., 31, 1148-1160 (1988); Smith и др., J.Med.Chem., 31 875-855 (1988)).
Промежуточные продукты, где L обозначает азот, присоединенный непосредственно к кольцу, могут быть получены сначала преобразованием гидрокси-группы в тозилатную или другую подобную удаляемую группу и последующим замещением азидом. Азид можно восстанавливать известными методами и затем алкилировать с получением желаемой функциональной группы (см., например, Smith и др., J.Med.Chem., 31, 875-855 (1988)).
Соединения по настоящему изобретению, содержащие цис-Ohi-функциональную группу, получают путем синтеза этилового эфира (S)-индолин-карбоновой кислоты из соответствующей кислоты (см. Vincent и др., Drug Design and Discovery, т. 9, с. 11-28 (1992)) и восстановления этого промежуточного продукта гидрированием над 5% Pd/C в этаноле с получением сложного эфира октагидроиндол-2-карбоновой кислоты, обычно называемого Ohi-эфиром, как представлено ниже: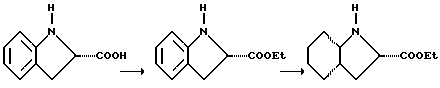
Соединения по настоящему изобретению, содержащие транс-Ohi -функциональную группу, получают по методу Vincent и др., Drug Desugn and Discovery, т.9, с.11-28 (1992)). Этот синтез представлен на схеме, показанной ниже: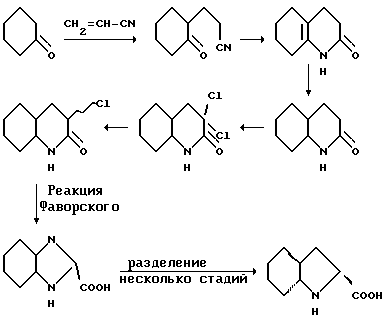
Соединение по настоящему изобретению, содержащее бициклическую систему с гетероатомом или без него, может быть получено по методу Teetz и др., Tetrahedron Letters, 25, 4479 (1984). Обычная схема получения следующая: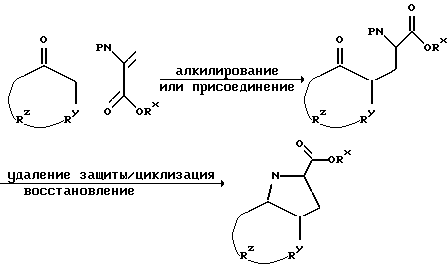
где P обозначает защитную группу и Rx - алкил.
Промежуточные соединения для введения N-замещенной глициновой функциональной группы (Y), используемые для получения соединений по настоящему изобретению, получают обычными методами.
Например, сложный галогенацетатный эфир, такой, как трет-бутилбромацетат, может быть преобразован в желаемое замещенное соединение путем обработки с помощью соответствующего первичного амина:
BrCH2COO-трет-бутил + RgNH2---> HNRgCH2COO-трет-бутил.
Трет-Бутил-бромацетат подвергают взаимодействию с соответствующим амином сами по себе или, предпочтительно, в нереакционноспособном растворителе, таком как спирт. Предпочтительно использовать мольный избыток амина для интенсификации реакции до ее полного протекания. Предпочтительно, реакционная смесь также содержит нереакционноспособный акцептор кислоты, такой как, по крайней мере, мольный эквивалент триэтиламина. Тогда как реагенты обычно объединяют при охлаждении (например, 0oC), реакцию обычно проводят при нагревании до комнатной температуры, после чего реакция завершается полностью за 24 часа. Хотя бромацетат является предпочтительным, для этого превращения можно использовать другие галогенацетаты, такие, как иодацетаты и хлорацетаты. Могут быть подобным образом использованы другие сложноэфирные группы. Трет-Бутиловый сложный эфир является предпочтительным, поскольку он может позже быть легко удален при последующей обработке анизолом и трифторуксусной кислотой.
Второй способ получения этих промежуточных продуктов представлен следующей реакционной схемой:
R0-CHO+H2NCH2COO-этил ---> R0-CH=NCH2COO этил ---> R0-CH2- NHCH2COO-этил,
где R0-CH2 обозначает Rg-группу, содержащую незамещенную метиленовую группу, соседнюю с местом присоединения к глициновой группе.
В вышеприведенной реакционной схеме, соответствующий альдегид смешивают с глициновым сложным эфиром в нереакционноспособном растворителе, таком как метанол или этанол. Если используют образовавшуюся соль сложного глицинового эфира, то может быть добавлен мольный эквивалент основания, такого как гидроксид калия, для получения свободного основания сложного аминоэфира. Взаимодействие альдегида со сложным глициновым эфиром приводит к образованию промежуточного основания Шиффа, которое затем можно восстанавливать in situ путем обработки восстановителем, таким как цианоборгидрид натрия. Образование основания Шиффа происходит обычно в течение менее часа; восстановление обычно заканчивается спустя 10-15 ч. Сложные метиловые или этиловые эфиры, пригодные в качестве этих групп, могут быть удалены (снятие защиты) путем обработки гидроксидом лития в водном диоксане. Использование соответствующего кетона вместо альдегида R0-CHO приводит к получению промежуточных продуктов, где метиленовая группа, связанная с глицинамином, замещена.
Альтернативно, и особенно для тех соединений, где Rg - Ar (т.е. без введения алкильной группы), предпочтительно получать промежуточное соединение P-X'-CONHAr обычными способами (например, путем взаимодействия активированной формы P-X'-COOH с ArNH2) и затем взаимодействия этого промежуточного соединения с алкилгалогенацетатом, как описано выше, с получением P-X'-CONHAr-CH2COO-алкила, который затем далее может быть преобразован обычным путем.
Многие целевые соединения по настоящему изобретению или промежуточные продукты для их получения могут быть взаимопреобразованы стандартными методами. Например, арильные соединения, которые замещены нитрогруппой, могут быть восстановлены (например, в присутствии гидросульфита натрия в нереакционноспособном растворителе, таком, как этанол, вода или их смесь). Когда нитросоединение кипятят с обратным холодильником в смеси вода/этанол в присутствии гидросульфита натрия, восстановление обычно полностью завершается за несколько часов. Полученный в результате амин может присутствовать в целевом продукте; если амин имеется в промежуточном продукте, то может быть желательным образование его в его целевую желаемую форму (например, ацилирование с получением ацилированного амина) или защита для избежания побочных реакций в процессе осуществления последовательности химических реакций. Если желательным соединением является свободный амин, то в этом случае особенно пригодной является Cbz-защитная группа. Другие превращения и взаимопревращения этого типа известны химикам-органикам.
Как ясно специалистам, вышеуказанные преобразования могут быть осуществлены при использовании исходных веществ, указанных выше, или в большинстве случаев также можно получать промежуточные ди- или трипептиды, содержащие одну и ту же соответствующую функциональную группу. В последних случаях может отпадать необходимость или потребность защиты различных групп; следовательно, порядок и тип химических реакций диктуется необходимостью и типом защитных групп, а также последовательностью осуществляемых реакций. Также специалисту понятно, что можно выбрать другие защитные группы для того, чтобы они служили целям защиты функциональной группы в процессе последовательности химических реакций, но также могут быть удалены при соответствующих условиях и в соответствующем порядке с учетом последовательности превращений. Например, в вышеприведенной схеме 1 G' включает заместители, где R обозначает -CN; эта нитрильная группа может быть преобразована в амидин или восстановлена до амина, которые могут быть необязательно далее преобразованы в гуанидины по настоящему изобретению.
Соединения по настоящему изобретению лучше всего выделять в виде солей присоединения кислот. Соли соединений формулы I, образованные из кислот, таких как указанные выше, пригодны в качестве фармацевтически приемлемых солей для введения в качестве антитромботических агентов и для приготовления составов этих агентов. Другие соли присоединения кислот могут быть получены и использованы для выделения и очистки пептидов. Например, также могут быть использованы соли с сульфокислотами, такими как метансульфокислота, н-бутансульфокислота, п-толуолсульфокислота и нафталинсульфокислота.
Соединение формулы I получают:
а) путем одновременного или последовательного удаления защитной группы (групп) P из соответствующего соединения формулы II:
(P)X-Y-NH-(CH2)r-G(P), (II)
где (P)X - радикал X, который может содержать одну или более защитных групп P, независимо выбранных из аминозащитной группы P для соединения формулы I, где X включает основной NH радикал, и карбоксизащитную группу P для соединения формулы I, где X включает карбоксиостаток и G(P) обозначает радикал G, который может содержать одну или более независимо выбранных аминозащитных групп P; или
б) для соединения формулы I, где R обозначает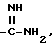
путем гидрирования соответствующего соединения формулы I, где
R обозначает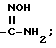
и затем, когда необходима соль соединения формулы I, образования соли с фармацевтически приемлемой кислотой.
Может быть предпочтительным проведение способа б) одновременно со способом а). Для соединения формулы I, в которой кислотнозащитной группой является трет-бутиловый сложный эфир и/или аминозащитными группами являются трет-бутилоксикарбонил, защитная (ые) группа (группы) могут быть удалены обработкой сильной кислоты, такой как трифторуксусная кислота или безводный хлороводород в инертном растворителе, таком, как диоксан или дихлорметан, в присутствии анизола. Для соединения формулы I, в которой кислотнозащитной группой является сложный бензиловый эфир и/или аминозащитной группой является бензилоксикарбонил, защитная (ые) группа (группы) могут быть удалены путем гидрирования, обычно осуществляемого в этанольном хлороводороде в присутствии палладия-на-угле в качестве катализатора.
Предпочтительным способом очистки соединений формулы I, при одновременном получении желаемой стабильной солевой формы, является способ, описанный в патенте США 5 250 660. Согласно этому способу, стабильные сульфаты или гидрохлориды получают путем очистки препаративной хроматографией с C18, обращенной фазой, при которой водный компонент содержит серную кислоту или соляную кислоту при pH 2,5 и органическим компонентом является ацетонитрил. pH кислой подвижной фазы доводят до величины примерно от pH 4 до примерно 6 с помощью анионообменной смолы в гидроксильной форме, например, как Bio-Rad AG-1X8. После установления pH раствор соли сульфата трипептида или гидрохлорида трипептида лиофилизуют с получением чистой соли в сухой порошкообразной форме. Например, согласно способу, сырой D-Phe-Pro-p-NHCH2C6H4C(NH)NH2-сульфат можно растворять в воде и раствор вводить в колонку размером 5 см х 50 см с фазой Vydac Cl8RP ВЭЖХ. Используют градиент 2-10% В (A = 0,01% H2SO4; B = ацетонитрил) в течение 10 ч. Многочисленные фракции собирают и те, в которых содержится продукт, определяемый с помощью аналитической R P ВЭЖХ, объединяют. pH объединенных фракций доводят до 4,0-4,5 с помощью смолы AG-1X8 в гидроксидной форме (Bio-Rad, 3300 Ragatto Blvd. , Richmond, 94804). Раствор фильтруют и фильтрат лиофилизуют с получением чистого D-, L-диамида в виде сульфатной соли.
Оптически активные изомеры диастереомеров по радикалу X также составляют часть настоящего изобретения. Такие оптические активные изомеры можно получать из соответствующих оптически активных предшественников путем вышеописанных способов, или путем разделения рацемических смесей. Это разделение можно осуществлять путем получения производных с хиральным реагентом и последующим хроматографированием или путем повторной кристаллизации. Удаление хирального вспомогательного соединения обычными методами приводит, по существу, к оптически чистым изомерам соединений по настоящему изобретению или к их предшественникам. Подробности в отношении разделений могут быть установлены из Jacques и др., Enantiomers, Racemares, and Resolutions, John Wiley and Sons, 1981.
Соединения, используемые в качестве исходных веществ в синтезе соединений по настоящему изобретению, хорошо известны и, если их нет в продаже, легко синтезируются стандартными способами, обычно используемыми для этой цели специалистами.
Следующие далее примеры относятся далее к описанию изобретения и к соответствующим сравнительным примерам, но они не ограничивают объем охраны изобретения.
Используемые в настоящем описании аббревиатуры имеют следующее значение:
Аминокислотные остатки: Arg = аргинил; Glu = глутамил; Cly = глицил; Pro = пролил; hPro = гомопролил; Azt = азетидин-2-карбонил; Phg = фенилглицил; Phe = фенилаланил; hPhe = гомофенилаланил; 1-Tiq = 1,2,3,4-тетрагидроизохинолин-1-карбонил; 3-Tiq = 1,2,3,4-тетрагидроизохинолин-3-карбонил; Cha = β- циклогексилаланил; hCha = α- амино -γ- циклогексилбутирил; NM1 = N-метилиндол-2-оил; Ohi = цис-октагидроиндол-2-оил; 1-Piq = пергидро-изохинолин-1-карбонил; 3-Piq = пергидроизохинолин-3-карбонил; Met = метионил; Met (O2) = S,S-диоксометионил. Agm - агматин; Boc - трет-бутилоксикарбонил; Bn - бензил; Cbz - бензилоксикарбонил; ДЦК - дициклогексилкарбодиимид; ДМФ - диметилформамид; Et - этил; ДМСО - диметилсульфоксид; EtOAc - этилацетат; Et2O - диэтиловый эфир; EtOH - этанол; Fmoc - 9-флуоренилметоксикарбонил; FAB-MS - масс-спектр, получаемый путем бомбардировки быстрыми атомами; FD-MS - масс-спектр с десорбцией поля; 1S-MS - масс-спектр, получаемый при применении пучка электронов; HRMS - масс-спектр высокого разрешения; HOBT = 1-гидроксибензотриазол-гидрат; ИК = инфракрасный спектр; RPHPLC - высокоэффективная жидкостная хроматография с обращенными фазами; Ph = фенил; TFA = ТФК = трифторуксусная кислота; ТГФ - тетрагидрофуран; ТСХ = тонкослойная хроматография.
Используют следующие параметры для высокоэффективной жидкостной хроматографии с обращенными фазами:
растворитель A: 0,05% водный раствор соляной кислоты (1,5 мл концентрированной соляной кислоты в 3 л воды);
растворитель B: ацетонитрил;
градиент: как указано в каждом примере;
Метод 1: колонка: 2,5 х 25 см, фаза Vydac C18; объемная скорость потока: 5 мл/мин;
Метод 2: колонка: 5 см х 25 см; фаза: Vydac C18; объемная скорость потока: 10 мл/мин;
Метод 3: колонка: 2,5 см x 50 см; фаза Vydac C18; объемная скорость потока: 10 мл/мин.
Если не указано ничего другого, установление и достижение pH осуществляют с помощью водных растворов кислоты или основания.
В примерах, где указывается 1H-ЯМР, продукт, полученный в реакции, охарактеризован данными протонного магнитного резонанса, подтверждающими, что получено указанное соединение; ИК без предоставления данных указывает, что получен удовлетворительный инфракрасный спектр. HRMS используют для подтверждения точной массы соединений, для которых не получен удовлетворительный элементный анализ, продукта по желаемому способу; указывается элементарный состав наблюдаемого иона (например, MH+).
Пример 1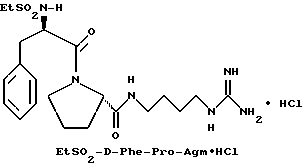
А) Получение Boc-D-Phe-Pro-OBn.
К раствору Boc-D-Phe-OH (89,1 г, 336 ммоль), Pro-OBn-гидрохлорида (81,2 г, 336 ммоль) HOBT (50 г, 370 ммоль) и N,N-диизопропилэтиламина (176 мл, 1,008 ммоль) в дихлорметане (600 мл) при 0oC добавляют 1-(3-диметиламинопропил)-3-этилкарбодиимид-гидрохлорид (71 г, 370 ммоль). После перемешивания в течение 18 ч смесь разбавляют диэтиловым эфиром (1 л) и промывают последовательно три раза 1н лимонной кислотой (250 мл), один раз водой (250 мл), три раза насыщенным водным раствором бикарбоната натрия (250 мл) и один раз насыщенным водным раствором хлорида натрия (250 мл). Органическую фазу сушат (Na2SO4), фильтруют и концентрируют в вакууме с получением 140 г (92,5%) бледно-желтой пены. FD/MS m/e: 452 (M+); 1H-ЯМР
Б) Получение p-Phe-Pro-OBn•TFA.
К перемешиваемому раствору Boc-D-Phe-Pro-OBn (68 г, 150 ммоль) в дихлорметане (50 мл) при 0oC добавляют анизол (20 мл), затем трифторуксусную кислоту (400 мл). После перемешивания в течение 3 ч растворители выпаривают в вакууме и вязкий маслянистый остаток растворяют в диэтиловом эфире (1,5 л) и охлаждают (72 ч). Белого цвета осадок отфильтровывают, промывают диэтиловым эфиром (300 мл) и сушат с получением 59,4 г (85%) белого порошка. 1H-ЯМР.
В) Получение EtSO2-D-Phe-Pro-OBn.
К перемешиваемому раствору D-Phe-Pro-OBn•ТФК (12 г, 25,7 ммоль) и триэтиламина (7 мл, 50,2 ммоль) в дихлорметане (200 мл) при -78oC добавляют этансульфонилхлорид (2,65 мл, 28,3 ммоль) по каплям через капельную воронку. Реакционный сосуд нагревают до 0oC и после перемешивания в течение 4 ч добавляют воду (10 мл). Органическую фазу промывают три раза 1н раствором соляной кислоты (100 мл), один раз насыщенным раствором хлорида натрия (100 мл) и затем растворитель удаляют в вакууме. Продукт очищают путем флэш-хроматографии на силикагеле, элюируя смесью этилацетата с гексаном (6:4). Содержащие продукт фракции (оценивают с помощью ТСХ) объединяют и концентрируют с получением 6,62 г (58%) желтого цвета масла, которое отверждается. 1H-ЯМР; FD-MS, m/e: 445 (M+); Анализ для C23H28N2O5S:
рассчитано, %: C 62,14; H 6,35; N 6,30.
найдено, %: C 61,87; H 6,37; N 6,18.
Г) Получение EtSO2-D-Phe-Pro-OH.
К перемешиваемому раствору EtSO2-D-Phe-Pro-OBn (4,5 г, 10,1 ммоль) в п-диоксане (150 мл) добавляют раствор моногидрата гидроксида лития (2,1 г, 50,5 ммоль) в воде (75 мл). После перемешивания в течение 16 ч объем раствора уменьшают в вакууме наполовину и раствор разбавляют водой (300 мл) и 0,1н раствором NaOH (100 мл).
Водную фазу затем промывают дважды диэтиловым эфиром (250 мл), подкисляют твердой лимонной кислотой и затем экстрагируют три раза этилацетатом (150 мл). Объединенные этил-ацетатные экстракты промывают насыщенным водным раствором хлорида натрия (200 мл), сушат (MgSO4), фильтруют и концентрируют с получением 3,6 г (90%) белого твердого вещества.
FD-MS, m/e: 355 (M+).
Анализ для C16H22N2O5S:
рассчитано, %: C 54,22; H 6,26; N 7,90.
найдено, %: C 54,40; H 6,42; N 7,85.
Д) Получение N,N-ди-Boc-S-метилизотиомочевины
К перемешиваемому раствору ди-трет-бутил-дикарбоната (100 г, 458 ммоль) в трет-бутаноле (300 мл) добавляют раствор сульфата бис-S-метилизотиомочевины (32,7 г, 117 ммоль) в воде (150 мл), затем раствор гидроксида натрия (19,2 г, 480 мл) в воде (150 мл). После перемешивания в течение 48 ч смесь концентрируют примерно до 1/3 первоначального объема в вакууме и разбавляют диэтиловым эфиром (500 мл). Органическую фазу промывают один раз водой (250 мл), три раза 1н раствором лимонной кислоты (250 мл) и вновь один раз водой (250 мл). Органическую фазу затем сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 42 г (62%) белого твердого вещества. 1H-ЯМР.
Е) Получение Ng,Ng'-ди-Boc-агматин
К перемешиваемому раствору 1,4-бутандиамина (23 г, 258 ммоль) в смеси 2: 1 диметилформамида с водой (300 мл) через капельную воронку добавляют раствор N,N'-ди-Boc-S-метилизотиомочевины (15 г, 52 ммоль) в диметилформамиде (100 мл). После перемешивания в течение 2 ч растворители удаляют в вакууме и остаток растворяют в 1н лимонной кислоте (250 мл), разбавляют водой (250 мл) и промывают этилацетатом (250 мл). Этилацетатную фазу снова экстрагируют 1н раствором лимонной кислоты (100 мл) и объединенные водные фазы подщелачивают с помощью карбоната натрия, насыщают твердым хлоридом натрия и экстрагируют дважды этилацетатом (250 мл). Объединенные этилацетатные экстракты промывают насыщенным водным раствором хлорида натрия (200 мл), сушат (MgSO4), фильтруют и концентрируют с получением 12,5 г (73%) вязкого сиропа. 1H-ЯМР.
Ж) Получение EtSO2-D-Phe-Pro-Agm(Boc)2.
К перемешиваемому раствору Ng,Ng'-ди-Boc-агматина (2 г, 6 ммоль) в дихлорметане (30 мл) добавляют EtSO2-D-Phe-Pro-OH (2,1 г, 6 ммоль), HOBT (810 мг, 6 ммоль) и N,N-диизопропилэтиламин (1,6 г, 12 ммоль) с последующим добавлением 1-(3-диметиламинопропил)-3-этилкарбодиимид-гидрохлорида (1,4 г, 73 ммоль). После перемешивания в течение 20 ч раствор разбавляют этилацетатом (300 мл) и промывают три раза 1н раствором лимонной кислоты (150 мл), один раз водой (150 мл) и дважды насыщенным водным раствором бикарбоната натрия. Органическую фазу затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток хроматографируют на силикагеле, элюируя возрастающим градиентом смеси этилацетата с гексаном (1:4) до этилацетата.
Содержащие продукт фракции (на основании ТСХ) объединяют и концентрируют с получением 2,4 г (60%) вязкого масла. 1H-ЯМР, FD-MS, m/e: 668 (MH+).
З) Получение EtSO2-D-Phe-Pro-Agm•HCl.
Перемешиваемую суспензию EtSO2-D-Phe-Pro-Agm(Boc)2 (1,6 г, 2,4 ммоль) в анизоле (1 мл) растворяют в трифторуксусной кислоте (20 мл) и продолжают перемешивание в течение 1 ч при комнатной температуре. Растворитель затем удаляют в вакууме и остаток распределяют между водой (100 мл) и диэтиловым эфиром (50 мл). Водную фазу промывают снова диэтиловым эфиром (50 мл) и затем частично концентрируют и лиофилизируют с получением 1,4 г сырой трифторацетатной соли. Половину этого вещества затем растворяют в воде и очищают с помощью R P HPLC (метод 1; 98/2 /A/B/); соотношение изменяют вплоть до 50/50 /A/B/, 60 минут) с получением 490 мг (81%) белого порошка. 1H-ЯМР; FD-MS, m/e: 467 (M+).
Анализ для C21H34N6O4S•HCl• H2O:
рассчитано, %: C 48,41; H 7,16; N 16,13; Cl 6,80.
найдено, %: C 48,01; H 6,81; N 16,15; Cl 6,97.
Пример 2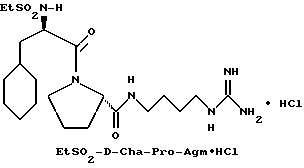
А) Поучение Boc-D-Cha-Pro-OBn.
Способом, по существу эквивалентным описанному в примере 1-А, Boc-D-Cha-Pro-ОВп получают из Boc-D-Cha-OH и Pro-OBn. HCl с выходом 91% (109 г), FD-MS, m/e 458 (M+).
Б) Получение D-Cha-Pro-OBn•TFA.
Способом, по существу эквивалентным описанному в примере 1-Б, D-Cha-Pro-OBn•TFA получают с выходом 130 г (116% от теоретически рассчитанного). 1H-ЯМР; FD-MS m/e: 359 (M+).
В) Получение EtSO2-D-Cha-Pro-OBn.
Способом, по существу эквивалентным описанному в примере 1-В, EtSO2-D-Cha-Pro-OBn получают с выходом 2,3 г (20%).
1H-ЯМР; FD-MS. m/e 450 (M+)
Анализ для C23H34N2O5S:
рассчитано, %: C 61,31; H 7,61; N 6,22.
найдено, %: C 61,55; H 7,59; N 6,28.
Г) Получение EtSO2-D-Cha-Pro-OH.
Способом, по существу эквивалентным описанному в примере 1-Г, EtSO2-D-Cha-Pro-ОН получают с выходом 0,78 г (48%).
1H-ЯМР, FD-MS, m/e: 361 (M+).
Д) Получение EtSO2-D-Cha-Pro-Agm(Boc)2.
Способом, по существу эквивалентным описанному в примере 1-Ж, 400 мг (40%) EtSO2-D-Cha-Pro-Agm(Boc)2 получают из EtSO2-D-Cha-Pro-OH и Ng,Ng'-ди-Boc-Agm. 1H-ЯМР; FD-MS, m/e. 674 (MH+).
Е) Получение EtSO2-D-Cha-Pro-Agm•HCl.
Способом, по существу эквивалентным описанному в примере 1-З, EtSO2-D-Cha-Pro-Agm•HCl получают с выходом 100 мг (45%). Продукт очищают с помощью RPHPLC (метод 1; 98/2 /A/B/, соотношение изменяют до 50/50 /A/B/, 60 мин). 1H-ЯМР; FD-MS, m/e 473 (M+),
Анализ для C21H40N6O4S•1,2HCl •H2O
рассчитано, %: C 47,20; H 8,15; N 15,73; Cl 7,96.
найдено, %: C 47,47; H 7,84; N 16,10; C 7,80.
Пример 3
А) Получение EtOCO-D-Phe-Pro-OH.
Способами, по существу эквивалентными, описанными в примерах 1-В и 1-Г, при использовании этихлорформиата вместе этансульфонилхлорида, получают 6,59 г (92%) EtOCO-D-Phe-Pro-OH.
1H-ЯМР; FD-MS, m/e 335 (M+);
Анализ для C17H22N2O5:
рассчитано, %: C 61,07; H 6,63; N 8,38.
найдено, %: C 60,88; H 6,72; N 8,14.
Б) Получение EtOCO-D-Phe-Pro-Agm•HCl.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 2,1 г (54%) EtOCO-D-Phe-Pro-Agm(Boc)2 из EtOCO-D-Phe-Pro-OH и Ng-Ng'-di-Boc-Agm. Затем способом, по существу эквивалентным описанному в примере 1-З, получают 390 мг (77%) EtOCO-D-Phe-Pro-Agm•HCl. Продукт очищают путем RPHPLC (метод 1; 98/2 /A/B/, изменяющееся до 50/50 /A/B/, 60 мин). 1H-ЯМР; FD-MS m/e 447 (M+).
Анализ для C22H34N6O4•0,9HCl •0,2ТФК•H2O
рассчитано, %: C 51,70; H 7,22; N 16,15; Cl 6,13.
найдено, % C 51,73; H 7,20; N 16,54; Cl 6,36.
Пример 4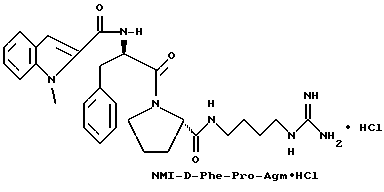
(N-[(1-Мeтил-1H-индoл-2-ил)кapбoнил]-D-фенилаланил-N- [4-[(аминоиминометил)амино]бутил]-L-пролинамид-моногидрохлорид).
А). Получение NMI-D-Phe-Pro-OH.
К раствору N-метил-индол-2-карбоновой кислоты (2,6 г, 14,9 ммоль) в безводном тетрагидрофуране (45 мл) добавляют пентафторфенол (3 г, 16,5 ммоль), затем 1-(3-диметиламинопропил)- 3-этилкарбодиимид (3,2 г, 16,5 ммоль). Смесь кипятят с обратным холодильником при перемешивании в течение 3,5 ч и затем охлаждают до комнатной температуры. К этой смеси добавляют раствор D-Phe-Pro-OBn•TFA (7 г, 14,9 ммоль) и N,N-диизопропилэтиламина (4 г, 30 ммоль) в тетрагидрофуране (25 мл). После перемешивания еще в течение 2 ч, растворители удаляют в вакууме и остаток растворяют в этилацетате (500 мл), после чего полученный раствор промывают три раза 0,1 н водным раствором бисульфата натрия (250 мл) и три раза 1н водным раствором карбоната калия (250 мл). Органическую фазу сушат (Na2SO4), фильтруют и концентрируют в вакууме с получением 6,5 г аморфного твердого вещества (смесь целевого продукта, загрязненная пентафторфенолом). Этот сырой продукт затем гидролизуют способом, по существу эквивалентным описанному в примере 1-Г, с получением 3,8 г (62%) не совсем белого твердого вещества. 1H-ЯМР; FD-MS, m/e 419 (M+).
Б) Получение NMI-D-Phe-Pro-Agm•HCl.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 900 мг (20%) NMI-D-Phe-Pro-Agm(Boc)2.
Затем способом, по существу эквивалентным описанному в примере 1-З, получают 144 мг (31%) NMI-D-Phe-Pro-Agm•HCl.
Сырой продукт растворяют в ледяной уксусной кислоте и очищают путем RPHPLC (метод 1, (A/B) от 90/10 до 40/60, 80 минут).
1H-ЯМР. FD-MS, m/e 532 (M+)
Анализ для C29H37N7O3•0,9HCl •0,6ТФК•0,5H2O:
рассчитано, %: C 56,46; H 6,29; N 15,27; Cl 4,97;
найдено, %: C 56,77; H 6,58; N 15,35; Cl 5,28.
Пример 5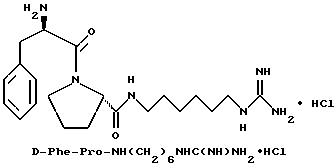
А) Получение Boc-D-Phe-Pro-OH.
К раствору Boc-D-Phe-Pro-OBn (145 г, 320 ммоль) в п-диоксане (660 мл) добавляют раствор моногидрата гидроксида лития (54 г, 1,280 ммоль) в воде (330 мл) при интенсивном перемешивании. Спустя 4 ч раствор концентрируют в вакууме примерно до 1/4 первоначального объема и разбавляют водой (350 мл) и 0,1н раствором гидроксида натрия (100 мл). Водную фазу промывают три раза диэтиловым эфиром (250 мл) и затем подкисляют до pH 3 твердой лимонной кислотой, в результате чего выпадает осадок. Твердое вещество фильтруют, промывают дважды водой и затем сушат в высоком вакууме, получая 91 г (78%) белого твердого вещества. 1H-ЯМР; FD-MS m/e 363 (M+).
Б) Получение Ng,Ng'-ди-Boc-6-аминогексилгуанидина
Способом, по существу эквивалентным описанному в примере 1-Е, получают 4,7 г (66%) Ng,Ng'-ди-Boc-6-амино гексилгуанидина из 1,6-гександиамина.
В) Получение Boc-D-Phe-Pro-NH(CH2)6NHC(NBoc)NH(Boc).
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 1,3 г (62%) Boc-D-Phe-Pro-6-NH(CH2)6NHC(NBoc)NH (Boc) из Boc-D-Phe-Pro-OH и Ng,Ng'-ди-Boc-аминогексил- гуанидина. 1H-ЯМР; FD-MS, m/e: 703 (M+).
Г) получение D-Phe-Pro-NH(CH2)6NHC(NH)NH2 •HCl.
Способом, по существу эквивалентным описанному в примере 1-З, получают приблизительно 100 мг D-Phe-Pro-NH(CH2)6NHC(NH)NH2•HCl. FD-MS, m/e 389 (M+)
Анализ для C21H34N6O2•0,9HCl •0,9ТФК•0,5H2O:
рассчитано, %: C 49,97; H 6,95; N 15,34;
найдено, %: C 49,60; H 7,13; N 15,23.
Пример 6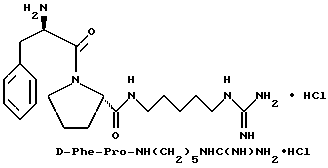
А) Получение Ng,Ng'-ди-Boc-5-аминопентилгуанидина
Способом, по существу эквивалентным описанному в примере 1-Е, получают 1,73 г (72%) Ng, Ng'-ди-Вос-5- аминопентилгуанидина из 1,5-пентандиамина. FD-MS, m/e: 345 (M+); 1H-ЯМР;
Б) Получение Boc-D-Phe-Pro-NH(CH2)5NHC(NBoc)NH(Boc)
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 1,9 г (92%) Boc-D-Phe-Pro-NH(CH2)5NHC(NBoc)NH(Boc) из Boc-D-Phe-Pro-OH и Ng, Ng'-ди-Boc-5-аминопентилгуанидина.
1H-ЯМР; FD-MS, m/e: 689 (M+).
В) Получение D-Phe-Pro-NH(CH2)5NHC(NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примере 1-З, получают примерно 100 мг D-Phe-Pro-NH(CH2)5NHC(NH)NH2 •HCl. Продукт очищают с помощью RPHPLC (метод 1; (A/B) от 98/2 до 40/60, 40 мин). FD-MS, m/e 389 (M+).
Анализ для C20H32N6O2•0,9HCl• 0,9ТФК•0,7H2O:
рассчитано, %: C 48,71; H 6,79; N 15,63;
найдено, %: C 48,34; H 6,68; N 16,01.
Пример 7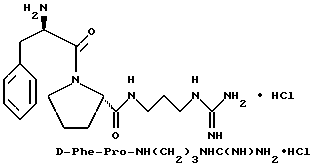
А) Получение Boc-D-Phe-Pro-NH(CH2)3NHC(NBoc)NH(Boc).
К раствору 1,3-диаминопропана (2,2 г, 30 ммоль) в диметилформамиде (25 мл) добавляют раствор N,N'-ди-Boc-S-метилизотиомочевины (2,9 г, 10 ммоль) в диметилформамиде (25 мл). После перемешивания в течение 1 ч смесь разбавляют дихлорметаном (400 мл) и промывают дважды смесью насыщенного водного раствора бикарбоната натрия и насыщенного водного раствора хлорида натрия (200 мл) и один раз насыщенным водным раствором хлорида натрия (250 мл). Органическую фазу сушат (MgSO4), фильтруют и частично концентрируют в вакууме до объема примерно 200 мл.
К этому раствору затем добавляют Boc-D-Phe-Pro-OH (3,6 г, 10 ммоль) HOBT (1,3 г, 10 ммоль) и N,N-диизопропил-этиламин (1,3 г, 10 ммоль), затем 1-(3-диметиламинопропил)-3-этилкapбoдиимид•HCl (2,1 г, 11 ммоль). После перемешивания в течение 16 ч растворители удаляют в вакууме и остаток обрабатывают этилацетатом (250 мл). Органическую фазу промывают три раза 1н раствором лимонной кислоты (200 мл), один раз водой (100 мл), два раза насыщенным водным раствором бикарбоната натрия (200 мл) и один раз насыщенным водным раствором хлорида натрия. Органическую фазу затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток после этого хроматографируют на силикагеле, элюируя ступенчато градиентом смеси этилацетата с гексаном (1:4) до этилацетата. Содержащие продукт фракции (оцениваемые с помощью ТСХ) концентрируют с получением 2,6 г (40%) вязкого бесцветного масла. 1H-ЯМР; FD-MS, m/e 661 (M+).
Б) Получение D-Phe-Pro-NH(CH2)3NHC(NH)NH2 •HCl.
Способом, по существу эквивалентным описанному в примере 1-З, получают 460 мг (71%) D-Phe-Pro-NH(CH2)3NHC(NH)NH2 •HCl. Продукт очищают путем RPHRLC (метод 1; (A/B) от 98/2 до 40/60, 40 мин).
1H-ЯМР; FD-MS, m/e: 361 (M+)
Анализ для C18H28N6O2•HCl• 1,1ТФК•1,1H2O:
рассчитано, %: C 44,66; H 6,20; N 15,47;
найдено, %: C 44,69; H 6,10; N 15,19.
Пример 8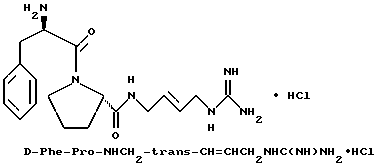
А) Получение Ng,Ng'-ди-Boc-4-амино-транс-2- бутенилгуанидина
Способом, по существу эквивалентным описанному в примере 1-Е, получают 2,4 г (42%) Ng,Ng'-ди-Boc-4-амино-транс-2- бутенилгуанидина из 1,4-диамино-транс-2-бутена.
Б) Получение Boc-D-Phe-Pro-NHCH2-trans-CH=CHCH2NHC (NBoc)NHBoc.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 2,7 г (55%) Boc-D-Phe-Pro-NHCH2-trans- CH=CHCH2NHC(NBoc)NHBoc из Boc-D-Phe-Pro-OH и Ng,Ng'-ди-Boc-4-амино-транс-2-бутенилгуанидина. 1H-ЯМР. FD-MS, m/e: 673 (M+).
В) Получение D-Phe-Pro-NHCH2-trans- CH=CHCH2NHC(NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примере 1-З, получают приблизительно 100 мг D-Phe-Pro-NHCH2-trans- CH=CHCH2NHC(NH)NH2•HCl. Продукт очищают путем RPHRLC (метод 1; (A/B от 98/2 до 40/60, 40 минут). 1H-ЯМР. FD-MS m/e 373 (M+)
Анализ для C19H28N6O2•HCl• 0,5ТФК•2,5H2O:
рассчитано, %: C 47,01; H 6,81; N 16,45;
найдено, %: C 47,36; H 6,53; N 16,70.
Пример 9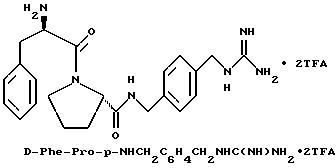
А) Получение p-H2NCH2C6H4CH2 NHC(NBoc)NHBoc
Способом, по существу эквивалентным описанному в примере 1-Е, получают 2,3 г (42%) p-H2NCH2C6H4CH2 NHC(NBoc)NHBoc из п-ксилолдиамино. 1H-ЯМР.
Б) Получение Boc-D-Phe-Pro-p-NHCH2-C6H4 CH2NHC(NBoc)NHBoc.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 2,8 г (63%) Boc-D-Phe-Pro-p-NHCH2-C6H4 CH2NHC(NBoc)NHBoc из Boc-D-Phe-Pro-OH и p-H2NCH2C6H4CH2NHC (NBoc) NHBoc.
1H-ЯМР. FD-MS, m/e: 723 (M+)
В) Получение D-Phe-Pro-p-NHCH2-C6H4CH2 NHC(NH)NH2•2TFA.
Способом, по существу эквивалентным описанному в примере 1-З, получают 725 мг (81%) целевой бис-ТФК-соли и далее не очищают путем RPHRLC. 1H-ЯМР; FD-MS, m/e: 423 (M+).
Анализ для C23H30N6O2•2,1ТФК •H2O:
рассчитано, %: C 48,05; H 5,05; N 12,36;
найдено, %: C 48,06; H 4,85; N 12,28.
Пример 10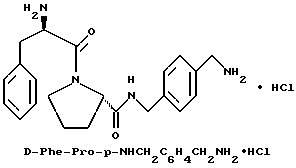
А) Получение N-Boc-n-(аминометил)бензиламина
К перемешиваемому раствору п-ксилолдиамина (10 г, 73 ммоль) в смеси диметилформамида с водой (1:1, 100 мл) добавляют ди-трет- бутил-дикарбонат (8 г, 37 ммоль). После перемешивания в течение 20 ч смесь концентрируют в вакууме и остаток распределяют между диэтиловым эфиром (200 мл) и 1н раствором лимонной кислоты (200 мл). Водную фазу промывают снова диэтиловым эфиром (200 мл) и затем подщелачивают с помощью твердого бикарбоната натрия и насыщают твердым хлоридом натрия. Водную фазу затем экстрагируют 4 раза этилацетатом (200 мл). Объединенные этилацетатные экстракты сушат (MgSO4), фильтруют и концентрируют с получением 2,1 г (24%) вязкого масла. 1H-ЯМР; FD-MS, m/e: 237 (MH+).
Анализ для C13H20N2O2:
рассчитано, %: C 66,07; H 8,53; N 11,85;
найдено, %: C 66,33; H 8,44; N 12,11.
Б) Получение Boc-D-Phe-Pro-p-NHCH2C6H4 CH2NHBoc.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 1,1 г (63%) Boc-D-Phe-Pro-p-NHCH2-C6H4 CH2NHBoc из Boc-D-Phe-Pro-OH и N-Вос-n-(аминометил) бензиламина.
1H-ЯМР; FD-MS, m/e: 581 (M+)
Анализ для C32H44N4O6:
рассчитано, %: C 66,19; H 7,64; N 9,65;
найдено, %: C 65,99; H 7,63; N 9,42.
В) Получение D-Phe-Pro-p-NHCH2C6H4CH2 NH2•HCl.
Способом, по существу эквивалентным описанному в примере 1-З, получают около 100 мг D-Phe-Pro-p-NHCH2-C6H4 CH2NH2•HCl. Продукт очищают путем RPHRLC (метод 1, (A/B) от 98/2 до 40/60 40 мин). 1H-ЯМР; FD-MS, m/e 381 (M+).
Анализ для C22H28N4O2•HCl• 1,1ТФК•H2O:
рассчитано, %: C 51,87; H 5,77; N 10,00;
найдено, % C 51,78; H 5,88; N 10,28.
Пример 11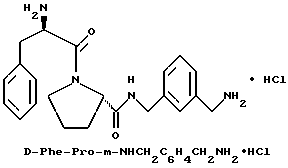
А) Получение N-Boc-м-(аминометил)бензиламина
Способом, по существу эквивалентным описанному в примере 10-А, получают 2,6 г (30%) N-Boc-м-(аминометил)бензиламина из м-ксилолдиамино. 1H-ЯМР; FD-MS, m/e: 237 (MH+).
Анализ для C13H20N2O2:
рассчитано, %: C 66,07; H 8,53; N 11,85;
найдено, %: 65,81; H 8,48; N 11,98.
Б) Получение Boc-D-Phe-Pro-m-NHCH2C6H4 CH2NHBoc.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 1,6 г (95%) Boc-D-Phe-Pro-m-NHCH2-C6H4 CH2NHBoc из Boc-D-Phe-Pro-OH и N-Boc-m-(аминометил) бензиламина.
1H-ЯМР; FD-MS, m/e: 581 (M+)
В) Получение D-Phe-Pro-m-NHCH2C6H4 CH2NH2•HCl
Способом, по существу эквивалентным описанному в примере 1-З, получают около 100 мг D-Phe-Pro-m-NHCH2-C6H4 CH2NH2•HCl.
1H-ЯМР; FD-MS, m/e: 381 (M+).
Анализ для C22H28N4O2•HCl• ТФК•H2O:
рассчитано, %: C 52,51; H 5,87; N 10,21;
найдено, %: C 52,13; H 6,21; N 10,48.
Пример 12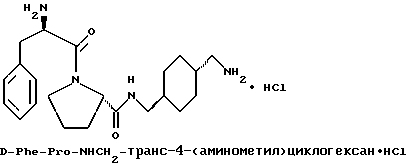
А) Получение N-Boc-транс-4-(аминометил)циклогексанкарбоновой кислоты
К раствору транс-4-(аминометил) циклогексанкарбоновой кислоты (50 г, 318 ммоль) в 1н растворе гидроксида натрия (334 мл, 334 ммоль) и трет-бутанола (400 мл) добавляют раствор ди-трет-бутил-дикарбоната (73 г, 334 ммоль) в тетрагидрофуране (50 мл). После перемешивания в течение 20 ч растворители удаляют в вакууме и остаток распределяют между водой (500 мл) и диэтиловым эфиром (250 мл). Водную фазу промывают снова диэтиловым эфиром (250 мл) и затем подкисляют твердой лимонной кислотой, в результате чего выпадает осадок белого цвета. Твердое вещество фильтруют, промывают дважды водой (100 мл) и сушат в вакууме с получением 48 г (59%) порошка белого цвета. 1H-ЯМР.
Б) Получение HOCH2-транс-4-(N-Boc-аминометил)циклогексана
К перемешиваемому раствору N-Boc-транс-4-(аминометил) циклогексанкарбоновой кислоты (15 г, 58 ммоль) в тетрагидрофуране (150 мл) при 0oC добавляют N-метилморфолин (5,9 г, 58 ммоль), а затем этилхлорформиат (6,3 г, 58 ммоль). После перемешивания в течение 30 мин, добавляют боргидрид натрия (6,5 г, 175 ммоль), и затем через капельную воронку в течение 5 мин добавляют метанол (300 мл). Смесь перемешивают в течение 1 ч и затем растворители удаляют в вакууме. Остаток растворяют в этилацетате (500 мл) и промывают дважды 1н раствором лимонной кислоты (250 мл), один раз водой (100 мл), дважды насыщенным водным раствором бикарбоната натрия (250 мл) и один раз насыщенным водным раствором хлорида натрия (250 мл). Органическую фазу сушат над сульфатом магния, фильтруют и концентрируют с получением 13 г (91%) целевого соединения. 1H-ЯМР.
В) Получение NH2CH2-транс-4-(N-Boc-аминометил) циклогексана
К перемешиваемому раствору HOCH2-транс-4-(N-Boc-аминометил) циклогексана (13 г, 53 ммоль) и трифенилфосфина (21 г, 80 ммоль) в тетрагидрофуране (300 мл) добавляют диэтилазодикарбоксилат (13,9 г, 80 ммоль) с последующим добавлением раствора дифенилфосфорилазида (22 г, 80 ммоль) в тетрагидрофуране (100 мл). После перемешивания в течение 16 ч растворители удаляют в вакууме и остаток хроматографируют на силикагеле, элюируя ступенчато градиентом смеси этилацетата с гексаном (1:3) до смеси этилацетата с гексаном (3:1). Содержащие продукт фракции (оцениваемые путем ТСХ) объединяют и концентрируют с получением 17,4 г сырого продукта (загрязненного соединением с более высоким Rf). Сырой азид растворяют в метаноле (200 мл) и этот раствор добавляют к перемешиваемой суспензии тонко размельченного Na2S•9H2O (51 г, 212 ммоль) и триэтиламина (1 г, 11 ммоль) в метаноле (100 мл). Полученную смесь кипятят с обратным холодильником в течение 16 ч, затем охлаждают до комнатной температуры и растворители удаляют в вакууме. Остаток разбавляют водой (250 мл) и подкисляют твердой лимонной кислотой. Водную фазу промывают дважды этилацетатом (250 мл), подщелачивают с помощью твердого бикарбоната натрия и насыщают твердым хлоридом натрия. Водную фазу затем экстрагируют три раза этилацетатом (200 мл) и объединенные экстракты сушат (MgSO4), фильтруют и концентрируют с получением 6,4 г (45%) вязкого масла. 1H-ЯМР.
Г) Получение N-Boc-D-Phe-Pro-NHCH2-транс-4-(N-Boc-аминометил)- циклогексана
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 4,5 г (74%) N-Boc-D-Phe-Pro-NHCH2-транс-4-(N-Boc- аминометил)циклогексана из Boc-D-Phe-Pro-OH и NH2CH2-транс-4-(N-Boc-аминометил)циклогексана. 1H-ЯМР; FD-MS, m/e 587 (M+).
Д) Получение D-Phe-Pro-NHCH2-транс-4-(аминометил) циклогексан•HCl
Способом, по существу эквивалентным описанному в примере 1-З, получают 588 мг (75%) D-Phe-Pro-NHCH2-транс-4-(аминометил)-циклогексан•HCl. В этом случае анализ с помощью RPHRLC показывает наличие очень чистой промежуточной ТФК-соли, но соль гигроскопична. Соль растворяют в 0,1н растворе HCl (20 мл), pH- значение доводят до 5, и образец лиофилизируют снова с получением стабильного твердого вещества белого цвета - гидрохлоридной соли.
1H-ЯМР; FD-MS, m/e: 387 (M+)
Анализ для C22H34N4O2•HCl• ТФК•2H2O:
рассчитано, %: C 50,30; H 7,04; N 9,78;
найдено, %: C 50,44; H 7,20; N 9,62.
Пример 13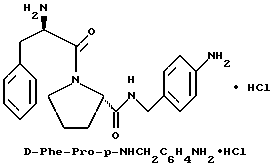
А) Получение Boc-D-Phe-Pro-p-NHCH2C6H4NH2
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 8 г Boc-D-Phe-Pro-p-NHCH2C6H4NO2 из Boc-D-Phe-Pro-OH и п-NO2-бензиламин-гидрохлорида•HCl. Промежуточный продукт растворяют в этаноле (250 мл) и нагревают до температуры кипения с обратным холодильником. К этому перемешиваемому раствору добавляют раствор Na2S2O4 (12,3 г, 70 ммоль) в воде (125 мл). После перемешивания при температуре кипения с обратным холодильником в течение 2 ч растворители удаляют в вакууме и остаток распределяют между этилацетатом (250 мл) и водой (250 мл). Водную фазу экстрагируют снова этилацетатом (250 мл), объединенные органические фазы сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 2,4 г (21%) слегка желтоватого твердого вещества.
1H-ЯМР; FD-MS, m/e: 466 (M+).
Анализ для C26H34N4O4:
рассчитано, %: C 66,93; H 7,34; N 12,01;
найдено, %: C 66,69; H 7,32; N 12,28.
Б) Получение D-Phe-Pro-p-NHCH2C6H4NH2 •HCl.
Способом, по существу эквивалентным описанному в примере 1-H, получают 180 мг (60%) D-Phe-Pro-p-NHCH2-C6H4 NH2•HCl. Продукт очищают путем RPHRLC (метод 1; (A/B) от 98/2 --- до 60/40, 60 мин). 1H-ЯМР; FD-MS, m/e: 366 (M+).
Анализ для C21H26N4O2•HCl• 0,6ТФК•0,5H2O:
рассчитано, %: C 55,51; H 6,00; N 11,66;
найдено, %: C 55,16; H 6,14; N 11,57.
Пример 14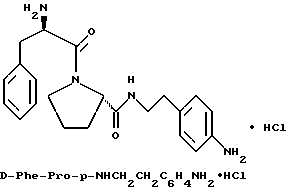
А) Получение Boc-D-Phe-Pro-p-NHCH2CH2C6H4NH2.
Способом, по существу эквивалентным описанному в примере 13-А, получают 3 г (23%) Boc-D-Phe-Pro-p-NHCH2CH2- C6H4NH2 из Boc-D-Phe-Pro-OH и п-NO2-фенетиламин-гидрохлорида.
1H-ЯМР; FD-MS, m/e: 480 (M+).
Анализ для C27H36N4O4:
рассчитано, %: C 67,48; H 7,55; N 11,66;
найдено, %: C 67,30; H 7,54; N 12,34.
Б) Получение D-Phe-Pro-p-NHCH2CH2C6 H4NH2•HCl.
Способом, по существу эквивалентным описанному в примере 1-З, получают 175 мг (58%) D-Phe-Pro-p-NHCH2CH2- C6H4NH2•HCl. Продукт очищают путем RPHRLC (метод 1; (A/B) градиент от 98/2 --- до 60/40, 60 мин). 1H-ЯМР; FD-MS, m/e: 380 (M+).
Анализ для C22H28N4O2•HCl• 0,7ТФК•0,7H2O:
рассчитано, %: C 55,18; H 6,15; N 11,00;
найдено, %: C 55,12; H 6,18; N 10,99.
Пример 15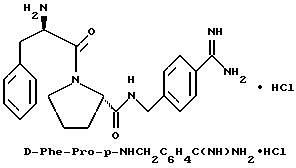
(D-Фенилаланил-N-[[(4-aминoиминoмeтил)фенил] метил] -L -пролин-амид-гидрохлорид)
А) Получение п-(аминометил)бензонитрил•ТФК
К перемешиваемой суспензии гидрида натрия (2,2 г, 56 ммоль, 60%-ная дисперсия в масле) в тетрагидрофуране (100 мл) добавляют 4-(бромметил)бензонитрил (10 г, 51 ммоль). К этой смеси добавляют медленно через капельную воронку раствор ди-трет-бутил-имино- дикарбоксилата (12,2 г, 56 ммоль). После перемешивания в течение 16 ч смесь разбавляют диэтиловым эфиром (300 мл) и промывают дважды водой (150 мл). Органическую фазу затем сушат (MgSO4), фильтруют и концентрируют. Полученное твердое вещество затем растворяют в минимальном количестве дихлорметана. Добавляют анизол (10 мл) и раствор охлаждают до 0oC. Раствор затем разбавляют трифторуксусной кислотой (200 мл) и дополнительно перемешивают в течение 1 часа. Растворитель затем удаляют в вакууме и маслянистый остаток интенсивно размешивают с диэтиловым эфиром (100 мл) и спустя 5 мин продукт затвердевает. Осадок фильтруют, промывают диэтиловым эфиром и сушат в вакууме, получая 11,3 г (90%) порошка белого цвета. ИК; 1H-ЯМР; FD-MS, m/e: 132 (M+).
Б) Получение Boc-D-Phe-Pro-p-NHCH2C6H4CN.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 7,4 г (78%) Boc-D-Phe-Pro-p-NHCH2C6H4CN из Boc-D-Phe-Pro-OH и п-(аминометил)бензонитрил. ТФК. В этом случае, продукт очищают путем перекристаллизации из диэтилового эфира. ИК; 1H-ЯМР; FD-MS, m/e: 476 (M+).
В) Получение Boc-D-Phe-Pro-p-NHCH2C6H4C(NH)NH2.
Газообразный сероводород барботируют через раствор Boc-D- Phe-Pro-p-NHCH2C6H4CN (2 г, 4,2 ммоль) в пиридине (25 мл) и триэтиламине (2,5 мл) в течение 30 мин. Реакционный сосуд затем закрывают и оставляют стоять при комнатной температуре в течение 2 дней. Раствор затем разбавляют водой (100 мл) и экстрагируют дважды этилацетатом (200 мл). Объединенную органическую фазу промывают дважды насыщенным водным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют в вакууме.
Остаток растворяют в ацетоне (50 мл), добавляют метилиодид (10 мл) и раствор кипятят с обратным холодильником в течение 2 ч. Растворители удаляют в вакууме, остаток растворяют в метаноле (20 мл), добавляют ацетат аммония (712 мг, 9,2 ммоль), и раствор кипятят с обратным холодильником в течение 12 часов. Растворитель снова удаляют в вакууме, остаток растворяют в 1н растворе лимонной кислоты (100 мл) и водную фазу промывают дважды этилацетатом (200 мл), затем подщелачивают твердым бикарбонатом натрия, насыщают твердым хлоридом натрия и экстрагируют дважды этилацетатом (200 мл). Объединенные этилацетатные экстракты сушат (MgSO4), фильтруют и концентрируют, получая 1,4 г (67%) вязкого масла. 1H-ЯМР; FD-MS, m/e: 494 (M+).
Г) Получение D-Phe-Pro-P-NHCH2C6H4C(NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примере 1-З, получают 7,7 г (57%) D-Phe-Pro-p-NHCH2C6H4C(NH)NH2•HCl. Продукт очищают путем RPHRLC (метод 3; градиент (A/B) от 98/2 - до 70/30, 300 мин). 1H-ЯМР; FD-MS, m/e: 394 (M+).
Анализ для C22H27N5O2•HCl• 1,4ТФК•1,5H2O:
рассчитано, %: C 49,76; H 5,12; N 11,70;
найдено, %: C 49,75; H 5,19; N 11,58.
Пример 16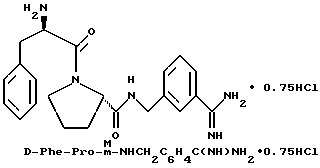
А) Получение м-(аминометил)-бензонитрил•ТФК
Способом, по существу эквивалентным описанному в примере 15-А, получают 10,8 г (86%) м-(аминометил)-бензонитрил•ТФК из м- (бромметил)бензонитрила. ИК; 1H-ЯМР; FD-MS, m/e: 132 (M+)
Б) Получение Boc-D-Phe-Pro-m-NHCH2C6H4CN.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 7,5 г (79%) Boc-D-Phe-Pro-m-NHCH2C6H4CN из Boc-D-Phe-Pro-OH и м-(аминометил)бензонитрил•ТФК. В этом случае, продукт очищают путем перекристаллизации из диэтилового эфира. ИК; 1H-ЯМР; FD-MS, m/e: 476 (M+).
Анализ для C27H32N4O4:
рассчитано, %: C 68,05; H 6,77; N 11,76;
найдено, %: C 68,27; H 6,82; N 11,96.
В) Получение Boc-D-Phe-Pro-m-NHCH2C6H4C(NH)NH2.
Способом, по существу эквивалентным описанному в примере 15-В, получают 1,1 г (53%) Boc-D-Phe-Pro-m-NHCH2C6H4C (NH)NH2. FD-MS, m/e 494 (M+)
Г) Получение D-Phe-Pro-m-NHCH2C6H4C(NH)NH2 •0,75HCl.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 0,65 г (63%) D-Phe-Pro-m-NHCH2C6H4C (NH)NH2•0,75HCl. Продукт очищают путем RPHRLC (метод 2; градиент (A/B) от 98/2 - до 75/25, 120 минут). FD/MS, m/e: 394 (M+).
Анализ для C22H27N5O2•0,75HCl •1,2ТФК•0,5H2O:
рассчитано, %: C 51,72; H 5,33; N 12,36; Cl 4,69;
найдено, %: C 51,79; H 4,93; N 11,96; Cl 4,82.
Пример 17
(D-Гомопролил-N-[[4-(аминоиминометил)фенил] метил] -L-пролин- амид-гидрохлорид)
А) Получение Cbz-D-hPro-ОН.
D-hPro-OH (5,0 г, 38,7 ммоль) растворяют в тетрагидрофуране (100 мл) и воде (30 мл). pH-Значение раствора устанавливают равным 9,5 с помощью 2н раствора гидроксида натрия и прибавляют по каплям бензилхлорформиат (5,5 мл, 38,7 ммоль) и pH поддерживают равным 9,5 с помощью 2н раствора гидроксида натрия. Реакционную смесь перемешивают еще в течение 1 ч при комнатной температуре. Органический растворитель выпаривают в вакууме, добавляют диэтиловый эфир (100 мл) и воду (50 мл) к полученному остатку. Водный слой отделяют, pH-значение раствора доводят до 2,8 с помощью 3н соляной кислоты и добавляют этилацетат (150 мл). Органический слой отделяют и сушат (MgSO4); фильтрат концентрируют в вакууме с получением 9,6 г (95%) прозрачного масла. 1H-ЯМР;
FD-MS, m/e: 264 (MH+).
Б). Получение Cbz-D-hPro-Pro-OH.
Cbz-D-hPro-OH (9,5 г, 36 ммоль) растворяют в этилацетате (100 мл) и раствор охлаждают до 0oC. К полученному раствору добавляют 2,4,5-трихлорфенол (7,1 г, 36 ммоль) и 1,3-дициклогексилкарбодиимид (7,4 г, 36 ммоль). Реакционную смесь перемешивают в течение 1 ч при 0oC и в течение 1 ч при комнатной температуре. Осадок фильтруют и фильтрат концентрируют в вакууме до получения масла. Масло растворяют в пиридине (100 мл), добавляют Pro-OH (4,2 г, 36 ммоль) и триэтиламин (5,0 мл, 36 ммоль). Реакционную смесь перемешивают при комнатной температуре (24 ч). Растворитель из реакционной смеси удаляют в вакууме с получением масла. Остаток растворяют в воде (100 мл), добавляют диэтиловый эфир (50 мл) и pH доводят до 9,5 с помощью 1н раствора гидроксида натрия. Водный слой экстрагируют дважды диэтиловым эфиром. Водный слой отделяют, pH доводят до 2,8 с помощью 3н соляной кислоты и добавляют этилацетат (150 мл). Органический слой отделяют, сушат (MgSO4) и фильтрат упаривают в вакууме с получением аморфного твердого вещества (11,4 г, 88%). FD-MS 361 (M+).
Анализ для C19H24N4O5:
рассчитано, %: C 63,32; H 6,71; N 7,77;
найдено, %: C 63,42; H 6,84; N 7,96.
В) Получение Cbz-D-hPro-Pro-p-NHCH2C6H4CN.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 2,2 г (84%) Cbz-D-hPro-Pro-p-NHCH2C6H4CN из Cbz-D-hPro-Pro-OH и p-NH2CH2C6H4CN•TFA.
1H-ЯМР; FD-MS, m/e. 474 (M+).
Анализ для C27H30N4O4:
рассчитано, %: C 68,34; H 6,37; N 11,81;
найдено, %: C 68,36; H 6,47; N 11,57.
Г) Получение D-hPro-Pro-p-NHCH2C6H4C(NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примере 15-В, получают Cbz-hPro-Pro-p-NHCH2C6H4C(NH)NH2 (28 ммоль, выход теоретический). Это сырое вещество затем растворяют в уксусной кислоте (350 мл) и через раствор в течение 30 мин барботируют газообразный HBr. После перемешивания в течение еще 1 ч, растворитель удаляют в вакууме и остаток растворяют в воде (200 мл) и полученный раствор промывают дважды этилацетатом (100 мл). Водную фазу затем доводят до pH 4 с помощью ионообменной смолы (Bio Rad AG1-X8, в основной форме) и лиофилизуют, получая рыхлое твердое вещество белого цвета. Продукт снова растворяют в воде (25 мл) и очищают путем препаративной RPHRLC (метод 3; градиент (A/B) от 98/2-- до 70/30, 300 мин), получая 5 г (41%) hPro-Pro-p-NHCH2 C6H4C(NH)NH2•0,9HCl•0,9HBr•0,5H2O.
1H-ЯМР; FD-MS, m/e: 357 (M+)
Анализ для C19H27N5O2•0,9HCl•0,9HBr•0,5H2O:
рассчитано, %: C 48,34; H 6,36; N 14,83; Cl 6,76; Br 15,23;
найдено, %: C 48,66; H 6,36; N 14,62; Cl 7,14; Br 14,90.
Пример 18
(N-[[4-(Аминоиминометил)фенил] метил] -1-[[(4aS, 8aS)-декагидро-1 (R)-изохинолинил]карбонил]-L-пролинамид-дигидрохлорид).
А) Получение Cbz-D-1-Piq-Pro-OH.
Раствор 1-изохинолинкарбоновой кислоты (50 г, 0,288. моль) в этаноле (150 мл) и 60 мл 5н HCl восстанавливают в присутствии 5% Rh/Al2O3 (14 г) и при давлении водорода 52 бара (750 пси) в аппарате для высокого давления при 50oC в течение 17 ч. Реакционную смесь фильтруют через слой диатомовой земли и фильтрат концентрируют в вакууме. Твердое вещество растирают с водой, фильтруют и сушат с получением DL-пергидро-1-изохинолинкарбоновой кислоты (DL-1-Piq-OH) (30 г, 48%); FD/MS 184 (MH+).
DL-1-Piq-OH (30,2 г, 137 ммоль) растворяют в тетрагидрофуране (150 мл) с водой (150 мл). pH раствора устанавливают равным 9,8 с помощью 5н раствора NaOH и прибавляют по каплям бензилхлорформиат (21,6 мл, 151 ммоль), поддерживая pH равным 9,5 с помощью 2н раствора NaOH. Реакционную смесь перемешивают еще в течение 2 ч при комнатной температуре. Органический растворитель упаривают в вакууме и к остатку добавляют диэтиловый эфир (150 мл) и воду (50 мл). Водный слой отделяют, pH раствора доводят до 2,5 с помощью 5н HCl и добавляют этилацетат (200 мл). Органический слой отделяют и сушат (MgSO4) и фильтрат концентрируют в вакууме, получая прозрачное масло. Масло растворяют в диэтиловом эфире (150 мл) и раствор оставляют стоять при комнатной температуре в течение 24 ч. Осадок фильтруют и сушат с получением 2-Cbz-DL-пергидро-1- изохинолинкарбоновой кислоты (Cbz-DL-1-Piq-OH) (32 г, 75%); FD-MS m/e 318 (MH+).
Cbz-DL-1-Piq-OH (31,8 г, 100 ммоль) растворяют в ДМФ (100 мл) и охлаждают до 0oC. К реакционной смеси добавляют трет-бутиловый сложный эфир пролина (17,1 г, 100 ммоль), 1-гидроксибензотриазол (13,5 г, 100 ммоль) и ДЦК (20,6 г, 100 ммоль).
Реакционную смесь перемешивают в течение 3 ч при 0oC и 24 ч при комнатной температуре. Осадок фильтруют и фильтрат концентрируют в вакууме с получением масла. Масло растворяют в этилацетате (200 мл) и воде (100 мл). Органический слой отделяют и промывают последовательно 1н раствором бикарбоната натрия, 1,5н раствором лимонной кислоты и водой. Органический слой сушат (MgSO4) и фильтрат упаривают с получением масла, которое высушивают с получением 2-Cbz-DL-пергидро-1-изохинолин- карбонил-L-пролил-трет-бутилового сложного эфира (Cbz-DL-1-Piq-Pro-O-t-Bu)
(47,0 г, 100%); FD-MS m/e: 470 (MH+).
Cbz-DL-1-Piq-Pro-O-t-Bu (47,0 г, 100 ммоль) помещают в круглодонную колбу, содержащую трифторуксусную кислоту (100 мл), CH2Cl2 (35 мл), анизол (5 мл), и перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь концентрируют в вакууме без нагревания и к остатку добавляют диэтиловый эфир (100 мл) и воду (100 мл). pH раствора доводят до 9,8 с помощью 5н раствора NaOH. Водный слой отделяют, pH раствора доводят до 2,5 с помощью 5н HCl и добавляют этилацетат (200 мл). Органический слой отделяют и сушат над сульфатом магния, фильтрат концентрируют в вакууме, получая прозрачное масло. Масло растворяют в диэтиловом эфире (700 мл) и к раствору добавляют (L)-(-)-α-метилбензиламин. Раствор оставляют стоять при комнатной температуре в течение 5 дней. Полученное твердое вещество фильтруют и промывают диэтиловым эфиром. Фильтрат промывают 1,5н раствором лимонной кислоты и водой. Органический слой сушат (MgSO4) и фильтрат упаривают с получением масла. Масло растворяют в диэтиловом эфире (400 мл) и оставляют стоять при комнатной температуре в течение 48 ч. Полученное твердое вещество фильтруют, промывают диэтиловым эфиром и сушат, получая 2-Cbz-D-пергидро-1-изохинолинкарбонил-L-пролин (Cbz-D-1-Piq-Pro-OH) (5,86 г, 36%); FAB-MS; 415 (MH+);
[α]D = -34,2o (c=0,5, метанол).
Б) Получение N-Boc-п-(аминометил)бензонитрила
К перемешиваемой суспензии гидрида натрия (4,6 г, 115 ммоль, 60%-ная дисперсия в масле) в тетрагидрофуране (150 мл) добавляют 4-(бромметил)-бензонитрил (20,5 г, 105 ммоль). К этой смеси добавляют (медленно через капельную воронку) раствор ди-трет- бутил-иминодикарбоксилата (25 г, 115 ммоль). После перемешивания в течение 16 ч смесь разбавляют диэтиловым эфиром (500 мл) и промывают трижды водой (250 мл). Органическую фазу затем сушат (MgSO4), фильтруют и концентрируют, получая 40,2 г сырого твердого вещества.
Полученное твердое вещество (28,3 г, 85 ммоль) затем растворяют в тетрагидрофуране (150 мл) и добавляют раствор гидроксида натрия (3,4 г, 85 ммоль) в метаноле (300 мл). После перемешивания в течение ночи раствор концентрируют примерно на 1/2 объема и добавляют воду с целью вызвать осаждение продукта. Осадок фильтруют и сушат в вакууме, получая 18,5 г (94%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 232 (M+).
Анализ для C13H16N2O2:
рассчитано, %: C 67,22; H 6,94; N 12,06;
найдено, %: C 67,19; H 7,16; N 11,82.
В) Получение п-(BocNHCH2)C6H4C(NH)NHCbz.
Способом, по существу эквивалентным описанному в примере 15-В, N-Boc-п-(аминометил)бензонитрил (32,7 г, 140 ммоль) превращают в п-(BocNHCH2)C6H4C(NH)NH2. Остаток от этого превращения растворяют в диметилформамиде (700 мл) и к раствору добавляют N,N-диизопропилэтиламин (72 г, 560 ммоль). К этому перемешиваемому раствору прибавляют по каплям бензилхлорформиат (48 г, 280 ммоль). После перемешивания в течение 16 ч добавляют воду (100 мл) и затем растворители удаляют в вакууме. Остаток распределяют между водой (250 мл) и этилацетатом (500 мл). Фазы разделяют и органическую фазу промывают три раза насыщенным водным раствором хлорида аммония (250 мл), один раз водой (200 мл) и дважды насыщенным водным раствором бикарбоната натрия (250 мл). Органическую фазу затем сушат (MgSO4), фильтруют и концентрируют, а продукт перекристаллизовывают из диэтилового эфира, получая 14 г (26%) белого твердого вещества. 1H-ЯМР; FD-MS, m/e: 384 (M+).
Г) Получение п-H2NCH2C6H4C(NH)NHCbz •2HCl.
К раствору п-(BocNHCH2)C6H4C(NH)NHCbz (11 г, 28,7 ммоль) в дихлорметане (125 мл) при 0oC добавляют анизол (10 мл), затем трифторуксусную кислоту (125 мл). После перемешивания в течение 2 ч растворители удаляют в вакууме и остаток растворяют в 1н HCl (50 мл) и раствор промывают дважды диэтиловым эфиром (50 мл). pH доводят до 3 с помощью ионообменной смолы (Bio Rad AG1-X8, в основной форме) и раствор лиофилизуют, получая 9,2 г (90%) порошка белого цвета. 1H-ЯМР; FD-MS, m/e: 284 (M+).
Д) Получение Cbz-1-Piq-Pro-p-NHCH2C6H4C (NH)NHCbz.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 4,4 г (79%) Cbz-1-Piq-Pro-p-NHCH2C6H4C(NH)NHCbz из Cbz-1-Piq-Pro-OH и p-H2NCH2C6H4C(NH)NHCbz•2HCl. В этом случае реакцию проводят в диметилформамиде в связи со сложностями, связанными с растворимостью p-H2NCH2C6H4C(NH)NHCbz•2HCl.
1H-ЯМР; FD-MS, m/e: 681 (MH+).
Анализ для C39H45N5O6:
рассчитано, %: C 68,91; H 6,67; N 10,30;
найдено, %: C 68,71; H 6,93; N 10,38.
Е) Получение 1-Piq-Pro-p-NHCH2C6H4C(NH)NH2•2HCl.
К раствору Cbz-1-Piq-Pro-p-NHCH2C6H4C(NH)NHCbz. (4,2 г, 6,1 ммоль) в этаноле (20 мл) добавляют 1н HCl (18,3 мл, 18,3 ммоль) и воду (100 мл). К этому перемешиваемому раствору добавляют 5%-ный палладий-на-угле (1 г) и через раствор в течение 2 ч барботируют газообразный водород. Смесь затем продувают азотом и после этого фильтруют через прокладку (слой) диатомовой земли. Фильтрат затем концентрируют в вакууме, снова растворяют остаток в воде (25 мл) и очищают путем RPHRLC (метод 2; градиент (A/B) от 98/2 -- до 60/40, 300 минут), получая 1,3 г (53%)
1-Piq-Pro-p-NHCH2C6H4C(NH)NH2•2HCl.
1H ЯМР FD/MS, m/e 412 (M+).
Анализ для C23H33N5O2•1,9HCl• 2,5H2O:
рассчитано, %: C 52,53; H 7,65; N 13,32; Cl 12,81;
найдено, %: C 52,63; H 7,36; N 13,47; Cl 12,95.
Пример 19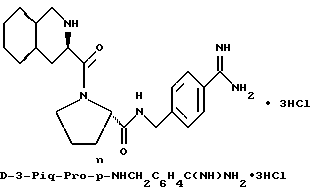
А) Получение Cbz-D-3-Piq-Pro-OH.
Д)-Фенилаланин (50 г, 302 ммоль) подвергают взаимодействию с 37%-ным раствором формальдегида (120 мл) и концентрированной HCl (380 мл) при кипячении с обратным холодильником. Спустя 30 мин добавляют дополнительные 50 мл формальдегида и реакцию продолжают в течение 3 часов. Реакционную смесь охлаждают до -10oC и осадок фильтруют. Твердое вещество сушат в вакууме, получая D-1,2,3,4-тетрагидро-3-изохинолинкарбоновую кислоту (24,2 г, 45%); FD-MS 178 (MH+).
Раствор D -1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты (17 г, 96 ммоль) в воде (200 мл) и 20 мл 5н HCl гидрируют в присутствии 5% Rh/Al2O3 (8,5 г) при давлении 138 бар (2000 пси) водорода в аппарате высокого давления при 120oC в течение 16 ч. Реакционную смесь фильтруют через прокладку (слой) из диатомовой земли и фильтрат подвергают сушке вымораживанием, получая D-пергидро-3-изохинолинкарбоновую кислоту (D-3-Piq-ОН) (21 г, 100%); FD-MS: 184 (MH+).
D-3-Piq-OH (21,0 г, 95,8 ммоль) растворяют в тетрагидрофуране (75 мл) и воде (50 мл). pH раствора доводят до 10,0 с помощью 5н раствора NaOH и прибавляют по каплям бензилхлорформиат (16,4 мл, 115 ммоль), поддерживая pH при 9,5 с помощью 2н раствора NaOH. Реакционную смесь перемешивают в течение дополнительного 1 ч при комнатной температуре. Органический растворитель упаривают в вакууме и к остатку добавляют диэтиловый эфир (100 мл) и воду (50 мл). Водный слой отделяют, pH раствора устанавливают равным 3,0 с помощью 3н HCl и добавляют этилацетат (250 мл). Органический слой отделяют и сушат (MgSO4). Фильтрат концентрируют в вакууме, получая прозрачное масло 2-Cbz-D-пергидро-3-изохинолинкарбоновую кислоту (Cbz-D-3-Piq-OH) (25,8 г, 85%); FD-MS: 318 (MH+).
Cbz-D-3-Piq-OH (17,2 г, 54 ммоль) растворяют в
ДМФ (50 мл) и охлаждают до 0oC. К полученному раствору добавляют трет-бутиловый сложный эфир пролина (9,2 г, 54 ммоль), 1-гидроксибензотриазол (7,3 г, 54 ммоль) и ДНК (11,1 г, 54 ммоль). Реакционную смесь перемешивают в течение 3 ч при 0oC и 24 ч при комнатной температуре. Осадок фильтруют и фильтрат концентрируют в вакууме с получением масла. Масло растворяют в этилацетате (200 мл) с водой (100 мл). Органический слой отделяют и промывают последовательно 1н раствором бикарбоната натрия, водой, 1,5н раствором лимонной кислоты и водой. Органический слой сушат (MgSO4) и фильтруют. Фильтрат упаривают с получением масла, которое высушивают, получая 2-Cbz-D-пергидро-3-изохинолинкарбонил-L- пролин-трет-бутиловый эфир (Cbz-D-3-Piq-Pro-O-Bu) (23,8 г, 94%). FAB-MS: 471 (MH+).
Cbz-D-3-Piq-Pro-O-t-Bu (31,2 г, 66,3 ммоль) помещают в круглодонную колбу, содержащую трифторуксусную кислоту (100 мл), анизол (5 мл), и перемешивают при комнатной температуре (1 ч). Реакционную смесь концентрируют в вакууме без нагревания и добавляют к остатку диэтиловый эфир (150 мл) и воду (100 мл). pH раствора доводят до 9,8 с помощью 5н раствора NaOH. Водный слой отделяют, pH раствора доводят до 2,8 с помощью 3н HCl и добавляют этилацетат (200 мл). Органический слой отделяют, сушат (MgSO4) и фильтруют. Масло растворяют в диэтиловом эфире (300 мл) и раствор оставляют стоять при комнатной температуре в течение 24 ч. Полученное твердое вещество фильтруют, промывают диэтиловым эфиром и сушат, получая 2-Cbz-пергидро-3-изохинолинкарбонил-L-пролин (Cbz-D-3-Piq-Pro-OH) (13,5 г, 49%); FAB-MS: 415 (MH+).
Анализ для C23H30N2O5:
рассчитано, %: C 66,65; H 7,29; N 6,76;
найдено, %: C 66,90; H 7,33; N 6,81.
Б) Получение Cbz-D-3-Piq-Pro-p-NHCH2C6H4C(NH)NHCbz.
Способом, по существу эквивалентным описанному в примере 18-Д, получают 1,6 г (49%) Cbz-D-3-Piq-Pro-p-NHCH2C6H4C(NH)NHCbz из Cbz-D-3-Piq-Pro-OH и p-H2NCH2C6H4C(NH)NHCbz• 2HCl. FD-MS, m/e 680 (M+)
В) Получение D-3-Piq-Pro-p-NHCH2C6H4C(NH)NH2 •3HCl.
Способом, по существу эквивалентным описанному в примере 17-В, получают 150 мг D-3-Piq-Pro-p-NHCH2C6H4C(NH)NH2•3HCl. Продукт очищают путем RPHRLC (метод 2; градиент (A/B) от 98/2 до 60/40, 240 минут). 1H-ЯМР; FD-MS, m/e: 412 (M+)
Анализ для C23H33N5O2•3HCl• 0,5H2O:
рассчитано, %: C 52,13; H 7,04; N 13,22;
найдено, %: C 52,35; H 7,23; N 12,95.
Пример 20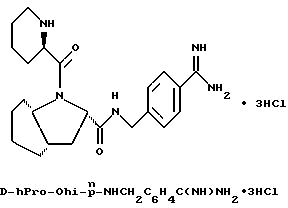
((S-цис)-N-[[4-(Аминоиминометил)фенил] метил] -октагидро-1-D- гомопролил-1H-индол-2-карбоксамид-тригидрохлорид)
А) Получение Cbz-D-hPro-Ohi-OH.
Газообразный HCl барботируют через перемешиваемую суспензию (S)-индолин-2-карбоновой кислоты (20 г, 110 ммоль) в этаноле (500 мл). Когда кислота полностью растворится, раствор доводят до температуры кипения с обратным холодильником. Спустя 16 ч, раствор охлаждают и растворитель удаляют в вакууме. Остаток растирают с диэтиловым эфиром и полученное не совсем белое твердое вещество отделяют путем фильтрации, промывают гексаном и сушат в течение ночи в вакууме при 30oC с получением гидрохлорида этилового эфира (S)-индолин-2-карбоновой кислоты (25,7 г, 78%).
Твердое вещество растворяют в этаноле (800 мл), добавляют 5%-ный Pd/C (25 г) и полученную суспензию гидрируют в аппарате Парра в течение 8 ч (4,1 бара, 60 пси). Раствор фильтруют и растворитель удаляют в вакууме. Остаток растворяют, растирают с диэтиловым эфиром и путем фильтрации получают 18,8 г (73%) не совсем белого твердого вещества (цис-Ohi-OEt•HCl).
Способом, по существу эквивалентным описанному в примере 1-А, получают 13,5 г (93%) Cbz-D-hPro-cis-Ohi-OEt из Cbz-D-hPro-ОH и цис-Ohi-OEt•HCl. 1H-ЯМР; FD-MS, m/e 442 (M+)
Анализ для C25H34N2O5:
рассчитано, %: C 67,85; H 7,74; N 6,33;
найдено, %: C 67,59; H 7,72; N 6,48.
Способом, по существу эквивалентным описанному в примере 1-Г, получают 12,5 г (102%) Cbz-D-hPro-cis-Ohi-OH.
1H-ЯМР; FD-MS, m/e: 414 (M+).
Анализ для C23H30N2O5:
рассчитано, %: C 66,65; H 7,29; N 6,76;
найдено, %: C 66,4; H 7,30; N 6,86.
Б) Получение Cbz-D-hPro-Ohi-p-NHCH2C6H4C(NH)NHCbz.
Способом, по существу эквивалентным описанному в примере 18-Д, получают 3,3 г (67%) Cbz-D-hPro-Ohi-p-NHCH2C6H4C(NH)NHCbz из Cbz-D-hPro-Ohi-OH и p-H2NCH2C6H4C(NH)NHCbz• 2HCl.
1H-ЯМР; FD-MS, m/e: 681 (MH+).
В) Получение D-hPro-Ohi-p-NHCH2C6H4C(NH)NH2 •3HCl.
Способом, по существу эквивалентным описанному в примере 18-Е, получают 2,2 г (66%) D-hPro-Ohi-p-NHCH2C6H4C(NH)NH2• 3HCl. Продукт очищают путем RPHRLC (метод 2; градиент (A/B) от 98/2 -- до 60/40, 3000 мин). 1H-ЯМР; FD-MS, m/e: 412 (M+).
Анализ для C23H33N5O2•3HCl• 0,5H2О:
рассчитано, %: C 52,13; H 7,04; N 13,22;
найдено, %: C 51,98; H 7,04; N 13,35.
Пример 21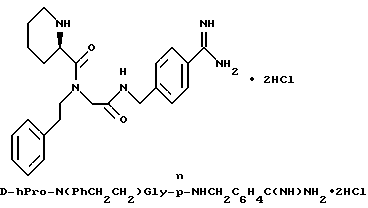
(D-Гомопролил-N (α)- (2-фенилэтил)-N-[[4-(аминоиминометил)фенил]- метил] -глицинамид-дигидрохлорид)
А) Получение Cbz-D-hPro-N(PhCH2CH2)Gly-OH.
К раствору фенилэтиламина (58 мл, 461 ммоль) и триэтиламина (21 мл, 154 ммоль) в этаноле (200 мл) при 0oC добавляют раствор трет-бутилбромацетата (30 г, 154 ммоль) в этаноле (50 мл) в течение 1 ч. Охлаждающую баню оставляют и раствор оставляют нагреваться до комнатной температуры. После перемешивания в течение ночи растворители удаляют в вакууме и остаток растворяют в 1н растворе лимонной кислоты. Водный раствор промывают дважды диэтиловым эфиром, подщелачивают твердым бикарбонатом натрия и затем экстрагируют три раза этилацетатом (20 мл). Объединенные этилацетатные экстракты сушат (MgSO4), фильтруют и оставляют стоять в течение 24 ч. Образовавшийся осадок фильтруют, промывают диэтиловым эфиром и высушивают с получением 10,5 г белого твердого вещества. Маточный раствор концентрируют до объема примерно 100 мл и затем разбавляют диэтиловым эфиром (400 мл). После стояния в течение 30 минут раствор фильтруют, получая дополнительные 23,5 г белого твердого вещества. Общее количество составляет 34 г (94%) N(PhCH2CH2)Gly-O-t-Bu. 1H-ЯМР; FD-MS, m/e 235 (M+)
Способом, по существу эквивалентным описанному в примере 1-А, получают 10,8 г (56%) Cbz-D-hPro-N(PhCH2CH2)Gly-O-t-Bu из Cbz-D-hPro-OH и N(PhCH2CH2)Gly-O-t-Bu.
1H-ЯМР. FD-MS, m/e: 480 (M+).
Анализ для C28H36N2O5:
рассчитано, %: C 69,98; H 7,55; N 5,83;
найдено, %: C 69,68; H 7,56; N 5,77.
Способом, по существу эквивалентным описанному в примере 18-А, для удаления защиты от Cbz-DL-1-Piq-Pro-O-t-Bu, получают 9,2 г (100%) Cbz-D-hPro-N(PhCH2CH2)Gly-OH.
1H-ЯМР; FD-MS, m/e: 425 (M+).
Анализ для C24H28N2O5:
рассчитано, %: C 67,91; H 6,65; N 6,60;
найдено, %: C 68,19; H 6,68; N 6,71.
Б) Получение Cbz-D-hPro-N(PhCH2CH2)Gly-p-NHCH2- C6H4C(NH)NHCbz.
Способом, по существу эквивалентным описанному в примере 18-Д, получают 3,2 г (55%) Cbz-D-hPro-N(PhCH2CH2)Gly-p- NHCH2C6H4C(NH)NHCbz из Cbz-D-hPro-N(PhCH2CH2)Gly-OH и p-H2NCH2C6H4C(NH)NHCbz•HCl. 1H-ЯMP; FD-MS, m/e 690 (M+)
Анализ для C40H43N5O6:
рассчитано, %: C 69,65; H 6,28; N 10,15;
найдено,%: C 69,80; H 6,46; N 10,14.
В) Получение D-hPro-N(PhCH2CH2)Gly-p-NHCH2 C6H4C(NH)NH2•2HCl.
Способом, по существу эквивалентным описанному в примере 19-Е, получают 770 мг (54%) D-hPro-N(PhCH2CH2)Gly-p- NHCH2C6H4C(NH)NH2•2HCl. Продукт очищают путем RPHRLC (метод 2; градиент (A/B) от 98/2 -- до 85/15, 120 минут).
1H-ЯМР; FD-MS, m/e: 423 (MH+).
Анализ для С24H31N5O•2HCl:
рассчитано, %: C 58,30; H 6,73; N 14,16;
найдено, %: C 58,05; H 6,60; N 12,28.
Пример 22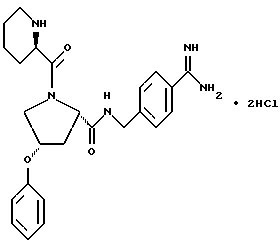
D-hPro-Pro(4-cis-PhO)-p-NHCH2C6H4C(NH)NH2 •2HCl
(цис-D-Гомопролил-N-[[4-(аминоиминометил)-фенил] метил] -4-фенокси- L-пролинамид-дигидрохлорид).
А) Получение Cbz-D-hPro-Pro(4-cis-PhO)-OH.
К раствору Cbz-Pro(4-транс-OH)-Et (58,8 г, 200 ммоль), трифенилфосфина (65,6 г, 250 ммоль) и фенола (23,5 г, 250 ммоль) в тетрагидрофуране (500 мл) при 0oC добавляют (прибавление по каплям в течение 1 ч) раствор диэтилазодикарбоксилата (40 мл, 250 ммоль) в ТГФ (50 мл). Охлаждающую баню затем удаляют и раствор оставляют нагреваться до комнатной температуры в течение 16 ч. После этого растворитель удаляют в вакууме и остающийся янтарного цвета сироп растирают с диэтиловым эфиром. Белое твердое вещество удаляют путем фильтрации и фильтрат концентрируют. Остаток затем хроматографируют на силикагеле (1 кг), элюируя ступенчатым градиентом от гексана до смеси 1:1 этилацетат с гексаном. Содержащие чистый продукт фракции (оценивают путем ТСХ) объединяют и концентрируют в вакууме с получением 36,3 г (50%) Cbz-Pro-(4-цис-фенокси)OEt в виде бесцветного сиропа. 1H-ЯМР; FD-MS, m/e: 369 (M+).
Анализ для C21H23NO5:
рассчитано, %: C 68,28; H 6,28; N 3,79;
найдено, %: C 68,38; H 6,30; N 3,89.
К раствору Cbz-Pro(4-цис-фенокси)-OEt (25 г, 67,7 ммоль) в этаноле (400 мл) добавляют 5%-ный Pd/C (5 г). После барботирования водорода через раствор в течение 3 ч раствор фильтруют через прокладку (слой) диатомовой земли, добавляют 3 мл концентрированной HCl и раствор концентрируют в вакууме. Остаток суспендируют в диэтиловом эфире при интенсивном перемешивании и затем фильтруют и высушивают, получая 14,2 г (77%) Pro(4-цис-фенокси)-OEt•HCl в виде белого твердого вещества. 1H-ЯМР; FD-MS, m/e 235 (M+).
Анализ для C13H18NO3Cl:
рассчитано, %: C 57,46; H 6,68; N 5,15;
найдено, %: C 57,68; H 6,78; N 5,18.
Способом, по существу эквивалентным описанному в примере 1-А, получают 19,4 г (100%) Cbz-D-hPro-Pro-(4-цис-фенокси)-OEt из Cbz-D-hPro-ОH и Pro-(4-цис-фенокси)-OEt•HCl.
1H-ЯМР; FS-MS, m/e: 480 (M+).
Анализ для C27H32N2O6:
рассчитано, %: C 67,48; H 6,71; N 5,83;
найдено, %: C 67,71; H 6,79; N 5,89.
Способом, по существу эквивалентным описанному в примере 1-Г, получают 16 г (100%) Cbz-D-hPro-(4-цис-фенокси)-OH.
1H-ЯМР; FD-MS, m/e: 452 (M+).
Анализ для C25H28N2O6:
рассчитано, %: C 66,36; H 6,24; N 6,19;
найдено, %: C 66,22; H 6,18; N 6,17.
Б) Получение Cbz-D-hPro-Pro(4-cis-PhO)-p-NHCH2- C6H4C(NH)NHCbz.
Способом, по существу эквивалентным описанному в примере 18-Д, получают 4,55 г (75%) Cbz-D-hPro-Pro(4-cis-PhO)-p- NHCH2C6H4C(NH)NHCbz из Cbz-D-hPro-Pro(4-cis-PhO)-OH и p-H2NCH2C6H4C(NH)NHCbz•2HCl. 1H-ЯМР; FD-MS, m/e: 718 (M+)
В) Получение D-hPro-Pro(4-cis-PhO)-p-NHCH2- C6H4C(NH)NH2•2HCl.
Способом, по существу эквивалентным описанному в примере 18-Е, получают 873 мг (40%) D-hPro-Pro(4-cis-PhO)-p- NHCH2C6H4C(NH)NH2•2HCl. Продукт очищают путем RPHRLC (метод 2; градиент (A/B) от 98/2 -- до 85/15, 120 мин). 1H-ЯМР; FD-MS, m/e: 451 (MH+).
Анализ для C25H31N5O3•2HCl:
рассчитано, %: C 57,47; H 6,37; N 13,40;
найдено, %: C 57,22; H 6,29; N 13,47.
Пример 23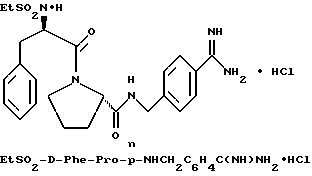
(N-(Этилсульфонил)-D-фенилаланил-N-[[4-(аминоиминометил)фенил] метил] - L-пролинамид-гидрохлорид)
А) Получение p-NH2CH2-C6H4CN•HCl.
Газообразный хлороводород при 0oC и в течение 10 мин барботируют через перемешиваемый раствор N-Boc-п-аминометил-бензонитрила (15 г, 64,6 ммоль) в этилацетате (400 мл). Охлаждающую баню удаляют и после перемешивания в течение 1,5 ч растворитель удаляют в вакууме и остаток суспендируют в диэтиловом эфире, фильтруют, промывают диэтиловым эфиром и сушат с получением 10,1 г (93%) белого твердого вещества, ИК; 1H-ЯМР; FD-MS, m/e: 132 (M+).
Анализ для C8H9N2Cl:
рассчитано, %: C 56,98; H 5,38; N 16,61; Cl 21,02;
найдено, %: C 56,36; H 5,46; N 16,22; Cl 21,31.
Б) Получение EtSO2-D-Phe-Pro-p-NHCH2-C6H4CN.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 1,5 г (80%) EtSO2-D-Phe-Pro-p-NHCH2- C6H4CN из EtSO2-D-Phe-Pro-OH и p-NH2CH2-C6H4CN•HCl.
ИК; 1H-ЯМР; FD-MS, m/e: 468 (M+).
Анализ для C24H28N4O4:
рассчитано, %: C 61,52; H 6,02; N 11,90;
найдено, %: C 61,23; H 6,13; N 11,80.
В) Получение EtSO2-D-Phe-Pro-p-NHCH2- C6H4C(=NOH)NH2•HCl.
К раствору EtSO2-D-Phe-Pro-p-NHCH2-C6H4CN (1 г, 2,1 ммоль) в абсолютном этаноле (35 мл) добавляют N, N-диизопропилэтиламин (0,47 мл, 2,7 ммоль), затем гидроксиламингидрохлорид (185 мг, 2,7 ммоль) и раствор доводят до кипения с обратным холодильником. Спустя 16 ч раствор охлаждают и растворители удаляют в вакууме. 250 мг этого вещества используют в следующей стадии, а оставшееся вещество очищают путем RPHRLC (метод 1, градиент (A/B) от 90/10 до 60/40, в течение 200 мин). ИК; 1H-ЯМР; FD-MS, m/e: 501 (M+).
Анализ для C24H31N5O5•1,2HCl• H2O:
рассчитано, %: C 51,17; H 6,12; N 12,42; Cl 7,55;
найдено, %: C 51,04; H 5,81; N 12,39; Cl 7,18.
Г) Получение EtSO2-D-Phe-Pro-p-NHCH2- C6H4C(=NH)NH2•HCl.
К раствору EtSO2-D-Phe-Pro-p-NHCH2- C6H4C(= NOH)NH2•HCl (250 мг, 0,52 ммоль) в этаноле (40 мл) и воде (19 мл) добавляют 1н раствор HCl (1 мл), затем 250 мг 5%-ного палладия-на-угле. Перемешиваемую суспензию помещают в атмосферу водорода на 18 часов и затем фильтруют, концентрируют и очищают путем RPHRLC (метод 1; градиент (A/B) от 90/10 до 60/40, в течение 200 мин), получая 140 мг (52%) EtSO2-D-Phe-Pro-p- NHCH2-C6H4C(=NH)NH2•HCl.
1H-ЯМР; FD-MS, m/e: 486 (M+).
Анализ для C24H31N5O4•HCl• 1,5H2О:
рассчитано, %: C 52,50; H 6,42; N 12,75;
найдено, %: C 52,56; H 6,19; N 12,59.
Еще 5 г продукта получают способом, описанным в примере 15, и очищают путем RPHPLC (метод 3; градиент (A/B): 98/2 в течение 60 мин, до 60/40 - в течение 300 мин).
Пример 24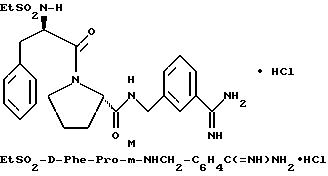
А) Получение EtSO2-D-Phe-Pro-m-NHCH2-C6H4C(=NOH)NH2.
Способом, по существу эквивалентным описанному в примере 23, EtSO2-D-Phe-Pro-m-NHCH2-C6H4C(= NOH)NH2 получают, используя м-Br-CH2-C6H4C вместо п-Br-CH2-C6H4CN. 140 мг (13%) этого кристаллического промежуточного продукта оставляют, а остаток вещества используют в стадии Б). 1H-ЯМР; FD-MS, m/e: 502 (M+)
Анализ для C24H31N5O5:
рассчитано, %: C 57,47; H 6,23; N 13,96;
найдено, %: C 57,28; H 6,21; N 13,66.
Б) Получение EtSO2-D-Phe-Pro-m-NHCH2- C6H4C(=NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примерах 23-В) и 23-Г), получают 0,27 г (28%, 2 стадии) EtSO2-D-Phe-Pro-m- NHCH2-C6H4C(=NH)NH2•HCl. 1H-ЯМР; FD-MS, m/e: 486 (M+).
Анализ для C24H31N5O4•1,1HCl• 2H2O:
рассчитано, %: C 51,32; H 6,48; N 12,47; Cl 6,94;
найдено, %: C 51,33; H 6,09; N 12,20; Cl 6,66.
Пример 25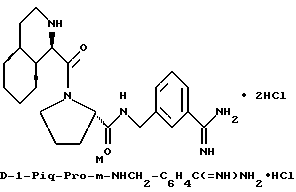
Способом, по существу эквивалентным описанному в примере 23, получают 0,86 г D-1-Piq-Pro-m-NHCH2- C6H4C(=NH)NH2•HCl из Cbz-D-1-Piq-Pro-OH и м-NH2CH2-C6H4CN•HCl.
1H-ЯМР; FD-MS, m/e: 412 (M+)
Анализ для C23H33N5O2•2,5HCl• 0,5H2O:
рассчитано, %: C 53,99; H 7,19; N 13,69;
найдено, %: C 54,19; H 7,02; N 13,81.
Пример 26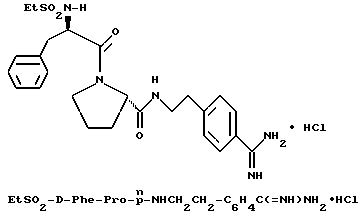
А) Получение метил-п-циано-транс-циннамата
К перемешиваемой суспензии NaH (6,1 г 60%-ной масляной суспензии 153 ммоль) и п-цианобензальдегида (20 г, 153 ммоль) в тетрагидрофуране (250 мл) при 0oC добавляют через капельную воронку раствор триметилфосфоноацетата (28 г, 153 ммоль) в тетрагидрофуране (50 мл). После перемешивания в течение 48 ч растворитель удаляют в вакууме и сырой остаток растворяют в этилацетате (500 мл). Этилацетатный раствор промывают один раз водой, три раза насыщенным водным раствором NaHSO3 и один раз солевым раствором. Органическую фазу затем сушат (MgSO4), фильтруют и концентрируют в вакууме, получая 28 г (98%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 187 (M+).
Б) Получение метил-п-циано-дигидроциннамата
К раствору метил-п-циано-транс-циннамата (13,6 г, 73 ммоль) в толуоле (485 мл) добавляют 5%-ный Pd/BaSO4 (2,7 г). После воздействия газообразного водорода под давлением 4 бара (60 пси) в течение 9 ч, раствор фильтруют, концентрируют в вакууме и хроматографируют на силикагеле, элюируя ступенчато градиентом от гексана до гексана с 30% этилацетата. Содержащие продукт фракции объединяют и концентрируют с получением 10,6 г (77%) бесцветного масла. ИК; 1H-ЯМР; FD-MS, m/e: 189 (M+)
В) Получение п-цианодигидрокоричной кислоты
Способом, по существу эквивалентным описанному в примере 1-Г, при использовании 1,1 эквивалента LiOH•H2O, получают 5,1 г (58%) п-циано-дигидрокоричной кислоты из метил-п-циано-дигидроциннамата. ИК; 1H-ЯМР; FD-MS, m/e: 175 (M+).
Г) Получение Boc-p-NHCH2CH2-C6H4CN.
К раствору п-циано-дигидрокоричной кислоты (6,7 г, 38,2 ммоль) и триэтиламина (5,9 мл, 42 ммоль) в трет-бутаноле (150 мл) добавляют дифенилфосфорилазид (11,6 г, 42 ммоль) и раствор доводят до температуры кипения с обратным холодильником. После перемешивания в течение ночи, раствор охлаждают и растворитель удаляют в вакууме. Остаток растворяют в этилацетате и промывают полученный раствор три раза с помощью 1н раствора лимонной кислоты, один раз насыщенным раствором соли, дважды насыщенным водным раствором бикарбоната натрия и затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток затем хроматографируют на силикагеле, элюируя с помощью смеси от гексана с 10% этилацетата до гексана с 50% этилацетата. Содержащие продукт фракции, как оценивают с помощью ТСХ, объединяют и концентрируют с получением 5,4 г (57%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e 246 (M+)
Анализ для C14H18N2O2:
рассчитано, %: C 68,27; H 7,37; N 11,37;
найдено, %: C 68,39; H 7,50; N 11,40.
Д) Получение p-NH2CH2CH2- C6H4CN•HCl.
Способом, по существу эквивалентным описанному в примере 23-А, получают 3,6 г (98%) p-NH2CH2CH2- C6H4CN•HCl. 1H-ЯМР; FD-MS, m/e: 147 (MH+)
Анализ для C9H11N2Cl:
рассчитано, %: C 59,18; H 6,07; N 15,34; Cl 19,41;
найдено, %: C 58,90; H 6,16; N 15,20; Cl 19,30.
Е) Получение EtSO2-D-Phe-Pro-p-NHCH2CH2- C6H4CN.
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 1,5 г EtSO2-D-Phe-Pro-p-NHCH2CH2- C6H4CN из EtSO2-D-Phe-Pro-OH и p-NH2CH2CH2-C6H4CN•HCl. ИК; 1H-ЯМР; FD-MS, m/e: 482 (M+).
Ж) Получение EtSO2-D-Phe-Pro-p-NHCH2CH2- C6H4C(=NOH)NH2•HCl.
К перемешиваемому раствору EtSO2-D-Phe-Pro-p- NHCH2CH2-C6H4CN (1 г, 2,07 ммоль) и N, N-диизопропилэтиламина (0,45 мл, 2,59 ммоль) добавляют гидроксиламингидрохлорид (180 мг, 2,59 ммоль) и раствор доводят до температуры кипения с обратным холодильником. Спустя 18 ч (при этой температуре), раствор охлаждают, растворитель удаляют в вакууме и остаток растворяют в уксусной кислоте (15 мл) и очищают путем RPHRLC (метод 2; градиент (A/B) от 90/10 до 60/40, в течение 200 минут). Фракции, содержащие чистый EtSO2-D-Phe-Pro-p-NHCH2CH2- C6H4C(= NOH)NH2•HCl, как определено с помощью аналитической RPHRLC, объединяют и pH устанавливают вышеописанным образом и лиофилизируют, получая 0,35 г (31%) EtSO2-D-Phe-Pro-p-NHCH2CH2- C6H4C(= NOH)NH2•HCl.
1H-ЯМР; FD-MS, m/e 516 (M+)
Анализ для C25H33N5O5S•HCl•H2O:
рассчитано, %: C 52,67; H 6,36; N 12,28; Cl 6,22;
найдено, %: C 52,40; H 6,10; N 12,35; Cl 6,51.
З) Получение EtSO2-D-Phe-Pro-p-NHCH2CH2- C6H4C(=NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примере 23-Г, получают 0,98 г (50%) EtSO2-D-Phe-Pro-p-NHCH2CH2- C6H4C(=NH)NH2•HCl из EtSO2-D-Phe-Pro-p-NHCH2CH2- C6H4C(=NOH)NH2•HCl.
1H-ЯМР; FD-MS, m/e: 500 (M+).
Анализ для C25H33N5O4S•2,6HCl• H2O:
рассчитано, %: C 49,03; H 6,19; N 11,44;
найдено, %: C 48,87; H 5,79; N 11,15.
Пример 27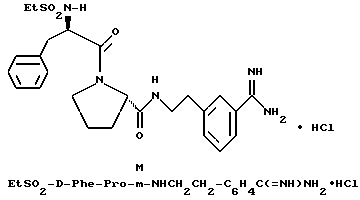
А) Получение EtSO2-D-Phe-Pro-m-NHCH2CH2- C6H4C(=NOH)NH2.
Способом, по существу эквивалентным описанному в примерах 26-А: 26-Д и 24-А, получают 0,15 г EtSO2-D-Phe-Pro-m-NHCH2CH2- C6H4C(=NOH)NH2 из м-цианобензальдегида. 1H-ЯМР; FD-MS, m/e: 516 (M+).
Анализ для C25H33N5O5S:
рассчитано, %: C 58,23; H 6,45; N 13,50;
найдено, %: C 57,99; H 6,57; N 13,28.
Б) Получение EtSO2-D-Phe-Pro-m-NHCH2CH2- C6H4C(=NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примере 24-Б, получают 0,21 г (20%) EtSO2-D-Phe-Pro-m-NHCH2CH2- C6H4C(=NH)NH2•HCl из EtSO2-D-Phe-Pro-m-NHCH2CH2- C6H4C(=NOH)NH2.
1H-ЯМР; FD-MS, m/e: 500 (M+).
Анализ для C25H33N5O4S•2,1HCl•0,7H2O:
рассчитано, %: C 51,00; H 6,25; N 11,89;
найдено, %: C 50,79; H 5,86; N 11,54.
Пример 28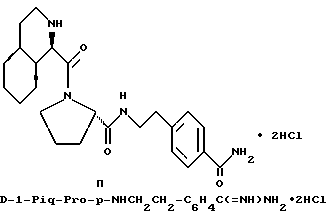
Способом, по существу эквивалентным описанному в примере 23, получают 0,85 г 1-Piq-Pro-p-NHCH2CH2- C6H4C(=NH)NH2•2HCl из Cbz-D-1-Piq-Pro-OH и p-NH2CH2CH2-C6H4CN•HCl.
1H-ЯМР; FD-MS, m/e: 426 (M+).
Анализ для C24H35N5O2•2HCl• 2H2O:
рассчитано, %: C 53,93; H 7,73; N 13,10;
найдено, %: C 53,94; H 7,60; N 13,06.
Пример 29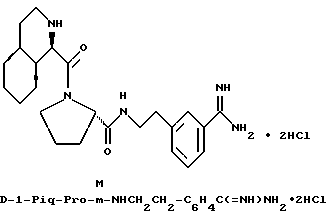
Способом, по существу эквивалентным описанному в примере 23, получают 0,8 г 1-Piq-Pro-m-NHCH2CH2- C6H4С(=NH)NH2•2HCl из Cbz-D-1-Piq-Pro-OH и m-NH2CH2CH2-C6H4CN•HCl.
1H-ЯМР; FD-MS, m/e: 416 (M+).
Анализ для C24H35N5O2•2HCl• 2H2O:
рассчитано, %: C 53,93; H 7,73; N 13,10;
найдено, %: C 53,62; H 7,57; N 13,18.
Пример 30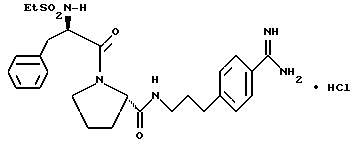
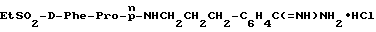
А) Получение п-HOCH2CH2CH2-C6H4CN
К перемешиваемому раствору метил-п-циано-дигидроциннамата (10 г, 53 ммоль) в тетрагидрофуране (150 мл) добавляют LiBH4 (1,15 г, 53 ммоль) и раствор кипятят с обратным холодильником. Спустя 2 ч раствор охлаждают и прикапывают буфер фосфата натрия (pH 7). По окончании выделения газа, добавляют этилацетат и воду и слои разделяют. Водную фазу экстрагируют однократно этилацетатом и объединенные этилацетатные фазы промывают рассолом, затем сушат (MgSO4), фильтруют и концентрируют с получением 8,1 г (95%) вязкого бесцветного масла. ИК; 1H-ЯМР; FD-MS, m/e: 161 (M+).
Б) Получение п-Br-CH2-CH2CH2-C6H4CN.
К перемешиваемому раствору п-HOCH2CH2CH2- C6H4CN (8,1 г, 50 ммоль) в тетрагидрофуране (100 мл) добавляют трифенилфосфин (14,4 г, 55 ммоль), затем тетрабромид углерода (18,2 г, 55 ммоль). После перемешивания в течение 18 часов, растворитель удаляют в вакууме и остаток хроматографируют на силикагеле, элюируя ступенчато градиентом от гексана до гексана с 20% этилацетата. Содержащие продукт фракции, как установлено по ТСХ, объединяют и концентрируют с получением 7,3 г (65%) вязкого бесцветного масла. ИК; 1H-ЯМР; FD-MS, m/e: 223 (M+).
Анализ для C10H10BrN:
рассчитано, %: C 53,60; H 4,50; N 6,25;
найдено, %: C 53,90; H 4,67; N 6,24.
В) Получение п-Boc2CH2CH2CH2- C6H4CN
К перемешиваемой суспензии NaH (1,4 г 60%-ной масляной дисперсии, 34 ммоль) в ДМФ (100 мл) прибавляют по каплям через капельную воронку раствор ди-трет-бутил-иминодикарбоксилата (7,4 г, 34 ммоль) в ДМФ (20 мл). После окончания выделения газа добавляют раствор п-Br-CH2CH2-CH2-C6H4CN (7 г, 31 ммоль) в ДМФ через капельную воронку и раствор нагревают до 70oC. После перемешивания в течение 12 ч при этой температуре, раствор охлаждают и растворитель удаляют в вакууме. Остаток растворяют в диэтиловом эфире и промывают 3 раза водой. Органическую фазу сушат над сульфатом магния, фильтруют и концентрируют, остаток хроматографируют на силикагеле, элюируя ступенчато градиентом от гексана до гексана с 20% этилацетата. Содержащие продукт фракции объединяют и концентрируют, получая 9,38 г (84%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 361 (M+).
Анализ: для C20H28N2O4:
рассчитано, %: C 66,64; H 7,83; N 7,77;
найдено, %: C 66,40; H 7,81; N 7,57.
Г) Получение п-NH2CH2CH2CH2- C6H4CN•HCl
Способом, по существу эквивалентным описанному в примере 23-А, получают 4,3 г (84%) п-NH2CH2CH2CH2- C6H4CN•HCl. ИК; 1H-ЯМР; FD-MS, m/e: 160 (M+).
Д) Получение EtSO2-D-Phe-Pro-p-NHCH2CH2CH2- C6H4C(=NOH)NH2•HCl.
Способом, по существу эквивалентным описанному в примерах 1-Ж и 26-Ж, получают 0,32 г EtSO2-D-Phe-Pro-p-NHCH2CH2CH2- C6H4С(= NOH)NH2•HCl из EtSO2-D-Phe-Pro-OH и p-NHCH2CH2CH2- C6H4CN•HCl.
1H-ЯМР; FD-MS, m/e: 530 (M+).
Анализ для C26H35N5O5S•1,2HCl• H2O:
рассчитано, %: C 52,88; H 6,51; N 11,84;
найдено, %: C 52,71; H 6,26; N 11,76.
Е) Получение EtSO2-D-Phe-Pro-p-NHCH2CH2CH2- C6H4C(=NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примере 23-Г, получают 0,13 г (67%) EtSO2-D-Phe-Pro-p-NHCH2CH2CH2- C6H4C(=NH)NH2•HCl из EtSO2-D-Phe-Pro-p-NHCH2CH2CH2- C6H4C(= NOH)NH2•HCl. 1H-ЯМР; FD-MS, m/e: 514 (M+). Анализ для C26H35N5O4S•1,5HCl• 2H2O:
рассчитано, %: C 51,67; H 6,75; N 11,59;
найдено, %: C 51,36; H 6,46; N 11,28.
Пример 31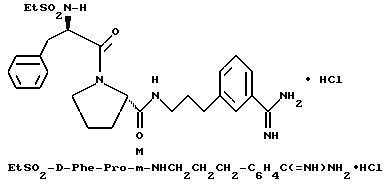
А) Получение EtSO2-D-Phe-Pro-m-NHCH2CH2CH2- C6H4C(=NOH)NH2•HCl.
Способом, по существу эквивалентным описанному в примерах 1-Ж и 26-Ж, получают 0,32 г EtSO2-D-Phe-Pro-m-NHCH2CH2CH2- C6H4C(= NOH)NH2•HCl из м-цианодигидрокоричной кислоты. 1H-ЯМР; FD-MS, m/e: 530 (M+).
Анализ для C26H35N5O5S•HCl• 1,1H2O:
рассчитано, %: C 53,30; H 6,57; N 11,95; Cl 6,05;
найдено, %: C 52,97; H 6,19; N 11,96; Cl 6,13.
Б) Получение EtSO2-D-Phe-Pro-m-NHCH2CH2CH2- C6H4C(=NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примере 23-Г, получают 0,12 г (62%) EtSO2-D-Phe-Pro-m-NHCH2CH2CH2- C6H4C(=NH)NH2•HCl из EtSO2-D-Phe-Pro-m-NHCH2CH2CH2- C6H4C(=NOH)NH2•HCl.
1H-ЯМР; FD-MS, m/e: 514 (M+).
Анализ для C26H35N5O4S•1,5HCl• H2O:
рассчитано, %: C 53,26; H 6,62; N 11,94;
найдено, %: C 53,19; H 6,25; N 12,00.
Пример 32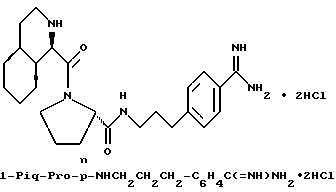
Способом, по существу эквивалентным описанному в примере 23, получают 0,66 г (48%) 1-Piq-Pro-p-NHCH2CH2CH2- C6H4C(= NH)NH2•HCl из 1-Piq-Pro-OH и p-NH2CH2CH2CH2-C6H4CN•HCl.
1H-ЯМР. FD-MS, m/e: 440 (M+).
Анализ для C25H37N5O2•2,1HCl• H2O:
рассчитано, %: C 56,21; H 7,75; N 13,11;
найдено, %: C 56,36; H 7,44; N 12,79.
Пример 33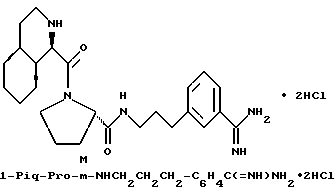
Способом, по существу эквивалентным описанному в примере 23, получают 0,64 г (46%) 1-Piq-Pro-m-NHCH2CH2CH2- C6H4C(= NH)NH2•HCl из 1-Piq-Pro-OH и
1H-ЯМР; FD-MS, m/e: 440 (M+).
Анализ для C25H37N5O2•2HCl• H2O:
рассчитано, %: C 56,60; H 7,79; N 13,20;
найдено, %: C 56,92; H 7,55; N 13,26.
Пример 34
А) Получение Boc-п-NHCH2C6H4NO2
К перемешиваемому раствору 4-нитробензиламин-гидрохлорида (15 г, 79 ммоль) и N,N-диизопропилэтиламина (14 мл, 79 ммоль) в дихлорметане (200 мл) добавляют ди-трет-бутилдикарбонат (17 г, 79 ммоль). Спустя 48 ч растворитель удаляют в вакууме и остаток растворяют в этилацетате (500 мл) и промывают дважды 1М раствором лимонной кислоты, один раз водой и один раз насыщенным водным раствором бикарбоната натрия. Органическую фазу сушат (MgSO4), фильтруют и концентрируют в вакууме с получением не совсем белого твердого вещества, которое перекристаллизуют из смеси хлороформа с гексаном. Три партии кристаллов объединяют, промывают гексаном и сушат в вакууме, получая 11,5 г (58%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 252 (M+).
Анализ для C12H16N2O4:
рассчитано, %: C 57,13; H 6,39; N 11,10;
найдено, %: C 57,27; H 6,60; N 11,13.
Б) Получение п-BocNHCH2C6H4NH2
К перемешиваемому раствору п-BocNHCH2C6H4NO2 (7,5 г, 29,7 ммоль) и NiCl2•6H2O (17,7 г, 74,3 ммоль) в метаноле (150 мл) при 0oC добавляют NaBH4 (5,6 г, 149 ммоль) маленькими порциями в течение 30 мин. По окончании добавления NaBH4 и спустя еще 15 мин растворитель упаривают в вакууме и остаток растворяют в концентрированном растворе гидроксида аммония и экстрагируют дважды дихлорметаном. Объединенные органические экстракты промывают солевым раствором, сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 6,4 г (97%) белого твердого вещества.
ИК; 1H-ЯМР: FD-MS, m/e: 222 (M+).
Анализ для C12H18N2O2:
рассчитано, %: C 64,84; H 8,16; N 12,60;
найдено, %: C 65,10; H 8,42; N 12,76.
В) Получение N,N-ди-Cbz-S-метилизотиомочевины
К перемешиваемой суспензии сульфата бис-S-метилизотиомочевины (20 г, 144 ммоль) в дихлорметане (200 мл) добавляют 5н раствора гидроксида натрия (16 мл). Раствор охлаждают до 0oC и прибавляют по каплям бензилхлорформиат (41 мл, 288 ммоль). В то же самое время добавляют 2н раствор гидроксида натрия в таком количестве, чтобы поддерживать pH раствора около 11. Охлаждающую баню затем удаляют и после нагревания до комнатной температуры фазы разделяют и водную фазу экстрагируют дихлорметаном (250 мл). Объединенные органические фазы затем промывают дважды с помощью 0,1 н HCl (250 мл) и один раз солевым раствором (250 мл). Органическую фазу затем сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 41 г (79%) вязкого бесцветного сиропа. 1H-ЯМР.
Г) Получение п-BocNHCH2C6H4NHC(NCbz)NHCbz
К перемешиваемому раствору п-BocNHCH2C6H4NH2 (5 г, 22,5 ммоль) в тетрагидрофуране (50 мл) добавляют N,N'-ди-Cbz-S-метилизотиомочевину (8,9 г, 24,7 ммоль). Спустя 48 ч растворитель удаляют в вакууме и остаток растворяют в хлороформе. Добавляют силикагель и растворитель удаляют в вакууме с получением не совсем белого порошка, который затем в сухом состоянии вносят в силикагелевую колонку. Содержимое колонки затем элюируют ступенчато градиентом от смеси гексана с 5% этилацетата до смеси гексана с 30% этилацетата. Содержащие продукт фракции (определяют по ТСХ) объединяют и концентрируют в вакууме с получением 7,6 г (63%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 532 (M+).
Анализ для C29H32N4O6:
рассчитано, %: C 65,40; H 6,06; N 10,52;
найдено, %: C 65,66; H 6,35; N 10,59.
Д) Получение HCl•p-NH2CH2C6H4NHC(NCbz)NHCbz
Способом, по существу эквивалентным описанному в примере 23-А, получают 4,7 г (89%) HCl•p-NH2CH2C6H4NHC(NCbz)NHCbz, в виде белого твердого вещества из p-BocNHCH2C6H4NHC(NCbz)NHCbz. ИК; 1H-ЯМР; FD-MS, m/e: 433 (M+).
Е) Получение EtSO2-D-Phe-Pro-p- NHCH2C6H4NHC(NH)NH2-HCl.
Способом, по существу эквивалентным описанному в примере 1-Ж и в примере 18-Е, получают 1,1 г EtSO2-D-Phe-Pro-p- NHCH2C6H4NHC(NH)NH2•HCl из EtSO2-D-Phe-ProOH и HCl•p-NH2CH2C6H4NHC (NCbz)NHCbz.
Анализ для C24H32N6O4S•HCl• H2O:
рассчитано, %: C 51,73; H 6,36; N 15,14;
найдено, %: C 52,32; H 5,99; N 14,79.
Пример 35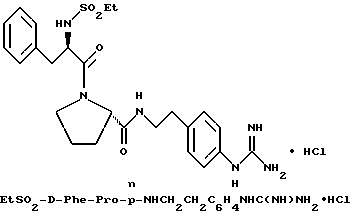
Способами, по существу эквивалентными описанным в примере 34, получают 1,8 г EtSO2-D-Phe-Pro-p-NHCH2CH2C6H4- CNH(NH)NH2•HCl из 4-нитрофенетиламин. HCl. ИК; 1H-ЯМР; FD-MS, m/e: 515 (M+).
HRMS (FAB), m/e, для C25H35N6O4S: рассчитано: 515,2441;
найдено: 515,2483.
Пример 36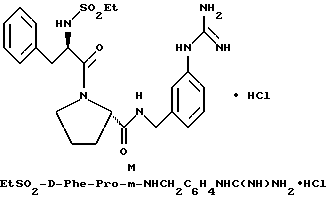
Способами, по существу эквивалентными описанным в примерах 30-В и 34-Б до 34-Е, получают 1,8 г EtSO2-D-Phe-Pro-m- NHCH2C6H4NHC(NH)NH2•HCl из 3-нитробензилбромида. ИК; 1H-ЯМР; FD-MS, m/e: 501 (M+) HRMS (FAB), m/e для C24H33N6O4S: рассчитано: 502,2284;
найдено: 501,2280.
Пример 37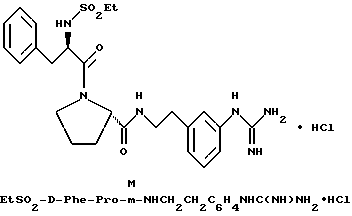
Способами, по существу эквивалентными описанным в примерах 26-Г, 26-Б (используя 5%-ный Pd/C вместо Pd/BaSO4 и этилацетат вместо толуола) и 34-Г до 34-Е, получают 0,85 г EtSO2-D-Phe-Pro-m- NHCH2CH2C6H4NHC(NH)NH2•HCl из 3-нитрокоричной кислоты.
1H-ЯМР; FD-MS, m/e: 515 (MH+).
Анализ для C25H34N6O4S•2,2HCl• 0,5H2O:
рассчитано, %: C 49,73; H 6,21; N 13,92;
найдено, %: C 49,45; H 5,82; N 13,55.
Пример 38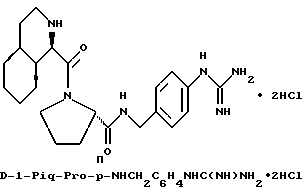
Способами, по существу эквивалентными описанным в примере 34, получают 0,94 г D-1-Piq-Pro-p-NHCH2C6H4NHC(NH)NH2•2HCl. Целевой продукт очищают путем RPHRLC (метод 2, градиент (A/B) от 98/2 до 70/30, 180 мин). 1H-ЯМР; FD-MS, m/e: 427 (MH+).
Анализ для C23H34N6O2•2HCl:
рассчитано, %: C 55,31; H 7,26; N 16,82; Cl 14,20;
найдено, %: C 55,05; H 7,23; N 16,55; Cl 14,24.
Пример 39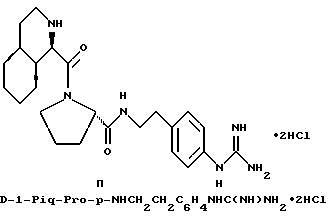
Способами, по существу эквивалентными описанным в примере 35, получают 1,03 г D-1-Piq-Рго-р-NHCH2CH2C6H4NHC(NH)NH2 •2HCl. Целевой продукт очищают путем RPHRLC (метод 2; градиент (A/B) от 98/2 до 70/30, 180 мин). 1H-ЯМР; FD-MS, m/e: 441 (M+).
Анализ для C24H36N6O2•2HCl• 1,5H2O:
рассчитано, %: C 53,33; H 7,65; N 15,55; Cl 13,12;
найдено, %: C 53,41; H 7,45; N 15,37; Cl 13,48.
Пример 40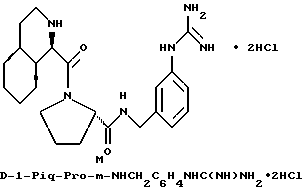
Способами, по существу эквивалентными описанным в примере 36, получают 1,04 г D-1-Piq-Pro-m-NHCH2C6H4NHC(NH)NH2•2HCl. Целевой продукт очищают путем RPHPLC (метод 2, градиент (A/B) от 98/2 до 70/30, 180 мин). 1H-ЯМР; FD-MS, m/e: 427 (M+).
Анализ для C23H34N6O2•2HCl•H2O:
рассчитано, %: C 53,38; H 7,40; N 16,24; Cl 13,70;
найдено, %: C 53,25; H 7,50; N 16,23; Cl 13,88.
Пример 41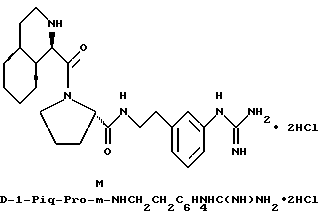
Способами, по существу эквивалентными описанным в примере 37, получают 0,96 г D-1-Piq-Pro-m-NHCH2CH2C6H4NHC(NH)NH2 •2HCl. Целевой продукт очищают путем RPHPLC (метод 2, градиент (A/B) от 98/2 до 70/30, 180 мин). 1H-ЯМР; FD-MS, m/e: 441 (M+).
Анализ для C24H36N6O2•2,1HCl• 1,5H2O:
рассчитано, %: C 52,97; H 7,61; N 15,44; Cl 13,68;
найдено, %: C 52,80; H 7,57; N 15,46; Cl 13,35.
Пример 42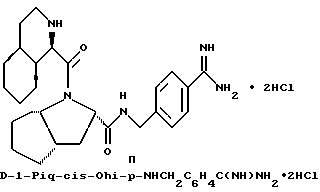
А) Получение Cbz-DL-1-Piq-cis-Ohi-OEt.
Способом, по существу эквивалентным описанному в примере 1-А, получают 16,6 г (100%) Cbz-DL-1-Piq-cis-Ohi-OEt из Cbz-DL-1-Piq-OH и цис-Ohi-OEt•HCl. 1H-ЯМР; FD-MS, m/e: 496 (M+).
Анализ для C29H40N2O5:
рассчитано, %: C 70,13; H 8,12; N 5,64;
найдено, %: C 69,96; H 8,23; N 5,73.
Б) Получение Cbz-D-1-Piq-cis-Ohi-p- NHCH2C6H4C(NH)NHCbz.
Способами, по существу эквивалентными описанным в примерах 1-Г и 18-Д, Cbz-D-1-Piq-cis-Ohi-p-NHCH2C6H4C(NH)NHCbz и Cbz-L-1-Piq-cis-Ohi-p-NHCH2C6H4C(NH)NHCbz получают из Cbz-D,L-1-Piq-Pro-OH и p-H2NCH2C6H4C(NH)NHCbz•2HCl. Эти диастереомеры разбавляют путем хроматографии на силикагеле, используя градиент этилацетат/гексан. Фракции, содержащие основной диастереомер (Rf= 0,31, этилацетат), объединяют и концентрируют с получением 1,3 г Cbz-L-1-Piq-cis-Ohi-p-NHCH2C6H4C(NH)NHCbz. Фракции, содержащие "следовый" диастереомер (Rf=0,19, этилацетат), объединяют и концентрируют с получением 1,5 г Cbz-D-1-Piq-cis-Ohi-p-NHCH2C6H4C(NH)NHCbz в виде белой пены. 1H-ЯМР; FD-MS, m/e: 735 (MH+).
Анализ для C43H51N5O6:
рассчитано, %: C 70,37; H 7,00; N 9,54;
найдено, %: C 70,20; H 7,22; N 9,36.
В) Получение D-1-Piq-cis-Ohi-p-NHCH2C6H4C(NH)NH2•2HCl.
Способом, по существу эквивалентным описанному в примере 18-Е, получают 0,61 г (63%) D-1-Piq-cis-Ohi-p- NHCH2C6H4C(NH)NH2•2HCl из Cbz-D-1-Piq-cis-Ohi-p-NHCH2C6H4C(NH)NHCbz. Используемый в этом случае ВЭЖХ-градиент составляет от 98/2 (A/B) до 70/30 (A/B) в течение 120 мин. 1H-ЯМР; FAB-MS, m/e: 466,4 (MH+).
Анализ для C27H39N5O2•2HCl• 1H2O:
рассчитано, %: C 58,27; H 7,79; N 12,58;
найдено, %: C 58,66; H 7,56; N 12,78.
Пример 43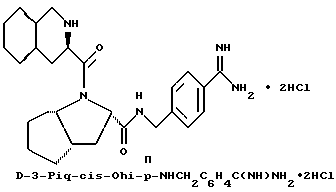
А) Получение D-3-Piq-cis-Ohi-p-NHCH2C6H4C(NH)NH2•2HCl
Способами, по существу эквивалентными описанным в примерах 1-А, 1-Г, 18-Д и 18-Е, получают 1,3 г 3-Piq-cis-Ohi-p- NHCH2C6H4C(NH)NH2•HCl. Используемый в этом случае ВЭЖХ-градиент составляет от 98/2 (A/B) до 70/30 (A/B) в течение 120 мин. 1H-ЯМР; FAB-MS, m/e: 466,4 (MH+).
Анализ для C27H39N5O2•2HCl.
рассчитано, %: C 60,22; H 7,67; N 13,00;
найдено, %: C 59,95; H 7,73; N 12,89.
Пример 44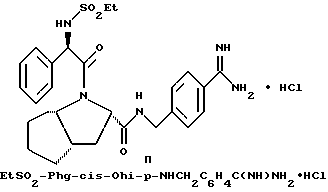
((S-цис)-N-[[4-(Аминоиминометил)фенил] метил] -1-[N-(этилсульфонил)- D-фенилглицил]-1H-индол-2-карбоксамид-гидрохлорид).
А) Получение Boc-D-Phg-cis-Ohi-OEt
Способом, по существу эквивалентным описанному в примере 1-А, получают 14,9 г (58%) Boc-D-Phg-cis-Ohi-OEt из Boc-D-Phg-OH и хлорангидрида этилового эфира (S)-цис-октагидроиндол-2-карбоновой кислоты. 1H-ЯМР; FD-MS, m/e: 430 (M+).
Анализ для C24H34N2O5:
рассчитано, %: C 66,95; H 7,96; N 6,51;
найдено, %: C 66,69; H 8,02; N 6,40.
Б) Получение D-Phg-cis-Ohi-OEt•HCl.
В охлаждаемый (0oC), перемешиваемый раствор Boc-D-Phg-цис-Ohi-OEt в этилацетате пропускают путем барботирования в течение 10 мин газообразный HCl. После перемешивания в течение 2 ч оставляют нагреваться до комнатной температуры, растворитель удаляют в вакууме. Полученное твердое вещество суспендируют в диэтиловом эфире и затем выделяют путем фильтрации, получая 10,7 г (97%) D-Phg-цис-Ohi-OEt•HCl. 1H-ЯМР; FD-MS, m/e: 331 (M+).
Анализ для C19H27N2O3Cl:
рассчитано, %: C 62,20; H 7,41; N 7,64;
найдено, %: C 62,42; H 7,36; N 7,85.
В) Получение EtSO2-D-Phg-cis-Ohi-OEt.
К раствору D-Phg-cis-Ohi-OEt•HCl HCl (10 г, 27 ммоль) и N,N-диизопропилэтиламина (10,7 мл, 61 ммоль) в ТГФ (200 мл) при -78oC прикапывают из капельной воронки раствор этансульфонилхлорида (3,9 г, 30 ммоль) в ТГФ (20 мл). Охлаждающую баню затем удаляют и раствор оставляют нагреваться до комнатной температуры. По истечении 18 ч раствор концентрируют в вакууме. Остаток растворяют в этилацетате (20 мл), промывают дважды 1н раствором лимонной кислоты (200 мл), насыщенным водным раствором бикарбоната натрия (200 мл) и солевым раствором (200 мл). Органическую фазу затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме с получением 11,2 г (97%) желтой пены. 1H-ЯМР; FD-MS, m/e: 422 (M+).
Анализ для C21H30N2O5S:
рассчитано, %: C 59,69; H 7,16; H 6,63;
найдено, %: C 59,94; H 7,08; N 6,78.
Г) Получение EtSO2-Phg-cis-Ohi-p- NHCH2C6H4C(NH)NH2.
Способами, по существу эквивалентными описанным в примерах 1-Г, 18-Д и 18-Е, получают 0,62 г EtSO2-Phg-цис-Ohi-п- NHCH2C6H4C(NH)NH2•2HCl. ВЭЖХ-градиент, используемый в этом случае, составляет от 90/10 (A/B) до 60/40 (A/B) в течение 120 мин.
1H-ЯМР; FAB-MS, m/e: 526,3 (MH+).
Анализ для C27H35N5O4S•HCl:
рассчитано, %: C 57,69; H 6,45; N 12,46;
найдено, %: C 57,47; H 6,48; N 12,20.
Пример 45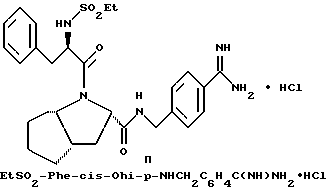
((S-цис)-N-[[4-(Аминоиминометил)фенил] метил] -1-[N-(этилсульфонил)- D-фенилаланил]-1H-индол-2-карбоксамид-гидрохлорид)
А) Получение EtSO2-Phe-cis-Ohi-p- NHCH2C6H4C(NH)NH2•2HCl
Способами, по существу эквивалентными описанным в примере 44, получают 1,5 г EtSO2-Phe-cis-Ohi-p-NHCH2C6H4C(NH)NH2 •HCl из Boc-D-Phe-OH.
Анализ для C28H37N5O4S•HCl•H2O:
рассчитано, %: C 56,51; H 6,94; N 11,77;
найдено, %: C 56,24; H 6,55; N 11,72.
Пример 46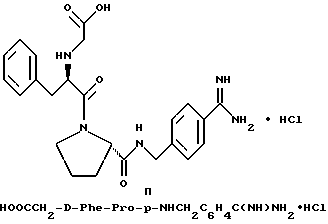
(N-(Карбоксиметил)-D-фенилаланил-N-[[4-(аминоиминометил)фенил] метил] - L-пролинамид-гидрохлорид)
А) Получение N-(t-BuOOCCH2)-N-Boc-D-Phe-Pro-OBn.
К раствору D-Phe-Pro-OBn•HCl (20 г, 51 ммоль) в ДМФ (100 мл) добавляют трет-бутил-бромацетат (9,9 г, 56 ммоль) за один раз, и в течение 30 мин прибавляют по каплям N,N-диизопропилэтиламин (17,4 мл, 101 ммоль). Эту смесь оставляют перемешиваться при комнатной температуре в течение 18 ч. Затем за один раз добавляют ди-трет-бутилдикарбонат (16,6 г, 76 ммоль) и N,N-диизопропилэтиламин (13,2 мл, 76 ммоль) и реакционную смесь дополнительно перемешивают в течение 24 ч. Растворитель удаляют в вакууме и остаток распределяют между этилацетатом (1 л) и 1 М водным раствором лимонной кислоты (500 мл). Слои разделяют и органическую фазу промывают один раз 1М водным раствором лимонной кислоты, два раза насыщенным водным раствором бикарбоната натрия и один раз солевым раствором (500 мл каждого из промывных средств). Органическую фазу сушат (MgSO4), фильтруют и концентрируют в вакууме. Янтарного цвета масло очищают путем хроматографии на силикагеле, элюируя градиентом этилацетат/гексан (от гексана до смеси гексана с 30% этилацетата). Содержащие продукт фракции объединяют и концентрируют с получением 19,0 г (66%) бесцветного масла, медленно кристаллизующегося при стоянии. 1H-ЯМР; FD-MS, m/e: 566 (M+).
Анализ для C32H42N2O7:
рассчитано, %: C 67,82; H 7,47; N 4,94;
найдено, %: C 68,06; H 7,33; N 5,17.
Б) Получение N-(t-BuOOCCH2)-N-Boc-D-Phe-Pro-OH.
К раствору N-(t-BuOOCCH2)-N-Boc-D-Phe-Pro-OBn (18,5 г, 33 ммоль) в этилацетате (250 мл) добавляют 5% Pd/C в качестве катализатора (5 г). Этот раствор в течение нескольких часов дегазируют в вакууме и помещают в атмосферу водорода на 2 ч при перемешивании. Источник водорода удаляют, добавляют диатомовую землю и суспензию фильтруют через слой диатомовой земли. Фильтрат концентрируют в вакууме с получением 13,2 г (84%) белой пены. 1H-ЯМР; FD-MS, m/e: 476 (M+).
Анализ для C25H36N2O7:
рассчитано, %: C 63,01; H 7,61; N 5,88; найдено, %: C 63,23; H 7,73; N 5,59;
В) Получение N-(t-BuOOCCH2)-N-Boc-D-Phe-Pro-p- NHCH2C6H4C(NH)NHCbz.
Способом, по существу эквивалентным по примеру 18-Д, получают 2,7 г (90%) N-(t-BuOOCCH2)-N-Boc-D-Phe-Pro-p- NHCH2C6H4C(NH)NHCbz из N-(t-BuOOCCH2)-N-Boc-D-Phe-Pro-OH-OH и p-H2NCH2C6H4C(NH)NHCbz•2HCl. 1H-ЯМР;
FD-MS, m/e: 743 (MH+).
Анализ для C41H51N5O8:
рассчитано, %: C 66,38; H 6,93; N 9,44;
найдено, %: C 66,08; H 6,92; N 9,16.
Г) Получение HOOCCH2-D-Phe-Pro-p- NHCH2C6H4C(NH)NH2•HCl.
В охлаждаемый (0oC) раствор N-(t-BuOOCCH2)-N-Boc-D-Phe- Pro-p-NHCH2C6H4C(NH)NHCbz (2,2 г, 3 ммоль) в диоксане (100 мл) пропускают путем барботирования газообразной HCl в течение 10 мин. После перемешивания в течение 3 часов смесь оставляют нагреваться до комнатной температуры, диоксан удаляют в вакууме. Остаток растворяют в смеси из абсолютного этанола (150 мл), воды (75 мл) и 1н HCl (6 мл). Туда же добавляют 5%-ный Pd/C (1 г). После дегазации с использованием вакуума, эту смесь помещают в атмосферу водорода на 16 ч при перемешивании и при комнатной температуре. Добавляют диатомовую землю и полученную суспензию фильтруют через слой диатомовой земли. Фильтрат концентрируют в вакууме с получением остатка, который немедленно очищают путем ВЭЖХ (метод 2; градиент (A/B) от 98/2 до 70/30, в течение 2 ч). Содержащие чистый продукт фракции объединяют и лиофилизируют с получением 1,1 г (72%) белого твердого вещества. 1H-ЯМР; FAB-MS, m/e: 452,3 (MH+).
Анализ для C24H29N5O4•2HCl:
рассчитано, %: C 54,97; H 5,96; N 13,35;
найдено, %: C 55,21; H 6,11; N 13,39.
Пример 47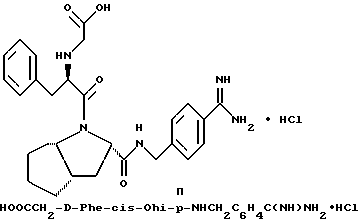
А) Получение трет-BuOOCCH2-D-Phe-cis-Ohi-OEt.
К раствору D-Phe-cis-Ohi-OEt•HCl (30 г, 79 ммоль) в ацетонитриле (400 мл) добавляют N,N-диизопропилэтиламин (41 мл, 236 ммоль) и трет-бутил-бромацетат (14 мл, 87 ммоль). Этот раствор нагревают до 50oC и выдерживают при этой температуре 3 ч. После охлаждения до комнатной температуры, раствор концентрируют в вакууме. Остаток растворяют в этилацетате (300 мл) и этот раствор промывают дважды насыщенным водным раствором хлорида аммония (200 мл), дважды насыщенным водным раствором бикарбоната натрия (200 мл) и дважды солевым раствором (200 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме с получением оранжевого масла, которое очищают путем хроматографии на силикагеле, элюируя градиентом от гексана до 1: 1 гексана с этилацетатом. Содержащие продукт фракции (установлено путем ТСХ) объединяют и концентрируют с получением 33,2 г (92%) бесцветного масла. 1H-ЯМР; FD-MS, m/e: 458 (M+).
Анализ для C26H38N2O5:
рассчитано, %: C 68,10; H 8,35; N 6,11;
найдено, %: C 68,37; H 8,47; N 5,90.
Б) Получение N-(t-BuOOCCH2)-N-Boc-D-Phe-cis-Ohi-OH.
К раствору трет-BuOOCCH2-D-Phe-cis-Ohi-OEt (30 г, 65 ммоль) в ТГФ (200 мл) добавляют N, N-диизопропилэтиламин (17 мл, 98 ммоль) и ди-трет-бутил-дикарбонат (15,7 г, 72 ммоль). Этот раствор доводят до легкого кипения с обратным холодильником и выдерживают при этой температуре в течение 16 ч. Нагревание прекращают, и после охлаждения раствор концентрируют в вакууме. Остаток растворяют в этилацетате (400 мл) и промывают дважды 1М раствором лимонной кислоты (200 мл), дважды насыщенным водным раствором бикарбоната натрия (200 мл) и дважды солевым раствором (200 мл). Органический раствор сушат (MgSO4), фильтруют и концентрируют в вакууме с получением желтого масла. Часть этого масла (24,8 г, 44 ммоль) растворяют в 300 мл диоксана. Туда же добавляют раствор, содержащий 2,05 г LiOH•H2O (49 ммоль) в 150 мл воды. Смесь перемешивают в течение 5 ч при комнатной температуре, после чего добавляют 100 мл насыщенного водного раствора хлорида аммония. Растворители удаляют в вакууме и остаток распределяют между насыщенным водным раствором бикарбоната натрия и диэтиловым эфиром. Слои разделяют и водный слой подкисляют до pH 3 с помощью лимонной кислоты. Подкисленный водный раствор экстрагируют 3 раза диэтиловым эфиром (200 мл) и эти экстракты объединяют, сушат над сульфатом магния, фильтруют и концентрируют с получением 24,3 г N-(t-BuOOCCH2)-N-Boc-D-Phe-cis-Ohi-OH в виде белой пены. 1H-ЯМР; FD-MS, m/e: 530 (M+).
Анализ для C29H42N2O7:
рассчитано, %: C 65,64; H 7,98; N 5,28;
найдено, %: C 65,39; H 8,04; N 5,39.
В) Получение HOOCCH2-D-Phe-cis-Ohi-p- NHCH2C6H4C(NH)NH2•HCl
Способом, по существу эквивалентным описанному в примерах 18-Д и 46-Г, получают 1,2 г (67%) HOOCCH2-D-Phe-cis-Ohi-p- NHCH2C6H4C(NH)NH2•HCl. 1H-MP; FAB-MS, m/e: 506,3 (MH+).
Анализ для C28H35N5O4•2HCl•H2O:
рассчитано, %: C 57,24; H 6,52; N 11,92;
найдено, %: C 57,40; H 6,30; N 11,69.
Пример 48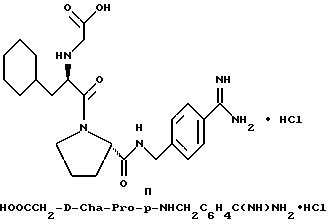
(N-Карбоксиметил)-D-циклогексилаланил-N-[[4-(аминоиминометил) фенил] метил]-L-пролинамид)
А) Получение HOOCCH2-D-Cha-Pro-p- NHCH2C6H4C(NH)NH2•HCl
Способом, по существу эквивалентным описанному в примере 46, получают 0,92 г HOOCCH2-D-Cha-Pro-p- NHCH2C6H4C(NH)NH2•HCl. 1H-ЯМР, FD-MS, m/e: 458 (M+).
Анализ для C24H35N5O4•HCl• 0,5H2O:
рассчитано, %: C 57,30; H 7,41; N 13,92;
найдено, %: C 57,52; H 7,29; N 13,83.
Пример 49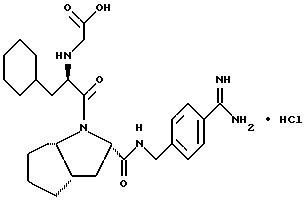
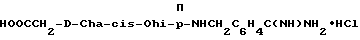
((S-цис)-N-[[4-(Аминоиминомeтил)фенил] метил] -1-[N-(карбоксиметил)- D-циклогексилаланил]-1H-индол-2-карбоксамид-гидрохлорид).
А) Получение HOOCCH2-D-Phe-cis-Ohi-p- NHCH2C6H4C(NH)NH2•HCl
Способами, по существу эквивалентными описанным в примерах 1-А и 1-Г, 18-Д, 44-Б, 47-А (используя бензилбромацетат) и 18-Е, получают 0,75 г HOOCCH2-D-Cha-cis-Ohi-p- NHCH2C6H4C(NH)NH2•HCl исходя из Boc-D-Cha и цис-Ohi-OEt•HCl. Для очистки этого вещества применяют ВЭЖХ, метод 2, используя градиент (A/B) от 98/2 до 70/30 в течение 3 ч. 1H-ЯМР; FAB-MS, m/e: 512,3 (MH+).
Анализ для C28H41N5O4•HCl:
рассчитано, %: C 61,36; H 7,72; N 12,78;
найдено, %: C 61,08; H 7,47; N 12,53.
Пример 50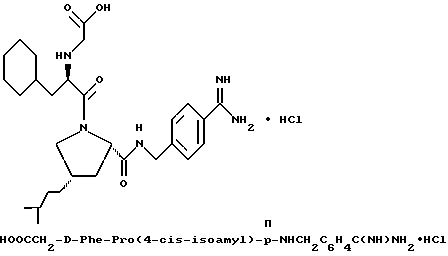
А) Получение Cbz-Pro(4-транс-OH)-OEt
К раствору Cbz-Pro(4-транс-OH)-OH (33 г, 124 ммоль) в этаноле (500 мл) добавляют п-толуолсульфокислоту (1 г) и раствор кипятят с обратным холодильником. Спустя 16 ч раствор охлаждают до комнатной температуры и растворитель удаляют в вакууме. Остаток растворяют в этилацетате (400 мл) и промывают дважды насыщенным водным раствором бикарбоната натрия и дважды насыщенным водным раствором хлорида натрия. Этилацетатный раствор сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 34,5 г (95%) бесцветного масла. 1H-ЯМР; FD-MS, m/e: 293 (M+).
Анализ для C15H19NO5:
рассчитано, %: C 61,42; H 6,53; N 4,77;
найдено, %: C 61,20; H 6,65; N 4,73.
Б) Получение Cbz-Pro(4-оксо)-OEt
Cbz-Pro-(4-транс-OH)-OEt (32,7 г, 111 ммоль) растворяют в дихлорметане (500 мл) при механическом перемешивании в круглодонной колбе емкостью 1 л. К этому раствору добавляют молекулярные сита 3  (100 г) и пиридинийхлорформиат (60 г, 278 ммоль) достаточно маленькими порциями при поддерживании интенсивного перемешивания. После перемешивания в течение 12 ч при комнатной температуре добавляют диэтиловый эфир (200 мл) и темного цвета суспензию декантируют от смолистого остатка и пропускают через колонку с силикагелем (200 г). Остаток промывают дважды дихлорметаном (200 мл) и объединенные промывные жидкости также пропускают через слой диоксида кремния. Фильтрат вымывают путем пропускания через колонку с силикагелем смесью 1:1 этилацетата с гексаном (4 л) и собирают фракции по 500 мл. Все содержащие продукт фракции, как установлено по ТСХ, объединяют и концентрируют в вакууме с получением 23,8 г (74%) бесцветного масла. 1H-ЯМР; FD-MS, m/e: 291 (M+).
(100 г) и пиридинийхлорформиат (60 г, 278 ммоль) достаточно маленькими порциями при поддерживании интенсивного перемешивания. После перемешивания в течение 12 ч при комнатной температуре добавляют диэтиловый эфир (200 мл) и темного цвета суспензию декантируют от смолистого остатка и пропускают через колонку с силикагелем (200 г). Остаток промывают дважды дихлорметаном (200 мл) и объединенные промывные жидкости также пропускают через слой диоксида кремния. Фильтрат вымывают путем пропускания через колонку с силикагелем смесью 1:1 этилацетата с гексаном (4 л) и собирают фракции по 500 мл. Все содержащие продукт фракции, как установлено по ТСХ, объединяют и концентрируют в вакууме с получением 23,8 г (74%) бесцветного масла. 1H-ЯМР; FD-MS, m/e: 291 (M+).
Анализ для C15H17NO5:
рассчитано, %: C 61,85; H 5,88; N 4,81;
найдено, %: C 61,57; H 5,82; N 4,71.
В) Получение Cbz-Pro-(4-изобутилметилиден)-OEt
Трет-бутоксид калия (34 г, 288 ммоль) суспендируют в тетрагидрофуране (800 мл) в высушенной в сушильном шкафу двугорлой круглодонной колбе, снабженной трубкой для ввода азота, стержневой магнитной мешалкой и капельной воронкой. К этой суспензии частями добавляют изоамилтрифенилфосфонийбромид (120 г, 288 ммоль). После перемешивания в течение 30 мин через капельную воронку прибавляют по каплям в течение 1 ч раствор Cbz-Pro(4-оксо)-OEt (70 г, 240 ммоль) в тетрагидрофуране (150 мл). После перемешивания еще 2 часа добавляют насыщенный водный раствор NH4Cl (100 мл). Этот раствор разбавляют этилацетатом (750 мл) и слои разделяют. Органический слой промывают два раза с 1н раствором лимонной кислоты, два раза насыщенным водным раствором бикарбоната натрия и два раза насыщенным водным раствором хлорида натрия. Органический раствор сушат над сульфатом магния, фильтруют и концентрируют с получением масла желтого цвета. Это масло очищают путем флэш-хроматографии на силикагеле, элюируя смесью 2:1 гексана с этилацетатом. Содержащие продукт фракции (определено с помощью ТСХ) объединяют и концентрируют в вакууме с получением 37 г (45%) бесцветного масла. 1H-ЯМР; FD-MS, m/e: 345 (M+).
Анализ для C20H27NO4:
рассчитано, %: C 69,54; H 7,88; N 4,05;
найдено, %: C 69,74; H 7,85; N 3,99.
Г) Получение Pro(4-цис-изоамил)-OEt•HCl
К раствору Cbz-Pro(4-изобутилметилиден)-OEt (37 г, 107 ммоль) в этаноле (500 мл) добавляют 5%-ный Pd/C (5 г). Через этот раствор барботируют азот в течение 5 мин и затем в течение 3 ч барботируют газообразный водород. Раствор фильтруют через слой диатомовой земли. Затем через раствор вплоть до насыщения барботируют газообразный хлороводород и после этого раствор концентрируют в вакууме с получением 26 г (97%) янтарного цвета масла.
1H-ЯМР; FD-MS, m/e: 214 (M+).
Анализ для C12H24ClNO2:
рассчитано, %: C 57,70; H 9,68; N 5,61;
найдено, %: C 57,46; H 9,50; N 5,82.
Д) Получение HOOCCH2-D-Phe-Pro-(4-цис-изоамил)-п- NHCH2C6H4C(NH)-NH2•HCl
Способами, по существу эквивалентными описанным в примерах 1-А и 1-Г, 18-Д, 44-Б, 47-А (используя бензилбромацетат) и 18-Е, получают 0,27 г HOOCCH2-D-Phe-Pro(4-цис-изоамил)-п- NHCH2C6H4(NH)NH2•HCl, исходя из Boc-D-Cha и Pro(4-цис-изоамил)-OEt•HCl. Для очистки этого соединения используют ВЭЖХ, метод 2, применяя градиент (A/B) от 98/2 до 50/50 в течение 3 ч. 1H-ЯМР; FAB-MS, m/e: 528,4 (MH+).
Анализ для C29H45N5O4•1,9HCl:
рассчитано, %: C 58,34; H 7,92; N 11,73; Cl 11,28;
найдено, %: C 58,30; H 7,85; N 11,83; Cl 11,27.
Пример 51
(D-Циклогексилаланил-N-[[4-(аминоиминометил)фенил] метил] -L- пролинамид-дигидрохлорид)
А) Получение Boc-D-Cha-Pro-p-NHCH2C6H4C(NH)NHCbz
Способами, по существу эквивалентными описанным в примерах 1-А, 46-Д и 18-Д, получают 32,5 г (94%) Boc-D-Cha-Pro-p- NHCH2C6H4C(NH)NHCbz, исходя из Boc-D-Cha-Pro-OH. 1H-ЯМР; FD-MS, m/e: 634 (M+).
Анализ для C35H47N5O6:
рассчитано, %: C 66,33; H 7,47; N 11,05;
найдено, %: C 66,30; H 7,47; N 11,26.
Б) Получение D-Cha-Pro-p-NHCH2C6H4C(NH)NHCbz•2HCl
Способом, по существу эквивалентным описанному в примере 23-А, получают 9,6 г (101%) D-Cha-Pro-p-NHCH2C6H4C(NH)NHCbz•2HCl из Boc-D-Cha-Pro-p-NHCH2C6H4C(NH)NHCbz. 1H-ЯМР; FD-MS, m/e: 534 (M+).
Анализ для C30H41N5O4Cl2:
рассчитано, %: C 59,40; H 6,81; N 11,54; Cl 11,69;
найдено, %: C 59,54; H 6,80; N 11,77; Cl 11,21.
В) Получение D-Cha-Pro-p-NHCH2C6H4C(NH)NH2 •2HCl
Способом, по существу эквивалентным описанному в примере 18-Е, получают 0,74 г (62%) D-Cha-Pro-p-NHCH2C6H4C(NH)NH2• 2HCl. Для очистки этого соединения используют ВЭЖХ, метод 2, применяя градиент (A/B) от 98/2 до 75/25 в течение 2,5 ч. 1H-ЯМР; FAB-MS, m/e: 400,3 (M+).
Анализ для C22H33N5O2•2,1HCl:
рассчитано, %: C 55,50; H 7,43; N 14,71; Cl 15,64;
найдено, %: C 55,64; H 7,50; N 14,65; Cl 15,81.
Пример 52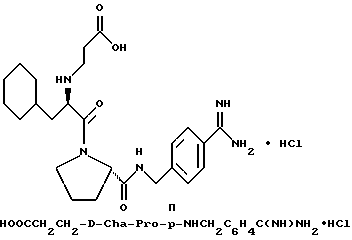
(N-(2-Карбоксиэтил)-D-циклогексилаланил-N-[[4-(аминоиминометил) фенил] метил]-L-пропиламид-гидрохлорид)
А) Получение HOOCCH2CH2-D-Cha-Pro-p- NHCH2C6H4C(NH)NH2•HCl
D-Cha-Pro-p-NHCH2C6H4C(NH)NHCbz•2HCl (2,5 г, 4,1 ммоль) суспендируют в этилацетате (100 мл) при перемешивании. К суспензии добавляют раствор 1 М KHCO3 (100 мл) и смесь перемешивают до полного растворения твердого вещества. Смесь переносят в делительную воронку и слои разделяют. Органический слой сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 1,24 г свободного основания в виде твердого белого вещества. Это твердое вещество растворяют в этаноле (100 мл). Добавляют бензилакрилат (0,41 г, 2,6 ммоль) и раствор перемешивают в течение 2 дней при комнатной температуре. Затем к этому раствору добавляют воду (50 мл), 1н HCl (4,6 мл) и 5%-ный Pd/C (0,5 г) и перемешиваемую суспензию дегазируют и помещают в атмосферу водорода. Спустя 16 ч добавляют диатомовую землю и суспензию фильтруют через слой диатомовой земли. Фильтрат концентрируют до объема 25 мл в вакууме и остаток очищают с помощью препаративной ВЭЖХ с обращенными фазами, метод 2, используя градиент (A/B) от 98/2 до 75/25 в течение 2,5 ч. Содержащие чистый продукт фракции (как определяют путем аналитической ВЭЖХ) объединяют, концентрируют и лиофилизуют с получением 0,27 г (23%) HOOCCH2CH2-D-Cha-Pro-p- NHCH2C6H4C(NH)NH2•HCl. FAB-MS, m/e: 472,4 (MH+).
Анализ для C25H37N5O4•1,9HCl:
рассчитано, %: C 55,50; H 7,25; N 12,94; Cl 12,45;
найдено, %: C 55,26; H 7,26; N 13,21; Cl 12,85.
Пример 53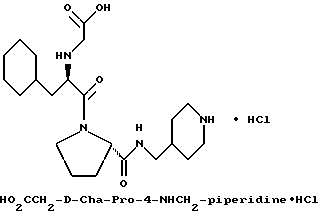
А) Получение Boc-4-(аминометил)пиридина
Способом, по существу эквивалентным описанному в примере 34-А, получают 19 г (87%) Boc-4-(аминометил)-пиридина из 4-(аминометил)пиридина. 1H-ЯМР.
Б) Получение 4-BocNHCH2-N-Cbz-пиперидина
4-BocNHCH2-пиридин (10 г, 48 ммоль) растворяют в этаноле (280 мл) и добавляют 5%-ный Pd/C (10 г). Суспензию встряхивают в атмосфере водорода (4,1 бара, 60 пси) в течение ночи при 60oC. Затем катализатор фильтруют и раствор концентрируют в вакууме с получением 9,0 г серого твердого вещества. 3,2 г твердого вещества растворяют в ТГФ (75 мл) и добавляют водный раствор (75 мл) карбоната калия (4,2 г, 30 ммоль). К этому перемешиваемому раствору добавляют бензилхлорформиат (2,3 мл, 16 ммоль). Спустя 15 мин раствор концентрируют в вакууме примерно до 1/2 первоначального объема и затем разбавляют этилацетатом. Органическую фазу отделяют и промывают солевым раствором, затем сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 4,6 г (76%) твердого белого вещества. 1H-ЯМР; FD-MS, m/e: 349 (M+).
В) Получение 4-NH2CH2-N-Cbz-пиперидин•HCl
Способом, по существу эквивалентным описанному в примере 23-А, получают 3 г (84%) 4-NH2CH2-N-Cbz-пиперидин•HCl из 4-BocNHCH2-N-Cbz-пиперидина. ИК; 1H-ЯМР; FD-MS, m/e: 249 (MH+).
Г) Получение N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-OH.
N-(t-BuO2CCH2)-N-Boc-D-Phe-Pro-OH (13 г, 27 ммоль) растворяют в этаноле (750 мл) и добавляют PtO2 (13 г). Суспензию встряхивают в атмосфере водорода при давлении 4,1 бара (60 пси) при 40oC в течение 16 ч. Катализатор затем фильтруют и фильтрат концентрируют в вакууме с получением 11,7 г (90%) белой пены. ИК; 1H-ЯМР; FD-MS, m/e: 483 (M+).
Анализ для C25H42N2O7:
рассчитано, %: C 62,22; H 8,77; N 5,80;
найдено, %: C 62,99; H 8,96; N 5,48.
Д) Получение HO2CCH2-D-Cha-Pro-4-NHCH2- пиперидин-гидрохлорида
Способами, по существу эквивалентными описанным в примерах 1-Ж и 46-Г, получают 1,1 г HO2CCH2-D-Cha-Pro-4-NHCH2- пиперидин-гидрохлорида из N-(t-BuO2CCH2)-N-Boc-D-Cha-Pro-OH и HCl•4-NH2CH2-N-Cbz-пиперидина. Продукт очищают путем RPHRLC, метод 2, используя градиент (A/B) от 98/2 до 70/30 в течение 2 ч. ИК; 1H-ЯМР; FD-MS, m/e: 423 (M+).
Анализ для C22H38N4O4•2HCl• 1,5H2O:
рассчитано, %: C 50,57; H 8,29; N 10,72;
найдено, %: C 50,31; H 8,46; N 10,93.
Пример 54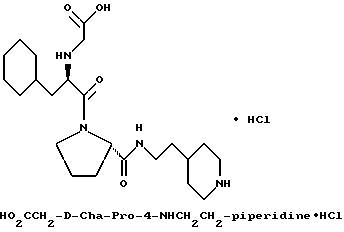
Получение HO2CCH2-D-Cha-Pro-4-NHCH2CH2- пиперидин-гидрохлорида
Способами, по существу эквивалентными описанным в примере 52, получают 0,59 г HO2CCH2-D-Cha-Pro-4-NHCH2CH2- пиперидин-гидрохлорида из 4-аминоэтилпиридина. Продукт очищают путем RPHRLC, метод 2, используя градиент (A/B) от 98/2 до 70/30 в течение 2 ч. ИК; 1H-ЯМР; FD-MS, m/e: 437 (M+).
Анализ для C23H40N4O4•2,5HCl• 1,5H2O:
рассчитано, %: C 49,80; H 8,27; N 10,10;
найдено, %: C 49,95; H 8,08; N 10,34.
Пример 55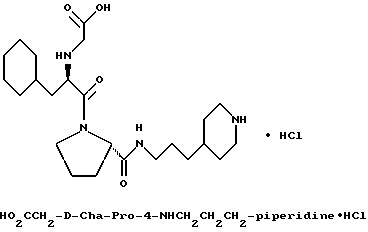
А) Получение 4-гидроксипропил-N-Cbz-пиперидина
Способами, по существу эквивалентными описанным в примере 53-Б, получают 28 г (67%) 4-гидроксипропил-N-Cbz-пиперидина из 4-гидроксипропилпиридина. 1H-ЯМР.
Б) Получение 4-(NH2CH2CH2CH2)-N-Cbz- пиперидин-гидрохлорида
Способами, по существу эквивалентными описанным в примерах 30-Б, 30-В и 30-Г, получают 7,3 г 4-(NH2CH2CH2CH2)- N-Cbz-пиперидин-гидрохлорида из 4-гидроксипропил-1-N-Cbz-пиперидина. 1H-ЯМР; FD-MS, m/e: 276 (M+).
В) Получение HO2CCH2-D-Cha-Pro-4- NHCH2CH2CH2-пиперидин•HCl
Способами, по существу эквивалентными описанным в примерах 53-Г и 53-Д, получают 0,39 г HO2CCH2-D-Cha-Pro-4- NHCH2CH2CH2-пиперидин•HCl из N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-OH и 4-аминопропил-N-Cbz-пиперидин•HCl. Продукт очищают путем RPHPLC, метод 2, используя градиент (A/B) от 98/2 до 70/30 в течение 2 ч. ИК; 1H-ЯМР; IS-MS, m/e: 451,4 (MH+).
Анализ для C24H42N4O4•2HCl• H2O:
рассчитано, %: C 53,23; H 7,56; N 10,35;
найдено, %: C 53,43; H 8,63; N 10,19.
Пример 56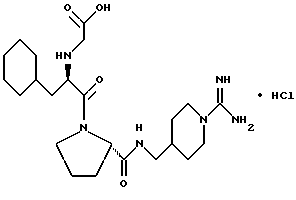
HO2CCH2-D-Cha-Pro-4-NHCH2-1-амидинопиперидин•HCl
(N-(Карбоксиметил)-D-циклогексилаланил-N-[[1-(аминоиминометил)- гексагидропиридин-4-ил]метил]-L-пролинамин-гидрохлорид)
Получение HO2CCH2-D-Cha-Pro-5-NHCH2-1- амидинопиперидин•HCl
Способами, по существу эквивалентными описанным в примерах 34-Г, 23-А, 1-Ж (используя N-(трет-BuO2CCH2)-N-Boc-D- Cha-Pro-OH), 18-Д и 1-3, получают 0,35 г HO2CCH2-D-Cha- Pro-4-NHCH2-1-амидинопиперидин-гидрохлорида из 4-BocNHCH2пиперидина. Целевой продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 75/25 в течение 150 минут). ИК; 1H-ЯМР; FAB-MS, m/e: 465 (MH+).
Анализ для C23H40N6O4•2HCl:
рассчитано, %: C 51,39; H 7,88; N 15,63; Cl 13,19;
найдено, %: C 51,66; H 7,98; N 15,80; Cl 13,48.
Пример 57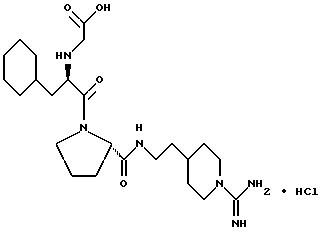
HO2CCH2-D-Cha-Pro-4-NHCH2CH2-1- амидинопиперидин•HCl
Получение HO2CCH2-D-Cha-Pro-4-NHCH2CH2- 1-амидинопиперидин•HCl
Способами, по существу эквивалентными описанным в примерах 34-Г, 23-А, 1-Ж (используя N-(трет-BuO2CCH2)-N-Boc-D- Cha-Pro-OH), 18-Д и 1-3, получают 0,34 г HO2CCH2-D-Cha- Pro-4-NHСH2CH2-1-амидинопиперидин•HCl из 4-Boc-NHCH2CH2-пиперидина. Целевой продукт очищают с помощью RPHRLC, метод 2, используя градиент (A/B) от 98/2 до 75/25, 150 мин, ИК; 1H-ЯМР; FAB-MS, m/e: 479,4 (MH+).
Анализ для C24H42N6O4•2HCl:
рассчитано, %: C 52,26; H 8,07; N 15,24; Cl 12,86;
найдено, %: C 52,56; H 8,15; N 15,37; Cl 13,07.
Пример 58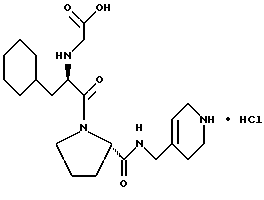
HO2CCH2-D-Cha-Pro-4-NHCH2-3,4-дегидро- пиперидин•HCl
А) Получение 4-BocNHCH2-N-метил-пиридиний-иодида
К перемешиваемому раствору 4-BocNHCH2-пиридина (20 г, 96 ммоль) в ацетонитриле (200 мл) добавляют иодметан (8,9 мл, 144 ммоль).
Спустя 16 ч раствор концентрируют в вакууме с получением 33,8 г (96%) вязкого светло-желтого масла. FD-MS, m/e: 223,1 (M+).
Б) Получение 4-BocNHCH2-N-Fmoc-3,4-дегидро-пиперидина
К перемешиваемому раствору 4-BocNHCH2-метилпиридинийиодида (7,7 г, 34 ммоль) в 1,2-дихлорэтане (100 мл) добавляют 1,8-бис(диметиламино)нафталина (1,5 г, 6,8 ммоль), затем 2-хлорэтилхлорформиата (5,3 г, 37 мл). Раствор кипятят с обратным холодильником и спустя 2 ч охлаждают до комнатной температуры и растворитель удаляют в вакууме, а остаток быстро хроматографируют через колонку с силикагелем, элюируя смесью гексана с 20% этилацетата. Органические растворители удаляют в вакууме и остаток растворяют в метаноле (300 мл) и кипятят с обратным холодильником в течение 20 мин. После этого добавляют насыщенный водный раствор бикарбоната натрия (100 мл) и растворители удаляют в вакууме. Остаток растворяют в воде (200 мл) и промывают этот раствор дважды гексаном, затем насыщают твердым NaCl и экстрагируют несколько раз этилацетатом.
Объединенные этилацетатные экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме с получением слегка желтоватого масла, которое растворяют в дихлорметане (75 мл). К этому перемешиваемому раствору затем добавляют N, N-диизопропилэтиламин (2,1 мл, 12,2 ммоль), после этого - 9-флуоренилметилхлорформиат (3,2 г, 12,2 ммоль). Спустя 2 ч растворитель удаляют в вакууме и остаток растворяют в этилацетате (250 мл) и промывают дважды 1н раствором лимонной кислоты, один раз солевым раствором, дважды насыщенным водным раствором бикарбоната натрия и, наконец, один раз солевым раствором. Органическую фазу после этого сушат над сульфатом магния, фильтруют и концентрируют в вакууме и остаток хроматографируют через колонку с силикагелем, элюируя ступенчато градиентом от гексана с 5% этилацетата до гексана с 50% этилацетата. Содержащие продукт фракции (определено путем ТСХ) объединяют и концентрируют с получением 4 г (27%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 435 (M+).
В) Получение N-(t-BuO2CCH2)-N-Boc-D-Cha-Pro-4- NHCH2-N-Fmoc-3,4-дегидро-пиперидина
Способами, по существу эквивалентными описанным в примерах 23-А и 1-Ж (используя N-(t-BuO2CCH2)-N-Boc-D-Cha-Pro-OH), получают 2,5 г N-(t-BuO2CCH2)-N-Boc-D-Cha-Pro-4- NHCH2-N-Fmoc-3,4-дегидро-пиперидина из 4-BocNHCH2-N-Fmoc- 3,4-дегидро-пиперидина. ИК; 1H-ЯМР; FD-MS, m/e: 799 (M+).
Г) Получение N-(t-BuO2CCH2)-N-Boc-D-Cha-Pro-4- NHCH2-3,4-дегидро-пиперидина
N-(t-BuO2CCH2)-N-Boc-D-Cha-Pro-4-NHCH2-N- Fmoc-3,4-дегидро-пиперидин (1,5 г, 1,9 ммоль) растворяют в морфолине (25 мл) и после перемешивания в течение 5 ч растворитель выпаривают в вакууме. Остаток растворяют в этилацетате и полученный раствор промывают дважды насыщенным водным раствором бикарбоната натрия, сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток затем растворяют в небольшом объеме хлороформа и хроматографируют на силикагеле, элюируя градиентом (A/B) от 5% до 10% A/B (А=9:1 метанол/концентрированный NH4OH; В = хлороформ). Содержащие продукт фракции, согласно ТСХ, объединяют и концентрируют в вакууме с получением 890 мг (82%) белого твердого вещества. 1H-ЯМР; FD-MS, m/e: 576 (MH+).
Д) Получение 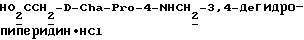
Через раствор N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro- 4-NHCH2-3,4-дегидро-пиперидина (820 мг, 1,4 ммоль) и анизола (1 мл) в диоксане (25 мл) при 0oC в течение 10 мин барботируют газообразный HCl. После перемешивания в течение 12 часов растворитель удаляют в вакууме и остаток растворяют в воде (50 мл) и полученный раствор промывают дважды диэтиловым эфиром. Водную фазу затем концентрируют до объема примерно 20 мл в вакууме и остаток очищают путем RPHRLC (метод 2; градиент (A/B) от 98/2 до 70/30, 2 ч). Содержащие продукт фракции, как определено с помощью аналитической RPHRLC, объединяют, частично концентрируют в вакууме и лиофилизуют с получением 442 мг (68%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 423 (MH+).
Анализ для C22H36N4O4•2HCl• 1,5H2O:
рассчитано, %: C 50,57; H 8,29; N 10,72;
найдено, %: C 50,31; H 8,46; N 10,93.
Пример 59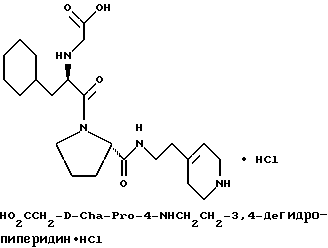
Получение HO2CCH2-D-Cha-Pro-4-NHCH2CH2- 3,4-дегидро-пиперидин•HCl
Способами, по существу эквивалентными описанным в примере 58, получают 73 мг HO2CCH2-D-Cha-Pro-4-NHCH2CH2- 3,4-дегидро-пиперидин-гидрохлорида из 4-BocNHCH2CH2-пиридина. Целевой продукт очищают путем RPHRLC метод 2 (градиент (A/B) от 98/2 до 70/30, 2 ч). ИК; 1H-ЯМР; IS-MS, m/e: 435,2 (MH+).
Анализ для C23H38N4O4•2,3HCl• 3H2O:
рассчитано, %: C 48,26; H 8,15; N 9,79; Cl 14,24;
найдено, %: C 48,31; H 7,93; N 9,66; Cl 14,56.
Пример 60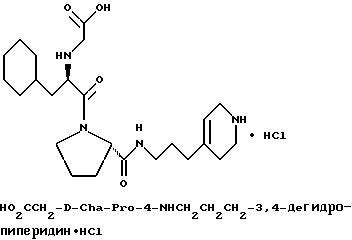
Получение HO2CCH2-D-Cha-Pro-4- NHCH2CH2CH2-3,4-дегидро-пиперидин•HCl
Способами, по существу эквивалентными описанным в примере 58, получают 205 мг HO2CCH2-D-Cha-Pro-4- NHCH2CH2CH2-3,4-дегидро-пиперидин-гидрохлорида из 4-BocNHCH2CH2CH2-пиридина. ИК; 1H-ЯМР; IS-MS, m/e: 449,2 (MH+).
Анализ для C24H40N4O4•2,3HCl• H2O:
рассчитано, %: C 52,37; H 3,11; N 10,18;
найдено, %: C 51,64; H 7,72; N 10,31; Cl 14,69.
Пример 61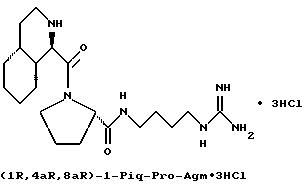
(N-[4-[(Аминоиминометил)амино] бутил] -1-[[(4aR,8AR)-декагидро-1- (R)-изохинолинил]карбонил]-1-пропиламид-тригидрохлорид)
Rf в этом примере определяют путем тонкослойной хроматографии на силикагеле (силикагель 60 F-254) в следующих системах (по объему):
(А) хлороформ : метанол : уксусная кислота = 135:15:1
(Б) этилацетат : уксусная кислота : абсолютный этанол = 90:10:10
(В) этилацетат : гексан = 70:30
(Г) хлороформ
А) N-Метоксикарбонилфенетиламин
К перемешиваемому раствору фенетиламина (75,2 мл, 0,6 моль) и триэтиламина (83 мл, 0,6 моль) в ТГФ (500 мл) медленно добавляют метилхлороформиат (46,2 мл, 0,6 моль), растворенный в ТГФ (50 мл). После реакции перемешивают еще 1 ч при комнатной температуре, добавляют диэтиловый эфир (2 л) и 1 н HCl (800 мл). Органический слой промывают водой, сушат над сульфатом магния, фильтруют и фильтрат концентрируют в вакууме с получением в виде прозрачного масла чистого целевого соединения (102 г, 95%).
Б) 2-Метоксикарбонил-DL-1,2,3,4-тетрагидроизохинолин-1-карбоновая кислота
К раствору N-метоксикарбонил-фенетиламина (102 г, 0,57 моль) в трифторуксусной кислоте (300 мл) добавляют глиоксиловую кислоту (63 г, 0,68 моль) и смесь нагревают до температуры кипения с обратным холодильником. После кипячения в течение 4 ч с обратным холодильником, реакционную смесь охлаждают до комнатной температуры, растворитель удаляют в вакууме и к остатку добавляют диэтиловый эфир (800 мл) с водой (100 мл). Реакционную смесь доводят до pH 12 с помощью 5н раствора NaOH и отделяют водный слой. К водному слою добавляют диэтиловый эфир (500 мл) и раствор подкисляют до pH 2,5 с помощью 5н HCl. Органический слой отделяют, сушат над сульфатом магния, фильтруют и фильтрат концентрируют в вакууме, получая в виде масла чистое целевое соединение (107 г; 80%). FAB-MS: 236 (MH+).
В) Трет-бутиловый эфир 2-метоксикарбонил-DL-1,2,3,4- тетрагидроизохинолин-1-карбоновой кислоты
К перемешиваемому охлаждаемому (0oC) раствору 2-метоксикарбонил-DL-1,2,3,4-тетрагидроизохинолин-1-карбоновой кислоты (2) (105 г, 0,45 моль) в CH2Cl2 (200 мл) добавляют трет-бутанол (52 мл, 0,54 моль) и ДЦК (92 г, 0,45 моль). Спустя 2 ч при 0oC и 24 ч при комнатной температуре, растворитель удаляют в вакууме и к остатку добавляют этилацетат (800 мл) с 1н раствором бикарбоната натрия (300 мл). Органический слой отделяют, промывают водой, 1,5 н раствором лимонной кислоты и водой. Органический слой сушат над сульфатом магния, фильтруют и фильтрат концентрируют в вакууме с получением в виде масла чистого целевого соединения (106 г; 81%); FAB-MS: 292 (MH+) ТСХ: Rf(A)= 0,61; элементный анализ для C16H21NO4:
рассчитано, %: C 65,96; H 7,27; N 4,81;
найдено, %: C 66,24; H 7,28; N 4,73.
Г) Трет-бутиловый эфир 2-метоксикарбонил-(1RS,4aSR,8aSR)- пергидроизоиндолин-1-карбоновой кислоты
Раствор трет-бутилового эфира 2-метилкарбонил-DL-1,2,3,4- тетрагидроизохинолин-1-карбоновой кислоты (105 г, 0,36 моль) в трет-бутаноле (800 мл) восстанавливают в присутствии 5% Rh/Al2O3 (52,5 г) при давлении водорода 55 бар (800 пси) в аппаратуре высокого давления при 50oC и в течение 24 ч. Реакционную смесь фильтруют через слой диатомовой земли, и фильтрат концентрируют в вакууме. Полученное масло сушат с получением чистого целевого соединения (96,5 г, 90%); FD-MS: 298 (MH+); ТСХ: Rf(B)=0,63.
Д) Этиловый эфир 2-метоксиркабонил-(1RS, 4aRS,8aRS)- пергидроизохинолин-1-карбоновой кислоты
К раствору трет-бутилового эфира 2-метоксикарбонил-(1RS,4aSR,8aSR)- пергидроизохинолин-1-карбоновой кислоты (81,2 г, 273 ммоль) в этаноле (500 мл) добавляют этоксид натрия (21%-ный в этаноле) (88,4 мл, 273 ммоль) и реакционную смесь кипятят с обратным холодильником в течение 24 ч. Органический растворитель выпаривают в вакууме, добавляют этилацетат (400 мл) и воду (100 мл) к полученному остатку. Органический слой отделяют, промывают дважды водой, сушат над сульфатом магния, фильтруют и фильтрат концентрируют в вакууме с получением в виде масла чистого целевого соединения (70 г, 95%); FAB-MS: 270 (MH+); TCX: Rf(A)=0,61.
Е) 2-Метоксикарбонил-(1RS, 4aRS, 8aRS)-пергидроизохинолин-1-карбоновая кислота
К раствору продукта стадии Д) (70 г, 260 ммоль) в ТГФ (250 мл) добавляют 2н раствор NaOH (156 мл, 312 ммоль) и реакционную смесь перемешивают при комнатной температуре (30 ч). Органический растворитель выпаривают в вакууме, к полученному остатку добавляют диэтиловый эфир (400 мл) и воду (100 мл). Водный слой отделяют и добавляют к нему этилацетат (400 мл). pH раствора доводят до 2,0 с помощью 5н HCl. Органический слой сушат (MgSO4), фильтруют и фильтрат концентрируют в вакууме с получением прозрачного масла. Масло кристаллизуют из гексана (200 мл) с получением чистого целевого соединения (46,4 г, 74%); FAB-MS: 242 (MH+); TCX: Rf(A)=0,36; элементный анализ для C12H19NO4:
рассчитано, %: C 59,74; H 7,94; N 5,81;
найдено, %: C 59,95; H 7,88; N 5,54.
Значения ЯМР получают путем эксперимента гомонуклеарного расщемления, COSY, HMQC и DEPT.
Ж) 2-Cbz-(1RS,4aRS,8aRS)-пергидроизохинолин-1-карбоновая кислота
К перемешиваемому раствору продукта стадии Е) (46 г, 191 ммоль) в безводном CH3CN (200 мл), при комнатной температуре и в инертной атмосфере, добавляют раствор иодтриметилсилана (62,4 мл, 440 ммоль) в CH3CN (60 мл). Реакционную смесь перемешивают при 55oC в течение 30 мин и охлаждают до комнатной температуры. Реакцию подавляют путем добавления воды (100 мл), затем добавляют метабисульфит натрия (1 г). pH реакционной среды устанавливают равным 10,0 с помощью 5н раствора NaOH и прибавляют по каплям бензилхлорформиат (27,3 мл, 191 ммоль), поддерживая pH 10 с помощью 2н раствора NaOH. После реакции перемешивают дополнительно 30 минут при комнатной температуре, органический растворитель выпаривают в вакууме и добавляют диэтиловый эфир (200 мл). Реакционную смесь затем оставляют стоять при комнатной температуре в течение 2 ч и добавляют этилацетат (200 мл). Водный раствор подкисляют до pH 2,5 с помощью 5н HCl; органический слой отделяют, сушат (MgSO4), фильтруют и фильтрат концентрируют в вакууме с получением чистого целевого соединения в виде масла (39,5 г, 65%); FAB-MS: 318 (MH+).
Элементный анализ для C18H23NO4:
рассчитано, %: C 68,12; H 7,30; N 4,41;
найдено, %: C 66,37; H 7,52; N 4,37.
З) 2-Cbz-(1RS,4aRS,8aRS)-пергидроизохинолин-1-карбонил-Pro-O-трет-Bu
К перемешиваемому, охлажденному до 0oC раствору продукта стадии Ж) (39 г, 123 ммоль) в ДМФ (200 мл) добавляют трет-бутиловый сложный эфир пролина (21,1 г, 123 ммоль), 1-гидроксибензотриазол (16,6 г, 123 ммоль) и ДЦК (25,3 г, 123 ммоль). Реакционную смесь перемешивают в течение 2 ч при 0oC и 24 ч при комнатной температуре. Полученный осадок фильтруют и фильтрат концентрируют в вакууме с получением масла. Масло растворяют в этилацетате (200 мл) с водой (100 мл). Органический слой промывают последовательно 1н раствором бикарбоната натрия, водой, 1,5н раствором лимонной кислоты и водой. Органический слой сушат (MgSO4), фильтруют и фильтрат выпаривают с получением аморфного твердого вещества, которое является целевым соединением в виде смеси диастереомеров (52,7 г, 91%). FAB-MS: 471 (MH+).
И) 2Cbz-(4AR,8aR)-пергидроизохинолин-1(R)-карбонил-Pro-OH
К перемешиваемому раствору продукта стадии З) (52,4 г, 111 ммоль) в CH2Cl2 (20 мл) добавляют трифторуксусную кислоту (70 мл) и анизол (5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч и концентрируют в вакууме без нагревания. Остаток разбавляют диэтиловым эфиром (400 мл), водой (100 мл) и pH раствора доводят до 10,0 с помощью 5н раствора NaOH. Водный слой отделяют и добавляют этилацетат (100 мл). pH раствора доводят до 2,5 с помощью 5н HCl; органический слой отделяют, сушат над сульфатом магния, фильтруют и фильтрат концентрируют в вакууме с получением прозрачного масла. Масло растворяют в диэтиловом эфире (500 мл) и к раствору добавляют (L)-(-)-альфа-метилбензиламин. Раствор выдерживают при комнатной температуре 24 ч. Полученное твердое вещество фильтруют, промывают диэтиловым эфиром и сушат. Твердое вещество суспендируют в этилацетате, промывают полученную суспензию 1,5н раствором лимонной кислоты и водой. Органический слой сушат над сульфатом магния, фильтруют и фильтрат выпаривают с получением целевого соединения в виде масла (20,2 г, 44%) FAB-MS: 415 (MH+); [α]D = 3,2o (c = 0,5, метанол); элементный анализ для C23H30N2O5:
рассчитано, %: C 66,65; H 7,30; N 6,76;
найдено, %: C 66,38; H 7,36; N 6,63.
К) 2-Cbz-(4aR,8aR)-пергидроизохинолин-1(R)-карбонил-Pro- NH-(CH2)4-NH-Boc
В колбе 1 продукт стадии И) (1,06 г, 2,55 ммоль) растворяют в ДМФ (10 мл), охлаждают до -15oC и добавляют N-метилморфолин (0,28 мл, 2,55 ммоль), затем добавляют изобутилхлорформиат (0,33 мл, 2,55 ммоль). Реакционную смесь перемешивают при -15oC в течение 2 мин.
В колбе 2 N-Boc-1,4-диамино-бутан (0,48 г, 2,55 ммоль) растворяют в ДМФ (10 мл), охлаждают до 0oC и к раствору добавляют N-метилморфолин (0,28 мл, 2,55 ммоль). Реакционную смесь перемешивают в течение 2 мин при 0oC.
Содержимое колбы 2 добавляют в колбу 1 и реакционную смесь перемешивают в течение 4 ч при -15oC и в течение 24 ч при комнатной температуре. К реакционной смеси добавляют 1н раствор бикарбоната натрия (1 мл) и растворитель удаляют в вакууме с получением масла. Остаток растворяют в этилацетате (200 мл) и промывают последовательно 1,5 н раствором лимонной кислоты, водой, 1н раствором бикарбоната натрия (100 мл) и водой. Органический раствор сушат (MgSO4), фильтруют и концентрируют досуха в вакууме, получая сырое целевое соединение в виде твердого вещества (1,47 г, 99%) FAB-MS: 585 (MH+); ТСХ: Rf(A)=0,70.
Л) (4aR,8aR)-пергидроизохинолин-1(R)-карбонил-Pro-Agm•3HCl
К перемешиваемому раствору продукта стадии К) (1,4 г, 2,4 ммоль) в CH2Cl2 (2 мл) добавляют трифторуксусную кислоту (25 мл) и анизол (2,5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 30 мин и концентрируют в вакууме без нагревания. Реакционную смесь разбавляют диэтиловым эфиром (100 мл) и суспензию декантируют. Полученное масло порошкуют дважды в диэтиловом эфире и высушивают. Высушенное масло растворяют в ТГФ (20 мл), добавляют триэтиламин (0,66 мл, 4,8 ммоль) и к смеси добавляют бис-Cbz-S-метилтиомочевину (0,859 г, 2,4 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 48 ч. Органический растворитель выпаривают в вакууме и остаток растворяют в этилацетате (200 мл) и промывают последовательно 1н раствором бикарбоната натрия (100 мл) и водой. Органический раствор сушат (MgSO4), фильтруют и концентрируют фильтрат досуха в вакууме с получением сырого твердого вещества (1,5 г, 79%); ТСХ: Rf(Г)=0,33. Сырое твердое вещество (1,5 г, 1,93 ммоль), растворенное в этаноле (50 мл) с водой (10 мл) и 1н HCl (5,8 мл, 5,8 ммоль), гидрируют в присутствии 5%-ного Pd/C в качестве катализатора (2,5 г) при комнатной температуре и под давлением. Катализатор удаляют путем фильтрования и фильтрат концентрируют в вакууме с получением масла. Масло растворяют в трифторуксусной кислоте (10 мл), добавляют тиоанизол (1,0 мл) и к смеси добавляют трифторметансульфокислоту (1,0). Реакционную смесь перемешивают при комнатной температуре в течение 0,5 ч и добавляют этиловый эфир (100 мл). Отделившуюся в результате отстаивания жидкость декантируют и полученное масло дважды растирают с диэтиловым эфиром и сушат в вакууме с получением сырого твердого вещества (1,3 г). Твердое вещество (1,3 г) растворяют в 0,05%-ном растворе HCl и вносят в колонку размером 5 х 25 см с неподвижной фазой Vydac C18-смолой. Для элюирования пептида из колонки используют градиент возрастающих концентраций CH3CN (от 2% до 25%). Фракции собирают и объединяют на основе профильной аналитической RPHRLC и лиофилизируют с получением чистого целевого соединения (0,139 г, 15%); FAB-MS 393 (MH+).
Элементный анализ для C20H36N6O2• 5HCl•3H2O:
рассчитано, %: C 38,28;
найдено, %: C 38,34.
Пример 62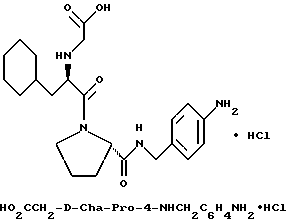
Получение HO2CCH2-D-Cha-Pro-4- NHCH2C6H4NH2•HCl
Способами, по существу эквивалентными описанным в примерах 1-Ж (используя N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-OH), 23-Г и 58-Д, получают 0,17 г HO2CCH2-D-Cha-Pro-4- NHCH2C6H4NH2•HCl из 4-нитробензиламин-гидрохлорида. Целевой продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 2 ч). ИК; 1H-ЯМР; FAB-MS, m/e; 431,3 (MH+).
Анализ для C23H34N4O4•2,2HCl• 1,5H2O:
рассчитано, %: C 51,37; H 7,35; N 10,42; Cl 14,50;
найдено, %: C 50,87; H 6,72; N 10,41; Cl 14,18.
Пример 63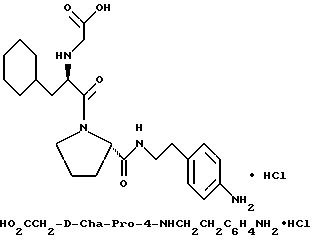
Получение HO2CCH2-D-Cha-Pro-4- NHCH2CH2C6H4NH2•HCl.
Способами, по существу эквивалентными описанным в примере 62, получают 0,19 г HO2CCH2-D-Cha-Pro-4- NHCH2CH2C6H4NH2•HCl из 4-нитрофенетиламина-гидрохлорида. Целевой продукт очищают путем RPHRLC, метод 2 (градиент A/B от 98/2 до 70/30, 2 часа). ИК; 1H-ЯМР; FAB-MS, m/e: 445,3 (MH+).
Анализ для C24H36N4O4•2,2HCl• 0,5H2О:
рассчитано, %: C 54,00; H 7,40; N 10,50; Cl 14,61;
найдено, %: C 53,65; H 7,59; N 10,24; Cl 14,33.
Пример 64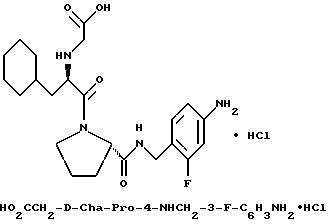
А) Получение 4-Boc2NCH2-3-F-C6H3NO2
К перемешиваемому раствору 2-фтор-4-нитротолуола (-5 г, 32 ммоль) в тетрахлориде углерода (160 мл) добавляют N-бром-сукцинимид (5,7 г, 32 ммоль), затем перекись бензоила (0,78г, 3,2 ммоль) и раствор кипятят с обратным холодильником. Спустя 12 ч нагревание удаляют и смесь разбавляют тетрахлоридом углерода (100 мл) и промывают водой. Органическую фазу затем разбавляют этилацетатом (300 мл), сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток растворяют в тетрагидрофуране (50 мл) и добавляют к перемешиваемому раствору NaH (60%-ная дисперсия в масле; 1,3 г, 32 ммоль) в ди-трет-бутилиминодикарбоксилат (6,9 г, 32 ммоль) в ТГФ (100 мл). После перемешивания в течение ночи растворитель удаляют в вакууме и остаток хроматографируют на силикагеле, элюируя ступенчатым градиентом от гексана до смеси гексана с 20% этилацетата. Содержащие продукт фракции (определяют путем ТСХ) объединяют и концентрируют в вакууме с получением 39 г (33%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 370 (M+).
Анализ для C17H23N2O6:
рассчитано, %: C 55,13; H 6,26; N 7,56;
найдено, %: C 55,27; H 6,23; N 7,44.
Б) Получение HO2CCH2-D-Cha-Pro-4-NHCH2-3- F-C6H3NH2•HCl
Способами, по существу эквивалентными описанным в примерах 23-А, 1-Ж (используя N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-ОH), 23-Г и 58-Ц, получают 0,44 г HO2CCH2-D-Cha-Pro-4-NHCH2- 3-F-C6H3NH2•HCl. Целевой продукт очищают, используя RPHRLC, метод 2 (градиент A/B от 98/2 до 70/30, 2 ч); ИК; 1H-ЯМР; FAB-MS, m/e: 449,3 (MH+).
Анализ для C23H33N4O4F•1,3HCl:
рассчитано, %: C 55,70; H 6,97; N 11,30; Cl 9,29;
найдено, %: C 55,38; H 6,97; N 11,05; Cl 9,31.
Пример 65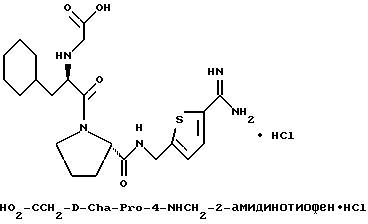
(N-(Карбоксиметил)-D-циклогексилаланил-N-[[5-(аминоиминометил)тиофен- 2-ил]метил]-L-пролинамид-гидрохлорид)
А) Получение 2-циано-5-формилтиофена
В высушенную в сушильном шкафу трехгорлую колбу емкостью 1 л вносят диизопропиламин (9 мл, 66 ммоль) и ТГФ (150 мл) в атмосфере азота. Колбу охлаждают до внутренней температуры -78oC (сухой лед с ацетоном). К этому перемешиваемому раствору шприцом добавляют н-бутиллитий (1,6 мл в гексане, 41,3 мл, 66,1 ммоль) и смесь оставляют при перемешивании в течение 5 мин. К этому раствору добавляют раствор 2-тиофенкарбонитрила (6,55 г, 60 ммоль) в ТГФ (30 мл) в течение 10 мин. Полученный ярко-красного цвета раствор перемешивают при -78oC в течение 45 мин, после чего добавляют шприцом диметилформамид (23,3 мл, 300 ммоль). Эту смесь перемешивают в течение 2 ч при -78oC, затем добавляют лимонную кислоту (около 10 г), а после этого - воду (60 мл). Летучие растворители удаляют в вакууме и остаток распределяют между диэтиловым эфиром и рассолом (200 мл каждого). Слои разделяют и водную фазу промывают один раз диэтиловым эфиром. Объединенную органическую фазу промывают один раз солевым раствором, сушат над сульфатом магния, фильтруют и концентрируют в вакууме с получением желтого цвета твердого вещества, которое очищают путем хроматографии на силикагеле, используя градиент этилацетат/гексан (от гексана до гексана с 50% этилацетата). Фракции, содержащие чистый продукт, объединяют и концентрируют в вакууме, получая 6,9 г (84%) 2-циано-5-формил-тиофена. 1H-ЯМР.
Б) Получение 2-циано-5-(гидроксиметил)тиофена
К раствору 2-циано-5-формил-тиофена (6,9 г, 50 ммоль) в этаноле (100 мл) добавляют порциями боргидрид натрия (1,9 г, 50 ммоль). После перемешивания в течение 5 мин растворитель удаляют в вакууме и остаток распределяют между этилацетатом и рассолом. Слои разделяют и органическую фазу промывают один раз 1М раствором лимонной кислоты и один раз солевым раствором, затем сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 6,1 г (88%) 2-циано-5-(гидроксиметил)тиофена. 1H-ЯМР; FD-MS, m/e: 140 (M+).
Анализ для C6H5NOS:
рассчитано, %: C 51,78; H 3,62; N 10,06;
найдено, %: C 51,54; H 3,62; N 9,86.
В) Получение 2-циано-5-(бромметил)тиофена
К раствору 2-циано-5-(гидроксиметил)тиофена (6,0 г, 43 ммоль) в ТГФ (50 мл) добавляют трифенилфосфин (15,7 г, 47 ммоль) и тетрабромид углерода (12,3 г, 47 ммоль). После перемешивания в течение ночи в атмосфере азота при комнатной температуре растворитель удаляют в вакууме и остаток растворяют в хлороформе, затем адсорбируют на силикагеле и вносят в силикагелевую колонку. Продукт элюируют, используя градиент этилацетат/гексан. Фракции, содержащие чистый продукт (как установлено путем ТСХ), объединяют и концентрируют в вакууме с получением 6,5 г (75%) 2-циано-5-(бромметил)тиофена. 1H-ЯМР; FD-MS, m/e: 203 (M+).
Анализ для C6H4NSBr:
рассчитано, %: C 35,66; H 1,99; N 6,93;
найдено, %: C 35,71; H 2,03; N 6,95.
Г) Получение 2-циано-5-(аминометил)тиофен•HCl
К охлажденному до 0oC раствору 2-циано-5-(бромметил)-тиофена (6,0 г, 30 ммоль) в ТГФ (50 мл) в атмосфере азота добавляют порциями NaH (60%-ная дисперсия в масле; 1,3 г, 33 ммоль). К этой перемешиваемой суспензии в течение 30 минут раствор ди-трет-бутилиминодикарбоксилата (7,1 г, 33 ммоль) в ТГФ (50 мл). После перемешивания в течение 3 ч, добавляют насыщенный водный раствор хлорида аммония (100 мл). Летучие растворители затем удаляют в вакууме и остаток распределяют между этилацетатом и водой. Слои разделяют и органическую фазу промывают дважды солевым раствором, сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 10,5 г (100%) 2-циано-5-Boc2NCH2-тиофена, который при стоянии кристаллизуется. Это твердое вещество растворяют в этилацетате (200 мл) и охлаждают до 0oC, используя баню льда с водой. Через раствор в течение 10 мин барботируют сухой газообразный HCl и смесь перемешивают в течение 2 ч, в течение которых она нагревается до комнатной температуры. Растворители удаляют в вакууме и полученное твердое вещество суспендируют в диэтиловом эфире и отделяют путем фильтрования. Белого цвета твердое вещество сушат в течение ночи в вакууме, получая 5,2 г (100%) 2-циано-5-(аминометил)тиофен•HCl. 1H-ЯМР; FD-MS, m/e: 139 (M+).
Анализ для C6H7N2SCl:
рассчитано: %: C 41,26; H 4,04; N 16,04;
найдено, %: C 41,19; H 4,12; N 15,82.
Д) Получение N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro- 4-NHCH2-2-цианотиофена
Способом, по существу эквивалентным описанному в примере 1-Ж (используя N-(тpeт-BuO2CCH2)-N-Boc-D-Cha-Pro-OH), получают 4,6 г (93%) N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro- 4-NHCH2-2-цианотиофена из 2-циано-5-(аминометил)тиофен•HCl. ИК; 1H-ЯМР; FD-MS, m/e: 602 (M+).
Е) Получение N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro- 4-NHCH2-2-C(NH)NHBoc-тиофена
Через раствор N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro- 4-NHCH2-2-цианотиофена (1,5 г, 2,5 ммоль) и триэтиламина (4,5 мл) в пиридине (45 мл) в течение 5 мин пропускают путем барботирования газообразный сероводород и затем сосуд закрывают и оставляют стоять в течение ночи. На следующее утро через раствор в течение 5 мин барботируют азот и растворитель удаляют в вакууме. Остаток растворяют в этилацетате и полученный раствор промывают один раз водой и один раз солевым раствором, после чего сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток затем растворяют в толуоле и полученный раствор концентрируют в вакууме, повторяя операцию два раза.
Остаток затем растворяют в ацетоне (100 мл) и добавляют иодметан (5 мл). После перемешивания в течение ночи при комнатной температуре растворители удаляют в вакууме. Полученную золотистую пену затем растворяют в метаноле (20 мл), добавляют NH4OAc (0,39 г, 5 ммоль) и раствор кипятят с обратным холодильником. Спустя 1 ч растворитель удаляют в вакууме и остаток растворяют в ТГФ (10 мл). К этому перемешиваемому раствору добавляют раствор K2CO3 (1,73 г, 12,5 ммоль) в воде (10 мл), затем добавляют ди-трет-бутил-дикарбонат (2,2 г, 10 ммоль). После перемешивания в течение 1 ч суспензию разбавляют этилацетатом (400 мл) и промывают водой, потом солевым раствором. Органическую фазу после этого концентрируют в вакууме и хроматографируют остаток на силикагеле, элюируя ступенчатым градиентом от гексана с 10% этилацетата до гексана с 75% этилацетата. Содержащие продукт фракции, как установлено путем ТСХ, объединяют и концентрируют в вакууме, получая 1,1 г (61%) белой пены. 1H-ЯМР; FD-MS, m/e: 720 (M+).
Анализ для C36H57N5O8S:
рассчитано, %: C 60,06; H 7,98; N 9,73;
найдено, %: C 59,76; H 8,07; N 9,52.
Ж) Получение HO2CCH2-D-Cha-Pro-4-NHCH2- 2-амидинотиофен•HCl
Способом, по существу эквивалентным описанному в примере 58-Д, получают 500 мг HO2CCH2-D-Cha-Pro-4-NHCH2- 2-амидинотиофенгидрохлорида. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 2 ч). ИК; 1H-ЯМР; FAB-MS, m/e: 464,2 (MH+).
Анализ для C22H33N5O4S•2HCl• H2O:
рассчитано, %: C 47,65; H 6,73; N 12,63; Cl 12,79;
найдено, %: C 47,53; H 6,57; N 12,59; Cl 12,67.
Пример 66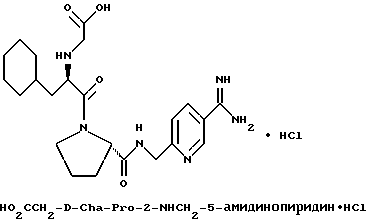
(N-(Карбоксиметил)-D-циклогексилаланил-N-[[5-(аминоиминометил)- пиридин-2-ил]метил]-L-пролинамид-гидрохлорид).
А) Получение N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-2- NHCH2-5-цианопиридина
Способами, по существу эквивалентными описанным в примере 64-А, примере 23-А и примере 1-Ж (используя N-(трет-BuO2CCH2)- N-Boc-D-Cha-Pro-OH), получают 4,4 г N-(трет-BuO2CCH2)-N- Boc-D-Cha-Pro-2-NHCH2-5-цианопиридина из 2-метил-5-цианопиридина. ИК; 1H-ЯМР; FD-MS, m/e: 597 (M+).
Б) Получение HO2CCH2-D-Cha-Pro-2-NHCH2-5- амидинопиридин•HCl
Способами, по существу эквивалентными описанным в примерах 65-Е и 65-Ж, получают 130 мг HO2CCH2-D-Cha-Pro-2- NHCH2-5-амидинопиридин-гидрохлорида из N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-2-NHCH2- 5-цианопиридина. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 2 ч. ИК; 1H-ЯМР; FAB-MS, m/e: 459,3 (MH+); HRMS (FAB), m/e: для C23H35N6O4:
рассчитано: 459,2720;
найдено: 459,2707.
Пример 67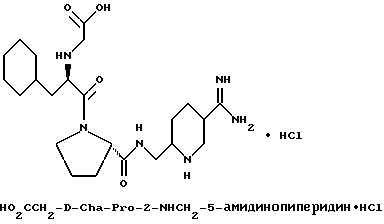
Получение HO2CCH2-D-Cha-Pro-2-NHCH2-5- амидинопиперидин-гидрохлорида
N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-2-NHCH2- 5-цианопиридин (1,2 г, 2 ммоль) получают способами, по существу эквивалентными описанным в примерах 23-А, 23-Б и 1-Б. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 2 ч). Фракции, содержащие второстепенный (minor) продукт, как установлено путем аналитической RPHRLC, объединяют, частично концентрируют в вакууме и лиофилизируют с получением 93 мг (9% бледно-зеленого твердого вещества. ИК; 1H-ЯМР; IS-MS, m/e: 465,5 (MH+).
HRMS (FAB), m/e: для C23H41N6O4:
рассчитано: 465,3189;
найдено: 465,3191.
Пример 68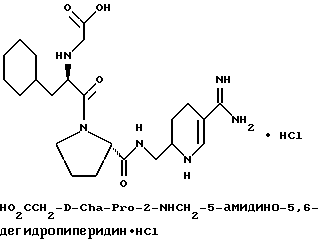
(N-(Карбоксиметил)-D-циклогексилаланил-N-[[5-(аминоиминометил)- 1,2,3,4-тетрагидро-пиридин-2-ил]метил]-L-пролинамид-гидрохлорид).
Получение HO2CCH2-D-Cha-Pro-2-NHCH2-5-амидино-5,6- дегидро-пиперидин-гидрохлорида
N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-2-NHCH2- 5-цианопиридин (1,2 г, 2 ммоль) получают способами, по существу эквивалентными описанным в примере 23-А, примере 23-Б и примере 2-Е. Продукт очищают путем RPHRLC, метод 2 (градиент (A-B) от 98/2 до 70/30, 2 ч). Фракции, содержащие главный продукт, как установлено путем аналитической RPHRLC, объединяют, частично концентрируют и лиофилизируют с получением 422 мг (39%) белого твердого вещества ИК; 1H-ЯМР; IS-MS, m/e: 463,3 (MH+).
Анализ для C23H38N6O4•2,9HCl• 2H2O:
рассчитано, %: C 45,71; H 7,49; N 13,91; Cl 17,01;
найдено, %: C 45,51; H 6,83; N 13,66; Cl 16,83.
Пример 69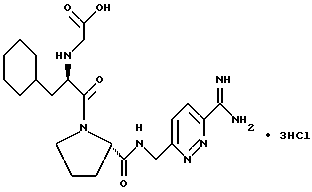
HO2CCH2-D-Cha-Pro-3-NHCH2-6-амидино- пиридазин•3HCl
(N-(Карбоксиметил)-D-циклогексилаланил-N-[[6-(аминоиминометил)- пиридазин-3-ил]метил]-L-пролинамид-тригидрохлорид).
А) Получение 3-метил-6-циноапиридазина
К перемешиваемому раствору 3-метил-пиридазина (11 г, 118 ммоль) в дихлорметане (200 мл) добавляют AlCl3 (0,05 г), затем триметилсилилцианид (21 г, 211 ммоль). Спустя 20 мин через капельную воронку добавляют раствор п-толуолсульфонилхлорида (38 г, 201 ммоль) в дихлорметане (50 мл) и раствор выдерживают при перемешивании в течение ночи. На следующее утро растворитель удаляют в вакууме и остаток суспендируют в этаноле при перемешивании в течение 15 мин и после этого фильтруют с получением твердого вещества белого цвета. Твердое вещество растворяют в ТГФ (200 мл) и к этому перемешиваемому раствору добавляют 1,8-диазабицикло[5.4.0] ундец-7-ен (16 мл, 105 ммоль). Спустя 1 ч растворитель удаляют в вакууме и остаток распределяют между гексаном и насыщенным водным раствором хлорида аммония. Фазы разделяют и водную фазу подщелачивают с помощью твердого карбоната натрия, затем экстрагируют три раза этилацетатом. Объединенные этилацетатные фазы сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 9 г (64%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 119,1 (M+).
Б) Получение HO2CCH2-D-Cha-Pro-3-NHCH2- 6-амидино-пиридазин-гидрохлорида
Способами, по существу эквивалентными описанным в примере 66, получают 90 мг HO2CCH2-D-Cha-Pro-3-NHCH2- 6-амидино-пиридазин-гидрохлорида из 3-метил-6-цианопиридазино. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B от 98/2 до 70/30, 2 ч). ИК; 1H-ЯМР; FAB-MS, m/e: 460,3 (MH+).
Анализ для C22H33N7O4•3HCl• 2H2O:
рассчитано, %: C 43,68; H 6,66; N 16,21;
найдено, %: C 44,04; H 6,45; N 15,57.
Пример 70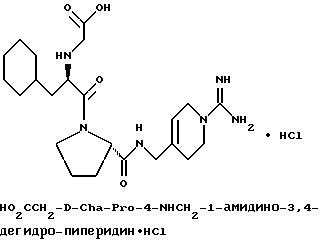
(N-(Карбоксиметил)-D-циклогексилаланил-N-[[1-(аминоиминометил)- 1,2,3,6-тетрагидропиридин-4-ил]метил]-L-пролинамид-гидрохлорид)
А) Получение N,N'-Boc2-тиомочевины
К перемешиваемой суспензии NaH (60%-ная дисперсия в масле; 9,4 г, 234 ммоль) в ТГФ (500 мл) при 0oC добавляют тиомочевину (4,0 г, 52 ммоль). Спустя 30 минут охлаждающую баню удаляют и реакционную смесь перемешивают в течение 30 мин при комнатной температуре. Снова сосуд (колбу) охлаждают до 0oC и через капельную воронку добавляют раствор ди-трет-бутил-дикарбоната (25 г, 115 ммоль) в ТГФ (100 мл). После перемешивания в течение 30 мин при 0oC и дополнительно 2 ч при комнатной температуре добавляют насыщенный водный раствор бикарбоната натрия. Раствор затем концентрируют примерно до половины первоначального объема в вакууме и добавляют этилацетат. Органическую фазу затем промывают насыщенным водным раствором бикарбоната натрия, затем солевым раствором и после этого сушат (MgSO4), фильтруют и концентрируют с получением 11,9 г (83%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 276 (M+).
Анализ для C11H20N2O4S:
рассчитано, %: C 47,81; H 7,30; N 10,14;
найдено, %: C 47,69; H 7,28; N 10,34.
Б) Получение N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro- 4-NHCH2-1-(N, N'-Boc2-амидино)-3,4-дегидро-пиперидина
К перемешиваемому раствору N-(трет-BuO2CCH2)-N-Boc- D-Cha-Pro-4-NHCH2-3,4-дегидро-пиперидина (0,6 г, 1 ммоль) и триэтиламина (0,35 г, 3,4 ммоль) в диметилформамиде (10 мл) добавляют N,N-Boc2-тиомочевины (0,28 г, 1 ммоль), затем HgCl2 (0,28 г, 1 ммоль). Спустя 4 ч растворитель удаляют в вакууме и остаток растворяют в этилацетате и промывают полученный раствор два раза солевым раствором. Органическую фазу затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Продукт очищают путем хроматографии на силикагеле, элюируя градиентом от 20% этилацетат/гексан до 75% этилацетат/гексан. Фракции, содержащие продукт, как установлено с помощью ТСХ, объединяют и концентрируют в вакууме с получением 800 мг (94%) белой пены. ИК; 1H-ЯМР; FD-MS, m/e: 820 (MH+).
Анализ для C42H70N6O10:
рассчитано, %: C 61,59; H 8,61; N 10,26;
найдено, %: C 61,81; H 8,79; N 10,45.
В) Получение HO2CCH2-D-Cha-Pro-4-NHCH2- 1-амидино-3,4-дегидро-пиперидино-гидрохлорида
Способами, по существу эквивалентными описанным в примере 58-Д, получают 0,22 г (55%) HO2CCH2-D-Cha-Pro-4-NHCH2- 1-амидино-3,4-дегидро-пиперидино-гидрохлорида из N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-4-NHCH2-1- (N, N'-Boc2-амидино)-3,4-дегидро-пиперидина. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 2 ч). ИК; 1H-ЯМР; FAB-MS, m/e: 463,3 (MH+).
Анализ для C23H38N6O4•2,2HCl• 2H2O:
рассчитано, %: C 47,73; H 7,70; N 14,52; Cl 13,47;
найдено, %: C 47,49; H 7,64; N 14,55; Cl 13,48,
Пример 71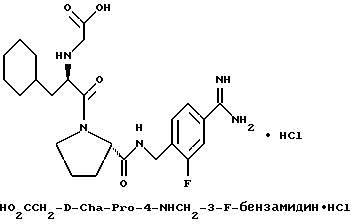
(N-(Карбоксиметил)-D-циклогексилаланил-N-[[4-(аминоиминометил)- 2-фторфенил]метил]-L-пролинамид-гидрохлорид)
Получение HO2CCH2-D-Cha-Pro-4-NHCH2-3-F- бензамидин•HCl
Способами, по существу эквивалентными описанным в примере 66, получают 0,27 г HO2CCH2-D-Cha-Pro-4-NHCH2-3-F- бензамидин-гидрохлорида из 3-F-4-метил-бензонитрила. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/0, 2 ч). ИК; 1H-ЯМР; FAB-MS, m/e: 476,3 (MH+).
Анализ для C24H34N5O4F•2HCl• 1,5H2O:
рассчитано, %: C 50,09; H 6,83; N 12,17; Cl 12,32;
найдено, %: C 49,89; H 6,65; N 12,17; Cl 12,42.
Пример 72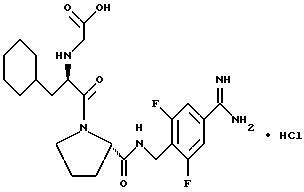
HO2CCH2-D-Cha-Pro-4-NHCH2-3,5-F2- бензамидин•HCl
(N-(Карбоксиметил)-D-циклогексилаланил-N-[[4-(аминоиминометил)- 2,6-дифторфенил]метил]-L-пролинамид-гидрохлорид)
Получение HO2CCH2-D-Cha-Pro-4-NHCH2-3,5- F2-бензамидин-гидрохлорида
Способами, по существу эквивалентными описанным в примере 65, получают 0,28 г HO2CCH2-D-Cha-Pro-4-NHCH2-3,5- F2-бензамидин•HCl из 3,5-F2-бензонитрила. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 150 мин). ИК; 1H-ЯМР; FAB-MS, m/e: 494,2 (MH+).
Анализ для C24H33N5O4F2•2HCl• 1,5H2O:
рассчитано, %: C 48,57; H 6,45; N 11,80; Cl 11,95;
найдено, %: C 48,26; H 6,17; N 11,89; Cl 11,90.
Пример 73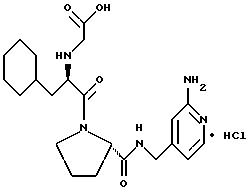
HO2CCH2-D-Cha-Pro-4-NHCH2-2- аминопиридин•HCl
А) Получение 4-метил-2-фталимидопиридина
К перемешиваемому раствору 4-метил-2-аминопиридина (50 г, 460 ммоль) в уксусной кислоте (1 л) добавляют фталевый ангидрид (68 г, 460 ммоль) и реакционную смесь нагревают до температуры кипения с обратным холодильником. Спустя 12 ч кипячения с обратным холодильником, добавляют уксусный ангидрид (43 мл, 460 ммоль) и раствор продолжают перемешивать при кипячении с обратным холодильником в течение дополнительных 48 ч. Растворитель затем удаляют в вакууме и твердый остаток суспендируют в толуоле и раствор концентрируют в вакууме, повторяя эту операцию дважды. Твердое вещество затем суспендируют в этилацетате при интенсивном перемешивании и фильтруют. После повторения этой операции промывки в этилацетате твердое вещество высушивают в течение ночи в вакууме с получением 46,6 г (42%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 238 (M+).
Анализ для C14H10N2O2:
рассчитано, %: C 70,58; H 4,23; N 11,76;
найдено, %: C 70,42; H 4,29; N 11,70.
Б) Получение N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-4- NHCH2-2-фталимидопиридина
Способами, по существу эквивалентными описанным в примерах 64-А, 23-А и 1-Ж (используя N-(трет-BuO2CCH2)-N-Boc- D-Cha-Pro-OH), получают 2,4 г N-(трет-BuO2CCH2)-N-Boc- D-Cha-Pro-4-NHCH2-2-фталимидопиридина из 4-метил-2-фталимидопиридина. ИК; 1H-ЯМР; FD-MS, m/e: 717,7 (M+).
В) Получение HO2CCH2-D-Cha-Pro-4-NHCH2- 2-аминопиридин-гидрохлорида
К перемешиваемому раствору N-(трет-BuO2CCH2)-N-Boc- D-Cha-Pro-4-NHCH2-2-фталимидопиридина (1,6 г, 2,2 ммоль) в этаноле (25 мл) добавляют гидразингидрат (0,52 мл, 10,4 ммоль). Спустя 1 ч растворители удаляют в вакууме и остаток растворяют в этилацетате и полученный раствор концентрируют в вакууме, повторяя эту операцию два раза. Остаток преобразуют способом, по существу эквивалентным описанному в примере 58-Д, получая 380 мг белого твердого вещества (37%). Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 150 мин). ИК; 1H-ЯМР; FAB-MS, m/e: 432,3 (MH+).
Анализ для C22H33N5O4•2,1HCl• H2O:
рассчитано, %: C 50,23; H 7,11; N 13,31;
найдено, %: C 50,05; H 7,08; N 13,54.
Пример 74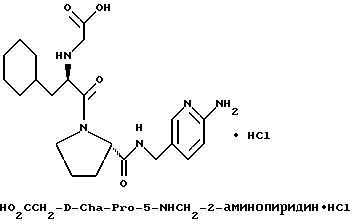
Получение HO2CCH2-D-Cha-Pro-5-NHCH2-2- аминопиридин-гидрохлорида
Способами, по существу эквивалентными описанным в примере 73, получают 0,88 г HO2CCH2-D-Cha-Pro-5-NHCH2-2- аминопиридин-гидрохлорида из 5-метил-2-аминопиридина. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 150 мин). ИК; 1H-ЯМР; FAB-MS, m/e: 432,3 (MH+).
Анализ для C22H33N5O4•2HCl• H2O:
рассчитано, %: C 50,58; H 7,14; N 13,40;
найдено, %: C 50,79; H 7,20; N 13,58.
Пример 75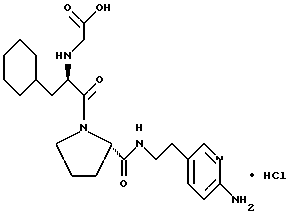
HO2CCH2-D-Cha-Pro-5-NHCH2CH2-2- аминопиридин•HCl
А) Получение 5-метил-2-Boc2N-пиридина
К перемешиваемому раствору 5-метил-2-аминопиридина (10,5 г, 100 ммоль) в дихлорметане (200 мл) при 0oC добавляют N,N-диизопропилэтиламин (25,8 г, 200 ммоль), затем ди-трет-бутил-дикарбонат (55 г, 250 ммоль) и, наконец, 4-(N, N-диметиламино) пиридин (12,2 г, 100 ммоль). Охлаждающую баню удаляют и раствор перемешивают в течение ночи. Смесь затем разбавляют этилацетатом (600 мл) и промывают три раза насыщенным водным раствором NH4Cl, один раз солевым раствором, дважды насыщенным водным раствором бикарбоната натрия и один раз снова солевым раствором. Органическую фазу затем сушат (MgSO4), фильтруют, концентрируют в вакууме и хроматографируют на силикагеле, элюируя градиентом от 10% этилацетата/гексан до 75% этилацетата/гексан. Фракции, содержащие продукт, как установлено путем ТСХ, объединяют и концентрируют в вакууме, получая 12,8 г (42%) белого твердого вещества. ИК; 1H-ЯМР; FD-MS, m/e: 308 (M+).
Анализ для C16H24N2O4:
рассчитано, %: C 62,32; H 7,84; N 9,08;
найдено, %: C 62,51; H 8,11; N 9,37.
Б) Получение 6-BrCH2-2-Boc2N-пиридина
Способом, по существу эквивалентным описанному в примере 64-А, получают примерно 11,6 г 5-BrCH2-2-Boc2N-пиридина (который загрязнен исходным материалом) из 5-метил-2-Boc2N-пиридина. 1H-ЯМР; FD-MS, m/e: 386,3 (M+).
Анализ для C16H23N2O4Br:
рассчитано, %: C 49,62; H 5,99; N 7,23;
найдено, %: C 49,86; H 6,00; N 7,07.
В) Получение 5-NCCH2-2-Boc2N-пиридина
К перемешиваемому раствору слегка загрязненного 5-BrCH2-2-Boc2N-пиридина (9,7 г, 25 ммоль) в диметилформамиде (150 мл) добавляют 18-краун-6 (1,32 г, 5 ммоль), затем KCN (1,95 г, 30 ммоль). После перемешивания в течение 6 ч растворитель удаляют в вакууме и остаток хроматографируют на силикагеле, элюируя ступенчатым градиентом от гексана до смеси 40% этилацетата с гексаном. Фракции, содержащие продукт, как установлено путем ТСХ, объединяют и концентрируют в вакууме с получением 2,6 г (31%, при процедуре в две операции) твердое белое вещество. ИК; 1H-ЯМР; FD-MS, m/e: 333,4 (M+).
Анализ для C17H23N3O4:
рассчитано, %: C 61,25; H 6,95; N 12,60;
найдено, %; C 61,09; H 6,92; N 12,53.
Г) Получение HO2CCH2-D-Cha-Pro-5-NHCH2CH2- 2-аминопиридин•HCl
К перемешиваемому раствору 5-NCCH2-2-Boc2N-пиридина (2,5 г, 7,5 ммоль) в метаноле (150 мл) добавляют CoCl2 (0,97 г, 7,5 ммоль) и воду (0,81 г, 45 ммоль). Спустя 5 мин небольшими порциями в течение 15 мин добавляют NaBH4 (2,84 г, 75 ммоль). Спустя дополнительные 5 мин растворитель удаляют в вакууме и остаток растворяют в концентрированном водном растворе NH4OH и экстрагируют несколько раз этилацетатом. Объединенные этилацетатные экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Затем, способом, по существу эквивалентным описанному в примерах 1-А и 73-В, остаток превращают, получая 1,2 г (33%) HO2CCH2-D-Cha-Pro-5-NHCH2CH2-2- аминопиридин•HCl. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 60/40, 150 мин). ИК; 1H-ЯМР; FAB-MS, m/e: 446,3 (MH+).
HRMS (FAB): для C23H36N5O4:
рассчитано: 446,2767;
найдено: 446,2769.
Пример 76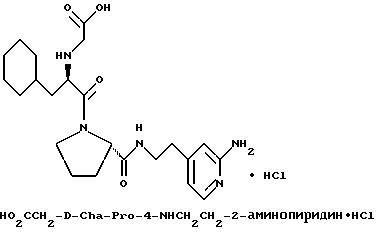
А) Получение 4-BocNHCH2CH2-2-CN-пиридина
К раствору 4-BocNHCH2CH2-пиридина (2,22 г, 10 ммоль) в ацетоне (50 мл) добавляют через капельную воронку в течение 10 мин раствор м-хлор-надбензойной кислоты (5,2 г, 30 ммоль) в ацетоне (50 мл). После перемешивания в течение ночи, растворитель удаляют в вакууме и остаток распределяют между водой (100 мл) и диэтиловым эфиром (100 мл). Органическую фазу отделяют и экстрагируют три раза водой. Объединенную водную фазу затем насыщают твердым NaCl и экстрагируют три раза дихлорметаном (100 мл). Объединенные дихлорметановые экстракты промывают один раз солевым раствором, сушат (Na2SO4), фильтруют и концентрируют в вакууме до небольшого объема и затем добавляют диэтиловый эфир. Белого цвета осадок фильтруют (2,0 г) и высушивают в вакууме.
Половину выделенного твердого вещества (4,2 ммоль) растворяют в дихлорметане (10 мл) и к этому перемешиваемому раствору добавляют триметилсилилцианид (0,84 мл, 6,3 ммоль), затем N,N-диметилкарбамоилхлорид (0,58 мл, 6,3 ммоль). После перемешивания в течение ночи, медленно добавляют 1 М водный раствор бикарбоната калия (1 мл) и смесь распределяют между этилацетатом и водой. Органическую фазу затем промывают три раза солевым раствором, сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 0,6 г (58%) янтарного цвета масла, которое кристаллизуется при стоянии.
ИК; 1H-ЯМР; FD-MS, m/e: 247 (M+).
Анализ для C13H17N3O2:
рассчитано, %: C 63,31; H 7,02; N 16,99;
найдено, %: C 63,31; H 7,02; N 16,71.
Б) Получение 4-BocNHCH2CH2-2-CbzNH-пиридина
К перемешиваемому раствору 4-BocNHCH2CH2-2-CN-пиридина (0,5 г, 2 ммоль) в метаноле (2,4 мл) добавляют 5н раствора NaOH (1,6 мл, 8 ммоль) и раствор кипятят с обратным холодильником. Спустя 24 ч раствор охлаждают до комнатной температуры и перемешивают в течение дополнительных 48 ч. Затем pH доводят до 7 с помощью 1н HCl и растворители удаляют в вакууме.
Остаток суспендируют в толуоле (50 мл) и кипятят с обратным холодильником. К этому перемешиваемому раствору добавляют последовательно триэтиламин (0,36 мл, 2,6 ммоль), бензиловый спирт (0,27 мл, 2,6 ммоль) и дифенилфосфориламид (0,72 г, 2,6 ммоль). После перемешивания при кипячении с обратным холодильником в течение ночи, раствор оставляют охлаждаться и после этого разбавляют этилацетатом (200 мл) и промывают дважды насыщенным водным раствором NH4Cl и дважды солевым раствором. Органическую фазу затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток затем хроматографируют на силикагеле, элюируя ступенчато градиентом от гексана до смеси гексана с 50% этилацетата. Содержащие продукт фракции, как определяют путем ТСХ, объединяют и концентрируют в вакууме с получением 0,37 г (50%) белого твердого вещества.
1H-ЯМР; FD-MS, m/e: 371,2 (M+).
Анализ для C20H25N3O4:
рассчитано, %: C 64,67; H 6,78; N 11,31;
найдено, %: C 64,90; H 7,07; N 11,06.
В) Получение HO2CCH2-D-Cha-Pro-4-NHCH2CH2- 2-аминопиридин•HCl
Способами, по существу эквивалентными описанным в примерах 23-А, 1-Ж (используя N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-OH), 23-Г и 58-Д, получают 48 мг HO2CCH2-D-Cha-Pro-4- NHCH2CH2-2-аминопиридин-гидрохлорида из 4-BocNHCH2CH2-2-CbzNH-пиридина. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 150 мин). 1H-ЯМР; FAB-MS, m/e: 446,4 (MH+).
Анализ для C23H35N5O4•1,5HCl• H2O:
рассчитано, %: C 53,30; H 7,49; N 13,51;
найдено, %: C 53,64; H 7,27; N 13,80.
Пример 77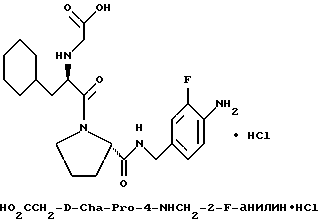
Получение HO2CCH2-D-Cha-Pro-4-NHCH2-2-F- анилин-гидрохлорида
Способом, по существу эквивалентным описанному в примере 64, получают 0,55 г HO2CCH2-D-Cha-Pro-4-NHCH2-2-F- анилин•HCl из 3-F-4-NO2-толуола. Продукт очищают путем RPHRLC, метод 2 (градиент (A/B) от 98/2 до 60/40, 180 мин). ИК; 1H-ЯМР; FAB-MS, m/e: 449,3 (MH+).
Анализ для C23H33N4O4F•0,9HCl• H2O:
рассчитано, %: C 55,32; H 7,25; N 11,22; Cl 6,39;
найдено, %: C 55,49; H 6,93; N 11,15; Cl 6,23.
Пример 78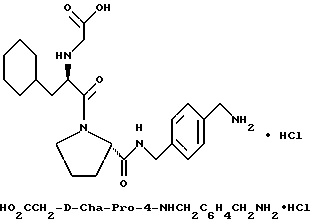
Получение HO2CCH2-D-Cha-Pro-4- NHCH2C6H4CH2NH2•HCl
Способом, по существу эквивалентным описанному в примере 10, используя N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-OH вместо Boc-D-Phe-Pro-OH, получают 0,53 г HO2CCH2-D-Cha-Pro-4- NHCH2-С6H4CH2NH2•HCl. Продукт очищают путем RPHRLC метод 2 (градиент (A/B) от 98/2 до 70/30, 150 мин). ИК; 1H-ЯМР; FD-MS, m/e: 445,4 (MH+).
Анализ для C24H36N4O4•2,2HCl• 0,5H2O:
рассчитано, %: C 54,00; H 7,40; N 10,50; Cl 14,61;
найдено, %: C 54,18; H 7,54; N 10,31; Cl 14,86.
Пример 79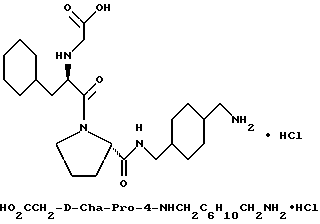
Получение HO2CCH2-D-Cha-Pro-4- NHCH2C6H10CH2NH2•HCl
Способом, по существу эквивалентным описанному в примере 12, используя N-(трет-BuO2CCH2)-N-Boc-D-Cha-Pro-OH вместо Boc-D-Phe-Pro-OH, получают 0,04 г HO2CCH2-D-Cha-Pro-4- NHCH2C6H10CH2NH2•HCl. Продукт очищают путем RPHPLC, метод 2 (градиент (A/B) от 98/2 до 70/30, 150 мин). 1H-ЯМР; FD-MS, m/e: 451 (MH+).
Анализ для C24H42N4O4•2,7HCl• 0,5H2O:
рассчитано, %: C 51,65; H 8,25; N 10,04;
найдено, %: C 51,47; H 7,87; N 9,97.
Пример 80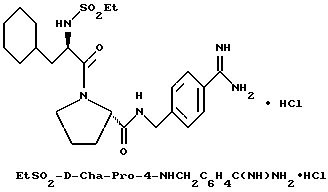
(N-(Этилсульфонил)-D-циклогексилаланил-N-[[4-(аминоиминометил) фенил] метил]-L-пролин-амидгидрохлорид)
Получение EtSO2-D-Cha-Pro-4- NHCH2C6H4C(NH)NH2•HCl.
Способом, по существу эквивалентным описанному в примере 18, используя EtSO2-D-Cha-ProOH вместо Cbz-D-1-Piq-ProOH, получают 3,6 г EtSO2-D-Cha-Pro-4-NHCH2C6H4C(NH)NH2 •HCl. Продукт очищают путем RPHPLC, метод 2 (градиент (A/B) от 90/10 до 50/50, 180 мин). ИК; 1H-ЯМР; FAB-MS, m/e: 492,3 (MH+).
Анализ для C24H37N5O4S•HCl:
рассчитано, %: C 54,58; H 7,25; N 13,26; Cl 6,71;
найдено, %: C 54,31; H 7,31; N 13,37; Cl 6,71.
Пример 81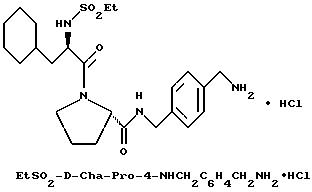
Получение EtSO2-D-Cha-Pro-4- NHCH2C6H4CH2NH2•HCl
Способом, по существу эквивалентным описанному в примере 10, используя EtSO2-D-Cha-ProOH вместо Boc-D-Phe-ProOH, получают EtSO2-D-Cha-Pro-4-NHCH2C6H4CH2NH2 •HCl. Продукт очищают путем RPHPLC, метод 2 (градиент (A/B) от 90/10 до 50/50, 180 мин). ИК; 1H-ЯМР; FAB-MS, m/e: 479,4 (MH+).
Анализ для C24H38N4O4S•HCl•H2O:
рассчитано, %: C 54,07; H 7,75; N 10,51; Cl 6,65;
найдено, %: C 54,13; H 7,44; N 10,51; Cl 6,78.
Пример 82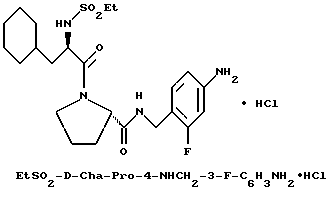
Получение EtSO2-D-Cha-Pro-4-NHCH2-3-F- C6H3NH2•HCl.
Способом, по существу эквивалентным описанному в примере 64, используя EtSO2-D-Cha-ProOH вместо 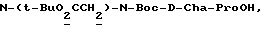 получают 0,53 г EtSO2-D-Cha-Pro-4-NHCH2-3-F-C6H3NH2• HCl. Продукт очищают путем RPHRLC метод 2 (градиент (A/B) от 90/10 до 50/50, 180 мин). ИК; 1H-ЯМР; FAB-MS, m/e: 483,3 (MH+).
получают 0,53 г EtSO2-D-Cha-Pro-4-NHCH2-3-F-C6H3NH2• HCl. Продукт очищают путем RPHRLC метод 2 (градиент (A/B) от 90/10 до 50/50, 180 мин). ИК; 1H-ЯМР; FAB-MS, m/e: 483,3 (MH+).
Анализ для C23H35N4O4SF•1,1HCl• 0,5H2O:
рассчитано, %: C 51,95; H 7,03; N 10,54; Cl 7,33;
найдено, %: C 52,09; H 6,94; N 10,39; Cl 7,24.
Пример 83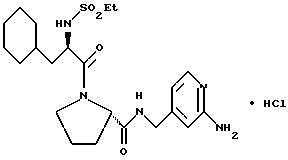
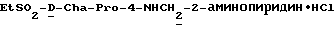
Получение EtSO2-D-Cha-Pro-4-NHCH2-2- аминопиридин-гидрохлорида
Способом, по существу эквивалентным описанному в примере 73, используя EtSO2-D-Cha-ProOH вместо 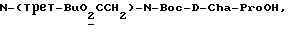 получают 0,22 г EtSO2-D-Cha-Pro-4-NHCH2-2-аминопиридин•HCl из 4-метил-2-аминопиридина. Продукт очищают путем RPHPLC, метод 2 (градиент (A/B) от 90/10 до 50/50, 180 мин). 1H-ЯМР; FAB-MS; m/e: 466,4 (MH+).
получают 0,22 г EtSO2-D-Cha-Pro-4-NHCH2-2-аминопиридин•HCl из 4-метил-2-аминопиридина. Продукт очищают путем RPHPLC, метод 2 (градиент (A/B) от 90/10 до 50/50, 180 мин). 1H-ЯМР; FAB-MS; m/e: 466,4 (MH+).
Анализ для C22H35N5O4S•1,1HCl:
рассчитано, %: C 52,25; H 7,19; N 13,85; Cl 7,71;
найдено, %: C 52,49; H 6,96; N 13,96; Cl 7,76.
Пример 84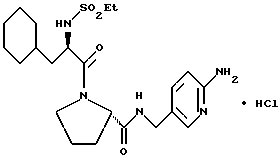
EtSO2-D-Cha-Pro-5-NHCH2-2-аминопиридин•HCl
Получение EtSO2-D-Cha-Pro-5-NHCH2-2- аминопиридин-гидрохлорида
Способом, по существу эквивалентным описанному в примере 73, используя EtSO2-D-Cha-ProOH вместо N-(t-BuO2CCH2)-N-Boc-D-Cha-ProOH, получают 0,24 г EtSO2-D-Cha-Pro-5-NСH2-2-аминопиридин•HCl из 5-метил-2-аминопиридина. Продукт очищают путем RPHPLC, метод 2, (градиент (A/B) от 90/10 до 50/50, 180 мин). 1H-ЯМР; FAB-MS, m/e: 466,4 (MH+).
Анализ для C22H35N5O4S•1,15HCl:
рассчитано, %: C 52,06; H 7,18; N 13,80; Cl 8,03;
найдено, %: C 52,38; H 6,97; N 14,20; Cl 8,46.
Пример 85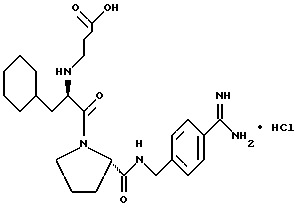
HOOCCH2CH2CH2-D-Cha-Pro-p- NHCH2C6H4C(NH)NH2•HCl
(N-(3-Карбоксипропил)-D-циклогексилаланил-N-[[4-(аминоиминометил) фенил] метил]-L-пролин-амид-гидрохлорид)
А) Получение Cbz-MeOOCCH=CHCH2-D-Cha-Pro-p- NHCH2C6H4C(NH)NHCbz
К раствору D-Cha-Pro-p-NHCH2C6H4C(NH)NHCbz•2HCl (2,5 г, 4,1 ммоль в ДМФ (50 мл) добавляют N,N-диизопропилэтиламин (2,2 мл, 12,2 ммоль) и метил-3-бромкротонат (0,95 г, 4,5 ммоль). После перемешивания в течение 48 ч, добавляют Cbz-Cl (0,7 мл, 5 ммоль) и дополнительный N,N-диизопропилэтиламин (0,85 мл, 5 ммоль). После перемешивания дополнительно 16 ч, летучие части удаляют в вакууме. Остаток распределяют между этилацетатом (100 мл) и насыщенным водным раствором хлорида аммония (100 мл). Слои разделяют и органическую фазу промывают один раз насыщенным водным раствором хлорида аммония (50 мл), дважды насыщенным водным раствором бикарбоната натрия (50 мл) и дважды солевым раствором (50 мл). Органическую фазу сушат (MgSO4), фильтруют и концентрируют в вакууме. Сырой остаток очищают путем флэш-хроматографии (градиент этилацетат/гексан), получая 660 мг (22%) белой пены. 1H-ЯМР; FD-MS, m/e: 766 (M+).
Анализ для C43H51N5O8:
рассчитано, %: C 67,43; H 6,71; N 9,14;
найдено, %: C 67,22; H 6,57; N 8,98.
Б) Получение HOOCCH2CH2CH2-D-Cha-Pro-p- NHCH2C6H4C(NH)NH2•HCl
К раствору Cbz-MeOOCCH= CHCH2-D-Cha-Pro-p- NHCH2C6H4C(NH)NHCbz (0,5 г, 0,65 ммоль) в этаноле (5 мл) добавляют 1н раствор гидроксида натрия (0,65 мл). После перемешивания в течение 5 ч при комнатной температуре, добавляют 1н HCl (3 мл), 10%-ный Pd/C (0,5 г), воду (15 мл) и этанол (25 мл). Перемешиваемую суспензию дегазируют в вакууме, затем помещают в атмосферу водорода на 18 ч. Добавляют диатомовую землю и суспензию фильтруют. Фильтрат концентрируют в вакууме и остаток очищают путем RPHRLC (метод 2; градиент (A/B) от 98/2 до 75/25, в течение 150 мин). Фракции, содержащие чистый продукт, объединяют и лиофилизируют с получением 46 мг (13%). 1H-ЯМР; FAB-MS, m/e: 486,3 (MH+).
Анализ для C26H39N5O4•2,1HCl:
рассчитано, %: C 55,15; H 7,35; N 12,19; Cl 13,24;
найдено, %: C 55,55; H 7,37; N 12,19; Cl 13,58.
Пример 86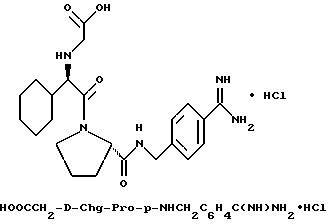
(N-(Карбоксиметил)-D-циклогексилглицил-N-[[4-(аминоиминометил) фенил] метил]-L-пролин-амид-гидрохлорид).
А) Получение D-циклогексилглицина
Способом, по существу эквивалентным описанному в примере 53-Б, получают 16,1 г (16%) D-циклогексилглицина, исходя из D-фенилглицина. 1H-ЯМР; FD-MS, m/e: 117 (M+).
Анализ для C8H15NO2:
рассчитано, %: C 61,12; H 9,62; N 8,91;
найдено, %: C 61,23; H 9,56; N 8,73.
Б) Получение Boc-D-циклогексилглицина
Способом, по существу эквивалентным описанному в примере 17-А (используя ди-трет-бутилдикарбонат), получают 22 г (90%) Boc-D-циклогексилглицина. 1H-ЯМР; FD-MS, m/e: 258 (M+),
Анализ для C13H23NO4:
рассчитано, %: C 60,68; H 9,01; N 5,44;
найдено, %: C 60,91; H 9,18; N 5,38.
В) Получение Boc-Pro-p-NHCH2C6H4C(NH)NHCbz
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 20,5 г (76%) Boc-Pro-p-NHCH2C6H4C(NH)NHCbz из Boc-Pro-OH и NH2CH2C6H4C(NH)NHCbz•2HCl. 1H-ЯМР; FD-MS, m/e: 481 (M+).
Анализ для C26H32N4O5:
рассчитано, %: C 64,98; H 6,71; N 11,66;
найдено, %: C 64,76; H 6,78; N 11,62.
Г) Получение Pro-p-NHCH2C6H4C(NH)NHCbz•2HCl
Способом, по существу эквивалентным описанному в примере 23-А, получают 18,4 г (100%) Pro-p-NHCH2C6H4C(NH)NHCbz• 2HCl. 1H-ЯМР; FD-MS, m/e 381 (M+).
Анализ для C21H26N4O3Cl2:
рассчитано, %: C 55,63; H 5,78; N 12,36;
найдено, %: C 54,19; H 6,27; N 12,15.
Д) Получение Boc-D-Chg-Pro-p-NHCH2C6H4C(NH)NHCbz
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 3,6 г (97%) Boc-D-Chg-Pro-p-NHCH2C6H4C(NH)NHCbz. 1H-ЯМР, FD-MS, m/e: 619 (M+).
Анализ для C34H45N5O6:
рассчитано, %: C 65,89; H 7,32; N 11,30;
найдено, %: C 67,59; H 8,07; N 10,99.
Е) Получение Cbz-t-BuOOCCH2-D-Chg-Pro-p- NHCH2C6H4C(NH)NHCbz
Способами, по существу эквивалентными описанным в примерах 23-А и 85-А (используя трет-бутил-бромацетат и бензил-хлорформиат), получают 1,6 г (45%) FD-MS, m/e: 769 (M+) N-Cbz-N-(t-BuOOCCH2)-D-Chg-Pro-p- NHCH2C6H4C(NH)NHCbz. 1H-ЯМР;
Анализ для C43H53N5O8:
рассчитано, %: C 67,26; H 6,96; N 9,12;
найдено, %: C 67,50; H 6,97; N 9,11.
Ж) Получение HOOCCH2-D-Chg-Pro-p- NHCH2C6H4C(NH)NH2•HCl
Способами, по существу эквивалентными описанным в примерах 23-А (используя диоксан в качестве растворителя) и 18-Е, получают 411 мг (61%) HOOCCH2-D-Chg-Pro-p- NHCH2C6H4C(NH)NH2•HCl. 1H-ЯМР; FAB-MS, m/e: 444,3 (MH+).
Анализ для C23H33N5O4•2,5HCl:
рассчитано, %: C 51,67; H 6,69; N 13,10;
найдено, %: C 51,84; H 6,50; N 13,15.
Пример 87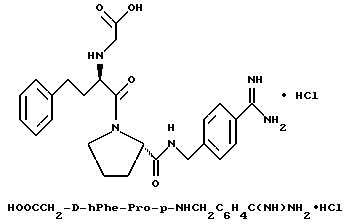
(N-(Карбоксиметил)-D-гомофенилаланил-N-[[4-(аминоиминометил) фенил] метил]-L-пролинамид-гидрохлорид)
А) Получение HOOCCH2-D-hPhe-Pro-p- NHCH2C6H4C(NH)NH2•HCl
Способом, по существу эквивалентным таковому, описанному в примере 86, получают 335 мг HOOCCH2-D-hPhe-Pro-p- NHCH2C6H4C(NH)NH2•HCl, исходя из Boc-D-hPhe-OH. 1H-ЯМР; FAB-MS, m/e: 466,3 (MH+).
Анализ для C25H31N5O4•2,1HCl• H2O:
рассчитано, %: C 53,61; H 6,32; N 12,50; Cl 13,29;
найдено, %: C 53,58; H 6,08; N 12,59; Cl 13,67.
Пример 88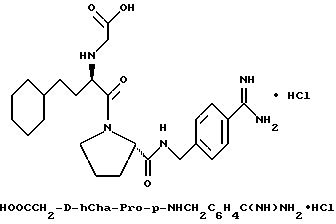
(N-(Карбоксиметил)-D-гомоциклогексилаланил-N-[[4-(аминоиминометил) фенил]метил]-L-пролин-амид-гидрохлорид)
А) Получение Boc-D-hCha-OH
Способом, по существу эквивалентным описанному в примере 53-Г, получают 5,1 г (100%) Boc-D-hCha-OH из Boc-D-hPhe-OH. 1H-ЯМР; FD-MS, m/e: 240 (M+).
Анализ для C15H27NO4:
рассчитано, %: C 63,13; H 9,54; N 4,91;
найдено, %: C 63,38; H 9,39; N 5,12.
Б) Получение HOOCCH2-D-hCha-Pro-p- NHCH2C6H4C(NH)NH2•HCl
Способами, по существу эквивалентными описанным в примере 86, получают 135 мг HOOCCH2-D-hCha-Pro-p- NHCH2C6H4C(NH)NH2•HCl. 1H-ЯМР; FAB-MS, m/e: 472,3 (MH+).
Анализ для C25H37N5O4•2,2HCl• 0,5H2O:
рассчитано, %: C 53,54; H 7,22; N 12,49; Cl 13,91;
найдено, %: C 53,29; H 7,01; N 12,46; Cl 14,30.
Пример 89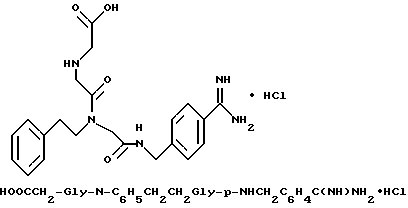
А) Получение HOOCCH2-Gly-N- C6H5CH2CH2Gly-p- NHCH2C6H4C(NH)NH2•HCl
Способами, по существу эквивалентными описанным в примерах 1-Ж, 1-Г, 23-А, 85-А и 18-Е, получают 365 мг N-HOOCCH2-Gly-N- C6H5CH2CH2-Gly-p- NHCH2C6H4C(NH)NH2•HCl. 1H-ЯМР; FAB-MS, m/e: 426,2 (MH+).
Анализ для C22H27N5O4•2,2HCl• 1,5H2O:
рассчитано, %: C 49,60; H 6,09; N 13,15; Cl 14,64;
найдено, %: C 47,79; H 5,71; N 13,31; Cl 14,49.
Пример 90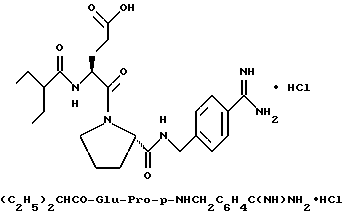
А) Получение Boc-(γ-OBn)-Glu-Pro-p- NHCH2C6H4C(NH)NHCbz
Способом, по существу эквивалентным описанному в примере 1-Ж, получают 2,7 г (64%) Boc-(γ-OBn)-Glu-Pro-p- NHCH2C6H4C(NH)NHCbz исходя из Boc-(γ-OBn)-Glu-OH и Pro-p-NHCH2C6H4C(NH)NHCbz•2HCl. 1H-ЯМР; FD-MS, m/e: 700 (M+).
Анализ для C38H45N5O8:
рассчитано, %: C 65,22; H 6,48; N 10,01;
найдено, %: C 65,00; H 6,56; N 10,06.
Б) Получение (γ-OBn)-Glu-Pro-p-NHCH2C6H4C(NH)NHCbz•2HCl
Способом, по существу эквивалентным описанному в примере 23-А, получают 2,38 г (98%) (γ-OBn)-Glu-Pro-p- NHCH2C6H4C(NH)NHCbz•2HCl. 1H-ЯМР; FD-MS, m/e: 600 (M+).
Анализ для C33H39N5O6Cl2:
рассчитано, %: C 58,93; H 5,84; N 10,41;
найдено, %: C 58,64; H 6,00; N 10,63.
В) Получение (C2H5)2CHCO-(γ-OBn)-Glu-Pro-p- NHCH2C6H4C(NH)NHCbz
К перемешиваемой смеси ( γ- OBn)-Glu-Pro-p- NHCH2C6H4C(NH)NHCbz•HCl (1,3 г, 2 ммоль) в ТГФ/H2O (по 50 мл каждого) добавляют K2CO3 (1,38 г, 10 ммоль) и 2-этилбутирилхлорид (0,3 г, 2,2 ммоль). После перемешивания в течение 10 мин летучие составные части удаляют в вакууме. Полученный остаток распределяют между водой и этилацетатом (100 мл каждого). Слои разделяют и органическую фазу промывают дважды, каждый раз насыщенным водным раствором хлорида аммония и солевым раствором, сушат (MgSO4), фильтруют и концентрируют в вакууме с получением 1,45 г (100%) соединения. 1H-ЯМР; FD-MS, m/e: 698 (M+).
Анализ для C39H47N5O7:
рассчитано, %: C 67,13; H 6,79; N 10,04;
найдено, %: C 67,11; H 6,70; N 9,74.
Г) Получение (C2H5)2CHCO-Glu-Pro-p- NHCH2C6H4C(NH)NH2•HCl
Способом, по существу эквивалентным описанному в примере 18-Е, получают 425 мг (47%) (C2H5)2CHCO-Glu-Pro-p- NHCH2C6H4C(NH)NH2•HCl. Для очистки продукта используют ВЭЖХ, метод 2 (градиент A/B от 98/2 до 75/25, в течение 150 мин). 1H-ЯМР; FAB-MS, m/e: 474,3 (MH+).
Анализ для C24H35N5O5•1,5HCl• 1,1H2O:
рассчитано, %: C 51,10; H 6,91; N 12,41; Cl 9,43;
найдено, %: C 51,10; H 6,81; N 12,41; Cl 9,62.
Пример 91
А) Получение (C2H5)2CHCO-Met(O2)- Pro-p-NHCH2C6H4C(NH)NH2•HCl
Способом, по существу эквивалентным описанному в примере 90, получают 530 мг (C2H5)2CHCO-Met(O2)-Pro- p-NHCH2C6H4C(NH)NH2•HCl. 1H-ЯМР;
FAB-MS, m/e: 508,2 (MH+).
Анализ для C24H37N5O5S•1,1HCl:
рассчитано, %: C 52,63; H 7,01; N 12,79; Cl 7,12;
найдено, %: C 52,42; H 7,03; N 12,80; Cl 6,99.
Пример 92
(N-(Метилсульфонилацетил)-L-циклогексилаланил-N-[[4-(аминоиминометил) фенил]метил]-L-пролин-амид-гидрохлорид).
Получение MeSO2CH2CO-D-Cha-Pro-p- NHCH2C6H4C(NH)NH2•HCl
Способом, по существу эквивалентным описанному в примере 1-Ж и в примере 18-Е, получают 550 мг MeSO2CH2CO-D-Cha-Pro-p- NHCH2C6H4C(NH)NH2•HCl. 1H-ЯМР; FAB-MS, m/e: 520,5 (MH+).
Анализ для C25H37N5O5S•1,2HCl• H2O:
рассчитано, %: C 51,64; H 6,97; N 12,04; Cl 7,32;
найдено, %: C 51,58; H 6,84; N 12,18; Cl 7,61.
Соединения изобретения способны селективно ингибировать тромбин относительно других протеиназ и неферментных протеинов, вызывающий свертывание крови, без заметной интерференции со способностью лизировать естественную коагуляцию в организме (соединения оказывают низкое ингибирующее воздействие на фибринолиз). Далее, такая селективность позволяет использовать их с тромболитическими агентами без заметной интерференции с тромболизом и фибринолизом. Кроме того, соединения по настоящему изобретению могут быть перорально эффективными.
В одном из своих аспектов изобретение относится к способу ингибирования тромбина у млекопитающих, который включает введение млекопитающему, нуждающемуся в лечении, эффективной (ингибирующей тромбин) дозы соединения формулы I.
Ингибирование тромбина, согласно настоящему способу, включает в качестве приемлемого как медицинское терапевтическое, так и/или профилактическое лечение.
Далее, изобретение относится к лечению, в случае человека или животного, состояний, которые требуют ингибирования тромбина. Соединения изобретения могут быть пригодны в случае животных, включая человека, для лечения или профилактики тромбоза и гиперкоагулируемости в крови и тканях. Болезненные состояния, при которых соединения обладают возможной полезностью для лечения или профилактики, представляют собой тромбоз и гиперкоагулируемость в крови и тканях. Эти болезненные состояния, при которых используются соединения для лечения и/или профилактики, включают венозный тромбоз и эмболию легких, артериальный тромбоз, такой как ишемия миокарда, инфаркт миокарда, нестабильная стенокардия, апоплексический удар за счет тромбоза и периферический артериальный тромбоз. Далее, соединения используются для профилактики атеросклеротических заболеваний, таких как коронарное артериальное заболевание, церебральное артериальное заболевание и периферическое артериальное заболевание. Далее, соединения могут использоваться вместе с тромболитическими агентами при инфаркте миокарда. Далее, соединения используются для лечения или профилактики для реокклюзии после тромболиза, чрескожной транслюминальной ангиопластики (PTCA) и операций, связанных с коронарным шунтированием. Далее, соединения могут быть полезными при профилактике ретромбоза после микрохирургии. Далее, соединения могут быть пригодными при антикоагулянтной обработке в связи с искусственными органами и клапанами сердца. Далее, соединения могут использоваться при антикоагулянтной обработке при гемодиализе и диссеменированной внутрисосудистой коагуляции. Дальнейшее возможное применение заключается в промывке катетеров и механических приспособлений, используемых в случае пациентов ин виво, и в качестве антикоагулянта для консервации ин витро крови, плазмы и других продуктов крови. Далее, соединения могут быть полезны при других заболеваниях, где коагуляция крови может являться фундаментальным сопутствующим процессом или источником вторичной патологии, таких как рак, включая метастаз, и воспалительные заболевания, включая артрит и диабеты. Антикоагулянтное соединение вводится перорально или парентерально, например, путем внутривенного вливания (вв), внутримышечной инъекции (вм) или подкожной инъекции (пк).
Специфическая доза соединения, вводимого согласно настоящему изобретению, с целью достижения терапевтического и/или профилактического эффектов естественно должна определяться индивидуальными обстоятельствами заболевания, включающими, например, вводимое соединение, скорость введения и состояние, подвергаемое лечению.
Типичная суточная доза для каждого из вышеуказанных полезных применений составляет примерно от 0,01 до 1000 мг/кг. Дозировочный режим может для профилактических целей отличаться, например, суточная доза может быть введена за один раз или может быть удобным введение многократных доз, 3 или 5 раз в день. В критических тревожных ситуациях соединение по изобретению вводят путем внутривенного вливания со скоростью примерно 0,01-20 мг/кг/час и, предпочтительно, около 0,1-5 мг/кг/час.
Способ по настоящему изобретению также осуществляется в сочетании с лизирующим тромб (сгусток крови) агентом, например таким, как плазминогенный тканевый активатор (t-PA), модифицированный t-PA, стрептокиназа или урокиназа. В случаях, когда происходит образование тромба и блокируется артерия или вена, либо частично, либо полностью, обычно используют лизирующий тромб агент. Соединение по изобретению может быть введено перед или вместе с лизирующим агентом или после него, предпочтительно, кроме того вместе с аспирином с целью предотвращения возобновления образования тромба.
Способ по настоящему изобретению также осуществляется в сочетании с антагонистом тромбоцитного гликопротеинового рецептора (Пб/Ша), который ингибирует агрегацию тромбоцитов. Соединение изобретения может быть введено перед или вместе с Пб/Ша-антагонистом или после него с целью предотвращения возобновления образования тромба.
Способ по настоящему изобретению также осуществляется в сочетании с аспирином. Соединение по изобретению может быть введено раньше или вместе с аспирином или после его использования с целью предотвращения возобновления образования тромба. Как констатировано выше, соединение по настоящему изобретению предпочтительно вводят в сочетании с лизирующим тромб агентом и аспирином.
Настоящее изобретение также относится к фармацевтическим составам для применения в вышеописанном способе лечения. Фармацевтические составы по изобретению включают эффективное ингибирующее тромбин количество соединения формулы I в сочетании с фармацевтически приемлемым носителем, наполнителем или разбавителем. Для перорального применения, антитромботическое соединение готовят в виде желатиновых капсул с лекарством или таблеток, которые могут содержать эксципиенты, такие как связующие, смазки, разрыхлители и т.п. Для парентерального введения, антитромботическое средство готовят в фармацевтически приемлемом разбавителе, например, в виде физиологического солевого раствора (0,9%-ный), 5%-ного раствора декстрозы, раствора Рингера и т.п.
Соединение по настоящему изобретению может входить в состав единичных дозировочных препаратов, содержащих дозу примерно 0,1-1000 мг. Предпочтительно, соединение находится в виде фармацевтически приемлемой соли, такой как, например, сульфат, ацетат или фосфат. Пример единичной дозировочной формы; 5 мг соединения настоящего изобретения в виде фармацевтически приемлемой соли в стерильной стеклянной ампуле на 10 мл. Другой пример единичной дозировочной формы содержит около 10 мг соединения по настоящему изобретению в виде фармацевтически приемлемой соли в 20 мл изотонического солевого раствора, содержащегося в стерильной ампуле.
Соединения можно вводить разными путями, включающими пероральное, ректальное, чрескожное, подкожное, внутривенное, внутримышечное введение и введение через нос. Соединения по настоящему изобретению, предпочтительно, готовят в виде состава до введения. Следовательно, следующим объектом настоящего изобретения является фармацевтическая композиция, содержащая эффективное количество соединения формулы I или его фармацевтически приемлемой соли или сольвата в сочетании с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
Активный ингредиент в такого рода композициях составляет 0,1-99,9 мас.% в расчете на композицию. Под выражением "фармацевтически приемлемый" подразумевают, что носитель, разбавитель или наполнитель должен быть совместим с другими ингредиентами композиции и не причинять вреда пациенту.
Настоящие фармацевтические композиции готовят известными способами с использованием хорошо известных и легко доступных ингредиентов. При получении композиций по настоящему изобретению активный ингредиент обычно смешивают с носителем или разбавляют носителем или включают в носитель, который может быть в форме капсулы, пакета, бумаги или другой тары. Когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким материалом, который выступает в виде разбавителя, наполнителя, или среды для активного ингредиента. Так, композиции могут быть в виде таблеток, пилюль, порошков, лепешек, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как в твердой или в жидкой среде), мягких или твердых желатиновых капсул, свечей, стерильных растворов для инъекций, стерильных упакованных пудр и т.п. Композиции по настоящему изобретению могут быть составлены таким образом, чтобы происходило быстрое, непрерывное или замедленное высвобождение активного ингредиента после введения пациенту путем хорошо известных специалисту применяемых процедур.
Следующие примеры составов служат только для иллюстрации,
но никоим образом не ограничивают объема охраны изобретения. Под "активным ингредиентом", естественно, понимают соединение, соответствующее формуле I, или его фармацевтически приемлемую соль или сольват.
Состав 1
Твердые желатиновые капсулы получены с использованием следующих ингредиентов, мг/капсула:
Активный ингредиент - 250
Крахмал, высушенный - 200
Стеарат магния - 10
Всего - 460
Состав 2
Таблетка получена с использованием указанных далее ингредиентов, мг/таблетка:
Активный ингредиент - 250
Микрокристаллическая целлюлоза - 400
Диоксид кремния - 10
Стеариновая кислота - 5
Всего - 665
Компоненты смешивают и прессуют с получением таблеток, каждая весом 665 мг.
Состав 3
Аэрозольный раствор получен в виде содержащего следующие компоненты, мас.%:
Активный ингредиент - 0,25
Этанол - 25,75
Propellant 22 (хлордифторметан) - 70,00
Всего - 100,00
Активное соединение смешивают с этанолом и смесь добавляют к части Propellant 22, охлаждают до -30oC и переносят в загрузочное устройство. Необходимое количество затем вводят в контейнер из нержавеющей стали и разбавляют остатком пропеллента. Затем контейнер снабжают клапанными элементами.
Состав 4
Таблетки, каждая из которых содержит 60 мг активного ингредиента, готовят следующим образом:
Активный ингредиент - 60 мг
Крахмал - 45 мг
Микрокристаллическая целлюлоза - 35 мг
Поливинилпирролидон (в виде 10%-ного водного раствора) - 4 мг
Натриевая соль карбоксиметилкрахмала - 4,5 мг
Стеарат магния - 0,5 мг
Тальк - 1 мг
Всего - 150 мг
Активный ингредиент, крахмал и целлюлозу просеивают через сито N 45 меш U. S. и тщательно смешивают. Водный раствор, содержащий поливинилпирролидон, смешивают с полученным порошком и смесь затем пропускают через сито N 14 меш U.S. Полученные таким образом гранулы высушивают при 50oC и пропускают через сито N 18 меш U.S. Натриевую соль карбоксиметилкрахмала, стеарат магния и тальк, предварительно пропущенные через сито N 60 меш U.S., затем добавляют к гранулам, которые после смешения прессуют на машине для изготовления таблеток с получением таблеток, каждая из которых весит 150 мг.
Состав 5
Капсулы, содержащие каждая 80 мг активного ингредиента, готовят следующим образом, мг:
Активный ингредиент - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Всего - 200
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито N 45 меш U. S. и заполняют этой смесью твердые желатиновые капсулы в количестве по 200 мг.
Состав 6
Свечи, содержащие каждая 225 мг активного ингредиента, готовят следующим образом, мг:
Активный ингредиент - 225
Глицериды насыщенных жирных кислот - 2000
Всего - 2225
Активный ингредиент пропускают через сито N 60 меш U.S. и суспендируют в предварительно расплавленных при использовании минимального необходимого тепла глицеридах насыщенных жирных кислот. Смесь затем разливают в формы для изготовления свечей номинальной емкостью 2 г и оставляют охлаждаться.
Состав 7
Суспензии, содержащие каждая 50 мг активного ингредиента на дозу 5 мл, готовят следующим образом:
Активный ингредиент - 50 мг
Натриевая соль карбоксиметилцеллюлозы - 50 мг
Сироп - 1,25 мл
Раствор бензойной кислоты - 0,10 мл
Отдушка - Достаточное количество
Краситель - Достаточное количество
Очищенная вода до общего объема - 5 мл
Активный ингредиент пропускают через сито N 45 меш U.S. и смешивают с натриевой солью карбоксиметилцеллюлозы и сиропом с получением мягкой пасты. Добавляют при перемешивании раствор бензойной кислоты, отдушку и краситель, разбавленные частью воды. После этого добавляют достаточное количество воды для достижения необходимого объема.
Состав 8
Состав для внутривенного введения может быть получен следующим образом:
Активный ингредиент - 100 мг
Изотонический солевой раствор - 1000 мл
Раствор вышеуказанных ингредиентов обычно вводят внутривенно пациенту со скоростью 1 мл/мин.
Соединения формулы I по изобретению являются перорально активными и селективно ингибируют действие тромбина у млекопитающих.
Способность соединений по настоящему изобретению быть эффективными и перорально активными ингибиторами тромбина оценивается в одном или нескольких из следующих испытаний.
Ингибирование тромбина демонстрируется ин витро ингибированием амидазной активности тромбина, как определяется в испытании, в котором тромбин гидролизует хромогенный субстрат, N-бензоил-L-фенилаланил-L-валил-L-аргинил-п-нитроанилид.
Испытание проводят путем смешения 50 мкл буфера (0,03 М ТРИС, 0,15 М NaCl, pH 7,4) с 25 мкл раствора бычьего тромбина или человеческого тромбина (0,21 мг/мл тромбостатного бычьего тромбина, Paeke-Davis, или очищенного человеческого тромбина, Enzyme Research Laboratories, South bend, India, в количестве около 8 N1T ед./мл, в том же самом буфере) и 25 мкл испытуемого соединения в растворителе (в 50%-ном водном метаноле, по объему). Добавляют 150 мкл водного раствора хромогенного субстрата (по 0,25 мг/мл) и скорости-гидролиза субстрата измеряют путем контролирования протекания реакций при 405 нм по высвобождению п-нитроанилина. Стандартные кривые строят путем вычерчивания кривой зависимости концентрации свободного тромбина от скорости гидролиза. Скорости гидролиза, наблюдаемые с испытуемыми соединениями, затем превращают в значения "свободного тромбина" в соответствующих тестах путем использования стандартных кривых. Связанный тромбин (связанный с испытуемым соединением) рассчитывают путем вычитания количества свободного тромбина, наблюдаемого в каждом опыте, из известного начального количества тромбина, используемого в опыте. Количество свободного ингибитора в каждом опыте рассчитывают путем вычитания числа молей связанного тромбина из числа молей добавленного ингибитора (испытуемого соединения).
Ka - величина представляет собой константу предположительного равновесия для реакции между тромбином и испытуемым соединением: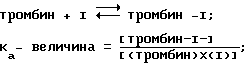
Kass-величину рассчитывают для ряда концентраций испытуемых соединений и среднее значение выражают в единицах литр на моль.
Следуя по существу вышеописанным методикам для человеческого тромбина и используя другую человеческую кровь, коагуляционную систему сериновых протеаз и протеаз фибринолитической системы с соответствующими хромогенными субстратами, идентифицированными ниже, оценивают селективность соединений настоящего изобретения в отношении фактора коагуляции сериновых протеаз и в отношении фибринолитической системы сериновых протеаз, также, как в существенной мере отсутствие у них интерференции с сериновыми протеазами фибринолитической системы. Ингибиторы тромбина, предпочтительно, не должны затрагивать фибринолиз, индуктируемый урокиназой, плазминогенным тканевым активатором (t-PA) и стрептокиназой. Это может быть важным для терапевтического использования таких агентов в качестве дополнения к тромболитической терапии за счет стрептокиназы, t-PA или урокиназы и для применения таких агентов в качестве эндогенного, не затрагивающего фибринолиз (в отношении t-PA и урокиназы) антитромботического агента. В дополнение к отсутствию интерференции с амидазной активностью фибринолитических протеаз, такая щадящая фибринолитическая система может быть исследована путем использования сгустков человеческой плазмы и их лизиса за счет соответствующих фибринолитических плазминогенных активаторов.
Человеческие факторы X, Xa, IXa, XIa и XIIa были получены от Enzyme Research Laboratories, South Bend, India, человеческая урокиназа получена от Leo Pharmaceuticals, Denmark; и рекомбинантный активированный протеин C (aPC) получен от Eli Lilly and Co. , по существу, согласно патенту США 4981952. Хромогенные субстраты - N-бензолп-Ile-Glu-Gly-Aig-п-нитроанилид (для фактора Xa); N-Cbz-D-Arg-Gly-Arg-п-нитроанилид (для испытания фактора IXa как субстрата фактора Xa); пироглутамил-Pro-Arg-п-нитроанилид (для фактора XIa и для aPC); H-D-Pro-Arg-п-нитроанилид (для фактора XIIa); и пироглутамил-Gly-Arg-п-нитроанилид (для урокиназы) - приобретены у Kabi Vitrum, Стокгольм, Швеция, или у Midwest Biotech, Fishers, Indiana. Бычий трипсин получен от Worthington Biocemicals, Freehold, New Jersy, а калликреин человеческой плазмы получен от Kabi Vitrum, Стокгольм, Швеция. Хромогенный субстрат H-D-Pfo-Phe-Arg-п-нитроанилид для калликреина плазмы приобретен у Kabi Vitrum, Стокгольм, Швеция. N-Бензоил-Phe-Val-Arg-п-нитроанилид, субстрат для человеческого тромбина и для трипсина, синтезирован согласно методике, описанной выше для соединений настоящего изобретения, используя известные методы сочетания пептидов из имеющихся в продаже реагентов, или получен от Midwest Biotech, Fishers, Indiana.
Человеческий плазмин получен от Boehringer Mannheim Indianapolis, Indiana; nt-PA в качестве единственного с цепной активностью эталона (chain activity reference) приобретен у American Diagnostica, greenwich, Connecticut; модифицированный t-PA6 (mt-PA6) получен от Eli Lilly and Company по известной в уровне техники методике (см. Burck и др. J. Biol. Chem. 265, 5120-5177 (1990)). Хромогенный субстрат плазмина H-D-Val-Leu-Lys-п-нитроанилид и субстрат тканевого плазминогенного активатора (t-PA) H-D-Ile-Pro-Arg-п-нитроанилид получен от Kabi Vitrum, Стокгольм, Швеция.
В вышеописанных хромогенных субстратах трехбуквенные символы Ile, Glu, Gly, Pro, Arg, Phe, Val, Leu и Lys используют для указания соответствующей группы аминокислоты: изолейцин, глутаминовая кислота, глицин, пролин, аргинин, фенилаланин, валин, лейцин и лизин, соответственно.
В нижеследующей таблице 1 приводятся Kass-величины, полученные с указанным соединением, соответствующим формуле (I).
Следует заметить, что, неожиданно, соединения, в которых X обозначает D-циклогексилаланильный фрагмент, обладают повышенной эффективностью в отношении ингибирования тромбина и показывают особенно удивительно повышенную эффективность в отношении ингибирования фактора Xa, когда сравнивают, например, с соответствующими соединениями, в которых X обозначает D-фенилаланильный фрагмент.
Материалы
Собачью плазму получают от бодрствующих собак смешанной породы (обоего пола, Hazelton-LRe, Kolamazoo, Мичиган, США) путем венепункции в 3,8%-ном цитрате. Фибриноген получают из свежей собачьей плазмы, а человеческий фибриноген получают из in-date ACD человеческой крови во фракции 1-2, согласно предшествующим методикам и описаниям: Smith, Biochem. J., 185, 1-11 (1980); и Smith и др., Biochemistry, 11, 2958-2967 (1972). Человеческий фибриноген (98%-ной чистоты и без плазмина) получают из Americam Diagnostica, Greenwich, Connecticut. Маркировка радиоактивным изотопом препаратов фибриногена 1-2 осуществляется, как сообщалось ранее: Smith и др., Biochemistry, 11, 2958-2967 (1972). Урокиназа получена от Leo Pharmaceuticals, Denmark, в виде 2200 Ploug единиц/пузырек. Стрептокиназа получена от Hoechst Roussel Pharmaceuticals, Сомервиль, Нью-Джерси.
Методы - Воздействия на лизис сгустков (тромбов) человеческой плазмы за счет t-PA
Сгустки человеческой плазмы получают в микропробирках путем добавления 50 мкл тромбина (73 N1H ед./мл) к 100 мкл человеческой плазмы, которая содержит меченый изотопом иода-125 фибриноген с радиоактивностью 0,0229 микрокюри. Лизис сгустка изучают путем покрытия сгустков 50 мкл урокиназы или стрептокиназы (50, 100 или 1000 ед./мл) и инкубации в течение 20 часов при комнатной температуре. После инкубации пробирки подвергают центрифугированию в микроцентрифуге Бекмана. 25 мкл надосадочной жидкости добавляют к 1,0 мл 0,03 М ТРИС/0,15 М NaCl-буферу для гамма-подсчета. Контроли по подсчету в отношении 100%-ного лизиса получают путем невключения тромбина (и замены буфера). Ингибиторы тромбина оценивают по возможной интерференции с фибринолизом за счет включения соединений в покрывные растворы в концентрациях 1,5 и 10 ед/мл. Грубые приближения значений IC50 оценивают путем линейной экстраполяции из данных точек к величине, которая должна означать 50% лизиса, для этой особой концентрации фибринолитического агента.
Антикоагулянтная активность
Материалы
Плазму собаки и плазму крысы получают от бодрствующих собак смешанной породы (обоего пола, Hazelton - LRe, Kalamazoo, Мичигина, США) или от анестезированных самцов крыс Sprague - Dawley (Harlan Sprague - Dawley, Inc., Индианаполиси, Индиана, США) путем венепункции в 3,8%-ном цитрате. Фибриноген получают из in-date ACD человеческой крови в виде фракции 1-2, согласно предшествующим методикам и описаниям: Smith, Biochem. J., 185, 1-11 (1980); и Smith и др., Biochemistry, 11, 2958-2967 (1972). Человеческий фибриноген также получен в виде 98%-ной чистоты и без плазмина, от American Diagnostica, Гринвич, Коннектикут. Реагенты коагуляции ACTIN, тромбопластин и человеческая плазма получены от Baxter Healthcare Corp., Dade Division, Майами, Флорида. Бычий тромбин, полученный от Parke-Davis (Ann. Дейтройт, Мичигин), используют для опытов по коагуляции в плазме.
Методы - Определения антикоагуляции
Методики осуществления испытаний по коагуляции такие, как описанные ранее; Smith и др., Trombosis Research, 50, 163-174 (1988). Прибор для определения коагуляции CoAScreener (American Labor. Inc) используют для всех измерений испытаний по коагуляции. Протромбиновое время (PT) измеряют путем добавления 0,05 мл солевого раствора и 0,05 мл реактива тромбопластин-C к 0,05 мл испытуемой плазмы. Активированное парциальное тромбопластиновое время (APTT) измеряют путем инкубации 0,06 мл испытуемой плазмы с 0,05 мл реактива Actin в течение 120 с с последующим добавлением 0,05 мл 0,02 М раствора CaCl2. Тромбиновое время (TT) измеряют путем добавления 0,05 мл солевого раствора и 0,05 мл тромбина (10 N1H д./мл) к 0,05 мл испытуемой плазмы. Соединения формулы I добавляют к человеческой плазме и плазме животных в виде большого ряда концентраций с целью определения пролонгированных эффектов при испытаниях в отношении APTT, PT и TT. Линейные экстраполяции осуществляют для оценки концентраций, необходимых для удвоения времени образования сгустков для каждого теста.
Животные
Самцов крыс Sprague Dawley (350-425 г, Horlan Sprague Dawley Inc., Индианаполис, 1N) анестезируют с помощью ксилазина (20 мг/кг, подкожно) и кетамина (120 мг/кг, подкожно) и содержат на нагреваемой водной поверхности (37oC). Яремную вену (вены) канюлируют, чтобы можно было осуществлять вливания (инфузии).
Модель артериовенозного шунта
Левую яремную вену и правую каротидную артерию канюлируют с помощью длиной 20 см тюбинга из полиэтилена ПЭ 60. В длиной 6 см центральный отрезок более широкого тюбинга (ПЭ 190) в просвет вставляют хлопковую нить (5 см) между более длинными отрезками до полного контура артериовенозного шунта. Кровь циркулирует через шунт в течение 15 минут, прежде чем нить осторожно удаляют и взвешивают. Вес влажной нити вычитают из общего веса нити и тромба (см. J.R. Smith, Br. J. Pharmacol., 77, 29 (1982)).
Когда соединение примера 48 сравнивают с D-MePhe-Pro-Arg-H (обсуждено выше на с. 2), в модели артериовенозного шунта, антитромботическая эффективность найдена в 9 раз большей во время непрерывной внутривенной инфузии. В отношении снижения веса тромба до той же самой степени (примерно до 20% от контроля), соединение примера 48 пролонгирует тромбиновое время плазмы примерно в 3 раза, тогда как тромбиновое время плазмы удлиняется более чем в 20 раз, в процессе инфузии стандартного соединения. Протромбиновое время и APTT удлиняется только примерно до 120% от контроля (несколько секунд) в течение инфузии соединения примера 48.
FeCl3 - Модель артериального повреждения
Каротидные артерии изолируют через срединобрюшинный и шейный разрез. Под каждую артерию помещают термопару и температуру сосуда непрерывно регистрируют на диаграммном ленточном самописце. Вокруг каждой каротидной артерии, непосредственно выше термопары, помещают манжетку тюбинга (0,058ID x 0,077OD x 4 мм, Baxter Med. Grade Silicone), продольно срезанного (cut longitudinally). Гексагидрат FeCl3 растворяют в воде и концентрацию (20%) выражают только с точки зрения действительного веса FeCl3. Для повреждения артерии и стимулирования тромбоза, 2,85 мкл этого раствора пипеткой вводят в манжетку артерии выше термопарного зонда. Артериальная окклюзия характеризуется быстрым падением температуры. Время окклюзии регистрируется в минутах и означает время, проходящее между введением FeCl3 и быстрым падением температуры в сосуде (см. K.D. Kurz, Thromb. Res., 60, 269 (1990)).
Модель спонтанного тромболиза
Ин витро данные говорят о том, что пептидные ингибиторы тромбина ингибируют тромбин и другие сериновые протеазы, такие, как плазмин и тканевый плазминогенный активатор. С целью оценки, ингибируют ли соединения фибринолиз ин виво, определяют скорость спонтанного тромболиза путем имплантации целиком меченого сгустка крови в легочный кровоток.
Крысиную кровь (1 мл) быстро смешивают с бычьим тромбином (4 IU, Parke-Davis) и меченым 125I человеческим фиброгеном (5 микрокюри, ICN), тотчас же вносят в силастиковый тюбинг и инкубируют при 37oC в течение 1 ч. Подвергнутый "старению" тромб удаляют из тюбинга, разрезают на сегменты в 1 см, промывают 3 раза 1н раствором соли и каждый сегмент подвергают подсчету импульсов в гамма-счетчике. Сегмент с известным счетом импульсов засасывают в катетер, чтобы потом имплантировать в яремную вену. Тонкий конец катетера вводят по соседству с правым предсердием и сгусток выталкивают в легочный кровоток. Спустя час после имплантации, сердце и легкие извлекают и раздельно подсчитывают число импульсов. Тромболиз выражают в процентах, где: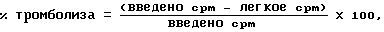
(cpm = отсчетов в минуту).
Фибринолитическое растворение имплантированного сгустка происходит в зависимости от времени (см. J. R. Clozel, Cardiovas. Pharmacol. 12, 520 (1988)).
Параметры коагуляции
Тромбиновое время плазмы (TT) и активированное парциальное тромбопластиновое время (APTT) измеряют с помощью фиброметра. Кровь отбирают из яремного катетера и собирают в шприц, содержащий цитрат натрия (3,8%-ный; 1 часть на 9 частей крови). Для измерения TT, плазму крысы (0,1 мл) смешивают с солевым раствором (0,1 мл) и бычьим тромбином (0,1 мл; 30 ед./мл в ТРИС-буфере; Parke Davis) при 37oC. Для APTT, плазму (0,1 мл) и раствор APTT (0,1 мл, Organon Teknika) инкубируют в течение 5 мин при 37oC и для инициирования коагуляции добавляют 0,025 М раствор CaCl2 (0,01 мл). Опыты повторяют два раза и берут среднее значение.
Индекс биодоступности
Измерение биоактивности, тромбинового времени (TT) плазмы, служит в качестве замены испытания классификационного соединения относительно предположения, что приросты в TT являются результатом ингибирования тромбина только за счет классификационного соединения. Время, протекающее от воздействия ингибитора тромбина относительно TT, определяют после внутривенного введения лекарственного средства анестезированным крысам и после оральной обработки бодрствующих крыс. За счет ограничений объема крови и числа точек, необходимых для определения времени, протекающего от времени обработки до времени, когда ответ возвращает к значениям до обработки, используют две популяции крыс. Каждый образец популяции означает чередующиеся последовательные временные точки. Среднее TT выше прошедшего времени используют для расчета площади под кривой (AUC). Индекс биодоступности рассчитывают по нижеуказанной формуле и выражают в виде процента относительной активности.
Площадь под кривой (AUC) прошедшего времени TT плазмы определяют и устанавливают для дозы. Этот индекс биодоступности выражает "% относительной активности" и рассчитывается как:
p.o. = перорально; i.v. = внутривенно).
Соединения
Растворы соединений готовят ежедневно свежими в 1н солевом растворе и инъецируют в виде болюса или вливают, начиная на 15 мин раньше и продолжая на всем протяжении экспериментальной пертурбации, которая составляет 15 мин в модели артериовенозного шунта и 60 мин в FeCl3-модели артериального повреждения и в модели спонтанного тромбоза. Инъекцируемый объем болюса составляет 1 мл/кг внутривенно и 5 мл/кг перорально, а объем инфузии составляет 3 мл/ч.
Статистика
Результаты выражают в виде значений +/-SEM. Для выявления статистически значимых различий используют рутинный анализ расхождений и затем применяют тест Dunnett для определения, какие значения являются различными. Уровень значимости отклонения от нулевого предположения адекватных значений составляет P < 0,05.
Животные
Кобелей (гончие; в возрасте от 18 месяцев до 2-х лет; весом 12-13 кг; Marshall Farms, North Rose, Нью-Йорк 14516) привязывают на ночь и спустя 240 минут после дозировки кормят пищей по рецепту согласно Purina (Purina Mills, St. Lous, Missouri). Воду дают в достаточном количестве для выживания. Поддерживают комнатную температуру при 24,9-28oC; относительная влажность составляет 45-50%; и освещают 600-1800 ч.
Фармакокинетическая модель
Испытуемое соединение готовят непосредственно перед дозировкой путем растворения в стерильном 0,9%-ном солевом растворе до получения препарата с концентрацией 5 мг/мл. Собаки получают одноразовую дозу 2 мг/кг испытуемого соединения перорально. Образцы крови (4,5 мл) отбирают из латеральной подкожной вены в часы 0,25, 0,5, 0,75, 1, 2, 3, 4 и 6 после дозирования. Образцы собирают в цитратированные трубки Vacutainer и держат на льду до концентрирования плазмы путем центрифугирования. Образцы плазмы преобразуют с помощью динитрофенилгидразина и анализируют с помощью ВЭЖХ (высокоэффективной жидкостной хроматографии) (колонка с Zorbax SB-C8), элюируя метанолом с 500 ммоль ацетата натрия, доводя до pH 7 с помощью фосфорной кислоты (60:40 по объему). Концентрацию в плазме испытуемого соединения регистрируют и используют для расчета фармакокинетических параметров: константа скорости элиминирования, Ke; общий клиренс, CIt; объем распределения (васкуляризации), VD; время максимальной концентрации в плазме испытуемого соединения, Tmax; максимальная концентрация испытуемого соединения при Tmax, Cmax; период полураспада плазмы, t0,5; площадь под кривой, A.U.C. и абсорбированная доля испытуемого соединения, F.
Модель коронарного артериального тромбоза на кролике
Хирургическое препарирование и оснащение инструментами в отношении собак такие, как описано Jackson и др., Circulation, 82, 930-940 (1990). Собак смешанных пород (в возрасте 6-7 месяцев, обоего пола, Hazelton-LRE, Kalamazoo MI, США) анестезируют пентобарбиталом натрия (30 мг/кг внутривенно, i. v. ), интубируют и вентилируют комнатным воздухом. Приливно-отливный объем и скорости дыхания регулируют за счет поддержания в крови PO2, PO2 и pH в обычных пределах. Подкожные игольчатые электроды вставляют для регистрации биопотенциала-П на электрокардиографе.
Левую яремную вену и общую каротидную артерию изолируют через левый медиолатеральный шейный разрез. Артериальное кровяное давление (ABP) измеряют непрерывно с помощью предварительно откалиброванного трансдуктора (датчика) Миллара (модель MPC-500, Millar Instruments, Хьюстон, TX, США), вставленного в каротидную артерию. Яремную вену канюлируют для отбора крови в процессе эксперимента. В дополнение, феморальные вены обеих задних лап канюлируют для введения испытуемого соединения.
Левую торакотомию осуществляют в пятой межреберной полости и сердце подвешивают в перикардиальной рамке. Величиной 1-2 см сегмент левой огибающей коронарной артерии (LCX) изолируют близко к первой главной диагональной вентрикулярной ветви, 26-го калибра с тонким игольчатым концом проволочный анодный электрод (покрытая тефлоном® 30-го калибра серебряно-платинированная медная проволока) длиной 3-4 мм вставляют в LCX и вводят в контакт с внутренней поверхностью артерии (закрепляют до окончания эксперимента). Стимулирование цикла завершают путем помещения катода в подкожный (s.c.) участок. Регулируемое пластиковое закрывающее приспособление помещают вблизи LCX, выше области электрода. Предварительно калиброванный электромагнитный поточный зонд (Carolina Medical Electronics, King, U.S.A.) помещают вблизи LCX, ближайшей к аноду, для измерения коронарного кровотока (CBF). Закрывающее приспособление (окклюдер) регулируют для достижения 40-50%-ного ингибирования ответа гиперемичного кровотока, наблюдаемого спустя 10 секунд после механической окклюзии LCX. Все гемодинамические и ЭКГ измерения регистрируют и анализируют с помощью системы банка данных (модель M3000, Modular. Instruments, Malven, PA, США).
Образование тромба и режимы введения соединения
Электролитического повреждения внутренней стороны достигают путем приложения к аноду постоянного тока 100 мкА (DC). Ток поддерживают в течение 60 минут и затем прерывают для проверки, окклюдирован сосуд или нет. Образование тромба происходит спонтанно, до тех пор, пока LCX полностью не окклюдируется (определяют в виде нулевого CBF и по увеличению S-т-сегмента). Введение соединения начинают после того, как окклюдирующий тромб существует уже в течение 1 часа. Инфузию в течение 2 ч соединений настоящего изобретения в дозах 0,5-1 мг/кг/ч начинают одновременно с инфузией тромболитического агента (например, тканевый плазминогенный активатор, стрептокиназа, AP SAC). Реперфузию осуществляют в течение 3 ч после введения испытуемого соединения. Реокклюзию коронарных артерий после успешного тромболиза определяют как нулевой CBF, который персистирует в течение ≥ 30 мин.
Гематология и определение шаблонного времени кровотечения
Подсчеты клеток цельной крови, гемоглобин и величины гематокритных чисел определяют на образце объемом 40 мкл цитратированной (3,8%) крови (1 часть цитрата на 9 частей крови) с помощью гематологического анализатора (Cell-Dyn 900, Sequoia-Turner, Mount View, VA, USA). Шаблонное время гингивального (десневого) кровотечения определяют с помощью устройства Simplate II для измерения времени кровотечения (Organon Teknika Durham, N.C., США). Устройство используют для того, чтобы сделать 2 горизонтальных разреза в десне любой верхней или нижней левой челюсти собаки. Каждый разрез имеет ширину 3 мм и глубину 2 мм. Делают разрезы и используют секундомер с остановом для определения, какое по длительности происходит кровотечение. Ватный тампон используют для впитывания крови, когда она медленно вытекает (просачивается) из разреза. Шаблонное время кровотечения представляет собой время от надреза до прекращения кровотечения. Времена кровотечения принимают: точно до введения испытуемого соединения (0 мин), 60 мин при вливании, по окончании введения испытуемого соединения (120 мин) и в конце эксперимента.
Все данные анализируют путем рутинного анализа расхождений (ANOVA) с последующим Student - Neuman - Kuelspost - hoc t-тестом для определения уровня достоверности. Повторяемые измерения ANOVA используют для определения значимых различий между временными точками в процессе экспериментов. Значения определяют для того, чтобы статистическое различие находилось минимально на уровне p < 0,05. Все значения представляют среднее число ± SEM. Все исследования проводят в соответствии с руководящими принципами American Physiological Society. Далее, подробное рассмотрение методик описывается Jackson и др., J. Cardiovasc. Pharmacol., 21, 587-599 (1993).
В табл. 2 дана антикоагуляция человеческой плазмы.
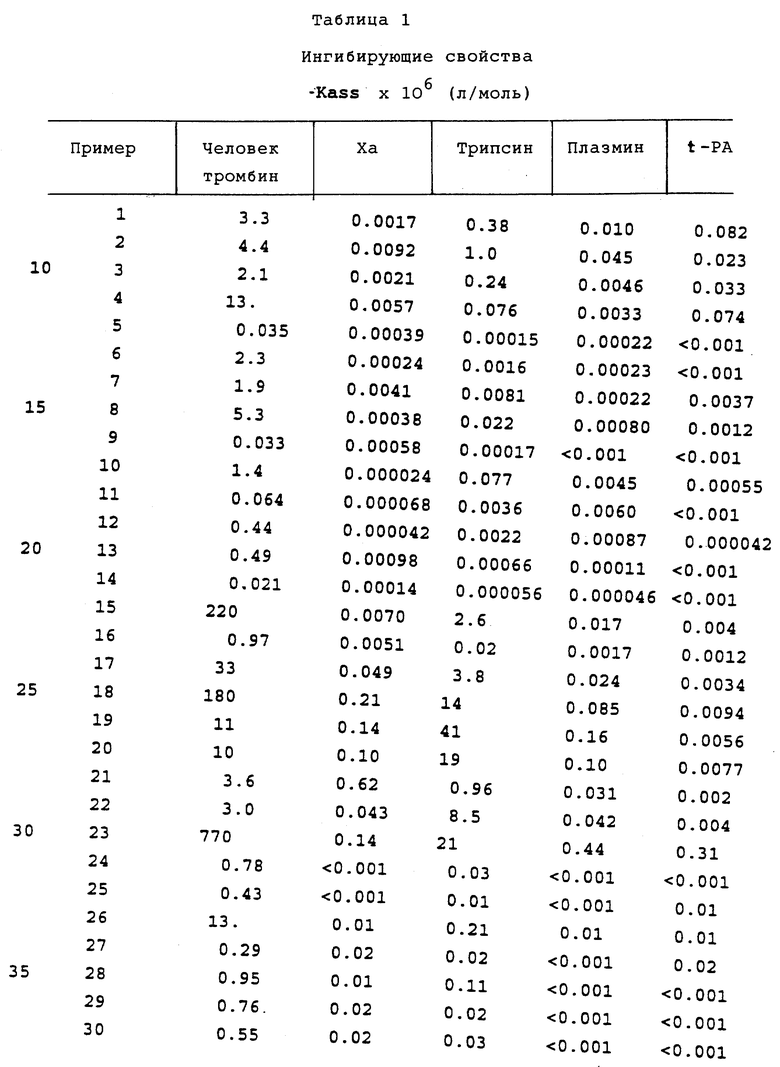


Пептидное производное общей формулы I
X - Y - NH - (CH2)r - G
или его фармацевтически приемлемая соль, или его сольват, где значения X, Y, r, G указаны в п.1 формулы изобретения, которое может найти применение при ингибировании тромбина у млекопитающего. Описывается также способ их получения, фармкомпозиция и способ ингибирования тромбина у млекопитающего. 4 с. и 2 з.п. ф-лы, 2 табл.
X - Y - NH - (CH2)r - G,
или его фармацевтически приемлемая соль, или его сольват,
где Х - D-гомопролинил,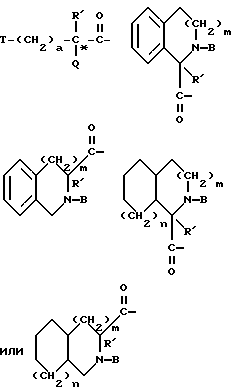
Т - С3 - С8циклоалкил, С1 - С8алкил;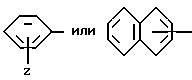
а = 0 или 1;
Q - NH-A;
А - водород, С1 - С4алкил, R''SO2-(где R'' - С1 - С4алкил), R''OC(O)-(где R'' - С1 - С4алкил), R''C(O)-(где R'' - 9- или 10-членная незамещенная или замещенная конденсированная бициклическая ароматическая гетероциклическая группа, имеющая один атом азота, которая может быть замещена С1 - С4алкилом) или -(СН2)g - СООН;
g = 1, 2 или 3;
В - водород;
R' - водород;
m = 1;
n = 1;
Z - водород;
Y обозначает
где Rg - С1 - С6алкил, С3 - С8циклоалкил или -(CH2)p-L-(CH2)q-T';
Pp - водород, С1 - С6алкил, С3 - С8циклоалкил или -(СН2)р-L-(CH2)q-T'; где р = 0, 1, 2, 3 или 4; L - связь, -0-, -S- или -NH-; q = 0, 1, 2 или 3 и T' - водород, С1 - С4алкил, С3 - С8циклоалкил или Ar, где Ar - фенил; Ry - -СН2- и Rz, взятый с Rу и тремя соседними атомами углерода, образует насыщенное карбоциклическое кольцо из 5 - 8 атомов;
r = 1 или 2;
G - -(CH2)s-R, где s = 0-5, -CH=CH-(CH2)t-R, где t = 0-3,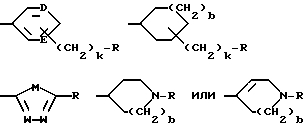
где D и E каждый - СН или один из D и Е - N, а другой - СН; k = 0 или 1; b = 1; М - S; каждый W - CH и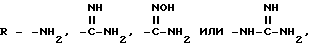
или его фармацевтически приемлемая соль, или фармацевтически приемлемый сольват указанного соединения, или его соль при условии, что А не является водородом, С1 - С4алкилом или третбутилоксикарбонилом, когда G - -(СН2)s-R или -СН= СН-(СН2)t-R, и R - -C(=NH)NH2 или -NH-C(=NH)NH2, Y - незамещенный пролинил (Rp - водород) и Т -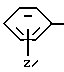
и при условии, что R не является амино или гуанидино, когда r = 1 и s = 0; и, кроме того, при условии, что А не обозначает водород, С1 - С4алкил, метилсульфонил или -(СН2)g-СООН, когда G - -(СН2)s-R, в котором R - -C(= NH)NH2 или -NH-C(=NH)NH2, Y обозначает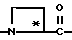 незамещенный пролинил (Rр - водород) или 4-гидроксипролинил (Rр - ОН), R' - водород, Т - циклогексил и Q - -NH-A; и, кроме того, при условии, что R''SO2- не является арилсульфонилом, когда G - -(CH2)s-R, в котором R - -NH-C(=NH)NH2, Y - незамещенный пролинил (Rр - водород) или 4-метилтиопролинил (Rр - SCH3) и Q - -NH-A; и при условии, что А не обозначает R''SO2-, когда G обозначает
незамещенный пролинил (Rр - водород) или 4-гидроксипролинил (Rр - ОН), R' - водород, Т - циклогексил и Q - -NH-A; и, кроме того, при условии, что R''SO2- не является арилсульфонилом, когда G - -(CH2)s-R, в котором R - -NH-C(=NH)NH2, Y - незамещенный пролинил (Rр - водород) или 4-метилтиопролинил (Rр - SCH3) и Q - -NH-A; и при условии, что А не обозначает R''SO2-, когда G обозначает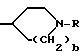
Т - С1 - С8алкил или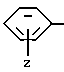
и Q - -NH-A; и, кроме того, из соединений формулы I исключены соединения, где Х - D-гомопролинил.

в которых Т - С3 - С8циклоалкил, С1 - С8алкил,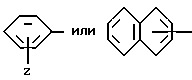
а = 0 или 1;
Q - -NH-A;
А - водород, С1 - С4алкил, R''SO2-(где R'' - С1 - С4алкил), R''OC(O)-(где R'' - С1 - С4алкил), R''C(O) - (где R'' - С1 - С4алкил) или -(СН2)g-СООН;
g = 1, 2 или 3;
В - водород;
R' - водород;
m = 1;
n = 1;
Z = водород;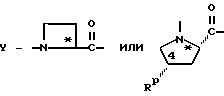
в которых Rр - водород;
r = 1 или 2;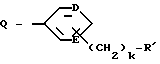
где D и Е каждый - СН, замещение является 1, 4-; k = 0 и R' - -C(=NH)NH2 или -NH2-C(=NH)-NH2 (т.е. G - 4-амидинофенил или 4-гуанидинофенил); или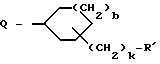
где k = 0 и R' - -C(=NH)NH2 или -NH2-C(=NH)-NH2, или k = 1 и R' - -C(= NH)NH2; или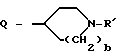
где R' - -C(=NH)NH2; и, кроме того, где алкил сам по себе или как часть другого заместителя является метилом, этилом, н-пропилом, изопропилом, н-бутилом, третбутилом, изобутилом или втор-бутилом;
С3 - С8циклоалкил является циклопропилом, метилциклопропилом, циклобутилом, циклопентилом, циклогексилом, 4-метилциклогексилом или циклооктилом;
9- или 10-членное гетероциклическое кольцо является индолилом, хинолинилом или изохинолинилом; и, кроме того, где любая гетероароматическая группа, перечисленная для определения R'' является независимо незамещенной или замещена одним заместителем, который приводит к устойчивой структуре, независимо выбранным из С1 - С4алкила.
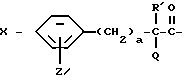
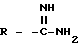
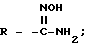
| ПРОИЗВОДНЫЕ ГЛИЦИНА ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ ТОРМОЗИТЬ СВЯЗЫВАНИЕ ФИБРИНОГЕНА У ФИБРИНОГЕННОГО РЕЦЕПТОРА ТРОМБОЦИТОВ | 1990 |
|
RU2024549C1 |
| Устройство для изготовления труб из термопластичного ленточного материала | 1979 |
|
SU887224A1 |
| US 4346078 A, 24.08.71982 | |||
| Способ получения полигидроурацилов | 1970 |
|
SU449079A1 |

