Изобретением предлагается способ химического ацилирования, в частности способ ацилирования в водной среде для получения такого антибиотика, как гидрат дигидрохлорида цефепима, известного также под полным названием 7-/2-(2-аминотиазол-4-ил)-(Z)-метоксииминоацетамидо/-3- /(1-метил-1-пирролидинио)метил/цеф-3-ем -4-карбоксилата. Изобретением кроме того дается устойчивая кристаллическая соль син-изомера тиазолильного промежуточного соединения, которое может быть использовано в синтезе применимых антибактериальных средств широкого профиля, и способ получения таких промежуточных соединений.
Известно большое число цефалоспориновых антибиотиков, содержащих боковую цепь 2-(2-аминотиазол-4-ил)-(Z)-2-метоксииминоуксусной кислоты, присоединяемой к 7-аминогруппе цефалоспорановой кислоты хорошо известными методами ацилирования. Во многих случаях процесс ацилирования включает в себя обязательную защиту аминогруппы и активацию карбоновой кислоты боковой цепи. Следовательно, в литературе приведено большое число аминозащитных групп для 2-аминогруппы тиазольного цикла и большое число активирующих групп для карбоновой кислоты. Поиск новейших защитных групп и активирующих групп для получения целевого антибиотика все еще является объектом многочисленных публикаций, что связано со стоимостью и токсичностью определенных активирующих групп. Таким образом, все еще существует необходимость получения антибиотиков широкого профиля на основе простой, устойчивой, кристаллической, экономичной и неядовитой боковой цепи с целевой геометрической (Z)-конфигурацией, легко соединяющейся с 7-аминогруппой цефалоспоринового ядара. Ниже следует перечень литературы, относящейся к отдельным представителям тиазольной боковой цепи.
В патенте США N 4203899 (Ochiai и др.), раскрываются соединения формулы:
R где R1 представляет аминогруппу, защищенную аминогруппу, гидроксил или защищенный гидроксил; R5 представляет гидроксил или защищенный гидроксил и W представляет гидроксил, С1-С4-алкоксигруппу, галоген или ОМ, где М щелочной металл.
где R1 представляет аминогруппу, защищенную аминогруппу, гидроксил или защищенный гидроксил; R5 представляет гидроксил или защищенный гидроксил и W представляет гидроксил, С1-С4-алкоксигруппу, галоген или ОМ, где М щелочной металл.
В заявке на патент Великобритании GB-2144424, 1985, раскрыто получение ряда производных пиридиний-цефалоспорина различными способами, в том числе использованием соединения формулы
R4HN или его соли, где R1 означает водород или атом галогена, R2 означает атом водорода или С1-С6-алкил и R4 представляет водород или аминозащитную группу, или активированного производного этого соединения.
или его соли, где R1 означает водород или атом галогена, R2 означает атом водорода или С1-С6-алкил и R4 представляет водород или аминозащитную группу, или активированного производного этого соединения.
В заявке на Европейский патент ЕР-160546, опублик. 1985, также раскрывается получение ряда производных цефалоспорина различными способами, в том числе использованием замещенных производных оксиминотиазолилуксусной кислоты формулы
R8HN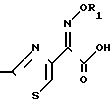 или ее реакционноспособного производного, где R8 представляет атом водорода или защитную группу для аминогруппы. Приемлимые примеры подобных реакционноспособных производных, которые приводятся в заявке, включают смешанные ангидриды кислот, галоидангидриды кислот, активные сложные эфиры, активные амиды и азиды кислот.
или ее реакционноспособного производного, где R8 представляет атом водорода или защитную группу для аминогруппы. Приемлимые примеры подобных реакционноспособных производных, которые приводятся в заявке, включают смешанные ангидриды кислот, галоидангидриды кислот, активные сложные эфиры, активные амиды и азиды кислот.
В патенте США N 4385181 (Farge и др.), 1983, раскрываются сложные тиоэфиры формулы
R1HN где R1 представляет водород или защитный радикал, Ro представляет водород, алкил, винил, цианометил или защитный радикал и R представляет алкил, 1-2-амино-2-карбоксиэтил, фенил или большое число разнообразных гетроциклических радикалов, перечисленных в столбцах 4-8, а также их син- и анти-изомеры и их смеси.
где R1 представляет водород или защитный радикал, Ro представляет водород, алкил, винил, цианометил или защитный радикал и R представляет алкил, 1-2-амино-2-карбоксиэтил, фенил или большое число разнообразных гетроциклических радикалов, перечисленных в столбцах 4-8, а также их син- и анти-изомеры и их смеси.
Помимо цитированных ссылок существует также большое число ссылок, в которых приводятся различные защитные группы для 2-аминозаместителя и еще большее число активирующих (отходящих групп фрагмента карбоновой кислоты, которые могут быть использованы при ацилировании производного 7-аминоцефалоспорина.
Однако наиболее здесь уместен патент Чехословакии N 238950, опублик. 1987, (Chemical Abstracts, т. 110, стр. 544 (1989)), раскрывающий соединение настоящего изобретения формулы
NH · HCl и согласно которому предполагается, что соединение имеет син-конфигурацию. Единственное доказательство получения соединения в патенте заключается в приводимом содержании хлора в 99-105% от теоретического значения.
· HCl и согласно которому предполагается, что соединение имеет син-конфигурацию. Единственное доказательство получения соединения в патенте заключается в приводимом содержании хлора в 99-105% от теоретического значения.
В связи с работой по созданию новых синтетических методов получения антибиотиков создатели изобретения, также как и другие исследователи в данной области испытывали потребность в простых, удобных, экономичных, кристаллических, устойчивых и неядовитых исходных продуктах для использования в изготовлении антибиотиков. Первоначальные попытки получить и использовать хлорангидрид 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты без применения защитных групп оказались безуспешными. Однако создателями изобретения в настоящее время обнаружено, что соединение по изобретению может быть получено в специфических условиях реакции. Открытие было дополнительно подтверждено после того, как создатели изобретения не смогли воспроизвести результаты патента Чехословакии. Целевой син-изомер гидрохлорида хлорангидрида, необходимый для получения целевого антибиотика, получить повторно не удалось. Более того, дополнительными опытами подтверждено, что, следуя указаниям прототипа, невозможно получить гидрохлорид целевого син-изомера хлорангидрида кислоты, по-существу не содержащего анти-изомера и имеющего спектр протонного ядерного магнитного резонанса, который приводится здесь.
Широкий спектр цефепимых антибиотиков приводится Aburaki и др. в патенте США N 4406899, 1983, а также способы их получения, описываемые двумя схемами реакции, в которых реагенты и продукты требуют использования блокирующих и деблокирующих групп. Согласно схеме реакции, иллюстрируемой реальными примерами, полученный продукт требует стадии хроматографической очистки для разделения смеси Δ2 и Δ3 -изомеров, а полученный в результате цефепим находится в цвитерионной форме. Однако цвитеррионная форма цефепима неустойчива при комнатной и повышенной температуре.
В патенте США N 4910301 (Murray A.Kaplan и др.), 1990, раскрываются термостойкие кристаллические соли цефепима в сухой порошковой форме, обладающие прекрасной устойчивостью при комнатной температуре и более высокой устойчивостью при повышенной температуре по сравнению с цвиттерионной формой, описанной Aburaki и др. в патенте США N 4406899.
В патенте США N 4868294 (Brundidge и др.), 1989, описано получение 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилатных солей, по существу не содержащих Δ2 -изомера, и их применение в способе ацилирования в водной среде для получения антибиотика цефепима в виде сульфатной соли.
В патенте США N 4754031 (Angerbauer и др.), 1988, описан способ получения нескольких цефалоспориновых антибиотиков, и в их числе цефепима в цвиттерионной форме. Хотя в этом способе не применяют защитных групп, тем не менее для активации реакции ацилирования в водной среде применяют ангидрид, а для получения цвиттерионной формы цефепима применяют хроматографические стадии очистки.
В патенте США N 4943631 (Brian E.Looker), 1990, описан улучшенный способ получения антибиотика цефепима в виде гидройодида. Образование нежелательного Δ2 -изомера регулируется применением в качестве промежуточного соединения сульфоксида цефалоспорина. Однако, приводимый в патенте способ остается дорогостоящим и неэффективным, поскольку включает две дополнительные стадии по сравнению со способом прототипа и в способе продолжают применять защитные группы, требующие процедур по блокированию и деблокированию. Кроме того, в качестве метода очистки способ требует колоночной хроматографии, что непрактично в промышленных масштабах.
Получение кристаллической сульфатной соли и цвиттериона, описанные ранее, происходят с применением по-существу одного способа ацилирования в водной среде и с применением различных блокирующих деблокирующих групп и активных сложных эфиров. Во всех случаях рекомендуемая кристаллическая форма гидрата дигидрохлорида цефепима должна быть получена через очищенную цвиттерионную форму цефепима. Таким образом, существует необходимость в создании простой и недорогой методики прямого ацилирования, не включающей дополнительных стадий реакции и удаление защитных групп, стадий регулирования стереохимии и этапов хроматографирования, и, что более важно, методики ацилирования, в которой целевой антибиотик гидрат дигидрохлорид цефепима получают по-существу без примеси антиизомера и Δ 2-изомера.
Изобретением дается способ химического ацилирования, в частности, способ ацилирования в водной среде для получения такого антибиотика, как гидрат дигидрохлорида цефепима, по-существу не содержащего анти-изомера и Δ 2 -изомера. Изобретением кроме того дается устойчивый кристаллический син-изомер 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхло-рид, гидрохлорида, по-существу не содержащий анти-изомера, который применяют в способе ацилирования для получения цефепиновых антибиотиков широкого профиля.
На фиг. 1 приведен спектр протонного ядерного магнитного резонанса син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетил- хлорид, гидрохлорида примера 10 в уксусной кислоте-d4 (100 МГц); на фиг. 2 спектр ядерного магнитного резонанса (протонного) продукта примера 12 в уксусной кислоте-d4 (100 МГц); на фиг. 3 спектр протонного ядерного магнитного резонанса продукта примера 13 в уксусной кислоте-d4 (100 МГц); на фиг. 4 спектр протонного ядерного магнитного резонанса продукта примера 14 в уксусной кислоте-d4 (100 МГц).
Изобретением дается способ ацилирования в водной среде для N-ацилирования 7-амино-3-/(1-метил-1-пирролидинио)метил /цеф-3-ем-4-карбоксилата син-изомером 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида, по-существу не содержащего анти-изомера, с получением термостойкого кристаллического гидрата дигидрохлорида цефепима, по-существу не содержащего анти-изомера и Δ 2 -изомера и представленного формулой Y, где n 1 или 2.
H2N
 · 2HCl·ZH2O
· 2HCl·ZH2O
Преимущества настоящего способа ацилирования в водной среде станут очевидными и могут быть оценены специалистом при объединении всех преимуществ и рассмотрении их в совокупности. Изъятие обычных амино- и карбоксилзащитных групп и соответственно изъятие дополнительных химических стадий, необходимых для блокирования и деблокирования, является несомненным преимуществом с точки зрения эффективности способа в целом и с точки зрения снижения стоимости за счет расхода реагентов по сравнению со способами-прототипами. В настоящем способе кроме того дается и обеспечивается контроль над стереохимической конфигурацией метоксииминоизомера и двойной Δ 3 -связью цефалоспоринового ядра без необходимости разделять нежелательные цефалоспориновые побочные продукты хроматографией и без необходимости применять определяющие стереохимию сульфоксидные промежуточные соединения, подобные описанным в патенте США N 4043631. Другое преимущество изобретения заключается в получении и применении незащищенного кристаллического гидрохлорида син-изомера 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида формулы III, которое позволяет избежать применения необычных, а иногда и сложных отходящих групп, описанных в прототипах. Использование простого хлорид-иона в качестве отходящей группы позволяет избежать применения потенциально токсичных отходящих групп, таких как 2-меркаптобензотиазол. Еще одно преимущество рекомендуемого воплощения настоящего способа ацилирования в водной среде заключается в получении целевого термостойкого кристаллического гидрата дигидрохлорида цефепима непосредственно из реакционной смеси процесса ацилирования без необходимости получать и выделять сульфат или цвиттерион цефепима. Основное преимущество рекомендуемого воплощения настоящего способа ацилирования в водной среде заключается в получении целевого антибиотика без необходимости применять какие-либо силилирующие средства или какие-либо растворимые силилированные производные. Настоящий способ с успехом осуществляют без применения защитных групп, регулирующих стереохимию групп, солюбилизирующих силилированных групп или хроматографии с получением водорастворимого кристаллического гидрата дигидрохлорида цефепима, по-существу не содержащего анти-изомера и Δ 2 -изомера, с высоким выходом непосредственно из водно-органической реакционной смеси.
Настоящим изобретением дается также устойчивый кристаллический син-изомер-2-(2-аминотиазол-4-ил)-2-метоксииминоацет- илхлорид, гидрохлорида, по-существу не содержащий антиизомера и представленный формулой III
NH · HCl
· HCl
В результате по-существу отсутствия анти-изомера соединение III может быть превращено в широкий спектр цефалоспоринов, в свою очередь по-существу не содержащих анти-изомера, без необходимости хроматографического разделения син- и антиизомеров. Вследствие повышенной устойчивости соединение IIi может быть выделено и сохранено, а при желании соединение III может быть превращено в концевые продукты в различных растворителях, что является преимуществом при получении целевых антибиотиков, по существу не содержащих Δ 2 -изомера. Другое преимущество промежуточного соединения формулы III состоит в том, что оно не требует блокирования (защиты) аминогруппы перед ацилированием или деблокирования (удаления защитной группы) аминогруппы после ацилирования, влияя, таким образом, на эффективность способа. Еще одно преимущество хлорангидрида формулы III заключается в его применении в способе ацилирования для получения широкого спектра цефалоспоринов. В отличие от других способов, например, приведенных в патенте США N 4406899 (Aburaki и др.) промежуточное соединение формулы III обладает простой и неядовитой отходящей группой (хлорид-ион), не требующих особых предосторожностей при ее удалении из целевого антибиотика, как в случае с большинством других известных отходящих групп. Кроме того, некоторые известные промежуточные соединения, содержащие другие отходящие группы, синтезируются с трудом, а другие промежуточные соединения, содержащие отходящие группы типа 2-меркаптобензотиазола, как обнаружено, являются токсичными (Chem. Abstracts, 1989, т. III(3), 19243 р.)
Схема реакции I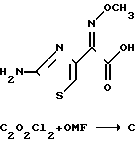 O
O (CH
(CH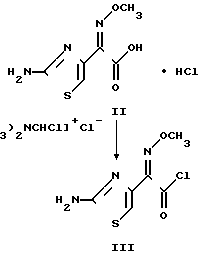

 cин-Изомер гидрохлорида хлорангидрида формулы IIl может быть получен из син-изомера кислоты формулы I, что отражено схемой реакции I. Кислоту формулы I вначале известными методами превращают в соответствующий гидрохлорид формулы II, после чего при желании выделяют в виде безводного кристаллического соединения формулы II. Образование гидрохлорида с успехом осуществляют использование по меньшей мере одного молярного эквивалента газообразного хлористого водорода в инертном органическом растворителе, таком как: толуол, ацетонитрил, дихлорметан, ацетон, бензол, ксилол, циклогексан, гексан, диоксан или диэтиловый эфир в температурном интервале от -10 до 50оС. Реакцию рекомендуется проводить в толуоле, дихлорметане или ацетонитриле, и образовавшийся в результате гидрохлорид формулы ll может быть выделен или использован in Situ. При проведении реакции в ацетонитриле образовавшийся гидрохлорид формулы ll имеет тенденцию прочно удерживать растворитель. Поэтому желательно использование гидрохлорида кислоты формулы ll из ацетонитрила возможно скорее на следующей стадии, чтобы избежать замещения сольвата атмосферной влагой. Наиболее предпочтительно проведение реакции в толуоле или дихлорметане при температуре от 0оС до комнатной температуры.
cин-Изомер гидрохлорида хлорангидрида формулы IIl может быть получен из син-изомера кислоты формулы I, что отражено схемой реакции I. Кислоту формулы I вначале известными методами превращают в соответствующий гидрохлорид формулы II, после чего при желании выделяют в виде безводного кристаллического соединения формулы II. Образование гидрохлорида с успехом осуществляют использование по меньшей мере одного молярного эквивалента газообразного хлористого водорода в инертном органическом растворителе, таком как: толуол, ацетонитрил, дихлорметан, ацетон, бензол, ксилол, циклогексан, гексан, диоксан или диэтиловый эфир в температурном интервале от -10 до 50оС. Реакцию рекомендуется проводить в толуоле, дихлорметане или ацетонитриле, и образовавшийся в результате гидрохлорид формулы ll может быть выделен или использован in Situ. При проведении реакции в ацетонитриле образовавшийся гидрохлорид формулы ll имеет тенденцию прочно удерживать растворитель. Поэтому желательно использование гидрохлорида кислоты формулы ll из ацетонитрила возможно скорее на следующей стадии, чтобы избежать замещения сольвата атмосферной влагой. Наиболее предпочтительно проведение реакции в толуоле или дихлорметане при температуре от 0оС до комнатной температуры.
Соль кислоты формулы II затем с успехом обрабатывают хлорирующим средством, наиболее предпочтительно оксалилхлоридом в сочетании с диметилформамидом с образованием устойчивого кристаллического син-изомера соединения III. Как показано применение других хлорирующих средств может привести к изомеризации с образованием нежелательного анти-изомера или смеси син- и анти-изомера. Кроме того, применение таких хлорирующих средств, как пентахлорид фосфора может привести к хлорированию в 5-положении тиазольного цикла с последующим нежелательным загрязнением антибиотика. Создатели настоящего изобретения обнаружили, что помимо получения гидрохлорида кислоты формулы II решающую роль в способе получения син-изомера соединения III, по-существу не содержащего анти-изомера, играет правильный подбор хлорирующего средства и условий реакции, таких как растворитель и температура.
Способы хлорирования, обычно применяемые для активации кислоты, хорошо известны. Пентахлорид фосфора, являющийся наиболее широко применяемым хлорирующим средством, для хлорирования соединения II не пригоден, поскольку приводит также и к изомеризации метоксииминогруппы с образованием нежелательного анти-изомера соединения III. Это четко показано в нижеследующих примерах 12, 13, 14 и 16. Другой известный способ хлорирования заключается в применении оксалилхлорида в сочетании с диметилформамидом. Однако, создателями настоящего изобретения обнаружено, что способ с применением оксалилхлорида, в котором в качестве катализатора применяют диметилформамид, не приводит к образованию значительных количеств целевого син-изомера соединения III. Это также четко показано в нижеследующем примере 15. После интенсивных исследований создатели настоящего изобретения выяснили, что применение диметилформамида в количестве менее эквимолярного количества относительно количества оксалилхлорида оказывает неблагоприятное воздействие на образование целевого син-изомера гидрохлорида хлорангидрида формулы III. Наиболее предпочтительно, если молярное количество диметилформамида будет превышать молярное количество диметилформамида. Изобретателями кроме того обнаружено, что применение избыточных количеств диметилформамида также неблагоприятно как для реакции, так и для устойчивости целевого продукта. Таким образом, изобретателями открыт способ регулирования неустойчивости продуктов реакции либо избытком хлорид-иона, образуемого оксалилхлоридом, либо избытком диметилформамида, играющего решающую роль при образовании устойчивого кристаллического син-изомера соединения III, по-существу не содержащего анти-изомера. В случае, если превращение соединения формулы II в соединение формулы III неполно, в выделенном в качестве продукта соединении III будет содержаться небольшое количество оставшегося син-изомера кислоты формулы II. Присутствие в полученном в качестве продукта соединении III некоторых количеств непрореагировавшего соединения II и небольших количеств анти-изомера соединения III не влияет на последующую реакцию ацилирования с целью успешного получения целевого антибиотика, по-существу не содержащего анти-изомера этого антибиотика.
Создателями настоящего изобретения обнаружено также, что температура реакции и растворитель для нее имеют существенное значение. Рекомендуется проводить реакцию в инертном органическом растворителе, таком как: дихлорметан, хлороформ или ацетонитрил при температуре ниже -10оС. Наиболее предпочтительно проведение реакции в дихлорметане и температурном интервале от -15 до -40оС.
Применение син-изомера гидрохлорида хлорангидрида формулы III для получения полезных антибиотиков широкого профиля путем обычной реакции ацилирования иллюстрируется схемой реакции 2. Более конкретно, схема реакции 2 иллюстрирует применение хлорангидрида формулы III для получения антибиотика широкого профиля цефепима, по-существу нес содержащего анти-изомера и Δ 2 -изомера. Кроме того, гидрохлорид хлорангидрида формулы III может быть использован для синтеза цефалоспориновых антибиотиков, характеризующихся син-конфигурацией 2-(2-аминотиазол-4-ил)-2-метоксииминоацет- ила, присоединенного к 7-аминогруппе цефалоспоринового ядра, таких как: цефодизим, цеменоксим, цефотаксим, цефпиром, цеподоксим, цефхином, цефтерам, цефтиофур, цефетамет и цефузонам.
Кроме того, для подтверждения того, что гидрохлорид хлорангидрида прототипа является анти-изомером, а не целевым син-изомером, создатели настоящего изобретения заменили в реакции ацилирования, иллюстрируемой схемой реакции 2, син-изомер соединения формулы III продуктом прототипа, полученным, например, в примере 14. Конечный цефалоспориновый продукт, полученный по методикам примеров 17 и 18, сравнивался с антибиотиком цефепимом, полученным способом настоящего изобретения. Как видно из сравнения (пример 19) анти-цефепим, полученный согласно указаниям прототипа, не идентичен полезному син-цефепиму широкого профиля, синтезированному использованием настоящего изобретения.
В применяемом здесь и в формуле изобретения значении выражение "по-существу не содержащий" означает, что соединение содержит менее примерно 5% нежелательного изомера. Предпочтительно, если соединение содержит менее примерно 1% нежелательного изомера.
Согласно способу изобретения N-ацилированием соединения формулы IV син-изомером гидрохлорида хлорангидрида формулы III получают антибиотик широкого профиля гидрат дигидрохлорида цефепима, по-существу не содержащий анти-изомера и Δ 2-изомера, что иллюстрируется схемой реакции 2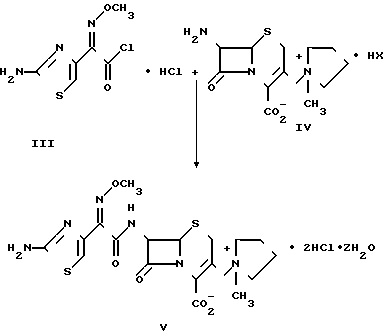
7-Амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилатная соль, по-существу не содержащая Δ2 -изомера и представленная формулой IV, где НХ НCl, Hl или H2SO4, может быть получена по общей методике, описанной S.P. Brundidge и др. в патенте США N 4868294.
Приемлимые растворители, которые могут быть использованы в способе ацилирования в водной среде, относятся к водно-органическим растворителям, таким как вода с водорастворимым органическим растворителем, например метанолом, этанолом, изопропанолом, бутанолом, ацетоном, тетрагидрофураном, ацетонитрилом, диоксаном, диметилацетамидом, диметилформамидом и т.п. Значение рН реакционной смеси контролируют титрованием приемлимом органическим и неорганическим основанием, таким как: гидроокись натрия, карбонат натрия, бикарбонат натрия, гидроокись калия, гидроокись аммония, первичный амин, вторичный амин, третичный амин и т.п. с нейтрализацией образующейся в результате соляной кислоты. Рекомендуемые органические основания, которые могут быть при этом использованы, включают, например: диэтиламин, триэтиламин, диизопропилэтиламин, N-метилморфолин, 2,6-лутидин, N,N-диметиланилин, N,N-диэтиланилин и т.п. К наиболее рекомендуемым основаниям относятся гидроокись аммония, триэтиламин или N-метилморфолин.
Ацилирование с успехом проводят при рН 5-7,5, предпочтительно при рН 6,2-6,8. Способ настоящего изобретения может быть осуществлен в температурном интервале от -50оС до комнатной температуры, предпочтительно при температуре от -10 до -40оС. По окончании ацилирования реакционную смесь подкисляют соответствующей кислотой, предпочтительно серной кислотой до рН 1,8-2,6 с образованием сульфатной соли целевого цефепимового антибиотика, по-существу не содержащего анти-изомера и Δ 2 -изомера. При желании сульфатная соль цефепима может быть превращена в другие соли цефепима, например, гидрат дигидрохлорида по методике Kaplan и др. в патенте США N 4910301.
Одним из аспектов настоящего изобретения является способ ацилирования для получения антибиотика широкого профиля цефепима, по-существу не содержащего анти-изомера и Δ 2 -изомера, и способ состоит в реакции 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилатной соли, по-существу не содержащей Δ 2 -изомера, с син-изомером 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорид, гидрохлоридом, по-существу не содержащим анти-изомера, в водном или предпочтительно смешанном водно-органическом растворителе при тщательно регулируемом значении рН в интервале 5-7,5.
Другим аспектом изобретения дается устойчивый кристаллический син-изомер гидрохлорида 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида, по-существу не содержащий анти-изомера, формулы
H2N · HCl
· HCl 
И еще одним аспектом настоящего изобретения дается способ получения устойчивого кристаллического син-изомера гидрохлорида 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида, по-существу не содержащего анти-изомера, и способ заключается в реакции безводного кристаллического гидрохлорида син-изомера 2-(2-аминотиазол-4-ил)-2-метоксииминоук-сусной кислоты со смесью, состоящей из по меньшей мере одного молярного эквивалента оксалилхлорида и по меньшей мере одного молярного эквивалента или небольшого избытка диметилформамида относительно количества оксалилхлорида, в инертном органическом растворителе при температуре ниже -10оС с образованием устойчивого кристаллического син-изомера гидрохлорида 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида, по-существу не содержащего анти-изомера.
Завершение N-ацилирования соединения формулы IV подтверждается известными и доступными методами детектирования, например тонко-слойной хроматографией, жидкостной хроматографией высокого давления и спектральными методами. Согласно рекомендуемому способу изобретения по окончании реакции добавляют достаточное количество кислоты, такой как соляная кислота, серная кислота и т. п. с целью гарантировать кристаллизацию целевой соли цефепима, после чего разбавляют водорастворимым растворителем, таким как метилэтилкетон, ацетон, изопропанол, бутанол и т.п. для индуцирования или завершения кристаллизации. Рекомендуется водно-органическую реакционную смесь обрабатывать достаточным количеством серной кислоты с кристаллизацией сульфатной соли цефепима, по-существу не содержащей анти-изомера и Δ2-изомера. Сульфат цефепима затем может быть превращен в рекомендуемый моногидрат дигидрохлорида по методике, описанной Kapla и др. в патенте США N 4910301. Полученный настоящим способом сульфат цефепима может быть нейтрализован слабым основанием, предпочтительно слабо щелочной ионообменной смолой, применяемой для таких целей, предпочтительно продажной ионообменной смолой, такой как: Амберлит LA-2, Дауэкс WGR, Био-Рад AG3-X4A, Амберлит 1RA 93, Амберлит IRA 35 и т.п. с образованием водного или водно-органического раствора, содержащего цвиттерионную форму цефепима. Раствор затем обрабатывают достаточным количеством соляной кислоты, возможно в присутствии водорастворимого органического растворителя с целью вызвать кристаллизацию рекомендуемого кристаллического моногидрата дигидрохлорида цефепима. Наиболее предпочтительно, если водно-органическая реакционная смесь, полученная при ацилировании в водной среде настоящим способом, обрабатывается достаточным количеством соляной кислоты с целью индуцирования и гарантировать кристаллизацию указанного антибиотика, а именно, гидрата дигидрохлорида цефепима при добавлении такого водорастворимого органического растворителя, как ацетон. Количество добавляемого водорастворимого органического растворителя должно быть достаточным для полного завершения кристаллизации указанного антибиотика, и обычно составляет 2-9 объема от количества воды в водно-органической реакционной среде с образованием в результате термостойкого кристаллического гидрата дигидрохлорида цефепима, по-существу не содержащего анти-изомера и Δ2 -изомера.
При желании получить только моногидрат дигидрохлорида цефепима водно-органическую реакционную смесь от ацилирования в водной среде предпочтительно обрабатывают достаточным количеством соляной кислоты и разбавляют приемлимым количеством вод растворимого органического растворителя вышеприведенным методом с целью гарантирования кристаллизации целевой моногидратной формы. Или же, если хотят получить устойчивый дигидрат дигидрохлорида цефепима, водно-органическая реакционная смесь может быть с успехом обработана соляной кислотой в большей эквивалентной концентрации и таким количеством водорастворимого органического растворителя, чтобы поддерживать кристаллизацию в точке помутнения перед добавлением дополнительного количества органического растворителя для завершения кристаллизации. Однако для специалиста очевидно, что если тщательно не регулировать стадию выделения из водно-органической реакционной смеси, возможно образование смеси кристаллических моногидратной и дигидратной форм цефепима. В любом случае получение только одного из желаемых гидратов может быть осуществлено из любого гидрата или из смеси гидратов путем перекристаллизации, описанной здесь.
Кристаллический моногидрат дигидрохлорида цефепима, полученный настоящим способом, может быть использован для получения устойчивого кристаллического дигидрата дигидрохлорида цефепима перекристаллизацией при регулируемых концентрациях растворителя и соляной кислоты и продолжительности выдерживания в точке помутнения (начальная кристаллизация) приведенным здесь способом. Или кристаллический дигидрохлорид дигидрат, полученный настоящим способом, также может быть использован для приготовления устойчивого кристаллического моногидрата дигидрохлорида цефепима в других регулируемых условиях, приведенных здесь. Таким образом, способ настоящего изобретения может быть использован для получения как целевого моногидрата, так и дигидрата указанного антибиотика.
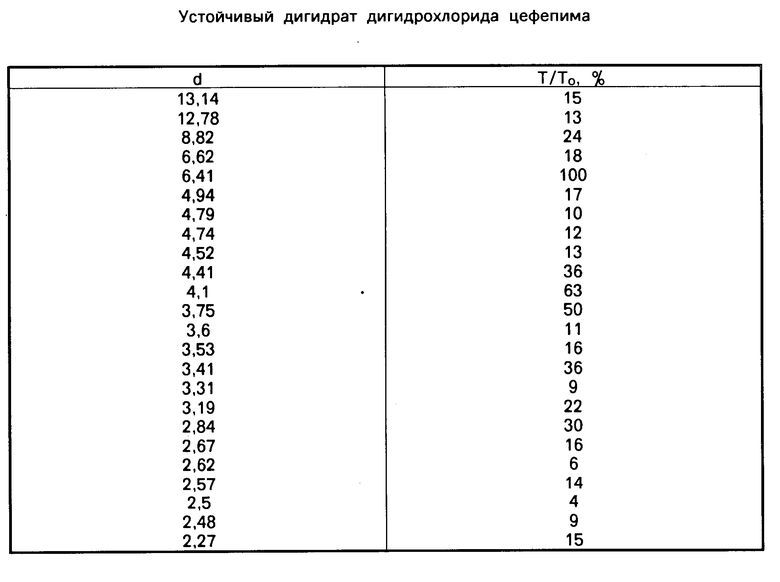
В отличие от лабильного дигидрата дигидрохлорида цефепима, описанного в патенте США N 4910301, который легко теряет второй моль воды, кристаллический дигидрат дигидрохлорида цефепима, который может быть получен настоящим способом, как найдено, обладает хорошо выраженным кристаллическим строением, при котором сохраняется второй моль воды. Новая кристаллическая форма дигидрата (иглоподобные кристаллы), как найдено, очень устойчива и ее кристаллическая морфология не меняется в различных условиях, например на воздухе при 70оС более двух месяцев, в вакууме над R2O5 при 50оС в течение 48 ч, в сушильном шкафу при 70оС в течение 96 ч и в условиях высокой и низкой относительной влажности. Кристаллический дигидрат обладает характеристическими пиками в инфракрасном спектре поглощения при 3574 см-1 и 3432 см-1, как показано FT-ИК диффузионной отражательной спектроскопией с КВr и в кювете для образца на 13 мм с помощью спектрометра Николе 20SX. Такой устойчивый к нагреванию и влаге кристаллический дигидрат цефепима кроме того охарактеризован порошковой диффракцией рентгеновских лучей, результаты которой приведены в таблице, где "d" относится к межплоскостным расстояниям и "T/To" относится к относительным процентным итенсивностям. Данные рентгено-структурного анализа получены использованием рентгеновского диффрактометра модели Ригаку Гейгефлекс и при излучении от никелевого источника с медным фильтром (Кα ) с длиной волны в 1,5425  .
.
Таким образом, одним из воплощений изобретения дается способ получения антибиотика гидрата дигидрохлорида цефепима, по-существу не содержащего анти-изомера и Δ2 -изомера, способ заключается в реакции 7-амино-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата, по-существу не содержащего Δ2 -изомера, с син-изомером гидрохлорида 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида, по-существу не содержащего анти-изомера, в смешанном водно-органическом растворителе при рН 5-7,5.
Рекомендуемое воплощение настоящего изобретения кроме того включает получение син-изомера гидрохлорида 2-(2-амино-тиазол-4-или)-2-метоксииминоа- цетилхлорида, по-существу не содержащего анти-изомера, реакцией безводной соли хлорангидрида син-изомера-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты со смесью, содержащей по меньшей мере один молярный эквивалент оксалилхлорида или по меньшей мере один молярный эквивалент или небольшой избыток по отношению к оксалилхлориду диметилформамида, в инертном органическом растворителе при температуре ниже -10оС.
Более предпочтительным воплощением настоящего изобретения дается способ получения антибиотика, гидрата гидрохлорида цефепима, по-существу не содержащего анти-изомера и Δ2 -изомера, способ заключается в реакции 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата, по-существу не содержащего Δ2 -изомера, с син-изомером гидрохлорида 2-(2-аминотиазол-4-ил)-2-метоксииминоацетил- хлорида, по-существу не содержащим анти-изомера, в водно-органическом растворителе и дополнительно включает добавление к конечной реакционной смеси достаточного количества соляной кислоты и водорастворимого органического растворителя.
Наиболее предпочтительным воплощением изобретения дается способ получения антибиотиков, моногидрата дигидрохлорида цефепима и дигидрата дигидрохлорида цефепима непосредственно из водно-органической реакционной смеси настоящего способа.
Применимость цефепима показана в патенте США N 4406899 (Aburaki и др.). Устойчивая дигидратная форма цефепима, полученная настоящим способом, обладает антибиотическими свойствами вышеупомянутого цефепима по патенту США N 4406899, и находит применение в качестве антибиотика аналогичным образом.
Очевидно, что описание и примеры служат иллюстративным целям и не предназначены для ограничения объема изобретения.
П р и м е р 1. Син-2-(2-Амино-тиазол-4-ил)-2-метоксииминоуксусная кислота, гидрохлорид.
В суспензию 25 г (124,25 ммоля) 2-(2-аминотиазол-4-ил)2-метоксииминоуксусной кислоты в толуоле (250 мл) при 20-28оС пропускают газообразный НСl, НСl вводят под поверхностью реакционной смеси двумя порциями по 8,1 г (222,2 ммоля) и 4,8 г (131,7 ммоля) с перемешиванием 30 мин между добавлением порций. После выдерживания 1 ч при 20оС продукт отфильтровывают в атмосфере азота, промывают толуолом (50 мл) и гексаном (250 мл) и высушиванием в вакууме при 20-25оС получают 28,68 г (97%) заглавного соединения.
П р и м е р 2. син-2-(2-Аминотиазол-4-ил)-2-метоксииминоацетилхлорид гидрохлорид.
К раствору 0,77 мл (10 ммолей) диметилформамида в дихлорметане (40 мл) при 5оС добавляют 0,89 мл (10 ммолей) 98-го оксалилхлорида в дихлорметане (4,1 мл). Добавлением по каплям поддерживают температуру 4-5оС. К полученной суспензии, охлажденной до -27оС, добавляют 2,37 г (10 ммолей) гидрохлорида 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты примера 1. Суспензию перемешивают 2,5 ч при -25оС. Фильтрованием в атмосфере азота и промыванием дихлорметаном (50 мл) и гексаном (100 мл) получают 1,78 г (69,5%) белого кристаллического заглавного соединения после высушивания в вакууме при 20оС.
Полученным хлорангидридом ацилируют гидрохлорид дефинилметилового эфира 7-аминодезацетоксицефалоспорановой кислоты в растворе пиридина с образованием дающего одно пятно (ТСХ) продукта, совпадающего и неразделимого с аутентичным образцом целевого дезацетоксицефалоспоринового эфира.
П р и м е р 3. син-2-(2-Аминотиазол-4-ил)-2-метоксииминоацетилхлорид гидрохлорид.
К раствору 1,55 мл (20 ммолей) диметилформамида в дихлорметане (80 мл) при 5оС добавляют 1,78 мл (20 ммолей) оксалилхлорида 98%-ной чистоты в дихлорметане (8,2 мл). Время прибавления при 5-8оС составляет 5 мин. Полученную суспензию перемешивают 10 мин при 5оС, затем охлаждают до -30оС и добавляют гидрохлорид 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (4,75 г, 20 ммолей) примера 1. Суспензию перемешивают 2,5 ч при температуре от -25 до -30оС. Фильтрованием в атмосфере азота и промыванием дихлорметаном (75 мл) и гексаном (100 мл) получают после высушивания при 20оС в вакууме 3,57 г (69,7%) кристаллического заглавного соединения.
Порцией полученного гидрохлорида хлорангидрида (сухого) ацилируют в растворе пиридина гидрохлорид дифенилметилового эфира 7-аминодезацетоксицефалоспорановой кислоты и получают дающий одно пятно (ТСХ) продукт, совпадающий и неразделимый с аутентичным образцом целевого дазацетоксицефалоспоринового эфира.
П р и м е р 4. Получение 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацетамидо /-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата (цефепима)


В 9 мл смеси ацетон-вода (2:1) с триэтиламином при рН 6,5 и 20оС растворяют моногидройодид 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбок-силат (0,85 г, 2 ммоля) (получен по методике, приведенной в патенте США N 4714760 (S.P.Brundidge и др.)/. Добавляют гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида (0,56 г, 2,2 ммоля) (получение см. пример 3) с применением триэтиламина для выдерживания рН в интервале 5-7. Анализ полученного раствора жидкостной хроматографией высокого давления показал 58%-ый выход целевого цефалоспорина (цефепима). Подкислением серной кислотой до рН 2,2 получают 0,63 г заглавного антибиотика в виде его сульфатной соли (выход по активности 51%), охарактеризованной в патенте США N 4406899 (Aburaki и др.), 1983, и патенте США N 4910301 (Kaplan и др.), 1990.
П р и м е р 5. син-2-(2-Аминотиазол-4-ил)-2-метоксиминоацетилхлорид, гидрохлорид.
К раствору 9,75 мл (125,9 ммоля) диметилформамида в дихлорметане (450 мл) при 5оС по каплям прибавляют раствор 11,21 мл (125,9 ммоля) оксалилхлорида (98%) в дихлорметане (15 мл). Прибавление при 5-7оС заканчивается за 10 мин. К полученной взвеси, охлажденной до -25оС, одной порцией добавляют 28,5 г (119,9 ммоля) гидрохлорида син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты. Взвесь перемешивают 3,5 ч при -25 до -30оС, фильтруют в атмосфере азота, промывают дихлорметаном (100 мл) и гексаном (400 мл) и высушивают при 20-25оС в вакууме. Выход кристаллического заглавного соединения 30,7 (72,5%).
Полученным хлорангидридом ацилируют в растворе пиридина гидрохлорид дифенилметилового эфира 7-аминодезацетоксицефалоспорановой кислоты и получают по-существу одно пятно (ТСХ) целевого дезацетоксицефалоспоринового эфира при сравнении с аутентичным образцом.
Заглавный хлорангидрид (200 мг, 0,8 ммоля) гидролизуют в воде. 1Н-ЯМР спектр идентичен спектру исходной син-кислоты.
П р и м е р 6. син-2-(2-Аминотиазол-4-ил)-2-метоксииминоацетилхлорид гидрохлорид.
К раствору 8,13 мл (105 ммолей) диметилформамида в дихлорметане (350 мл) при 5оС по каплям прибавляют 9,34 мл (105 ммолей) оксалилхлорида (98%-ой чистоты) в дихлорметане (5 мл). Максимальная температура в ходе прибавления 7оС. Образовавшуюся суспензию перемешивают 10 мин при 5оС и затем охлаждают до -27оС. Одной порцией добавляют гидрохлорид 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (23,8 г, 100 ммолей). Суспензию перемешивают 2,5 ч при от -25 до -30оС, фильтруют в атмосфере азота, промывают дихлорметаном (25 мл) и гексаном (125 мл) и после высушивания при 20оС в вакууме получают 21,39 г (83,5%) кристаллического гидрохлорида хлорангидрида.
Вычислено, С 28,14; Н 2,76; N 16,41; S 12,52.
С6Н7N3O2SCl2.
Найдено, С 28,25; Н 2,93; N 16,32; S 12,67.
1Н-ЯМР (ДМСО-d6) δ 3-93 (CH3), 7,04 (Н5).
П р и м е р 7. син-2-(2-Аминотиазол-4-ил)-2-метоксииминоуксусная кислота, гидрохлорид.
В суспензию 87 г (432,4 ммоля) син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксус-ной кислоты в толуоле (870 мл) при 22оС двумя порциями вводят газообразный НСl: 17,5 г (480 ммолей) за 30 мин и 15 г (410 ммолей) за 20 мин с перемешиванием 20 мин между введением порций. Взвесь перемешивают 1,5 ч при 25оС, фильтруют в атмосфере азота, промывают толуолом (100 мл) и гексаном (400 мл) и после высушивания в вакууме при 20-25оС получают 100,2 г (97,5) основного соединения.
Вычислено, C 30,32; Н 3,39; N 17,68; S 13,49; Cl 14,92.
С6Н8N3O3SCl.
Найдено, С 30,51; Н 3,39; N 17,54; S 13,37; Сl 14,90.
П р и м е р 8. син-2-(2-Аминотиазол-4-ил)-2-метоксииминоацетилхлорид гидрохлорид.
К раствору 32,4 мл (419,7 ммолей) диметилформамида в дихлорметане (400 мл) при 5оС по каплям прибавляют 37,4 мл (419,7 ммоля) 98%-ного оксалилхлорида. Полученную суспензию охлаждают до -25оС и добавляют при -25оС к суспензии 95 г (399,7 ммоля) гидрохлорида син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты примера 7. Суспензию перемешивают 2,5 ч при температуре от -25 до -28оС, фильтруют в атмосфере азота, промывают дихлорметаном (100 мл) и гексаном (500 мл) и после высушивания в вакууме при 20-25оС получают 84,3 г (82,3%) кристаллического заглавного соединения.
Вычислено, С 28,14; Н 2,76; N 16,41; S 12,52.
С6Н7N3О2SCl2.
Найдено, С 27,90; Н 3,10; N 16,14; S 12,27.
1Н-ЯМР (ДМСО-d6) δ 3,95 (СН3), 7,04 (Н5).
П р и м е р 9. Получение 7-(2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацетамидо /-3-(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата (цефепима).
К раствору 240 мл ацетона и 80 мл воды добавляют 20 г гидройодида 7-амино-3-/(1-метил-1-пирролидинио)метил/-цеф-3-ем-4-карбоксилата (0,047 моля) и смесь перемешивают. Использованием автоматического прибора для титрования модели Радиометр АВV80, настроенного на конец титрования при рН 6,5 и заполненного N-метилморфолином, 4 порциями с интервалом в 5 минут, поддерживая рН при 6,5, добавляют гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида (20 г, 0,0785 моля) (получение см. пример 5). По окончании прибавления мелкую взвесь перемешивают еще 20 минут при комнатной температуре. Добавлением 21 мл 6 н. Н2SO4 значение рН реакционной смеси понижают до 2,65. Происходит осаждение заглавного соединения. Взвесь затравливают и перемешивают 20 минут при комнатной температуре. Добавлением 16 мл 6 н. Н2SO4устанавливают рН взвеси 1,8 и перемешивание продолжают еще 60 мин. Взвесь фильтруют в вакууме и промывают 70 мл смеси вода-ацетон (1:1) и затем 70 мл ацетона с получением 24,09 г (88,5% стехиом. выход по массе) заглавного соединения, идентичного соединению примера 4 и цефепиму, охарактеризованному в патенте США N 4406899 (Aburaki и др.), 1983, и в патенте США N 4910301 (Каplan и др.), 1990.
П р и м е р 10. Получение син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхло-рида гидрохлорида.
К раствору диметилформамид (8,76 мл, 0,113 моля) в дихлорметане (375 мл) при 5оС по каплям прибавляют оксалилхлорид (9,64 мл, 0,111 моля), поддерживая температуру при 5-6оС. Суспензию перемешивают 10 мин и затем охлаждают до -25оС. В атмосфере сухого азота в течение 11 мин порциями прибавляют гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксиими-ноуксусной кислоты (25 г) и взвесь перемешивают 2,5 ч при -25оС. Продукт фильтруют в атмосфере сухого азота и фильтровальный пирог промывают дихлорметаном (80 мл). Высушиванием продукта при 20-25оС в вакууме над Р2O5 получают 23,88 г (88,6%) заглавного соединения в виде бледно-желтого кристаллического вещества.
Вычислено, С 28,14; Н 2,76; N 16,41; S 12,52; Сl 27,68.
С6Н7N3O2SCl2.
Найдено, С 28,06; Н 2,71; N 16,26; S 12,30; Cl 27,23.
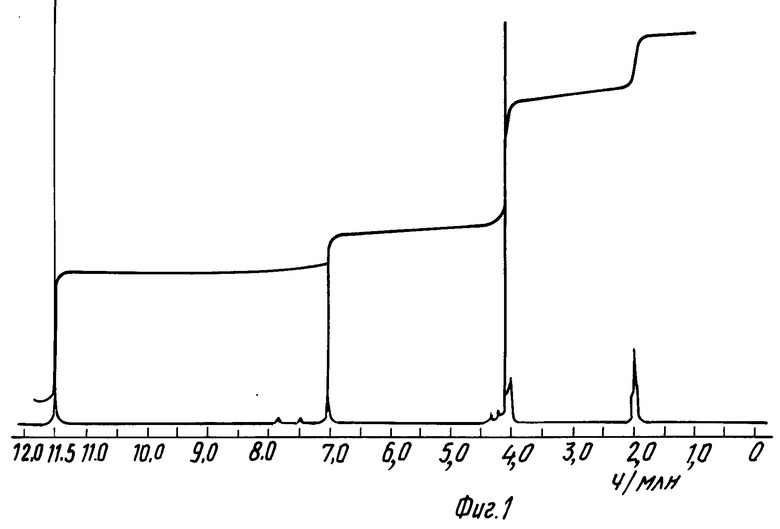
Продукт вышеприведенного опыта охарактеризован спектром протонного ядерного магнитного резонанса (1Н-ЯМР) в уксусной кислоте-d4, приведенном на фиг. 1.
1Н-ЯМР (СD4CO2D) δ: 4,14 (СН3), 7,1 (Н5).
Остаточный уровень гидрохлорида кислоты по СН3 (4,11) интегралу составляет 5,1% Следовый уровень изомерного Н5 виден при 7,67 ч/млн.
П р и м е р 11. син-2-(2-Аминотиазол-4-ил)-2-метоксииминоацетилхлорид гидрохлорид.
К раствору диметилформамида (17,92 мл, 231,9 ммоля) в дихлорметане (375 мл) при 5оС добавляют оксалилхлорид (19,76 мл, 220,8 ммоля). Время прибавления при 5-6оС составляет 15 мин. Полученную суспензию перемешивают 10 мин при 5-6оС и затем охлаждают до -25оС. Добавляют гидрохлорид 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (25 г, 105,2 ммоля). Полученный раствор засевают заглавным соединением с образованием взвеси продукта. Суспензию перемешивают 2,5 ч при -25оС, фильтруют в атмосфере сухого азота, промывают дихлорметаном (150 мл) и после высушивания в вакууме при 20-25оС получают 9,61 г (35,7%) кристаллического заглавного соединения.
Порцией сухого гидрохлорида хлорангидрида ацилируют в растворе пиридина гидрохлорида дифенилметилового эфира 7-аминодезацетоксицефалоспорановой кислоты и получают продукт с по-существу единственным пятном (ТСХ), совпадающим и неразделимым с аутентичным образцом целевого дезацетоксицефалоспоринового эфира.
П р и м е р 12. Получение 2-(2-аминотиазол -4-ил)-2-метоксииминоацетилхлорида гидрохлорида.
Экспериментальная методика примера 1 патента Чехословакии N 238950 воспроизведена следующим образом.
В 30 мл бензола при 21оС образуют взвесь син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (4 г) со значением КF 0,06% Добавляют одну каплю диметилформамида с последующим прибавлением одной порцией 5 г порошкообразного пентахлорида фосфора. Температура повышается до 34оС примерно за 2 мин и ее затем в течение 1 мин повышают до 40оС с полным растворением реагентов. Раствор оставляют охлаждаться и при 36оС образуется осадок. После перемешивания 30 минут температура достигает 22оС. Светло-желтый осадок отфильтровывают в атмосфере сухого азота и промывают 30 мл бензола и 20 мл гептана. Выход составляет 2,88 г после высушивания в вакууме 18 ч при 20-25оС над Р2О5.
Продукт вышеприведенного опыта охарактеризован спектром протонного ядерного магнитного резонанса (1Н-ЯМР) в уксусной кислоте-d4, приведенном на фиг. 2, где Н5 проявляется при 7,56 ч/млн и СН3 при 4,34 ч/млн. Такой спектр отвечает заглавному соединению с конфигурацией анти-изомера, а не син-изомера, как указано в патенте Чехословакии.
П р и м е р 13. Получение 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида гидрохлорида.
Экспериментальная методика примера 2 патента Чехословакии N 238950 воспроизведена следующим образом.
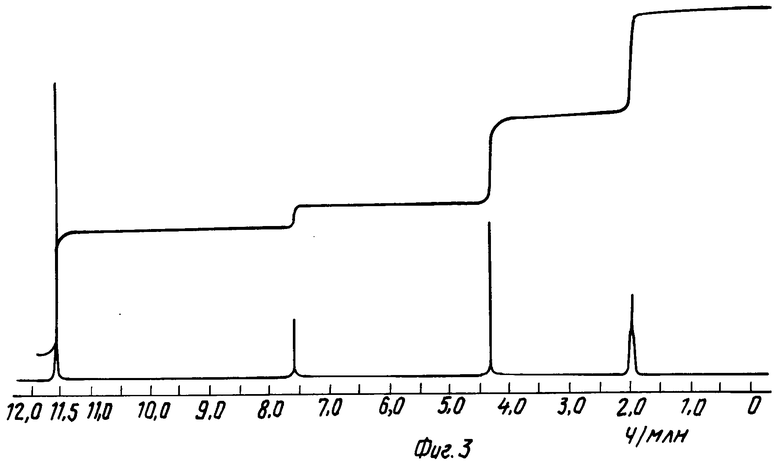
В 20 мл ацетонитрила, в котором устанавливают значение KF в 0,22% образуютвзвесьсин-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (4 г), имеющей значение KF 0,06% При 20оС добавляют одну каплю диметилформамида, а после добавления 6 г порошкообразного пентахлорида фосфора температура повышается до 40оС с полным растворением реагентов. Раствор охлаждают до 20оС, а при 33оС образуется осадок. После перемешивания 30 мин, продукт собирают в атмосфере сухого азота и промывают 30 мл бензола и 20 мл гептана. После сушки при 20-25оС в вакууме над Р2О5 в течение 18 ч получают 1,86 г продукта.
Продукт вышеприведенного опыта охарактеризован спектром протонного ядерного магнитного резонанса (1Н-ЯМР) в уксусной кислоте-d4, приведенным на фиг. 3, где Н5 проявляется при 7,56 ч/млн и СН3 при 5,31 ч/млн. 1Н-ЯМР спектр отвечает заглавному соединению с конфигурацией анти-изомера, а не син-изомера, как указано в патенте Чехословакии.
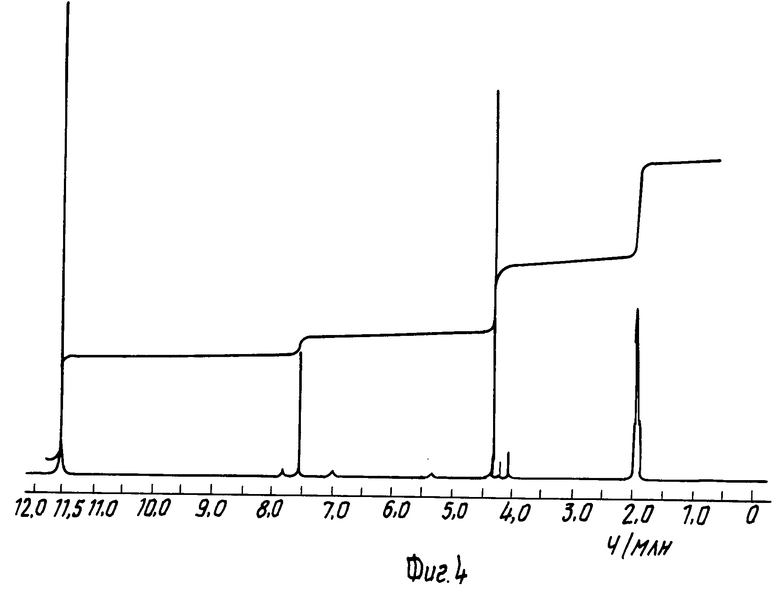
П р и м е р 14. Получение 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида гидрохлорида.
Экспериментальная методика примера 3 патента Чехословакии N 238950 воспроизведена следующим образом.
К 30 мл дихлорметана добавляют концентрированную соляную кислоту (0,16 мл) и после охлаждения до -10оС порциями прибавляют 6,5 г пентахлорида фосфора. После нагревания до 0оС одной порцией добавляют 4 г син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусную кислоту со значением KF 0,06% при этом температура повышается до 2оС. Полное растворение наступает через 9 мин при 0оС. Спустя 40 мин начинает образоваться осадок. Образовавшуюся взвесь перемешивают 2,8 ч при 2-3оС, фильтруют в атмосфере сухого азота, промывают 30 мл бензола и 20 мл гептана и после сушки 18 ч в вакууме над Р2О5 и при 20-25оС получают 3,42 г светложелтого порошка.
Продукт вышеприведенного опыта охарактеризован спектром протонного ядерного магнитного резонанса (1Н-ЯМР) в уксусной кислоте-d4, приведенном на фиг. 4, где Н5 проявляется при 7,56 ч/млн и СН3 при 4,31 ч/млн. 1Н-ЯМР спектр отвечает заглавному соединению с конфигурацией анти-изомера, а не син-изомера, как указано в патенте Чехословакии.
П р и м е р 15. Попытка получения 2-(2-аминотиазол-4-ил)-2-метоксииминоацетил- хлорида гидрохлорида.
Общая методика, приведенная в примере 7 патента США N 4203899, для предотвращения защищенной аминотиазолуксусной кислоты в соответствующий хлорангидрид применена незащищенной аминотиазолуксусной кислоте следующим образом.
В 30,5 мл бензола суспендируют гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты и охлаждают до 20оС. Добавляют оксалилхлорид (2,09 мл, 0,024 моля) с последующим прибавлением диметилформамида (0,5 мл, 0,0065 моля). Температура повышается до 22оС при интенсивном выделении газа. В течение 20 мин при 20оС газообразование прекращается и взвесь перемешивают 2 ч при 20±2оС. Взвесь концентрируют в вакууме с удалением растворителя и полученный желтый продукт сушат 16 ч в вакууме над Р2О5 и при 20-25оС. Выход 2,59 г.
Продукт вышеприведенного опыта охарактеризован спектром протонного ядерного магнитного резонанса в уксусной кислоте-d4, в котором Н5 проявляется при 7,6 ч/мин, а СН3 при 4,37 ч/млн. Спектр продукта отвечает заглавному соединению с конфигурацией анти-изомера.
П р и м е р 16. Попытка получения 2-(2-аминотиазол-4-ил)-2-метоксииминоацетил- хлорида гидрохлорида.
Общая методика, приведенная в примере 59 патента США N 4203899, для превращения защищенной аминотиазолуксусной кислоты в соответствующий хлорангидрид применена к незащищенной аминотиазолуксусной кислоте следующим образом.
В 25 мл дихлорметана суспендируют гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (2,38 г, 0,01 моля) и после охлаждения до 4оС добавляют 2,08 г (0,01 моля) пентахлорида фосфора. При охлаждении льдом температура повышается до 6оС и после охлаждения вновь до 4оС взвесь перемешивают 1 ч. Осадок фильтруют в атмосфере сухого азота, промывают дихлорметаном (10 мл) и после сушки в вакууме при 20-25оС получают 1,4 г бледно-желтого твердого вещества.
Продукт вышеприведенного опыта охарактеризован спектром протонного ядерного магнитного резонанса (1Н-ЯМP) в уксусной кислоте-d4, в котором Н5 проявляется при 7,61 ч/млн, а СН3 при 4,34 ч/млн. Спектр продукта отвечает заглавному соединению с конфигурацией анти-изомера. Кроме того продукт загрязнен непревращенной кислотой (1Н-ЯМР имеет Н5 при 7,07 ч/млн и СН3 при 4,06 ч/млн), что дополнительно подтверждено совмещением с исходной кислотой.
П р и м е р 17. Ацилирование 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата, Нl-соли использованием 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида гидрохлорида (анти-форма примера 14).
К предварительно охлажденному раствору 9 мл ацетона и 3,4 мл воды при 10оС добавляют Нl-соль 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбокси- лата (1,13 г, 2,26 ммоля). При 0оС пятью порциями прибавляют гидрохлорид 2-(2-аминотиазол-4-ил)-2-метоксииминоацетил- хлорида (1,09 г, 4,21 ммоля) (получение см. пример 14) вместе с триэтиламином (0,37 мл, 2,66 ммоля) для поддержания рН при 6-7. Реакционную смесь перемешивают 15 мин при комнатной температуре. Анализ полученного раствора жидкостной хроматографией высокого давления (С18колонка с градиентом 2-25% ацетонитрила в 0,005 М NH4H2PO4) показал наличие (по плодащи пика) 72,4% антицефепима со временем удерживания 13,08 мин и отсутствие поддающегося обнаружению син-цефепима с ожидаемым временем удерживания примерно 8,5 минут. Подкислением серной кислотой до рН 1,9 получают 1,48 г анти-цефепима в виде сульфатной соли. Идентификация продукта подтверждена 1Н-ЯМР спектроскопией (ДМСО-d6), согласно которой продукт содержит 0,58 моля соли триэтиламина.
П р и м е р 18. Ацилирование 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата, Нl-соли использованием 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида гидрохлорида (анти-изомер примера 14).
К предварительно охлажденному раствору 108 мл ацетона и 40,5 мл воды добавляют при 10оС Нl-соль 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата (13,5 г, 0,0317 моля). Добавлением 2,7 мл 14% NH4OH устанавливают во взвеси рН 7. При температуре 10оС в течение 60 минут порциями прибавляют гидрохлорид 2-(2-аминотиазол-4-ил)-2-метоксииминоацетил- хлорида (13,05 г, 0,015 моля) получен по методике примера 14/ с применением 14% NH4OH (27 мл) для поддержания рН 6,3-7 в течение первого получаса прибавления и рН 6,1-6,6 в течение второго получаса прибавления. Реакционную смесь перемешивают 30 минут при комнатной температуре. Реакционную смесь фильтруют и промывают 6 мл смеси ацетон-вода (2:1) и затем 6 н. Н2SO4 (15 мл), которую медленно прибавляют к фильтрату с установлением рН 1,87-1,9. После перемешивания 1 ч нерастворимую часть отфильтровывают и фильтровальный пирог промывают 21 мл смеси ацетон-воды (2:1) и затем 30 мл ацетона. К фильтрату в течение 30 мин добавляют 1 л ацетона и полученную смесь перемешивают 40 мин при 5-8оС. Продукт отфильтровывают, дважды промывают 24 мл смеси ацетон-вода (4:1), 60 мл ацетона и после сушки в вакууме получают 20,64 г (116% от стехиом. массы) анти-цефепима в виде сульфатной соли (95,4% чистоты согласно ЖХВД). 1Н-ЯМР спектр отвечает строению антицефепима, содержащему 3 моля аммонийных солей.
П р и м е р 19. Сравнение продукта примера 9 (син-изомер цефепима) с продуктом примера 17 (анти-изомер цефепима) показало следующие отличия в физических показателях.
Хроматография высокого давления изомеров цефепима осуществлена на колонке Уотерс М. Бондапак С18 (3,9 х 300 мм) с применением в качестве системы растворителей 1000 мл воды, содержащей 2,88 г (0,013 моля) натриевой соли гептансульфоновой кислоты, с установлением рН 4 добавлением уксусной кислоты и 100 мл ацетонитрила, при скорости потока 2 мл/мин. Продукты анализируют визуально с помощью детектора с переменной длиной волны формы Уотерс модели 450, установленного на 254  , и получают следующие результаты.
, и получают следующие результаты.
Время удерживания, мин
син-Изомер цефепима
(пример 9) 10,5
анти-Изомер цефепима
(пример 17) 37,8
Спектры 1Н-ЯМР син- и анти-изомеров цефепима по метоксимовой группе получены для дигидрохлоридов на ЯМР-спектрометре Брукер АМХ-400 FT с применением в качестве растворителя дейтерированного диметилсульфоксида. Указываемые химические сдвиги соотносятся с ДМСО при 2,49 ч/млн. Система нумерации, приведенная далее в формуле и таблице, дается только для удобства.
син- и анти-цефепим
NH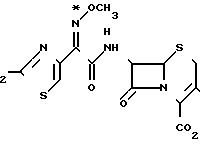
 Волнистой линией обозначены син- и анти-изомеры по метоксииминогруппе.
Волнистой линией обозначены син- и анти-изомеры по метоксииминогруппе.
Сравнение химических сдвигов в 1Н-ЯМР спектрах (м.д.)
Обозначение син-Цефепим анти-Цефепим
С2-Н2 4,04; 3,65 4,02,3,65
С6-Н 5,33 5,31
С7-Н 5,88 5,85
С11-Н2 4,6; 4,31 4,59; 4,3
С12-Н3 2,93 2,93
С13-Н4 3,7; 3,4 3,6; 3,3
С14-Н4 2,10 2,1
С18-Н4 6,88 7,57
С20-Н3 3,92 4,05
NH 9,83 9,56
NH2 8,6 8,7
1Н-ЯМР спектры двух изомеров цефепима по метоксииминогруппе отличаются значительно. СН(18) тиазольного цикла син-(Z)-метоксимового изомера при 6,88 смещается в высокое поле при 7,57 ч/млн для анти-(Е)-метоксимового изомера СН(18).
П р и м е р 20. син-2-(2-Аминотиазол-4-ил)-2-метоксииминоуксусная кислота, гидрохлорид.
син-2-(2-Аминотиазол-4-ил)-2-метоксии- миноуксусную кислоту (95,3 г, 424 ммоля) в дихлорметане (570 мл) размельчают в смесителе (15 мин под азотом). Полученную мелкую суспензию разбавляют дихлорметаном (100 мл) и переносят под азотом в реактор Бучи на 1 л с рубашкой. В реакторе создают давление азота в 5 psi (0,35 кг/см2) и смесь перемешивают со скоростью 375 об/мин и охлаждении до -2оС. В головное пространство реактора со скоростью 0,2 г в мин вводят хлористый водород (15,4 г, 424 ммоля). Наблюдается повышение температуры на 2оС. Смесь перемешивают еще 30 мин при 0оС, фильтруют и промывают под азотом дихлорметаном (350 мл). Высушиванием твердого продукта 18 ч в вакууме получают 110,9 г (111% нескорректированный выход) заглавного соединения в виде снежно-белого порошка.
Вычислено, С 30,32; Н 3,39; N 17,68; S 13,49; Cl 14,91.
С6Н8N3SCl.
Найдено, С 29,37; Н 3,17; N 16,34; S 12,70; Сl 16,99.
1Н-ЯМР (ДМСО-d6) δ 4,05 (с, 3Н, СН3), 5,9 (с, 15 мол. остаточного СН2Сl2), 7,1 (с, 1Н, С-5 Н). Сигналы также видны при 4,18 (с, 3Н, СН3) и 7,7 (с, 1Н, С-5 Н), что соответствует примерно 2% анти-изомера.
П р и м е р 21. син-2-(2-Амино-тиазол-4-ил)-2-метоксииминоуксусная кислота, гидрохлорид.
син-2-(2-Аминотиазол-4-ил)-2-метоксии- миноуксусную кислоту (25 г, 124 ммоля) в ацетонитриле (125 мл) титруют под азотом 1,39 М раствором НСl в ацетонитриле (89,2 мл, 123,9 ммоля) и выдерживают при 10-15оС. Смесь перемешивают еще 30 мин при 10-15оС, фильтруют и промывают под азотом ацетонитрилом (200 мл). Сушкой твердого продукта 3 ч вакууме при 45оС получают 29,5 г (97,4%), нескорректированный выход) заглавного соединения в виде снежно-белого порошка.
1Н-ЯМР (СD3OD) δ 2,05 (с, 13% мас./мас. остаточный ацетонитрил), 4,1 (с, 3Н, СН3), 7,1 (с, 1Н, С-5Н). Сигналы кроме того видны при 4,2 (с, 3Н, СН3) и 7,8 (с, 1Н, С-5Н), соответствующие прим. 0,5% анти-изомера.
П р и м е р 22. син-2-(2-Аминотиазол-4-ил)-2-метоксииминоацетилхлорид гидрохлорид.
Гидрохлорид 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (56,24 г, 210 ммолей), содержащий примерно 11 мас./мас. остаточного ацетонитрила, в дихлорметане (450 мл) размельчают 3 мин под азотом в смесителе, затем охлаждают до -35оС и переносят под азотом в течение 5 мин в хорошо перемешиваемую взвесь реактива Вильсмайера, также охлажденную до -35оС. Взвесь реактива Вильсмайера получают добавлением при 0оС оксалилхлорида (28,2 г, 221 ммоль) порциями к раствору диметилформамида (16,89 г, 231 ммоль) в дихлорметане с последующим охлаждением до -35оС. В ходе прибавления температура реакции повышается до -28оС. После прибавления реакционную смесь засевают продуктом. После перемешивания еще 2,5 ч в температурном интервале от -28 до -35оС смесь фильтруют и фильтровальный пирог промывают под азотом дихлорметаном (200 мл). Через пирог 30 мин пропускают азот, после чего сушат 12 ч при комнатной температуре в вакууме. Заглавное соединение получено в виде снежно-белого порошка (42,9 г, выход 72%). 1Н-ЯМР (CD3OD)δ 4-06 (c, 3Н, СН3), 7,12, (с, 1Н, С-5Н). Сигналы кроме того видны при 7,18, соответствующий прим. 5% гидрохлорида кислоты, при 7,8 (с, С-5Н), соответствующим прим. 0,5% анти-изомера. После дериватизации с диэтиламином в ацетонитриле ЖХВД показала заглавное соединение (син-изомер в виде его диэтиламидного производного) со временем удерживания 9,6 мин, гидрохлорид кислоты со временем удерживания 2,8 мин и анти-изомер (в виде его диэтиламинного производного) с временем удерживания 16,4 минуты. Отношение син-изомер: гидрохлорид кислоты: анти-изомер составляет 90:5:<1.
П р и м е р 23. син-2-(2-Аминотиазол-4-ил)-2-метоксииминоацетилхлорид гидрохлорид.
В смесителе 20 мин размельчают под азотом син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусную кислоту (84,7 г, 421 ммоль) в дихлорметане (570 мл). Полученную мелкую суспензию разбавляют дихлорметаном (100 мл) и под азотом переносят в реактор Бучи на 1 л с рубашкой. В реакторе создают давление азота в 5 psi (0,35 кг/см2) и смесь перемешивают со скоростью 375 об./мин и охлаждении до -2оС. В головное пространство реактора со скоростью 0,2 г в минуту вводят хлористый водород (15,3 г, 421 ммоля). Наблюдается повышение температуры на 2оС. Смесь перемешивают еще 30 мин при 0оС, размельчают 3 минуты в смесителе, охлаждают до -35оС и под азотом переносят за 5 мин в хорошо перемешиваемую взвесь реактива Вильсмайера, также охлажденную до -35оС. Взвесь реактива Вильсмайера получают добавлением порциями при 0оС оксалилхлорида (56,1 г, 439 ммоля) к раствору диметилформамида (33,8 г, 462 ммоля) в дихлорметане (880 мл) с последующим охлаждением до -35оС. В ходе прибавления температура реакции повышается до -28оС. После прибавления реакционную смесь засевают продуктом. После дополнительного выдерживания в температурном интервале от -28 до -35оС в течение 2,5 ч смесь фильруют и фильтровальный пирог промывают под азотом дихлорметаном (350 мл). Через фильтровальный пирок 30 мин пропускают азот и затем твердый продукт сушат 12 ч в вакууме при комнатной температуре. Заглавное соединение получено в виде снежно-белого порошка (95,2 г, нескорректированный выход 89%).
Вычислено, C 28,14; Н 2,76; N 16,41; S 12,52; Сl 27,68.
С6Н7N3O2SCl2.
Найдено, С 28,11; Н 2,62; N 16,20; S 12,22; Сl 26,74.
1H-ЯМР (СDOD) δ: 4,06 (с, 3Н, СН3), 7,12 (с, 1Н, С-5Н). Сигналы кроме того видны при 7,18 (с, С-5Н), соответствующий прим. 4% гидрохлорида кислоты, и при 7,8 (с, С-5Н), соответствующий прим. 2% анти-изомера. После дериватизации с диэтиламином анализ ЖХВД показал заглавное соединение (в виде его диэтиламидного производного) со временем удерживания 9,6 мин, исходную кислоту со временем удерживания 2,8 мин и анти-изомер (в виде его диэтиламидного производного) со временем удерживания 16,4 мин. Отношение син-изомер исходная кислота-анти-изомер составляет 90:4:2.
П р и м е р 24. Получение 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацетамидо /-3-/(1-метил-1-пирролидинил)метил/цеф-3-ем-4-карбоксилата дигидрохлорид гидратов.
В 60 мл воды постепенно растворяют 7-амино-3-/(1-метил-1-пирролидинио)метил /цеф-3-ем-4-карбоксилат, гидройодид (15,01 г, 35,29 ммоля), поддерживая рН ниже 6,5 использованием триэтиламина. Добавляют ацетон (120 мл) и полученный водно-ацетоновый раствор охлаждают в температурном интервале от -15 до -20оС. Поддерживая рН ниже 7,5 и выше 5 с помощью автотитратора модели Радиометр АВV80, заполненного триэтиламином и настроенного на конец титрования при рН 6,5, добавляют гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида (14,6 г активного, 45,4 ммоля), продолжающееся 2 ч и 7 мин. Реакционную смесь оставляют постепенно нагреваться от -12оС до 5оС до момента завершения ацилирования (согласно ЖХВД), после чего реакционную смесь фильтруют.
Фильтрат подкисляют 12 н. соляной кислотой (17,7 мл, 0,212 моля), после чего для инициирования кристаллизации добавляют ацетон (210 мл). Взвесь перемешивают 1,25 ч и затем разбавляют дополнительным количестивом ацетона (195 мл). Взвесь охлаждают до 0-5оС, перемешивают 0,75 ч и фильтруют. После вакуумного фильтрования влажный фильтровальный пирог промывают ацетоном и сушат примерно сутки в вакууме при 45оС. Дигидрохлоридную гидратную солевую форму заглавного соединения выделяют с 95,5%-ной чистотой (12,8 г, стехиометрический выход по массе 63,8%).
П р и м е р 25. Получение 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацетамидо /-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата дигидрохлорид гидратов.
К холодному (-30оС) раствору ацетона (120 мл) и воды (40 мл) одновременно и по отдельности в сухом виде в течение 30 мин прибавляют гидройодид 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-кар- боксилата (15 г, 35 ммолей) и гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксиими-ноацетилхлорида (10,49 г активного, 40,9 ммоля). С помощью автотитратора модели Радиометр АВV80, заполненного триэтиламином и настроенного на конец титрования при рН 6,5, значение рН реакционной смеси поддерживают в интервале 5,5-7 с охлаждением от -20 до -40оС. По окончании прибавления реагентов полученную мелкую непрозрачную взвесь нагревают до 0-5оС и перемешивают до полного растворения твердых веществ (20 мин), и к этому времени высокоэффективная жидкостная хроматография указывает на завершение ацилирования. Затем реакционную смесь фильтруют.
Фильтрат подкисляют 12 н. соляной кислотой (17,6 мл, 0,212 моля), затем для инициирования кристаллизации добавляют ацетон (150 мл). Взвесь перемешивают 1 ч, затем в течение 30 мин разбавляют дополнительным количеством ацетона (315 мл), перемешивают 30 мин при комнатной температуре и охлаждают 1 ч при 0-5оС. После вакуумного фильтрования продукт промывают ацетоном (250 мл) и сушат в вакууме при 45оС. Заглавное соединение выделено в виде его дигидрохлорида, гидрата чистотой (17,68 г, стехиометрический выход по массе 87,7%). Содержание воды методом Карла Фишера 4,45%
П р и м е р 26. Получение 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацет-амидо /-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилат дигидрохлорид гидратов.
В 60 мл постепенно при комнатной температуре растворяют гидройодид 7-амино-3-/(1-метил-1-пирролидинио)метил/-цеф-3-ем-4-карбоксилата (14,75 г, 34,7 ммоля), поддерживая рН ниже 6,5 использованием триэтиламина. Красно-оранжевый раствор обесцвечивают при 0-5оС активированным углем (3 г). Уголь отфильтровывают и полученный светло-янтарный раствор хранят при 0-5оС. Угольный пирог промывают 22,5 мл воды. Промывной раствор смешивают с 68 мл ацетона и полученный водно-ацетоновый раствор охлаждают до -30оС. Установив температуру в пределах от -20оС до -30оС и поддерживая рН в интервале 5,5-6,5 с помощью автотитратора модели Радиометр АВV80, заполненного триэтиламином и настроенного на конец титрования при рН 6,5, добавляют устойчивым прикапыванием обесцвеченный раствор 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-кар- боксилат к охлажденному водно-ацетоновому раствору от промывания угольного пирога при одновременном добавлении гидрохлорида син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида (9,06 г активного, 35,4 ммоля) и 180 мл ацетона (устойчивый поток). По окончании прибавления реагентов реакционную смесь нагревают до 0-5оС и перемешивают до полного растворения твердых веществ. После окончания ацилирования (согласно ВЭЖХ) реакционную смесь фильтруют.
Фильтрат подкисляют 12 н. соляной кислотой (17,6 мл, 0,212 моля), затем при комнатной температуре в течение часа разбавляют ацетоном (690 мл) и выдерживают один час при комнатной температуре. Продукт фильтруют под вакуумом, промывают ацетоном (250 мл) и сушат 15 ч в вакууме примерно при 45оС. Заглавное соединение выделено чистотой 89,6% (15,44 г, стехиометрический выход по массе 76,4%). Содержание воды методом Карла Фишера 4,07%
П р и м е р 27. Получение 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацетамидо /-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилат дигидрохлорид гидратов
К холодному (-30оС) раствору ацетона (120 мл) и воды (40 мл) одновременно или раздельно в сухом виде в течение 30 мин прибавляют гидрохлорид 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-кар- боксилата (14,34 г, 43 ммоля) и гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксиими-ноацетилхлорида (11,01 г активного, 43 ммоля). С помощью автотитратора модели Радиометр АВV80, заполненного триэтиламином и настроенного на конец титрования при рН 6,5, в реакционной смеси поддерживают рН в интервале 5,5-7 с охлаждением смеси в пределах от -20 до -40оС. По окончании прибавления реагентов полученную мелкую непрозначную взвесь нагревают до 0-5оС и перемешивают до полного растворения твердых веществ (20 мин), и в этот момент по данным высокоэффективной жидкостной хроматографии ацилирование завершается. Затем реакционную смесь фильтруют.
Фильтрат подкисляют 12 н. соляной кислотой (21,4 мл, 0,257 моля) и для инициирования кристаллизации добавляют ацетон (75 мл). Кристаллизационную взвесь перемешивают 1 час, после чего в течение 30 мин разбавляют ацетоном (505 мл). После перемешивания 30 мин при комнатной температуре взвесь охлаждают 1 ч до 0-5оС и фильтруют. Продукт промывают ацетоном (250 мл) и сушат в вакууме при 45оС. Заглавное соединение выделено в виде его дигидрохлорида, гидрата чистотой 93,5% (18,81 г, стехиометрический выход по массе 85,1%). Содержание воды методом Карла Фишера 4,2%
П р и м е р 28. Превращение моногидрата дигидрохлорида цефепима в дигидрат дигидрохлорид цефепима.
В 1200 мл деионизированной воды растворяют моногидрат, дигидрохлорид цефепима (300 г, чистота 99,9% по данным ВЭЖХ, КF 3,8%). Добавляют 6 н. соляную кислоту (132 мл, 1,5 эквивалента). Раствор фильтруют и промывают деионизированной водой (300 мл).
К профильтрованному раствору добавляют ацетон (1500 мл) и в течение 20 мин по каплям прибавляют дополнительное количество ацетона (4000 мл). Раствор выдерживают в точке помутнения до момента интенсивного образования кристаллов дигидрата (иглы под микроскопом, в точке помутнения возможно затравливание). В течение 25 мин прибавляют дополнительное количество ацетона (8000 мл) и густую взвесь перемешивают один час при 25оС.
Принадлежность кристаллов дигидрату (иглы) подтверждена сравнением кристаллов под микроскопом с аутентичным образцом. Взвесь фильтруют и промывают ацетоном (2 х 1500 мл). Фильтровальный пирог сушат 15 ч вакууме при 40оС. Выход дигидрата дигидрохлорида цефепима 305,10 г (98,6%), чистота согласно ВЭЖХ 99% КF 6,5%
П р и м е р 29. Превращение дигидрата дигидрохлорида цефепима в моногидрат дигидрохлорид цефепима.
В 75 мл депонизированной воды растворяют дигидрат дигидрохлорид цефепима (15 г, чистота согласно ВЭЖХ 99,2% КF 6,4%). Добавляют 6 н. соляную кислоту (0,9 мл, 0,2 эквивалента) и раствор фильтруют через фильтр на 0,45 мкм.
К профильтрованному раствору по каплям за 20 минут прибавляют ацетон (200 мл) с образованием замутненного раствора (в этот момент возможно затравливание). Без выдерживания в данном состоянии по каплям за 40 мин прибавляют еще ацетон (400 мл) и взвесь охлаждают один час в бане со льдом до 0-5оС.
Принадлежность кристаллов моногидратной форме подтверждена сравнением под микроскопом с аутентичным образцом. Взвесь фильтруют и промывают ацетоном (2 х 60 мл). Фильтровальный пирог сушат в вакууме 15 ч при 40оС. Выход моногидрата дигидрохлорида цефепима 13,28 г (91,8%) и строение кристаллов, как подтверждено, такое же, что и описанное в патенте США N 4910301 (Каplan и др.).
П р и м е р 30. Получение 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацетамидо /-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилат дигидрохлорид гидратов.
К холодному (-22оС) раствору ацетона (120 мл) и воды (40 мл) в течение 25 мин одновременно или раздельно в сухом виде прибавляют гидройодид 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-кар- боксилата (14,67 г активного, 0,0345 моля) и гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида (9,93 г активного, 0,0388 моля). С помощью автотитратора модели Радиометр АВV80, заполненного триэтиламином и настроенного на конец титрования при рН 6,5, в реакционной смеси поддерживают рН в интервале 5-7 при охлаждении от -20 до -30оС. По окончании прибавления реагентов полученную непрозрачную взвесь нагревают до 0-5оС до полного растворения твердых веществ, и в этот момент по данным высокоэффективной жидкостной хроматографии ацилирование завершается. Реакционную смесь фильтруют и фильтрат делят на две равные порции.
Метод А. Одну порцию полученного фильтрата подкисляют 12 н. соляной кислоты (8,8 мл, 0,106 моля) и разбавляют ацетоном (114 мл) до помутнения фильтрата. Фильтрат затравливают кристаллами дигидрата дигидрохлорида цефепима (0,5 г) и взвесь нагревают примерно 3 ч при 40оС. Смесь охлаждают 1 ч при 0-5оС и фильтруют. Продукт промывают ацетоном и сушат в вакууме при комнатной температуре. Дигидрохлорид заглавного соединения кристаллизован с чистотой в 93,3% (7,34 г, стехиометрический выход по массе 67,3%). Содержание воды анализом по Карлу Фишеру -4,1% и FT-ИК (Диффузивное отражение с КВr) анализ с пиками поглощения при 3574 см и 3432 см-1 указывают на то, что продукт является смесью моногидрата (гранулоподобные кристаллы) и дигидрата (иглоподобные кристаллы) заглавного соединения.
Метод В.
Вторую порцию фильтрата подкисляют 12 н. соляной кислотой (8,8 мл, 0,106 моля) и в течение 1 ч разбавляют ацетоном (206 мл). Взвесь выдерживают до начала кристаллизации, после чего охлаждают до 0-5оС и выдерживают 1 ч. Взвесь фильтруют, продукт промывают ацетоном и сушат в вакууме при 45оС. Дигидрохлорид заглавного соединения кристаллизован чистотой в 95,3% (8,6 г, стехиометрический выход по массе 85,3%). Содержание воды анализом по Карлу Фишеру 4,6% и FT-ИК (Диффузивное отражение с КВr) анализ показывают, что продукт является моногидратной формой заглавного соединения с содержанием дигидрата менее 0,4%
П р и м е р 31. Получение 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацетамидо /-3-/(1-метил-1-пирролидинил)метил/цеф-3-ем-4-карбоксилат дигидрохлорид дигидрата.
К холодному (-22оС) раствору ацетона (120 мл) и воды (40 мл) в течение 25 мин одновременно или раздельно в сухом виде прибавляют гидройодил 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-кар- боксилата (14,61 г активного, 0,0344 моля) и гидрохлорид син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида (9,94 г активного, 0,0388 моля). С помощью автотитратора модели Радиометр АВV 80, заполненного триэтиламином и настроенного на конец титрования при рН 6,5, в реакционной смеси поддерживают рН в интервале 5-7 при охлаждении от -20 до -30оС. По окончании прибавления реагентов полученную непрозрачную взвесь нагревают до 0-5оС до полного растворения твердых веществ, и в этот момент по данным высокоэффективной жидкостной хроматографии ацилирование завершается. Реакционную смесь фильтруют и фильтрат делят на две равные порции.
Метод А.
Одну порцию полученного фильтрата подкисляют 12 н. соляной кислотой (11,7 мл, 0,404 моля) и разбавляют ацетоном до помутнения фильтрата. Фильтрат затравливают кристаллами дигидрата дигидрохлорида цефепима (0,3 г) и взвесь нагревают примерно 1 ч при 50оС. Смесь охлаждают до комнатной температуры, разбавляют ацетоном и перемешивают в течение 15 ч. Шлам снова нагревают при 40оС в течение 1 ч и разбавляют ацетоном. Суммарное количество ацетона в 280 мл используют для кристаллизации продукта. После постепенного охлаждения в течение 1 ч до 0-5оС смесь фильтруют, промывают ацетоном (125 мл) и сушат в вакууме при 45оС.
Основное соединение в виде цефепима дигидрохлориддигидрата выделяют в количестве 97,8% (8,19 г, стехиометрический выход по массе 80,9%). Содержание воды анализом по Карлу Фишеру 6,5% и FT-ИК (Диффузивное отражение с КВr) анализ с пиками поглощения при 3574 и 3432 см-1указывают на то, что продукт представляет собой дигидратную форму (иглоподобные кристаллы) заглавного соединения.
Метод В. Вторую порцию фильтрата подкисляют 12 н. соляной кислотой (14,6 мл, 0,1752 моля) и добавляют ацетон при перемешивании до помутнения. Фильтрат затравливают кристаллами дигидрата дигидрохлорида цефепима (0,3 г) и перемешивают при комнатной температуре в течение 1,5 ч. Смесь дополнительно разбавляют ацетоном, перемешивают при комнатной температуре в течение 15 ч, а затем нагре- вают при 40оС в течение 1 ч. Дополнительно разбавляют смесь ацетоном до общего его объема в 231 мл, после чего охлаждают до 0-5оС и выдерживают 1 ч. Взвесь фильтруют, продукт промывают 125 мл ацетона и сушат в вакууме при 45оС. Дигидрат дигидрохлорид цефепима выделяют с чистотой в 96,3% (8,68 г, стехиометрический выход по массе 85,7%). Содержание воды анализом по Карлу Фишеру 6,7% и FT-ИК (Диффузивное отражение с КВr) анализ показывают, что продукт является дигидратной формой заглавного соединения (иглоподобные кристаллы).

Сущность изобретения: свободную от Δ2 -изомера кислотно-аддитивную соль 7-амино-3-[(1-метил-1-пирролидино)метил]-цеф-3-ем-4-карбоксилата ф-лы I, где НХ HCl, HJ или серная кислота, подвергают взаимодействию с гидрохлоридом 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида, по существу свободным от антиизомера, в водно-органическом растворителе при рН 5,0 7,5 с последующим выделением из полученной реакционной смеси целевого продукта в виде его кристаллического моно- или-дигидрата дигидрохлорида. Структура соединения ф-лы I:  13 з.п. ф-лы, 4 ил. 1 табл.
13 з.п. ф-лы, 4 ил. 1 табл.

где HX означает HCl, HJ или H2SО4,
подвергают взаимодействию с гидрохлоридом 2-(2-аминотиазол-4-ил)-2- метоксииминоацетилхлорида, по существу свободным от антиизомера, в водно-органическом растворителе при pH 5,0 7,5 с последующим выделением из полученной реакционной смеси целевого продукта в виде его кристаллического моно- или дигидрата дигидрохлорида.
минус 40oС.
| Патент США N 4943631, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

