Изобретение относится к ингибитору серинпротеазы, имеющему ацилгуанидиновую боковую цепь, к содержащим его фармацевтическим композициям, а также к использованию указанного ингибитора для лечения и профилактики заболеваний, связанных с тромбином.
Серинпротеазы представляют собой ферменты, которые среди прочего играют важную роль в каскаде коагуляции крови. Членами этой группы являются, например, тромбин, трипсин, факторы VIIa, IXa, Xa, XIa, XIIa и протеин С.
Тромбин является серинпротеазой, которая регулирует последнюю стадию в каскаде коагуляции. Главной функцией тромбина является расщепление фибриногена для генерирования мономеров фибрина, которые образуют нерастворимый гель путем поперечного сшивания. Кроме того, тромбин регулирует свое собственное продуцирование, активируя факторы V и VIII на ранних стадиях каскада. Он также оказывает важные воздействия на клеточном уровне, где он воздействует на специфичные рецепторы, вызывая агрегацию тромбоцитов, активацию клеток эндотелия и пролиферацию фибробласта. Таким образом, тромбин играет центральную регуляторную роль в гемостазе и образовании тромбов. Поскольку ингибиторы тромбина могут иметь широкий круг терапевтических применений, в этой области проводились интенсивные исследования.
При разработке синтетических ингибиторов серинпротеазы, и более конкретно тромбина, возрос интерес к малым синтетическим пептидам, которые распознаются протеолитическими ферментами так же, как природные субстраты. В результате были получены новые пептидоподобные ингибиторы, такие как ингибиторы переходного состояния тромбина и низкомолекулярный ингибитор тромбина Иногатран (Inogatran) (Thromb. Haemostas. 1995, 73:1325 (Abs. 1633), WO 93/11152 (Example 67)), который, как описано, должен быть сильным и селективным ингибитором тромбина. Родственные соединения описаны в WO 95/23609, в отличие от Иногатрана и его гомологов соединения, описанные в этой заявке, имеют ароматическую группу в агматиноподобной группе.
Не ослабевает поиск более эффективных и более селективных ингибиторов серинпротеазы для того, чтобы получить ингибиторы, которые могут вводиться в меньших дозах и дают меньше побочных действий, и эти действия менее тяжелы.
Был найден новый класс очень сильных ингибиторов серинпротеазы, в особенности являющихся селективными ингибиторами тромбина или Ха, имеющих формулу I: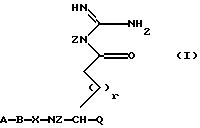
в которой А представляет Н, R1, R1-O-C(O)-, R1-SO2-, R2ООС-(CHR2)m- или N-защитную группу, где R1 выбран из групп (C1-12)алкил, (С3-8)циклоалкил и из (С6-14)арил, (C7-15)аралкил, арильные группы которых могут быть необязательно замещены (C1-6)алкокси или галогеном; каждая группа R2 независимо представляет Н или имеет те же значения, что и R1; m = 1, 2 или 3;
В представляет собой связь, D-1-Piq, или L- или D-аминокислоту, имеющую гидрофобную, основную боковую цепь, причем аминокислота может быть необязательно замещена N(C1-6)алкилом; или А и В вместе представляют остаток R3R4N-CHR5-C(O)-, в котором R3 и R4 независимо представляют R1 или N-защитную группу и R5 представляет гидрофобную или основную боковую цепь;
Х представляет L-аминокислоту с гидрофобной боковой цепью, представляющей (C1-12)алкил, необязательно замещенную одной или более (С6-14)арильными группами, или Х представляет циклическую аминокислоту, -NR2-CH2-C(O)- или фрагмент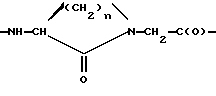
в котором n = 2, 3 или 4,
Q представляет Н или -C(O)Y, где Y представляет Н или 2-тиазол,
Z представляет Н,
r = 0 или 1, если Q представляет -C(O)Y, или
r = 0, 1, 2, 3 или 4, если Q представляет Н;
или их пролекарством, или их фармацевтически приемлемой солью.
Вещества по настоящему изобретению используются для лечения и профилактики опосредованных тромбином или связанных с тромбином заболеваний. Они включают ряд тромботических и протромботических состояний, в которых активирован коагуляционный каскад, включающих, но не ограничивающихся, глубокий тромбоз вен, легочную эмболию, тромбофлебит, закупорку артерий в результате тромбоза или эмболии, откупорку артерий во время или после пластической хирургии сосудов или тромболиза, рестеноз после повреждения артерий или инвазивных кардиологических процедур, послеоперационный венозный тромбоз или эмболию, острый или хронический атеросклероз, удар, инфаркт миокарда, рак и метастазы, и нейродегенеративные заболевания. Вещества по изобретению могут быть также использованы как антикоагулянты в экстракорпоральных потоках кровообращения, которые необходимы при диализе и хирургии. Вещества по изобретению могут быть также использованы как антикоагулянты in vitro.
Предпочтительные соединения по изобретению имеют формулу I, в которой Х представляет L-аминокислоту с гидрофобной боковой цепью или -NR2-CH2-C(O)-.
Другими предпочтительными соединениями формулы I являются те, у которых Х представляет циклическую аминокислоту или Х представляет -NR2-CH2-C(О)- или фрагмент
Особенно предпочтительными соединениями являются соединения, в которых А представляет R2OOC-(CHR2)m-, В представляет D-аминокислоту, имеющую гидрофобную боковую цепь, и Х представляет азетидинкарбоновую кислоту, пролин, пипеколиновую кислоту, 3,4-дегидропролин, 2-октагидроиндолкарбоновую кислоту.
Еще более предпочтительными соединениями являются соединения, в которых А представляет НООС-СН2- и В представляет D-Phe, D-Cha, D-Coa, D-Dpa, p-Cl-D-Phe, p-OMethyl-D-Phe, p-OEthyl-D-Phe, D-Nle, m-Cl-D-Phe, 3,4-ди-OMe-D-Phe или D-Chg.
Когда А представляет R1-SO2-, предпочтительно В представляет связь, D-1-Piq, или D-аминокислоту, имеющую гидрофобную боковую связь, и Х представляет 2-азетидинкарбоновую кислоту, пролин, пипеколиновую кислоту, 3,4-дегидропролин, 2-октагидроиндолкарбоновую кислоту или фрагмент
Более предпочтительными являются соединения, в которых А представляет этил-SO2- или бензил-SO2-, В представляет связь, D-Phe, D-Cha, D-Coa, D-Dpa, p-Cl-D-Phe, p-OMethyl-D-Phe, p-OEthyl-D-Phe, D-NIe, м-Cl-D-Phe, 3,4-ди-ОМе-D-Рhе, D-Chg.
Наиболее предпочтительными являются соединения формулы I, в которых Q представляет Н и r = 0, 1 или 2.
N-защитная группа, как она определена при определении группы А, является любой N-защитной группой, применяемой в пептидах. Подходящие N-защитные группы можно найти в Т.W. Green and P.G.M. Wuts: Protective Groops in Organic Synthesis, Second Edition (Wiley, NY, 1991) и в The Peptides, Analysis, Synthesis, Biology, Vol. 3, E. Gross and J. Meienhofer, Eds. (Academic Press, New York, 1981).
Термин "необязательно замещенный D,L-α-гидроксиацетил" означает группу формулы HO-CRaRb-C(O)-, в которой Ra и Rb независимо представляют Н, гидрофобную боковую цепь или Ra и Rb вместе образуют 5- или 6-членное кольцо, которое необязательно конденсировано с одним или двумя алифатическими или ароматическими 6-членными кольцами, причем 5- или 6-членное кольцо состоит из атомов углерода и необязательно одного гетероатома, выбранного из N, О и S. Предпочтительными D,L-α-гидроксиацетильными группами являются 2-гидрокси-3-циклогексилпропионил- и 9-гидроксифтор-9-карбоксил.
Термин (С1-12)алкил означает разветвленную или неразветвленную алкильную группу, имеющую от 1 до 12 атомов углерода, такую как метил, этил, трет-бутил, изопентил, гептил, додецил и т.п. Предпочтительными алкильными группами являются (С1-6)алкил группы, имеющие 1-6 атомов углерода. Более предпочтительными в определении R6, R7 и R6 являются (С1-3)алкил группы, имеющие 1-3 атома углерода, такие как метил, этил, изопропил. Группа (C1-12)алкенил представляет разветвленную или неразветвленную ненасыщенную углеводородную группу, имеющую от 2 до 12 атомов углерода. Предпочтительными являются (С2-6)алкенил группы. Примерами являются этенил, пропенил, аллил и т.п.
Термин (C1-6)алкилен означает разветвленную или неразветвленную алкиленовую группу, имеющую от 1 до 6 атомов углерода, такую как -(СН2)m, где m = 1 - 6, -СН(СН3)-, -СН(СН3)-(СН2)- и т.д. Предпочтительными алкиленовыми группами в определении Y являются этилен и метилен.
Группа (C2-12)алкинил представляет разветвленную или неразветвленную углеводородную группу, содержащую тройную связь и имеющую от 2 до 12 атомов углерода. Предпочтительными являются (С2-6)алкинил группы, такие как этинил и пропинил.
Группа (С6-14)арил представляет ароматическую группу из 6-14 атомов углерода. Арильная группа может, кроме того, содержать один или несколько гетероатомов, таких как N, S или О. Примерами арильных групп являются фенил, нафтил, изохинолил, инданил и т.п. Наиболее предпочтительной является фенильная группа.
Группы (С7-15)аралкил и (C8-16)аралкенил являются соответственно алкильными и алкенильными группами, замещенными одной или несколькими арильными группами, с общим числом атомов углерода соответственно от 7 до 15 или от 8 до 16.
Термин (C1-6)алкокси означает алкокси группу, имеющую 1-6 атомов углерода, алкильная часть которой может иметь значения, определенные выше.
Термин (С3-8)циклоалкил означает циклоалкильную группу, имеющую 3-8 атомов углерода, а именно циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил. Предпочтительными циклоалкильными группами являются циклопентил и циклогексил.
Термин "галоген" означает фтор, хлор, бром или йод.
Термин "эфирные производные" означает любое подходящее эфирное производное, предпочтительно (C1-4)алкиловые эфиры, такие как метиловый, этиловый или трет-бутиловый эфиры.
Термины 1- и 3-Tiq обозначают соответственно 1,2,3,4-тетрагидроизохинолин-1- или -3-карбоновую кислоту; 1- и 3-Piq представляют соответственно 1- и 3-карбоксипергидроизохинолин; Atc представляет 2-аминотетралин-2-карбоновую кислоту; Aic представляет аминоинданкарбоновую кислоту; Phe представляет фенилаланин; Cha представляет циклогексилаланин; Dpa представляет дифенилаланин; Соа представляет циклооктилаланин; Chg представляет циклогексилглицин; Nle представляет норлейцин.
Термин "гидрофобная боковая цепь" означает (C1-12)алкил, необязательно замещенный одним или несколькими (С3-8)циклоалкил группами или (С6-14)арил группами (которые могут содержать гетероатом, например азот), такими как циклогексил, циклооктил, фенил, пиридинил, нафтил, тетрагидронафтил и т.п., причем гидрофобная боковая цепь может необязательно быть замещена заместителями, такими как галоген, трифторметил, низший алкил (например, метил или этил), низший алкокси (например, метокси), фенилокси, бензилокси и т.п.
Термин "замещенный" означает замещенный одним или несколькими заместителями.
Аминокислоты, имеющие основную боковую цепь, представляют, например, аргинин и лизин, предпочтительно аргинин, но не ограничены этим. Термин аминокислоты, имеющие нейтральную боковую цепь, относится к таким аминокислотам, как метионинсульфон и т.п.
Циклические аминокислоты представляют, например, 2-азетидинкарбоновую кислоту, пролин, пипеколиновую кислоту, 1-амино-1-карбокси-(С3-8)циклоалкан (предпочтительно С4, С5 или С6), 4-пиперидинкарбоновую кислоту, 4-тиазолидинкарбоновую кислоту, 3,4-дегидропролин, азапролин, 2-октагидроиндолкарбоновую кислоту и т.п. Предпочтительными являются 2-азетидинкарбоновая кислота, пролин, пипеколиновая кислота, 4-тиазолидинкарбоновая кислота, 3,4-дегидропролин и 2-октагидроиндолкарбоновая кислота.
Термин "пролекарство" означает соединение, в котором амидиновая группа соединения формулы I защищена, например, гидрокси или (C1-6)алкоксикарбонильной группой.
Изобретение, кроме того, включает способ получения соединения формулы I, включающий сочетание подходящих защищенных аминокислот или аналогов аминокислот с последующим удалением защитных групп.
Соединения по формуле I могут быть получены обычным для таких соединений способом.
Для этой цели подходящие производные аминокислот или пептиды с защищенным N в α-положении (и с защищенной боковой цепью, если присутствует реакционно-способная боковая цепь) активируют и вводят в реакцию сочетания с подходящими производными аминокислот или пептидами с защищенным карбоксилом либо в растворе, либо на твердом носителе. Защиту α-аминогрупп обычно осуществляют уретановыми группами, такими как лабильные под действием кислот трет-бутилоксикарбонильная (Воc) группа, бензилоксикарбонильная (Z) группа и их замещенные аналоги или лабильная под действием оснований 9-фторенилметилоксикарбонильная (Fmoc) группа. Z группа может быть также удалена каталитическим гидрирированием. Другие подходящие аминозащитные группы включают Nps, Bmv, Врос, Msc и др. Хороший обзор аминозащитных групп дан в The Peptides, Analysis, Synthesis, Biology, Vol. 3, E. Gross and J. Meienhofer, Eds. (Academic Press, New York, 1981). Защиту карбоксильных групп осуществляют путем образования сложных эфиров, например эфиров, лабильных под действием оснований, подобных метиловым или этиловым эфирам, или эфиров, лабильных под действием кислот, подобных трет-бутиловым эфирам, или эфиров, лабильных при гидрогенолизе, подобных бензиловым эфирам. Защита группы боковой цепи ацилгуанидина может быть осуществлена при использовании вышеупомянутых групп.
Активация карбоксильной группы подходящих защищенных аминокислот или пептидов может осуществляться азидом, смешанным ангидридом, активным эфиром или карбодиимидным методом, особенно с добавлением каталитических и подавляющих рацемизацию соединений, таких как 1-гидроксибензотриазол, N-гидроксисукцинимид, 3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазин, N-гидрокси-5-норборнен-2,3-дикарбоксимид, что описано в The Peptides, Analysis, Synthesis, Biology (см. выше) и Pure and Applied Chem. 59(3), 331-344 (1987). Сочетание ацилгуанидиновой группы с С-концом подходящих N-α-защищенных производных аминокислот или пептидов может быть осуществлено вышеописанными методами сочетания.
В подходящем способе для получения соединений согласно изобретению, где Q представляет -C(О)Y, защищенную глютаминовую кислоту (Glu) сочетают с группой Y, снимают защиту, после чего сочетают N-конец с А-В-Х-частью молекулы для получения соединения формулы A-B-X-Glu-Y. По отдельной процедуре S-метилизомочевину защищают и превращают в защищенный гуанидин. Защищенный гуанидин сочетают с А-В-Х-Glu-Y и после снятия защиты получают соединение формулы I.
Соединения по изобретению, которые могут быть в форме свободного основания, могут быть выделены из реакционной смеси в форме фармацевтически приемлемой соли. Фармацевтически приемлемые соли могут быть также получены путем реакции свободного основания формулы I с органической или неорганической кислотой, такой как хлористый водород, бромистый водород, йодистый водород, серная кислота, фосфорная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, метансульфоновая кислота, фумаровая кислота, янтарная кислота, щавелевая кислота, лимонная кислота, бензойная кислота и аскорбиновая кислота.
Соединения согласно настоящему изобретению могут иметь один или несколько хиральных атомов углерода и могут поэтому быть получены в виде чистого энантиомера или в виде смеси энантиомеров, или в виде смеси, содержащей диастереомеры. Способы получения чистых энантиомеров хорошо известны, например кристаллизация солей, полученных из оптически активных кислот и рацемической смеси, или хроматография с использованием хиральных колонок. Для диастереомеров могут быть использованы колонки с прямой фазой или с обращенной фазой.
Соединения согласно изобретению могут вводиться энтерально или парентерально и для человека предпочтительно в суточной дозе 0,001-100 мг на кг веса тела, предпочтительно 0,01-10 мг на кг веса тела. Эти соединения в смеси с фармацевтически приемлемыми вспомогательными веществами, описанными, например, в стандартной ссылке Gennaro et al. Remington's Pharmaceutical Sciences (18th ed., Mack Publishing Company, 1990, см. в особенности Part 8: Pharmaceutical Preparations and Their Manufacture), могут быть спрессованы в твердые стандартные дозы, такие как пилюли, таблетки, или могут быть превращены в капсулы или свечи. Посредством фармацевтически пригодных жидкостей соединения могут быть также применены в форме раствора, суспензии, эмульсии, например, для использования как препарат для инъекций или как спрей, например, для использования в виде носового спрея.
Для изготовления дозированных единиц, например таблеток, предполагается использовать обычные добавки, такие как наполнители, красители, полимерные связующие и тому подобное. В общем, может быть использована любая фармацевтически приемлемая добавка, которая не нарушает функцию активного соединения.
Подходящие носители, с которыми могут вводиться композиции, включают лактозу, крахмал, производные целлюлозы и т.п. или их смеси, используемые в подходящих количествах.
Изобретение дополнительно поясняется нижеследующими примерами.
Термин "Glu (гуанидин)" означает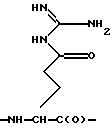
Bzl - бензил; Вос - трет-бутилоксикарбонил; Cbz - бензилоксикарбонил; Ас - ацетил; РАс - фенилацетил; Glu - глютаминовая кислота; Cha - циклогексилаланин; Pro - пролин; Phe - фенилаланин; 1-Piq - 1-карбоксипергидроизохинолин; Nal - 2-нафтилаланин; Asp - аспарагиновая кислота.
Времена удерживания (Rt ЖХ), если не указано другое, приведены для жидкостной хроматографии с обращенной фазой на колонке Supelcosil LC-18-DB (2,1 мм • 25 см).
ПРИМЕР 1
HOOC-CH2-D-Cha-(N-циклопентил)-Gly-β-Ala-гуанидин
(а). N-трет-бутилоксикарбонил-S-метилизотиомочевина
Полусульфат S-метилизотиомочевины (10 г) суспендировали в дихлорметане (100 мл). К суспензии добавляли при перемешивании 4 н. раствор гидроокиси натрия (10 мл). Реакционную смесь помещали в ледяную баню, добавляли по каплям дитрет-бутилдикарбонат (15,7 г) в дихлорметане (100 мл) вместе с 2 н. раствором гидроокиси натрия для поддержания рН около 11. После того, как добавление завершилось, реакционную смесь оставляли при перемешивании на ночь при комнатной температуре. Отделяли дихлорметановый слой и водный слой экстрагировали дихлорметаном. Объединенные органические слои промывали водой, сушили над сульфатом натрия, фильтровали и выпаривали под вакуумом. Остаток очищали хроматографией на силикагеле (элюент: гептан/этилацетат 4/1 об./об. %), получая N-трет-бутилоксикарбонил-S-метилизотиомочевину (9,38 г).
ТСХ: Rf=0,75, силикагель, этилацетат/гептан 3/1 об./об.%.
(b). N-бензилоксикарбонил-N'-трет-бутилоксикарбонил-S-метилизотиомочевина
N-трет-бутилоксикарбонил-S-метилизотиомочевину (2 г) растворяли в дихлорметане (20 мл). К раствору при перемешивании добавляли 4 н. раствор гидроокиси натрия (2 мл). Реакционную смесь помещали в ледяную баню; N-бензилоксикарбонилоксисукцинимид (2,62 г) в дихлорметане (20 мл) добавляли по каплям вместе с 2 н. раствором гидроокиси натрия для поддержания рН около 11. После того, как добавление завершилось, реакционную смесь оставляли при перемешивании на ночь при комнатной температуре. Отделяли дихлорметановый слой и водный слой дважды промывали дихлорметаном. Объединенные органические слои промывали водой и сушили над сульфатом натрия, фильтровали и выпаривали под вакуумом. Остаток очищали хроматографией на силикагеле (элюент: гептан/этилацетат 3/2 об. /об. %), получая N-бензилоксикарбонил-N'-трет-бутилоксикарбонил-S-метилизотиомочевину (3,15 г).
ТСХ: Rf=0,78, силикагель, этилацетат/гептан 1/1 об./об.%.
(с). N-Cbz-N1-(трет-Вос)-гуанидин
N-бензилоксикарбонил-N1-трет-бутилоксикарбонил-S-метилизотиомочевину (3,15 г) растворяли в метанольном растворе аммиака (2,4 М, 50 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь упаривали под вакуумом и полученный остаток очищали хроматографией на силикагеле (элюент: дихлорметан/метанол 95/5 об. /об. %), получая N-Cbz-N'-(трет-Вос)-гуанидин (1,83 г).
ТСХ: Rf= 0,80, силикагель, дихлорметан/метанол 9/1 об./об.%.
(d). N-(трет-Вос)-гуанидин гидрохлорид
10% палладий на древесном угле (250 мг) и 3,12 мл 2 н. раствора хлористого водорода добавляли к раствору N-Cbz-N'-(трет-Вос)-гуанидина (1,8 г) в метаноле (50 мл). Смесь гидрировали при атмосферном давлении при комнатной температуре в течение 1 ч. Палладиевый катализатор удаляли фильтрацией и растворитель удаляли выпариванием при пониженном давлении, получая количественно N-(трет-Вос)-гуанидин гидрохлорид.
ТСХ: Rf=0,10, силикагель, дихлорметан/метанол 9/1 об./об.%.
(e). Cbz-β-Ala-(N-трет-Вос)-гуанидин
Cbz-β-Ala-OH (171 мг) растворяли в сухом диметилформамиде (5 мл). После добавления триэтиламина (212 мкл) реакционную смесь помещали под азот и охлаждали до -15oС. Затем добавляли изобутилхлороформат (99 мкл) и смесь перемешивали в течение 15 мин при -15oС. К холодной смеси добавляли N-(трет-Вос)-гуанидин гидрохлорид (150 мг) и триэтиламин (106 мкл). Реакционную смесь перемешивали в течение 1 ч при -15oС и затем выдерживали при комнатной температуре в течение 45 мин. Триэтиламин гидрохлорид отфильтровывали и фильтрат выпаривали до сухого остатка. Остаток очищали хроматографией на силикагеле (элюент: дихлорметан/метанол 95/5 об./об.%), получая Cbz-p-Ala-(N-трет-Вос)-гуанидин (256 мг).
ТСХ: Rf=0,50, силикагель, дихлорметан/метанол 9/1 об./об.%.
(f). H-β-Ala-(N-трет-Вос)-гуанидин гидрохлорид.
10% палладий на древесном угле (100 мг) и 300 мкл 4 М раствора хлористого водорода добавляли к раствору Cbz-β-Ala-(N-трет-Вос)-гуанидина (220 мг) в диметилформамиде (5 мл). Смесь гидрировали при атмосферном давлением при комнатной температуре в течение 1 ч. Палладиевый катализатор удаляли фильтрацией и растворитель удаляли выпариванием при пониженном давлении, получая количественно H-β-Ala-(N-трет-Вос)-гуанидин гидрохлорид.
ТСХ: Rf=0,10, силикагель, дихлорметан/метанол 9/1 об./об.%.
(g). N-цикпопентил-Glу-ОМе
Циклопентанон (15,6 г) добавляли к раствору H-Gly-OMe•HCI (23,2 г) в 200 мл метанола. Смесь перемешивали в течение 15 минут и добавляли цианборогидрид натрия (7 г). Величину рН доводили до 6. Реакционную смесь перемешивали в течение 16 час при комнатной температуре. Для завершения реакции добавляли циклопентанон (1 г) и продолжали перемешивание. Протекание реакции контролировали по ТСХ. Когда весь исходный материал исчезал, смесь подкисляли до рН 2 и перемешивали в течение 30 минут. Растворитель удаляли и остаток разбавляли водой. Раствор промывали эфиром, рН доводили до 12 6 н. гидроокисью натрия и экстрагировали дихлорметаном. Объединенные органические слои промывали насыщенным раствором хлористого натрия, сушили над сульфатом натрия и выпаривали под вакуумом, получая 16 г масла.
Rf=0,46 в смеси этилацетат/пиридин/уксусная кислота/вода 63/20/6/11 (об. %) на окиси кремния.
(h). N-(трет-бутилоксикарбонилметил)-D-Cha-OMe
Трет-бутилбромацетат (17 г) добавляли при перемешивании к раствору H-D-Cha-OMe•HCl (26 г) в 300 мл ацетонитрила. Величину рН смеси доводили до 8,5 диизопропилэтиламином. Смесь перемешивали в течение 16 часов при комнатной температуре и выпаривали под вакуумом. Остаток растворяли в дихлорметане и раствор промывали водой, сушили над сульфатом натрия и выпаривали под вакуумом. Хроматография на силикагеле в смеси гексан/этилацетат 9/1 (об./об.%) дала 20 г N-(трет-бутилоксикарбонилметил)-D-Cha-OMe.
Rf=0,46 в смеси этилацетат/пиридин/уксусная кислота/вода 15,75/5/1,5/2,75 (об.%) на окиси кремния.
(i). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OMe
Величину рН раствора N-(трет-бутилоксикарбонилметил)-D-Cha-OMe (20 г) и дитрет-бутилдикарбоната (17 г) доводили до 8,5 диизопропилэтиламином. Смесь перемешивали в течение 16 часов при комнатной температуре. Растворитель удаляли под вакуумом. К остатку добавляли дихлорметан и воду. Органический слой отделяли, промывали холодным 1 н. хлористым водородом, водой, 5% гидрокарбонатом натрия и водой. Органический слой сушили над сульфатом натрия и фильтрат упаривали до аморфного твердого N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OMe с выходом 28 г.
Rf= 0,60 в смеси этилацетат/пиридин/уксусная кислота/вода 252/20/6/11 (об.%) на окиси кремния.
(j). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OH
Раствор N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OMe (28 г) в 420 мл смеси диоксан: вода 9/1 (об.%) обрабатывали избытком 1 н. гидроокиси натрия для поддержания рН на уровне 13 в течение 90 минут при комнатной температуре. После подкисления смесь выливали в воду и экстрагировали дихлорметаном. Органический слой промывали водой и сушили над сульфатом натрия. Фильтрат упаривали, получая 24 г N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OH.
Rf=0,23 в смеси дихлорметан/метанол 9/1 (об.%) на окиси кремния.
(k). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-(N-циклопентил)-Gly-OMe
N-циклопентил-Gly-OMe (10,2 г) и 2-1Н-бензотриазол-1-ил-1,1,3,3-тетраметилуронийтетрафторборат (TBTU, 21,2 г) добавляли к раствору N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OH (24 г) в 300 мл N, N-диметилформамида. Величину рН доводили до 8,5. Смесь оставляли на ночь при перемешивании при комнатной температуре и концентрировали выпариванием. К остатку добавляли воду и этилацетат. Органический слой отделяли и промывали 1 н. хлористым водородом, водой, 5% гидрокарбонатом натрия и водой и сушили над сульфатом натрия. Фильтрат выпаривали и остаток хроматографировали на силикагеле в смеси гексан/этилацетат 8/2 (об./об.%) в качестве элюента. Фракции, содержащие N-трет-бутилоксикарбонилметил-N-Вос-D-Сhа-N-циклопентил-Сlу-ОМе, разливали тонким слоем и выпаривали. Выход 17 г.
Rf=0,57 в смеси гексан/этилацетат (об.%) на окиси кремния.
(l). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-(N-циклопентил)-Gly-OH
N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-(N-циклопентил)-Gly-OMe (17 г) омыляли в смеси диоксан/вода 1/1 (об.%, 150 мл) и разбавляли гидроокисью натрия, получая 15 г аморфного твердого вещества. Хроматография на силикагеле со смесью дихлорметан/метанол 95/5 (об.%) в качестве элюента дала 13 г N-трет-бутилоксикарбонилметил-N-Вос-О-Сhа-N-циклопентил-Glу-ОН.
Rf=0,30 в смеси дихлорметан/метанол 9/1 (об.%) на окиси кремния.
(m). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-(N-циклопентил)-Gly-β-Ala-(N-трет-трет-Вос)-гуанидин
N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-(N-циклопентил)-Gly-OH (310 мг) растворяли в сухом диметилформамиде (10 мл). После добавления триэтиламина (168 мкл) реакционную смесь помещали под азот и охлаждали до -15oС. Затем добавляли изобутилхлороформат (79 мкл) и смесь перемешивали в течение 15 мин при -15oС. H-β-Ala-(N-трет-Вос)-гуанидин гидрохлорид (154 мг) растворяли в сухом диметилформамиде и добавляли по каплям к охлажденному смешанному раствору ангидрида, поддерживая рН на уровне 8,5 добавлением триэтиламина. Реакционную смесь перемешивали 1 час при -15oС и затем 1 час при 0oС. Реакционную смесь упаривали до сухого остатка. Остаток растворяли в этилацетате и затем промывали водой, рассолом, сушили над сульфатом натрия и концентрировали под вакуумом. Остаток очищали хроматографией на окиси кремния (элюент: дихлорметан/метанол 95/5 об. %), получая N-(трет-бутилоксикарбонилметил) -N-Boc-D-Cha-(N-циклопентил)-Gly-β-Ala-(N-трет-Вос)-гуанидин (268 мг).
ТСХ: Rf=0,80, силикагель, дихлорметан/метанол 9/1 (об.%).
(n). HOOC-CH2-D-Cha-(N-циклопентил)-Gly-β-Ala-гуанидин
N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-(N-циклопентил)-Gly-β-Ala-(N-трет-Вос)-гуанидин (265 мг) обрабатывали 90% смесью трифторуксусная кислота/вода (10 мл) в течение 2 ч при комнатной температуре. Реакционную смесь концентрировали под вакуумом, остаток растворяли в воде и напрямую подвергали препаративной ЖХВР в колонке Supelcosil LC-18-DB, используя градиентную элюирующую систему от 20% А/80% В до 20% А/20% В/60% С за 40 мин при скорости потока 15 мл/мин (А: 0,5 М фосфатный буфер, рН 2,1; В: вода; С: ацетонитрил/вода 6/4). Выход: 185 мг HOOC-CH2-D-Cha-(N-циклопентил)-Gly-β-Ala-гуанидина.
Rt (ЖХ): 32,22 мин, от 20% А/80% В до 20% А/20% B/60% С за 40 мин.
ПРИМЕР 2
HOOC-CH2-D-Cha-Pro-Glu (гуанидин)-2-тиазолил
(a). H-D-Cha-OMe•HCl
К охлажденному (-20oС) и сухому метанолу (195 мл) добавляли по каплям тионилхлорид (28 мл). Добавляли H-D-ChaOH•HCl и реакционную смесь нагревали при кипячении с обратным холодильником в течение 5 часов. Смесь концентрировали под вакуумом и переиспаряли с метанолом (3 раза). Остаток кристаллизовали из смеси метанол/диэтиловый эфир, получая Н-D-Cha-OMe•HCl в виде кристаллического порошка (40,9 г).
ТСХ: Rf= 0,66, силикагель, н-бутанолуксусная кислота/вода 10/1/3 (об.%).
(b). N-(трет-бутилоксикарбонилметил)-D-Cha-OMe
Трет-бутилбромацетат (36 г) добавляли при перемешивании к раствору H-D-Cha-OMe•HCl (40,9 г) в 400 мл ацетонитрила. Величину рН смеси доводили до 8,5 диизопропилэтиламином. Смесь перемешивали в течение 16 часов при комнатной температуре и выпаривали под вакуумом. Остаток растворяли в дихлорметане и раствор промывали водой, сушили над сульфатом натрия и выпаривали под вакуумом. Хроматография на силикагеле в смеси гептан/этилацетат 9/1 (об. %) давала 64 г N-(трет-бутилоксикарбонилметил)-D-Cha-OMe.
ТСХ: Rf= 0,25, силикагель, этилацетат/гептан 1/1 (об.%).
(c). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OMe
Величину рН раствора N-(трет-бутилоксикарбонилметил)-D-Cha-OMe (64 г) и дитрет-бутилдикарбоната (40,3 г) доводили до 8,5 диизопропилэтиламином. Смесь перемешивали в течение 16 часов при комнатной температуре. Растворитель удаляли под вакуумом. К остатку добавляли дихлорметан и воду. Органический слой отделяли, промывали холодным 1 н. хлористым водородом, водой, 5% гидрокарбонатом натрия и водой. Органический слой сушили над сульфатом натрия и выпаривали фильтрат, получая аморфный твердый N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OMe с выходом 59,6 г.
ТСХ: Rf=0,50, силикагель, этилацетат/гептан 1/1 (об.%).
(d). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OH
Раствор N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OMe (59,6 г) в 900 мл смеси диоксан/вода 9/1 (об.%) обрабатывали избытком 6 н. гидроокиси натрия, чтобы поддерживать рН 12, в течение 6 часов при комнатной температуре. После подкисления смесь выливали в воду и экстрагировали дихлорметаном. Органический слой промывали водой и сушили над сульфатом натрия. Фильтрат упаривали, получая 54 г N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OH.
ТСХ: Rf=0,60, силикагель, дихлорметан/метанол 9/1 (об.%).
(e). N-трет-бутилоксикарбонилметил)-N-Boc-D-Cha-Pro-Obzl
К холодному (0oС) раствору N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-OH (13,5 г) в N,N-диметилформамиде (150 мл) добавляли последовательно 1-гидроксибензотриазол (7,09 г), дициклогексилкарбодиимид (7,61 г), H-Pro-OBzl•HCI (9,31 г) и триэтиламин (6 мл). Смесь перемешивали при 0oС в течение 1 часа и затем оставляли на ночь при комнатной температуре. Смесь охлаждали до -20oС и дициклогексамочевину удаляли фильтрацией. Фильтрат упаривали до сухого остатка. Остаток растворяли в этилацетате и промывали последовательно 5% гидрокарбонатом натрия, водой, 3% лимонной кислотой и рассолом, сушили над сульфатом натрия и концентрировали под вакуумом. Остаток хроматографировали на силикагеле в смеси гептан/этилацетат 3/1 (об.%) в качестве элюента. Фракции, содержащие N-трет-бутилоксикарбонилметил-N-Вос-D-Сhа-Рrо-OBzl разливали тонким слоем и выпаривали. Выход 15 г.
ТСХ: Rf= 0,70, силикагель, этилацетат/гептан 1/1 (об.%).
(f). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-Pro-OH
10% палладий на древесном угле (750 мг) добавляли к раствору N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-Pro-OBzl (15 г) в метаноле (150 мл). Смесь гидрировали при атмосферном давлением при комнатной температуре в течение 1 ч. Палладиевый катализатор удаляли фильтрацией и растворитель удаляли выпариванием при пониженном давлении, получая 11,2 г N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-Pro-OH.
ТСХ: Rf= 0,65, силикагель, этилацетат/пиридин/уксусная кислота/вода 213/20/61/11 (об.%).
(g). Boc-Glu(О-трет-Bu)-N-МеОМе
К раствору Boc-Glu(O-трет-Вu)-ОН (15 г) в дихлорметане (150 мл) добавляли N, O-диметилгидроксиламин гидрохлорид (5,3 г) и [2-1Н-бензотриазол-1-ил-1,1,3,3-тетраметилуронийтетрафторборат] (15,87 г) и рН доводили до 8-8,5 добавлением триэтиламина. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Смесь последовательно промывали холодным 0,3 М раствором соляной кислоты, водой, 5% водным раствором гидрокарбоната натрия и водой. Органический слой сушили над сульфатом натрия, фильтровали и выпаривали. Остаток очищали хроматографией на силикагеле (элюент: дихлорметан/метанол 98/2 об.%), получая Boc-Glu(O-трет-Bu)-N-MeOMe (17,5 г).
ТСХ: Rf=0,85, силикагель, дихлорметан/метанол 9/1 (об.%).
(h). Boc-Glu(O-трет-Вu)-2-тиазолил
К холодному (-78oС) раствору н-бутиллития (88,9 ммоль) в диэтиловом эфире (90,7 мл) при перемешивании добавляли по каплям раствор 2-бромтиазола (14,6 г) в диэтиловом эфире (75 мл). После перемешивания раствора при -78oС в течение 30 мин раствор добавляли по каплям к раствору Boc-Glu(O-трет-Вu)-N-МеОМе (14 г) в сухом тетрагидрофуране (150 мл). Смесь перемешивали при -78oС в течение 1 ч, затем смесь выливали в охлажденный льдом 5% водный раствор лимонной кислоты. Смеси давали нагреться до комнатной температуры, после чего слои разделяли. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали водой, насыщенным водным раствором хлористого натрия, сушили над сульфатом натрия, фильтровали и выпаривали. Остаток очищали хроматографией на окиси кремния (элюент: дихлорметан/этилацетат 9/1 об.%), получая Вос-Сlu(O-трет-Вu)-2-тиазолил (6,83 г).
ТСХ: Rf= 0,92, силикагель, дихлорметан/этилацетат 7/3 (об.%).
(i). H-Glu-(2-тиазолил)•TFA
Boc-Glu(O-трет-Вu)-2-тиазолил (450 мг) растворяли в 3 мл трифторуксусной кислоты (TFA), 1 мл дихлорметана и 150 мкл анизола и перемешивали в течение 1 ч при комнатной температуре. После удаления растворителя выпариванием сырой амин выделяли в виде желтого масла с количественным выходом и использовали непосредственно для получения N-Boc-N-(трет-бутилоксикарбонилметил)-D-Cha-Pro-Glu-(2-тиазолила).
ТСХ: Rf= 0,10, силикагель, этилацетат/пиридин/уксусная кислота/вода 63/20/6/11 об.%.
(j). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-Pro-Glu-(2-тиазолил)
N-Boc-N-(трет-бутилоксикарбонилметил)-D-Cha-Pro-OH (590 мл) растворяли в сухом диметилформамиде (15 мл). После добавления диизопропилэтиламина (416 мкл) реакционную смесь помещали под азот и охлаждали до -15oС. Постепенно добавляли изобутилхлороформат (158 мкл) и смесь выдерживали при перемешивании при -15oС в течение 15 мин. H-Glu-(2-тиазолил)•ТFА (300 мг) растворяли в сухом диметилформамиде (10 мл) и добавляли по каплям к холодному раствору смешанного ангидрида, поддерживая рН на уровне 8,5 добавлением диизопропилэтиламина. Реакционную смесь перемешивали в течение 30 мин при -15oС. Реакционную смесь выпаривали до сухого остатка. Остаток растворяли в этилацетате и последовательно промывали водой, 5% водным раствором гидрокарбоната натрия, водой, рассолом, сушили над сульфатом натрия и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (элюент: дихлорметан/метанол 95/5 об.%), получая N-Boc-N-трет-бутилоксикарбонилметил-D-Cha-Pro-Glu-(2-тиазолил) (171 мг).
ТСХ: Rf=0,92, силикагель, дихлорметан/этилацетат 9/1 (об.%).
(k). N-(трет-бутилоксикарбонил)-S-метилизотиомочевина
Полусульфат S-метилизотиомочевины (10 г) суспендировали в дихлорметане (100 мл). К суспензии при перемешивании добавляли 4 н. раствор NaOH. Реакционную смесь помещали в баню со льдом и по каплям добавляли ди-трет-бутилдикарбонат (15,7 г) вместе с 2 н. раствором гидроокиси натрия для поддержания рН около 11. После завершения реакции реакционную смесь перемешивали в течение ночи при комнатной температуре. Дихлорметановый слой отделяли и водный слой экстрагировали дважды дихлорметаном. Объединенные органические слои промывали водой и сушили над сульфатом натрия, фильтровали и выпаривали под вакуумом. Остаток очищали хроматографией на окиси кремния (элюент: гептан/этилацетат 4/1 oб.%), получая выход 9,38 г.
ТХС: Rf=0,75, силикагель, этилацетат/гептан 3/1 (об.%).
(l) N-бензилоксикарбонил-N'-трет-бутилоксикарбонил-S-метилизомочевина
N-трет-бутилоксикарбонил-S-метилизотиомочевину (2 г) растворяли в дихлорметане (20 мл). К раствору при перемешивании добавляли 4 н. раствор гидроокиси натрия (2 мл). Реакционную смесь помещали в ледяную баню и по каплям добавляли N-бензилоксикарбонилоксисукцинимид (2,62 г) в дихлорметане (20 мл) вместе с 2 н. раствором гидроокиси натрия для поддержания рН около 11. После того, как добавление завершилось, смесь оставляли на ночь при перемешивании при комнатной температуре. Дихлорметановый слой отделяли и водный слой дважды промывали дихлорметаном. Объединенные органические слои промывали водой и сушили над сульфатом натрия, фильтровали и выпаривали под вакуумом. Остаток очищали хроматографией на силикагеле (элюент: гептан/этилацетат 3/2 об.%), получая 3,15 г.
ТСХ: Rf=0,78, силикагель, этилацетат/гептан 1/1 (об.%).
(m). N-(Cbz)-N'-(трет-Вос)-гуанидин
N-(бензилоксикарбонил)-N'-(трет-бутилоксикарбонил)-S-метилизомочевину (3,15 г) растворяли в метанольном растворе аммиака (2,4 М, 50 мл). Реакционную смесь оставляли при перемешивании на ночь при комнатной температуре. Смесь выпаривали под вакуумом и получившийся остаток очищали хроматографией на силикагеле (элюент: дихлорметан/метанол 95/5 об.%), получая 1,83 г.
ТСХ: Rf=0,80, силикагель, дихлорметан/метанол 9/1 ( об.%).
(n). N-(трет-Вос)-гуанидин гидрохлорид
К раствору N-(Cbz)-N'-(трет-Вос)-гуанидина (1,8 г) в метаноле (50 мл) добавляли 10% палладий на древесном угле (250 мг) и 3,12 мл 2 н. раствора хлористого водорода. Смесь гидрировали под атмосферным давлением при комнатной температуре в течение 1 ч. Палладиевый катализатор удаляли фильтрацией и растворитель удаляли выпариванием при пониженном давлении, получая количественно N-(трет-Вос)-гуанидин гидрохлорид.
ТСХ: Rf=0,10, силикагель, дихлорметан/метанол 9/1 ( об.%).
(о). N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-Pro-Glu(N-трет-Вос)-гуанидин-2-тиазолил
К охлажденному (0oС) раствору N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-Pro-Glu-2-тиазолила (170 мг) в дихлорметане (5 мл) добавляли последовательно дициклогексилкарбодиимид (123 мг), N-(трет-Вос)-гуанидин, НСl (75 мг) и триэтиламин (53 мкл). Смесь перемешивали при 0oС в течение 1 часа и затем еще час выдерживали при комнатной температуре. Смесь охлаждали до -20oС и удаляли фильтрацией дициклогексилмочевину. Фильтрат упаривали до сухого остатка. Остаток хроматографировали на силикагеле в смеси дихлорметан/метанол 95/5 об. % в качестве элюента. Фракции, содержащие N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-Pro-Glu(N-трет-Вос)-гуанидин-2-тиазолил разливали тонким слоем и выпаривали. Выход: 173 мг.
ТСХ: Rf=0,50, силикагель, дихлорметан/метанол 9/1 (об.%).
(р). HOOC-CH2-D-Cha-Pro-Glu(гуанидин)-2-тиазолил
N-(трет-бутилоксикарбонилметил)-N-Boc-D-Cha-Pro-Glu(N-трет-Вос)-гуанидин-2-тиазолил обрабатывали 90% смесью трифтор-уксусной кислоты с водой (10 мл) в течение 2,5 ч при комнатной температуре. Реакционную смесь концентрировали под вакуумом, остаток растворяли в воде и напрямую подавали на колонку препаративной ЖХВР DeltaPak RP-C18, используя систему градиентного элюирования от 20% А/80% В до 20% А/45% В/35% С за 40 мин при скорости потока 50 мл/мин (А: 0,5 М фосфатный буфер, рН 2,1, В: вода; С: ацетонитрил/вода 6/4). Выход: 22 мг НООС-СН2-D-Сhа-Рrо-Сlu(гуанидин)-2-тиазолила.
Rt (ЖХ): 35,04 мин, А 20%, В 80%, С 0% до А 20%, В 20%, С 60% за 40 мин.
ПРИМЕР 3
(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-Glu (гуанидин)-2-тиазолил
(a). 2-Cbz-(4aR,8aR)-пергидроизохинолин-1(R,S)-карбоновая кислота
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбоновую кислоту синтезировали так, как описано в ЕР 0643073, Пример 1.
ТСХ: Rf= 0,85, этилацетат/пиридин/уксусная кислота/вода 63/20/6/11 на окиси кремния.
(b). 2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R,S)-карбонил-Рrо-O-(трет-Вu)
К холодному раствору (0oС) 2-Cbz-(4aR,8aR)-пергидроизохинолин-1(R,S)-карбоновой кислоты (500 мг) в диметилформамиде (5 мл) добавляли последовательно DCC1 (1,3-дициклогексилкарбодиимид, 342 мг), НОВТ (1-гидроксибензотриазолгидрат, 319 мг), Н-Рrо-О-(трет-Вu) (270 мг) и триэтиламин (0,55 мл). Реакционную смесь перемешивали при 0oС в течение 1 ч и затем оставляли на ночь выдерживаться при комнатной температуре. Реакционную смесь охлаждали до -20oС и DCU (1,3-дициклогексилмочевину) удаляли фильтрацией. Фильтрат концентрировали под вакуумом и остаток растворяли в этилацетате. Этот раствор тщательно промывали 5% водным
раствором гидрокарбоната натрия, 3% водным раствором лимонной кислоты, водой и рассолом, сушили над сульфатом натрия и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (элюент: гептан/этилацетат 4/1 об. %), получая 2-Cbz-(4aR,8aR)-пергидроизохинолин-1 (R,S)-карбонил-Pro-O-(трет-Bu) (634 мг).
TCX: Rf= 0,90, этилацетат/пиридин/уксусная кислота/вода 63/20/6/11 на окиси кремния.
(с). 2-Cbz-(4aR,8aR)-пергидроизохинолин-1(R,S)-карбонил-Рrо-ОН
2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R,S)-карбонил-Рrо-O-трет-бутиловый эфир (600 мг) смешивали со смесью дихлорметана (1 мл), трифторуксусной кислоты (3 мл), анизола (0,15 мл) в течение 1 ч при комнатной температуре. Реакционную смесь концентрировали под вакуумом при низкой температуре и остаток растворяли в воде при рН 9,5. Водную фазу промывали диэтиловым эфиром, после чего водный слой подкисляли до рН 2,5 2 М раствором соляной кислоты. Водный слой экстрагировали этилацетатом и органическую фазу промывали рассолом, сушили над сульфатом натрия и концентрировали под вакуумом, получая 2-Cbz-(4aR,8aR)-пергидроизохинолин-1(R,S)-карбонил-Рrо-ОН (588 мг).
TCX: Rf=0,54, этилацетат/пиридин/уксусная кислота/вода 60/3/1/2 на окиси кремния.
(d). (4aR,8aR)-пергидроизохинолин-1(R, S)-карбонил-Pro-Glu-2-тиазолил
Сочетание 2-Cbz-(4aR, 8aR)-пергидроизохинолин-1(R, S)-карбонил-Рrо-ОН и H-Glu-(2-тиазолил)•ТFА в смешанный ангидрид, гуанидизацию, снятие защиты и очистку делали в соответствии с методикой, описанной в Примере 2. Выход: 64,7 мг.
Rt (ЖХ): 28,93 мин, от 20% А, 80% В к 20% А, 20% В и 60% С за 40 мин.
ПРИМЕР 4
EtSO2-D-Cha-Pro-Glu(гуанидин)-2-тиазолил
(a). Boc-D-Cha-Pro-OPAc
Boc-D-Cha-Pro-OPAc получали подобно тому, как описано в Примере 1, используя Boc-D-Cha-OH и Н-Рrо-ОРАс.
ТСХ: Rf=0,5, дихлорметан/метанол 95/5 на окиси кремния.
(b). EtSO2-D-Cha-Pro-OPAc
Boc-D-Cha-Pro-OPAc (3,8 г) растворяли в 50% TFA/дихлорметан (25 мл) и перемешивали в течение 30 минут при комнатной температуре. Реакционную смесь упаривали под вакуумом. Сырой амин растворяли в дихлорметане (50 мл) и добавляли этансульфонилхлорид (0,8 мл) при -78oС. Для поддержания рН на уровне 8 в ходе реакции добавляли триэтиламин. Смесь перемешивали в течение 3 ч при комнатной температуре, после чего добавляли воду (25 мл). После дополнительного перемешивания в течение 30 мин при комнатной температуре реакционную смесь концентрировали под вакуумом. Остаток растворяли в диэтиловом эфире и промывали 1 н. раствором соляной кислоты, водой, 5% раствором гидрокарбоната натрия и рассолом, сушили над сульфатом натрия, фильтровали и упаривали под вакуумом. Растирание сырого продукта с метанолом дает этил-SO2-D-Сhа-Рrо-ОРАс (3,0 г).
ТСХ: Rf=0,6, дихлорметан/метанол 95/5 на окиси кремния.
(с). Этил-SO2-D-Сhа-Рrо-ОН
К раствору этил-SO2-D-Сhа-Рrо-ОРАс (10 г) в тетрагидрофуране (250 мл) добавляли 1 М раствор тетрабутиламмонийфторида в тетрагидрофуране (84 мл). Реакционную смесь перемешивали в течение 30 мин при комнатной температуре и выливали в воду (1 л). Водный раствор экстрагировали этилацетатом. Объединенные органические слои тщательно промывали 1 н. раствором соляной кислоты и водой, сушили над сульфатом натрия и концентрировали под вакуумом. Остаток очищали кристаллизацией из смеси этилацетат/диизопропиловый эфир, получая этил-SO2-D-Cha-Pro-OH (6,0 г).
ТСХ: Rf=0,2, этилацетат/пиридин/уксусная кислота/вода 163/20/6/11 на окиси кремния.
(d). Этил-SO2-D-Cha-Pro-Glu(гуанидин)-2-тиазолил
Сочетание этил-SO2-D-Cha-Pro-OH и H-Glu-(2-тиазолил)•TFA в смешанный ангидрид, гуанидизацию, снятие защиты и очистку делали в соответствии с методикой, описанной в Примере 2. Выход: 83 мг.
Rt (ЖХ): 28,33 мин, от 20% А, 60% В и 20% С к 20% А и 80% С за 30 мин.
ПРИМЕР 5
N-Ac-D-Phe-2-Nal-Glu(гуанидин)-2-тиазолил
N-Ac-D-Phe-2-Nal-OMe получали подобно тому, как описано в Примере 2, используя N-Ac-D-Phe-OH и H-2-Nal-OMe.
(а). N-Ac-D-Phe-2-Nal-OH
Раствор N-Ac-D-Phe-2-Nal-OMe (1,55 г) в 20 мл смеси диоксан/вода 9/1 (об.%) обрабатывали избытком 6 н. гидроокиси натрия, поддерживая рН на уровне 12, в течение 1 часа при комнатной температуре. После подкисления смесь выливали в воду и экстрагировали дихлорметаном. Органический слой промывали водой и сушили над сульфатом натрия. Фильтрат упаривали и получали 1,7 сырого N-Ac-D-Phe-2-Nal-OH.
ТСХ: Rf= 0,5, силикагель, этилацетат/пиридин/уксусная кислота/вода 63/20/6/11 (об.%).
(b). N-Ac-D-Phe-2-Nal-Glu(гуанидин)-2-тиазолил
Сочетание N-Ac-D-Phe-2-Nal-OH и H-Glu-тиазолил•ТFА в смешанный ангидрид, гуанидизацию, снятие защиты и очистку делали в соответствии с методикой, описанной в Примере 2. Выход: 218 мг.
Rt (ЖХ): 29,97 мин, от 20% А, 60% В и 20% С к 20% А и 80% С за 30 мин.
ПРИМЕР 6
N-Me-D-Cha-Pro-Glu(гуанидин)-2-тиазолил
Воc-(N-Me)-D-Cha-Pro-OH получали по методике, подобной той, что описана в Примере 2, используя Воc-(N-Me)-D-Cha-Pro-OH и H-Pro-OBzl.
Н-(N-Me)-D-Cha-Pro-Glu(гуанидин)-2-тиазолил
Сочетание Воc-(N-Me)-D-Cha-Pro-OH и H-Glu-(2-тиазолил)•TFA в смешанный ангидрид, гуанидизацию, снятие защиты и очистку делали в соответствии с методикой, описанной в Примере 2. Выход: 85 мг.
Rt (ЖХ): 32,27 мин, от 20% А и 80% В к 20% А, 20% В и 60% С за 40 мин.
ПРИМЕР 7
N-Ac-D-Phe-Phe-Glu(гуанидин)-2-тиазолил
Названное в заголовке соединение получали по методике, аналогичной описанной в Примере 5. Выход: 112 мг.
Rt (ЖХ): 42,97 мин, от 20% А, 80% В до 20% А, 20% В и 60% С за 40 мин.
ПРИМЕР 8
N-Me-D-Phe-Pro-Glu(гуанидин)-2-тиазолил
Воc-(N-Me)-D-Phe-Pre-OMe получали по такой же методике, какая описана в Примере 2, используя Воc-(N-Me)-D-Cha-OH и Н-Рrо-ОМе. Воc-(N-Me)-D-Phe-Pro-OH получали по такой же методике, какая описана в Примере 5, используя Воc-(N-Me)-D-Phe-Pro-OMe.
Rt (ЖХ): 28,0 мин, от 20% А, 80% В до 20% А, 20% В и 60% С за 40 мин.
N-Me-D-Phe-Pro-Glu(гуанидин)-2-тиазолил
Сочетание Воc-(N-Me)-D-Phe-Pro-OH и Н-Glu-(2-тиазолил)•TFA в смешанный ангидрид, гуанидизацию, снятие защиты и очистку делали в соответствии с методикой, описанной в Примере 2. Выход: 79,3 мг.
Rt (ЖХ): 28,28 мин, от 20% А и 80% В к 20% А, 20% В и 60% С за 40 мин.
ПРИМЕР 9
BzlSO2-norLeu(цикло)-Gly-Glu(гуанидин)-2-тиазолил
norLeu(цикло)-Glu обозначает фрагмент структуры с формулой
(а). Boc-norLeu(цикло)-ОН
К раствору L-α-амино-ε-капролактама (10 г) в смеси диоксан/вода (2/1 об. %) (30 мл) при перемешивании добавляли 1 н. раствор гидроокиси натрия (7,8 мл) и вслед за этим ди-трет-бутилкарбонат (18,8 г). Смесь перемешивали в течение 16 ч при комнатной температуре и концентрировали под вакуумом. Остаток растворяли в этилацетате и промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и выпаривали под вакуумом. Сырой продукт растирали с гексаном, фильтровали и сушили под вакуумом, получая Boc-norLeu(цикло)-ОН (16 г).
ТСХ: Rf=0,85, этилацетат/гептан 1/1 на окиси кремния.
(b). Boc-norLeu(цикло)-Glu-OMe
Boc-norLeu(цикло)-ОН (10 г) растворяли в дихлорметане (100 мл). При -20oС медленно добавляли 1 М раствор бис-(триметилсилил)амида в смеси ТГФ/циклогексан (1/1 об.%) (1 г-экв) и смесь перемешивали в течение 30 мин. После этого добавляли метилбромацетат (4 мл) и смесь перемешивали в течение 2 ч при комнатной температуре. Дополнительно вводили бис-(триметилсилил)амид в смеси ТГФ/циклогексан (1/1 об.%), чтобы довести реакцию до конца. Смесь разбавляли дихлорметаном и промывали 0,1 н. раствором соляной кислоты, водой, 5% водным раствором бикарбоната натрия и рассолом, сушили над сульфатом натрия, фильтровали и выпаривали под вакуумом. Остаток очищали хроматографией на окиси кремния (элюент: гептан/этилацетат 6/4 об.%), получая Boc-norLeu(цикло)-Glu-OMe (12 г).
ТСХ: Rf=0,55, этилацетат/гептан 6/4 на окиси кремния.
(c). BzlSO2-norLeu(цикло)-Glu-OMe
Boc-norLeu(цикло)-Glu-OMe (3 г) растворяли в 50% смеси ТГФ/дихлорметан (30 мл) и перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь упаривали под вакуумом. Сырой амин растворяли в дихлорметане (25 мл) и при 0oС медленно добавляли раствор бензилсульфонилхлорида (2,25 г) в дихлорметане (10 мл). Для поддержания рН на уровне 8 во время реакции добавляли триэтиламин. Смесь перемешивали в течение 1 ч при комнатной температуре, после чего смесь концентрировали под вакуумом. Остаток растворяли в этилацетате и промывали 5% раствором гидрокарбоната натрия, водой и рассолом, сушили над сульфатом натрия, фильтровали и упаривали под вакуумом. Остаток очищали хроматографией на окиси кремния (элюент: дихлорметан/этилацетат 95/5 об.%), получая ВzlSO2-norLeu(цикло)-Glu-OMe (3,9 г).
ТСХ: Rf=0,40, дихлорметан/этилацетат (9/1) на окиси кремния.
(d). BzlSO2-norLeu(цикло)-Glu-OH
Раствор BzlSO2-norLeu(цикло)-Glu-OMe (3,9 г) в 100 мл смеси диоксан/вода (9/1) обрабатывали избытком 1 н. гидроокиси натрия, чтобы поддерживать рН на уровне 13, в течение 2 часов при комнатной температуре. После подкисления смесь выливали в воду и экстрагировали дихлорметаном. Органический слой промывали водой и сушили над сульфатом натрия. Фильтрат упаривали, получая 3,6 г указанного в заголовке соединения.
ТСХ: Rf= 0,60, этилацетат/пиридин/уксусная кислота/вода (63/20/6/11) на окиси кремния.
(e). BzlSO2-norLeu(цикло)-Gly-Glu(гуанидин)-2-тиазолил
Сочетание ВzlSO2-nоrLеu(цикло)-Glu-OH и H-Glu-(2-тиазолил)•ТFА в смешанный ангидрид, гуанидизацию, снятие защиты и очистку делали в соответствии с методикой, описанной в Примере 2. Выход: 34,6 мг.
Rt (ЖХ): 21,42 мин, oт 20% А, 60% В и 20% С к 20% А и 80% С за 30 мин.
ПРИМЕР 10
Получение ацилгуанидинов на оксимной смоле Кайзера
К суспензии 4,0 г оксимной смолы Кайзера (NovaBiochem, 1,10 ммоль/г) в 80 мл смеси дихлорметан/диметилформамид (3:2 об.%) добавляли Boc-β-Ala-OH (3,3 г, 17,6 ммоль), N-гидроксибензотриазол (3,0 г, 22,0 ммоль) и N,N-диизопропилкарбодиимид (3,4 мл, 22,0 ммоль). Суспензию встряхивали в течение 16 часов при комнатной температуре. Смолу отфильтровывали и промывали смесью дихлорметан/диметилформамид (3: 2 об.%), диметилформамидом, 2-пропанолом и дихлорметаном (три раза каждым). Непрореагировавшие оксимные группы блокировали обработкой смолы 80 мл смеси уксусный ангидрид/диизопропилэтиламин/N-метилпирролидон (3: 1:12 об.%) в течение 30 минут при комнатной температуре. Реакционную смесь удаляли фильтрацией и смолу промывали N-метилпирролидоном, смесью 2-пропанол/дихлорметан (1:3 об.%) и дихлорметаном (три раза каждым). Полученную производную смолу (4,96 г) сушили под вакуумом. Порции этой смолы по 50 мг переносили в 24 реактора автомата органического синтеза. Смолу подвергали набуханию путем ее промывки смесью дихлорметан/диметилформамид (3:2 об.%) и дихлорметаном (два раза).
i) Снятие защиты Воc-группы
Смолу предварительно обрабатывали 1 мл 25% трифторуксусной кислоты в дихлорметане и затем в течение 30 мин подвергали реакции с 2 мл 25% трифторуксусной кислоты при периодическом пробулькивании азота. Смолу промывали дихлорметаном, 2-пропанолом и дихлорметаном (два раза каждым).
(ii) Образование первого блока конструкции (А)
Смолу подвергали набуханию путем ее промывки смесью дихлорметан/диметилформамид (3:2 об.%). К смоле добавляли 1 мл 0,2 М раствора блока конструкции A (Boc-Pro-OH, Boc-(N-метил)-Gly-OH, Boc-Gly-OH или Boc-Phe-OH) в дихлорметане, 0,5 мл 0,44 М TBTU в диметилформамиде и 0,5 мл 0,44 М диизопропилэтиламина в дихлорметане. Смолу выдерживали при комнатной температуре в течение 60 мин при периодическом пробулькивании азота. После удаления растворителя смолу промывали смесью дихлорметан/диметилформамид (3:2 по объему), диметилформамидом, 2-пропанолом и дихлорметаном (два раза каждым).
iii) Снятие защиты Вос-группы
Вос-группу блока конструкции А удаляли, используя методику, описанную в i).
iv) Образование второго блока конструкции (В)
Второй блок конструкции соединяли по такой же методике, как описана в iii), используя 0,2 М раствора блока конструкции В (Boc-D-Cha-OH или Boc-D-Phe-OH) в смеси дихлорметан/диметилформамид (3/2 по объему).
v) Снятие зашиты Воc-группы
Вос-группу блока конструкции В удаляли, используя методику, описанную в i), за исключением того, что предварительную обработку проводили трифторуксусной кислотой.
vi) Образование третьего блока конструкции (С)
Смолу промывали смесью дихлорметан/диметилформамид (3:2 по объему) и затем дважды промывали 2 мл 0,11 М раствора диизопропилэтиламина в смеси дихлорметан/диметилформамид (3: 2 по объему). К смоле добавляли 1 мл смеси дихлорметан/диметилформамид (3: 2 по объему), 0,5 мл 0,44 М раствора сульфонилхлоридов С (фенилсульфонилхлорид, 4-хлорфенилсульфонилхлорид или 7-метоксинафтилсульфонилхлорид) в смеси дихлорметан/диметилформамид (3:2 по объему) и 0,5 мл 0,44 М раствора диизопропилэтиламина в дихлорметане. Реакцию со смолой проводили в течение 60 минут при комнатной температуре при периодическом пробулькивании азота. После удаления реакционной смеси смолу промывали смесью дихлорметан/диметилформамид (3:2 по объему), диметилформамидом, 2-пропанолом и дихлорметаном (2 раза каждым).
vii) Отщепление продукта от смолы
Смолу дважды промывали диметилформамидом перед добавлением 1,8 мл 0,2 М раствора гуанидин•НСl в диметилформамиде и 0,2 мл диизопропилэтиламина. Смолу выдерживали при комнатной температуре в течение 64 часов при периодическом пробулькивании азота. Смолу отфильтровывали и фильтрат собирали в пробирку. Смолу дважды промывали диметилформамидом. Собранные фильтраты упаривали до сухого остатка.
Характеристика
Все компоненты характеризовали по жидкостной хроматографии с обращенной фазой на колонке Supelcosil LC-18-DB (4,6 мм • 25 см), используя следующие условия: Поток: 1,0 мл/мин, Буферы: А - вода, В - ацетонитрил/вода (9:1 по объему), С - 0,5 М фосфатный буфер с рН 2,1, Градиент 1: 0->30 мин 55% А-25% В-20% С->15% А-65% В-20% С. Детектирование вели по УФ-спектру на 210 нм. Времена удерживания в минутах приведены в таблице 1. Кроме того, все компоненты анализировали методом электрораспылительной ионизационной масс-спектрометрии. Таблица 1 показывает значения М+Н, определенные в положительной фазе. Для всех соединений обнаруженная масса хорошо согласуется с ожидаемыми значениями.
ПРИМЕР 11
Кроме того, при использовании способа по настоящему изобретению были получены следующие соединения:
-HOOC-CH2-D-Cha-(1-аминоциклогексилкарбокси)-β-Ala-гуанидин;
-(НООС-СН2)2-В-Cha-Рrо-β-Аlа-гуанидин;
-НОOC-CH2-N-Me-D-Cha-Pro-β-Alа-гуанидин;
-НООС-СН2-N-аллил-D-Chа-Рrо-β-Аlа-гуанидин;
-НООС-СН2-N-пропаргил-D-Сhа-Рrо-β-Аlа-гуанидин;
-НООС-СН2-N-бензил-D-Сhа-Рrо-β-Аlа-гуанидин;
-НООС-СН2-N-циклопропил-D-Сhа-Рrо-β-Аlа-гуанидин,
-HOOC-CH2-N-циклoбyтил-D-Cha-Pro-β-Ala-гyaнидин;
-НООС-СН2-N-циклопентил-D-Сhа-Рrо-β-Аlа-гуанидин;
-НООС-СН2-N-циклогексил-D-Сhа-Рrо-β-Аlа-гуанидин;
-2-пропилпентаноил-Аsр(ОМе)-Pro-β-Ala-гуанидин;
-2-пропилпентаноил-Аsр-Рrо-β-Аlа-гуанидин;
-Этил-SO2-D-Сhа-Рrо-β-Аlа-гуанидин;
-Этил-SO2-D-Рhе-Рrо-β-Аlа-гуанидин;
-[2-(Bzl-SO2-NH)-пиридинил-1-СН2-СО]-β-Ala-гуанидин;
-[3-(Bzl-SO2-NH)-пиридин-2-он-1-СН2-СО]-β-Ala-гуанидин;
-Bzl-SO2-norLeu(цикло)-Gly-β-Ala-гуанидин;
-N-Me-D-Phe-Pro-Glu(гуанидин)-СООН;
-N-Me-D-Phe-Pro-Glu(гуанидин)-(2-оксазолил);
-N-Me-D-Cha-Pro-Glu(гуанидин)-СООН;
-N-Me-D-Cha-Pro-Glu(гуанидин)-(2-тиазолил),
-N-Me-D-Cha-Pro-Glu(гуанидин)-(2-оксазолил);
-HOOC-CH2-D-Phe-Pro-Glu(гуанидин)-СООН;
-HOOC-CH2-D-Phe-Pro-Glu(гуанидин)-(2-тиазолил);
-HOOC-CH2-D-Phe-Pro-Glu(гуанидин)-(2-оксазолил);
-HOOC-CH2-D-Cha-Pro-Glu(гуанидин)-СООН;
-HOOC-CH2-D-Cha-Pro-Glu(гуанидин)-(2-оксазолил);
-Этил-SO2-D-Рhе-Рrо-Сlu(гуанидин)-СООН;
-Этил-SO2-D-Phe-Pro-Glu(гуанидин)-(2-тиазолил);
-Этил-SO2-D-Рhе-Рrо-Сlu(гуанидин)-(2-оксазолил);
-Этил-SO2 -D-Сhа-Рrо-Сlu(гуанидин)-СООН;
-Этил-SO2-D-Сhа-Рrо-Сlu(гуанидин)-(2-оксазолил);
-1-Piq-Pro-Glu(гуанидин)-СООН;
-1-Piq-Pro-Glu(гуанидин)-(2-оксазолил);
-HOOC-CH2-D-Cha-(N-циклопентил)-Gly-Clu(гуанидин)-(2-тиазолил);
N-Me-D-Phe-(N-циклопентил)-Gly-Glu(гуанидин)-(2-тиазолил);
-2-пропилпентаноил-Аsр(ОМе)-Pro-Glu(гуанидин)-(2-тиазолил);
-2-гидрокси-3-циклогексилпропионил-Рrо-Сlu(гуанидин)-(2-тиазолил);
-1-Piq-(N-циклопентил)-Gly-Glu(гуанидин)-(2-тиазолил);
-Дифенилпропионил-Pro-Glu(гуанидин)-(2-тиазолил);
- соединения, в которых остаток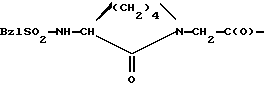
сочетается с -Glu(гуанидин)-СООН, -Glu(гуанидин)-(2-тиазолилом) или с Glu(гуанидин)-(2-оксазолилом);
-9-гидроксифтор-9-карбоксил-Рrо-β-Аlа-гуанидин;
-9-гидроксифтор-9-карбоксил-(N-циклопентил)-Gly-β-Ala-гуанидин;
-9-гидроксифтор-9-карбоксил-азетидин-2-карбоксил-β-Аlа- гуанидин.
ПРИМЕР 12
Антитромбиновый тест
Тромбин (Фактор IIа) представляет фактор в каскаде коагуляции. Антитромбиновую активность соединений по настоящему изобретению определяли путем спектроскопического измерения скорости гидролиза хромогенного субстрата S-2238, на который влияет тромбин. Этот тест на антитромбиновую активность в буферной системе использовали для определения значения IC50 для испытуемых соединений.
Среда анализа. Буфер Трометамин-NаСl-полиэтиленгликоль 6000 (TNP).
Стандартное вещество: 12581 (Kabi), Носитель: TNP-буфер. Солюбилизации могут способствовать диметилсульфоксид, метанол, этанол, ацетонитрил или трет-бутиловый спирт, которые не вызывают неблагоприятных эффектов в концентрациях до 2,5% на конечную реакционную смесь.
Методика
Реагенты:
1. Трометамин-NaCl (TN) буфер. Состав буфера: Трометамин (Tris) 6,057 г (50 ммоль), NaCl 5,844 г (100 ммоль), вода до 1 л. рН раствора доводят до 7,4 при 37oС добавлением НСl (10 ммоль/л).
2. TNP-буфер: Полиэтиленгликоль 6000 растворяли в TN-буфере до концентрации 3 г/л.
3. Раствор S-2238: Одну ампулу S-2238 (25 мг, Kabi Diagnostica, Sweden) растворяли в 20 мл TN-буфера до концентрации 1,25 мг/мл (2 ммоль/л).
4. Раствор тромбина: Тромбин человека (16000 nKat на ампулу, Centraal Laboratorium voor Bloedtransfusie, Amsterdam, The Netherlands) растворяли в TNP-буфере, чтобы получить исходный раствор 835 nKat/мл. Непосредственно перед использованием этот раствор разбавляли TNP-буфером до концентрации 3,34 nKat/Мл.
Все используемые ингредиенты имели квалификацию "для анализа".
Для приготовления водных растворов использовали сверхчистую воду (качества Milli-Q).
Приготовление растворов испытуемого и стандартного соединений
Испытуемое и стандартное соединения растворяли в воде Milli-Q до концентраций исходных растворов 10-2 моль/л. Каждую концентрацию дробно разводили носителем до концентраций 10-3, 10-4 и 10-5 моль/л. Разведения, включая исходное разведение, использовали при анализе (конечные концентрации в реакционной смеси: 3•10-3; 10-3; 3•10-4; 10-4; 3•10-5; 10-5; 3•10-6 и 10-6 моль/л соответственно).
Процедура
При комнатной температуре 0,075 мл и 0,025 мл растворов испытуемого соединения или стандартного соединения, или носителя поочередно вводили пипеткой в ячейки микротитровального планшета, и эти растворы разбавляли соответственно 0,115 мл и 0,0165 мл TNP-буфера. Аликвоты 0,030 мл раствора S-2238 добавляли в каждую ячейку и планшет предварительно подогревали и предварительно инкубировали при встряхивании в инкубаторе (Amsterdam) в течение 10 мин при 37oС. После прединкубации начинали гидролиз S-2238, добавляя в каждую ячейку 0,030 мл раствора тромбина. Планшет инкубировали (при встряхивании в течение 30 сек) при 37oС. Спустя 1 мин после начала инкубации в течение 90 мин через каждые 2 мин измеряли поглощение каждого образца при 405 нм, используя кинетический микротитровальный планшет-ридер (Twinreader plus, Flow Laboratories).
Все данные передавались на персональный компьютер IBM с помощью LOTUS-MEASURE. Для каждой концентрации соединения (выраженной в моль/л реакционной смеси) и для контроля строили графики зависимости поглощения от времени реакции в мин.
Оценка реакции: Для каждой конечной концентрации максимальное поглощение рассчитывали из графика опыта. Значение IС50 (конечная концентрация, выраженная в мкмоль/л, вызывающая 50% ингибирование максимального поглощения контроля) рассчитывали методом logit transformation analysis согласно Hafner et. al. (Arzneim.-Forsch./Drug Res. 1977, 27(11): 1871-3).
В таблице 2 представлены данные о значениях IС50 для соединений по изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПРОТЕАЗЫ СЕРИНА | 1997 |
|
RU2178419C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1997 |
|
RU2172321C2 |
| ИНГИБИТОРЫ ТРОМБИНА | 1996 |
|
RU2178796C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,5-А]ПИРИДИНА КАК ИНГИБИТОРЫ СЕРИНПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2175327C2 |
| СИНТЕТИЧЕСКИЕ ПОЛИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167163C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ LH АГОНИСТОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2248979C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1998 |
|
RU2183642C2 |
| СПОСОБ УСКОРЕННОГО ПЕПТИДНОГО СИНТЕЗА В РАСТВОРЕ | 2002 |
|
RU2237673C2 |
| ПРОИЗВОДНЫЕ 16-ГИДРОКСИ-11-(ЗАМЕЩЕННЫЙ ФЕНИЛ)-ЭСТРА-4,9-ДИЕНА, СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2187510C2 |
| ИНГИБИТОР СЕРИНОВЫХ ПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1999 |
|
RU2232760C2 |


Изобретение относится к соединениям формулы I (значения радикалов определены в описании), их пролекарствам и их фармацевтически приемлемым солям. Соединения обладают высокой антикоагуляционной активностью и могут использоваться при лечении и предупреждении заболеваний, связанных с тромбином. Описана также фармацевтическая композиция, включающая соединение по п. 1. 2 c. и 9 з.п. ф-лы, 2 табл.
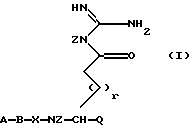

в которой А - Н, R1, R1-O-C(O)-, R1-SO2-, R2OOC-(CHR2)m-, или N-защитная группа, где R1 выбран из групп (C1-12) алкил, (С3-8) циклоалкил, (C6-14) арил, (C7-15)аралкил, арильные группы которых могут быть необязательно замещены (C1-6)алкокси или галогеном;
каждая группа R2 независимо - Н или имеет те же значения, что и R1;
m = 1, 2 или 3;
В представляет собой связь, D-1-Piq, или L- или D-аминокислоту, имеющую гидрофобную или основную боковую цепь, причем аминокислота может быть необязательно замещена N (C1-6) -алкилом,
или А и В вместе - остаток R3R4N-CHR5-C(О)-, в котором R3 и R4 независимо представляют R1, или N-защитная группа, R5 - гидрофобная или основная боковая цепь;
Х - L-аминокислота с гидрофобной боковой цепью, представляющей (C1-12)алкил, необязательно замещенный одной или более (С6-14)арилгруппами, или Х - циклическая аминокислота, -NR2-CH2-C(O) - или фрагмент
в котором n = 2, 3 или 4;
Q - Н или -C(O)Y, где Y - Н или 2-тиазол;
Z - Н, r = 0 или 1, если Q представляет C(O)Y, или r = 0, 1, 2, 3 или 4, если Q представляет Н;
или его пролекарство, или его фармацевтически приемлемая соль.
4. Соединение по п. 3, отличающееся тем, что А - R1-SO2- или R2OOC- (CHR2)m-; В - связь, D-1-Piq или D-аминокислота, имеющая гидрофобную боковую цепь, и Y - 2-тиазол.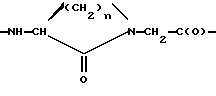
8. Соединение по п.7, отличающееся тем, что А - этил-SО2- или бензил-SО2-; В - связь, D-Phe, D-Cha, D-Coa, D-Dpa, p-Cl-D-Phe, p-OMe-D-Phe, p-OEt-D-Phe, D-NIe, m-Cl-D-Phe, 3,4-ди-OMe-D-Phe или D-Chg.
Приоритет по пунктам и признакам:
13.02.1996 по пп.1-3, 10 и 11;
19.02.1996 по пп.1-3, 10 и 11 (уточнение радикалов);
23.08.1996 по пп.1-11 (уточнение радикалов).
| Способ обезуглероживания электролита топливного метанольно-воздушного элемента | 1977 |
|
SU686642A3 |
| ПРОИЗВОДНЫЕ ГЛИЦИНА ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ ТОРМОЗИТЬ СВЯЗЫВАНИЕ ФИБРИНОГЕНА У ФИБРИНОГЕННОГО РЕЦЕПТОРА ТРОМБОЦИТОВ | 1990 |
|
RU2024549C1 |
| Реверсивный феррорезонансный преобразователь двухфазного напряжения в трехфазное | 1972 |
|
SU454651A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| US 5332726 A, 26.07.1994. | |||
