Настоящее изобретение относится к некоторым аза циклогексапептидным соединениям и способам их получения.
Аза циклогексапептидные соединения, в настоящем изобретении - cоединение I (Seq ID Nos. 1-15), отличаются тем, что имеют атом азота, присоединенный к циклогексапептидному кольцу по 5-му атому углерода компонента 4-гидрокси-орнитина (в дальнейшем-"C-5-орн") и могут быть представлены формулой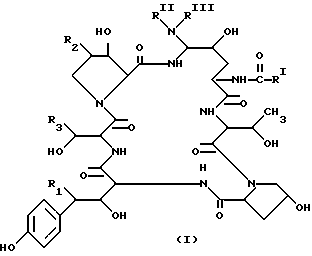
где
R1 - H или OH;
R2 - H, CH3 или OH;
R3 - H, CH3, CH2CN, CH2CH2NH2 или CH2CONH2;
RI - C9-C21 алкил, C9-C21 алкенил, C1-C10 алкоксифенил или C1-C10 алкоксинафтил;
RII - H, C1-C4 алкил, C3-C4алкенил, (CH2)2OH, (CH2)2-4NRIVRV, CO(CH2)1-4NH2;
RIII - H, C1-C4 алкил, C3-C4алкенил, (CH2)2-4OH, (CH2)2-4NRIVRV или
RII и
RIII, взятые вместе, - (CH2)4-, -(CH2)5-, -(CH2)2O(CH2)2-, -(CH2)2NH(CH2)2-,
RIV - H или C1-C4алкил,
RV - H или C1-C4алкил;
и их аддитивные соли кислоты.
Использованные здесь выражения "алкил", "алкенил" или "алкокси" подразумевают радикалы как с разветвленными, так и с нормальными цепями.
Соединения в настоящем изобретении главным образом получены в виде смесей стереоизомерных форм, в которых обычно преобладает одна форма. Подбор условий с целью получения целевого изомера в качестве преобладающего не выходит за рамки обычных навыков опытного синтетика. Соединение в предпочтительной стереоизомерной форме, указанной здесь как "нормальная" форма, показаны в рабочих примерах с пунктирными линиями связей, лежащих за плоскостью в положении "C-5-орн". Обозначение "эпи" применяется к тем соединениям, в которых группа в положении "C-5-орн" находится над плоскостью.
Фармацевтически применимые соли, получаемые в качестве солей связанных кислот, образуются с такими кислотами, как хлорoводородная, бромоводородная, фосфорная, серная, малеиновая, лимонная, уксусная, винная, сукциновая, щавелевая, малоновая, глутаминовая и им подобными, и включают другие кислоты, дающие фармацевтически применимые соли, перечисленныe в Journal of Pharmaceutical Science, 66, 2, (1977).
Характерные ядра для аза производных в настоящем изобретении (cоединение I) и идентификатор последовательности (Seguence ID) для этих соединений представлены в нижеследующей табл. 1. Поскольку пептидные ядра являются неизменными, независимо от заместителей RI, RII или RIII, и поскольку номер идентификатора последовательности назначен для вариаций ядер, то амины и соли имеют те же идентификаторы последовательностей.
Одно из соединений, особенно выделяющееся из остальных, поскольку способно бороться с грибковыми инфекциями, обозначено как cоединение I-6, где RII - H, RIII - CH2CH2NH2 и RI - 9,11-диметилтридецил (ДМТД), и которое может быть отмечено как cоединение I-6-1 (Seq. ID N 6).
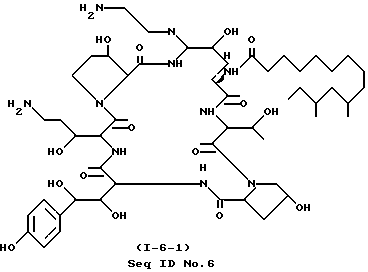
Вышеназванное обозначение I-6-1 относится к первому соединению, в котором расположение ядра - I-6. Поскольку во всех соединениях в настоящем изобретении заместитель в "C-5-орн" - азот, заместители по указанному атому азота могут варьироваться, и все соединения, которые имеют одинаковые R1, R2 и R3, будут относится к Seq ID N 6.
Соединения растворимы в низших спиртах и полярных апротонных растворителях, таких как диметилформамид (ДМФА), диметилсульфоксид (ДМСО) и пиридин. Они нерастворимы в растворителях типа диэтилового эфира и ацетонитрила.
Соединения по настоящему изобретению используется как антибиотики, особенно как фунгициды, или как средства борьбы с простейшими организмами. В качестве фунгицидов они используются для борьбы как с волокнистыми грибами, так и дрожжами. Эти соединения особенно приспособлены для использования при лечении грибковых инфекций млекопитающих, особенно вызванных видами Candida, такими как C. albicans, C. tropicalis и C. preudotropicalis, видами Cryptococcus, такими как C. neoformans и, видимо, Aspergillus: A. fumidatus, A. flavus, A. niger. Они также используются для лечения или профилактики пневмонии Pneumocystis carinii, к которой пациенты с пониженным иммунитетом особенно восприимчивы, о чем будет сказано в дальнейшем.
Соединения по настоящему изобретению могут быть получены из циклопептидов формулы
путем серии превращений, в которых атом кислорода в положении "C-5-орн" (которое также может быть названо как гемиаминальное положение) в конечном итоге замещается атомом азота. Исходными материалами могут быть натуральные или модифицированные натуральные продукты, как это будет впоследствии описано. Если R1-водород, а не гидроксил, аза соединения могут быть получены в результате другой серии реакций. Метод, применимый для получения соединений, в которых R1 может быть как водородом, так и гидроксилом, описан первым.
Идентификаторы последовательностей исходных материалов показаны в табл. 2.
Соединения А-4 и А-7 описаны в литературе (J. Antibiontcs 45, 1855-60 Dec. 1992) как пневмокандин Bo и пневмокандин Ao, если RI=ДМТД.
Когда в соединении A-1 R1 и R2 представлены одним из возможных заместителей и R3 - H, CH3 или -CH2CONH2(Seq ID Nos. 16, 19, 22, 25-27 и 30), оно может быть непосредственно использовано в первом методе. Если R3 - CH2CN или -CH2CH2NH2, то группа -CH2CONH2 сначала может быть превращена в -CH2CN или -CH2CH2NH2, как это последовательно описано, и все модифицированные соединения (Seq ID Nos. 17-18, 20-21, 23-24, 28-29) могут использоваться в первом методе [см. т. 2], или иначе, соединение, в котором R3-CH2CONH2, может быть использовано для получения соединения с азотом в гемиаминальном положении, и затем -CH2CONH2-группа полученного продукта может быть превращена в -CH2CN или -CH2CH2NH2.
Если R1, R2 и R3 в исходном материале такие же, что и в продукте, то может быть использована следующая последовательность превращений:
* Положение "C-5-орн" или гемиаминальное положение.
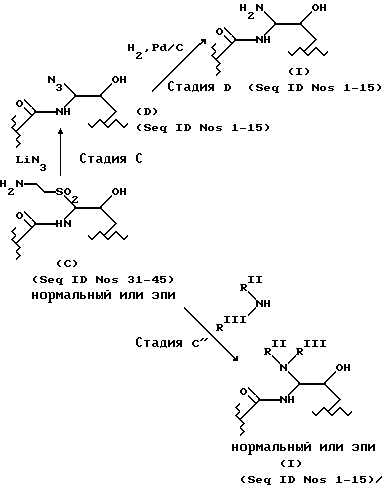
На стадии A исходный материал, соединение A (Seq ID Nos. 16-30), алкилтиол или арилтиол и кислота реагируют в апротонном растворителе в условиях отсутствия влаги в течение времени, достаточного для протекания реакции с образованием cоединения B (Seq ID Nos. 31-45), показанного в табл. 3. Было установлено, что этиламинотиол хорошо подходит для проведения этой стадии. Соединения B получены из соединений A (табл. 2).
Для cтадии A подходящими кислотами являются сильные органические и минеральные кислоты. Примерами сильных органических кислот являются сульфокамфарная кислота, п-толуолсульфокислота и метансульфокислота. Минеральные кислоты включают в себя хлороводородную и бромoводородную кислоту. Предпочтительной является сульфокамфарная кислота.
Подходящими растворителями являются ДМФА, ДМСО, 1-метил-2-пирролидон и гексаметилтриамид фосфорной кислоты (ГМАФ). Предпочтительны ДМФА и ДМСО.
Реакция в общем случае протекает при комнатной температуре в течение 1-10 дней.
При проведении реакции циклогексапептидное соединение, тиол и кислота перемешиваются в подходящем растворителе до окончания превращения. Реакционную смесь разбавляют водой и проводят обращеннофазную флэш-хроматографию, используя в качестве элюента 10-40%-ный раствор ацетонитрила в воде, содержащий 0,1% трифторуксусной кислоты. Трифторуксусная кислота в дальнейшем именуется ТФУК. Фракции, содержащие целевой продукт, могут быть сконцентрированы и лиофилизированы, и лиофилизированный материал может быть очищен препаративной высокоэффективной жидкостной хроматографией (ВЭЖХ).
Подходящие колонки для ВЭЖХ продаются под торговыми марками или торговыми названиями "ZORBAX" (DuPont), "DeltaPak" (Waters), Bio-Rad (Bio-Rad), "LICHROPREP" RP18 (E. Merck). Конкретные типы колонок указаны в рабочих примерах.
На стадии B соединение C, сульфон (Seq ID Nos. 31-45) получают окислением соединения B. Подходящими окислителями являются "OXONE" (KHSO5 : KHSO4 : K2SO4= 2: 1:1, Aldrich Chemicals), м-хлор-пероксибензойная кислота и пероксиуксусная кислота. Идентификатор последовательности (Seq ID) для cоединения C такой же, как и для cоединения B, поскольку атом, присоединенный к гемиаминальному углероду по-прежнему остается атомом серы. Таким образом, идентификаторы последовательностей (Seq ID) для сульфонов таковы (см. табл. 4).
Окисление тиоэфира (cоединение B) до сульфона (cоединение C) проводится примерно двумя молями окислителя. При использовании одного моля окислителя продуктом реакции является сульфоксид, который может быть далее превращен в сульфон. При образовании аза соединений в качестве промежуточных продуктов могут использоваться сульфоксиды, но предпочтительнее использование сульфонов. Используют незначительный избыток окислителя сверх двухмольного количества.
Реакция проводится в водной среде, преимущественно в смеси ацетонитрила с водой. Предпочтительнее примерно одинаковое содержание компонентов, хотя можно использовать смеси с их соотношением от 1:9 до 9:1.
При проведении реакции окислитель добавляется к раствору cоединения B (Seq ID Nos. 31-45) в смеси ацетонитрил: вода = 1:1, и смесь оставляет стоять при комнатной температуре на время, достаточное для завершения реакции образования cоединения C, обычно от 30 мин до 1 ч.
По завершении реакции соединение выделяют из реакционной смеси разбавлением водой и хроматографированием. Для стадии очистки подходящей является обращеннофазная колоночная флэш-хроматография (C18). Предпочтительным элюентом является 30-45%-ный ацетонитрил в воде (содержащий 0.1% ТФУК) с шагом изменения состава 5%. Нужные фракции лиофилизируют для выделения целевого промежуточного сульфона, cоединения C (Seq ID Nos. 31-45). Промежуточный продукт является неустойчивым, поэтому выделение должно проводиться как можно быстрее.
Соединение C может быть превращено в соединение, имеющее атом азота, непосредственно присоединенный к положению "C-5-орн". Как показано на схеме синтеза, реакция cоединения C с азидом щелочного металла приводит к вступлению азидной группы в то положение, тогда как реакция в аммиаком или амином дает аминогруппу в положение "C-5-орн" (cоединение I). Соединение D - важный промежуточный продукт для получения большинства веществ по настоящему изобретению. Хотя cоединение D имеет атом азота в положении "C-5-орн", но поскольку оно не является продуктом, для него установлен отдельный номер идентификатора последовательности. Номера идентификаторов последовательности (Seq ID Nos.) для cоединения D находятся в табл. 5.
Азид может быть получен добавлением азида щелочного металла к раствору сульфона (cоединения C; Seq ID Nos. 31-45) в апротонном растворители при перемешивании при комнатной температуре в течение времени, достаточного для завершения реакции образования азида, определяемого методом ВЭЖХ. После этого реакционная смесь может быть разбавлена водным раствором кислоты, например ТФУК, и затем целевой азид (соединение D) может быть выделен из реакционной смеси хроматографически. Для этого подходящей является обращеннофазная колоночная флэш-хроматография (C18) с использованием 10-25% ацетонитрила в воде (содержащего 0.1% ТФУК) с шагом изменения состава 5%.
Азид (соединение D) может быть восстановлен до соединения, имеющего свободную аминогруппу, которое представлено среди продуктов в настоящем изобретении (Seq ID Nos. 1-15).
Восстановление может быть проведено путем смешивания азида (соединение I) с палладием, нанесенным на уголь (Pd/C), в растворителе типа ледяной уксусной кислоты и насыщения водородом под давлением, равным давлению в питающем баллоне, в течение 10-20 ч. Для выделения продукта сначала отфильтровывают катализатор, а фильтрат лиофилизируют для получения аминосоединения (Seq ID 1-15), в котором аминогруппа является первичной.
Полученный таким образом амин может быть превращен в замещенный амин, как это описано далее.
Соединение I, в котором - NRIIRIII представлен группой -NHCH2CH2NH2 или гомологами -NH(CH2)2-4NRIVRV, может быть полученo из сульфонa по методу, согласно которому диамин H2(CH2)2-4NRIVRV реагирует с сульфоном (cоединение C, Seq ID Nos. 31-45).
Реакция проводится в указанном ранее апротонном растворителе при комнатной температуре. Используемый избыток амина составляет порядка 10 моль. Реакция проводится в течение времени от одного до нескольких часов.
При проведении реакции соответствующий амин добавляется в раствор сульфона в безводном апротонном растворителе, и реакционная смесь перемешивается при комнатной температуре до получения cоединения I (Seq ID Nos. 1-15), в котором заместитель в положении "C-5-орн" - NRIIRIII. Целевое соединение может быть выделено разбавлением реакционной смеси водным раствором ТФУК с последующим хроматографическим разделением. Для этого подходящей является обращеннофазная колоночная флэш-хроматография (C18) с использованием 10-25% ацетонитрила в воде (содержащего 0.1% ТФУК) с шагом изменения состава 5%. Соответствующие фракции могут быть лиофилизированы для выделения продукта в виде трифторацетата.
Трифторацетат может быть превращен в гидрохлорид путем растворения в воде и пропускания через колонку Bio-Rad AG2-X8 (Cl-).
Если в формуле (I) R1 - водород, cоединение I' (Seq ID Nos. 1-3, 15), то атом азота может быть непосредственно введен в гемиаминальное положение путем реакции образования азида, который затем восстанавливают в амин, который в свою очередь может быть частично алкилирован или ацилирован для получения конечного продукта.
Реакция показана на приведенной ниже схеме синтеза.

Хотя в некоторых натуральных циклогексапептидах R1-водород, более часто R1-гидроксил. Таким образом, для некоторых соединений соединение A' на схеме синтеза получено на первой стадии из соответствующего соединения, в котором R1-гидроксил.
Получение восстановленного соединения может быть выполнено при перемешивании соответствующего гидроксисоединения в растворе LiClO4 в диэтиловом эфире при комнатной температуре, добавлении ТФУК и последующем прибавлении триэтилсилана. После этого смесь подвергают быстрому перемешиванию (4-10 ч) до тех пор, пока исходное гидроксисоединение более не обнаруживается аналитической ВЭЖХ. После этого реакционную смесь выливают в дистиллированную воду, чтобы получить восстановленный продукт в виде осадка, который потом отделяется обычными методами. Полученный таким образом восстановленный продукт может использоваться для получения азида в очищенном либо неочищенном виде.
Продукты, в которых R1-водород, могут быть получены прибавлением модифицированного циклогексапептида к предварительно приготовленному раствору НN3, НN3 может быть получен из азида натрия и ТФУК. Реакция протекает при комнатной температуре с получением продукта, содержащего азидную группу, и который может быть выделен обычными методами и очищен ВЭЖХ.
Очищенный азид может быть восстановлен водородом на палладии, нанесенном на уголь, в аминосоединение способом, подобным вышеописанному.
Амины, полученные, как это указано выше, и имеющие первичную аминогруппу -NH2, затем могут быть алкилированы обычными способами для получения замещенной аминогруппы. Короче говоря, алкилирование может проводиться взаимодействием соответствующего алкилгалогенида с амидом (cоединение I, NRIIRIII= NH2); Seq ID Nos. 1-15) в апротонном растворителе в присутствии основания для получения монозамещенного амина (cоединение I, NRIIRIII, где RII-C1-C4 алкил, C3-C4 алкенил, (CH2)2-4OH, (CH2)2-4NRIVRV). Последние могут быть выделены из реакционной смеси обычными методами.
Амины, полученные, как это описывается выше, и имеющие первичную аминогруппу -NH2, могут быть ацилированы обычными способами для получения замещенной аминогруппы. Предполагается, что ацильная группа -CO(CH2)1-4NH2. Поскольку ацилирующая кислота содержит первичную аминогруппу, то до проведения ацилирования она защищена заместителем типа бензилоксикарбонильной группы. Предпочтительным является использование активированного эфира типа пентафторфенилового. Ацилирование можно проводит в апротонном растворителе в присутствии основания, такого как ди(изо-пропил)этиламин, при комнатной температуре в течение времени от одного до нескольких часов до получения продукта ацилирования. Продукт может быть выделен разбавлением реакционной смеси метанолом и очищен ВЭЖХ (cоединение I, NRIIRIII=-NHCO(CH2)1-4NH2).
Аминосоединение, в котором аминогруппа в гемиаминальном положении является полностью замещенной, т.е. в которой ни RII, ни RIII не являются атомами водорода,
предпочтительнее получать из сульфона (cоединение B, Seq ID Nos. 31-45) взаимодействием с соответствующим замещенным амином NHRIIRIII. Реакция может быть проведена путем добавления амина к перемешиваемому раствору сульфона в течение времени, достаточного для протекания реакции. Продукт может быть выделен и очищен препаративной ВЭЖХ с лиофилизацией соответствующих компонентов.
Изобретение также охватывает соли связанных кислот. Соединение при обычном способе его выделения получается в виде соли с какой-либо кислотой (связанной кислотой). Обычно это соль ТФУК. Полученная таким образом соль может быть растворена в воде и пропущена через анионообменную колонку, несущую целевой анион. Элюат, содержащий целевую соль, может быть сконцентрирован для получения соли в виде твердого продукта.
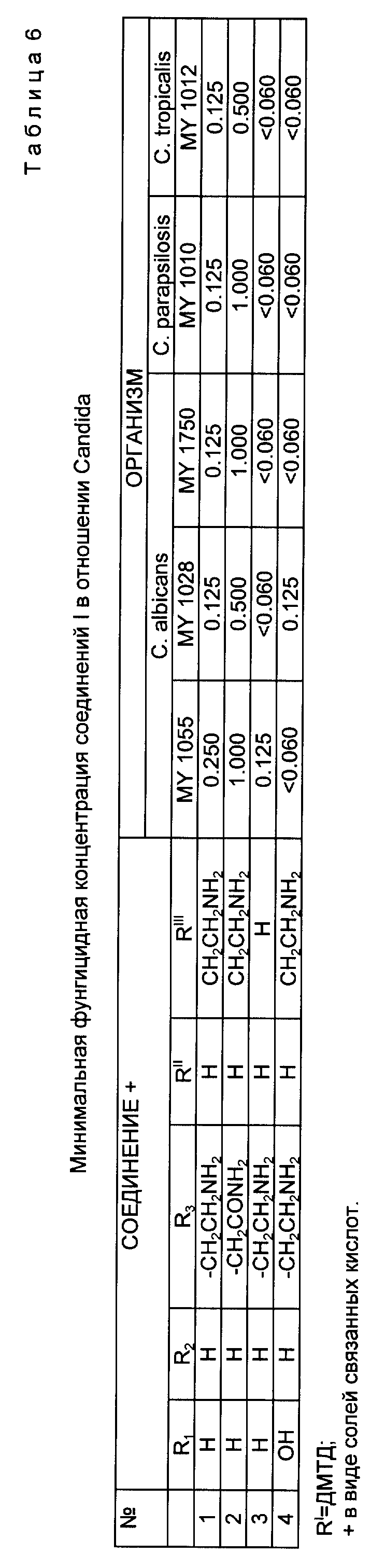
Соединения по настоящему изобретению активны против многих грибков, в частности против видов Candida. Фунгицидные свойства могут быть проиллюстрированы минимальной фунгицидной концентрацией (МФК), определенной для некоторых организмов вида Candida в опыте по разбавлению культуры микроорганизмов, выполненном в среде азотной грибковой основы с добавкой 1% декстрозы (Yeast Nitrogen Base (DIFCO) medium with 1% dextrose (YNBD).
В представленном опыте соединения с начальной концентрацией 5 мг/мл были солюбилизированы в 100%-ном ДМСО. Приготовленный раствор лекарства разбавлением водой был доведен до концентрации 512 мкг/мл, так что окончательное содержание ДМСО было около 10%. Затем раствор распределяется из многоканального дозатора в первую колонку 96-ячеечной тарелки (каждая ячейка содержала 0.075 мл YNBD), так что полученная концентрация лекарства составила 256 мкг/мл. Соединения из первой колонки разбавлялись от ряда к ряду в 2 раза, при этом концентрация лекарства изменялась от 256 до 0.12 мкг/мл.
С использованием спектрофотометра при 600 нм было установлено, что культуры микроорганизмов возрастом 4 ч, подлежащие испытанию, соответствуют 0.5 стандарта Мак Фарланда. Эта суспензия была разбавлена в YNBD в соотношении 1:100 до достижения в ячейке концентрации клеток 1 - 5 • 104, образующих колонию организмов (ОКО)/мл. Аликвоты суспензии (0.075 мл) были привиты в каждую ячейку микротитровой тарелки, что выразилось в конечной концентрации клеток 5 - 25 • 103 ОКО/мл и конечной концентрации лекарства от 128 до 0.06 мкг/мл. Каждый опыт включал один ряд контрольных ячеек, не содержащих лекарства, и один ряд контрольных ячеек, не содержащих клеток.
После 24 ч инкубации микротитровые тарелки осторожно встряхнули на вибростоле для перемешивания суспензии клеток. Для перенесения 1.5 мкл образцов из каждой ячейки 96-ячеечной микротитровой тарелки в одиночные ячейки, содержащие декстрозный агар Sabouraud (SDA), был использован микродозатор MIC-2000. Привитые ячейки с SDA подвергли инкубации в течение 24 ч при 35oC. Результаты приведены в табл. 6.
Активность соединений против грибков in vitro может быть продемонстрирована для тех же соединений в опыте in vivo.
Организмы из посеянной предыдущим вечером культуры Candida albicans MY 1055 суспендируют в стерильном соляном растворе и определяют концентрацию клеток гемацитометрическим подсчетом и доводят до 3.75 • 105 клеток/мл. Затем 0.2 мл этой суспензии вводят в вену, расположенную на хвосте мыши, так что конечная прививка составляет 7.5 • 104 клеток/мышь.
Опыт продолжают внутрибрюшинным (I.P.) введением водного раствора cоединения I с различной концентрацией дважды в день в течение последующих 4 дней самкам мыши DBA/2 весом от 18 до 20 г, которые были предварительно инфицированы Candida albicans способом, описанным ранее. Контрольной группе мышей вводят (I. P. ) дистиллированную воду. Через 7 дней мышей умерщвляют углекислым газом, обе почки удаляют в асептических условиях и помещают в стерильные полиэтиленовые пакеты, содержащие 5 мл стерильного солевого раствора. Почки в пакетах раздробляют до гомогенного состояния, разбавляют стерильным солевым раствором и аликвоты наносят на поверхность тарелок SDA. Тарелки подвергают инкубации при 35oC в течение 48 ч, и грибковые колонии пронумеровывают для подсчета образующих колонию организмов (ОКО) на 1 г почек. Соединения (1), (2), (3) и (4) дали более чем 99%-ный эффект снижения концентрации выделяемых ОКО Candida при 0.09 и 0.375 мг/кг (I.P.) дважды в день в течение 4 последовательных дней.
Соединения по настоящему изобретению также полезны для ингибирования или облегчения течения инфекционных заболеваний, вызванных Pneumocystis carini у пациентов с пониженным иммунитетом. Эффективность соединений, являющихся предметом настоящего изобретения, в терапевтических или противоинфекционных целях может быть продемонстрирована в опытах на крысах с подавленным иммунитетом.
В представленном опыте была определена эффективность соединения I-6-1 (R1= OH; R2= H; R3=CH2CH2NH2; RI=ДМТД; RII=H; RIII=CH2CH2NH2). У крыс Спрага-Доули (Sprague-Dawley) весом около 250 г иммунитет был подавлен добавлением дексазона в питьевую воду (2.0 мг/л). Крыс держали на низкопротеиновой диете в течение 7 недель, чтобы вызвать развитие пневмоцистозной пневмонии из скрытой инфекции. Перед применением лекарства две крысы были умерщвлены для подтверждения наличия пневмонии Pneumocystis carini. Было найдено, что обе крысы заражены. Пяти крысам, весящим около 150 г, дважды в день в течение 4 дней вводили подкожно (sc) cоединение I-6-1 в 0.25 мл растворителя (дистиллированная вода). Также проводился контрольный опыт с чистым растворителем. Все животные продолжали получать дексазон в питьевой воде и находились на низкопротеиновой диете в течение всего периода лечения. По окончании лечения все животные были умерщвлены, легкие были удалены и обследованы, степень заболевания была определена микроскопическим исследованием окрашенных срезов. По результатам исследований cоединение I-6-1 понизило содержание цист P. carini у 5 крыс по меньшей мере на 90% при дозировке 0.075 мг/кг и выживании всех крыс.
Выдающиеся свойства используются наиболее эффективно в том случае, если соединение вводится в новую фармацевтическую композицию, содержащую подходящий носитель, в соответствии с общепринятыми технологиями составления лекарств.
Новые композиции содержат как минимум терапевтическую противогрибковую или антипневмоцистическую дозу активного соединения. В общем случае композиция содержит по меньшей мере 1 мас.% cоединения I. Концентрированные композиции, используемые для разбавления, могут содержать его 90% и более. Композиции включают в себя формы, пригодные для перорального, местного, парентерального (включая внутрибрюшинное, подкожное, внутримышечное и внутривенное) введение, прием через нос, в виде свеч и вдыханием. Композиции могут быть упакованы тонко смешанными с компонентами, пригодными в качестве благоприятной среды.
Композиции, предназначенные для перорального применения, могут быть жидкими или твердыми. Для приготовления жидких составов терапевтический агент может быть смешан с жидким носителем, таким как вода, гликоли, масла, спирты, и т. п. , а для твердых препаратов, таких как капсулы и таблетки, - с твердыми носителями, такими как крахмалы, сахара, каолин, этилцеллюлоза, карбонаты кальция и натрия, фосфат кальция, тальк, лактоза, обычно с замасливателем, таким как стеарат кальция вместе с разрыхлителями и т.п. Из-за легкости их приема таблетки и капсулы представляются наиболее многообещающими дозированными пероральными формами. Наиболее перспективным представляется приготовление дозированных форм с целью облегчения приема и единообразия дозировки. Композиции в виде дозированных форм составляют предмет настоящего изобретения.
Композиции могут быть составлены для инъекций и могут иметь форму суспензий, растворов или эмульсий в маслообразных или водных растворителях, таких как 0.85% хлорид натрия или 5% декстроза в воде, и могут содержать суспендирующие, стабилизирующие и/или диспергирующие агенты. Буферные агенты, а также солевой раствор или глюкоза могут добавляться для того, чтобы сделать растворы изотоническими. Соединениями также могут быть солюбилизированы в спирте/пропиленгликоле или полиэтиленгликоле для капельных внутривенных вливаний. Эти композиции также могут быть представлены дозированными формами в ампулах или многодозовых упаковках, преимущественно с добавлением консерванта. С другой стороны, активные ингредиенты могут быть в форме порошков для разбавления подходящим растворителем перед введением.
Термин "дозированная форма" используется в описании и формуле и обозначает физически дискретные порции, каждая из которых содержит заранее определенное количество активного ингредиента вместе с фармацевтическим носителем, рассчитанное для произведения желаемого терапевтического эффекта. Примерами таких дозированных форм являются таблетки, капсулы, пилюли, пакеты порошка, брикеты, мерные дозы в ампулах или многодозовые упаковки и т.п. Порционная дозировка в настоящем изобретении содержит обычно от 100 до 200 мг одного из соединений.
Если соединение предназначено для противогрибкового использования, то может быть использован любой метод введения. Для лечения грибковых инфекций обычно используется пероральное или внутривенное введение.
Если соединение должно использоваться в борьбе с пневмоцистическими инфекциями, желательно непосредственно вводить его в легкие или бронхи. По этой причине предпочтительнее ингаляция. Для ингаляционного введения упаковку соединения по настоящему изобретению удобно производить в виде аэрозольного пульверизатора, представленного упаковками под давлением или распылителями. Предпочтительной системой для ингаляции является дозированный аэрозоль, который может быть представлен в виде суспензии или раствора cоединения I в подходящих распыляемых носителях, таких как фтороуглероды или углеводороды.
Хотя соединения в настоящем изобретении могут использоваться в качестве таблеток, капсул, местных препаратов, ингаляционных порошков, свеч и т.п., растворимость в воде этих соединений делает их пригодными для использования в составах для инъекций, а также в жидких композициях и аэрозольных пульверизаторах.
Следующие примеры иллюстрируют изобретение, но не могут быть истолкованы как ограничения.
Примеры 1-3 иллюстрируют получение продуктов по первому из описанных методов, а именно - через сульфон. Этот метод может использоваться при получении любого из соединений, но должен использоваться для достижения хорошего выхода, если R1=OH.
Пример 4 и последующие иллюстрируют получение продуктов прямым замещением азота на кислород в гемиаминальном положении "5-орн". Этот метод предпочтителен, если R1, RII и RIII-H.
Пример 3 иллюстрирует использование в качестве исходного материала соединениe, в котором R3 был восстановлен до -CH2CH2NH2 из натурального продукта, в котором R3 - CH2CONH2. Аналогично для соединений, в которых R3-CH2CN, также может использоваться частично модифицированное соединение.
Примеры 9 и 10 иллюстрируют проведение превращения гемиаминального кислорода в азот и превращение -CH2CN в -CH2CH2NH2.
Пример 1.

Часть A. Получение промежуточного 1-[4-гидрокси-5-(эпи)-аминоэтилтио-N2-(10,12-диметил-1-оксотетрадецил)орнитин] -5-(3-гидроксиглутамин)-6-(3-гидроксипролин)ехинокандина B(Seq ID N 34).
Раствор 500 мг (0,47 ммоль) пневмокандина Bo (Seq ID N 19), 5.34 г (47 ммоль) гидрохлорида 2-амино-этантиола и 109 мг (0.47 ммоль) (1S)-(+)-10-сульфокамфарной кислоты в 40 мл безводного ДМФА перемешивают при 25oC в течение 6 дней. Реакционную смесь разбавляют 40 мл воды и флэш-хроматографируют на "LICHRO-PREP" RP18 (40-63 мкм, 15.0 г), набитом в колонку с использованием 10%-ного ацетонитрила в воде. Колонку элюируют 10-40%-ным ацетонитрилом в воде, собирают две фракции по 120 мл при каждом шаге изменения концентрации в 10%. Из двух фракций в 40%-ном водном ацетонитриле получают 185 мл материала, который очищают препаративной ВЭЖХ "ZORBAX" C8 (21.2 x 250 мм), элюируя 40-45%-ным ацетонитрилом в воде с добавкой 0.1% ТФУК и получают 128 мг трифторацетата 1-[4-гидрокси-5-(эпи)-аминоэтилтио- N2-(10,12-диметил-1-оксотетрадецил)-орнитин] -5-(3- гидроксиглутамин)-6-(3-гидроксипролин)ехинокандина B в виде белого безводного твердого вещества.
ПМР (400 МГц, CD3OD) δ 1.34 (d, J=6.3 Hz, 3H), 2.89 (m, 1H), 4.72 (d, J= 4.9 Hz, 1H)
Масс-спектр (Li), m/e 1131 (MH+Li)+
Часть B. Получение промежуточного сульфона (Seq ID 34).
К перемешиваемому раствору тиосоединения (444 мг, 0.358 ммоль), полученного как это описано в части A, в 15 мл раствора ацетонитрила, разбавленного водой в соотношении 1:1, добавляют "OXONE" (324 мг эквивалентных 1,06 ммоль кислого персульфата калия). После 45 мин раствор разбавляют таким же объемом воды и быстро хроматографируют, используя обращеннофазную (C18) флэш-хроматографическую колонку, элюируя 35-43%-ным ацетонитрилом в воде с добавкой 0,1% ТФУК с шагом изменения концентрации 2%. Фракции, содержащие продукт, лиофилизируют и получают 357 мг (выход 86%) эпи-сульфона.
ПМР (400 МГц, CD3OD) δ 3.48 (m, 2H), 3.55 (m, 1H), 3.71 (m, 1H), 3.91 (dd, 1H), 4.00 (m, 1H), 5.17 (dd, 1H), 6.76 (d, 2H), 7.16 (d, 2H).
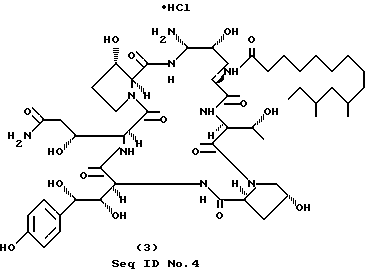
Часть C. Получение продукта формулы (I); соединение 1-4 (Seq ID N 4).
К перемешиваемому раствору 1.2 г (0,945 ммоль) эпи-сульфона, полученного, как это описано в части B, в 20 мл безводного ДМФА, добавляют этилендиамин (568 мг, 9.45 ммоль). После 1 ч анализ реакционной смеси с применением ВЭЖХ (RP-C18, 40% CH3CN/H2O (0,1% ТФУК) показал полное превращение в два полярных продукта в соотношении 37:63. За проведением обращеннофазной колоночной (C18) флэш-хроматографии с 10-40%-ным ацетонитрилом в воде с добавкой 0,1% ТФУК в качестве элюента и шагом изменения концентрации 5% следует лиофилизация соответствующих фракций с образованием 200 мг (выход 21%) нормального продукта в форме (бис)-трифторацетата.
ПМР (400 МГц, CD3OD) δ 1.14 (d, J = 6.2, Hz, 3H), 2.72 (dd, J=15.4 и 3.8 Hz, 1H), 4.10 (m,), 5.04 (dd, J=8,7 и 3.2 Hz, 1H), 5.09 (dd, J = 8.5 и 4.2 Hz, 1H), 5.18 (br s, 1H), 6.74 (d, J = 8.6 Hz, 2H), 7.12 (d, J=8.6 Hz, 2H), 7.47 (d, J = 8,6 Hz, 1H), 7.71 (d, J = 10.0 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 8.71 (d, J = 8.7 Hz, 1H).
Масс-спектр (Li), m/e 1113.5 (MLi)+
(Бис)-трифторацетат, полученный ранее, растворяют в воде, и раствор пропускают через колонку Bio-Rad AG2-X8 (Cl-), промываемую водой. Элюат, содержащий продукт, лиофилизируют с образованием вышеуказанных соединений в виде (бис)-гидрохлорида.
Лиофилизация фракций, содержащих основное вещество, дает эпи-продукт.
ПМР (400 МГц, CD3OD) δ/ 3.02 (m, 1H), 3.14 (m, 3H), 4.16 (m, 1H), 5.10 (dd, 1H), 6.76 (d, 2H), 7.14, (d, 2H).
Масс-спектр (Li), m/e 1113.9 (MLi)+.
Пример 2.
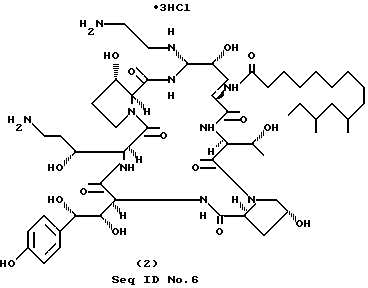
Часть A. Получение промежуточного сульфона (Seq ID N 36).
Исходное вещество, cоединение A-6RI=ДМТД (Seq ID N 21), получают, как это описано для таких соединений в разделе "Получение исходных материалов".
Соединение А-6 превращают затем в эпи-тиосоединение B-6(Seq ID N 36) способом, аналогичным описанному в части A примера 1.
К перемешиваемому раствору 285 мг (0.241 ммоль) соединения B-6 в 14 мл раствора ацетонитрила, разбавленного водой в соотношении 1:1, добавляют "OXONE" (162 мг эквивалентных 0.530 ммоль кислого персульфата калия). После 45 мин раствор разбавляют таким же объемом воды и хроматографируют. За проведением обращеннофазной колоночной (C18) флэш-хроматографии с 30-45%-ным ацетонитрилом в воде с добавкой 0,1% ТФУК в качестве элюента и шагом изменения концентрации 5% следует лиофилизация содержащих продукт фракций с образованием 212 мг эпи-сульфона (соединение C-6-Seq ID N 36).
Выход 84%.
ПМР (400 МГц, CD3OD) δ 3.08 (m, 2H), 3.46 (t, J = 6.6 Hz, 2H), 3.68 (m), 5.05 (m), 6.77 (d, J = 8.5 Hz, 2H), 7.15 (d, 8.5 Hz, 2H).
Масс-спектр (Li), m/e 1039.9.
Часть B. Получение продукта формулы (2) (cоединение 1-6; RII=H; RIII=2 аминоэтил); Seq ID N 6.
К перемешиваемому раствору cоединения C-6, полученного как это описано в части A (418 мг, 0.305 ммоль) в 10 мл безводного ДМФА, добавляют этилендиамин (183 мг, 3.05 ммоль). После 1 ч анализ реакционной смеси с применением ВЭЖХ (RP-C18, 35% CH3CN/H2O (0,1% ТФУК) показал полное превращение в два полярных продукта в соотношении 36:64. Реакционную смесь разбавляют раствором, содержащим 190 мл воды и 0,4 мл ТФУК, и хроматографируют. За проведением обращеннофазной колоночной (C18) флэш-хроматографии с 10-25% ацетонитрилом в воде с добавкой 0,1% ТФУК в качестве элюента и шагом изменения концентрации 5% следует лиофилизация соответствующих фракций с образованием 111 мг продукта в форме (трис)-трифторацетата. Выход 21%.
ПМР (400 МГц, CD3OD) δ 1.17 (J=6.2 Hz), 2.44 (dd, J = 7.0 и 13.2 Hz, 1H), 4.10 (m), 2.7-3.0 (m, 4H), 3.06 (t, J = 7.0 Hz, 2H), 3.82 (m, 3H), 3.97 (dd, J = 11.2 и 3.2 Hz, 1H), 4.03 (m, 2H), 4.70 (d, J = 2.3 Hz, 1H), 5.00 (d, J = 3.3 Hz, 1H), 6,75 (d, J = 8,6 Hz, 2H), 7.11 (d, J = 8.6 Hz, 2H).
Масс-спектр (Li), m/e 1099,9 (MLi)+.
(Трис)-трифторацетат, полученный ранее, растворяют в воде и раствор пропускают через колонку Bio-Rad AG2-X8 (Cl-), промываемую водой. Элюат, содержащий продукт, лиофилизируют с образованием 93 мг вышеуказанного соединения в виде (трис)-гидрохлорида.
Пример 3.
A. Получение азида (Seq ID N 49).
К перемешиваемому раствору 297 мг (0,257 ммоль) эписульфона (пример 1, часть B) в 10 мл безводного ДМФА добавляют азид лития (126 мг, 257 ммоль). После 1 ч анализ реакционной смеси с применением ВЭЖХ (RP-C18, 40% CH3CN/H2O (0,1% ТФУК) показал полное превращение в единственный, значительно менее полярный продукт в соотношении 37:63. За проведением обращеннофазной колоночной (C18) флэш-хроматографии с 30-65%-ным ацетонитрилом в воде в качестве элюента и шагом изменения концентрации 5% следует лиофилизация соответствующих фракций с образованием сырого азида. С помощью препаративной ВЭЖХ (C18, 40-45% CH3CN/H2O (0,1% ТФУК) с шагом изменения концентрации 5% получено азидное соединение D-4 (Seq ID N 49).
ПМР (400 МГц, CD3OD) δ 1,14 (d, J = 6,1 Hz, 3H), 2,50 (dd, J = 15,6 и 9,9 Hz, 1H), 2,84 (dd, J = 15,6 и 3,3 Hz, 1H), 3,95 (dd, J = 11,2 и 3,1 Hz, 1H), 4,05 (m, 2H), 4,56 (m, 3H), 4,98 (dd, J = 8,5 и 3,5 Hz, 1H), 5,10 (dd, J = 8,3 и 4,2 Hz, 1H), 5,26 (dd, J = 8,5 и 2,2 Hz, 1H), 6,74 (d, J = 8,6 Hz, 2H), 7,12 (d, J = 8,6 Hz, 2H), 7,44 (d, J = 8,3 Hz, 1H), 7,76 (d, J = 9,9 Hz, 1H), 8,26 (d, J = 8,1 Hz, 1H), 8,83 (d, J = 8,7 Hz, 1H), 9,00 (d, J = 8,5 Hz, 1H).
ИК-спектр (Nujol), 2110 см-1.
Часть B. Получение амина.
Смесь азидного соединения D-4, полученного, как это описано в части A (137 мг, 0,126 ммоль), и 10%-ный Pd/C (137 мг) в ледяной уксусной кислоте (10 мл) насыщают водородом под давлением, равным давлению в питающем баллоне, в течение 14 ч. Катализатор удаляют фильтрованием, и фильтрат лиофилизируют для получения сырого амина. Очистка с помощью препаративной ВЭЖХ (C18, 35-41% CH3CN/H2O (0,1% ТФУК) с шагом изменения концентрации 3%), за которой следует лиофилизация соответствующих фракций, дает аза соединение I-1, RII, RIII = H (Seq ID N 1) в виде трифторацетата.
Выход 48%.
ПМР (400 МГц, CD3OD) δ 1,13 (d, J = 6,1 Hz, 3H), 2,49 (dd, J = 15,6 и 9,8 Hz, 1H), 2,81 (dd, J = 15,6 и 3,4 Hz, 1H), 3,97 (dd, J = 11,1 и 3,1 Hz, 1H), 4,03 (m, 1H), 4,11 (m, 1H), 4,47 (dd, J = 11,7 и 5,5 Hz, 1H), 4,57 (m, 2H), 5,00 (m, 1H), 5,10 (m, 1H), 5,14 (d, J = 2,2 Hz, 1H), 6,74 (d, J = 8,6 Hz, 2H), 7,12 (d, J = 8,6 Hz, 2H), 7,42 (d, J = 8,3 Hz, 1H), 8,89 (d, J = 8,8 Hz, 1H).
Масс-спектр m/z 1071,0 (MLi)+.
Трифторацетат растворяют в воде и раствор пропускают через колонку Bio-Rad AG2-X8 (Cl-), промываемую водой. Элюат, содержащий продукт, лиофилизируют с образованием 66 мг соединения I-4, RII, RIII = H в виде гидрохлорида.
В следующих опытах pастворитель A - 95% воды, 5% ацетонитрила, 0,1% ТФУК, а pастворитель B - 95% ацетонитрила, 5% воды, 0,1% ТФУК. Используемые выражения "в вакууме" и "на роторе" относятся к удалению растворителя с помощью роторного испарителя.
Пример 4.

A. Получение промежуточного азида. Соединение D (Seq ID N 46).
Пневмокандин B0 (cоединение A-4; Seq ID N 19) (5,00 г, 4,69 ммоль) растворяют в 2М LiClO4 в диэтиловом эфире при комнатной температуре. Добавляют при перемешивании ТФУК (2,50 мл), затем к раствору прибавляют триэтилсилан (5,00 мл). Гетерогенную смесь интенсивно перемешивают в течение 6 ч, после чего аналитическая ВЭЖХ (C18 "ZORBAX", 45% pастворителя A, 55% pастворителя B, 0,1% ТФУК, 1,5 мл/мин) не обнаружила присутствия исходного пневмокандина B0 в количествах, превышающих следовые. Смесь выливают в 200 мл дистиллированной воды, фильтруют и высушивают на воздухе. Влажный твердый продукт перемешивают с диэтиловым эфиром, фильтруют и высушивают на воздухе; получают 5,6 г сырого моновосстановленного пневмокандина B0 (cоединение A-1, Seq ID N 16).
Выделенный сырой продукт в твердом виде добавляют к заранее приготовленному раствору HN3, полученному растворением при охлаждении NaN3 (3,06 г, 47,0 ммоль) в 100 мл ТФУК. После перемешивания при комнатной температуре в течение 30 мин реакционную смесь выливают в 350 мл дистиллированной воды и перемешивают в течение 15 мин. Осадок отфильтровывают, растворяют в метаноле и отгоняют растворитель в роторном испарителе. Остаточная вода удаляется азеотропной отгонкой со 100%-ным этанолом. Конечный твердый продукт сушат в глубоком вакууме для удаления летучих примесей. Смесь очищают двумя равными порциями с помощью препаративной ВЭЖХ (C18 "DELTAPAK", 60 мл/мин, фракции 48 мл) использованием изменяющейся концентрации элюента от 70% A/ 30% B до 50% A/ 50% B. Соответствующие фракции (определяются по УФ-поглощению при λ = 220/ и 277 нм) объединяют. Недостаточно очищенные фракции также объединяют и процесс повторяют, как это описано выше. Таким способом получают 1,78 г (выход 35%) азида D-1 (Seq ID N 46).
ПМР (400 МГц, CD3OD) δ 7,02 (d, 2H), 6,69 (d, 2H), 5,30 (d, 1H), 5,11 (d, 1H), 4,98 (d, 1H), 2,74 (dd, 1H), 1,13 (d, 3H).
Масс-спектр (Li), m/z 1081 (MH+Li)+.
B. Получение амина формулы (4). Соединение I-1 (RII, RIII = H; Seq ID N 1).
Очищенный азид (1,50 г), соединение D-1, полученный, как это описано выше, растворяют в 40 мл метанола. Добавляют 33%-ную водную уксусную кислоту (15 мл), затем - 0,20 г 10%-ного Pd/C, затем продувают реакционный сосуд азотом. Вытесняют азот водородом и интенсивно перемешивают смесь в атмосфере водорода в течение 3 ч. Суспензию фильтруют через стеклянный фильтр (dпор = 0,2 мкм) и чистый раствор упаривают досуха на роторном испарителе. Осадок растворяют примерно в 20 мл дистиллированной воды, вымораживают и лиофилизируют. Получают 1,47 г (95%) целевого аминосоединения (Seq ID N 1) в виде белого твердого вещества.
ПМР (400 МГц, CD3OD) δ 7,02 (d, 2H), 6,69 (d, 2H), 5,09 (d, 1H), 5,01 (d, 1H), 2,77 (dd, 1H), 1,15 (d, 3H).
Масс-спектр (Li), m/z 1055 (MH+Li)+.
Пример 5.

A. Получение промежуточного бензилоксикарбонильного соединения (Seq ID N 1).
В 1 мл ДМФА растворяют амин формулы (4) (пример 4) (200 мг, 0.180 ммоль) и пентафторфенил N-бензилоксикарбонил-3-аминопропанат. Добавляют ди(изо-пропил)этиламин (0.035 мл, 198 ммоль) и перемешивают смесь при комнатной температуре в течение 1 ч. Реакционную смесь разбавляют 2 мл метанола и очищают препаративной ВЭЖХ (C18 "DELTAPAK", фракция 48 мл) с использованием изменяющейся концентрации элюента от 70% A/30% B до 48% A/52% B. Соответствующие фракции (определяются по УФ-поглощению при λ = 220 и 277 нм) объединяют, вымораживают и лиофилизируют. Получают 100 мг (44%) целевого промежуточного продукта.
ПМР (400 МГц, CD3OD) δ 7.32 (d, 5H), 7.01 (d, 2H), 6.99 (d, 2H), 5.64 (bd, 1H), 1.18 (d, 3H).
Масс-спектр (Li), m/z 1259 (MLi)+.
B. Получение 3-аминопропаноильного соединения формулы (5);
Соединение I-1 RII=H, RIII=CO(CH2)2NH2(Seq ID N 1).
Бензилоксикарбонильное соединение (см. часть A) (94 мг, 0.075 ммоль) растворяют в смеси 3 мл метанола, 1 мл воды и 0.2 мл уксусной кислоты. Добавляют 10%-ный Pd/C (48 мг) и продувают реакционный сосуд азотом. Затем продувают сосуд водородом и интенсивно перемешивают смесь в течение 2 ч под давлением водорода 1 атм. Удалением летучих веществ в роторном испарителе получают твердый продукт. Продукт растворяют, примерно, в 4 мл 50%-ного водного ацетонитрила, вымораживают и лиофилизируют. Получают 80 мг (91%) целевого соединения формулы (5) в виде белого твердого вещества.
ПМР (400 МГц, CD3OD) δ 7.01 (d, 2H), 6.69 (d, 2H), 6.67 (d, 1H), 5.10 (d, 1H), 4.99 (d, 1H), 3,12 (m, 2H), 1,91 (s, 3H), 1.17 (d, 3H).
Масс-спектр (Li), m/z 1125 (MLi)+.
Пример 6.
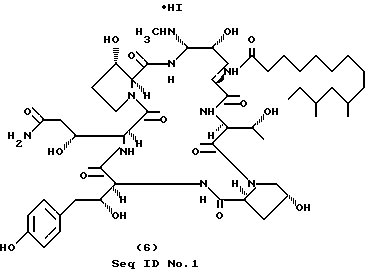
Получение N-метиламиносоединения формулы (6); cоединение I-1 (RII=H; RIII=CH3) (Seq ID N 1)
Амин формулы (5) примера 5 (45.6 мг, 0.135 ммоль) растворяют в 0.5 мл сухого ДМФА. Добавляют 0.021 мл (0.473 ммоль) иодометана, затем - 0.0824 мл (0.473 ммоль) ди(изо-пропил)этиламина. После 24-часового перемешивания при комнатной температуре отгонкой на роторном испарителе удаляют летучие вещества и сырой продукт анализируют масс-спектрометрически.
Масс-спектр (Li), m/z 1068 (MLi)+.
Пример 7.

A. Получение промежуточного нитрильного (N-цианометильного) cоединения I-1; RII=H; RIII=CH2CN (Seq ID N 1).
Аминосоединение, полученное как описано в примере 4 (4540 мг, 0.451 ммоль), растворяют в 3 мл сухого ДМФА. Добавляют 0.063 мл (0.902 ммоль) бромацетонитрила, который был предварительно очищен путем пропускания через небольшой слой MgSO4-NaHCO3, после чего добавляют ди(изо-пропил)этиламин (0.157 мл, 0.902 ммоль). Прозрачную реакционную смесь перемешивают в течение 12 ч, после чего разбавляют небольшим объемом воды. Раствор очищают препаративной ВЭЖХ (C18 "DELTAPAK", изменение концентрации от 70% A/30%B до 47% A/ 53%B, фракции 48 мл). Соответствующие фракции, определяемые по УФ-поглощению при 220 и 277 нм, собирают, вымораживают и лиофилизируют. Получают 338 мг (62%) целевого промежуточного цианометильного соединения в виде нерастворимого в воде твердого вещества.
ПМР (400 МГц, CD3OD) δ 7.01 (d, 2H), 6.69 (d, 2 H), 5.12 (dd, 1H), 5.01 (dd, 1H) 3,80 (s, 2H), 2.76 (dd, 1H), 1.15 (d, 3H).
Масс-спектр (Li), m/z 1094 (MH+Li)+.
B. Получение N-аминоэтильного соединения формулы (7).
Соединение I-1; RII=H; RIII=(CH2)2NH2 (Seq ID N 1).
Нитрильное (цианометильное) соединение, полученное ранее (300 мг, 0.249 ммоль), растворяют в 5 мл метанола, затем добавляют 237 мг (0.997 ммоль) гексагидрата хлорида никеля (II). Тремя порциями добавляют к раствору 189 мг (4.99 ммоль) боргидрида натрия. Сразу же образуется черный осадок, после чего смесь перемешивают 15 мин при комнатной температуре. Гетерогенную смесь разбавляют примерно 20-40 мл воды и добавляют примерно 10-15 мл 2 н. HCl. Перемешивание продолжают еще в течение 45 мин до тех пор, пока черный осадок не растворится с образованием сине-зеленого раствора. Очистка проводится препаративной ВЭЖХ (C18 "DELTAPAK", изменение концентрации от 70%A/ 30%B до 55%A/ 45%B, фракции 48 мл). Соответствующие фракции, определяемые по УФ-поглощению при 220 и 277 нм, собирают, вымораживают и лиофилизируют. Получают 180 мг (55%) целевого продукта. Вещество растворяют в 30 мл воды и пропускают через ионообменную колонку (в форме Cl-), промываемую дистиллированной водой. Раствор вымораживают и лиофилизируют. Получают 149 мг (степень извлечения 94%) целевого аминоэтильного соединения формулы (7) в виде белого твердого вещества.
ПМР (400 МГц, CD3OD) δ 7.01 (d, 2H), 6/69 (d, 2H), 5.11 (dd, 1H), 5.07 (dd, 1H), 1.14 (d, 3H).
Масс-спектр (Li), m/z 1098 (MH+Li)+.
Пример 8.
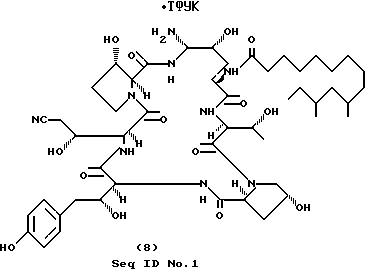
А. Получение промежуточного азида. (Seq ID N 47).
Нитрил пневмокандина Bo (2.00 г, 1.91 ммоль) растворяют в 24 мл 2М раствора LiClO4 в диэтиловом эфире. Добавляют 2.00 мл триэтилсилана, затем 1 мл ТФУК, смесь интенсивно перемешивают в течение 6 ч при комнатной температуре. Смесь выливают в 300 мл воды, перемешивают и фильтруют. Вещество с фильтра растворяют в минимальном количестве метанола, и растворитель отгоняют на роторном испарителе. Остаточную воду удаляют азеотропной перегонкой со 100%-ным этанолом, и остаток оставляют на ночь в глубоком вакууме для удаления летучих примесей. Получают моновосстановленный по месту бензильного углерода продукт. Полученное сырое твердое вещество и азид натрия (1.26 г, 19.4 ммоль) помещают в круглодонную колбу, снабженную мешалкой и помещенную в охлаждающую баню. Медленно добавляют 50 мл ТФУК, охлаждающую баню убирают и смесь перемешивают 2 ч. Смесь выливают в 300 мл воды и фильтруют. Твердое вещество растворяют в метаноле, растворитель отгоняют на роторном испарителе, выдерживают в глубоком вакууме для удаления летучих примесей. Сырой материал очищают препаративной ВЭЖХ (C18"DELTAPAK", изменение концентрации от 55% A/45% B до 45%А/В, фракции 56 мл). Соответствующие фракции, определяемые по УФ-поглощению при 220 и 277 нм, собирают, вымораживают и диофилизируют. Получают 0.59 г (29%) целевого промежуточного азида.
ПМР (400 МГц, CD3OD) δ 7.00 (d 2H), 6.69 (d, 2H), 5.34 (d, 1H), 5.07 (dd, 1H), 5.00 (m, 1H), 2.88 (dd, 1H), 1.17 (d, 3H).
Масс-спектр (Li), m/z 1036 (M-N2+Li)+.
В. Получение соединения формулы (8) (Seq ID N 48).
Очищенный азид (часть А)(0.15 г, 0.142 ммоль) растворяют в смеси 4 мл метанола, 1 мл воды и 0,5 мл уксусной кислоты. К раствору добавляют 50 мг 10%-ного Pd/C. Реакционный сосуд продувают N2, затем H2. Смесь интенсивно перемешивают в течение 5 ч при комнатной температуре под давлением H2 1 атм. Последующая фильтрация через стеклянный фильтр (dпор=0.2 мкм) и удаление летучих примесей на роторном испарителе позволяет получить 0.124 г (80%) целевого соединения формулы (8) в виде белого твердого вещества.
ПМР (400 МГц, CD3OD) δ 7.00 (d, 2H) 6.69 (d, 2H), 5.04 (d, 1H), 5.01 (m, 1H), 2.79 (dd, 1H), 1.18 (d, 3H).
Масс-спектр (Li), m/z 1037 (MH+Li)+.
Пример 9.
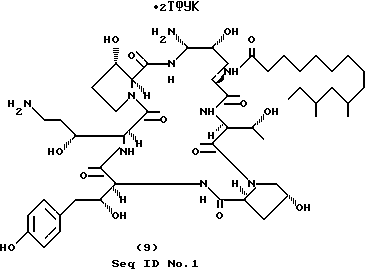
Получение аминосоединения формулы (9) (Seq ID N 3).
Очищаемый азид-нитрил из примера 8 (часть А) (44 мг, 0.0416 ммоль) растворяют в 1.5 мл метанола, после чего добавляют CoCl2•H2O (59 мг, 0.25 ммоль). Затем порциями осторожно добавляют NaBH4(8 х 12 мг, 2.50 ммоль). Черную гетерогенную реакционную смесь перемешивают 30 мин при комнатной температуре. Реакцию прекращают путем добавления примерно 1.5 мл 2 н. HCl и достаточного для растворения осадка количества уксусной кислоты. Мутный раствор разбавляют 3 мл воды и очищают препаративной ВЭЖХ (C18 "ZORBAX", изменение концентрации от 70% А/30% В до 60%А/40% В, 15 мл/мин, фракции 15 мл). Соответствующие фракции, определяемые по УФ-поглощению при 210 и 277 нм, собирают, вымораживают и лиофилизируют. Получают 38 г (72%) соединения формулы (9) в виде белого твердого вещества.
ПМР (400 МГц, CD3OD) δ 6.99 (d, 2H), 6.70 (d, 2H), 5.11 (d, 1H), 5.0 (m, 2H), 1.17 (d, 3H).
Масс-спектр (Li), m/z 1041 (MH+Li)+.
Пример 10.
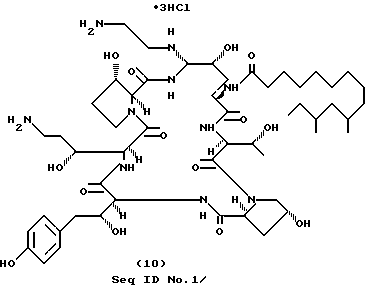
А. Получение промежуточного бис-нитрильного соединения (cоединение I-2, RII=H; RIII=CH2CN; RI=ДМТД) (Seq ID N 2).
Амино-нитрильное соединение из примера 8 (часть В) (500 мг, 0.459 ммоль) растворяют в 3 мл сухого ДМФА. Добавляют 0.064 мл (0.917 ммоль) бромацетонитрила, который был предварительно очищен путем пропускания через небольшой слой MgSO4- NaHCO3, после чего добавляют ди(изо-пропил)этиламин (0.155 мл, 0.917 ммоль). Реакционную смесь перемешивают в течение 18 ч. Смесь разбавляют водой и очищают препаративной ВЭЖХ (C18 "DELTAPAK", 60 мл/мин, изменение концентрации от 70% А/30%В до 50% В, фракции 48 мл). Соответствующие фракции, определяемые по УФ-поглощению при 220 и 277 нм, собирают, вымораживают и лиофилизируют. Получают 198 мг (36%) целевого соединения I-1; RII=H; RIII= CH2CN.
ПМР (400 МГц, CD3OD) δ 7.00 (d,2H), 6.69 (d, 2H), 5.08 (dd, 1H), 5.01 (dd, 1H), 3.73 (s, 2H), 2.79 (dd, 1H), 1.18 (d, 3H).
Масс-спектр (Li) m/z 1076 (MH+Li)+.
В. Получение соединения формулы (10) (SeQ ID N 3).
Бис-нитрил (часть А) (184 мг, 0.155 ммоль) растворяют в 3 мл метанола. Также в метаноле растворяют 148 мг (0.621 ммоль) NiCl2• 6H2O, и тремя порциями добавляют 117 мг (3.1 ммоль) NaBH4. Через 5 минут добавляют 148 мг (0.621 ммоль) CoCl2•H2O и перемешивают еще 1 мин. Дополнительно прибавляют еще 117 мг NaBH4 и продолжают перемешивание еще в течение 15 мин. Последние 60 мг NaBH4 добавляются для того, чтобы довести реакцию до завершения. Смесь разбавляют водой, подкисляют 2 н. HCl и перемешивают до растворения черного осадка. Очистка препаративной ВЭЖХ (C18 "ZORBAX", 15 мл/мин, изменение концентрации от 70% А/30% В до 55% А/ 45% В, фракции 22.5 мл, 220, 277 нм) после лиофилизации дает твердое вещество. Твердое вещество растворяют в воде и пропускают через ионообменную колонку (в форме Cl-), вымораживают и лиофилизируют. Получают 81.1 мг (44%) целевого соединения формулы (10) (cоединение 1 - 3; Seq ID N 3) в виде белого твердого вещества.
ПМР (400 МГц, CD3OD) δ 7.00 (d, 2H), 6.70 (d, 2H), 3 - 3,3 (m, 6H), 1.18 (d, 3H).
Масс-спектр (Li), m/z 1084 (MH + Li)+.
Примеры 11 - 14.
В действиях, выполняемых аналогично тому, что описано в примере 4, соответствующие натуральные или модифицированные натуральные циклопептидные продукты, полученные, как это описано в дальнейшем при приготовлении исходных материалов, при проведении раздельных операций растворяют в растворе LiClO4 в диэтиловом эфире и при перемешивании прибавляют туда ТФУК и триэтилсилан в течение 5 - 10 ч. Затем выливают смесь в воду, фильтруют и перемешивают твердый продукт с диэтиловым эфиром, высушивают на воздухе и получают циклопептид, в котором R1 восстановлен до H.
Моновосстановленное соединение добавляют при охлаждении к предварительно полученному из NaN3 и ТФУК раствору HN3, перемешивают при комнатной температуре от 30 мин до 1 ч и затем выливают в воду для получения азида, который выделяют способом, описанным ранее.
Азид обрабатывают водородом, как это описано ранее, используя Pb/C В качестве катализатора, и выделяют продукт из фильтрата после отделения катализатора.
Продукты, получаемые этим способом, приведены в табл. 7.
Примеры 15 - 17.
В действиях, выполняемых аналогично тому, что описано в gримере 7, соединения из примеров 11, 13 и 14 растворяют в ДМФА и добавляют к раствору очищенный бромацетонитрил, а затем - ди(изопропил)амин, смесь перемешивают в течение 12 - 18 ч для получения нитрильного (N-цианометильного) соединения. В дальнейшем его очищают препаративной ВЭЖХ.
Нитрил растворяют в метаноле и химически восстанавливают с использованием хлорида никеля (II) и боргидрида натрия для получения аминоэтильного замещенного соединения. Полученные соединения приведены в табл. 8.
Примеры 18 - 21.
В результате операций, выполняемых аналогично тому, что описано в примерах 1, 2 и 3, могут быть получены соединения, содержащие перечисленные в табл. 9 заместители.
Примеры 22 - 25.
В результате операций, выполняемых аналогично тому, что описано в примере 1 получаются следующие соединения (см. табл. 10).
Пример 26.
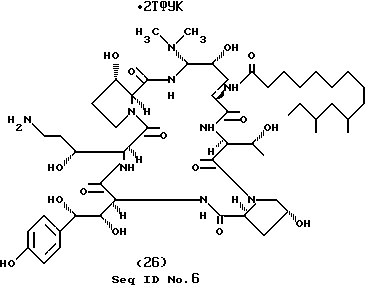
Вышеуказанное соединение получают способом, аналогичным описанному в примере 2 (часть B), замещая диметиламин этилендиамином для получения соединения с М.м. 1334.43.
Пример 27.

Вышеуказанное соединение получают способом, аналогичным описанному в примере 26, замещая пиперидин диметиламином для получения соединения в М.м. 1374.
Пример 28.
1000 спрессованных таблеток, каждая из которых содержит 500 мг соединения формулы (2) [cоединение 1 - 6 (RII=H; RIII=2-аминоэтил); Seq ID N 6], получаемых из следующих ингредиентов:
Соединение - Масса, г
Соединение из примера 2 - 500
Крахмал - 750
Гидрат двухосновного фосфата кальция - 5000
Стеарат кальция - 2.5
Тонкоизмельченные ингредиенты хорошо смешивают и гранулируют с 10%-ной крахмальной пастой. Гранулы сушат спрессовывают в таблетки.
Пример 29.
1000 твердых желатиновых капсул, каждая из которых содержит 500 мг того же соединения, получаемых из следующих ингредиентов:
Соединение - Масса, г
Соединение из примера 2 - 500
Крахмал - 250
Лактоза - 750
Тальк - 250
Стеарат кальция - 10
Однородная смесь ингредиентов, полученная смешением, используется для наполнения твердых желатиновых капсул, состоящих из двух частей.
Пример 30.
Аэрозольная композиция может быть получена исходя из следующих ингредиентов:
Соединение - Содержание в одной емкости
Соединение из примера 2 - 24 мг
Лецитин NF, жидкий концентрированный - 1.2 мг
Трихлор(фтор)метан, NF - 4.026 г
Дихлор(дифтор)метан, NF - 12.15 г
Пример 31.
250 мл раствора для инъекций могут быть получены с использованием общепринятых методов из следующих ингредиентов:
Соединение - Масса, г
Декстроза - 12.5 г
Вода - 250 мл
Соединение из примера 4 - 400 мг
Ингредиенты смешивают и затем стерилизуют для использования.
Получение исходных материалов.
A-4, если RI=ДМТД, может быть произведен путем культивирования Zalerion arboricola АТСС 206868 в питательной среде с использованием маннитола в качестве первичного источника углерода, как это описано в пат. США N 5, 021, 341 от 4 июня 1991 г.
A-7, если RI=ДМТД, может быть произведен путем культивирования Zalerion arboricola АТСС 20868 в питательной среде как это описано в пат. США N 4, 931, 352 от 5 июня 1990 г.
A-10, если RI - линолеил, может быть произведен путем культивирования Aspergillus nidulans NRRL 11440 в питательной среде, как это описано в пат. США N 4, 288, 549 от 8 сентября 1981 г.
A-11, если RI-11-метилтридецил, может быть произведен путем культивирования Aspergillus sydowi в питательной среде, как это описано в J. Antibiotcs XL (N 3), p. 28 (1987).
A-12 может быть произведен путем культивирования Zalerion arboricola АТСС 20958 11440 в питательной среде, как это описано в заявке сер. N 07/630, 457, поданной 19 декабря 1990 г. (Atty Docket N 18268).
Соединения, в которых R1 - водород, могут быть получены, как это описано в примере 4, часть A.
Соединения, в которых R3-CH2CN, такие как A-2, A-5 и A-8, могут быть произведены путем реакции соединения, имеющего карбоксильную группу в соответствующем положении с избытком цианурхлорида в апротонном растворителе. При этом могут использоваться молекулярные сита. По завершении реакции сита, если они использовались, удаляют, и фильтрат концентрируют для получения нитрильного соединения, как это более полно описано в заявке сер. N 936, 434 от 3 сентября 1992 г.
Соединения, в которых R3-CH2CH2NH2, такие как A-3, A-6 и A-9, могут быть произведены путем как химического, так и каталитического восстановления нитрила. Наиболее подходящим для этого является проведение реакции с использованием большого мольного избытка боргидрида натрия с хлоридом кобальта, как это более полно описано в заявке сер. N 936, 558 от 3 сентября 1992 г.
Исходные материалы, в которых RI - различные группы из встречающихся в натуральных продуктах, могут быть получены деацилированием липофильных групп натуральных продуктов, подвергая эти продукты действию деацилирующих энзимов в питательной среде до тех пор, пока не произойдет полное деацилирование. Указанные энзимы предварительно получают культивированием микроорганизмов семейства Pseudomondaceae или Actinoplanaceae как это описано в Experentia 34, 1670 (1978) или пат. США 4, 293, 482, выделяют деацилированный циклопептид и затем ацилируют деацилированный циклопептид, смешивая с подходящим активным эфиром RICOX для получения cоединения A с требуемой ацильной группой.



Изобретение относится к некоторым аза циклогексапептидным соединениям, имеющим атом азота, присоединенный к циклогексапептидному кольцу по 5-му атому углерода компонента 4-гидрокси-орнитина ("C-5-орн"), формулы I, где R1-H или OH; R2 - H, CH3 или OH; R3 - H, CH3, CH2CN, CH2CH2NH2 или CH2CONH2; RI - C9-C21-алкил, C9-C21-алкенил, C1-C10-алкоксифенил или C1-C10алкоксинафтил; RII - H, C1-C4-алкил, C3-C4-алкенил, (CH2)2-4OH, CO(CH2)1-4NH2, (CH2)2-4NRIVRV; RII и RIII, взятые вместе, -(CH2)4-, -(CH2)5-, -(CH2)2O(CH2)2-, -(CH2)2NH(CH2)2-; RIV - H или алкил; RV - H или алкил и их аддитивные соли. Соединения I активны против многих грибков, в частности против видов Candida. Соединения по настоящему изобретению также полезны для ингибирования или облегчения течения инфекционных заболеваний, вызванных Pneunocystis carini у пациентов с пониженным иммунитетом. 4 c. и 7 з.п. ф-лы, 10 табл.
 \
\

где R1 = H или OH;
R2 = H;
R3 = CH2CN, CH2CH2NH2 или CH2CONH2;
RI = (C9 - C21)-алкил;
RII = H, (C1 - C4)-алкил;
RIII = H, (CH2)nNH2;
n = 2 или 3,
или их аддитивные соли кислоты.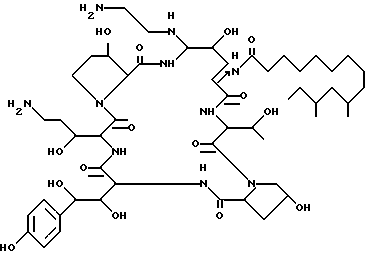
| Способ получения циклопептида | 1968 |
|
SU535902A3 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

