Изобретение относится к органической химии, а именно к усовершенствованному способу получения 3-алкоксикарбонил-4-гидрокси-2-метил-2H-1,2-бензотиазин-1,1- диоксидов формулы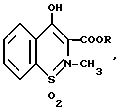
где
R - CH3, C2H5.
Эти соединения используются в качестве интермедиатов в синтезе высокоэффективных нестероидных противовоспалительных препаратов класса "оксикамов" (пироксикам, изоксикам, судоксикам и др.). Они могут также применяться как УФ-сорбенты для стабилизации полимеров.
Известен способ получения соединения I (R=CH3) взаимодействием N-метилсахарина с 2 - 2,5 моль метилового эфира монохлоруксусной кислоты в среде диметилформамида или диметилсульфоксида с последующей обработкой интермедиата (без выделения) 3,5 - 4,5 моль метилата натрия при 40 - 50oC. Общая продолжительность процесса 6 ч. В этих условиях выход сырого продукта составляет 28% [1].
Недостатками этого способа являются: использование более, чем 100%-ного избытка галогенацетата; низкий выход целевого продукта; применение в качестве исходного продукта N-метилсахарина, который получают метилированием сахарина диазометаном или окислением 2-метил-1,2-бензизотиазолтиона перекисью водорода в ледяной уксусной кислоте.
Известен трехстадийный способ получения этил-4-гидрокси-2- метил-2H-1,2-бензотиазин-3-карбоксилат-1,1-диоксида, заключающийся во взаимодействии сахаринилнатрия с этиловым эфиром монохлоруксусной кислоты в среде диметилформамида в течение 1 ч при 130oC с выделением этилового эфира 3-оксо-1,2-бензотиазолин-2-уксусной кислоты 1,1-диоксида (II) с выходом 82%, изомеризации последнего под действием 2,5 моль этилата натрия в среде этанола при 57 - 62oC (2 часа) с последующим разложением натриевой соли и выделением соединения (III) экстракцией хлористым метиленом и растиранием в бензоле, метилированием продукта (III) иодистым метилом в диметилформамиде, содержащем метилат натрия, с последующей перекристаллизацией. Этил-4-гидрокси-2-метил-2H-1,2-бензотиазин-3- карбоксилат-1,1-диоксид (1, R = C2H5) получают с выходом 71%. В расчете на исходный сахаринат натрия выход целевого продукта 40,2% [2].
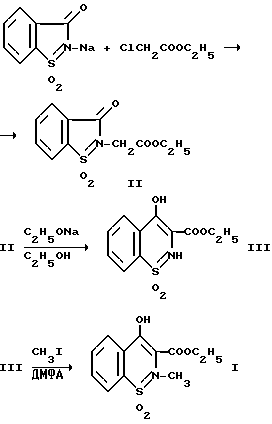
Недостатками способа, являющегося прототипом предлагаемого изобретения, являются: многостадийность; низкий выход конечного продукта в расчете на исходный сахаринат натрия (40,2%); использование сахарината натрия, синтезируемого взаимодействием сахарина с алкоксидами натрия; большой расход хлористого метилена в качестве экстрагента (5 л на 1 моль продукта) на стадии выделения интермедиата (III); длительность процесса: дополнительный расход времени необходим также на выделение двух интермедиатов (разложение, экстракция, сушка).
Технической задачей изобретения является упрощение технологии процесса и увеличение выхода целевого продукта.
Поставленная задача решается тем, что в качестве исходного продукта используют сахарин и целевой продукт получают без выделения интермедиатов.
Реакция осуществляется путем взаимодействия сахарина с алкиловыми эфирами монохлоруксусной кислоты в присутствии эквимольного количества безводного поташа в среде диметилформамида при 120oC, изомеризации промежуточного продукта и алкилирования иодистым метилом. Выход целевого продукта 70 - 73%. Процесс ведут в одном реакторе.
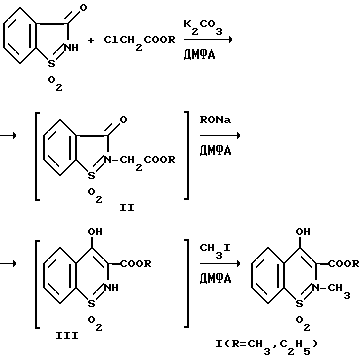
Общая продолжительность процесса превращения сахарина в продукт (1) составляет 3 - 4 ч.
Использование диметилсульфата и метилового эфира бензолсульфокислоты позволяет получить из сахарина целевой продукт с достаточно высоким выходом (65 и 62 соответственно).
Для увеличения выхода целевого продукта был проведен поиск дегидрогалогенирующего реагента в реакции сахарина с эфирами монохлоруксусной кислоты. В присутствии оснований (триэтиламин, пиридин, этилат и ацетат натрия, гидроксид калия, углекислый натрий) выходы целевого продукта не превышают 30%. Оптимальным дегидрогалогенирующим агентом является углекислый калий (в эквимольном соотношении), позволяющий получить целевой продукт (1) с выходом 73%.
Предлагаемый способ получения соединения (1) имеет следующие преимущества перед прототипом: одностадийность способа; использование сахарина вместо сахарината натрия в качестве исходного соединения; экономичность процесса; значительное сокращение продолжительности процесса; увеличение выхода целевого продукта (73% по сравнению с 40,2% - прототип).
Пример 1. Этил-4-гидрокси-2-метил-2H-1,2-бензотиазин-3- карбоксилат-1,1-диоксид (1, R = C2H5).
В четырехгорловую круглодонную колбу, снабженную механической мешалкой, обратным холодильником, термометром и капельной воронкой, загружают 30 г (0,16 моль) сахарина, 1,3 г (0,08 моль) безводного углекислого калия, 20 г (0,16 моль) 2-этил-1-хлорацетата в 250 мл диметилформамида. Реакционную смесь нагревают при перемешивании при 120oC в течение 1 ч. Реакционную смесь охлаждают до 25oC и быстро добавляют горячий раствор (50 - 60oC) этилата натрия, полученного из 11,3 г (0,49 моль) металлического натрия и 200 мл этанола, в 75 мл диметилформамида. Реакционная смесь разогревается до 50 - 60oC и при этой температуре смесь выдерживают в течение 1 ч. Добавление этилата натрия сопровождается появлением красного окрашивания, которое через 10 - 15 мин переходит в ярко-оранжевое, и выпадением осадка. Реакционную смесь охлаждают до 15oC и медленно добавляют 69,8 г (0,49 моль) иодистого метила. Оранжевая окраска переходит в бледно-желтую, и выпадает обильный осадок. Реакционную смесь перемешивают в течение 1 ч при 30oC и выливают в смесь льда и 17 мл конц. HCl, выпавший осадок отфильтровывают, промывают водой, высушивают. Получают 33 г (73%) продукта (1) с т.пл. 135,5 - 138oC. Литер. данные: т.пл. 136 - 138oC /C.R.Rasmussen, J. Org. Chem. 39, 1554, (1974)3.
Найдено, %: C 50,80; H 4,71; N 4,80; S 11,50.
C12H13NO5S.
Вычислено, %: C 50,87; H 4,63; N 4,94; S 11,32.
ИК-спектр ( ν, , см-1, KBr): 1640, 1605, 1565. Спектр ПМР (CDCl3, δ м.д. ): 1,4 (CH3), 2,94 (N-CH3), 4,43 (-O-CH2-), 7,45 (C6H4), 12,11 (OH).
Пример 2. К смеси 18,3 г (0,1 моль) сахарина, 6,9 г (0,05 моль) K2CO3 и 150 мл диметилформамида быстро приливают 12,3 г (0,1 моль) этилового эфира монохлоруксусной кислоты. Нагревают 1 ч при 120oC. Реакционную смесь охлаждают до 25oC и приливают к ней горячий раствор (50 - 60oC) этилата натрия, полученного из 6,9 г (0,3 моль) металлического натрия и 120 мл этанола, в 50 мл диметилформамида. Наблюдается саморазогревание реакционной массы до 50oC и при этой температуре реакционную смесь выдерживают в течение 1 ч. Смесь охлаждают до 15oC и прикапывают 25,2 (0,2 моль) диметилсульфата. Затем при 30oC смесь выдерживают в течение 1 ч. Раствор обрабатывают смесью льда с 12,5 мл конц. HCl. Выпавший осадок отфильтровывают, промывают водой. После высушивания получают 18,4 г (65%) этил-4-гидрокси-2-метил-2H-1,2-бензотиазин-3-карбоксилат-1,1- диоксида.
Пример 3. Смесь из 5 г (0,027 моль) сахарина, 1,9 г (0,013 моль) поташа, 3,4 г (0,027 моль) этилового эфира монохлоруксусной кислоты и 50 мл диметилформамида нагревают 1 ч при 120oC. К реакционной смеси, охлажденной до 25oC, быстро приливают горячий раствор (50 - 60oС) этилата натрия, полученного из 1,9 г (0,08 моль) металлического натрия и 8 мл этанола. Выдерживают реакционную смесь при 50 - 60oС в течение 1 ч. Смесь охлаждают до 15oC и прикапывают 17,2 г (0,2 моль) метилового эфира бензолсульфокислоты. Нагревают реакционную смесь 2 ч при 30oC. После обработки, аналогичной примеру 1, получают 4,7 г (62%) этил-4-гидрокси-2- метил-2H-1,2-бензотиазин-3-карбоксилат-1,1-диоксида.
Пример 4. Метил-4-гидрокси-2-метил-2H-1,2-бензотиазин-3- карбоксилат-1,1-диоксид (1, R=CH3).
Смесь 15 г (0,08 моль) сахарина, 5,65 г (0,04 моль) поташа, 8,7 г (0,08 моль) 2-метил-1-хлорацетата и 80 мл диметилформамида нагревают 1 ч при 120oC, охлаждают до 25oC и быстро приливают горячий раствор (50 - 60oС) метилата натрия, полученного из 5,7 г (0,25 моль) металлического натрия и 50 мл метанола, в 40 мл диметилформамида. Температура реакционной смеси повысилась до 50oC. Смесь нагревают в течение 1 ч при 50 - 60oC, охлаждают до 15oC и медленно прибавляют 34,8 г (0,25 моль) иодистого метила. Перемешивают 1 ч при 30oC. После обычной обработки получают 15 г (70%) метил-4-гидрокси-2-метил-2H-1,2-бензотиазин- 3-карбоксилат-1,1-диоксида с т.пл. 165 - 165,5oC. Лит. данные: т. пл. 163 - 165oC (пат. США N 4024136, кл. C 07 D 279/02, 1977).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ N-(2- ПИРИДИЛ) -2- МЕТИЛ-4- ГИДРОКСИ-2Н -1,2- БЕНЗОТИАЗИН -3- КАРБОКСАМИД -1,1- ДИОКСИДА | 1990 |
|
SU1764296A1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-МЕТИЛ-5-ГЕПТЕН-2-ОНА | 1995 |
|
RU2078075C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРАЗИНОВ, СОДЕРЖАЩИХ О-ГИДРОКСИФЕНИЛЬНУЮ ГРУППУ | 1991 |
|
RU2026295C1 |
| СПОСОБ ПОЛУЧЕНИЯ КУБОВЫХ КРАСИТЕЛЕЙ И ПИГМЕНТОВ, СОДЕРЖАЩИХ ПЕРИЛЕНОВЫЙ ФРАГМЕНТ | 1997 |
|
RU2128200C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(ХЛОРМЕТИЛ)СИЛАТРАНА | 1992 |
|
RU2096412C1 |
| ПРОИЗВОДНЫЕ 1-МЕТИЛ-5-ХЛОРПИРАЗОЛА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2186772C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,2-БИС-(3,5-ДИБРОМ-4-ГИДРОКСИФЕНИЛ)ПРОПАНА | 1992 |
|
RU2034823C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАМЕТИЛСИЛАНА | 1993 |
|
RU2044739C1 |
| ГИПОХОЛЕСТЕРИНЕМИЧЕСКОЕ СРЕДСТВО | 1992 |
|
RU2034540C1 |
| Способ получения 2,2-диалкил-1,3-диокса-6-(2-оксиалкил)-6-аза-2-силациклооктанов | 1983 |
|
SU1143748A1 |
Использование: в химии гетероциклических веществ, в частности в способе получения промежуточных нестероидных веществ для синтеза противовоспалительных препаратов. Сущность изобретения: продукт - 3-алкоксикарбонил-4-окси-2-метил - 2Н-1,2-бензотиазин-1,1-диоксиды. Выход 73%. Реагент 1: сахарин. Реагент 2: эфир монохлоруксусной кислоты. Условия реакции: в среде диметилформамида при повышенной температуре в присутствии углекислого калия при эквимолекулярном соотношении реагентов. Полученную реакционную смесь непосредственно обрабатывают алкоголятом натрия и затем метилирующим агентом. 1 з.п. ф-лы.
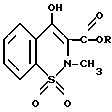
где R - метил или этил,
из эфира монохлоруксусной кислоты в среде диметилформамида при повышенной температуре с последующей изомеризацией под действием алкоголята натрия и N-метилированием продукта изомеризации, отличающийся тем, что эфир монохлоруксусной кислоты подвергают взаимодействию с эквимолярным количеством сахарина в присутствии углекислого калия, полученную реакционную массу непосредственно обрабатывают алкоголятом натрия и затем метилирующим агентом.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Патент США N 4483982, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| C.R.Rosmussen | |||
| J | |||
| Org | |||
| Chem | |||
| ПРИБОР ДЛЯ ЗАПИСИ И ВОСПРОИЗВЕДЕНИЯ ЗВУКОВ | 1923 |
|
SU1974A1 |
| Машина для изготовления проволочных гвоздей | 1922 |
|
SU39A1 |

