Изобретение относится к серии новых амидо и карбамидо соединений, обладающих антигиперхолестериновой активностью, которые поэтому могут быть использованы при лечении и профилактике гиперхолестеремии, атеросклероза и тому подобных заболеваний. Изобретение также охватывает методы и композиции, использующие такие соединения, а также способы их получения.
Считается, что среди причин ишемической сердечной недостаточности (которая может быть результатом ангины, инфаркта миокарда и тому подобное) атеросклероз является наиболее важным. Как полагают, поверхность клеток под эндодермальным клеточным слоем кровяных сосудов аккумулирует эфиры холестерина, и это является главной причиной атеросклероза.
Ингибиторы ацил-CoA: холестерин ацилтрансферазы (именуемые здесь АСАТ) ингибируют синтез эфиров холестерина на поверхности клеток и ингибируют образование и развитие атеросклероза, вызываемого накоплением эфиров холестерина.
Кроме того, установлено, что имеется связь между атеросклерозом и гиперхолестеринемией. Холестерин, содержащийся в пище, абсорбируется в виде свободного холестерина в клетках слизистой кишечного тракта. Затем он этерифицируется с помощью АСАТ и попадает в кровь. Поэтому ингибитор АСАТ препятствует повышению концентрации холестерина в крови путем ингибирования абсорбции пищевого холестерина в кровь.
По этой причине соединения по настоящему изобретению, обладающие способностью ингибировать активность АСАТ, являются полезными при лечении и профилактике атеросклероза.
Соединения по настоящему изобретению обладают (9H-ксантен-9-ил)метильной группой, 6,11-дигидробенз[b, e] окзепин-11-ильной группой, (1-фенилциклоалкил)метильной группой, n-алкоксифенильной группой или алкильной группой, присоединенными к амидной или карбамидной группе. Соединение, содержащее (9H-ксантен-9-ил)метильную группу, описано в публикации WO 93/06096 и EP 337375. Соединения, содержащие 6,11-дигидробенз[b, e] окзепин-11-ильную группу, описаны в публикации EP 497201. Соединения, содержащие (1-фенилциклоалкил)метильную группу, описаны в публикации EP 293880. Соединения, содержащие n-алкоксифенильную группу, описаны в публикации EP 42194. Соединения, содержащие алкильную группу, описаны в публикации EP 283742. Соединения дифенилмочевины описаны в WO 92/03413. Другие в чем-то подобные соединения описаны в публикации EP 439059 и 477778.
Было обнаружено, что соединения по настоящему изобретению, и особенно те, которые содержат (9H-ксантен-9-ил)метильную группу, обладают значительно лучшей ингибирующей АСАТ активностью, чем известные ранее соединения, указанные выше, и/или обладают намного лучшей оральной абсорбционной способностью.
Таким образом, объектом настоящего изобретения является серия новых амидо и карбамидо производных, преимущественно настоящее изобретение касается таких соединений, обладающих полезной антигиперхолестериновой активностью.
Настоящее изобретение относится к соединениям формулы (I)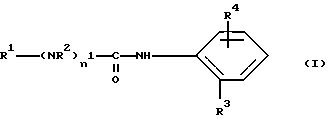
где
R1 означает алкильную группу, имеющую от 1 до 20 атомов углерода, или группу формулы (II), (III), (IV) или (V)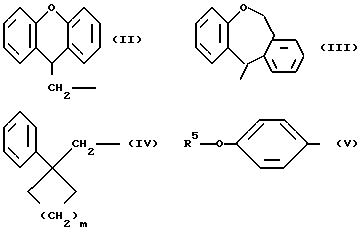
где
R5 означает алкильную группу, имеющую от 1 до 15 атомов углерода;
m означает целое число от 1 до 4;
любое ароматическое кольцо в указанной группе, представленной R1, является незамещенным;
R2 означает атом водорода или алкильную группу, имеющую от 1 до 10 атомов углерода;
R3 означает алкильную группу, имеющую от 1 до 10 атомов углерода,
алкоксигруппу, имеющую от 1 до 10 атомов углерода,
алкилтиогруппу, имеющую от 1 до 10 атомов углерода,
алкилсульфинильную группу, имеющую от 1 до 10 атомов углерода,
алкилсульфонильную группу, имеющую от 1 до 10 атомов углерода, алкоксиалкильную группу, в которой алкоксильная часть имеет от 1 до 6 атомов углерода и алкильная часть имеет от 1 до 4 атомов углерода;
R4 означает группу формулы (VI), (VII), (VIII), (IX), (X) или (XI)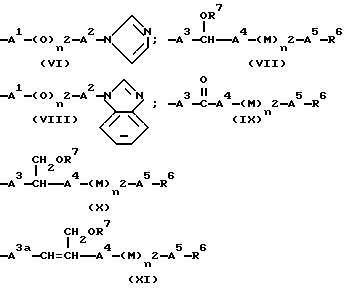
где
A1 означает одинарную связь или алкиленовую группу, имеющую от 1 до 4 атомов углерода;
A2 означает одинарную связь или алкиленовую группу, имеющую от 1 до 6 атомов углерода;
A3, A3a, A4 и A5 независимо выбраны из группы, содержащей одинарную связь и алкиленовую группу, имеющую от 1 до 10 атомов углерода, которая может быть насыщенной или может включать углерод-углеродную двойную связь, при условии, что общее число атомов углерода в A3, A4 и A5 и в A3a, A4 и A5 не превышает 10;
R6 означает алкильную группу, имеющую от 1 до 6 атомов углерода, циклоалкильную группу, имеющую от 3 до 9 атомов углерода в одном или более алифатических карбоциклических кольцах, указанные кольца являются незамещенными или имеют или фенильную группу, замещенную по крайней мере атомом галогена; и
в группах формул (VI) или (VIII) имидазолильная и бензимидазольная группы могут быть незамещенными или замещенными по крайней мере одной C1-C4-алкильной группой,
R7 означает атом водорода, бензильную группу, фосфоногруппу или группу формулы (XII)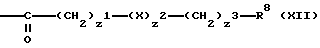
где
X1 является 0 или 1;
Z2 является 0, 1 или 2;
X является кислородом или атомом серы или сульфинилом, сульфонилом или фениленом группой, при условии, что когда Z2 является 2, по крайней мере один X является фениленовой группой;
Z3 является 9 или целым числом от 1 до 4;
R8 является карбоксигруппой, фенильной группой, группой формулы
-NR9R10,
где
R9 и R10 являются независимо выбранными из группы, содержащей атом водорода или алкильную группу, имеющую от 1 до 4 атомов углерода;
или гетероциклической группой, имеющей от 5 до 6 атомов в кольце, из которых 1 или 2 являются гетероатомами, выбранными из группы, содержащей атом кислорода или азота, указанная гетероциклическая группа является незамещенной или замещенной у атома углерода, атом кислорода или алкильной группой, имеющей от 1 до 4 атомов углерода;
Указанные группы формулы (CH2)z1 и (CH2)z2 являются незамещенными или замещенными у атомов углерода алкильной группы, имеющей от 1 до 4 атомов углерода, или группой формулы -NR9R10, где R9 и R10 определены выше;
n1 равно 0 или 1;
n2 равно 0 или 1;
M означает кислород, атом серы, сульфинильную группу или сульфонильную группу.
При условии, что, когда R4 означает указанную группу формулы (VII), (IX), (X) или (XI), R1 не означает указанную алкильную группу, и что, когда n2 является 1, A4 не означает одинарную связь, и что, когда n1 является 0, R3 является этилом и R4 является 2-ацетилом, R1 не означает метильную группу;
или и к их фармацевтически приемлемым солям.
Изобретение относится также к композиции для лечения или профилактики гиперхолестеринемии или атеросклероза, которая содержит эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым носителем или разбавителем.
Изобретение, кроме того, относится к методу лечения или профилактики гиперхолестеринемии или атеросклероза у млекопитающих, в частности у человека, которые заключаются в назначении указанному млекопитающему эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Способ получения этих соединений и его солей также составляет часть настоящего изобретения и описывается дальше более подробно.
В соединениях по настоящему изобретению, где R1 означает алкильную группу, это может быть прямоцепочечная или разветвленная алкильная группа, имеющая от 1 до 20 атомов углерода, и примеры включают группы метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, гексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, гептил, 1-метилгексил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 1-пропилбутил, 4,4-диметилпентил, октил, 1-метилгептил, 2-метилгепил, 3-метилгептил, 4-метилгепил, 5-метилгептил, 6-метилгептил, 2-пропилпентил, 2-этилгексил, 5,5-диметилгексил, нонил, 3-метилоктил, 4-метилоктил, 5-метилоктил, 6-метилоктил, 1-пропилгексил, 2-этилгептил, 2,2-диметилгептил, децил, 1-метилнонил, 3-метилнонил, 8-метилнонил, 3-этилоктил, 1,1-диметилоктил, 2,2-диметилоктил, ундецил, 4,8-диметилнонил, додецил, тридецил, 1,1-диметилундецил, тетрадецил, 2,2-диметилдодецил, пентадецил, 3,7,11-триметилдодецил, гексадецил, 4,8,12-триметилтридецил, 1-метилпентадецил, 14-метилпентадецил, 13,13-диметилтетрадецил, гептадецил, 15-метилгексадецил, октадецил, 1-метилгептадецил, нонадецил, икозил и 3,7,11,15-тетраметилгексадецил. Среди них мы предпочитаем такие группы, которые имеют от 3 до 20 атомов углерода, особенно от 4 до 20 атомов углерода, более предпочтительно такие, которые имеют от 10 до 16 атомов углерода, и еще более предпочтительно такие, которые имеют от 11 до 14 атомов углерода, в частности, группы ундецил, 1,1-диметилундецил и 2,2-диметилундецил.
Когда R1 означает группу формулы (II), (III), (IV) или (V)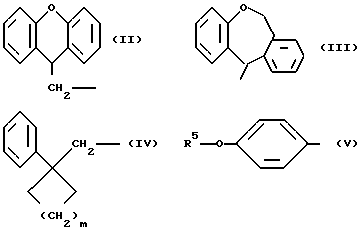
любые ароматические кольца могут быть незамещенными или они могут быть замещены одним или более заместителями α , например:
алкильной группой, имеющей от 1 до 4 атомов углерода, такой как группа метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил, из которых группы метил, этил, пропил и изопропил являются предпочтительными;
алкоксильной группой, имеющей от 1 до 4 атомов углерода, такой как группа метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, из которых группы метокси и этокси являются предпочтительными; и
атомами галогена, такими как атомы фтора, хлора, брома и иода, из которых атомы фтора, хлора и брома являются предпочтительными.
В случае групп формулы (II), предпочтительными заместителями являются алкильные и алкоксильные группы, имеющие 1 или 2 атома углерода и атомы галогена, особенно метильные и метоксильные группы и атомы хлора и брома, более предпочтительно метоксигруппа и атомы хлора и брома. Любые из этих заместителей, особенно предпочтительные и более предпочтительные заместители, могут быть у любого замещаемого атома углерода (9H-ксантен 9-ил)метильной группы, представленной формулой (II), но предпочтительно представлены на 2- или 3-атомных углерода. Нет также особых ограничений по числу заместителей, исключениям подвергаемого числа замещаемых положений и возможности стерических затруднений, однако, когда группа замещена, предпочтительными являются от 1 до 3 заместителей, о один заместитель является предпочтительным. Однако незамещенная группа является более предпочтительной.
В случае групп формулы (III) предпочтительными заместителями являются алкильные и алкоксильные группы, имеющие 1 или 2 атома углерода и атомы галогена, особенно метильные и метоксильные группы и атомы хлора и брома. Любые из этих заместителей, особенно предпочтительные заместители, могут быть у любого замещающего атома углерода 6,11-дигидробенз[b, e]окзепин-11-ильной группы, представленной формулой (III), но предпочтительно представлены на 2-атомах углерода. Нет также особых ограничений по числу заместителей, за исключением подвергаемого числа замещаемых положений и возможности стерических затруднений, однако, когда группа замещена, предпочтительными являются от 1 до 3 заместителей, а один заместитель является предпочтительным. Однако незамещенная группа является более предпочтительной.
В случае групп формулы (IV) предпочтительными заместителями являются алкильные и алкоксильные группы, имеющие 1 или 2 атома углерода и атомы галогена, особенно метильные и метоксильные группы и атомы хлора и брома, более предпочтительно метоксигруппа и атомы хлора и брома. Любые из этих заместителей, особенно предпочтительные и более предпочтительные заместители, могут быть у любого замещаемого атома углерода бензольного кольца, представленного формулой (IV), но предпочтительно представлены на 2-, 3- или 4-атомах углерода. Нет также особых ограничений по числу заместителей, за исключением подвергаемого числа замещаемых положений и возможности стерических затруднений, однако, когда группа замещена, предпочтительными являются от 1 до 3 заместителей, а один заместитель является предпочтительным. Однако незамещенная группа является более предпочтительной. В группе формулы (IV) предпочтительно 2 или 3.
В случае групп формулы (V) предпочтительными заместителями являются алкильные и алкоксильные группы, имеющие 1 или 2 атома углерода и атомы галогена, но предпочтительно группа не имеет больше заместителей на бензольном кольце в дополнение к группе формулы R5-0-. R5 представляет алкильную группу, которая может быть прямой или разветвленной алкильной группой, имеющей от 1 до 15 атомов углерода, и примеры включают группы метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, 2-метиобутил, неопентил, 1-этилпропил, гексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-биметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, гептил, 1-метилгексил, 2-метилгексил, 4-метилгексил, 5-метилгексил, 1-пропилбутил, 4,4-диметилпентил, октил, 1-метилгептил, 2-метилгепил, 3-метилгептил, 4-метилгептил, 5-метилгептил, 6-метилгептил, 1-пропилпентил, 2-этилкексил, 5,5-диметилгексил, нонил, 3-метилоктил, 4-метилоктил, 5-метилоксил, 6-метилоктил, 1-пропилгексил, 2-этилгептил, 6,6-диметилгептил, децил, 1-метилнонил, 3-метилнонил, 8-метилнонил, 3-этилоктил, 3,7-диметилоктил, 7,7-диметилоктил, ундецил, 4,8-диметилнонил, додецил, тридецил, тетрадецил, пентадецил и 3,7,11-триметилдодецил. Среди них мы предпочитаем такие группы, которые имеют от 3 до 20 атомов углерода, особенно от 4 до 15 атомов углерода, более предпочтительно такие, которые имеют от 9 до 12 атомов углерода.
Из всех значений R1 предпочтительны такие соединения, где R1 означает группу формулы (I):
Когда R2 и R3 означают алкильную группу, это может быть прямоцепочечная или разветвленная алкильная группа, имеющая от 1 до 10 атомов углерода, и примеры включают группы метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, 2-метиобутил, неопентил, 1-этилпропил, 1,1-диметилпропил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, гептил, 1-метилгексил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 1-пропилбутил, 1,1-диметилпентил, октил, 1-метилгептил, 2-метилгептил, 3-метилгептил, 4-метилгептил, 5-метилгептил, 6-метилгептил, 1-пропилпентил, 2-этилгексил, 1,1-диметилгексил, нонил, 1-метилоктил, 2-метилоктил, 3-метилоктил, 6-метилоктил, 1-пропилгексил, 1,1-диметилгептил, децил, 1-метилнонил, 3-метилнонил, 8-метилнонил, 3-этилоктил, 1,1-диметилоктил и 7,7-диметилоктил. Из них предпочтительны в случае R2 такие, которые имеют от 4 до 8, более предпочтительно от 5 до 8 атомов углерода, и из этой группы более предпочтительны группы с прямой цепочкой. Альтернативно R2 является предпочтительно атомом водорода. Предпочтительными группами для R3 являются такие, которые имеют от 1 до 8, более предпочтительно от 1 до 8, более предпочтительно от 1 до 6, атомов углерода, которые могут быть группами с прямыми или разветвленными цепочками, например группы метил, этил, изопропил, изобутил, трет-бутил, 1,1-диметилбутил, 1,1-диметилпентил и 1,1-диметилгексил, более предпочтительно группы изопропил и трет-бутил.
Когда R3 означает алкоксигруппу, это может быть прямоцепочечная или разветвленная алкильная группа, имеющая от 1 до 10 атомов углеродов, и примеры включают группы метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентилокси, изопентилокси, 2-метилбутокси, неопентилокси, 1-этилпропокси, 1,1-диметилпропокси, гексилокси, 4-метилпентилокси, 3-метилпентилокси, 2-метилпентилокси, 1-метилпентилокси, 3,3-диметилбутокси, 2,2-диметилбутокси, 1,1-диметилбукокси, 1,2-диметилбутокси, 1,3-диметилбутокси, 2,3-диметилбутокси, 2-этилбутокси, гептилокси, 1-метилгексилокси, 2-метилгексилокси, 3-метилгексилоси, 4-метилгексилокси, 5-метилгексилокси, 1-пропилбутокси, 1,1-диметилпентилокси, октилокси, 1-метилгептилокси, 2-метилгептилокси, 3-метилгентилокси, 4-метилгептилокси, 5-метилгептилокси, 6-метилгептокси, 1-пропилпентилокси, 2-этилгексилокси, 1,1-диметилгептилокси, нонилокси, 1-метилоктилокси, 2-метилоктилокси, 3-метилоктилокси, 6-метилоктилокси, 1-пропилгексиллокси, 1-этилгептилокси, 1,1-диметилгептилокси, децилокси, 1-метилонилокси, 3-метилнонилокси, 8-метилнонилокси, 3-этилоктилокси, 1,1-диметилоктилокси и 7,7-диметилоктилокси, из которых предпочтительны такие, которые имеют от 3 до 8 углерода. Из этих групп особенно предпочтительными являются изопропокси и трет-бутокси группы.
Когда R3 означает алкилтио группу, это может прямоцепочечная или разветвленная алкилтио группа, имеющая от 1 до 10 атомов углерода, и примеры включают группы метилтио, этилтио, пропилтио, изопропилтио, бутилтио, изобутилтио, втор-бутилтио, трет-бутилтио, пентилтио, изопентилтио, 2-метилбутилтио, неопентилтио, 1-этилпропилтио, 1,1-диметилпропилтио, гексилтио, 4-метилпентилтио, 3-метилпентилтио, 2-метилпентилтио, 1-метилпентилтио, 3,3-диметилбутилтио, 2,2-диметилбутилтио, 1,1-диметилбутилтио, 1,2-диметилбутилтио, 1,3-диметилбутилтио, 2,3-диметилбутилтио, 2-этилбутилтио, гентилтио, 1-метилгексилтио, 2-метилгексилтио, 3-метилгексилтио, 4-метилгексилтио, 5-метилгексилтио, 1-пропилбутилтио, 1,1-диметилпентилтио, октилтил, 1-метилгептилтио, 2-метилгептилтио, 3-метилгептилтио, 5-метилгептилтио, 6-метилгептилтио, 1-пропилпентилтио, 2-этилгексилтио, 1,1-диметилгексилтио, нонилтио, 1-метилоктилтио, 2-метилоктилтио, 3-метилоктилтио, 6-метилоктилтио, 1-пропилгексилтио, 1-этилгептилтио, 1,1-диметилгептилтио, децилтио, 1-метилнонилтио, 3-метилнонилтио, 8-метилнонилтио, 3-этилоктилтио, 1,1-диметилоктилтио и 7,7-диметилоктилтио, из которых предпочтительны такие, которые от 1 до 4 атомов углерода. Из этих групп особенно предпочтительны метилтио, изопропилтио и трет-бутилтио группы.
Когда R3 означает алкилсульфинильную группу, это может быть прямоцепочечная или разветвленная алкилсульфинильная группа, имеющая от 1 до 10 атомов углерода, и примеры включают группы метилсульфинил, пропилсульфинил, изопропилсульфинил, бутилсульфинил, изобутилсульфинил, втор-бутилсульфинил, трет-бутилсульфинил, пентилсульфинил, изопентилсульфинил, 2-метилбутилсульфинил, неопентилсульфинил, 1-этилпропилсульфинил, 1,1-диметилпропилсульфинил, гексилсульфинил, 4-метилпентилсульфинил, 3-метилпентилсульфинил, 2-метилпентилсульфинил, 1-метилпентилсульфинил, 3,3-диметилбутилсудьфинил, 2,2-диметилбутилсульфинил, 1,1-диметилбутилсульфинил, 1,2-диметилбутилсульфинил, 1,3-диметилбутилсульфинил, 2,3-диметилбутилсульфинил, 2-этилбутилсульфинил, гептилсульфинил, 1-метилгексилсульфинил, 2-метилгексисульфинил, 3-метилгексилсульфинил, 4-метилгексилсульфинил, 5-метилгексилсульфинил, 1-пропилбутилсульфинил, 1,1-диметилпентилсульфинил, октилсульфинил, 1-метилгептилсульфинил, 2-метилгептилсульфинил, 3-метилгептилсульфинил, 4метилгептилсульфинил, 5-метилгептисульфинил, 6-метилгептисульфинил, 1-пропилпентилсульфинил, 2-этилгексисульфинил, 1,1-диметилгексилсульфинил, нонилсульфинил, 1-метилоктилсульфинил, 2-метилоктилсульфинил, 3-метилоктилсульфинил, 6-метилокстилсульфинил, 1-пропилгексилсульфинил, 1-этилгептилсульфинил, 1,1-диметилгептилсульфинил, децилсульфинил, 1-метилнонилсульфинил, 3-метилнонилсульфинил, 8-метилнонилсульфинил, 3-этилоктилсульфинил, 1,1-диметилоктилсульфинил и 7,7-диметилоктилсульфинил, из которых предпочтительны такие, которые имеют одного до четырех атомов углерода. Из этих групп особенно предпочтительны группы метилсульфинил, изопропилсульфинил и трет-бутилсульфинил.
Когда R3 означает алкилсульфонильную группу, это может быть прямоцепочечная или разветвленная алкилсульфонильная группа, имеющая от 1 до 10 атомов углерода, и примеры включают группу метилсульфонил, этилсульфонил, пропилсульфонил, изопропилсульфонил, бутилсульфонил, изобутилсульфонил, втор-бутилсульфонил, трет-бутилсульфонил, пентилсульфонил, изопентилсульфонил, 2-метилбутилсульфонил, неопентилсульфонил, 1-этилпропилсульфонил, 1,1-диметилпропилсульфонил, гексилсульфонил, 4-метилпропенсульфонил, 3-метилпентилсульфонил, 2-метилпентилсульфонил, 1-метилпентилсульфонил, 3,3-диметилбутилсульфонил, 2,2-диметилбутилсульфонил, 1,1-диметилбутилсульфонил, 1,2-диметилбутилсульфонил, 1,3-диметилбутилсульфонил, 2,3-диметилбутилсульфонил, 2-этилбутилсульфонил, гептилсульфонил, 1-метилгексилсульфонил, 2-метилгексилсульфонил, 3-метилгексилсульфонил, 4-метилгексилсульфонил, 5-метилгексилсульфонил, 1-пропилбутилсульфонил, 1,1-диметилпентилсульфонил, октилсульфонил, 1-метилгептилсульфонил, 2-метилгептилсульфонил, 3-метилгептилсульфонил, 4-метилгептилсульфонил, 5-метилгептилсульфонил, 6-метилгептилсульфонил, 1-пропиленгептилсульфонил, 2-этилгексилсульфонил, 1,1-диметилгексилсульфонил, 1-метилоктилсульфонил, 2-метилоктилсульфонил, 3-метилоктилсульфонил, 6-метилоктилсульфонил, 1-пропилгексилсульфонил, 1-этилгептилсульфонил, 1,1-диметилгептилсульфонил, децилсульфонил, 1-метилнонилсульфонил, 3-метилнонилсульфонил, 8-метилнонилсульфонил, 3-этилоктилсульфонил, 1,1-диметилоктилсульфонил и 7,7-диметилоктилсульфонил, из которых предпочтительны такие, которые имеют от 1 до 4 атомов углерода. Из этих групп особенно предпочтительны группы метилсульфонил, изопропилсульфонил и трет-бутилсульфонил.
Когда R3 означает фенилтио, фенилсульфинил или фенилсульфонил группу, фенильная группа может быть незамещенной или может быть замещена одним или более заместителями, выбранными из группы, содержащей заместители, определенные и показанные в примерах далее. Нет также особых ограничений по числу заместителей, за исключением подвергаемого числа замещаемых положений и возможности стерических затруднений, однако, когда группа замещена, предпочтительными являются от 1 до 3 заместителей, а 1 заместитель является предпочтительным. Особые примеры замещенных и незамещенных групп включают группы фенилтио, 4-метилфенилтио, 2-метилфенилтио, 3-метилфенилтио, 4-пропилфенилтио, 2-метоксифенилтио, 3-метоксифенилтио, 4-этоксифенилтио, 3-фторфенилтио, 4-хлорфенилтио, 3-бромфенилтио, фенилсульфилин, 4-метилфенилсульфинил, 2-метилфенилсульфинил, 3-этилфенилсульфинил, 4-пропилфенилсульфинил, 2-метоксифенилсульфинил, 3-метоксифенилсульфинил, 4-этоксифенилсульфинил, 3-фторфинилсульфинил, 4-хлорфенилсульфинил, 3-бромфенилсульфинил, фенилсульфонил, 4-метилфенилсульфонил, 2-метилфенилсульфонил, 3-этилфенилсульфонил, 4-пропилфенилсульфонил, 2-метоксифенилсульфонил, 3-метоксифенилсульфонил, 4-этоксифенилсульфонил, 3-фторфенилсульфонил, 4-хлорфенилсульфонил и 3-бромфенилсульфонил, предпочтительно группы фенилтио, 4-метилфенилтио, 2-метилфенилтио, 4-хлорфенилтио, фенилсульфонил, 4-метилфенилсульфонил, 2-метилфенилсульфонил и 4-хлорфенилсульфонил.
Когда R3 означает алкоксиалкильную группу, они содержат алкокси группу, имеющую от 1 до 6, предпочтительно от 1 до 4, атомов углерода, который может быть заместителем у алкильной группы, имеющей от 1 до 4 атомов углерода. Примеры таких групп включают алкокси группы, имеющие от 1 до 6 атомов углерода, включая группы, которые могут быть представлены R3, и алкильные группы, имеющие от 1 до 4 атомов углерода, входящие inter alia, в группы 6, которые могут быть представлены R1. Конкретные примеры таких алкоксиалкильных групп включают метоксиметил, этоксиметил, изопропоксиметил, трет-бутоксиметил, 1-метоксиэтил, 2-метоксиэтил, 1-этоксиэтил, 2-этоксиэтил, 1-изопропоксиэтил, 2-изопропоксиэтил, 1-трет-бутоксиэтил, 2-трет-бутоксиэтил, 1-метоксипропил, 2-метоксипропил, 3-метоксипропил, 1-этоксипропил, 2-этоксипропил, 3-этоксипропил, 1-изопропоксибутил, 2-изопропоксибутил, 3-изопропоксибутил, 4-изопропоксибутил, 1-трет-бутоксибутил, 2-трет-бутоксибутил, 3-трет-бутоксибутил, 4-трет-бутоксибутил и 1,1-диметил-2-метоксиэтил. Из них более предпочтительными группами являются такие, в которых алкокси группа, имеющая от 1 до 4 атомов углерода, являются заместителями у метильной группы, предпочтительно группы метоксиметил, этоксиметил, изопропоксиметил и трет-бутоксиметил, и более предпочтительными группами являются группы метоксиметил и изопропоксиметил.
R4 означает группу формулы (VI), (VII), (VIII), (IX), (X) или (XI):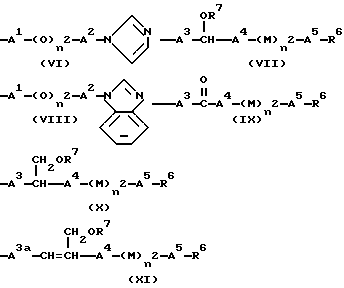
В случае групп формул (VI) и (VIII), где A1 означает алкиленовую группу, имеющую от 1 до 4 атомов углерода, это может быть прямоцепочечная или разветвленная группа, предпочтительно имеющая от 1 до 4 атомов углерода. Две "свободные" вакансии могут быть у того же самого атома углерода (в этом случае группу иногда называют "алкилдиеновой" группой ) или, когда имеется 2 или более атомов углерода, у разных атомов углерода. Прямоцепочечные группы являются предпочтительными. Примеры таких групп включают группы метилен, этилен, пропилен, 1-этилэтилен, триметилен и тетраметилен, из которых группы метилен, этилен и пропилен являются наиболее предпочтительным. Альтернативно A1 может представлять одинарную связь, однако предпочтительны такие соединения, где A1 представляет алкиленовую группу, предпочтительно метиленовую группу.
В случае групп формул (VI) и (VIII) A2 означает одинарную связь или алкиленовую группу, имеющую от 1 до 6 атомов углерода. Это может быть прямоцепочечная или разветвленная группа, предпочтительно имеющая от 2 до 4 атомов углерода. Две "свободные" вакансии могут быть у того же самого атома углерода или у разных атомов углерода. Прямоцепочечные группы являются предпочтительными. Примеры таких групп включают группы этилен, триметилен, 1-метилэтилен, тетраметилен, 1-метилтриметилен, 2-метилтриметилен, 3-метилтриметилен, 1-метилпропилен, 1,1-диметилэтилен, пентаметилен, 1-метилтетраметилен, 2-метилтетраметилен, 3-метилтетраметилен, 4-метилтетраметилен, 1,1-диметилтриметилен, 2,2-диметилтриметилен, 3,3-диметилтриметилен, гексаметилен, 1-метилпентаметилен, 2-метилпентаметилен, 3-метилпентаметилен, 4-метилпентаметилен, 5-метилпентаметилен, 1,1-диметилтетраметилен, 2,2-диметилтетраметилен, 3,3-диметилтетраметилен и 4,4-диметилтетраметилен. Из них предпочтительны группы этилен, триметилен, 1-метилэтилен и тетраметилен. В соединениях, в которых n2 является 1, предпочтительно, чтобы A2 не являлся одинарной связью и алкиленовой группой иной, чем метилен.
Также в случае формул (VI) и (VIII) имидазолильная и бензимидазолильная группы могут быть незамещенными или замещенными по крайней мере одним заместителем, выбранным из группы, содержащей заместители β , определенные выше. Примеры таких заместителей β включают:
алкильные группы, имеющие 1 до 4 атомов углерода, такие как определены; подтверждены примерами выше для заместителей, предпочтительно группы метил и этил, и
фенильные группы, которые являются незамещенными или замещены по крайней мере одним заместителем, выбранным из группы, содержащей указанные заместители (которые могут быть такими, как определены и подтверждены примерами выше), например группами фенил, 2-хлорфенил, 4-хлорфенил, 2-метоксифенил, 3-метоксифенил и 2-метилфенил, более предпочтительно группы фенил и 2-хлорфенил.
Однако незамещенные группы имидазолил и бензимидазолил и они же, замещенные группами метил или этил, являются предпочтительными.
Также, в случае групп формул (VII), (IX), (X) и (XI), A3, A3a, A4 и A5 могут каждый означать алкиленовую группу, имеющую от 1 до 10 атомов углерода, которая может иметь по крайней мере одну двойную связь. Когда группа содержит 2 или более двойных связей, группы, представленные A3, A3a, A4 и A5, могут быть одинаковы или отличаться друг от друга. Примеры таких групп включают насыщенные алкиленовые группы, имеющие от 1 до 10 атомов углерода, такие как группы метилен, этилен, триметилен, 1-метилэтилен, тетраметилен, 1-метиотриметилен, 2-метилтриметилен, 3-метилтриметилен, 1-метилпропилен, 1,1-диметилэтилен, пентаметилен, 1-метилтетраметилен, 2-метилтетраметилен, 3-метилтетраметилен, 4-метилтетраметилен, 1,1-диметилтриметилен, 2,2-диметилтриметилен, 3,3-диметилтриметилен, гексаметилен, 1-метилпентаметилен, 2-метилпентаметилен, 3-метилпентаметилен, 4-метилпентаметилен, 5-метилпентаметилен, 1,1-диметилтетраметилен, 2,2-диметилтетраметилен, 3,3-диметилтертраметилен, 4,4-диметилтетраметилен, гексаметилен, 1-метилгексаметилен, 2-метилгексаметилен, 5-метилгексаметилен, 3-этилпентаметилен, октаметилен, 2-метилгептаметилен, 5-метилгептаметилен, 2-этилгексаметилен, 2-этил-3-метилпентаметилен, 3-этил-2-метилпентаметилен, нонаметилен, 2-метилоктаметилен, 7-метилоктаметилен, 4-этилгептаметилен, 3-этил-2-метилгексаметилен, 2-этил-1-метилгексаметилен, декаметилен, 2-метилнонаметилен, 8-метилнонаметилен, 5-этилоктаметилен, 3-этил-2-метилгептаметилен и 3,3-диэтилгептаметилен; и прямоцепочечные или разветвленные алкиленовые группы, имеющие от 2 до 10 атомов углерода, такие как 2-пропилен, 1-метил-2-пропилен, 2-метил-2-пропилен, 2-этил-2-пропилен, 2-бутилен, 1-метил-2-бутилен, 2-метил-2-бутилен, 1-этил-2-бутилен, 2-пептенилен, 1-метил-2-пентенилен, 2-метил-2-пентенилен, 3-пентенилен, 1-метил-3-пентенилен, 2-метил-3-пентинилен, 1-метил-4-пентенилен, 2-метил-2-пентенилен, 2-гексенилен, 3-гексенилен, 4-гексенилен, 5-гексенилен, гептенилен, октенилен, нонилен и децен. Альтернативно любая одна или более из этих групп могут означать одинарную связь.
A3 и A3a каждый предпочтительно означают одинарную связь или алкиленову группу, имеющую от 1 до 7 атомов углерода, более предпочтительно от 1 до 4 атомов углерода и наиболее предпочтительно алкиленовую группу, имеющую от 1 до 7, более предпочтительно от 1 до 4 атомов углерода, в то время как A4 представляет одинарную связь или алкиленовую группу, имеющую от 1 до 7 атомов углерода. Кроме того, наиболее предпочтительна одинарная связь или метиленовая, или этиленовая группа.
A5 предпочтительно означает связь или алкиленовую группу, имеющую от 1 до 6 атомов углерода, которая может быть незамещенной или может быть замещена алкильной группой, имеющей от 1 до 4 атомов углерода, и примеры включают группы метилен, метилметилен, этилен, триметилен, 1-метилэтилен, тетраметилен, 1-метилтриметилен, 2-метилтриметилен, 3-метилтриметилен, 1-метилпропилен, 1,1-диметилэтилен, пентаметилен, 1-метилтетраметилен, 2-метилтеттраметилен, 3-метилтетраметилен, 4-метилтетраметилен, 1,1-диметилтриметилен, 2,2-диметилтриметилен, 3,3-диметилтриметилен, гексаметилен, 1-метилпентаметилен, 2-метилпентаметилен, 3-метилпентаметилен, 4-метилпентаметилен, 5-метилпентаметилен, 1,1-диметилтетраметилен, 2,2-диметилтетраметилен, 3,3-диметилтетраметилен 4,4-диметилтетраметилен, из которых предпочтительны группы метилен, этилен и пропилен.
В тех случаях, когда группа формулы (VII), (IX), (X) или (XI) включает две или более группы, представленные A3, A3a, A4 и 5, общее число атомов углерода, содержащегося в этих группах, не должно превышать 10.
Также в случае групп формул (VII), (IX), (X) и (XI), где R6 означает алкильную группу, которая может быть прямоцепочечной или разветвленной группой, имеющей от 1 до 4 атомов углерода, и примеры включают группы метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, и трет-бутил, из которых предпочтительны метил, изопропил и трет-бутил.
Когда R6 означает циклоалкильную группу, она имеет от 3 до 9 кольцевых атомов углерода и одно или более, предпочтительно одно или два и более предпочтительно одно, углеводородное кольцо и примеры включают группы циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, норинанил и норборнил, предпочтительно группы циклобутил, циклопентил, циклогексил, и циклогептил и более предпочтительно циклопентил и циклогексил. Эти циклоалкильные группы могут быть незамещенными или они могут иметь в своем кольце по крайней мере один заместитель, выбранный из группы, содержащей заместитель α , определенный и показанный в примерах выше. Примеры таких замещенных групп включают группы циклопентил, циклогексил, циклогептил, 4-метилциклогексил, 4-этилциклогексил, 4-пропилциклогексил, 4-трет-бутилциклогексил, 4-метоксициклогексил, 4-этоксициклогексил, 4-этоксициклогексил и борнил.
Когда R6 означает арильную группу, она имеет от 6 до 10 атомов углерода, более предпочтительно от 6 до 10 атомов углерода, в одном или более предпочтительно в одном или двух и более предпочтительно в одном карбоциклическом кольце и примеры незамещенных групп включают группы фенил, 1-нафтил, и 2-нафтил, предпочтительно группу фенил. Такие группы могут быть незамещенными или они могут иметь в кольце по крайней мере один заместитель, выбранный из группы, содержащей заместители α , определенные и подтвержденные в примерах выше. Примеры таких заместителей включают группы фенил, 2-метилфенил, 2-метоксифенил, 2-хлорфенил и 3-хлорфенил.
По определению R7, R9 и R10 могут быть атомами водорода или алкильными группами, имеющими от 1 до 4 атомов углерода. В случае алкильных групп это могут быть прямоцепочечные или разветвленные алкильные группы и примеры включают группы метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил. Из них предпочтительны такие алкильные группы, которые имеют от 1 до 2 атомов углерода, и наиболее предпочтительна метильная группа. Альтернативно предпочтительно, чтобы R9 и R10 каждый означал атом водорода. При комбинации R9 и R10 в группе -NR9R10 предпочтительно, чтобы R9 и R10 оба означали атомы водорода или метильные группы.
Гетероциклическая группа, которая может быть представлена R8, может иметь от 5 до 6 кольцевых атомов, из которых от 1 до 2 являются гетероатомами, выбранными из группы, содержащей атомы азота и кислорода. Примеры таких групп включают группы фурил, пиранил, тетрагидрофурил, тетрагидропиранил, пирролил, имидазолил, пиридил, пиразинил, пиримидинил, пирролидинил, имидазолидинил, имидазолинил, пиразолидинил, пиперидил, пиперазинил, диоксоленил и морфолинил (особенно морфолино). Предпочтительно являются группы морфолино, имидазолил и диоксоленил. Такие группы могут быть незамещенными или могут быть замещены у атома углерода атомом кислорода или алкильной группой, имеющей от 1 до 4 атомов углерода. Примеры алкильных заместителей описаны выше. Из замещенных групп особенно предпочтителен необязательно алкилзамещенный 2-оксо-1,3-диоксолен-4-ил группы и наиболее предпочтителен 5-метил-2-оксо-1,3-диоксолен-4-ил.
Примеры групп, которые могут быть представлены R7, включают атом водорода и группы карбамолил, бензил, бензоил и фосфоно [-PO(OH)2]и группы формул (XIII) - (XXXV); которые представлены на фиг. 1 и 2.
n1 является предпочтительно 0.
Из всех групп, представленных R1, предпочтительны (9H-ксантен-9-ил)метил, 6, 11-дигидробенз[b,e]окзепин-11-ил, 4-децилоксифнил и (1-фенилциклоалкил)метильная группы.
Из всех групп, обозначенных R4, предпочтительны имеющие формулы (VIa), (VIIa) и (IXa)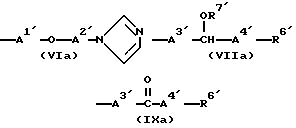 ,
,
в которых R6 представляет циклопентильную группу, циклогексильную группу, циклогептильную группу, фенильную группу, 2-метилфнильную группу, 2-хлорфенильную группу или 4-хлорфенильную группу;
R7' представляет 3-карбоксипропионильную группу, 2-карбоксибензоильную группу или 2-аминоацетильную группу;
A1' представляет метиленовую группу;
A2' представляет алкиленовую группу, имеющую от 2 до 4 атомов углерода;
A3' представляет одинарную связь или алкиленовую группу, имеющую от 1 до 3 атомов углерода, которые могут быть заторможены двойной связью (в частности метиленовая или этиленовая группа);
A4' представляет одинарную связь или алкиленовую группу, имеющую от 1 до 5 атомов углерода, которые могут быть заторможены двойной связью;
Из всех, представленных R7, предпочтительны 3-карбоксипропионил, 2-карбоксибензоил и 3-аминоацетильная группа.
Из всех групп, входящих в заместители α, предпочтительными являются метил и метокси группы и атомы фтора, хлора и брома.
Из всех групп, входящих в заместители β, , предпочтительными являются группы метил, этил, пропил и фенил.
R2 предпочтительно означает атом водорода или группу гексил или гептил.
R3 предпочтительно означает группу метил, этил, изопропил, трет-бутил, метоксиметил, изопропоксиметил, трет-бутилтио, изопропилтио, метилтио или фенилтио.
R4 может быть в любом положении бензольного кольца, образующего часть соединения формулы (I). Однако особенно предпочтительно такие, которые находились бы в орто-положении по отношении к аминогруппе мета-положении по отношению к R3 или в мета-положении по отношению в аминогруппе и в пара-положении по отношению к R3.
Предпочтительными классами соединений по настоящему изобретению являются;
(A) такие соединения формулы (I) и их соли, определенные выше, в которых R1 означает группу формул (II) и (IV)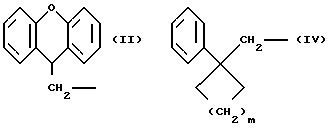 ,
,
(в которой ароматические кольца являются незамещенными или замещенными по крайней мере одним заместителем, выбранным из группы, содержащей заместители α , определенные выше, и m определенно выше) и n1 является 0, и более предпочтительны
(B) такие соединения формулы (I) и их соли, определенные выше, в которых R1 означает группу формулы (I) и ароматические кольца являются незамещенными.
Также предпочтительны классы соединений формулы (I) и их соли, определенные выше, в которых:
(C) R3 означает алкильную группу, имеющую от 1 до 10 атомов углерода, алкилтио группу, имеющую от 1 до 10 атомов углерода, или алкоксильную группу, имеющую от 1 до 10 атомов углерода, более предпочтительно.
(D) R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтио группу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода.
Также предпочтительны такие классы соединений формулы (I) и их соли, определенные выше, в которых:
(E) R4 означает группу формулы (VI), (VII) или (X), как определено выше, в которой M представляет атом кислорода, более предпочтительно
(F) в случае, когда n2 является 1, R4 означает группу формулы (VI), в которой общее число атомов углерода в A1 и A2 является от 2 до 4, или
(F') в случае, когда n2 является 0, R4 означает группу формулы (VI), в которой общее число атомов углерода в A1 и A2 является от 1 до 3, или
(G) R4 означает группу формулы (VII), в которой общее число атомов углерода в A3, A4 и A5 является от 1 до 6, и R6 означает алкильную группу, имеющую от 1 до 6 атомов углерода, или циклоалкильную группу, имеющую от 1 до 6 атомов углерода, более предпочтительно.
(H) R4 определено в (G) выше и R7 означает атом водорода или группу формулы (XVI), (XXIV), (XXV) или (XXX), еще более предпочтительно.
(I) R4 определено в (H) выше и R6 означает незамещенную группу, или
(J) R4) означает группу формулы (X), в которой общее число атомов углерода в A3, A4 и A5 является от 1 до 6, и R6 означает алкильную группу, имеющую от 1 до 6 атомов углерода, или циклоалкильную группу, имеющую от 3 до 7 атомов углерода, более предпочтительно.
(K) R4 определено в (J) выше и R7 означает атом водорода или группу формулы (XVI), (XXIV), (XXV) или (XXX) еще более предпочтительно
(L) R4 определено в (K) выше и R6 означает незамещенную циклогексильную группу.
Особенно предпочтительными являются такие соединения формулы (I) и его соли, указанные выше, в которых любые сочетания определений (A) - (J) также используются. Например, предпочтительны такие, в которых RI такое, как в (A) или в (B), и R3 такое, как в (A) или в (B), и R3 такое, как в (C) или в (D), особенно (A) + (C) или (B) + (D) и более предпочтительно (B) + (D) + (E) и еще более особенно (B) + (D) + (E) + [(F или (F') или (G) или (J)]. Даже более предпочтительными являются (B) +(D) + (E) + [(F) или (F')] и (B) + (D) + (E) + (H) и (B) + (D) + (E) + (K). Из них особенно предпочтительными являются (B) + (D) + (E) + [(F) или (F')] или (B) + (D) + (E) + (J) и (B) + (D) + (E) + (L).
В самых предпочтительных соединениях настоящего изобретения RI означает (9H-ксантен-9-ил)метильную группу; nI является 0; R3 означает группу метилтио, изопропилтио, изопропил или трет-бутил; R4 означает группу формулы (VIa), (VIIa) или (IXa) или (IXa), в которой R6' означает циклопентильную группу, циклогексильную группу, циклогептильную группу, фенильную группу, 2-метилфенильную группу, 2-хлорфенильную группу или 4-хлорфенильную группу; R7' означает 3-карбоксипропионильную группу, 2-карбокси-бензоильную группу или аминоацетильную группу; AI' представляет метиленовую группу; A2' представляет алкиленовую группу, имеющую от 2 до 4 атомов углерода; A3' представляет одинарную связь или алкиленовую группу, имеющую от 1 до 3 атомов углерода, которые могут быть заторможены двойной связью (в частности, метиленовая или этиленовая группа); A4' представляет одинарную связь или алкиленовую группу, имеющую от 1 до 5 атомов углерода, которые могут быть заторможены двойной связью.
Место присоединения R4 к безольному в соединении формулы (I) находится в орто-положении по отношению к аминогруппе и в мета-положении по отношению к R3 или в мета-положении по отношению к аминогруппе и в пара-положении по отношению к R3.
Когда соединение по настоящему изобретению содержит основную группу в своей молекуле, например аминогруппу или имидазолильную группу, оно может образовать аддитивную соль кислоты. Примеры такой соли добавления кислоты: соли с минеральными кислотами, особенно галогенводородных кислот (таких как фтористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота или хлористоводородная кислота), азотной кислоты, угольной кислоты, серной кислоты или фосфорной кислоты; соли с низким алкилсульфоновыми кислотами, такими как метансульфоновая кислота, трифторметансульфоновая кислота или этансульфоновая кислота; соли с арилсульфоновыми кислотами, такими как бензолсульфоновая кислота или n-толуолсульфоновая кислота; соли с органическими карбоновыми кислотами, такими как уксусная кислота, фумаровая кислота, виноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, бензойная кислота, миндальная кислота, аскорбиновая кислота, молочная кислота, глюконовая кислота или лимонная кислота; и соли с аминокислотами, такими как глутаминовая кислота или аспарагиновая кислота.
Конкретные примеры соединений по настоящему изобретению даны в следующих формулах от (I-I) до (I-5), в которых группы заместителей определены в соответствующих формулах табл. 1-5, например табл.1 относится к формуле (I-I), табл. 2 относится к формуле (1-2), и так далее до табл. 5, которая относится к формуле (1-5). В формулах указаны номера подходящих периферических положений, приведенных на фиг. 3-6. В табл. 1-5 используется следующая аббревиатура:
Bimd - Бензимидазол
Bu - Бутил
cBu - Циклобутил
iBu - Изобутил
tBu - Трет-бутил
Bz - Бензил
Et - Этил
Hp - Гептил
cHp - Циклогептил
Hx - Гексил
cHx - Циклогексил
Imd - Имидазолил
Me - Метил
cOc - Циклооктил
Ph - Фенил
Pn - Пентил
cPn - Циклопентил
Pr - Пропил
cPr - Циклопропил
iPr - Изопропил
G - 1,1-Диметилундецил
J - Ундецил
K - 2,2-Диметилдодецил
Из соединений, указанных в табл.1-5, предпочтительными являются соединения, приведенные в табл. 6.
Наиболее предпочтительными соединениями являются соединения N:
I - I. N-[2трет-Бутил-5-(5циклогексил-3-гидроксипентил)-фенил]-2- (9H-ксантен-9-ил)ацетамид;
1-25. N [2-трет-Бутил-5-(4-циклогексил-3-гидроксибутил)-фенил]-2- (9H-ксантен-9-ил)ацетамид;
1 - 49. N-[2-трет-Бутил-5-(6-циклогексил-3-гидроксигексил)-фенил] -2- (9H-ксантен-9-ил)ацетамид;
1 - 73. N-[2-трет-Бутил-5-(7-циклогексил-3-гидроксигептил) фенил]-2-(9H-ксантен-9-ил)ацетамид;
1 - 131. N-[2-трет-Бутил-5-(3-циклогексил-3-гидроксипропил)- фенил]-2-(9H-ксантен-9-ил)ацетамид;
1 - 179. N-[2-трет-Бутил-5-(2-циклогексил-1-гидроксиэтил)- фенил]-2-(9H-ксантен-9-ил)ацетамид;
1 - 684. N-[2-трет-Бутил-5-(6-циклопентил-1-гидроксиэтил)- фенил]-2-(9H-ксантен-9-ил)ацетамид;
1 - 1102. I-(2-(4-трет-Бутил-3-[2-(9H-ксантен-9-ил)-ацетамидо] фенил)этил)-2-циклогексилэтил натрия сукцинат;
1 - 1111. I-(2-(4-трет-Бутил-3-[2-(9H-ксантен-9-ил)-ацетамидо] фенил)этил)-3-циклогексилпропил натрия сукцинат;
1-1129. Натрий I-(2-(4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил)этил) -5-циклогексилпентил сукцинат;
1-1474. N-{ 2-[3-(I-Имидазолил)пропокси]метил-6-метилтиофенил} -2-(9H-ксантен-9-ил)ацетамид гидрохлорид;
1-1475. N-{ 2-[3-(I-Имидазолил)пропокси]метил-6-метилтилфенил} -2-(9H-ксантен-9-ил)ацетамид;
1-1480. N-{ 2-[3-(I-Имидазолил)пропокси] метил-6-трет-бутилфенил} -2-(9H-ксантен-9-ил)ацетамид гидрохлорид;
1-1552. Натриевая соль α -1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил) -2-циклогексил этил карбоксиметилтиоацетата;
1-1565. N-(2-трет-Бутил-5-{ 3-[2-(1-имидазолил)ацетокси] -4-циклогексилбутил}фенил]-2-(9H-ксантен-9-ил)ацетамид гидрохлорид;
1-1567. Натриевая соль N-(2-трет-бутил-5-{3- [2-(карбоксиметокси)ацетокси] -4-циклогексилбутил} фенил) -2-(9H-ксантен-9-ил)ацетамид;
1-1933. Натриевая соль N-(2-трет-бутил-5-{7-циклогексил-3-[2(карбоксиметокси)ацетокси] гептил}фенил)-2-(9H0ксантен-9-ил)ацетамид;
1-1977. N-[2-трет-Бутил-5-[4-циклогексил-2-(гидроксиметил)- бутил]фенил] -2-(9H-ксантен-9-ил)ацетамид;
1-2389. N-{ 2-трет-Бутил-5-[4-(2-циклогексилэтокси)-3-гидрокситутил]фенил} -2-(9H-ксантен-9-ил)ацетамид;
1-2390. N-[2-трет-Бутил-5-(5-циклогексилокси-3-гидроксипентил) фенил]-2-(9H-ксантен-9-ил)ацетамид;
1-2713. N-[2-трет-Бутил-5-{ 4-(2-циклогексилэтокси)-3-[2-(1- имидазолил)ацетокси]бутил}фенил]-2-(9H-ксантен-9-ил)ацетамид гидрохлорид;
1-2768. Натриевая соль N-(2-трет-бутил-5-{3-[2-карбоксиметокси)ацетокси] -4-циклогексилоксипентил} фенил)-2-(9H-ксантен-9-ил)ацетамид;
1-2920. N-{ 2-трет-Бутил-5-[(2-этил-1-имидазолил)метил] фенил} -2-(9H-ксантен-9-ил)ацетамид;
и их фармацевтически приемлемые соли.
Соединения по настоящему изобретению могут быть получены различными способами, например, как показано в следующих реакционных схемах I-XXVI; приведенных на фиг.7-32.
В вышеуказанных формулах:
R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, A1, A2, A3, A4, A5 и m определены выше;
R11 означает алкильную группу, имеющую от 1 до 6 атомов углерода (предпочтительно метильная или этильная группа);
R12 означает алкилсульфонильную или арилсульфонильную группу, в которых арильная группа определена выше, и алкильная группа имеет от 1 до 6 атомов углерода (предпочтительно метансульфонильная или n-толуолсульфонильная группа);
A3b означает алкиленовую группу, имеющую от 1 до 8 атомов углерода (при условии, что число атомов углерода в углеродной цепочке меньше, чем в A3 на один атом углерода);
R4a имеет те же значения, что указаны для R4 (при условии, что он может необязательно иметь защищающую группу);
R4b означает алкиленовую группу, имеющую от 1 до 10 атомов углерода, которая может быть насыщенной или может включать углерод-углеродную двойную связь (при условии, что число атомов углерода в углеродной цепочке меньше, чем в A4 на один атом углерода);
X1означает гидроксильную группу или атом галогена (предпочтительно хлор);
X2 означает атом галогена (предпочтительно бром или иод), алкилсульфонилокси группу, имеющую от 1 до 6 атомов углерода (предпочтительно метансульфонилокси группа) или арилсульфонилокси группу, в которой арильная часть определена выше;
W1 означает гидроксизащитную группу (предпочтительно триалкилсилильную группу, в частности трет-бутилдиметилсилильную группу), метоксиметильную группу, ацильную группу, аралкильную группу (в частности бензильную группу) или тетрагидропиранильную группу;
W2 означает гидроксизищитную группу (предпочтительно триалкилсилильную группу, в частности трет-бутилдиметилсилильную группу), аралкильную группу (в частности бензильную или тритильную группу), ацильную группу (в частности ацетильную группу) или тетрагидропиранильную группу;
W3 означает гидроксизащитную группу (предпочтительно триалкилсилильную группу, в частности трет-бутилдиметилсилильную группу; алкоксиалкильную группу, в частности метоксиметильную группу; или тетрагидропиранильную группу, наиболее предпочтительно трет-бутилдиметилсилильную группу);
Xs означает группу формулы
[R13S(O)p]C[R14S(O)q]
где
R13 и R14 одинаковы или различны и каждый означает алкильную группу, имеющую от 1 до 6 атомов углерода, или арильную группу, определенную выше, или R13 и R14, взятые вместе, означают алкиленовую группу, имеющую от 1 до 5 атомов углерода; и p и q являются одинаковыми или различными и каждый является 0,1 или 2), предпочтительно одну из следующих групп формулы (101), (102), (103), (104) или (105)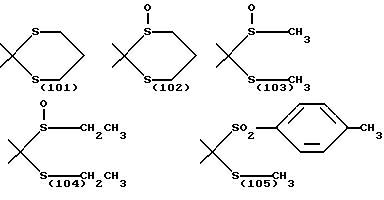 ,
,
R6a означает -(M)n2-A5-R6 , в которой M, n2, A5 и R6 определены выше;
R15 означает имидазолильную или бензимидазолильную группу, замещенную по крайней мере одним заместителем, выбранным из группы, содержащей заместитель, указанный выше;
s означает 0 или целое число от 1 до 7;
v означает целое число от 1 до 10.
Реакционная схема 1
Стадия 1: Конденсация
В этой стадии соединение формулы (4) получают взаимодействием соединения формулы (2) с соединением формулы (3) в инертном растворителе. Когда X1 означает гидроксигруппу, реакцию проводят в присутствии конденсирующего агента и основания; и когда X1 означает атом галогена, ее проводят в присутствии основания.
Когда X1 означает гидроксильную группу (стадия 1а), реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал по крайней мере в некоторой степени. Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликоль диметиловый эфир; кетоны, такие как ацетон или метилэтилкетон; нитросоединения, такие как нитроэтан или нитробензол; нитрилы, такие как ацетонитрил или изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфортриамид; и сульфоксиды, такие как диметилсульфоксид или сульфолан; более предпочтительны ароматический углеводород (в частности бензол), галогенированный углеводород (в частности метиленхлорид) или простой эфир (в частности тетрагидрофуран).
Нет также конкретных ограничений по природе используемого конденсирующего агента и примеры таких подходящих агентов включают: ди(низший алкил)азодикарбоксилат-трифенилфосфин, такой как диэтилазодикарбоксилат-трифенилфосфин; N-(низший алкил)-5-арилизоксазолий-3'-сульфонаты, такие как N-этил-5-фенилизоксазолий-3'-сульфонат, N,N'-дициклоалкилкарбодиимиды, такие как N, N'-дициклогексилкарбодиимид (DCC); 2-гало-1-(низший алкил)пиридиний галогениды, такие как 2-хлор-1-метилпиридиний йодиды; диарилфосфорилазиды, такие как дифенилфосфорилазид (DPPA); фосфорилхлориды, такие как диэтилфосфорилхлорид; производные имидазола, такие как N,N'-карбонилдиимидазол (CDI); и производные карбодиимида, такие как 1-этил-3-(3-диметиламинопропил)карбодиимид (EDAPC); предпочтительно N,N'-дициклогексилкарбодиимид, 2-хлор-1-метилпиридиний йодид и диэтил фосфорилхлорид.
Нет также конкретных ограничений по природе используемого основания при условии, что отсутствуют отрицательные воздействия на молекулы реагентов, и примеры включают органические основания, такие как триэтиламин, трибутиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N, N-диметиламино)пиридин, 4-пирролидинопиридин, N, N-диметиланилин, N,N-диметиланилин, N, N-диэтиланилин, 1,5-диазабицикло[4,3,0]нон-5-ен, 1,4-диазабицикло[2,2,2]октан (DABCO) и 1,8-диазабицикло[5,4,0]ундец-7-ен (DBU). Из них предпочтительны этиламин, диизопропилэтиламин или пиридин.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 150oC, более предпочтительно от 25 до 120oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 10 мин до 48 ч, более предпочтительно от 1 до 24 ч, будет обычно достаточно.
После завершения реакции желаемого соединение формулы (1) может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Когда X1 означает атом галогена (стадия 1b), реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры предпочтительных растворителей включают ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, дихлорэтан, хлорбензол или дихлорбензол; эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликоль диметиловый эфир; кетоны, такие как ацетон или метилэтилкетон; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфортриамид. Из них предпочтительны ароматические углеводороды (в частности бензол), галогенированные углеводороды (в частности метиленхлорид).
Нет также конкретных ограничений по природе используемого основания при условии, что оно не оказывает отрицательного воздействия на молекулы реагентов, и примеры включают органические основания, такие как триэтиламин, трибутиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино) пиридин, N, N-диметиланилин, N, N-диэтиланилин, 1,5-диазабицикло[4,3,0] нон-5-ен, 1,4-диазабицикли[2, 2, 2]октан(DABCO) и 1,8-диазабицикло[5, 4, 0] ундец-7-ен (DBU). Из них предпочтительны пиридин или N,N-диметиланилин.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, а основном, удобно проведение реакции при температуре от -78 до 50oC, более предпочтительно от -40 до 25oC. Время, необходимое для реализации, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 5 мин. до 24 ч., более предпочтительно от 10 мин. до 24 ч., будет обычно достаточно.
После завершения реакции желаемое соединение формулы (1) может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Желаемое соединение, если необходимо, может быть затем очищено таким обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадий 2: снятие защитной группы
По этой стадии соединение формулы (1a) получают взаимодействием формулы (4) со снимающим защитную группу агентом в инертном растворителе для удаления группы, представленной W1.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал по крайней мере в некоторой степени. Примеры подходящих растворителей включают; ароматические углеводороды, такие как бензол, толуол или ксилол; гологенированные углеводороды, такие как метилхлорид, хлороформ, дихлорэтан; простые эфиры, такие как диэтиловый эфир, татерагидрофуран, диоксан, диметоксиэтан; спирты, такие как метанол или этанол; и нитрилы, такие как ацетонитрил или изобутиронитрил. Из них предпочтительны ароматические углеводороды (в частности бензол), эфиры (в частности тетрагидрофуран) и спирты (в частности метанол).
Когда используемая защитная группа является силильной группой, такой как трет-бутилдиметилсилильная группа, они могут быть сняты с использованием неорганической кислоты, такой как соляная кислота, или реагента, способного к образованию иона фтора, такого как тетрабутиламмоний фторид. Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от комнатной до 70oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако, при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч. будет обычно достаточен.
Когда используемая защитная группа является метоксиметильной группой, она может быть удалена с пользованием неорганической кислоты, такой как соляная кислота, в органическом растворителе, таком как диоксан, метанол или этилацетат. Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изображению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от комнатной до 70oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч. будет обычно достаточен.
Когда используемая защитная группа является тетрагидропиранильной группой, она может быть снята с использованием неорганической кислоты, такой как соляная кислота, или органической кислоты, такой как n-толуолсульфокислота. Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, а основном, удобно проведение реакции при температуре от комнатной до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч будет обычно достаточен.
Когда используемая защитная группа является ацильной группой, такой как ацетильная группа, они могут быть сняты с использованием алкоксида щелочного металла, такого как метоксид натрия или метоксид калия, или гидроксида щелочного металла, такого как гидроксида натрия или гидроксида калия. Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от комнатной до 60oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч. будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетата; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Желаемое соединение, если необходимо, затем может быть очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадий 3: Этерификация
По этой стадии соединение формулы (1b) получают взаимодействие соединения формулы (1a) с ангидридом кислоты, моноэфиром, в частности бензил-, тре-бутил- или бензгидрилсложный эфир, или моноэфир, в частности бензил-, трет-бутил или бензгидрилсложный эфир, моногалогенангидрид карбоновой кислоты в качестве инертного растворителя. Когда используют ангидрид дикарбоновой кислоты, реакцию обычно проводят в присутствии основания; когда используют моноэфир дикарбоновой или карбоновой кислоты, реакцию обычно проводят в присутствии конденсирующего агента и основания; и, когда используют моноэфир дикарбоновой или карбоновой кислоты, реакцию обычно и предпочтительно осуществляют в присутствии конденсирующего агента или основания; и, когда используют моноэфир моногалогенангидрид дикарбоновой кислоты, реакцию обычно и предпочтительно проводят в присутствии основания.
Когда используют ангидрид кислоты (стадия 3a), реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; простые эфиры, такие как тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликоль диметиловый эфир; кетоны, такие как ацетон или метилэтилкетон; нитрилы, такие как ацетонитрил или изобутиронитрил; и пиридин или его замещенные производные, такие как пиридин или 2,6-лутидин. Из них предпочтительны ароматические углеводороды (в частности толуол или ксилол) и пиридин или его замещенные производные (в частности пиридин).
Нет также конкретных ограничений по природе используемого основания при условии, что отсутствуют отрицательные воздействия на молекулы реагентов, и примеры включают органические основания, такие как триэтиламин, трибутиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N, N-диметиламино)пиридин, N, N-диметиланилшин, N, N-диэтиланилин, 1,5-диазабицикло[4,3,0] нон-5-ен, 1,4-диазибицикло[2, 2, 2]октан (DABCO) или 1,8-диазибицикло [5, 4, 0]ундец-7ен (DBU), предпочтительно триэтиламин или пиридин.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 50 до 150oC, предпочтительно от 70 до 120oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 72 ч., предпочтительно от 1 до 30 ч., будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: подкисление реакционной смеси: фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение может быть, если необходимо, затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колончатая хроматография.
Когда используется моноэфир дикарбоновой или карбоновой кислоты (стадия 3b), реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры предпочтительных растворителей включают: ароматические углеводы, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; простые эфиры, такие как тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликоль диметиловый эфир; кетоны, такие как ацетон или метилэтилкетон; и нитрилы, такие как ацетонитрил или изобутиронитрил. Из них предпочтительны ароматические углеводороды (в частности бензол), простые эфиры (в частности тетрагирофуран) и галогенированные углеводороды (в частности, метиленхлорид).
Нет также конкретных ограничений по природе используемого конденсирующего агента, и примеры таких подходящих агентов включают ди(низший алкил)азодикарбоксилат-трифенилфосфин, такой как диэтилазодикарбоксилат-трифенилфосфин; N-(низший алкил)-5-арилизоксазолий-3'-сульфонаты, такие как N-этил-5-фенилизоксазолий-3'-сульфонат; N,N'-дициклоалкилкарбодиимиды, такие как N,N'-дициклогексилкарбодиимид; 2-галоген-1-(низший алкил) пиридиний галогениды, такие как 2-хлор-1-метилпиридиний йодиды; диарилфосфорилазиды, такие как дифенилфосфорилазид (DPPA); производные имидазола, такие как N,N'-карбонилдиимидазол (CDI); и карбодиимиды, такие как 1-этл-3-(3-диметиламинопропил)карбодиимид (EADAPC). Из них предпочтительны N,N'-дициклогексилкарбодиимид, 2-хлор-1-метилпиридиний йодид и 1-этил-3-(3-диметиламинопропил)карбодиимид.
Нет также конкретных ограничений по природе используемого основания, при условии, что отсутствуют отрицательные воздействия на молекулы реагентов, и примеры включают органические основания, такие как триэтиламин, трибутиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N, N-диметиламино)пиридин, N,N-лиметиланилин, N,N-диэтиланилин, 1,5-диазабицикло[4,3,0]нон-5-ен,1,4-диазобицикло[2,2,2]октан (DABCO) или 1,8-диазобицикло[5,4,0]ундец-7-ен (DBU). Из них предпочтителен триэтиламин или N-диизопропил-N-этиланилин.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -60oC до 120oC, предпочтительно от 0 до 70oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей в предпочтительных условиях, описанных выше, период времени от 10 мин до 48 ч, предпочтительно от 1 до 24 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение может быть, если необходимо, затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, в именно хроматография.
Дебензелирование полученного таким образом монобензилового эфира может быть осуществлено каталитическим восстановлением с использованием катализатора, такого как палладий на угле или палладиевая чернь, в штоке водорода или муравьиной кислотой с получением соединения карбоновой кислоты.
Снятие защитной группы полученного таким образом монотретбутилового эфира может быть осуществлено с использованием кислотного катализатора, такого как хлористый водород/диоксан, с получением соединения карбоновой кислоты.
Снятие защитной группы полученного таким образом монобензгидрилового эфира может быть осуществлено с использованием кислотного катализатора, такого как трифторуксусная кислота/анизол, с получением соединения карбоновой кислоты.
Когда используется моноэфир моногалогенангидрид кислоты (стадия 3c), реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ или дихлорэтан; сложные эфиры, такие как этилацетат, бутилацетат или диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликоль диметиловый эфир; кетоны, такие как ацетон или метилэтилкетон; и нитрилы, такие как ацетонитрил или изобутиронитрил. Из них предпочтительны ароматические углеводороды (в частности бензол) или галогенированные углеводороды (в частности метиленхлорид).
Нет также конкретных ограничений по природе используемого основания при условии, что отсутствуют отрицательные воздействия на молекулы реагентов, т любые, которые могут применяться в обычных реакциях, могут таким же образом использоваться и здесь. Примеры включают органические основания, такие как триэтиламин, трибутиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N, B-диметиламино)пиридин, N,N-диметилаланин, N,N-диэтилаланин, 1,5-диазабицикло[4,3,0] нон-5-ен, 1,4-диазобицикло[2,2,2]октан (DABCO)или 1,8-диазобицикло[5,4,0] ундец-7-ен (DBU). Из них предпочтительны триэтиламин или пиридин.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -40 до 100oC, предпочтительно от 0 до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 10 мин до 48 ч, предпочтительно от 1 до 24 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение может быть, если необходимо, затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Дебензелирование полученного таким образом монобензилового эфира может быть осуществлено каталитическим восстановлением с использованием катализатора, такого как палладий на угле, в токе водорода с получением соединения карбоновой кислоты.
Снятие защитной группы полученного таким образом моно-трет-бутилового эфира может быть осуществлено с использованием кислотного катализатора, такого как хлористый водород/диоксан, с получением соединения карбоновой кислоты.
Снятие защитной группы полученного таким образом монобензгидрилового эфира может быть осуществлено с использованием кислотного катализатора, такого как трифторуксусная кислота/анизол, с получением соединения карбоновой кислоты.
Когда 3 означает группу формулы -C-(=O)-CHR8-NR9R10 (где R9 и R10 каждый независимо означают алкильную группу), желаемое соединение формулы (Ib) может быть получено взаимодействием соединения формулы (Ia) с соединением формулы: HOC(= O)-CHR8-NR9R10 тем же методом, что описан в способе стадии 3b. Когда R7 означает группу формулы -C(=O)-CHR8-NR9R10 (где R9 и R10 означают атом водорода), желаемое соединение формулы (Ib) может быть получено взаимодействием соединения формулы (Ia) с соединением формулы: HO-C(= O)-CHR8-NR10-COOB и тем же методом, что описан в способе стадии 3b, и с последующим удалением трет-бутоксикарбонильной группы с использованием кислоты, такой как соляная кислота.
Когда R7 означает группу формулы -P(=O)(OH)2,желаемое соединение формулы (Ib) может быть получено взаимодействием соединения формулы (Ia) с соединением формулы: CIP(=O)(OCH2Ph)2 тем же методом, что описан в способе стадии 3c с последующим удалением бензильной группы каталитическим восстановлением.
Стадия 4: Окисление
В этой стадии соединение формулы (Ic) может быть получено взаимодействием соединения формулы (Ia) с окислительным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ или дихлорэтан; сложные эфиры, такие как этилформиат или этилацетат; простые эфиры, такие как тетрагидрофуран, диметоксиэтан или диоксан; кетоны, такие как ацетон, метилэтилкетон или метилизобутилкетон; и нитрилы, такие как ацетонитрил или изобутиронитрил. Из них предпочтительны галогенированные углеводороды (в частности метиленхлорид).
Нет также особых ограничений по природе используемого окислительного агента, и любой агент такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры предпочтительных окислительных агентов включают: неорганические оксиды металлов, включая оксиды марганца, такие как перманганат калия; соединения хромовой кислоты, такие как хромовая кислота-серная кислота, комплекс ангидрид хромовой кислоты-пиридин или пиридиний хлорхромат; и соединения церия, такие как аммоний церий нитрат (CAN); и реагенты, которые можно использовать при диметилсульфоксидном окислении (например диметилсульфоксид и дициклогексилкарбодиимид, оксалил хлорид, уксусный ангидрид, пентоксид фосфора или комплекс пиридин-серный ангидрид). Из них предпочтителен пиридиний хлорхромат или диметилсульфоксид/оксалилхлорид).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -78 до 50oC, более предпочтительно от -60 до 25oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 10 мин до 24 ч, более предпочтительно от 1 до 12 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединения; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 5: Конденсация
В этой стадии соединение формулы (1c) может быть получено взаимодействием соединения формулы (2) с соединением формулы (5) в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 1 реакционной схемы 1 и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 6: Восстановление
В этой стадии соединение формулы (1а) может быть получено взаимодействием соединения формулы (1с) в восстановительным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводы, такие как бензол, толуол или ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; и спирты, такие как метанол или этанол. Из них предпочтительны спирты (в частности метанол) и эфиры (в частности тетрагидрофуран).
Нет также особых ограничений по природе используемого восстановительного агента, и любой восстановительный агент такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры подходящих восстановительных агентов включают натрийборгидрид и диизобутилалюминийгидрид.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -78 до 50oC, более предпочтительно от -60 до 25oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 5 мин до 24 ч, более предпочтительно от 10 мин до 12 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединения, если необходимо, затем может быть очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Реакционная схема II
Стадия 7: Алкилирование
В этой стадии соединение формулы (8) может быть получено алкилированием соединения формулы XsH2 с соединением формулы (7).
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; нитросоединения, такие как нитроэтан; нитрилы, такие как ацетонитрил или изобутиронитрил; амиды, такие как диметилформамид, диметилацетамид или гексаметилфосфортриамид; и сульфоксиды, такие как диметилсульфоксид или сульфолан. Из них предпочтительны амиды (в частности диметилформамид) и сульфоксиды (в частности диметилсульфоксид).
Нет также особых ограничений по природе используемого основания при условии, что отсутствуют отрицательные воздействия на молекулы реагентов и любые, которые могут применяться в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включают гидриды щелочных металлов, такие как гидрид лития, гидрид натрия или гидрид калия; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, трет-бутоксид калия или метоксид лития; и органические основания металлов, такие как бутиллитий или диизопропиламид лития. Из них предпочтительны гидриды щелочных металлов (в частности гидрид натрия) или органические основания металлов (в частности бутиллитий или диизопропиламид лития).
Подходящие соединения формулы XsH2 включают такие серосодержащие соединения, как 1,3-дитиан, (метилтио) метил n-толуосульфон и (метилтио)метил метилсульфоксид.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 100oC, более предпочтительно от 0 до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 10 мин до 24 ч, более предпочтительно от 1 до 12 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающего с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение, сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученные таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 8: Алкилирование
В этой стадии соединение формулы (10) может быть получено алкилированием соединения формулы (8) с соединением формулы (9).
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или лиметоксиэтан; амиды, такие как диметилформамид, диметилацетамид или гексаметилфосфортриамид; и сульфоксиды, такие как деметилсульфоксид или сульфолан. Из них предпочтительны эфиры (в частности тетрагидрофуран или диметоксиэтан).
Нет также особых ограничений по природе используемого основания при условии, что отсутствуют отрицательные воздействия на молекулы реагентов и любые, которые могут применяться в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включают гидриды щелочных металлов, такие как гидрид лития, гидрид натрия или гидрид калия; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, трет-бутоксид калия или метоксид лития; и органические основания металлов, такие как бутиллитий или диизопропиламид лития. Из них предпочтительны гидриды щелочных металлов (в частности гидрид натрия) или органические основания металлов (в частности бутиллитий).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -78 до 100oC, более предпочтительно от -60 до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 10 мин до 24 ч, более предпочтительно от 30 мин до 6 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Когда желаемое соединение является неустойчивым, его можно использовать
Стадия 9: Десульфирование
В этой стадии соединение формулы (11) может быть получено сольволизом соединения формулы (10) в присутствии кислотного катализатора, соли ртути или соли серебра.
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал; по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; спирты, такие как метанол или этанол; кетоны, такие как ацетон или метилэтилкетон; и воду. Из них предпочтительны спирты (в частности метанол) или эфиры (в частности тетрагидрофуран).
Нет также особых ограничений по природе используемого кислотного катализатора, и любой кислотный катализатор такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры подходящих кислотных катализаторов включают: неорганические кислоты, такие как соляная кислота, бромистоводородная кислота, серная кислота, хлорная кислота или фосфорная кислота; органические кислоты, такие как метансульфоксилота, n-толуолсуотфокислота, трифторуксусная кислота три трифторметансульфоновая кислота. Из них предпочтительная концентрированная соляная кислота.
Когда в качестве соединения формулы XsH2 используют 1,3-дитиан, сольволиз можно проводить в присутствии хлорида ртути или нитрата серебра.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 25 до 150oC, более предпочтительно от 50 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 30 мин до 24 ч, более предпочтительно от 1 до 12 ч, будет обычно достаточно.
После завершения реакции делаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединения; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 10: Восстановление кето группы
В этой стадии соединение формулы (6) может быть получено взаимодействием соединения формулы (11) с восстанавливающим агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 6 реакционной схемы 1, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 10а: Восстановление нитро группы
Эта стадия включает получение соединения формулы (5) восстановлением соединения формулы (11) восстанавливающим агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 12 реакционной схемы III, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема III
Стали II: Введение защитной группы
В этой стадии соединение формулы (12) может быть получено взаимодействием соединения формулы (6) с защитным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: гелогенированные углеводороды, такие как метиленхлорид, хлороформ или дихлорэтан; простые эфиры, такие как тетрагидрофуран, диоксан или диметоксиэтан; и амиды, такие как формамид, диметилформамид или диметилацетамид. Из них предпочтительным галогенированные углеводороды (в частности метиленхлорид) и амиды (в частности диметилфорамид).
Нет также особых ограничений по природе используемых защитных групп, и любая защитная группа, обычно используемая в общеизвестных реакциях, может быть таким же образом применена и здесь. Примеры подходящих защитных групп включают трет-бутилдиметилсилил хлорид, дигидропиран или метоксиметилхлорид.
Например, защита с использованием указанных выше групп может быть осуществлена как описано в "Protective Croups in Organic synthesis, Second Edition", T.W. Green & P.G.M. Wut; John Wiley and Sons, Inc., New York (1991).
Реакцию можно осуществлять в широком интервале температуре, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависит от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 100oC, более предпочтительно от 0 до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 10 мин до 48 ч, более предпочтительно от 30 мин до 24 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 12: Восстановление нитро группы
Эта стадия включает получение соединения формулы (3) восстановлением соединения формулы (12) восстанавливающим агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: спирты, такие как метанол или этанол и воду. Из них предпочтительны спирты.
Нет также особых ограничений по природе используемого восстановительного агента, и любой восстановительный агент такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры подходящих восстановительных агентов включают цинк/уксусную кислоту, железо/соляную кислоту или олово/соляную кислоту, предпочтительно цинк/уксусную кислоту.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению, предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном удобно проведение реакции при температуре от -20 до 150oC, более предпочтительно от 0 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии что реакцию осуществляют в предпочтительных условиях, описанных выше периода времени от 10 мин до 24 ч., более предпочтительно от 20 мин до 12 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Реакционная схема IV
Стадия 13: Образование оксокарбоксилата
В этой стадии соединение формулы (14) может быть получено воздействием соединения формулы (13) с карбодиимидазолом и затем с калиевой солью моноэфира малоновой кислоты (предпочтительно R11 означает метильную или этильную группу) в присутствии эфирата магнийбромида.
Активированное соединение соответствующего соединения формулы (13) может быть получено способом, известным специалистам, например, аналогично способу, описанному в Synthesis 478 (1978).
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ или дихлорэтан; простые эфиры, такие как тетрагидрофуан, диоксан или диметоксиэтан; нитрилы, такие как ацетонитрил или изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфортриамид; и сульфоксиды, такие как диметилсульфоксид или сульфолан. Из них предпочтителен диэтиловый эфир, тетрагидрофуран или нитрилы, такое как ацетонитрил.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таки факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 60oC. Когда декарбоксилирование протекает медленно, температуре реакции позволяют подняться до интервала от 20 до 50oC для промотирования реакции. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени около 12 ч будет обычно достаточен при температуре от комнатной до 60oC.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 14: Алкилирование
В этой стадии соединение формулы (16) может быть получено взаимодействием соединения формулы (14) с соединением формулы (15) в присутствии основания.
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как дихлорметан; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; спирты, такие как метанол, этанол или трет-бутанол; нитросоединения, такие как нитроэтан; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфортриамид; и сульфоксиды, такие как диметилсульфоксид или сульфолан. Из них предпочтительны эфиры (в частности тетрагидрофуран), амиды (в частности диметилформамид) или спирты.
Нет также особых ограничений по природе используемого основания при условии, что отсутствуют отрицательные воздействия на молекулы реагентов и любые, которые могут применяться в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включают: неорганические основания, включая гидриды щелочных металлов, такие как гидрид лития, гидрид натрия или гидрид калия; или алкоксиды щелочных металлов такие как метоксид натрия, этоксид натрия, трет-бутоксид калия или метоксид лития; более предпочтительны алкоксиды щелочных металлов (в частности метоксид натрия, этоксид натрия или трет-бутоксид калия).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -30 до 100oC, более предпочтительно от 0 до 60oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 1 до 24 ч, более предпочтительно от 1 до 10 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 15: Декарбоксилирование
В этой стадии соединение формулы (11) может быть получено, подвергая соединение формулы (16) гидролизу в присутствии основания с последующим декарбоксилированием.
Эти реакции обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры предпочтительных растворителей включают: простые эфиры, такие как тетрагидрофуран, диоксан или диметоксиэтан; спирты, такие как метанол, этанол или трет-бутанол; и смеси спиртов и воды. Из них предпочтительны спирты или смесь спирта и воды.
Нет также особых ограничений по природе используемого основания при условии, что отсутствуют отрицательные воздействия на молекулы реагентов и любые, которые могут применяться в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включают: карбонаты щелочных металлов такие как карбонат натрия, карбонат калия или карбонат лития; гидроксиды щелочных металлов или гидроксиды щелочноземельных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид бария или гидроксид лития. Из них предпочтителен гидроксид калия.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 150oC, более предпочтительно от 25oC до 100oC.
Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 30 мин до 24 ч, предпочтительно от 1 до 10 ч, будет обычно достаточен.
После завершения гидролиза pH реакционной смеси доводят до значения pH около 5 путем добавления кислоты, такой как концентрированная соляная кислота или разбавленная серная кислота, для осуществления декарбоксилирования.
Реакция декарбоксилирования может осуществляться в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 50 до 150oC, более предпочтительно от 70 до 120oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 30 мин до 24 ч, предпочтительно от 1 до 4 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Реакционная схема V
Стадия 16: Конденсация
В этой стадии соединение формулы (18) может быть получено воздействием соединения формулы (2) с соединением формулы (17) в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии I реакционной схемы 1, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 17: Снятие защитной группы
По этой стадии соединение формулы (19) может быть получено взаимодействием соединения формулы (18) со снимающим защитную группу агентом.
Метод, выбранный для снятия защитной группы, будет изменяться в зависимости от природы защитной группы, однако такие методы являются хорошо известными и снятие защиты может быть осуществлено обычными способами.
Когда защитной группой является трет-бутилдиметилсилильная, она может быть удалена снимающим защиту агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры предпочтительных растворителей включают: простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; и спирты, такие как метанол или этанол.
Нет также особых ограничений по природе используемого снимающего защиту агента, и любые снимающие защиту агенты, обычно применяемые в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих снимающих защиту агентов включают соляную кислоту или тетрабутиламмонийфторид, предпочтительно соляную кислоту.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 48 ч будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающего с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 17: Ведение уходящей группы
По этой стадии соединение формулы (20), (93) или (95) может быть получено взаимодействием соединения формулы (19), (23) или (30) с соединением, содержащим алкилсульфонильную или арилсульфонильную группу (предпочтительно метансульфонильную или n-толуолсульфонильную группу) в присутствии основания.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол; галогенированные углеводороды, такие как метиленхлорид или хлороформ; и простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан.
Нет также особых ограничений по природе используемого снимающего защиту агента, и любые снимающие защиту агенты, обычно применяемые в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих снимающих защиту агентов включают соляную кислоту или тетрабутиламмонийфторид, предпочтительно соляную кислоту. Предпочтительны галогенированные углеводороды (в частности метиленхлорид или дихлорэтан).
Нет также особых ограничений по природе используемого основания, и любые основания, которые могут применяться в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включают органические основания, такие как триэтиламин, диизопропиламин, изопропилэтиламин, N-метилфорфолин, пиридин, 4-(N,N-диметиламино)пиридин, N,N-диметиланилин или N, N-диэтиланилин, из которых предпочтительны триэтиламин или диизопропиламин.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 50oC, более предпочтительно от 0 до 25oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 5 мин до 10 ч, более предпочтительно от 10 мин до 3 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученное таким образом желаемое соединение, если необходимо, может быть затем очищено такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 19: Введение имидазолильной группы
По этой стадии соединение формулы (Id), (94), (96) или (97) может быть получено взаимодействием соединения формулы (20), (93), (95) или (40) с имидазолом или бензимидазолом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликоль диметиловый эфир; нитрилы, такие как ацетонитрил или изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфортриамид; и сульфоксиды, такие как диметилсульфоксид или сульфолан. Из них предпочтительны амиды (в частности диметилформамид) или сульфоксиды (в частности диметилсульфоксид).
Реакция может быть иногда ускорена проведением ее в присутствии иодида натрия или иодида калия.
Реакцию можно осуществить в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 180oC, предпочтительно от 20 до 120oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч. более предпочтительно от 2 до 12 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: добавление воды к реакционной смеси и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Реакционная схема VI
Стадия 20: Конденсация
В этой стадии соединение формулы (22) может быть получено взаимодействием соединения формулы (2) с соединением формулы (21). Входящая в эту стадию реакция по существу та же, что и в стадии 1 реакционной схемы 1, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 21: Снятие защитной группы
По этой стадии соединение формулы (23) может быть получено взаимодействием соединения формулы (22) со снимающим защитную группу агентом.
Когда гидроксизащитной группой является трет-бутилдиметоксилильная группа, она может быть удалена обработкой неорганической кислотой, такой как соляная кислота, или соединением, способным к образованию аниона фтора, таким как тетрабутиламмоний фторидом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: спирты, такие как метанол; и простые эфиры, такие как тетрагидрофуран или диоксан.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 70oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 18 ч будет обычно достаточен.
Когда гидроксизащитной группой является алифатическая ацетильная группа, она может быть удалена обработкой основанием.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают; воду; органические растворители, включая спирты, такие как метанол, этанол или пропанол; и простые эфиры, такие как тетрагидрофуран или диоксан; или смеси воды и одного или более этих органических растворителей.
Нет также конкретных ограничений по природе используемого основания, и любые основания, обычно применяемые в обычных реакциях, могут таким же образом использоваться и здесь, за исключением тех групп, соединения которых не оказывают действия по удалению защитной группы. Примеры подходящих оснований включают: алкоксиды щелочных металлов, такие как метоксид натрия; карбонаты щелочных металлов, такие как натрий карбонат, калий карбонат или литий карбонат; гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия или гидроксид лития; и аммиак, который может быть в различных видах, таких как водный аммиак или концентрированный аммиак в метаноле.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 150oC для предотвращения побочных реакций. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 10 ч будет обычно достаточен.
Когда гидроксизащитной группой является группа алкоксиметил, тетрагидропиранил, тетрагидротиопиранил, тетрагидрофуранил, тетрагидротиофуранил или замещенная этильная группа, она может быть обычно удалена обработкой кислотой.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: спирты, такие как метанол или этанол; простые эфиры, такие как тетрагидрофуран или диоксан; и смеси воды и одного или более этих органических растворителей.
Нет также конкретных ограничений по природе используемого кислотного агента, и любые кислоты, обычно применяемые в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих кислот включают кислоты Бронстеда, включая неорганические кислоты, такие как соляная кислота или серная кислота; органические кислоты, такие как уксусная кислота или n-толуолсульфокислота; и сильные кислотные ионообменные смолы, такие как Dowex (торговая марка) 50W.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 10 мин до 18 ч будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 22: Окисление
В этой стадии соединение формулы (24) может быть получено взаимодействием соединения формулы (23) с окислительным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ или дихлорэтан; сложные эфиры, такие как этилформиат, этилацетат или диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; кетоны, такие как ацетон или метилэтилкетон; и нитрилы, такие как ацетонитрил или изобутиронитрил. Из них предпочтительны галогенированные углеводороды (в частности метиленхлорид) или эфиры (в частности тетрагидрофуран).
Нет также особых ограничений по природе используемого окислительного агента, и любой окислительный агент, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры предпочтительных окислительных агентов включают% оксиды марганца, такие как перманганат калия; оксиды хрома, такие как комплекс ангидрид хромовой кислоты-пиридин; и реагенты, которые можно использовать при диметилсульфоксидном окислении (такие как комплексы диметилсульфоксид и дициклогексилкарбодиимид, оксалилхлорид, уксусный ангидрид, пентаоксид фосфора или пиридин-серный ангидрид).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 60 до 40oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 2 до 16 ч будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Реакционная схема VII
Стадия 23% Конденсация
В этом стадии соединение формулы (26) может быть получено взаимодействием соединения формулы (25) с соединением формулы (2) в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 1 реакционной схемы 1, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 24: Замещение (X _→ Aco)
По этой стадии соединение формулы (27) может быть получено взаимодействием соединения формулы (26) с солью уксусной кислоты в присутствии йодида натрия.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как тетрагидрофуран, диаксан или диметоксиэтан; спирты, такие как трет-бутанол; кетоны, такие как ацетон или метилэтилкетон; нитросоединения, такие как нитрометан; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфортриамид; сульфоксиды, такие как диметилсульфоксид или сульфолан; и органические кислоты, такие как уксусная кислота, из них предпочтительны амиды (в частности диметилформамид) или сульфоксиды (в частности диметилсульфоксид).
Подходящие для использования в этой реакции ацетаты включают соли щелочных металлов уксусной кислоты, такие как ацетат натрия или ацетат калия.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном удобно проведение реакции при температуре от 0 до 150oC, более предпочтительно от 25 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч, более предпочтительно от 2 до 6 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 25: Снятие защитной группы
По этой стадии соединение формулы (28) может быть получено взаимодействием соединения формулы (27) с деацилирующим агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан: спирты, такие как метанол или этанол; и воду. Из них предпочтительны спирты (в частности метанол) или смесь воды и одного или более спиртов (в частности метиленхлорид).
Нет также особых ограничений по природе используемого деацилирующего агента, и любой деацилирующий агент, обычно используемый в общеизвестных реакциях, может быть таким же образом использован и здесь. Примеры подходящих деацилирующих агентов включают неорганические основания, включая карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия или карбонат лития; гидрокарбонаты щелочных металлов, такие как гидрокарбонат натрия, гидрокарбонат калия или гидрокабонат лития; и гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия или гидроксид лития; гидроксиды щелочноземельных металлов, такие как гидроксид бария; или алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, трет-бутоксид калия или метоксид лития. Из них предпочтительны гидроксиды щелочных металлов (в частности гидроксид натрия или гидроксид калия).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 100oC, более предпочтительно от 25 до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 10 мин до 24 ч, более предпочтительно от 30 мин до 12 ч будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 26: Окисление
В этой стадии соединение формулы (24а) может быть получено взаимодействием соединения формулы (28) с окислительным агентом. Входящая в эту стадию реакция по существу та же, что и в стадии 22 реакционной схемы VI, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема VIII
Стадия 27:
В этой стадии соединение формулы (29) может быть получено взаимдействием соединения формулы (24) с реагентом Виттига-Хорнера (Witting-Hjrner), такого как диэтоксифосфорилацетат в присутствии основания.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол: простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфортриамид; и сульфоксиды, такие как диметилсульфоксид или гексаметилфосфортриамид; и сульфоксиды, такие как диметилсульфоксид или сульфолан; более предпочтительны эфиры (в частности тетрагидрофуран) или амиды (в частности диметилформамид).
Нет также конкретных ограничений по природе используемого основания, и любые основания, обычно применяемые в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включают такие соединения, которые используются в стадии 7.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 100oC, более предпочтительно от 0 до 50oC. Время, необходимое для реакции, может также широко изменяться, в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени т 10 мин до 48 ч, более предпочтительно от 1 до 24 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение, сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Двойная связь, образующаяся в продукте, может быть, если желательно, каталитически восстановлена в токе водорода.
Реакцию восстановления обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают% сложные эфиры, такие как этилацетат; простые эфиры, такие как тетрагидрофуран, диоксан или диметоксиэтан; и спирты, такие как метанол или этанол; более предпочтительны спирты, такие как метанол или этанол; простые эфиры, такие как тетрагидрофуран или диоксан; жирные кислоты, такие как уксусная кислота, или смеси одного или более органического растворителя и воды.
Нет также особых ограничений по природе используемого катализатора, и любой катализатор, обычно используемый в общеизвестных реакциях, может быть таким же образом использован и здесь. Примеры подходящих кислотных катализаторов включают палладий на угле или никель Ренея.
Реакцию можно осуществлять в широком интервале температур, точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -10 до 50oC, более предпочтительно от 0 до 25oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 10 мин до 24 ч, более предпочтительно от 30 мин до 6 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает% фильтрование катализатора и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 28% Восстановление
В этой стадии соединение формулы (30) может быть получено взаимодействием соединения формулы (29) с восстановительным агентом. Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан: и спирты, такие как метанол или этанол. Из них предпочтительны эфиры (в частности тетрагидрофуран) или спирты (в частности метанол).
Нет также особых ограничений по природе используемого восстановительного агента, и любой восстановительный агент такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры подходящих восстановительных агентов включают натрийборгидрид и диизобутилалюминийгидрид.
Реакцию можно осуществлять в широком интервале температур, и точеная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20oC до 100oC, более предпочтительно от 0 до 80oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 10 минут до 24 ч, более предпочтительно от 1 до 10 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 29: Окисление
В этой стадии соединение формулы (31) может быть получено взаимодействием соединения формулы (30) с окислительным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: галогенированные углеводороды, такие как метиленхрорид или хлороформ; и простые эфиры, такие как тетрагидрофуран, диметоксиэтан или диоксан. Из них предпочтительны галогенированные углеводороды (в частности метиленхлорид).
Нет также особых ограничений по природе используемого окислительного агента, и любой агент такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры подходящих окислительных агентов включают: соединения хрома, такие как комплекс ангидрид хромовой кислоты-пиридин или пиридиний-хлорхормат; и реагенты, которые можно использовать при диметилсульфоксидном окислении ( такие как комплекс диметилсульфоксида и дициклогкесилкарбодиимид, оксалил хлорид, уксусный ангидрид, пентоксид фосфора или комплекс пиридин-серный ангидрид). Из них предпочтительны пиридинийхлорхормат или диметилсульфоксид/оксалилхлорид).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -78 до 40oC, более предпочтительно от -60 до 25oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 30 мин до 6 ч, более предпочтительно от 1 до 3 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Реакционная схема IX
Стадия 30: Алкилирование
В этой стадии соединение формулы (Ie) может быть получено взаимодействием соединения формулы (31) с желаемым реактивом Гриньяра.
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол; и простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -78 до 50oC, более предпочтительно от -60 до 25oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 10 мин до 24 ч более предпочтительно от 30 минут до 1 ч, будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 31: Этерификация
В этой стадии соединение формулы (If) может быть получено этерификацией соединения формулы (Ie) в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 3 реакционной схемы 1, и может быть осуществлена с использованием тех де реагентов и в тех же реакционных условиях.
Стадия 32: Окисление
В этой стадии соединение формулы (Ig) может быть получено взаимодействием соединения формулы (Ie) с окислительным агентом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 29 реакционной схемы VIII, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема X
Стадия 33: Алкилирование
В этой стадии соединение формулы (Ih) может быть получено взаимодействием соединения формулы (24) с желаемым реактором Гриньяра в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 30 реакционной схемы IX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 34: Этерификация
В этой стадии соединение формулы (Ii) может быть получено этерификацией соединения формулы (Ih) в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 3 реакционной схемы 1, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 35: Окисление
В этой стадии соединение формулы (Ij) может быть получено взаимодействием соединения формулы (Ih) с окислительным агентом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 22 реакционной схему VI, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема XI
Стадия 36:
В этой стадии соединение формулы (Ik) может быть получено взаимодействием соединения формулы (24) с реагентом Виттига-Хорнера (Witting-Horner) в присутствии основания.
Реакция обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликоль диметиловый эфир; амиды, такие как диметилформамид, диметилацетамид или гексаметилфосфортриамид; и сульфоксиды, такие как диметилсульфоксид или сульфолан. Из них предпочтительны эфиры (в частности тетрагидрофуран) или амиды (в частности диметилформамид).
Нет также конкретных ограничений по природе используемого основания, и любые основания, обычно применяемые в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включает такие основания, которые используются в стадии 7.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 100oC, более предпочтительно от 0 до 70oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 10 мин до 24 ч, более предпочтительно от 30 мин до 12 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Двойная связь, образующаяся в продукте, может быть, если желательно, каталитически восстановлена в токе водорода.
Реакцию восстановления обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: сложные эфиры, такие как этилацетат; простые эфиры, такие как тетрагидрофуран, диоксан или диметоксиэтан; спирты, такие как метанол или этанол; и жирные кислоты, такие как уксусная кислота; вода, более предпочтительны спирты, такие как метанол или этанол, простые эфиры, такие как тетрагидрофуран или диоксан, жирные кислоты, такие как уксусная кислота, или смесь одного или более органического растворителя и воды.
Нет также особых ограничений по природе используемого катализатора, и любой катализатор, обычно используемый в общеизвестных реакциях, может быть таким же образом использован и здесь. Примеры подходящих кислотных катализаторов включают палладий на угле, платину и никель Ренея.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобном проведение реакции при температуре от -10 до 50oC, более предпочтительно от 0 до 25oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 20 мин до 24 ч, более предпочтительно от 30 мин до 6 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: фильтрование катализатора и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 37: Восстановление
В этой стадии соединение формулы (Im) может быть получено восстановлением соединения формулы (Ik) восстанавливающим агентом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 6 реакционной схемы 1, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 38: Этерификация
В этой стадии соединение формулы (In) может быть получено этерификацией соединения формулы (Im) в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 3 реакционной схемы 1, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема XII
Стадия 39: Изоцианирование (карбоновой кислоты)
В этой стадии соединение формулы (33) может быть получено взаимодействием соединения формулы (32) с дифенилфосфорилазидом в присутствии основания.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол ил кислот; галогенированные углеводороды, такие как метиленхлорид или дихлорэтан; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликоль диметиловый эфир; нитрилы, такие как ацетонитрил или изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфортриамид. Из них предпочтительны ароматические углеводороды (в частности бензол или тооуол) или эфиры (в частности тетрагидрофуран).
Нет также конкретных ограничений по природе используемого основания, и любые основания, обычно применяемые в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включают основания, такие как триэтиламин, диизопропилэтиламин, N-метилфорфолин, пиридин, 4-(N, N-диметиламино)пиридин, N,N-диметиланилин или N,N-диэтиланилин. Из них предпочтительны триэтиламин или диизопропилэтиламин.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов,_ как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 50 до 150oC, более предпочтительно от 70 до 120oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 30 мин до 24 ч, более предпочтительно от 1 до 12 ч, будет обычно достаточен.
После завершения реакции может быть обнаружено, что желаемое соединение является слишком неустойчивым для очистки и поэтому его можно использовать в последующей реакции без очистки.
Стадия 40: Присоединение
в этой стадии соединение формулы (34), в котором R2 означает атом водорода, может быть получено взаимодействием соединения формулы (33) с указанным соединением формулы (3), (5), (17), или (21).
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид или дихлорэтан; и простые эфиры, такие как тетрагидрофуран, диоксан или диметоксиэтан. Из них предпочтительны ароматические углеводороды (в частности бензол или толуол). \\2 Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 150oC, более предпочтительно от 25 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанные выше, период времени от 10 мин до 24 ч, более предпочтительно от 1 до 6 ч, будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя. Полученный таким образом желаемый продукт, если необходимо, может быть затем очищен такими обычными способами, как перекристаллизация, переосаждение или различные хроматографические способы, а именно колоночная хроматография.
Стадия 41: Изоцианирование (амина)
В этой стадии соединение формулы (36) может быть получено взаимодействием соединения формулы (35) с фосгеном, трихлорметилхлормиатом или бис(трихлорметил)карбонатом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: галогенированные углеводороды, такие как метиленхлорид, хлороформ или дихлорэтан; и простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан или диэтиленгликоль. Из них предпочтителен метиленхлорид.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -40 до 50oC. более предпочтительно от -20 до 0oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 10 мин до 24 ч, более предпочтительно от 1 до 6 ч, будет обычно достаточен.
После завершения реакции желаемое соединение предпочтительно используется в последующей реакции без очистки ввиду неустойчивости соединения.
Стадия 42: Присоединение
В этой стадии соединение формулы (34) может быть получено взаимодействием соединения формулы (36) амином формулы: R1R2NH в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 40 реакционной схемы XII, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 43: Снятие защитной группы
В этой стадии соединение формулы (1p) может быть получено взаимодействием соединения формулы (34) со снимающим защиту агентом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 2 реакционной схемы 1, и может быть осуществлена с использованием тех же реагентом и в тех же реакционных условиях. Если желательно, вторичная гидроксильная группа соединения формулы (34) может быть модифицирована по способу, описанному в стадии 3.
Способы получения указанных исходных соединений формул (15), (17), (21) и (25) пояснены далее.
Реакционная схема XIII
Стадия 44: Восстановление
В этой стадии соединение формулы (38) может быть получено взаимодействием соединения формулы (37) с восстановительным агентом.
Нет также особых ограничений по природу используемого восстановительного агента, и любой восстановительный агент такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры подходящих восстановительных агентов включают дибораны, такие как диборан или комплекс диборан-диметилсульфид.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают простые эфиры, такие как тетрагидрофуран, диоксан или 1,2-диметиоксиэтан.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 20 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 1 до 24 ч будет обычно достаточно.
Альтернативно к этой реакции соединение формулы (37) подвергают взаимодействию с хлорформиатом, таким как этилхлорформиат, с получением смешанного ангидрида, и затем продукт может быть восстановлен натрий боргидридом.
Растворитель, используемый в синтезе смешанного ангидрида, предпочтительно является эфиром, таким как 1,2-диметоксиэтан или тетрагидрофуран.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 30oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 30 мин до 6 ч будет обычно достаточно.
После завершения реакции ангидрид может быть выделен из реакционной смеси различными хорошо известными методами, например фильтрованием реакционной смеси на Целите (торговая марка). Фильтрат затем добавляют к смеси воды и тетрагидрофурана, содержащей боргидрид натрия, для осуществления восстановления.
Реакцию восстановления можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 30 мин до 3 ч будет обычно достаточно.
Стадия 45: Введение уходящей группы
По этой стадии соединение формулы (15) может быть получено взаимодействием соединения формулы (38) с алкилсульфонилгалогенидом в инертном растворителе в присутствии основания. Входящая в эту стадию реакция по существу та же, что и в стадии 13 реакционной схемы V, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Сульфонаты, полученные в этой стадии, если желательно, подвергают взаимодействию с йодидом натрия или йодидом калия с получением йодида.
Подходящим растворителем для этой реакции является кетон, такой как ацетон или метилэтилкетон.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 80oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 10 мин до 8 ч будет обычно достаточно.
Реакционная схема XIV
Стадия 46: Введение уходящей группы
По этой стадии соединение формулы (40) может быть получено взаимодействием соединения формулы (39) с алкилсульфонил или арилсульфонилгалогенидом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 45 реакционной схемы XIII, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях. Эта стадия применяется для получения соединения, в котором A1 является отличным от одинарной связи.
Далее, сульфонаты, полученные в этой стадии, если желательно, преобразуют в соответствующие йодиды, аналогично способу получения, описанному в стадии 45.
Стадия 46(a): Получение спиртового соединения
По этой стадии соединение формулы (41) может быть получено взаимодействием соединения формулы (39) с желаемым диольным соединением (HO-A2-OH) или галогеноспиртовым соединением формулы (hal-A2-OH) в присутствии основания. Эта стадия применяется для получения соединения, в котором A1 является одинарной связью.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как тетрагидрофуран или 1,2-диметоксиэтан; и амиды, такие как диметилформамид или диметилацетамид.
Нет также конкретных ограничений по природе используемого основания, и любые основания, обычно применяемые в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включают карбонаты щелочных металлов, такие как карбонат калия.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 20 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 2 до 12 ч будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; отгонку растворителя; добавление воды и несмешивающегося с водой органического растворителя, такого как диэтиловый эфир; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя.
Стадия 47: Получение спиртового соединения
По этой стадии с соединение формулы (41) может быть получено взаимодействием соединения формулы (40) с желаемым диольным соединением в присутствии основания. Эта стадия применяется для получения соединения, в котором является отличным от одинарной связи.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как тетрагидрофуран или 1,2-диметоксиэтан; и амиды, такие как диметилформамид или диметилацетамид.
Нет также конкретных ограничений по природе используемого основания, и любые основания, обычно применяемые в обычных реакциях, могут таким же образом использоваться и здесь. Примеры подходящих оснований включают карбонаты щелочных металлов, такие как гидрид натрия или гидрид калия.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 60oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление вод и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя.
Стадия 48: Введение защитной группы
По этой стадии соединение формулы (42) может быть получено взаимодействием соединения формулы (41) с защищающим агентом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 11 реакционной схемы III, и может быть осуществлена с использованием тех же реагентов (за исключением метоксиметилхлорида) и в тех же реакционных условиях.
Стадия 49: Восстановление нитрогруппы
В этой стадии соединение формулы (17) может быть получено восстановлением соединения формулы (42) восстанавливающим агентом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 12 реакционной схемы III, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема XIV
Стадия 50: Восстановление
В этой стадии соединение формулы (44) может быть получено восстановлением соединения формулы (43) восстанавливающим агентом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 44 реакционной схемы III, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 51: Введение защитной группы
По этой стадии соединение формулы (45) может быть получено взаимодействием соединения формулы (414) с защищающим агентом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 11 реакционной схемы III, и может быть осуществлена с использованием тех же реагентов (за исключением метоксиметилхлорида) и в тех же реакционных условиях.
Стадия 52: Введение нитро группы
В этой стадии соединение формулы (21) может быть получено восстановлением соединения формулы (45) с восстанавливающим агентом в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 12 реакционной схемы III, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема XVI
Стадия 53: Алкилирование
В этой стадии соединение формулы (47) может быть получено алкилированием соединения формулы (46) с диметилсульфоксидом в присутствии N-хлорсуккцинимидом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают галогенированные углеводороды, такие как метиленхлорид или хлороформ.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 60oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: фильтрование нерастворимого материала, если таковой имеется; добавлением галогенированного углеводородного растворителя, такого как метиленхлорид; промывание органической фазы насыщенным водным раствором гидрокарбоната натрия; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя.
Стадия 54: Окисление
В этой стадии соединение формулы (48) может быть получено взаимодействием соединения формулы (47) с окислительным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: галогенированные углеводороды, такие как метиленхлорид или хлороформ; и спирты, такие как метанол или этанол.
Нет также особых ограничений по природе используемого окислительного агента, и любой агент такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры предпочтительных окислительных агентов включают м-хлорпербензойную кислоту.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 60oC (предпочтительно при комнатной температуре). Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч будет обычно достаточно.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы насыщенным водным раствором гидрокарбоната натрия; и затем отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как бензойный сульфат магния; и отгонку растворителя.
Стадия 55: Галогенирование
В этой стадией соединение формулы (25) может быть получено взаимодействием соединения формулы (48) с галогенводородом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают галогенированные углеводороды, такие как метиленхлорид, хлороформ или 1,2-дихлорэтан.
Используемым галогенводородом, например, может быть хлористый водород или бромистый водород.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 60oC. Время, необходимое для реакции при температуре от -20 до 60oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако, при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, желаемое соединение может выть выделено собиранием осадка фильтрованием.
Исходный материал формулы (2), используемый в настоящем изобретении, может быть получен синтезом галогенангидрида из коммерчески доступной карбоновой кислоты, либо из карбоновой кислоты, которая может быть получена известным специальным способом. В частности соединение, имеющее (9Н-ксантен-9-ил)метильную группу, может быть получено аналогично способу, описанному в патентной заявке Японии Hei 4-243357. 6,11-Дигидробенз[b,e]окзепин-11-карбоновая кислота известна из EP 0497201 (1992). Альтернативно исходные соединения, используемые в настоящем изобретении, могут быть получены следующим образом.
Реакционная схема XVII
Стадия 56:
В этой стадии соединение формулы (50) может быть получено взаимодействием соединения формулы (49) с оксалилхлоридом для получения хлорангидрида кислоты и затем взаимодействием этого продукта с диазометаном.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают диэтиловый эфир или тетрагидрофуран.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -20 до 60oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над сушильным агентом, таким как безводный сульфат магния; и отгонку растворителя.
Стадия 57:
В этой стадии соединение формулы (5) может быть получено взаимодействием соединения формулы (50) со спиртом в присутствии металла, такого как бензоат серебра.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают спирты, такие как метанол или этанол.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 40 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 3 ч будет обычно достаточен.
После завершения реакции желаемое соединение может быть выделено из реакционной смеси обычными методами. Например, один подходящий способ включает: полную нейтрализацию реакционной смеси; фильтрование нерастворимого материала, если таковой имеется; добавление воды и несмешивающегося с водой органического растворителя, такого как этилацетат; промывание органической фазы водой; отделение органической фазы, содержащей желаемое соединение; сушку экстракта над осушающим агентом, таким как безводный сульфат магния; и отгонку растворителя.
Стадия 58:
По этой стадии соединение формулы (2а) может быть получено гидролизом формулы (51) в инертном растворителе. Входящая в эту стадию реакция по существу та же, что и в стадии 25 реакционной схемы VII,и может быть осуществлена с использованием тех же реагентов и в этих же реакционных условиях.
Реакционная схема XVIII
Стадия 59: Восстановление литийалюминийгидридом
Эта стадия включает получение спиртового соединения взаимодействия соединения формулы (49) с восстановительным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагирофуран, диоксан, диметоксиэтан и диэтиленогликоль диметиловый эфир (особенно тетрагидрофуран).
Нет также особых ограничений по природе используемого восстановительного агента и любой восстановительный агент такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры подходящих восстановительных агентов включают литийалюминийгидрид.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 70oC, более предпочтительно от 40 до 70oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 15 мин до 2 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь охлаждают на ледяной бане; добавляют воду и насыщенный водный раствор гидрокарбоната натрия и далее несмешивающийся с водой органический растворитель, например диэтиловый эфир; отфильтровывают нерастворимый материал, если таковой имеется; фильтрат промывают водой, водным раствором гидрокарбоната натрия и водным раствором хлорида натрия, в указанной последовательности; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт можно, если желательно, затем очистить такими обычными способами, как перекристаллизация или различными хроматографическими способами, а именно колоночной хроматографией.
Стадия 60: Введение уходящей группы
Эта стадия включает получение алкил- или арилсульфонатного соединения путем взаимодействия спиртового соединения, полученного как описано в стадии 59, с реагентом, используемым для введения алкил- или арилсульфонильных групп (предпочтительно метансульфонильной группы) в присутствии основания в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 18 реакционной схемы V, и может быть осуществлена с использованием тех же реагентов и в тех реакционных условиях.
Стадия 61: Йодирование
Эта стадия включает получение соединения (52) взаимодействием алкил- или арилсульфонатного соединения, полученного как описано в стадии 60, с йодирующим агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: кетоны, такие как ацетон, метилэтилкетон или метилизобутилкетон (особенно метилизобутилкетон).
Нет таких особых ограничений по природе используемого йодирующго агента, и любой йодирующий агент, обычно используемый в общеизвестных реакциях, может быть таким же образом использован и здесь. Примеры подходящих йодирующих агентов включают йодид натрия.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 30 до 150oC, более предпочтительно от 60 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 48 ч, более предпочтительно от 5 до 20 ч, будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такой методики является следующим : реакционную смесь освобождают от растворителя дистилляцией; остаток смешивают с водой; водную смесь экстрагируют несмешивающимся с водой растворителем, например бензолом, диэтиловым эфиром, этилацетатом или тому подобное; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в качестве остатка. Полученный таким образом продукт, если желательно, очищается далее общеизвестными методами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Стадия 62: Синтез предшественника альдегида
Стадия включает получение алкилированного предшественника алкилированного альдегида взаимодействием соединения формулы (52) и соединением формулы X3H2 в присутствии основания в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 7 реакционной схемы 11, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 63: Гидролиз
Эта стадия включает получение соединения формулы (53) гидролизом предшественника альдегида, полученного, как описано в стадии (57) 62, в присутствии соединения тяжелого металла.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: смеси воды с нитрилом, таким как ацетонитрил, или спирт, такой как метанол или этанол.
Используемым соединением тяжелого металла может быть, например, хлорид ртути или нитрат серебра.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 50 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 1 до 5 ч будет обычно достаточно.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь охлаждают на ледяной бане; добавляют несмешивающийся с водой растворитель, например бензол, этиловый эфир, этилацетат или тому подобное; отфильтровывают нерастворимый материал; фильтрат промывают водой, водным раствором ацетата натрия, водным раствором хлорида аммония и водным раствором хлорида натрия в указанной последовательности; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт можно, если желательно, затем очистить такими обычными способами, как перекристаллизация или различными хроматографическими способами, а именно колоночной хроматографией.
Стадия 64: Окисление (для получения карбоксильной группы)
Эта стадия включает получение соединения (2а) взаимодействием соединения формулы (53) с окислительным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают смесь воды с трет-бутанолом.
Нет также особых ограничений по природе используемого окислительного агента, и любой агент такого рода, используемый в обычных реакциях, может быть таким же образом использован и здесь. Примеры подходящих окислительных агентов включают хлорид натрия.
Реакцию можно осуществить в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 50oC, более предпочтительно при комнатной температуре. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 30 мин до 5 ч, более предпочтительно от 1 до 3 ч, будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь экстрагируют несмешивающимся с водой растворителем, например метиленхлоридом; экстракт промывают водой; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт можно, если желательно, затем очистить таким обычными способами, как перекристаллизация, или различными хроматографическими способами, а именно колоночной хроматографией.
Реакционная схема XIX
Стадия 65: Реакция Виттига
Эта стадия включает получение соединения формулы (54) взаимодействием соединения формулы (Ig) с метоксиметилтрифенилфосфонийхлоридом в присутствии основания. +Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликоль диметиловый эфир (особенно тетрагидрофуран).
Нет также особых ограничений по природе используемого основания, и любое основание, обычно используемое в общеизвестных реакциях, может быть таким же образом использовано и здесь. Пример подходящего основания включает бутиллитий.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 10 до 40oC и, когда в качестве основания применяется бутиллитий, более предпочтительно при температуре от 0 до 5oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 5 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь выливают в водный раствор хлорида аммония; водную смесь экстрагируют несмешивающимся с водой растворителем, например диэтиловым эфиром, этилацетатом и тому подобное; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт можно, если желательно, затем очистить обычными способами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Стадия 66: Гидролиз
Эта стадия включает получение соединения формулы (55) путем взаимодействия соединения формулы (54) с кислотой.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликоль диметиловый эфир (особенно тетрагидрофуран).
Нет также особых ограничений по природе используемой кислоты, и любая кислота, обычно используемая в общеизвестных реакциях, может быть таким же образом использована и здесь. Пример подходящей кислоты включает соляную кислоту.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции от 10 до 100oC, более предпочтительно при температуре от 30 до 80oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь выливают в воду: водную смесь экстрагируют несмешивающимся с водой растворителем, например бензолом, диэтиловым эфиром, этилацетатом или тому подобное; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт может быть использован в дальнейшей реакции без очистки или может, если желательно, быть очищен обычными способами, такими как перекристаллизация или различные хроматографические способы, а именно колоночной хроматографией.
Стадия 67: Восстановление
Эта стадия включает получение соединения формулы (56) взаимодействием соединения формулы (55) с восстановительным агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 10 реакционной схемы 11, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема XX
Стадия 68: Галогенирование
Эта стадия включает получение соединения формулы (57) взаимодействием соединения формулы (23) с галогенирующим агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: галогенированные углеводороды, такие как метиленхлорид и хлороформ (особенно метиленхлорид).
используемыми галогенирующими агентами могут быть, например, сочетание трифенилфосфина и тетрабромуглерод.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 10 до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако, при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч, более предпочтительно от 2 до 5 ч, будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами, например реакционную смесь промывают водой и затем растворитель отгоняют из органической фазы, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт, если желательно, может быть затем очищен обычными способами, такими как перекристаллизация или различные хроматографические методы, а именно колоночной хроматографией.
Стадия 69: Образование фосфониевого соединения
Эта стадия включает получение соединения формулы (58) взаимодействием соединения формулы (57) с трифенилфосфином.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают углеводороды, такие как бензол, толуол или ксилол (особенно толуол).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 50 до 150oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно, температуры реакции и природы реагентов и используемых растворителей. Однако, при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 5 до 24 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. В качестве такого метода является следующий: реакционную температуру поднимают до комнатной температуры; остаток собирают фильтрацией и промывают толуолом или гексаном и затем высушивают. Полученный таким образом продукт, если желательно, может быть затем очищен обычными способами, такими как перекристаллизация.
Такая же стадия также включена в получение соединения формулы (84) из соединения формулы (15а). (Реакционная схема XXI)/
Стадия 70: Реакция Гриньяра
Эта стадия включает получение соединения формулы (61) реакцией Гриньяра между соединением формул (59) и (60).
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетерагидрофуран, диоксан, диметоксиэтан и диэтиленгликоль диметиловый эфир (особенно диэтиловый эфир).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 40oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 10 мин до 2 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: водный раствор хлорида аммония добавляют к реакционной смеси; водную смесь экстрагируют несмешивающимся с водой растворителем, например бензолом, диэтиловым эфиром, этилацетатом или тому подобное; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт, если желательно, может быть затем очищен обычными способами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Стадия 71: Окисление Сверна (Swern)
Эта стадия включает получение соединения формулы (62) взаимодействием соединения формулы (61) с окислительным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают галогенированные углеводороды, такие как метиленхлорид, хлороформ и дихлорэтан (особенно метиленхлорид).
Используемым окислительным агентом может быть, например, сочетание оксалилхлорида и диметилсульфоксида.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -100 до -50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 10 мин до 1 ч будет обычно достаточен. После этого реакционную смесь охлаждают до -78oC и затем добавляют основание (предпочтительно триэтиламин), после чего температуре позволяют подняться до комнатной за период от 10 до 30 мин.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь выливают в воду; водную смесь экстрагируют несмешивающимся с водой растворителем, например бензолом, диэтиловым эфиром, этилацетатом или тому подобное; экстракт промывают разбавленной соляной кислотой и водой в указанном порядке; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт может обычно быть использован в дальнейшей реакции без очистки или может, если желательно, быть затем очищен обычными способами, такими как перекристаллизация или различные хроматографические способы, а именно колоночной хроматографией.
Стадия 72? Реакция Виттига
Эта стадия включает получение соединения формулы (63) посредством реакции Виттига с использованием соединений формул (58) и (62) в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 65 реакционной схемы XIX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 73: Дебензилирование и восстановление двойной связи
Эта стадия включает получение соединения формулы (64) восстановлением соединения формулы (63) с восстанавливающим агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: спирты, такие как метанол, этанол, пропанол, изопропанол, бутанол, изобутанол или трет-бутанол (особенно этанол); и простые эфиры, такие как диэтиловый или тетрагидрофуран.
Нет также особых ограничений по природе используемого восстанавливающего агента, и любое основание, обычно используемое в общеизвестных реакциях, может быть таким же образом использовано и здесь. Пример подходящего восстанавливающего агента включает водород в присутствии палладиевого катализатора.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 10 до 40oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 10 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами, например катализатор отфильтровывают и затем отгоняют растворитель из фильтрата, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт можно, если желательно, затем очистить обычными способами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Реакционная схема XXI
Стадия 74: Защита гидроксильной группы
Эта стадия включает получение соединения формулы (65) взаимодействием соединения формулы (61) с реагентом, защищающим гидроксильную группу.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя, при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают диметилформамид.
Примеры подходящих защитных групп включают метоксиметильную и трет-бутилдиметилсилильную группы. Условия реакции будут зависеть от природы вводимой защитной группы, например:
(I) Введение метоксиметильной группы
Желаемое соединение может быть получено взаимодействием соединения формулы (61) с метоксиметилхлоридом в присутствии органического третичного амина, такого как триэтиламин в подходящем растворителе, например диметилформамиде.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 50 до 100oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 10 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такой методики является следующим: реакционную смесь освобождают от растворителя дистилляцией; к остатку добавляют ледяную воду; водную смесь экстрагируют несмешивающимся с водой растворителем, например бензолом, диэтиловым эфиром, этилацетатом или тому подобное; экстракт промывают разбавленной соляной кислотой и водой в указанном порядке; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в качестве остатка. Полученный таким образом продукт, если желательно, можно очистить далее общеизвестными методами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
(II) Введение трет-бутилдиметилсилильной группы
Желаемое соединение может быть получено взаимодействием соединения формулы (61) с трет-бутилдиметилсилилхлоридом в присутствии органического третичного амина, такого как триэтиламин (если необходимо, при дополнительном присутствии 4-(N,N-диметиламино)пиридина в подходящем растворителе, например диметилформамиде.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 10 до 60oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 30 мин до 5 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такой методики является следующим: реакционную смесь освобождают от растворителя дистилляцией; к остатку добавляют ледяную воду; водную смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное; экстракт промывают разбавленной соляной кислотой и водой в указанном порядке; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в качестве остатка. Полученный таким образом продукт, если желательно, можно очистить далее общеизвестными методами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Стадия 75: Дебензилирование
Эта стадия включает получение соединения формулы (66) восстановлением соединения формулы (65) с восстанавливающим агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 73 реакционной схемы XX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 76: Окисление Сверна (Swern)
Эта стадия включает получение соединения формулы (67) взаимодействием соединения формулы (66) с окислительным агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 71 реакционной схемы XX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 77: Реакция Виттига
Эта стадия включает получение соединения формулы (68) посредством реакции Виттига с использованием соединений формул (67) и (84) в присутствии основания и в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 65 реакционной схемы XIX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 78a и 78b: Восстановление
Эта стадия включает получение соединений формул (69) и (85) взаимодействием соединения формулы (68) в восстановительным агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 73 реакционной схемы XX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях, за исключением того, что реакцию по стадии 78a предпочтительно проводят в диэтиловом эфире при комнатной температуре в течение периода времени от 30 мин до 1 ч, а по стадии 78b предпочтительно проводят в этаноле при комнатной температуре за период от 5 до 10 ч.
Реакционная схема XXII
Стадия 79: Восстановление
Эта стадия включает получение соединений формулы (71) взаимодействием соединения формулы (70) с восстановительным агентом.
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: галогенированные углеводороды, такие как метиленхлорид, хлороформ: тетрахлоруглерод, дихлорэтан. хлорбензол и дихлорбензол (особенно метиленхлорид).
Нет также особых ограничений по природе используемого восстановительного агента, и любой восстановительный агент, обычно используемый в общеизвестных реакциях, может быть таким же образом использован и здесь. Пример подходящего восстановительного агента включает диизобутиламмонийгидрид.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 10 до 40oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 30 мин до 5 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такой методики является следующим: реакционную смесь гасят добавлением метанола при охлаждении льдом; к реакционной смеси добавляют разбавленную соляную кислоту для разбавления нерастворенного материала; смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в качестве остатка. Полученный таким образом продукт, если желательно, можно затем очистить общеизвестными методами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Стадия 80: Окисление Сверна (Swern)
Эта стадия включает получение альдегидного соединения путем взаимодействия соединения формулы (71) с окислительным агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 71 реакционной схемы XX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 81: Реакция Виттига
Эта стадия включает получение соединения формулы (72) взаимодействием альдегидного соединения, полученного как описано в стадии 80, с реагентом Виттига, с использованием трифенилфосфонийбромида в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 65 реакционной схемы XIX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 82: Образование диольной группы
Эта стадия включает получение диольного соединения взаимодействием соединения формулы (72) с реагентом для образования диольной группы.
Реакцию обычно и предпочтительно осуществляют в присутствии подходящего растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Пример подходящего растворителя включает смесь ацетонитрила и воды.
Примером подходящего окислительного агента, который может быть использован, является тетраоксид осмия (который может быть использован в сочетании с морфолин-N-оксидом в качестве окислительного агента).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 100oC, более предпочтительно от 10 до 40oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 5 до 12 ч, более предпочтительно от 1 до 24 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такой методики является следующим: реакционную смесь выливают в воду; водную смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное; затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт, если желательно, можно затем очистить общеизвестными методами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Стадия 83: Образование бензилиденовой группы
Эта стадия включает получение соединения формулы (73) взаимодействием диольного соединения, полученного, как описано в стадии 82, с бензилиден диметилацеталем в присутствии кислотного катализатора.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают галогенированные углеводороды, такие как метиленхлорид, хлороформ, тетрахлоруглерод или дихлорэтан (особенно метиленхлорид).
Нет также особых ограничений по природе используемого кислотного катализатора, и любой кислотный катализатор, обычно используемый в общеизвестных реакциях, может быть таким же образом использован и здесь. Примером подходящего кислотного катализатора является n-толуолсульфоновая кислота.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 10 до 40oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, периода времени от 1 до 5 ч будет обычно достаточно.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такой методики является следующим: к реакционной смеси добавляют водный раствор гидрокарбоната натрия; водную смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное; экстракт промывают водой; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в качестве остатка. Полученный таким образом продукт может быть использован в следующей стадии без очистки или, если желательно, может быть затем очищен общеизвестными методами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Стадия 84: Восстановление
Эта стадия включает получение соединения формул (74) и (75) взаимодействием соединения формулы (73) с восстановительным агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 79 реакционной схемы XXII, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема XXIII
Стадия 85: Окисление Сверна (Swern)
Эта стадия включает получение соединения формулы (76) путем взаимодействия соединения формулы (74) с окислительным агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 71 реакционной схемы XX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 86: Реакция Виттига
Эта стадия включает получение соединения формулы (77) посредством реакции Виттига, используя соединения формул (76) и (58) (см. реакционную схему XX), в присутствии основания в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 65 реакционной схемы XIX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 87: Окисление Сверна (Swern)
Эта стадия включает получение соединения формулы (78) путем взаимодействия соединения формулы (75) с окислительным агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 71 реакционной схемы XX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 88: Реакция Виттига
Эта стадия включает получение соединения формулы (79) посредством реакции Виттига, используя соединения формул (78) и (58), в присутствии основания в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 65 реакционной схемы XIX, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Желаемое соединение по изобретению может быть получено восстановительным дебензилированием и восстановлением двойной связи соединения формулы (77) или (79), используя те же способы, что описаны в стадии 73.
Реакционная схема XXIV
Стадия 85: Эпоксидирование
Эта стадия включает получение соединения формулы (80) путем взаимодействия соединения формулы (31) с триметилсульфоксонийиодидом в присутствии основания.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликоль диметиловый эфир (особенно тетрагидрофуран); и сульфоксиды, такие как диметилсульфоксид.
Нет также особых ограничений по природе используемого основания, и любое основание, обычно используемое в общеизвестных реакциях, может быть таким же образом использовано и здесь. Пример подходящего основания включает гидрид натрия.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 20 до 60oC. Время, необходимое для реакции, может также широко меняться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 30 мин до 5 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь разбавляют несмешивающимся с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное; органическую фазу промывают водой; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Полученный таким образом продукт можно, если необходимо, затем очистить такими обычными способами, как перекристаллизация или различными хроматографическими способами, а именно колоночной хроматографией.
Стадия 90: Расщепление эпоксидной группы
Эта стадия включает получение соединения формул (82) и (83) путем взаимодействия соединения формулы (80) с соединением формулы (81) в присутствии кислоты Льюиса.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он можно растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают галогенированные углеводороды, такие как метиленхлорид, хлороформ, тетрахлоруглерод или дихлорэтан (особенно метиленхлорид).
Нет также особых ограничений по природе используемой кислоты Льюиса, и любая кислота Льюиса, обычно используемая в общеизвестных реакциях этого типа, может быть таким же образом использована и здесь. Пример подходящей кислоты Льюиса включает эфират трифторида бора.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 10 до 60oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 5 мин до 15 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь освобождают от растворителя дистилляцией; концентрата выливают в воду; водную смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное; и затем растворитель отгоняют из экстракта, оставляя продукты формул (82) и (83) в качестве остатка. Продукты можно, если желательно, очистить далее общеизвестными методами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Если необходимо, гидроксильная группа соединений формул (82) и (83) может быть ацилирована и каждое из этих полученных ацилированных соединений может быть отделено хроматографически с последующим деацетилированием с получением соединения (82) или (83) в индивидуальном виде.
Стадия 90: Реакций Гриньяра
Эта стадия включает получение соединения формулы (61) взаимодействием соединения формулы (61а) с реактивом Гриньяра (R6A4bMgBr) в присутствии йодида меди, что обеспечивает альтернативный способ получения соединения (61), использованного в качестве исходного продукта в реакционной схеме XXI.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликоль диметиловый эфир (особенно тетрагидрофуран).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от -100до -50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 5 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь охлаждают на ледяной бане; добавляют водный раствор хлорида аммония, и затем перемешивают смесь; затем смесь экстрагируют несмешивающимся с водой растворителем, таким как диэтиловый; эфир; экстракт промывают водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия в указанном порядке; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Продукт, если желательно, может быть затем очищен обычными способами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Реакционная схема XXV
Стадия 92: Снятие защитной группы.
Когда гидроксизащитной группой является трет-бутилдиметилсилильная группа, она может быть снята обработкой соединением, способным к образованию аниона фтора, таким как тетрабутиламмоний фторид или соляная кислота.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают простые эфиры, такие как тетрагидрофуран, или спирты, такие как метанол.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 60oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 10 до 18 ч будет достаточен.
Когда используемая защитная группа является метоксиметильной группой, она может быть удалена обработкой кислотой.
Нет также особых ограничений по природе используемой кислоты, и любая кислота, обычно используемая в общепринятых реакциях, может быть таким же образом использована и здесь. Примеры подходящих кислот включают: кислоты Бронстеда, включая галогенводородные кислоты, такие как хлористоводородная кислота или бромистоводородная кислота, и органические кислоты, такие как уксусная кислота или n-толуолсульфокислота; и сильные кислотные ионообменные смолы, такие как Dowex (торговая марка) 50W.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере некоторой степени. Примеры подходящих растворителей включают спирты, такие как метанол или этанол; простые эфиры, такие как тетрагидрофуран или диоксан; и смеси одного или более растворителей с водой.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от 0 до 50oC. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 19 мин до 18 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь выливают в воду; водную смесь экстрагируют несмешивающимися с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Продукт может быть обычно использован в дальнейшей реакции без очистки или может, если желательно, быть очищен обычными способами, такими как перекристаллизация или различные хроматографические способы, а именно колоночной хроматографией.
Стадия 93: Реакция инверсии Мицунобу (Mitsunobu)
Эта стадия включает получение соединения формулы (87) взаимодействием соединения формулы (86) с бензойной кислотой в присутствии трифенилфосфина и диэтил азодикарбоксилата.
реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликоль диметиловый эфир (особенно тетрагидрофуран).
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительно реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно приведение реакции при температуре от 0 до 40oC. Время, необходимое для реакции может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь выливают в воду; водную смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное; разбавленный водный раствор промывают водным раствором гидрокарбоната натрия; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка, Продукт, если желательно, может быть затем очищен обычными способами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
По следующей стадии соединение формулы (88) может быть получено из соединения формулы (87) тем же способом, что описано в стадии 12 (реакционная схема III), с последующей обработкой, как описано в стадии I (реакционная схема I).
Стадия 94:
Эта стадия включает получение соединения формулы (89) взаимодействием соединения формулы (88) с основанием.
Нет также конкретных ограничений по природе используемого основания, и любое основание, обычно используемое в общеизвестных реакциях, может быть таким же образом использовано и здесь. Примеры подходящих оснований включают алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, трет-бутоксид калия или метоксид лития, предпочтительно этоксид натрия.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают спирты, такие как метанол, этанол, пропанол, изопропанол, бутанол, изобутанол, трет-бутанол или изоамиловый спирт.
Реакцию можно осуществлять в широком интервале температур, и точная температура не является критической по изобретению. Предпочтительная реакционная температура будет зависеть от таких факторов, как природа растворителя и используемый исходный продукт реакции. Однако, в основном, удобно проведение реакции при температуре от комнатной до точки кипения используемого растворителя. Время, необходимое для реакции, может также широко изменяться в зависимости от многих факторов, а именно температуры реакции и природы реагентов и используемых растворителей. Однако при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, период времени от 1 до 24 ч будет обычно достаточен.
После завершения реакции продукт может быть выделен из реакционной смеси обычными методами. Пример такого метода следующий: реакционную смесь выливают в воду и затем нейтрализуют; смесь экстрагируют несмешивающимися с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное; и затем растворитель отгоняют из экстракта, оставляя желаемый продукт в виде остатка. Продукт, если желательно, может быть затем очищен обычными способами, такими как перекристаллизация или различные хроматографические способы, а именно колоночная хроматография.
Стадия 95: Введение уходящей группы
Эта стадия включает получение соединения формулы (90) взаимодействием соединения формулы (41) с алкилсульфонилгалогенидом 9предпочтительно метансульфонилхлоридом) в присутствии основания.
Входящая в эту стадию реакция по существу та же, что и в стадии 18 реакционной схемы Y, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 96: Введение имидазолильной группы
Эта стадия включает получение соединения формулы (91) взаимодействием соединения формулы (90) с имидазолом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 19 реакционной схемы Y, и может быть осуществлена с использованием тех же реагентов и тех же реакционных условиях.
Стадия 97: Восстановление нитрогруппы
Эта стадия включает получение соединения формулы (92) взаимодействием соединения формулы (91) с восстановительным агентом в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии 12 реакционной схемы III, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия 98: Конденсация
Эта стадия включает получение соединения формулы (1d) взаимодействием соединения формулы (92) с соединением формулы (2) (реакционную схему I) в инертном растворителе.
Входящая в эту стадию реакция по существу та же, что и в стадии I реакционной схемы I, и может быть осуществлена с использованием тех же реагентов и в тех же реакционных условиях.
Такие соединения, которые содержат группу, представленную формулой R7, в соответствии с настоящим изобретением могут быть получены с использованием соединения, полученного восстановительным дебензилированием и восстановлением двойной связи соединений формул (77) и (79) или с использованием соединений формул (56), (64). (82), (83) и (89), которые могут быть получены стадиями, описанными выше, теми же методами, что описаны в стадии 3.
Когда R7 включает оксикарбонильную группу, реакция может быть осуществлена следующим образом.
Добавляют раствор трихлорметилхлорформиата в тетерагидрофуране при охлаждении льдом к тетрагидрофурановому раствору, содержащему пиридин, и полученную смесь перемешивают при комнатной температуре в течение подходящего периода времени, например 1 ч. Смесь затем вновь охлаждают на льду и добавляют раствор соединения формулы (1a), (1e), (1h), (1m), (56), (64), (82), (83) или (89) в тетрагидрофуране и смесь опять перемешивают, например, 1 ч. Тетрагидрофуран удаляют дистилляцией и остаток растворяют в метиленхлориде. Затем добавляют раствор желаемого спирта в метиленхлориде. К полученной таким образом смеси добавляют 4-(N,N-диметиламино)пиридин и полученную смесь перемешивают при комнатной температуре, например, 1 ч.
После завершения реакции реакционную смесь можно разбавить несмешивающимся с водой растворителем, таким как бензол, диэтиловый эфир, этилацетат или тому подобное. Разбавленную смесь затем промывают водой и насыщенным водным раствором хлорида натрия в указанном порядке, и растворитель отгоняют из органической фазы с получением продукта. Продукт, если желательно, может быть затем очищен хроматографически или перекристаллизацией.
Соединение, где карбоксильная группа, представленная R7, является солью щелочного металла, может быть получено следующим образом.
К раствору производного карбоновой кислоты добавляют основание в растворителе. Количество основания предпочтительно составляет от 0,9 до 1 моль на моль производного карбоновой кислоты. Нет также конкретных ограничений по природу используемого основания, и любое основание, обычно используемое в общеизвестных реакциях, может быть таким же образом использовано и здесь. Примеры подходящих оснований включают: гидрокарбонат щелочных металлов, такие как гидрокарбонат натрия, гидрокарбонат калия или гидрокарбонат лития; карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия или карбонат лития; гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия или гидроксид лития; алкоксиды щелочных металлов, такие же методы натрия, этоксид натрия, трет-бутоксид калия или метоксид лития. Если необходимо, полученную смесь растворяют до получения раствора (используя ультразвук, если необходимо), после чего растворитель отгоняют с получением желаемой соли щелочного металла.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; спирты, такие как метанол этанол или изопропанол; сложные эфиры, такие как этилацетат или метилацетат; галогенированные углеводороды, такие как метиленхлорид или хлороформ; вода; а также смеси воды и одного или более водорастворимого органического растворителя (например диоксан, диметоксиэтан, метанол и изопропиловый спирт). Когда используют водную смесь растворителя, ее лиофилизация, как таковая или после отгонки органического растворителя, дает желаемую соль щелочного металла.
Соль неорганической кислоты, такой как соляная кислота, или органической кислоты, такой как малеиновая кислота, соединения по настоящему изобретению, обладающего аминогруппой или гетероциклической группой, содержащей основной атом азота, такой как имидазольная группа, может быть получена следующим образом.
Такое соединение растворяют в подходящем растворителе и затем добавляют от 1 до 10 эквивалентов желаемой кислоты. Полученный остаток собирают фильтрацией с получением желаемой соли. Если осадка нет, растворитель отгоняют с получением желаемой соли. Если полученная соль образует стеклообразный материал, соль вновь растворяют в воде или в смеси воды и водорастворимого органического растворителя, такого как диоксан, диметоксиэтан, метанол или изопропиловый спирт, и раствор затем лиофилизуют как таковой или после отгонки водорастворимого органического растворителя. Следовательно, очищенное соединение может быть получено в виде соли в форме или порошка, или пены. Продукт может быть, если необходимо, очищен перекристаллизацией.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. Нет особых ограничений по природе используемого растворителя при условии, что он не обладает отрицательным действием на реакцию и что он может растворять исходный материал, по крайней мере в некоторой степени. Примеры подходящих растворителей включают: простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; спирты, такие как метанол, этанол или изопропанол; сложные эфиры, такие как этилацетат или метилацетат; галогенированные углеводороды, такие как метиленхлорид или хлороформ.
Соединения по настоящему изобретению обладают превосходной ингибирующей активностью по отношению к ацил-CoA: холестерин ацилтранферазы, хорошо абсорбируются после орального введения и менее токсичны, чем ранее известные соединения. Поэтому они полезны при лечении и предупреждении атеросклероза.
Активность соединений по настоящему изобретению проиллюстрирована в следующих испытаниях.
β - Липопротеин очень низкой плотности ( β - VLDL)
Кровь получают [используя антикоагулирующую этилендиаминтетрауксусную кислоту (5 мМ)] от японских белых кроликов, питаемых по 2% по весу холестериновой диетой в течение 2 недель. β - VLDL (d<1,006 г/мл) выделяют из плазмы ультрацентрифугированием по методу Хатча и Лиса [Hatch, ET. and Lees Adv. Lipid Res. , 6, 1 - 68 (1968)] и диализируют в 10 мМ натрий фосфатном буфере (pH 7,4), содержащем 150 мМ хлорида натрия при 4oC.
Получение макрофагов мышей (Мф)
Перитонеальные клетки от нестимулированных самок мышей DDY (вес тела 20 - 30 г), харвестируют в растворе фосфатного буфера (PBS) как описано Эдельсон и Кох [Edelson, PJ. and Cohn, ZA., 1976, IN VITRO Methods in Cell-Medi ated and Tumor Ymmunity, eds, Bloon, BR and David, JR., (Academic, New-York), 333 - 340]. Жидкость от мышей собирают и клетки отделяют центрифугированием при 400 xg в течение 10 мин при 4oC и промывают один раз PBS. Клетки вновь суспендируют в модифицированной по Dulbecco среде Eagle (DMEM), содержащей 10% (по объему) сыворотку новорожденного теленка (FCS), пенициллин (100 ед./мл) и стрептомицин (100 μ л/мл ) при конечной концентрации 3•106 клеток на мл. Аликвоты (1 мл) суспензий этих клеток диспергируют на пластиковые чашки Петри (35•10 мм) и затем инкубируют в инкубаторе CO2 (5% CO2/95% воздуха) при 37oC в течение 2 ч. Каждую чашку промывают дважды PBS без сыворотки для удаления неслипшихся клеток. Клетки промывают дважды по 2 мл PBS и используют в эксперименте.
Ингибирование ACAT В Мф
Ингибирование ACAT в Мф определяют по способу, описанному Brown et al. [Brown, MS. , Goldstein JL., Krieger, M., Ho, YK. and Anderson, RGW. (1979) J. Cell Biol., 82, 597 - 613]. Переацилирование холестерина вызывается добавлением β - VLDL (конечная концентрация 50 μ л/мл холестерина), комплекса [14-C] олеат-альбумина (конечные концентрации: 0,2 мМ олеата и 0,6 мг/мл альбумина), и испытуемое соединение растворяют в этаноле в монослое Мф, препарат инкубируют при 37oC в течение 3 ч в инкубаторе CO2. Клетки промывают три раза PBS, и клеточную жидкость экстрагируют 1 мл гексана/изопропанолом (3:2 по объему). Экстракт жидкости упаривают в токе азота. [14C] олеат холестерина разделяют тонкослойной хроматографией на силикагеле с использованием 85: 15: 1 по объему смеси гексана, диэтилового эфира и уксусной кислоты в качестве разделяющих растворителей. Активность ACAT в Мф определяют измерением радиоактивности и скорость ингибирования (%) рассчитывают сравнением контрольной активности с таковой исследуемого соединения при данных концентрациях. Результаты показаны в следующей табл. 7.
Соединение A является ранее известным соединением, имеющим формулу (A):
Оно описано в WO 93/06096.
Соединения по настоящему изобретению могут быть назначены любым известным способом, известным для ранее известных соединений, имеющих тот же тип активности. Например, они могут быть назначены орально, приемлемо в виде таблеток, капсул, гранул, порошков или сиропов. Эти лекарственные составы могут быть получены общепринятыми способами добавлением подходящих добавок, таких как растворители, связующие, разрыхлители, коррогенты (модификаторы лекарственных веществ), смазочные агенты и стабилизаторы. Дозировки могут варьироваться в зависимости от состояния пациента и возраста: однако дневная доза от 1 до 500 мг на кг веса тела, предпочтительно от 1 до 100 мг на кг веса тела, может, в основном, даваться взрослому человеку в единичной дозе или в виде раздробленной дозы.
Настоящее изобретение далее иллюстрируется следующими неограничивающими примерами. В этих примерах номера соединений соответствуют номерам табл. 1 - 5. Получение некоторых исходных материалов, используемых в этих примерах, иллюстрируются последующими приготовлениями. Некоторые фармацевтические препараты, включающие соединения по настоящему изобретению, далее показаны в последующих составах.
Пример 1. N-{ 2-[3-(1-Имидазолил)пропокси]метил-6-метил-тиофенил}-2-(9H-ксантен-9-ил) ацетамид
(соединение N 1 - 1475)
I (i) N-{2-[3-(1-Мезилокси)пропокси]метил-6-метилтиофенил}-2-(9H-ксантен-9-ил) ацетамид
212 мг (0,48 моль) N-[2-(3-гидроксипропокси)метил-6-метилтиофенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 4) суспендируют в 15 мл метиленхлорида. Суспензию охлаждают на ледяной бане и затем добавляют 102 мг (0,89 ммоль) метансульфонилхлорида и затем 94 мг (0,90 ммоль) триэтиламина. Температуре реакционной смеси позволяют подняться до комнатной температуры, после чего смесь перемешивают дополнительно 1 ч. По окончании этого времени реакцию останавливают добавлением воды. Смесь затем разбавляют метиленхлоридом, после чего органический слой промывают разбавленным водным раствором соляной кислоты и затем водой. Растворитель затем удаляют отгонкой при пониженном давлении с получением 251 мг (количественный выход) указанного в заголовке соединения в виде кристаллов. Эти кристаллы используют в следующей стадии без очистки.
I(ii) N-{2-[3-(1-Имидазолил)пропокси]метил-6-метилтиофенил}-2-(9H-ксантен-9-ил) ацетамид
151 мг сырых кристаллов N-{2-[3-(мезилокси)пропокси]метил-6-метилтиофенил}-2-(9H-ксантен-9-ил) ацетамид [полученный как описано в стадии (i) выше] суспенжируют в 3 мл диметилформамида. К суспензии добавляют 162 мг (2,39 ммоль) имидазола и смесь перемешивают в течение 5,5 ч при 90oC. По окончании этого времени реакции позволяют вернуться к комнатной температуре, после чего разбавляют этилацетатом и промывают несколько раз водой. Растворитель затем удаляют отгонкой при пониженном давлении, и полученный осадок подвергают колоночной хроматографии сквозь 15 г силикагеля. Фракции, элюированные смесями метиленхлорида и метанола, колеблющимися от 100:2 до 100:7 по объему, собирают. Перекристаллизация из смеси метиленхлорида и метанола приводит к получению 162 мг (выход 68%) указанного в заголовке соединения в виде кристаллов, плавящихся при 176,5 - 177oC.
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3267, 1649, 1515, 1481, 1260, 757.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ , ppm: 1,88 (2H, квинтет, J = 6,5 Гц); 2,28 (3H, синглет); 2,73 (2H, дублет, J = 6,5 Гц); 3,17 (2H, триплет, J = 6,5 Гц); 3,93 (2H, триплет, J = 6,5 Гц); 4,10 (2H, синглет); 4,65 (1H, триплет, J = 6,5 Гц); 6,7 - 7,7 (14H, мультиплет).
Пример 2. N-{2-[3-(1-Имидазолил)пропокси]метил-6-метилтиоценил}-2-(9H-ксантен-9-ил) ацетамид гидрохлорид (соединение N 1 - 1474)
100 мг N-{2-[3-(1-имидазолил)пропокси]метил-6-метилтиофенил}-2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 1) растворяют в смеси метанола метиленхлорида. К раствору добавляют избыток концентрированной соляной кислоты, и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток растворяют в метаноле и добавляют диэтиловый эфир с получением 85 мл указанного в заголовке соединения в виде кристаллов, плавящихся при 175 - 184oC.
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3405, 3269, 1649, 1516, 1482, 1261, 758.
Элементный анализ:
Рассчитано для:
C29H29N3O3S•HCl•1/2H2O:
Вычислено: %: C 63•90%, H 5,73; N 7,71; Cl 6,50.
Найдено, %: C 63,86; H 5,78; N 7,69; Cl 6,35.
Пример 3. N-{2-[3-(1-Имидазолил)пропокси]метил-6-метилтиофенил}-2-(9H-ксантен-9-ил) ацетамид (соединение N 1 - 1477)
Следуя способу, подобному описанному в примере 1, но используя N-[5-(3-гидроксипропокси)метил-2-метилтиофенил] -2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 4) в качестве исходного продукта, в соответствующем количестве, что используется в этом примере, получают указанное в заголовке соединение в виде маслянистого вещества.
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 2923, 1679, 1574, 1480, 1254, 757.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ ppm: 1,88 - 2,4 (2H, мультиплет); 2,05 (3H, синглет); 2,75 (2H, дублет, J = 6,5 Гц); 3,43 (2H, триплет, J = 6,5 Гц); 3,9 - 4,3 (2H, мультиплет); 4,45 (2H, синглет); 4,70 (1H, триплет, J = 6,5 Гц); 6,8 - 7,6 (12H, мультиплет); 8,0 - 8,2 (1H, уширенный синглет); 8,3 - 8,5 (1H, уширенный синглет).
Пример 4. N-{5-[3-(1-Имидазолил)пропокси]метил-2-метилтиофенил}-2-(9H-ксантен-9-ил) ацетамид гидрохлорид (соединение N 1 - 1476)
Следуя способу, подобному описанному в примере 2, но используя N-{5-[3-(1-имидазолил)пропокси] метил-2-метилтиофенил} -2-(9H- ксантен-9-ил)ацетамид (полученный, как описано в примере 3) в качестве исходного продукта, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 85 - 86oC (после перекристаллизации из смеси метиленхлорида и этилацетата).
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3036, 1660, 1526, 1479, 1458, 1258, 760.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ ppm: 2,1 - 2,35 (2H, уширенный синглет); 2,21 (3H, синглет); 2,80 (2H, дублет, J = 7 Гц); 3,49 (2H, уширенный синглет); 4,3 - 4,6 (2H, уширенный синглет); 4,47 (2H, синглет); 4,69 (1H, триплет, J = 7 Гц); 6,9 - 7,4 (12H, мультиплет); 8,11 (1H, синглет); 8,27 (1H, синглет); 9,0 - 9,15 (1H, уширенный синглет).
Пример 5. N-[2-Этил-6-(1-гидроксипропил)фенил] -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1434).
9,6 мг (0,18 моль) метоксида натрия добавляют к метанольному раствору, содержащему 37 мг (0,060 ммоль) N-(2-этил-6-{1-[2-(9H- ксантен-9-ил)ацетокси] пропил} фенил-2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в приготовлении 55). Смесь затем перемешивают в течение 4 ч при 60oC. По окончании этого времени реакционный раствор разбавляют диэтиловым эфиром. К реакционной смеси добавляют воду, и указанное в заголовке соединение распределяют между органическим растворителем и водой. Органический слой затем отделяют и промывают водой 3 раза. Растворитель удаляют отгонкой при пониженном давлении, и полученный остаток подвергают колоночной хроматографии через 5 г силикагеля. Элюирование смесью метиленхлорида и этилацетата, колеблющейся от 10:1 до 5: по объему, приводит к 21 мг (выход 89%) указанного в заголовке соединения в виде кристаллов, плавящихся при 167 - 168oC (после перекристаллизации из смеси диэтилового эфира и гексана.
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 1655, 1635, 1520, 1482, 1460, 1260, 756.
Спектр ядерного магнитного резонанса (CDCl3, 270 Мгц), δ , ppm: 0,78 (3H, триплет, J = 7 Гц), 1,06 (3H, триплет, J = 7,5 Гц); 1,55 - 1,8 (2H, мультиплет); 2,33 (2H, квартет, J = 7 Гц), 2,79 (2H, дублет, J = 7 Гц), 4,34 (1H, триплет, J = 7 Гц), 4,72 (2H, триплет, J = 7 Гц), 6,95 - 7,5 (11H, мультиплет).
Пример 6. N-[2-(3Гидрокси-3-фенил-1-пропенил)-6-метоксиметилфенил] -2- (9H-ксантен-9-ил)ацетамид (соединение N 1-1443
10 мл тетрагидрофурановой суспензии, содержащей 152 мг (0,392 моль) N-(2-формил-6-метоксиметилфенилфенил)-2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в приготовлении 53), 199 мг (0,432 ммоль) фенацилтрифенилфосфоний бромида и 48 мг (0,471 ммоль) триэтиламина, нагревают в течение 6 ч при кипячении с обратным холодильником. По окончании этого времени смеси позволяют достичь комнатной температуры. Добавляют 4 мл метанола и затем постепенно добавляют к смеси 155 мг (4,10 ммоль) боргидрида натрия, которую затем перемешивают в течение 1 ч. Реакционный раствор разбавляют этилацетатом и промывают некоторое время водой. Растворитель удаляют отгонкой при пониженном давлении. Остаток подвергают колоночной хроматографии через 20 г силикагеля. Элюирование 4:1 по объему смесью метиленхлорида и этилацетата приводит к 126 мг (выход 66%) указанного в заголовке соединения в виде кристаллов, плавящихся при 204,5 - 206,5oC (после перекристаллизации из метанола).
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3258, 1652, 1482, 1457, 1263, 752.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 2,72 (2H, дублет, J = 7 Гц); 3,10 (3H, синглет); 3,94 (2H, синглет); 4,70 (1H, триплет, J = 7 Гц); 5,34 (1H, дублет, J = 7 Гц); 6,32 (1H, дублет из дублетов, J = 16 & 7 Гц); 6,53 (1H, дублет, J = 16 Гц); 7,1 - 7,6 (16H, мультиплет).
Пример 7. N-[2-(3-Гидрокси-3-фенилпропил)-6-метоксиметил-фенил] - 2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1444) 35 мг 10% массе палладия на угле добавляют к 15 мл тетрагидрофуранового раствора, содержащего 111 мг (0,226 ммоль) N-[2(3-гидрокси-3-фенил-1-пропенил)-6-метоксиметилфенил]-2-(9H-ксантен- 9-ил)ацетамида (полученного, как описано в примере 6), и смесь энергично перемешивают в течение 15 ч в токе водорода. Реакционный раствор затем фильтруют с помощью фильтра Цеолита (торговая марка), и катализатор промывают тетрагидрофураном. Фильтрат и промывные воды объединяют и растворитель удаляют из объединенного раствора отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 15 г силикагеля. Элюирование 4:1 по объему смесью метиленхлорида и этилацетата приводит к 86 мг (выход 77%) указанного в заголовке соединения в виде кристаллов, плавящихся при 143,5 - 146oC (после перекристаллизации из метанола и гексана).
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3370, 3280, 1652, 1515, 1480, 1457, 1260, 757.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ , ppm: 1,7 - 2,1 (2H, мультиплет); 2,2 - 2,6 (2H, мультиплет); 2,53 (2H, дублет, J = 7 Гц); 3,07 (3H, синглет); 3,96 (2H, синглет); 4,2 - 4,6 (1H, мультиплет); 4,59 (1H, триплет, J = 7 Гц); 6,9 - 7,7 (16H, мультиплет).
Пример 8. N-[2-Этил-6-(3-оксо-6-фенилгексил)фенил] -2-(9H-ксантен-9-ил)ацетамид (8а) (соединение N 1-1445); и N-[2-Этил-6-(3-гидрокси-6-фенилгексил)фенил]-2-(9H-ксантен- 9-ил) ацетамид (8b) (соединение N 1-1446)
93 мг 10% по массе палладия на угле добавляют к 5 мл метанольного раствора, содержащего 154 мг N-[2-этил-6-(3-оксо- 6-фенил-1-гексенил)фенил]-2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в примере 68), и смесь энергично перемешивают в течение 3 ч в токе водорода. По окончании этого времени реакционный раствор затем фильтруют с помощью фильтра Целита (торговая марка), и катализатор промывают несколько раз этилацетатом. Фильтрат и промывные воды объединяют и растворитель удаляют из объединенного раствора отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 10 г силикагеля. Элюирование 2:1 по объему смесью диэтилового эфира и гексана приводит к 87 мг (выход 56%) 3-оксо соединения и 53 мг (выход 34%) 3-гидрокси соединения (8 b), оба в виде кристаллов. (8a) N-[2-Этил-6-(3-оксо060фенилгексил)фенил-2-(9H-ксантен- 9-ил)ацетамид:
Плавящийся при 113 - 114oC (после перекристаллизации из смеси метиленхлорида и диэтилового эфира).
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3246, 1709, 1653, 1528, 1480, 1459, 1262, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,09 (3H, триплет, J = 7,5 Гц); 1,78 (2H, квартет, J = 7 Гц); 2,26 (2H, триплет, J = 7,5 Гц); 2,38 - 2,55 (6H, мультиплет); 2,62 (2H, триплет, J = 6 Гц); 2,84 (2H, дублет, J = 7 Гц); 4,73 (1H, триплет, J = 7 Гц); 6,94 - 7,43 (16H, мультиплет).
(8b) N-[2-Этил-6-(3-гидрокси-6-фенилгексил)фенил] -2- (9H-ксантен-9-ил)ацетамид:
Плавящийся при 131 - 132oC (после перекристаллизации из смеси диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3274, 1648, 1522, 1480, 1459, 1260, 754.
Спектр ядерного магнитного резонанса (CDCl3, 237 МГц), δ , ppm: 1,09 (3H, триплет, J = 7,5 Гц); 1,31 - 1,79 (6H, мультиплет); 2,18 - 2,47 (4H, мультиплет); 2,56 (2H, триплет, J = 7,5 Гц); 2,71 (1H, дублет из дублетов, J = & 14,5 Гц); 2,76 (1H, дублет из дублетов, J = 7 & 14,5 Гц); 3,18 - 3,30 (1H, мультиплет); 4,70 (1H, триплет, J = 7 Гц); 6,97 - 7,42 (16H, мультиплет).
Пример 9. N-{2-трет-Бутил-5-[5-(4-фторфени)-3-гидрокси-1- пентил]фенил} -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1319)
Следуя способу, подобному описанному в примере 6, но используя N-{2-трет-бутил-5-[5-(4-фторфенил)-3-оксо-1-пентенил] фенил} - 2-(9H-ксантен-9-ил)ацетамид в качестве исходного продукта, получают указанное в заголовке соединение в виде маслянистого вещества. Исходный продукт получают согласно способу, описанному в примере 68, но используя 4-фтор-1-(4-диэтоксифосфорил-3-оксобутил)бензол и N-(2трет-бутил-5-формилфенил)-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 15).
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3450, 1673, 1600, 1573, 1508, 1477, 1456.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,17 (9H, синглет); 1,85 - 2,0 (2H, мультиплет); 2,4 - 2,5 (1/3H, мультиплет); 2,65 - 2,8 (2H, мультиплет); 2,72 (5/3H, дублет, J = 7Гц); 4,2 - 4,35 (1H, мультиплет); 4,77 (1H, триплет, J - 7 Гц); 6,22 (1H, дублет из дублетов, J = 7 & 16 Гц); 6,53 (1H, дублет, J = 16 Гц); 6,9 - 7,6 (15H, мультиплет).
Пример 10. N-{2-трет-Бутил-5-[5-(4-фторфенил)-3- гидроксипентил)фенил} -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1318)
Следуя способу, подобному описанному в примере 7, но подвергая N-{2-трет-бутил-5-[5-(4фторфенил)-3-гтдрокси-1-пентенил] фенил} -2- (9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 9) каталитическому восстановлению, указанное в заголовке соединение получают в виде пенообразного вещества.
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3450, 1677, 1600, 1575, 1509, 1479, 1456.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ , PPm: 1,17 (9H, синглет); 1,4-1,65 (2H, мультиплет); 1,7-1,9 (2H, мультиплет); 2,3-2,9 (6H, мультиплет); 3,4-3,7 (1H, мультиплет); 4,74 (1H, триплет, J=7 Гц); 6,9-7,5 (15H, мультиплет).
Пример 11. N-[2-Этил-6-(3-гидрокси-1-фенилпропил)фенилъ-2-(9Н-ксантен-9-ил)ацетамид (соединение N 1-1440)
Следуя способу, подобному описанному в примере 8, но используя N-[2-этил-6-(3-оксо-3-фенил-1-пропенил)фенил-2-(9Н-ксантен-9-ил) ацетамид в качестве исходного продукта, в соответствующем количестве, что используется в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 192-193oC (после перекристаллизации из смеси этилацетата и гексана). Исходный продукт получают согласно способу, описанному в примере 6, но используя N-(2-этил-6-формилфенил)-2-(9Н-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 29).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3242, 1657, 1522, 1480, 1459, 1254, 760.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,09 (1H, триплет, J=7,5 Гц), 1,82-1,93 (2H, мультиплет), 2,23-2,48 (4H, мультиплет), 2,68 (2H, дублет, J= 7 Гц), 4,36 (1H, триплет, J=6 Гц), 4,69 (1H, триплет, J=7 Гц), 6,96-7,44 (16H, мультиплет).
Пример 12. N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил]-2-(9Н-ксантен-9-ил) ацетамид (соединение N 1-25).
0,18 мл I M раствора циклогексилметилмагний бромида в диэтиловом эфире добавляют к 2 мл тетрагидрофурана и смесь охлаждают до -78oC. 2 мл тетрагидрофуранового раствора, содержащего 50 мг (0,12 ммоль) N-[2-трет-бутил-5-(3-оксопропил)-фенил] -2-(9Н-ксантен-9-ил)ацетамида (полученного, как описано в приготовлении 19), добавляют затем по каплям к этому раствору за период в 5 мин. Смесь затем перемешивают 40 мин при этой температуре, после чего температуре позволяют вернуться постепенно к 0oC. Реакцию затем останавливают добавлением насыщенного водного раствора хлорида аммония. Добавляют диэтиловый эфир, что дает возможность указанному в заголовке соединению распределиться между органическим растворителем и водой. Органический слой отделяют и промывают водой, после чего растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 10 г силикагеля. Элюирование 100:15 по объему смесью метиленхлорида и этилацетата приводит к 41 мг (вывод 67%) указанного в заголовке соединения в виде кристаллов, плавящихся при 145-146oC (после перекристаллизации из смеси метиленхлорида и диэтилового эфира).
Инфракрасный абсорбционный спектр (KBr), ν , см-1: 3343, 3244, 1654, 1529, 1478, 1458, 1252, 760, 749.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm 0,76-1,87 (15H, мультиплет); 1,16 (9H, сигнлет); 2,20-2,84 (4H, мультиплет); 3,55-3,83 (1H, мультиплет); 4,74 (1H, триплет, J-7 Гц); 6,92-7,45 (11H, мультиплет).
Пример 13. N-[2-трет-Бутил-5-(4-циклогексил-3-оксобутил)фенил]-2(9H-ксантен-9- ил)-ацетамид (соединение N 1-1263)
1 мл метиленхлоридного раствора, содержащего 16 мг (0,13 ммоль) оксалилхлорида охлаждают до -78oC. 1,5 мл метиленхлоридного раствора, содержащего 20 мг (0,25 ммоль) диметилсульфоксида, добавляют затем к этому раствору. Полученную смесь затем перемешивают 5 мин, после чего 1 мл метиленхлоридного раствора, содержащего 56 мг (0,11 ммоль) N-[2-трет-бутил-5(4-циклогексил-3-гидроксибутил)фенил] -2-(9H-ксантен-9-ил) ацетамида (полученного, как описано в примере 12), добавляют затем по каплям к этому раствору за период в 5 мин при той же температуре. Смесь затем перемешивают 15 мин и к ней по каплям добавляют 1 мл метиленхлоридного раствора, содержащего 64 мг (0,64 ммоль) триэтиламина. Смесь перемешивают 10 мин, и затем реакционной смеси позволяют вернуться к комнатной температуре. Реакцию затем разбавляют диэтиловым эфиром и промывают разбавленной водной соляной кислотой и затем водой. Растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 10 г силикагеля. Элюирование 100: 8 по объему метиленхлорида в этилацетате приводит к 51 мг (выход 91%) указанного в заголовке соединения в виде кристаллов, плавящихся при 154-155oC (после перекристаллизации из смеси диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3233, 1711, 1637, 1529, 1481, 1456, 1261, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ppm: 0,80-1,91 (11H, мультиплет); 1,15 (9Н, синглет); 2,29 (2Н, дублет, J=6,5 Гц); 2,36-2,90 (6Н, мультиплет); 4,74 (1Н, триплет, J=7 Гц); 6,91-7,45 (11Н, мультиплет).
Пример 14. N-[2-трет-Бутил-5-(2-циклогексил-1-гидроксиэтил)фенил] -2(9H-ксантен-9-ил) ацетамид (соединение N 1-179)
8 мл тетрагидрофуранового раствора, содержащего 100 мг (0,25 ммоль) N-(2-трет-бутил-5-формил)-2-(9H-ксантен-9-ил)-ацетамида (полученного, как описано в приготовлении 15) охлаждают на ледяной бане. Затем к раствору добавляют по каплям 0,38 мл (0,38 ммоль) 1 М раствора циклогексилметилмагнийбромида в диэтиловом эфире. Полученную смесь затем перемешивают 20 мин при этой температуре, затем разбавляют диэтиловым эфиром и промывают разбавленной водной соляной кислотой и затем водой. Растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 20 г силикагеля. Элюирование смесь метиленхлорида и этилацетата, изменяющейся от 7:1 до 2:1 по объему, приводит к получению 95 мг (выход 76%) указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3425, 1678, 1508, 1479, 1458, 1250.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ppm: 0,80-1,95 (13H, мультиплет); 1,18 (9H, синглет); 2,32-2,54 (0,5H, мультиплет); 2,70 (1,5H, дублет, J= 7 Гц); 4,41-4,54 (0,25H, мультиплет); 4,67-4,83 (0,75H, мультиплет); 4,75 (1H, триплет, J=7 Гц); 7,01-7,49 (11H, мультиплет).
Пример 15. N-[2-Этил-6-(1-гидрокси-2-фенилпропил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1441)
Следуя способу, подобному описанному в примере 14, но используя N-(2-этил-6-формилфенил)-2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 29) и циклогексилметилмагнийбромид в качестве исходных продуктов в соответствующем соотношении, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 128-129oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3240, 1648, 1524, 1480, 1459, 1260, 754.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,03 (3H, триплет, J= 7,5 Гц); 1,81-1,96 (1H, мультиплет); 2,07-2,23 (1H, мультиплет); 2,28 (2H, квартет, J=7,5 Гц); 2,54 (2H, триплет, J=7,5 Гц); 2,56 (1H, дублет из дублетов, J=7 и 15 Гц); 2,62 (1H, дублет из дублетов, J=7 и 15 Гц); 4,42 (1H, триплет, J=7 Гц); 4,67 (1H, триплет, J=7 Гц); 7,01-7,42 (16H, мультиплет).
Пример 16. N-[2-трет-Бутил-5-(6-фенил-1-гидроксигексил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1283)
Следуя способу, подобному описанному в примере 12, но используя 5-фенилпентилмагний йодид и N-(2-трет-бутил-5-формилфенил -2(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 15) в качестве исходных продуктов в соответствующем соотношении, используемом в этом примере, получают указанное в заголовке соединение в пенообразном виде.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3395, 3280, 1653, 1522, 1480, 1459, 1256, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ppm: 1,17 (9H, мультиплет); 1,25-1,85 (8H, мультиплет); 2,42 (0,4H, дублет, J=7 Гц); 2,61 (2H, триплет, J=8 Гц); 2,69 (1,6H, дублет, J=7 Гц); 4,26-4,34 (0,2H, мультиплет); 4,63 (0,8H, триплет, J=6,5 Гц); 4,73 (1H, триплет, J=7 Гц); 6,97-7,45 (16H, мультиплет).
Пример 17. N-[2-трет-Бутил-5-(4-циклогексил-1-гидроксибутил)фенил}-2-)(9H-ксантен-9-ил) ацетамид (соединение N 1-227)
Следуя способу, подобному описанному в примере 14, но используя 3-циклогесилпропилмагнийиодид и N-(2-трет-бутил-5-формилфенил)-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 15) в качестве исходных продуктов, в соответствующем соотношении, используемом в этом примере, получают указанное в заголовке соединение в пенообразном виде.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3395, 3280, 1655, 1522, 1480, 1459, 1256, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,80-0,94 (2H, мультиплет); 1,11-1,80 (15H, мультиплет); 1,17 (9H, синглет); 2,43 (0,4H, дублет, J= 7 Гц); 2,70 (1,6H, дублет, J=7 Гц); 4,28-4,34 (0,2H, мультиплет); 4,60-4,68 (0,8 мультиплет); 4,73 (1H, триплет, J= 7 Гц); 6,97-7,94 (11H, мультиплет).
Пример 18. N-[2-трет-Бутил-5-(6-циклогексил-1-гидроксигексил)фенил]-2-(9Hксантан-9-ил) ацетамид (соединение N 1-275)
Следуя способу, подобному описанному в примере 14, но пользуясь 5-циклогексилпентилмагнийиодид и N-(2-трет-бутил-5-формилфенил)-2-(9H-ксантил (полученный, как описано в приготовлении 15) в качестве исходных продуктов в соответствующем соотношении, используемом в этом примере, получают указанное в заголовке соединение в пенообразном виде.
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3395, 3290, 1653, 1522, 1480, 1459, 1256, 756.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,77-0,94 (2H, мультиплет); 1,06-1,80 (19H, мультиплет); 1,17 (9H, синглет); 2,43 (0,4H, дублет, J = 7 Гц); 2,70 (1,6H, дублет, J = 7 Гц); 4,28-4,35 (0,2H, мультиплет); 4,63- (0,8H, триплет, J = 6,5 Гц); 4,74 (1H, триплет, J = 7 Гц); 6,96-7,46 (11H, мультиплет).
Пример 19. N-[2-трет-Бутил-5-(оксо-6-фенилгексил)фенил]-2-(9H-ксантиен-9-ил) ацетамид (соединение N 1-1325)
Следуя способу, подобному описанному в примере 13, но используя N-[2-трет-бутил-5-(6-фенил-1-гидроксигексил)фенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 16) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 119-120oC (после перекристаллизации из смеси гексана и этилацетата).
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3240, 1686, 1646, 1524, 1482, 1459, 1258, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270МГц), δ ppm: 1,19 (9H, синглет); 1,45 (2H, квинтет, J=7,5 Гц); 1,70 (2H, квинтет, J = 7,5 Гц); 1,78 (2H, квинтет, J= 8 Гц), 2,40-2,45 (0,2H, мультиплет); 2,60-2,80 (0,2H, мультиплет); 2,65 (2H, триплет, J = 7,7 Гц); 2,74 (1,8H, дублет, J=7Гц); 2,94 (1,8= 7 Гц); 4,74 (1H, триплет, J = 7 Гц); 7,05-7,43 (14,1H, мультиплет); 7,72 (1H,дублет из дублетов, J = 2& 8 Гц); 8,05 (0,9H, дублет, J = 2 Гц).
Пример 20 N-[2-трет-Бутил-5-(6-циклогексил-1-оксогексил)фенил]-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-1262)
Следуя способу, подобному описанному в примере 13, но используя N-[2-трет-бутил-5-(6-циклогексил-1-гидроксигексил)фенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 18) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заготовке соединение в виде кристаллов, плавящихся при 133,5-134oC (после перекристаллизации из смеси гексана и диизопропилового эфира).
Инфракрасный абсорбционный спектр (КВг), νmax , см-1: 3235, 1986, 1644, 1524, 1482, 1459, 1258, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,80-0,96 (2H, мультиплет); 1,06-1,78 (17H, мультиплет); 1,20 (9H, синглет); 2,33-2,47 (0,2H, мультиплет); 2,65-2,75 (0,2H, мультиплет); 2,74 (1,8H, дублет, J - 7 Гц); 2,93 (1,8H, триплет, J = 7 Гц); 4,75 (1H, триплет, J = 7 Гц); 7,07-7,43 (9, 1H, мультиплет); 7,73 (1H, дублет из дублетов, J = 2& 8 Гц); 8,06 (0,9H, синглет).
Пример 21. N-[2-трет-Бутил-5-(6-циклогексил-5-оксогексил)фенил]-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-1268)
0,66 мл (7,65 ммоль) оксалилхлорида и 30 мг N,N-диметилформамида добавляют по очереди к 10 мл метиленхлоридной суспензии, содержащей 368 мг (1,53 ммоль) 2-(9H-ксантен-9-ил)-уксусной кислоты. Суспензию перемешивают на ледяной бане в течение 30 мин и затем при комнатной температуре в течение 1 ч. По истечении этого времени растворитель и избыток реагентов удаляют отгонкой при пониженном давлении. Остаток вновь растворяют в 10 мл метиленхлорида, и полученный раствор помещают на ледяную баню. Затем к раствору добавляют 2 мл метиленхлорида раствора, содержащего 2-трет-бутил-5-(6-циклогексил-5-оксогексил)анилин и 0,56 мл пиридина, и смесь перемешивают 10 мин. Реакционный раствор затем разбавляют диэтиловым эфиром, промывают разбавленным водным раствором гидроксида натрия и разбавленной водной соляной кислотой и затем водой, в этом порядке. Растворитель затем удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 40 г силикагеля. Элюирование 100:20 по объему смесью гексана и этилацетата приводят к получению 663 мг (выход 86%) указанного в заготовке соединения в виде кристаллов, плавящихся при 126-127oC (после перекристаллизации из смеси диизопропилового эфира и гексана) пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1709, 1663, 1518, 1478, 1459, 1256, 762.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,81-1,02 (2H, мультиплет); 1,05-1,93 (13H, мультиплет); 1,18 (9H, синглет); 2,27 (2H, дублет, J = 7 Гц); 2,28-2,75 (6H, мультиплет); 4,74 (1H, триплет, J = 7 Гц); 6,88-7,45 (11H, мультиплет).
Пример 22. N-[2-трет-Бутил-5-(6-гексил-5-гидроксигексил)фенил]-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-587)
37 мл (0,99 ммоль) боргидрата натрия добавляют к 10 мл метанольной суспензии, содержащей 544 мг (0,986 ммоль) N-[2-трет-бутил-5-(6-гексил-оксогексил)фенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 21), на ледяной бане. Ледяную баню затем удаляют и реакционную смесь перемешивают 1 ч при комнатной температуре. По истечении этого времени реакционный раствор разбавляют диэтиловым эфиром, промывают насыщенным водным раствором хлорида аммония и затем водой несколько раз. Растворитель затем удаляют отгонкой при пониженном давлении с получением 523 мг (выход 97%) указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3450, 1678, 1509, 1479, 1458, 1250.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,77-1,85 (19H, мультиплет); 1,16 (9H, синглет); 2,23-2,37 (0,5H, мультиплет); 2,45 (0,5H, дублет, J = 7 Гц); 2,60 (1,5H, триплет, J = 7 Гц); 2,70 (1,5H, дублет, J = 7 Гц); 3,63-3,79 (1H, мультиплет); 4,74 (1H, триплет, J = 7 Гц); 6.89-7,45 (11H, мультиплет).
Пример 23. N-[2-трет-бутил-5-(4-циклогексил-2-оксобутил)фенил]-2-(9H-ксантен- 9-ил)ацетамид (соединение N 1-1265)
Следуя способу, подобному описанному в примере 21, но используя 2-трет-бутил-5-(4-циклогексил-2-оксобутил)анилин в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 143-135oC (после перекристаллизации из смеси ацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3379, 2922, 1701, 1663, 1480, 1458, 1258, 761.
Спектр ядерного магнитного резонанса (CDCl3, 270 Мгц), δ ppm: 0,7-1,0 (2H, мультиплет); 1,0-1,3 (3H, мультиплет); 1,16 (9H, синглет); 1,3-1,7 (8H, мультиплет); 2,2-2,75 (4H, мультиплет); 3,39 (2/5H, синглет); 3,66 (8/5H, синглет); 4,73 (1H, триплет, J = 7 Гц); 6,9 - 7,45 (11H, мультиплет).
Пример 24. N-[2-трет-Бутил-5-(4-циклогексил-2-гидроксибутил)фенил]-2-(9H-ксантен -9-ил-ацетамид (соединение N 1-373)
Следуя способу, подобному описанному в примере 22, но используя N-[2-трет-бутил-5-(4-циклогексил-2-оксобутил)фенил] -2-(9H-ксантен-9 -ил)ацетамид (полученный, как описано в примере 23) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3430, 1678, 1600, 1575, 1480, 1457.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,8-1,0 (2H, мультиплет); 1,0-1,8 (13H, мультиплет); 1,70 (9H, синглет); 2,3-2,9 (4H, мультиплет); 3,7-3,85 (1H, мультиплет); 4,74 (1H, триплет, J = 7 Гц); 6,9-7,5 (11H, мультиплет).
Пример 25. N-[2-трет-Бутил-5-(5-циклогексил-3-оксопентил)фенил] -2- (9H-ксантен-9-ил)ацетамид (соединение N 1-1264)
Следуя способу, подобному описанному в примере 21, но используя 2-трет-бутил-5-(5-циклогексил-3-оксопентил)анилин (полученный по способу, подобному описанному в приготовлении 7) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 3460, 1708, 1678, 1480, 1458.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ , ppm: 0,7-1,0 (2H, мультиплет); 1,1-1,8(IIH, мультиплет); 1,15 (9H, синглет), 2,3-2,9 (8H, мультиплет); 4,74 (IH, триплет, J =7 Гц); 6,9-7,5 (IIH, мультиплет).
Пример 26. N-[2-трет-Бутил-5-(5-циклогексил-3-гидроксипентил)фенил]-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-1)
Следуя способу, подобному описанному в примере 22, но используя N-[2-трет-Бутил-5-(5-циклогексил-3-оксопентил)фенил] - -2-(9H-ксантен-9-ил)ацетамид(полученный, как описано в примере 23) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 148-149oC (после перекристаллизации из смеси ацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 3371, 2921, 2849, 1658, 1518, 1480, 1458, 1259, 761.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ, ppm: 0,7-1,0 (2H, мультиплет); 1,0-1,85 (15H, мультиплет); 1,16 (9H, синглет); 2,3-2,9 (4H, мультиплет); 3,4-3,7(IH, мультиплет); 4,74 (IH, триплет, J=7Гц); 6,9-7,5 (IIH, мультиплет).
Пример 27. N-[2-трет-Бутил-5-(5-циклогексил-4-оксопентил)фенил]-2-(9H-ксантен-9-ил) ацетамид(соединение N 1-1269)
Следуя способу, подобному описанному в примере 21, но используя 2-трет-бутил-5-(5-циклогексил-4-оксопентил)фенил анилин в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заготовке соединение в виде кристаллов, плавящихся при 193-194,5oC (после перекристаллизации из смеси метиленхлорида и диэтилового эфира). Исходный продукт получают согласно способу, подобному описанному в приготовлении 7, но используя 2-трет-бутил-5-(5-циклогексил-4-оксопентил)-1-нитробензол (полученный по способу, подобному описанному в приготовлении 9).
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 3233, 1703, 1640, 1536, 1480, 1459, 1258, 756.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ, ppm: 0,8-1,0(2H, мультиплет); 1,05-1,4 (3H, мультиплет); 1,17 (9H, синглет); 1,6-2,0 (8H, мультиплет); 2,2-2,75 (8H, мультиплет); 4,74 (IH, триплет, J = 7Гц); 6,9-7,45 (IIH, мультиплет).
Пример 28. N-[2-трет-Бутил-5-(5-циклогексил-4-гидроксипентил)фенил]-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-495)
Следуя способу, подобному описанному в примере 22, но используя N-[2-трет-бутил-5-(5-циклогексил-4-оксопентил)фенил]-2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 27) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 157-158oC (после перекристаллизации из смеси диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3238, 1639, 1531, 1482, 1459, 1257, 756.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ, ppm: 0,75-1,05 (2H, мультиплет); 1,1-1,9 (15H, мультиплет); 1,16 (9H, синглет); 2,25-2,4 (1/2H, мультиплет); 2,45 (I/2H, уширенные дублеты, J = 8 Гц); 2,61 (3/2H, триплет, J = 7 Гц); 2,70 (3/2H, дублет, J =7 Гц); 3,65-3,75 (IH, уширенный синглет); 4,75 (IH, триплет, J =7 Гц); 6,9-7,45 (IIH, мультиплет).
Пример 29. N-[2-трет-Бутил-5-(5-циклогексил-2-оксопентил) фенил] -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1266)
Следуя способу, подобному описанному в примере 21, но используя 2-трет-бутил-5-(5-циклогексил-2-оксопентил)анилин в качестве исходного продукта, в соответствующем количестве, используемого в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 146-147oC (после перекристаллизации из смеси диизопропилового эфира и гексана). Исходный продукт получают согласно способу, подобному описанному в приготовлении 7, но используя 2-трет-бутил-5-(5-циклогексил-2-оксопентил)-1-нитробензол (полученный по способу, подобному описанному в приготовлении 9).
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 3386, 1711, 1663, 1519, 1480, 1458, 1259, 764.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ, ppm: 0,77-0,94 (2H, мультиплет); 1,08-1,28 (6H, мультиплет); 1,16 (9H, синглет); 1,50-2,72 (7H, мультиплет); 2,28-2,40 (0,4H, мультиплет); 2,45 (2H, триплет, J = 7 Гц); 2,70 (I,6H, дублет, J =7 Гц); 3,38 (0,4H, синглет); 3,65 (I,6H, синглет); 4,73 (IH, триплет, J = 7 Гц); 6,94-7,40 (IIH, мультиплет).
Пример 30. N-[2-трет-Бутил-5-(5-циклогексил-2-гидроксипентил) фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-397)
Следуя способу, подобному описанному в примере 22, но используя N-[2-трет-бутил-5-(5-циклогексил-2-оксопентил)фенил]-2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 29) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение виде кристаллов, плавящихся при 109-111oC (после перекристаллизации из диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 3246, 1643, 1527, 1482, 1458, 1259, 760.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ, ppm: 0,80-0,97 (2H, мультиплет); 1,12-1,77 (15H, мультиплет); 1,17 (9H, мультиплет); 2,35-2,55 (0,8H, мультиплет); 2,60 (0,8H, дублет из дублетов, J = 9 & 14 Гц); 2,70 (1,6H, дублет, J = 7 Гц); 2,80 (IH, дублет из дублетов, J = 4 & 14 Гц); 3,55-3,87 (IH, мультиплет); 4,74 (IH, триплет, J = 7 Гц); 6,97-7,41 (IIH, мультиплет).
Пример 31. N-[2-трет-Бутил-5-(6-циклогексил-2-оксогексил)фенил] -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1267)
Следуя способу, подобному описанному в примере 21, но используя 2-трет-бутил-5-(6-циклогексил-2-оксогексил)анилин в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта. Исходный продукт получают согласно способу, подобному описанному в приготовлении 7, но используя 2-трет-бутил-5-(6-циклогексил-2-оксогексил) -1-нитробензол (полученный по способу, подобному описанному в приготовлении 9).
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 3280, 1713, 1655, 1518, 1480, 1459, 1256, 758.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ, ppm: 0,75-0,90 (2H, мультиплет); 1,10-1,30 (8H, мультиплет); 1,17 (9H, синглет); 1,50-2,70 (7H, мультиплет); 2,31 -2,42 (0,4H, мультиплет); 2,47 (2H, триплет, J = 7 Гц); 2,70 (1,6H, дублет, J = 7 Гц), 3,38 (0,4H, синглет), 3,65 (1,6H, синглет); 4,73 (IH, триплет, J = 7 Гц); 6,94-7,40 (IIH, мультиплет).
Пример 32 N-[2-трет-Бутил-5-(6-циклогексил-2-гидроксигексил)фенил] -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-421)
Следуя способу, подобному описанному в примере 22, но используя N-[2-трет-бутил-5-(6-циклогексил-2-оксогексил)фенил] -2-(9-ксантен-9-ил) ацетамид (полученный, как описано в примере 31) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в вид пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 3380, 3275, 1656, 1522, 1480, 1459, 1256, 758.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ, ppm: 0,8-0,94 (2H, мультиплет); 1,12-1,75 (17H, мультиплет); 1,17 (9H, синглет); 2,33-2,55 (0,8, мультиплет); 2,60(0,8H, дублет из дублетов, J = 9 & 14 Гц); 2,70 (1,6H, дублет, J = 7 Гц); 2,80 (0,8H, дублет из дублетов, J = 3 & 14 Гц); 3,52-3,88 (IH, мультиплет); 4,74 (IH, триплет, J = 7 Гц); 6,97-7,41 (IIH, мультиплет).
Пример 33. N-[2-трет-Бутил-5-(4-циклогептил-2-оксобутил)фенил]-4-децилоксибензамид (соединение N 4-117)
Следуя способу, подобному описанному в примере 21, но используя 4-децилоксибензойную кислоту и 2-трет-бутил-5-(4-циклогексил-2-оксобутил)анилин в качестве исходных продуктов в соответствующем соотношении, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 98-99oC (после перекристаллизации из смеси этилацетат и гексана). 2-трет-Бутил-5-(4-циклогептил-3-оксобутил)анилин, используемый в качестве исходного продукта, получают согласно способу, подобному описанному в приготовлении 7, но используя 2-трет-бутил-5-(4-циклогептил-3-оксобутил)-1-нитробензол (полученный по способу, подобному описанному в приготовлении 6).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3314, 1715, 1640, 1502, 1251, 766.
Спектр ядерного магнитного резонанса (CLCl3, 270 МГц), δ: 0,88 (3H, триплет, J = 7 Гц; 1,05 - 1,7 (26H, мультиплет); 1,43 (9H, синглет); 1,75 - 1,9 (2H, мультиплет); 1,9 - 2,1 (1H, мультиплет); 2,31 (2H, дублет, J = 7 Гц); 2,7 - 2,8 (2H, мультиплет); 2,8 - 2,9 (2H, мультиплет); 4,03 (2H, триплет, J = 7 Гц); 6,95 -7,05 (3H, мультиплет); 7,32 (1H, дублет, J=8Гц); 7,60 (1H, синглет); 7,78 (1H, синглет); 7,85 (2H, дублет, J = 8 Гц).
Пример 34. N-[2-трет-Бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -4- децилоксибензамид (соединение N 4-17)
Следуя способу, подобному описанному в примере 22, но используя N-[2-трет-бутил-5-(4-циклогептил-3-оксобутил) фенил] -4-децилоксибензамид (полученный, как описано в примере 31) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заготовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3480, 1664, 1607, 1503, 1468.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ : 0,88 (3H, триплет, J=7ГЦ); 1,05 - 1,9 (33H,мультиплет);1,43 (9H, синглет); 2,6-2,9(9H, мультиплет): 3,65 - 3,8 (1H, мультиплет); 4,02 (2H, триплет, J= 7 Гц); 6,98 (2H, дублет, J= 8Гц); 7,02 (1H, дублет, J=8Гц); 7,33 (1H, дублет, J=8Гц); 7,61 (1H, синглет); 7,78 (1H, синглет); 7,85 (2H, дублет, J=8 Гц).
Пример 35, N-[2-трет-Бутил-5-(3-фенил-3-оксопропил)фенил]-2-(9H-ксантен-9-ил) ацетамид (Соединение N 1-1326)
0,58 мл (4,20 ммоль) триэтиламина и 0,31 мл (2,10 ммоль) диэтилфосфорилхлорида добавляют к 20 мл бензольной суспензии, содержащей 505 мг (2,10 ммоль) 2-(9H-ксантен-9-ил)уксусной кислоты и затем смесь перемешивают в течение 1 ч. По истечении этого времени к реакционной суспензии добавляют 5 мл бензольного раствора, содержащего 587 мг (2,09 ммоль) 2-трет-бутиль-5-(3-фенил-3-оксопропил)анилина (полученного по способу, подобному описанному в приготовлении 7), и затем 10 мг 4-пирролидинопиридина, и полученную смесь нагревают с обратным холодильником в течение 4,5 ч. Реакционную смесь затем разбавляют диэтилацетатом, промывают разбавленным водным раствором соляной кислоты, водой и затем насыщенным водным раствором карбоната натрия и насыщенным водным раствором хлорида натрия, в данном порядке. Растворитель затем удаляют отгонкой при пониженном давлении и полученный остаток подвергают колоночной хроматографии на силикагеле. Элюирование 4:1 по объему смесью метиленхлорида и циклогексана дает 934 мг (выход 86%) указанного в заголовке соединения в виде кристаллов, плавящихся при 116-118oC (после перекристаллизации из диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3216, 1686, 1640, 1481, 1457, 1254.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ppm: 1,17 (27/4 H, синглет); 1,22 (9/4H, синглет); 2,46 (9/4H, дублет, J = 7 Гц); 2,65 -2,85 (1H, мультиплет); 2,71 (3/2H, дублет, J=7Гц); 3,04 (3/2H, триплет, J=7 Гц); 3,05-3,02 (1/2H, мультиплет); 3,32 (3/2H, триплет, J=7Гц); 4,74 (1H, триплет, J= 7 Гц); 6,7-6,7 (14H, мультиплет); 7,99 (2H, дублет, J=7Гц).
Пример 36. N-[2-трет-Бутил-5-(3-гидрокси-3-фенилпропил)фенил]-2-(9H-ксантен -9-ил)ацетамид (Соединение N 1-1300)
Следуя способу, подобному описанному в примере 22, но используя N-[2-трет-бутил-5-(3-фенил-3-оскопропил)фенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 35) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке в виде кристаллов, плавящихся при 136-137oC (после перекристаллизации из диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3267, 1658, 1525, 1479, 1255.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ppm: 1,16 (15/2H, синглет); 1,18 (3/2H, синглет); 1,8-2,8 (6H, мультиплет); 4,5-4,8 (2H, мультиплет); 6,9-7,5 (16H, мультиплет).
Пример 37э N-[2-трет-Бутил-5-(3-оксо-4-фенилбутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (Соединение N 1-1327)
Следуя способу, подобному описанному в примере 37, но используя 2-трет-бутил-5-(3-оксо-4-фенилбутил) анилин ( полученный по способу, подобному описанному в приготовлении 7) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 122-123oC (после перекристаллизации из диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1 : 3224, 1716, 1640, 1456, 1257.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ppm: 1,15 (27/4H, синглет): 1,18 (9H, синглет); 2,41 (1/2H, дублет, J=7 Гц); 2,5-2,6 (1/2H, мультиплет); 2,69 (3-2H, дублет, J=7ГЦ): 2,7-2,9 (3/2H, мультиплет); 3,67 (1/2H, синглет); 3,71 (3/2H, синглет); 4,74 (1H, триплет, J=7 Гц); 6,85-7,4 (16H, мультиплет); 1,8-2,8 (6H, мультиплет): 4,5-4,8 (2H, мультиплет); 6,9-7,5 (16H, мультиплет).
Пример 38. N-[2-трет-Бутил-5-(3-гидрокси-4-фенилбутил)фенил]-2-(9H-ксантен-9-ил)ацетамид
(Соединение N 1-1301)
Следуя способу, подобному описанному в примере 22, но используя соединение, полученное по примеру 37, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3403, 3278, 1655, 1458, 14116, 1256.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,16 (27/4H, синглет); 1,20 (9/4H, синглет); 1,75-2,0 (2H, мультиплет); 2,3-2,95 (6H, мультиплет); 3,7-4,0 (1H, мультиплет); 4,74 (1H, триплет, J=7 Гц); 6,9-7,45 (16H,мультиплет).
пример 39. N-[2-трет-Бутил-5-(4-циклогептил-3-гидроксибутил)фенил]-2-(9H-ксантен -9-ил)ацетамид (Соединение N 1-949)
Следуя способу, подобному описанному далее в примере 40, но используя N-[2-трет-бутил-5-(4-циклогептил-3-трет-бутилдиметилсилилоксибутил) фенил] -2-(9H-ксантен -9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении II) в качестве исходного продукта в соответствующем количестве, используя в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3400, 1668, 1596, 1475, 1458.
Спектр ядерного резонанса (CDCl3, 270 МГц), δ ppm: 1,0-1,8 (17H,мультиплет); 1,16 (9H, синглет); 2,3-2,9 (4H, мультиплет); 3,6-3,8 (1H,мультиплет); 4,74 (1H, триплет, J=7 Гц); 7,5 (11H, мультиплет).
Пример 40. N-[2-трет-Бутил-5-(3-циклогексил-2- гидроксипропил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-349)
1,5 мл 2 H водной соляной кислоты добавляют к 15 мл тетрагидрофуранового раствора, содержащего 813 мг (1,30 ммоль) N-[2-трет-бутил-5-(3-циклогексил-2-трет-бутилдиметилсилоксипропил) фенил]-2=(9H-ксантен-9-ил) ацетамида ( полученного, как описано в приготовлении II), и смесь перемешивают 2,5 ч при 50oC. Реакционной смеси позволяют вернуться к комнатной температуре. Добавление смеси этилацетата и воды дает возможность указанному в заголовке соединению распределяться между органическим растворителем и водой. Органический слой отделяют и промывают насыщенным водным раствором гидрокарбоната натрия и затем насыщенным водным раствором хлорида натрия. Растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 50 г силикагеля. Элюирование смесью этилацетата и гексана, изменяющейся от 1:2 до 2:3 по объему, приводит к получению 650 мг (выход 98%) указанного в заголовке соединения в виде кристаллов, плавящихся при 177-178oC (после перекристаллизации из изопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3393, 3223, 1641, 1537, 1482, 1457, 1257Ю 760.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ,ppm: 0,82-1,04 (2H, мультиплет); 1,12-1,87 (11H, мультиплет); 1,17 (9H, мультиплет); 2,35-2,50 (0,08H, мультиплет); 2,57 (0,8H, дублет из дублетов, J=8 & 14Гц); 2,70 (1,6H, дублет, J=7 Гц); 2,79 (0,8H, дублет из дублетов, =4 J & 8 Гц); 3,67 -3,99 (1H, мультиплет); 4,74 91H, J=7 Гц); 6,97-7,41 (1H, мультиплет).
Пример 41. N-[5-(4-Циклогексил-3-гидроксибутил)-2-изопропилфенил] -2- (9H-ксантен-9-ил)ацетамид (соединение N 1-26)
Следуя способу, подобному описанному в примере 40, но используя N-[5-(4-циклогексил-3-трет-бутилдиметилсилилоксибутил)-2- изопропилфенилъ-2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении II) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 144-145oC (после перекристаллизации из смеси этилацетат и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3282, 2919, 1644, 1528, 1481, 1260, 757.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , pp m: 0,8-1,9 (15H, мультиплет); 1,05 (6H, дублет, J=7 Гц); 2,5-2,9 (5H, мультиплет), 3,65-3,85 (1H, мультиплет); 4,69 (1H, триплет, J=7 Гц); 7,0-7,5 (11H, мультиптет).
Пример 42. N-[2-трет-Бутил-5-(3-циклогексил-3-гидроксипропил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (Соединение N 1-131)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(3-циклогексил-3-трет-бутилдиметилсилилоксипропил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении II) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3400, 3270, 1653, 1522, 1480, 1457, 1256, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ б ppm: 0,9001,89 (13H, мультиплет); 1,16 (9H, синглет); 2,30-2,87(4H, мультиплет); 3,28-3,45 (1H, мультиплет); 4,74 (1H, триплет, J=7 Гц); 6,95-7,42 (11H, мультиплет).
Пример 43. N-[2-трет-Бутил-5-(6-циклогексил-3-гидроксигексил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (Соединение N 1-49)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(6-циклогексил-3-трет-бутилдиметилсилилоксигексил) фенилъ-2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении II) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3390, 3280, 1655, 1522, 1480, 1459, 1256, 758.
Спектр ядерного магнитного резонанса (CDCL3, 270 МГц), δ , ppm: 0,80-0,95 (2H, мультиплет); 1,10-1,82 (17H, мультиплет); 1,16 (9H, синглет); 2,30-2,82 (4H, мультиплет); 3,50-3,69 (1H, мультиплет); 4,74 (1H, триплет, J=7 Гц); 6,95-7-41 (11H, мультиплет).
Пример 44. N-[2-трет-Бутил-5-(7-циклогексил-3-гидроксигептил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (Соединение N 1-73)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(7-циклогексил-3-трет-бутилдиметилсилилоксигептил) фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении II) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3390, 3270, 1653, 1522, 1480, 1459, 1256, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,77-0,93 (2H, мультиплет); 1,10-1,81 (19H, мультиплет); 1,16 (9H, синглет); 2,30-2.83 (4H, мультиплет0; 3,47-369 (1H, мультиплет); 4,74 (1H, триплет, J= 7 Гц); 6,95-7,42 (11H, мультиплет).
Пример 45. N-[2-трет-Бутил-5-(6-циклогексил-4-нидроксигексил) фенил]-2-(9H-ксантен-9-ил)ацетамид (Соединение N 1-519)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(6-циклогексил-4-трет-бутилдиметилсилилоксигексил- фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении II) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3450, 1659 (broad0, 1600, 1575, 1478, 1453.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,7-1,0 (2H, мультиплет); 1,0-1,9 917H, мультиплет); 1,17 (9H, синглет); 2,2-2,8 (4H, мультиплет); 3,5-3,7 (1H, мультиплет); 4,76 (1H, триплет, J=7 Гц); 6,9-7,5 (11H, мультиплет). N-[2[трет-Бутил-5-(5-циклогексид-5-гидроксипентил)фенил]- 2-(9H-ксантен-9-ил)ацетамид (Соединение N 1-577)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(5-циклогексил-5-трет-бутилдиметилсилилоксипентил) фенил] -2-(9Hксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении II) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 147-148oC (после перекристаллизации из смеси метиленхлорида и диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3336, 1657, 1519, 1480, 1458, 1260, 761.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,93-1,86 (17H, мультиплет); 1,16 (9H, синглет); 2,27-2,35 (0,4H, мультиплет); 2,45 (0,4H, дублет J=7 Гц); 2,60 (1,6H, триплет, J=7 Гц); 2,70 (1,6H, дублет, J= 7 Гц); 3,32-3,41 (1H, мультиплет); 4,74 (1H, триплет, J=7 Гц); 6,93-7,41 (11H, мультиплет).
Пример 47. N-[2-трет-Бутил-5-(4-циклопентил-3-гидроксибутил) фенил]-2-(9H-ксантен-9-ил)ацетамид (Соединение N 1-747)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(4-циклопентил-3-трет-бутилдиметилсилилоксибутил) фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении II) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 142-143oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2951, 1654, 1529, 1478, 1253, 760.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,0-1,3 (2H, мультиплет); 1,16 (9H, синглет(; 1,3-2,0 (11H, мультиплет); 2,2-2,9 (4H, мультиплет); 3,5-3,8 (1H, мультиплет); 4,74 (1H, триплет, J=7 Гц); 6,9-7,5 (11H, мультиплет).
Пример 48. N-[2-трет-Бутил-5-(6-циклопентил-5-гидроксигексил) фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-839)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(6-циклопентил-5-трет-бутилдиметилсилилоксигексил) фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении II) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 132-133oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2949, 1649, 1523, 1479, 1458, 1258, 760.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,0-1,3 (2H, мультиплет); 1,17 (9H, синглет); 1,3-2,0 (15H, мультиплет); 2,25-2,35 (2/5H, мультиплет); 2,45 (2/5H, дублет, J=7 Гц); 2,60 (8/5H, триплет, J=8 Гц); 2,69 (8/5H, дублет, J=7 Гц); 3,6-3,7 (1H, мультиплет); 4,74 (1H, триплет, J=7 Гц); 6,9-7,5 (11H, мультиплет).
Пример 4. N-[2-трет-Бутилтио-6-(3-гидрокси-3-фенилпропил)фенил] - 2-(9H-ксантен-9-ил)ацетамид
(соединение N 1-1442)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутилтио-6-(3-трет-бутилдиметилсилилокси-3-фенилпропил)фенил] -2- (9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 11) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 145-147oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2959, 1662, 1481, 1456, 1259, 762.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,13 (9H, синглет); 1,99 (2H, квартет, J=7 Гц); 2,60 (2H, триплет, J=8 Гц); 2,75 (2H, дублет, J= 7 Гц); 4,51 (1H, триплет, J=6 Гц); 4,70 (1H, триплет, J=7 Гц); 7,0-7,5 (16H, мультиплет).
Пример 50. N-[2-Метил-6-(3-гидрокси-3-фенилпропил)фенил] -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1438)
Следуя способу, подобному описанному в примере 40, но используя N-[2-метил-6-(3-трет-бутилдиметилсилилокси-3-фенилпропил)фенил] -2-(9H-ксантен- 9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 11) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 193-194oC (после перекристаллизации из смеси метиленхлорида, метанола и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3235, 1648, 1523, 1479, 1458, 754.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ , ppm: 1,5-2,8 (6H, мультиплет); 2,05 (3H, синглет); 4,2-4,5 (1H, мультиплет); 4,68 (1H, триплет, J=7 Гц); 6,9-7,56 (11H, мультиплет),
Пример 51. N-[2-Метил-6-(3-оксо-3-фенилпропил)фенил]-2-(9H-ксантен-9-ил)ацетамид соединение N 1-1437)
Следуя способу, подобному описанному в примере 13, но используя N-[2-метил-6-(3-гидрокси-3-фенилпропил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 50) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение, плавящееся при 175-176oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3280, 1684, 1653, 1480, 1457, 1259, 762.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ, ppm: 2,10 (3H, синглет); 2,62 (2H, триплет, J=7 Гц), 2,85 (2H, дублет, J=7 Гц), 3,23 (2H, триплет, J= 7 Гц); 4,75 (1H, триплет, J=7 Гц); 6,9-8,05 (16H, мультиплет); 8,1-8,3 (1H, уширенный синглет).
Пример 52. N-[2-Метил-6-(3-гидроксибутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1435)
Следуя способу, подобному описанному в примере 40, но используя N-[2-метил-6-(3-трет-бутилдимесилилоксибутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 11) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 167-168oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3244, 1653, 1530, 1481, 1457, 1260, 758.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ , ppm: 1,05 (3H, дублет, J= 7 Гц); 1,4-1,9 (2H, мультиплет); 2,06 (3H, синглет); 2,32 (2H, триплет, J=7 Гц); 2,70 (2H, дублет, J=7 Гц); 3,1-3,6 (1H, мультиплет); 4,68 (1H, триплет, J=7 Гц); 6,8-7,9 (11H, мультиплет).
Пример 53. N-[2-Метил-6-(3-оксобутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1436)
Следуя способу, подобному описанному в примере 13, но используя N-[2-метил-6-(3-гидроксибутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный по способу, описанному в примере 52) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 167-168oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3281, 1711, 1648, 1480, 1260, 754.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ , ppm: 1,95 (3Н, синглет); 2,03 (3Н, синглет); 2,1-2,9 (4Н, мультиплет); 2,75 (2Н, дублет, J= 7 Гц); 4,68 (1Н, триплет, J=7 Гц); 6,75-7,5 (11Н, мультиплет); 8,0-8,3 (1Н, уширенный синглет).
Пример 54. N-[4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]-фенил]-6-фенилгексил гидросукцинат (соединение N 1-1337)
1 мл пиридинового раствора, содержащего 276 мг (0,504 ммоль) N-[2-трет-бутил-5-(6-фенил-1-гидроксигексил)фенил] -2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в примере 16), 58 мг (0,58 ммоль) янтарного ангидрида и 64 мг (0,52 ммоль) 4-(N,N-диметиламино)-пиридина, перемешивают в течение 1 ч при 100oC. По истечении этого времени реакционной раствор разбавляют этилацетатом, промывают 2 н. водной соляной кислотой один раз, водой дважды и насыщенным водным раствором хлорида натрия. Растворитель затем удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии на силикагеле. Элюирование 9:1 по объему смесью метиленхлорида и метанола приводит к 294 мг (выход 90%) указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 3260, 1734, 1715, 1651, 1480, 1459, 1254, 760.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,10-1,44 (4H, мультиплет); 1,16 (9H, синглет); 1,53-1,98 (4H, мультиплет); 2,38-2,80 (8H, мультиплет); 4,74 (1H, триплет, J=7 Гц); 5,67 (1H, триплет, J=7 Гц); 7,04-7,51 (16H, мультиплет).
Пример 55. N-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]-фенил}-6-фенилгексил гидросукцинат (соединение N 1-1336)
4 мл метанольного раствора, содержащего 165 мг (0,255 ммоль) 1-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}-6-фенилгексил гидросукцината (полученного, как описано в примере 54), охлаждают на ледяной бане и затем по каплям добавляют 0,1 н. водный раствор гидроксида натрия. Реакционный раствор конденсируют упариванием при пониженном давлении, поддерживая температуру ниже 15oC. К остатку добавляют 20 мл толуола и затем добавляют достаточное количество этанола до тех пор, пока раствор станет гомогенным. Растворитель затем отгоняют при пониженном давлении. С целью удаления остаточной воды к остатку добавляют толуол и смесь вновь отгоняют. Эту процедуру повторяют несколько раз и получают 170 мг (количественный выход) указанного в заголовке соединения в виде пенообразного продукта.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,03-1,80 (8H, мультиплет); 1,06 (9H, мультиплет); 2,10-2,59 (7,2H, мультиплет); 2,80-2,98 (0,8H, мультиплет); 4,61 (0,8H, триплет, J=7 Гц); 4,60-4,70 (0,2H, мультиплет); 5,27-5,33 (0,2H, мультиплет); 5,53 (0,8H, триплет, J=7 Гц); 6,85-7,32 (11H, мультиплет).
Пример 56. 1(2-{ 4-трет-Бутил-3-[2-9H-ксантен-9-ил)ацетамидо] -фенил} этил)-2-циклогексил гидросукцинат (соединение N 1-1109)
4 мл метанольного раствора, содержащего 74 мг (0,14 ммоль) N-[2-трет-бутил-5-(4-циклогенсил-3-гидроксибутил)фенил] -2-(9H-ксантен-9-ил) ацетамида (полученного, как описано в примере 12), 15 мг (0,15 ммоль) янтарного ангидрида, 18 мг (0,15 ммоль) 4-(N,N-диметиламино)пиридина и 112 мг (1,41 ммоль) пиридина, перемешивают в течение 9 ч при 125oC. По истечении этого времени реакционной смеси позволяют вновь достичь комнатной температуры. Реакционную смесь затем разбавляют диэтиловый эфиром и промывают разбавленной водной соляной кислотой и затем водой. Растворитель затем удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 20 г силикагеля. Элюирование с использованием метода градиентного элюирования смесями метиленхлорида и метанола, колеблющимися от 100: 5 до 10: 1, приводит к получению 73 мг (выход 84%) указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 1722, 1671, 1479, 1458, 1250.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,75-2,00 (15Н, мультиплет); 1,14 (9Н, синглет); 2,19-2,80 (8Н, мультиплет); 4,72 (1Н, триплет, J= 7 Гц); 4,92-5,20 (1Н, мультиплет); 6,87-7,47 (11Н, мультиплет).
Пример 57. 1-(2-{4-трет-Бутил-3-[2-[(9H-ксантен-9-ил)ацетамидо]-фенил} этил)- 2-циклогексил натрий сукцинат (соединение N 1-1102)
Следуя способу, подобному описанному в примере 55, но используя 1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил}этил)- 2-циклогексилэтил гидросукцинат (полученный, как описано в примере 56) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1720, 1655, 1578, 1524, 1480, 1459, 1414, 1255, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,68-1,86 (15H, мультиплет); 1,09 (9H, синглет); 2,14-2,74 (8H, мультиплет); 4,70 (1H, триплет, J= 7 Гц); 4,73-5,05 (1H, мультиплет); 6,79-7,43 (11H, мультиплет).
Пример 58 1-(2-{ 4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] -фенил} этил)-3- циклогексилпропил гидросукцинат (соединение N 1-1118)
Следуя способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(5-циклогексил-3-гидроксипентил)-фенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 26) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 1728, 1672, 1479, 1457, 1418.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,7-1,0 (2H, мультиплет); 1,0-2,0 (15H, мультиплет); 1,15 (9H, синглет); 2,3-2,8 (8H, мультиплет); 4,73 (1H, триплет, J=7 Гц); 4,9-5,0 (1H, мультиплет); 6,9-7,5 (11H, мультиплет),
Пример 59. 1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]-фенил} этил)- 3-циклогексилпропил натрий сукцинат (соединение N 1-1111)
Следуя способу, подобному описанному в пример 55, но используя 1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)- 3-циклогексилпропил гидросукцинат (полученный, как описано в примере 58) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 3100-3500 (broad), 1716, 1672, 1573, 1455, 1412.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,65-0,9 (2H, мультиплет); 0,9-1,85 (15H, мультиплет); 1,10 (9H, синглет); 2,0-2,75 (8H, мультиплет); 4,69 (1H, триплет, J=7 Гц); 4,8-4,9 (1H, мультиплет); 6,9-7,4 (11H, мультиплет).
Пример 60. 1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]-фенил} этил)- 3-циклопентилэтил гидросукцинат (соединение N 1-1183)
Следуя способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(4-циклопентил-3-гидроксибутил)-фенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в приготовлении 47) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 1725, 1671, 1478, 1456, 1417.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,9-1,2 (2H, мультиплет); 1,14 (9H, синглет); 1,3-2,0 (11H, мультиплет); 2,0-3,8 (8H, мультиплет); 4,72 (1H, триплет, J=7 Гц); 5,0-5,15 (1H, мультиплет); 6,9-7,5 (11H, мультиплет).
Пример 61. 1-(2-{ 4-трет-Бутил-3-[-(9H-ксантен-9-ил)ацетамидо]-фенил} этил)- 3-циклопентилэтил натрий сукцинат (соединение N 1-1176)
Следуя способу, подобному описанному в примере 55, но используя 1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил}этил)- 2-циклопентилэтил гидросукцинат (полученный, как описано в примере 60) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 3100-3500 (broad), 1711, 1672, 1576, 1478, 1454.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,9-1,15 (2H, мультиплет); 1,09 (9H, синглет); 1,3-1,8 (11H, мультиплет); 2,2-3,15 (8H, мультиплет); 4,68 (1H, триплет, J=7 Гц); 4,7-5,0 (1H, мультиплет); 6,8-7,4 (11H, мультиплет).
Пример 62. N-{ 5-[3-(1-Имидазолил)пропокси] -2-метилтиофенил}-2-(9H-ксантен- 9-ил)ацетамид (соединение N 1-1479)
Следуя способу, подобному описанному в примере 1, но используя N-[5-(3-гидроксипропокси)-2-метилтиофенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 54) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединения в виде кристаллов, плавящихся при 127-129oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3297, 1658, 1576, 1480, 1456, 1257, 756.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ ppm: 1,95 (3H, синглет); 2,0-2,5 (2H, мультиплет); 2,75 (2H, дублет, J=7 Гц); 3,95 (2H, триплет, J= 5 Гц); 4,18 (2H, триплет, J=6 Гц); 4,70 (1H, триплет, J=7 Гц); 6,51 (0,6H, дублет, J= 2 Гц); 5,65 (0,4H, дублет, J=2 Гц); 7,6-7,9 (11H, мультиплет); 8,0-8,4 (2H, мультиплет).
Пример 63. N-{ 5-(3-(1-Имидазолил)пропокси] -2-метилтиофенил}-2-(9H-ксантен-9-ил) ацетамид гидрохлорид (соединение N 1-1478)
Следуя способу, подобному описанному в примере 2, но используя N-{5-[3-(1-имидазолил)пропокси] -2-метилтиофенил} -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 62) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 155-159oC (после перекристаллизации из смеси метанола и диэтилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3262, 2921, 1658, 1574, 1482, 1259, 751.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,98 (3H, синглет); 2,3-2,6 (2H, уширенный синглет); 2,75 (2H, дублет, J=7 Гц); 4,0-4,2 (2H, уширенный синглет); 4,5-4,8 (3H, мультиплет); 6,55 (1H, уширенный синглет); 7,0-7,5 (11H, мультиплет); 8,11 (1H, уширенный синглет); 8,22 (1H, уширенный синглет); 9,60 (1H, уширенный синглет).
Пример 64. N-[2-трет-Бутил-5-(5-циклогептил-3-оксопентил)фенил]-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-1271)
Следуя способу, подобному описанному в примере 21, но используя 2-трет-бутил-5-(5-циклогептил-3-оксопентил)анилин (полученный, как описано в приготовлении 6) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 136-137oC (после перекристаллизации из смеси метиленхлорида, диэтилового эфира и гесана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1701, 1665, 1520, 1480, 1458, 1300.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,06-1,73 (15H, мультиплет); 1,16 (9H, синглет); 2,38-2,59 (8H, мультиплет); 4,74 (1H, триплет, Y = 7 Гц); 6,91-7,44 (11H, мультиплет).
Пример 65. N-[2-трет-Бутил-5-(5-циклогептил-3-гидроксипентил) фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-959)
Следуя способу, подобному описанному в примере 22, но используя N-[2-трет-бутил-5-(5-циклогептил-3-оксопентил) фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 64) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке в виде кристаллов, плавящихся при 136-137oC (после перекристаллизации из смеси метиленхлорида, диэтилового эфира и гесана).
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 3380, 1659, 1518, 1480, 1459, 1260, 760.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,09-1, 86 (19H, мультиплет); 1,16 (9H, синглет); 2,30-2,85 (4H, мультиплет); 3,46-3,67 (1H, мультиплет); 4,74 (1H, триплет, Y = 7 Гц); 6,95-7,42 (11H, мультиплет).
Пример 66. 1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] -фенил} этил)-2-циклогептилэтил гидросукцинат соединение N 1-1221)
Следуя способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(4-циклогептил-3-гидроксибутил)фенил] -2-(9H-ксантен- -9-ил)ацетамид (полученный, как описано в примере 39) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 1726, 1670, 1575, 1478, 1458.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,05-2,0 (17H, мультиплет); 1,14 (9H, синглет); 2,2-2,8 (8H, мультиплет); 4,73 (1H, триплет Y = 7 Гц); 4,9-5,2 (1H, мультиплет); 6,9-7,5 (11H, мультиплет).
Пример 67. 1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]- фенил} этил)-2-циклогептилэтил натрий сукцинат (соединение N 1-1220)
Следуя способу, подобному описанному в примере 55, но используя 1-(2-{ 4-третбутилд-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил}этил-2- -циклогептилэтил гидросукцинат (полученный, как описано в примере 66) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (CHCl3, νmax , см-1: 3100-3500 (broad), 1711, 1674, 1652, 1576, 1480, 1457.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,0-1,9 (17H, мультиплет); 1,09 (9H, синглет); 2,2-2,7 (8H, мультиплет); 4,70 (1H, триплет J = 7 Гц); 4,70-5,0 (1H, мультиплет); 6,8-7,5 (11H, мультиплет).
Пример 68. N-[2-Этил-6-(3-оксо-6-фенил-1-гексенил)фенил]- -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1447)
49 мг (1,13 ммоль) гидрида натрия (в виде 55% по массе суспензии в минеральном масле) промывают дважды, каждый раз гексаном, и суспендируют в 6 мл диметилформамида. К суспензии прибавляют 2 мл диметилформамидного раствора, содержащего 289 мг (0,97 ммоль) 5-диэтотоксифосфорид-1-(4-оксопентил)бензола, при охлаждении льдом. Реакционную смесь затем немедленно возвращают в комнатной температуре, после чего перемешивают 30 мин. К реакционной смеси добавляют 300 мг (0,808 ммоль) N-(2-этил-6-формил)-2-(9H-ксантен-9-ил)ацетамида, вновь охлаждая льдом. Смесь затем перемешивают 1 ч при температуре ледяного охлаждения и затем 3 ч при комнатной температуре. По истечении этого времени реакционной раствор выливают в ледяную воду и экстрагируют диэтиловым эфиром. Органический экстракт промывают водой. Растворитель удаляют отгонкой при пониженном давлении, а полученный остаток перекристаллизовывают из смеси метиленхлорида и диэтилового эфира с получением 220 мг (выход 53%) указанного в заголовке соединения в виде кристаллов, плавящихся при 168-170oC.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3287, 1651, 1518, 1480, 1459, 1260.
Спектр ядерного магнитного резонанса. (CDCl3, 270 МГц), δ , ppm: 0,92 (1/2H, триплет, J = 7 Гц); 1,02 (5/2H, триплет, J = 7 Гц); 1,98 (2H, квинтет, J = 7 Гц); 2,15 (1/3H, дублет, J = 7 Гц); 2,23 (2H, квартет, J = 7 Гц); 2,56 (2H, триплет, J = 7 Гц); 2,26 (2H, триплет, J = 7 Гц); 2,77 (5/3H, дублет, J = 7 Гц); 4,70 (1H, триплет, J = 7 Гц); 6,5-6,7 (2H, мультиплет); 6,9-7,5 (16H, мультиплет).
Пример 69. 3-[4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]- -фенил]1-циклогексилпропил гидросукцинат (соединение N 1-1100)
Следуя способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(3-циклогептил-3-гидроксипропил)-фенил] -2-(9H- -ксантен-9-ил)ацетамид (полученный, как описано в примере 42) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax б см-1: 2930, 1732, 1713, 1481, 1458, 1254, 761.
Пример 70. Натрий-3-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]-фенил}-1- -циклогексилпропил сукцинат (соединение N 1-1093)
Следуя способу, подобному описанному в примере 55, но используя 3-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}-1- -циклогексилпропил гидросукцинат (полученный, как описано в примере 69) в качестве исходного продукта в соответствующем количестве, используемом в этот примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2929, 1726, 1657, 1578, 1481, 1457, 1255, 759.
Пример 71. 1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]- -фенил} этил)-5-циклогексилпентил гидросукцинат (соединение N 1-1136)
Следует способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(7-циклогексил-3-гидроксигептил)-фенил] -2-(9H- -ксантен-9-ил)ацетамид (полученный, как описано в примере 44) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1733, 1713, 1481, 1458, 1253, 760.
Пример 72. Натрий 1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)- -ацетамидо] фенил}этил-5-циклогексилпентил сукцинат (соединение N 1-1129)
Следуя способу, подобному описанному в примере 70, но используя 1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-5- -циклогексилпентил гидросукцинат (полученный, как описано в примере 71) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1726, 1656, 1578, 1524, 1481, 1458, 1415, 1364, 1300, 1255, 759.
Пример 73. 1-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]- -фенил}-2-циклогексилэтил гидросукцинат (соединение N 1-1022)
Следуя способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(2-циклогексил-1-гидроксиэтил)фенил] -(9H-ксантен- -ил)ацетамид (полученный, как описано в примере 14) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 1736, 1714, 1481, 1458, 1255, 761.
Пример 74. Натрий 1-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]-фенил}-2- -циклогексилэтил сукцинат (соединение N 1-1023)
Следуя способу, подобному описанному в примере 70, но используя 1-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил}- -2-циклогексилэтилд гидросукцинат (полученный, как описано в примере 73) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1728, 1658, 1578, 1481, 1458, 1256, 758.
Пример 75. 1-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]- -фенил}-3-циклогексилпропил гидросукцинат (соединение N 1-2865)
Следуя способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(3-циклогексил-1-гидроксипропил)фенил] -2-(9H-ксантен- -9-ил)ацетамид (полученный, как описано в примере 99) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1736, 1714, 1481, 11458, 1255, 759.
Пример 76. Натрий 1-{4-трет-бутил-3-(9H-ксантен-9-ил)-ацетамидо] фенил} -3-циколгексилпропил сукцинат (соединение N 1-1026)
Следуя способу, подобному описанному в примере 70, но используя 1-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} -3- -циклогексилпропил гидросукцинат (полученный, как описано в примере 75) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1728, 1658, 1578, 1481, 1458, 1255, 758.
Пример 77. 1-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}-6- -циклогексилгексил гидросукцинат (соединение N 1-1053)
Следуя способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(6-циклогексил-1-гидроксигексил)фенил] -2-(9H-ксантен- -9-ил)ацетамид (полученный, как описано в примере 18) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 2852, 1736, 1713, 1652, 1480, 1458, 1245, 1165, 759.
Пример 78. Натрий 1-{4-трет-бутил-3-[2-(9H-ксантен-4-ил)ацетамидо]-фенил}- -6-циклогексилгексил сукцинат (соединение N 1-1046)
Следуя способу, подобному описанному в примере 70, но используя 1-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}-6- -циклогексилгексил гидросукцинат (полученный, как описано в примере 77) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 2852, 1728, 1656, 1567, 1480, 1458, 1416, 1255, 757.
Пример 79. (R)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил} этил)- -2-циклогексилэтил гидросукцинат (соединен N 1-1109)
Раствор 337 мг (0,471 ммоль) (R)-1-(2-{4-трет-бутил-3-[2-(9H- -ксантен-9-ил)ацетамидо] фенил} этил)-2-циклогексилэтил бензил гидросукцината (полученного, как описано в приготовлении 25) в 10 мл этилацетата энергично перемешивают в токе водорода в присутствии 173 мг 10% по массе палладия на угле. Реакционную смесь затем фильтруют и катализатор промывают этилацетатом. Фильтрат и промывные воды объединяют и растворитель удаляют отгонкой при пониженном давлении. Остаток очищают колоночной хроматографией через 15 г силикагеля, используя 1: 19 по объему смесь метанола и метиленхлорида в качестве элюента, с получением указанного в заголовке соединения в виде пенообразного продукта.
[α]
Пример 80. Натрия (R)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен- -9-ил)-ацетамидо]фенил}этил)-2-циклогексиэтил сукцинат (соединение N 1-1102)
Следуя способу, подобному описанному в примере 70, но используя (R)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил}этил)- -2-циклогексиэтил гидросукцинат (полученный, как описано в примере 79) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1721, 1655, 1576, 1526, 1482, 1459, 1416, 1256, 760.
[α]
Пример 81. (S)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил} этил)- -2-циклогексилэтил гидросукцинат (соединение N 1-1109)
Следуя способу, подобному описанному в примере 54, но используя (S)-N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2- -(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 102) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
[α]
Пример 82. Натрий (S)-1-(2-{4-трет бутил-3-[2-(9H-ксантен-9-ил)- -ацетамидо]фенил}этил)-2-циклогексилэтил сукцинат (соединение N 1-1102)
Следуя способу, подобному описанному в примере 70, но используя (S)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо)фенил} этил)- -2-циклогексилэтил гидросукцинат (полученный, как описано в примере 81) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1721, 1655, 1578, 1482, 1459, 1416, 1256, 760.
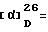 = +5.4o (C = 1.10, метанол).
= +5.4o (C = 1.10, метанол).
Пример 83. 1-(2-{ 4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-4-циклогексилбутил гидросукцинат (соединение N 1-1127)
Следуя способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(6-циклогексил-3-гидроксигексил)фенил] -2-(9H- -ксантен-9-ил)ацетамид (полученный, как описано в примере 43) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1733, 1713, 1481, 1458, 1254, 760.
Пример 84. Натрий 1-(2-этил-{4-трет-бутил-3-[2-(9H-ксантен-9- -ил)-ацетамидо]фенил}этил)-4-циклогексилбутил сукцинат (соединение N 1-1120)
Следуя способу, подобному описанному в примере 70, но используя 1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-4- -циклогексилбутил гидросукцинат (полученный, как описано в примере 83) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2933, 1736, 1481, 1458, 1254, 761.
Пример 85. 3-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил}-1-(циклогексилоксиметил)пропил гидросукцинат (соединение N-1-2441)
Следуя способу, подобному описанному в примере 56, но используя N-[2-трет-бутил-5-(4-циклогексилокси-3-гидроксибутил)фенил] -2-(9H- -ксантен-9-ил)ацетамид (полученный, как описано в примере 215) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2922, 1736, 1481, 1458, 1254, 761.
Пример 86. Натрий 3-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил} -1- -[(циклогексилокси)метил]пропил сукцинат (соединение N 1-2657)
Следуя способу, подобному описанному в примере 70, но используя 3-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}-1- -(циклогексилоксиметил)пропил гидроксукцинат (полученный, как описано в примере 85) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2932, 1728, 1656, 1578, 1481, 1458, 12256, 760.
Пример 87. (S)-1-(2-{4-трет-Бутил-3-[2-(9H-косантон-9-ил)ацетамидо]фенил}этил)- 3-циклогексилпропил гидросукцинат (соединение B1-1118)
Следуя способу, подобному описанному в примере 56, не используя (S)-N-[2-трет-бутил-5-(5-циклогексил-3-гидроксицентил)фенил] -2- (9H-ксантен-9-ил)ацетамид полученный, как описано в примере 114) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный адсорбционный спектр (KBr), νmax , см-1: 2923, 2851, 1733. 1713, 1652, 1480, 1458, 1414, 1253, 760.
Пример 88. Натрий (S)-1-(2-{ 4-трет-бутил-3-х2-(9H-консантен-9-ил)ацетамидо]фенил}этил)-3- циклогексилпропил сукцинат (соединение N 1-1111)
Следуя способу, подобному описанному в примере 70, не используя (S)-1-(2-{ 4-трет-ьутил-3-[3-(9H-ксантен-9-ил)ацетамидо] фенил} этил-3- циклогексилпропил гидросукцинат (полученный, как описано в примере 87) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 2851, 1726, 1656, 1577, 1480, 1458, 1414, 1254, 759. [α]
Пример 89.(R)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]-фенил} этил)-3- циклогескилпропил гидросукцинат (соединение N 1-1118)
Следуя способу, подобному описанному в примере 56, но используя (R)-N_ [2-трет-бутил-5-(5-циклогексил-3-гидроксипентил)фенил] -2-(9H-ксантен- 9-ил)ацетамид (полученный, как описано в примере 115) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 2851, 1733, 1713, 1652, 1480, 1458, 1414, 1253, 760.
Пример 90. Натрий (R)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)-ацетамидо]фенил}этил)-3-цикло гексилпропил сукцинат (соединение N 1-1111)
Следуя способу, подобному описанному в примере 70, но используя (R)-1-(2-{ 4-трет-бутил-3-[2(9H-ксантен-9-ил)ацетамидо] фенил} этил)-3-циклогексилпропил гидросукцинат (полученный, как описано в примере 89) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 2851, 1726, 1656, 1577, 1480, 1458, 1414, 1254, 759.
Пример 91. 1-(2-{3-Изопропил-2-[2-(9H-ксантен-9-ил)ацетамидо]-фенил}этил)-4-цикло гексилбутил гидросукцинат (соединение N 1-2866)
Следуя способу, подобному описанному в приготовлении 25, но используя N-[2-изопропил-6-(6-циклогексил-3-гидроксигексил) фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 125) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают бензиловый эфир указанного в заголовке соединения. Его затем дебензилируют способом, подобным описанному в примере 79, с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr, νmax , см-1: 2924, 2851, 1718, 1655, 1578, 1479, 1458, 1406, 1255.
Пример 92. Натрий 1-(2-{ 3-изопропил-2-[2-(9H-ксантен-9-ил)ацетамидо] фенил}этил)-4-циклогексилбутил сукцинат (соединение N 1-2867)
Следуя способу, подобному описанному в примере 70, но используя 1-(2-{ 3-изопропил-2-[2-(9H-ксантен-9-ил)ацетамидо] фенил}этил) -4-циклогексилбутил гидросукцинат (полученный, как описано в примере 91) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 2851, 1732, 1655, 1578, 1479, 1458, 1255.
Пример 93. 2-(2-{ 4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-3-циклогексилпропил гидросукцинат (соединение N 1-2349)
Следуя способу, подобному описанному в приготовлении 25, но используя N-[2-трет-бутил-5-[4-циклогексил-3-(гидроксиметил)бутил] фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 207) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают бензиловый эфир указанного соединения. Его затем дебензилируют способом, подобным описанному в примере 79, с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 2851, 1732, 1655, 1649, 1639, 1524. 1479, 1458, 1255.
Пример 94. Натрий 2-(2-{40трет-Бутил-3-[2-(9H- ксантен-9-ил)ацетамидо] фенил}этил)-3-циклогексилпропил сукцинат (соединение N 1-2367)
Следуя способу, подобному описанному в примере 70, не используя 2-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил) -3-циклогексилпропил гидросукцинат (полученный, как описано в примере 93) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2922, 2851, 1724, 1655, 158, 1479, 1458, 1414, 1255.
Пример 95. N-[2-трет-Бутил-5-(7-циклогептил-5-оксогептил)фенил]-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-2868)
Следуя способу, подобному описанному в примере 21, но используя 2-трет-бутил-5-(7-циклогептил-5-оксогептил)анилин (полученный по способу, подобному описанному в приготовлении 7) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2922, 2855, 1711. 1657, 1520, 1479, 1458, 1414, 1363, 1255.
Пример 96. N-[2-трет-Бутил-5-(7-циклогептил-5-гидроксигептил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1013)
Следуя способу, подобному описанному в примере 22. но используя N-[2-трет-бутил-5-(7-циклогептил-5-оксогептил)фенил]-2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 95) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1:2924, 2855, 1655, 1576, 1524, 1479, 1458, 1414, 1363, 1300, 1255.
Пример 97. N-[2-трет-Бутил-5-(3-циклогептил-3-оксопропил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2869)
Следуя способу, подобному описанному в примере 21, но используя 2-трет-бутил-5-(3-циклогептил-3-оксопропил)анилин (полученный по способу, подобному описанному в приготовлении 7) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 155-156o (после перекристаллизации из смеси метиленхлорида и гексана).
инфракрасный абсорбционный спектр (KBr), νmax , см-1: 32187, 2927, 2857, 1703, 1649, 1520, 1479, 1458, 1255, 759.
Пример 98. N-[2-трет-Бутил-5-(3-циклогептил-3-гидроксипропил)фенил] -2 -(9H-ксантен-9-ил)ацетамид (Соединение N1-943)
Следуя способу, подобному описанному в примере 22, но используя N-[2-трет-бутил-5-(3-циклогептил-3-оксопропил)фенил]-2 -(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 97) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3400, 3272, 2922, 2855, 1655, 1577, 1522, 1480, 1458, 1255, 759.
Пример 99. N-[2-трет-Бутил-5-(3-циклогепсил-1-гидроксипропил)фенил] -2 -(9H-ксантен-9-ил)ацетамид (Соединение N1-203)
Следуя способу, подобному описанному в примере 14, но используя 2-циклогексилэтилмагний йодид в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3390, 3284, 2923, 1656, 1577, 1520, 1481, 1481, 1458, 1256, 758.
Пример 100. (R)-N-[2-трет-Бутил-5-(4-циклогепсил-3-гидроксибутил)фенил] -2 -(9H-ксантен-9-ил)ацетамид (Соединение N1-25)
Следуя способу, подобному описанному в примере 40, но используя (R)-N-{ 2-трет-бутил-5-[4-циклогексил-3-(трет -бутилдиметилсилилокси)бутил]фенил}-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 23) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 140-141oC (после перекристаллизации из изопропилового эфира).
Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3340, 3230, 165, 1532, 1478, 1459, 1254, 760. [α]
Пример 101. (S)-N-[2-трет-Бутил-5-(4-циклогексил-3 -бензоилоксибутил)-фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1547)
Следуя способу, подобному описанному в примере 21, но используя (S)-[2трет-Бутил-5-(4-циклогексил-3-бензоилоксибутил)анилин (полученный, как описано в приготовлении 24) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде стеклообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3280, 1715, 1655, 1520, 1480, 1457, 1275, 1258, 1115, 909. [α]
Пример 102. (S)-N-[2-трет-Бутил-5-(4-циклогексил-3 -гидроксибутил)-фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-25)
13,3 мл 0,58 М этанольного раствора этоксида натрия добавляют к 1,62 г (2,57 ммоль) (S)-N-[2-трет-бутил-5-(4-циклогексил-3 -бензоилоксибутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 101), и полученную смесь перемешивают при комнатной температуре в течение 8 ч., после, чего ее нагревают с обратным холодильником в течение 2 ч. По истечении этого времени растворитель удаляют отгонкой при пониженном давлении, и полученный остаток смешивают и затем экстрагируют этилацетатом. Экстракт промывают водой и насыщенным водным раствором хлорида натрия, в этом порядке. Органическую фазу сушат над безводным сульфатом магния и растворитель удаляют отгонкой при пониженном давлении. Остаток очищают колоночной хроматографией через 150 г силикагеля, используя 2:8 по объему смесь ацетонитрила и бензола в качестве элюента. Концентрированный элюат перекристаллизовывают из диизопропилового эфира и получают 1,14 г (выход 85%) указанного в заголовке соединения, плавящегося при 141-142oC.
Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3350, 3240, 1655, 1532, 1478, 1459, 1254, 760. [α]
Пример 103. N-[(9H-Ксантен-9-ил)метил] -N'-[2-трет-бутил-5-(4 -циклогексил-3-гидробутил)фенил]мочевина (соединение N 1-39)
Следуя способу, подобному описанному в примере 40, но используя N-[(9H-Ксантен-9-ил)метил] -N'-[2-трет-бутил-5-(4 -циклогексил-3-гидробутил)фенил] мочевину (полученный, как описано в приготовлении 20) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 156-157oC (после перекристаллизации из смеси метиленхлорида и гексана).
Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3343, 2923, 2851, 1641, 1558, 1481, 1458, 1297, 1256, 1188, 756.
Пример 104. N-[2-трет-Бутил-5-(4-циклогексил-3-гидроксибутил)фенил]-2,2 -диметилдодеканамид (соединение N 5-19)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-Бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2,2 -диметилдодеканамид (полученный, как описано в приготовлении 46) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3395, 3304, 2923, 1651, 1509, 1479, 1448, 1363, 823.
Пример 105. N-[2-трет-Бутил-5-(5-циклогексил-1 -гидроксипентил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-251)
Следуя способу, подобному описанному в примере 14, но используя 4-циклогексилбутилмагний бромид в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 121-122oC (после перекристаллизации из смеси метиленхлорида, диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 2922, 28521, 1655, 1643, 1578, 1527, 1481, 1458, 1410, 1257
Пример 106. N-[2-трет-Бутил-5-(4-циклогексил-3 -гидроксибутил)фенил]-6,11-дигидродибенз[b,e]оксепин-11-карбоксамид (соединение N 2-19)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-Бутил-5-(4-циклогексил-3 -гидроксибутил)фенил]-6,11-дигидродибенз[b,e] оксепин-11-карбоксамид (полученный, как описано в приготовлении 47) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3407, 3278, 2922, 2872, 2851, 1655, 1519, 1497, 1448, 699.
Пример 107. N-[2-трет-Бутил-5-(4-циклогексил-3 -гидроксибутил)фенил-2-(1-фенилциклопентил)ацетамид (соединение N 3-13)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-Бутил-5-[4-циклогексил-3-(трет-бутил-диметилсилилокси) бутил] фенил]-2-(1-фенилциклопентил)ацетамид (полученный, как описано в приготовлении 48) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3407, 3278, 2922, 2872, 2851, 1655, 1519, 1497, 1448, 699.
Пример 108. N-[2-(1,1-Диметил-2-метоксиэтил)-6-(4-циклогексил-3-гидроски- бутил]фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2870)
Следуя способу, подобному описанному в примере 14, но используя циклогексилметилмагний бромид и N-[2-(1,1-диметил-2-метоксиэтил)-6-3-(оксипропил)фенил] -2-(9H- ксантен-9-ил)-ацетамид (полученный, как описано в приготовлении 26) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3260, 1667, 1509, 1480, 1459, 1254, 1096, 963, 758.
Пример 109. (S)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]-фенил}этил)-2- циклогексилэтил дигидрофосфат (соединение N 1-2871)
Следуя способу, подобному описанному в примере 79, но используя (S)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)-2- циклогексилэтил)дибензил дигидрофосфат (полученный, как описано в приготовлении 49) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде стеклообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3260, 1659, 1526, 1480, 1459, 1254, 1188, 1096, 988, 758.
Пример 110. Натрий (S)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-2- циклогексилэтил гидрофосфат (соединение N 1-1099)
Следуя способу, подобному описанному в примере 70, но используя (S)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-2- циклогексилэтил дигидрофосфат 9полученный, как описано в примере 109) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1717, 1651, 1576, 1480, 1459, 1404, 1256, 1134, 1077, 909, 760.
Пример 111. (S)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)-2- циклогексилэтил гидрофталат (соединение N 1-2872)
Следуя способу, подобному описанному в примере 54, но используя (S)-N-[2-трет-бутил-5-(4-циклогексил-3-гидроскибутил)фенил[-2-(9H- ксантен-9-ил)ацетамид (полученный, как описано в примере 102) и фталевый ангидрид в качестве исходного продуктов в соответствующем соотношении, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 1717, 1651, 1576, 1480, 1459, 1404, 1256, 1134, 1077, 909, 760.
Пример 112. Натрий (S)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)-2- циклогексилэтил фталат (соединение N 1-1104)
Следуя способу, подобному описанному в примере 70, но используя (S)-1-(2-{4-трет-бутил-3-[-2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)-2- циклогексилэтил (полученный, как описано в примере 111) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3260, 1705, 1653, 1574, 1480, 1459, 1397, 1258, 1079, 909, 758.
Пример 113. (S)-N-[2-трет-Бутил-5-(5-циклогексил-3-бензоилоксипентил)-фенил]-2- (9H-ксантен-9-ил)ацетамид (соединение N 1-1857)
Следуя способу, подобному описанному в примере 21, но используя (S)-2-трет-бутил-5-(5-циклогексил-3-бензоилоксипентил)анилин в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3260, 2923, 2851, 1717, 1653, 1519, 1481, 1457, 1275, 1257, 1115, 759, 712.
[α]
Пример 114. (S)-N-[2-трет-Бутил-5-(5-циклогексил-3-гидроксипентил)-фенил]-2- (9H-ксантен-9-ил)ацетамид (соединение N 1-1)
Следуя способу, подобному описанному в примере 102, но используя (S)-N-[2-трет-бутил-5-(5-циклогексил-3-бензоилоксипентил)фенил] -2-(9H- ксантен-9-ил)ацетамид (полученный, как описано в примере 113) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 136-137oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3377, 2921, 1660, 1516, 1481, 1458, 1423, 1412, 1260, 756.
[α]
Пример 115. (R)-N-[2-трет-бутил-5-(5-циклогексил-3-гидроксипентил)-фенил]-2- (9H-ксантен-9-ил)ацетамид (соединение N 1-1)
Следуя способу, подобному описанному в примере 40, но используя (R)-N-{ 2-трет-бутил-5-[5-циклогексил-3-(трет- бутилдиметилсилилокси)пентил] фенил} -2-(9H-ксантен-9-ил)ацетамид (полученный по способу, описанному в приготовлении 11) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 136-137oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3377, 2921, 2849, 1660, 1516, 1481, 1458, 1423, 1412, 1260, 756.
[α]
Пример 116. (S)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]-фенил}этил)-2- циклогексилэтил (соединение N 1-2873)
Следуя способу, подобному описанному в примере 79, но используя (S)-[1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ид)ацетамидо] фенил} этил)-2- циклогексилэтид] бензил малонат (полученный, как описано в примере 27) в качестве исходного продукта в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3260, 1729, 1632, 1480, 1459, 1302, 1254, 1156, 910, 760.
Пример 117. Натрий (S)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)- ацетамидо]фенил}этил)-2-циклогексилэтил малонат (соединение N 1-1101)
Следуя способу, подобному описанному в примере 70, но используя (S)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-2- циклогексилэтил гидромалонат (полученный, как описано в примере 116) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3250, 1717, 1653, 1605, 1480, 1459, 1300, 1256, 1256, 1156, 909, 758.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,70-0,90 (2H, мультиплет); 1,13-1,80 (13H, мультиплет); 1,08 (9H, синглет); 2,26-2,67 (4H, мультиплет); 3,03 (0,2H, дублет, J=16 Гц); 3,20 (0,2H, дублет, J= 16 Гц); 3,23 (1,6H, синглет); 4,70 (1H, триплет, J=7 Гц); 4,80-4,84 (0,2H, мультиплет); 4,92-5,00 (0,8H, мультиплет); 6,82 (11H, мультиплет).
Пример 118. N-[(1-Фенилциклопентил)метил] -N'-[2-трет-бутил-5-(4- циклогексил-3-гидроксибутил)фенил]мочевина (соединение N 3-19)
Следуя способу, подобному описанному в примере 40, но используя N-[2-(1-фенилциклопентил)метил] -N'-[2-трет-бутил-5-(3-трет- бутилдиметилсилилокси-4-циклогексилбутил)фенил] мочевину (полученную, как описано в приготовлении 22) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3344, 2922, 2852, 1641, 1558, 1448, 1418, 1364, 1253, 1073, 763, 701.
Пример 119. Глицин (S)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)- 2-циклогексилэтиловый эфир гидрохлорид (соединение N 1 - 1105)
0,21 мл 4 н. раствора хлористого водорода в диоксане добавляют к 374 мл (0,547 ммоль) 11-трет-бутоксикарбонилглицина (S)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)- 2-циклогексилэтилового эфира и полученную смесь перемешивают в течение ночи. Реакционную смесь разбавляют затем диэтиловым эфиром, после чего добавляют насыщенный водный раствор гидрокарбоната натрия. Смесь перемешивают и органический слой отделяют и затем промывают водой; растворитель далее удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 15 г силикагеля, используя метод ступенчатого элюирования, смесью этилацетата и метанола, изменяющейся от 100:0 до 100:3 по объему, что дает 277 мг свободного основания, соответствующего указанного в заголовке соединения в виде пенообразного продукта. 3 мл 4 н. раствора хлористого водорода в диоксане добавляют к свободному основанию и из смеси удаляют растворитель отгонкой при пониженном давлении, с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 2853, 1747, 1655, 1576, 1522, 1481, 1458, 1365, 1254.
Пример 120. N, N-Диметилглмцин (S)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)- 2-циклогексилэтиловый эфир (соединение N 1-2874)
Следуя способу, подобному описанному в приготовлении 25, но используя (S)-N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2-(9H- ксантен-9-ил)ацетамид (полученный, как описано в примере 102) и N, N-диметилглицингидрохлорид в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 2853, 1736, 1655, 1578, 1522, 1479, 1458, 1255, 1194.
Пример 121. N,N-Диметилглицин (S)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)- 2-циклогексилэтиловый эфир гидрохлорид (соединение N 1 - 1106)
Следуя способу, подобному описанному в примере 119, но используя N,N-диметилглицин (S)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)- 2-циклогексилэтиловый эфир (полученный, как описано в примере 120) и 4 н. раствор хлористого водорода в диоксане в качестве исходных продуктов, в соответствующем соотношении, используемом в этом приготовлении, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 2853, 1743, 1655, 1576, 1522, 1481, 1458, 1414, 1363, 1255.
Пример 122. (S)-1-(2-{4-трет-Бутил-3-{2-(9H-ксантен-9-ил)ацетамидо]-фенил}этил)- 2-циклогексилэтил гидроглутарат (соединение N 1-2875)
Следуя способу, подобному описанному в приготовлении 56, но используя (S)-N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 102) и глутаровый ангидрид в качестве исходных продуктов, в соответствующем соотношении, используемом в этом приготовлении, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 2853, 1730, 1709, 1655, 1649, 1641, 1578, 1524, 1479, 1458, 1254.
Пример 123. Натрий (S)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)-ацетамидо]фенил}этил)- 2-циклогексилэтил глютарат (соединение N 1-1103)
Следуя способу, подобному описанному в примере 70, но используя (S)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)- 2-циклогексилэтил гидроглютарат (полученный, как описано в примере 122) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 29224, 2853, 1720, 1655, 1576, 152226, 1479, 1458, 1410, 1302, 1254.
Пример 124. N-[2-Изопропил-6-(6-циклогексил-3-оксо-2-гексенил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2976)
Суспензию, содержащую 239 г (0,620 ммоль) N-(2-изопропил-6-формилфенил)-2-(9H-ксантен-9-ил)ацетамида (полученный, как описано в приготовлении 29), 347 мг (0,682 ммоль) (5-циклогексил-2-оксопентил)трифенилфосфоний бромида и 66 мг (0,652 ммоль) триэтиламина в 10 мл тетрагидрофурана, нагревают с обратным холодильником в течение 4 ч. По истечении этого времени реакционную смесь охлаждают до комнатной температуры и затем разбавляют диэтиловым эфиром; разбавленную смесь затем промывают разбавленной соляной кислотой. Органическую фазу отделяют и промывают водой до нейтральной реакции, после чего высушивают и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 25 г силикагеля, используя 4:1:1 по объему смесь гексана, метиленхлорида и этилацетата в качестве элюента, с получением 223 мг (выход 67%) указанного в заголовке соединения в виде кристаллов, плавящегося при 190 - 1191oC (после перекристаллизации из смеси метиленхлорида и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 2851, 1655, 1634, 1578, 1518, 1479, 1458, 1300, 1255.
Пример 125. N-[2-Изопропил-6-(6-циклогексил-3-гидроксигексил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1387)
Следуя способу, подобному описанному в примере 8, но используя N-2-изопропил-6-(6-циклогексил-3-оксо-2-гексенил)фенил- 2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 124) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, исходный продукт каталитически гидрируют, после чего кетогруппу восстанавливают до гидроксильной группы тем же способом, что описан в примере 9, с получением указанного в заголовке соединения в виде кристаллов, плавящихся при 142 - 143oC (после перекристаллизации из смеси диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 29224, 22851, 1655, 1634, 1578, 1518, 1479, 1458, 1300, 1255.
Примеры 126 - 152.
Повторяют способ, описанный в примере 35 или 21, за исключением того, что в качестве исходного продукта используют соответствующее производное анилина, в соответствующем количестве, используемом в этом примере, с получением соединений, определенных в следующей таблице 8. Номера соединений (N соед.), указанные в табл. 8, являются номерами, используемыми для соединений табл. 1 - 5. В этих и последующих таблицах используется следующая аббревиатура:
Эфир - Диэтиловый эфир
Спектр - Инфракрасный абсорбционный спектр
MI - Метиленхлорид
iPr2O - Диизопропиловый эфир
Hxa - Гексан
AcOE - Этилацетат
AT - Ацетон
Другие аббревиатуры определены здесь по отношению к табл. 1 - 5.
В колонке "т. пл.", когда продукт не является кристаллическим, указана его физическая форма, например "масло".
Примеры 159 - 161. Повторяют способ, описанный в примере 14, за исключением того, что в качестве исходного продукта используют N-(2-изопропил-6-формилфенил)-2-(9H-ксантен-9-ил)ацетамид, в соответствующем количестве, подобном используемому в этом примере, с получением соединений, представленных в табл. 10.
Примеры 162 - 188. Повторяют способ, описанный в примере 22, за исключением того, что в качестве исходного продукта используют соединение, полученное в соответствующем примере 126 - 152 в соответствующем количестве, подобном используемому в этом примере, с получением соединений, представленных в следующей таблице 11, восстановлением кетогруппы.
1) определенное методом Нуйола
2) вызванное полиморфизмом
Пример 189. (S)-N-[2-трет-Бутил-5-(6-циклогексил-3 -гидроксигексил)-фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-49)
Следуя способу, подобному описанному в примере 40, но используя (S)-N-[2-трет-бутил-5-[6-циклогексил-3-(трет-бутилдиметилсилилокси-гексил] фенил} -2-(9H-ксантен-9-ил)ацетамид в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 125-127oC (после перекристаллизации из диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1658, 1518, 1480, 1458, 1256, 763, 756.
[α]
Пример 190. (R)-N-[2-трет-Бутил-5-(6-циклогексил-3 -гидроксигексил)-фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-49)
190 (I) (R)-N-[2-трет-Бутил-5-(6-циклогексил-3 -бензоилоксигексил)фенил] -2-(9H-ксантен-9-ил)ацетамид.
Следуя способу, подобному описанному в приготовлении 24 (II), но используя (S)-N-[2-трет-бутил-5-(6-циклогексил-3 -гидроксифенил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 189) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
190 (II) (R)-N-[2-трет-Бутил-5-(6-циклогексил-3 гидроксигексил)фенил]-2-(9H-ксантен-9-ил)ацетамид
5 мл I н. раствора гидроксида натрия добавляют к раствору 460 мг (0,70 ммоль) (R)-N-[2-трет-бутил-5-(6-циклогексил-3 -бензоилоксигексил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в стадии (I) примера 189) и полученную смесь перемешивают в течение 2 ч. По истечении этого времени из реакционной смеси удаляют растворитель отгонкой при пониженном давлении, разбавляют затем диэтиловым эфиром, и остаток сначала разбавляют этилацетатом и затем подкисляют 2 н. водной соляной кислотой. Органическую фазу отдаляют и затем промывают водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, в этом порядке. Смесь высушивают над безводным сульфатом натрия и растворитель далее удаляют отгонкой при пониженном давлении. Полученный остаток очищают перекристаллизацией из диизопропилового эфира с получением 180 мг (выход 47%) указанного в заголовке соединения в виде кристаллов, плавящихся при 128-129oC.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1658, 1515, 1480, 1458, 1255, 758.
[α]
Пример 191. (S)-N-[2-трет-Бутил-5-(7-циклогексил-3 -гидроксигептил)-фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-73)
Смесь 1,20 г (1,96 ммоль) (S)-N-{ 2-трет-бутил-5-[7-циклогексил-3- (метоксиметокси)гептил]фенил}-2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в приготовлении 43) в 12 мл 4 н. раствора хлористого водорода в диоксане перемешивают 30 мин. и затем растворитель удаляют отгонкой при пониженном давлении, pH раствора устанавливают до 7 добавлением водного раствора гидрокарбоната натрия, и полученную смесь дважды экстрагируют этилацетатом. Объединенные экстракты промывают насыщенным водным раствором хлорида натрия. Промытые экстракты высушивают и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографии на силикагеле, используя 4: 1 по объему смесь гексана и этилацетата в качестве элюента, с получением 0,9 г указанного в заголовке соединения в виде масла. Масло растирают в гексане и полученный кристаллический продукт собирают с получением 0,83 г (выход 75%) указанного в заголовке продукта в виде кристаллов.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1656, 1524, 1479, 158, 1255, 759.
[α]
Пример 192. (R)-N-[2-трет-Бутил-5-(7-циклогексил-3 -гидроксигептил)-фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-73).
Следуя способу, подобному описанному в примере 190 (I), но используя (S)-N-[2-трет-бутил-5-(7-циклогексил-3 гидроксигептил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 191) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают (R)-бензоилпроизводное исходного продукта. Его гидролизуют способом, подобным описанному в примере 190 (II), с получением указанного в заголовке соединения в виде кристаллов, плавящихся при 100-102oC (после перекристаллизации из гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1656, 1524, 1479, 1458, 1255, 759.
[α]
Пример 193. N-{2-[3-(2-Этил-1-имидазолил)пропил]оксиметил -6-метил-тиофенил}-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2906)
Следуя способу, подобному описанному в примере 1, но используя 2-этилимидазол в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 159-160oC (после перекристаллизации из смеси гексана и метиленхлорида).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3266, 1676, 1649, 1600, 1576, 1518, 1493, 1481, 1457, 1433, 1366, 1258.
Пример 194. N-{2-[3-(1-Имидазолил)пропил]оксиметил-6-метилсульфонил-метил} -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1989) и N-{2-[3-(1-имидазолил)пропил]оксиметил-6-метилсульфинил-фетил} -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1990)
224 мг (0,71 ммоль) м-хлорбензойной кислоты (55% чистоты) добавляют в течение 3 мин. к раствору 200 мг (0,40 ммоль) N-{2-[3-(1-имидазолил)пропокси]метил-6-метилтиофенил} -2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в примере 1) в 15 мл метиленхлорида, и полученную смесь перемешивают в течение 2,5 ч. По истечении этого времени растворитель удаляют отгонкой при пониженном давлении, и полученный остаток разбавляют этилацетатом. Разбавленный раствор промывают насыщенным водным раствором гидрокарбоната натрия и водой, в этом порядке. Органическую фазу сушат и растворитель удаляют отгонкой при пониженном давлении. Остаток очищают колоночной хроматографией через 12 г силикагеля, используя способ градиентного элюирования смесями метиленхлорида и метанола в соотношениях от 100:1 до 100:6 по объему в качестве элюента, а получением 114 г (выход 54%) менее полярного сульфонильного производного (соединение N 1-1989) и 87 г (выход 42%) более полярного сульфинильного производного (соединение N 1-1990) соответственно, оба в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1:
(соединение N 1-1989) 3249, 1679, 1600, 1577, 1507, 1480, 1458, 1309, 1255, 1127, 961, 760.
(соединение N 1-1990) 3222, 3165, 3108, 1680, 1600, 1576, 1509, 1480, 1457, 1256, 1110, 1096, 1080, 1052, 1034.
Пример 195 N-{2-[3-(1-Имидазолил)пропил]оксиметил-6 -изопропилфенил}-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2907)
Следуя способу, подобному описанному в примере 1, но используя N-[2-изопропил-6-[(3-гидроксипропил)оксиметил] фенил]-2 -(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 31) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 131-132oC (после перекристаллизации из смеси метиленхлорида, диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2960, 1670, 1480, 1460, 1250, 1080.
Пример 196. N-{2-[3-(1-Имидазолил)пропил]оксиметил-6 -изопропилфенил}-2-(9H-ксантен-9-ил)ацетамид гидрохлорид (соединение N 1-1481)
0,39 мл 4 н. раствора хлористого водорода в диоксане добавляют к раствору 192 мг (0,39 ммоль) N-{2-[3-(1-имидазолил)пропил]оксиметил -6-изопропилфенил}-2-(9H-ксантен-9-ил)-ацетамида (полученного, как описано в примере 195) в 2 мл метиленхлорида, и из полученной смеси удаляют растворитель отгонкой при пониженном давлении. Полученный остаток растворяют в небольшом количестве диоксана и, после добавления воды, полученную смесь лиофилизуют с получением 206 мг указанного в заголовке соединения в виде порошка.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 2960, 1670, 1480, 1460, 1250, 1090.
Пример 197. N-{ 2-Изопропил-6-[3-(1-имидазолил)пропил] фенил]-2 -(9H-ксантен-9-ил)ацетамид (соединение N 1-2028)
Следуя способу, подобному описанному в примере 1, но используя N-[2-изопропил-6-(3-гидроксипропил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 30) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 194-195oC (после перекристаллизации из смеси диэтилового эфира, метиленхлорида и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3272, 2963, 1645, 1525, 1480, 1457, 1257, 757.
Пример 198. N-(2-Изопропил-6-[3-(1-имидазолил)пропил]фенил}-2-(9H- ксантен-9-ил)ацетамид гидрохлорид (соединение N 1-2027)
Следуя способу, подобному описанному в примере 196, но используя N-(2-изопропил-6-[3-(1-имидазолил)пропил] фенил} -2-(9H-ксантен- 9-ил)ацетамид (полученный, как описано в примере 197) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 2960, 1660, 1570, 1480, 1458, 1250.
Пример 199. N-(2-Изопропил-6-[2-(1-имидазолил)этил]оксиметилфенил}-2-(OH-ксантен-9-ил) ацетамид (соединение N 1-2908).
Следуя способу, подобному описанному в примере 1, но используя N-[2-изопропил-6-(2-гидроксиэтил)оксиметилфенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в приготовлении 32) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 2960, 1675, 1480, 1460, 1250, 1150, 1080.
Пример 200. N-(2-Изопропил-6-[2-(1-имидазолил)этил]оксиметилфенил}- 2-(9H-ксантен-9-ил)ацетамид гидрохлорид (соединение N 1-1484)
Следуя способу, подобному описанному в примере 196, но используя N-(2-изопропил-6-[2-(1-имидазолил)этил]косиметилфенил}-2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 199) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбирующий спектр (CHCl3), νmax , см-1: 2960, 1670, 1480, 1458, 1250, 1120, 1090.
Пример 201. N-[2-Изопропил-6-(1-имидазолилметил)фенил]-2-(9H-rcfynty -9-илацетамид (соединение N 1-2024)
Суспензию 500 г (1,23 ммоль) N-(2-изопропил-6-хлорметилфенил)- 2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в приготовлении 28 (iv)), 847 мг (12,3 ммоль) имидазола и 277 мг (1,9 ммоль) йодида натрия в 10 мл N, N-диметилформамида перемешивают при 90oC в течение 2 ч, и затем температуру понижают до комнатной. Полученную смесь разбавляют этилацетатом. Разбавленную смесь промывают несколько раз водой и один раз насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом натрия, и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток растирают в смеси диэтилового эфира и гексана, и полученный закристаллизовавшийся продукт собирают с получением 471 мг (выход 88%) указанного в заголовке соединения в виде кристаллов, плавящегося при 193-195" (после перекристаллизации из смеси метанола, диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2961, 1671, 1508, 1475, 1458, 1250, 1236, 758.
Пример 202. N-[2-Изопропил-6-(1-имидазолилметил)фенил] -2-(9H-ксантен- 9-ил)ацетамид гидрохлорид (соединение N 1-2023)
Следуя способу, подобному описанному в примере 196, но используя N-(2-изопропил-6-(1-имидазолилметил)фенил]-2-(9H0ксантен-9-ил) ацетамид (полученный, как описано в примере 201) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 2960, 1670, 1480, 1458, 1250, 1120, 1090.
Пример 203. N-[2-Изопропил-6-(1-бензимидазолилметил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2001)
Следуя способу, подобному описанному в примере 201, но используя бензимидазол в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 244-245oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2961, 1677, 1499, 1480, 1458, 1258, 753, 737.
Пример 204. N-[2-Изопропил-6-(1-бензимидазолилметил)фенил]2-(9h-ксантен-9-ил)ацетамид гидрохлорид (соединение N 1-2002)
Следуя способу, подобному описанному в примере 196, но используя N-[2-изопропил-6-(1-бензимидазолилметил)фенил] -2-(9H-ксантен- 9-ил)ацетамид (полученный, как описано в примере 203) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (CHCl3), νmax , см-1: 2960, 1670, 1576, 1510, 1480, 1458, 1250.
Пример 205. N-(2-Метил-6-[3-(1-имидазолил)пропил]оксиметилфенил}- -2-(9H-ксантен-9-ил)ацетамид гидрохлорид (соединение N 1-2909)
Следуя способу, подобному описанному в примере 35, но используя 2-метил-6-{ [3-(1-имидазолил)пропил] оксиметил} анилин (полученный, как описано в приготовлении 45) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, проводят ацилирование с получением производного амида исходного соединения, которое преобразуют в указанный в заголовке гидрохлорид в виде кристаллов, плавящихся при 109-110oC (после перекристаллизации из смеси этанола и диэтилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1652, 1632, 1576, 1520, 1481, 1481, 1458, 1258, 1086, 763.
Пример 206. N-[2-Изопропил-6-(4-циклогексил-3-гидроксибутил)фенил]-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-1385)
Следуя способу, подобному описанному в приготовлении 19, окисляют N-[2-изопропил-6-(3-гидроксипропил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 30) с получением N-[2-изопропил-6-(3-оксопропил)фенил] -2-(9H-ксантен-9-ил)ацетамида, который затем взаимодействует с реактивом Гриньяра подобно способу, описанному в примере 14, с получением указанного в заголовке соединения в виде кристаллов, плавящихся при 148oC (после перекристаллизации из смеси диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3262, 2923, 1648, 1523, 1481, 1457, 1259, 757.
Пример 207. N-(2-Трет-Бутил-5-[4-циклогексил-3-(гидроксиметил)бутил] фенил}-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1984)
Следуя способу, подобному описанному в примере 22,восстанавливают N-[2-трет-бутил-5-(4-циклогексил-3-формилбутил)фенил] -2- (9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 33) с получением указанного в заголовке соединения в виде кристаллов, плавящихся при 130-131oC (после перекристаллизации из смеси диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2922, 2851, 1736, 1655, 1603, 1578, 1524, 1479, 1458, 1416, 1363, 1255.
Пример 208. N9[2-трет-Бутил-5-(5-циклогексил-1-бензилоксиметил-1- пентенил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1955)
Суспензию 1,36 г (1,87 ммоль) [4-трет-бутил-3-[2-(9H-ксантен-9-ил) ацетамидо] фенил]метилтрифенилфосфоний бромида (полученного, как описано в приготовлении 34) в 15 мл тетрагидрофурана подвергают ледяному охлаждению, и добавляют по каплям в течение более 3 мин 2,10 мл (2, 10 ммоль) тетрагидрофуранового раствора натрий гексаметилдисилазида. Через 35 мин к смеси добавляют раствор 511 мг (1,86 ммоль) 2-бензилокси-5-циклогексил-2-пентанона (полученного, как описано в приготовлении 35) в 5 мл тетрагидрофурана. Полученную смесь перемешивают в течение 10 мин при ледяном охлаждении, 35 мин при комнатной температуре и, наконец, при 50oC в течение 4,5 ч. По истечении этого времени температуру реакционной смеси понижают до комнатной температуры и затем добавляют насыщенный водный раствор хлорида аммония с целью остановить реакцию. Реакционную смесь затем разбавляют этилацетатом и разбавленную смесь промывают водой и насыщенным водным раствором хлорида натрия в данном порядке. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 100 г силикагеля, используя 1:4 по объему смесь этилацетата и гексана в качестве элюента, с получением 781 мг (выход 65%) указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3260, 1651, 1518, 1480, 1457, 1364, 1256, 1096, 1071, 754.
Пример 209. N-(2-трет-Бутил-5-[5-циклогексил-2-(гидроксиметил)- пентил] фенил}-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1979)
Раствор 1,54 г (2,30 ммоль) N-[2-трет-бутил-5-(5-циклогексил- 2-бензилоксиметил-1-пентил)фенил]-2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в примере 208) в 15 мл этанола энергично перемешивают в токе водорода в присутствии 1,54 г 10% по массе палладия на угле при комнатной температуре в течение 260 мин и затем при 40oC в течение 2 ч. По истечении этого времени реакционную смесь фильтруют и фильтрат концентрируют отгонкой при пониженном давлении. Концентрат колоночной хроматографией через 75 г силикагеля, используя 2:3 по объему смесью этилацетата и гексана в качестве элюента. Фракции, содержащие желаемое соединение с небольшим количеством примеси, объединяют и концентрируют при пониженном давлении. Концентрат вновь очищают колоночной хроматографией в тех же условиях, что указаны выше, с получением 1,19 г (выход 90%) указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3260, 1651, 1518, 1480, 1457, 1364, 1256, 1096, 1071, 754.
Пример 210. N-[2-трет-Бутил-5-(7-циклогексил-2-бензилоксиметил-1-гептенил)фенил]-2- (9H-ксантен-9-ил)ацетамид (соединение N 1-1957)
Следуя способу, подобному описанному в примере 208, но используя 1-бензилокси-7-циклогексил-2-гептанон в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 110oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3222, 2925, 2853, 1641, 1536, 1481, 1457, 1258, 964, 759, 753.
Пример 211. N-(2-трет-Бутил-5-[7-циклогексил-2-(гидроксиметил)гептил] фенил}-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1982)
Следуя способу, подобному описанному в примере 209, но используя N-[2-трет-бутил-5-(7-циклогексил-2-бензилоксиметил-1-гептенил)фенил] - 2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 210) в качестве исходного продукта, в соответствующем количестве, используемого в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3396, 3269, 29243, 2851, 1655, 1523, 1481, 1458, 1256, 758.
Пример 212. N-[2-трет-Бутил-5-(4-циклогексил-2-бензилоксиметил-1-бутенил)фенил] -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1954)
Следуя способу, подобному описанному в примере 208, но используя 1-бензилокси-4-циклогексил-2-бутанон в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 2922, 2851, 1643, 1576, 1514, 1479, 1456, 1410, 1365, 1255.
Пример 213. N-[2-трет-Бутил-5-[4-циклогексил-2-(гидроксиметил)бутил] фенил]2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1977)
Следуя способу, подобному описанному в примере 209, но используя N-[2-трет-бутил-5-(4-циклогексил-2-бензилоксиметил-1-бутенил) фенил]2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 212) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2922, 2851, 1655, 1578, 1560, 1524, 1479, 1458, 1419, 1363, 1255.
Пример 214. N-{2-трет-Бутил-5-[4-циклогексилокси-2- -(гидроксиметил)бутил] фенил} -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2186)
Следует способу, подобному описанному в примере 208, но используя 1-бензилокси-4-циклогексилокси-2-бутанон (полученный, как описано в приготовлении 37), его подвергают реакции Виттига. Полученный продукт затем взаимодействует по способу, подобно описанному в примере 209, что дает указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax см-1: 3260, 1653, 1522, 1480, 1459, 1364, 1256, 1096, 1034, 760.
Пример 215. N-[2-трет-Бутил-5-(4-циклогексилокси-3- -гидроксибутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединен N 1-2387)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(4-циклогексилокси-3-трет- бутилдиметилсилилоксибутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный по способу, описанному в приготовлении 11) в качестве исходного продукта, в соответствующем количестве, используемого в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3270, 2932, 1655, 1522, 1481, 1458, 1256, 1118, 1096, 760.
Пример 216. N-[2-трет-бутил-5-(5-циклогексилокси-3-бензилокси-1- -пентенил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединен N 1-1945)
Следуя способу, подобному описанному в примере 208, но используя 1-бензилокси-4-циклогексилоксибутаналь (полученный, как описано в примере 38) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 1655, 1520, 1480, 1459, 1364, 1256, 1096, 758.
Пример 217. N-[2-трет-Бутил-5-(5-циклогексилокси-3- -гидроксипентил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2390)
Следуя способу, подобному описанному в примере 209, восстанавливают N-2-трет-бутил-5-(5-циклогексилокси-3-бензилокси-1- -пентенил)фенил-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 216) с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный адсорбционный спектр (пленка), νmax , см-1: 3250, 1655, 1522, 1480, 1459, 1354, 1256, 1190, 1094, 758.
Пример 218. N-[2-трет-Бутил-5-(4-циклогексилметилокси-3- -гидроксибутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2388)
и
N-{ 2-трет-бутил-5-[3-циклогексилметилокси-3- -(гидроксиметил)пропин] фенил}-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2189(
(I) 4 μл эфирата трифторида бора добавляют к раствору 73 мг (0,165 ммоль) N-[2-трет-бутил-5-2-(оксиран-2-ил)этил] -2-(9H-ксантен- -9-ил)ацетамида (полученного, как описано в приготовлении 39) и 41 мг (0,359 моль) циклогексилметанола в 2 мл метиленхлорида и полученную смесь перемешивают в течение 280 мин. По истечении этого времени ее концентрируют отгонкой при пониженном давлении. Полученное таким образом бесцветное масло в виде остатка растворяют в 2 мл этилацетата, 0,2 мл пиридина и 0,1 мл уксусного ангидрида добавляют к раствору, и полученную смесь оставляют на 45 мин. Реакционную смесь затем разбавляют этилацетатом и разбавленную смесь промывают 2 н. водной соляной кислотой, водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, в этом порядке. Ограниченную фазу сушат над безводным сульфатом магния и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 10 г силикагеля, используя 1:3 по объему смесь этилацетата и гексана в качестве элюента, давая 62 мг (выход 63%) смеси ацетильных производных, содержащей соединение N 1-2188 и соединение N 1-2189 в качестве смолообразного продукта.
(II) 15 мг (0,28 ммоль) метоксида натрия добавляют к раствору 72 мг (0,12 ммоль) смеси продуктов, полученных, как описано в стадии (I) выше, в 2 мл метанола и полученную смесь перемешивают при 50oC в течение 70 мин. По истечении этого времени реакционную смесь концентрируют отгонкой при пониженном давлении, и концентрат разбавляют отгонкой при понижении давлении, и концентрат разбавляют этилацетатом. Разбавленную смесь промывают насыщенным водным раствором хлорида аммония и насыщенным водным раствором хлорида натрия, в этом порядке. Ограниченную фазу сушат над безводным сульфатом магния и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 40 г силикагеля, используя 2:3 по объему смесь этилацетата и гексана в качестве элюента, давая смесь, содержащую менее полярное соединение, которое является соединением N 1-2388, и более полярное соединение, которое является соединением N 1-2189. Смесь отделяют и очищают колоночной хроматографией в тех же условиях, что приведены выше, с получением 44 мг (выход 66%) соединения N 1-2388 и 20 мг (выход 30%) соединения N 1-2189, оба в виде смолообразного продукта.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1:
(соединение N 1-2388) 3390, 3270, 1653, 1522, 1480, 1458, 1256, 1121, 909, 758;
(соединение N 1-2389) 3400, 3270, 1653, 1522, 1480, 1458, 1256, 1117, 909, 758.
Пример 219. N-{2-трет-Бутил-5-[4-(2-циклогексилэтокси)-3- -гидроксибутил]фенил}-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2389)
Следуя способу, подобному описанному в примере 218, но используя 2-циклогексилэтанол в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде стеклообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3270, 1655, 1522, 1480, 1459, 1256, 1094, 758.
Пример 220. α -(S)-1-(2-{4-трет-Бутен-3-[2-(9H-ксантен-9-ил) ацетамидо] фенил}этил)-2-циклогексилэтид L-аспартат гидрохлорид (соединение N 1-1548)
Следуя способу, подобному описанному в приготовлении 25, но используя дихлоргексиламиновую соль α -трет-бутоксикарбонил-L- -аспартата и (S)-N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2-(9H- ксантен-9-ил)ацетамид (полученный, как описано в примере 102) в качестве исходных продуктов, в соответствующем соотношении, используемом в этом примере, получают свободное основание, соответствующее указанному в заголовке соединению, 3 мл 4 н. раствора хлористого водорода в диоксане добавляют к 292 мг (0,366 ммоль) свободного основания и полученную смесь оставляют при комнатной температуре на 20 ч. По истечении этого времени растворитель удаляют отгонкой при пониженном давлении, и получают 198 мг указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3240, 1746, 1651, 1618, 1482, 1459, 1254, 909, 760.
Пример 221. N-Лизина-(S)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)- 2-циклогексилэтиловый эфир дигидрохлорид (соединение N 1-1549)
Следуя способу, подобному описанному в приготовлении 25, но используя дициклогексиламиновую соль N,N'-ди-трет-бутоксикарбонил-L-лизина и (S)-N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 102) в качестве исходных продуктов, в соответствующем соотношении, используемом в этом примере, получают производное эфирного производного L-лизина. Подвергают снятию защитной группы способом, описанным в примере 220, с получением указанного в заголовке соединения в виде пенообрзаного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3420, 1740, 1642, 1526, 1482, 1457, 1256, 760.
Пример 222. L-Глутамина-(S)-1-2-{ 2-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил}этил- 2-циклогексилэтиловый эфир гидрохлорид (соединение N 1-1550)
Следуя способу, подобному описанному в приготовлении 25, но используя N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 12) и α -бензил N-бензилоксикарбонил-L-глутамат, получают производное глутамата. 293 мг (0,333 ммоль) этого производного глутамата растворяют в 10 мл метанола и затем добавляют 0,88 мл раствора, полученного разбавлением 4 н. раствора хлористого водорода в диоксане в десять раз по отношению к первоначальному объему. Полученную смесь энергично перемешивают в атмосфере водорода и в присутствии 60 мг 10% по массе палладия на угле в течение 1 ч. По истечении этого времени катализатор отфильтровывают и промывают метанолом. Фильтрат и промывные воды объединяют в растворитель удаляют отгонкой при пониженном давлении с получением 220 мг указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3230, 1732, 1651, 1520, 1480, 1458, 1418, 1256, 758.
Пример 223. α -1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил} этил)-2- циклогексилэтил карбоксиметилтиоацетат (соединение N 1-1551)
Раствор 200 мг N-[4-трет-бутил-5-(4-циклогексил-3-{9-[(бензилоксикарбонил)метилтио] ацетокси} бутил)фенил] -2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в приготовлении 40) в 10 мл метанола смешивают с 0,44 мл муравьиной кислоты и полученную смесь энергично перемешивают при нагревании при 50oC в течение 3 ч в присутствии 200 мг палладиевой черни. По истечении этого времени реакционной смеси дают остыть до комнатной температуры. Реакционную смесь затем фильтруют с помощь фильтра Целита (торговая марка) и катализатор промывают матанолом. Фильтрат и промывание воды объединяют, и растворитель удаляют отгонкой при пониженном давлении.
Полученный остаток очищают колоночной хроматографией через 10 г силикагеля, используя метод градиентного элюирования смесью метиленхлорида и метанола, изменяющийся от 100:5 до 100:15 по объему в качестве элюента, с получением 121 мг (выход 69%) указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1726, 1602, 1577, 1481, 1458, 1416, 1256, 760.
Пример 224. Натриевая соль α -1-(2-{4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-2- циклогексилэтил карбоксиметилтиоацетата (соединение N 1-1552)
Следуя способу, подобному описанному в примере 70, но используя α -1-(2-{ 4-трет-бутил-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-2- циклологексилэтил карбоксиметилтиоацетат (полученный, как описано в примере 223) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1722, 1641, 1602, 1481, 1457, 1393, 1292, 1257, 760.
Пример 225. α-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-2-циклогексилэтил карбоксиметилсульфонилацетат (соединение N 1-1553)
Следуя способу, подобному описанному в примере 223, но используя N-[2-трет-бутил-5-(4-циклогексил-3-{2-[(бензилоксикарбонил)метилсульфонил] ацетокси}бутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 41), получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 2925, 1733, 1633, 1480, 1458, 1398, 1320, 1255, 760.
Пример 226. Натриевая соль α -1-(2-{4-трет-бутил-3-[2-9H-ксантен-9-ил)ацетамидо] фенил} этил)-2-циклогексилэтил карбоксиметилсульфонилацетата (соединение N 1-1554)
Следуя способу, подобному описанному в примере 70, но используя α-1-(2-{ 4-трет-бутил-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил-2 -циклогексилэтил карбоксиметилсульфонилацетат (полученный, как описано в примере 225) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 1732, 1641, 1480, 1458, 1383, 1318, 1256, 760.
Пример 227. α -1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетемидо]фенил} этил)-2-циклогексилэтил карбоксиметилсульфинилацетат (соединение N 1-1555)
Следуя способу, подобному описанному в примере 223, но используя N-[2-трет-бутил-5-(4-циклогексил-3-{ 2-[(бензилоксикарбонил) метилсульфонил]ацетокси} бутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 41), получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 2923, 1729, 1605, 1481, 1458, 1414, 1385, 1298, 1256, 1034, 760.
Пример 228. Натриевая соль α-1-(2-{4-трет-бутил-3-[2-(9H-ксанрен-9-ил)ацетамидо] фенил} этил) -2-циклогексилэтил карбоксиметилсульфинилацетата (соединение N 1-1556)
Следуя способу, подобному описанному в примере 70, но используя α - 1-(2-{ 4-трет-бутил-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил) -2-циклогексилэтил карбоксиметилсульфинилацетат (полученный, как описано в примере 225) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 2852, 1727, 1640, 1615, 1481, 1458, 1384, 1295, 1257, 1033, 760.
Пример 229. α-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил} этил) -2-циклогексилэтил 4-(морфолинометил)бензоат (соединение N 1-1557)
Следуя способу, подобному описанному в приготовлении 25, но используя N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2- (9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 12) и 4-морфолинометилбензойную кислоту в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, и плавящихся при 123-125oC (после перекристаллизации из диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 1713, 1650, 1480, 1457, 1274, 1258, 1116, 1097, 869, 758.
Пример 230. N-[2-трет-Бутил-5-(3-карбоксиметоксикарбонилокси-4- циклогексилбутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1558)
Следуя способу, подобному описанному в примере 79, N-{2-трет-бутил-5-[3-(бензоилоксикарбонил)метоксикарбонилокси-4-циклогексил- бутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 42), дебензилируют с получением указанного в заголовке соединения в виде кристаллов, плавящихся при 143-145oC (после перекристаллизации из смеси этилацетата и гексана)
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 2924, 2852, 1745, 1629, 1481, 1458, 1366, 1255, 1118, 1097, 760.
Пример 231. Натриевая соль N-[2-трет-бутил-5-(3- карбоксиметоксикарбонилокси-4-циклогексилбутил)фенил]-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-1559)
Следуя способу, подобному описанному в примере 70, но используя N-[2-трет-бутил-5-(3-карбоксиметоксикарбонилокси-4-циклогексил-бутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 230) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединения в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 2924, 2852, 1742, 1626, 1577, 1518, 1481, 1458, 1423, 1257, 760.
Пример 232. N-[2-трет-Бутил-5-[3-(4-карбоксиметилфенокси)карбонилокси-4-циклогексилбутил) фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1560)
Следуя способу, подобному описанному в приготовлении 42, но используя 4-(бензилоксикарбонил)метилфенол и N-[2-трет-бутил-5-(4- циклогексил-3-гидроксибутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 12) в качестве исходных продуктов, в соответствующем соотношении, используемом в этом примере, получают карбонатное производное. Его дебензилируют способом, подобным описанному в примере 79, с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 2924, 2853, 1757, 1710, 1509, 1481, 1458, 1255, 1218, 1193.
Пример 233. Пример 233. Натриевая соль N-[2-трет-бутил-5-[3-(4- карбоксиметилфенокси)карбонилокси-4-циклогексилбутил)фенил] -2- (9H-ксантен-9-ил)ацетамид (соединение N 1-1561)
Следуя способу, подобному описанному в примере 70, но используя N-[2-трет-бутил-5-[3-(4-карбоксиметилфенокси)карбонилокси-4-циклогексилбутил) фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 230) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 2924, 2852, 1756, 1657, 1600, 1577, 1509, 1481, 1458, 1256, 1217, 1198.
Пример 234. N-[2-тpет-бутил-5-[3-(1-карбоксиэтокси)карбонилокси-4- циклогексилбутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1562)
Следуя способу, подобному описанному в приготовлении 42, но используя бензил L-лактат и N-[2-трет-бутил-5-(4-циклогексил-3- гидроксибутил)фенил] -2-(9H-каснтен-9-ил)ацетамид (полученный, как описано в примере 12) в качестве исходных продуктов, в соответствующем соотношении, используемом в этом примере, получают карбонатное производное. Его дебензилируют способом, подобным описанному в примере 79, с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1744, 1480, 1458, 1259, 761.
Пример 235. Натриевая соль N-[2-треб-бутил-5-[3-(1- карбокси)карбонилокси-4-циклогексилбутил)фенил] -2-(9H-ксантен- 9-ил)ацетамид (соединение N 1 -1563)
Следуя способу, подобному описанному в примере 70, но используя N-[2-трет-бутил-5-[3-(1-карбоксиэтокси)карбонилокси-4- циклогексилбутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 234) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3270, 1728, 1614, 1577, 1522, 1481, 1458, 1415, 1366, 1259.
Пример 236 N-{2-трет-Бутил-5-[3-(5-метил-2-оксо1,3-диоксолен-4-ил)- метоксикарбонилокси-4-циклогексилбутил] фенил} -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1564)
Следуя способу, подобному описанному в приготовлении 42, но используя (5-метил-2-оксо-1.3-диоксолен-4-ил)метиловый спирт и N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил]-2- (9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 12 в качестве исходных продуктов, в соответствующем соотношении, используемом в этом примере, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1822, 1745, 1657, 1480, 1458, 1257, 1226, 788, 761.
Пример 237. N-(2трет-Бутил-5-{ 3-[2-(1-имидазолил)ацетокси] -4- циклогексилбутил} фенил)-2-(9H-ксантен-9-ил)ацетамид гидрохлорид (соединение N 1-1565)
(i) N-(2-трет-Бутил-5-{3-[2-(1-имидазолил)ацетокси]-4- циклогексилбутил} фенил)-2-(9H-ксантен-9-ил)ацетамид
Суспензию, содержащую422 мг (0,80 ммоль) N-[2-трет-бутил-5- (4циклогексил-3-гидроксибутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 12), 311 мг (1,51 ммоль) дициклогексилкарбодиимида, 142 μл (1,76 ммоль) пиридина, 20 мг (0,16 ммоль) 4-(N,N-диметиламино)пиридин и 156 мг (0,96 ммоль) гидрохлорида 2-(1-имидазолил) уксусной кислоты в 10 мл метиленхлорида, перемешивают в течение 3 дней. По истечении этого времени нерастворимые продукты отфильтровывают и фильтрат промывают водой и насыщенным водным раствором хлорида натрия, в указанном порядке, после чего высушивают над безводным сульфатом натрия. Растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 75 г силикагеля, используя 1:4 по объему смесь этилацетата и гексана в качестве элюента, с получением 507 мг (количественный выход) указанного в заголовке соединения в виде бесцветного пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2926, 2853, 1748, 1655, 1510, 1480, 1458, 1256, 1080, 758, 731.
(ii) N-(2-трет-Бутил-5-{ 3-[2-(1-имидазолил)ацетокси]-4- циклогексилбутил} фенил)-2-(9H-ксантен-9-ил)-ацетамида [полученный, как описано в стадии (i) выше] в 3 мл диэтилового эфира и образовавшиеся кристаллы собирают фильтрацией и промывают диэтиловым эфиром. Кристаллы очищают жидкой хроматографией высокого разрешения через ODS (120  30 мм • 250 мм), используя 3: 1 по объему смесь ацетонитрила и воды в качестве элюента, с получением геля. Этот гель кристаллизуют растиранием в диэтиловом эфире с получением 170 мг (выход 85%) указанного в заголовке соединения в виде бесцветных кристаллов, плавящихся при 125 - 133oC.
30 мм • 250 мм), используя 3: 1 по объему смесь ацетонитрила и воды в качестве элюента, с получением геля. Этот гель кристаллизуют растиранием в диэтиловом эфире с получением 170 мг (выход 85%) указанного в заголовке соединения в виде бесцветных кристаллов, плавящихся при 125 - 133oC.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 2853, 1746, 1655, 1576, 1522, 1482, 1459, 1258, 1230, 758.
Примеры 238 - 243. Повторяют способ, описанный в приготовлении 25, за исключением того, что в качестве исходного продукта используют соответствующее соединение, полученное в соответствующих примерах 213, 209 или 211 в соответствующем количестве, подобном используемому в этом примере, с получением соответствующего производного бензилового эфира, которое затем дебензилируют способом, подобным описанному в примере 79, с получением соединений по примерам 238, 240 и 242. Соединение примеров 238, 240 и 242 затем преобразуют в их натриевые соли согласно способу, подобному описанному в примере 70 (примеры 239, 241 и 243). Подробности приведены в следующей табл. 12. Аббревиатура объясняется выше ранее.
Пример 244. N-(2-трет-Бутил-5-{3-[2-(карбоксиметокси)ацетокси]-4- циклогексилбутил}фенил)-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1566)
Следуя способу, подобному описанному в приготовлении 25, но используя бензил N-[2-(трет-бутил-5-(4цтклогексил-3- гидроксибутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 12) и 2-(бензилоксикарбонилметокси)уксусную кислоту в качестве исходных продуктов, в соответствующем соотношении, используемом в этом примере, получают производное бензилового эфира. Его затем дебензилируют способом, подобным описанному в примере 79, с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1744, 1652, 1480, 1458, 1253, 1217, 1142, 760.
Пример 245. Натриевая соль N-(2трет-бутил-5-{3-[2- (карбоксиметокси)ацетокси] -4-(циклогексилбутил} фенил-2- (9H-ксантен-9-ил)ацетамид (соединение N 1-1556)
Следуя способу, подобному описанному в примере 70, но используя N-(2-трет-бутил-5-{ 3-[2-(карбоксиметокси)ацетокси] -4- циклогексилбутил}фенил)-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 244) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3249, 1741, 1642, 1615, 1577, 1523, 1481, 1458, 1421, 1365, 1338, 1300, 1255, 1137.
Пример 246. (S)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)-2- циклогексилбутил гидросукцинат (соединение N 1-1127)
Следуя способу, подобному описанному в приготовлении 25, но используя (S)-N-[2-трет-бутил-5-(6-циклогексил-3- гидроксигексил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 189) в качестве исходного продукта, в соответствующем соотношении, используемом в этом примере, получают производное бензилового эфира, Его затем дебензилируют способом, подобным описанному в примере 79, с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1732, 1713, 1656, 1480, 1458, 1253, 1167, 760.
Пример 247. Натриевая соль (S)-1-(2-{4-трет-бутил-3-[2- (9H-ксантен-9-ил)ацетамидо]фенил}этил)-4-циклогексилбутил сукцинат (соединение N 1-1120)
Следуя способу, подобному описанному в примере 70, но используя (S-1-(2-{ 4трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил}этил)-4- циклогексибутил гидросукцинат (полученный, как описано в примере 246) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3270, 1726, 1656, 1578, 1524, 1481, 1458, 1415, 1365, 1301, 1256.
Пример 248. (R)-1-(2-{ 4-трет-Бутил-3-[2-(9H-ксантен-9- ил)ацетамидо] фенил}этил)-4-циклогексилбутил гидросукцинат (соединение N 1-1127)
Следуя способу, подобному описанному в приготовлении 79, но используя (R)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил}этил)- 4-циклогексилбутил бензилсукцинат 9полученный, как описано в приготовлении 25), дебензилируют с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1733, 1713, 1655, 1480, 1458, 1253, 1166, 760.
Пример 249. Натриевая соль (R)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен- 9-ил)ацетамидо]фенил}этил)-4-циклогексилбутил сукцинат (соединение N 1-1120)
Следуя способу, подобному описанному в примере 70, но используя (R)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)- 4-циклогексилбутил гидросукцинат (полученный, как описано в примере 248) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3270, 1728, 1656, 1577, 1523, 1481, 1416, 1365, 1301, 1255.
Пример 250. (S)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9- ил)ацетамидо]фенил}этил)-5-циклогексилпентил гидросукцинат (соединение N 1-1136)
Следуя способу, подобному описанному в приготовлении 25, но используя (S)-N-[2-трет-бутил-5-(7-циклогексил-3-гидроксигептил)фенил] -2- (9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 191) в качестве исходного продукта, в соответствующем соотношении, используемом в этом примере, получают производное бензилового эфира. Его затем дебензилируют способом, подобным описанному в примере 79, с получением указанного в заголовке соединения в виде кристаллов, плавящихся при 93-95oC (после перекристаллизации из гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1732, 1713, 1610, 1480, 1458, 1252, 1166, 760.
Пример 251. Натриевая соль (S)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен- 9-ил)ацетамидо]фенил}этил)-5-циклогексилпентил сукцинат (соединение N 1-1129)
Следуя способу, подобному описанному в примере 70, но используя (S)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил}этил)-5- циклогексилпентил гидросукцинат (полученный, как описано в примере 250) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3274, 1726, 1656, 1578, 1523, 1481, 1458, 1416, 1365, 1300, 1256.
Пример 252. (R)-1-(2-{4-трет-Бутил-3-[2-(9H-ксантен-9- ил)ацетамидо]фенил}этил-5-циклогексилпентил гидросукцинат (соединение N 1-1136)
Следуя способу, подобному описанному в приготовлении 25, но используя (R)-N-[2-трет-бутил-5-(7-циклогексил-3-гидроксигептил)фенил]-2- (9H-кесантен-9-ил)ацетамид (полученный, как описано в примере 192) в качестве исходного продукта, в соответствующем соотношении, используемом в этом примере, получают производное бензилового эфира. Его затем дебензилируют способом, подобным описанному в примере 79, с получением указанного в заголовке соединением в виде кристаллов, плавящихся при 95-97oC (после перекристаллизации из гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1732, 1712, 1627, 1480, 1457, 1251, 1221, 1165, 752.
Пример 253. Натриевая соль (R)-1-(2-{4-трет-бутил-3-[2-(9H-ксантен- 9-ил)ацетамидо]фенил}этил)-5-циклогексилпентил сукцинат (соединение N 1-1129)
Следуя способу, подобному описанному в примере 70, но используя (R)-1-(2-{ 4-трет-бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} этил)-5- циклогексилпентил гидросукцинат (полученный, как описано в примере 252) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3273, 1727, 1656, 1578, 1523, 1481, 1458, 1416, 1365, 1300, 1255.
Пример 254. N-{2-трет-Бутил-5-[3-(4-(карбоксиметилбензоилокси)-4- циклогексилбутил]фенил}-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1568)
Следуя способу, подобному описанному в примере 79, N-(2-трет-бутил-5- { 3-[(4-бензилоксикарбонилметил)бензолокси-4-циклогексилбутил] фенил}-2- (9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 44) дебензилируют с получением указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2924, 2853, 1713, 1639, 1612, 1578, 1522, 1458, 1418, 1365, 1275, 1255, 1180, 1117.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц, δ , ppm: 0,81-2,06 (15H, мультиплет); 1,13 (9H, синглет); 2,29-2,45 (4/3H, мультиплет); 2,58-2,76 (8/3H, мультиплет); 3,72 (2H, синглет); 4,72 (1H, триплет, J=7 Гц); 5,23-5,38 (1H, мультиплет); 6,88-7,45 (13H, мультиплет); 8,00 (2H, дублет, J=8 Гц).
Пример 255. Натриевая соль N-{2-трет-бутил-5-[3-(4- (карбоксилметилбензоилокси)-4-циклогексилбутил] фенил} -2-(9H-ксантен- 9-ил)ацетамид (соединение N 1-1569)
Следуя способу, подобному описанному в приготовлении 42, N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2- (9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 12) взаимодействует с дифенилметил-4-гидроксибензоатом с получением карбонатного производного. ,24 мг полученного таким образом производного растворяют в 5 мл метиленхлорида и добавляют к полученному раствору 1 мл анизола и 5 мл трифторуксусной кислоты. Полученную таким образом смесь оставляют стоять 1 ч после чего растворитель удаляют при пониженном давлений. Полученный остаток кристаллизуют растиранием в смеси гексана и диизопропилового эфира. Кристаллы собирают фильтрацией и затем перекристаллизовывают из диизопропилового эфира с получением 403 мг (выход 80%) указанного в заголовке соединения, плавящегося при 139-141oC.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1759, 1715, 1694, 1641, 1605, 1577, 1508, 1481, 1458, 1422, 1366, 1257, 1215.
Пример 258. N-[2-трет-Бутил-5-(4-циклогексил-3- карбамоилоксибутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1572)
64 μл (0,54 ммоль) трихлорацетилизоцианата добавляют к раствору 272 мг (0,52 ммоль) N-[2-трет-бутил-5-(4-циклогексил-3- гидроксибутил)фенил]-2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в примере 12) в 6 мл бензола, и полученную смесь перемешивают в течение 10 мин. По истечение этого времени добавляют метанольный раствор карбоната кальция при перемешивании. Реакционную смесь смешивают с водой и органическую фазу отделяют и высушивают. Растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 50 г силикагеля, используя 15: 85 по объему смесь этилацетата и метиленхлорида в качестве элюента, давая 135 мг (выход 46%) указанного в заголовке соединения в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3389, 2924, 1715, 1575, 1480, 1458, 1333, 1256, 760, 733.
Пример 259. N-[2-трет-Бутил-5-(3-гидроксидодецил)фенил] -2- (9H-ксантен-9-ил)ацетамид (соединение N 1-2910)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(3-трет-бутилсилилоксидодецил)фенил] -2- (9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 11) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде аморфного твердого продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2953, 2926, 2855, 1655, 1578, 1524, 1479, 1458, 1414, 1363, 1300, 1255.
Пример 260. N-[2-трет-Бутил-5-(4-циклогексил-3-гидробутил)фенил]- 2,2-диметилпропанамид (соединение N 5-118)
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутил-5-(3-трет-бутилдиметилсилилокси-4-циклогексил)фенил] - 2,2-диметилпропанамид (полученный по способу, подобному описанному в приготовлении 11) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пены.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2957, 2922, 2853, 1655, 1508, 1481, 1448, 1416, 1363, 1200, 1165.
Пример 261. N-(2-трет-Бутил-5-{ 4-циклогексил-3-[(4- карбоксифенил)карбонилокси] бутил} фенил)--2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1573)
Следуя способу, подобному описанному в примере 79, но используя N-(2-трет-бутил-5-{ 4-циклогексил-3-[(4- бензилоксикарбонилфенил)карбонилокси] бутил} фенил)-2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 25) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пены.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1718, 1699, 1650, 1481, 1458, 1410, 1256, 1118, 760.
Пример 262. Натриевая соль N-(2-трет-бутил-5-{4-циклогексил-3-[(4-карбоксифенил)карбонилокси]бутил}фенил) -2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1574)
Следуя способу, подобному описанному в примере 70, но используя N-(2-трет-бутил-5-[4-циклогексил-3-[(4-карбоксифенил)карбонилокси] бутил} фенил)- 2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 261) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2923, 1715, 1481, 1397, 1273, 1259, 1117, 760, 742.
Пример 263. N-(2-трет-Бутил-5-{7-циклогексил-3-[2-(карбоксиметокси) ацетокси]гептил}фенил)-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1932)
Следуя способу, подобному описанному в примере 79, но используя N-(2-трет-бутил-5-{ 7-циклогексил-3-[2-(бензилоксикарбонилметокси)ацетокси] гептил} фенил)-2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 25) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1727, 1655, 1649, 1578, 1522, 1482, 1459, 1420, 1256, 1138, 758.
Пример 264. Натриевая соль N-(2-трет-бутил-5-{ 7-циклогексил-3-[2-(карбоксиметокси)ацетокси] гептил} фенил)-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1933)
Следуя способу, подобному описанному в примере 70, но используя N-(2-трет-бутил-5-{ 7-циклогексил-3-[2-(карбоксиметокси)ацетокси] гептил} фенил)- 2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 263) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1740, 1651, 1611, 1518, 1256, 1140, 876, 759.
Пример 265. N-{ 2-[3-(1-Имидазолил)пропоксиметил]-6-метоксифенил}-2-(9H-ксантен-9-ил)ацетамид (соединение N 1-2077)
Следуя способу, подобному описанному в примере 35, но используя 2-[3-(1-имидазолил)пропоксиметил]-6-метоксианилин (полученный по способу, подобно описанному в приготовлении 45) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 155-156oC (после перекристаллизации из этилацетата).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1676, 1553, 1479, 1471, 1462, 1367, 1261, 1246, 1081, 760.
Пример 266. N-{2-[3-(1-Имидазолил)пропоксиметил]-6-метокси-фенил]-2-(9H-ксантен-9-ил) ацетамид гидрохлорид (соединение N 1-2076)
Следуя способу, подобному описанному в примере 2, но используя N-{2-[3(1-имидазолил)пропоксиметил] -6-метоксифенил]-2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 265) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3397, 3274, 1652, 1527, 1483, 1458, 1286, 1260, 1080, 761.
Пример 267. N-(2-трет-Бутил-5-{3-циклогексил-3-[2-(карбоксиметокси)ацетокси] пропил} фенил)- 2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1848)
Следуя способу, подобному описанному в примере 79, но используя N-(2-трет-бутил-5-{ 3-циклогексил-3-[2-(бензилоксикарбонилметокси)ацетокси] пропил} фенил)-2-(9H-ксантен-9-ил)ацетамид (полученный по способу, подобному описанному в приготовлении 25) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 128-129oC (после перекристаллизации из диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3292, 1750, 1656, 1481, 1458, 1255, 1215, 1139, 760.
Пример 268. Натриевая соль N-(2-трет-бутил-5-{ 3-циклогексил-3-[2-(карбоксиметокси)ацетокси] пропил} фенил)- 2-(9H-ксантен-9-ил)ацетамид (соединение N 1-1849)
Следуя способу, подобному описанному в примере 70, но используя N-(2-трет-бутил-5-{ 3-циклогексил-3-[2-(карбоксиметокси)ацетокси] пропил} фенил-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 267) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде пены.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3419, 3278, 1741, 1651, 1614, 1481, 1458, 1421, 1256, 1222, 1138, 760.
Пример 269. N-[2-трет-Бутил-5-(1-имидазолилметил)фенил] -2-(9H- ксантен-9-ил)ацетамид (соединение N 1-2922)
Следуя способу, подобному описанному в приготовлении 21(I), но используя N-(2-трет-бутил-5-гидроксиметилфенил)-2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в приготовлении 14) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают метансульфонильное производное. Его преобразуют в указанное в заголовке соединение в виде кристаллов, плавящихся при 222-223oC (после перекристаллизации из смеси метанола, метиленхлорида и гексана), следуя способу, подобному описанному в примере 1(II).
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , pppm: 1,16 (9H, синглет); 2,70 (2H, дублет, J=7 Гц); 4,69-4,80 (1H, мультиплет); 5,09 (2H, синглет); 6,79-7,57 (15H, мультиплет).
Пример 270. N-[2-трет-Бутил-5-(1-имидазолилметил)фенил]-2-(9H-ксантен-9-ил)ацетамид гидрохлорид (соединение N 1-2919)
Следуя способу, подобному описанному в примере 196, но используя N-[2-трет-бутил-5-(1-имидазолилметил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 269) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,16 (9H, синглет); 2,75 (2H, дублет, J=7 Гц); 4,68-4,73 (1H, мультиплет); 5,35 (2H, синглет); 7,01-7,46 (13H, мультиплет); 7,62 (1H, уширенный синглет); 9,01 (1H, уширенный синглет).
Пример 271. N-{ 2-трет-Бутил-5-[(2-этил-1-имидазолил)метил]фенил}-2-(9H-ксантен-9-ил) ацетамид (соединение N 1-2921)
Следуя способу, подобному описанному в примере 269, но используя 2-этилимидазол вместо имидазола в соответствующем количестве, получают указанное в заголовке соединение в виде аморфного твердого продукта.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,16 (9H, синглет); 1,21-1,37 (3H, мультиплет); 2,52-2,71 (4H, мультиплет); 4,70-4,80 (1H, мультиплет); 5,03 (2H, синглет); 6,72-7,43 (14H, мультиплет).
Пример 272. N-{ 2-трет-Бутил-5-[(2-этил-1-имидазолил)метил]фенил}-2-(9H-ксантен-9-ил) ацетамид гидрохлорид (соединение N 1-2920)
Следуя способу, подобному описанному в примере 196, но используя N-[2-трет-бутил-5-(2-этил-1-имидазолилметил)фенил] -2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в примере 271) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде порошка.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,15 (9H, синглет); 1,48 (1H, триплет, J=7,5 Гц); 2,75 (2H, дублет, J=7 Гц); 3,07-3,16 (2H, мультиплет); 4,70 (1H, триплет, J=7 Гц); 5,16 (2H, синглет); 6,81 (1H, дублет, J=7,5 Гц 6,93-7,42 (12H, мультиплет); 7б56 (1H, уширенный синглет).
Приготовление 1
2-[(3-Гидроксипропокси)метил]-6-метилтио-1-нитробензол
I(I) 2-Мезилоксиметил-6-метилтио-1-нитробензол
76 мл (0,66 ммоль0 мезилхлорида и затем 69 мг (0,68 ммоль) триэтиламина добавляют к 5 мл метиленхлоридного раствора, содержащего 1-- мг (0,50 ммоль) 2-гидроксиметил-6-метилтио-1-нитробензола на ледяной бане. Смесь перемешивают при температуре ледяной бани в течение 75 мин. По источении этого времени к реакционной смеси добавляют воду и смесь экстрагируют этилацетатом. Органический экстракт отделяют 2 H, водной соляной кислотой и затем водой. Растворитель удаляют отгонкой при пониженном давлении с получением указанного в заголовке соединения. Этот продукт используют в следующей стадии без очистки.
I(II) 2-[(3-Гидроксипропокси)]-6-метилтио-1-нитробензол
33 мг (0,76 ммоль) гидрида натрия (в виде 55% по массе суспензии в минеральном масле) промывают дважды гексаном и затем к суспензии добавляют 1 мл диметилформамида. Полученного суспензию охлаждают на ледяной бане и затем прибавляют 0,8 мл (11 моль) 1,3-пропандиола. Смесь перемешивают в течение 30 мин при этой температуре и затем 30 мин при комнатной температуре. К полученному раствору 1 мл диметилформамидного раствора, содержащего весь 2-мезилоксиметил-6-метилтио-1-нитробензол, полученный, как описано в стадии (i), при ледяном охлаждении, и смесь перемешивают 2 ч при температуре ледяного охлаждения и затем 1,5 ч при комнатной температуре. По истечении этого времени к реакционному раствору добавляют воду и смесь экстрагируют этилацетатом. Органический экстракт промывают несколько раз водой и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 10 г силикагеля, используя смеси метиленхлорида и этилацетат в отношении, изменяющемся от 10:1 до 8:1 по объему, в качестве элюента с получением 95 мг (выход 74%) указанного в заголовке соединения в виде маслянистого продукта.
Инфракрасный абсорбционный спектр (CDCl3), νmax , см-1: 3530, 1590, 1525, 1357.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ , ppm: 1,83 (2H, квинтет, J=6 Гц); 2,46 (3H, синглет); 3,61 (2H, триплет, J=6 Гц); 3,73 (2H, триплет, J=6 Гц); 4,57 (2H, синглет); 7,25-7,6 (3H, мультиплет).
Приготовление 2
2-[(3-трет-Бутилдиметилсилилоксипропокси)метил]-6 метилтио-1-нитробензол
927 мг (6,15 ммоль) трет-бутилдиметилсилил хлорида, 622 мг (6,15 ммоль) триэтиламина и 60 мг (0,49 ммоль) 4-(N, N-диметиламино)пиридина добавляют к 20 мл метиленхлоридного раствора, содержащего 1,044 г (4,06 ммоль) 2-[(3-гидроксипропокси)метил] -6-метилтио-1-нитробензола (полученного, как описано в приготовлении 1). Смесь перемешивают в течение ночи при комнатной температуре. По истечении этого времени реакционный раствор разбавляют метиленхлоридом и промывают разбавленной водной соляной кислотой и затем водой. Затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 40 г силикагеля. Элюирование смесями метиленхлорида и гексана в отношении, изменяющемся от 1:1,5 до 1:1 по объему, приводит к получению 1,26 г (выход 83%) указанного в заголовке соединения в виде маслянистого продукта.
Инфракрасный абсорбционный спектр (CHCl), νmax , см-1: 1590, 1527, 1358.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ , ppm: 0,04 (6H, синглет); 0,88 (9H, синглет); 1,78 (2H, квинтет, J=6 Гц); 2,46 (3H, синглет); 3,53 (2H, триплет, J= 6 Гц); 3,68 (2H, триплет, J=6 Гц); 4,54 (2H, синглет); 7,25-7,5 (3H, мультиплет).
Приготовление 3.
N-{ 2-[(3-трет-Бутилдиметилсилилоксипропокси)метил] -6-метил-тиофенил} - 2-(9H-ксантен-9-ил)ацетамид
(i) 2-[(3-трет-Бутилдиметилсилилоксипропокси)метил]-6- метилтиоанилин
3,53 мг (54 ммоль) цинка и 0,5 мл уксусной кислоты добавляют к 20 мл метанольного раствора, содержащего 1,25 г (3,37 ммоль) 2-[(3-трет-бутилдиметилсилилоксипропокси)метил] - 6-метилтио-1-нитробензола (полученного, как описано в приготовлении 2) на ледяной бане. Смесь затем перемешивают 50 мин, после чего разбавляют этилацетатом. Смесь затем фильтруют с помощью фильтра Цеолита (торговая марка).
Нерастворимый осадок промывают этилацетатом. Фильтрат и промывные воды объединяют, концентрируют упариванием при пониженном давлении приблизительно до 5 мл и опять разбавляют этилацетатом; затем добавляют насыщенный водный раствор гидрокарбоната натрия. Желаемое соединение распределяют между органическим растворителем и раствором гидрокарбоната. Органический слой, содержащий осадок и водный слой, фильтруют, вновь используют фильтр Цеолита, и нерастворимый продукт промывают этилацетатом. Органические слои объединяют и промывают водой. Растворитель затем удаляют отгонкой при пониженном давлении с получением 1,10 г (выход 96%) указанного в заголовке соединения в виде маслянистого продукта.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ , ppm: 0,04 (6H, синглет); 0,88 (9H, синглет), 1,89 (2H, квинтет, J=6 Гц); 2,31 (3H, синглет); 3,51 (2H, триплет, J= 6 Гц); 3,68 (2H, триплет, J=6 Гц); 4,48 (2H, синглет); 4,9 (2H, уширенный синглет); 6,60 (1H, триплет, J=7,5 Гц); 6,98 (1H, дублет дублетов, J=7,5 b 1,5 Гц); 7,35 (1H, дублет дублетов, J=7,5 т 1,5 Гц).
(II) N-{ 2-[3-трет-Бутилдиметилсилилоксипропокси)метил]- 6-метил-тиофенил]-2-(9H-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в примере 21, вес 2-[3(3-трет-бутилдиметилсилилоксипропокси)метил]-6-метилтиоанилин, полученный, как описано в стадии (I) выше, ацилируют, получая указанное в заголовке соединение в виде кристаллов, плавящихся при 185,5-186oC (после перекристаллизации из смеси метиленхлорида и метанола) с выходом 77%.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3263, 1648, 1517, 1260, 1097.
Спектр ядерного магнитного резонанса (CDCl3, 60 МГц), δ , ppm: 0,01 (6H, синглет); 0,84 (9H, синглет); 1,67 (2H, квинтет, J=6 Гц); 2,30 (3H, синглет); 2,85 (2H, дублет, J=7 Гц); 3,28 (2H, триплет, J=6 Гц); 3,63 (2H, триплет, J=6 Гц); 4,09 (2H, синглет); 4,72 (1H, триплет, J=7 Гц); 6,9-7,5 (11H, мультиплет).
Приготовление 4.
N-[2-[(3-Гидроксипропокси)метил] -6-метилтиофенил] -2- (9H-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в примере 40, N-{2-[(3-трет-бутилдиметилсилилоксипропокси)метил] -6-метилтиофенил} -2- (9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 3) подвергают снятию защитной силильной группы с получением указанного в заголовке соединения в виде кристаллов, плавящихся при 207-208oC (после перекристаллизации из ацетона) с количественным выходом.
Инфракрасного абсорбционный спектр (KBr), νmax , см-1: 3398, 1649, 1514, 1482, 1259.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,71 (2H, квинтет, J=5,5 Гц); 1,86 (1H, триплет, J=5,5 Гц); 2,37 (3H, синглет); 2,80 (2H, дублет, J=7 Гц); 3,32 (2H, триплет, J=5,5 Гц); 3,69 (2H, квартет, J=5,5 Гц); 4,09 (2H, синглет); 4,73 (1H, триплет, J=7 Гц); 7,0-7,5 (11H, мультиплет).
Приготовление 5.
Этил-5-циклогептил-3-оксовалерат
343 мг (2,12 ммоль) карбонилдиимидазола добавляют к 4 мл ацетонитрилового раствора, содержащего 300 мг (1,76 ммоль) 3-циклогептилпропионовой кислоты. Затем смесь перемешивают 1 ч при 40oC для получения активного эфира.
Отдельно 273 мг (1б06 ммоль) комплекса магний бромида с диэтиловым эфиром добавляют к 4 мл тетрагидрофурановой суспензии, содержащей 360 мг 92,12 ммоль) этилкалиймалоната, на ледяной бане. Затем смесь перемешивают 1 ч при температуре ледяной бани и затем 1 ч при комнатной температуре. По истечении этого времени ацетонитриловый раствор, содержащий активный эфир, который получен как описано выше, добавляют по каплям при комнатной температуре за период времени более 5 мин к полученной суспензии, которая содержит магниевую соль моно-эфира малоновой кислоты. Когда прибавление по каплям завершается, смесь перемешивают затем 1 ч при 60oC. Реакционный раствор далее разбавляют диэтиловым эфиром и промывают насыщенным водным раствором гидрокарбоната натрия, разбавленным водной соляной кислотой, водой и насыщенным водным раствором хлорида натрия, в этом порядке. Затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 20 г силикагеля. Элиюрирование одним метиленхлоридом приводит к получению 376 мг (выход 87%) указанного в заголовке соединения в виде маслянистого продукта.
Инфракрасный абсорбционный спектр (чистый), νmax см-1: 1733, 1710, 1305, 1230, 1030.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,06-1,73 (15H, мультиплет); 1,26 (3H, триплет, J=7 Гц); 2,54 (2H, триплет, J=7,5 Гц); 3,44 (2H, синглет); 4,20 (2H, квартет, J=7 Гц).
Приготовление 6.
2-трет-Бутил-5-(5-циклогептил-3-оксопентил)-1-нитробензол 454 мг (4,06 ммоль) трет-бутоксида калия добавляют к 10 мл тетрагидрофуранового раствора, содержащего 1,00 г (4,16 ммоль) этил-5-циклогептил-3-оксовалерата (полученного, как описано в приготовлении 5). Затем смесь перемешивают 10 мин для получения соответствующей калиевой соли. 1,1 г (3,47 ммоль) 2-трет-бутил-5-йодметил-1- нитробензола (полученного, как описано в приготовлении 51) добавляют к смеси при охлаждении на ледяной бане, и смесь перемешивают 2 ч при температуре ледяной бани. Реакционный раствор разбавляют диэтиловым эфиром и промывают водой. Растворитель отгонкой при пониженном давлении, и полученный остаток растворяют в 1 о мл этанола. Добавляют 1,73 мл 2 н. водного раствора гидроксида натрия и смесь нагревают в течение 4 ч при кипячении с обратным холодильником. Реакционную смесь охлаждают на ледяной бане, добавляют достаточное количество водной соляной кислоты для достижения значения смеси pH 5,0 и смесь нагревают затем с обратным холодильником в течение 2 ч при кипячении с обратным холодильником. По истечении этого времени реакционный раствор разбавляют диэтиловым эфиром и промывают водой. Растворитель удаляют отгонкой при пониженном давлении и полученный остаток подвергают колоночной хроматографии через 70 г силикагеля. Элюирование 2:1 по объему смесью метиленхлорида и гексана приводит к получению 850 кг (выход 68%) указанного в заголовке соединения в виде маслянистого продукта.
Инфракрасный абсорбционный спектр (чисты), νmax см-1: 1703, 1525, 1362.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,06-1,23 (2H, мультиплет); 1,25-1,71 (13H, мультиплет); 1,38 (9H, синглет);2,39 (2H, триплет, J= 7,5 Гц); 2,73 (2H, триплет, J=7Гц); 2,88 (2H, триплет, J= 7Гц); 7,12(1H,дублет, J=2Гц); 7,26 (1H, дублет дублетов, J= 2 и 8 Гц); 7,44 (1H, дублет, J=8 Гц).
Приготовление 7
2-трет-Бутил-5-(5-циклогептил-3-оксопентил)анилин
3,08 мг (47,1 ммоль) порошка цинка и затем 0,32 мл уксусной кислоты добавляют к 16 мл метанольного раствора, содержащего 847 мг (2,36 ммоль) 2-трет-бутил-5-(5-циклогептил-3-оксопентил)-1-нитробензола (полученного, как описано в приготовлении 6) на ледяной бане. Смесь затем перемешивают 1 ч. По истечении этого времени добавляют дополнительно 0,32 мл уксусной кислоты и смесь дополнительно перемешивают 1 ч. Реакционную смесь затем фильтруют с помощью фильтра Целита. Нерастворимый осадок промывают этилацетатом. Фильтрат и промывные воды объединяют, концентрируют до 10 мл упариванием при пониженном давлении и разбавляют этилацетатом для распределения желаемого соединения между органическим растворителем и водой. Для удаления нерастворимого материала растворитель фильтруют, вновь используя фильтр Целита, и нерастворимый продукт промывают опять этилацетатом. Органические слои объединяют и промывают насыщенным водным раствором гидрокарбоната натрия и затем водой. Растворитель затем удаляют отгонкой при пониженном давлении для получения 789 мг (количественный выход) указанного в заголовке соединения в виде маслянистого продукта.
Инфракрасный абсорбционный спектр (чистый), νmax , см-1: 3490, 3375, 1702, 1618, 1419.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ,ppm: 1,05-1,22 (2H, мультиплет); 1,23-1,71 (13H, мультиплет); 1,39 (9H, синглет); 2,39 (2H, триплет, J=7,5 Гц); 2,65-2,81 (4H, мультиплет); 3,65-3,83 (2H, уширенный синглет); 6,47 (1H, дублет, J=2 Гц); 1,05-1,22 (2H, мультиплет); 6,55 (1H, дублет дублетов, J=2 и 8 Гц); 7,14 (1H, J=8 Гц).
Приготовление 8
1-[2-(4-Метилфенил)сульфонил-2-метилтиоэтил] гексан 990 мг (22,7 ммоль) гидрида натрия (в виде 55% по массе суспензии с минеральном масле) промывают дважды гексаном и суспендируют в 40 мл диметилформамида. К этой суспензии прибавляют 4,35 мг (20,1 ммоль) метилтиометил-n-толилсульфона и через 5 мин добавляют 3,1 кг(22,2 ммоль) циклогексилметилбромида. Смеси позволяют достичь комнатной температуры, после чего перемешивают в течение 5 ч.
По истечении этого времени добавляют разбавленный водный раствор аммоний хлорида для остановки реакции. Реакционную смесь экстрагируют диэтиловым эфиром и экстракт промывают насыщенным водным раствором хлорида натрия. Растворитель затем удаляют отгонкой при пониженном давлении. Полученный остаток подвергают коленчатой хроматографии через 100 г силикагеля. Элюирование 1:3 по объему смесью этилацетата и гексана приводит к получению 5,57 г (выход 89%)указанного в заготовке соединения в виде кристаллов, плавящихся при 65-66oC (после перекристаллизации из смеси диизопропилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1595, 1445, 1300, 1146, 1084, 961, 816, 760.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,74-1,34 (5H, мультиплет); 1,46 (1H, дублет дублетов. J=3 и 132 и 13 Гц); 1,50-1,74 (6H, мультиплет); 1,92 (1H, дублет дублетов, J=3 и 9 и 9 и 13 Гц); 2,23 (3H, синглет); 2,46 (3H, синглет); 3,75 (1H, дублет дублетов, J=3 и 12 Гц); 7,35 (2H, дублет, J=8 Гц); 7,82 (2H, дублет, J=8 Гц).
Приготовление 9
2-трет-Бутил-5-(3-циклогексил-2-оксопропил)-1-нитробензол
9(I) 2-трет-Бутил-5-[3-циклогексил-2-(4-метилфенил)- сульфонил-2-метилтиопропил]-1-нитробензол
3,45 мл (5,52 ммоль) 1,6 М гексанового раствора бутиллития добавляют по каплям в течение периода времени более 5 мин к 10 мл тетрагидрофуранового раствора, содержащего 1,73 г (5,52 ммоль) 1-[2-(4-метилфенил)сульфонил-2-меттлтиоэтил]гексана (полученного, как описано в приготовлении 8), поддерживая температуру - 78oC. Через 15 мин добавляют по каплям 12 мл диметилформамидного раствора, содержащего 1,68 г (5,26 ммоль) 5-йодметил-2-трет-бутил-1-нитробензола (полученного, как описано в приготовлении 51). Реакционной смеси позволяют достичь комнатной температуры, после чего перемешивают в течение 80 мин. По истечении этого времени добавляют разбавленный водный раствор аммоний хлорида для остановки реакции. Добавляют воду и диэтиловый эфир и продукт распределяют между органическим растворителем и водой. Органический слой отделяют и промывают насыщенным водным раствором хлорида натрия. Растворитель затем удаляют отгонкой при пониженном давлении с получением указанного в заголовке соединения. Так как это соединение неустойчиво, его используют в следующей стадии без очистки.
9(i) 2-трет-Бутил-5-(3-циклогексил-2-оксопропил)-1-нитробензол
Весь 2-трет-бутил-5-[3-циклогексил-2-(4-метилфенил)сульфонил-2- метилтиопропил]-1-нитробензол, полученный, как описано в стадии (i) выше, растворяют в 50 мл метанола, Добавляют 5 мл концентрированной водной соляной кислоты и полученный раствор нагревают 2 ч при кипячении с обратным холодильником. По истечении этого времени растворитель удаляют при пониженном давлении. Полученный остаток растворяют в диэтиловом эфире и растворитель промывают водой, насыщенным водным раствором гидрокарбоната натрия, водой и насыщенным водным раствором хлорида натрия, в том порядке. Растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 150 г силикагеля. Элюирование 1:5 по объему смесью этилацетата и гексана приводит к получению 1,20 г (выход 73%) указанного в заголовке соединения в виде маслянистого продукта.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax см-1: 1717, 1532, 1449, 1370, 814.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц). δ ppm: 0,83-0,98 (2H, мультиплет); 1,04-1,46 (3H, мультиплет); 1,39 (9H, синглет); 1,59-1,73 (5H, мультиплет); 1,76-1,92 (1H, мультиплет); 2,35 (2H, дублет, J=7 Гц); 3,67 (2H, синглет); 7,14 (1H, дублет, J=2 Гц); 7,26 (1H, дублет дублетов, J= 2 и 8Гц); 7,50 (1H, дублет, J=8 Гц).
Приготовление 10
5-(3-Циклогексил-2-трет-бутилдиметилсилилоксипропил)-2-трет-бутил-1- нитробензол
10 (I) 5-(3-Циклогексил-2-гидроксипропил)-2-трет-бутил-1-нитробензил
Следуя способу, подобному описанному в примере 22, но используя 1,70 г (5,36 ммоль) 2-трет-бутил-5-(3-циклогексил-2-оксопропил) -1-нитробензола (полученного, как описано в приготовлении 9), получают 1,74 г указанного в заголовке соединения в виде маслянистого продукта. Продукт используют в следующей стадии без очистки.
Инфракрасный абсорбционный спектр (жидкая пленка) νmax , см-1: 3560, 3410, 1532, 1449, 1368, 812.
Спектр ядерного магнитного резонанса (CDCL3, 270 МГц), δ ,ppm: 0,80-1,04 (2H, мультиплет); 1,07-1,55 (6H, мультиплет); 1,39 (9H, синглет); 1,61-1,83 (5H, мультиплет); 2,64 (1H, дублет дублетов, J=8 и 14 Гц); 2,78 (1H, дублет дублетов, J= 4 и 14 Гц); 3,89-4,00 (1H, мультиплет); 7,18 (1H, дублет, J= 2Гц); 7,30 (1H, дублет дублетов, J= 2 и 8 Гц); 7,47 (1H, дублет, J=8 Гц).
10(i) 5-(3-Циклогексил-2-трет-бутилдиметилсилилоксипропил)- 2-трет-1-нитробензол
973 мг (6,46 ммоль) трет-бутилдиметилсилилхлорида, 0,90 мл (6,46 ммоль) триэтиламина и 67 мг (0,55 ммоль) 4-(N,N-диметиламино) пиридина добавляют к 10 мл диметилформамидного раствора, содержащего 1,74 г 5-(3-циклогексил-2-гидроксипропил)-2-трет-бутил-1-нитробензола [полученного, как описано в стадии (i) выше] . Смесь затем перемешивают в течение 1,5 ч при комнатной температуре и затем 2,5 ч при 40oC. По истечении этого времени реакционный раствор выливают в воду, экстрагируют 3:1 по объему смесью гексана и этилацетата. Органический слой промывают разбавленной водной соляной кислотой, водой, насыщенным водным раствором гидрокарбоната натрия и насыщением водным раствором хлорида натрия. Затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 75 г силикагеля. Элюирование 1: 9 по объему смесью диэтилового эфира и гексана приводит к получению 2,22 г (выход 96% за 2 стадии) указанного в заголовке соединения в виде маслянистого продукта.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 1534, 1368, 1254, 1067, 835.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: -0,26 (3H, синглет); -0,05 (3H, синглет); 0,78-0,97 (2H, мультиплет); 0,81 (9H, синглет); 1,10-1,40 (6H, мультиплет); 1,38 (9H, синглет); 1,62-1,75 (5H, мультиплет); 2,60 (1H, дублет дублетов, J = 7 и 13 Гц); 2,78 (1H, дублет дублетов, J = 5 и 13 Гц); 3,85-3,94 (1H, мультиплет); 7,12 (1H, дублет, J = 2 Гц). 7,23 (1H, дублет дублетов, J = 2 и 8 Гц); 7,42 (1H, дублет, J = 8 Гц).
Приготовление II
N-[2-трет-Бутил-5-(3-циклогексил-2-трет-бутилдеметилсилилоксипропил) фенил]-2-(9H-ксантен-9-ил)ацетамид
11 (I) 2-трет-Бутил-5-(3-циклогексил-2-трет-бутилдиметилсилилокси -пропил)анилин
Следуя способу, подобному описанному в примере 7, но используя 1,70 г (5,36 ммоль) 5-(3-циклогексил-2-трет-бутилдиметилсилилилоксипропил)-2-трет-бутил -1-нитробензола (полученного, как описано в приготовлении 10) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде маслянистого продукта.
11 (II) N-[2-трет-Бутил-5-(3-циклогексил-2-трет -бутилдиметилсилилоксипропил)фенил]-2-(9H-ксантен-9-ил)ацетамид.
Следуя способу ацилирования, подобному описанному в примере 21, но используя 2-трет-бутил-5-(3-циклогексил-2-трет -бутилдиметилсилилоксипропил)анилин [полученный, как описано в стадии (I) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 148-150oC (после перекристаллизации из смеси диизопропилового эфира и гексана). Выход в обоих стадиях количественный.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3232, 1641, 1533, 1482, 1256, 759.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: -0,37 (0,6H, синглет); -0,20 (2,4H, синглет); -0,10 (0,6H, сингет); -0,02 (2,4H, синглет); 0,75-0,97 (2H, мультиплет); 0,78 (1,8H, синглет); 0,82 (7,2H, синглет); 1,06-1,47 (6H, мультиплет); 1,16 (9H, синглет); 1,60-1,77 (5H, мультиплет); 2,37-2,76 (4H, мультиплет); 3,68-2,76 (0,2H, мультиплет); 3,92 (0,8H, квинтет, J = 6 Гц); 4,75 (1H, триплет, J = 7 Гц); 6,90-7,41 (11H, мультиплет).
Приготовление 12
2-трет-Бутил-5-гидросксиметил-1-нитробензол
10 мл тетрагидрофуранового раствора, содержащего 3,12 г (28,8 моль) этилхлорформиата, добавляют в течение 10 мин к 60 мл тетрагидрофуранового раствора, содержащего 6,0 г (26,9 ммоль) 4-трет-бутил-3-нитробензойной кислоты и 3,12 г (30,9 ммоль) триэтиламина на ледяной бане. Реакционный раствор перемешивают 45 мин при этой температуре, после чего фильтруют с помощью фильтра Целита. Нерастворимый осадок промывают тетрагидрофураном, и фильтрат и промывные воды объединяют. Объединенный раствор добавляют по каплям к смешанному раствору, который состоит из 40 мл тетрагидрофурана и 40 мл воды, содержащей 3,76 г (9,95 ммоль) боргидрида натрия, в течение 25 мин на ледяной бане. Реакционную смесь затем перемешивают 2 ч. при температуре ледяной бани, после чего концентрируют упариванием при пониженном давлении. Максимально возможно удаляют тетрагидрофуран при пониженном давлении и остаток распределяют между диэтиловым эфиром и водой. Продукт экстрагируют из водного слоя диэтиловым эфиром. Органические слои объединяют и промывают дважды водой и затем один раз насыщенным водным раствором хлорида натрия. Растворитель затем удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 100 г силикагеля. Элюирование смесями этилацетата и гексана в отношении, изменяющемся от 20:80 до 30: 70 по объему, приводит к получению 5,24 г (выход 93%) указанного в заголовке соединения в виде маслянистого продукта.
Спектр ядерного резонанса (CDCl3, 270 МГц), δ , ppm: 1,40 (9H, синглет); 4,69 (2H, дублет, J = 5 Гц). 7,33 (1H, синглет); 7,41 (1H, дублет, J = 9,5 Гц). 7,53 (1H, дублет, J = 9,5 Гц).
Приготовление 13
2-трет-Бутил-5-(2-трет-бутилдиметилсилилоксиметил)-1-нитробензол
4,15 мг (27,5 ммоль) трет-бутилдиметилсилилхлорида, 3,85 мл (27,6 ммоль) триэтиламина и 815 мг (0,503 ммоль) 4-(N, N-диметиламино)пиридина добавляют к 50 мл метиленхлоридного раствора, содержащего 5,24 г (25,0 ммоль) 2-трет-бутил-5-гидроксиметил-1-нитробензола (полученного, как описано в приготовлении 12) на ледяной бане. Реакционной смеси дают вернуться к комнатной температуре, после чего перемешивают в течение 40 мин. По истечении этого времени реакционный раствор 1: 1 по объему элюируют гексана и диэтилового эфира и промывают водой, разбавленной водой соляной кислотой, опять водой, насыщенным водным раствором гидрокарбоната натрия, в этом порядке. Затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 100 г силикагеля. Элюирование 1:1 смесью метиленхлорида и гексана приводит к получению 8,04 г (выход 99%) указанного в заголовке соединения в виде маслянистого продукта.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,09 (6H, синглет); 0,93 (9H, синглет); 1,38 (9H, синглет); 4,69 (2H, синглет); 7,2-7,6 (3H, мультиплет).
Приготовление 14
N-(2-трет-Бутил-5-гидрокситметилфенил)-2-(9H-ксантен-9 -ил)ацетамид
14 (I) 2-трет-Бутил-5-(2-трет-бутилдиметилсилилоксиметил)анилин
Следуя способу, подобному описанному в примере 7, но используя 2-трет-бутил-5-(2-трет-бутилдиметилсилилоксиметил)-1-нитробензол (полученный, как описано в приготовлении 13) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде маслянистого продукта с количественным выходом.
14 (II) N-[2-трет-Бутил-5-(2-трет-бутилдиметилсилилоксиметил)фенил]-2- (9H-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в примере 21, но используя 2-трет-бутил-5-(2-трет-бутилдиметилсилилоксиметил)анилин [полученный как описано в стадии (I) выше] в качестве исходного продукта, указанное в заголовке соединение в виде кристаллов с выходом 84%.
14 (III) N-(2-трет-Бутил-5-гидроксиметилфенил)-2-(9H-ксантен-9 -ил)ацетамид.
Следуя способу, подобному описанному в примере 40, но используя N-[2-трет-бутилдиметилсилилоксиметил)фенил] -2- (9H-ксантен-9-ил)ацетамид [полученный, как описано в стадии (II) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 137-138oC (после перекристаллизации из смеси этилацетата и гексана) с количественным выходом.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1664, 1541, 1478, 1460, 1250.
Спектр ядерного резонанса (CDCl3, 270 МГц), σ , ppm: 1,17 (9H, синглет); 2,41 (2/5H, уширенный дублет, J = 7 Гц); 2,71 (8/5H, дублет, J = 7 Гц); 4,35-4,45 (2/5H, синглет); 4,73 (1H, триплет, J = 7 Гц); 7,05-7,55 (11H, мультиплет).
Приготовление 15
N-(2-трет-Бутил-5-формилфенил)-2-(9H-ксантен-9-ил)ацетамид
1,24 г (3,08 ммоль) N-(2-трет-бутил-5-гидроксиметилфенил)-2 -(9H-ксантен-9-ил)ацетамида (полученного, как описано в приготовлении 14) добавляют к 12 мл метиленхлоридной суспензии, содержащей 1,06 г (4,92 ммоль) пиридинхлорхромата. Смесь затем перемешивают в течение 1,75 ч. По истечении этого времени реакционную суспензию разбавляют диэтиловым эфиром, фильтруют через колонку, используя 50 мл абсорбента Флоризил (торговая марка), и элюируют 1: 1 по объему смесью метиленхлорида и диэтилового эфира. Затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 50 г силикагеля. Элюирование 1:9 по объему смесью диэтилового эфира и метиленхлорида приводит к получению указанного в заголовке соединения, содержащего небольшие количества примесей. Перекристаллизация этой формы из смеси этилацетата и гексана приводит к 442 мг указанного в заголовке соединения в виде кристаллов, плавящихся при 172,5-174oC (после перекристаллизации из смеси этилацетата и гексана). Маточный раствор концентрируют и подвергают колоночной хроматографии через 100 г силикагеля. Элюируя 4: 6 по объему смесью этилацетата и гексана приводит к получению 643 мг указанного в заголовке соединения. Общий выход 88%.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 1702, 1642, 1482, 1459, 1260, 760.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,21 (6H, синглет); 2,74 (2H, дублет, J = 7 Гц); 4,75 (1H, триплет, J = 7 Гц); 7,07-7,15 (4H, мультиплет); 7,21-7,29 (2H, мультиплет); 7,39 (2H, дублет дублетов, J = 2 и 8 Гц); 7,49 (1H, дублет, J = 8 Гц); 7,66 (1H, дублет дублетов, J = 2 и 8 Гц); 0,08 (1H, уширенный синглет); 9,98 (1H, синглет).
Приготовление 16
N-[2-трет-Бутил-5-(2-этоксикарбонилэтенил)фенил] -2 -(9H-ксантен-9-ил)ацетамид
153 мг (3,51 ммоль) гидрида натрия (в виде 55% по массе суспензии в минеральном масле) промывают дважды гексаном; затем добавляют 5 мл диметилформамида. Суспензию охлаждают на ледяной бане и затем прибавляют 1 мл диметилформамидного раствора, содержащего 673 мг (3,00 ммоль) 2-диэтоксифосфорилацетата. Когда пенообразование закончилось, реакционную смесь перемешивают далее 40 мин при комнатной температуре. Смесь затем охлаждают опять на ледяной бане и в течение 5 мин добавляют 1,0 г (2,5 ммоль) N-(2-трет-бутил-5-формилфенл)-2-(9H-ксантел-9-ил)ацетамида. Смесь перемешивают 20 мин. при температуре ледяной бани и 10 мин при комнатной температуре. По истечению этого времени реакционную смесь разбавляют диэтиловым эфиром, выливают и ледяную воду и экстрагируют диэтиловым эфиром. Органический экстракт промывают водой. Растворитель удаляют отгонкой при пониженном давлении. Полученный остаток перекристаллизовывают из смеси метиленхлорида, диэтилового эфира и гексана с получением 804 мг указанного в заголовке соединения в виде кристаллов, плавящихся при 180-181oC. Маточной раствор концентрируют и перекристаллизовывают из той же самой смеси растворителей с получением дополнительных 263 мг указанного в заголовке соединения. Всего получают 1,067 г (выход 90%) указанного в заголовке соединения.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3235, 1711, 1645, 1523, 1480, 1458, 1257, 116, 756.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,18 (9H, синглет); 136 (3H, триплет, J = 7 Гц); 2,33-2,46 (0,4H, мультиплет); 2,72 (0,4H, дублет, J = 7 Гц); 4,28 (2H, квартет, J = 7 Гц); 4,74 (1H, триплет, J = 7 Гц); 6,42 (1H, дублет, J = 16 Гц); 7,03-7,45 (10H, мультиплет); 7,63 (1H, дублет, J = 16 Гц); 7,70 (1H, синглет).
Приготовление 17
N-[2-трет-Бутил-5-(2-этоксикарбонилэтил)фенил] -2-(9H-ксантен-9 -ил)ацетамид
Следуя способу каталитического восстановления, подобному описанному в примере 8, но используя N-[2-трет-Бутил-5-(2-этоксикарбонилэтил)фенил]-2-(9H-ксантен-9 -ил)ацетамид (полученный, как описано в приготовлении 16) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают с выходом 95% указанное в заголовке соединение в виде кристаллов, плавящихся при 160,5-11,5oC (после перекристаллизации из смеси метиленхлорида и диэтилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3223, 1736, 1640, 1539, 1481, 1261, 1192, 760.
Спектр ядерного резонанса (CDCl3, 270 МГц), δ , ppm: 1,16 (9H, синглет); 1,26 (3H, триплет, J = 7 Гц); 2,35-2,50 (1H, мультиплет); 2,58-2,75 (3,5H, мультиплет); 2,86-2,98 (1,5H, мультиплет); 4,16 (2H, квартет, J = 7 Гц); 4,74 (1H, триплет, J = 7 Гц); 6,91-7,45 (11H, мультиплет).
Приготовление 18
N-[2-трет-Бутил-5-(3-гидрокситпропил)фенил]-2-(9H- ксантен-9-ил)ацетамид
5,96 мл (5,96 ммоль) 1 M гексановго раствора диизобутилалюминий-гидрида добавляют по каплям в течение 10 мин к 14 мл тетрагидрофуранового раствора, содержащего 702 мг (1,49 ммоль) N-[2-трет-бутил-5- (этоксикарбонилэтил)фенил] -2-(9H-ксантен-9-ил)ацетамида (полученного, как описано в приготовлении 17), предварительно охлажденного до -78oC. Смесь перемешивают 75 мин при этой температуре и затем 45 мин при комнатной температуре. По истечении этого времени реакционный раствор выливают в смесь разбавленную водной соляной кислоты и льда и затем экстрагируют диэтиловым эфиром. Органический экстракт промывают водой, и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 30 г силикагеля. Элюирование 5:4 по объему смесью метиленхлорида и этилацетата приводит к 427 мг (выход 67%) указанного в заголовке соединения в виде кристаллов, плавящихся при 185-186oC (после перекристаллизации из смеси метиленхлорида и диэтилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3417, 3212, 1659, 1530, 1481, 1457, 1254, 765, 757.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ppm: 1,16 (9H, синглет); 1,67-1,98 (2H, мультиплет); 2,33-2,53 (1H, мультиплет); 2,61-2,76 (3H, мультиплет); 3,53-3,75 (2H, мультиплет); 4,74 (1H, триплет, J=7 Гц); 6,92-7,46 (11H, мультиплет).
Приготовление 19
N-[2-трет-Бутил-5-(3-оксопропил)фенил]-2-(9H-касантен-9-ил) ацетамид
2 мл метиленхлоридного раствора, содержащего 155 мг (1,98 ммоль) диметилсульфоксида, добавляют по каплям в течение 5 мин к 2 мл метиленхлоридного раствора, содержащего 126 мг (0,99 ммоль) оксалилхлорида, который предварительно охлаждают до -78oC. Смесь затем перемешивают 5 мин при этой температуре, после чего добавляют 15 мл метиленхлоридного раствора, содержащего 354 мг (0,825 ммоль) N-[2-трет-бутил-5-(3-гидроксипропил)фенил] -2-(9H-ксантен-9-ил) ацетамида (полученного, как описано в приготовлении 18), и смесь перемешивают 15 мин при этой температуре. К реакционной смеси добавляют 501 мг (4,95 ммоль) триэтиламина, и смесь перемешивают 5 мин при этой же температуре. Затем реакционной смеси дают вернуться к комнатной температуре, после чего разбавляют диэтиловым эфиром и промывают разбавленной водой соляной кислотой и затем водой. Растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 20 г силикагеля. Элюирование 100:5 по объему смесью метиленхлорида и этилацетата приводит к 310 кг (выход 88%) указанного в заголовке соединения в виде кристаллов, плавящихся при 176-177oC (после перекристаллизации из смеси метиленхлорида и диэтилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3230, 1729, 1641, 1534, 1481, 1458, 1260, 759.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,16 (9H, синглет); 2,36-3,00 (6H, мультиплет); 4,74 91H, триплет, J= 7 Гц); 6,92-7,45 11H, мультиплет); 9,84 (1H, синглет).
Приготовление 20
N-[(9H-Ксантен-9-ил)метил] =N'-(2-трет-бутил-5-[4-циклогексил-3- (трет-бутилдиметилсилилокси)бутил]фенил}мочевина
Раствор 240 мг (1,0 ммоль) 2-(9H-ксантен-9-ил)уксусной кислоты, 275 мг (1,00 ммоль) дифенилфосфорилазида и 139 μл (1,00 ммоль) триэтиламина в 3 мл бензола нагревают с обратным холодильником при перемешивании в течение 2,5 ч, после чего добавляют раствор 459 мг (1,10 ммоль) 2-трет-бутил-5-[4-циклогексил-3-(трет-бутилдиметилсилилокси)бутил] анилина в 0,5 мл бензола. Полученную смесь нагревают с обратным холодильником в течение 3 ч. По истечении этого времени реакционную смесь разбавляют этилацетатом, и разбавленную смесь промывают разбавленным водным раствором соляной кислоты, водой, затем насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, в данном порядке. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 25 г силикагеля, используя 95: 5 по объему смеси метиленхлорида и этилацетата в качестве элюента с получением 414 мг (выход 65%) указанного в заголовке в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см -1: 3341, 2953, 2926, 2854, 1637, 1561, 1481, 1458, 1256, 1075, 835, 755.
Приготовление 21
(2-Фенилциклопентил) уксусная кислота
21 (i) (1-Фенилцилопентил) метанол
Раствор 1,15 г (6,04 ммоль) (1-фенилцилопентил) карбоновой кислоты в 10 мл тетрагидрофурана добавляют по каплям в течение 20 мин к суспензии 344 мг (9,06 ммоль) литийалюминийгидрида в 20 мл тетрагидрофурана при ледяном охлаждении, и полученную смесь перемешивают 30 мин при 60oC. По истечении этого времени реакционную смесь вновь охлаждают льдом и затем прибавляют 0,3 мл воды, 10 мл 2 н. водного раствора гидроксида натрия и 1 мл воды, в данной последовательности, и полученную смесь разбавляют диэтиловым эфиром. Образующийся белый гелеобразный продукт отфильтровывают и фильтр промывают водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, в данном порядке. Органическую фазу высушивают над безводным сульфатом натрия, и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток подвергают колоночной хроматографии через 50 г силикагеля, используя метиленхлорид в качестве элюента с получением 1,06 г (выход 99%) указанного в заготовке соединения в виде бесцветных кристаллов.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3310, 2952, 2929, 2873, 1496, 1446, 1059, 1032, 766, 699, 567,
21 (II) (1-Фенилциклопентил)метилметансульфонат
659 мг (6,07 ммоль) метансульфонилхлорила и затем 838 μл (6,01 ммоль) триэтиламина добавляют при охлаждении на ледяной бане к раствору 1,00 г (5,67 ммоль) (1-фенилцилопентил)метанола) полученного, как описано в стадии (I) выше] в 18 мл метиленхлорида, и полученную смесь перемешивают 1 ч. По истечении этого времени реакционную смесь разбавляют диэтиловым эфиром и разбавленную смесь промывают 2 н. водной соляной кислотой, водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, в данном порядке. Органическую фазу высушивают над безводным сульфатом натрия и растворитель отгоняют с получением 1,44 г (выход количественный) указанного в заготовке соединения в виде бесцветных кристаллов.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2959, 2943, 2874, 1337, 1180, 1166, 975, 960, 853, 845, 770, 750, 702, 526, 505.
21 (III) (1-Фенилциклопенитил)метилиодид
5,00 г (33,3 ммоль) йодида натрия добавляют к раствору 1,00 г (3,93 ммоль) (1-фенилциклопентил)метилметансульфоната [полученного, как описано в стадии (II) выше] в 10 мл метилизобутилкетона, и полученную смесь нагревают с обратным холодильником при перемешивании в течение 18 ч. По истечении этого времени растворитель удаляют упариванием при пониженном давлении и полученный остаток распределяют между диэтиловым эфиром и водой. Органическую фазу промывают водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, в данном порядке. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 50 г силикагеля, используя 1:1 по объему смеси метиленхлорида и гексана в качестве элюента с получением 504 мг (выход 45%) указанного в заготовке соединения в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 2956, 2872, 1496, 1446, 1210, 1188, 760, 699, 546.
21 (IV) 2-(1-Фенилциклопентил)метил-1,3-дитиан 720 μл 1,6 М гексанового раствора бутиллития добавляют по каплям при -78oC к раствору 126 мг (1,05) 1,3-дитиана в смеси 1,5 мл тетрагидрофурана и 720 μл гексаметилфосфортриамида при перемешивании, и полученную смесь перемешивают 10 мин при охлаждении на бане из льда и соли. По истечении этого времени раствор 200 мг (0,699 ммоль) (1-фенилциклопентил)метил йодида [полученного, как описано в стадии (III) выше] добавляют по каплям к смеси при -78oC. Полученную смесь затем перемешивают 20 мин при охлаждении на ледяной бане с солью. Для остановки реакции в реакционной смеси добавляют насыщенный водный раствор хлорида аммония, после чего экстрагируют диэтиловым эфиром. Экстракт промывают насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 15 г силикагеля, используя 3:2 по объему смеси гексана и метиленхлорида в качестве элюента с получением 100 мг (выход 51%) указанного в заголовке соединения в виде бесцветного пенообразного продукта.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,57 - 2,10 (12 H, мультиплет); 2,61 - 2,85 (4H, мультиплет); 3,58 (1H, триплет, J= 5,9 Гц); 7,18-7,37 (5H, мультиплет).
21 (V) 2-(1-Фенилциклопентил)ацетальдегид
263 мг (0,969 ммоль) хлорида ртути и 120 мг (1,29 ммоль) карбоната кальция триэтиламина добавляют к суспензии 90 мг (0,323 ммоль) 2-(1-фенилцилопентил)метил-1,3-дитиана [полученного, как описано в стадии (IV) выше] в смеси 4 мл ацетонитрила и 0,6 мл воды, и полученную смесь перемешивают 3 ч при 90oC. По истечении этого времени реакционную смесь охлаждают и разбавляют этилацетатом. Нерастворимый материал отфильтровывают с помощью фильтра Целита (торговая марка). Фильтрат затем промывают IM водным раствором ацетата натрия, насыщенным водным раствором хлорида аммония и насыщенным водным раствором хлорида натрия, в данном порядке. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 5 г силикагеля, используя метиленхлорида в качестве элюента, с получением 58,4 мг (выход 96%) указанного в заголовке соединения в де бесцветного пенообразного продукта.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ ppm: 1,76-1,99 (8H, мультиплет); 2,64 92H, дублет J = 3?3 Гц); 7,1807,34 (5H, мультиплет); 9,43 (1H, триплет, J = 3,3 Гц).
21 (VI) 2-(1-Фенилциклопентил)уксусная кислота
1 мл водного раствора из 39 мг (0,402 ммоль) сульфаминовой кислоты и затем 1 мл водного раствора из 37,6 мг (0,416 ммоль) хлорида натрия добавляют по каплям к раствору 58,4 мг (0,310 ммоль) 2-(1-фенилкицлопентил)ацетальдегида [полученного, как описано в стадии (V) выше] в 1 мл трет-бутанола при комнатной температуре, и полученную смесь перемешивают 1 ч при комнатной температуре, после чего экстрагируют метиленхлоридом. Экстракт промывают водой и затем высушивают над безводным сульфатом натрия. Растворитель удаляют отгонкой при пониженном давлении с получением бесцветных кристаллов. Их перекристаллизовывают из смеси этилацетата и гексана с получением 57 мг (выход 90%) указанного в заголовке соединения в виде пластинчатых кристаллов.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 2967, 2945, 2869, 1706, 1426, 1400, 1204, 1195, 911, 773, 698.
Приготовление 22
N-[(1-Фенилциклопентил)метил] -N'-[2-трет-бутил-5- (3-трет-бутил-диметилсилилокси-4-циклогексилбутил)фенил]мочевина
Следуя способу, подобному описанному в приготовлении 20, но используя 2-(1-фенилциклопентил)уксусную кислоту (полученную, так описано в приготовлении 21) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде пенообразного продукта.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,59 (6H, синглет); 0,92 (9H, синглет); 1,17-1,93 (23 H, мультиплет); 1,27 (9H, синглет); 2,39-2,62 (2H, мультиплет); 3,31(3H, дублет, J = 5 Гц);3,80 - 3,82 (1H, мультиплет); 5.74 (1H, синглет); 6,84 - 7,28 (8H, мультиплет).
Приготовление 23
(R)-N-(2-трет-Бутил-5-[4-циклогексил-3-(трет-бутилдиметилсилилокси)бутил] фенил}-2-(9H-ксантен-9-ил)ацетамид
23(I) (S)-2-Циклогексил-1-(бензоилоксиметил)этиловый спирт
Раствор реактива Гриньяра, полученный из 9,79 г (60,0 ммоль) циклогексилбромида и 1,46 г (60,0 ммоль) магния в 85 мл тетрагидрофурана, добавляют по каплям в течение 10 мин к суспензии 1,90 г (9,98 ммоль) йодида меди в 50 мл тетрагидрофурана при -75oC. Смесь оставляют на 10 мин, и затем добавляют по каплям раствор (S)-бензоилоксиметилоксирана, [α]D = 4,82o (C = 1, толуол), в 20 мл тетрагидрофурана в течение 15 мин. Полученную смесь перемешивают 3 ч. при той же температуре и затем 30 мин. при 0oC. По истечении этого реакцию останавливают добавлением насыщенного водного раствора хлорида аммония. Реакционную смесь затем смешивают с 20 мл концентрированного водного аммиака, после чего экстрагируют диэтиловым эфиром. Экстракт промывают насыщенным водным раствором хлорида аммония, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, в данном порядке. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 250 г силикагеля, используя 1:4 по объему смеси этилацетата и метиленхлорида в качестве элюента с получением 10,0 г (выход 82%) указанного в заголовке производного спирта в виде бесцветного масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 3460, 1451, 1364, 1102, 1048, 1028, 737, 699.
[α]
23 (II) (S)-[3-Бензилокси-2-(трет-бутилдиметилсилилоксипропил) циклогексан
Следуя способу, подобному описанному в приготовлении 10 (II), но используя (S)-2-циклогексил-1-(бензоилоксиметил)этиловый спирт [полученный, как описано в стадии (I) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде масла с количественным выходом.
Инфракрасный абсорбционный спектh (жидкая пленка), νmax , см-1: 1451, 1362, 1252, 1125, 1028, 970, 835, 776.
[α]
23 (iii) (S)-2-трет-Бутилдиметилсилилокси-3-циклогексилпропиловый спирт
Раствор 3,46 г (9,55 ммоль) (S)-[3-бензилокси-2-(трет-бутилдиметилсилилоксипропил)циклогексана [полученного, как описано в стадии (II) выше] в 50 мл этанола перемешивается в течение 320 мин. в токе водорода в присутствии 580 мг 10% по массе палладия на угле. По истечении этого времени реакционную смесь фильтруют и катализатор промывают этанолом. Фильтрат и промывные воды объединяют, и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографии через 180 г силикагеля, используя 1:4 по объему смесь этилацетата и гексана в качестве элюента, с получением 2,54 г (выход 98%) указанного в заголовке спиртового производного в виде бесцветного масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax 3400, 1449, 1254, 1080, 969, 837, 776.
[α]
23 (IV) (S)-2-трет-Бутилдиметилсилилокси-3-циклогексилпропаналь 1,5 мл (21,1 ммоль) диметилсульфоксида добавляют по каплям в течение 2 мин. к раствору 1,0 мл (11.5 ммоль) оксалилхлорида в 20 мл метиленхлорида при -78oC. Смесь оставляют стоять 10 мин., после чего добавляют по каплям в течение 5 мин. раствор 2,54 г (9,32 ммоль) (S)-2-трет-бутилдиметилсилилокси-3-циклогексилпропилового спирта [полученного, как описано в стадии (III) выше] в 12 мл метиленхлорида. Полученную смесь перемешивают 20 мин. при этой же температуре, после чего добавляют 6,5 мл (46,6 ммоль) триэтиламина. Смесь оставляют стоять 7 мин., и охлаждающую баню убирают. После еще 10 мин. реакцию останавливают добавлением водного раствора хлорида аммония. Реакционную смесь экстрагируют диэтиловым эфиром. Экстракт промывают дважды водой и один раз насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении с получением 2,55 г (выход количественный) указанного в заголовке альдегидного соединения в виде бесцветного масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 1738, 1472, 1449, 1256, 1115, 1007, 940, 839, 778.
23 (V) (S)-2-трет-Бутил-5-(3-трет-бутилдиметилсилилокси-4-циклогексил -1-бутенил)-1-нитробензол
8,6 мл (8,6 ммоль) 1M тетрагидрофуранового раствора гексаметилдисилазида натрия добавляют в течение 20 мин. к суспензии 5,04 г (8,67 ммоль) (4-трет-бутил-3-нитрофенил)метилтрифенилфосфоний йодида (полученного, как описано в приготовлении 52) в 80 мл тетрагидрофурана при -78oC, и смесь оставляют стоять 1 ч. Затем сразу весь добавляют раствор 2,33 Г (8,61 ммоль) (S)-2-трет-бутилдиметилсилилокси-3-циклогексилпропанола [полученного, как описано в стадии (IV) выше] в 7 мл тетрагидрофурана, и полученную смесь перемешивают 1 ч. при этой же температуре. Охлаждающую баню убирают и смесь перемешивают далее 4 ч. Реакцию останавливают добавлением насыщенного водного раствора хлорида аммония, после чего реакционную смесь экстрагируют 1:3 по объему смесью этилацетата и гексана. Экстракт промывают водой и насыщенным водным, раствором хлорида натрия, в данном порядке, после чего высушивают над безводным сульфатом магния. Растворитель удаляют отгонкой при пониженном давлении и полученный остаток очищают колоночной хроматографией через 150 г силикагеля, используя 1:19 по объему смесь диэтилового эфира и гексана в качестве элюента, с получением фракций, содержащих небольшое количество примесей. Эти фракции вновь очищают колоночной хроматографией через 150 г силикагеля, используя 1:4 по объему метиленхлорида и гексана в качестве элюента, с получением 2,78 г (выход 72%) указанного в заголовке олефинового производного в виде бесцветного масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 1534, 1368, 1254, 1096, 1073, 1003, 970, 938, 837,776.
23 (vi) (R)-2-трет-Бутил-5-(3-трет-бутилдиметилсилилокси-4-циклогексилбутил)-1- нитробензол
Раствор 2,75 г (6,17 ммоль) (S)-2-трет-бутил-5-(3-трет- бутилдиметилсилилокси-4-циклогексил-1-бутенил)-1-нитробензола [полученного, как описано в стадии (v) выше] в 30 мл диэтилового эфира энергично перемешивают в течение 20 мин. при 0oC в токе водорода в присутствии 303 мг 10% по массе палладия на угле. Реакционную смесь затем обрабатывают способом, подобным описанному в стадии (III) выше, с получением 2,65 г (выход 96%) указанного в заголовке нитробензольного производного в виде бесцветного масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax, см-1: 1532, 1472, 1449, 1368, 1254, 1077, 1024, 1005, 978, 835, 774.
[α]
23 (VII) (R)-N-{2-трет-Бутил-5-[4-циклогексил-3-(трет-бутил- диметилсилилокси)бутил]фенил}-2-(9H-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в приготовлении 3, но (R)-2-трет-бутил-5-(3-трет-бутилдиметилсилилокси-4-циклогексилбутил) -1-нитробензол [полученный, как описано в стадии (VI) выше] преобразуют в анилиновое производное. Его ацилируют способом, подобным описанному в примере 21, с получением указанного в заголовке соединения с выходом 94% в виде кристаллов, плавящихся при 173 - 174,5oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3460, 1640, 1538, 1482, 1459, 1256, 1079, 835, 758.
[α]
Приготовление 24
(S)-2-трет-Бутил-5-(бензоилокси-4-циклогексилбутил)анилин
24 (I) (R)-2-трет-Бутил-5-(3-гидрокси-4-циклогексилбутил)-1-нитробензол
Следуя способу, подобному описанному в примере 40, но использ3я (R)-2-трет-бутил-5-(3-трет-бутилдиметилсилилокси-4-циклогексилбутил) -1-нитробензол [полученный, как описано в приготовлении 23 (VI) ] в качестве исходного продукта, в соответствующей пропорции, подобной используемой в примере 40, получают указанное в заголовке спиртовое производное с 98% выходом в виде бесцветного масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax 3360, 1532, 1449, 1368, 1254, 1063, 1048, 834, 810.
[α]
24 (II) (S)-2-трет-Бутил-5-(3-бензоилокси-4-циклогексилбутил)-1-нитробензол
Раствор 734 мл (4,21 ммоль) диэтилазодикарбоксината в 5 мл тетрагидрофурана добавляют по каплям в течение 5 мин. к раствору 1,16 г (3,48 ммоль) (R)-2-трет-бутил-5-(3-гидрокси-4-циклогексилбутил)-1- нитробензола [полученного, как описано в стадии (I) выше], 1,10 г (4,21 ммоль) трифенилфосфина и 512 мг (4,19 ммоль) бензойной кислоты в 12 мл тетрагидрофурана при охлаждении льдом. Температуре реакционной смеси дают постепенно подняться до комнатной, после чего полученную смесь перемешивают 12 ч. По истечении этого времени реакционную смесь разбавляют этилацетатом. Разбавленную смесь промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, в данном порядке. Органическую фазу высушивают над безводным сульфатом магния и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографии через 150 г силикагеля, используя 1:9 по объему смеси диэтилового эфира и гексана в качестве элюента с получением 1.19 г (выход 78%) указанного в заголовке бензильного производного в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax, см-1: 1715, 1532, 1451, 1368, 1273, 1113, 1069, 1026, 712.
[α]
24 (III) (S)-2-трет-Бутил-5-(3-бензоилокси-4-циклогексилбутил)анилин
следуя способу, подобному описанному в приготовлении 3, но используя (S)-2-трет-бутил-5-(3-бензоилокси-4-циклогексилбутил)-1-нитробензол [полученный, как описано в стадии (III) ] в качестве исходного продукта, в соответствующем количестве, подобном используемому в примере 40, получают указанное в заголовке соединение в виде сырого продукта. Продукт используют в последующей реакции без дальнейшей очистки.
Приготовление 25
(R)-1-(2-{ 4-трет-Бутил-3-(9Н-ксантен-9-ил)ацетамидо]-фенил} этил)-2-циклогексилэтил бензилсукцинат
Суспензию 249 мг (0,474 ммоль) (R)-N-[2-трет-бутил-5-(4- циклогексил-3-гидроксибутил)фенил]-2-(9Н-ксантен-9-ил)ацетамида (полученного, как описано в примере 100), 218 мг (1,05 ммоль) бензилгидросукцината, 76 мг (0,62 ммоль) 4-(N,N-диметиламино)пиридина и 256 мг (1,34 ммоль) гидрохлорида 1-этил-3-(3'-диметиламинопропил)карбодиимида в 5 мл тетрагидрофурана перемешивают 17 ч при комнатной температуре. По истечении этого времени реакционную смесь разбавляют этилацетатом, и разбавленную смесь промывают 2 н. водной соляной кислотой, водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, в данном порядке. Органическую фазу высушивают над безводным сульфатом магния и затем растворитель удаляют над безводным сульфатом магния и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 20 г силикагеля, используя 1:9 по объему смеси этилацетата и гексана в качестве элюента с получением 352 мг (количественный выход) указанного в заготовке соединения в виде стеклообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 1734, 1659, 1480, 1459, 1416, 1256, 1215, 1158, 870, 758.
Приготовление 26
N-[2-1,1-Диметил-2-метокси)этил-6-(3-оксопропил)фенил] -2-(9Н- ксантен-9-ил)ацетамид
26(I) N-[2-(1,1-Диметил-2-метокси)этил-6-[3-(трет-бутил- диметилсилилокси)пропил]фенил]-2-(9Н-ксантен-9-ил)-ацетамид.
Следуя способу, подобному описанному в примере 21, не используя 2-(1,1-диметил-2-метокси)этил-6-[3-(трет-бутилдиметилсилилокси) пропил]анилин в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке амидное производное в виде бесцветных кристаллов.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3249, 1649, 1526, 1480, 1459, 1258, 1102, 8837, 754.
26(II)N-[2-(1,1-Диметил-2-метокси)этил-6-(3-оксопропил)фенил) -2-(9Н-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в примере 40, N-[2-(1,1-диметил-2-метокси)этил-6-[3-(трет-бутилдиметилсилилокси) пропил] фенил]-2-(9Н-ксантен-9-ил)ацетамид [полученный, как описано в стадии (I) выше] подвергают снятию силильной защитной группы, после чего его окисляют способом, подобным описанному в приготовлении 19, с получением указанного в заголовке соединения в виде кристаллов, плавящихся при 112-113,5oC.
Инфракрасный абсорбционный спектр (KBr), νmax ,см-1: 3225, 1725, 1648, 1534, 1482, 1459, 1262, 1107, 756.
Приготовление 27
(S_ -1-(2-{ 4-трет-Бутил-3-[2-(9Н-ксантен-9-ил)ацетамидо]фенил}этил- 2-циклогексилэтил бензилсукцинат
Следуя способу, описанному в приготовлении 25, но используя (S)-N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил 2-(9Н-ксантен-9-ил)ацетамид (полученный, как описано в примере 102) и бензилгидромалонат в качестве исходных продуктов, в соответствующем соотношении, используемом в этом приготовлении, указанное в заголовке соединение получают в виде пенообразного продукта.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,78-1,02 (2Н, мультиплет); 1,10-1,90 (13Н, мультиплет); 1,16 (9Н, синглет); 2,40-2,71 (4Н, мультиплет); 3,38 (2Н, синглет); 4,74 (1Н, триплет, J=7 Гц); 5,05-5,16 (1Н, мультиплет); 5,19 (2Н, синглет); 6,90-7,40 (16н, мультиплет).
Приготовление 28
N-(2-Изопропил-6-гидроксиметилфенил)-2-(9Н-кмсантен-9-ил) ацетамид
Способ А
28(I) 2-Изопропил-метилтиометиланилин
15,51 г (116 ммоль) N-сукцинамида добавляют в течение 20 мин к раствору 11,2 мг (82,9 ммоль) 2-изопропилаланилина и 7,22 г (116 ммоль) диметилсульфида в 200 мл метиленхлорида при поддерживании внутренней температуры между 10 и 20oC. Через 15 мин 11,73 г (116 ммоль) триэтиламина добавляют к смеси при нагревании с обратным холодильником в течение 9 ч. По истечении этого времени растворитель удаляют отгонкой при пониженном давлении. Полученный остаток смешивают с диэтиловым эфиром и появившиеся нерастворимые продукты отфильтровывают. Фильтрат концентрируют упариванием при пониженном давлении, и концентрат очищают колоночной хроматографией через 300 г силикагеля, используют способ градиентного элюирования смесями гексана и метиленхлорида, изменяющимися от 4: 1 до 0:1, в качестве элюента с получением 10,45 г (выход 65%) указанного в заголовке сульфидного производного в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 3447, 3354, 1623, 1460, 1447, 1280, 1268, 1242, 1049, 747.
28(II) 2-Изопропил-6-метилсульфонилметиланилин
12,9 г (52,3 ммоль) м-хлорбензойной кислоты (70% чистоты) добавляют в течение 15 мин к суспензии 10,21 г (52,3 ммоль) 2-изопропил-6-метилтиометиланилина [полученного, как описано в стадии (I) выше] и 5,31 (50 ммоль) карбоната натрия в 200 мл метиленхлорида при охлаждении льдом. Реакционную смесь перемешивают 1,5 ч при той же температуре, после чего ее разбавляют диэтиловым эфиром, и разбавленную смесь промывают насыщенным водным раствором гидрокарбоната натрия, водным раствором сульфита натрия и водой, в данном порядке. Затем растворитель удаляют отгонкой при пониженном давлении, и полученный остаток перекристаллизовывают из смеси диэтилового эфира и диизопропилового эфира с получением 7,74 г указанного в заголовке соединения в качестве первой партии продукта. Маточный раствор концентрируют упариванием при пониженном давлении, и концентрат повторно перекристаллизовывают по той же самой смеси растворителей с получением дополнительно 0,75 мг указанного в заголовке S-оксидного производного. Указанное в заголовке соединение плавится при 91-91,5oC (после перекристаллизации из смеси метиленхлорида и диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3465, 3367, 1644. 1461, 1437, 1417, 1018, 948, 752.
28 (III) 2-Изопропил-6-хлорметиланилин гидрохлорид
Газообразный хлористый водород вводят в течение 35 мин в раствор 7,74 г 2-изопропил-6-метилсульфонилметиланилина [полученного, как описано в стадии (II) выше] в 80 мл 1,2-дихлорэтана, нагретого до 50oC, с помощью газоотводной трубки. Реакционную смесь охлаждают затем до комнатной температуры и добавляют 50 мг гексана. Выпавшие кристаллы собирают фильтрацией и промывают гексаном с получением 7,78 г (выход 96%) указанного в заголовке гидрохлорида анилина в виде порошка.
28 (IV) N-(2-Изопропил-6-хлорметилфенил)-2-(9Н- ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в примере 21, но используя 1,0 г (4,17 ммоль) 2-(9Н-ксантен-9-ил)уксусной кислоты, получают хлорид кислоты. Весь данный хлорид кислоты растворяют в 30 мл метиленхлорида и затем добавляют 917 мг (4,17 ммоль) 2-изопропил-6-хлорметиланилин гидрохлорида [полученного, как5 описано в стадии (III) выше], и полученную смесь охлаждают до -78oC. Раствор 1,18 мг (9,17 ммоль) N-диизопропил-N-этиламина в 5 мл метиленхлорида затем добавляют по каплям в этой смеси. Температуре смеси дают постепенно подняться до 0oC в течение 2 ч, после чего добавляют раствор 0,30 г (2,32 ммоль) N-диизопропил-N-этиламина в 1 мл метиленхлорида. Смесь затем перемешивают 20 мин при той же температуре. По истечении этого времени реакционную смесь разбавляют метиленхлоридом и разбавленную смесь промывают 2 н. водной соляной кислотой, один раз насыщенным водным раствором гидрокарбоната натрия и затем один раз насыщенным водным раствором хлорида натрия. Органическую фазу концентрируют упариванием при пониженном давлении до объема около 10 мл и концентрат разбавляют 10 мл диэтилового эфира. Выпавшие кристаллы собирают с получением 0,73 г указанного в заголовке соединения в качестве первой партии продукта. Маточный раствор затем концентрируют упариванием при пониженном давлении и концентрат смешивают с диэтиловым эфиром для высаживания 0,46 г второй партии продукта. Концентрат, полученный из маточного раствора второй партии продукта, очищают колоночной хроматографией через 15 г, используя 50:1 по объему смеси метиленхлорида и этилацетата в качестве элюента, с получением дополнительных 0,19 г указанного в заголовке соединения в виде кристаллов. Общий выход 1,38 г (выход 82%). Указанное в заголовке соединение плавится при 199,5-202oC (после перекристаллизации из этилацетата и диэтилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3218, 1656, 1598, 1576, 1530, 1482, 1456, 1409, 1359, 1262.
28(V) N-(2-Изопропил-6-ацетоксиметилфенил)-2-(9Н-ксантен- 9-ил)ацетамид
Суспензию 189 мг (0,467 ммоль) N-(2-изопропил-6-хлорметилфенил) -2-(9Н-ксантен-9-ил)ацетамида [полученного, как описано в стадии (IV) выше], 153 мг (186 ммоль) ацетата натрия и 105 мг (0,70 ммоль) йодида натрия в 2 мл N,N-диметилформамида перемешивают 3 ч при 50oC. Реакционную смесь разбавляют этилацетатом, и разбавленную смесь несколько раз промывают водой. Органическую фазу высушивают над безводным сульфатом магния, и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 7 г силикагеля, используя способ градиентного элюирования смесями метиленхлорида и этилацетата, изменяющимися от 50:1 до 12: 1, в качестве элюента с получением 126 мг (выход 63%) указанного в заголовке ацетильного производного в виде кристаллов, плавящихся при 159-160oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3249, 1746, 1647, 1600, 1577, 1525, 1480, 1458, 1261, 1217.
28(VI) N-(2-Изопропил-6-гидроксиметилфенил)-2-(9Н- ксантен-9-ил)ацетамид
Раствор 79 мг (1,98 ммоль) гидроксида натрия в 1 мл воды добавляют к раствору 567 мг (1,32 ммоль) N-(2-изопропил-6-ацетоксиметилфенил)-2-(9H-ксантен-9-ил)ацетамида [полученного, как описано в стадии (v) выше в 15 мл метанола, и полученную смесь перемешивают в течение 1 ч при комнатной температуре. По истечении этого времени реакционную смесь распределяют между этилацетатом и водой. Органическую фазу промывают насыщенным водным раствором хлорида натрия и растворитель удаляют отгонкой при пониженном давлении с получением 511 мг (количественный выход) указанного в заготовке соединения, плавящегося при 155-156oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3248, 1660, 1646, 1528, 1480, 1457, 1255, 1045.
Способ B
28(vii) N-(2-Изопропил-6-гидроксиметилфенил)-2-(9H-ксантен-9-ил) ацетамид
4,24 г (25 ммоль) нитрита серебра добавляют к раствору 10,14 г (25,0 ммоль) N-(2-изопропил-6-хлорметилфенил)-2-(9H-ксантен-9-ил) ацетамида [полученного, как описано в стадии (iv) выше] в смеси 500 мл ацетона, 100 мл тетрагидрофурана и 200 мл воды при 60oC. По истечении этого времени реакционную смесь концентрируют упариванием при пониженном давлении и концентрат разбавляют этилацетатом. Разбавленную смесь промывают водой и насыщенным водным раствором хлорида натрия, в данном порядке. Органическую фазу высушивают над безводным сульфатом магния и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через силикагель, используя 3:1 по объему, смесью метиленхлорида и этилацетата в качестве элюента с получением 6,41 г (выход 66%) указанного в заголовке соединения в виде кристаллов.
Приготовление 29
N-(2-Изопропил-6-формилфенил)-2-(9H-ксантен-9-ил)ацетамид
Способ A
Следует способу, подобному описанному в приготовлении 19, но используя N-(2-изопропил-6-гидроксиметилфенил)-2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в приготовлении 28) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 201-202oC (после перекристаллизации из смеси метиленхлорида, диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3263, 2966, 1695, 1647, 1585, 1516, 1516, 1481, 1458, 1408, 1396, 1663, 1257.
Способ B
300 мг (4,0 ммоль) триметиламина N-оксида добавляют при охлаждении льдом к раствору 406 мг (1,0 ммоль) N-(2-изопропил-6-хлорметилфенил)-2-(9H-ксантен-9-ил)ацетамида [полученного, как описано в стадии (IV) выше] в смеси 6 мл диметилсульфоксида и 2 мл метиленхлорида. После завершения прибавления температуре дают постепенно достичь комнатной и полученную смесь перемешивают 15 ч при комнатной температуре. По истечении этого времени реакционную смесь разбавляют этилацетатом и разбавленную смесь промывают водой и насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через силикагель, используя метиленхлорид в качестве элюента с получением 173 г (выход 45%) указанного в заголовке соединения в виде кристаллов.
Приготовление 30
N-[2-Изопропил-6-(3-гидроксипропил)фенил]-2-(9H-ксантен-9-ил) ацетамид
30(I) N-[2-Изопропил-6-(2-этоксикарбонилэтенил)фенил] -2- (9H-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в приготовлении 16, но используя N-(2-изопропил-6-формилфенил)-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 29) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 217-218oC (после перекристаллизации из смеси этилацетата и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3275, 2969, 1709, 1666, 1638, 1517, 1480, 1460, 1313, 1253, 1178, 761.
30(II) N-[2-Изопропил-6-(3-гидрокси-1-пропилен)фенил] -2- (9H-ксантен-9-ил)ацетамид
68 μл эфирата трифторида бора добавляют при -78oC к раствору 228 мг (0,5 ммоль) N-[2-изопропил-6-(2-этоксикарбонилэтилен) фенил] -2-(9H-ксантен-9-ил)ацетамида [полученного, как описано в стадии (I) выше] в 6 мл метиленхлорида. Полученную смесь перемешивают в течение 30 мин, после чего в течение 5 мин добавляют по каплям 2,0 мл 1,0 М раствора диизобутилалюминийгидрида. Температуре смеси дают подняться до комнатной и затем к реакционной смеси добавляют 10 мл 10% мас./объем водного раствора виноградной кислоты для остановки реакции. Реакционную смесь затем разбавляют метиленхлоридом. Разбавленную смесь промывают насыщенным водным раствором гидрокарбоната натрия и затем насыщенным водным раствором хлорида натрия. Органическую фазу сушат над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток растирают в смеси диэтилового эфира и гексана для того, чтобы вызвать кристаллизацию. Кристаллы собирают фильтрацией с получением 185 мг (выход 85%) указанного в заголовке спиртового производного, плавящегося при 225-227oC (после перекристаллизации из смеси ацетонитрила и диизопропилового эфира).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3285, 2962, 1653, 1480, 1458, 1256, 756.
230(III) N-[2-изопропил-6-(3-гидроксипропил)фенил] -2-(9H- ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в примере 8, но используя N-[2-изопропил-6-(3-гидрокси-1-пропенил)фенил] -2-(9H-ксантен-9-ил) ацетамид [полученный, как описано в стадии (II) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 218-219oC (после перекристаллизации из смеси диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3405, 3236, 2963, 1652, 1518, 1482, 1458, 1260, 758.
Приготовление 31
N-{ 2-Изопропил-6-[(3-гидроксипропил)оксиметил]фенил}-2- (9H-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в приготовлении 1, не используя N-(2-изопропил-6-гидроксиметилфенил)-2-(9H-ксантен-9-ил) ацетамид (полученный, как описано в приготовлении 28) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 150-151oC (после перекристаллизации из смеси диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax см-1:
Приготовление 32
N-[2-Изопропил-6-(2-гидроксиэтил)оксиметилфенил] -2- (9H-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в приготовлении 1 (II), но используя N-(2-изопропил-6-хлорметилфенил)-2-(9H-ксантен-9-ил)ацетамид [полученный, как описано в приготовлении 28 (IV)] и этиленгликоль в качестве исходных продуктов, в соответствующем соотношении, используемом в этом приготовлении, получают указанное в заголовке соединение, плавящееся при 153-154oC (после перекристаллизации из смеси диэтилового эфира и гексана) с выходом 52%.
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3384, 3255, 2962, 2869, 1650, 1522, 1478, 1457, 1258, 1116, 752.
Приготовление 33
N_ [2-трет-Бутил-5-(4-циклогексил-3-формилбутил)фенил] -2- (9H-ксантен-9-ил)ацетамид
33(i) N-[2-трет-Бутил-5-(4-циклогексил-3-(метоксивинилиден) бутил)фенил] -2-(9H-ксантен-9-ил)ацетамид
1,51 мл (2,42 ммоль) 1,6 М гексанового раствора бутиллития добавляют по каплям в течение 3 мин к суспензии 828 мг (2,42 ммоль) метоксиметилтрифенилфосфонийхлорида в 10 мл тетрагидрофурана при охлаждении льдом. Полученную смесь затем перемешивают 30 мин при той же температуре, после чего к смеси добавляют 422 мг (0,805 ммоль) N-[2-трет-бутил-5-(4-циклогексил-3-оксобутил)фенил] -2-(9H-ксантен-9- ил) ацетамида (полученного, как описано в примере 13). Температуре смеси дают подняться до комнатной и затем реакционную смесь перемешивают дополнительно 1 ч. Для остановки реакции к реакционной смеси добавляют насыщенный водный раствор хлорида аммония, после чего экстрагируют диэтиловым эфиром. Экстракт промывают дважды водой и один раз насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 25 г силикагеля, используя способ градиентного элюирования смесями метиленхлорида и этилацетата, изменяющимися от 100:3 до 100:5 по объему, в качестве элюента. Объединенные элюаты концентрируют упариванием при пониженном давлении, и концентрат перекристаллизовывают из смеси диэтилового эфира и гексана с получением 278 г (выход 62%) указанного в заголовке винилэфирного производного в виде кристаллов, плавящихся при 117-118oC.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 2924, 2847, 1670, 1639, 1578, 1541, 1529, 1481, 1458, 1257, 1213.
33 (i) N-[2-трет-Бутил-5-(4-циклогексил-3-(метоксивинилиден) бутил)фенил]-2-(9H-ксантен-9-ил)ацетамид
2 мл 2 н. водной соляной кислоты добавляют к раствору 278 мг (0,503 ммоль) N-[2-трет-бутил-5-(4-циклогексил-3-(метоксивинилиден) бутил)фенил]-2-(9H-ксантен-9-ил)ацетамида [полученного, как описано в стадии (I) выше] в 8 мл тетрагидрофурана, и полученную смесь перемешивают 4 ч при 50oC. По истечении этого времени реакционной смеси дают остыть до комнатной температуры, после чего разбавляют диэтиловым эфиром. Разбавленную смесь промывают водой до нейтральной среды и затем насыщенным водным раствором хлорида натрия. Органическую фазу затем высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 15 г силикагеля, используя способ градиентного элюирования смесями метиленхлорида и этилацетата, изменяющимися в отношениях от 100:3 до 100:5 по объему, в качестве элюента. Объединенные элюаты концентрируют упариванием при пониженном давлении, и концентрат перекристаллизовывают из смеси диэтилового эфира и гексана с получением 227 г (выход 83%) указанного в заголовке соединения, плавящегося при 121-122oC.
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 2924, 2851, 1726, 1655, 1639, 1576, 1527, 1479, 1458, 1419, 1363, 1298, 1257.
Приготовление 34
{ A-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо] фенил} метилметилтрифенилфосфоний бромид
34 (I) N-[2-трет-Бутил-5-бромметилфенил]-2-(9H-ксантен-9-ил) ацетамид
3,19 г (9,63 ммоль) тетрабромида углерода добавляют к раствору 2,47 г (6,40 ммоль) N-(2-трет-бутил-5-гидроксиметилфенил)-2- (9H-ксантен-9-ил)ацетамида (полученного, как описано в приготовлении 14) и 2,04 г (7,77 ммоль) трифенилфосфина в 20 мл метиленхлорида, и полученную смесь перемешивают 4 ч при комнатной температуре. По истечении этого времени реакционную смесь помещают в колонку, содержащую 100 г силикагеля, и колонку элюируют 1: 9 по объему смесью диэтилового эфира и метиленхлорида. Элюаты объединяют и концентрируют упариванием при пониженном давлении с получением 2,52 г (выход 85%) указанного в заголовке бромида в виде кристаллов, плавящихся при 229-231oC (после перекристаллизации из этилацетата).
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3216, 1640, 1535, 1487, 1457, 1414, 1366, 1262, 1236, 763.
34 (II) { 4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил} метилметилтрифенилфосфоний бромид
Раствор 2,52 г (5,42 ммоль) N-[2-трет-бутил-5-бромметилвенил]-2-(9H-ксантен-9-ил)ацетамида [полученного, как описано в стадии (I) выше] и 1,58 г (6,02 ммоль) трифенилфосфина в 25 мл толуола нагревают с обратным холодильником 5 ч при энергичном перемешивании. По истечении этого времени дают остыть до комнатной температуры и образовавшийся осадок собирают фильтрацией и распыляют. Осадок промывают толуолом и гексаном и высушивают над безводным сульфатом натрия с получением 3,71 натрия с получением 3,71 г (выход 90% указанного в заголовке соединения в виде порошка.
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3420, 1669, 1482, 1459, 1439, 1256, 1111, 834, 753, 691.
Приготовление 35
1-Бензилокси-5-циклогексил-2-пентанон
35 (I) 4-Циклогексил-1-(бензилоксиметил)бутанол
Раствор 1,01 г (6,73 ммоль) 2-бензилоксиацетальдегида в 6 мл диэтилового эфира добавляют по каплям в течение 3 мин к 14 мл (8,6 ммоль) 0,6 М раствора в диэтиловом эфире 3-циклогексилпропилмагний бромида при ледяном охлаждении, и полученную смесь перемешивают 30 мин при той же температуре и затем 10 мин при комнатной температуре. Для остановки реакции к реакционной смеси добавляют насыщенный водный раствор хлорида аммония. Реакционную смесь затем разбавляют диэтиловым эфиром и разбавленную смесь промывают водой и насыщенным водным раствором хлорида натрия, в указанном порядке. Органическую фазу высушивают над безводным сульфатом магния и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 150 г силикагеля, используя 1:3 по объему смеси этилацетата и гексана в качестве элюента, с получением 1,27 г (выход 68%) указанного в заголовке спиртового производного в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 3470, 3031, 1497, 1451, 1364, 1206, 1100, 1028, 735, 699.
35 (II) 1-Бензилокси-5-циклогексил-2-пентанон
Следуя способу, подобному описанному в приготовлении 19, но использ4уя 4-циклогексил-1-(бензилоксиметил)бутанол [полученный, как описано в стадии (I) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 3032, 1721, 1497, 1451, 1260, 1210, 1102, 1028, 737, 699.
Приготовление 36
3-Циклогексилокси-1-(бензилоксиметил)пропиловый спирт
и
4-циклогексилокси-2-бензилоксибутиловый спирт
36 (i) 4-Циклогексилоксипропиловый спирт
14,0 мл (14,0 ммоль) 1 М гексанового раствора диизобутилалюминий гидрида добавляют по каплям в течение 5 мин к раствору 1,01 г (647 ммоль) 1,5-диоксаспиро[5,5] ундекана в 5 мл метиленхлорида, и полученную смесь перемешивают 1 ч при комнатной температуре. По истечение этого времени реакционную смесь охлаждают льдом и добавляют по каплям в течение 5 мин 20 мл метанола для остановки реакции. Смесь перемешивают 20 мин при комнатной температуре и затем добавляют 2 н. водный раствор соляной кислоты для растворения осадка. Затем смесь экстрагируют диэтиловым эфиром. Экстракт промывают водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия в этом порядке. Органическую фазу высушивают над безводным сульфатом магния и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 25 г силикагеля, используя 3:1 по объему смеси диэтилового эфира и гексана в качестве элюента, с получение 774 мг (выход 76%) указанного в заголовке спиртового производного в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1; 3390, 1451, 1366, 1258, 1092, 984, 967, 930, 889.
36 (II) 1-Циклогексилоксипропаналь
Следуя способу, подобному описанному в приготовлении 19, но используя 3-циклогексилоксипропиловый спирт (полученный, как описано в стадии (I) выше) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 2728, 1727, 1451, 1364, 1258, 1212, 1100, 1025, 984,889.
36 (III) 4-Циклогексилокси-1-бутен
3,3 мл 1,6 М гексанового раствора бутиллития добавляют по каплям в течение 4 мин к суспензии 1,88 мг (5,26 ммоль) метилтрифенилфосфоний бромида в 19 мл тетрагидрофурана при -20oC. Реакционную смесь оставляют на 25 мин, после чего добавляют по каплям в течение 3 мин раствор 747 мг (4,75 ммоль) 3-циклогексилоксипропана (полученного, как описано в стадии (1) выше) в 5 мл тетрагидрофурана. Реакционную смесь затем перемешивают 30 мин при комнатной температуре. Для остановки реакции к реакционной смеси добавляют насыщенный водный раствор хлорида аммония, после чего экстрагируют диэтиловым эфиром. Экстракт промывают водой и насыщенным водным раствором хлорида натрия в указанном порядке. Органическую фазу высушивают над безводным сульфатом магния и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 25 г силикагеля, используя 1:1 по объему смеси метиленхлорида и гексана в качестве элюента, с получением 462 мг ( выход 63) указанного в заголовке олефинового производного в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка) νmax , см-1: 3079, 1642, 1451, 1364, 1258, 1107, 992, 957, 913.
36 (IV) 4-Циклогексилокси-2-гидроксибутиловый спирт 1,9 мл (0,15 ммоль) 2% мас/объем раствора тетраоксида осмия добавляют к раствору 462 мг (3,00 ммоль) 4-циклогексилокси-1-бутена [полученного, как описано в стадии (III) выше] и 702 мг (5,99 ммоль) N-метилморфолин-N-оксида в смеси 20 мл ацетонитрила и 5 мл воды, и полученную смесь перемешивают 12 ч при комнатной температуре. Для остановки к реакционной смеси добавляют водный раствор сульфита натрия, после чего экстрагируют этилацтатом. Экстракт промывают водой, 2 н. водной соляной кислотой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия в указанном порядке. Органическую фазу высушивают над безводным сульфатом магния и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 15 г силикагеля, используя этилацетат в качестве элюента, с получением 443 мг (выход 78%) указанного в заголовке производного диола в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 3990, 1451, 1366, 1258, 1092, 994, 951, 872, 791.
36 (v) 2-Фенил-4-(2-циклогексилэтил)-1,3-диоксолан
43 мг (1,23 ммоль) n-толуолсульфоновой кислоты моногидрата добавляют при охлаждении льдом к раствору 473 мг (2.51 ммоль) 4-циклогексилокси-2-гидроксибутилового спирта [полученного, как описано в стадии (IV) выше] и 1,14 мл (7,56 ммоль) диметилацеталя бензальдегида в 10 мл метиленхлорида, и полученную смесь перемешивают 2,5 ч при комнатной температуре. Для остановки реакции к реакционной смеси добавляют насыщенный водный раствор гидрокарбоната натрия, после чего экстрагируют диэтиловым эфиром. Экстракт промывают насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом магния и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 50 г силикагеля, используя 1:9 по объему смесь диэтилового и метилхлорида в качестве элюента, с получением 578 мг (выход 83%) указанного в заголовке производного 1,3-диоксолана в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 1453, 1403, 1366, 1219, 1096, 1026, 914, 758, 699.
36 (VI) 3-Циклогексилокси-1-(бензилоксиметил)пропиловый спирт и 4-циклогексилокси-2-бензилоксибутиловый спирт.
4,5 мл (4,5 ммоль) 1 М гексанового раствора диизобутилалюминий гидрида добавляют по каплям в течение 5 мин к раствору 560 мг (2,03 ммоль) 2-фенил-4-(2-циклогексилэтил)-1,3-диоксолана [полученного, как описано в стадии (V)выше) в 5 мл метиленхлорида, и полученную смесь перемешивают 50 мин. Для остановки реакции к реакционной смеси добавляют 0,8 мл метанола, и образовавшийся осадок растворяют добавлением 2 н. водный соляной кислоты, после чего экстрагируют диэтиловым эфиром. Экстракт промывают водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом магния и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищает колоночный хроматографией через 60 г силикагеля, используя 1:4 по объему смесь этилацетата и метиленхлорида в качестве элюента, с получением 409 мг (выход 72%) менее полярного спирта, 3-циклогексилокси-1-(бензилоксиметил)пропилового спирта и 133 мг (выход 24%) более полярного вторичного спирта, 4-циклогексилокси-2-бензилоксибутилового спирта соответственно, оба в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1:
3-Циклогексилокси-1 (бензилоксиметил)пропиловый спирт 3450, 1453, 1364, 1206, 1100, 1026, 737, 699.
4-Циклогексилокси-2-бензилоксибутиловый спирт 3440, 1453, 1208, 1092, 1938, 737, 699.
Приготовление 37
1-Бензилокси-4-циклогексилокси-2-бутанон
Следуя способу, подобному описанному в приготовлении 19, но используя 3-циклогексилокси-1-(бензилоксиметил)пропиловый спирт (полученный, как описано в приготовлении 36) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка, νmax , см-1: 3032, 1725, 1453, 1366, 1258, 1209, 1195, 1026, 739, 698.
2-Бензилокси-4-циклогексилоксибутаналь
Следуя способу, подобному описанному в приготовлении 19, но используя 4-циклогексилокси-2-бензилоксибутиловый спирт (полученный, как описано в приготовлении 36) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают в заголовке соединение в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax , см-1: 3032, 1734, 1453, 1366, 1209, 1104, 1026, 951, 739, 699.
Приготовление39
N-{ 2-трет-Бутил-5-[2-(оксиран-2-ил)этил] фенил} -2-(9N-ксантен-9-ил)ацетамид
Суспензию 690 мг (15,8 ммоль) гидрида натрия (в виде 55% по массе дисперсии в минеральном масле, предварительно промытой ге5ксаном) в 10 мл диметилсульфоксида перемешивают 30 мин, после чего добавляют 2,66 г (12,1 ммоль) триметилсульфоксоний иодида. Полученную смесь перемешивают 1 ч при 40oC и затем прибавляют по каплям в течение 5 мин раствор 4,50 г (10,5 ммоль) N-[2-трет-бутил-5-(3-оксопропил)фенил] -2-(9N-ксантен-9-мл)ацетамида (полученного, как описано в приготовлении 19) в 25 мл тетрагидрофурана. Реакционную смесь затем перемешивают 1 ч, после чего реакционную смесь затем перемешивают 1 ч, после чего реакционную смесь затем перемешивают 1 ч, после чего реакционную смесь охлаждают до комнатной температуры и затем разбавляют этилацетатом. Разбавленную смесь промывают несколько раз водой и один раз насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом магния, и затем растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографии через 250 г силикагеля, используя 9:1 по объему смесь метиленхлорида и этилацетата в качестве элюента с получением 3,00 г (выход 65%) указанного в заголовке соединения в виде кристаллов, плавящихся при 156-157oC (после перекристаллизации из этилацетата и гексана)
Инфракрасный абсорбционный спектр (KBr), νmax , см-1:3266, 2965, 1655, 1576, 1481, 1458, 1404, 1302. 1254, 829, 756.
Приготовление 40
N-(2-трет-Бутил-5-{ 4-циклогексил-3-[2-(бензилоксикарбонилметилтио) ацетокси]бутил}фенил-2-(9N-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в приготовлении 25, но используя N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2-(9N- ксантен-9-ил)ацетамид (полученный, как описано а примере 12) и 2-(бензилоксикарбонилметилтио)уксусную кислоту в качестве исходных продуктов, в соответствующем соотношении, используемом в этом приготовлении, получают указанное в заголовке соединенные в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3275, 2923, 1733, 1660, 1481, 1457, 1256, 1153, 1119, 760.
Приготовление 41
N-(2-трет-Бутил-5-{4-циклогексил-3- [2-(бензилоксикарбонилметилсульфонил)ацетокси] бутил}фенил) - 2-(9H-ксантен-9-ил)ацетамид
и
N-(2-трет-Бутил-5-{4-цтклогексил-3- [2-(бензилоксикарбонилметилсульфинил)ацетокси] бутил}фенил) - 2-(9H-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в примере 194, но используя N-(2-трет-бутил-5-{ 4-цикдлгексил-3- [2-(бензилоксикарбонилметилтио)ацетокси]бутил} фенил) -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в приготовлении 40) в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанные в заголовке соединения в виде пенообразных продуктов.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1:
N-(2-трет-бутил-5-{ 4-циклогексил-3- [2-бензилоксикарбонилметилсульфонил)ацетокси] бутил] фенил) -2-(9H-ксантен-9-ил)ацетамид 3274, 2924, 1738, 1660, 1481, 1458, 1340, 1294, 1256, 1117, 760;
N-(2-трет-бутил-5-{4-циклогексил-3- [2-(бензилоксикарбонилметилсульфинил)ацетокси] бутил}фенил)-2-(9H-ксантен-9-ил)ацетамид 3271, 2923, 1732, 1660, 1481, 1457, 1257, 1118, 1058, 759.
Приготовление 42
N-{ 2-трет-Бутил-5-[3-(бензилоксикарбонилметоксикарбонилокси) -4-циклогексилбутил]фенил}-2-(9H-ксантен-9-ил)ацетамид
Раствор 55 μл (0,46 ммоль) трихлорметил хлорформиата в 1 мл тетрагидрофурана добавляют по каплям раствор 74 μл (0,91 ммоль) пиридина в 1 мл тетрагидрофурана при охлаждении льдом и затем температуре дают постепенно достичь комнатной. Смесь перемешивают 1 ч при комнатной температуре, после чего опять охлаждают и добавляют по каплям 400 мг (0,76 ммоль) N-[2-трет-бутил-5- (4-циклогексил-3-гидроксибутил)фенил]-2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 12) в 3 мл тетрагидрофурана. Смесь перемешивают 1 ч при той же температуре и затем растворитель удаляют отгонкой при пониженном давлении с получением пенообразного продукта в виде осадка. Этот осадок растворяют в 1 мл метиленхлорида и добавляют по каплям раствор 151 мг (0,91 ммоль0 бензил - гидроксиацетата в 1,5 мл метиленхлорида и затем 110 мг (0,91 ммоль) 4-(N, N-диметиламино)пиридина при охлаждении льдом. Смесь перемешивают при комнатной температуре 1 ч и затем реакционную смесь разбавляют метиленхлоридом, после чего ее промывают водой и насыщенным водным раствором хлорида натрия, в этом порядке. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 75 г силикагеля, используя 1: 9 по объему смесью метиленхлорида и этилацетата в качестве элюента, с получением 522 г (выход 96%) указанного указанного в заголовке соединения в виде бесцветного пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3274, 2924, 2853, 1750, 1655, 1480, 1458, 1422, 1256, 1194, 758.
Приготовление 43
N-{ 2-трет-Бутил-5-[7-циклогексил-3-(метоксиметокси) гептил] фенил}-2-(9H-ксантен 9-ил)ацетамид
43 (I) (S)-[6-Бензилокси-5-(метоксиметокси)гексил] циклогексан 25,6 мл (184 ммоль) триэтиламина и затем 9,3 мл (122 ммоль) метоксиметилхлорида добавляют к раствору 10 г (34.4 ммоль) (S) -(I-бензилокметил-5-циклогексил)пентилового спирта [полученного подобно способу, описанному в приготовлении 23 (I), но используя циклогексилропилмагний бромид] в 200 мл N, N-диметилформамида, и полученную смесь перемешивают при 80oC 5,5 ч. По истечении этого времени реакционную смесь концентрируют упариванием при пониженном давлении и концентрат смешивают с ледяной водой, после чего экстрагируют три раза этилацетатом. Объединенные экстракты промывают насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом натрия и растворитель отгоняют при пониженном давлении. Полученный остаток очищают колоночной хроматографией через силикагель, используя 9:1 по объему смесь тегсана и этилацетата в качестве элюента, с получением 10,2 г (выход 89%) указанного в заголовке метоксиметильного производного в виде масла.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ ppm: 0,73-1,80 (19H, мультиплет); 3,39 (3H, синглет); 3,51 (2H, дублет, J = 5Гц); 3,69-3,85 (IH, мультиплет); 4,56 (2H, синглет); 4,68(IH, дублет, J=7 Гц); 4,79 (IH, дублет, J = 7 Гц); 7,23-7,42 (5H, мультиплет).
43 (II) (S)-6-Циклогексил-2-(метоксиметилокси)гексиловый спирт
Следуя способу, подобному описанному в приготовлении 23 (III), но используя (S)-[6-бензилокси-5-(метоксиметокси)гексил] циклогексан [полученный, как описано в стадии (I) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде масла.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ ppm: 0,75-1,8 (19H, мультиплет); 2,9-3,2 (IH, уширенный синглет); 3,43 (3H, синглет); 3,45-3,65 (3H, мультиплет); 4,68 (IH, дублет, J = 7 Гц); 4,74 (IH, дублет, J = 7 Гц);
43 (III) (S)-2-трет-Бутил-5-(3-метоксиметилокси-7-циклогексил-1- гептенил)-1-нитробензол
(S)-6-Циклогексил-2-(метоксиметилокси)гексиловый спирт [полученный, как описано в стадии (II) выше] окисляют способом, подобным описанному в приготовлении 23 (IV) с получением альдегидного производного. Его подвергают реакции Виттига способом, подобным описанному в приготовлении 23(V), с получением указанного в заголовке олефинового производного в виде масла.
43 (IV) (S)-2-трет-Бутил-5-(3-метоксиметилокси-7- циклогексилгептил)анилин.
Раствор 2,2 г (5,27 ммоль) (S)-2-трет0бутил-5-(3-метоксиметилокси-7-циклогексил-1-гептенил) -1-нитробензола [полученного, как описано в стадии (III) выше] в 40 мл этанола энергично перемешивают в токе водорода в течение 6 ч в присутствии 1,5 г 10% по массе палладия на угле. Катализатор затем отфильтровывают и промывают этанолом. Фильтрат и промывные воды объединяют и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через силикагель, используя 9:1 по объему смесь гексана и этилацетата в качестве элюента, с получением 1,16 г (выход 57% указанного в заголовке производного анилина в виде масла.
Спектр ядерного магнитного резонанса (CDCI3, 270 МГц), δ ppm: 0,75-1,9 (21H, мультиплет); 1,40 (9H, сигнлет); 2,43-2,70 21H, мультиплет); 3,41 (3H, синглет); 3,59 (IH, квинтет, J = 6 Гц); 4,68 (2H, синглет); 6,51 (IH, синглет); 6,58(IH, дублет, J= 8 Гц); 7,13 (IH, дублет, J= 8 Гц).
43 (V) (S)-N-(2-трет-Бутил-5-[7-цтклогексил -3-(метоксиметокси) гептил] фенил}-2-(9H-ксантен 9-ил)ацетамид
Следуя способу, подобному описанному в примере 35, но используя (S)-2-трет-бутил-5-(3-метоксиметилокси-7-циклогексилгептил)анилин [полученный, как описано в стадии (IV) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 71-73oC (после перекристаллизации из гексана). Инфракрасный абсорбционный спектр (KBr) νmax , см-1: 3221, 1639, 1577, 1535, 1482, 1456, 1263, 1255, 1034. Спектр ядерного магнитного резонанса (CDCL3, 270 МГц), δ ppm: 0,75-1,90 (21H, мультиплет); 2,25-8,80 (4H, мультиплет); 3,42 (3H, синглет); 3,45-3,70 (IH, мультиплет); 4,60-4,82 (3H, мультиплет); 6,90-7,46 (IIH, мультиплет).
Приготовление 44
N-{2-трет-Бутил-5-[3-(4-бензилоксикарбонилметилбензоилокси) -4-циклогексилбутил]фенил}-2-(9H-ксантен-9-ил)ацетамид
44 (I) 4-Гидроксиметилфенилуксусная кислота
Раствор 9,00 г (39,3 ммоль) n-бромметилфенилуксусной кислоты, 3,14 г (78,6 ммоль) гидроксида натрия и 3,54 г (43,2 ммоль) ацетата натрия в 40 мл воды перемешивают в течение 4 ч при температуре 100oC. По окончании этого времени его охлаждают до комнатной температуры и затем подкисляют 2 н. водной соляной кислотой. Затем раствор экстрагируют этилацетатом. Экстракт промывают насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом натрия, а растворитель удаляют отгонкой при пониженном давлении. Полученный осадок перекристаллизовывают из смеси этилацетата и метиленхлорида с получением 3,60 г (выход 55%) указанного в заголовке соединения, плавящегося при 136-137oC.
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3304, 1703, 1520, 1421, 1408, 1360, 1288, 1234, 1200, 1188, 1009.
44 (II) Бензил 4-гидроксиметилфенилацетат 2,52 г (23,8 ммоль) карбоната натрия и 8,13 г (47,6 ммоль) бензилбромида добавляют к раствору 3,95 г (23,8 ммоль) 4-гидроксиметилфенилуксусной кислоты [полученной, как описано в стадии (I) выше] в 40 мл N, n-диметилформамида, и полученную смесь перемешивают в течение 3,5 ч. По окончании этого времени реакционную смесь разбавляют диэтиловым эфиром, и разбавленную смесь промывают водой, а затем насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом натрия, а растворитель удаляют отгонкой при пониженном давлении. Полученный осадок очищают колоночной хроматографией через 200 г силикагеля, используя метод градиентного элиминирования, смесью метиленхлорида и этилацетата, изменяющейся от 100: 5 до 100:3 по объему, что дает 5,03 г (выход 82%) указанного в заголовке соединения в виде масла.
Инфракрасный абсорбционный спектр (жидкая пленка), νmax см-1: 2941, 2876, 1734, 1516, 1499, 1456, 1423, 1377, 1336, 1259, 1221, 1149.
44 (III) Бензил 4-формилфенилацетат
Следуя способу, подобному описанному в приготовлении 19, но используя бензил 4-гидроксиметилфенилацетат [полученный, как описано в стадии (II) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 54-55oC (после перекристаллизации из смеси диэтилового эфира и гексана).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 1726, 1689, 1607, 1578, 1458, 1425, 1383, 1338, 1225, 1192, 1169.
44 (IV) Бензил 4-карбоксифенилацетат
Следуя способу, подобному описанному в приготовлении 21 (vi), но используя бензил 4-формилфеилацетат [полученный, как описано в стадии (III) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 123-124oC (после перекристаллизации из смеси метиленхлорида и этилацетата).
Инфракрасный абсорбционный спектр (KBr), νmax , см.-1: 1718, 1703, 1685, 1610, 1450, 1429, 1323, 1292, 1271, 1182, 1151.
44 (V) N-(2-трет-Бутил-5-[3-(4-бензилоксикарбонилметилбензоилокси) -4-циклогексилбутил]фенил}-2-(9H-ксантен-9-ил)ацетамид
Следуя способу, подобному описанному в приготовлении 25, но используя бензил 4-карбоксифенилацетат [полученный, как описано в стадии (VI) выше] и N-[2-трет-бутил-5-(4-циклогексил-3- гидроксибутил)фенил] -2-(9H-ксантен-9-ил)ацетамид (полученный, как описано в примере 12) в качестве исходных продуктов, в соответствующем соотношении, используемом в этом приготовлении, получают указанное в заголовке соединение в виде пенообразного продукта.
Инфракрасный абсорбционный спектр (KBr), νmax, см-1: 2924, 2851, 1736, 1715, 1686, 1655, 1612, 1578, 1522, 1479, 1458, 1273, 1257.
Приготовление 45
2-Метил-6-{[3-(1-имидазолил)пропил]оксиметил}анилин
45 (I) 2-Метил-6-мезилоксиметил-2-нитробензол
Следуя способу, подобному описанному в приготовлении I (I), но используя 2-метил-6-гидроксиметил-1-нитробензол в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке метансульфонил производное в виде кристаллов, плавающих при 48-50oC.
45 (II) 2-Метил-6-[(3-гидроксипропил)оксиметил]-1-нитробензол
Следуя способу, подобному описанному в приготовлении I (II), но используя 2-метил-6-мезилоксиметил-1-нитробензол [полученный, как описано в стадии (i) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке спиртовое производное в виде масла.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,84 (2H, квинтет, J=6 Гц); 2,35 (3H, сиглент); 3,62 (2H, триплет, J=6 Гц); 3,75 (2H, широкий синглет); 4,56 (2H, синглет); 7,2-7,43 (3H, мультиплет),
45 (III) 2-Метил-6-[(3-мезилоксопропил)оксиметил]-1- нитробензол
Следуя способу, подобному описанному в примере 1 (I), но используя 2-метил-6-[(3-гидроксипропил)оксиметил]-1-нитробензол [полученный, как описано в стадии (II) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке метансульфонил производное в виде масла.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 2,01 (2H, квинтет, J= 6 Гц); 2,35 (3H, синглет); 3,03 (3H, синглет); 3,56 (2H, триплет, J=6 Гц); 4,32 (2H, триплет, J=6 Гц); 4,56 (2H, синглет); 7,20-7,42 (3H, мультиплет).
45 (IV) 2-Метил-6-{[3-(1-имидазолил)пропил]оксиметил]-1- нитробензол
Следуя способу, подобному описанному в примере I (II), но используя 2-метил-6-[(3-мезилоксипропил)оксиметил] -1-нитробензол [полученный, как описано в стадии (III) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом примере, получают указанное в заголовке имидазолил производное в виде масла.
Спектр ядерного магнитного резонанса (CDCl3, 270 Мгц), δ , ppm: 1,99 (2H, квинтет, J=6 Гц); 2,37 (3H, синглет); 3,38 (3H, триплет, J=6 Гц); 4,04 (2H, триплет, J= 6 Гц); 4,54 (2H, синглет); 6,91 (1H, синглет); 7,05 (1H, синглет); 7,2-7,42 (3H, мультиплет); 7,48 (1H, синглет).
45 (V) 2Метил-6-{[3-(1-имидазолил)пропил]оксиметил}анилин
Следуя способу, подобному описанному в приготовлении 43 (IV), но используя 2-метил-6-{ [3-(1-имидазолил)пропил] оксиметил} -1-нитробензол [полученный, как описано в стадии (IV) выше] в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке анилиновое производное в виде масла.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 2,01 (2H, квинтет, J=6 Гц); 2,19 (3H, синглет); 3,38 (2H, триплет, J=6 Гц); 4,04 (2H, уширенный синглет); 4,52 (2H, синглет); 6,65 (1 H, триплет, J=7 Гц); 6,85 (1H, синглет); 6,93 (1H, дублет, J=7 Гц); 7,04 (1H, синглет); 7,06 (1H, дублет, J=7 Гц); 7,40 (1 H, синглет).
Приготовление 46
N-{ 2-трет-Бутил-5-[4-циклогексил-3-(трет-бутилдиметилсилилокси) бутил} -2,2-диметилдодеканамид
Следуя способу, подобному описанному в примере 21, но используя 2,2-диметилдодекановую кислоту и 2-трет-бутил-5-[4-циклогексил-3- (трет-бутилдиметилсилилокси)бутил] анилин [полученный, как описано в приготовлении II (I) выше] в качестве исходных продуктов, в соответствующем соотношении, используемом в этом примере, получают указанное в заготовке соединение в виде пенообразного продукта.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,05 (6H синглет); 0,83 (9H, синглет); 0,85-0,96 (6H, мультиплет); 1,11-1,35 (30H, мультиплет); 1,40 (9H, синглет); 1,58-1,78 (6H, мультиплет); 2,53-2,67 (2H, мультиплет); 3,80 (1H, nhbgktn, J=5 Гц); 6,94 (1H, дублет, J=8 Гц); 7,29 )1H, дублет, J=8 Гц); 7,38 (1H, синглет); 7,56 (1H, синглет).
Приготовление 47
N-{2-трет-Бутил-5-[4-циклаогексил-3-(трет-бутилдиметилсилокси) бутил]фенил}-6, 11-дигидробенз-[b, e]оксипенил-11-карбоксамид
Следуя способу, подобному описанному в примере 21, но используя 6, 11-дигидробенз[b, e] оксепин-11-карбоновую кислоту и 2-трет-бутил--5-[4-циклогексил-3-(трет-бутилдиметилсилилокси)бутил] анилин [полученный, как описано в приготовлении II (I) выше] в качестве исходных продуктов, в соответствующем соотношении, используемой в этом примере, получают указанное в заголовке соединение в виде бесцветных кристаллов.
Инфракрасный абсорбционный спектр (KBr), νmax , см-1: 3276, 2925, 1648, 1520, 1447, 1256, 1075, 835, 774, 759.
Приготовление 48
N-{ 2-трет-Бутил-5-[4-циклогексил-3-(трет-бутилдиметилсилилокси) бутил] фенил}--2-(фенил-циклопентил)ацетамид
Следуя способу, подобному описанному в примере 21, но используя 2-(1-фенилциклопентил)уксусную кислоту и 2-трет-бутил-5- [4-циклогексил-3-(трет-бутилдиметилсилилокси)бутил] анилин [полученный, как описано в приготовлении II (I) выше] в качестве исходных продуктов, в соответствующем соотношении, используемом в виде пенообразного продукта.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 0,06 (6H, синглет); 0,85-0,96 (6H, мультиплет); 0,91 (9H, синглет); 1,07 (9H, синглет); 1,18-1,41 (12H, мультиплет); 1,57-2,19 (5H, мультиплет); 2,55-2,60 (2H, мультиплет); 2,70 (1H, синглет); 3,02 (1Нб синглет); 3,78 (1H, триплет, J=6 Гц); 6,33 (1H, синглет); 6,88-7,45 (8H, мультиплет).
Приготовление 49
(S)-1-(2-{ 4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил} этил)-2-циклогексилэтил дибензил фосфат
0,86 мл (1,72 ммоль) 2 M тетрагидрофуранового раствора третбутилмагнийхлорида добавляют в течение 2 мин к раствору 420 мг (0,799 ммоль) (S)-N-[22-трет-бутил-5-(4-циклогексил-3-гидроксибутил] -2-(9H-ксантен-9-ил) оцетамида (полученного, как описано в примере 102) в 5 мл тетрагидрофурана, и полученную смесь перемешивают еще 10 мин. Затем добавляют раствор 556 мг (1,87 ммоль) дибензилфосфорилхлорида в 2 мл тетрагидрофурана, после чего смесь перемешивают 1 ч. Для остановки реакции к реакционной смеси добавляют насыщенный водный раствор хлорида аммония, затем разбавляют водой. Разбавленную смесь экстрагируют этилацетатом. Экстракт промывают насыщенным водным раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом магния, и растворитель удаляют отгонкой при пониженном давлении. Полученный остаток очищают колоночной хроматографией через 50 г силикагеля, используя 1: 9 по объему смесью этилацетата и метиленхлорида в качества элюента получая 505 мг (выход 80%) указанного в заголовке соединения в виде смолообразного продукта.
Инфракрасный абсорбционный спектр (пленка), νmax , см-1: 3255, 1655, 1522, 1480, 1459, 1256, 999, 758, 696.
Приготовление 50
(К)-2-(2{4-трет-Бутил-3-[2-(9H-ксантен-9-ил)ацетамидо]фенил} этил)-2-циклогексилбутил бензил сукцинат
0,09 мг Диэтилазодикарбоксилата добавляют при охлаждении на ледяной бане к раствору 260 мг (0,47 ммоль) (S)-N-[2-трет-бутил-5-(6-циклогекссил-3-гидроксигексил)фенил] -2-(9H- ксантен-9-ил)ацетамида (полученного, как описано в примере 189), 117 мг (0,56 ммоль) бензил гидросукцината и 148 мг (0,56 ммоль) трифенилфосфина в 5 мл тетрагидрофурана, и полученную смесь перемешивают 1 ч при комнатной температуре. По истечении этого времени реакционную смесь перемешивают с насыщенным водным раствором гидрокарбоната натрия, после чего экстрагируют этилацетатом. Экстракт промывают насыщенным водным раствором хлорида натрия затем высушивают над безводным сульфатом натрия. Растворитель удаляют отгонкой при пониженном давлении, и полученный остаток очищают колоночной хроматографией через силикагель, используя способ градиентного элюирования, смесью этилацетата и гекссана в отношении от 1:10 до 1: 5 по объему в качестве элюента, с получением фракций, содержащих небольшие количества примесей. Хроматографию повторяют в тех же условиях и получают 258 мг (выход 74%) указанного в заголовке соединения в виде пенообразного продукта.
[α]D = + 5.05o (с=1.11, CHCl3)/
Приготовление 51
2-трет-Бужил-5-(йодметил)-1-нитробензол
51(i) 2-трет-Бутил-5-(метансульфонилоксиметил)-1-нитробензол
Следуя способу, подобному описанному в приготовлении 21 (ii), но используя 2-трет-бутил-5-гидроксиметил-1-нитробензол (полученный, как описано в приготовлении 12) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заготовке метансульфонилпроизводное.
51 (ii) 2-трет-Бутил-5-(йодметил)-1-нитробензол 1,65 г (11 моль) йодида натрия добавляют к раствору 2,00 г (6,96 ммоль) 2-трет-бутил-5-(метансульфонилоксиметил)-1-нитробензола [полученного, как описано в стадии (i) выше] в 40 мл ацетона, и смесь перемешивают при 50oC в течение 20 мин. По истечении этого времени реакционную смесь охлаждают до комнатной температуры и фильтруют. Остаток промывают этилацетатом и объединенные фильтрат и промывание жидкости концентрируют упариванием при пониженном давлении. Концентрат растворяют в этилацетате, и полученный раствор промывают водой, водным раствором трисульфата натрия и насыщенным водным раствором хлориды натрия, в данной последовательности. Органическую фазу высушивают над безводным сульфатом натрия и растворитель удаляют отгонкой при пониженном давлении с получением 2,18 г (выход 98%) указанного в заголовке соединения в виде кристаллов, плавящихся при 98-99oC (после перекристаллизации из смеси метиленхлорида, диэтилового эфира и гексана).
Инфракрасный абсорбированный спектр (KBr), νmax см-1: 1530, 1370, 1250, 1169, 1061, 886, 839, т 807, 627.
Спектр ядерного магнитного резонанса (CDCl3, 270 МГц), δ , ppm: 1,39 (H, синглет); 4,39 (2H, синглет); 7,32 (1H, дублет, J=2 Гц); 7,44 (1H, дублет дублетов, J= 2&8 Гц); 7,48 (1H, дублет, J=8 Гц).
Приготовление 52
(4-трет-Бутил-3-нитрофенил)метилтрифенилфосфоний йодид
Следуя способу, подобному описанному в приготовлении 34 (II), но используя 2-трет-бутил-5-(йодметил)-1-нитробензол (полученный, как описано в приготовлении 51) в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде порошка. Продукт используют в следующей стадии без дополнительной очистки.
Приготовление 53
N-[2-Метоксиметил-6-формилфенил]-2-(9H-ксантен-9-ил)ацетамид
53 (i) N-[2-Метоксиметил-6-(гидроксиметил)фенил] -2-(9H-ксантен-9 ил)ацетамид
Следуя способу, подобному описанному в приготовлении 14, но используя 2-метоксиметил-6-(трет-бутилдиметилсилилоксиметил)-1-нитробензол в качестве исходного продукта, в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке амидное производное в виде кристаллов, плавящихся при 194-194,5oC (после перекристаллизации из смеси метилхлорида и этилацетата).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3256, 1647, 1596, 1577, 1523, 1484, 1457, 1362, 1301, 1263, 1098, 1066, 752.
53(II) N-[2-Метоксиметил-6-формилфенил]-2-(9H-ксантен-9-ил)ацетамид.
Следуя способу, подобному описанному в приготовлении 19, но используя N-[2-метоксиметил-6-(гидроксиметил)фенил] -2-(9H-ксантен-9-ил)ацетамид [полученный, как описано в стадии (I) выше] в качестве исходного продукта в соответствующем количестве, используемом в этом приготовлении, получают указанное в заголовке соединение в виде кристаллов, плавящихся при 202,5-203oC (после перекристаллизации из смеси метиленхлорида и ацетона).
Инфракрасный абсорбционный спектр (KBr), νmax см-1: 3270, 1698, 1645, 1592, 1578, 1518, 1482, 1456, 1254, 1250, 1112, 755.
Приготовление 54
N-[5-(3-гидроксипропокси)-2-метилтиофенил]-2-(9H-ксантен-9-ил) ацетамид
После процедуры, подобной той, что описана в приготовлении 14, но с использованием 5-(3-трет-бутилдиметилсилилоксипропил)окси-2-метилтио-1-нитробензола в качестве исходного материала, в относительном количестве, аналогично использованному в указанном приготовлении, целевое соединение получают в виде кристаллов с температурой плавления 136-137oC (после рекристаллизации из смеси этилацетата, диэтилового эфира и гексана).
Спектр инфракрасного поглощения (KBr), νmax: 3297, 1657, 1600, 1574, 1530, 1481, 1457, 1299, 1260, 1237, 1189.
Приготовление 55
N-(2-этил-6-{ 1-[2-(9H-ксантен-9-ил)ацетокси]пропил}фенил)-2 (9H-ксантен-9-ил)ацетамид
После процедуры, подобной той, что описана в примере 21, но с использованием 2,2 эквивалента 2-(9H-ксантен-ил)ацетилхлорида и 1 эквивалента 2-этил-6-(1-гидроксипропил)-анилина, получают целевое соединение в виде пеноподобного материала.
Спектр инфракрасного поглощения (KBr), νmax: 2968, 2932, 2876, 1726, 1657, 1601, 1576, 1479, 1458, 1363, 1342, 1255.
Лекарственный состав 1
Препарат в виде твердой капсулы.
100 мг одного из следующих активных соединений: 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния, помещают в твердую желатиновую капсулу обычного отдельного типа, и капсулы промывают и затем высушивают. Используемыми активными соединениями являются полученные, как описано в примерах 2, 4, 12, 26, 57 и 59.
Лекарственный состав 2
Препарат в виде мягкой капсулы
Готовят смесь одного из соединений, полученных, как описано в примерах 2, 4, 12, 26, 57 или 59, с маслом соевых бобов. Смесь выливают в желатин, используя наполняющий насос для получения мягких капсул, каждая из которых содержит 100 мг активного ингредиента. Капсулы промывают и высушивают. Другие капсулы готовят, используя масло хлопковых семян вместо соевых бобов. Если желательно, могут быть также использованы другие пищевые масла.
Лекарственный состав 3
препарат в виде таблетки
100 г активного соединения, 0,2 мг коллоидного диоксида кремния, 5 мг стеарата магния, 275 мг качественной кристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы смешивают и затем формуют в таблетки обычными методами. Используемыми активными соединениями являются полученные, как описано в примерах 2, 4, 12, 26, 57 и 59. Когда желательно, осуществляют покрытие таблеток.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АМИДОВ И КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АСАТ-ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 1996 |
|
RU2128165C1 |
| ПЕПТИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2106357C1 |
| ГЕКСАГИДРОНАФТАЛИНОВЫЕ СЛОЖНОЭФИРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2104997C1 |
| АЗАСТЕРОИДНЫЕ СОЕДИНЕНИЯ | 1991 |
|
RU2070204C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СЛОЖНЫЕ ЭФИРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2086541C1 |
| АНАЛОГИ ЛИПИДА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2076107C1 |
| ПРОИЗВОДНЫЕ ИЗОКСАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2165415C2 |
| ПИПЕРИДИЛОКСИ- ИЛИ ХИНУКЛИДИНИЛОКСИ-ИЗОКСАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СВОЙСТВАМИ УМЕНЬШАТЬ НАРУШЕНИЯ ПОЗНАВАТЕЛЬНОЙ СПОСОБНОСТИ | 1993 |
|
RU2079496C1 |
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2097383C1 |
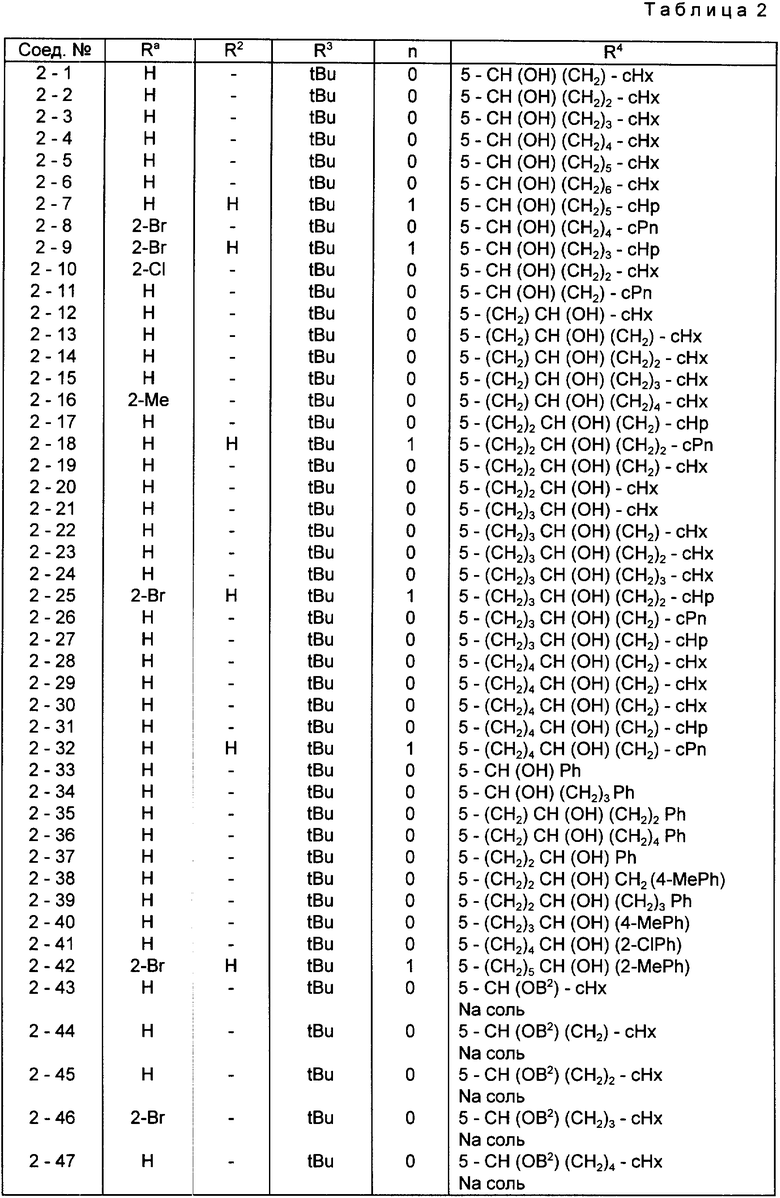
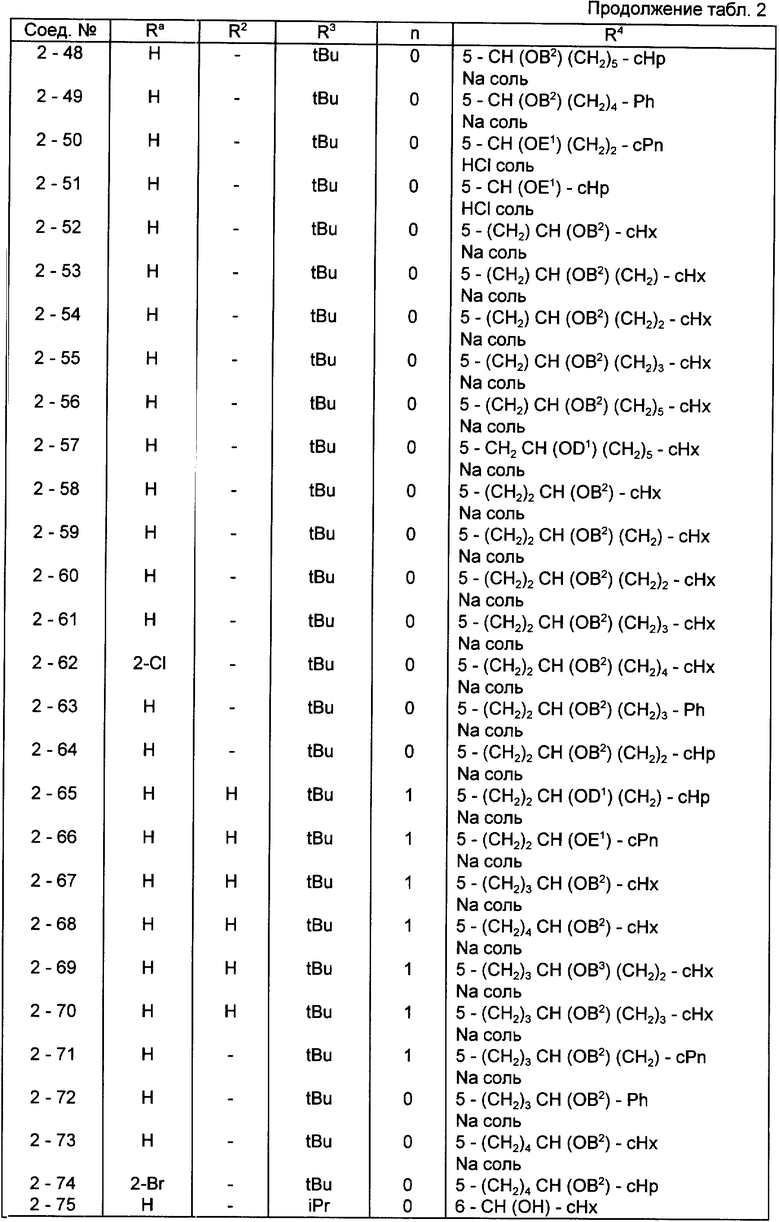
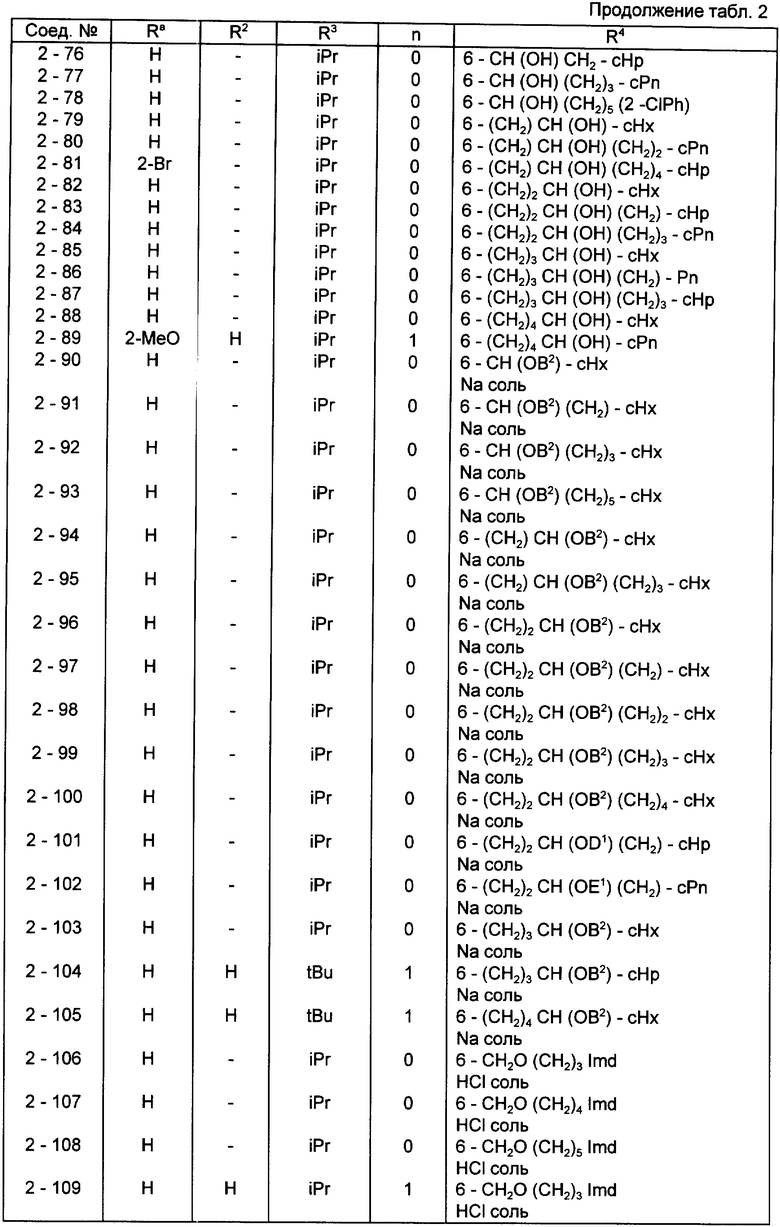
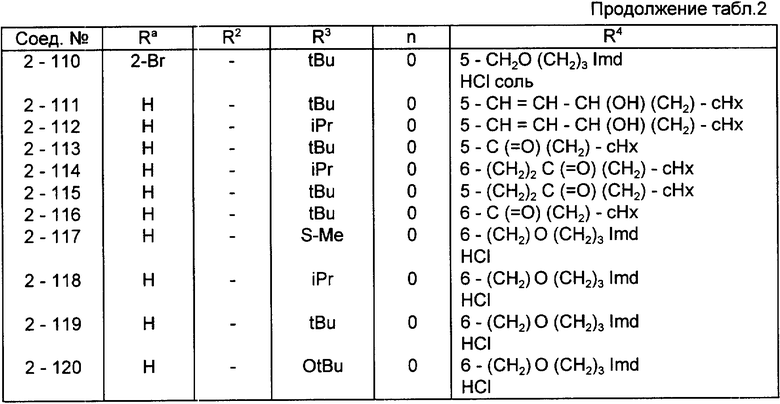
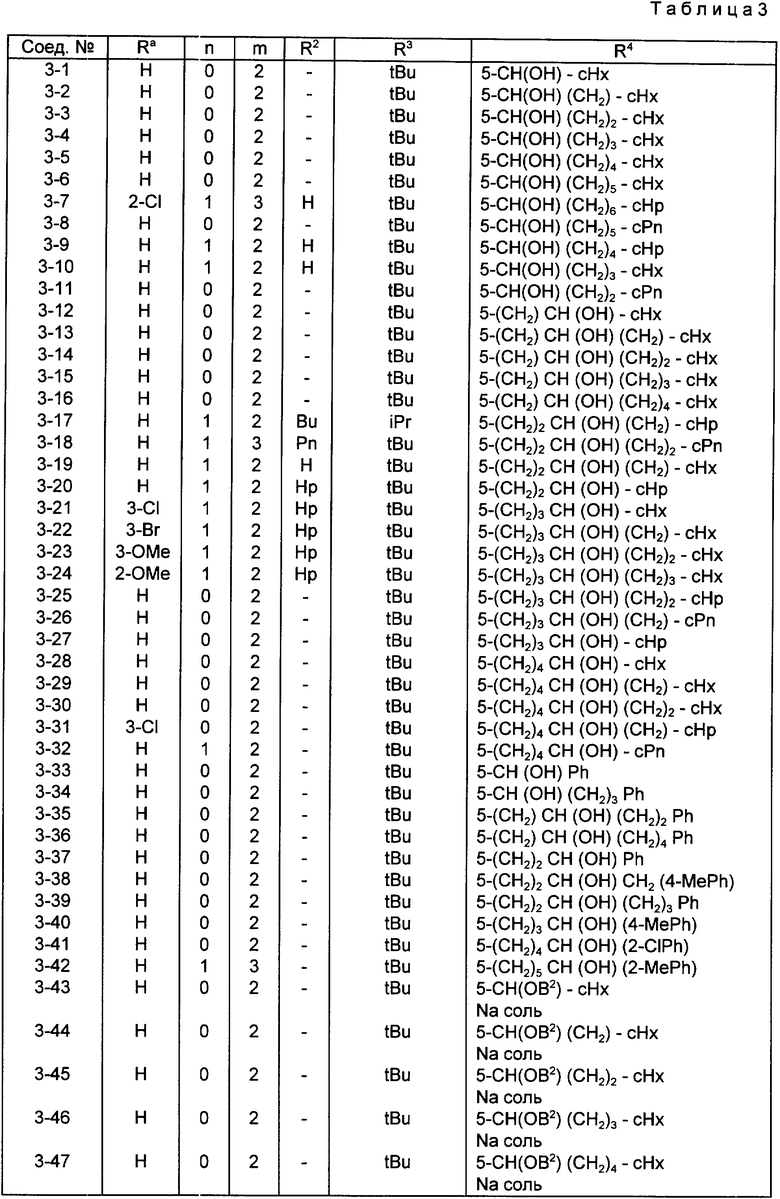
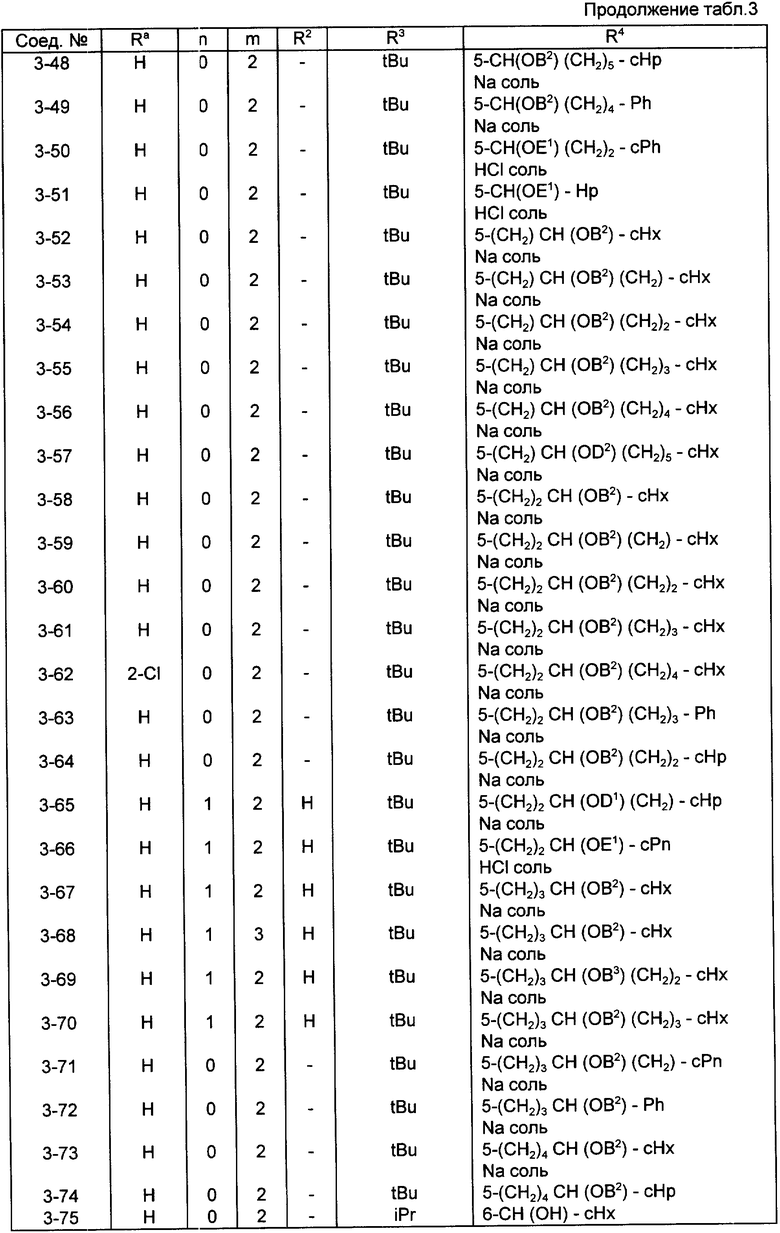
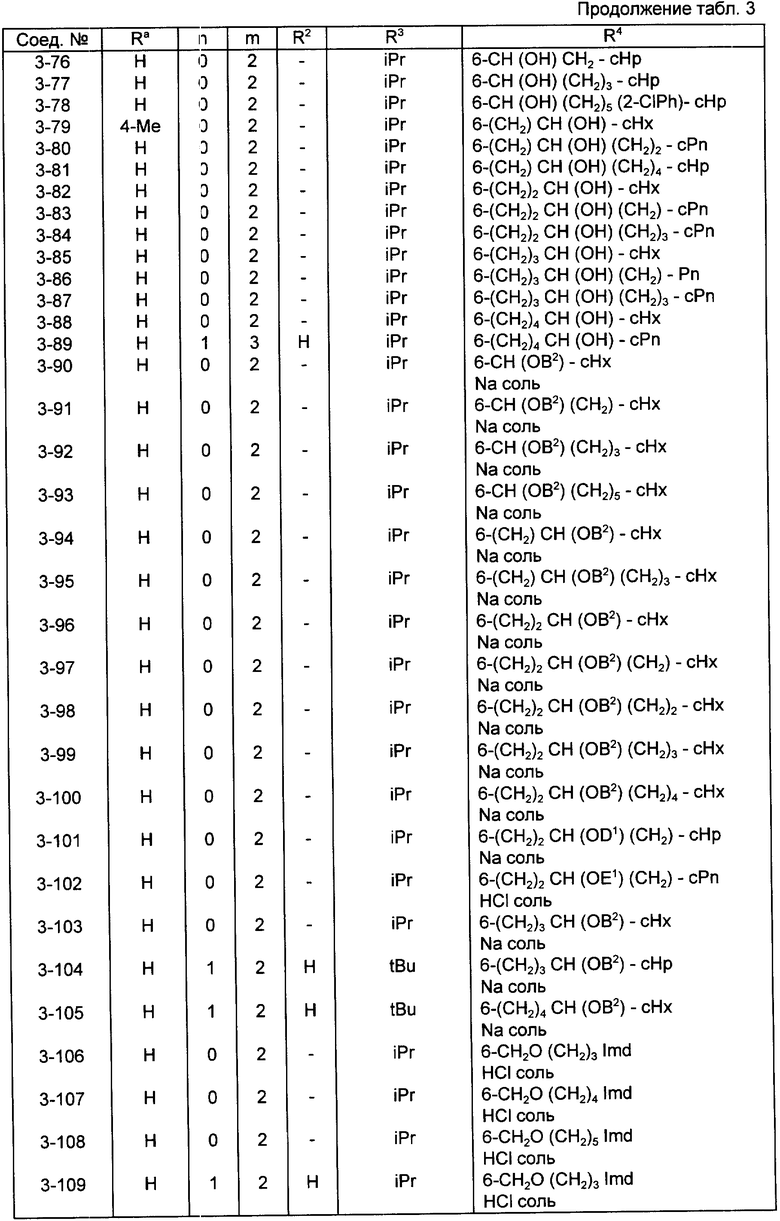
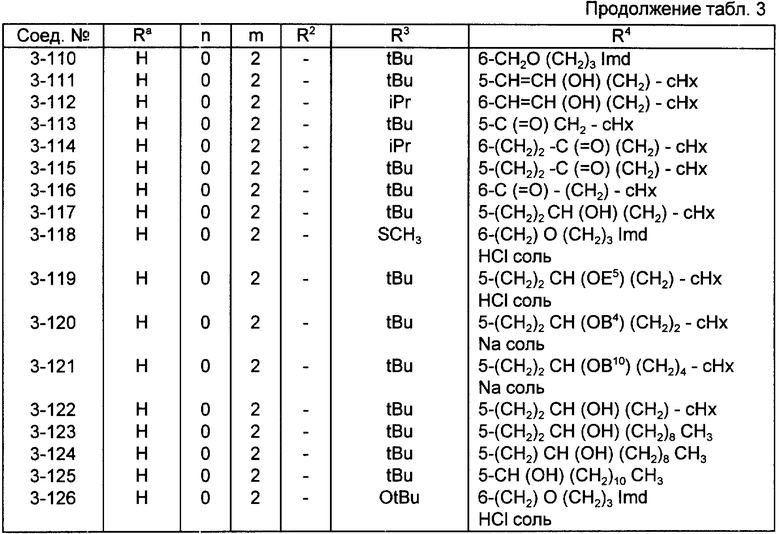
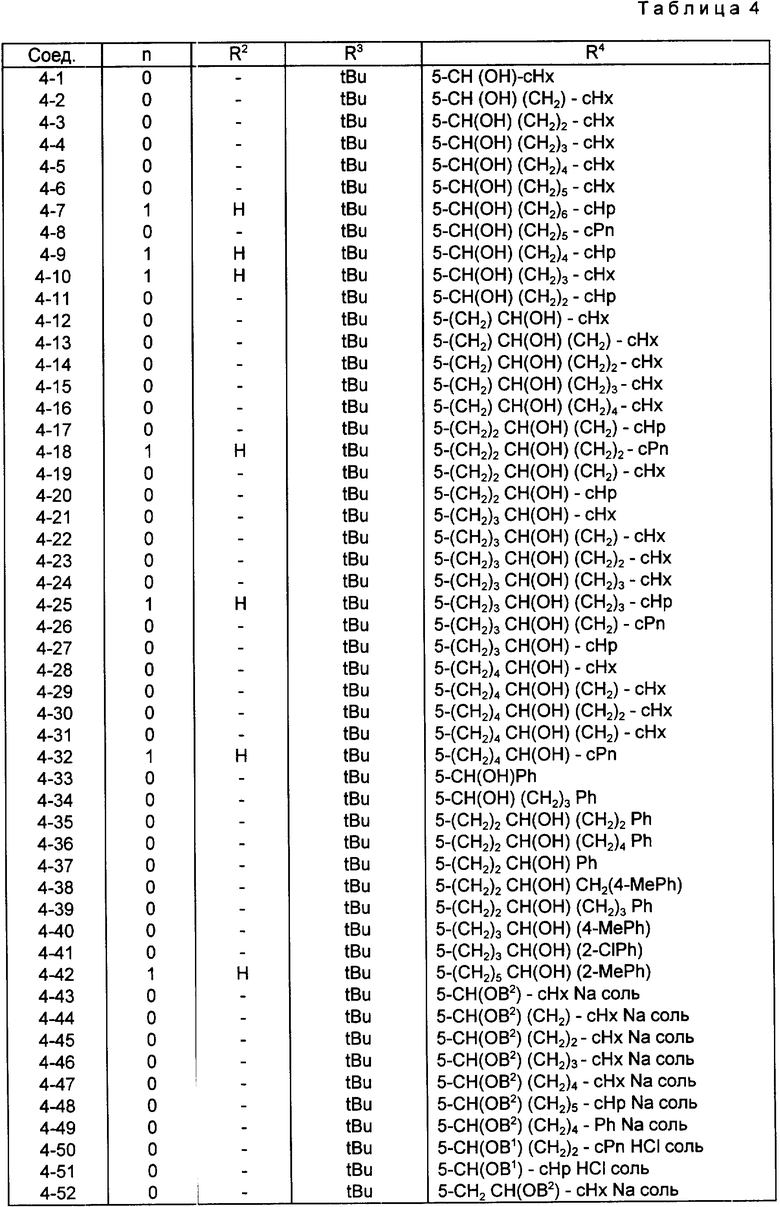
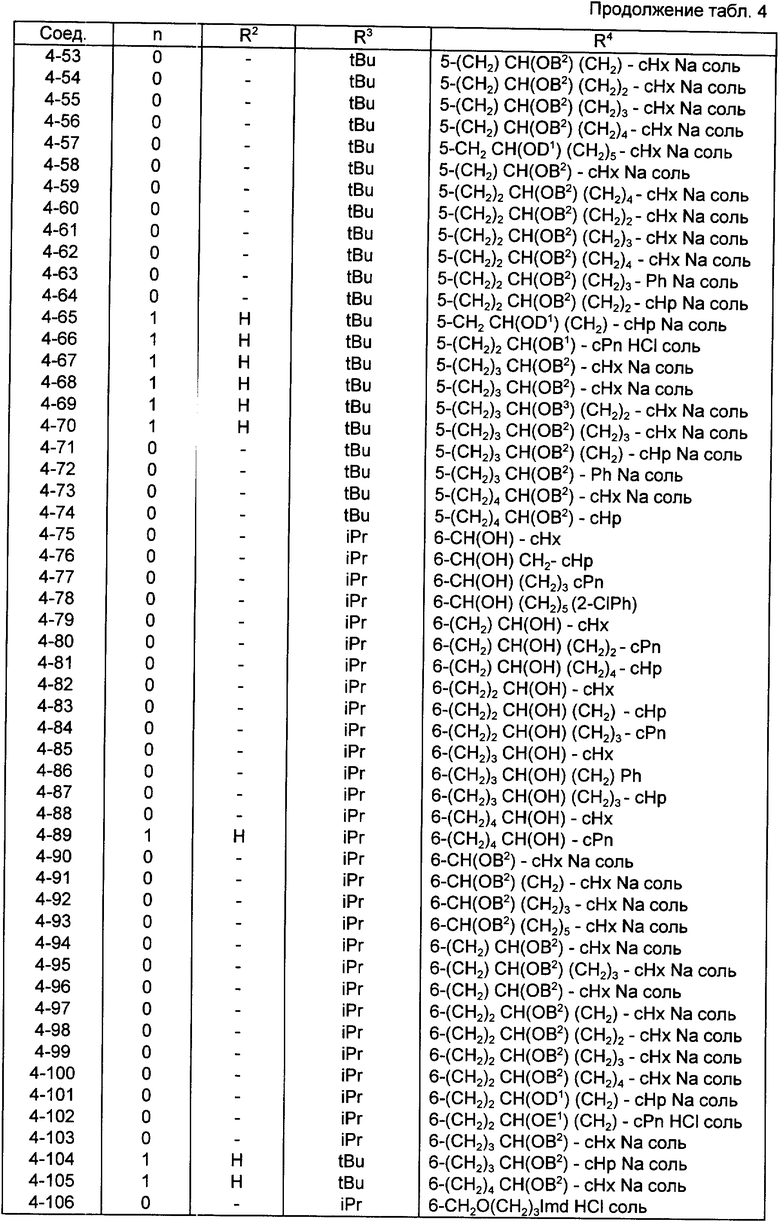
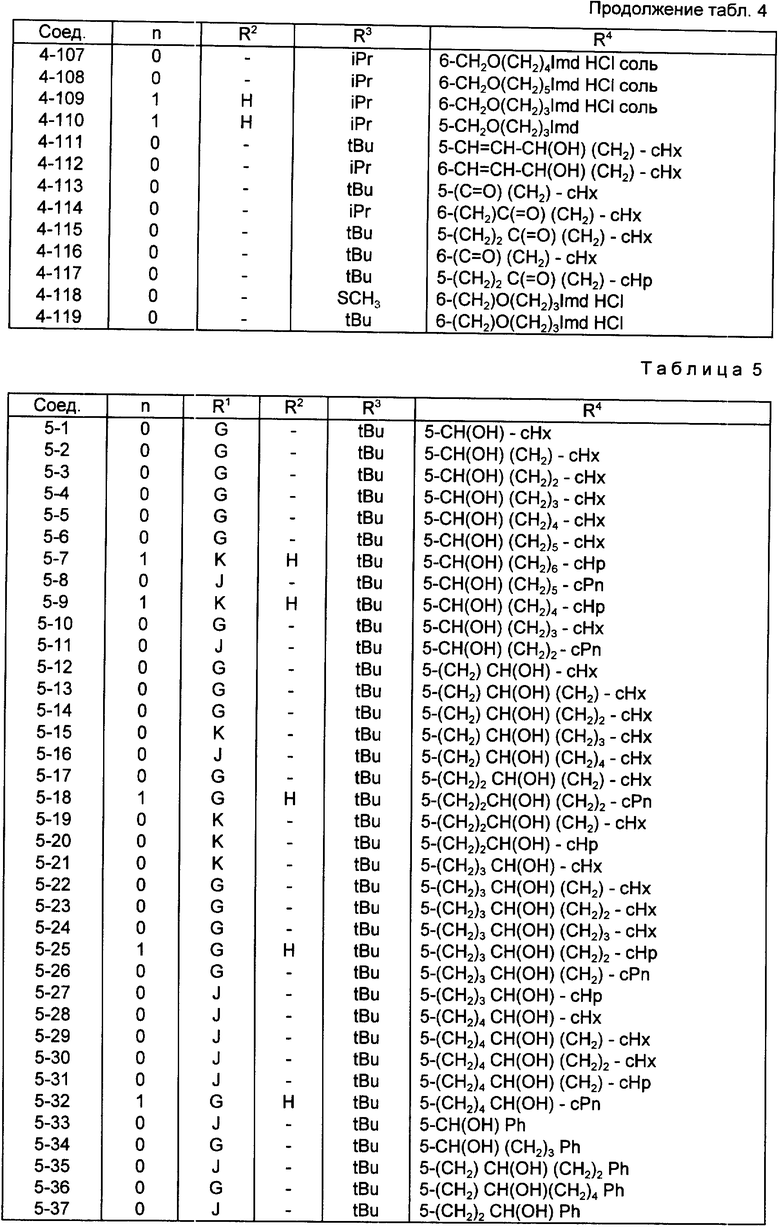
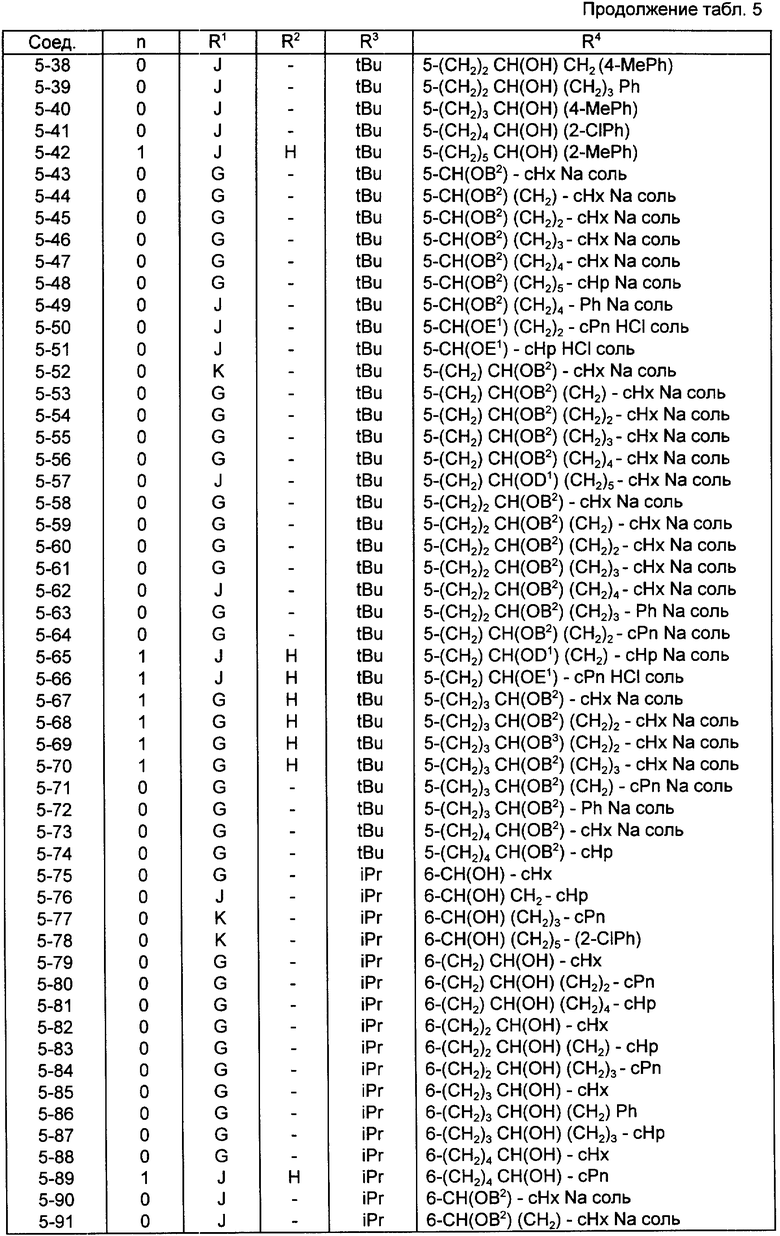
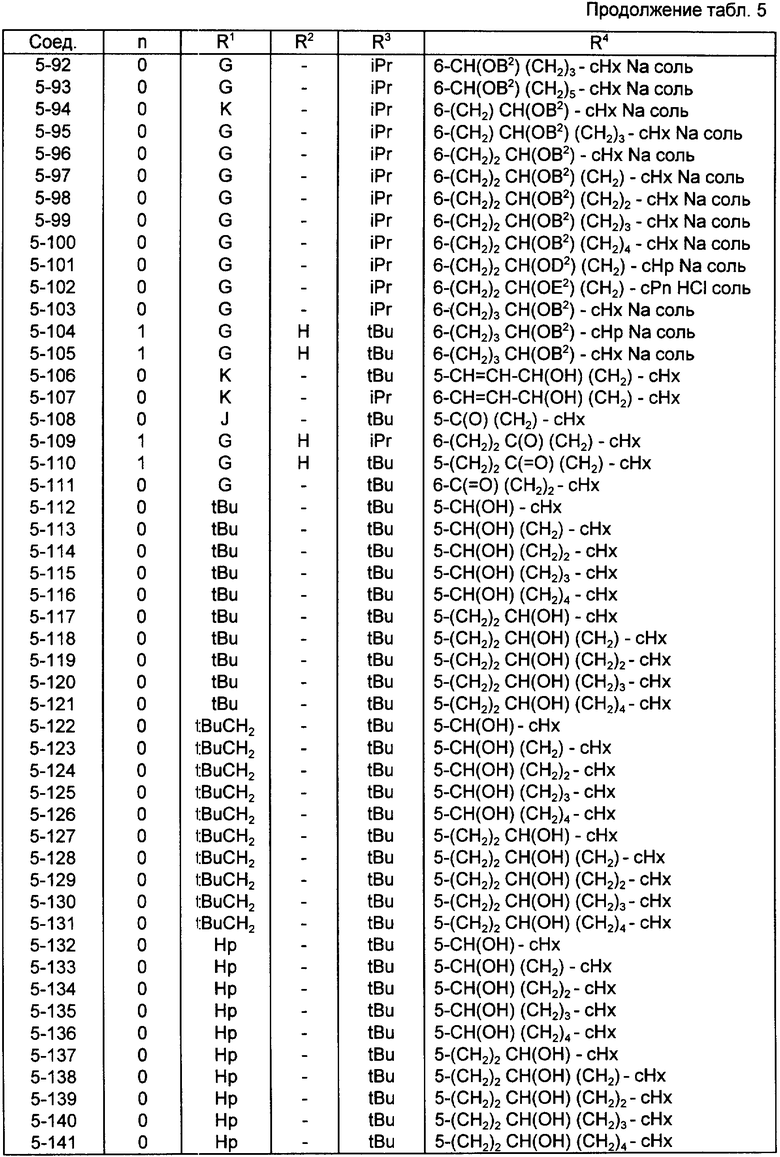


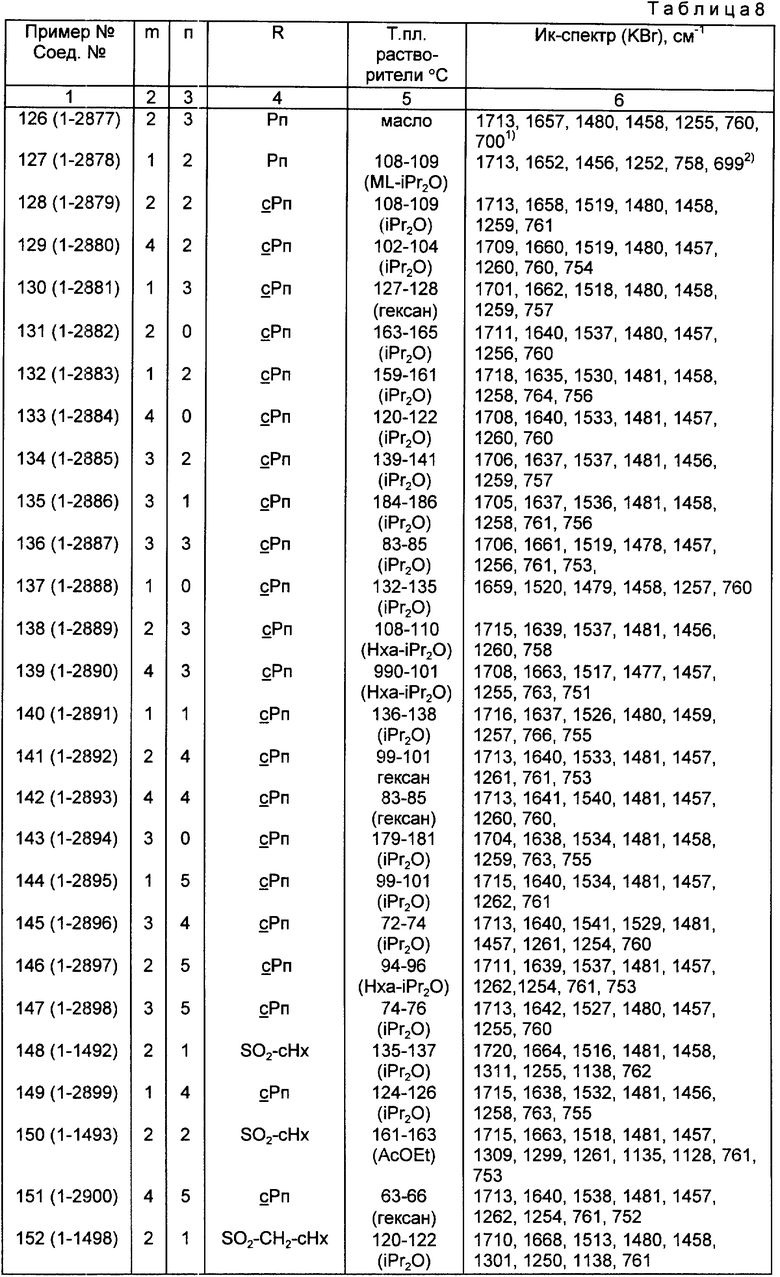
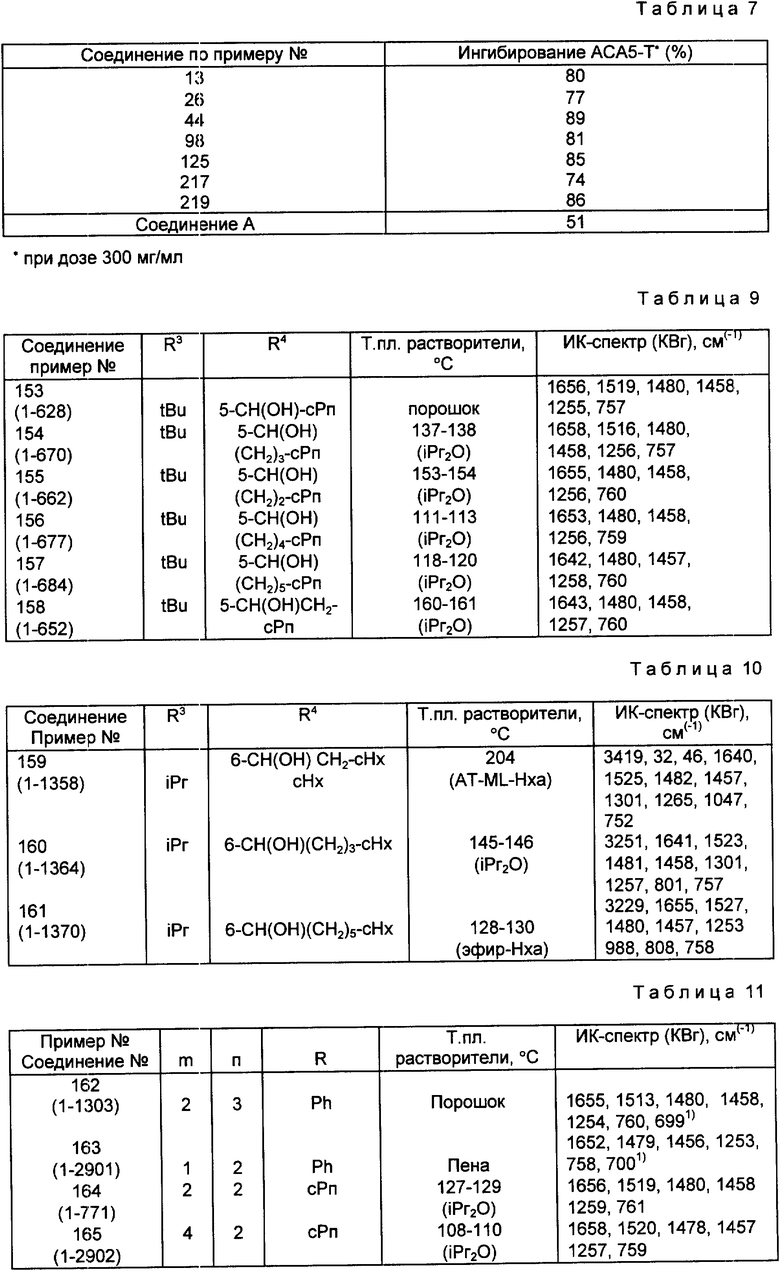
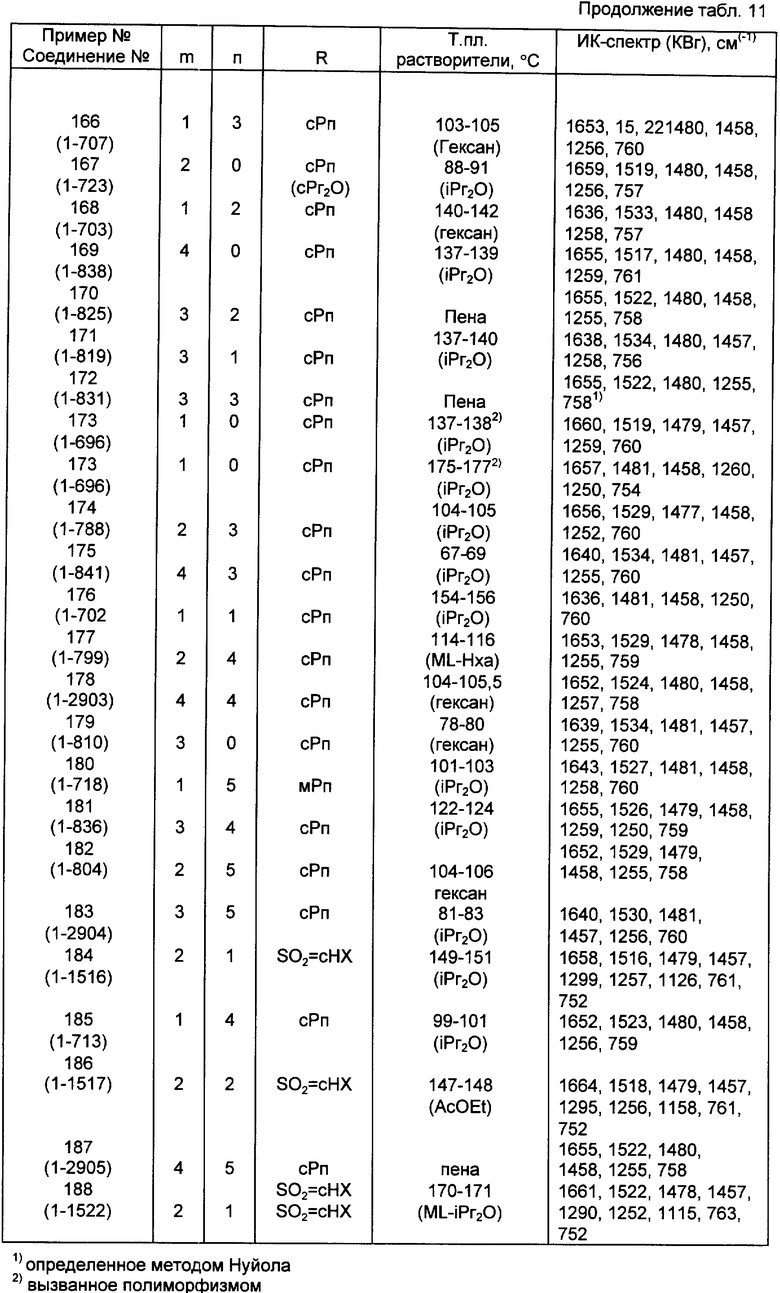
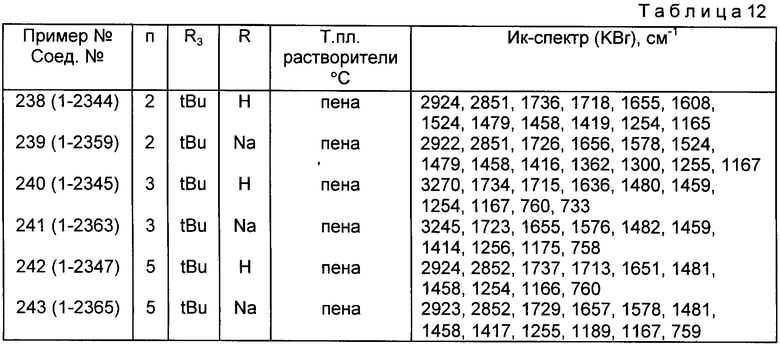
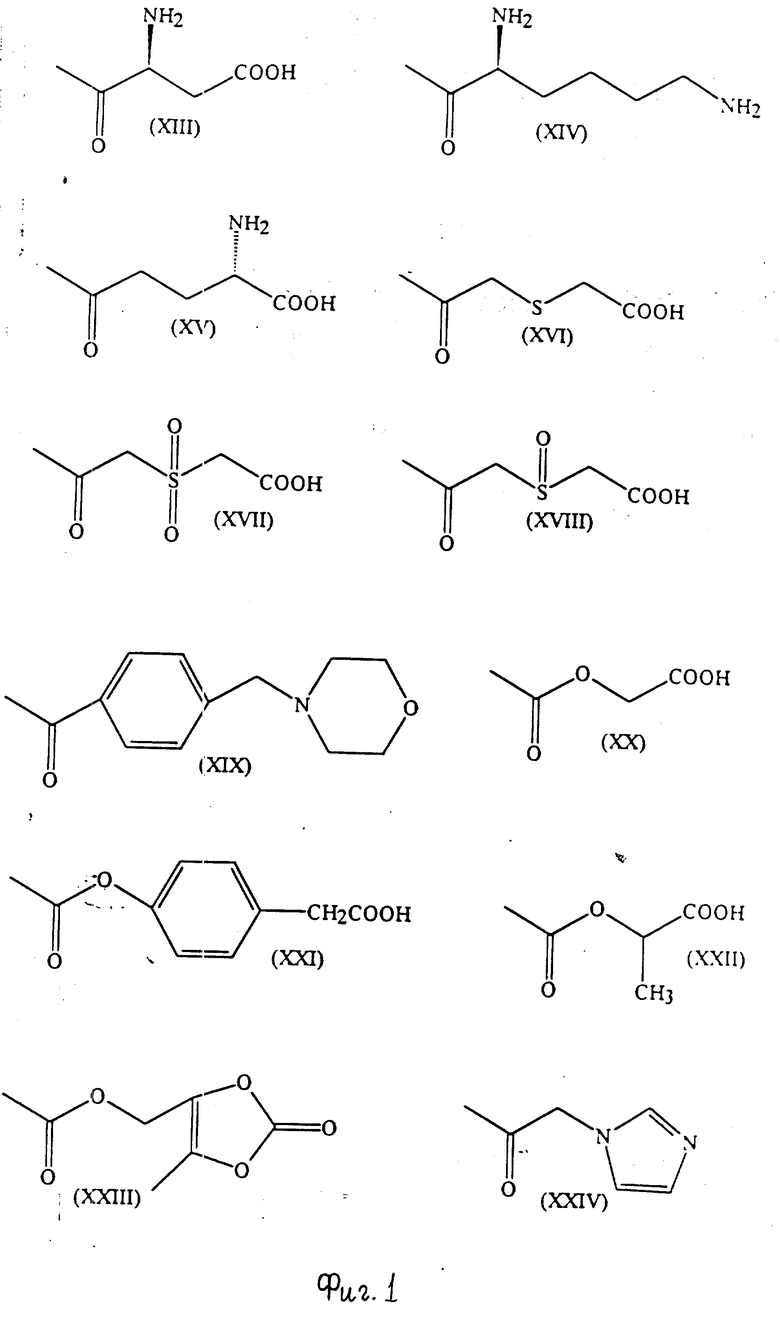
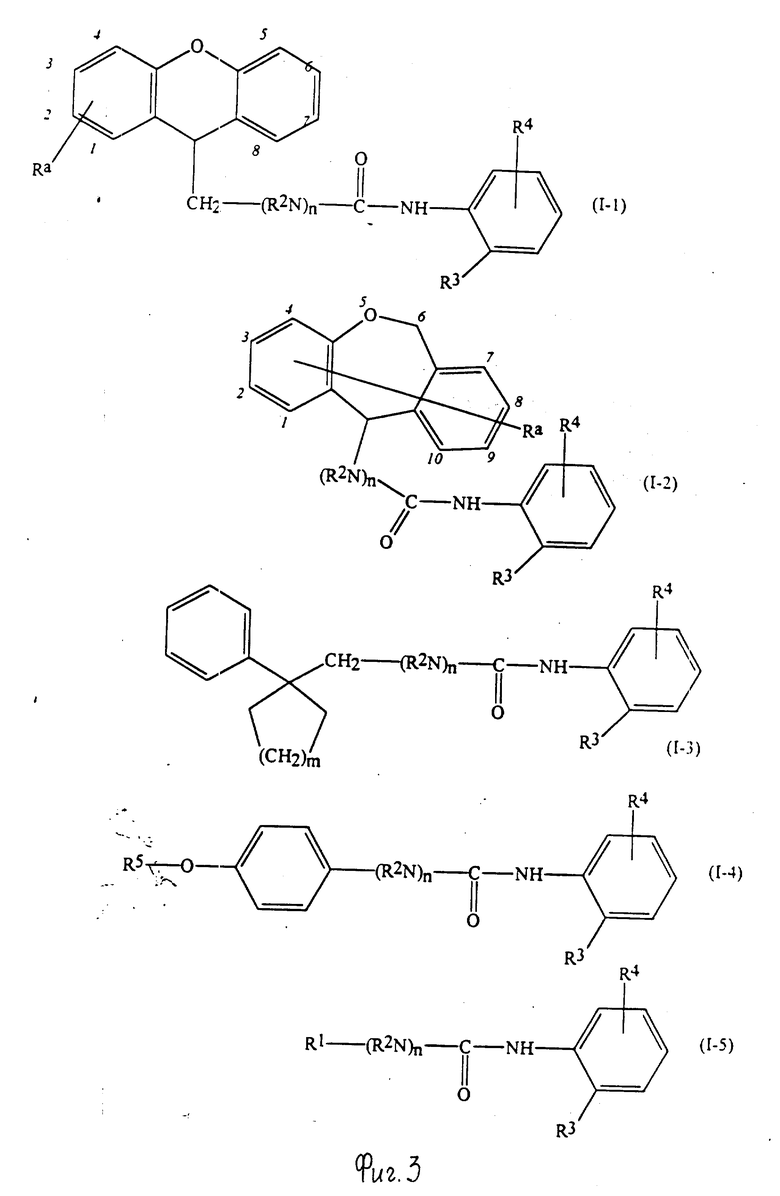
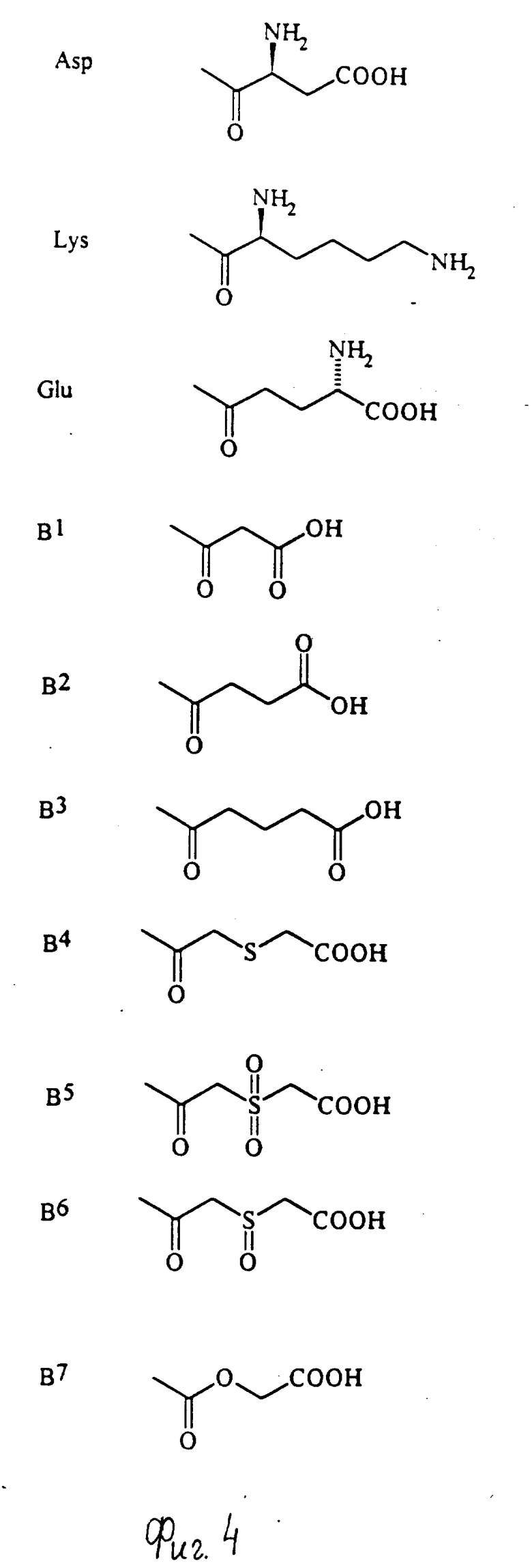
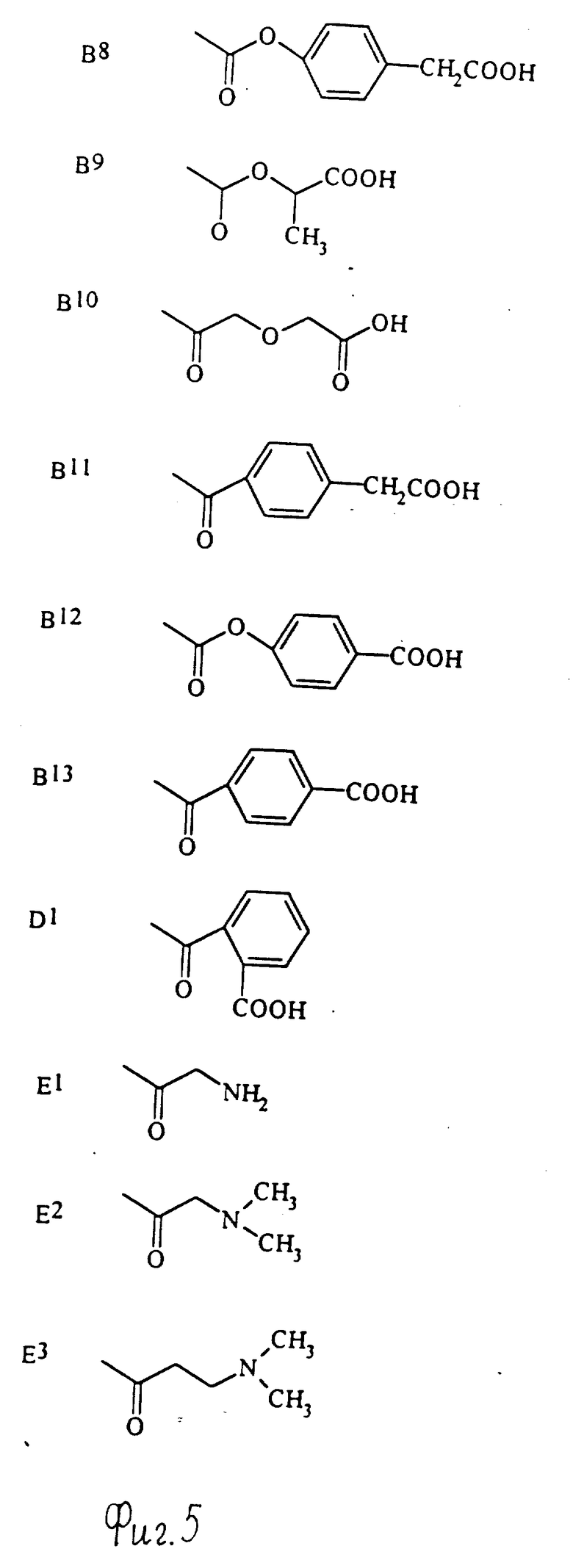
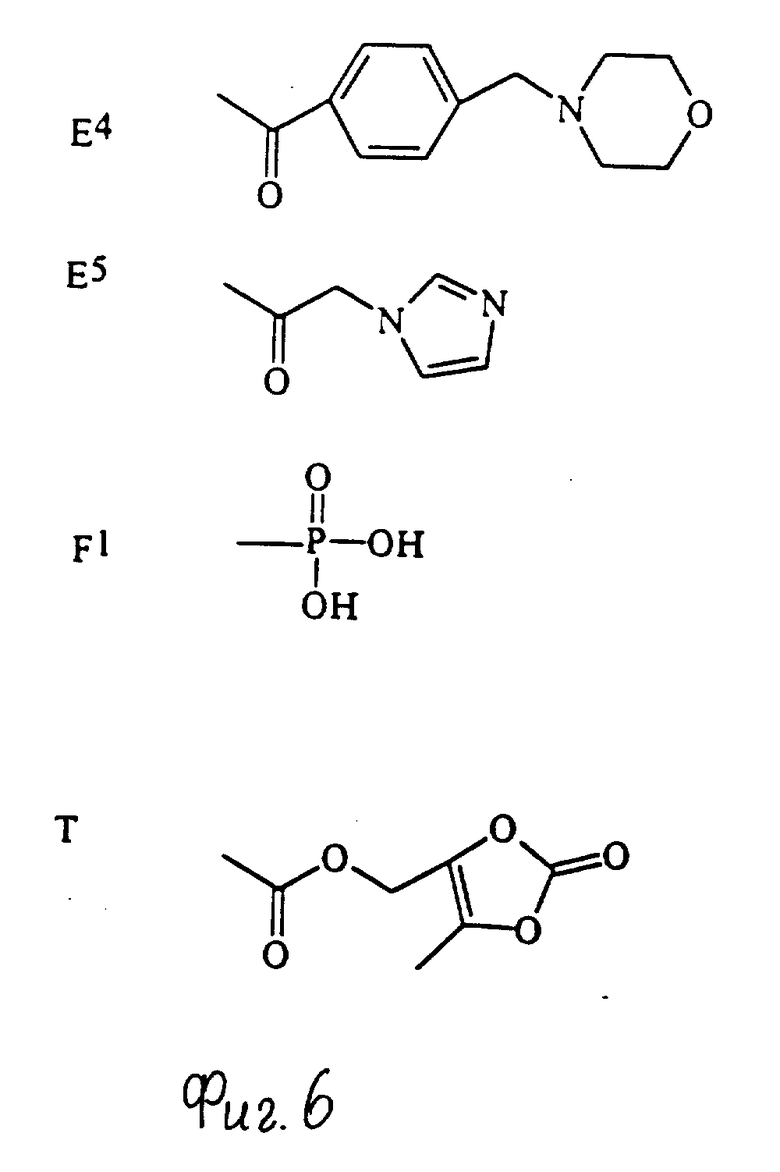
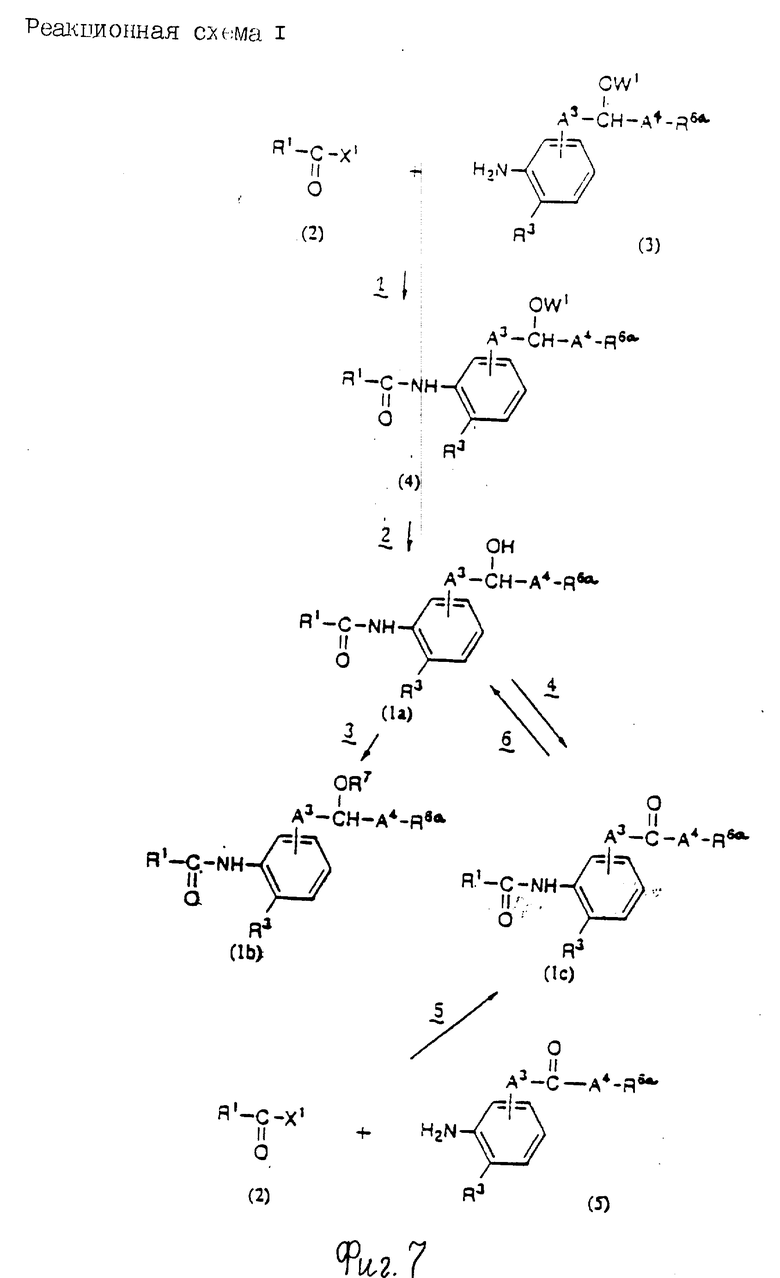
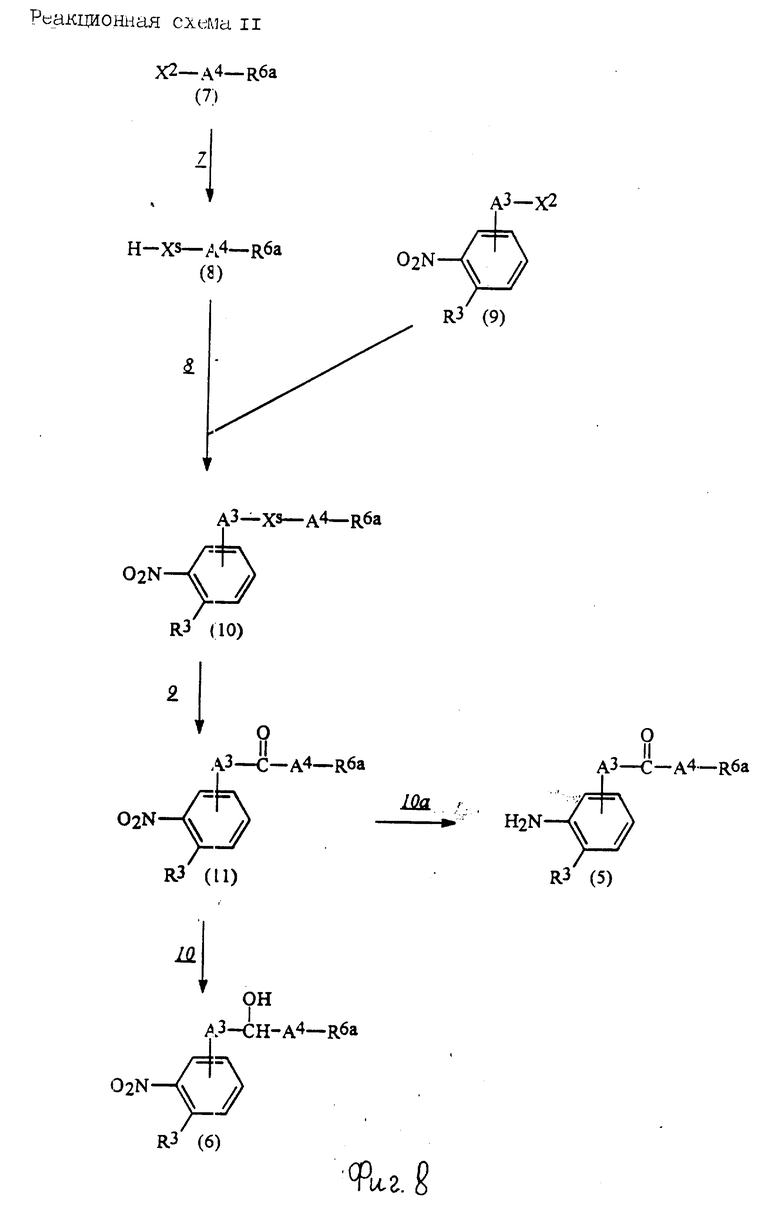
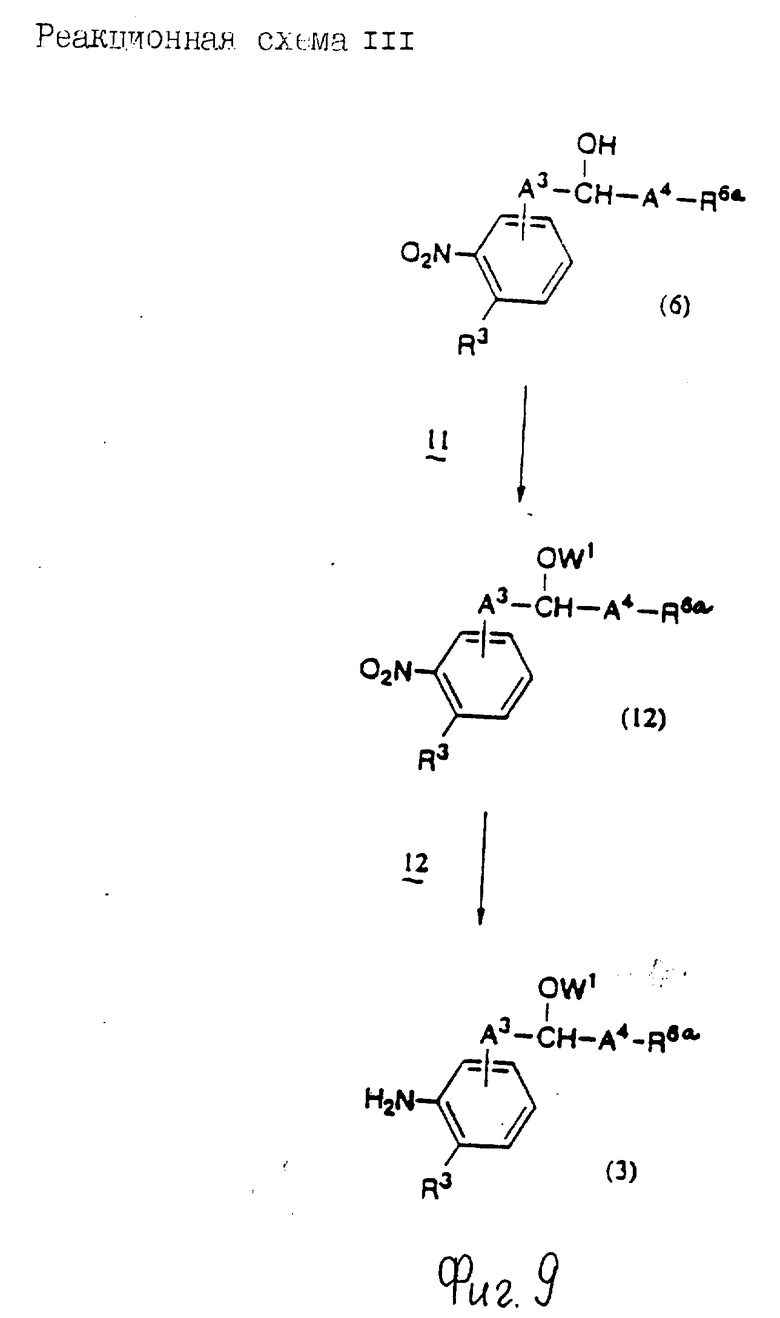
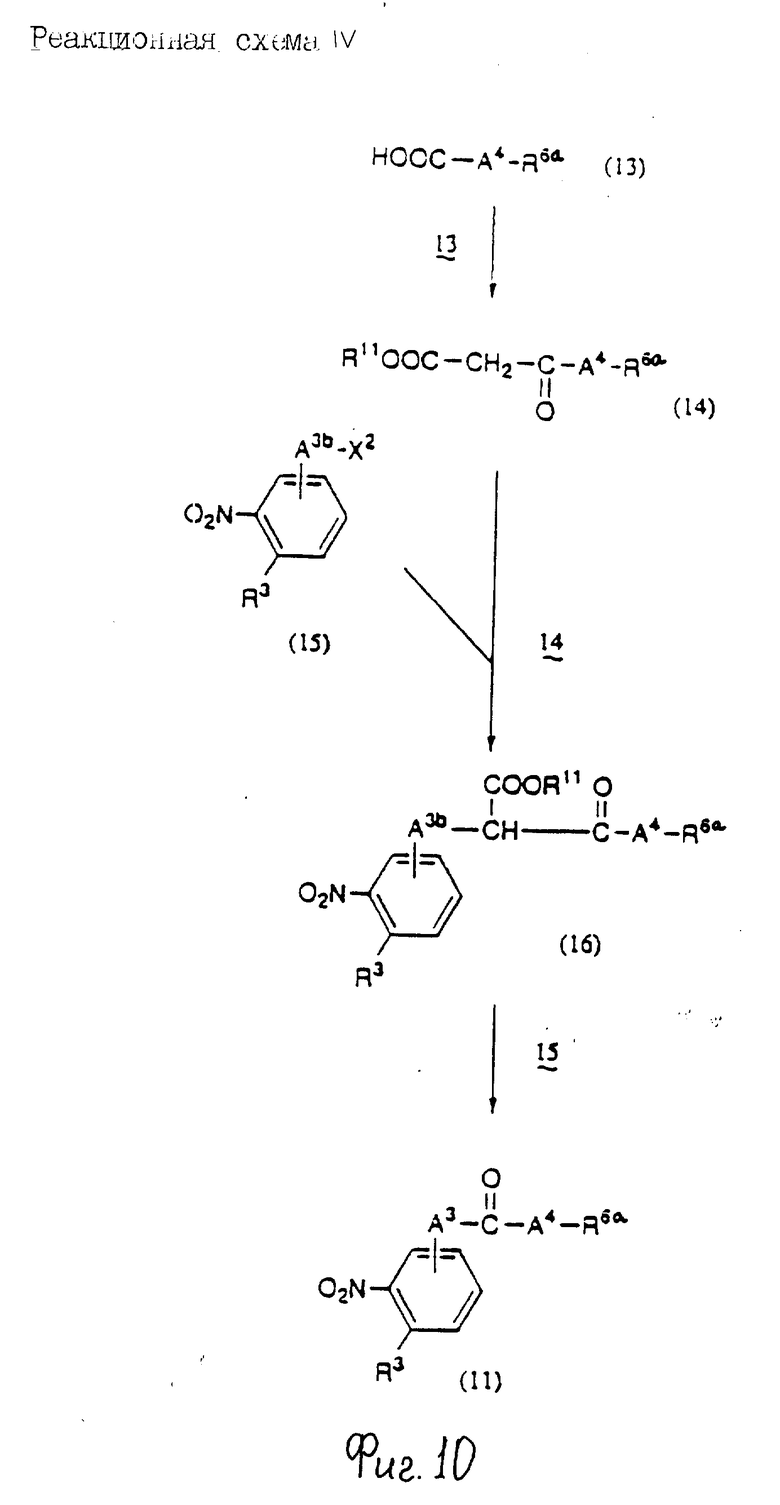
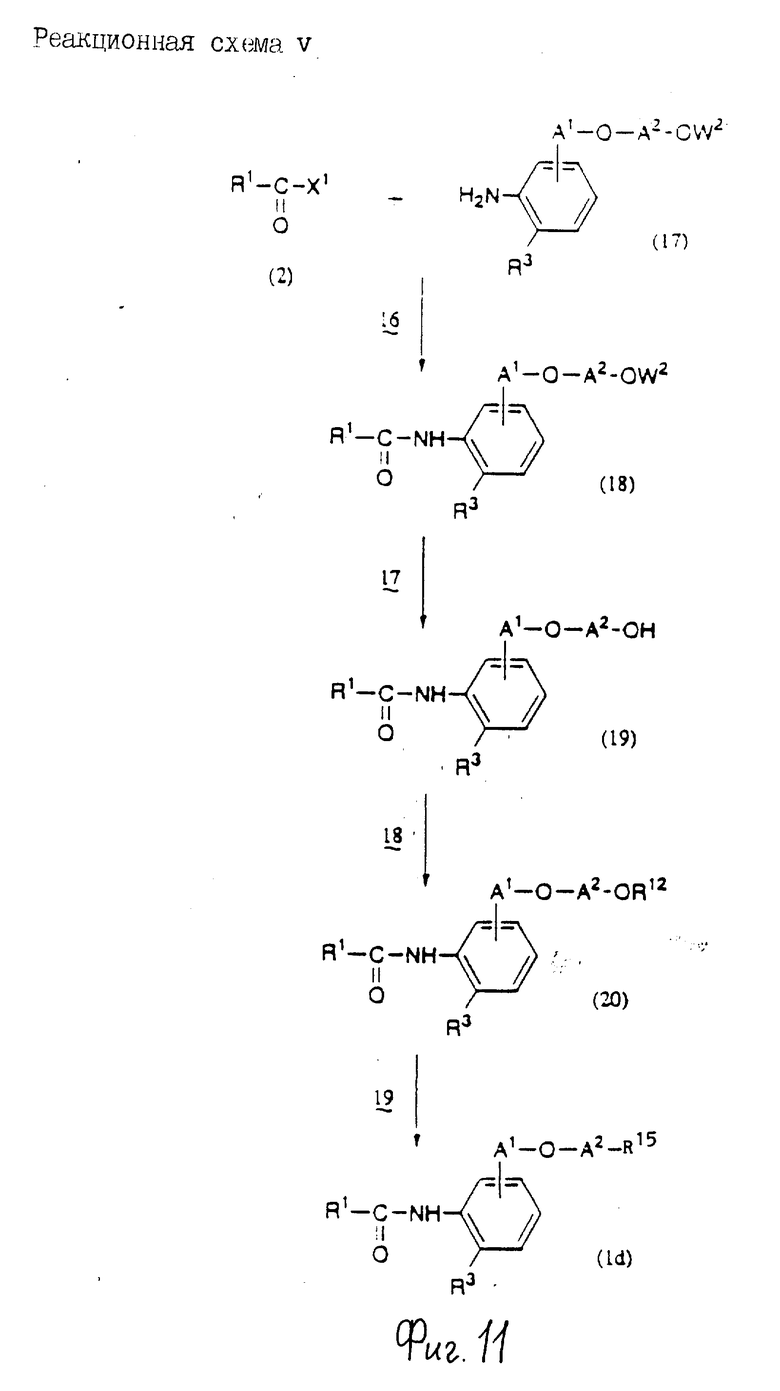
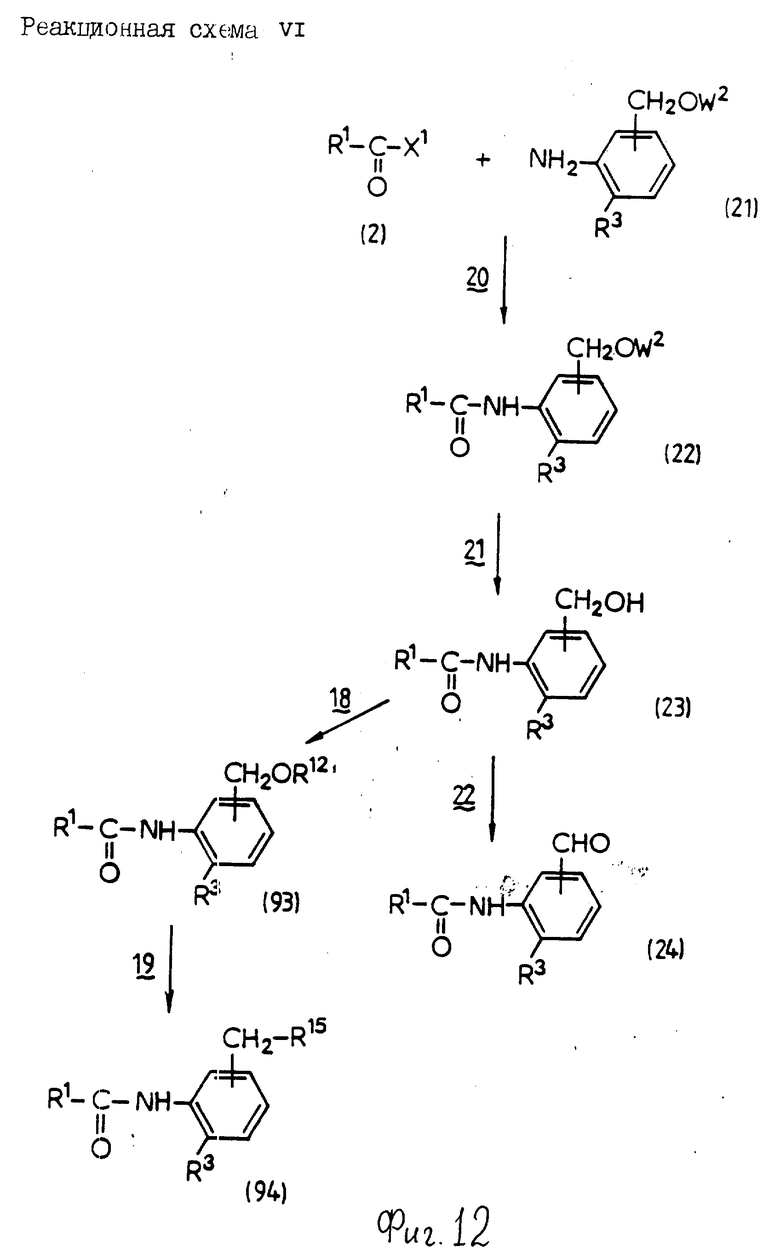
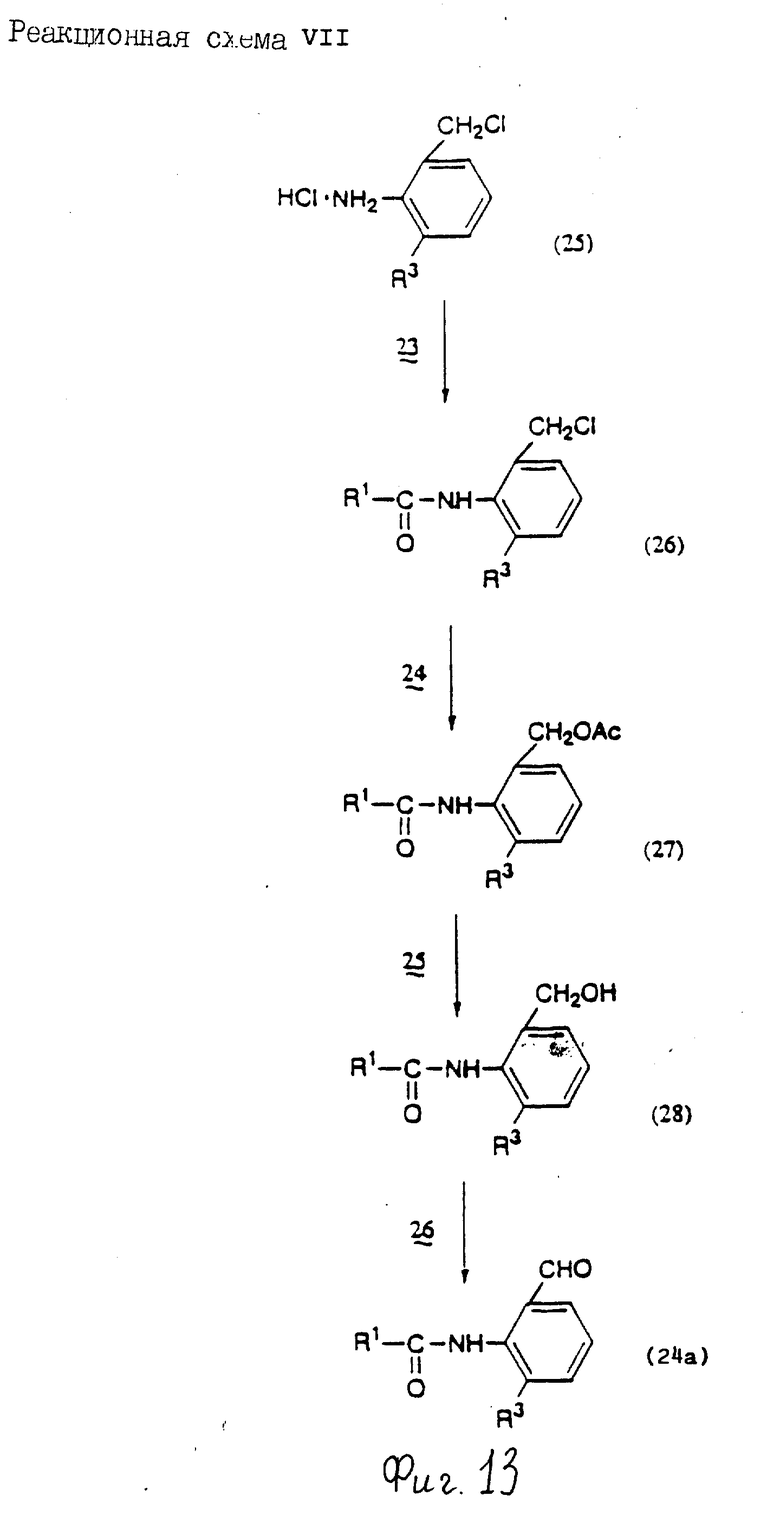
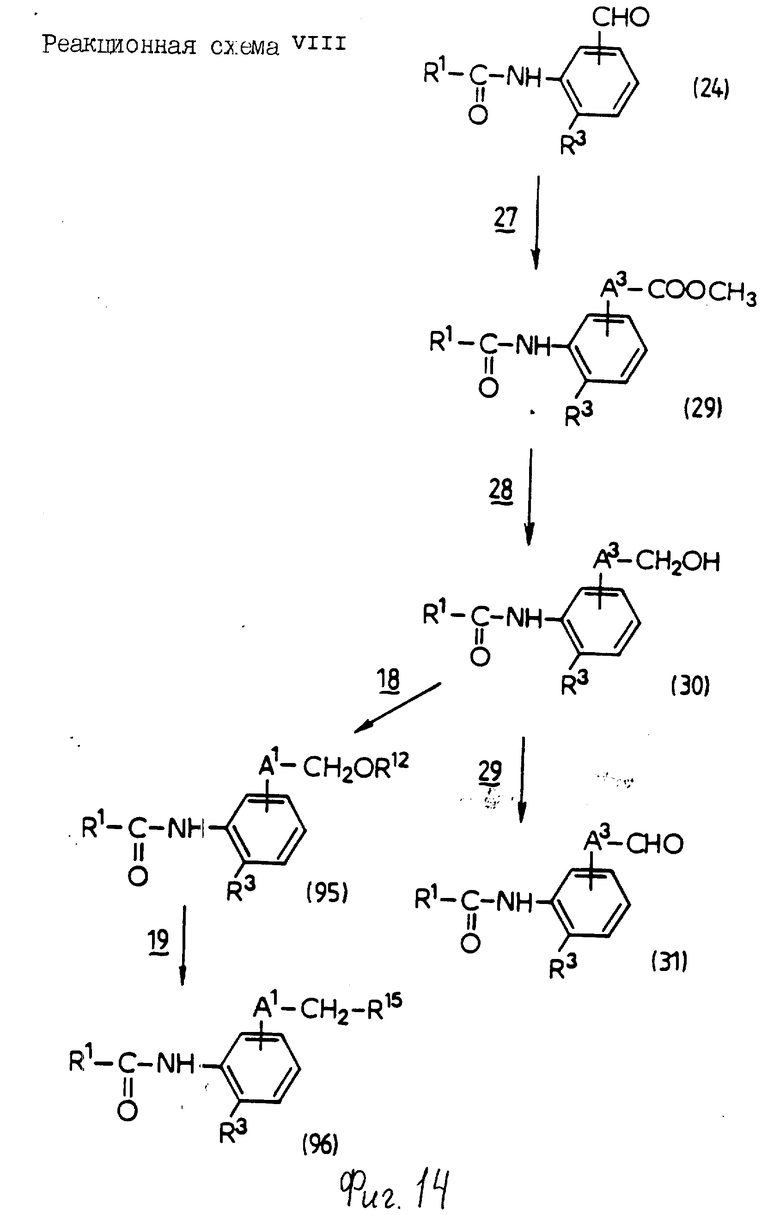
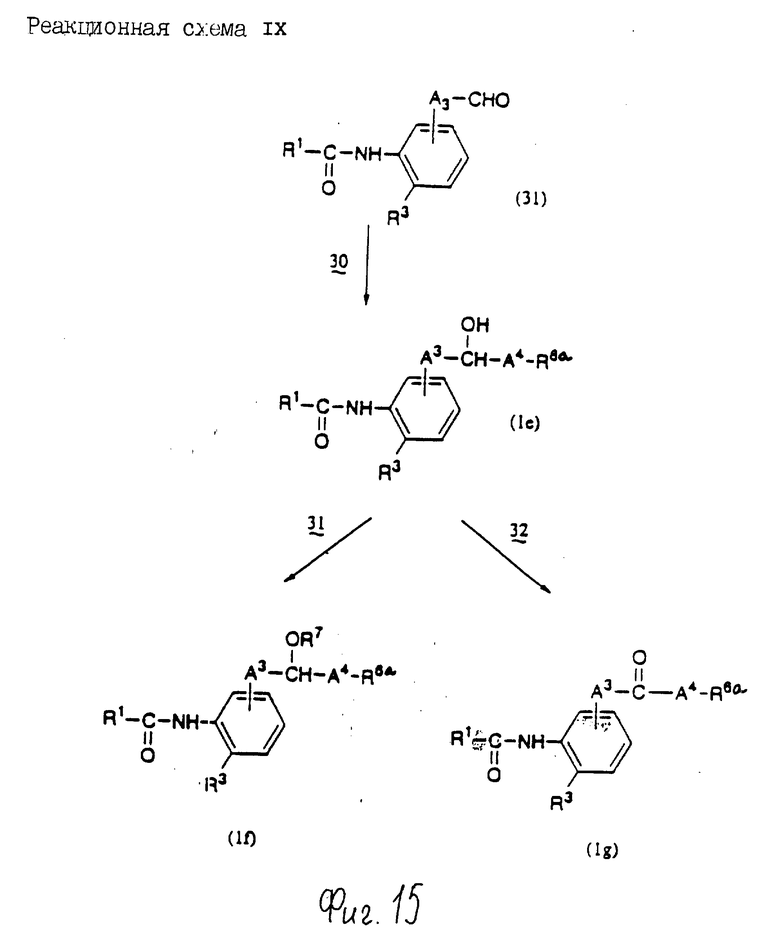
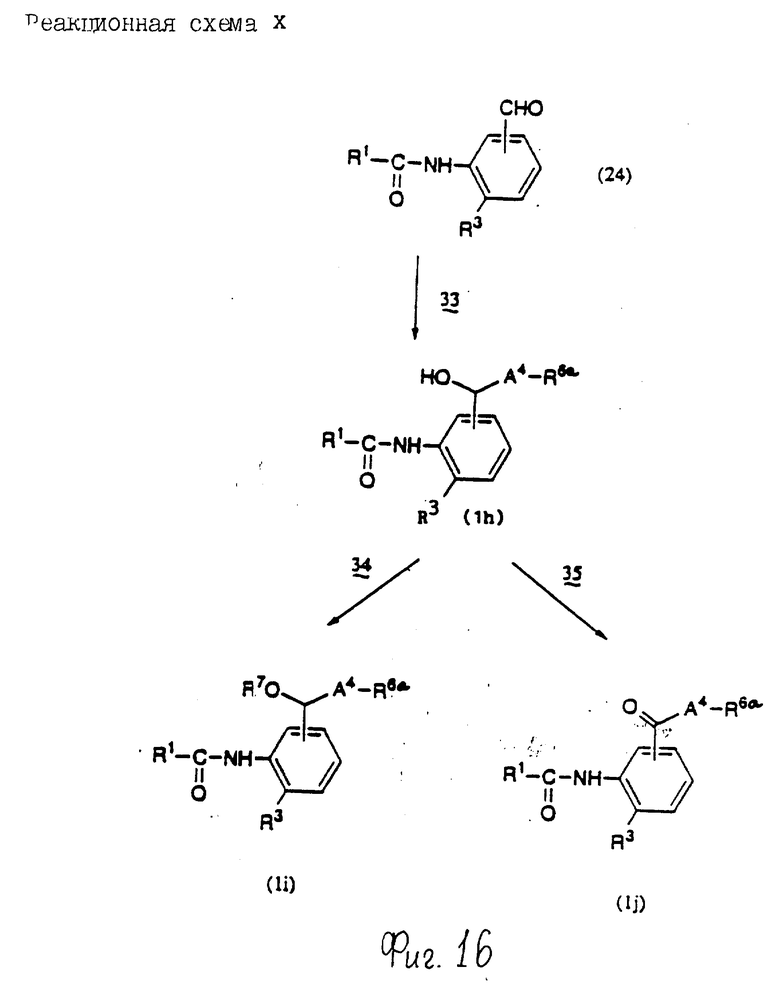
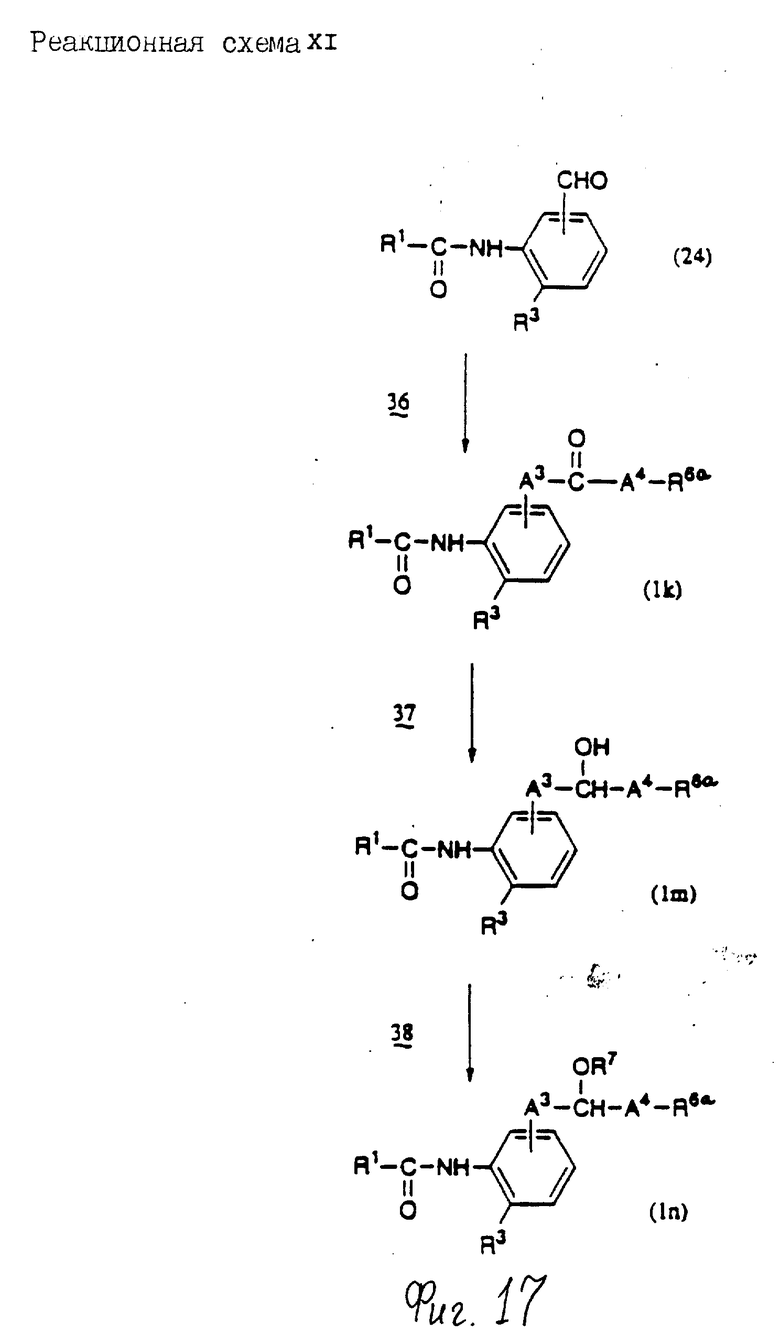
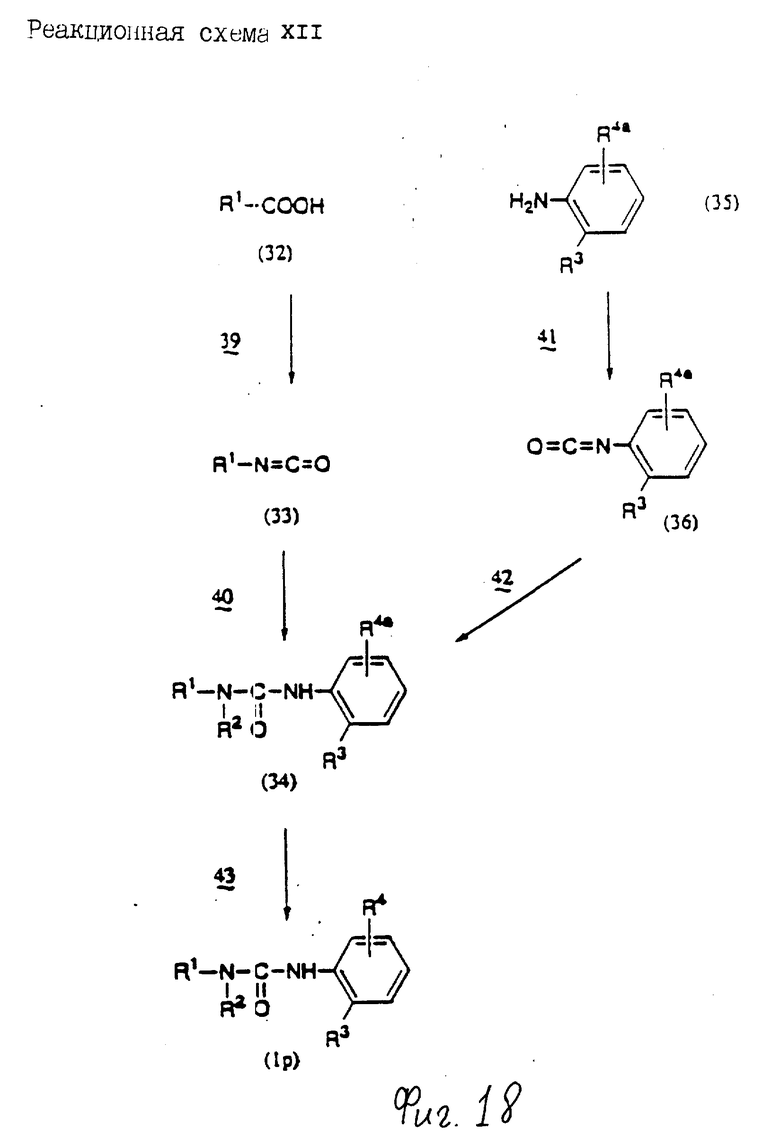
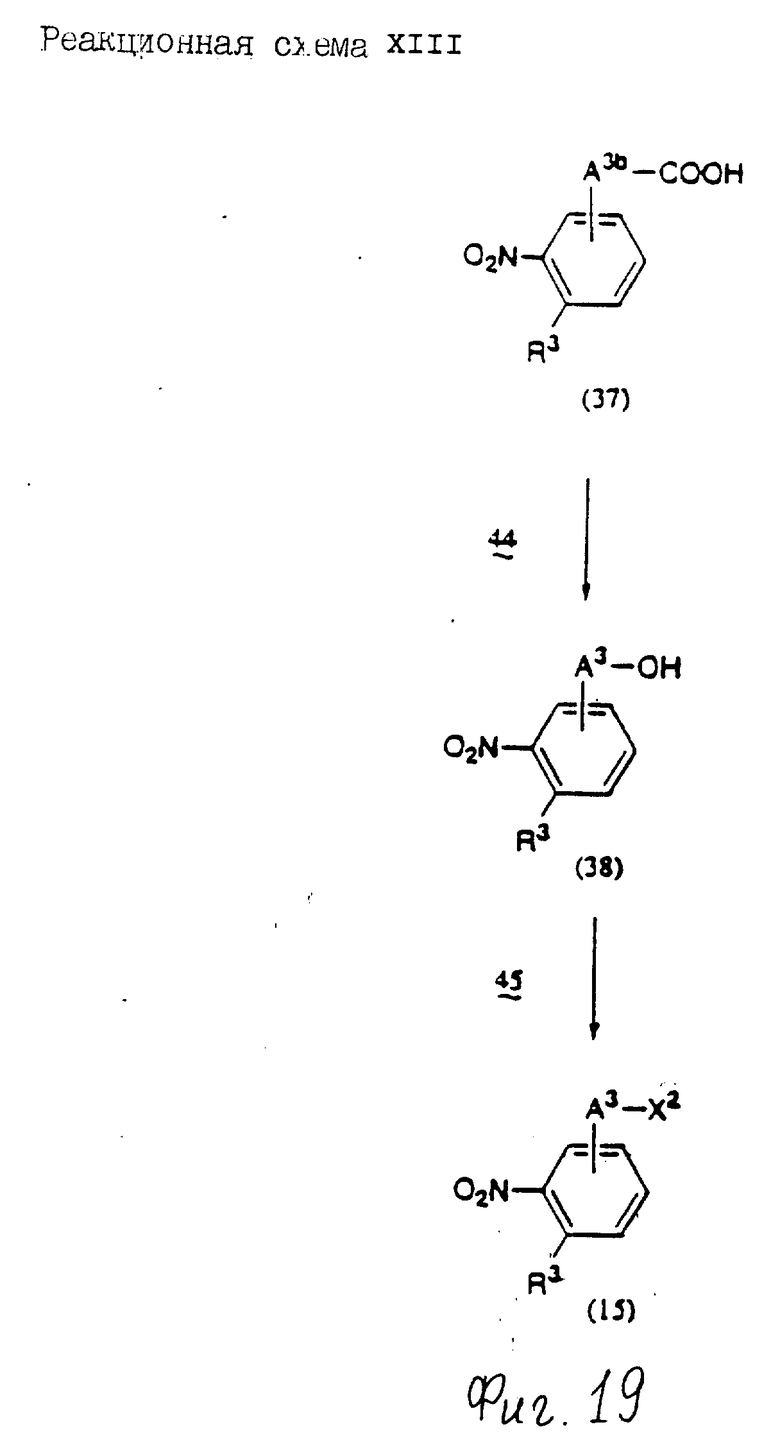
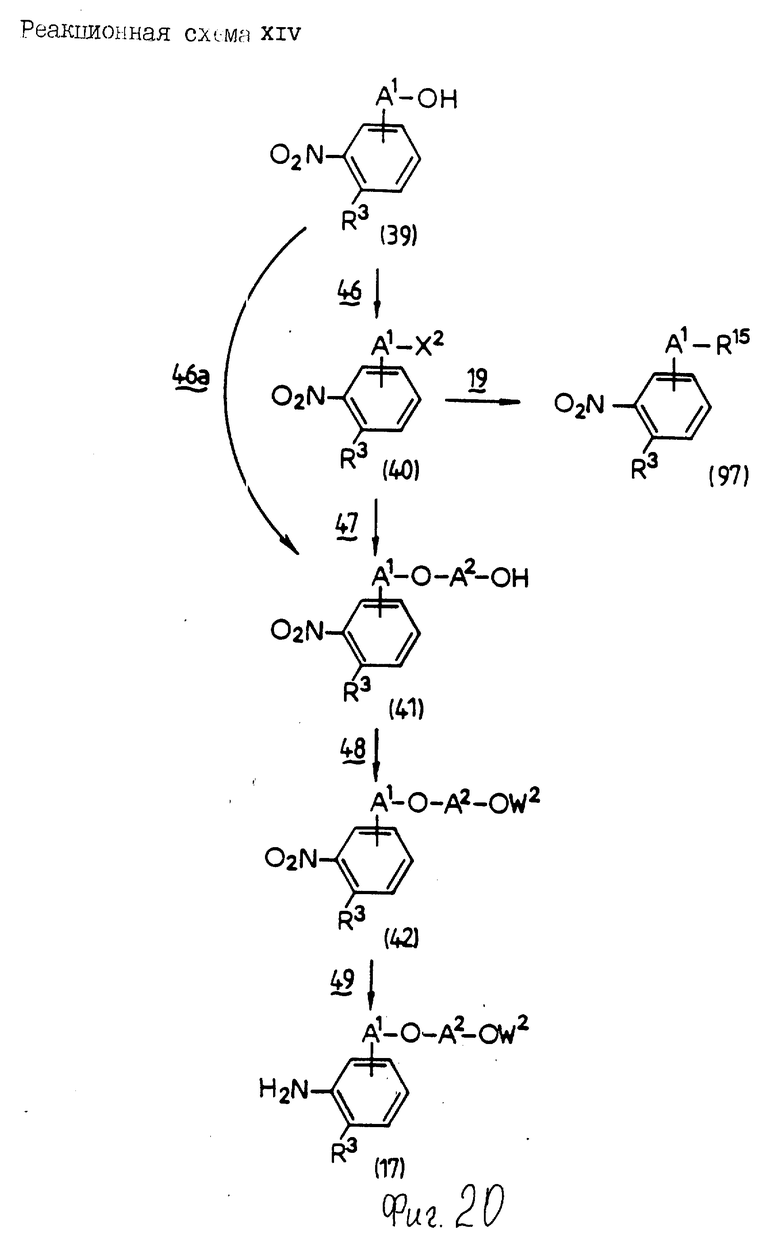
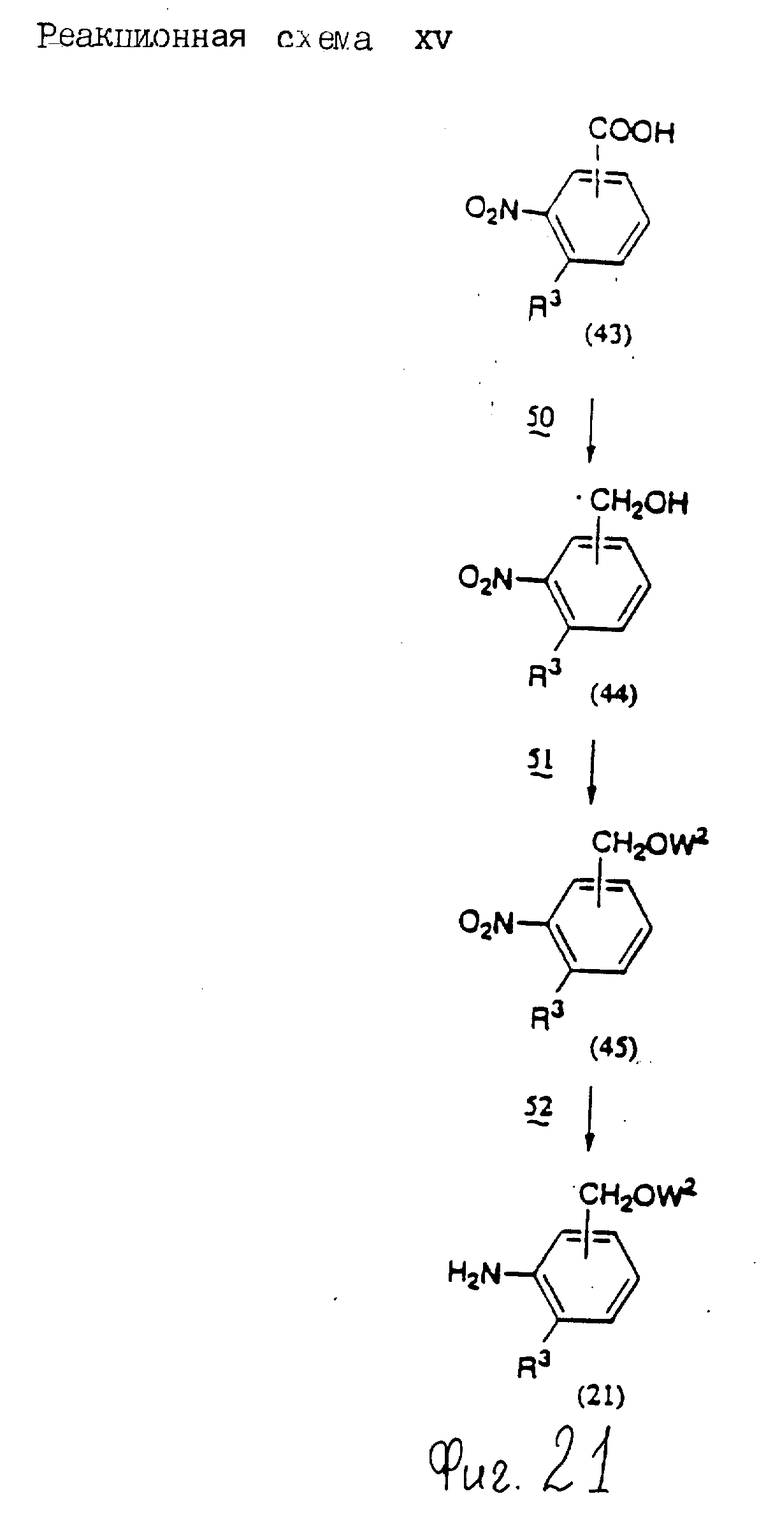
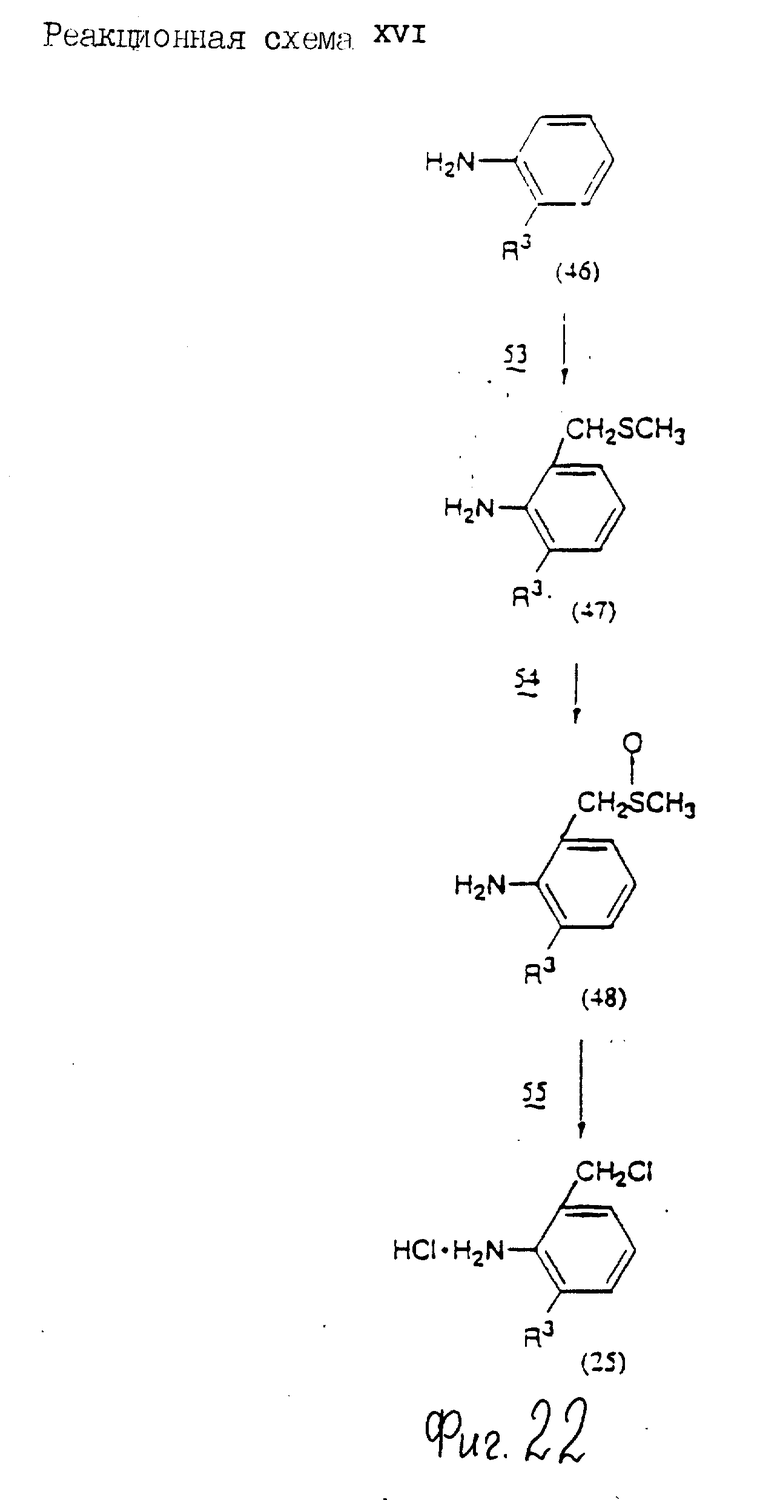
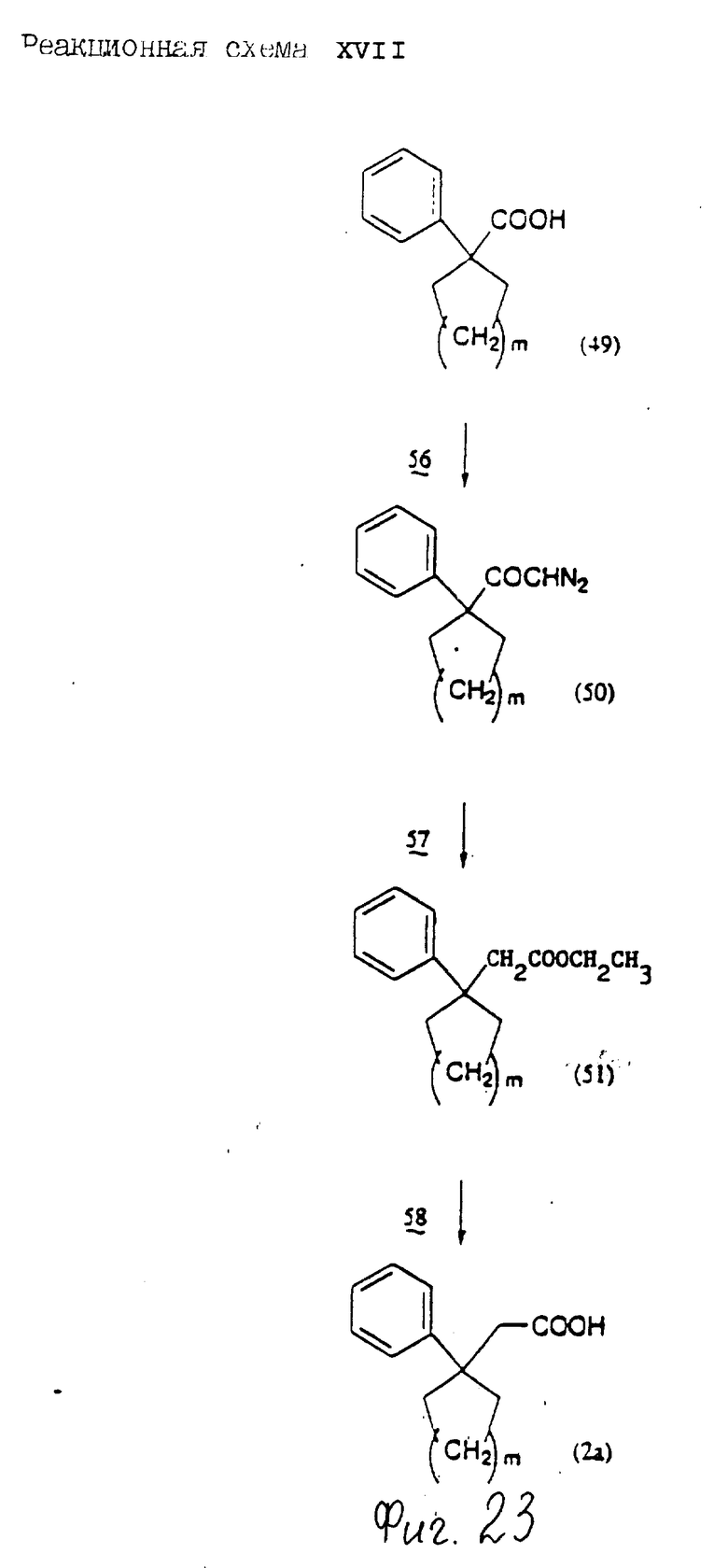
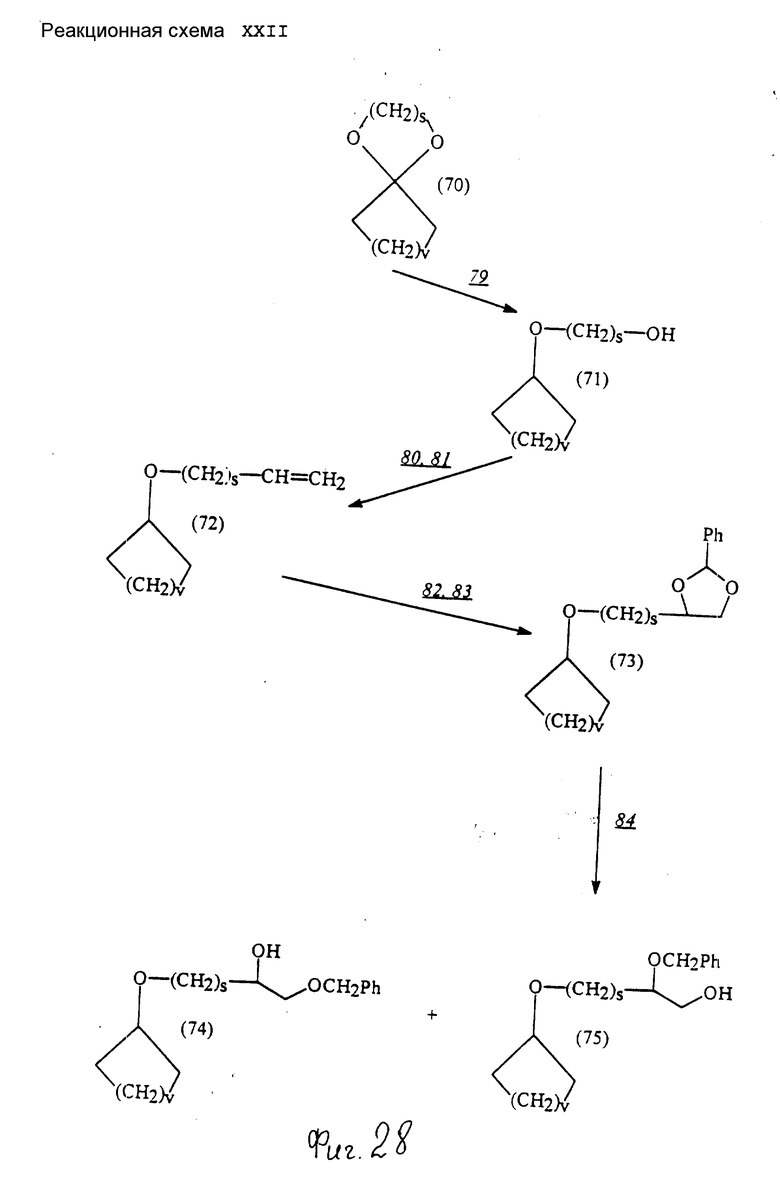
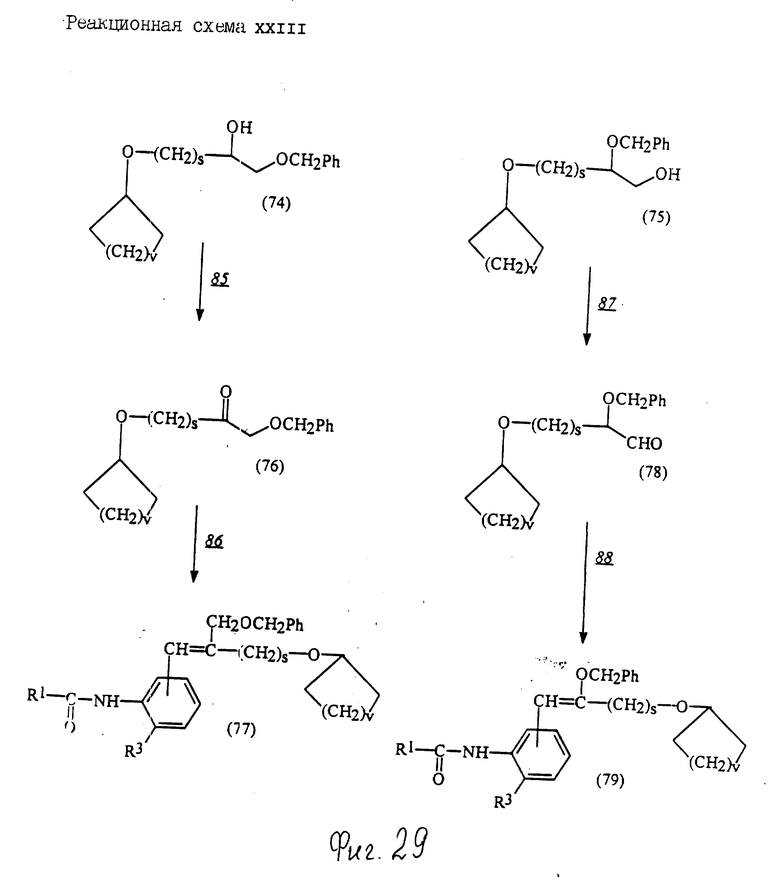
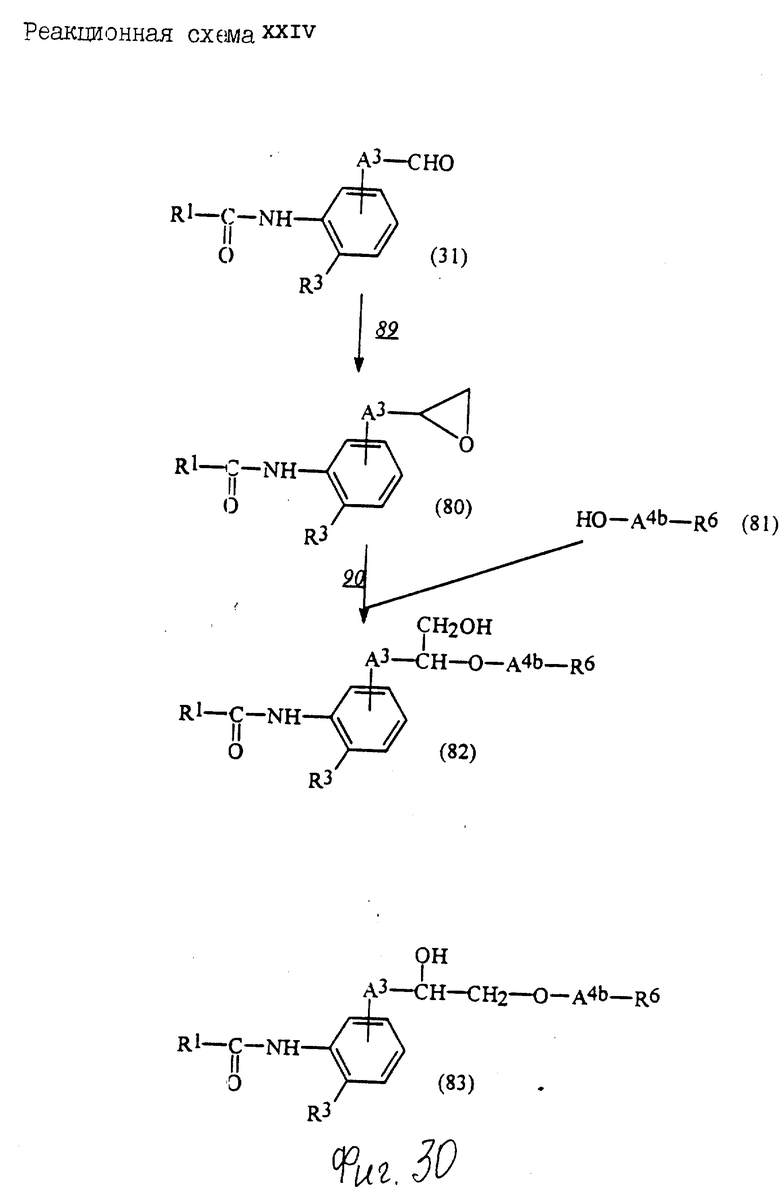
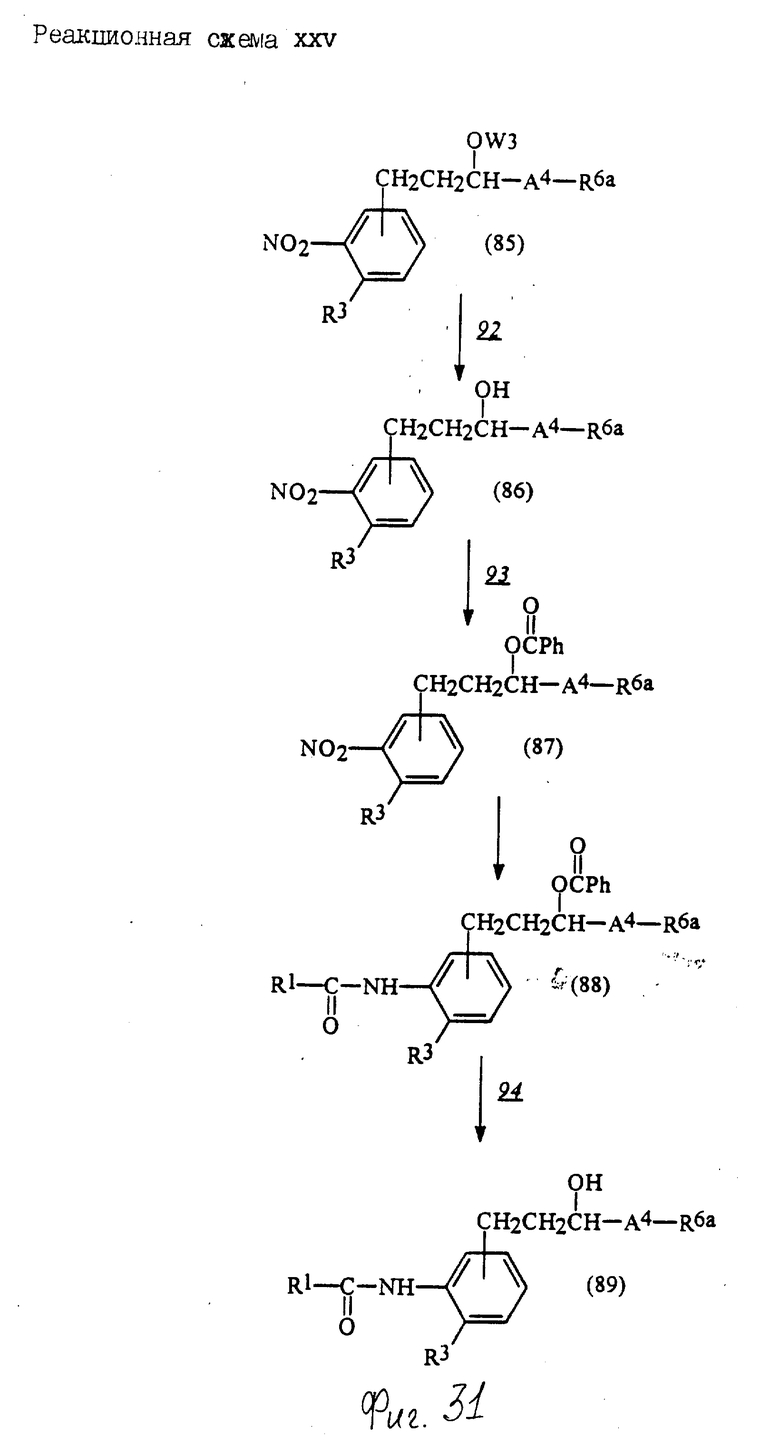
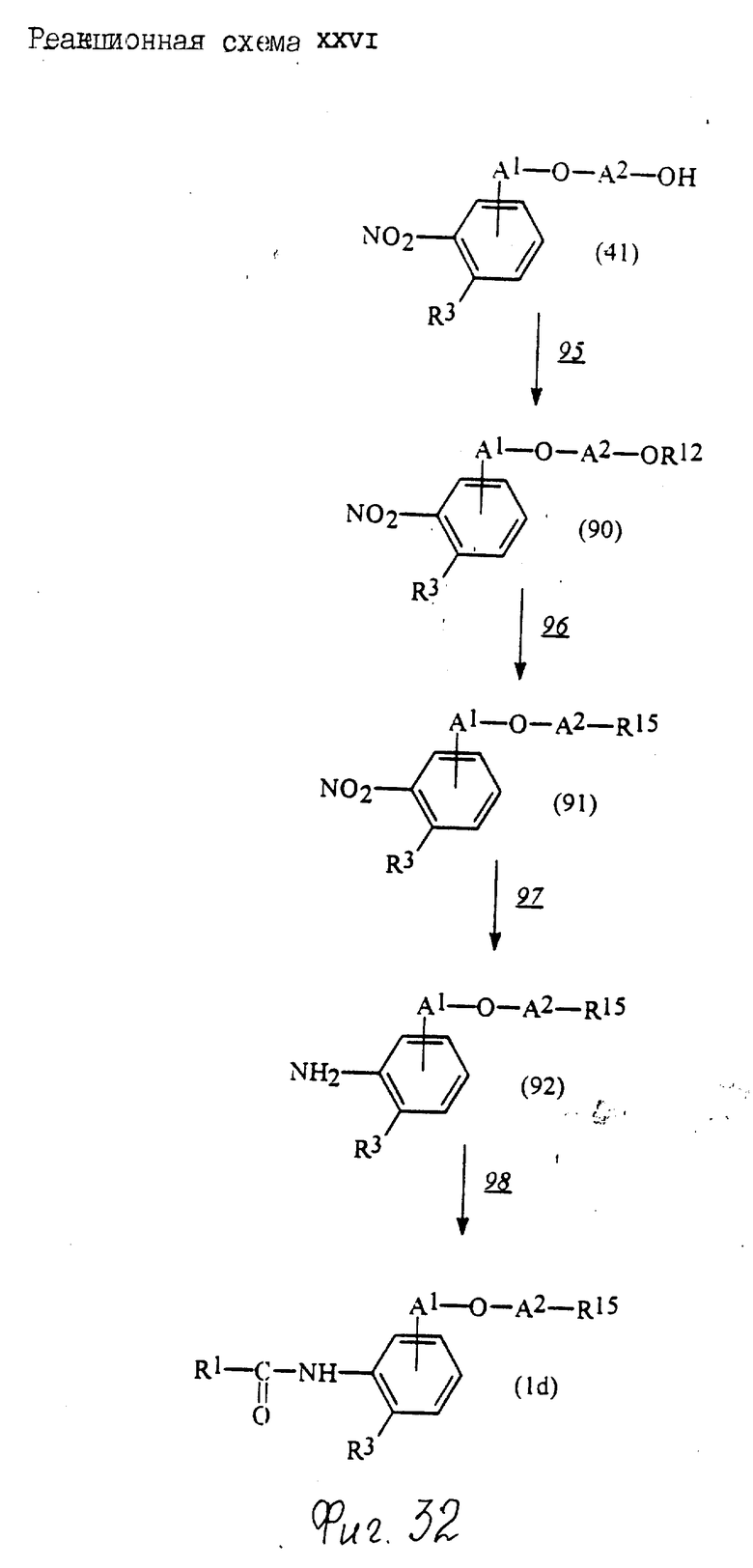
Предложены соединения формулы (I):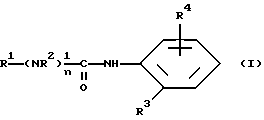
где R1 означает алкильную группу; или группу формулы (II), (III), (IY) или (Y):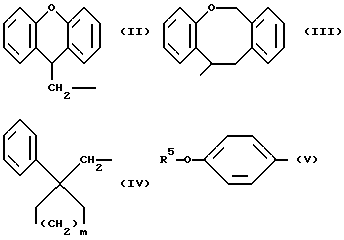
и где R2, R3, R4 и R5 являются водородом или различными оорганическими группами; и их фармацевтически приемлемые соли; а также способы их получения и их использование при лечении и профилактике гиперхолестеремии и атеросклероза. 3 с. и 29 з.п. ф-лы, 32 ил., 12 табл.
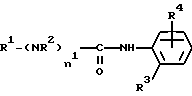
где R1 означает алкильную группу, имеющую от 1 до 20 атомов углерода, или группу формулы II, III, IV или V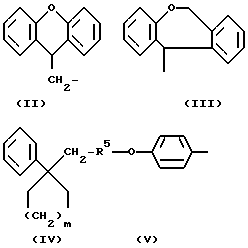
где R5 означает алкильную группу, имеющую от 1 до 15 атомов углерода; m обозначает целое число от 1 до 4; и любое ароматическое кольцо в указанной группе, рапределенной R1, является незамещенным;
R2 означает атом водорода или алкильную группу, имеющую от 1 до 10 атомов углерода;
R3 означает
алкильную группу, имеющую от 1 до 10 атомов углерода,
алкоксигруппу, имеющую от 1 до 10 атомов углерода;
алкилтиогруппу, имеющую от 1 до 10 атомов углерода,
алкилсульфинильную группу, имеющую от 1 до 10 атомов углерода,
алкилсульфонильную группу, имеющую от 1 до 10 атомов углерода, или
алкоксиалкильную группу, в которой алкоксильная часть имеет от 1 до 6 атомов углерода и алкильная часть имеет от 1 до 4 атомов углерода;
R4 означает группу формулы VI, VII, VIII, IX, X или XI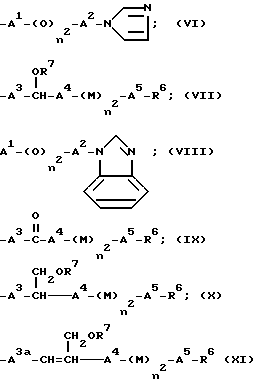
где A1 означает одинарную связь или алкиленовую группу, имеющую от 1 до 4 атомов углерода;
A2 означает одингарную связь или алкиленовую группу, имеющую от 1 до 6 атомов углерода;
A3, A3 a, и A5 независимо выбраны из группы, содержащей одинарную связь и алкиленовые группы, имеющие от 1 до 10 атомов углерода, которые могут быть насыщенными или могут включать углерод-углеродную двойную связь, при условии, что общее число атомов углерода в A3, A4 и A5 и в A3 a, A4 и A5 не превышает 10;
R6 означает алкильную группу, имеющую от 1 до 6 атомов углерода, циклоалкильную группу, имеющую от 3 до 9 атомов углерода в одном или более алифатических карбоциклических кольцах, указанные кольца являются незамещенными, или фенильную группу, замещенную по крайней мере одним атомом галогена, и в группах формулы VI или VIII имидазольная и бензимидазольная группа могут быть незамещенными или замещенными по крайней мере одной С1 - С4-алкильной группой;
R7 означает атом водорода, бензильную группу, фосфоногруппу или группу формулы XII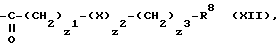
где z1 является 0 или 1;
z2 является 0, 1 или 2;
Х является кислородом, или атомом серы, или сульфинилом, сульфонилом или фениленовой группой при условии, что, когда Z2 является 2, по крайней мере один Х является фениленовой группой;
Z3 является 0 или целым числом от 1 до 4; и
R8 является карбоксигруппой, фенильной группой, группой формулы
-NR9R1 0,
где R9 и R1 0 является независимо выбранными из группы, содержащей атом водорода или алкильную группу, имеющую от 1 до 4 атомов углерода,
или гетероциклической группой, имеющей от 5 до 6 атомов в кольце, из которых 1 или 2 являются гетероатомами, выбранными из группы, содержащей атом кислорода или азота, указанная гетероциклическая группа является незамещенной или замещенной у атома углерода атомом кислорода или алкильной группой, имеющей от 1 до 4 атомов углерода; и
указанные группы формул  являются незамещенными или замещенными у атома углерода алкильной группой, имеющей от 1 до 4 атомов углерода, или группой формулы -NR9R1 0, где R9 и R1 0 определены выше;
являются незамещенными или замещенными у атома углерода алкильной группой, имеющей от 1 до 4 атомов углерода, или группой формулы -NR9R1 0, где R9 и R1 0 определены выше;
n1 является 0 или 1;
n2 является 0 или 1;
M означает атом кислорода, или сульфонильную группу, при условии, что, когда R4 означает указанную группу формулы VII, IX, X или XI, R1 не означает указанную алкильную группу, и что когда n2 является 1, A4 не означает одинарную связь, и что, когда n1 является 0, R3 является этилом и R4 является 2-ацетилом, R1 не означает метильную группу,
или их фармацевтически приемлемые соли.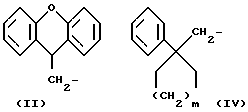
и n1 равно 0.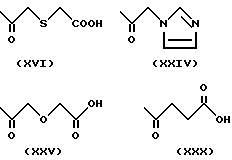
11. Соединение формулы I и его соли по п.1, где R4 означает группу формулы VII, как указано в п.1, в которой общее число атомов углерода в A3, A4 и A5 составляет от 1 до 6, где R6 означает незамещенную циклогексильную группу.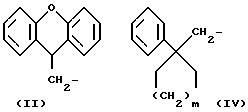
n1 равно 0 и R3 означает алькильную группу, имеющую от 1 до 10 атомов углерода, алкилтиогруппу, имеющую от 1 до 10 атомов углерода, или алкоксильную группу, имеющую от 1 до 10 атомов углерода.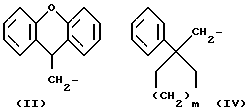
n1 равно 0 и R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода или алкоксильную группу, имеющуюот 1 до 6 атомов углерода.
R1 означает группу формулы II, а ароматические кольца являются незамещенными;
R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода, и
R4 означает группу формулы IV, VII, или Х, как указано в п.1, в которой М означает атом кислорода.
R1 означает группу формулы II, а ароматические кольца являются незамещенными;
R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода;
R4 означает группу формулы VI, VII или Х, как указано в п.1, в котором М означает атом кислорода; и
в случае, когда n2 равно 1, R4 означает группу формулы VI, как указано в п.1, в которой общее число атомов углерода в А1 и А2 составляет от 2 до 4.
R1 означает группу формулы II, а ароматические кольца являются незамещенными;
R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода;
R1 означает группу формулы VI, VII или Х, как указанов п.1, в котором М означает атом кислорода; и
в случае, когда r2 равно 0, R4 означает группу формулы VI, как указано в п.1, в которой общее число атомов углерода в А1 и А2 составляет от 1 до 3.
R1 означает группу формулы II, а ароматические кольца являются незамещенными;
R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода; и
R4 означает группу формулы VII, как указано в п.1, в которой общее число атомов углерода в А3, А4 и А5 составляет от 1 до 6,
R6 означает алкильную группу, имеющую от 1 до 6 атомов углерода или циклоалкильную группу, имеющую от 3 до 7 атомов углерода.
R1 означает гурппу формулы II, а ароматические кольца являются незамещенными;
R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода; и
R4 означает группу формулы Х, как указано в п.1, в которой общее число атомов углерода в А3, А4 и А5 составляет от 1 до 6, и
R6 означает алкильную группу, имеющую от 1 до 6 атомов углерода, или циклоаклильную группу, имеющую от 3 до 7 атомов углерода.
R1 означает группу формулы II, а ароматические кольца являются незамещенными;
R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода;
R4 означает группу формулы VII, как указано в п.1, в которой общее число атомов углерода в А3, А4 и А5 составляет от 1 до 6
R7 означает атом водорода или группу формулы XVI, XXIV, XXV или ХХХ;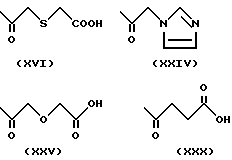
25. Соединение формулы I и его соли по п.1, где:
R1 означает группу формулы II, а ароматические кольца являются незамещенными;
R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода;
R4 означает группу формулы Х, как указано в п.1, в которой общее число атомов углерода в А3, А4 и А5 составляет от 1 до 6, и
R7 означает атом водорода или группу формулы XVI, XXIV, XXV или ХХХ, как указано в п.24.
R1 означает группу формулы II, а ароматические кольца являются незамещенными;
R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода;
R4 означает группу формулы VII, как указано в п.1, в которой общее число атомов углерода в А3, А4 и А5 составляет от 1 до 6, и
R6 означает незамещенную циклогексильную группу.
R1 означает группу формулы II, а ароматические кольца являются незамещенными;
R3 означает алкильную группу, имеющую от 1 до 6 атомов углерода, алкилтиогруппу, имеющую от 1 до 6 атомов углерода, или алкоксильную группу, имеющую от 1 до 6 атомов углерода;
R4 означает группу формулы Х, как указано в п.1, в которой общее число атомов углерода в А3, А4 и А5 составляет от 1 до 6, и
R6 означает незамещенную циклогексильную группу.
R1 означает (9Н-ксантен-9-ил) метильную гурппу;
n1 равно 0;
R3 означает группу метилтио-, изопропилтио-, изопропил или трет-бутил;
R4 означает группу формулы VIa, VIIa или IXa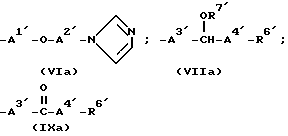
в которой R6 означает циклопентильную группу, циклогексильную группу, циклогептильную группу, фенильную группу, 2-хлорфенильную группу или 4-хлорфенильную группу;
R7' означает 3-карбоксипропионильную группу, 2-карбоксибензольную группу или аминоацетильную группу;
А1' означает метиленовую группу;
А2' оначает алкиленовую группу, имеющую от 2 до 4 атомов углерода;
А3' означает одинарную связь или алкиленовую группу, имеющую от 1 до 3 атомов углерода, которая может быть прервана двойной связью (в частности, метиленовая или этиленовая группа);
А4' означает одинарную связь или алкиленовую группу, имеющую от 1 до 5 атомов углерода, которая может быть прервана двойной связью;
место присоединения R4 к бензольному кольцу соединения формулы I находится в орто-положении по отношению к аминогруппе и мета-положении по отношению к R3 или в мета-положении по отношению к аминогруппе и пара-положении по отношению к R3.
N-[2-трет-бутил-5-(5-циклогексил-3-гидроксипентил)фенил] -2-(9Н-ксантен-9-ил)ацетамид;
N-[2-трет-бутил-5-(4-циклогексил-3-гидроксибутил)фенил] -2-(9Н-ксантен-9-ил)ацетамид;
N-[2-трет-бутил-5-(6-циклогексил-3-гидроксигексил)фенил] -2-(9Н-ксантен-9-ил)ацетамид;
N-[2-трет-бутил-5-(7-циклогексил-3-гидроксигептил)фенил] -2-(9Н-ксантен-9-ил)ацетамид;
N-[2-трет-бутил-5-(3-циклогексил-3-гидроксипропил) фенил] -2-(9Н-ксантен-9-ил)ацетамид;
N-[2-трет-бутил-5-(2-циклогексил-1-гидроксиэтил) фенил]-2-(9Н-ксантен-9-ил)ацетамид;
N-[2-трет-бутил-5-(6-циклопентил-1-гидроксиэтил) фенил]-2-(9Н-ксантен-9-ил)ацетамид;
1-(2-{ 4-трет-бутил-3-[2-(9Н-ксантен-9-ил)ацетамидо] фенил} этил)-2-циклогексилэтил натрия сукцинат;
1-(2-{ 4-трет-бутил-3-[2-(3Н-ксантен-9-ил)ацетамидо] фенил}этил)-3-циклогексидпропил натрия сукцинат;
натрий 1-(2-{ 4-трет-бутил-3-[2-(9Н-ксантен-9-ил)ацетамидо]фенил}этил-5-циклогексилпентил сукцинат;
N-{ 2-[3-(1-имидазолил)пропокси] метил-6-метилтиофенил}-2-(9Н-ксантен-9-ил)ацетамид гидрохлорид;
N-{ 2-[3-(1-имидазолил)пропокси]метил-6 метилтиофенил}-2-(9Н-ксантен-9-ил)ацетамид;
N-{ 2-[3-(1-имидазолил)пропокси] метил-6-трет-бутилфенил} -2-(9Н-ксантен-9-ил)ацетамид гидрохлорид;
натриевая соль α-1-(2-{ 4-трет-бутил-3-[2-(9Н-ксантен-9-ил) ацетамидо] фенил} этил)-2-циклогексил этил карбоксиметилтиоацетата;
N-[2-трет-бутил-5-{ 3-[2-(1-имидазолил) ацетокси] -4-циклогексилбутил} фенил]-2-(9Н-ксантен-9-ил)ацетамид гидрохлорид;
натриевая соль N-(2-трет-бутил-5-{ 3-[2-карбоксиметокси) ацетокси]-4-циклогексилбутил} фенил)-2-(9Н-ксантен-9-ил)ацетамид;
натриевая соль N-2(2-трет-бутил-5-{7-циклогексил-3-[2-(карбоксиметокси)ацетокси]гептил} фенил)-2-(9Н-ксантен-9-ил)ацетамид;
N-[2-трет-бутил-5-[4-циклогексил-2-(гидроксиметил) бутил]фенил]-2-(9Н-ксантен-9-ил)ацетамид;
N-[2-трет-бутил-5-{ 4-(2-циклогексилэтокси)-3-гидроксибутил] фенил] -2-(9Н-ксантен-9-ил) ацетамид;
N-[2-трет-бутил-5-(5-циклогексилокси-3-гидроксипентил) фенил]-2-(9Н-ксантен-9-ил) ацетамид;
N-(2-трет-бутил-5-{4-(2-циклогексилэтокси)-3-[2-(1-имидазолил] ацетокси] бутил}фенил)-2-(9Н-ксантен-9-ил) ацетамид гидрохлорид;
натриевая соль N-(2-трет-бутил-5-{3-[2-(карбоксиметокси) ацетокси]-4-циклогексилоксипентил} фенил)-2-(9Н-ксантен-9-ил) ацетамид;
N-{ 2-трет-бутил-5-[(2-этил-1-имидазолил) метил] фенил}-2-(9Н-ксантен-9-ил)ацетамид;
и его фармацевтически приемлемые соли.
