Изобретение относится к конъюгатам терапевтически применяемых антрациклинов с такими носителями, как поликлональные и моноклональные антитела или белки или пептиды естественного или синтетического происхождения; к способам их получения, к содержащему их фармацевтическому составу и к их применению при лечении определенных опухолей у млекопитающих. Изобретение также относится к новым производным антрациклина и к их получению.
В последние годы было синтезировано много высокоцитотоксичных антрациклинов. Например, такие, которые несут морфолино или морфолино-замещенное кольцо, связанное в положении C-3' сахарной части, продемонстрировали многообещающую противоопухолевую активность на экспериментальных опухолях мышей или крыс (см. Bioactive molecules, 55 - 101, vol. 6, Edited by J. Wikkiam Zown, Elveiser 1988).
Сущность настоящего изобретения заключается в создании конъюгатов антрациклина с носителями, такими как моноклональные или поликлональные антитела или белки или пептиды, или с другими носителями синтетического происхождения с целью извлечения преимущества из высокой эффективности антрациклинов, например, морфолино-производных, для улучшения их терапевтической эффективности и снижения их токсичности.
Конъюгаты согласно настоящему изобретению, характеризующиеся наличием чувствительной к кислоте ацеталевой связи, имеют общую формулу (I)
(A-O-W-Z-)a-T
в которой
часть A-O - обозначает остаток любого антрациклина формулы A-O-H, содержащего по крайней мере одну первичную или вторичную гидроксильную группу; а обозначает целое число от 1 до 30;
W обозначает остаток формулы (2):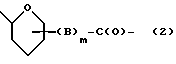
в которой
B обозначает C1-C6 алкиленовую. группу, возможно гетеропрерванную, а m обозначает 0 или 1; C1-C6 алкиленовая группу может представлять собой C1-C6 алкиленовую группу, такую как этилен метилен, этилиден или н-пропилен. Гетероатом, прерывающий алкиленовую группу, может представлять собой кислород или азот. Предпочтительно B обозначает -CH2-O-CH2-, -CH2-NH-CH2-, -CH2-, -C3H6- или -C6H12-; Z обозначает спейсерную группу, а T - носитель.
Предпочтительными группами Z являются:
I) -NH-, а T-Z обозначает остаток носителя формулы T-H2, несущий по крайней мере одну свободную аминогруппу, или
II) -NH-(D)d-N= CH-, где d обозначает целое число от 0 до 2, (D) обозначает -NH-CO(CH2)nCO-NH- ( n равно 2 или 4), а T-Z содержит остаток носителя формулы T-CHO, несущего по крайней мере одну формильную группу.
III) пиперазинилкарбонильная часть или группа формулы
-NH-(D)a-NH-CO-,
где
(D) и a имеют вышеуказанные значения, а T-Z содержит остаток носителя формулы T-/COOH/a, в которой a имеет вышеуказанные значения.
В вышеприведенной формуле (1) гликозид антрациклина A-O-H предпочтительно является производным от соединений формулы (3):
в которой:
R1 обозначает атом водорода, окси- или метокси-группу;
а) R2 обозначает оксигруппу, а R4 или R5 обозначают атом водорода, а другое из R4 и R5 обозначает оксигруппу, или R4 обозначает атом йода и R5 обозначает атом водорода, или оба и R4 и R5 обозначают атомы водорода или
б) R2 обозначает атом водорода и R4 или R5 обозначает оксигруппу и другое из R4 и R5 обозначает атом водорода; и R3 обозначает аминогруппу или обозначает атом азота, заключенный в кольцо морфолино (MO) или 3-циано-4-морфолино (CM) или 2-метокси-4-морфолино (MM), в котором атом азота связан в C-3'
Антрациклин может быть связан с носителем через гидроксильную группу в положении C-14 или C-4' соединения антрациклина. В одном варианте осуществления носитель, связанный с антрациклином в положении C-14, формула (3'), обозначает часть A-O-: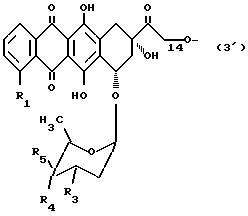
в которой
R1 и R2 имеют вышеуказанные значения, а R4 или R5 обозначает оксигруппу, или R4 обозначает атом водорода или йода, а R5 обозначает атом водорода.
В другом варианте осуществления носитель, связанный с антрациклином через оксигруппу в C-4', формула (3''), обозначает часть A-O-: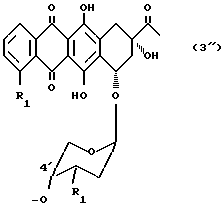
в которой
R1 и R3 имеют вышеуказанные значения.
Носитель обычно выбирают из поликлонального антитела или его фрагмента, содержащего антигенсвязывающий центр, способный связываться с опухолеспецифическим антигеном; моноклонального антитела или его фрагмента, содержащего антигенсвязывающий центр, способный связываться с антигеном, предпочтительно или селективно экспрессируемого на популяциях опухолевых клеток; пептида или белка, способного предпочтительно или селективно связываться с опухолевой клеткой; и полимерного носителя.
Часть носителя T-NH2 или T-/COOH/a конъюгатов таким образом предпочтительно производят из поликлональных антител, направленных против опухолеспецифических антигенов; или из моноклональных антител, связывающихся с антигенами, предпочтительно или селективно экспрессируемых на популяциях опухолевых клеток; или из природных или рекомбинантных пептидов или белков или факторов роста, предпочтительно или селективно связываемых с опухолевыми клетками; или из природных или синтетических полимерных носителей, таких как полилизин, полиглутамовая кислота, полиаспарагиновая кислота и их аналоги и производные, или таких как текстран или другие полимерные углеводные аналоги и их производные; или из синтетических сополимеров, таких как производные из N-(2-оксипропил)метакриламида (ОПМА) (см. S. Kopecek, Macromole cules. H. Benoit and P. Rempp., Ed.: 505 - 520 (1982). Pergamon Press. Oxford, England; или из поли(аминокислотных)сополимеров, таких как поли(GluNa, Ala, Tyr), которые полезны в качестве конъюгируемых носителей лекарственного препарата, обеспечивающих направленную доставку препарата к легочной ткани (P. Lcencan et al., Journal of Bioactive and Compatible Polymers, Vol, 4, июль 1989 г.).
Часть носителя также может быть произведена из частей вышеупомянутых носителей, таких как Fab или F(alo')2 фрагментов антител, или таких как части вышеупомянутых пептидов или белков, полученных с помощью технологии рекомбинантной ДНК.
Представительными примерами вышеупомянутых антител и соответствующих возможных терапевтических применений являются:
анти T-клеточное антитело T101 (Rayston, I. et al., J. Immunal 1980, 125, 725),
анти CD5 антитело OKTI (Орто) ATCC CRL 8000 (хронические лимфолейкозы);
антитело OKT9 против трансферринового рецептора (Орто) ATCC CRL 8021 (овариальные и другие опухоли);
антимеланомное антитело MAb 9.2.27 (Bumol, T.F. et al., Proc. Nat.i. Acad. Sci. USA 1982, 79, 1245 (меланомы);
антитело противораковых маркеров, такое как:
против карциннэмбрионального антигена 1116 NS-3d AR CC CRL 8019;
против альфа-фетопротеина OM 3-1.1 ATCC HB 134 (также гепатомы); 791T/36/Emoleton, M.S. et al., Br.S. Cancer 1981, 43, 582/ (также остеогенная саркома);
B 72.3 (патент США N 4.522.918 (1985)) (колоректальные карционмы и другие опухоли);
антитело против оварительного рака OVB 3 ATCC HB 9147;
антитело против рака груди (HMGF-антиген) (Aboud-Pira E. et al., Cancer ReS. 1988, 48, 3188);
против рака мочевого пузыря I 3.10 (Jn.D.S.et al., Eur. S. Urol. 1987, 13, 198).
Иллюстративными примерами вышеуказанных факторов роста и белков природного или рекомбинантного происхождения являются FGF, EGF, PDGE, TGF-α, α-MS, Интерлейкины, интерфероны, TNF меланотропин (MH) и т.д.
Носитель T-CHO предпочтительно является производным из поликлональных или моноклональных антител, имеющих углеводную часть, предпочтительно размещенную в области Fc, селективно окисленную в альдегидные группы с помощью химических или ферментных методов описано в патенте США N 4671958 (9 июня 1987 г. ). Носитель T-CHO также может быть произведен в результате формилирования или окисления подходящих полимерных носителей, или в результате окисления до альдегидных групп углеводных остатков подходящих гликопротеинов.
Изобретение далее обеспечивает создание способа получения соединения формулы (1), который включает:
а) превращение производного формулы 4:
A1-O-W-OH,
в которой
A1-O- представляет собой остаток любого антрациклина, несущего по крайней мере одну первичную или вторичную гидроксильную группу и имеющего аминогруппу относящейся к сахару части, которая защищена или заменена производным морфолино, а имеет вышеуказанные значения, в активированное производное формулы 5:
A1-O-W-L,
в которой
A1-O и W имеют те же самые значения, что и вышеуказанные, а L обозначает активирующую группу для получения амидной связи, такую как N-оксисукцинамидо, N-оксисульфосукцинамидо или 2,4-динитрофенокси, или 2,3,4,5,6-пентафторфенокси или трет-бутокси карбонилокси; и
б) (I) конденсирование полученных соединений формулы (5), как определено выше, с помощью соединения формулы T-NH2, как указано ранее; или
(II) введение в реакцию активированного соединения формулы (5) с производным формулы H2-(D)d-NH2, таким как гидразин (d = 0) или сукциновый (d = 1 и n = 2) или адипиновый (d = 1 и n = 4) дигидразид, возможно со снятием защиты полученного производного формулы (6):
A1-O-W-NH-(D)d-NH2,
в которой A1-O-, W, d и (D) имеют вышеуказанные значения, и его конденсирование с помощью соединения формулы T-CHO или T-/COOH/a как определено ранее, или
(III) введение в реакцию соединения формулы (5) с 1,4-пиперазином и конденсирование полученного соединения формулы (7):
в которой
A1-O и W имеют вышеуказанные значения, с соединением формулы T/COOH/a, как определено выше, возможно в присутствии конденсирующего агента, для получения конъюгата формулы (1), в которой непрореагировавшие, возможно присутствующие активированные карбоксильные группы могут быть подавлены с помощью фармацевтически приемлемого амина.
Например, методом активирования для превращения производных формулы (4) и производные N-оксисукцинимидила формулы (5) на стадии (А) настоящего процесса является реакция производного формулы 4 с N-оксисукционимидом или с его водорастворимой солью 3-замещенного сульфоната натрия в присутствии N, N'-дициклогексилкарбодиимида в таком растворителе, как этилацетат или N,N-диметилформамид. В таком случае в формуле (5) - L обозначает остаток: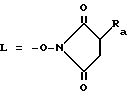
в которой
Ra обозначает атом водорода или группу сульфата натрия.
Методы конденсации для получения конъюгатов формулы (1), начиная от вышеупомянутых производных формулы (5) и носителя формула T-NH2 осуществляются в условиях, способных создать ковалентные связи амидного типа, совместимые со структурой носителя. Предпочтительные условия заключают в себе применение буферированных водных растворов с pH 7 - 9,5, температурой 4 - 37oC, в течение времени от нескольких часов до нескольких дней.
Например, условия для конденсации (б) (I) между соединениями формулы (5) и антителами T-NH2 таковы: водный фосфат натрия 0,1М и водный хлорид натрия 0,1М при pH 8, содержащий моноклональное антитело с концентрацией 1 мг/мл, обработанный 30-кратным молярным избытком 10%-ного (вес/объем) раствора вещества 6 в N,N-диметилформамиде, в течение 24 ч при 20oC. Конъюгат очищают гельфильтрацией на колонке Sephadex G-25 (Pharmacia Fine Chemical, Piscataway, Нью-Джерси), элюируют соляным раствором с фосфатным буфером (PBS).
Способы получения коньюгатов формулы (1),конденсации вышеупомянутых производных 6 с носителем формулы T-CHO осуществляется в условиях, способных создавать ковалентные связи типа гидразона, совместимые со структурой носителя. Предпочтительные условия включают применение буферированных водных растворов при pH 4-7,5, температуре 4-37oC, в течение времени от нескольких часов до нескольких дней.
Условия для связи (б) (II)между соединениями формулы (6) и антителами T-CHO таковы: водный ацетат натрия 0,1M и водный хлорид натрия 0,1M при pH 6, содержащий моноклональное антитело при концентрации 1 мг/мл, обрабатывали 30-кратным молярным избытком раствора 5 вес.%, объем веществом формулы (8) в том же самом буфере с раствором 50% (вес/объем) вещества 8 в том же самом буфере, в течение 24 ч при 20oC. Коньюгат очищают гель-фильтрацией согласно вышеизложенному.
Способ получения коньюгата формулы (1) путем конденсирования вышеуказанных производных 6 или 7 носителя формулы T(COOH)a осуществляется в условиях, способных образовывать ковалентные связи амидного типа, совместимые со структурой носителя.
Предпочтительные условия влекут за собой применение буферсодержащих водных растворов при pH 7-9,5, температуре 4-37oC в течение от нескольких часов до нескольких дней. Другие условия включают в себя применение сухого диметилформамида или диметилсульфоксида при комнатной температуре в течение 1-3 ч. Предпочтительное условие для конденсации между соединениями формулы 6 или 7 и активированным носителем формулы T(CO-E)a, в которой E обозначает подходящую активирующую карбонильную группу, такую как n-нитрофенил, - это сухой полярный растворитель, такой как диметилформамид, содержащий от 5 до 50 мг/мл соединения 6 или 7, обработанного с помощью эквивалентного количества соединения T/CO-E/a в течение 24 ч при комнатной температуре.
В данном случае период "от нескольких часов до нескольких дней" может составлять от 4 ч до 5 дней.
Производные общей формулы 4,5,6 и 7 и их получение являются новыми и находятся в рамках настоящего изобретения. Соединения 4,5,6 и 7 являются как полезными промежуточными соединениями, так и/или терапевтически активными противоопухолевыми агентами. Производные формулы (4) получают с помощью способа, который включает в себя:
а) конденсирование возможно защищенного антрациклина формулы A-O-H, как определено выше, при условии, что аминогруппа сахарной части антрациклина A-O-H представлена в форме защищенной аминогруппы, с дигидропиран-карбоксильным производным формулы 8:
в которой
B и W имеют вышеуказанные значения, а Rx обозначает защитную группу.
б) удаление всех или каждой из защитных групп из полученных промежуточных соединений, с тем, чтобы образовать производное формулы (4):
A-O-W-OH
в которой W имеет вышеуказанные значения, при условии, что аминогруппа из сахарной части антрациклинового остатка A-O-представляется в форме свободной аминогруппы; и
(1) превращение свободной аминогруппы сахарной части антрациклина формулы 4' в морфолино-производное или
(II) защита указанной свободной аминогруппы указанного антрациклины формулы 4'.
Промежуточное производное формулы 4' и его получение также обладают новизной и образуют часть настоящего изобретения. Производные являются как полезными промежуточными соединениями, так и или терапевтически активные противоопухолевые вещества. Преобразование антрациклинов общей формулы (4), как они охарактеризованы ранее, может быть осуществлено обработкой соединением формулы (8), определенной выше.
Изобретение также обеспечивает создание способа для получения производных формулы 4', причем он включает:
а) конденсирование возможно защищенного антрациклина вышеуказанной формулы A-OH при условии, что аминогруппа сахарной части антрациклина A-O-H находится в виде защищенной аминогруппы, с дигидропиранкарбоксильным производным формулы (8), как описано выше; и
б) удаление всей или каждой защитной группы из полученного промежуточного соединения.
Предпочтительные условия для осуществления реакции типично защищенных антрациклинов A-O-H с соединением 8 влечет за собой применение сульфокислоты, такой как n-толуолсульфокислота в качестве катализатора, в безводном амолярном растворителе, таком как хлористый метилен, при комнатной температуре в течение времени от нескольких часов до одного дня, с последующей обработкой слабой щелочью для деблокирования защитных групп.
Производные 8 представляют собой смесь анантиомеров. Однако реакция этерификации с гидроксильной группой антрациклинов, проводимая с кислотным катализатором, дает только два диастереоизомера, обозначаемые как x и y, имеющие неопределенную пространственную конфигурацию кольца (R или S) как в положении ацетали C-2", так и в другом положении, несущем группу -(B)m-C(O)ORx.
Полученное соединение формулы A-O-W-OP x преобразуют в соединение формулы (4), как определено выше, гидролизом и снятием защиты. Некоторые из исходных соединений формулы 8 являются новыми находятся в рамках настоящего изобретения.
Предпочтительные производные формулы 8 включают:
8A) 2-этоксикарбонил-3,4-дигидро-2H-пиран /m=0, Rx = C2H5/,
8B) этил 2-(3,4-дигидро-2H-пиран-2-ил)метилоксиацетат /B=CH2-O-CH2, m=1, Rx=C2H5/;
8C) метил 2-(3,4-дигидро-2H-пиран-2-ил)метилоксиацетат /B=CH2-O-CH2, m= 1, Rx=CH3/;
8D) этил 2-(3,4-дигидро-2H-пиран-2-ил)метилтиоацетат /B=CH2-NH-CH2, m=1, Rx=C2H5/.
8Е) этил 2-(3,4-дигидро-2Н-пиран-2-ил)метиламиноацетат /В=СН2-NH-CH2, m= 1, Rx=C2H5/.
Соединения общей формулы 8 могут быть получены несколькими путями. Например, они могут быть получены легко из имеющейся 3,4-дигидро-2H-пиран-2-карбоновой кислоты, соли натрия (CA S 16698-52-5) формулы H или из соответствующего производного метанола (CAS 3749-36-8) формулы M:
Более конкретно, соединение 8A получают путем введения в реакцию соединения H с иодистым этилом в безводном полярном растворителе, таком как диметилформамид, как описано в "Macromo-Iecules", т.12, 1, с.5-9, январь-февраль 1979 г. Метилоксиацетат 8B может быть получен путем введения в реакцию спирта M с соляной кислотой в сухом диметилформамиде при 100oC в присутствии двух эквивалентов гидроокиси натрия или калия, после этого охлаждением смеси и ее обработкой иодистым этилом.
Соединения формулы 8C-E могут быть получены методом конденсации Вюрца йодо-производного M, полученные путем замещения сложного эфира сульфокислоты M иодистым натрием, и этил 3-иодопропионата, этил 4-иодобутаноата или этил 7-иодогептаноата. Метиламиноацетат 8Г получают кипячением с обратным холодильником сложного эфира сульфокислоты спирта M с этилглицином в сухом толуоле в течение 6 ч.
Исходные антрациклины формулы (3) для использования в настоящем изобретении включают те, которые несут свободную гидроксильную группу, в положении C-4' или C-14, также как: даунорубицин (3a: R1=OCH3, R2=R5=H, R3=H2, R4=OH),
4-деметоксидаунорубицин (3b: R1=R2=R5=H, R3=NH2, R4=OH);
4'-эпидаунорубицин (3c: R1=OCH3, R2=R4=H, R3=NH2, R5=OH);
4'-дезоксидоксорубицин (3d: R1=OCH3, R2=OH, R4=R5H, R3=NH2);
4'-дезокси-4'-иододоксорубицин (3e: R1=OCH3, R2=OH, R5=H, R4=S, R3=NH2),
и те, которые как первичные, так и вторичные оксигруппы, также как доксорубицин (3f: R1=OCH3, R2=R4=OH, R5=H, R3=NH2),
4'-эпидоксорубицин (3g: R1=OCH3, R2=R5=OH, R4=H, R3=NH2), они при этом описаны в предшествующих патентах, см.: F. Arcamone "DOXORUNICIN", Medicinal Chenistry Vol17, New York. Y., 1981.
Например, антрациклины, первоначально несущие гидроксильную группу, в частности, такие, которые обозначены соединениями 3a-e преобразуют в их N-трифторацетиловые аналоги, которые затем вводят в реакцию с производными сложного эфира дигидропиранкарбоновой кислоты общей формулы (8), как описано ранее, для получения соединений 4(a-e) (A-Z), в которых (a-e) обозначают остаток антрациклинов формулы 3''(a-e) и 3'(d), (A-Z) обозначает остаток общей формулы 2, полученный в результате конденсации соединения формулы (8) с гидроксильной группой в положении C-4' или C-14 антрациклина.
Антрациклины, несущие обе гидроксильные группы в положениях C-4' и C-14, в частности те, которые представлены соединениями 3(f, g), могут быть селективно сконденсированы на гидроксильной группе C-14 с соединениями формулы (8), после временной защиты другой гидроксильной группы в положении C-4', для получения производных общей формулы 4 и 4', обозначенные как 4(f, g)(A-Z) и 4'(f, g)(A-Z), в которых (f, g) обозначает остаток антрациклина формулы 3'(f, g) (пиранилирование в C-14) или 3''(f, g) (пиранилирование в C-4') и (A-Z) имеют то же самое значение, что и указанного выше.
Производные 4 и 4', как изложено выше, в которых остаток -O-W связан в C-14-положении гидроксильной группы, получают путем введения в реакцию соединения формулы (8), а тех же самых условиях, что и описаны ранее, с производными антрациклина, несущим замещенную гидроксильную группу в C-4', общей формулы (9):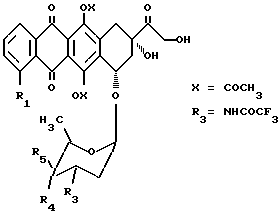
в которой R1 имеет вышеуказанное значение, R4 или R5 обозначает ацетокси-группу, а другое из R4 и R5 обозначает атом водорода.
Например, N-трифторацетиловые производные общей формулы 3 (R3=NHCOCF3), такие как 3(f, g), вначале защищают в положениях C-9 и C-14 в виде 9, 14-этилортоформата после реакции с триэтилортоформиат, согласно процедуре, описанной H. Umezawa et al., J. Antib. Vol XXXIII, N 12, 1581 (1980), затем ацилируют на фенольных и C-4'-гидроксильных группах с использованием ангидрида уксусной кислоты и пиридина, после чего разблокируют в положении гидроксильной группы C-14 с помощью водной соляной кислоты для получения производных 6, 11, 4'-три-O-ацетил-N-трифторацетилдоксорубицина формулы (9), которые конденсируют с соединением формулы 8 в тех же самых вышеизложенных условиях, для получения, после разблокирования фенольных групп с помощью морфолина в метаноле и разблокирования гидроксильной группы в положении C-4' с помощью метилата натрия в метаноле, соединения 10, И наконец, обработка соединения 10 водным едким натром 0,1H дает производное 4'. Способ иллюстрируется схемой 1.
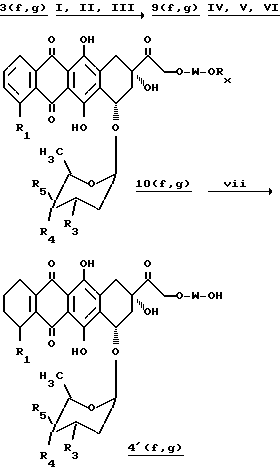
R3=NHCOCF3;
R4=H и R5=OH или R4=OH и R5=H
Реагенты и условия:
I: CH(C2H5O)3, pTSA, CH2Cl2; II: (CH3CO)2O, пиридин; III: 0,1H HCl-THF; IV: 8. pTSA, CH2Cl2, V: морфолин, CH3OH, VI: NaOCH3, CH3OH; VII: 0,1HNaOH.
Согласно другому аспекту настоящего изобретения обеспечивается получение морфолино (MO) и производного морфолино (CM) и (MM) общей формулы 4(MO), 4(CM) и 4(MM) от производных общей формулы 4'(R3=NH2), замещенных в положениях C-4' или C-14, получаемых согласно вышеизложенному, согласно стандартным процедурам, описанным в предшествующих патентах.
В частности, получение соединений 4-морфолино 4(a-g)(A-Z)(MO) и 3-циано-4-морфолино 4-(a-g)(A-Z)(CM) происходит в соответствии с процедурой, описанной E. M. Acton et al. b J. Med. Chem. 1984, 27, 638: производные 2-метокси-4-морфолино 4(a-g)(A-Z)(MM) получают согласно процедуре, описанной в патенте США N 4672057, от 9 июня 1987 г. Необходимо подчеркнуть, что остаток - W, в соединениях формулы 4'(a-g)(A-Z) не вредит получению вышеупомянутых производных морфолино.
Согласно способу получения производных морфолино (MO), соединения 4'(a-g)(A-Z), растворенные в воде, вначале вводят в реакцию с 2-оксиэтил-диальдегидом (II)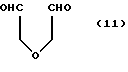
а затем с цианборгидридом натрия для получения производного морфолино.
В результате изменения условий реакции при восстановлении цианборгидрида, в частности, путем добавления цианида натрия извлекают производные цианморфолино (CN).
Получение производных 2-метокси-4-морфолино (MM) осуществляют обработкой соединений 4'(a-g)(A-Z), растворенных в воде, с помощью 1-метокси-2'-оксидиацетальдегида (12)
а затем с помощью цианбергидрида натрия.
Конъюгаты формулы I настоящего изобретения представляют собой ценные терапевтические вещества, так как они содержат ацеталевую связь, которая выделяет исходное лекарственное вещество A-O-H после гидроний-ион-катализованного гидролиза или после ферментативного расширения in vivo.
Известно, что в злокачественных опухолях имеет место высокая скорость гликолиза по сравнению с нормальной тканью. Это вызывает увеличение производства лактата и таким образом уменьшение pH в опухоли (см.: H.M.Pauen et al, Z.Naturforsch TeiI B, 23 (1968) 1461). Также соединения формулы 4,4', 6 и 7 могут выделять цитотоксичный антрациклин внутри тканей опухоли.
Изобретение представляет двухуровневую специфичность действия соединений, первый из них заключается в предпочтительной локализации конъюгата в опухолевой ткани с помощью распознавания антигена, а второй заключается в предпочтительном высвобождении лекарственного средства в его активной форме в опухолевой ткани с помощью предпочтительного кислотного расщепления.
Конъюгаты, полученные согласно описанным способам, характеризуются различными физико-химическими методиками.
Сохранение первоначального молекулярного веса и отсутствие образования агрегатов оценивается процедурами хроматографической гель-фильтрации (Ju., D. S.. et al., J, Urol. 140, 415, 1988) с одновременным и независимым детектированием антрациклина и антитела при различных длинах волн и с помощью методов гель-электрофореза.
Общее распределение заряда полученных соединений оценивается с помощью хроматографических ионообменных методов.
Концентрация антрациклина оценивается с помощью спектрофотометрического титрования относительно стандартной калибровочной кривой, полученной из исходного антрациклина.
Концентрация белка оценивается с помощью колориметрических анализов, таких как анализ с помощью бицинконовой кислоты (Smith P.K. et al., Anal. Biochim. 150, 76, 1985) или анализ с красителем Брэдфорда (Bradfora, M,M., AnaI. Biochim. 72, 248, 1976).
Сохранение связывающей активности антигена антител после процедур конъюгации оценивается с помощью метода твердофазного иммуноферментного анализа (Yu, D. S. et al., S.Urol. 140, 415, 1988) и с помощью цитофлуориметрических методов (Galiego J. et al., Int. j. Cancar 33, 737, 1984).
Оценка сохранения цитотоксичности конъюгатов по сравнению с исходным лекарственным средством оценивается испытанием ингибирования забора 3H-тимидина с помощью мишеневых клеток, после достаточного длительного времени инкубации для объединения максимального цитотоксического эффекта (DiIImann, R.O. et al, Cancer ReS, 48, 6097, 1988).
Оценка селективной цитотоксичности конъюгатов по отношению к антиген-положительной по сравнению с антиген-отрицательной клеточной линии осуществляется с помощью теста на ингибирование поглощения 3H-тимидина антиген-положительными относительно антиген-отрицательных клеточных линий, после краткого времени инкубации (DiIImann, R.O. et al., Cancer ReS. 48, 6096, 1988). Кислотная чувствительность конъюгата оценивается вышеупомянутыми методами хроматографии после инкубации соединений в соответствующих буферированных растворах.
В качестве альтернативы, для оценки стабильности в плазме используют радиоизотопное мечение конъюгатов в частности, относящейся к антителу (125I), и/или в части, относящейся к антрациклину (14C), и аналитические методы ВЭЖХ.
Терапевтический эффект соединений и улучшение их терапевтической эффективности по сравнению с исходным лекарственным средством оцениваются в животных моделях пересаженных опухолей человека. Голые мыши, несущие ксенотрансплантаты опухолей человека, подвергаются обработке соответствующими дозами конъюгатов, чистого лекарственного средства, антитела и физической смеси лекарственного средства и антитела, при эквивалентных дозах, и рост опухоли регистрируют и сравнивают в различных группах лечения.
Конъюгаты иммуноглобулина готовятся в качестве лекарственного средства в виде фармацевтических составов с фармацевтически приемлемым носителем или разбавителем. Любой соответствующий носитель или разбавитель может быть использован. Пригодные носители и разбавители включают физиологические соляные растворы и растворы декстрозы Рингера.
Конюъгаты согласовано изобретению полезны в качестве противоопухолевых средств. Млекопитающее, например, человек или животное, следовательно, может подвергаться лечению с помощью метода, включающего введение ему фармакологические эффективного количества конъюгата формулы I, как определено выше. Состояние человека или животного может быть улучшено таким образом.
Тонкослойную хроматографию осуществляют с помощью кизель-гелевых планшетов фирмы Мерк F254, с использованием следующих элюирующих систем (объем/объем):
система A: мектиленхлорид:метанол (98:2),
система B: метиленхлорид:метанол:уксусная кислота:вода (80:20:7:3),
система C: метиленхлорид:метанол (95:5),
система D: метиленхлорид:ацетон (4:1),
система E: метиленхлорид:метанол:уксусная кислота (80:20:1),
система F: метиленхлорид:ацетон (9:1),
система G: метиленхлорид:ацетон (95:5),
система H: метиленхлорид:метанол (90:10).
Изобретение дополнительно иллюстрируется прилагаемыми чертежами, на которых:
фиг. 1 представляет собой график, на котором показаны результаты твердофазного иммуноферментного анализа, изложенного в примере 30, следующем ниже, для оценки связывания с клеточной линией меланомы человека иммуноконъюгата рецептора антитрансферрина согласно изобретению I3 (линия X-X-X) и антитела ОКТ9 исходного рецептора противочеловечьего трансферрина (линия  ). В графике оптическое поглощение на 495 нм (ось y) приведено относительно концентрации антитела в нг/мл.
). В графике оптическое поглощение на 495 нм (ось y) приведено относительно концентрации антитела в нг/мл.
На фиг. 2 изображен график, показывающий результаты оценки, изложенной в нижеследующем примере 31, селективной цитотоксичности иммуноконюъгата 13 рецептора антитрасферрина согласно изобретение (линия  ) и исходного чистого лекарственного средства антрициклина (линия
) и исходного чистого лекарственного средства антрициклина (линия  ). В графике (3H) включение тимидина в виде процентного значения для контроля (ось y) приводится относительно концентрации антрациклина в нМ (ось X).
). В графике (3H) включение тимидина в виде процентного значения для контроля (ось y) приводится относительно концентрации антрациклина в нМ (ось X).
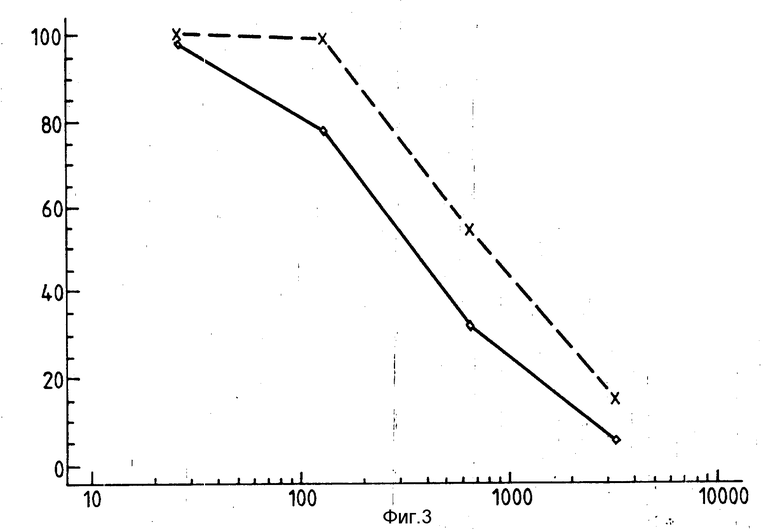
Фиг. 3 представляет собой график, на котором показаны результаты оценки, приведенной в нижеследующем примере 32, ингибирования цитотоксичности конъюгата 13 рецептора антитрансферрина. Линия X-X-X = иммуноконъюгату 13 + антитело ОКТ9, а линия  = иммуноконъюгату 13. Введение (3Н) тимидина в качестве контрольного % (ось y) приводится относительно концентрации антрациклина в нМ.
= иммуноконъюгату 13. Введение (3Н) тимидина в качестве контрольного % (ось y) приводится относительно концентрации антрациклина в нМ.
На фиг. 4 изображено ингибирование в зависимости от дозы цитотоксичности иммуноконъюгата 13 различными количествами избытка неконъюгированного антитела ОКТ9, как описано в нижеследующем примере. На графике включение (3Н) тимидина в виде процентного значения контроля (ось y) приведено относительно избытка свободного антитела (ось x).
На фиг. 5 показаны результаты оценки, приведенные в нижеследующем примере 34, селективной цитотоксичности конъюгата 17 FGF (линия X-X-), не относящегося к делу контроля иммуноконъюгата 14 (линия  ) и свободного антрациклина (линия
) и свободного антрациклина (линия  ). На графике включение (3H) тимидина в виде процентного значения контроля (ось y) приведено относительно концентрации антрациклина в нМ (ось x).
). На графике включение (3H) тимидина в виде процентного значения контроля (ось y) приведено относительно концентрации антрациклина в нМ (ось x).
Пример 1. Получение этил 2-(3,4-дигидро-2H-пиран-2-ил) метилоксиацетата (8B)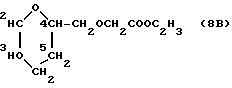
К 2-метанол-3,4-дигидро-2H-пирану (формула M, 4,2 г, 35 ммолей), растворенному в безводном метаноле (100 мл), добавляли едкий натр (1,6 г, 40 ммолей) и слегка нагревали. Затем растворитель удаляли в вакууме и остаток растворяли в диметилсульфоксиде (100 мл). К этому раствору, нагретому до 110oC, добавляли 2-хлор-натрий-ацетат (6 г, 1,5 ммоля) в диметилсульфоксиде (100 мл) в течение 2 ч при энергичном перемешивании. После отстаивания в течение получаса реакционную смесь охлаждали, добавляли триэтиламин (5 мл) и иодистый этил (5 мл) и хранили всю ночь при комнатной температуре. Затем реакционную смесь разбавляли водой и экстрагировали простым этиловым эфиром. После стандартной обработки целевое очищали хроматографическим путем на кремневой кислоте, элюируя со смесью петролейного эфира-простого эфира (80 : 10 объем/объем). Получали 1,68 г соединения 5 в виде масла, выход 26%. Тонкослойная хроматография на плате из Кизельгеля F254 (фирма Мерк) с использованием в качестве элюирующей системы смеси петролейного эфира: простого эфира 1 : 1 по объему. Rf = 0,7.
1H ЯМР (200 МГц, CDCl3) δ : 1,26 (t, J = 7,1 Гц, 3H, CH2CH3); 1,6 - 2,2 (m, 4H, CH2-3, CH2-4); 3,64 (d, J = 5,2 Гц, 2H, CH2O); 4,00 (m, 1H, H-2); 4,13 (m, 2H, OCH2C=O); 4,19 (g, J = 7,1 Гц, 2H, CH2CH3); 4,65 (m, 1H, H-5); 6,35 (ddd, J = 6,2, 2,0, 2,0 Гц, 1H, H-6).
Пример 2.
Получение 4'-эпи-4'-O-(2-карбокситетрагидропиран-6-ил) даунорубицина: X и y изомеры (2''R, 6''R и 2''S, 6''S). 4'c(A): A1-O=3''c(R3 = NH2);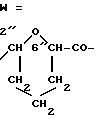
4'-эпи-N-трифторацетилдаунорубицин (3c : R3 = NHCOCF3) (3,1 г, 5 ммолей) растворяли в безводном хлористом метилене (500 мл) и обрабатывали 3,4-дигидро-2H-пиран-2-этоксикарбонилом (8A, 3,4 г, 25 ммолей), полученным согласно описанию в "Macromolecules Vol 12, N 1, с. 5 - 9, янв.-февр. 1979 г., и п-толуолсульфокислотой (100 мг) при комнатной температуре в атмосфере азота. Спустя час хроматографический контроль показал образование двух продуктов, имеющих, соответственно, значение Rf в 0,53 и 0,36 в системе A.
Реакционную смесь промывали водным раствором 5%-ного двууглекислого натрия и воды, затем органическую фазу отделяли, высушивали на безводном сульфате натрия, концентрировали до малого объема при пониженном давлении и хроматографировали на колонке с кремневой кислотой для получения производных 4'-эпи-4'-O-(2-карбоэткоситетрагидропуран-6-ил)-N-трифторацетил даунорубицина от целевого соединения, имеющих соответственно значение Rf в 0,53 (1,5 г, выход 40%) и 0,36 (1,28 г, выход 32%) в системе A.
Соединение Rf = 0,53; FD-MS: м/э 663 (М+).
1H ЯМР (200 МГц, CDCl3) среди прочего δ : 1,31 (COOCH2CH3, J = 7,1 Гц), 1,33 (5'-CH3, J = 6,4 Гц), 2,41 (COCH3), 3,40 (4'-H, J = 9,7 Гц), 4,07 (4-CH3O), 4,55 (6''-H), 4,96 (2''-H0, 5,30 (7-H), 5.38 (1'-H), 7,92 (NHCOOCF3).
Соединение Rf = 0,36,
FD-MS м/э 663 (М+),
1H ЯМР (200 МГ, CDCl3), среди прочего, δ : 1,28 (COOCH2, CH3, J = 7,1 Гц), 1,38 (5'-CH3, J = 6,4 Гц), 2,42 (COCH3), 3,54 (4'-H, J = 8,7 Гц); 4,07 (4-CH3O), 4,42 (6''-H), 5,10 (2''-H), 5,29 (7-H) 5,48 (1'-H), 6,62 (NHCOCF3).
Соединение Rf = 0,53 (1,4 г, 1,79 моля) растворяли водным раствором 0,2 н. гидроокиси натрия в атмосфере азота и хранили при 0oC в течение 8 ч. Затем водный раствор настраивали до pH с помощью водного 1 н. хлористого водорода и экстрагировали повторно хлористым метиленом. Органическую фазу промывали водой, отделяли, сушили на безводном сульфате натрия и выпаривали при пониженном давлении до получения x, одного из двух изомеров (x и y) целевого соединения 4'c (A), (0,9 г, выход 79%).
P = 0,60 (система B), FDMS: м/э 639 (м+).
Соединение Rf = 0,36 (1,1 г, 1,4 ммоля) гидролизовали согласно вышеописанному для получения, после стандартной обработки, другого изомера у целевого соединения 4'c (A), (0,72 г, выход 80%). Rf = 0,45 (система B). FD-MS: м/э 639 (М+).
Пример 3.
Получение 4'-эпи-4'-O-(2-карбокситетрагидропиран-6-ил)-3'- деамино-3'(4-морфолино)даунорубицин: изомер x (один из двух 2''R, 6''R или 2''S, 6''S).
4c(A)(MO)" A-O = 3''c (R3 = MO);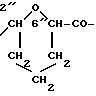
соединение 4'c(A) (x-изомер)(1,2 г, 1,8 ммоля), полученное согласно изложенному в примере 2, растворяли в воде (200 мл), доводили pH до 7,5 с помощью бикарбоната натрия, к нему добавляли 2-оксиэтил-диальдегид (1,3 г), растворенный в воде (20 мл). Спустя 1 ч раствор цианборгидрида натрия (100 мг) в воде (6 мл) был добавлен при перемешивании при комнатной температуре. Спустя 30 мин реакционную смесь доводили до pH 6 с помощью водного раствора 5%-ной уксусной кислоты и экстрагировали с помощью н-бутилового спирта. Органическую фазу промывали водой и растворитель удаляли при пониженном давлении. Остаток очищали хроматографическим путем на колонке кремневой кислоты с использованием системы метиленхлорид:метанол:уксусная кислота (90: 8: 2) по объему) для получения, после стандартной обработки, целевого соединения 4c(A)(MO) (0,6 г, выход 45%).
Rf = 0,70 (система B). FD-MS : м/э 724 (M+).
1H ЯМР (200 МГц, CDCl3), среди прочего δ : 1,29 (5'-CH3, J=6,3 Гц), 2,42 (COCH3), 2,4-2,8 (CH2-N-CH2), 2,98 (3'-H), 3,65 (CH2O-CH2), 3,40 (4'-H, J= 9,0 Гц), 4,08 (4-CH3O), 4,30 (6"-H), 5,00 (2"-H), 5,27 (7-H), 5,51 (1'-H).
Пример 4.
Получение производного N-оксисукцинимидила 4'-эпи-4'- 0-(2-карбокситетрагидропиран-6-ил)-3'-деамино-3'(4-морфолино) даунорубицина: X-изомер (один из двух 2"R, 6"R или 2"S, 6"S). 5c(A)(MO): A-O=3"c (R3=MO);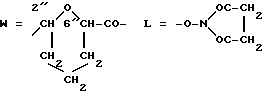
Соединение 4c(A)(MO)(0,55 г, 0,7 ммоля), полученное согласно изложенному в примере 3, растворяли в безводном диметилформамиде (25 мл) и обрабатывали N-оксисукцинимидом (95 мг) и N,N-дициклогексилкарбодинимидом (0,15 г). Смесь хранения при 4oC в течение двух дней, затем растворитель удаляли при пониженном давлении. Остаток собирали небольшим количеством этилацетата, отфильтровывали и растворитель удаляли при пониженном давлении. Эту процедуру выполняли через четыре раза с целью удаления дициклогексилмочевины. И, наконец, остаток обрабатывали простым эфиром, из которого кристаллизовали 0,45 г, выход 72%, чистого и целевого производного 5c(A)(MO).
Rf=0,45 (система B). FD-MS м/э 821 (M+).
1H ЯМР (200 МГц, CDCl3) среди прочего δ : 2,42 (COCH3), 2,4-2,6 (CH2-N-CH2), 2,82 (CO-CH2-CH2-CO), 3,5-3,7 (CH2-O-CH2), 4,08 (4-CH3O), 5,13 (2"-H), 5,28 (7-H, 6"-H), 5,52 (1'-H).
Пример 5.
Получение 6,11,4'-три-O-ацетил-N-трифторацетил-доксорубицина(9).
N-трифторацетил-дооксорубицин (3f: R3= NHCOCF3) (6,4 г, 10 ммолей) суспендировали в безводном метиленхлориде (750 мл) и к нему добавляли триэтилортоформиат (150 мл) и п-толуолсульфокислоту (3 г), при перемешивании при комнатной температуре. Спустя 3 ч хроматографический контроль показал исчезновение исходного материала. После стандартной обработки водой органическую фазу отделяли, сушили на безводном сульфате натрия и отфильтровывали. Растворитель удаляли в вакууме для получения сырого продукта, который растворяли в смеси пиридина (50 мл) и уксусного ангидрида (50 мл) и к нему добавляли небольшое количество диметиламинопиридина (0,5 г). Спустя 2 ч, реакционную смесь выливали в воду и лед (2000 мл). Осадок собирали на спеченном стекле, промывали водой, растворяли тетрагидрофураном (400 мл) и отрабатывали с помощью 0,1 н. водного хлористого водорода (50 мл) всю ночь при комнатной температуре. Затем добавляли хлористый метилен и органическую фазу промывали водой, 5%-ным бикарбонатом натрия и дважды водой. После обычной обработки остаток очищали хроматографическим путем на колонке с кремниевой кислотой с использованием системы растворителя метиленхлорид:ацетон (90:10 по объему) для получения 5 г, выход 65%, целевого соединения 9. Rf=0,18 (система D). FD-MS м/э 765 (M+).
1H ЯМР (200 МГц, CDCl3) δ : 1,20 (d, J=6,4 Гц, H, 5'-CH3), 1,9-2,- (m, 2H, 2'-CH2), 2,1-2,4 (m, 2H, 8-CH2), 2,19, 2,49, 2,53 (S, 9H, COCH3 x 3), 2,95 (bt, J=4,2, 1H, CH2-CH), 3,13 (bm, 2H, 10-CH2), 3,99 (S, 3H, 4-CH3O), 4,20 (q, J=6,6 Гц, 1H, 5'-H), 4,35 (m, 1H, 3'-H), 4,44 (bs, 1H, 9-OH), 4,74 (m, 2H, CH2-OH), 5,1-5,3 (m, 3H, 7-H, 1'-H, 4'-H), 6,40 (bd, J=6,4 Гц, 1H, NH), 7,3-7,8 (m, 3H, 1-H, 2-H, 3-H).
Пример 6.
Получение 14-0-(2-карбокситетрагидропира-6-ил)доксорубицина, изомеров x и y (2"R, 6"R и 2"S, 6"S).
4'f(A):A1-O=3'f (R3=NH2);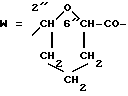
6,11,4'-три-О-ацетил-N-трифторацетилдоксорубицин (10f) (5 г, 6,5 ммолей), полученный согласно описанному в примере 5, растворяли в безводном хлористом метилене (600 мл) и обрабатывали 2-этоксикарбонил-3,4-дигидро-2Н-пираном (8A) (5г, 36 молей) и п-толуолсульфокислотой (500 мг) при комнатной температуре в атмосфере азота. Спустя 2 ч реакционную смесь промывали водным раствором 5%-ного двууглекислого натрия и воды, затем органическую фазу отделяли, сушили на безводном сульфате натрия, концентрировали до малого объема при пониженном давлении и к ней добавляли петролейный эфир. Осадок растворяли в метаноле (500 мл), к нему добавляли морфолин (8 мл) и хранили при комнатной температуре в течение 2 ч. После этого к реакционной смеси добавляли уксусную кислоту (10 мл) и экстрагировали хлористым метиленом. Органическую фазу концентрировали до малого объема и хроматографировали на колонке с кремниевой кислотой, используя, в качестве системы элюирования, смесь хлористого метилена-ацетона (95:5 по объему) для получения N-трифторацетил 14-О-(2-карбонилтетрагидропиран-6-ил)доксорубицина (10) (4,2 г, выход 82%). Rf = 0,41 (система D); FD-MS м/э 795 (M+).
1H ЯМР (200 M, CDCl3) среди прочего δ : 1,3 (5'-CH3, COOCH2CH3, 3,65 (4'-H, J= 2,1 Гц), 4,08 (4-OCH3), 4,21 (COOCH2CH3, J=7,0 Гц), 4,48 (6"-H), 4,88 (9-COCH-O), 5,04 (2"-H), 5,27 (7-H), 5,50 (I'H), 6,69 (NH).
Соединение 10 f растворяли при 0oC с помощью 0,1 н. едкого натра (500 мл) в атмосфере азота. Спустя 1 ч реакционную смесь доводили до pH 6 уксусной кислотой, экстрагировали н-бутанолом и промывали водой. Органическую фазу отделяли и растворитель удаляли в вакууме. Остаток собирали с небольшим количеством хлористого метилена и к нему добавляли простой эфир для получения целевого продукта 4,f(A) (изомеры x, y), 2,9 г (выход 66%).
Rf = 0,5 (система B); FD-MS м/э 671 (M+).
Пример 7.
Получение 14-0-(2-карбокситетрагидропиран-6-ил)-3'-деамино-3'/2(S)-метокси -4-морфолино/доксорубицина: изомеры X и y (2"R, 6"P и 2"S, 6"S).
4f(А)(ММ): А-О=3'f(R3=ММ);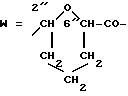
14-0-(2-карбоксиметрагидропиран-6-ил)доксорубицин (4,f(A), 1,3 г, 2 ммоля), полученный как описано в примере 6, растворяли в воде (300 мл), и к нему добавляли 1-метокси-2,2'-оксидиацетальдегид (12) (3,4 г), растворенный в смеси ацетонитрила (20 мл) и воды (20 мл). Смесь доводили до pH 7,5-8,0 с помощью триэтиламина и оставляли в условиях перемешивания при комнатной температуре на 1 ч. Затем добавляли цианборангидрид натрия (0,126 г), растворенный в воде (10 мл), и реакционную смесь хранили при комнатной температуре в течение 15 мин. После этого добавляли ацетон (10 мл) и к реакционной смеси добавляли небольшое количество уксусной кислоты и ее экстрагировали н-бутанолом. Органическую фазу промывали водой и растворитель удаляли в вакууме. Остаток хроматографировали на колонке с кремневой кислотой, используя в качестве элюирующего растворителя смесь хлористого метилена: уксусной кислоты: метанола (100:1:7 по объему). Целевое соединение 4 (А)(ММ) (0,30 г, выход 26%) извлекали в результате обычной обработки.
Rf = 0,8 (система E), FG-MS м/э 771 (М+).
1H ЯМР (200 МГц, CDCL3) среди прочего δ : 1,34 (5'-CH3), 3,39 (CH3-O "морфолино"), 4,07 (4-OCH3), 4,47 (6''-H), 4,58 (CH-OCH3 "морфолино"), 4,85 (9-COCH2-O-), 5,04 (2''-H), 5,27(7-H), 5,59 (1'-H).
Пример 8.
получение N-оксисукцинимидил-производного 14-0-(2-карбокситетрагидропиран-6-мл)-3'-деамино-3'/2(S)-метокси -4-морфолино/доксорубицина: изомеры x и y (2''R, 6''R и 2'S, 6''S).
5f(A)(MM): А-0-3'f (R3=ММ);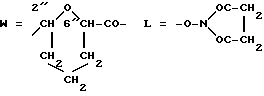
Соединение 4f(А)(ММ)(0,150 мг), полученное согласно изложенному в примере 7, растворяли в безводном диметилформамиде (25 мл) и обрабатывали W-гидроксискцинимидом (95 мг) и N, N'-дициклогексилкарбодиимидом (0,15 г), следуя процедуре, описанной в примере 4. Целевое соединение 5f(A)(MM) получали с выходом 85% (0,15 г).
Rf = 0,20 (система А); FD-MS м/э 868 (M+).
Пример 9.
Получение 14-0-(2-карбоксиметилоксиметил-тетрагидропиран-6-ил) доксорубицина: смесь изомера 2''R, 6''R; 2''S, 6''S; 2''S, 6''R; 2''R, 6''S.
4'f(B): A1-0=3'f (R3=NH2);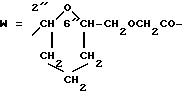
6,11,4'-три-O-ацетил-N-трифторацетил-доксорубицин (9f, 0,65 г, 0,85 ммоля), полученный согласно описанному в примере 5, растворяли в безводном хлористом метилене (50 мл) и обрабатывали соединением 8В (0,7 г, 3,5 ммолей) и безводной п-толуолсульфокислотой (0,045 г) в атмосфере азота при комнатной температуре. Спустя 10 мин добавляли водный насыщенный раствор двууглекислого натрия (20 мл). Органическую фазу отделяли и дважды промывали водой, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Маслянистый остаток растворяли в метаноле (50 мл) и обрабатывали вначале морфолином (1,2 мл) в течение 2 ч, а затем метилатом натрия (0,1 г) в течение 0,5 ч.
После этого реакционную смесь доводили до pH 7,5 с помощью водного 0,1 н. хлористого водорода и экстрагировали хлористым метиленом. После стандартной обработки остаток хроматографировали на кремневой кислоте, используя хлористый метилен в качестве элюирующего агента, для получения 0,4 г (выход 56%) защищенного N-трифторацетильного и (2-этоксикарбонильного) производного целевого продукта. Rf=0,31 (система D).
FD-MS: м/э 839 (76,M+); 639 (84); 201 (100).
0,4 г (0,47 ммоля) вышеописанного N-трифторацетильного 2-этоксикарбонильного производного обрабатывали водным едким натром 0,1H (100 мл) при 0oC в атмосфере азота в течение 90 мин.
После этого раствор доводили до pH 5 уксусной кислотой, экстрагировали водой, насыщенной н-бутанолом, и промывали н-бутанолом, насыщенным водой. Растворитель удаляли при пониженном давлении; остаток собирали смесью хлористого метилена-метанола, и целевой продукт 4'f(B), 0,32 г (выход 84%), осаждали добавлением этилового эфира и собирали на спеченной стеклянной воронке. Rf=0,31 (система B).
FD-MS: м/э 716 (23, М+); 321 (100).
Пример 10.
получение 14-0-(2-карбоксиметилоксиметил-тетрагидропиран-6-ил)-3'-деамино -3'(4-мофолино)доксорубицина: смесь изомеров 2''R, 6''R; 2''S, 6''S; 2''S, 6''R; 2''R, 6''S.
4f(B)(MO): A-o=3'f (R3=MO);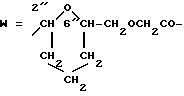
Продукт 4'f (B) (0,3 г) растворяли в сухом диметилформамиде (40 мл) и обрабатывали триэтиламином (0,3 мл) и бис (2-простым иодэтиловым эфиром) (11,2 мл) при 4-5oC.
Спустя 24 ч раствор подкисляли уксусной кислотой и растворитель удаляли при пониженном давлении. Остаток обрабатывали водным 0,1н. едким натром при 0oC в атмосфере азота. Спустя 20 мин раствор подкисляли уксусной кислотой и экстрагировали н-бутанолом, промывали водой. Растворитель удаляли при пониженном давлении и сырой материал очищали на колонке с кремневой кислотой, элюируя смесью хлористого метилена/метанола (90:5 по объему) для получения целевого соединения 4f (B) (MO) (0,05 г, выход 23%). Rf = 0,48 (система B).
FAB-MS: 757 (M+).
Пример 11.
Получение N-оксисукцинимидильного производного 14-0-/2-карбоксиметилоксиметил-тетрагидропиран-6-ил/-3'-деамино-3' (4-морфолино) доксорубицина: смесь изомеров 2"R, 6"R; 2"S, 6"S; 2"S, 2"S; 2"R, 6"S.
5f (B) (MO): A-O=3'f (R3=MO); .
.
Продукт 4f (B) (MO) (0,03 г, 0,037 ммоля) растворяли в сухом диметилформамиде (3 мл) и к нему добавляли N-гидроксисукцинамид (0,01 г) и дициклогиксилкарбодиимид (0,02 г). Смесь хранили при комнатной температуре в течение 8 ч, после этого растворитель удаляли в вакууме и остаток подвергали хроматографии на колонке с крепневой кислотой, используя в качестве элюента хлористый метилен: метанол (95:5), для получения целевого соединения 5f (B) (MO) (0,0278 г, выход 85%). Rf = 0,52 (система H).
FAB-MS: 852 (13, MH+); 200 (100).
Пример 12.
Получение 14-О-(2-карбоксиметилоксиметил-тетрагидропиран-6-ил -3'-деамино-3'/2(S)-метокси-4-морфолиноо/-доксорубицина: смесь изомеров 2"R, 6"R; 2"S, 6"S; 2"S, 2"S; 2"R, 6"S.
4f(B) (ММ): A-O=3'f (R3=ММ); .
.
14-О-(2-карбоксиметилоксиметил-тетрагидропиран-6-ил) доксорубицин (4'f (B), 0,3 г), полученный согласно описанному в примере 9, превращали в целевое соединение 4f (B) (ММ) путем обработки 21-метокси-2,2'-оксидиацетальдегидом и восстановлением натрийцианборгидридом, согласно тому же процессу, описанному в примере 7.
Rf = 0,50 (система B). FAB-MS: 787.
Пример 13.
Получение N-оксисукцинимидильного производного 14-О-/2-карбоксиметилоксиметил-тетрагидропиран-6-ил/-3'-диамино-3'/2 (S)-метокси-4-морфолино)доксорубицина: смесь изомеров 2"R, 6"R; 2"S, 6"S; 2"S, 6"R; 2"R, 6"S.
5f(B) (ММ): A-O=3'f (R3=ММ); .
.
К продукту 4f(B) (ММ), полученному в примере 12 (0,03 г, 0,037 ммоля), растворенному в диметилформамиде (3 мл), добавляли N-гидроксисукцинимид и его превращали в целевое соединение 5f(B) (ММ) (пример 8); Rf = 0,55 (система H). FAB-MS: 882.
Пример 14.
Получение 14-0-(6-пиперазинкарбонилтетрагидропиран-2-ил)-3'-деамино-3 '/2(S)-метокси-4-морфолини/доксорубицина: изомеры x и y(2''R, 6''R; и 2''S, 6''S).
7f(A)(MM): A-0 = 3'f(R3=MM):
N-оксисукцинимицильное производное 14-0-(2-карбокситетрагидропиран-6-ил)-3'-диамино-3'/2 (S)-метокси-4-морфолинил/-доксорубицина 5 (A) (MM), 100 мг/, полученное согласно описанному в примере 8, растворяли в безводном тетрагидрофуране (20 мл), охлаждали при 0oC, и к нему добавляли раствор 1,4-пиперазина 50 мг) в безводном тетрагидрофуране (2 мл). Реакционную смесь перемешивали при 0oC в течение 15 мин, затем разбавляли хлористым метиленом (100 мл), промывали водой ( 3х50 мл). Органическую фазу отделяли, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Сырой продукт подвергали хроматографии на колонке с кремневой кислотой, используя в качестве системы элюирования смесь хлористого метилена/метанола (80/20 по объему). Целевое соединение 7f (A) (MM) (80 мг) осаждали простым этиловым эфиром.
Тонкослойная хроматография на Кизельгелевом планшете (фирма Мерк) F254, система элюирования хлористый метилен/метанол (70/30 по объему), Rf=0,55. FD-MS: Z -средняя молекулярная масса 839.
Примеры 15.
Получение 14-0-(6-гидразинокарбонилтетрагидропиран-2-ил)-3'-деамино-3'/2 (S)- метокси-4-морфолини/доксорубицина: изомеры x и y (2''R, 6''R и 2''S, 6''S). 6f(A)(MM): A-0=3'f,
N-оксисукцинимидильное производное 14-0-(2-карбокситетрагидропиран-6-ил)-3'-деамино-3'/2 (S)-метокси-4-морфолинил/- доксорубицина 4 (A)(MM), 100 мг/, полученное согласно изложенному в примере 8, растворяли в безводном тетрагидрофуране (20 мл), и к нему добавляли IM раствор гидрата гидразина в изопропаноле (5 мл). Смесь хранили при 0oC в течение 1 ч. После этого добавляли хлористый метилен и смесь промывали три раза холодной водой. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли под вакуумом. Остаток очищали на колонке с кремневой кислотой, используя смесь хлористого метилена/метанола (99/1 по объему). Целевое соединение 6f(A)(MM) (95мг) осаждали простым этиловым эфиром.
Тонкослойная хроматография на Кизельгелевой планшете (фирмы Мерк) F254, система элюирования хлористый метилен/метанол (95/5 по объему) Rf= 0,23. FD-MS: Z-средняя молекулярная масса 785.
1H ЯМР (200 МГц, CDCl3: δ 1,36 (d, J=6,6 Гц, 3H, 5'-CH3)$; 1,2-2,2 (m, 9H 3''-CH2, 4''-CH2, 5''-CH2, 2'-CH2, 8ax-H); 2,3-2,7 (m, 6H, 3'-H, 8e-H, NCH2CH2, NCH2CH2O, NCH2-CH(OCH3)O); 2,9-3,4 (m, 2H, 10-CH2); 3,33 (S, 3H, NCH2CH(OCH3)O); 3,55, 3,90 (два m, 2H, NCH2CH2O); 3,68 (m, 1H, 4'-H); 3,95 (m, 1H, 5'-H); 4,08 (s, 3H, 4-OCH3); 4,38 (m, 1H, 6"-H); 4,49 (m, 1H, NCH2CH(OCH3)O); 4,6-4,9 (m, 2H, 14-C2); 4,97 (m, 1H, 2"-; 5,29 (m, 1H, 7-); 5,54 (m, 1H, 1'-H; 7,39 (d, S = 7,6 Гц, 1H, 3-H; 7,59 (bd, S = 6,8 Гц, 1H, CON; 7,78 (t, S = 7,6 Гц, 1H, 2-H); 8,3 (d, S = 7,6 Гц, 1H, 1-H); 13,29 (S, 1H, 11-OH); 13,98 (S, 1H, 6-OH).
Пример 16.
Получение конъюгата 11 против меланомы человека. A-O=3'f (R3=MM;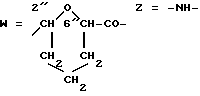 .
.
Раствор 10-2 М соединения 5 f(A)(ММ) (пример 8) в диметилформамиде (38 мкл) добавляли к 1 мл раствора 2 мг/мл очищенного моноклонального антитела EpI против меланомы человека. (P. Giacomini, O. Segatto, P.G. Natali, Int. J. Cancer 39, 1987, 729 в соляном растворе с фосфатным буфером с pH 7,5. Реакционную смесь перемешивали всю ночь при комнатной температуре о и осветляли центрифугированием. Продукт изолировали гельфильтрационной хроматографией на колонне Sephadex G-25 (PD-10, фирма Фармация), элюируя с использованием соляного раствора с фосфатным буфером с pH 7,5. Удаленную пиковую фракцию собирали (2 мл) и анализировали спектрофотометрически на содержание антрациклина при 480 нм. Содержание белка анализировали с помощью колориметрического белкового анализа (BCA, Pierce. Конъюгат 11 содержал 0,98 мг/мл антитела с соотношением антрациклин:белок 8,7:1. Физико-химический профиль продукта определяли гель-фильтрацонным анализом с использованием ВЭЖХ с детектированием на двух волнах (280 и 480 нм) и с помощью электрофореза в полиакриламидном геле с додецилсульфатом натрия. С помощью ВЭЖХ молекулярный вес продукта был примерно 160 кД, а абсорбция антрациклина на 480 мн соответствовала тому же самому молекулярному весу. Доказательство образования ковалентной связи было получено с помощью электрофореза в полиакриламидном геле с додецилсульфатом натрия, где как абсорбция антрациклина на 480 нм, так и реакция белка с красителем Кумаси находится на молекулярном весе в 160 кД.
Пример 17.
Получение конъюгата 12 против карциномы толстой кишки. A-O=3'f (R3=ММ);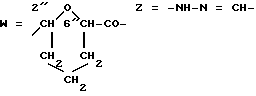 .
.
Раствор антитела B72.3 (патент США 4522918, 1985) с концентрацией 2,6 мг/мл в 0, Ю1М фосфатном буфере, pH (1 мл) обрабатывали с помощью 0,Ю1 мл раствора 0,1М NaIO4 в воде, при 4oC в темноте. Спустя 1 ч продукт очищали гель-фильтрационной хроматографией на колонке Seppphaoex G-25 (PD-10, Фармация), элюируя с 0,1М фосфатным буфером, pH 6. Содержащую белок фракцию (1,7 мг, 23 мл) обрабатывали с помощью 30 молярных эквивалентов раствора 10-2М соединения 6f(A) (ММ). (Пример 15) в диметилформамиде. Спустя 24 ч при 37oC в темноте, реакционную смесь центрифугировали и очищали гель-фильтрационной хроматографией на колонке Sephadex G-25 (PD-10, фармация), элюируя с помощью 0,1M фосфатного буфера, pH 7,3. Содержащую балок пиковую фракцию собирали (2 мл) и анализировали согласно изложенному в примере 16. Конъюгат I2 содержал 0,45 мг/мл антитела с соотношением антрациклин/белок в 1,3/1 и давал аналитический профиль, аналогичный конъюгату, полученному в примере 16.
Пример 18.
Получение конъюгата I3 против рецептора трансферрина. A-O=3'f (R3=MM);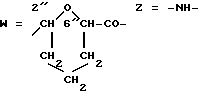
Раствор 10E-2M соединения 5f(A)(MM)(описано в примере 8) в N,N-деметилформамиде (37 мкл) добавляли к 1 мл раствора в количестве 2 мг/мл очищающего моноклонального антитела ОКТ9 мыши против рецептора трансферрина (Sutnerlame), R, Delia, L., Schneider, C, Newman, R, Kemshead, S., Greaves, M. , Proc. Nat. Acad. Sci. USA, 78 (1981), 4515; ATCC GRL 8021) в 0,1M NaH2PO4, 0,1M NaCl, pH 8 в буфере. Реакционную смесь перемешивали с вечера всю ночь при комнатной температуре в темноте и осветляли центрифугированием. Целевой конъюгат I3 изолировали гель-фильтрационной хроматографией на колонке Sephadex G-25 (P-10, Фармация, Кат. N 042-04200M). Выделенную пиковую фракцию собирали и анализировали на содержание антрациклина (спектрофотометрически на 480 нм) и в отношении содержания белка (реагент BCA для анализа белка, P erce, Кат. N 23225). Конъюгат содержал 1,94 мг/мл антитела и 80,9 мкг/мл антрациклина, с молярным соотношением антрациклин:белок, равным 9,5. Аналитический профиль продукта оценивали как в примере 16, с подобными результатами.
Пример 19.
Получение конъюгата I4 против рака толстой кишки. A-O=3'f (R3=MM);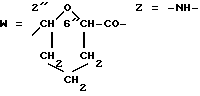
Конъюгат формулы I4, содержащий 0,74 мг/мл белка и 27,5 мкг/мл антрациклина, с соотношением антрациклин/белок, равным 9,1, получали действуя как в примере 18 и используя вместо ОКТ9 раствор антитела B72.2 (2,3 мг/мл)/Schiom, J et al. , патент США 4 522 918 (1985)/ и 43 мкл раствора 5f(A)(MM).
Пример 20.
Получение конъюгата I5 против эпидермального фактора роста. A-O=3'f (R3= MM);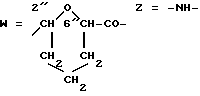
Конъюгат I5, содержащий 0,49 мг/мл белка и 14,3 мкг/мл антрациклина, с соотношением антрациклин/белок, равным 6,9, получали действуя согласно примеру 18 и используя вместо ОКТ9 раствор антитела против эпидермального фактора роста (1,15 мг/мл) (BioMacor, Кат. N 6080, Клон 29,2) и 13 мкл раствора соединения 5f (A) (MM).
Пример 21. Получение конъюгата 16 против рецептора трансферрина. A - 0 = 3'f (R3 = MM); 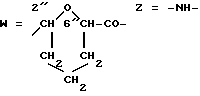
Целевое соединение 16, содержащее 0,34 мг/мл белка и 5,3 мкг/мл антрациклина, с соотношением антрациклин/белок, равным 3,9, получали действуя как в примере 18 и используя вместо ОКТ9 раствор антитела В3/25 против рецептора трансферрина (0,5 мг/мл) (Boehringer, Кат. N 1118 048) и 9,4 мкл раствора 5f (A) (MM).
Пример 22. получение конъюгата 17FGF. A - 0 = 3'f (R3=MM); 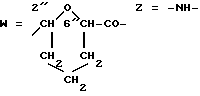
Соединение 17, содержащее 0,77 мг/мл белка, и 117 мкг/мл антрациклина, с соотношением антрациклин/белок, равным 3,4, получался действуя как в примере 18 и используют вместо ОКТ9 раствор рекомбинантного FGF человека (1,35 мг/мл) (Barr et al., J. Biol. Chem. 263 (1988), 16471) и 48 мкл раствора 5f (A) (MM).
Пример 23. получение конъюгата 18 против рецептора трансферрина. A - 0 = 3'f (R3 = MO);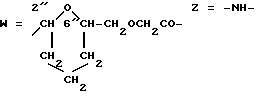
Конъюгат 18, содержащий 0,46 мг/мл белка и 13 мкг/мл антрациклина, с соотношением антрациклина/белок, равным 4,90, получали действуя как в примере 18 и используя 37 мкл раствора 10E-2 M 5f (B) (MO) (пример II).
Пример 24.
получение конъюгата 19 против рака толстой кишки. A - 0 = 3'f (R3 = MO); 
Конъюгат формулы 19, содержащий 0,46 мг/мл белка и 8,38 мкг/мл антрациклина, с соотношением антрациклин: белок, равным 4,5, получали действуя как в примере 18 и используя вместо ОКТ9 антитело B72.3 против рака толстой кишки и 43 мкл 10E-2M раствора 5f (B) (MO) (пример II).
Пример 25.
Получение конъюгата 110 против рецептора трансферрина. A - 0 = 3' f (R3 = MO); 
Соединение 110, содержащее 0,67 мг/мл белка и 20,3 мкг/мл антрациклина, с соотношением антрациклин/белок, равным 7,6, было получено действуя как в примере 18 и используя 37 мкл 10E-2 M раствора 5 f (B) (MM) (пример 13).
Пример 26.
Получение конъюгата 111 против рака толстой кишки. A - 0 = 3' f (R3 = MO);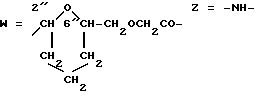
Действуя как в примере 18 и используя вместе ОКТ9 антитело B72,3 против рака толстой кишки и 43 мкл 10E-2 M раствора соединения 5 f (B) (MO), получали целевое соединение 111, содержащее 0,55 мг/мл белка и 7,4 мкг/мл антрациклина, при соотношении антрациклин/белок, равном 3.2.
Пример 27.
Получение конъюгата поли (Glu, Na, Ala, Tyr) (1:1:1) 112. A - 0 = 3' f (R3 = MM);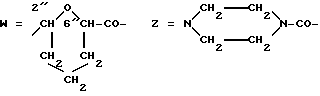
Поли (Glu, Na, Ala, Tyr) (1:1:1), средневесовая молекулярная масса 25000 - 40000 (Sigma), (0,2 г), растворяли в воде (5 мл) при перемешивании при комнатной температуре. Соответствующую свободную кислоту осаждали из водного раствора при pH 3 с помощью 0,1н, HCl. Поли(Glu-ОН, Ala, Tyr) (0,17 г), извлеченный и высушенный под вакуумом, растворяли в сухом диметилформамиде (10 мл) и к нему добавляли 14-0-(6-пиперазинкарбонилтетрагидропиран-2-ил)-3'-деамино -3'/2(S)-метокси-4-морфолини/доксорубицин/7 (A) (MO), 0,035 г/(описано в примере 14) и N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (ЭЭДХ) (0,08 г). Добавляли другую аликвотную пробу ЭЭДХ (0,08 г) после 3 ч. Реакционную смесь перемешивали с вечера всю ночь при комнатной температуре, затем выливали в простой этиловый эфир (300 мл). Осадок суспендировали в воде (10 мл) и обрабатывали 0,1н, NaOH (14 мл); раствор доводили до pH 8,5 с помощью 0,1n, HCl и пропускали на колонке Sephadek G10. Водный раствор лиофилизовали для получения 0,16 г целевого соединения 112.
Содержание антрациклина (объем/объем), рассчитанное как 3'-деамино-3'/2 (S)-метокси-4-морфолинил/доксорубицингидрохлорид: 10%.
Пример 28.
Получение конъюгата поли- L-глутамоновой кислоты 113. A - 0 = 3' f (R3 = MM); 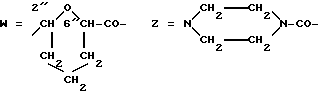
Поли-L-глутамоновую кислоту, средневесовая молекулярная масса 2000 - 15000 (Sigma) (0,1 г) и 14-0-(6-пиперазинкарбонилтетрагидропиран-2-ил) -3'-деамино-3'/2 (S)-метокси-4-морфолинил/доксорубицин/7 f (A) (MM), 0,03 г/ (описано в примере 14) растворяли в безводном диметилформамиде (2 мл) и перемешивали в течение 3 ч. после этого добавляли N-этокси-карбонил-2-этокси-1,2-дигидро-хинолин (ЭЭДХ) (0,03 г). Смесь выдерживали при перемешивании всю ночь, затем выливали в смесь простого этилового эфира и петролейного эфира. Осадок собирали на фильтре из спеченного стекла, промывали простым этиловым эфиром и растворяли в 2,5%-ном водном растворе двууклекислого натрия (8 мл). Раствор пропускали через колонку обратной фазы RP-8, 40-63 мкм (Мерк) (30х1,8 см) и элюировали смесью воды и ацетонитрила. Элюат, содержащий конъюгат, лиофилизовали, затем собирали на фильтре из спеченного стекла, промывали метанолом и простым этиловым эфиром для получения целевого соединения I13 (65 мг). В результате спектроскопического анализа, конъюгат содержит 16% (весовых %) 3'-деамино-3'/2(S)-метокси-4-морфолинил/доксорубицинаидрохлорида.
Пример 29.
Получение конъюгата поли-L-глутамовой кислоты I14. A-O=33'f (R3=MM); .
.
Следуя той же самой процедуры, которая описана в примере 28, поли-L-глутамовую кислоту, средневесовая молекулярная масса 2000-15000 (Sigma) (0,1 г) и 14-0-(6-гидразинокарбонилтетрагидропиран-2-ил)-3' [2(S)-метокси-4-морфолини] доксорубицин [6а f (A)(MM), 0,05 г] (описано в примере 15), вводили в реакцию в диметилформамиде в присутствии ЭЭДХ. После обработки и очистки на RP-8, 40-63 мкм (Мерк) колонке, собирали 50 мг целевого соединения I14.
В результате спектроскопического анализа конъюгата содержит 12% (вес/вес) 3'-деамино-3'/2(S)-метокси-4-морфолинил/ -доксорубицингидрохлорида.
Пример 30.
Оценка связывания клеток конъюгата I3 против рецептора трансферрина на клеточной линии меланомы человека.
Ссылка: Matsui, M. et al., J. Immunology, 141 (1988) 1410 Bumol, T.F. et al, Antibody-Mediated Delivery Systems, Rodwell, J.D. Ed (1988( 55, Harper, J.R. et al., Anal. Biochem 113 (1980) 51.
Клетки меланомы человека М10 (АТТС СР 8021) в среде P PMI 1640 (PBI 12/167B), 10% фетальная телячья сыворотка Jlow Laboratorie 29101-54), 1% глутамина, помещали в 96-ячеечные микротитрационные планшеты (Cost ar 3596) (10000 клеток/ячейку). Спустя 16 ч при 37oC, клетки промывали соляным раствором с фосфатным буфером, 30 фетальной телячьей сыворотки и инкубировали с разными концентрациями конъюгата I3 (пример 18), или антитела OKT9, в соляном растворе с фосфатным буфером, 3% фетальной телячьей сыворотки, 100 мкл, 5-1000 кг/мл в течение 1 ч при 37oC. После трех промывок соляным раствором с фосфатным буфером в каждую ячейку добавляли 100 мкл добавляли 100 мкл свежеприготовленного раствора субстрата o-финилендиаминдигидрохлорида (Sigma P6912) (0,5 мг/мл) и H2O2 (0,015%), в H2O. После инкубирования в 30' при 37oC реакцию останавливали добавлением 25 мкл 4,5 H H2SO4 в каждую ячейку, и поглощение считывали на 495 нм с помощью считывающего устройства Bio Rad EIA, модель 2550. Конъюгат давал хорошее сохранение активности в связывании клеток по сравнению с родственным антителом, как показано на фиг. 2.
Пример 31.
Оценка селективной цитотоксичности конъюгата I3 против рецептора трансферрина.
Ссылка: Di II mag, R.O. al., Cancar R es., 48 (1988) 6097, Ahmad, A. et al., Anticancer Res. 10 (1990) 837.
Клетки меланомы человека M10 (АТСС CRU 8021) помещали в 96-ячеечные планшеты (Cest аг 3596) и 200 мкл RPMI 1640 (PBI 12/167B), содержащем 10% фетальной телячьей сыворотки (Flow Laboratories 29101-54), 1% глутамин, (10000 клеток/ячейку) при 37oC. Спустя 18 ч культуральную среду удаляли и добавляли 100 мкл свежей среды. Добавляли по три ячейки пятьдесят микролитров различных концентраций конъюгата 13 и относительных контрольных образцов в соляном растворе с фосфатным буфером, 3% фетальной телячьей сыворотки. Планшеты инкубировали в течение 24 ч при 37oC, затем добавляли 0,8 микроКюри/ячейку (3H)тимидина (NEN DuPont NET 355) в 20 мкл среды и инкубирование продолжали при 37oC в течение 7 ч. Клетки собирали с помощью харвестера клеток Multi Mash 2000 фирмы Дайнатэк на бумажном фильтре (Whatman 1827842), фильтры сушили на воздухе, помещали в сосуды со сцинтилляционной жидкостью (Kontron Analytical Supertron 56920-04010) и подсчитывали в сцинтилляционном счетчике (Коп гоп Instrument Betamatic).
Все конъюгаты были существенно менее цитоксичны, чем исходное лекарство, не специфический конъюгат против рецептора трансферрина давал эффект, в пять раз превышающий не относящийся к нему контрольный конъюгат, как показывает сравнение между IC50 обоих соединений.
Результаты типичного эксперимента показаны на фиг.2.
Пример 32.
Ингибирование цитотоксичности иммуноконъюгата 13 против рецептора трансферрина с помощью неконъюгированного антитела.
Ссылка: Chacendhary, K.K. et al., Nature 339 (1989) 394, Batra, J.K. et al. : Mol. and Cell Bid II (1991) 2200, Siegal, C.B. et al., J. Bid. Chem. 265 (1990) 16313.
Клетки меланомы человека M10 (АТСС CRL 8021) культивировали в 96-ячеечных планшетах (Costar 3596) и 200 мкл RPMI 1640 (PBI 12/167B), содержащем 10% фетальной телячьей сыворотки (Flow Laboratories 29101-54), 1% глутамина (10000 клеток на ячейку) при 37oC. Спустя 18 ч культуральную среду удаляли и добавляли 50 мкл свежей среды. Затем в строенные ячейки добавляли 50 мкл раствора антитела ОКТ 9 в полной культуральной среде (конечная концентрация в ячейках от 7000 до 56 мкл/мл). Контрольные ячейки обрабатывали с помощью 50 мкл полной культуральной среды. Планшеты инкубировали в течение 1 ч при 4oC, затем добавляли 50 мкл различных концентраций конъюгата I3, в полной культуральной среде (конечная концентрация антитела в ячейках от 70 до 0,56 мкл/мл, что соответствует 1:100 молярному избытку свободного антитела, добавляемого в предшествующей стадии). Спустя 24 ч при 37oC добавляли 0,8 микроКюри/ячейку (3H)тимидина (NEN DuPont NET 355) в 20 мкл среды, и инкубирование продолжали при 37oC в течение 7 ч. Клетки собирали и оценивали включение радиоактивности как в примере 31.
Как показано на фиг.3, добавление свободного антитела ингибирует цитотоксичность иммуноконъюгата в диапазоне доз содержания антрациклина, подтверждая таким образом опосредованный эффект на рецептор описываемых здесь соединений.
Результаты из типичного эксперимента показаны на фиг.3.
Пример 33.
Ингибирование в зависимости от величины дозы цитотоксичности иммуноконъюгата против рецептора трансферрина I3 с помощью неконъюгированного антитела.
Ссылка: Chaundhary, v.K. et al., Nature 339 (1989) 394, Batra, J.K. et al. : Mol. and Cell Bid II (1991) 2200, Siegal, C.B. et al., J.. Biol. 265 (1990) 16318.
Клетки M10 культивировали и инкубировали в течение 18 ч, как описано в примере 32. После этого культуральную среду удаляли и добавляли 50 мкл свежей среды. Затем в тройные ячейки добавляли 50 мкл раствора антитела ОКТО в полной культуральной среде (конечная концентрация в ячейках от 2120 до 211,2 мкг/мл). Концентрацию ячейки обрабатывали с помощью полной культуральной среды. Планшеты инкубировали в течение 1 ч при 4oC. Затем добавляли 50 мкл конъюгата I3 в полной культуральной среде (конечная концентрация антитела в ячейках 2,12 мкг/мл). Ячейки инкубировали, собирали урожай и оценивали включение радиоактивности как в примере 31.
Как показано на фиг.4, ингибирование в зависимости от дозы цитотоксичности конъюгата осуществляется предварительной обработке клеток различными избытками неконъюгированного антитела, что подтверждает цитотоксичность через посредство рецептора этих
Пример 34.
Оценка селективной цитотоксичности конъюгата I7 FGF.
Действуя как в примере 31 и используя конъюгат формулы I7, показывали селективную цитотоксичноть конъюгата FGF (фиг.5).
Пример 35.
Используя протокол, описанный в примере 31 и различные сочетания антрациклинов, линкеров и носителей, обеспечивающих направленную доставку препарата, получали различные конъюгаты, и их цитотоксичность проверяли на различных клеточных линиях, используя не связывающие конъюгаты в качестве контролей. Как показано в табл.1, специфические конъюгаты существенно более эффективны при ингибировании роста опухолевых клеток по сравнению с контрольными, не связывающими, конъюгатами. Соотношение между IC50 может быть взято в качестве показателя селективности соединений. Таким образом, от 7 до 38% специфического конъюгата имеет такую же цитотоксичность, что и 100% доза контрольного конъюгата. Отмеченные величины селективности были подтверждены экспериментами, проведенными с различными антрациклинами, линкерами, мишеневыми клетками и для различных длительностей инкубирования.
Пример 36.
Оценка соединения I14.
Соединение I14 изучали "in vivo" против лейкемий мыши P388, стойких к доксорубицину, в сравнении с 3'-деамино-3'-/2(s)-метокси-4-морфолино/доксорубицином (ДММ) и доксорубицином. Данные приведены в табл.2.
Конъюгат показал подобную противоопухолевую активность, но при более высоких дозах, свободного лекарственного средства.
| название | год | авторы | номер документа |
|---|---|---|---|
| СВЯЗУЮЩИЙ АГЕНТ ДЛЯ БИОАКТИВНЫХ ПРЕПАРАТОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2116087C1 |
| АНТРАЦИКЛИНОВЫЕ ГЛИКОЗИДЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ДИИОДОПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1991 |
|
RU2100366C1 |
| ПРОИЗВОДНЫЕ 3'-АЗИРИДИНО-АНТРАЦИКЛИНА, СПОСОБЫ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2149163C1 |
| N-ФЕНИЛАЛКИЛАЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ α -АМИНОКАРБОКСАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАКОЛОГИЧЕСКИ АКТИВНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПРЕДУПРЕЖДЕНИЯ РАЗВИТИЯ БОЛЕЗНИ | 1992 |
|
RU2097371C1 |
| АКРИЛОИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ПИРРОЛА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2094430C1 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНА | 1995 |
|
RU2159619C2 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРСВЯЗАННЫЕ АНТРАЦИКЛИНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2145965C1 |
| АНТРАЦИКЛИН ГЛИКОЗИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2081878C1 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНОНА И ИХ ИСПОЛЬЗОВАНИЕ ПРИ АМИЛОИДОЗЕ | 1995 |
|
RU2167661C2 |
| АНТРАЦИКЛИНОВЫЙ ГЛИКОЗИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2118328C1 |




Использование: в медицине, а именно в терапии. Сущность изобретения: конъюгат представляет собой соединение формулы (1) [A-O-W-Z]a - T, в которой часть A-O представляет собой остаток любого антрациклина формулы A-O-H, несущего, по крайней мере, одну первичную или вторичную гидроксильную группу; а - обозначает целое число от 1 до 30; N - остаток формулы (2)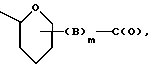
в которой B обозначает C1 - C6 алкиленовую группу, возможно разорванную, m равно 0 или 1; Z обозначает спейсерную группу, а T - часть носителя. Предложенные конъюгаты могут найти применение в качестве противоопухолевых средств. 12 с. и 13 з.п.ф-лы, 2 табл., 5 ил.
(A - O - W - Z)a - T (1),
в которой часть A - O - представляет собой остаток любого антрациклина формулы A - O - H, несущего, по крайней мере, одну первичную или вторичную гидроксильную группу; а обозначает целое число от 1 до 30; W обозначает остаток формулы (2)
в которой B обозначает C1 - C6 алкиленовую группу, возможно, в данном случае прерванную гетероатомом, m означает 0 и 1; Z обозначает спейсерную группу, а T - часть носителя.
NH-(D)d-N = CH- ,
где d означает целое число от 0 до 2, (D) означает -NH - CO(CH2)nCO - NH (n равно 2 или 4), а T - Z содержит остаток носителя формулы T - CHO, несущий по крайней мере одну формильную группу или T(COOH)a, где a имеет значение, приведенное в п.1, или Z означает пиперазилкарбонильную часть или группу формулы
NH - (D)d - NH - CO-,
в которой (D) и d имеют вышеуказанные значения, а T - Z содержит остаток носителя формулы T - (COOH)a, как он определен выше.
в которой R1 обозначает атом водорода, окси- или метоксигруппу; R2 обозначает оксигруппу и R4 или R5 обозначают атом водорода, а другое из R4 и R5 обозначает оксигруппу; R4 обозначает атом йода и R5 обозначает атом водорода, или оба R4 и R5 обозначают атомы водорода или R2 обозначает атом водорода и R4 или R5 обозначают оксигруппу, а другое из R4 или R5 обозначают атом водорода; R3 обозначает аминогруппу или обозначает атом азота, включенный в кольцо морфолино.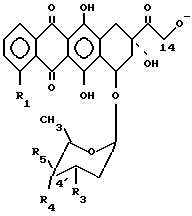
в которой R1 и R3 имеют значения, определенные в п.3, а R4 или R5 обозначают атом водорода, а другое из R4 и R5 обозначают оксигруппу, или R4 обозначает атом водорода или йода, а R5 - атом водорода.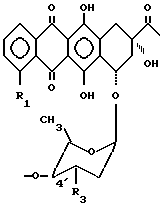
в которой R1 и R3 имеют указанные в п.2 значения.
CH2 - O - CH2-, -CH2 - NH - CH2, -CH2-, -C3H6- или C6H1 2-.
A' - O - W - OH (4)
в которой A' - O - обозначает остаток антрациклина, несущий, по крайней мере, одну первичную или вторичную гидроксильную группу и имеющий аминогруппу части сахара, защищенную или замещенную морфолиногруппой, а имеет значение, приведенное в п.1, в активированное производное формулы (5)
A' - O - W - L,
в которой A' - O- и W имеют вышеуказанные значения, а L обозначает активирующую группу для создания амидной связи; и конденсирование полученных соединений формулы (5), как определено выше, с соединением формулы T - H2 по п.2, или введение в реакцию активированного соединения формулы (5) с производными формулы NH2 - (D)d - NH2, в которой D и d имеют значения, приведенные в п. 2, возможно со снятием защитной группы с полученного производного формулы (6)
A' - O - W - NH - (D)d - NH2 (6),
в которой A' - O-, W, d и (D) имеют значения, указанные в п.2, и его конденсирование с соединением формулы T - CHO или T - (COOH)a, как определено в п.2, или введение в реакцию соединения (5) с 1,4-пиперазином и конденсирование полученного соединения формулы (7)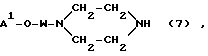
в которой A' - O- и W имеют вышеуказанные значения, с соединением формулы T(COOH)a, как определено выше, возможно в присутствии агента конденсации, для получения конъюгата формулы (1), в котором непрореагировавшие, возможно присутствующие активированные карбоксильные группы, могут быть блокированы с фармацевтически приемлемым амином.
4'-эпи-4'-О-(2-карбокситетрагидропиран- 6-ил)-3'-деамино-3'-(4-морфолино)-даунорубицина;
14-О-(2-карбокситетрагидропиран-6-ил)-3'-деамино-3'- (2-метокси-4-морфолино)-доксорубицина; и
14-О-(2-карбокситетрагидропиран-6-ил)-3'-деамино-3'- (2-метокси-4-морфолино)-доксорубицина.
в которой B и m имеют значения, указанные в п.1, а Rx обозначает защитную группу, удаление каждой защитной группы с полученного промежуточного соединения для получения таким образом производного формулы (4) A - O - W - OH (4), в которой A - O- и W имеют значения, указанные в п.1, с условием, что аминогруппа части сахара остатка антрациклина A - O- представлена в форме свободной аминогруппы и превращение свободной аминогруппы части сахара антрациклина формулы (4) в производную морфолино, или защиту указанной свободной аминогруппы указанного антрациклина формулы (4).
4'-эпи-4'-О-(2- карбокситетрагидропиран-6-ил)-даунорубицина;
14-О-(2-карбокситетрагидропиран-6-ил)-доксорубицина и
14-О-(2-карбоксиметилоксиметил-тетрагидропиран-6-ил)-доксорубицина.
4'-эпи-4'-О-(2-сукцинимидокарбонил- тетрагидропиран-6-ил)-3'-деамино-3'-(4-морфолино)-даунорубицина;
14-О-(2-сукцинимидокарбонил-тетрагидропиран-6-ил)-3'-деамино-3'-(2- метокси-4-морфолино)-доксорубицина; и
14-О-(2-сукцинимидокарбонилметилоксиметил-тетрагидропиран-6-ил)-3'-деамино- 3'-(4-морфолино)-доксорубицина.
2-этоксикарбонил-3,4-дигидро-2Н-пирана;
этил-2-(3,4-дигидро-2Н-пиран-2-ил)метилоксиацетата;
метил-2-(3,4-дигидро-2Н-пиран-2-ил)метилоксиацетата;
этил- 2-(3,4-дигидро-2Н-пиран-2-ил)метиламиноацетата.
| EP, А, 0014853, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |

