Изобретение относится к способу ацилирования 7-аминогруппы цефалоспоранового кольца.
7-ACA (7-амино-3-ацетоксиметил-3-цефем-4-карбоновая кислота) формулы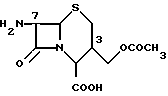
является хорошо известным соединением, которое предложено в качестве исходного сырья в различных синтезах, в частности в синтезах многих цефалоспоринов.
Целый ряд наиболее важных цефалоспоринов получают через следующие стадии:
а) осуществление ацилирования 7-аминогруппы цефалоспоранового кольца аминотиазолилуксусной кислотой, возможно замещенной, у которой защищена амминогруппа;
б) снятие защиты аминогруппы и
в) необязательно замещение 3-ацетоксиметильной группы цефалоспоринового кольца нуклеофильным агентом.
Последовательность указанных стадий может по желанию меняться. Например, последовательность стадий может быть а), б), в); или а), в), б); или также в), а), б), как описано в Journal of Antibiotics, дек. 1978: 1262-1271 и в BE-A-823861 Такедой (относительно антибиотика цефотиам).
В каждом случае ацилирование 7-аминогруппы цефалоспоринового кольца осуществляется аминотиазолилуксусной кислотой, возможно замещенной, у которой аминогруппа, защищена, с последующим снятием защиты.
Известные защитные группы, применяемые в практике, требуют использования дорогостоящего исходного сырья (такого как тритил или BOC), критических условий для введения реагентов и критических кислотных условий для удаления защиты.
Создание аминотиазольного кольца непосредственно на аддукте имеет тот недостаток, что оно требует применения высокоопасных реагентов, таких как дикетены и ангидриды.
И вот неожиданно было найдено, что некоторые чрезвычайно важные преимущества достигаются в том случае, если аминозащитной группой является фенилацетильная группа или феноксиацетильная группа: эти защитные группы дешевы, их легко ввести, и они совместимы с условиями активирования карбонила, необходимыми для реакции с 7-ACA.
Применение фенил- или феноксиуксусных кислот для защиты аминогруппы аминотиазолилуксусной кислоты (чтобы активировать ее карбоксильную группу) крайне важно, потому что такая защита позволяет иметь весьма устойчивый аддукт, который крайне трудно удалить химически, так что указанный аддукт благодаря такой защите может быть подвергнут последующей химической обработке без вовлечения защитной группы.
Далее неожиданно оказалось, что упомянутые защитные группы допускают их избирательное удаление в исключительно мягких условиях простым гидролизом в водном растворе, в основном при комнатной температуре, в присутствии пенициллин-G-амидазы или пенициллин-V-амидазы, которые катализируют гидролиз амидной связи N-фенилацетил- или N-феноксиацетилмминотиазодбной части молекулы с гораздо более высокой скоростью чем та, с которой гидролизуется амидная связь, присутствующая в положении 7 конечного продукта реакции.
Энзимы пенициллина-G-амидаза и пенициллин-V-амидаза сами по себе известны, и их применение описано, например, в GB-A 1480850 и GB-A 1473100: было известно, что используя такие энзимы можно получить ферментативно (вместе химического пути) 6-APA, исходя из пенициллина-G- или пенициллина-V, и 7-ADCA, исходя из цефалоспорина-G или цефалоспорина-V.
Поэтому использование N-фенил- или N-феноксиуксусной защитной группы, избирательно снимаемой энзиматическим гидролизом в водном растворе, или без удаления ацетоэфирной группы, присутствующей в молекуле, или значительного гидролиза амидной связи в положении 7, представляет собой очень важно экономическое и экологическое преимущество в сравнении со способами ацилирования 7-аминогруппы цефалоспоранового кольца, в частности для введения защиты и снятия защиты аминогруппы в известных синтезах 7-аминотиазолилцефалоспоринов.
Особую важность представляет также тот факт, что реакция происходит в водной среде и получаются высокие выходы.
Изобретение относится к способу ацилирования 7-аминогруппы цефалоспоранового кольца, предусматривающий получение 7-ACA аминотиазолилзащищенного аддукта ацилированием указанной аминогруппы аминотиазолилуксусной кислотой, возможно замещенной, у которой аминогруппа защищена, с последующим снятием защиты аминогруппы, отличающемуся тем, что аминозащитная группа выбрана из группы, состоящей из фенилацетильной и соответственно из феноксиацетильной группы и что снятие защиты осуществляют гидролизом в водном растворе при температуре 0-50oC и при pH 5-9 в присутствии энзима, выбранного из группы, состоящей из пенициллин-G-амидазы и пенициллин-V-амидазы соответственно.
В частности, было найдено, что упомянутый гидролиз можно проводить при температуре 15-35oC и при pH 6-8.
Изобретение также относится к новой аминотиазолилуксусной кислоте и ее α-замещенным производным, у которых аминогруппа защищена фенилкарбонильной группой, выбранной из группы, состоящей из фенилацетильной или феноксиацетильной группы; и также к 7-ACA аминотиазолилзащищенному аддукту, в котором защита аминогруппа осуществляется фенилкарбонильной группой, выбранной из группы, состоящей из фенилацетильной и феноксиацетильной группы.
Частью изобретения являются также новые, возможно замещенные α-аминотиазолилуксусной кислотой и 3-замещенные, N-фенилацетил- и N-феноксиацетилфалоспорины.
Пример 1. Получение 7-/2-(аминотиазол-4-ил)/ ацетамидоцефалоспорановой кислоты.
А) Получение N-фенилацетиламинотиазолилуксусной кислоты (защищенный ацилирующий агент).
К 18,6 г (0,1 моля) этилового эфира аминотиазолилуксусной кислоты в 100 мл органического негидроксилированного растворителя (метиленхлорид, THF, диоксан, ацетонитрил и др.) в присутствии 1,2 мольных эквивалента органического основания, например триэтиламина, прибавляют при перемешивании при 0-5oC 15,4 г (0,1 моля) хлорангидрида фенилуксусной кислоты в течение 15 мин.
Реакция заканчивается через несколько часов при комнатной температуре (тонкослойная хроматография на кремнеземе в этилацетат/гексане 7:3 Rf: около 0,7). Сосуд затем охлаждают подо льдом, а органическую фазу промывают последовательно разбавленной хлористоводородной кислотой, водным раствором бикарбоната и водой. Раствор сушат и затем выпаривают под вакуумом. Маслянистый остаток затвердевает самопроизвольно. Продукт может быть кристаллизован из гексан/этилацетата. Однако в этом нет необходимости, потому что сырой продукт можно использовать непосредственно на следующей стадии. Эта стадия состоит в обработке раствора эфира, полученного выше, водным раствором NaOH; последний получают, растворяя 1 г NaOH в каждых 4 г эфира, подлежащего гидролизу. При смещении растворов имеет место легкое разогревание. Реакционную смесь перемешивают 5-6 ч, этого времени достаточно для окончания гидролиза, за развитием которого следят посредством тонкослойной хроматографии.
Когда реакция закончена, смесь выпаривают под вакуумом для удаления большей части растворителя, поддерживая температуру кипения на низком уровне, чтобы избежать гидролиза амидной связи. Реакционную смесь затем разбавляют водой при перемешивании и охлаждают, и подкисляют HCl. Осадок, состоящий из N-фенилацетиламинотиазолилуксусной кислоты (далее обозначаемой как соединение "X") собирают посредством фильтрации и затем промывают водой на фильтре и наконец сушат, получая 22 г (79%).
Б) Получение 7-амидо-((2-N-фенилацетиламино-4-тиазолил)-2-ацетил)-3-ацетоксиметил-3-цефем- 4-карбоновой кислоты.
13,8 г (0,05 моля) кислоты "X", полученной согласно разделу А, суспендированной в 30 мл метиленхлорида 9,5 г оксалилхлорида (0,075 моля) и несколькими каплями DMC при комнатной температуре при перемешивании. Когда выделение газа практически прекращается, прибавляют еще 3 г оксалилхлорида, нагревают слегка орошая, в течение 30 мин. Реакционную смесь затем концентрируют под вакуумом. К остатку, имеющему интенсивно зеленый цвет, прибавляют 50 мл метиленхлорида и полученную смесь прибавляют по каплям при перемешивании и под атмосферой азота при -10oC, к смеси 12,2 (0,045 моля) 7-АСА и 40 мл триэтиламина в 100 мл метиленхлорида.
Когда прибавление заканчивается, температуре дают подняться до комнатной и затем реакционную смесь промывают холодной водой и разбавленной кислотой.
Высушенную органическую фазу выпаривают, получая маслянистый остаток красного цвета. Этот материал, хорошо высушенный под вакуумом, может самопроизвольно затвердевать.
Однако, если к нему добавить небольшой объем этанола при 50-60oC, это позволит выделить кристаллическое твердое вещество (соединение "X"), которое собирают на фильтре. 16,7 г (67%). Подобные выходы получают, когда обрабатывают суспензию 7-АСА в метиленхлориде 2 молярными эквивалентами BSA до полного растворения с последующей добавкой триэтиламина и затем хлорангидрида кислоты.
Указанное соединение может быть получено также следующим образом.
К 3,8 г (0,05 моля) кислоты, полученной в соответствии с разделом А, в 300 мл THF в присутствии 6,2 г (0,06 моля) N-метиленформолина по каплям прибавляют 5,4 г этилового эфира хлормуравьиной кислоты. Через 3 часа прибавляют 13,6 г (0,05 моля) 7-АСА в 50 мл метиленхлорида и 50 мл триэтиламина при комнатной температуре.
Через 12 ч (всегда при комнатной температуре), реакционную смесь концентрируют под вакуумом, затем разбавляют метиленхлоридом и повторно промывают разбавленной HCl и водой и наконец сушат. Красноватый остаток, полученный выпариванием растворителя, затем обрабатывают этанолом, получая таким образом твердый 7-АСА аминотиазолил-защищенный аддукт (соединение "Y"), которое отфильтровывают 14 г (59%).
В) Энзиматический гидролиз
5,3 г соединения "Y", полученного в соответствии с разделом Б, суспендируют в 120 мл воды и суспензию обрабатывают NaOH при pH 8, при 36oC. Вся твердая фаза растворяется в течение 10-15 мин. Затем добавляют 500 единиц энзима PGA и за развитием гидролиза следят посредством жидкостной хроматографии высокого давления, так как не наблюдается прямого соответствия между израсходованным основанием и развитием гидролиза. Когда исчезает исходный материал, иммобилизованный энзим отфильтровывают, реакционную смесь концентрируют под вакуумом объемом 50 мл, затем охлаждают и подкисляют до pH 3,6. Осадок собирают и смывают с фильтра абсолютным этанолом. Продукт представляет собой 7-/2(аминотиазол-4-ил/ацетамидоцефалоспорановую кислоту, которая может быть кристаллизована из водного раствора этанола 3,3 г (80%).
Пример 2. Получение 7/((2-амино-4-тиазолили)ацетил)амино-3- ///1-/2-(диметил-амино)-этил/-1H-тетразол-5-ил/тио/метил-8-оксо -5-тиа-1-аза-бицикло/4,2,0/окт-2-ен-2-карбоновой кислоты. 2 HCl (цефотиам)
5 г (12,1 ммоля) 7-/2(аминотиазол-4-ил)/ацеамидоцефалоспорановой кислоты, полученной в примере 1-В (выше), растворяют в 40 мл H2O с 2,3 г (13,2 ммоля) 1-(2-диметил-аминоэтил)-1H-тетразол -5-тиола (соединение "Z") и 2,18 г (26 ммолей) NaHCO3 при 70oC. Смесь оставляют реагировать при 70oC на 2 ч, следя развитием посредством тонкослойной хроматографии.
Смесь затем охлаждают до 20oC и раствор, при pH 7 элюируют на XAD 1180.
Продукт элюируют смесью H2O метанол, выпаривая под вакуумом обогащенную фракцию, и цефотиам кристаллизуют добавлением изопропанола и концентрированной HCl. Затем проводят фильтрацию и сушку под вакуумом.
Молярный выход: 67%.
Альтернативный синтез.
20 ммолей 7-/2-аминотиазол-4-ил)/ ацетамидоцефалоспорановой кислоты из примера 1-В обрабатывают 20 ммолями упомянутого соединения "Z" в 150 мл ди-THF /H2O 1/1 и 40 ммолями NaHCO3 при 60oC под азотом в течение 4 ч.
В конце реакции раствор концентрируют под вакуумом, устанавливая pH на 7, и экстрагируют 3 раза с помощью 40 мл CH2Cl2. Остаток водной суспензии концентрируют дальше и подкисляют до pH 3,0 концентрированной HCl, затем охлаждают до 0oC и фильтруют. Твердую фазу очищают в изопропаноле при 50oC в течение 30 мин и затем охлаждают, отфильтровывают и сушат.
Молярный выход 58%.
Пример 3. Получение цефотиама.
А) Получение 7-амино-цеф-3-ем-3-1-(2-диметил-аминоэтил)- 1H-тетразол-5-тиол-4-карбоновой кислоты.
15,3 г (88,3 ммоля) 1-(2-диметил-апиноэтил) -1H-тетразол-5-тиола (соединение "Z") прибавляют к 100 мл трифторуксусной, охлажденной до -5oC. Затем загружают 24 г 7-АСА (88,1 ммоля) малыми количествами в течение 30 мин при температуре от 0 до -5oC. За исключением 7-АСА следят посредством тонкослойной хроматографии. В конце реакции трифторуксусную кислоту выпаривают под вакуумом при 45oC. К концентрату добавляют 50 мл THE.
Полученный осадок затем отфильтровывают под вакуумом. Твердое вещество регенерируют водой при pH 7 бикарбонатом натрия, извлекая основу титульного соединения в виде белого твердого вещества, которое фильтруют под вакуумом и сушат.
Молярный выход 76%.
Альтернативный синтез.
15,3 г (88,3 ммоля) упомянутого соединения "Z" прибавляют к 100 мл метансульфоновой кислоты, охлажденной до +5oC. Затем загружают 24 г 7-АСА (88,1 ммоля) малыми количествами при температуре от 0 до +5oC.
В конце реакции добавляют 100 мл изопропиловой кислоты для кристаллизации диметансульфонатной соли титульного соединения.
Затем проводят фильтрацию, и твердое вещество помещают в 80 мл воды, устанавливая pH с помощью NaHCO3. Прибавляют 30 мл изопропанола и осуществляют кристаллизацию титульного соединения при 15oC.
Выход 78%.
ЯМР ( δ в DMSO - d6): 2,58 (6H, с, N-диметил); 3,22 (2H, т, этеро-N-метилен); 3,52 (2H, к AB, 2CH2); 4,24 (2H, к 3CH2); 4,60 (2H, м, CH2-N-диметил); 4,76 (1H, д, 6CH); 4,94 (1H, д, 7CH).
Б) Получение N-фенилацетилзащищенного 3-замещенного цефалоспорина.
10 ммолей защищенного ацилирующего агента "X" в 10 мл безводного DME и 29 мл CH2Cl2 обрабатывают 10 ммолями N-метилморфолина при -30oC. Затем прибавляют 10 ммолей этилового эфира хлормуравьиной кислоты, поддерживая температуру при -30oC в течение 30 минут. Смесь затем выливают в 10-ммолярный раствор соединения, полученного в соответствии с разделом А), растворяли в 10 мл DMF, 20 мл CH2Cl2 и 10 ммолях N-метилморфолина. Реакция конденсации заканчивается за 1 ч, растворитель выпаривают под вакуумом, а остаток извлекают небольшим количеством воды и доводят pH до 3,5 концентрированной HCL. Затем отфильтровывают кристаллическое твердое вещество и очищают его изопропаноле при 4oC. Молярный выход составляет 65%.
Альтернативный синтез.
20 ммолей защищенного ацилирующего агента "Х" подвергают реакции с 20 ммолями соединения, полученного в соответствии с разделом А), в 200 мл CH3CN/H2O 1/1 и 50 ммолях NaHCO3 при 65oC в течение 3 ч в инертной атмосфере азота.
В конце реакции осуществляют концентрирование под вакуумом, устанавливая pH на 7, и затем дважды экстрагируют 45 миллилитрами CH2CH2. Затем водную суспензию концентрируют дальше и доводят pH до 3,5 и фильтруют.
Твердое вещество может быть подвергнуто энзиматическому гидролизу в том виде, как оно есть, или его можно очистить нагреванием в изопропиловом спирте, отфильтровать и высушить.
Выход: 55%.
ЯМР ( δ в DMCO-d6): 2,62 (6H, с, N диметил); 3,40 (2H, т, этеро-N-метилен); 3,50 (2H, к, AB, 2CH2); 3,65 (2H, с, CH2-Ph); 3,76 (2H, с, CH2-CO); 4,25 (2H, к, АВ, 3CH2); 4,65 (2H, м, CH2-N-диметил); 5,05 (1H, д, 6CH); 5,65 (1H, к, 7CH); 6,92 (1H, с, тиазол 5H); 7,30 (5H, м, Ph).
В) Энзиматический гидролиз.
10 г (15,5 ммолей) N-фенилацетилзащищенного цефалоспорина, полученного, как описано в вышеприведенном разделе Б), диспергируют в 100 мг H2O при комнатной температуре и затем переводят обратно в раствор содой 1н. при pH 7,5.
К этому раствору прибавляют 10 г PG-амидазы, иммобилизованной на Eupergit C (170/U/g), следя за энзиматическая гидролизом с NaOH 1н. по автоматическому титратору.
По расходу основания отмечают соответствующее ему образование цефотиама (за этим также следят посредством тонкослойной хроматографии). Энзим отфильтровывают, когда реакция заканчивается, и раствор пропускают через адсорбент - смолу XAD 1180.
Элюат из смолы, содержащий цефотиам, высушивают, собирают 4-н HCl и к нему добавляют изопропанол для кристаллизации дигидрохлорида цефотиама. Затем проводят фильтрацию и сушку под вакуумом.
Моляный выход, 72%.
Пример 4. Получение цефотаксима.
А) Получение N-фенацетиламинотиазол-L-метоксиаминоуксусной кислоты.
10 г метоксииминотиазолуксусной кислоты в 100 мл метиленхлорида и 20 мл триэтиламина прибавляют при 0oC при перемешивании к 1,2-мольным эквивалентам хлорангидрида фенилуксусной кислоты. Через несколько часов добавляют 10 мл триэтиламина и затем еще 0,5 эквивалентов хлорида. Реакция заканчивается за ночь. Органическую фазу промывают водой, затем сушат и концентрируют. Остаток взмычивают с небольшим количеством этанола, который удаляет возможный остаток фенилуксусной кислоты.
Выход около 80%.
1Н ЯМР DMSO - d6 3,75 (2H, с, PhCH2); 3,90 (3H, с, NOCH3); 7,30 (5H, м, Ph); 7,50 (1H, с, SCH); 12,75 (1H, с, COOH).
Б) Получение N-фенилацетилцефотаксима
2 г продукта, полученного в соответствии с примером 4-А, в 60 мл безводного THF прибавляют при температуре около 5oC к 0,65 г дициклогезилкарбомида, соответствующим 0,5 молярным эквивалентам. Смесь перемешивают при указанной температуре в течение 30 мин, затем еще 30 мин при комнатной температуре.
Осадок дициклогезилмочевины, образующийся за это время, отфильтровывают, органическую фазу охлаждают до -20oC.
Затем в течение нескольких минут добавляют раствор 0.85 г 7-АСА в 15 мл метиленхлорида и 2 мл триэтиламина. Затем температуре дают подняться и поддерживают ее такой в течение 3 ч. Раствор выпаривают, извлекая затем остаток диоксаном, причем этот остаток состоит из смеси N-фенацентилцефотаксима и N-фенацетиламинотиазол- α -метоксииминоуксусной кислоты, осаждающей с диэтиламинной солью последнее соединение в виде соли. Затем фильтруют и сушат. Остаток кристаллизуют из этилацетата, получая желаемый N-фенацетилцефотаксим.
С другой стороны, разделение можно осуществить путем вываривания с небольшим количеством метиленхлорида остатка, состоящего из смеси двух указанных соединений, в которой N-фенацетиламинотиазол- α -метоксииминоуксусная кислота является менее растворимой, с последующей кристаллизацией остатка после выпаривания CH2Cl2 из этилацетата.
Молярный выход 71%.
1H-ЯМР DMCO-d6: 2,05 (3H, с, COCH3); 3,55 (2H, к АВ, 2CH2); 3,75 (2H, с, PhCH2); 3,90 (3H, с, NOCH3); 4,65-5,0 (2H, к AB, 3 CH2); 5,18 (1H, д, 6 CH); 5,85 (1H, к, 7 CH); 7,3 (5H, м, Ph); 7,4 (1H, с, S CHC); 9,75 91H, д, CONH); 12,8 (1H, с, COOH).
В) Энзиматический гидролиз
0,5 г N-фенацетилцефотаксима растворяют в 2 мл ацетонитрила и прибавляют, при перемешивании, к 25 мл буфера при pH 8.
Добавляют приблизительно 400 U1 PGA на Eupergit и за ходом гидролиза следят посредством тонкослойной хроматографии при 37oC. Реакцию заканчивают за 40-50 мин. Затем проводят фильтрацию с последующим концентрированием в очень малом объеме и подкисление при 0oC. Цефотаксим выделяют фильтрацией и затем снова растворяют после охлаждения в небольшом количестве этилацетата и фильтруют для удаления остатка фенилуксусной кислоты.
Молярный выход 65%.
Пример 5. Проводя реакцию, как описано в примере 4, нашли, что возможно получать другие важные цефалоспорины, такие как цефтазимид, цефменоксим, цефиксим, цефтриаксон, цефодизим, цефтибутен, цефтизоксим, цефепим.
7-АСА аминотиазолилзащищенные аддукты, возможно 3-замещенные, являющиеся источниками цефалоспоринов, в которых аминозащитная группа представляет собой фенилацетильную группу, введенные в раствор при pH 7,5, с CH3CN или без него, при 10%, подвергнуты действию PGA при постоянном pH, поддерживаемым с помощью основания. Показано, насколько и в этом случае PGA избирателен в его гидролитическим действии по отношению 7-амидной связи цефалоспорина. В конце энзиматического гидролиза PGA собирают фильтрацией, концентрируют под вакуумом, слегка подкисляют и экстрагируют органическим растворителем, не смешивающимся с водой: фенилуксусной кислотой. Водную фазу концентрируют до небольшого объема и извлекают вышеупомянутые цефалоспорины.
Очевидно, что в случае некоторых цефалоспоринов уксуснокислотная часть в боковой цепи была предварительно защищена как трет-бутиловый эфир, чтобы затем быть удаленной на конечной стадии путем легкого кислотного гидролиза.
Та же методика, что и раскрытая для N-защищенной фенилуксусной серии, действительная и для осуществления защиты феноксиуксусной части молекулы, в каковом случае применяют пеницилин-V-амидазу.
Использование: в химии антибиотиков цефалоспоранового ряда. Сущность изобретения: способ ацилирования 7-аминогруппы цефалоспоранового ряда, в соответствии с которым получают аминотиазолилзащищенный аддукт 7-амино-3-ацетоксиметил-3-цефем-4-карбоновой кислоты ацилированием указанной аминогруппы аминотиазолилуксуной кислотой, у которой аминогруппа защищена фенилацетильной группой или феноксиацетильной группой с последующим снятием защиты аминогруппы водным гидролизом в присутствии G-амидазы или соответственно пенициллин-V-амидазы. Раскрыт также новый N-фенилацетил-3-замещенный цефалоспорин, используемый в указанном способе. 2 с.и 1 з.п.ф-лы.
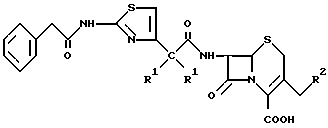
где R1 - оба водород или вместе образуют группу =N - CH3;
R2 - OCOCH3 или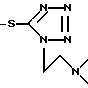 ,
,
| FR, патент, 2249889, кл.C 07 D 501/04, 1980 | |||
| GB, патент, 2161476, кл.C 07 D 501/06, 1986. |
