По данной заявке заявляется приоритет на основании заявки США N 60/007283, поданной 6 ноября 1995 г. , и данная заявка является частичным продолжением данной ранней заявки.
В международной PCT заявке WO 91 13872 раскрываются производные диоксино [2,3-e] индола следующей формулы, в которой R1 представляет H, алкил, CO2R2, CONHR2, циано, галоид, CHO и др. , R2 представляет H, алкил, (CH2)mY;
Y представляет циклоалкил или циклоалкенил, (замещенный) фенил, пиридил, нафтил, индолил; m представляет от 0 до 6; A и B представляют O, CH2, S; и X представляет CH2(CH2)mNR2R2, в качестве серотонергических и допаминергических агентов, полезных для лечения ЦНС и сердечно-сосудистых нарушений.
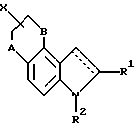
Патент США 5318988 раскрывает 2-аминометил-хроманы следующей ниже формулы, полезных для лечения заболеваний центральной нервной системы. В данной группе соединений A, B и D являются идентичными или различными и представляют водород, галоген, циано, азидо, нитро, ди- или трифторметил, ди- или трифторметокси, гидроксил или карбоксил, с прямой или разветвленной цепью алкил, алкенил, ацил, алкокси или алкоксикарбонил, или моно- или дизамещенный или незамещенный амино, амидо или сульфонамидо, или A может быть определен, как указано, а B и D, взятые вместе, образуют 5 - 7-членное насыщенное, частично ненасыщенное, или ароматическое карбоциклическое кольцо, или гетероциклическое кольцо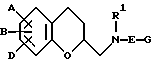
имеющее вплоть до двух атомов серы, азота или кислорода, не обязательно одну или две карбонильные функциональные группы в кольце и необязательно кольцо, замещенное алкилом, разветвленным алкилом или циклоалкилом; E представляет прямую связь или представляет с прямой или разветвленной цепью алкилен, алкенилен или алкинилен; G представляет арил, имеющий 6 - 10 атомов углерода, или 5 - 7-членное, насыщенное или ненасыщенное гетероциклическое кольцо, которое не связано с помощью азота и имеет вплоть до 3 гетероатомов из ряда, включающего атомы азота, кислорода или серы, с которым может быть также необязательно сконденсировано дополнительное насыщенное, частично ненасыщенное или ароматическое 6-членное кольцо или циклоалкил или мостиковое бикарбоциклическое кольцо.
В патентах США N N 5126366, 5166367, 5189171, 5235055 и N 5245051 описывается ряд антипсихотических агентов формулы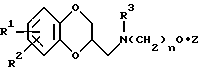
в которой Z представляет аминофенокси, кумарин, карбостирил, хинолин или хроман; R1 и R2 представляют независимо водород, алкил, алкокси, аралкокси, алканоилокси, гидрокси, галоид, амино, моно- или диалкиламино, алканамидо, или алкансульфонамидо, или R1 и R2 вместе представляют метилендиокси, этилендиокси или пропилендиокси; R3 представляет водород или алкил; n = 2, 3 или 4.
В соответствии с данным изобретением предоставляется группа новых антипсихотических агентов формулы I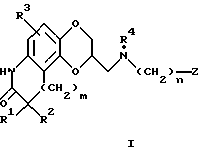
в которой R1 и R2 представляют независимо водород, алкил с 1-6 атомами углерода, фенил или бензил; или R1 и R2, взятые вместе, представляют бензилиден, необязательно замещенный группой R3, определенной ниже, или алкилиден с 1-6 атомами углерода, или R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют карбонильный фрагмент или циклоалкильную) группу, имеющую 3 - 6 атомов углерода;
R3 представляет водород, гидрокси, галоид, трифторме-тил, трифторметокси, алкил с 1-6 атомами углерода, алкокси с 1-6 атомами углерода, арилалкокси с 7-12 атомами углерода, алканоилокси с 2-6 атомами углерода, амино, моно- или диалкиламино, в котором каждая алкильная группа имеет 1-6 атомов углерода, алканоамидо с 2-6 атомами углерода или алкансульфонамидо с 1-6 атомами углерода;
R4 представляет водород или алкил с 1-6 атомами углерода;
m = 0, 1 или 2;
n = 0, 1, 2, 3, 4, 5 или 6;
Z представляет водород, гидрокси, алкил с 1-6 атомами углерода, алкенил с 2-6 атомами углерода, алкинил с 2-6 атомами углерода, алкокси с 1-6 атомами углерода, циклоалкил с 3-8 атомами углерода, полициклический алкил с 7-15 атомами углерода, фенил, необязательно замещенной группой R3, определенной выше, фенокси, необязательно замещенный R3, определенным выше, нафтил, необязательно замещенный R3, определенным выше, или нафтилокси, необязательно замещенный R3, определенным выше, гетероарил или гетероарилокси, в котором гетероциклическое кольцо гетероальной или гетероарилоксигруппы выбрано из тиофена, фурана, пиридина, пиразина, пиримидина, индола, индазола, имидазола, хромана, кумарина, карбостирила, хинолина, бензизоксазола, бензоксазола, пиразола, пиррола, тиазола, оксазола или изоксазола, и гетероциклическое кольцо является необязательно замещенным заместителем R3, определенным выше;
или их фармацевтически приемлемые соли.
Из данных соединений предпочтительными представителями являются соединения, в которых R1 и R2 представляют водород или вместе образуют бензилиден, необязательно замещенный заместителем R3, определенным выше, или взятые вместе с углеродом, к которому они присоединены, образуют карбонильный фрагмент, R3 и n имеют значения, определенные выше, R4 представляет водород, m = 0 или 1, и Z представляет водород, гидрокси, алкил с 1-6 атомами углерода, циклоалкил с 3-8 атомами углерода, полициклический алкил с 7-15 атомами углерода, фенил, необязательно замещенный R3, определенным выше, фенокси, необязательно замещенный R3, определенным выше, нафтил, необязательно замещенный R3, определенным выше, или нафтилокси, необязательно замещенный R3, определенным выше, гетероарил или гетероарилокси, гететроциклическое кольцо гетероарильной или гетероарилоксигрупп выбрано из тиофена, фурана, пиридина, пиримидина, индола, индазола, хромана, кумарина, карбостирила, хинолина, бензизоксазола, бензоксазола, и гетерокольцо необязательно замещено R3, определенным выше.
Наиболее предпочтительными являются те соединения, в которых R1, R2 и R4 представляют водород, m представляет 0, и R3, и Z имеют значения, определенные в предыдущем абзаце. Данное изобретение относится как к R-, так и к S-стереоизомерам бензодиоксанметанамина, а также к смесям R- и S-стереоизомеров. На протяжении данной заявки наименования продуктов данного изобретения, в которых абсолютная конфигурация бензодиоксанметанамина не указывается, охватывают индивидуальные R- и S-энантиомеры, а также их смеси.
Фармацевтически приемлемыми солями являются соли, производимые из таких органических и неорганических кислот, как уксусная, молочная, лимонная, винная, янтарная, фумаровая, малеиновая, малоновая, миндальная, яблочная, соляная, бромистоводородная, фосфорная, азотная, серная, метансульфоновая, толуолсульфоновая и аналогичные известные приемлемые кислоты.
Аминометил-2,3,8,9-тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-оны получаются, как описано ниже. В частности, соответствующим образом замещенный нитрогваякол алкилируется аллилбромидом в присутствии подходящего основания, такого, как гидрид натрия, а затем по схеме (1) (см. в конце описания) демитилируется реагентом, таким как гидроокись натрия. Получающийся в результате 4-нитро-2-аллилоксифенол затем алкилируется глицидилтозилатом или эпигалоидгидрином в присутствии основания, такого как гидрид натрия, и нагревается в высоко кипящем растворителе, таком как мезитилелен или ксилол, для осуществления, как перегруппировки аллильной группы, так и циклизации диоксанового кольца. Получающийся первичный спирт превращается в тозилат с помощью реакции с п-толуолсульфонилхлоридом, в присутствии пиридина, или альтернативно в галогенид с помощью реакции с четырехбромистым углеродом или четыреххлористым углеродом в сочетании с трифенилфосфином. Аллильная боковая цепь превращается в уксуснокислотный фрагмент с помощью окислительного расщепления с помощью перманганата калия, а нитрогруппа восстанавливается в амин с помощью водорода и палладия на угле и осуществляется циклизация в лактам водной соляной кислотой. Замещение тозилата или галогенида замещенным подходящим образом амином в некоторых высоко кипящих растворителях, таких как диметилсульфоксид, дает целевое соединение данного изобретения.
Метилтозилат оксиндолдиоксана, описанный на схеме (1), может также получаться, как дано на схеме (1a); замещенный соответствующим образом салицилальдегид алкилируется аллилбромидом в присутствии подходящего основания, такого как гидрид натрия. Альдегидный фрагмент затем превращается в фенол с помощью обработки m-хлорпероксибензойной кислотой с последующим расщеплением промежуточного формиатного сложного эфира основной окисью алюминия в метаноле. Получающийся в результате 2-аллилоксифенол затем алкилируется глицидилтозилатом или эпигалоидгидрином в присутствии основания, такого как гидрид натрия, и нагревается в высококипящем растворителе, таком как мезитилен или ксилол для осуществления перегруппировки аллильной группы. Циклизация в бензодиоксанметанол завершается с помощью обработки бикарбонатом натрия в этаноле. После превращения спирта в тозилат с помощью п-толуолсульфонилхлорида в пиридине аллильная боковая цепь окислительно расщепляется в уксуснокислотный фрагмент с помощью перманганата калия, а нитрогруппа вводится с помощью обработки азотной кислотой в дихлорэтане. Восстановление нитрогруппы и циклизация в пактам проводятся, как описано на схеме (1). Когда R3 представляет галоген, для восстановления предпочитается катализатор, такой как окись платины или платина на сульфидированном углероде.
Метилтозилат оксиндолдиоксана может также получаться из замещенного соответствующим образом бензодиоксанметанола, как показано на схеме (1b) в конце описания. После превращения спирта в тозилат, как описано выше, нитрогруппа вводится с помощью обработки азотной кислотой в дихлорметане и восстанавливается водородом в присутствии подходящего катализатора, такого как окись платины или платина на сульфидированном углероде. Оксиндол получается с помощью модификации процедуры Гассман'а и др. (J. Amer. Chem. Soc. , 96, 5512 (1974)), и получающийся в результате тиометиловый эфир расщепляется с помощью обработки с использованием никеля Ренея.
Альтернативно, аллильная боковая цепь может превращаться в остаток пропилового спирта с помощью гидроборирования смесью боран/ТГФ с последующей обработкой перекисью водорода, как показано выше. Первичный спирт может окисляться в карбоновую кислоту подходящим окисляющим агентом, таким как перманганат калия, и циклизоваться в пактам, как описано ранее, с помощью водной соляной кислоты. Замещение тозилата или галогенида замещенным подходящим образом амином в некоторых высококипящих растворителях, таких как диметилсульфоксид, как описано выше, дает соединения изобретения, в которых m = 1. Аналогичная технология, по которой пропиловый спирт превращается в бромид с помощью обработки четырехбромистым углеродом и трифенилфосфином, замещается цианидом с помощью обработки цианидом натрия в диметилформамиде, и гидролизуется в гомологическую кислоту, может применяться для получения соединений изобретения, в которых m = 2, при условии, что бензодиоксанметанол подходящим образом защищен на протяжении данной процедуры.
Соединения изобретения, в которых R1 и R2 объединены с образованием бензилиденового или алкилиденового остатка, могут получаться с помощью конденсации лактамов, описанных выше, с соответствующим ароматическим или алифатическим альдегидом. Соединения изобретения, в которых R1 и R2 представляют алкил, могут получаться с помощью алкилирования промежуточных карбоновых кислот или их соответствующих эфиров в стандартных условиях. Соединения изобретения, в которых R1 и R2 объединены с образованием карбонила (т. е. изатины) могут получаться с помощью окисления соответствующих оксиндолов. Соответствующие нитрогваяколы являются известными соединениями или могут быть получены любым обычным специалистом в данной области. Альтернативно, 4-нитро-2-аллил-фенолы, используемые в процессе (1), описанном выше, могут быть получены из соответствующим образом 5- или 6-замещенного салицилальдегида с помощью процедуры (3), описанной ниже, или из соответствующим образом 3- или 4-замещенного салицилальдегида с помощью процедуры (4), описанной ниже, в соответствии с которой [2-(триметилсилил)этокси] метилхлорид (SEMCI) применяется в качестве гидроксизащищающей группы во время превращения альдегида в формиатный эфир с помощью метахлорнадбензойной кислоты с последующим гидролизом в гидроксигруппу. Замещенные амины R4-NH(CH2)n-Z, являются известными соединениями или могут свободно получаться специалистами в данной области. Соединения изобретения могут разделяться на их энантиомеры с помощью общепринятых методов или, предпочтительно, они могут получаться непосредственно с помощью замены (2R)-(-)-глицидил 3-нитробензолсульфонатом или тозилатом (в случае S-/бензодиоксанметанамина) или (2S)-(+)-глицидил 3-нитробензолсульфонатом или тозилатом (в случае R-энантиомера) вместо эпигалоидгидрина или рацемического глицидилтозилата в описанных выше процедурах.
Соединения данного изобретения являются агонистами авторецепторов допамина, т. е. они служат для модулирования синтеза и высвобождения нейротрансмиттерного допамина. Эти соединения также действуют в качестве частичных агонистов при постсинаптическом допаминовом D2-рецепторе, способных функционировать в качестве или агонистов, или антагонистов в зависимости от уровня допаминэргической стимуляции. Они также служат для модулирования допаминэргической нейротрансимиссии, и поэтому они полезны для лечения нарушений допаминэргической системы, таких как шизофрения, шозоаффективные расстройства, болезнь Паркинсона, синдром Тоуретт'а и гиперпролактинемия, и при лечении пристрастия к лекарствам или каким-либо веществам, такого как злоупотребление и /или пристрастие/ этанолом или кокаином, или родственных заболеваний.
Действие соединений данного изобретения на синтез допамина установлено по методу авторов Walters и Roth, Naunyn-Schmiedeberg's Arch. Pharmacol. 296; 5-14, 1976, по которому крысам (самцы, Sprague-Dawley, Charles River, 200-350 г) вводился носитель или испытываемое лекарство за десять минут до введения гамма-бутиролактона (GBL, ; 750 мг/кг, и. п. для ингибирования потока допаминэргического импульса) и за 20 минут до NSD-1015 (100 мг/кг, и. п. для предотвращения превращения допа в допамин). Через 30 минут после введения NDS-1015 всех крыс обезглавливали, и извлекали nucleus accumbens и стриатум для анализа. После экстракции ткани перхлорной кислотой экстракты помещались на колонки из окиси алюминия для сбора концентрата допа и других катехинов. Данный элюат затем подвергался анализу B HPLC (высоко-эффективная жидкостная хроматография или жидкостная хроматография высокого давления) с использованием электрохимической детекции для количественных уровней присутствия допа. Агонисты допаминовых авторецепторов в используемых выше условиях ингибируют накопление допа. При испытании на данной модели соединение Примера 1, характерный представитель других соединений изобретения, ингибирует накопление допа на 67.5% при дозе 10 мг/кг, п. к. (подкожно).
Антипсихотическая активность соединений изобретения дополнительно устанавливалась с помощью определения способности соединений снижать локомоторную активность мышей по методу Martin и Bendensky, J. Pharmacol. Exp. Therap. 229: 706-711, 1984, по которому мышам (самцы, CF-1, Charles River, 20-30 г) инъецировали носитель или различные дозы каждого лекарства, и локомоторная активность измерялась в течение 30 минут при использовании автоматизированных мониторов ИК активности (Омнитек - 8 х 8 дюйм открытое поле), расположенных в затемненном помещении. Величины ЕД50 вычислялись по результатам подсчета горизонтальной активности, полученным через 10 - 20 минут после введения дозы лекарства с использованием анализа нелинейной регрессии с обратным предсказанием. Результаты данного испытания с соединениями изобретения приводятся ниже.
Сродство по отношению к допаминовому D2 рецептору устанавливалось с помощью видоизменения стандартной экспериментальной процедуры испытания авторов Seemen и Schaus, European Journal of Pharmacology 203: 105-109, 1991/, по которой гомогенизированная строительная ткань головного мозга крыс инкубируется с 3H-хинпиролом и различными концентрациями испытываемого соединения, фильтруется и промывается, и осуществляется подсчет с помощью сцинтилляционного счетчика Betaplate. Результаты данного испытания с соединениями, характерными для данного изобретения, даются ниже.
Результаты стандартных процедур испытания, описанных в двух предыдущих абзацах, даны в таблице.
Таким образом видно, что соединения данного изобретения обладают сильным сродством к допаминовым рецепторам и заметно влияют на синтез нейротрансмиттерного допамина. Следовательно, они полезны для лечения допаминэргических расстройств, таких как шизофрения, шизоаффективные нарушения, болезнь Паркинсона. Синдром Тоуретт'а, гиперлактинемия и пристрастие к лекарствам или каким-либо веществам.
Соединения данного изобретения могут назначаться для приема орально или парентерально, в чистом виде или в сочетании с общепринятыми фармацевтическими носителями. Применимые твердые носители могут включать одно или более веществ, которые могут также действовать как вкусовые агенты, смазочные агенты, солюбилизаторы, суспендирующие агенты, наполнители, агенты, способствующие скольжению (проглатыванию), компрессионные вспомогательные средства, связующие или дезинтегрирующие таблетки агенты или инкапсулирующие материалы. В порошках носитель представляет тонко измельченное твердое вещество, которое находится в смеси с тонко измельченным активным ингредиентом. В таблетках активный ингредиент смешан с носителем, имеющим необходимые свойства сжатия, в подходящих пропорциях, и он сжат так, чтобы ему была придана желаемая форма и размер. Порошки и таблетки предпочтительно содержат до 99% активного ингредиента. Подходящие твердые носители включают, например, фосфат кальция, стеарат магния, тальк, сахара, лактозу, декстрин, крахмал, желатин, целлюлозу, метилцеллюлозу, натриевую карбоксиметилцеллюлозу, поливинилпирролидин, низкоплавкие воски и ионообменные смолы.
Жидкие носители могут использоваться при получении растворов, суспензий, эмульсий, сиропов и эликсиров. Активный ингредиент данного изобретения может растворяться или суспендироваться в фармацевтически приемлемом жидком носителе, таком как вода, органические растворители, смеси их или фармацевтически приемлемые масла или жиры. Жидкий носитель может содержать другие подходящие фармацевтические добавки, такие как солюбилизаторы, эмульгаторы, буферные добавки, консерванты, подслащивающие агенты, вкусовые и ароматизирующие агенты, суспендирующие, загущающие, красящие агенты, регуляторы вязкости, стабилизаторы или осмо-регуляторы. Подходящие примеры жидких носителей для орального и парентерального назначения включают воду (особенно содержащую добавки, как указаны выше, например, производные целлюлозы, предпочтительно раствор натриевой карбоксиметилцеллюлозы), спирты (включая одноатомные и многоатомные спирты, например, гликоли) и их производные, и масла (например, фракционированное кокосовое масло и арахисовое масло). Для парентерального назначения носителем может быть также масляный сложный эфир, такой как этилолеат и изопропилмиристат. Стерильные жидкие носители используются в стерильных композициях в форме жидкости для парентерального назначения.
Жидкие фармацевтические композиции, которые представляют стерильные растворы или суспензии, могут использоваться, например, с помощью внутримышечных, интраперитональных или подкожных инъекций. Стерильные растворы могут также назначаться внутривенно. Для орального назначения композиция может быть или в твердой или в жидкой форме.
Предпочтительно фармацевтическая композиция представляет форму единичной дозы, например, в виде таблеток или капсул. В таких формах композиция подразделена на единичные дозы, содержащие соответствующие количества активного ингредиента; формы единичных доз могут представлять композиции в упаковке, например, порошки в пакетиках, ампулы, пузырьки, предварительно заполненные шприцы или саше, содержащие жидкости. Форма единичной дозы может быть, например, в виде самой капсулы или таблетки, или она может представлять соответствующее число каких-либо таких композиций в форме упаковки.
Дозировка, используемая при лечении конкретного психоза, должна определяться субъективно лечащим врачом. Переменные факторы, которые принимаются во внимание, включают конкретный вид психоза и степень его, возраст и характер ответной реакции пациента. На основе профиля активности и действенности соединений данного изобретения, сравнимых с клинически используемым антипсихотическим рисперидоном, считается, что стартовая доза примерно 5 мг в день с постепенным нарастанием ежедневной дозы примерно до 75 мг в день обеспечит желаемый дозировочный уровень для человека.
Следующие ниже примеры иллюстрируют получение характерных представителей соединений данного изобретения.
Промежуточное соединение 1
3-Аллилокси-4-метокси нитробензол
97.5 г (0.51 моля) натриевой соли 5-нитрогваякола растворялось в одном литре ДМФ, и добавлялось 1,5 эквивалента аллилбромида. Реакционная смесь нагревалась до 65oC в течение двух часов, после чего большая часть темной окраски исчезала, и ТСХ (1: 1 метиленхлорид/гексан) показывала потерю исходного материала. Растворитель концентрировался в вакууме, и остаток промывался водой. Продукт отделялся фильтрованием и сушился в вакууме. Данная процедура давала 112 г бледножелтого твердого вещества. Образец перекристаллизовы-вался из метанола, давая т. пл. 93-94oC.
Промежуточное соединение 2
2-Аллилокси-4-нитрофенол
К одному литру диметилсульфоксида добавлялось 750 мл 2 н. водной гидроокиси натрия, и смесь нагревалась до 65oC. Бледно-желтый 3-аллилокси-4-метоксинитробензол, полученный выше, добавлялся порциями на протяжении 30- минутного периода, и затем температура поднималась до 95oC и поддерживалась в течение 3 часов, после чего исходный материал расходовался. Смесь оставлялась охлаждаться и выливалась в смесь 1 л льда и 2 л 2 н. HCl. Отделялось 73 г сырого, но гомогенного (с помощью ТСХ 1: 1 метиленхлорид/гексан) желаемого продукта в виде светло-коричневого твердого вещества с помощью фильтрования. Данный материал впоследствии растворялся в 1: 1 смеси гексан/метиленхлорид и фильтровался через силикагель, давая 68 г бледно-желтого твердого вещества, которое после перекристаллизации из смеси этилацетата и гексана давало т. пл. 61-62oC. Водные маточные жидкости от первоначальной кристаллизации, указанной выше, экстрагировались 2 литрами этилацетата. Данное вещество сушилось над сульфатом натрия, фильтровалось и выпаривалось до темного масла. Колоночная хроматография на двуокиси кремния с использованием 1: 1 смеси метиленхлорид/гексан давала дополнительно 12 г целевого соединения в виде желтого твердого вещества. Элюирование 2%-ным метанолом в трихлорметане давало 12 г темного масла, которое медленно кристаллизовалось в вакууме. Данное вещество оказалось продуктом Кляйзена, 3-аллил-4-нитрокатехином.
Промежуточное соединение 3
2-(2-Аллилокси-4-нитрофеноксиметил)-оксиран
20 г (0.50 моля) 60% гидрида натрия в минеральном масле помещалось в двух-литровую колбу и промывалось 500 мл гексана. Добавлялся 1 л ДМФ, а затем 77 г (0,40 моля) 2-аллилокси-4-нитрофенола, полученного на предыдущей стадии. Добавление фенола осуществлялось порциями в атмосфере аргона. После перемешивания смеси в течение 30 минут при комнатной температуре в атмосфере аргона добавлялось 108 г (0.48 моля) (R)-глицидилтозилата, и смесь нагревалась при 70-75oC в атмосфере азота на протяжении ночи. После охлаждения ДМФ удалялся в вакууме и заменялся одним литром метиленхлорида. Данная смесь промывалась 500 мл порциями 2 н. HCl, насыщенного бикарбоната натрия и насыщенного солевого раствора и сушилась над сульфатом натрия. Смесь фильтровалась, концентрировалась до масла в вакууме и хроматографировалась на силикагельной колонке с использованием 1: 1 смеси гексана и метиленхлорида в качестве элюента. Это давало 43 г продукта, загрязненного следами двух исходных веществ, а затем 21 г чистого продукта в виде бледно-желтого твердого вещества. Нечистый материал перекристаллизовывался из 1.2 л 10% этилацетат/гексан, давая 34 г чистого (гомогенный на силикагеле ТСХ с 1: 1 смесью гексан: метиленхлорид) (R)-2-(2-аллилокси-4-нитрофеноксиметил)оксирана (т. пл. 64oC).
Элементный анализ для C12H13NO5
Вычислено: C 57.37, H 5.21, N 5.58.
Найдено: C 57.50, H 5.21, N 5.43.
Промежуточное соединение 4.
(3-Аллил-7-нитро-2,3-дигидро-бензо(1,4)диоксин-2-ил)-метанол
(R)-2-(2-Аллилокси-4-нитрофеноксиметил)-оксиран (20 г, 80 ммолей), полученный выше, нагревался при 155oC в мезитилене в течение 24 часов в атмосфере азота. Отфильтровывание черного твердого вещества, которое образовалось, дало 1,5 г очень полярного вещества. Выпаривание растворителя в вакууме с последующей колоночной хроматографией на силикагеле с метиленхлоридом в качестве элюента давало 10 г выделенного исходного вещества и 7,5 г желаемого перегруппированного (S)-(8-аллил-7-нитро-2,3-дигидро-бензо(1,4)диоксин-2-ил)-метанола, который медленно кристаллизовался при стоянии в вакууме (т. пл. 67oC. Выход в расчете на выделенный исходный материал составил 75%.
Элементный анализ для C12H13NO5
Вычислено: C 57.37, H 5.21, N 5.58
Найдено: C 57.26, H 5.20, N 5.35
Промежуточное соединение 5.
Толуол-4-сульфоновой кислоты аллил-7-нитро-2,3-дигидро-бензо(1,4)-диоксин-2-илметиловый эфир
9.55 г (38.0 ммоля) (S)-(8-аллил-7-нитро-2,3-дигидро-бензо(1,4)-диоксин-2-ил)-метанола растворялось в 465 мл пиридина, добавлялось 465 мл пиридина, 29.0 г (152 ммоля) п-толуолсульфонилхлорида, и смесь перемешивалась при комнатной температуре в атмосфере азота на протяжении ночи. Затем добавлялась вода для погашения избытка тозилхлорида, и растворитель удалялся в вакууме и заменялся метиленхлоридом. Данный раствор промывался 2 н. HCl, насыщенным бикарбонатом натрия и насыщенным солевым раствором, и сушился над сульфатом магния. Фильтрование, выпаривание в вакууме и колоночная хроматография на силикагеле с 1: 1 смесью гексан: метиленхлорид в качестве элюента давали 12,6 г (92) (R)-аллил-7-нитро-2,8-дигидро-бензо(1,4)диоксан-2-илметилового эфира толуол-4-сульфоновой кислоты, который медленно кристаллизовался при стоянии, давая рыжевато-коричневое твердое вещество (т. пл. 60-62oC).
Элементный анализ для C17H19NO7
Вычислено: C 56.29, H 4.72, N 3.45
Найдено: C 56.13, H 4.58, N 3.44
Промежуточное соединение 6.
(6-Нитро-3-(толуол-4-сульфонилоксиметил)-2,3-дигидро-бензо(1,4)- диоксин-5-ил)-уксусная кислота
Перманганат калия (11.7 г, 0.074 моля) помещался в колбу, которая была оборудована механической мешалкой, капельной воронкой и ледяной ванной. В колбу добавлялось 150 мл воды и тетрабутиламмонийхлорид /1.0 г, 3.7 ммоля/ при перемешивании. Через капельную воронку медленно добавлялся (R)-аллил-7-нитро-2,3-бензо(1,4)диоксин-2-илметиловый эфир толуол-4-сульфоновой кислоты, полученный выше, растворенный в 100 мл бензола, и реакционная смесь перемешивалась дополнительно в течение 30 минут в ледяной ванне. Ледяная ванна затем удалялась, и смесь перемешивалась в течение 24 часов при комнатной температуре. К смеси при хорошем перемешивании в ледяной ванне добавлялось 30 г бисульфита натрия, и смесь подкислялась концентрированной HCl до тех пор, пока величина pH не будет менее 3. Подкисленный прозрачный желтый раствор затем экстрагировался этилацетатом, и объединенные экстракты сушились над безводным сульфатом магния. Концентрированный остаток хроматографировался на силикагельной колонке с использованием этилацетата в качестве элюента, давая 6.3 г (60%) (R)-(6-нитро-3-(толуол-4-сульфонилоксиметил)-2,3-дигидро- бензо(1,4)-диоксин-5-ил)-уксусной кислоты в виде бледно-желтого твердого вещества. Кристаллизация из метиленхлорида давала светло-желтое твердое вещество с т. пл. 158-159oC.
Элементный анализ для C18H17NO9S•1/4 H2O
Вычислено: C 50.52, H 4.12, N 3.27
Найдено: C 50.51, H 3.83, N 3.12.
Промежуточное соединение 7
2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
Карбоновая кислота (6.0 г, 0.0142 моля): полученная выше, измельчалась в тонко-дисперсный порошок. К данному порошку добавлялось 300 мл воды и 5 мл 2.5 н. NaOH до тех пор, пока pH не будет 8, и гетерогенный раствор перемешивался в течение 30 минут до тех пор, пока твердое вещество не диспергировалось равномерно. Затем добавлялся 1.0 г 10% палладия на угле, и смесь гидрировалась во встряхивателе Парра в течение 24 часов при давлении водорода 52 фунт/кв. дюйм /3.656 кг/кв. см/. Катализатор отфильтровывался и промывался водой. Объем фильтрата затем уменьшался наполовину, и фильтрат подкислялся 15 мл концентрированной HCl при перемешивании в ледяной ванне, и осаждался белый твердый кислотный продукт, (R)-(6-амино-3-(толуол-4-сульфонилоксиметил)-2,3-дигидро- бензо(1,4)диоксин-5-ил)-уксусная кислота. Данный гетерогенный раствор затем нагревался при 50oC в течение 24 часов. По прохождении данного времени ТСХ (5% метанол/метиленхлорид на силикагеле) показала, что аминокислота медленно замещалась лактамом, и реакционная смесь становилась сразу прозрачной, а затем начинало осаждаться целевое соединение в виде белого твердого вещества. После того, как смесь охлаждалась до комнатной температуры и перемешивалась в течение дополнительного часа, белое твердое вещество отфильтровывалось, промывалось диэтиловым эфиром и сушилось в вакууме при комнатной температуре. Продукт (R)-2(толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-3-он (т. пл. 225-227oC) был чистым без дополнительной перекристаллизации и весил 4.2 г (79%).
Элементный анализ для C18H17NO6
Вычислено: C 57.59, H 4.57, N 3.73
Найдено: C 57.34, H 4.55, N 3.69.
Пример 1
2-/Бензиламино-метил/-2,3,8,9-тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0.80 г, 2.13 ммоля) и 99.5% бензиламин /1.42 г, 11.72 ммоля/ объединялись в 15 мл сухого ДМСО, при этом через раствор барботировался сильный ток азота. Смесь нагревалась до 75oC в течение 3 часов. Реакционная смесь охлаждалась и бралась в 400 мл этилацетата. Смесь промывалась шестью 100 мл порциями воды. Объединенные водные смывные жидкости обратно экстрагировались шестью 50 мл порциями этилацетата. Органические фракции объединялись, промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались, давая коричневое масло. Данное масло подвергалось хроматографии на силикагельной колонке с использованием 0,75% смеси метанол/метиленхлорид для удаления загрязняющих примесей. Смесь 1% метанол/метиленхлорид элюировала желаемый продукт, который получался в виде масла (1.85 г, 65%). Данное масло кристаллизовалось из изопропанола с добавлением раствора фумаровой кислоты (0.76 г, 6.57 ммоля) в горячем изопропаноле, давая 2,21 г (S) энантиомера целевого соединения в виде светло-желтого твердого четвертьгидрата монофумарата, т. пл. 202oC.
Элементный анализ для C18H18N2O3• C4H4O4• 0,25 H2O
Вычислено: C 61.32, H 5.26, N 6.50
Найдено: C 61.31, H 5.01, N 6.42.
Пример 2
2-(Бензиламино-метил)-1-бензилиден-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
Целевое соединение выделялось в виде побочного продукта после продолжительного нагревания реакционной смеси, описанной в примере 1. Бензиламин, применяемый в данной реакции, содержал, как это впоследствии определялось, приблизительно 0,5% бензальдегида. Целевое соединение свободно отделялось с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента смеси 0.75% метанол/метиленхлорид. Продукт конденсации получался в виде оранжевого масла и кристаллизовался путем добавления раствора фумаровой кислоты в горячем изопропаноле, давая 0.30 г ярко-оранжевого твердого четвертьгидрата полуфумарата (S) конфигурации, т. пл. 206oC.
Элементный анализ для C25H22N2O3• 0.50 C4H4O4• 0.25 H2O
Вычислено: C 70.35, H 5.36, N 6.08
Найдено: C 70.31, H 5.13, N 6.04.
Пример 3
2-{ [3-(Индол-3-ил)-пропиламино] -метил} -2,3,8,9-тетрагидро-7H-1,4- диоксино-[2,3-e] -индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (1.5 г, 4.2 ммоля) в ДМСО (80 мл) медленно добавлялся с помощью капельной воронки к 3-(3-аминопропил)индолу /1.1 г, 6.3 ммоля/ в ДМСО (50 мл), и смесь нагревалась при 75oC в течение 17 часов. Большинство ДМСО удалялось при пониженном давлении, и остаток затем распределялся между водой и раствором дихлорметан/изопропанол (3: 1). Отделенный органический слой сушился над безводным сульфатом натрия, фильтровался, концентрировался в вакууме и подвергался колоночной хроматографии на силикагеле с использованием сначала смеси этилацетат/гексан (7: 3), затем этилацетата и, наконец, 5% метанола в этилацетате в качестве элюентов. Ожидаемый продукт отделялся, обрабатывался 0.25М этанольной фумаровой кислотой и осаждался минимальным количеством гексана, давая 70 мг (S) энантиомера целевого соединения в виде рыжевато-коричневой твердой фумаратной соли. Масс спектр (м/е), 377 /M+/.
Пример 4
2-{ [2-(1H-Индол-3-ил)-этиламино] -метил} -2,3,8,9-тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (1.2 г, 3.2 ммоля) и триптамин (1.5 г, 9.6 ммоль) в ДМСО (50 мл) помещались в трехгорлую колбу, снабженную конденсором, термометров и барботером азота, погруженным в раствор. Реакционная смесь нагревалась при 75oC в течение 5 часов, охлаждалась до комнатной температуры и распределялась между водой и этилацетатом. Этилацетатный слой отделялся, сушился над безводным сульфатом натрия, концентрировался в вакууме и подвергался колоночной хроматографии на силикагеле с использованием сначала этилацетата, и затем 2,5%, 5%, 10% метанола в этилацетате в качестве элюентов. Свободное основание желаемого продукта получалось (0,85 г, 2.3 ммоля) в виде масла, которое растворялось в 50 мл смеси этанола и диэтилового эфира (1: 1) в виде раствора и обрабатывалось 10.3 мл 0.25М фумаровой кислоты в этаноле. Добавление гексана давало 0.30 г (S) энантиомера целевого соединения в виде не совсем белого твердого три четверти гидрата полуфумамата, т. пл. 175-176oC.
Элементный анализ для: C21H21N3O3• 1/2 C4H4O4•3/4 H2O
Вычислено: C 63.51, H 5.68, N 9.66
Найдено: C 63.51, H 5.75, N 9.47
Пример 5
2-[(3-Гидрокси-пропиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0.37 г, 1.0 ммоля) в 3-амино-1-пропаноле (20 мл) нагревался при 75oC в течение 15 часов. В течение данного периода гетерогенная реакционная смесь начинала светлеть. Реакционная смесь охлаждалась до комнатной температуры и распределялась между раствором дихлорметан/IPA (3: 1) и солевым раствором. Отделенный водный слой экстрагировался смесью дихлорметан/IPA (3: 1), и объединенные экстракты промывались насыщенным раствором бикарбоната натрия и водой для удаления аминопропанола, сушились над безводным сульфатом натрия и концентрировались в вакууме. Остаток хроматографировался на колонке из основной окиси алюминия с использованием 5% метанола в дихлорметане в качестве элюента, давая свободное основание (0.2 г, 72%) ожидаемого продукта. Свободное основание растворялось в этаноле (10 мл) обрабатывалось избытком 0.25% этанольной фумаровой кислоты и осаждалось гексаном, давая 0.090 г (S) энантиомера целевого соединения в виде бледно-желтого твердого четвертьгидрата полуфумарата, т. пл. 192-193oC.
Элементный анализ для: C14H18N2O4• 1/2 C4H4O4•1/4 H2O
Вычислено: C 56.38, H 6.06, N 8.22
Найдено: C 56.31, H 6.13, N 8.00
Пример 6
2-(Бензиламино-метил)-2,3,9,10-тетрагидро-7H-1,4- диоксино[2,3-f] хинолин-8-он
1 М Раствор ВНз•ТГФ (11.00 мл, 11.00 ммолей) помещался в 100 мл круглодонную колбу, снабженную вводом азота, капельной воронкой и термометром. Раствор охлаждался до 0oC в ванне из льда и воды. К данному охлажденному раствору добавлялся 8-аллил-7-нитро-2,3-дигидро-бензо[1,4] -диоксин-2-илметиловый эфир (R)-толуол-4-сульфоновой кислоты (2.25 г, 5.56 ммоля) в 10 мл сухого ТГФ по каплям на протяжении 10-минутного периода. Реакционная смесь оставлялась достигать комнатной температуры, а затем перемешивалась на протяжении ночи. Затем она охлаждалась до 0oC, и к ней добавлялось по каплям 2.42 мл абсолютного этанола и 6.16 мл /18,5 ммоля/ 3 н. раствора гидроокиси натрия. Через несколько минут на протяжении 20-минутного периода добавлялось 4.2 мл 30%-ного водного раствора перекиси водорода. Смесь нагревалась до 48oC в течение трех часов. Смесь затем охлаждалась до 0oC, и к ней добавлялось 6.08 г карбоната калия. Смесь перемешивалась в течение 0.5 часа, а затем оставлялась стоять на протяжении ночи. На следующее утро твердое вещество исчезало. Реакционная смесь разбавлялась водой и экстрагировалась этилацетатом. Органический слой промывался солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался, давая прозрачное вязкое масло /1.20 г, 51%/ 8-(3-гидрокси-пропил)-7-нитро-2,3-дигидро-бензо[1,4] диоксин-2-илметилового эфира (R)-толуол-4-сульфоновой кислоты.
В 50 мл 3-горлую колбу, оборудованную магнитной мешалкой, термометром, вводом азота и капельной воронкой, добавлялся перманганат калия (1.24 г, 9.25 ммоля), вода (15 мл) и тетра-н-бутиламмонийхлорид (0.17 г). Пурпурный раствор охлаждался до 0oC, и к нему по каплям добавлялся 8-(3-гидрокси-пропил)-7-нитро-2,3-дигидро-бензо[1,4] -диоксин-2-илметиловый эфир (R)-толуол-4-сульфоновой кислоты (1,20 г, 2,84 ммоля), полученный выше, в 10.3 мл бензола. Реакционная смесь перемешивалась при комнатной температуре на протяжении ночи, и затем добавлялось 4.40 г бисульфита натрия. Окраска смеси исчезала после 5-минутного перемешивания. После дополнительного 10-минутного перемешивания добавлялся 4 н. изопропанольная HCl для доведения pH до примерно 1. Смесь разбавлялась водой и экстрагировалась этилацетатом. Органический слой промывался четыре раза 50 мл солевого раствора, сушился над сульфатом магния, фильтровался и концентрировался, давая прозрачное желтое масло (0,95 г, 77%) 3-[6-нитро-3-(толуол-4-сульфонилоксиметил)-2,3-дигидро-бензо[1,4] диоксин-5- ил] -пропионовой кислоты.
3-[6-Нитро-3-(толуол-4-сульфонилоксиметил)-2,3-дигидро- бензо-[1,4] диоксин-5-ил] -пропионовая кислота (0,95 г, 2.17 ммоля) бралась в 8 мл изопропанола и переносилась в сосуд Парра. К данному раствору добавлялась вода (50 мл), 2.5 н. раствор гидроокиси натрия (0,83 г) и метанол (30 мл). Раствор продувался сильной струей азота, и к нему добавлялся 10%-ный палладий на угле (0.32 г). Смесь гидрировалась в аппарате Парра при 57 фунт/кв. дюйм (4.008 кг/см) водорода. Через двадцать часов смесь фильтровалась, и катализатор промывался водой. К водному фильтрату добавлялось 2.3 мл концентрированной HCl, и раствор нагревался на протяжении ночи при 55oC. Когда раствор охлаждался, осаждалось 0,45 г (R)-2-(тозилат-метил)-2,3,9,10-тетрагидро-7H-1,4-диоксино[2,3-f] хинолин-8-она и собиралось с помощью фильтрования и сушилось в вакууме.
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,9,10-тетрагидро-7H-1,4- диоксино[2,3-f] хинолин-8-он (0,45 г, 1.16 ммоля) и 99.5% бензиламина /0.68 г, 6.33 ммоля/ объединялись в 15 мл сухого ДМСО и нагревались до 80oC в течение 7 часов в атмосфере азота. К данному раствору добавлялось 150 мл воды, и смесь экстрагировалась этилацетатом. Органический слой промывался солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался, давая светло-коричневое твердое вещество. Данное твердое вещество подвергалось колоночной хроматографии на силикагеле с использованием 0.75% метанол/метиленхлорид для удаления загрязняющих примесей. Смесь 4% метанол/метиленхлорид элюировала желаемый продукт, который получался в виде белого твердого вещества (0.29 г, 77%). Данное вещество кристаллизовалось из изопропанола с добавлением раствора фумаровой кислоты (0.11 г, 0.94 ммоль) в горячем изопропаноле, давая 0.32 г /S/ энантиомера целевого соединения в виде легкого белого твердого монофумарата, т. пл. 219oC.
Элементный анализ для C19H20N2O3•C4H4O4
Вычислено: C 62,72, H 5.49, N 6.36
Найдено: C 62.34, H 5.32, N 6.19
Пример 7
(2-[(4-Метилбензиламино)-метил)-2,3,8,9-тетрагидро-7H-1,4- диоксино-[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0,80 г, 2.13 ммоль) и 4-метилбензиламин /1.42 г, 11.7 ммоля/ объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 4 часов в атмосфере азота. Реакционная смесь в атмосфере азота. Реакционная смесь охлаждалась и распределялась между 70 мл этилацетата и 200 мл деионизированной воды. Органический слой промывался солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался, давая коричнево-оранжевое масло. Данное масло подвергалось хроматографии на силикагеле с использованием смеси 0.75% метанол/метиленхлорид для удаления примесей. Смесь 2% метанол/метиленхлорид элюировала желаемый продукт, который изолировался в виде масла (0.40 г, 58). Масло кристаллизовалось из изопропанола с добавлением раствора фумаровой кислоты (0.16 г, 1.36 ммоля) в горячем изопропаноле, давая 0.39 г (S) энантиомера целевого соединения в виде не совсем белой твердой фумаратной соли, т. пл. 204-205oC, которая была загрязнена дополнительной половиной эквивалента фумаровой кислоты.
Элементный анализ для: C25H26N2O9•1.5 C4H4O4
Вычислено: C 60.24, H 5.26, N 5.62
Найдено: C 60.18, H 5.26, N 5.79
Пример 8
2-Аминометил-2,3,8,9-тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (3,0 г, 8.0 ммолей) и азид натрия (1.6 г, 24.0 ммоля) помещались в 300 мл ДМФ, и реакционная смесь нагревалась при 45oC в течение 15 часов. Большинство ДМФ удалялось, и остаток распределялся между дихлорметанолом и водой. Дихлорметановый слой отделялся,
сушился над безводным сульфатом магния и концентрировался. Остаток был достаточно чистым без дополнительной очистки и идентифицировался как желаемый продукт, (S)-2-азидометил-2,3,8,9-тетрагидро-7H-1,4-диоксино-[2,3-e] индол-8-он. Аликвотное количество азида (0,8 г, 3.2 ммоля) в этаноле (50 мл) гидрировалось с использованием 10% палладия на угле (100 мг) в течение 15 часов. Получающаяся смесь подкислялась 4 н. изопропанольной HCl до тех пор, пока величина pH не становилась менее 3. Катализатор затем отфильтровывался, и фильтрат концентрировался. Остаток растворялся в 90%-ном водном этаноле и осаждался диэтиловым эфиром, давая (S) энантиомер целевого соединения в виде белого твердого 1.25 гидрата гидрохлорида (0,7 г, 85%), т. пл. 278-280oC.
Элементный анализ для: C11H12N2O3•HCl•1.25 H2O
Вычислено: C 47.32, H 5.59, N 10.03
Найдено: C 47.48, H 5.44, N 10.08.
Пример 9
2-(Циклогексилметиламино-метил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0.80 н. , 2.13 ммоля) и циклогексилметиламин (1.33 г, 11.7 ммоль) объединялись в 15 мл ДМСО и нагревались до 80oC в течение 6 часов в атмосфере азота. Реакционная смесь оставлялась охлаждаться и распределялась между 100 мл этилацетата и 150 мл деионизированной воды. Органический слой промывался солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался, давая черное масло. Данное масло подвергалось колоночной хроматографии на силикагеле с использованием смеси 0,75% метанол/метиленхлорид для удаления примесей. Смесь 2% метанол/метиленхлорид элюировала желаемый продукт, который был в виде масла (0,38 г, 56%) после концентрирования в вакууме. Данное масло кристаллизовалось из изопропанола с добавлением раствора фумаровой кислоты (0,15 г, 1.3 ммоля) в горячем изопропаноле, давая 0.39 г (S) энантиомера целевого соединения в виде не совсем твердого белого полугидрата монофумарата, т. пл. 187-188oC.
Элементный анализ для: C18H24N2O3• C4H4O4•0.5 H2O
Вычислено: C 59,85, H 6.62, H 6.34
Найдено: C 59,81, H 6.61; N 6.28
Пример 10
2-[(2-Пиридин-3-ил-этиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- Диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0.80 г, 2.1 ммоля) и 3-(2-аминоэтил)пиридин (1.30 г, 10,6 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 9 часов в атмосфере азота. Реакционная смесь оставлялась охлаждаться, а затем распределялась между метиленхлоридом и деионизированной водой. Водный слой экстрагировался дополнительным метиленхлоридом. Органические фракции объединялись, промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались, давая оранжевое масло. Данное масло подвергалось хроматографии на силикагельной колонке с использованием смеси 0.75% метанол/метиленхлорид для удаления примесей. Элюирование смесью 3-5% метанол/метиленхлорид давало желаемый продукт, который получался в виде бежевого твердого вещества (0,48 г, 69%) после концентрирования в вакууме. Данное вещество перекристаллизовывалось из изопропанола с добавлением 4 н. изопропанольной HCl, давая 0,27 г (S) энантиомера целевого соединения в виде светло-желтого твердого 0.75 гидрата дигидрохлорида, т. пл. 174-176oC.
Элементный анализ для: C18H19N3O3•2 HCl•0.75 H2O
Вычислено: C 52.50, H 5.51, N 10.00
Найдено: C 52.19, H 6.29, N 10.00
Пример 11
2-{ [(3-Диметиламино-фенокси)-пропиламино] -метил} -2,3,8,9- тетрагидро-7H-1,4-Диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0.80 г, 2.13 ммоля) и 3-(3-диметиламинофенокси)пропиламин (2.07 г, 10.6 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 4 часов в атмосфере азота. Реакционная смесь бралась в 150 мл метиленхлорида и промывалась шесть раз 40 мл порциями воды. Органический слой промывался солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался в вакууме, давая коричневое масло. Данное масло подвергалось хроматографии на силикагельной колонке с использованием смеси 0.75% метанол/метиленхлорид для удаления примесей. Элюирование смесью 12% метанол/метиленхлорид давало желаемый продукт, который получался в виде масла (0.27 г, 32%) после концентрирования в вакууме. Масло кристаллизовалось из изопропанола с добавлением раствора фумаровой кислоты (0,086 г, 0.75 ммоля) в горячем изопропаноле, давая 0.17 г /S/ энантиомера целевого соединения в виде коричневого твердого монофумарата, т. пл. 136-138oC.
Элементный анализ для: C22H27N3O4•C4H4O4
Вычислено: C 60.81, H 6.08, N 8.18
Найдено: C 61.17, H 6.21, N 8.3
Пример 12
2-{ [(Тиофен-2-илметил)-амино] -метил} -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0.80 г, 2.13 ммоля) и тиофен-2-метиламин (1.45 г, 12.80 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 6 часов в атмосфере азота. Реакционная смесь бралась в 150 мл метиленхлорида и промывалась шесть раз 40 мл порциями воды. Органический слой промывался солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался в вакууме, давая темно-оранжевое масло. Данное масло подвергалось хроматографии на силикагельной колонке с использованием смеси 0.75% метанол/метиленхлорид для удаления примесей. Элюирование смесью 1-2% метанол/метиленхлорид давало желаемый продукт, который получался в виде масла (0.44 г, 54%) после концентрирования в вакууме. Неочищенный продукт кристаллизовался из изопропанола с добавлением раствора фумаровой кислоты (0,18 г, 1.5 ммоля) в горячем изопропаноле, давая 0,48 г (S) энантиомера целевого соединения в виде светло-желтого твердого четвертьгидрата монофумарата, т. пл. 210-211oC.
Элементный анализ для: C16H16N2O3• C4H4O4•0,25H2O
Вычислено: C 54.98, H 4.73, N 6.41
Найдено: C 55.03. H 4.70, N 6.23.
Пример 13
2-{ [3-(Хинолин-7-илокси)-пропиламино] -метил} -2,3,8,9-тетрагидро- 7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0,80 г, 2.13 ммоля) и 3-(хинолин-7-иолокси)пропиламин (2.15 г, 10.6 ммоль) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 6 часов в атмосфере азота. Реакционная смесь бралась в 150 мл метиленхлорида и промывалась шесть раз 40 мл порциями воды. Органический слой промывался солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался, давая темно-коричневое масло. Данное масло подвергалось хроматографии на силикагеле с использованием смеси 0.75% метанол/метиленхлорид для удаления примесей. Элюирование смесью 4-5% метанол/метиленхлорид давало желаемый продукт, который получался в виде бежевого твердого вещества (0.30 г, 35%) после концентрирования в вакууме. Неочищенное твердое вещество перекристаллизовывалось из изопропанола с добавлением 4 н. изопропанольной HCl, давая 0.14 г (S) энантиомера целевого соединения в виде светло-желтого твердого гидрата дигидрохлорида, т. пл. 176-177oC.
Элементный анализ для: C23H23N3O4•2HCl•H2O
Вычислено: C 55.65, H 5.48, N 8.46
Найдено: C 55.48, H 5.98, N 8.36
Пример 14
(2-{ [(Адамантан-1-илметил)-амино] -метил} -2,3,8,9-тетрагидро- 7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0,70 г, 1.87 ммоля) и 1-адамантанметиламин (1.55 г, 9.35 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 6 часов в атмосфере азота. Реакционная смесь бралась в 150 мл этилацетата и промывалась шесть раз 40 мл порциями воды. Органический слой промывался солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался в вакууме, давая темно-коричневое масло. Данное масло подвергалось колоночной хроматографии на силикагеле с использованием смеси 0.75% метанол/метиленхлорид в качестве элюента, давая 0,51 г (74%) желаемого продукта в виде бежевого твердого вещества. Сырой продукт перекристаллизовывался из изопропанола с добавлением раствора фумаровой кислоты (0.18 г, 1.5 ммоля) в горячем изопропаноле, лава2 0,49 г (S) энантиомера целевого соединения в виде не совсем белого твердого полугидрата монофумарата, т. пл. 201-202oC.
Элементный анализ для: C22H28N2O3• C4H4O4•0.5 H2O
Вычислено: C 63.25, H 6.73, N 5.67
Найдено: C 63.23, H 6.87, N 5.60
Пример 15
2-(Бензиламино-метил)-2,3,8,9-тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
(S)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (2.0 г, 5.3 ммоля), полученный таким же образом, как (R) энантиомер, описанный выше, с заменой (R)-глицидилтозилата (S)-глицидилтозилатом, и бензиламин (2,9 мл, 26.7 ммоля) помещались в 20 мл свежего ДМСО в атмосфере азота. Смесь затем нагревалась до 75-80oC при перемешивании в течение 3 часов. После охлаждения при комнатной температуре реакционная смесь распределялась между этилацетатом и солевым раствором. ДМСО слой экстрагировался этилацетатом, и объединенные этилацетатные экстракты промывались водой для удаления следов ДМСО, сушились над безводным сульфатом натрия и концентрировались в вакууме. Остаток хроматографировался на силикагельной колонке с использованием этилацетата в качестве элюента, давая свободное основание (1.4 г, 4.5 ммоля, 83%) ожидаемого продукта в виде затвердевшего масла при пониженном давлении. Свободное основание растворялось в 20 мл этанола, обрабатывалось 0.25 М этанольной фумаровой кислотой (10 мл) и осаждалось диэтиловым эфиром, давая целевое соединение в виде бледно-желтой твердой фумаратной соли, преимущественно в (R)-конфигурации, т. пл. 195-196oC. Данный образец, фармакологические результаты которого сообщались в данной заявке, по данным определения с помощью хиральной ВЭЖХ, содержал 9% (S)-энантиомера.
Элементный анализ для: C18H18N2O3•C4H4O4
Вычислено: C 61.97, H 5.20, N 6.57
Найдено: C 61.96, H 5.13, N 6.51
Пример 16
2-(Пентиламинометил)-2,3,8,9-тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (0.80 г, 2.13 ммоля и пентиламин (0,93 г, 10.6 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 6 часов в атмосфере азота. Реакционная смесь бралась в 150 мл метиленхлорида и промывалась шестью 40 мл порциями воды. Органический слой промывался солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался в вакууме, давая коричневое масло, которое хроматографировалось на колонке с силикагелем с использованием смеси 0.75% метанол/метиленхлорид для удаления примесей. Элюирование смесью 1-2% метанол/метиленхлорид давало желаемый продукт, который получался в виде масла (0.20 г, 32%) после концентрирования в вакууме. Продукт кристаллизовался из изопропанола с добавлением раствора фумаровой кислоты (0.090 г, 0.77 ммоля) в горячем изопропаноле, давая 0.10 г (S) энантиомера целевого соединения в виде не совсем белого твердого гемифумарата, т. пл. 238-239oC.
Элементный анализ для: C16H22N2O3•0.5 C4H4O4
Вычислено: C 62.05, H 6.94, N 8,04
Найдено: C 61.54, H 6.89, N 7.92
Пример 17
2-[(4-Метокси-бензиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (1.00 г, 2.66 ммоля) и 4-метоксибензиламин (1.4 мл, 10.7 ммоля) объединялись в 10 мл сухого ДМСО и нагревались до 85oC в течение 3,5 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 150 мл воды, и смесь экстрагировалась дважды 250 мл порциями 35% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая масло, которое подвергалось колоночной хроматографии на силикагеле с использованием смеси 2% метанол/метиленхлорид в качестве элюента. Свободное основание целевого соединения (0,51 г, 64%) получалось в виде масла после концентрирования в вакууме. Продукт кристаллизовался из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.44 г (S) энантиомера целевого соединения в виде не совсем белого твердого четвертьгидрата фумарата, т. пл. 205-205.5oC.
Элементный анализ для: C19H20N2O4• C4H4O4•0.25 H2O
Вычислено: C 59.93, H 5.36, N 6.08
Найдено: C 59.93, H 5.23, N 6.14
Пример 18
2-(Нафталин-1-ил-метиламино-метил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол (1.05 г, 2.80 ммоля) и 1-нафталинметиламин (2.05 мл, 14 ммолей) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 4 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 150 мл воды, и смесь экстрагировалась дважды 250 мл порциями 35% этилацетата в гексане. Объединенные органические экстракты промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая масло, которое подвергалось колоночной хроматографии на силикагеле с использованием смеси 1% метанол/метиленхлорид в качестве элюента. Свободное основание целевого соединения (0.27 г, 27%) получалось в виде масла после концентрирования в вакууме. Продукт кристаллизовался из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.25 г (S) энантиомера целевого соединения в виде светло-желтого твердого полугидрата фумарата, т. пл. 167-168oC.
Элементный анализ для: C22H20N2O3• C4H4O4• 0.5 H2O
Вычислено: C 64,32, H 5.19, N 5.77
Найдено: C 64.19, H 5.48, N 5.47
Пример 19
2-(4-Трифторметил-бензиламино-метил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (1.03 г, 2.75 ммоля) и 4-трифторметилбензиламин (1.60 мл, 11.2 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 3,5 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 150 мл воды, и смесь экстрагировалась дважды 250 мл порциями 35% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая масло, которое подвергалось колоночной хроматографии на силикагеле с использованием смеси 5% метанол/метиленхлорид в качестве элюента. Свободное основание целевого соединения (0.56 г, 54%), полученное таким образом, кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.06 г (S) энантиомера целевого соединения в виде не совсем белого твердого фумарата, т. пл. 211-212oC.
Элементный анализ для: C19H17F3N2O3• C4H4O4
Вычислено: C 55,87, H 4.28, N 5.67
Найдено: C 55,56, H 3.93, N 5.75
Пример 20
2-(4-Фтор-бензиламино)-метил-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] -индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (1.15 г, 3.07 ммоля) и 4-фторбензиламин (1.56 мл, 13.6 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 3,5 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 150 мл воды, и смесь экстрагировалась дважды 250 мл порциями 50% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая масло, которое хроматографировалось на силикагельной колонке с использованием смеси 0.75% метанол/метиленхлорид в качестве элюента. Свободное основание целевого соединения (0,62 г, 62%), полученное таким образом, кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.69 г (S) энантиомера целевого соединения в виде не совсем белого твердого фумарата, т. пл. 218-220oC.
Элементный анализ для: C18H17FN2O3•C4H4O4
Вычислено: C 59,46, H 4.76, N 65.30
Найдено: C 59.04, H 4.67, N 6.23
Пример 21
2-(4-фенил-бутиламино)-метил-2,3,8,9-тетрагидро-7H-1,4- диоксино-[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H- диоксино[2,3-e] индол-8-он (1.05 г, 2.80 ммоля) и фенилбутиламин (1,99 мл, 12,5 ммоля) объединялись в 15 мл сухого ДМСО и нагревались при 85oC в течение 4 часов в атмосфере азота. После охлаждения до комнатной температуры, добавлялось 200 мл воды, и смесь экстрагировалась дважды 300 мл порциями 50% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая масло, которое подвергалось хроматографии на силикагельной колонке с использованием смеси 1% метанол/CH2Cl2 в качестве элюента. Свободное основание целевого соединения (0.26 г, 30%), полученное таким образом, кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0,2 г энантиомера целевого соединения в виде рыжевато-коричневого, твердого полугидрата фумарата, т. пл. 185-186oC.
Элементный анализ для: C21H24N2O3• C4H4O4•0.5H2O
Вычислено: C, 62.88; H, 6.12; N, 5.87
Найдено: C, 62.88; H, 6.04; N, 5.79.
Пример 22
2-(4-фтор-бензиламино-метил)-9-(4-фтор-фенил-этилиден)-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
Целевое соединение выделялось как сопутствующий продукт после продолжительного нагревания реакционной смеси, описанной в Примере 20. Было определено, что 4-фторбензиламин, применяемый в данной реакции, содержит 4-фторбензальдегид. Целевое соединение отделялось от ранних фракций от хроматографии, описанной в Примере 20. Продукт конденсации был получен в виде оранжевого масла и кристаллизовался с добавлением раствора фумаровой кислоты в горячем этаноле, давая 0.06 г оранжевого твердого полугидрата полуфумарата (S) конфигурации, т. пл. 202oC.
Элементный анализ для: C25H20F2N2O3• 0,5C4H4O4•0,5H2O
Вычислено: C, 63.27; H, 4.39; N, 5.09
Найдено: C, 65.01; H, 4.53; N, 5.58
Пример 23
N-(3-{ 3-[(8-оксо-2,3,8,9-тетрагидро-7H-1,4-диоксино[2,3-e] индол-2- илметил)-амино} -пропокси} -фенил)-ацетамид
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (1.04 г, 2.77 ммоля) и 3-(3-ацетамидофенокси)пропиламин (2.6 г, 12.5 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 3.5 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 200 мл воды, и смесь экстрагировалась дважды 250 мл порциями 50% этилацетата в гексане. Объединенные органические фазы промывались cолевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая масло, которое подвергалось хроматографии на силикагельной колонке с использованием смеси 2% метанол/CH2Cl2 в качестве элюента. Полученное таким образом свободное основание целевого соединения (0.68 г, 62%) кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.56 г (S) энантиомера целевого соединения в виде светло-желтого твердого полугидрата полуфумарата, т. пл. 197-198oC.
Элементный анализ для: C22H25N3O5• 0.5C4H4O4•0,5H2O
Вычислено: C, 61.23; H, 5.93; N, 8.79
Найдено: C, 60.64; H, 6.06; N, 8.56
Пример 24
2-(Проп-2-иниламинометил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] -индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (1.03 г, 2.75 ммоля) и пропаргиламин (0,85 мл, 12.3 ммоля) объединялись в 20 мл сухого ДМСО и нагревались до 85oC в течение 3.5 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 100 мл воды, и смесь экстрагировалась дважды 200 мл порциями 50% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая масло, которое подвергалось хроматографии на силикагельной колонке с использованием смеси 1% метанол/CH2Cl2 в качестве элюента. Полученное таким образом свободное основание (0.50 г, 71%) кристаллизовалось из изопропанола с добавлением одного эквивалента фумаровой кислоты, давая 0,42 г (S) энантиомера целевого соединения в виде светло-желтого фумарата, т. пл. 167-168oC.
Элементный анализ для: C14H14N2O3•C4H4O4
Вычислено: C, 57,75; H, 4.85; N, 7,48
Найдено: C, 57.93; H, 5.16; N, 7.28
Пример 25
2-[(3-Трифторметил-бензиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (1.01 г, 2.70 ммоля) и 3-трифторметилбензиламин (1.75 мл, 12,0 ммолей) объединялись в 15 мл сухого ДМСО и нагревались до 85oC в течение 4 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 200 мл воды, и смесь экстрагировалась дважды 250 мл порциями 50% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая масло, которое подвергалось хроматографии на силикагельной колонке с использованием смеси 2% метанол/CH2Cl2 в качестве элюента. Свободное основание целевого соединения (0.20 г, 20%), полученное таким образом, кристаллизовалось из изопропанола с добавлением одного эквивалента фумаровой кислоты, давая 0,17 г (S) энантиомера целевого соединения в виде светло-желтого твердого полугидрата фумарата, т. пл. 158-160oC.
Элементный анализ для: C19H17F3N2O3• C4H4O4•0.5 H2O
Вычислено: C, 54.87; H, 4.41; N, 5.56
Найдено: C, 54.70; H, 4.09; N, 5.57
Пример 26
2-(Бензиламино-метил)-2,3-дигидро-7H-1,4-диоксино[2,3-e] индол-8,9-дион
0,60 г (1.94 ммоля) (S)-2-(бензиламино-метил)-2,3,8,9-тетрагидро- 7H-1,4-диоксино[2,3-e] индол-8-она, полученного, как описано в Примере 1, бралось в 200 мл н. NaOH и 150 мл метанола и перемешивалось без защиты от воздуха в течение 48 часов, нейтрализовалось и экстрагировалось этилацетатом. Концентрирование органической фазы давало 200 мг темного красного масла. Данное масло кристаллизовалось из изопропанола с добавлением раствора фумаровой кислоты (79 мг, 0.68 ммоля) в горячем изопропаноле, давая 0.12 г (S) энантиомера целевого соединения в виде темно-красного твердого вещества, т. пл. 177oC (разл. )
Элементный анализ для: C19H17F3N2O3• C4H4O4
Вычислено: C, 55,87; H, 4.28; N, 5,67
Найдено: C, 55,56; H, 3,93; N, 5.75
Пример 27
2-[(4-Трифторметокси-бензиламино)-метил] -2,3,8,9-тетрагидро-7H- 1,4-диоксино[2,3-e] индол-8-он
(R)-2-(Толуол-4-сульфонилоксиметил)-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он (1,0 г, 2.7 ммоля) и 4-трифторметоксибензиламин (2.0 г, 11 ммолей) объединялись в 30 мл сухого ДМСО и нагревались до 90oC в течение 4 часов в атмосфере азота. После охлаждения до комнатной температуры, смесь разбавлялась 400 мл смеси 1: 1 этилацетат/гексан и промывалась 400 мл насыщенного раствора бикарбоната натрия, двумя 250 мл порциями воды и насыщенным солевым раствором. Смесь сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме, давая масло, которое подвергалось хроматографии на силикагельной колонке с использованием смеси 1% метанол/СHCl3 в качестве элюента. Полученное таким образом свободное основание целевого соединения /0.49 г/ кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0,29 г (S) энантиомера целевого соединения в виде белого твердого фумарата, т. пл. 201-202oC.
Элементный анализ для: C19H17F3N2O3• C4H4O4
Вычислено: C, 54.12; H, 4.15; N, 5.49
Найдено: C, 55.80; H, 3.97; N, 5.36
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 8
(R)-2-(Толуол-4-сульфонилоксиметил)-6-метил-2,3- дигидробензо[1,4] диоксин
(S)-(6-метил)-2,3-дигидробензо(1,4 диоксин-2-ил)-метанол (10,1 г, 56,2 ммоля) растворялся в 375 мл пиридина. К данному раствору добавлялось 21.4 г (0.11 моля) п-толуолсульфонилхлорида и смесь перемешивалась при комнатной температуре в атмосфере азота в течение ночи. Реакционная смесь охлаждалась в ванне из смеси лед/вода, и к ней медленно добавлялось 400 мл воды. Смесь перемешивалась при комнатной температуре в течение 4 часов, и затем растворитель удалялся в вакууме, давая темно-коричневое масло. Оно растворялось в этилацетате и промывалось 2 н. HCl (водн. ), водой и насыщенным солевым раствором и сушилось над сульфатом магния. Фильтрование, выпаривание в вакууме и хроматография на силикагельной колонке с использованием 40% гексана в дихлорметане в качестве элюента давали 15.1 г (89%) целевого соединения в виде бесцветного масла.
1H (CDCl3) дублет, 7.80 δ (2H); дублет, 7.38 δ (2H); синглет, 6.61 δ (3H); подъем, 4.40-3.90 δ (5H); синглет, 2.40 δ (3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 9
(R)-2-(Толуол-4-сульфонилоксиметил)-6-метил-7-нитро-2,3- дигидробензо-[1,4] диоксин
(R)-2-(толуол-4-сульфонилоксиметил)-6-метил-2,3-дигидробензо[1,4] -диоксин (15.1 г, 50 ммолей) растворялся в 154 мл дихлорэтана и охлаждался до 0oC в ванне из смеси лед/вода. К этому охлажденному раствору в течение 10 минут по каплям добавлялся раствор азотной кислоты (уд. вес 1.49) в 38 мл дихлорэтана. Смесь перемешивалась при 0oC в атмосфере азота в течение 1 часа, после чего реакционная смесь охлаждалась путем добавления льда. Смесь разбавлялась метиленхлоридом и промывалась насыщенным водным раствором бикарбоната натрия, водой, насыщенным солевым раствором и сушилась над сульфатом магния. Фильтрование и выпаривание в вакууме давали 16.8 г целевого соединения в виде желтого твердого вещества. 1H (ДMCO-d6) дублет, 7.80 δ (2H); дублет, 7.45 δ (2H); синглет, 7.40 δ (1H); синглет, 6.98 δ (1H); мультиплет, 4.57 δ (1H); мультиплет, 4.40 δ (2H); мультиплет, 4.20 δ (1H); мультиплет, 4,10 δ (1H); синглет, 2.43 δ (3H); синглет, 2.40 δ (3H).
(R)-2-(толуол-4-сульфонилоксиметил)-6-метил-7-амино-2,3- дигидробензо[1,4] диоксин
(R)-2-(толуол-4-сульфонилоксиметил)-6-метил-7-нитро-2,3- дигидробензо[1,4] диоксин (16.8 г, 44,6 ммоля) добавлялся к суспензии 1.8 г 10% палладия на угле в 150 мл метанола. Смесь гидрировалась в течение ночи с использованием аппарата Парра при давлении азота 58 пси. Затем смесь фильтровалась через целит, и катализатор промывался этилацетатом. Фильтрат концентрировался в у вакууме, давая 11.7 г целевого соединения в виде светло-желтого твердого вещества. 1H (ДМСО-d6) дублет, 7.80 δ (2H); синглет, 6.40 δ (1H); синглет, 6.08 δ (1H); синглет, 4.40 δ (2H); мультиплет, 4.30 δ (2H); мультиплет, 4.10 δ (2H); мультиплет, 3.80 δ (1H); синглет, 2.40 δ (3H); синглет, 1.90 δ (3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 11
(R)-2-(толуол-4-сульфонилоксиметил)-6-метил-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
В трехгорлую колбу, снабженную капельной воронкой, термометром и вводом азота помещалось 5.56 г (41.4 ммоля) этилметилтиоацетата и 130 мл сухого метиленхлорида. Раствор охлаждался до -78oC с помощью ванны из смеси сухой лед/изопропанол, и к нему по каплям добавлялось 5,20 г (38,4 ммоля) сульфонилхлорида в течение 3-минутного периода. К получающейся в результате смеси, в течение 15 минут добавлялся раствор (R)-2-(толуол-4-сульфонилоксиметил)-6-метил-7- амино-2,3-дигидробензо[1,4] диоксина (11.7 г, 33,6 ммоля) и Proton Sponge (8,64 г, 40.3 ммоля) в 220 мл метиленхлорида. Смесь перемешивалась при -78oC в течение двух часов, затем в течение 10 минут по каплям добавлялось 5,2 г (40.3 ммоля) диизопропилэтиламина, и перемешивание продолжалось дополнительно в течение часа при -78oC, после чего реакционной смеси давалась возможность достичь комнатной температуры, при которой она перемешивалась в течение ночи в атмосфере азота. Получающийся в результате раствор разбавлялся до 500 мл метиленхлоридом и промывался насыщенным солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался в вакууме, давая коричневое масло. Оно растворялось в 140 мл ледяной уксусной кислоты и перемешивалось в течение 1.5 часов при комнатной температуре в атмосфере азота. Растворитель удалялся в вакууме и заменялся 500 мл метиленхлорида. Смесь промывалась 300 мл порциями насыщенного водного раствора биркабоната натрия и насыщенным солевым раствором, сушилась над сульфатом магния, фильтровалась и концентрировалась в вакууме до оранжевой пены. Пена растворялась в 175 мл ТГФ и добавлялась к суспензии примерно 100 г никеля Ренея (ник-Рен во взвешенном состоянии в виде шлама в воде), которая промывалась водой, 0,5%-ным водным раствором уксусной кислоты, снова водой и окончательно ТГФ. Реакционная смесь перемешивалась при комнатной температуре в течение 48 часов, затем раствор декантировался, и катализатор тщательно промывался ТГФ. Объединенные органические фракции уменьшались по объему до тех пор, пока не образовался осадок. Целевое соединение (5.15 г, 50%) выделялось в виде бежевого твердого вещества, т. пл. 233-235oC, с помощью фильтрации.
Элементный анализ для: C19H19NO6S • 0,25 H2O
Вычислено: C, 57.93; H, 4.99; N, 3.56
Найдено: C, 57,72; H, 4.96; N, 3.56
Пример 28
2-[(4-метил-бензиламино)-метил] -6-метил-2,3,8,9-тетрагидро-7H- 1,4-диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-метил-2,3,8,9- тетгагидро-7H-диоксино[2,3-e] индол-8-он (1.20 г, 3.10 ммоля) и 4-метилбензиламин (2.07 г, 17.0 ммолей) объединялись в 22 мл сухого ДМСО и нагревались до 85oC в течение 3.5 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 150 мл воды, и смесь экстрагировалась дважды 250 мл порциями 35% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая 0,38 г светло-желтого твердого вещества. Полученное таким образом свободное основание целевого соединения кристаллизовалось из этанола с добавлением фумаровой кислоты (0,24 г, 1.2 ммоля), давая 0.43 г (S) энантиомера целевого соединения в виде желтого твердого четвертьэтанолята фумарата, т. пл. 225-227oC.
Элементный анализ для: C20H22N2O3• C4H4O4• 0,25 C2H6O
Вычислено: C, 63,27; H, 5.78; N, 6.08
Найдено: C, 63,46; H, 5.79; N, 5.97
Пример 29
2-{ [(тиофен-2-илметил)-амино] -метил} -6-метил-2,3,8,9-тетрагидро- 7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-метил-2,3,8,9- тетрагидро-7H-1,4-диоксин[2,3-e] индол-8-он (1.00 г, 2.57 ммоля) и тиофен-2-метиламин (1,42 г, 12.6 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 90oC в течение 6 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 250 мл воды, и в осадок выпадало коричневое твердое вещество. Оно растворялось в метиленхлориде, сушилось над сульфатом магния, фильтровалось и концентрировалось в вакууме, давая хлопьевидное оранжевое твердое вещество, которое подвергалось хроматографии на силикагельной колонке с использованием смеси 1,5% метанол/CH2Cl2 в качестве элюента. Полученное таким образом свободное основание целевого соединения (0.27 г) кристаллизовалось из изопропанола с добавлением фумаровой кислоты (0.10 г, 0.90 ммоля), давая 0.28 г (S) энантиомера целевого соединения в виде светло-оранжевого твердого фумарата, т. пл. 211-212oC.
Элементный анализ для: C17H18N2O3S•C4H4O4
Вычислено: C, 56,49; H, 4.97; N, 6.27
Найдено: C, 56,21; H, 4.99; N, 6.41
Пример 30
6-метил-2-{ [(нафталин-1-илметил)-амино] -метил} -2,3,8,9-7H- тетрагидро-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-метил-2,3,8,9-тетрагидро- 7H-1,4-диоксино[2,3-e] индол-8-он (0.80 г, 2.1 ммоля) и 1-нафталинметиламин (1.57 г, 10.3 ммоля) объединялись в 15 мл сухого ДМСО и нагревались до 90oC в течение 6 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 250 мл воды, и смесь экстрагировалась дважды 250 мл порциями 35% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая светло-оранжевое масло. Масло подвергалось хроматографии на силикагельной колонке с использованием смеси 1,5% метанол/CH2Cl в качестве элюента, давая 0.39 г свободного основания целевого соединения в виде светло-желтого твердого вещества. Оно кристаллизовалось из изопропанола с добавлением фумаровой кислоты (0.13 г, 1.2 ммоля), давая 0.36 г (S) энантиомера целевого соединения в виде светло-желтого твердого четвертьэтанолята фумарата, т. пл. 194-195oC.
Элементный анализ для: C23H22N2O3•C4H4O4
Вычислено: C, 66.11; H, 5.34; N, 5.71
Найдено: C, 66.03; H, 5.34; N, 5.80
Пример 31
2-(бензиламино-метил)-6-метил-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-метил-2,3,8,9-тетрагидро- 7H-1,4-диоксино[2,3-e] индол-8-он (1,57 г, 4.04 ммоля) и бензиламин (2.18 г, 5.6 ммоля) объединялись в 10 мл сухого ДМСО и нагревались до 85oC в течение 3 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 150 мл воды, и смесь экстрагировалась дважды 250 мл порциями 35% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая оранжевое масло. Хроматография на силикагельной колонке с использованием смеси 2% метанол/CH2Cl2 в качестве элюента давала 1.08 г свободного основания целевого соединения в виде светло-бежевого твердого вещества. Оно кристаллизовалось из изопропанола с добавлением фумаровой кислоты (0.42 г, 3.7 ммоля), давая 0.49г (S) энантиомера целевого соединения в виде бежевого твердого фумарата, т. пл. 219-220oC.
Элементный анализ для: C19H20N2O3•C4H4O4
Вычислено: C, 62,72; H, 5.49; N, 6.36
Найдено: C, 62.44; H, 5.29; N, 5.57
Пример 32
2-[(4-фтор)-бензиламино-метил] -6-метил-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-метил-2,3,8,9-тетрагидро- 7H-1,4-диоксино[2,3-e] индол-8-он (0,28г, 0.72 ммоля) и 4-фторбензиламин (0.45 г, 3.6 ммоля) объединялись в 10 мл сухого ДМСО и нагревались до 90oC в течение 4 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 150 мл воды, и смесь экстрагировалась дважды 250 мл порциями 35% этилацетата в гексане. Объединенные органические фазы промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме, давая оранжевое полутвердое вещество. Хроматография на силикагельной колонке с использованием смеси 1% метанол/CH2Cl2 в качестве элюента давала свободное основание целевого соединения в виде коричневого масла. Оно кристаллизовалось из изопропанола с добавлением фумаровой кислоты (0.20 г, 1.7 ммоля), давая 0.06 г (S) энантиомера целевого соединения в виде бежевого твердого фумарата, т. пл. 233-234oC.
Элементный анализ для: C19H19FN2O3•C4H4O4
Вычислено: C, 60.26; H, 5.06; N, 6.11
Найдено: C, 59.96; H, 4.87; N, 6.14
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 12
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3-дигидробензо[1,4] диоксин
(S)-(6-фтор)-2,3-дигидробензо(1,4-диоксин-2-ил)метанол (17 г, 92 ммоля) растворялся в 1 литре пиридина. К данному раствору добавлялось 38 г (0.20 моля) п-толуолсульфонилхлорида, и смесь перемешивалась при комнатной температуре в атмосфере азота в течение трех дней. Реакционная смесь охлаждалась в ванне из смеси лед/вода и к ней медленно добавлялось 10 мл воды. Смесь перемешивалась при комнатной температуре в течение 2 часов, и затем растворитель удалялся в вакууме и заменялся 800 мл метиленхлорида. Данный раствор промывался дважды 500 мл 1 н. водным раствором HCl, насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором и сушился над сульфатом натрия. Фильтрование, выпаривание в вакууме и хроматографирование на силикагельной колонке с использованием 50% гексана в дихлорметане в качестве элюента давали 25.1 г (89%) целевого соединения в виде не совсем белого твердого вещества.
1H (CDCl3) дублет, 7.86 δ (2H); дублет, 7.32 δ (2H); дублет дублетов, 6,65 δ (1H); мультиплет, 6.58 δ (2H); мультиплет, 4.34 δ (1H); мультиплет, 4.20 δ (3H); мультиплет, 4.00 δ (1H); синглет, 2.43 δ (3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 13
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-7-нитро-2,3- дигидробензо[1,4] диоксин
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3-дигидробензо[1,4] диоксин (25,1 г, 74 ммоля) растворялся в 250 мл дихлорэтана и охлаждался до 0oC в ванне лед/вода. К данному охлажденному раствору в течение 15 минут по каплям добавлялся раствор азотной кислоты (удельн. вес 1.49) в 60 мл дихлорэтана. Смесь перемешивалась при 0oC в атмосфере азота в течение двух часов, после чего реакция гасилась путем добавления 500 г льда. Смесь разбавлялась до 700 мл метиленхлоридом и промывалась насыщенным водным раствором бикарбоната натрия, водой, насыщенным солевым раствором и сушилась над сульфатом натрия. Фильтрование и выпаривание в вакууме давали 25 г неочищенного продукта. Он подвергался хроматографии на силикагельной колонке с использованием смеси 1: 1 гексан/этилацетат в качестве элюента, давая 21 г целевого соединения в виде желтого твердого вещества.
1H (CDCl3) дублет, 7.80 δ (2H) дублет, 7.50 δ (1H); дублет, 7.38 δ (2H); дублет, 6.76 δ (1H) мультиплет, 4.40 δ (2H); мультиплет, 4.25 δ (2H); мультиплет, 4.15 δ (1H); синглет, 2.43 δ (3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 14
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-7-амино-2,3- дигидробензо-[1,4] диоксин
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-7-нитро-2,3- дигидробензо[1,4] диоксин (21 г, 55 ммолей) добавлялся к суспензии 2,0 г 10% палладия на угле в 250 мл метанола. К ней добавлялось 15 мл 4 н. изопропанольной HCl. Смесь гидрировалась в течение 20 часов с использованием аппарата Парра при давлении азота 50-60 фунт/кв. дюйм (3.515-4.219 кг/кв. см. ). Смесь затем фильтровалась через целит, и катализатор промывался дополнительным количеством метанола. Фильтрат концентрировался в вакууме, давая 21,4 г целевого соединения в виде серого твердого хлоргидрата.
1H (ДМСО-d6) дублет, 7.80, δ (2H); дублет, 7.47 δ (2H); дублет, 6.95 δ (1H); дублет, 6.85 δ (1H); мультиплет, 4.40 δ (1H); мультиплет, 4.25 δ (3H); мультиплет 4.00 δ (1H); синглет, 2.40 δ (3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 15
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
В трехгорлую колбу, снабженную капельной воронкой, термометром и вводом азота помещалось 6.15 мл (48.0 ммолей) этилметилтиоацетата и 65 мл сухого метиленхлорида. Раствор охлаждался до -78oC с помощью сухой ванны лед/изопропанол, и к нему в течение 5 минут по каплям добавлялся раствор 3.80 г (47.0 ммолей) хлорида серы в 30 мл метиленхлорида. Реакционная смесь поддерживалась при температуре -78oC в течение 30 минут. В течение 1 часа по каплям к смеси добавлялся раствор (R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-7-амино-2,3- дигидробензо[1,4] диоксина (15.7 г, 45.0 ммолей) и Proton Sponge. (11.7 г, 47.0 ммолей) в 150 мл метиленхлорида. Смесь перемешивалась при -78oC в течение 2 часов, затем в течение 10 минут по каплям добавлялся раствор 9,5 г (54 ммоля) диизопропилэтиламина в 20 мл дихлорметана и перемешивание продолжалось в течение дополнительного часа при -78oC, после чего реакционной смеси дозволялось достичь комнатной температуры, при которой она перемешивалась в течение 8 часов в атмосфере азота. Получающийся в результате раствор разбавлялся до 500 мл метиленхлоридом и промывался насыщенным солевым раствором, сушился над сульфатом магния, фильтровался и концентрировался в вакууме до образования коричневого масла. Оно растворялось в 200 мл ледяной уксусной кислоты и перемешивалось в течение 10 часов при комнатной температуре в атмосфере азота. Затем растворитель удалялся в вакууме и замещался 500 мл метиленхлорида. Смесь промывалась 300 мл порциями насыщенного водного раствора бикарбоната натрия и насыщенного солевого раствора, сушилась над сульфатом магния, фильтровалась и концентрировалась в вакууме, давая коричневое масло, которое подвергалось хроматографии на силикагельной колонке с использованием 2%-ной смеси метанола в метиленхлориде в качестве элюента. Полученное таким образом светло-коричневое твердое вещество (13.0 г, 66%) растворялось в 200 мл ТГФ и добавлялось к суспензии приблизительно 200 г никеля Ренея (ник-Рен во взвешенном состоянии в виде шлама в воде) в 600 мл ТГФ, которая промывалась водой, 0.5% водной уксусной кислотой, снова водой и окончательно ТГФ. Реакционная смесь перемешивалась при комнатной температуре в течение 8 часов, затем раствор декантировался, и катализатор тщательно промывался ТГФ. Объединенные органические фракции концентрировались в вакууме, и продукт подвергался хроматографии на силикагельной колонке с использованием метиленхлорида в качестве элюента. Целевое соединение (4.54 г, 40%) выделялось в виде не совсем белого твердого вещества, т. пл. 205-206oC.
Элементный анализ для: C18H16FNO6S • 0.25 H2O
Вычислено: C, 54.34; H, 4.18; N, 3.52
Найдено: C, 54.12; H, 4.24; N, 3.41
Пример 33
2-(бензиламино-метил)-6-фтор-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он (1.0 г, 2.5 ммоля) и бензиламин (1.3 г, 12.5 ммолей) объединялись в 30 мл сухого ДМСО и нагревались при 80-90oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры, смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0.5% метанол/CHCl3 в качестве элюента, давая 0,65 г свободного основания целевого соединения в виде бледно-желтого масла. Оно кристаллизовалось из метанола с добавлением одного эквивалента фумаровой кислоты, давая 0,62 г
(S) энантиомера целевого соединения в виде желтого твердого фумарата, т. пл. 205-207oC.
Элементный анализ для: C18H17FN2O3•C4H4O4
Вычислено: C, 59.46; H, 4.76; N, 6.30
Найдено: C, 59.34; H, 4.81; N, 6.18
Пример 34
6-фтор-2-[(4-фтор-бензиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9-тетрагидро- 7H-1,4-диоксино[2,3-e] индол-8-он (1.0 г, 2.5 ммоля) и 4-фторбензиламин (1,25 г, 10 ммолей) объединялись в 30 мл сухого ДМСО и нагревались при 80-90oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке использованием смеси 0,5% метанол/CHCl3 в качестве элюента, давая 0.55 г свободного основания целевого соединения в виде бледно-желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.50 г (S) энантиомера целевого соединения в виде белого твердого фумарата, т. пл. 205-207oC.
Элементный анализ для: C18H16F2N2O3• C4H4O4
Вычислено: C, 57.15; H, 4.36; N, 6.06
Найдено: C, 56.85; H, 4.31; N, 5.92
Пример 35
6-фтор-2-[(4-метил-бензиламино)-метил] -9-(4-метил-бензилиден)- 2,3,8,9-тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9-тетрагидро-7H- 1,4-диоксино[2,3-e] индол-8-он (1.0 г, 2.5 ммоля) и 4-метилбензиламин (1.2 г, 10 ммолей) объединялись в 30 мл сухого ДМСО и нагревались при 80oC в течение 4 часов без защиты от атмосферного воздуха. После охлаждения до комнатной температуры смесь концентрировалась в высоком вакууме с одновременным нагреванием при 100oC. Добавлялось 300 мл метиленхлорида, и раствор промывался 250 мл насыщенного водного раствора бикарбоната натрия и насыщенным солевым раствором, сушился над сульфатом натрия, фильтровался и концентрировался в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0.5% метанол/CHCl3 в качестве элюента, давая 0.51 г свободного основания целевого соединения в виде желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.49 г (S) энантиомера целевого соединения в виде оранжевого твердого моногидрата фумарата, т. пл. 239-241oC.
Элементный анализ для: C27H25FN2O3• C4H4O4•H2O
Вычислено: C, 64.35; H, 5.40; N, 4.84
Найдено: C, 64.65; H, 5.26; N, 4.60
Пример 36
6-фтор-2-[(4-метил-бензиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он (1.0 г, 2.5 ммолей) и 4-метилбензиламин (1.2 г, 10 ммолей) объединялись в 30 мл сухого ДМСО и нагревались при 80oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0.5% метанол/CHCl3 в качестве элюента и кристаллизовался из этанола с добавлением фумаровой кислоты, давая 0.57 г (S) энантиомера целевого соединения в виде не совсем белого твердого фумарата, т. пл. 203-204oC.
Элементный анализ для: C19H19FN2O3C4H4O4
Вычислено: C, 60.26; H, 5.06; H, 6.11
Найдено: C, 60.13; H, 4.90; N, 6.01
Пример 37
2-{ [3-(3-диметиламино-фенокси)-пропиламино] -метил} -6-фтор-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9-тетрагидро-7H- 1,4-диоксино[2,3-e] индол-8-он (1.0 г, 2.5 ммоля) и 3-(3-диметиламинофенокси)-пропиламин (1.92 г, 10 ммолей) объединялись в 30 мл сухого ДМСО и нагревались при 80-90oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 1% метанол/CHCl3 в качестве элюента, давая 0.25г свободного основания целевого соединения в виде желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.19 г (S) энантиомера целевого соединения в виде рыжевато-коричневого твердого фумарата, т. пл. 121-123oC.
Элементный анализ для: C22H26FN3O4•C4H4O4
Вычислено: C, 58.75; H, 5.69; N, 7.91
Найдено: C, 58.95; H, 5.73; N, 8.09
Пример 38
2-{ [(адамантан-1-илметил)-амино] -метил} -6-фтор-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он (1.0 г, 2.5 ммоля) и 1-адамантилметиламин (1.6 г, 10 ммолей) объединялись в 30 мл ДМСО и нагревались при 80-90oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры, смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0.5% метанол/CHCl3 в качестве элюента, давая 0.70 г свободного основания целевого соединения в виде бесцветного масла. Оно кристаллизовалось из этанола с добавлением 4 н. изопропанольной HCl, давая 0.54 г (S) энантиомера целевого соединения в виде белого твердого мелкокристаллического хлоргидрата, т. пл. более 260oC.
Элементный анализ для: C22H27FN2O3•HCl
Вычислено: C, 62.48; H, 6.67; N, 6.62
Найдено: C, 62,13; H, 6.82; N, 6.56
Пример 39
6-фтор-2-{ [3-(1H-индол-3-ил)-пропиламино] -метил} -2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9-тетрагидро-7H- 1,4-диоксино[2,3-e] индол-8-он (1.0 г, 2.5 ммоля) и 3-(3-аминопропил) индол (1.74 г, 10 ммолей) объединялись в 30 мл сухого ДМСО и нагревались при 80-90oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0.5% метанол/CHCl3 в качестве элюента, давая 0.33 г свободного основания целевого соединения в виде бледно-желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.29 г (S) энантиомера целевого соединения в виде желтого твердого фумарата, т. пл. 133oC.
Элементный анализ для: C22H22FN3O3•C4H4O4
Вычислено: C, 61.05; H, 5.12; N, 8.21
Найдено: C, 61.39; H, 5.40; N, 8.24
Пример 40
2-[(4-хлор-бензиламино)-метил] -6-фтор-2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9-тетрагидро-7H- 1,4-диоксино[2,3-e] индол-8-он (0.55 г, 1.4 ммоля) и 4-хлорбензиламин (1.4 г, 10 ммолей) объединялись в 40 мл сухого ДМСО и нагревались при 100oC в течение 6 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0.5% метанол/CHCl3 в качестве элюента, давая 0.13 г свободного основания целевого соединения в виде бледно-желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента (R)-миндальной кислоты, давая 0,06 г (S) энантиомера целевого соединения в виде персикового цвета твердой соли (R)-миндальной кислоты, т. пл. 187-188oC.
Элементный анализ для: C18H16ClFN2O3•C8H8O3
Вычислено: C, 60.65; H, 4.70; N, 5.44
Найдено: C, 60.48; H, 4.49; N, 5.37
Пример 41
6-фтор-2[((4-трифторметил-бензиламина)-метил] -2,3,8,9- тетрагидро-7H-диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9-тетрагидро- 7H-1,4-диоксино[2,3-e] индол-8-он (0,93 г, 2.37 ммоля) и 4-трифторметилбензиламин (1.65 мл, 11.6 ммоля) объединялись в 13 мл сухого ДМСО и нагревались при 85oC в течение 3.5 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 200 мл воды, и смесь экстрагировалась двумя 250 мл порциями смеси 1: 1 этилацетат/гексан, и объединенные экстракты промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 2% метанол/CH2Cl2 в качестве элюента, давая 0,70 г свободного основания целевого соединения в виде желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0,32 г (S) энантиомера целевого соединения в виде белого твердого трехчетвертного гидрата фумарата, т. пл. 192oC.
Элементный анализ для: C19H16F4N2O3• C4H4O4• 0.75 H2O
Вычислено: C, 52.53; H, 4.12; N, 5.33
Найдено: C, 52.27; H, 3.85; N, 5.28
Пример 42
6-фтор-2-[(4-фенил-бутиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-фтор-2,3,8,9-тетрагидро-7H- 1,4-диоксино[2,3-e] индол-8-он (0,85 г, 2.16 ммоля) и 4-фенилбутиламин (1.55 мл, 9.8 ммоля) объединялись в 20 мл сухого ДМСО и нагревались при 85oC в течение 3,5 часов в атмосфере азота. После охлаждения до комнатной температуры добавлялось 200 мл воды, и смесь экстрагировалась двумя 250 мл порциями смеси 1: 1 этилацетат и гексана, и объединенные экстракты промывались солевым раствором, сушились над сульфатом магния, фильтровались и концентрировались в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0,5% метанол/CH2Cl2 в качестве элюента, давая 0.40 г свободного основания целевого соединения в виде желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0,33 г (S) энантиомера целевого соединения в виде светло-желтого твердого четвертьгидрата полуфумарата, т. пл. 164oC.
Элементный анализ для: C21H23FN2O3• 0.5 C4H4O4• 0,25 H2O
Вычислено: C, 63.80; H, 5.94; N, 6.47
Найдено: C, 63.81; H, 5.75; N, 6.33
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 16
2-аллилокси-5-хлорфенол
К 14 г (0.35 моля) 60% дисперсии гидрида натрия в минеральном масле в двухлитровой колбе добавлялось 500 мл гексана. Смесь непродолжительно перемешивалась, твердое вещество осаждалось, и плавающая сверху жидкость декантировалась. Добавлялся ДМФ (800 мл) с последующим добавлением раствора 47 г (0.30 моля) 5-хлорсалицилальдегида в 50 мл ДМФ. Смесь перемешивалась при комнатной температуре в атмосфере азота в течение 30 минут, затем добавлялось 54,5 г (0.45 моля) аллилбромида. Смесь нагревалась при 65oC в атмосфере азота в течение 18 часов. Растворитель удалялся в вакууме и заменялся одним литром метиленхлорида. Данный раствор промывался водой и насыщенным солевым раствором и сушился над сульфатом натрия. Затем он фильтровался и добавлялось 150 г (0.50-0.75 моля) 57-86% м-хлорпероксибензойной кислоты, и смесь перемешивалась при комнатной температуре в течение ночи. Реакционная смесь фильтровалась, и фильтрат промывался тремя 400 мл порциями насыщенного водного раствора бикарбоната натрия и насыщенным солевым раствором, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток растворялся в 750 мл метанола и перемешивался со 150 г основной окиси алюминия в течение 15 часов. Затем смесь фильтровалась и выпаривалась в вакууме, и неочищенный продукт распределялся между 500 мл каждого из 1 н. водного NaOH и метиленхлорида. Органическая фаза экстрагировалась с дополнительными 500 мл основания, и объединенные основные экстракты промывались 500 мл метиленхлорида. Наконец, основные экстракты осторожно подкислялись концентрированной HCl и экстрагировались двумя 400 мл порциями метиленхлорида. Объединенные органические экстракты промывались насыщенным солевым раствором, сушились над сульфатом натрия, фильтровались и концентрировались в вакууме, давая 37.3 г целевого соединения в виде желтого масла.
1H (CDCl3) синглет, 6.90 δ (1H); квартет, 6.70 δ (2H); синглет, 6.20 δ (1H, OH); мультиплет, 5,97 δ (1H); квартет, 5.25 δ (2H); дублет, 4.50 δ (2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 17
(R)-2-(2-аллилокси-5-хлорфеноксиметил)-оксиран
К 4.0 г (79 ммолей) 60% дисперсии гидрида натрия в минеральном масле в однолитровой колбе добавлялось 300 мл гексана. Смесь непродолжительно перемешивалась, твердое вещество выпадало в осадок, и плавающая сверху жидкость декантировалась. Добавлялся ДМФ (500 мл) с последующим добавлением раствора 14.6 г (79 ммолей) 2-аллилокси-5-хлорфенола в 100 мл ДМФ. Смесь перемешивалась при комнатной температуре в атмосфере азота в течение 30 минут, затем добавлялось 18.0 г (79 ммолей) (R)-глицидилтозилата. Смесь нагревалась в атмосфере азота при 80oC в течение 24 часов. Растворитель выпаривался в вакууме и заменялся 750 мл метиленхлорида. Раствор промывался 500 мл порциями 2 н. водной HCl, насыщенным водным раствором бикарбоната натрия, насыщенным солевым раствором, сушился над сульфатом натрия, фильтровался и выпаривался в вакууме, давая 20 г неочищенной смолы. Она подвергалась хроматографии на силикагельной колонке с использованием метиленхлорида в качестве элюента, давая 9.2 г целевого соединения в виде бесцветного масла.
1H (CDCl3) синглет, 6.93 δ (1H); дублет, 6.90 δ (1H); дублет, 6.80 δ (1H); мультиплет, 6.05 δ (1H); дублет, 5.40 δ (1H); дублет, 5.25 δ (1H); дублет 4.55 δ (2H); дублет дублетов, 4.25 δ (1H); дублет дублетов 3,97 δ (1H); мультиплет, 3,35 δ (1H); триплет, 2.95 δ (2H); триплет, 2.75 δ (1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 18
(S)-(8-аллил-6-хлор-2,3-дигидро-бензо[1,4] диоксин-2-ил)-метанол
(R)-2-(2-аллилокси-5-хлорфеноксиметил)-оксиран (9.2 г, 38 ммолей) растворялся в 500 мл мезитилена и нагревался с обратным холодильником в атмосфере азота в течение 48 часов. Растворитель удалялся в вакууме и заменялся 500 мл этанола. Добавлялся бикарбонат натрия (50 г, 0.60 моля), и смесь перемешивалась при комнатной температуре в атмосфере азота в течение 24 часов. Смесь затем фильтровалась и концентрировалась в вакууме. Растворитель заменялся 500 мл метиленхлорида, и раствор промывался водой и насыщенным солевым раствором, сушился над сульфатом натрия, фильтровался и выпаривался в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием хлороформа в качестве элюента, давая 8.9 г целевого соединения в виде бесцветного масла.
1H (CDCl3) синглет, 6.80 δ (1H); синглет 6.73 δ (1H); мультиплет, 5.95 δ (1H); дублет, 5.10 δ (1H); дублет, 5.05 δ (1H); мультиплет, 4.25 δ (2H); мультиплет, 4.10 δ (1H); мультиплет, 3.85 δ (2H); мультиплет, 3.30 δ (2H); широкий синглет, 2.00 δ (1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 19
(R)-8-аллил-6-хлор-2,3-дигидро-бензо-(1,4) диоксин-2-ил-метиловый эфир толуол-4-сульфоновой кислоты
8.9 г (37 ммолей) (S)-8-аллил-6-хлор-2,3-дигидро-бензо[1,4] -диоксин-2-ил)метанола растворялось в 500 мл пиридина, добавлялось 14,3 г (75 ммолей) п-толуолсульфонилхлорида, и смесь перемешивалась при комнатной температуре в атмосфере азота в течение 3 дней. Затем добавлялась вода для гашения избыточного тозилхлорида, и растворитель удалялся в вакууме и замещался 500 мл метиленхлорида. Данный раствор промывался дважды 300 мл 2 н. водной HCl, насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, и сушился над сульфатом натрия. Фильтрование, выпаривание в вакууме и хроматография на силикагельной колонке смесью 1: 1 гексан/метиленхлорид в качестве элюента давали 10,9 г желаемого тозилата в виде бесцветного масла.
1H (CDCl3) дублет, 7.80 δ (2H); дублет, 7.30 δ (2H); синглет, 6.75 δ (1H); синглет, 6.70 δ (1H); мультиплет, 5.85 δ (1H); синглет, 5,08 δ (1H); дублет, 5.03 δ (1H); мультиплет, 4.40 δ (1H); мультиплет, 4.20 δ (2H); дублет дублетов, 4,05 δ (1H); мультиплет, 3.20 δ (2H); синглет, 2.45 δ (3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 20
(R)-(7-хлор-3-(толуол-4-сульфонилоксиметил)-2,3-дигидро-бензо(1,4)- диоксин-5-ил)-уксусная кислота
Перманганат калия (14 г, 87 ммолей) растворялся в 140 мл воды и помещался в водяную баню. К нему добавлялось 1.4 г (4.9 ммоля) хлористого тетра-н-бутиламмония, и затем в течение 30 минут по каплям добавлялся раствор 10.9 г (R)-8-аллил-6-хлор-2,3-дигидро-бензо[1,4] -диоксин-2-илметилового эфира толуол-4-сульфоновой кислоты (28 ммолей) в 100 мл бензола. Смесь перемешивалась при комнатной температуре в течение ночи. Затем добавлялся бисульфит натрия (17 г, 0.12 моля), и смесь подкислялась концентрированной HCl и экстрагировалась двумя 300 мл порциями этилацетата. Объединенные экстракты промывались водой и насыщенным солевым раствором и сушились над сульфатом натрия. Фильтрование, концентрирование в вакууме и хроматография на силикагельной колонке с использованием смеси 3% метанола в метиленхлориде давали 5.9 г целевого соединения в виде вязкого желтого масла.
1H (CDCl3) дублет, 7.75 δ (2H); дублет, 7.35 δ (2H); синглет, 6.78 δ (1H); синглет, 6.75 δ (1H); мультиплет, 4.40 δ (1H); мультиплет, 4.20 δ (3H); дублет дублетов, 4.05 δ (1H); коллапсированный AB квартет, 3.50 δ (2H); синглет, 2.45 δ (3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 21
(R)-(7-хлор-6-нитро-3-(толуол-4-сульфонилоксиметил)-2,3- дигидробензо-(1,4)диоксин-5-ил)-уксусная кислота
К смеси 3.0 г (7.3 ммоля) (R)-(7-хлор-3-(толуол-4-сульфонилоксиметил)-2,3- дигидробензо(1,4)диоксин-5-ил) -уксус ной кислоты в 50 мл дихлорэтана в ванне лед/вода добавлялся раствор 3,6 мл (85 ммолей) азотной кислоты (удельн. вес 1.49) в 50 мл дихлорэтана. Смеси давалось достичь комнатной темпера туры, и она перемешивалась в течение ночи. Для гашения реакции добавлялся лед, и смесь разбавлялась до 300 мл метиленхлоридом, промывалась водой и насыщенным солевым раствором, сушилась над сульфатом натрия. Концентрирование в вакууме давало 2.8 г целевого соединения в виде не совсем белого твердого вещества.
1H (ДМСО-d6) широкий синглет, 12.75 δ (1H); дублет, 7.80, δ (2H); дублет, 7.45 δ (2H); синглет, 7.15 δ (1H); мультиплет, 4.60 δ (1H); мультиплет 4.37 δ (2H); мультиплет, 4.15 δ (2H); синглет, 3.45 δ (2H); синглет, 2.40 δ (3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 22
(R)-2-(толуол-4-сульфонилоксиметил)-6-хлор-2,3,8,9-тетрагидро-7H- 1,4-диоксино[2,3-e] индол-8-он
К раствору 2.8г (6.1 ммоля) (R)-(7-хлор-6-нитро-3-(толуол-4-сульфонилоксиметил)-2,3- дигидро-бензо(1,4)диоксин-5-ил)-уксусной кислоты в 200 мл метанола добавлялось 100 мг оксида платины. Смесь гидрировалась в аппарате Парра при давлении 50 фунт/кв. дюйм (3.515 кг/кв. см) в течение 4 часов. Смесь фильтровалась через целит и концентрировалась в вакууме, давая 2.8 г неочищенной аминокислоты. Она повторно растворялась в 250 мл метанола и добавлялось 50 мл 4 н. изопропанольной HCl. Смесь нагревалась при 50oC в течение 24 часов. Растворитель удалялся в вакууме и добавлялось 400 мл этилацетата. Данный раствор промывался 200 мл воды, 200 мл насыщенного водного раствора бикарбоната натрия и насыщенным солевым раствором, сушился над сульфатом натрия, фильтровался и концентрировался в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 1% метанола в метиленхлориде в качестве элюента, давая 1,4 г целевого соединения в виде не совсем белого твердого вещества.
1H (ДМСО-d6) синглет, 10.63 δ(1H); дублет, 7.80 δ (2H); дублет, 7.45 δ (2H); синглет, 6.75 δ (1H); мультиплет, 4.55 δ (1H); мультиплет, 4.35 δ (1H); мультиплет, 4.20 δ (2H); дублет дублетов, 4.03 δ (2H); AB квартет, 3.25 δ (2H); синглет, 2.40 δ (3H).
Пример 43
6-хлор-2-[((4-хлор-бензиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] иидол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-хлор-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он (0.80 г, 2.0 ммоля) и 4-хлорбензиламин (1.1 г, 8.0 ммолей) объединялись в 25 мл сухого ДМСО и нагревались при 80-90oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0,5% метанол/CHCl3 в качестве элюента, давая 0,36 г свободного основания целевого соединения в виде бледно-желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0.28 г (S) энантиомера целевого соединения в виде рыжевато-коричневого твердого полугидрата фумарата, т. пл. 207-209oC.
Элементный анализ для: C18H16Cl2N2O3• C4H4O4• 0.5 H2O
Вычислено C, 52.39; H, 4.20; N, 5.55
Найдено: C, 52.37; H, 4.01; N, 5.61
Пример 44
6-хлор-2-[(4-метил-бензиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-хлор-2,3,8,9-тетрагидро-7H- диоксино[2,3-e] индол-8-он (0,80 г, 2.0 ммоля) и 4-метилбензиламин(1.0 г, 8.0 ммолей) объединялись в 25 мл сухого ДМСО и нагревались при 80-90oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0,5% метанол/CHCl3 в качестве элюента, давая 0.43 г свободного основания целевого соединения в виде бледно-желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0,32 г (S) энантиомера целевого соединения в виде белого твердого фумарата, т. пл. 218 - 219oC.
Элементный анализ для: C19H19ClN2O3•C4H4O4
Вычислено: C, 58.17; H, 4,88; N, 5.90
Найдено: C, 57.84; H, 4.53; N, 5.97
Пример 45
2-(бензиламино)-метил-6-хлор-2,3,8,9-тетрагидро-7H-1,4- диоксина[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-хлор-2,3,8,9- тетрагидро-7H-1,4-диоксино[2,3-e] индол-8-он (0.80 г, 2.0 ммоля) и бензиламин (0,86 г, 8,0 ммолей) объединялись в 25 мл сухого ДМСО и нагревались при 80-90oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0,5% метанол/CHCl3 в качестве элюента, давая 0,33 г свободного основания целевого соединения в виде бледно-желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0,28 г (S) энантиомера целевого соединения в виде белого твердого фумарата, т. пл. 192-193oC.
Элементный анализ для: C18H17ClN2O3•C4H4O4
Вычислено: C, 57,34; H, 4.59; N, 6.08
Найдено: C, 57,45; H, 4.48; N, 6.26
Пример 46
6-хлор-2-[(4-фтор-бензиламино)-метил] -2,3,8,9-тетрагидро-7H-1,4- диоксино[2,3-e] индол-8-он
(R)-2-(толуол-4-сульфонилоксиметил)-6-хлор-2,3,8,9-тетрагидро-7H- 1,4-диоксино[2,3-e] индол-8-он (0.80 г, 2.0 ммоля) и 4-фторбензиламин (1.0 г, 0.80 ммоля) объединялись в 25 мл сухого ДМСО и нагревались при 80-90oC в течение 4 часов в атмосфере аргона. После охлаждения до комнатной температуры смесь разбавлялась 500 мл смеси 1: 1 этилацетат/гексан и промывалась 250 мл насыщенного водного раствора бикарбоната натрия и двумя 250 мл порциями воды, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался хроматографии на силикагельной колонке с использованием смеси 0,5% метанол/CHCl3 в качестве элюента, давая 0,41 г свободного основания целевого соединения в виде бледно-желтого масла. Оно кристаллизовалось из этанола с добавлением одного эквивалента фумаровой кислоты, давая 0,29 г (S) энантиомера целевого соединения в виде желтого твердого четвертьгидрата фумарата, т. пл. 213-214oC.
Элементный анализ для: C18H16ClFN2O3• C4H4O4 • 0,25 H2O
Вычислено: C, 54.67; H, 4.27; N, 5.79
Найдено: C, 54.54; H, 4.08; N, 5,60
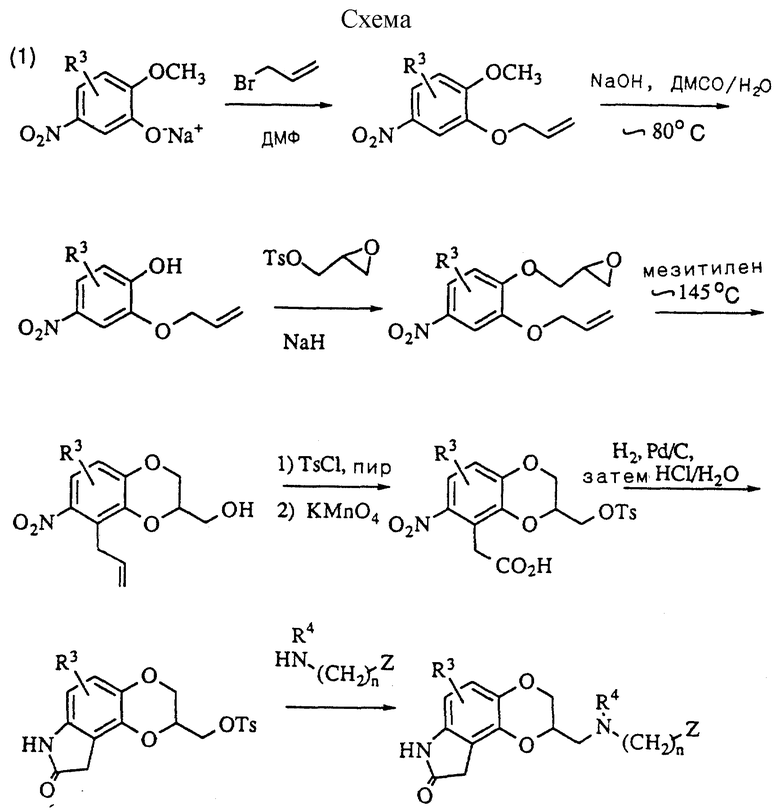
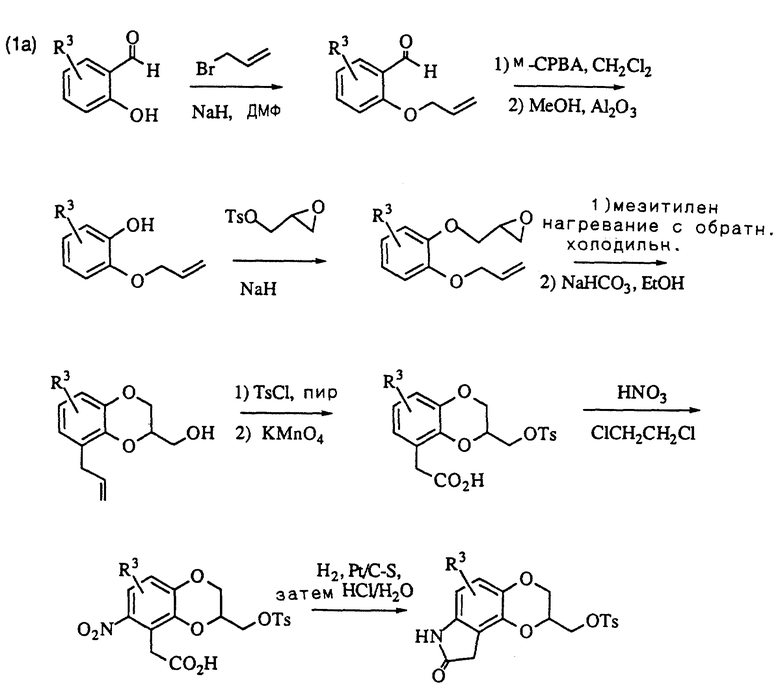
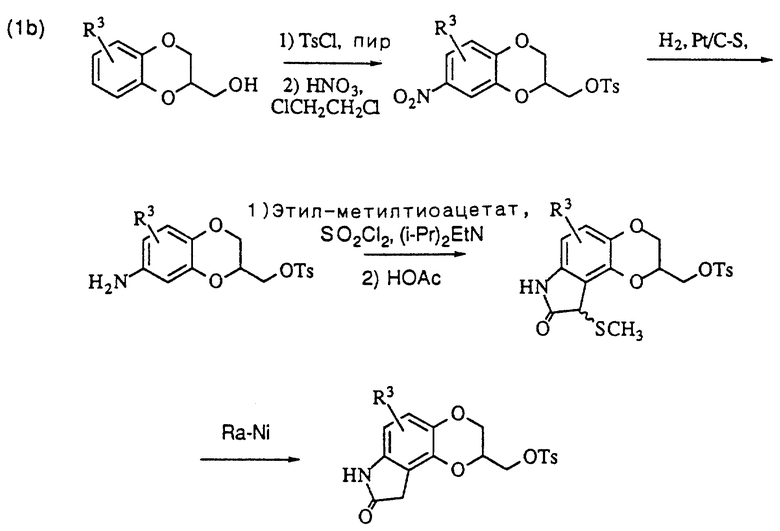


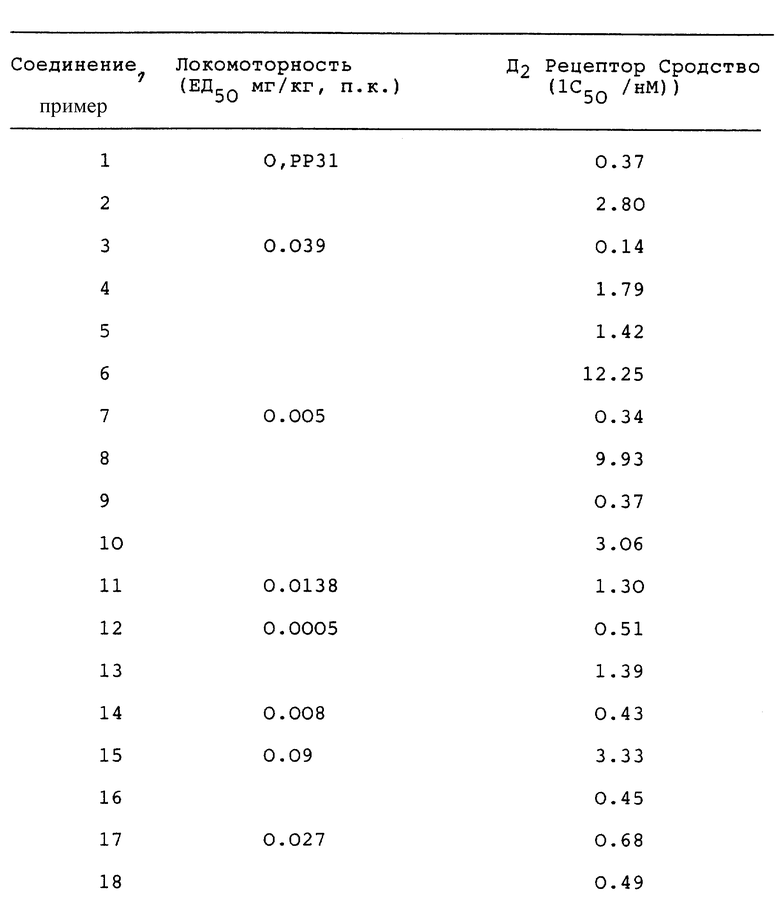

Описываются новые соединения -аминометил-2,3,8,9-тетрагидро-7Н-1-диоксино[2,3-е] -индол-8-оновые соединения формулы I, в которой R1 и R2 представляют собой независимо водород; или R1 и R2, взятые вместе, представляют бензилиден, необязательно замещенный радикалом R3, определенным ниже, или R1 и R2, взятые вместе с углеродом, к которому они присоединены, образуют карбонильный фрагмент; R3 - водород, галоид, трифторметил, трифторметокси, алкил с 1-6 атомами углерода, алкокси с 1-6 атомами углерода, амино, моно- или диалкиламино, в котором каждая алкильная группа имеет 1-6 атомов углерода, алканамидо с 2-6 атомами углерода; R4 - водород или алкил с 1-6 атомами углерода; m = одно из целых числе 0 или 1; n = 0-6 целое число; Z - водород, гидрокси, алкил с 1 - 6 атомами углерода, алкенил с 2-6 атомами углерода, алкинил с 1-6 атомами углерода, циклоалкил с 3-8 атомами углерода, полициклический алкил с 7-15 атомами углерода, фенил, необязательно замещенный R3, определенным выше, фенокси, необязательно замещенный R3, определенным выше, нафтил, необязательно замещенный радикалом R3, определенным выше, нафтилокси, необязательно замещенный радикалом R3, определенным выше, гетероарил или гетероарилокси, в которых гетероциклическое кольцо гетероарила или гетероарилокси группы выбрано из тиофена, фурана, пиридина, индола, хинолина и гетероциклическое кольцо является необязательно замещенным радикалом R3, определенным выше; или их фармацевтически приемлемые соли. Описывается также способ лечения. Новые соединения могут найти применение для лечения расстройств допаминэргической системы. 2 с. и 40 з. п. ф-лы, 1 табл.
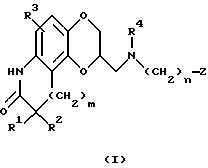
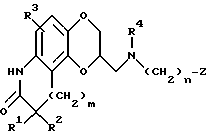
в которой R1 и R2 представляют собой независимо водород, или R1 и R2, взятые вместе, представляют бензилиден, необязательно замещенный радикалом R3, определенным ниже, или R1 и R2, взятые вместе с углеродом, к которому они присоединены, образуют карбонильный фрагмент;
R3 - водород, галоид, трифторметил, трифторметокси, алкил с 1-6 атомами углерода, алкокси с 1-6 атомами углерода, амино, моно- или диалкиламино, в котором каждая алкильная группа имеет 1-6 атомов углерода, алканамидо с 2-6 атомами углерода;
R4 - водород или алкил с 1-6 атомами углерода;
m = 0 или 1;
n = 0 - 6, целое число;
Z - водород, гидрокси, алкил с 1-6 атомами углерода, алкенил с 2-6 атомами углерода, алкинил с 1-6 атомами углерода, циклоалкил с 3-8 атомами углерода, полициклический алкил с 7-15 атомами углерода, фенил, необязательно замещенный R3, определенным выше, фенокси, необязательно замещенный R3, определенным выше, нафтил, необязательно замещенный радикалом R3, определенным выше, нафтилокси, необязательно замещенный радикалом R3, определенным выше, гетероарил или гетероарилокси, в которых гетероциклическое кольцо гетероарила или гетероарилоксигруппы, выбрано из тиофена, фурана, пиридина, индола, хинолина и гетероциклическое кольцо является необязательно замещенным радикалом R3, определенным выше,
или их фармацевтически приемлемые соли.
2-(бензиламино-метил)-1-бензилиден-2,3,8,9-тетрагидро-7Н-1,4-диоксино[2,3-е] индол-8-он,
2-(бензиламино-метил)-2,3,9,10-тетрагидро-7Н-1,4-диоксино[2,3-f] хинолин-8-он,
2-аминометил-2,3,8,9-тетрагидро-7Н-1,4-диоксино[2,3-е] индол-8-он,
2-[(2-пиридин-3-ил-этиламино)-метил] -2,3,8,9-тетрагидро-7Н-1,4-диоксино[2,3-е] индол-8-он,
2-(проп-2-иниламинометил)-2,3,8,9-тетрагидро-7Н-1,4-диоксино[2,3-е] индол-8-он,
2-{ [тиофен-2-илметил)-амино] -метил} -6-метил-2,3,8,9-тетрагидро-7Н-1,4-диоксино[2,3-е] индол-8-он,
6-фтор-2-[(4-метил-бензиламино)-метил] -9-(4-метил-бензилиден)-2,3,8,9-тетрагидро-7Н-1,4-диоксино[2,3-е] индол-8-он, или его фармацевтически приемлемую соль.
в которой R1 и R2 представляют собой независимо водород, или R1 и R2, взятые вместе, представляют бензилиден, необязательно замещенный радикалом R3, определенным ниже, или R1 и R2, взятые вместе с углеродом, к которому они присоединены, образуют карбонильный фрагмент;
R3 - водород, галоид, трифторметил, трифторметокси, алкил с 1-6 атомами углерода, алкокси с 1-6 атомами углерода, амино, моно- или диалкиламино, в которых каждая алкильная группа имеет 1-6 атомов углерода, алканамидо с 2-6 атомами углерода;
R4 - водород или алкил с 1-6 атомами углерода;
m = 0 или 1;
n = 0 - 6, целое число;
Z - водород, гидрокси, алкил с 1-6 атомами углерода, алкенил с 2-6 атомами углерода, алкинил с 1-6 атомами углерода, циклоалкил с 3-8 атомами углерода, полициклический алкил с 7-15 атомами углерода, фенил, необязательно замещенный R3, определенным выше, фенокси, необязательно замещенный R3, определенным выше, нафтил, необязательно замещенный R3, определенным выше, или нафтилокси, необязательно замещенный R3, определенным выше, гетероарил или гетероарилокси, в которых гетероциклическое кольцо гетероарильной или гетероарилоксигруппы выбрано из тиофена, фурана, пиридина, индола, хинолина и гетероциклическое кольцо является необязательно замещенным радикалом R3, определенным выше,
или его фармацевтически приемлемой соли.
Приоритет по пунктам и признакам:
06.11.1995 по пп. 1-16;
15.10.1996 по пп. 17-46 и признакам: R1 и R2 - бензилиден, необязательно замещенный группой R3, где R3 - трифторметоксигруппа, Z-алкенил с 2-6 атомами углерода, алкинил с 2-6 атомами углерода, а в качестве заболевания, которое можно лечить соединениями формулы I, шизоаффективные нарушения.
| WО 8113872 A1, 19.09.1991 | |||
| US 5235055 А, 10.08.1994 | |||
| Способ получения производных 3-хинолинкарбоновой кислоты или их солей | 1975 |
|
SU582766A3 |
| Каплан Т.И | |||
| и др | |||
| Клиническая психиатрия | |||
| - М., Медицина, 1994, с.264-269, 238-240. | |||

