Изобретение относится к технологии получения активного угля на основе косточек плодов и скорлупы орехов и может быть использовано в медицине при получении энтеросорбента для поглощения токсинов и выведения из организма радиоактивного йода, а также для адсорбции газов и паров.
Известен способ получения дробленого активного угля из скорлупы орехов, включающий сушку, частичное сжиганием, активирование смесью водяного пара, диоксида углероды и кислорода, отмывку угля кислотой, сушку и повторную активацию (см. Патент Японии 56-28846, кл. C 01 B, 31/10, B 01 J 20/20, 1981).
Недостатком известного способа является значительная сложность технологического процесса, многостадийность, образование кислотных отходов, низкий выход углеродного остатка, узкий спектр пористой структуры получаемых углей.
Наиболее близким к изобретению по технической сущности и количеству совпадающих признаков является способ получения активного дробленого угля, включающий карбонизацию скорлупы орехов и косточек плодов в интервале температур 200-700oC со скоростью нагрева более 100 град/мин, активацию продукта при 900oC водяным паром, углекислым газом и воздухами или их смесью (см. Патент США N 4616001, кл. B 01 J 20/20, C 01 B 31/10, 1979.).
Недостатком прототипа является невозможность получения активных углей с высокой адсорбционной емкостью по отношению к среднемолекулярным метаболитам, например, цианкобаламину, а также радиоактивному йоду.
Другим недостатком является низкий выход активного угля и плохая воспроизводимость его качественных характеристик, обусловленная трудностью регулирования процессов карбонизации и активации, наличием в газовой фазе воздуха (кислорода).
Известно, что взаимодействием углерода и кислорода протекает по экзотермической реакции с большим выходом тепла, которое надо вовремя отводить, исключая образования макропор C+O2_→ CO2.
Целью изобретения является повышение адсорбционной емкости активного угля по среднемолекулярным веществам, например цианкобаламину и радиоактивному йоду I131, а также увеличение выхода готового продукта.
Поставленная цель достигается предлагаемым способом, включающим карбонизацию плодовых косточек и скорлупы орехов, которую ведут при температуре 20-759oC со скоростью ее подъема 30-50 град/мин. активацию осуществляют смесью водяного пара и диоксида углерода при 820-870oC, а дробление о размера частиц 0,5-3,5 мм проводят после карбонизации, отмывку угля ведут водой при 70-100oC и соотношении уголь:вода, равном 1:15±5.
Отличие предлагаемого способа от прототипа состоит в том, что карбонизация плодовых косточек и скорлупы орехов ведут при температуре 200-750oC со скоростью ее подъема 30-50 град/мин. затем карбонизат дробят до размера частиц 0,5-3,5 мм и активируют при 820-870oC смесью водяного пара и диоксида углерода, а полученный уголь отмывают водой при соотношении уголь:вода, равном 1:15±5.
Осуществление карбонизации в интервале температур 20-750oC с скоростью 30-50 град/мин в практике производства дробленых косточковых углей неизвестно.
Неизвестны также и предложенные параметры процесса активации этих углей.
Сущность предлагаемого способа состоит в следующем.
Берут косточки, например слив, абрикосов, персиков, вишни или скорлупу грецкого, кокосового, миндального орехов и помещают их в печь (стационарную или вращающуюся), которую начинают нагревать (стац. печь) от комнатной (20oC) температуры до температуры 750oC (удаление летучих до 85-90%) со скоростью подъема 30=50 град/мин. Полученный карбонизат (с содержанием летучих не более 15%) дробят до частиц с размером 0,5-3,5 мм. Эти частицы помещают во вращающуюся печь, нагретую до 850oC и активируют их смесью водяного пара и диоксида углерода в течение 70-120 мин до обгара 40-60% Затем уголь выгружают и после остывания заливают 15 кратным объемом воды при 70-100oC, выдерживают его в воде 30-90 мин при тщательном перемешивании, после чего воду сливают, а у голь сушат и анализируют.
В предложенном способе пористая структура углеродного адсорбента формируется за счет постепенного, плавного удаления летучих веществ ароматического характера, которых в косточковом исходном сырье 70-78% При этом значительную роль в управлении пористой структурой играет размер косточек или скорлупы. Этот фактор обеспечивает сохранение в исходном продукте ароматических углеводородов, которые при нагревании выделяют системы тончайших каналов, обусловливающих при дальнейшей парогазовой активации специфическую микропористую структуру.
Изучалось влияние скорости нагрева, начальной и конечной температуры карбонизации и величины взятых частиц на эффективность поглощения получаемого угля по среднемолекулярным веществам, таки как цианкобаламин и радиоактивный йод.
В результате проведения многочисленных экспериментов было установлено, что на качество получаемого угля существенное влияние оказывает размер кусков углеродсодержащей массы, скорость подъема температуры при нагревании и температурный интервал нагрева.
Определяющими являются: использование дробленой косточки или ореховой скорлупы, желательно с размером частиц более 7-10 мм, скорость подъема температуры при карбонизации, которая должна находится в интервале 30-50 град/мин, а температурный интервал должен составлять 20-750oC. При этом выдержка должна составлять 5-15 мин.
Авторами было установлено, что карбонизация косточкового сырья при вышеуказанных параметрах обуславливает развитие пористого углеродного скелета с "длинными и разветвленными" каналами а правильно выбранный режим активации при более высоких температурах 820-870oC способствует расширению этих каналов до размеров 0,8-0,9 нм, что и обеспечивает высокую эффективность получаемого угля по цианкобаламину и радиоактивному йоду, которые в полном объеме должны удаляться из организма человека.
Дробление скорлупы орехов и косточек плодов после карбонизации до размера частиц 0,5-3,5 мм исключает при парогазовой активации образование вздутий, пузырей, больших раковин.
Применение в качестве активатора смеси пара и диоксида углерода позволяет регулировать необходимый размер пор (приблизительно равный 0,8-0,9 нм) по реакциям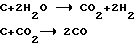
Отмывка угля относительно большим количеством воды (1:15±5) удаляет легкорастворимые соли калия и натрия, содержащиеся в исходном сырье, обеспечивая легкую доступность токсичным молекулам адсорбирующей поверхности микропор.
Следующие примеры поясняют сущность изобретения.
Пример 1. Берут 1,0 кг косточки абрикоса (с ядрами), с влажностью 0,8% выходом летучих 77,0% содержанием золы 0,9% и размером 10 х 20 мм. Косточку загружают во вращающуюся электропечь, нагретую по длине барабана 3 секциями до 250, 450 и 750oC. За счет изменения угла наклона печи и вращения обеспечивают продвижение косточек таким образом, что скорость подъема температуры составляет 40 град/мин.
Затем косточку выгружают, дробят до размера частиц 0,5-3,5 мм и помещают в другую вращающуюся печь, нагретую до 850oC.
В печь подают водяной пар и диоксид углерода в соотношении (80:20) и активируют 90 мин, после чего угол выгружают, остужают и заливают горячей (90oC) водой в соотношении 1:15, выдерживают при тщательном перемешивании 60-70 мин, воду сливают, уголь сушат и анализируют.
Пример 2. Берут 5,0 кг скорлупы грецкого ореха с размером частиц не менее 15х25 мм, влажностью 6% выходом летучих 73% содержанием золы 0,5% Скорлупу карбонизуют в стационарной электропечи, при 20-750oC со скоростью нагрева 50 град/мин, после 8 минутной выдержки при конечной температуре карбонизат выгружают, дробят до 0,5-3,5 мм и активируют во вращающейся печи при температуре 870oC в атмосфере водяной пар+диоксид углерода (70:30) в течение 90 мин, затем уголь выгружают и отмывают как в примере 1.
В табл. 1 представлены результаты экспериментов по влиянию скорости подъема температуры при карбонизации (20-750oC) на качество получаемого активного угля. Активация в данной серии опытов осуществлялась при 850oC смесью водяного пара и углекислого газа, размер частиц составлял 0,5-3,5 мм.
Из табл. 1 следует, что максимальное поглощение цианкобаламина, йода и высокий выход угля обеспечивается только при скоростях подъема, равных 30,40,50 град/мин.
Уменьшение или увеличение скорости подъема температуры существенно ухудшает качество получаемых углей.
Опытами было установлено, что наряду со скоростью подъема температуры при карбонизации на адсорбционную емкость и выход угля оказывает влияние как верхней, так и нижний пределы температурного интервала карбонизации, 20-750oC. При увеличении температуры начала карбонизации (20oC) уменьшается емкость углей по йоду примерно на 20-40% а при повышении верхнего предела температуры выше 750oC снижаются показатели емкости по цианкобаламину и выходов активного угля примерно на 25-30% при понижении (ниже 750oC) снижается показатель адсорбции цианкобаламина (примерно на 35%).
В табл. 2 приведены данные, иллюстрирующие влияние состава активирующей смеси на адсорбционную емкость и выхода активных углей. Карбонизация осуществлялась в интервале температур 20-750oC при скорости подъема 40 град/мин. Температура активации составляла 850oC.
Эксперименты показывают, что только водяной пар в смеси с диоксидом углерода делает возможность получать активные угли с высокой адсорбционной емкостью по среднемолекулярным метаболитам, обуславливают высокий выход угля и его однородность.
При этом отмечено, что лучшие результаты получают при соотношении пар: диоксид, равном (75±10:25±10).
Следует указать, что повышение температуры активации выше 870oC, равно как и снижение ее ниже 820oC способствует ухудшению показателей качества в среднем на 30-40%
В табл. 3 представлены данные по влиянию отмывки угля водой (1:15) на степень очистки от цианкобаламина и адсорбцию йода. Уголь приготовлен как в примере 12.
Из анализа данных табл. 3 следует, что отмывка конечного угля водой приводит к повышению адсорбционной емкости угля по исследованным веществам, а также, что наилучший результат получается при соотношении уголь:вода, равном 1:15±5, и увеличение расхода воды более, чем в 20 раз, нецелесообразно.
Из вышеизложенного следует, что каждый из признаков заявляемой совокупности в большей или меньшей степени влияет на достижение поставленной цели, а именно: повышение адсорбционной емкости по среднемолекулярным веществам, например, цианкобаламину, радиоактивному йоду увеличение выхода готового продукта, а вся совокупность является достаточной для характеристики заявляемого технического решения.
Способ является экологически чистым, не требует дефицитного и дорогостоящего оборудования, обеспечен сырьевой базой отходами предприятий по переработке косточковых фруктов и орехов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ УГЛЕРОДНОГО АДСОРБЕНТА | 2008 |
|
RU2372287C1 |
| СПОСОБ ПОЛУЧЕНИЯ АКТИВНОГО УГЛЯ ИЗ КОСТОЧЕК ПЛОДОВ И СКОРЛУПЫ ОРЕХОВ | 1997 |
|
RU2111923C1 |
| СПОСОБ ПОЛУЧЕНИЯ АКТИВНОГО УГЛЯ | 1993 |
|
RU2057067C1 |
| СПОСОБ ПОЛУЧЕНИЯ АКТИВНОГО УГЛЯ | 1995 |
|
RU2083491C1 |
| СПОСОБ ПОЛУЧЕНИЯ УГЛЕРОДНОГО ВОЛОКНИСТОГО АДСОРБЕНТА | 1992 |
|
RU2049168C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДРОБЛЕНОГО АКТИВНОГО УГЛЯ | 2002 |
|
RU2221745C2 |
| СПОСОБ ПОЛУЧЕНИЯ АКТИВНОГО УГЛЯ | 1998 |
|
RU2145938C1 |
| СПОСОБ ПОЛУЧЕНИЯ АКТИВНОГО УГЛЯ | 2007 |
|
RU2339573C1 |
| СПОСОБ ПОЛУЧЕНИЯ АКТИВНОГО УГЛЯ | 1993 |
|
RU2023663C1 |
| СПОСОБ ПОЛУЧЕНИЯ АКТИВИРОВАННОГО МОДИФИЦИРОВАННОГО УГЛЯ | 2016 |
|
RU2622660C1 |
Изобретение относится к получению дробленого активного угля. Сущность способа состоит в том, что скорлупу орехов или плодовую косточку карбонизуют при температуре 20 - 750oС со скоростью подъема температуры 30 - 50 град/мин, затем дробят до размера частиц 0,5 - 3,5 мм и активируют смесью водяного пара и диоксида углерода при 820 - 870oС. Для повышения адсорбционной емкости по среднемолекулярным веществам активный уголь отмывают водой в соотношении уголь : вода, равном 1 : 15 ± 5. Получаемый продукт имеет более высокую адсорбционную емкость по цианокобаламину и радиоактивному йоду, что очень важно для очистки организма человека от вредных факторов воздействия окружающей среды. 1 з.п.ф-лы, 3 табл.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US, патент, 4616001, кл | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| GB, заявка, 1545238, кл | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

