В течение прошедшего десятилетия была замечена и исследовалась взаимосвязь между поглощением (потреблением) моноаминов и множеством различных заболеваний и состояний. Например, хлоргидратная соль флуоксетина (dl-N-метил-3-фенил-3-[4-(трифторметил)фенокси] пропанамина) является селективным ингибитором перепотребления серотонина (5-гидрокситриптамина), полезным для лечения депрессии и, возможно, для лечения расстройств в питании, алкоголизма и других нарушений. Аналогичным образом хлоргидрат томоксетина [(-)dl-N-метил-3-фенил-3-(2-метилфенокси)пропанамин-хлоргидрат] является селективным ингибитором поглощения норэпинефрина, исследуемым в клинических условиях на его противодепрессантную активность. Эти соединения наряду со многими описываются в патентах США как являющиеся сильными блокаторами поглощения разнообразных физиологически активных моноаминов, включая серотонин, норэпинефрин и допамин.
Настоящее изобретение предоставляет способ получения новых замещенных в кольце 2-пиперазинил- или 2-гомопиперазинил-1,2,3,4-тетрагидронафталинов, которые являются селективными ингибиторами поглощения серотонина и не оказывают непосредственного воздействия на нейронные рецепторы. Следовательно, есть основание ожидать, что эти соединения будут давать меньше побочных эффектов, поскольку они не блокируют эффективно моноаминовые рецепторы или не ингибируют поглощение других моноаминов.
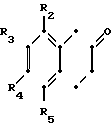
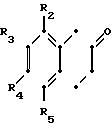
Более конкретно данное изобретение относится к способу получения производных тетрагидронафталинов, соединений общей формулы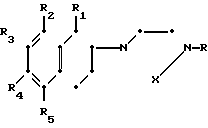 где R - водород или метил;
где R - водород или метил;
R1 - водород или метил;
Х - СН2СН2- или -СН2СН2СН2-;
R2 - водород, галоид, С1-С3-алкокси или С1-С3-алкил;
R3 - водород, галоид;
R4 - водород, галоид С1-С3-алкил или С1-С3-алкокси;
R5 - водород, С1-С3-алкил или С1-С3-алкокси;
При следующих условиях:
(а) если R1 является метилом, то R2 и R4 могут быть одновременно водородом;
(b) если R1 является водородом, один из R2 и R4 не является водородом;
(с) R5 может быть иным, чем водород, только когда R2 является отличным от водорода;
(d) R3 может быть галоидом, только когда R4 не является водородом,
и к их фармацевтически приемлемым кислотно-аддитивным солям.
Хотя все соединения настоящего изобретения являются полезными для лечения разнообразных расстройств, которые связаны с пониженной нейтротрансмиссией серотонина у млекопитающих (или в качестве промежуточных продуктов для таких соединений), некоторые из соединений являются предпочтительными. Так, Х предпочтительно представляет собой -СН2-СН2-. Также, когда R4 является иным, чем водород, R1 представляет собой предпочтительно метил, и когда R2 является отличным от водорода, R1 предпочтительно представляет собой водород.
Когда R2 является иным, чем водород, он предпочтительно представляет собой алкокси или галоид и, более предпочтительно, является метокси или хлором. Наиболее предпочтительно R2, когда не является водородом, представляет собой метокси. Предпочтительно также, когда R2является иным, чем водород, чтобы R5 также был иным, чем водород. В частности, когда R5 является отличным от водорода, он предпочтительно представляет собой галоид, и наиболее предпочтительно бром.
Когда R4 является иным, чем водород, он предпочтительно представляет собой галоид и, наиболее предпочтительно, хлор.
Соединения настоящего изобретения имеют асимметрический углерод, представленный атомом углерода, помеченным звездочкой в следующей ниже формуле: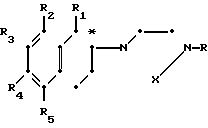
 Таким образом, каждое из соединений существует в виде его индивидуальных d- и l-стереоизомеров, а также в виде рацемической смеси таких изомеров. Соответственно, соединения настоящего изобретения включают не только dl-рацематы, но также и их соответствующие оптически активные d- и l-изомеры.
Таким образом, каждое из соединений существует в виде его индивидуальных d- и l-стереоизомеров, а также в виде рацемической смеси таких изомеров. Соответственно, соединения настоящего изобретения включают не только dl-рацематы, но также и их соответствующие оптически активные d- и l-изомеры.
Кроме того, когда R1 представляет собой метил, присутствует еще один асимметрический углерод, расположенный на R1 заместителе, что дает возможность существования дополнительного класса стереоизомеров.
Как упоминалось выше, изобретение включает фармацевтически приемлемые кислотно-аддитивные соли соединений, определенных приведенной выше формулой. Поскольку соединения данного изобретения являются аминами, они являются основными по своей природе и соответственно взаимодействуют с любой из ряда неорганических и органических кислот, образуя фармацевтически приемлемые кислотно-аддитивные соли. Поскольку свободные амины соединений данного изобретения являются в типичном случае маслами при комнатной температуре, предпочтительно превращать свободные амины в их соответствующие фармацевтически приемлемые кислотно-аддитивные соли для легкости в обращении с ними и в назначении для приема, поскольку последние являются обычно твердыми при комнатной температуре. Кислотами, обычно применяемыми для образования таких солей, являются неорганические кислоты, такие, как соляная кислота, бромистоводородная кислота, йодистоводородная, серная, фосфорная кислота и аналогичные, и органические кислоты, такие, как п-толуолсульфокислота, метансульфокислота, щавелевая кислота, п-бромфенилсульфокислота, угольная, янтарная, лимонная кислота, бензойная кислота, уксусная кислота и аналогичные. Примерами таких фармацевтически приемлемых солей являются сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, вторичный кислый фосфат, первичный кислый фосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, пропионат, деканоат, каприлат, акрилат, формат, изобутират, капроат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, бутен-1,4-диоат, гексин-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, сульфонат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, -гидроксибутират, гликоллат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и аналогичные. Предпочтительными фармацевтически приемлемыми кислотно-аддитивными солями являются соли, образуемые с минеральными кислотами, такими, как соляная и бромистоводородная кислоты, и соли, образуемые с органическими кислотами, такими, как малеиновая кислота.
Кроме того, некоторые из этих солей могут образовывать сольваты с водой или органическими растворителями, такими, как этанол. Такие сольваты также включены как соединения данного изобретения.
Следующие ниже соединения дополнительно иллюстрируют соединения, предусмотренные в объеме данного изобретения:
1-Метил-2-пироразинил-8-этокси-1,2,3,4-тетрагидронафталин
2-(N-метилпиперазинил)-8-этил-1,2,3,4-тетрагидронафталин
2-Пиперазинил-8-метилтио-1,2,3,4-тетрагидронафталин
2-Гомопиперазинил-8-этилтио-1,2,3,4-тетрагидронафталин
1-Метил-2-пиперазинил-6-этил-1,2,3,4-тетрагидронафталин
1-Метил-2-(N-метилпиперазинил)-6-этокси-1,2,3,4-тетрагидронафталин
1-Метил-2-гомопиперазинил-6-метилтио-1,2,3,4-тетрагидронафталин
1-Метил-2-пиперазинил-6-н-пропил-1,2,3,4-тетрагидронафталин
2-Пиперазинил-5-трифторметил-8-иод-1,2,3,4-тетрагидронафталин
2-(N-метилпиперазинил)-5-ацетил-8-хлор-1,2,3,4-тетрагидронафталин, 2-Гомопиперазинил-5-фторацетил-8-метилтио-1,2,3,4-тетрагидронафталин
2-(N-метилпиперазинил-(8-н-пропил-1,2,3,4-тетрагидронафталин
2-Пиперазинил-6-этилтио-1,2,3,4-тетрагидронафталин
2-(N-метилпиперазинил)-6-изопропил-1,2,3,4-тетрагидронафталин и аналогичные.
Соединения настоящего изобретения могут приготавливаться с помощью приемов, хорошо известных специалистам в данной области. Данные соединения предпочтительно синтезируются с помощью получения выбранных тетралонов, которые восстановительно аминируются пиперазином, N-метилпиперазином или соответствующими гомопиперазиновыми гомологами, давая выбранные соединения данного изобретения. Другие соединения данного изобретения доступны с помощью видоизменения кольцевых заместителей после стадий восстановительного аминирования.
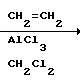
Схемами этих реакций являются следующие:
А. Синтез тетралонов
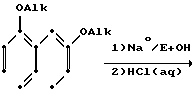
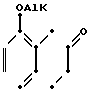

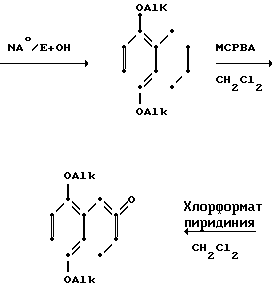
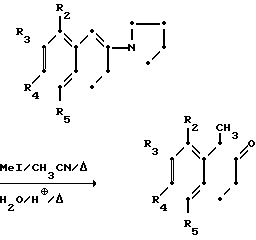
В. Воостановительное аминирование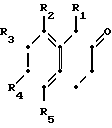
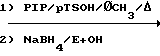
PIP = незамещенных или замещенный пиперазинильный или
гомопиперазинильный фрагмент
Как указывалось выше, тетралоны представляют собой промежуточные продукты, которые, когда они восстановительно аминируются пиперазином или гомопиперазиновым соединением, дают в результате соединения данного изобретения или соединения, которые имеют в сердцевине структуру соединений данного изобретения.
Тетралоны могут быть получены с помощью любого из широкого ряда разнообразных признанных методов. Например, они могут быть получены с помощью реакции Фриделя-Крафтса соответствующего замещенного в кольце фенилацетилхлорида с этиленом в присутствии хлористого алюминия.
1,7-Диалкоксинафталин может восстанавливаться натрием в соответствующий моноалкокситетралон.
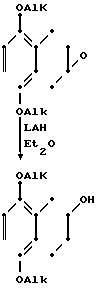
Еще один способ получения тетралона представляет собой, осуществляемый через 1,4-диалкоксинафталин. Нафталин восстанавливается натрием в 1,4-дигидронафталин, и последний окисляется в соответствующий эпоксид м-хлорнадбензойной кислотой. Эпоксид восстанавливается литийалюминийгидридом (LiAH), и получающийся в результате спирт окисляется в требуемый продукт с использованием хлорхромата пиридиния.
Когда R1 в соединениях данного изобретения представляет собой метил, метилзамещенный тетралон может быть получен из соответствующего незамещенного тетралона. Тетралон сначала обрабатывается пирролидином с получением соответствующего 1,2-дигидро-3-пирролидинилнафталина. Последний после обработки метилйодидом и кислотного гидролиза дает желаемый 1-метил-2-тетралон.
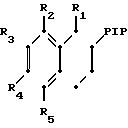
Тетралон может с помощью простого восстановительного аминирования с использованием незамещенного или замещенного пиперазина или гомопиперазина (Р1Р) превращаться в соединение данного изобретения или в соединение, полезное в качестве промежуточного продукта соединения данного изобретения. Тетралон сначала подвергается взаимодействию с Р1Р с образованием соответствующего енамина, после чего енамин восстанавливается боргидридом натрия с получением тетрагидронафталина.
Оптически активные изомеры рацематов изобретения также считаются частью данного изобретения. Такие оптически активные изомеры могут быть получены из их соответствующих оптически активных предшественников с помощью приемов, описанных выше, или с помощью расщепления рацемических смесей. Данное расщепление или разделение может осуществляться в присутствии расщепляющего агента, с помощью хроматографии или повторной кристаллизации. Особенно полезными расщепляющими агентами являются d- или l-винные кислоты, d- и l-дитолуилвинные кислоты и аналогичные.
Соединения, применяемые в качестве исходных материалов в синтезе соединений данного изобретения являются хорошо известными и легко синтезируются с помощью стандартных процедур, обычно применяемых специалистами в данной области.
Фармацевтически приемлемые кислотно-аддитивные соли данного изобретения в типичном случае образуются с помощью реакции 1,2,3,4-тетрагидронафталина данного изобретения с эквимолярным или избыточным количеством кислоты. Реагенты обычно объединяются во взаимно приемлемом для обоих растворителей, таком, как диэтиловый эфир или бензол, и соль обычно выпадает в осадок из раствора в течение периода в пределах примерно от одного часа до 10 дней и может отделяться с помощью фильтрования.
Следующие примеры далее иллюстрируют соединения настоящего изобретения и способы их синтеза. Примеры не предназначены для того, чтобы ограничивать объем данного изобретения в каком-либо отношении, и не должны рассматриваться как ограничительные.
Если не указано иначе, данные ЯМР, приводимые в примерах, относятся к свободным основаниям обсуждаемых соединений.
П р и м е р 1. Получение дихлоргидрата 2-(метилпиперазинил)-8-метокси-1,2,3,4-тетрагидронафталина.
А. 8-Метокси-2-тетралон.
К 1 л ацетона добавляют 50,0 г (0,31 моль) 1,7-дигидронафталина. К раствору затем добавлялись 95,0 г (0,69 моль) порошкообразного карбоната калия и 65 мл (0,69 моль) диметилсульфата. Смесь перемешивали при нагревании с обратным холодильником в атмосфере азота в течение примерно 18 ч. Смесь затем оставляли охлаждаться до комнатной температуры и разбавляли 2 л воды, после чего ее экстрагировали метиленхлоридом. Органические экстракты объединяли, промывали последовательно водой и насыщенным водным раствором хлористого натрия, сушили над сульфатом натрия и упаривали в вакууме, давая коричневое масло.
Масло перегоняли в вакууме, давая 52,51 г (90,1%) 1,7-диметоксинафталина в виде светло-оранжевого прозрачного масла; т.кип. 155-157оС/4 мм рт. ст.
ЯМР (СДСl3): 7,6-6,9 (м, 5Н), 6,7-6,6 (д., j = 7,2, 1Н), 3,88 (с., 3Н), 3,84 (с., 3Н).
Описанный выше продукт (52,5 г, 0,279 моль) растворяли в 450 мл этанола. К раствору затем добавляли 54,4 г (2,37 моль) натрия со скоростью, достаточной для поддержания мягкого кипения с обратным холодильником. Через смесь пропускали азот для удаления водорода, который образовывался. Смесь затем нагревали с обратным холодильником до тех пор, пока весь натрий не потреблялся, после чего ее охлаждали до комнатной температуры, разбавляли 300 мл воды с последующим добавлением 350 мл концентрированный НCl, а затем нагревалась на паровой бане в течение 30 мин. Смесь разбавляли водой до тех пор, пока все оставшееся твердое вещество не растворяли, а затем охлаждалась до комнатной температуры и экстрагиpовали простым эфиром. Органические экстракты объединяли, промывали водой, а затем насыщенным водным насыщенным хлористым натрием, сушили над сульфатом натрия и упаривали в вакууме, давая желтое масло. Масло растворяли в минимальном количестве эфира и добавляли к примерно 250 мл насыщенного водного бисульфита натрия. Двухфазная система перемешивали энергично в течение 18 ч.
Получающаяся в результате бесцветная суспензия фильтровали, и собранное твердое вещество промывали эфиром и сушили в вакууме. Твердое вещество затем добавляли примерно к 300 мл 50%-ного водного карбоната калия. Добавляли эфир и смесь энергично перемешивали до тех пор, пока все твердое вещество не растворялось. Двухфазную смесь затем разделяли, и водную часть экстрагировали эфиром. Объединенные эфирные фазы промывали последовательно водой и насыщенным водным хлористым натрием, сушили над сульфатом натрия и упаривали в вакууме, давая 32,8 г (67%) целевого соединения в виде бесцветной, кристаллической массы.
ЯМР (СДСl3): 7,2-7,0 (триплет, j = 7,2, 1Н), 6,8-6,6 (триплет, j = 7,2, 2Н), 3,76 (синглет, 3Н), 3,48 (синглет, 2Н), 3,14-2,92 (триплет, j = 7,2, 2Н), 2,62-2,46 (т., j = 7,2, 2Н).
В. Дихлоргидрат 2-(N-метилпиперазинил)-8-метокси-1,2,3,4-тетрагидронафталина.
Тетралон (10 г, 56,8 моль) растворяли в 200 мл толуола. К раствору затем добавлялись 13,0 мл (0,117 моль) N-метилпиперазина с последующим добавлением 25,1 г (0,13 моль) п-толуолсульфокислоты. Смесь перемешивали при нагревании с обратным холодильником при постоянном удалении воды. Через 2 ч смесь охлаждали до комнатной температуры, и летучие вещества удаляли в вакууме, давая 3-(N-метилпиперазинил)-5-метокси-1,2-дигидронафталин в виде красновато-оранжевой пульпы.
Пульпу растворяли в 200 мл этанола. К раствору добавляли 30 мл уксусной кислоты с последующим добавлением 10 г боргидрида натрия. Смесь перемешивали в течение 2 ч при комнатной температуре, после чего ее разбавляли 200 мл 10% -ной НCl, а затем перемешивали в течение дополнительного часа при комнатной температуре. Смесь разбавляли водой и экстрагировали эфиром. Водный слой затем подщелачивали добавлением гидроксида аммония и экстрагировали метиленхлоридом. Метиленхлоридные экстракты объединяли, сушили над сульфатом натрия и упаривали в вакууме, давая красно-коричневое масло. Данное масло растворяли в метиленхлориде и помещали на колону мгновенной хроматографии из кремнезема. Колонка элюировали метиленхлоридом, содержащим 3%-ным МеОН и следы гидроксида аммония, давая светло-оранжевое прозрачное масло. Масло растирали с гексаном. Получающуюся в результате смесь фильтровали, фильтрат упаривали в вакууме, давая 7,5 г светло-желтого твердого вещества. 1 г данного твердого вещества превращали в дихлоргидратную соль и кристаллизовали из метанола, получая 1,10 г целевого соединения в виде бесцветных кристаллов; т.пл. выше 200оС.
С16H24N2O˙2HCl.
Вычислено,%: С 57,66; Н 7,86; N 8,40.
Найдено,%: С 57,46; Н 7,80; N 8,31.
ЯМР (СДСl3): 7,12-6,88 (т., j = 7,2, 1Н), 6,76-6,48 (м., 2Н), 3,76 (с., 3Н), 3,20-2,36 (м., 12Н), 2,31 (с., 3Н), 2,22-1,92 (м., 2Н), 1,8-1,2 (м., 1Н).
МС: 260 (25), 216 (8), 202 (9), 189 (17), 188 (11), 174 (10), 162 (32), 160 (39), 100 (92), 70 (62), 53 (100).
П р и м е р 2. Получение дихлоргидрата 2-пиперазинил-8-метокси-1,2,3,4-тетрагид-ронафталина.
Тетралон (1,0 г, 5,68 ммоль), полученный в примере 1, обрабатывали в соответствии с процедурой, описанной в примере 1, с применением пиперазина вместо N-метилпиперазина, с получением 0,22 г желтого, вязкого масла. Данное масло обpабатывали газообразным HCl для получения 0,13 г целевого соединения в виде бесцветных кристаллов.
C15H22N2O ˙2HCl.
Вычислено,%: С 56,43; Н 7,58; N 8,77.
Найдено,%: С 56,22; Н 7,51; N 8,53.
ЯМР (СДСl3): 7,22-7,00 (триплет, j = 7,2, 1Н), 6,84-6,60 (мультиплет, 2Н), 3,91 (синглет, 3Н), 3,28-2,5, (мультиплет, 12Н), 2,45 (синглет, 1Н), 2,36-2,0 (мультиплет, 2Н), 1,96-1,40 (мультиплет, 1Н).
МС: 247 (23), 246 (100), 245 (8), 231 (5), 204 (65), 161 (78).
П р и м е р 3. Получение дималеата 2-пиперазинил-8-хлор-1,2,3,4-тетрагидро-нафталина.
А. 8-Хлор-2-тетралон.
Смесь 30,0 г (0,176 моль) о-хлорфенилуксусной кислоты и 40 мл тионилхлорида перемешивалась в течение 18 ч. Летучие вещества затем удаляли в вакууме, давая 32,76 г (99,0% ) о-хлорфенилацетилхлорида в виде прозрачной, бледно-желтой, подвижной жидкости.
ЯМР (СДСl3): 7,5-7,1 (м., 4Н), 4,2 (с., 2Н).
AlCl3 (46,5 г, 0,348 моль) суспендировали в 400 мл метиленхлорида. Смесь затем охлаждали до -78оС, и по каплям на протяжении 1 ч добавляли раствор 32,76 г (0,174 моль) полученного ранее хлорфенилацетилхлорида в 100 мл метиленхлорида. Баня из смеси сухой лед/ацетон, затем заменяли баней из ледяной воды. В реакционную смесь барботировали этилен, и в течение этого периода времени температура поднималась до 15оС. Добавление этилена прекращали и смесь оставляли перемешиваться при примерно 5оС в течение 4 ч. Затем к смеси добавляли лед для разложения любых оставшихся алюминиевых комплексов. По окончании экзотермической реакции смесь разбавляли водой (500 мл) и энергично перемешивали до тех пор, пока все твердое вещество не растворялось. Водную и органическую фазы разделяли, и органическую фазу промывали 3 раза 400 миллилитровыми порциями 1 н. НСl и два раза с использованием каждый раз 400 мл 10% водного бикарбоната натрия. Органическую фазу затем сушили над сульфатом натрия и упаривали в вакууме, давая бледно-оранжевый остаток. Остаток растворяли в смеси гексана и эфира (1:1) и наносили на колонку мгновенной хлоратографии из кремнезема, которую затем элюировали смесью гексана и эфира (1:1) давая светло-желтый остаток, который кристаллизовался из смеси гексана и эфира (4:1), давая 10,55 г (33,6%) целевого соединения.
ЯМР (СДСl3): 7,5-7,2 (м., 3Н), 3,7 (с., 2Н), 3,3-3,0 (т., j = 7, 2Н), 2,8-2,4 (т., j = 7, 2Н).
МС: 180 (60), 165 (9), 138 (100), 117 (52), 115 (50), 103 (48), 89 (20), 76 (25), 74 (18), 63 (30), 57 (9), 52 (28), 51 (20), 42 (6), 39 (32).
ИК (нуйол): 2950, 2927, 1708, 1464, 1450, 1169, 1141.
В. Дималеат 2-пиперазинил-8-хлор-1,2,3,4-тетрагидронафталина.
Указанный выше тетралон (0,5 г, 2,78 ммоль) обрабатывали пиперазином, и получающийся в результате продукт восстанавливался боргидридом натрия в соответствии с процедурой, описанной в примере 1. Продукт обрабатывали малеиновой кислотой и получали 0,12 г целевого соединения; т.пл. 180-181оС.
C14H19N2Cl ˙2C4H4O4.
Вычислено,%: С 54,72; Н 5,64; N 5,80.
Найдено,%: С 54,81; Н 5,67; N 5,89.
ЯМР (СДСl3): 7,4-6,8 (м., 3Н), 3,3-2,6 (м., 15Н), 2,55 (с., 1Н).
МС: 252 (10), 250 (35), 210 (28), 208 (100), 167 (17), 165 (39), 129 (40), 54 (68).
П р и м е р 4. Получение дималеата 2-(N-метилпиперазинил)-8-хлор-1,2,3,4-тет- рагидронафталина.
С использованием способа примера 3 0,5 г (2,78 ммоль) 8-хлор-2-тетралона обрабатывали N-метилпиперазином, и получающийся в результате продукт восстанавливали боргидридом натрия, продукт обрабатывали малеиновой кислотой, и получали 0,48 г целевого соединения в виде бесцветных кристаллов; т.пл. 199-201оС.
C15H21N2Cl ˙2C4H4O4.
Вычислено,%: С 55,59; Н 5,88; N 5,64.
Найдено,%: С 55,81; Н 6,02; N 5,59.
ЯМР (СДСl3): 7,4-6,8 (м., 3Н), 3,3-2,4 (м., 11Н), 2,3 (с., 3Н), 2,2-1,0 (м., -Н).
МС: 266 (18), 264 (52), 222 (5), 224 (12), 193 (32), 129 (30), 45 (100).
П р и м е р 5. Получение дималеата 2-(гомопиперазинил)-8-хлор-1,2,3,4-тетра-гидронафталина.
С применением процедуры примера 3, 2,0 г (11,1 ммоль) 8-хлор-2-тетралона подвергали взаимодействию с 2,2 г (22,2 ммоль) гомопиперазина, и получающийся в результате продукт восстанавливался боргидридом натрия, и восстановленный продукт обрабатывали малеиновой кислотой; получали 0,13 г целевого соединения в виде бесцветных кристаллов; т.пл. 146-148оС.
С15H21N2Cl ˙2C4H4O4.
Вычислено,%: С 55,59; Н 5,88; N 5,64.
Найдено,%: С 55,89; Н 6,02; N 5,37.
ЯМР (СДСl3): 7,24-6,80 (м., 3Н), 3,28-2,48 (м., 14Н), 2,47 (с., 1Н), 2,24-1,08 (м., 3Н).
МС: 266 (17), 265 (12), 264 (49), 224 (5), 222 (20), 220 (15), 210 (18), 209 (21), 208 (55), 207 (48), 206 (16), 196 (19), 194 (22), 165 (48), 129 (24), 98 (42), 72 (75), 54 (100).
П р и м е р 6. Получение дималеата 2-(N-метилпиперазинил)-8-фтор-1,2,3,4-тет- рагидронафталина.
А. 8-Фтор-2-тетралон.
о-Фторфенилуксусная кислота (35,9 г, 0,233 ммоль) перемешивали в 40 мл тионилхлорида в течение 24 ч при комнатной температуре. Летучий материал удалялся в вакууме, и получали желтую подвижную жидкость. Жидкость перегоняли в вакууме, и получали 27,45 г (68,5%) о-фторфенилацетилхлорида в виде бесцветной жидкости; т.кип. 85оС/4 мм рт.ст.
ЯМР (СДСl3): 7,6-6,9 (м., 4Н), 4,3-4,1 (д., j = 4, 2Н).
Хлористый алюминий (42,5 г (0,32 моль) перемешивали в 400 мл метиленхлорида, и получающийся в результате раствор охлаждали до -78оС. К раствору затем добавляли по каплям раствор 27,45 г (0,16 моль) полученного ранее ацилхлорида в 100 мл метиленхлорида на протяжении 1 ч. Баня из смеси сухой лед/ацетон заменяли баней из ледяной воды, и в колбу энергично барботировали этилен, при этом температуру поднимали с -50оС до +17оС. После завершения экзотермической реакции добавления этилена прекращали, и реакционную смесь перемешивали в течение 2 ч при температуре примерно 5оС, а затем в течение 2 ч при комнатной температуре.
К реакционной смеси затем осторожно добавляли лед. После окончания получающейся в результате экзотермии реакционную смесь разбавляли 500 мл холодной воды. Органическую и водную фазы разделяли, и органическую фазу промывали три раза 100 мл 1 н. HCl и два раза 100 мл насыщенного водного бикарбоната натрия. Органический слой затем сушили над сульфатом натрия и упаривали в вакууме с получением желтого остатка. Остаток растворялся в смеси гексана и эфира (1:1) и помещали на колонку мгновенной хроматографии из кремнезема. Колонку элюировали смесью гексана и эфира (1:1) давая желтый вязкий остаток. Остаток кристаллизовался из смеси гексана и эфира (4:1), и получали всего 5,85 г тетралона в виде бесцветного твердого вещества.
ЯМР (СДСl3): 7,4-6,7 (м., 3Н), 3,6 (с., 2Н), 3,2-2,9 (т., j = 6, 2Н), 2,7-2,4 (т., j = 6, 2Н).
МС: 164 (100), 149 (23), 140 (8), 138 (31), 136 (17), 135 (57), 134 (12), 133 (40), 123 (31), 122 (100), 120 (41), 115 (24), 109 (26), 107 (18), 101 (22), 96 (34), 89 (7), 83 (16), 75 (17), 63 (22), 57 (21), 51 (17), 39 (18).
ИК (КВr, таблетка): 3436, 3427, 3401, 1716, 1705, 1495, 1246, 886 см-1.
В. Дималеат 2-(N-метилпиперазинил)-8-фтор-1,2,3,4-тетрагидронафтилина.
Полученный выше тетралон (1,0 г, 6,1 ммоль) растворяли в 30 мл толуола. К раствору затем добавляли 1,4 мл (12,2 ммоль) N-метилпиперазина и 2,78 г (14,6 ммоль) п-толуолсульфокислоты. Смесь нагревали при кипячении с обратным холодильником в течение 18 ч при постоянном удалении воды, после чего смесь охлаждали до комнатной температуры. Растворитель удаляли в вакууме, давая 3-(N-метилпиперазинил)-5-фтор-1,2-дигидронафталин в виде красновато-оранжевого твердого вещества. Дигидронафталин растворялся в 20 мл этанола. К раствору добавлялось 1,5 мл уксусной кислоты с последующим добавлением общего количества примерно 0,5 г боргидрида натрия порциями. Получающаяся в результате смесь перемешивалась в течение 2 ч при комнатной температуре, псле чего смесь разбавляли 10%-ной водной HCl и перемешивали в течение дополнительного часа при комнатной температуре. Реакционную смесь добавляли водой и экстрагировали эфиром. Водный слой выливали на лед и подщелачивали добавлением гидроксида аммония. Затем ее экстрагировали метиленхлоридом. Метиленхлоридные экстракты объединяли, промывали насыщенным водным хлористым натрием, сушили над сульфатом натрия и упаривали в вакууме, давая коричневое вязкое масло.
Масло растворяли в метиленхлориде и помещали на колонку мгновенной хроматографии. Колонку элюировали 5% -ным метанолом в метиленхлориде со следами гидроксида аммония, и элюат упаривали в вакууме, получая 0,25 г продукта - свободного основания в виде светло-желтого, прозрачного вязкого остатка.
Остаток растворяли в метаноле, и раствор нагревали до кипячения. К раствору затем добавляли два эквивалента малеиновой кислоты в метаноле, и получившую в результате смесь нагревали до кипения. Раствор фильтровали и фильтрат охлаждали до комнатной температуры с образованием кристаллов. Раствор затем охлаждали до 0оС, и кристаллы отфильтровывали и сушили в вакууме, получая 0,37 г целевого соединения в виде бесцветных кристаллов; т.пл. 204-205оС.
C15H21N2F ˙2C4H4O4.
Вычислено,%: С 57,49; Н 6,08; N 5,83.
Найдено,%: С 57,32; Н 5,97; N 6,02.
ЯМР (СДСl3): 7,3-6,8 (м., 3Н), 3,3-2,4 (м., 12Н), 2,3 (с., 3Н), 2,2-1,4 (м., 3Н).
МС: 249 (17), 248 (100), 247 (13), 204 (27), 190 (14), 189 (11), 179 (21), 177 (68), 176 (22), 150 (51), 148 (58), 100 (76), 70 (83), 58 (93).
При применении способа, описанного подробно в примерах 3 и 6, получали соединения примеров 7-9.
П р и м е р 7. Получение дихлоргидрата 2-(N-метилпиперазинил)-8-метил-1,2,3,4-тет- рагидронафталина.
C16H24N2 ˙2HCl.
Вычислено,%: С 60,57; Н 8,26; N 8,83.
Найдено,%: С 60,31; Н 8,36; N 8,62.
ЯМР (СДСl3): 6,96 (с., 3Н), 3,12-2,40 (м., 12Н), 2,36 (с., 3Н), 2,28-2,0 (м., 2Н), 2,28 (с., 3Н), 1,92-1,40 (м., 1Н).
МС: 245 (15), 244 (81), 243 (8), 201 (10), 200 (20), 185 (19), 183 (10), 174 (17), 173 (51), 172 (23), 145 (63), 143 (63), 129 (58), 100 (88), 70 (62), 58 (100).
П р и м е р 8. Получение дималеата 2-(N-метилпиперазинил)-5,8-диметил-1,2,3,4-тетрагидронафталина.
C17H26N2 ˙2C4H4C4.
Вычислено,%: С 61,21; Н 6,99; N 5,71.
Найдено,%: С 61,06; Н 6,85; N 5,84.
МС: 260 (18), 258 (94), 243 (6), 214 (14), 200 (12), 187 (28), 186 (13), 144 (15), 143 (31), 100 (62), 72 (60), 58 (100).
П р и м е р 9. Получение дималеата 2-(N-метилпиперазинил)-8-бром-1,2,3,4-тет- рагидронафталина.
C15H21N2Br ˙2C4H4О4.
Вычислено,%: С 51,03; Н 5,40; N 5,17.
Найдено,%: С 51,32; Н 5,54; N 5,42.
ЯМР (СДСl3): 7,36-7,16 (дд. , j = 3,6, 7,2, 1Н), 7,0-6,76 (м., 2Н), 3,13-2,33 (м., 12Н), 2,28 (с., 3Н), 2,20-1,86 (м., 2Н), 1,76-1,12 (м., 1Н).
МС: 310 (40), 308 (40), 266 (11), 264 (10), 252 (13), 250 (10), 239 (35), 237 (35), 224 (10), 210 (12), 208 (12), 130 (70), 129 (88), 128 (65), 115 (30), 100 (92), 99 (50), 72 (75), 70 (95), 58 (100), 56 (69), 54 (98).
П р и м е р 10. Получение дихлоргидрата 2-(N-метилпиперазинил)-5,8-диметокси-1,2,3,4-тетрагидронафталина.
А. 5,8-Диметокси-2-тетралон.
К 1 л ацетона добавляли 50,0 г (0,31 моль) 1,4-дигидроксинафталина. К получающемуся в результате раствору добавляли 95,0 г (0,69 моль) порошкообразного карбоната калия и 65 мл (0,69 моль) диметилсульфата. Получающуюся в результате смесь перемешивали при нагревании с обратным холодильником в течение 18 ч, после чего они разбавляли 2 л воды, а затем экстрагировали метиленхлоридом. Органические экстракты объединяли, сушили над сульфатом натрия и упаривали в вакууме, получая черное масло. Масло перегоняли в вакууме, получая 12,5 г 1,4-диметоксинафталина в виде оранжевого кристаллического твердого вещества, т.кип. 155оC/5 мм рт.ст.
Диметоксинафталиновое соединение (66,5 ммоль) растворяли в 120 мл этанола. Получающуюся в результате смесь нагревали до кипения с обратным холодильником в атмосфере азота и добавляли порциями 11,7 г (0,51 моль) натрия. Перемешивание при нагревании с обратным холодильником в атмосфере азота продолжали до тех пор, пока все твердое вещество не растворялось. Смесь затем перемешивали в течение 15 мин при комнатной температуре, затем осторожно разбавляли 50 мл воды. Смесь выпаривали в вакууме для удаления этанола, а остаток разбавляли водой и экстрагировали эфиром. Органические экстракты объединяли, сушили над сульфатом натрия и упаривали в вакууме, давая 11,1 г 5,8-диметокси-1,4-дигидронафталина в виде желтого масла.
Дигидронафталин (58,4 ммоль) растворяли в 80 мл метиленхлорида. К раствору добавляли 12,9 г (0,063 моль) 85%-ной м-хлорнадбензойной кислоты (МСРВА) в 140 мл метиленхлорида по каплям на протяжении 10 мин. Во время добавления требовалось охлаждение для поддержания температуры примерно при 25оС. Смесь затем перемешивали при комнатной температуре в течение 45 мин, после чего ее промывали насыщенным водным раствором бикарбоната натрия для удаления любого количества м-хлорбензойной кислоты. Органическую фазу промывали водой, а затем насыщенным водным бикарбонатом натрия, сушили над сульфатом натрия и упаривали в вакууме, получая коричневую вязкую смолу. Смолу растворяли в эфире и помещали на кремнеземную колонку для мгновенной хроматографии. Колонку элюировали эфиром, и фракции 3-6 объединяли и упаривали в вакууме, получая прозрачное вязкое оранжевое масло. Масло растворяли в смеси гексана и эфира (1:1) и помещали на кремнеземную колонку мгновенной хроматографии. Колонку элюировали смесью гексана и эфира (2:1). Фракции 5-8 объединяли и упаривали в вакууме, получая 2,35 г 5,8-диметокси-2,3-оксо-1,2,3,4-тетрагидронафта-лина в виде бесцветных игл; т.пл. 128-129оС.
Оксосоединение (2,35 г, 11,4 ммоль) растворяли в 50 мл эфира и раствор добавляли по каплям к кипящей с обратным холодильником суспензии 1,53 г алюмогидрида лития (Li АН) в 100 мл эфира. Получающуюся в результате смесь нагревали с обратным холодильником в течение 5 ч, а затем охлаждали до 0оС. К смеси затем добавляли последовательно и осторожно 1,53 мл воды, 1,53 мл 15% -ной водной гидроокиси натрия и 4,59 мл воды. Смесь энергично перемешивали в течение 18 ч при комнатной температуре, а затем фильтровали через слой целита. Фильтр промывали эфиром, и фильтрат упаривали в вакууме, получая 2,1 г (88,6%) 2-гидрокси-5,8-диметокси-1,2,3,4-тетрагидронафталина в виде не совсем белого твердого вещества.
Тетрагидронафталин (2,1 г, 10,1 ммоль) растворяли в 20 мл метиленхлорида. Получающийся в результате раствор добавляли к раствору 3,27 г (15,2 ммоль) хлорхромата пиридиния в 60 мл метиленхлорида. Смесь перемешивали в течение 7 ч при комнатной температуре, а затем фильтровали через слой целита. Фильтрат упаривали в вакууме до темного вязкого остатка. Остаток растворяли в метиленхлориде и помещали на кремнеземную колонку мгновенной хроматографии. Колонку элюировали метиленхлоридом, и элюат упаривали в вакууме, получая 0,8 г (38,4%) 5,8-диметокси-2-тетралона в виде золотистого кристаллического твердого вещества.
В. Дихлоргидрат 2-(N-метилпиперазинил)-5,8-диметокси-1,2,3,4-тетрагидронаф-талина.
Диметокситетралон (0,4 г, 1,94 ммоль) растворяли в 10 мл толуола. К раствору добавляли 0,46 мл (4 ммоль) N-метилпиперазина с последующим добавлением 0,91 г (4,8 ммоль) п-толуолсульфокислоты. Смесь перемешивали в течение 3,5 ч при нагревании с обратным холодильником в атмосфере азота при постоянном удалении воды. Смесь затем охлаждали до комнатной температуры, и летучее вещество удаляли в вакууме, получая 3-(N-метилпиперазинил)-5,8-диметокси-1,2-дигидронафталин в виде оранжево-коричневого остатка.
Дигидронафталин растворяли в 10 мл этанола. К получающемуся раствору добавляли 0,3 г боргидрида натрия порциями. Смесь перемешивали в течение 18 ч при комнатной температуре, после чего ее разбавляли примерно 15 мл 10%-ной водной НСl. Получающуюся в результате смесь перемешивали в течение 45 мин при комнатной температуре, а затем разбавляли водой и экстрагировали эфиром. Водный слой подщелачивали гидроксидом аммония и экстрагировали метиленхлоридом. Метиленхлоридные экстракты объединяли, сушили над сульфатом натрия и упаривали в вакууме, получая темно-оранжевое масло.
Масло растворяли в метиленхлориде и помещали на кремнеземную колонку мгновенной хроматографии. Колонку элюировали 3%-ным метанолом в метиленхлориде со следами гидроксида аммония, и элюат упаривали в вакууме, получая 0,31 г свободного основания целевого соединения в виде слабо-оранжевого стеклянистого твердого вещества.
Твердое вещество растворяли в этаноле, и раствор насыщался газообразным хлористым водородом. По мере охлаждения раствора постепенно образовывалось твердое вещество, давая 0,14 г дихлоргидратной соли целевого соединения в виде бесцветных кристаллов; т.пл. выше 200оС.
C15H26N2O2 ˙2HCl.
Вычислено,%: С 56,20; Н 7,77; N 7,71.
Найдено,%: С 56,09; Н 7,65; N 7,78.
ЯМР (СДСl3): 6,48 (синглет, 2Н), 3,69 (синглет, 6Н), 3,16-2,32 (мультиплет, 12Н), 2,26 (синглет, 3Н), 2,28-1,92 (мультиплет, 2Н), 1,72-1,20 (мультиплет, 1Н).
МС: 291 (11), 290 (50), 289 (8), 275 (8), 246 (10), 232 (10), 231 (13), 219 (16), 218 (12), 191 (53), 190 (100), 164 (36), 99 (48), 43 (88).
П р и м е р 11. Получение дималеата 1-метил-2-(N-метилпиперазинил)-8-хлор-1,2,3,4-тетрагидронафталина.
А. 1-Метил-8-хлор-2-тетралон.
К 100 мл толоула добавляли 5,0 г (27,8 ммоль) 8-хлор-2-тетралона. К получающемуся раствору затем добавляли 3,5 г пирролидина и смесь нагревали до кипения с обратным холодильником в течение 3 ч, после чего растворитель удаляли в вакууме, получали 3-пирролидино-5-хлор-1,2-дигидронафталин в виде темного масла (около 6 г).
Дигидронафталин растворяли в 30 мл п-диоксана. К раствору добавляли 10 мл метилиодида, и смесь нагревали до кипения с обратным холодильником в течение 18 ч в атмосфере азота. К реакционной смеси затем добавляли 25 мл воды и 1 мл уксусной кислоты, и нагревание продолжали в течение 4 ч. Реакционную смесь затем охлаждали до комнатной температуры и растворитель удаляли в вакууме. Получающийся остаток суспендировали в воде и водную смесь экстрагировали эфиром. Органические экстракты объединяли, промывали насыщенным водным хлористым натрием, сушили над сульфатом натрия и упаривали в вакууме, получая темное масло. Масло растворяли в эфире и помещали на кремнеземную колонку мгновенной хроматографии. Колонку элюировали смесью гексана и эфира (1:1), содержащей следы гидроксида аммония. Элюат упаривали в вакууме, получая 3,14 г 1-метил-8-хлор-2-тетралона в виде подвижной оранжевой жидкости.
ЯМР (СДСl3): 7,4-7,0 (мультиплет, 3Н), 4,40-3,70 (квартет, j = 7,2, 1Н), 3,32-2,0 (мультиплет, 4Н), 1,52-1,36 (дублет, j = 7,2, 3Н).
В. 1-Метил-2-(N-метилпиперазинил)-8-хлор-1,2,3,4-тетрагидронафталин.
Описанный выше тетралон (1,0 г, 5,2 ммоль) растворяли в 30 мл толуола. К раствору добавляли 1,1 (10,3 ммоль) N-метилпиперазина и 0,75 мл метансульфокислоты, и смесь нагревали до кипения с обратным холодильником в течение 18 ч в атмосфере азота при постоянном удалении воды. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли в вакууме, получая 3-(N-метилпиперазинил)-4-метил-5-хлор-1,2-дигидронафталин в виде оранжевого вязкого остатка.
Дигидронафталин (примерно 5,2 ммоль) растворяли в 25 мл этанола. К получающемуся раствору затем добавляли 1,5 мл уксусной кислоты с последующим добавлением примерно 0,5 г боргидрида натрия порциями. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре, после чего добавляли 30 мл 10%-ной водной HCl, и смесь перемешивали в течение дополнительных 2 ч при комнатной температуре. Реакционную смесь разбавляли 100 мл воды и экстрагировали один раз эфиром. Водную часть подщелачивали гидроксидом аммония и экстрагировали метиленхлоридом. Метиленхлоридные экстракты объединяли, сушили над сульфатом натрия и упаривали в вакууме, получая вязкое оранжевое масло.
Масло растворяли в метиленхлориде и помещали на кремнеземную колонку мгновенной хроматографии. Колонку элюировали метиленхлоридом, содержащим 3% -ный метанол и следы гидроксида аммония, и элюат упаривали в вакууме, получая 0,41 г желтого стекла. Стекло начинало кристаллизоваться, и смесь растирали с гексаном. Бесцветное, твердое кристаллическое вещество отделяли с помощью фильтрования, после чего смесь охлаждали. Было найдено, что бесцветное твердое вещество не является желаемым продуктом.
Фильтрат затем упаривали в вакууме, получая 0,21 г бледно-желтого, прозрачного стекла. Стекло растворяли в метаноле и обрабатывали двумя эквивалентами малеиновой кислоты в метаноле при нагревании с обратным холодильником. Получающейся в результате смеси давали возможность медленно охлаждаться до комнатной температуры и получали 0,30 г целевого соединения в виде бесцветного кристаллического твердого вещества; т.пл. 174-175оС.
C16H23N2Cl ˙2C4H4O4.
Вычислено,%: С 56,41; Н 6,12; N 5,48.
Найдено,%: С 56,63; Н 6,01; N 5,49.
ЯМР (СДСl3): 7,24-6,8 (мультиплет, 3Н), 3,68-3,32 (кв., j = 7, 1Н), 3,0-2,32 (м. , 10Н), 2,30 (с., 3Н), 2,10-1,20 (м., 3Н), 1,24-1,08 (д., j = 7, 3Н).
МС: 280 (8), 278 (28), 236 (4), 207 (20), 135 (100), 100 (31), 70 (58), 58 (59), 53 (54), 43 (62).
П р и м е р 12. Получение дихлоргидрата 1-метил-2-пиперазинил-8-хлор-1,2,3,4-тет-рагидронафталина.
При использовании способа примера 11 1,94 г (10 ммоль) 1-метил-8-хлор-2-тетралона обрабатывали пиперазином с последующим восстановлением боргидридом натрия и обработкой HCl с получением 0,67 г целевого соединения в виде бесцветных кристаллов; т.пл. выше 200оС.
C15H21N2Cl ˙2HCl.
Вычислено,%: С 53,35; Н 6,86; N 8,29.
Найдено,%: С 53,13; Н 6,97; N 8,22.
ЯМР (СДСl3): 7,28-6,8 (м. , 3Н), 3,72-3,32 (м., 1Н), 3,16-2,16 (м., 10Н), 2,24 (с., 1Н), 2,15-1,44 (м., 3Н), 1,18-1,04 (д., j = 7, 3Н).
МС: 266 (6), 264 (18), 224 (19), 223 (10), 222 (59), 208 (14), 179 (31).
П р и м е р 13. Получение тозилата 1-метил-2-пиперазинил-8-метокси-1,2,3,4-тетрагидронафталина.
А. 1-Метил-8-метокси-2-тетралон.
К 75 мл толуола добавляли 3,52 г (20 ммоль) 8-метокси-2-тетралона с последующим добавлением 2,5 г пирролидина. Смесь нагревали до кипения с обратным холодильником в течение 3 ч, после чего растворитель выпаривали в вакууме, получая 3-пирролидино-5-метокси-1,2-дигидронаф- талин в виде темного масла.
Масло растворяли в 25 мл п-диоксана. К раствору затем добавляли 7,5 мл йодистого метила, и смесь перемешивали в течение 18 ч при нагревании с обратным холодильником в атмосфере азота. Смесь затем разбавляли 25 мл воды и 1 мл ледяной уксусной кислоты, после чего она перемешивалась в течение 3 ч при нагревании с обратным холодильником. Смесь затем охлаждали до комнатной температуры, и летучие вещества удаляли в вакууме. Получающийся остаток суспендировали в воде, и водную смесь экстрагировали эфиром. Органические экстракты объединяли, промывали насыщенным водным хлористым натрием, сушили над сульфатом натрия и упаривали в вакууме, получая 3,5 г коричневого масла. Масло растворяли в смеси гексана и эфира (1:1) и помещали на кремнеземную колонку мгновенной хроматографии. Колонку элюировали смесью гексана и эфира (1:1), и элюат упаривали в вакууме, получая 3,27 г (86,5%) целевого тетралона в виде светло-коричневого прозрачного масла.
В. 1-Метил-2-пиперазинил-8-метокси-1,2,3,4-тетрагидронафталин.
Указанный выше тетралон (3,27 г, 17,3 ммоль) растворяли в 150 мл толуола. К раствору затем добавляли 3,04 г (34,6 ммоль) пиперазина с последующим добавлением 7,2 г (38,2 ммоль) п-толуолсульфокислоты. Смесь перемешивали при нагревании с обратным холодильником в атмосфере азота при постоянном удалении воды. Спустя 2 ч реакционную смесь охлаждали до комнатной температуры, и летучие вещества удаляли в вакууме с получением тозилата 3-пиперазинил-4-метил-5-метокси-1,2-ди- гидронафталина в виде светло-желтого твердого вещества.
Твердое вещество суспендировали примерно в 250 мл этанола. К смесь добавляли порциями 2,4 г боргидрида натрия. Смесь перемешивали в течение 18 ч при комнатной температуре. Реакционную смесь затем разбавляли 10%-ной водной HCl, после чего ее дополнительно разбавляли водой, а затем экстрагировали эфиром. Водный слой подщелачивали гидроксидом аммония и экстрагировали смесью хлороформа и изопропилового спирта (3:1). Органические слои объединяли, сушили над сульфатом натрия и упаривали в вакууме, получая желтое масло. Масло растворяли в метиленхлориде и помещали на кремнеземную колонку мгновенной хроматографии. Колонку элюировали метиленхлоридом, содержащим 5% метанола и следы гидроксида аммония. Элюат выпаривали в вакууме, получая 0,35 г целевого соединения в виде желтого стекла.
ЯМР (СДСl3): 7,2-6,42 (м. , 3Н), 3,76 (с., 3Н), 3,6-3,2 (м., 1Н), 3,04-2,40 (м. , 13Н), 2,0-1,40 (м., 1Н), 1,28-1,16 (д., j = 7), 1,13-1,00 (д., j = 7).
П р и м е р 14. Получение дихлоргидрата 2-пиперазинил-6-хлор-1,2,3,4-тетрагидро-нафталина.
Смесь, состоящую из 6-хлор-2-тетралона (5,41 г), пиперазина (5,16 г) и 4А молекулярных сит (8 г) в 100 мл сухого толуола: нагревали с обратным холодильником при перемешивании в течение короткого периода времени в атмосфере азота. Толуол выпаривали и к остатку добавляли 100 мл /ТГФ и 10 мл метанола. Добавляли цианоборгидрид натрия (1,86 г), раствор перемешивали на холоду в атмосфере азота и порциями периодически над поверхностью добавляли газообразный HCl до тех пор, пока реакционный раствор не оставался кислым. После перемешивания при комнатной температуре в течение ночи, сита отфильтровывали, и фильтрат выпаривали досуха. Остаток распределяли между 1 н. HCl и простым эфиром; водную фазу отделяли и экстрагировали два раза эфиром. Раствор подщелачивали гидроксидом натрия. Масло, которое образовывалось, экстрагировали в эфир, и эфирный раствор сушили над сульфатом натрия. Выпаривание эфира давало твердое вещество (5,1 г). Раствор твердого вещества в этаноле обрабатывали избытком газообразного HCl. После охлаждения в течение некоторого периода времени кристаллы, которые отделялись, отфильтровывали и промывали этанолом. После перекристаллизации из метанола получали 2,64 г целевого соединения; т.пл. 264-265оС.
C14H21Cl3N2.
Вычислено,%: С 51,95; Н 6,54; N 8,65.
Найдено,%: С 52,17; Н 6,60; N 8,71.
П р и м е р 15. Получение дихлоргидрата 2-пиперазинил-6-бром-1,2,3,4-тетрагидро-нафталина.
С помощью способа, описанного в примере 14, целевое соединение приготавливали из 6-бром-2-тетралона; т.п. 258-260оС (этанол).
C14H21BrCl2N2.
Вычислено,%: С 45,68; Н 5,75; N 7,61.
Найдено,%: С 45,65; Н 6,01; N 7,85.
П р и м е р 16. Получение дихлоргидрата 2-пиперазинил-6-фтор-1,2,3,4-тетрагидро-нафталина.
С помощью способа, описанного в примере 14, с использованием молекулярных сит 3А вместо 4А, приготавливали целевое соединение из 6-фтор-2-тетралона; т.пл. 243-245оС (этанол).
C14H21Cl2FN2.
Вычислено,%: С 54,73; Н 6,89; N 9,12.
Найдено,%: С 54,48; Н 6,62; N 9,22.
П р и м е р 17. Получение дихлоргидрата 2-пиперазинил-6-метил-1,2,3,4-тетрагидро- нафталина.
С помощью в основном способа, описанного в примере 14, приготавливали целевое соединение из 6-метил-2-тетралона; т.пл. 266-268оС (метанол).
C15H23Cl2N2.
Вычислено,%: С 59,41; Н 7,98; N 9,24.
Найдено,%: С 59,61; Н 7,87; N 8,99.
П р и м е р 18. Получение дихлоргидрата 2-пипераззинил-6-метокси-1,2,3,4-тетра- гидронафталина.
В основном с помощью способа, описанного в примере 14, целевое соединение получалось из 6-метокси-2-тетралона; т.пл. 264-266оС (метанол).
C15H24Cl2N2O.
Вычислено,%: С 56,43; Н 7,58; N 8,77.
Найдено,%: С 56,26; Н 7,42; N 8,50.
П р и м е р 19. Получение дихлоргидрата 2-пиперазинил-6,7-дихлор-1,2,3,4-тетра- гидронафталина.
Смесь, состоящую из 6,7-дихлор-2-тетралона (4,30 г), пиперазина (3,44 г), и молекулярных сит 4А (4Г) в 100 мл толуола перемешивали на протяжении ночи в атмосфере азота и подогревали недолго. Сита отфильтровывали, и фильтрат освобождали от растворителя на испарителе. К остатку добавляли 75 мл ТГФ и 6 мл метанола. Раствор охлаждали на ледяной бане в атмосфере азота, и добавляли 1,24 г цианоборгидрида натрия. Периодически добавляли небольшие количества газообразного HCl до тех пор, пока раствор не оставался кислым. Раствор перемешивали на протяжении ночи при температуре окружающей среды, растворители выпаривали, и остаток растворяли в разбавленной HCl. Кислый раствор экстрагировали тремя порциями эфира, а затем подщелачивался гидроксидом натрия. Масло, которое отделялось, экстрагировалось эфиром, и эфирный раствор сушился сульфатом натрия. Остаток, остающийся после выпаривания эфира, растворялся в этаноле, и добавлялся избыток безводного HCl. Кристаллическое твердое вещество, выделившееся из этанола, плавилось примерно при 272-275оС. После перекристаллизации из этанола получали 1,1 г целевого соединения; т.пл. 278-282оС.
C14H20Cl4N2.
Вычислено,%: С 46,95; Н 5,63; N 7,84.
Найдено,%: С 47,22; Н 5,54; N 7,74.
П р и м е р 20. Получение дихлоргидрата 2-(N-метилпиперазинил)-6-бром-1,2,3,4-тет- рагидронафталина.
Смесь 6-бром-2-тетралона (4,50 г) N-метилпиперазина (4,0 г) и карбоната калия (55,52 г) в 75 мл ТГФ перемешивали в течение примерно 1 ч. Твердые вещества отфильтровывали, и к фильтрату добавляли цианоборгидрид натрия (1,24 г). К перемешиваемому раствору при комнатной температуре добавляли порциями достаточное количество эфирного HCl, чтобы подкислить раствор. Когда ЯМР-анализ образца реакционного раствора показывал отсутствие винильного протона промежуточного енамина, растворители выпаривались. Остаток распределяли между эфиром и 1 н. HCl и эфирную фазу отбрасывали. Водную фазу подщелачивали и органическое вещество, которое отделялось, экстрагировали в простой эфир и этилацетат. Экстракты сушили сульфатом натрия. Растворители выпаривали, и остаточное основное вещество в этаноле обрабатывали избытком соляной кислоты. Получалось 4,41 г целевого соединения, т.пл. 270-272оС (разл.).
C15H23N2BrCl2.
Вычислено,%: С 47,14; Н 6,07; N 7,33.
Найдено,%: С 46,99; Н 5,95; N 7,54.
П р и м е р 21. Получение дихлоргидрата 2-(N-метилпиперазинил)-6-хлор-1,2,3,4-тет- рагидронафталина.
При использовании в способе, в общем описанном в пример 20, 6-хлор-2-тетралона вместо 6-бром-2-тетралона, получали целевое соединение; т.пл. 269-271оС (этанол).
C15H23N2Cl3.
Вычислено,%: С 53,35; Н 6,86; N 8,29.
Найдено,%: С 53,11; Н 6,68; N 8,23.
П р и м е р 22. Получение дихлоргидрата цис-1-метил-2-пиперазинил-6-хлор-1,2,3,4-тетрагидронафталина.
Раствор 13,7 г 6-хлор-2-тетралона и 12,7 мл пирролидина в 250 мл сухого бензола нагревали с обратным холодильником с использованием ловушки для воды Дина-Старка в течение 17 ч. Бензол и избыток пирролидина удаляли на вращающемся испарителе. К остатку добавляли сухой толуол, а затем выпаривали оставляя кристаллический остаток.
ЯМР (СДСl3): 1,9 (м., 4Н), 2,17-2,54 (м., 2Н), 2,54-2,83 (м., 2Н), 3,25 (м., 4Н), 5,07 (с., 1Н), 6,62-7,42 (м., 3Н).
Твердое вещество растворяли в 100 мл сухого диоксана, добавляли 24 мл метилйодида и раствор нагревали с обратным холодильником в течение примерно 3,5 ч. Примерно через 0,5 ч начинало отделяться кристаллическое твердое вещество. Раствор, содержащий 1 мл уксусной кислоты в 50 мл воды, добавляли к реакционной смеси, и всю смесь нагревали с обратным холодильником в течение примерно 3,5 ч. Растворители выпаривали, и остаток распределялся между водой и эфиром. Эфирную фазу отделяли, экстрагировали небольшим количеством разбавленной соляной кислоты и сушили. Удаление растворителя давало примерно 12 г масла. Препаративная HPZ C давала 9,3 г 6-хлор-1-метил-2-тетралона.
ЯМР (СДСl3): 1,48 (д. , 3Н), 2,28-2,67 (м., 2Н), 2,78-3,2 (м., 2Н), 3,25-3,73 (м., 1Н), 7,0-7,35 (м., 3Н).
К 15 мл 4А молекулярных сит в 250 мл сухого толоула добавлялись пиперазин (6,37 г) и 6-хлор-1-метил-2-тетралон (8,6 г). При использовании ловушки для воды Дина-Старка смесь перемешивали и нагревали при кипении с обратным холодильником в атмосфере азота. Спустя 23 ч анализ ЯМР образца реакционного раствора показал присутствие тетралона. Добавляли 500 мг п-толуолсульфокислоты и смесь перемешивали и нагревали с обратным холодильником в течение дополнительных 26 ч. Добавляли дополнительно 10 г 4А сит, и нагревание с обратным холодильником продолжали в течение дополнительных 10 ч. Сита отфильтровывали и толуол выпаривали из фильтрата. Остаток растворялся в смеси 200 мл ТГФ и 20 мл метанола. К получающемуся в результате раствору, перемешиваемому в атмосфере азота при охлаждении на ледяной бане, добавляли цианоборгидрид натрия (2,68 г). К раствору добавляли газообразный HCl порциями до тех пор, пока раствор не оставался кислым. После перемешивания на протяжении ночи при комнатной температуре растворители удаляли на вращающемся испарителе, и остаток распределяли между эфиром и разбавленной соляной кислотой. Кислый раствор отделяли и подщелачивали на холоде гидроксидом натрия. Масло, которое отделялось, экстрагировали эфиром и эфирный раствор сушили сульфатом магния. Удаление эфира давало 8,1 г масла. Масло объединялось с 1 г продукта от предыдущей реакции, проведенной в небольшом масштабе. Раствор всего продукта в этаноле обрабатывали избытком газообразного хлористого водорода. Кристаллы, которые отделялись, отфильтровывали и дегидрировали на паровой бане со 100 мл метанола. Несколько сборов кристаллов, полученных из метанола, объединяли и перекристаллизовывали из этанола, давая 6,3 г целевого соединения. При сушке при 120оС в вакууме дихлоргидрат терял один моль хлористого водорода и давал следующее:
C15H22N2Cl2
Вычислено,%: С 59,80; Н 7,36; N 9,30.
Найдено,%: С 59,83; Н 7,11; N 9,29.
Образец дихлоргидратной соли обрабатывали гидроксидом натрия для получения свободного амина, который имел следующие данные анализа:
ЯМР (СДСl3): 1,14 (д., j = 7, 3Н), 1,63 (м., j = 6,4, 12, 1Н), 2,06 (м. , j = 3, 1, 1Н), 2,14 (широкий м., 1Н), 2,36 (м., j = 3, 4,4, 12, 1Н), 2,48-2,68 (м. , 4Н), 2,75 (м., 1Н), 2,85 (м., 1Н), 2,93 (т., j = 5,2, 4Н), 3.14 (м., j = 7, 4.4, 1,1Н), 6,97-7,12 (м., 3Н).
После стояния во влажной атмосфере в течение 60 ч образец дихлоргидрата давал дигидрат дихлоргидрата; т.пл. 244-246оС.
C15H27N2O2Cl3.
Вычислено,%: С 48,21; Н 7,28; N 7,50.
Найдено,%: С 48,30; Н 7,05; N 7,35.
Свободный амин получали из (±) рацемического кристаллического дихлоргидрата (32,7 г) с водой, эфиром и 40 мл 5 н. гидроксида натрия. Основную водную порцию экстрагировали дополнительно три раза эфиром. Объединенные эфирные экстракты промывали два раза водой и один раз насыщенным раствором хлористого натрия. Эфирный раствор сушили над сульфатом магния и выпаривали, получая 24,8 г свободного амина.
А. Получение (+) цис-1-метил-2-пиперазинил-6-хлор-1,2,3,4-тетрагидронафталина, соли (-) ди-п-толуол-L-винной кислоты (2:1).
(-) Ди-п-толуол-L-винную кислоту (18,9 г), растворенную в 42 мл метанола, добавляли к свободному амину (24,8 г) в 100 мл метанола при перемешивании. Осадок растирали десять раз с горячим метанолом, содержащим 10% воды (общий объем 11,25 л), получая 21,2 г соли (-) ди-п-толуол-L-винной кислоты, т.пл. 244-245оС.
Свободный амин получали и из соли (21,1 г) с эфиром, водой и 10 мл 5 н. гидроксида натрия. Объединенные эфирные экстракты промывали четыре раза водой и один раз насыщенным хлористым натрием. Эфирный раствор сушили над сульфатом магния, фильтровали и выпаривали, получая 11,3 г.
(-) Ди-п-толуол-L-винную кислоту (8,64 г), растворенную в 60 мл метанола, добавляли к амину (11,3 г) в 1400 мл метанола. Смесь подогревали в течение короткого периода времени на паровой бане и оставляли стоять при комнатной температуре. Осадок отфильтровывали и промывали метанолом, а затем простым эфиром, и его сушили в вакууме, получая 19,2 г соли (-) ди-п-толуол)-L-винной кислоты; т.пл. 250-251оС.
C50H60N4O8Cl2.
Вычислено,%: С 65,57; Н 6,60; N 6,12.
Найдено,%: С 65,61; Н 6,50; N 6,15.
В. Получение (+) цис-1-метил-2-пиперазинил-6-хлор-1,2,3,4-тетрагидронафталина, Д(-)-виннокислотной соли (1:1).
Д(-)-винную кислоту (5,78 г), растворенную в 100 мл метанола, добавляли к свободному амину, полученному из части А, растворенному в 450 мл метанола. Осадок, который быстро образовался, оставляли стоять в течение ночи при комнатной температуре. Его отфильтровывали и промывали метанолом, а затем простым эфиром. Осадок сушили воздухом, получая 14,5 г соли Д(-)-винной кислоты; т.пл. 203оС. Соль Д(-)-винной кислоты перекристаллизовывали с помощью растворения ее в 4,5 л метанола, содержащего 50 мл воды, при нагревании с обратным холодильником, после чего раствор концентрировался до 2,5 л.
После стояния на протяжении ночи при комнатной температуре осадок отфильтровывали и промывали метанолом, а затем простым эфиром. После сушки получали 11,3 г соли Д(-)-винной кислоты; т.пл. 208-209оС.
C19H27N2O6Cl.
Вычислено,%: С 55,01; Н 6,56; N 6,75.
Найдено,%: С 55,12; Н 6,71; N 6,68.
Удельное вращение в ДМСО при 25оС:
589 нм + 48,04 градуса,
365 нм + 176,94 градуса.
С. Дигидрат дихлоргидрата (+) цис-1-метил-2-пиперазинил-6-хлор-1,2,3,4-тетра- гидронафталина.
Свободное основание приготавливали из Д(-)-виннокислотной соли с использованием 50 мл 1 н. гидроксида натрия и эфира. Эфирные экстракты промывали два раза 25 мл 1 н. гидроксида натрия. Эти водно-щелочные экстракты экстрагировали три раза эфиром. Объединенные эфирные экстракты промывали два раза водой и один раз насыщенным раствором хлористого натрия. Эфирный раствор сушили над сульфатом натрия, фильтровали и выпаривали.
Дихлоргидратную соль приготавливали с помощью добавления метанольного раствора хлористого водорода к свободному амину в метаноле. Осадок отфильтровывали, промывали метанолом и сушили, получали 7,0 г; т.пл. 205оС.
Удельное вращение в воде при 25оС:
589 нм + 47,72 градусов,
365 нм + 175,84 градусов.
Образец дихлоргидратной соли гидратировали во влажной атмосфере в течение 40 ч и получали дигидрат дихлоргидрата; т.пл. 225-230оС.
C15H27N2O2Cl3.
Вычислено,%: С 48,21; Н 7,28; N 7,50.
Найдено,%: С 48,17; Н 7,01; N 7,58.
Удельное вращение в воде при 25оС:
589 нм + 45,83 градусов,
365 нм + 169,64 градусов
Д. (-)цис-1-Метил-2-пиперазинил-6-хлор-1,2,3,4-тетрагидронафталин, (+) ди-п-толуол-Д-виннокислотная соль (2:1).
Объединенные фильтраты от растираний (+)цис-1-метил-2-пиперазинил-6-хлор-1,2,3,4-тетрагидронафталина, соли (-)ди-п-толуоил-винной кислоты (2:1) объединяли и превращали в свободный амин (11,8 г) с использованием смеси эфира, воды и гидроксида натрия.
(+)Ди-п-толуоил-Д-винную кислоту (9,0 г), растворенную в 60 мл метанола, добавляли к амину в 1500 мл метанола. Смесь подогревали в течение короткого промежутка времени на паровой бане и оставляли стоять при комнатной температуре на протяжении ночи.
Осадок отфильтровывали и промывали метанолом, а затем эфиром. После сушки в вакууме получали 23,5 г соли (+)ди-п-толуоил-Д-винной кислоты; т.пл. 249-250оС.
C50H60N4O8Cl2.
Вычислено,%: С 65,57; Н 6,60; N 6,12.
Найдено,%: С 65,41; Н 6,41; N 6,12.
Е. (-)цис-1-Метил-2-пиперазинил-6-хлор-1,2,3,4-тетрагидронафталин, соль L (+)-винной кислоты (1:1).
Свободный амин приготавливали из данной соли (23,5 г) эфиром и 1 н. гидроксидом натрия. Водно-щелочную часть экстрагировали три раза дополнительными количествами эфира. Объединенные эфирные экстракты промывали три раза водой и один раз насыщенным раствором хлористого натрия. Эфирный раствор сушили над сульфатом натрия, фильтровали и выпаривали, получая 9,7 г свободного амина.
L(+)-Винную кислоту (5,53 г), растворенную в 100 мл метанола, добавляли к свободному амину в 500 мл метанола. Осадок, который образовывался после завершения добавления кислоты, оставляли стоять при комнатной температуре на протяжении ночи. Осадок отфильтровывали и промывали метанолом, а затем эфиром. После сушки воздухом получали 14,4 г соли L(+)-винной кислоты; т. пл. 204-206оС.
Данную соль перекристаллизовывали с помощью растворения ее в 4,5 л метанола при нагревании с обратным холодильником, после чего раствор концентрировали до 2 л, фильтровали и оставляли стоять при комнатной температуре на протяжении ночи.
Получающийся в результате осадок отфильтровывали и промывали метанолом, а затем эфиром. После сушки получали 11,1 г соли.
C19H27N2O6Cl.
Вычислено,%: С 55,01; Н 6,56; N 6,75.
Найдено,%: С 55,18; Н 6,79; N 6,76.
Удельное вращение в ДМСО при 25оС: 589 нм - 45,57 градусов,
365 нм - 169,75 градусов
F. Дигидрат дихлоргидрата (-)цис-1-метил-2-пиперазинил-6-хлор-1,2,3,4-тетра- гидронафталина.
Свободный амин продукта из раздела Е (11,1 г) приготавливали с помощью обработки его 55 мл 1 н. гидроксида натрия и эфира. Эфирный экстракт промывали два раза 25 мл 1 н. гидроксида натрия. Объединенные эфирные экстракты промывали два раза 25 мл воды и один раз насыщенным раствором хлористого натрия. Эфирный раствор сушили над сульфатом натрия, фильтровали и выпаривали.
Дихлоргидратную соль получали с помощью добавления метанольного хлористого водорода к свободному амину в метаноле. Получающийся осадок отфильтровывали и промывали метанолом, получая 7,5 г; т.пл. 216-218оС.
Удельное вращение в воде при 25оС:
589 нм - 44,73 градусов.
365 нм - 166,11 градусов.
Образец дихлоргидратной соли гидратировали в дигидрат, оставляя ее стоять во влажной атмосфере в течение 40 ч, и получали 7,5 г; т.пл. 230-235оС.
C15H27N2O2Cl3.
Вычислено,%: С 48,21; Н 7,28; N 7,50.
Найдено,%: С 48,20; Н 7,07; N 7,56.
Удельное вращение в воде при 25оС:
589 нм - 45,69 градусов.
365 нм - 167,63 градусов.
П р и м е р 23. Получение дихлоргидрата 1-метил-2-пиперазинил-1,2,3,4-тетрагидро- нафталина.
С использованием 1-метил-2-тетралона и пиперазина в способе, описанном в примере 22, получали целевое соединение; т.пл. 227-230оС (этанол).
C15H24N2Cl2.
Вычислено,%: С 59,41; Н 7,98; N 9,24.
Найдено,%: С 59,37; Н 7,73; N 9,52.
П р и м е р 24. Получение дихлоргидрата 1-метил-2-(N-метилпиперазинил)-1,2,3,4-тет-рагидронафталина.
Раствор 1-метил-2-тетралона (5,0 г) и N-метилпиперазина (6,25 г) в 140 мл толуола перемешивали и нагревали с обратным холодильником в атмосфере азота с использованием ловушки Дина-Старка для азеотропного удаления воды. Спустя 60 ч толуол выпаривали, и остаток растворяли в 90 мл ТГФ и 9 мл метанола. Раствор охлаждали при перемешивании в атмосфере азота, добавляли 2, г цианоборгидрида натрия и периодически порциями по мере необходимости добавляли газообразный HCl до тех пор, пока раствор не становился кислым. Переработка, выделение и обработка неочищенного основного продукта сухим HCl, как описано в примере 22, давали целевое соединение, т.пл. 262-265оС (разл. , этанол).
C16H26N2Cl2.
Вычислено,%: С 60,57; Н 8,26; N 8,83.
Найдено,%: С 60,84; Н 8,10; N 8,96.
П р и м е р 25. Получение дихлоргидрата 2-гомопиперазинил-6-хлор-1,2,3,4-тетра- гидронафталина.
К раствору 6-хлор-2-тетралона (7,22 г) и гомопиперазина (8,0 г) в 100 мл толуола добавляли 10 г 3А молекулярных сит. Смесь нагревали на паровой бане в течение 2 ч, сита отфильтровывали и толуол выпаривали из фильтрата. Остаток растворяли в смеси 100 мл ТГФ и 30 мл метанола. Получающийся в результате раствор охлаждали на ледяной бане при перемешивании в атмосфере азота, добавляли 2,51 г цианоборгидрида натрия, и при перемешивании и охлаждении порциями периодически добавляли газообразный хлористый водород до тех пор, пока раствор не становился кислым. После перемешивания на протяжении ночи при комнатной температуре растворители выпаривали и остаток распределяли между разбавленной соляной кислотой и эфиром. Кислотно-водный слой отделяли и подщелачивали гидроксидом натрия. Масло, которое образовывали, экстрагировали эфиром. Эфирный раствор сушили сульфатом магния, и эфир выпаривался. Остаток растворяли в этаноле и к раствору добавляли избыток газообразного HCl. Растворитель выпаривали. Остаток кристаллизовали медленно из смеси этанола и эфира и получали 3,22 г целевого соединения, т.пл. 181-183оС.
C15H23N2Cl3.
Вычислено,%: С 53,35; Н 6,86; N 8,29.
Найдено,%: С 53,40; Н 7,12; N 8,49.
П р и м е р 26. Получение дихлоргидрата 1-метил-2-пиперазинил-6-бром-1,2,3,4-тет- рагидронафталина.
4 г 1-метил-6-бром-1,2,3,4-тетрагдиронафтален-2-она и 2,89 г пиперазина нагревали при кипении с обратным холодильником на протяжении ночи в 100 мл толуола, содержащего 5 г 4А молекулярных сит и 50 мг п-толуолсульфокислоты. Реакционную смесь фильтровали и фильтрат выпаривали. К остатку добавлялись цианоборгидрид натрия (1,05 г), 100 мл ТГФ и 10 мл метанола. Периодически к раствору при перемешивании добавляли газообразный HCl при температуре ледяной бани до тех пор, пока раствор не становился кислым.
Данную реакционную смесь перемешивали при комнатной температуре на протяжении ночи и выпаривали. Остаток экстрагировали эфиром и водой. Кислотно-водную часть подщелачивали на холоду гидроксидом натрия и экстрагировали эфиром. Эфирный экстракт сушили над сульфатом магния, фильтровали и упаривали, давая масло. Дихлоргидратную соль получали с помощью добавления газообразного HCl к свободному основанию в этаноле на холоду. Перекристаллизация дихлоргидратной соли из метанола давала 0,45 г целевого соединения, т.пл. 243-245оС.
C15H23N2BrCl2.
Вычислено,%: С 47,14; Н 6,07; N 7,33.
Найдено,%: С 47,39; Н 7,31; N 7,15.
Как отмечалось выше, соединения данного изобретения полезны для селективного ингибирования поглощения серотонина. Поэтому еще одно воплощение данного изобретения заключается в способе ингибирования поглощения серотонина у млекопитающих, который включает введение млекопитающему, требующему повышенной нейротрансмиссии серотонина, фармацевтически эффективного количества соединения данного изобретения.
Термин "фармацевтически эффективное количество", используемый здесь, представляет количество соединения изобретения, которое способно ингибировать потребление (поглощение) серотонина. Конкретная доза назначаемого для приема соединения, согласно данному изобретению, будет определяться конечно конкретными обстоятельствами, присущими конкретному случаю, включающими, например, вводимое соединение, способ введения, и состояние, подвергаемое лечению. Типичная суточная доза обычно содержит примерно 0,01-20 мг/кг активного соединения данного изобретения. Предпочитаемые суточные дозы обычно составляют примерно 0,05-10 мг/кг, и в идеальном случае примерно 0,1-5 мг/кг.
Соединения могут вводиться с помощью разнообразных способов, включая оральный, ректальный, трансдермальный (через кожу), подкожный, внутривенный, внутримышечный и интраназальный (в нос). Особым признаком соединений данного изобретения является то, что они обладают продолжительным сроком действия и поэтому способны ингибировать потребление серотонина в течение продолжительного периода времени. Также характерным признаком соединений данного изобретения является то (как было найдено), что они проявляют низкую степень токсичности по отношению к млекопитающим. Наконец, соединения изобретения являются чрезвычайно селективными как ингибиторы поглощения серотонина по отношению к поглощению других моноаминов.
Было показано, что большое разнообразие физиологических функций подвержено влиянию серотонэргических невральных систем головного мозга. Считают, что как таковые, соединения данного изобретения обладают способностью лечить разнообразные нарушения у млекопитающих, связанные с этими невральными системами, такие как расстройства, депрессия, алкоголизм, боли, потеря памяти, беспокойство и курение. Поэтому настоящее изобретение представляет также способы лечения указанных выше расстройств при дозах, представленных выше для ингибирования поглощения серотонина у млекопитающих.
Следующий эксперимент был проведен для демонстрации способности соединений настоящего изобретения ингибировать поглощение серотонина. Данная общая процедура описана в работе авторов
Wong et al. Drug development Research 6: 397-403 (1985).
Самцов крыс Sprague-Dawley (110-150 г) от Харлан Индастриз Кумберленд, 1N) кормили досыта "Purina Chow" в течение по крайней мере 3 дней до того, как они были использованы в исследованиях. Крыс умерщвляли путем обезглавливания. Головной мозг всех особей удаляли и рассекали (вскрывали). Кору головного мозга гомогенизировали в 9 объемах среды, содержащей 0,32М сахарозы и 10 мМ глюкозы. После дифференциального центрифугирования при 1000 г в течение 10 мин и 17000 г в течение 28 мин отделялись неочищенные синаптосомальные препараты. Окончательные таблетки суспендировали в той же среде и хранили во льду до тех пор, пока они не будут использоваться в тот же день.
Синаптосомальное поглощение 3Н-серотониа (3Н-5-гидрокситриптамина, 3Н-5НТ) определяли следующим образом. Кортикальные синаптосомы (эквивалентные 1 мг протеина) инкубировали при 37оС в течение 5 мин в 1 мл Кребс-бикарбонатной среды, содержащей также 10 мМ глюкозы, 0,1 ипрониазида, 1 мМ аскорбиновой кислоты, 0,17 мМ ЕДТА и 50 нМ 3Н-5НТ. Реакционную смесь немедленно разбавляли 2 мл охлажденного льдом Кребсбикарбонатного буфера и фильтровали под вакуумом с использованием сборника клеток (Brandel, Gaithersburg, МД). Фильтры прополаскивали два раза приблизительно 5 мл охлажденного льдом 0,9%-ного физиологического раствора и переносили в счетную пробирку, содержащую 10 мл сцинтилляционной жидкости (PCS, Aruersham. Qrbington Heights, IL). Радиоактивность измеряли с помощью жидкостного сцинтилляционного спектрофотометра. Аккумулирование 3Н-5НТ при 4оС представлено фон и вычиталось из всех проб.
Повторяя эксперимент, приведенный выше для соединений аналогичной структуры, получали данные по активности для сравнения с активностью соединений настоящего изобретения (см.табл. 1 и 2).
Отличительной особенностью продуктов заявленного способа является наличие в 2-положений тетралина. В отличие от обычных структурных аналогов, содержащих в 2-положении моно- или диалкиламино группу или непиперазиновую циклическую аминогруппу, продукты способа согласно изобретению проявляют гораздо более превосходящую активность по ингибированию поглощения серотонина, о чем свидетельствуют данные, представленные в табл. 1 с результатами оценки различных соединений настоящего изобретения. В табл. 1 в первой колонке указан номер примера оцениваемого соединения; следующие 7 колонок указывают структуру оцениваемого соединения в сочетании с формулой, представленной в заголовке; в предпоследнем столбце указана солевая форма оцениваемого соединения; и последний столбец дает количество испытываемого соединения, выраженное в наномолярной концентрации, необходимой для ингибирования поглощения 3Н-5НТ на 50%, и приведенной в табл. 1 в виде величины IC50. Числа в скобках, если они указаны, представляют ингибирование в процентах при 1000 нМ.
Соединения данного изобретения преобразуются в готовую препаративную форму, включающую соединение изобретения и фармацевтически приемлемый носитель, разбавитель или эксципиент его.
Композиции предпочтительно формулируются в виде дозированных единичных форм, причем каждая дозировка обычно содержит примерно 0,1-500 мг, и предпочтительно примерно 1-250 мг, активного ингредиента. Термин "единичная дозированная форма" относится к физически дискретным единицам, подходящим в качестве разовых дозировок для людей и других млекопитающих, причем каждая единица содержит заданное количество активного материала, рассчитанного для обеспечения желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим носителем.
Следующие ниже примеры готовых препаратов являются лишь иллюстративными и никоим образом не предназначаются для ограничения объема изобретения.
Готовый препарат 1.
Твердые желатиновые капсулы приготавливают с использованием следующих ингредиентом:
Количество, мг/капсула
Дигидрат дихлоргидрата
цис-1-метил-2-пипера-
зинил-6-хлор-1,2,3,4-тет- рагидронафталина 250 Крахмал, высушенный 200 Стеарат магния 10 Всего: 460
Указанные выше ингредиенты смешиваются и заполняются в твердые желатиновые капсулы в количестве 460 мг.
Готовый препарат 2.
Таблетка приготавливается с использованием указанных ниже ингредиентов:
Количество, мг/таблетка
Дихлоргидрат
цис-1-метил-2-пипе-
разинил-1,2,3,4-тет- рагидронафталина 250
Целлюлоза, микро- кристаллическая 400
Двуокись кремния, дымная 10 Стеариновая кислота 5 Всего 665
Компоненты смешиваются и прессуются с образованием таблеток каждая массой 665 мг.
Готовый препарат 3.
Аэрозольный раствор приготавливают с содержанием следующих компонентов, мас.%:
Дихлоргидрат
2-пиперазинил-6-
хлор-1,2,3,4-тетра- гидронафталина 0,25 Этанол 29,75
Пропеллант 22 (хлордифторметан) 70,00
Активное соединение смешивают с этанолом, и смесь добавляют к части пропелланта 22, охлажденного до -30оС, и переносят в заполняющее устройство. Затем в контейнер из нержавеющей стали подают требуемое количество и разбавляют остатком пропелланта. Затем контейнер снабжается клапанным элементом.
Готовый препарат 4.
Таблетки, каждая содержащая 60 мг активного ингредиента, изготавливают следующим образом:
Дималеат 2-(N-метил-
пиперазинил)-8-хлор-
1,2,3,4-тетрагидро- нафталина 60 мг Крахмал 45 мг
Микрокристаллическая целлюлоза 35 мг
Поливинилпирролидон
(в виде 10%-ного раствора в воде) 4 мг
Натриевый карбокси- метил-крахмал 4,5 мг Стеарат магния 0,5 мг Тальк 1 мг Всего: 150 мг
Активный ингредиент, крахмал и целлюлозу пропускают через сито N 45 меш США и тщательно смешивают. Водный раствор, содержащий поливинилпирролидон, смешивают с полученным в результате порошком и смесь затем пропускают через сито США N 14 меш. Гранулы, полученные таким образом, сушат при 50оС и пропускают через сито США N 18 меш. Натриевый карбоксиметил-крахмал, стеарат магния и тальк, предварительно пропущенные через сито N 60 меш США, затем добавляют к гранулам, которые после смешения прессуются на таблетирующей машине, давая таблетки, каждая массой 150 мг.
Готовый препарат 5.
Капсулы, каждая содержащая 80 мг активного ингредиента, изготавливают следующим образом:
Дихлоргидрат 2-пипе-
разинил-5-бром-8-метокси-
1,2,3,4-тетрагидро- нафталина 80 мг Крахмал 59 мг
Микрокристал- лическая целлюлоза 59 мг Стеарат магния 2 мг Всего 200 мг
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито N 45 меш США и заполняют в твердые желатиновые капсулы в количестве по 200 мг.
Готовый препарат 6.
Медицинские свечи или суппозитории, каждая содержащая 225 мг активного ингредиента, изготавливают следующим образом:
гидрат дихлоргидрата 2-
(N-метилпиперазинил)-5-
формил-8-метокси-
1,2,3,4-тетрагидро- нафталина 225 мг
Глицериды насыщенной жирной кислоты 2000 мг Всего 2225 мг
Активный ингредиент пропускают через сито N 60 меш США и суспендируют в глицеридах насыщенной жирной кислоты, предварительно расплавленных с использованием минимально необходимого тепла. Смесь затем выливают в форму для свечей номинальной емкостью 2 г, и ее оставляют охлаждаться.
Готовый препарат 7.
Суспензии, каждая содержащая 50 мг активного ингредиента на дозу в 5 мл, приготавливают следующим образом:
Дихлоргидрат 2-
(N-метилпиперазинил)-5-
циано-8-метокси-
1,2,3,4-тетрагидро- нафталина 50 мг
Натриевая карбокси- метилцеллюлоза 50 мг Сироп 1,25 мл
Раствор бензойной кислоты 0,10 мл Корригент По желанию Краситель По желанию
Очищенная вода до общего количества 5 мл
Активный ингредиент пропускают через сито N 45 меш США и смешивают с натриевой карбоксиметилцеллюлозой и сиропом, образуя гомогенную пасту. Раствор бензойной кислоты, корригент и красящее вещество разбавляют частью воды и добавляют при перемешивании. Затем добавляют достаточное количество воды для получения требуемого объема.
Готовый препарат 8.
Готовый внутривенный препарат может быть получен следующим образом:
Дихлоргидрат 2-(N-метил-
пиперазинил)-8-метокси-
1,2,3,4-тетрагидро- нафталина 100 мг
Изотонический физиоло- гический раствор 1000 мл
Раствор указанных выше ингредиентов обычно вводят внутривенно со скоростью 1 мл/мин субъекту, страдающему от депрессии.
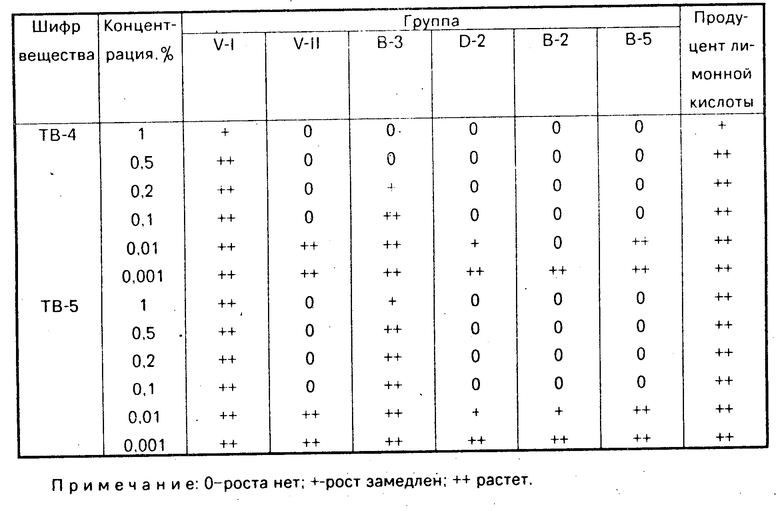

Сущность изобретения: продукт общей формулы I, где R - H, CH3 ; R1-H, CH3 ; Х - -CH2CH2- или -CH2-CH2-CH2 ; R2-H , Hal, C1-C3 -алкокси, C1-C3 -алкил; R3-H , Hal; R4-H , Hal, C1-C3 -алкил, C1-C3 -алкокси; R5-H , C1-C3 -алкил, C1-C3 -алкокси. Реагент 1: соответствующий тетралон 2. Реагент 2: соответствующий пиперазин или гомопиперазин 3. Условия реакции: полученный при взаимодействии соединений 2 и 3 продукт восстанавливают. Новые соединения - селективные ингибиторы поглощения серотонина, которые не оказывают непосредственного воздействия на нейронные рецепторы. 1 табл. Формула I: 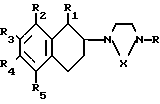
Способ получения производных 1,2,3,4-тетрагидронафталинов общей формулы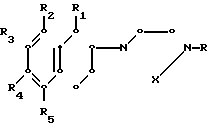
где R -водород или метил;
R1 - водород или метил;
Х - группа -СН2СН2 или -СН2СН2СН2-;
R2 - водород, галоид, С1 - С3-алкокси и С1 - С3-алкил;
R3 - водород или галоид;
R4 - водород, галоид, С1 - С3-алкил и С1 - С3-алкокси;
R5 - водород, С1 - С3-алкил и С1 - С3-алкокси, при условии, что, если R1 - метил, то R2 и R4 одновременно могут быть водородом, если R1 - водород, один из R2 и R4 - водородом, а другой отличен от водорода;
R5 может быть не водородом, только когда R2 отличен от водорода;
R3 может быть галоидом, когда R4 не водород;
или их фармацевтически приемлемой кислотно-аддитивной соли, отличающийся тем, что соединение общей формулы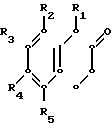

где R1 - R5 имеют указанные значения,
подвергают взаимодействию с соединением общей формулы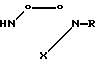
где R имеет указанные значения,
и восстанавливают полученный в результате продукт.
