Изобретение является частичным продолжением заявки Мерк 191391А, поданной 15 декабря 1993 г. Настоящая заявка относится к заявке Мерк 18996 с U. S-S-N. 08/059038 от 7 мая 1993 г., которая является частично продолженной заявкой заявки Мерк 185971В, которая в свою очередь является частично продолженной заявкой заявки Мерк 185971А, которая является частичной продолженной заявкой заявки США с серийным номером 07/7895 08 от 8 ноября 1991 г. Настоящая заявка относится к следующим патентным документам: заявке США с серийным номером 595913 от 11 октября 1990 г (дело Мерк 18236); заявке США с серийным номером 746460 от 16 августа 1991 г. (дело Мерк 18466); заявке Мерк, поданной 23 октября 1991 г., и заявке Мерк 18416.
Изобретение относится к соединениям, которые ингибируют протеазу, закодированную вирусом иммунодефицита человека, или их фармацевтически приемлемым солям, и такие соединения используются для профилактики инфицирования HIV, лечения инфицирования HIV и лечения приобретенного в результате синдрома иммунодефицита (AIDS). Настоящее изобретение также относится к фармацевтическим композициям, содержащим такие соединения, и способу применения настоящих соединений и других агентов для лечения AIDS и вирусного инфицирования HIV.
Область техники, к которой относится изобретение.
Ретровирус, обозначенный как вирус иммунодефицита человека (HIV), представляет собой этиологический агент комплексного заболевания, которое заключается в прогрессирующем разрушении иммунной системы (синдром приобретенного иммунодефицита) и вырождении центральной и периферической нервной системы. Такой вирус ранее был известен, как LAV, HTLV-111 или ARY. Общим признаком ретровирусной репликации является экстенсивная посттрансляционная обработка полипротеинов-предшественников вирусно закодированной протеазой, в результате чего вырабатываются зрелые вирусные белки, требующиеся для сборки и функционирования вируса. Ингибирование такого процесса предотвращает образование инфекционного в обычных условиях вируса. Так например, Kohl, N.E. с сотр., в Proc. Natl Acad. Sci.. 85, 4686 (1988) показали, что генетическая инактивация протеазы, закодированной HIV, приводит в результате к образованию незрелых, неинфекционных вирусных частиц. Эти результаты показывают, что ингибирование HIV протеазы представляет собой жизнеспособный способ лечения AIDS и профилактики или лечения заражения HIV.
Нуклеотидная последовательность HIV обнаруживает наличие ро1 гена в одной открытой рамке считывания (Rather, Z, с сотр., Nature, 313, 277 (1985)). Гомология аминокислотной последовательности обеспечивает доказательство того, что pol последовательность кодирует обратную транскриптазу, эндонуклеазу и HIV протеазу (Toh, H. с сотр., EMBO J., 4, 1267 (1985); Power M.D, с сотр. , Science, 231, 1567 (1986); Pearl, L.H. с сотр., Nature, 329, 351, (1987)). Заявители продемонстрировали тот факт, что соединения настоящего изобретения являются ингибиторами HIV протеазы.
Краткое изложение сущности изобретения.
Соединения формулы I, указанной в тексте описания, могут использоваться для ингибирования HIV-протеазы, профилактики инфицирования HIV, лечения инфицирования HIV и для лечения AlDS, причем они могут применяться как таковые, в виде фармацевтически приемлемых солей, ингредиентов фармацевтической композиции, необязательно в комбинации с другими антивирусными агентами, иммуномодуляторами, антибиотиками или вакцинами. Раскрываются также способы лечения AIDS, способы профилактики инфицирования HIV и способы лечения инфицирования HIV.
В заявке могут использоваться сокращения, представленные в конце описания.
Описание изобретения и предпочтительных реализаций.
Изобретение относится к соединениям формулы I, их комбинациям или их фармацевтически приемлемым солям, предназначенным для ингибирования HIV протеазы, профилактики или лечения заражения HIV и лечения приобретенного в результате синдрома иммунодефицита (AIDS). Соединения формулы I определены следующим образом: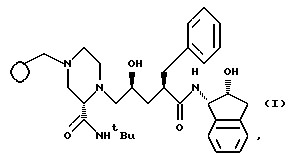
или их фармацевтически приемлемые соли
где  представляет собой устойчивый 8-10-членный бициклический гетероцикл, каждое из колец которого может быть насыщенным или ненасыщенным, причем указанный гетероцикл состоит из атомов углерода и 1-3 гетероатомов, выбранных из группы, состоящей из N, S или O, и указанный гетероцикл может быть незамещенным или замещенным OH, галогеном, C1-4алкилом, оксогруппой;
представляет собой устойчивый 8-10-членный бициклический гетероцикл, каждое из колец которого может быть насыщенным или ненасыщенным, причем указанный гетероцикл состоит из атомов углерода и 1-3 гетероатомов, выбранных из группы, состоящей из N, S или O, и указанный гетероцикл может быть незамещенным или замещенным OH, галогеном, C1-4алкилом, оксогруппой;
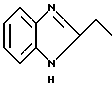
при условии, что  не является следующими группами:
не является следующими группами: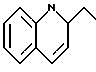
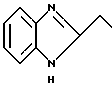
Одна из реализаций настоящего изобретения представляет собой соединения формулы I или их фармацевтически приемлемые соли, в которых  представляет собой устойчивый 8-10-членный бициклический гетероцикл, любое из колец которого может быть насыщенным или ненасыщенным, и указанный гетероцикл состоит из атомов углерода и 2-х гетероатомов, выбранных из группы, состоящей из N или O, причем гетероатомы находятся в различных кольцах.
представляет собой устойчивый 8-10-членный бициклический гетероцикл, любое из колец которого может быть насыщенным или ненасыщенным, и указанный гетероцикл состоит из атомов углерода и 2-х гетероатомов, выбранных из группы, состоящей из N или O, причем гетероатомы находятся в различных кольцах.
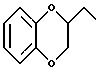
Вторая реализация изобретения представляет собой соединения формулы I, в которой  ограничен значениями:
ограничен значениями:
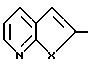
X представляет собой O или S,
или их фармацевтически приемлемые соли.
Третья реализация изобретения представляет собой соединения формулы I, в которой 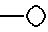 ограничен значением:
ограничен значением: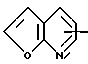
или их фармацевтически приемлемые соли.
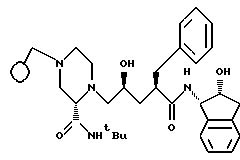
Другое воплощение настоящего изобретения представляет собой соединение A: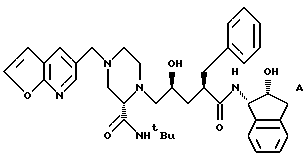
которое представляет собой N-(2(R)-гидрокси-1(S)-инданил)- 2(R)-фенилметил-4(S)-гидрокси-5(1-(4-(3-фуро[2.3-b] пиридил- метил)-2(S)-N'-(трет. -бутилкарбоксамидо)пиперазинил)пентанамид, или его фармацевтически приемлемую соль.
Соединения настоящего изобретения имеют хиральные центры и существуют в виде рацематов, рацемических смесей и индивидуальных диастереомеров или энантиомеров, причем все изомерные формы входят в настоящее изобретение. Рацемическая смесь охватывает смеси стереоизомеров в соотношении 50:50 и в других соотношениях.
В том случае, когда любая переменная (например,  ) встречается более одного раза в любой составляющей части соединения или в формуле I, ее обозначение в каждом случае не зависит от обозначения в любом другом случае. Кроме того, комбинации заместителей и/или переменных фрагментов допустимы лишь в тех случаях, когда такие комбинации обеспечивают в результате устойчивые соединения.
) встречается более одного раза в любой составляющей части соединения или в формуле I, ее обозначение в каждом случае не зависит от обозначения в любом другом случае. Кроме того, комбинации заместителей и/или переменных фрагментов допустимы лишь в тех случаях, когда такие комбинации обеспечивают в результате устойчивые соединения.
Используемый в тексте описания термин "алкил", если не указано особо, включает как разветвленные, так и прямоцепные (нормального строения) насыщенные алифатические углеводородные группы, содержащие указанное число углеродных атомов (Me - метил, Et - представляет собой этил, Pr - пропил, Bu - бутил); используемый в тексте термин "галоген" обозначает фтор, хлор, бром и иод.
Фармацевтически приемлемые соли соединений формулы I (в виде водо- или маслорастворимых или диспергируемых продуктов) включают традиционные нетоксичные соли или соли четвертичного аммония, которые получают, например, из неорганических или органических кислот или оснований. Примеры таких солей присоединения кислот включают ацетат, адипинат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, дигидрохлорид, дифосфат, додецилсульфат, этансульфонат, фумарат, глюкогептаноат, глютамат, глицерофосфат, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, 2-гидрокси- этансульфонат, лактат, малеат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат и ундеканоат. Основные соли включают соли аммония, такие соли щелочных металлов, как соли натрия и калия; такие соли щелочноземельных металлов, как соли кальция и магния, соли с органическими основаниями, такие как соли дициклогексиламина, N-метил-D-глюкамина, а также соли с такими аминокислотами, как аргинин, лизин и т.п. Кроме того, основные азотсодержащие группы могут быть четвертичными с помощью таких агентов, как низшие алкилгалогениды, такие как метил, этил, пропил и бутил хлорид, бромиды и иодиды; такие диалкилсульфаты, как диметил-, диэтил-, дибутил- и диамилсульфаты, такие длинноцепные галогениды, как децил, лаурил, миристил и стеарил хлориды, бромиды и иодиды, такие аралкилгалогениды, как бензил- и фенетилбромиды и т.п. Другие фармацевтически приемлемые соли включают этанолятсульфат и сульфатные соли.
Схемы I-II (см. в конце описания) показывают получение новых соединений изобретения. Примеры специально иллюстрируют приложение следующих ниже схем к конкретным соединениям.
Реакции амидного сочетания, используемые для получения соединений настоящего изобретения, обычно осуществляют карбодиимидным методом с помощью таких реагентов, как дициклогексилкарбодиимид или 1-этил-3-(3-диметиламинопропил)карбодиимид. Другие методы получения амидной или пептидной связи включают; но не ограничиваются ими, синтетические маршруты через хлорангидрид кислоты, азид, смешанный ангидрид или активированный сложный эфир. Обычно осуществляют реакцию жидкофазного амидного сочетания, однако вместо нее можно использовать твердофазный синтез с помощью классического метода Мэррифилда. Присоединение или удаление одной или более защитных групп является общепринятой процедурой.
Дополнительная родственная информация по сущности синтетических методов содержится в EPO 0337714 и EPO 0541168.
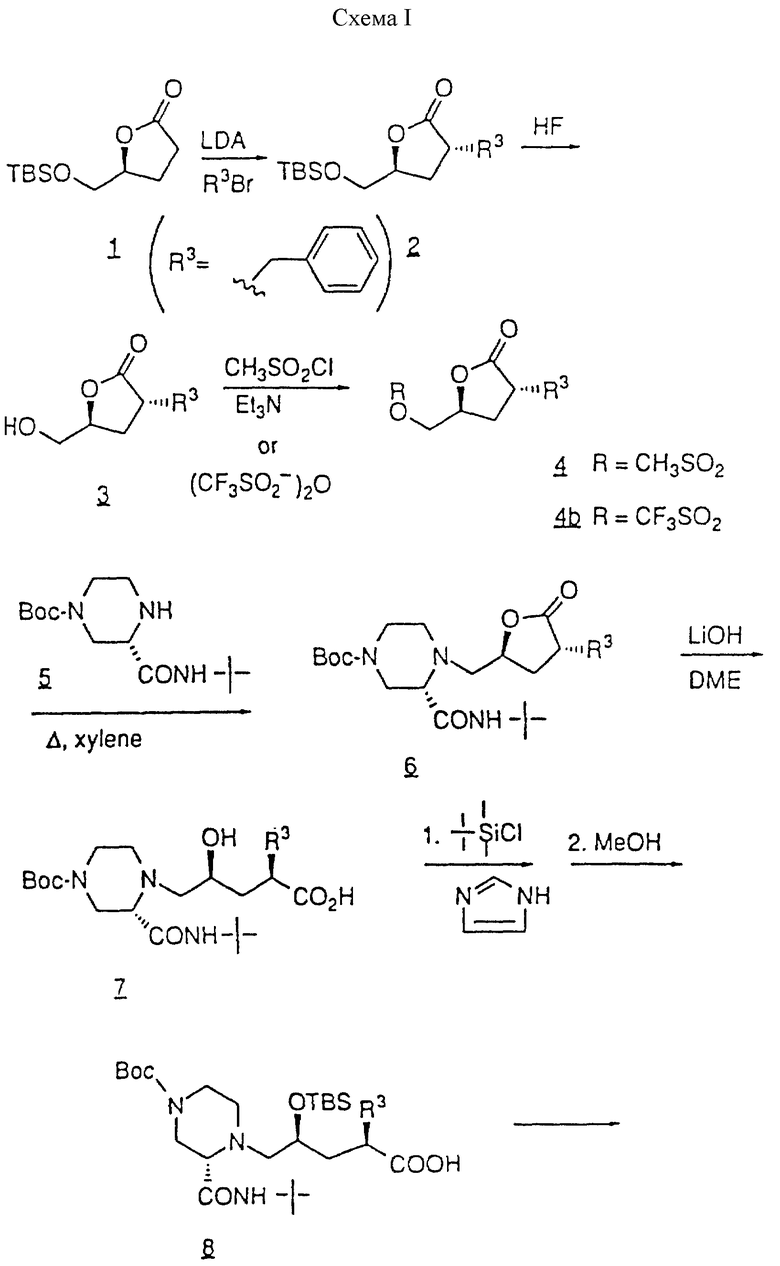
Один из методов получения соединений формулы I представлен Схемой I. Дигидро-5(S)-(трет. -бутилдиметилсилилоксиметил)- 3(2H)-фуранон (соединение 1, указанное ниже) получают стандартными методами, известными в данной области, из коммерчески доступного дигидро-5(S)-(гидроксиметил)-2(3H)-фуранона. После алкилирования соединения 1 с образованием соединения 2 защитную группу лактона 2 удаляют с помощью водного раствора HF с получением соединения 3.
Спиртовую группу соединения 3 активируют превращением в такую уходящую группу, как мезилат, тозилат или трифлат, в результате обработки спирта хлористым сульфонилом или (предпочтительно) серным ангидридом, например ангидридом трифторметансульфокислоты, в присутствии такого затрудненного аминного основания, как триэтиламин, диэтилизопропиламин или 2,6-лутидин, с получением такого соединения, как Соединение 4. Уходящую группу Соединения 4 замещают на амин 5, например 4-(1,1- диметилэтоксикарбониламино)-пиперазин-2(S)-карбоксамид, в таком растворителе, как DMF или ксилол, с получением такого соединения, как соединение 6. Трифторметансульфонилокси-группу можно замещать на амин при комнатной температуре в таком растворителе, как изопропанол или хлористый метилен, в результате обработки N,N- диизопропилэтиламином.
Соединение 6 гидролизуют с помощью водного раствора гидроксида лития или натрия и полученную в результате гидрокси кислоту 7 превращают в защищенную гидрокси кислоту 8. Гидроксильную группу для удобства защищают такой стандартной силил-защитной группой, как трет.-бутилдиметилсилил или трет-бутилдифенилсилил.
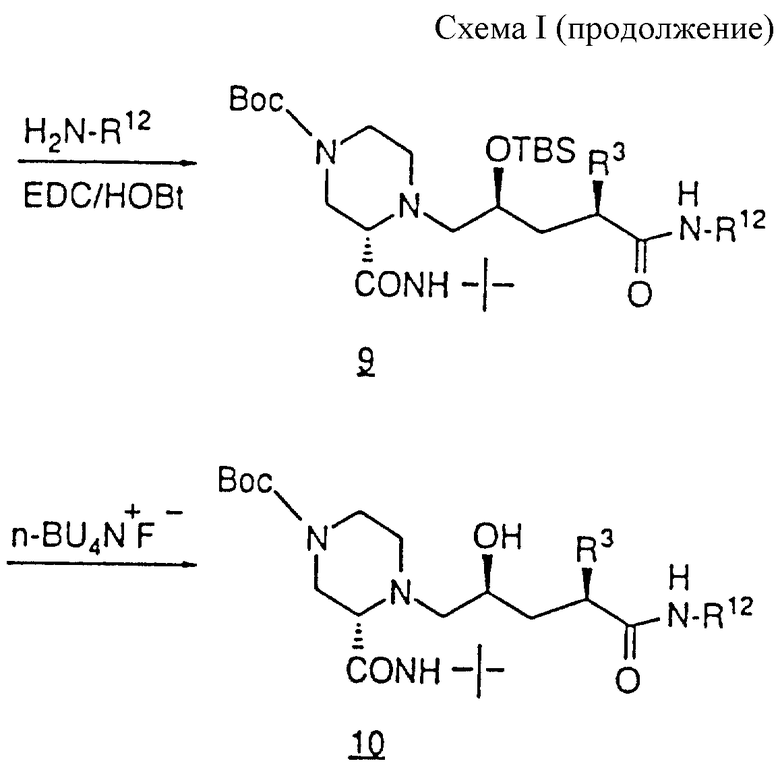
Защищенную гидрокси-кислоту 8 далее сочетают с желаемым R12 амином с образованием соединения 9, а силильную защитную группу удаляют с помощью фторид-иона с получением соединения 10.
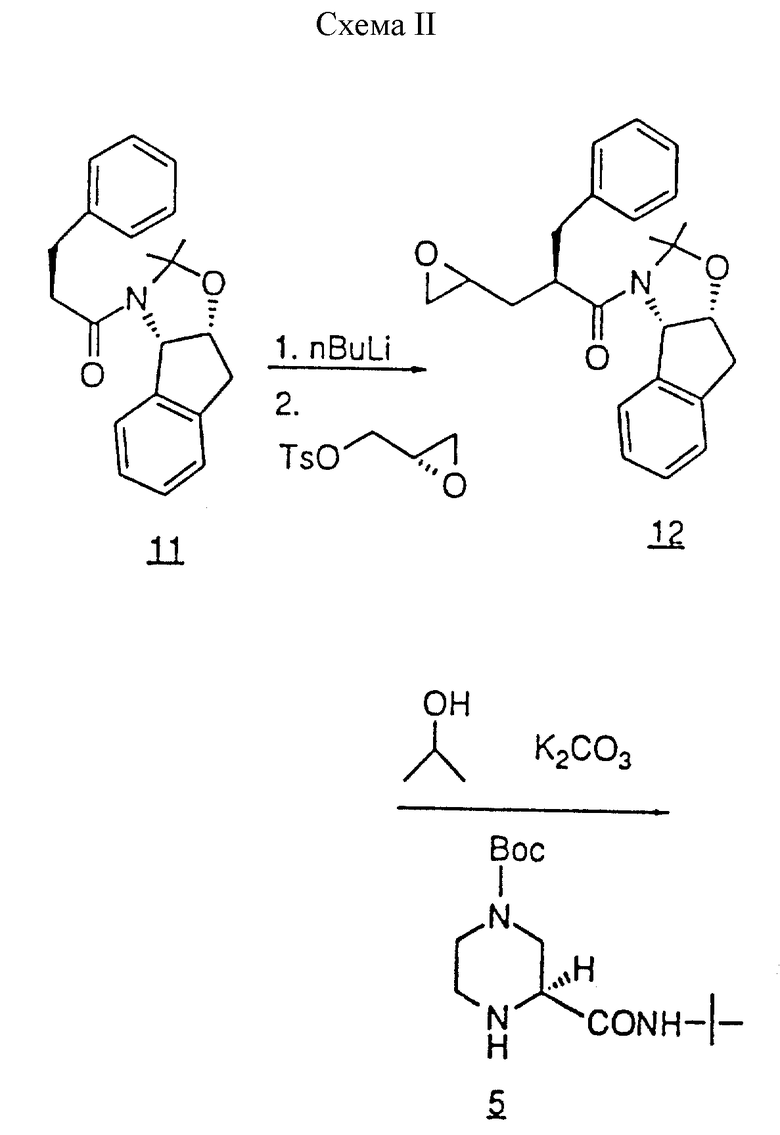
Одним из предпочтительных методов является синтез эпоксида 12 по реакции 11 в присутствии сильного основания. В качестве сильного основания может использоваться металлсодержащее основание, причем реакцию проводят в среде такого инертного безводного органического растворителя, как например циклические или ациклические углеводороды, включающие гексан, пентан, циклогексан и т.п. Подходящие сильные основания включают: LiN/(CH3)3Si/2, KN(CH3)3Si/2, NaN/(CH3)3Si/2, н-бутиллитий (н-BuLi), втор.-BuLi, трет.-BuLi, трет.-бутилат калия, диизопропиламид лития (LDA), изопропилциклогексиламид лития, пирролидид лития, тетраметилпиперидид лития, фениллитий, хлорид изопропилмагния, хлорид изобутилмагния, и другие аналогичные сильные основания, известные в данной области. Предпочтительными сильными основаниями являются н-BuLi, втор. -BuLi, LiN/(CH3)3Si/2 и LDA, причем н-BuLi и LiN/(CH3 )3Si/2 являются наиболее предпочтительными.
Предпочтительно на 1 молярный эквивалент соединения 11 используют 1-2 молярных эквивалента сильного основания.
Соединение 13 получают по реакции соединения 12 с N-трет.- бутил-4-(1, 1-диметилэтоксикарбониламино)пиперазин-2(S) карбоксамидом (5). Предпочтительно 1-3 молярных эквивалента амина (5) используют на молярный эквивалент эпоксида 12, причем более предпочтительным является соотношение V:IV, равное 1,05:1 молярному эквиваленту.
Эту реакцию можно проводить в любом подходящем растворителе, например в растворителе, выбранном из таких углеводородов, как толуол, таких простых эфиров, как диэтиловый эфир, таких спиртов, как метанол, этанол или изопропанол, таких нитрилов, как ацетонитрил, и таких сложных эфиров, как этилацетат, или их комбинаций, причем спирты являются наиболее предпочтительными растворителями, а изопропанол - наиболее предпочтительным растворителем. Температура реакции может поддерживаться в интервале от окружающей до температуры дефлегмации используемого растворителя, однако предпочтительно проводить реакцию при повышенной температуре, например в интервале 80- 90oC, наиболее предпочтительно при 83-85oC.
Активированные глицидолы могут быть получены способами, известными в данной области, как это описано, например, в статье J. Klunder с сотр., J. Org. Chem., 1989, 54, 1295-1304, и в ссылках, цитированных в данной работе.
Амидные соединения, такие как соединение 11, могут быть получены согласно стандартным методикам, известным специалистам в данной области, например по методике примера 10, с использованием соответствующих исходных материалов.
Защитные группы, например азот-защищающие группы, могут использоваться, когда это необходимо, для практической реализации настоящего изобретения. Так например, азот в положении 4 2-трет.- бутилкарбоксамид пиперазина может быть защищен такой группой, как BOC, CBZ, бензил, 4-метоксибензил, 2,4-диметоксибензил, трифторацетамид, триалкилсилил, и другими группами, известными в данной области.
Соединение формулы 15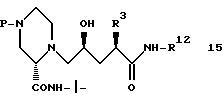
в которой P представляет собой такую азот-защищающую группу, как -BOC или -CBZ,
также получают согласно способу, описанному в Схеме I, предпочтительно используя 5-трифторметансульфонилоксиметиловый аналог лактона 4.
Соединение формулы 16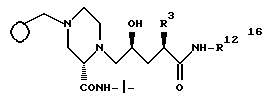
может быть получено различными путями из соединения 14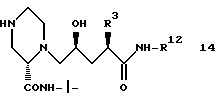
которое получают после удаления азот-защищающей группы из соединения 15 с использованием способов, хорошо известных в данной области, например путем каталитического гидрирования с целью удаления CBZ группы или обработки триметилсилилтрифлатом и 2,6- лутидином при 0oC в таком растворителе, как CH2Cl2, или обработкой с помощью 6 N HCl в изопропаноле с целью удаления группы BOC.
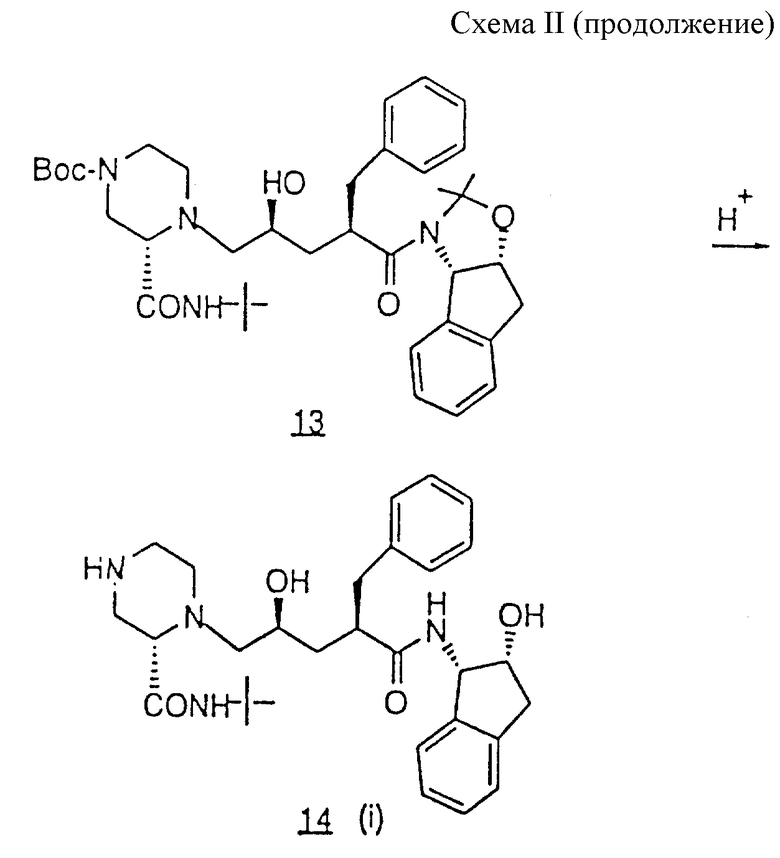
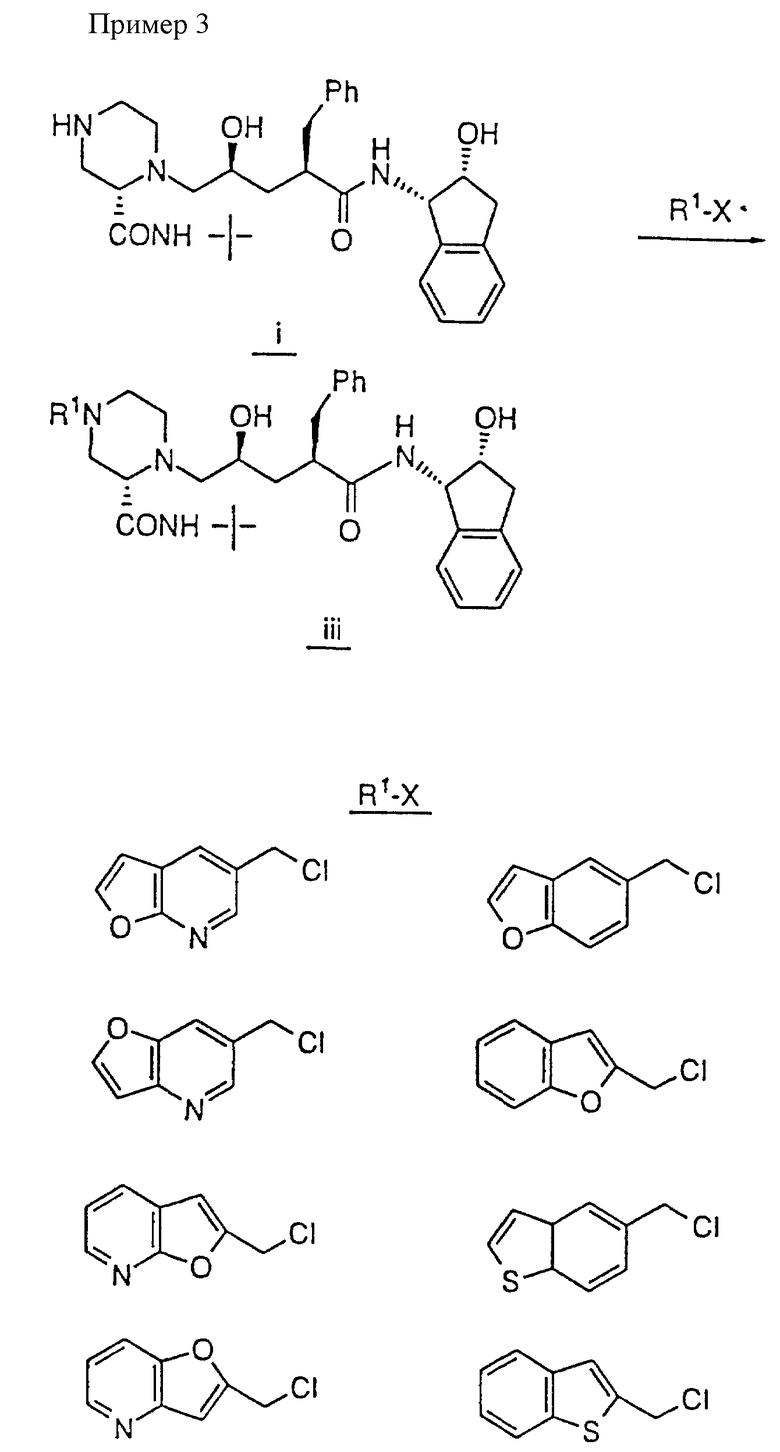
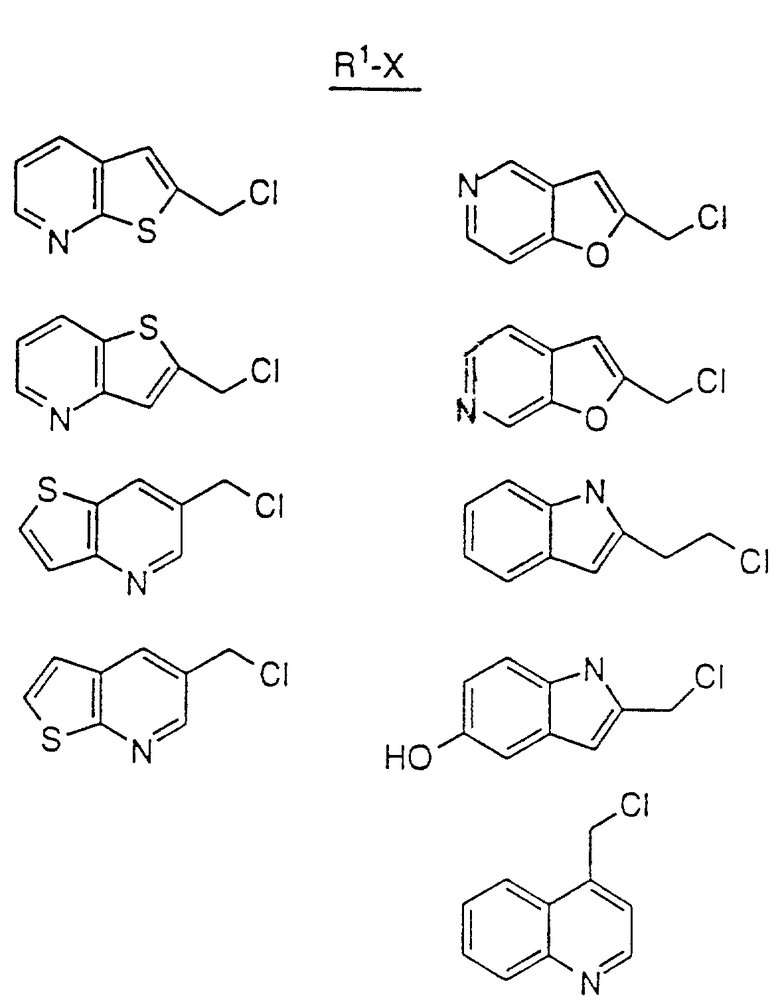
Азот в положении 4 пиперазинила в соединении 14 может быть проалкилирован соединением формулы R1-X в таком растворителе, как ДМФ в присутствии Et3N, при комнатной температуре, в том случае, когда X представляет собой Cl, Br или I. Методики таких процедур хорошо известны специалисту в данной области.
Соединения настоящего изобретения также проиллюстрированы в таблице примера 3, приведенного ниже.
Соединения настоящего изобретения полезны для подготовки и проведения анализов на скрининг антивирусных соединений. Так например, соединения настоящего изобретения весьма полезны для выделения энзимных мутантов, которые являются прекрасными инструментами скрининга на более мощные антивирусные соединения. Кроме этого, соединения настоящего изобретения могут использоваться для установления или определения сайта связывания других антивирусных агентов с HIV протеазой, например методом конкурентного ингибирования. Таким образом, соединения настоящего изобретения представляют собой коммерческие продукты, продаваемые для таких целей.
Соединения настоящего изобретения могут использоваться для ингибирования HIV протеазы, профилактики или лечения заражения вирусом иммунодефицита человека (HIV) и лечения таких последующих патологических состояний, как AIDS. Лечение AIDS или профилактика, либо лечение заражения HIV включает, но не ограничивается этим, лечение большого числа состояний HIV-инфицирования: AIDS ARC (AIDS родственный комплекс), как симптоматического, так и асимптоматического характера, а также острой или потенциальной подверженности действию HIV. Так например, соединения настоящего изобретения могут использоваться для лечения заражения HIV после подозрения на последующую подверженность действию HIV, например при переливании крови, трансплантации органов, замене жидкостной среды тела, наличия следов укуса, случайного укола иглой или воздействия на кровь пациента в ходе хирургического вмешательства.
В этих целях, соединения настоящего изобретения могут применяться орально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, внутриягодичные инъекции или вливания), путем ингаляционного распыления или ректально в рецептурах единичной дозировки, включающей традиционные нетоксичные фармацевтически приемлемые носители, стимуляторы и вспомогательные агенты.
Таким образом, в соответствии с настоящим изобретением дополнительно обеспечивается способ лечения и фармацевтические композиции для лечения заражения HIV и AIDS. Такое лечение включает применение на пациенте, нуждающемся в таком лечении, фармацевтической композиции, содержащей фармацевтический носитель - и терапевтически эффективное количество соединения настоящего изобретения или его фармацевтически приемлемой соли.
Такие фармацевтические композиции могут выпускаться в виде орально применяемых суспензий или таблеток; назальных спрэев; стерильных препаратов для инъекций, например в виде стерильных водных или масляный суспензий для инъекций, или свечей.
При оральном применении в виде суспензий такие композиции готовят согласно методам, широко известным в области приготовления фармацевтических рецептур, и они могут содержать микрокристаллическую целлюлозу для обеспечения массы, альгиновую кислоту или альгинат натрия в качестве суспендирующего агента, метилцеллюлозу в качестве усилителя вязкости и подслащивающие агенты и/или отдушки, известные в этой области. В виде таблеток немедленного выделения такие композиции могут содержать микрокристаллическую целлюлозу, дикальцийфосфат, крахмал, стеарат магния и лактозу и/или другие эксципиенты, связующие вещества, расширители, дезинтеграторы, разбавители и смазывающие вещества, известные в данной области.
При применении в виде назальных аэрозолей или путем ингаляции такие композиции готовят методами, хорошо известными в области фармацевтических рецептур, и они могут выпускаться в виде растворов в физиологическом растворе, с использованием бензилового спирта или других подходящих консервантов, промоторов адсорбции для усиления биоприменимости, фторуглеродов и/или других солюбилизирующих или диспергирующих агентов, известных в данной области.
Растворы или суспензии для инъекций могут формироваться согласно известным методам, с использованием нетоксичных, парентерально применимых разбавителей или растворителей, таких как маннит, 1,3-бутандиол, вода, раствор Рингера или изотонический раствор хлористого натрия или подходящих диспергирующих или смачивающих и суспендирующих агентов, таких как стерильные, мягкие, устойчивые масла, включая синтетические моно- или диглицериды, или жирные кислоты, включая олеиновую кислоту.
При ректальном применении в виде свечей такие композиции могут готовиться путем смешивания лекарства с таким нераздражающим эксципиентом, как масло какао, синтетические глицеридные сложные эфиры или полиэтиленгликоли, которые являются твердыми веществами при обычных температурах, но сжижаются и/или растворяются в ректальной полости с выделением лекарства.
Уровни дозировок порядка 0,02-5,0 или 10,0 г/день могут использоваться для лечения или профилактики указанных выше состояний, причем оральные дозировки в 2-5 раз выше. Так например, заражение HIV эффективно лечится путем применения 1,0-50 мг соединения на килограмм веса тела при приеме лекарства 1-4 раза в день. Согласно одному из предпочтительных режимов лечения, дозировки 100-400 мг каждые 6 часов применяли орально на каждом пациенте. Однако следует иметь в виду, что конкретный уровень дозировок и частота приема лекарства для каждого конкретного пациента может изменяться и будет зависеть от большого числа факторов, включая активность конкретного соединения, метаболическую стабильность и длительность действия такого соединения, возраст пациента, вес тела, общее состояние здоровья, пол, тип и время применения, скорость выделения, лекарственной комбинации, тяжести конкретного болезненного состояния и хозяина, подвергаемого терапии.
Настоящее изобретение также относится к комбинации соединений, ингибирующих HIV протеазу, с одним или более агентами, используемыми для лечения AIDS. Так например, соединения настоящего изобретения могут эффективно применяться как в период до воздействия, так и в период после воздействия, в комбинации с эффективными количествами AIDS антивирусных агентов иммуномодуляторами, обеззараживающими агентами или вакцинами, известными специалистам в данной области.
Следует иметь в виду, что сфера комбинаций соединений настоящего изобретения с AIDS антивирусными агентами, иммуномодуляторами, противоинфекционными агентами или вакцинами не ограничивается перечнем, представленным в приведенной в конце описания таблице, но, в принципе, включает любые комбинации с любым фармацевтическим составом, предназначенным для лечения AIDS.
Некоторые соединения, приведенные в таблице, представляют собой следующее:
Соединение B представляет собой 6-хлор-4-(S)-циклопропил-3,4- дигидро-4-((2-пиридил)этинил)квиназолин-2(1Н)-он;
Соединение C представляет собой(-)-6-хлор-4(S)-трифторметил- 1,2-дигидро-4(Н)-3,1-бензоксазин-2-он;
невирапин представляет собой N-циклопропил-5,11-дигидро-4- метил-6H-дипиридо/3,2-b/:2',3'-e//1,4/диазепин-6-он.
Соединения B и С синтезировали согласно способам ЕР 0569083, на содержание которого ссылаются в данном описании. Невирапин синтезирован Klunder J. M. с сотр., Med. Chem., 35, 1887 (1992); Нargrave K.D. с сотр., J. Med. Chem. , 34, 2231 (1991); Cohen K.A. с сотр., J. Biol. Chem., 260, 14670 (1991), причем на все эти три работы ссылаются в настоящем описании.
Предпочтительные комбинации одновременно и поочередно применяются в лечении ингибитора HIV протеазы и ненуклеозидного ингибитора HIV обратной транскриптазы. Необязательный третий компонент в комбинации представляет собой нуклеозидный ингибитор HIV обратной транскриптазы, такой как AZT, ddC или ddI. Предпочтительным ингибитором HIV протеазы является соединение A. Предпочтительные ненуклеозидные ингибиторы HIV обратной транскриптазы включают соединение В, соединение C или невирапин. Такие комбинации могут давать синергетические эффекты на ограничение распространения HIV. Предпочтительные комбинации включают:
(1) соединение A, совместно с предпочтительным ненуклеозидным ингибитором HIV обратной транскриптазы, и, необязательно, AZT или ddI, либо ddC;
(2) соединение A и любое соединение, выбранное из AZT, ddI или ddC.
Анализ на ингибирование микробиально экспрессированной HIV протеазы
Исследование ингибирования реакции экспрессии протеазы в Eschericia coli в присутствии пептидного субстрата (Val - Ser -Gln - Asn-(бетанафтил)Ala-Pro-Ile-Val, 0,5 мг/мл во время инициирования реакции) проводили в 50 мМ ацетата Na, pH 5,5 при 30oC в течение 1 часа. Различные концентрации ингибитора в 1,0 мкл ДМСО добавляли к 25 мкл пептидного раствора в воде. Реакцию инициировали добавлением 15 мкл 0,33 нМ протеазы (0,11 нг) в растворе 0,133 М ацетата Na, pH 5,5 и 0,1% альбумина бычьей сыворотки. Реакцию прекращали с помощью 160 мкл 5% фосфорной кислоты. Продукты реакции разделяли методом HPLC (VVDAC, широкопористый, 5 см C-18 обратимая фаза, ацетонитрильный градиент, 0,1% фосфорной кислоты). Степень ингибирования реакции определяли из высот пиков продуктов. HPLC независимо синтезированных продуктов, служивших количественными стандартами, подтвердила состав продуктов. Соединение A имело значение IC50 порядка 0,27 нМ.
Анализ на распространение клеток
Ингибирование распространения HIV в клеточной культуре измеряли согласно методу Nunberg J.H, с сотр., J.Virol. 65, 4887 (1991). B этом анализе МТ-4 Т-лимфоидные клетки инфицировали HIV-1 (дикий тип, если не указано особо) с использованием заранее определенного прививочного материала и культуры инкубировали в течение 24 часов. К этому времени ≤ 1% клеток были положительными согласно косвенной иммунофлуоресценции. Затем клетки тщательно промывали и распределяли в чашках для культур с 96-ю углублениями. В углубления добавляли серийные двукратные разбавления ингибитора и культуры выдерживали еще в течение 3 дней. Через 4 дня после заражения 100% клеток в контрольной культуре оказались инфицированными. Накопление HIV р24 прямо коррелировало с распространением вируса. Ингибиторную концентрацию клеточной культуры определяли как концентрацию ингибитора, выраженную в наномолях/литр, которая уменьшает распространение инфекции, по крайней мере на 95%, или ее обозначали как CIC95. Значение CIC95 для соединения A составило 25 нМ.
Ингибирование распространения вируса.
A. Приготовление суспензии клеток МТ-4, инфицированной HIV.
МТ клетки инфицировали в день 0 с концентрацией 250,000/ мл 1:1000 разбавлением HIV-1 штамма линии IIIb (конечная концентрация 125 пг p24/мл, что достаточно для образования ≤ 1% зараженных клеток на день 1 и 25-100% на день 4). Клетки инфицировали и выращивали в следующей среде: RPMI (Виттакер Био-Продактс), 10% инактивированной плодной телячьей сыворотки, 4 мМ глутамина (Гибко Лабс.) и 1:100 Пенициллин-Стрептомицин (Гибко Лабс.).
Смесь инкубировали в течение ночи при 37oC в атмосфере, содержащей 5% CO2.
B. Обработка ингибиторами.
Готовили матрицу наномолярного интервала концентраций парных комбинаций. В день 1 аликвоты в 125 мкл ингибиторов добавляли к равным объемам HIV-инфицированных МТ-4 клеток (50,000 на углубление) в микротитрическую пластину для клеточной культуры с 96-ю углублениями. Инкубацию продолжали в течение 3 дней при 37oC в атмосфере, содержащей 5% CO2.
C. Измерение распространения вируса.
С использованием многоканальной пипетки осажденные клетки ресуспендировали и 125 мкл собирали на отдельную микротитрическую пластину. Супернатант анализировали на наличие HIV р24 антигена.
Концентрацию HIV р24 антигена измеряли с помощью энзимного иммуноанализа, описанного ниже. Аликвоты р24, подлежащие измерению, добавляли в микроуглубления, покрытые моноклональным антителом, специфичным на антиген оболочки HIV. На этой и других последующих соответствующих стадиях микроуглубления промывали. Затем добавляли биотинилированное HIV-специфичное антитело, после чего добавляли конъюгат стрептавидин - пероксидаза хрена. При добавлении пероксида водорода и тетраметилбензидинового субстрата протекала цветная реакция. Интенсивность цвета была пропорциональна концентрации HIV р24 антигена.
Расчет степени синергии или усиленного ингибирования.
При наличии синергетического эффекта парные комбинации ингибиторов, как это было обнаружено, проявляют отчетливо усиленное ингибирование распространения вируса по сравнению с действием каждого ингибитора по отдельности или в сравнении с простым аддитивным ингибированием от каждого ингибитора.
Полученные данные обрабатывали следующим образом: фракционные ингибиторные концентрационные соотношения (FIC) рассчитывали согласно статье Elion с сотр. , J. Biol, Chem., 208, 477 (1954). Минимальную сумму FIC, демонстрирующую максимальный синергетический эффект, определяли для различных парных комбинаций. Чем меньше число, тем выше синергетический эффект.
Пример 1. Получение N-(2(R)-гидpoкcи-1(S)-инданил)- 2(R)-фенилметил-4(S)-гидрокси)-5-(1-(2(S)-N-(трет. -бутил- карбоксамидо)пиперазинил)-пентанамида, Соединение 14
Стадия 1: Получение дигидро-5-(S)-((трет.-бутилдифенилсилил)- оксиметил)-3(R)фенилметил-3(2Н)-фуранона.
Раствор диизопропиламида лития (LDA) получали добавлением 1,55 мл н-BuLi (2,5 М в гексане) к 0,55 мл (39 ммоля) диизопропиламина в 10 мл ТГФ при -78oC. Через 30 минут добавляли дигидро-5-(S)-((трет.-бутилдифенилсилил)-оксиметил)- 3(2H)-фуранона (1,38 г, 3,89 ммоля) в 5 мл ТГФ. Еще через 30 минут перемешивания добавляли бромистый бензил (0,68 г, 3,9 ммоля) и перемешивание продолжали в течение 3 часов, после чего реакцию прекращали путем добавления 10% водного раствора лимонной кислоты. Раствор экстрагировали этилацетатом (2 х 50 мл), который подвергали обратной промывке рассолом, сушили, фильтровали и концентрировали с получением масла. Продукт очищали методом хроматографии (SiO2, 20% EtOAc/ гексан) с получением целевого соединения.
Стадия 2: Получение дигидро-5(S)-(гидроксиметил)-3(R)- фенилметил-3(2H)-фуранона.
К 5,26 г дигидро-5(S)-((трет.-бутилдифенилсилил)оксиметил- 3(R)фенилметил-3(2H)-фуранона в 40 мл ацетонитрила добавляли 1,34 мл 49% водного раствора HF. Через 18 часов при комнатной температуре реакционную смесь концентрировали досуха и остаток распределяли между водой (50 мл) и этилацетатом (50 мл). Органический слой промывали рассолом, сушили, фильтровали и концентрировали с получением продукта в виде желтовато-коричневого твердого вещества (т.пл. 69-72oC).
Стадия 3: Получение дигидро-5(S)-((трифторметансульфонил)- оксиметил)-3(R)-фенилметил-3(2H)-фуранона.
К раствору 18,4 г (89,2 ммоля) дигидро-5(S)-(гидроксиметил) -3(R)-фенилметил-3(2Н)-фуранона в 350 мл хлористого метилена, охлажденного до 0oC, добавляли 13,51 мл 2,6-лутидина (115,98 ммоля), после чего прикапывали 16,51 мл трифторметансульфонового ангидрида (98,1 ммоля). Через 1,5 часа при 0oC реакционную смесь переливали в смесь 300 мл льда с рассолом и перемешивали в течение 0,5 часа. Затем водный слой экстрагировали хлористым метиленом (3 х 150 мл), органические слои промывали 10% HCl (2 х 75 мл), насыщали NaHCO3 (100 мл), водой (100 мл), сушили над MgSO4, фильтровали и концентрировали с получением твердого остатка. В результате очистки методом хроматографии однократного испарения (колонка 120 х 150 мм, градиентное элюирование смесями гексаны: EtOAc, 4:1-3:1) получали целевое соединение, т.пл. 53-54oC.
Стадия 4: Получение 4-(1,1-диметилэтоксикарбониламино)-1- (фенилметилкарбониламино)-пиперазин-2S-карбоновой кислоты
Целевое соединение получали, следуя методике, описанной Bigge C.F., Hays S. J., Novak P.M., Drummond J.T., Johnson Y., Bobovski T., Tetrahedron Lett, 1989, 30, 5193; используя в качестве исходного соединения 2(S)-пиперазинкарбоновую кислоту (см. Felder E., Maffei S., Pietra S., Pitre D., Helv, Chim. Acta, 1960, 117, 888.
Стадия 5: Получение N-трет. -бутил-4-(1,1- диметилэтоксикарбонил-амино)-1-(фенилметилкарбониламино) пиперазин-2(S)-карбоксамида.
К 9.90 г (27,16 ммоля) продукта со стадии 4,растворенного в 75 мл ДМФ и охлажденного до 0oC, добавляли 5,73 г (29,88 ммоля) ЕДС, 4,03 г (29,88 ммоля) HOBt, 3,14 мл (29,88 ммоля) трет.-бутиламина и, наконец, 4,16 мл (29,88 ммоля) триэтиламина. Реакционную смесь перемешивали в течение 18 часов и реакционный объем концентрировали наполовину. Затем смесь разбавляли 600 мл EtOAc и промывали 10% HCl (2 х 75 мл), насыщенным NaHCO3 (1 х 75 мл), водой (3 х 75 мл) и рассолом (1 х 50 мл), сушили над MgSO4 и концентрировали с получением твердого вещества. Такое твердое вещество обрабатывали смесью EtOAc:гексан (1:2) и фильтровали с получением целевого соединения в виде белого твердого вещества; т.пл. 134-135oC.
Стадия 6: Получение N-трет. -бутил-4-(1,1- диметилэтоксикарбониламино)пиперазин-2(S)-карбоксамида.
К 1,20 г (2,86 ммоля) N-трет.-бутил-4-(1,1- диметилэтоксикарбониламино)-1-(фенилметилкарбониламино)пиперазин-2-(S) - карбоксамида и 1,1 г (0,086 ммоля) 10% Pd/C добавляли 15 мл метанола. В реакционный сосуд вводили водород и реакционную смесь перемешивали в течение 2 часов, фильтровали через целит и промывали этанолом. Растворители удаляли в вакууме с получением целевого продукта в виде пены.
Спектр 1H-ЯМР (300 МГц, CDCl3) δ : 6,65 (широкая полоса, 1H), 4.10 (мультиплет, 1H), 3.81 (широкая полоса, 1H), 3.21 (двойной дублет, J = 18 и 7 Гц), 3.02-2.70 (мультиплет, 4H), 2.10-2.0 (широкая, 1H), 1.50 (синглет, 9H), 1.41 (синглет, 9H).
Стадия 7: Получение дигидро-5(S)-(4-(1,1- диметилэтоксикарбониламино)-2(S)-N-(трет. -бутилкарбоксамидо) -пиперазинил)метил)-3(R)-фенилметил-3(2Н)-фуранона.
К раствору 22,40 г (0,0662 моля) дигидро-5(S)- ((трифторметансульфонил)оксиметил)-3-(R)-фенилметил-3(2Н)-фуранона (полученного на стадии 3) и 18,0 г (0,063 моля) N-трет.-бутил-4- (1,1-диметилэтоксикарбониламино) пиперазин-2(S)-карбоксамида, растворенного в 180 мл изопропанола, добавляли 11,53 мл (0,062 моля) N,N-диизопропилэтиламина. Через 2,5 часа добавляли еще 1,2 г дигидро-5(S)-(трифторметансульфонил)оксиметил)-3(R)- фенилметил-3(2Н)-фуранона. Реакционную смесь анализировали методом тонкослойной хроматографии (ТСХ) через 3,5 часа и концентрировали до состояния густого масла. В результате обработки смесью EtOAc/гексаны (1:2, 200 мл) получали белое твердое вещество, которое отфильтровывали и отбрасывали. Масло очищали методом колонной хроматографии мгновенного испарения (колонка 120х150 мм, градиентное элюирование смесью EtOAc/гексаны 1:1, 2:1, 3:1 до 100% содержаний EtOAc) с получением целевого соединения.
Спектр 1H-ЯМР (400 МГц, CDCl3) δ : 7.34-7.17 (мультиплет, 5H), 6.31 (широкий синглет, 1H), 4.38 (широкий мультиплет, 1H), 3.96-3.92 (мультиплет, 1H), 3.79 (широкий мультиплет, 1H), 3.16 (двойной дублет, J = 13,6 и 4,4 Гц, 1H), 3.08-2.99 (мультиплет, 3H), 2.90-2.82 (мультиплет, 1H), 2.80 (двойной дублет, J=13.5 и 8.9 Гц, 1H), 2.78 (мультиплет, 1H), 2.67-2.61 (мультиплет, 1H), 2.58-2.49 (мультиплет, 1Н), 2.38-2.32 (мультиплет, 1H), 2.32-2.04 (мультиплет, 1H), 1.99-1.92 (мультиплет, 1H), 1.45 (синглет, 9Н), 1.29 (синглет, 9H).
Стадия 8: Получение 2(R)-фенилметил-4(S)-(трет.- бутилдиметилсилилокси)-5-(1-(4-(1,1-ди- метилэтоксикарбониламино)))-2(S)-N-(трет. - бутилкарбоксамидо)-пиперазинил)-пентанамида.
К 25,50 г (52,50 ммоля) дигидро-5(S)-(4-(1,1-диметил- этоксикарбониламино))-2(S)-N-(трет. - бутилкарбоксамидо)пиперазанил)-метил-3(R)-фенилметил-3(2Н)- фуранона, растворенного в 120 мл ДМФ, охлажденного до 0oC, добавляли раствор 60 мл воды и 1,512 г (63,01 ммоля) гидроксида лития. Через 0,5 часа реакцию прекращали добавлением 10% HCl до pH 6 и раствор концентрировали в вакууме. Остаток растворяли в 50 мл воды и экстрагировали EtOAc (4 х 75 мл) и органические слои промывали водой (1 х 20 мл), рассолом (1 х 20 мл). Водный слой подвергали обратной экстракции EtOAc (2 х 75 мл) и объединенные органические слои сушили над MgSO4 и концентрировали с получением желтого твердого вещества. Такой сырой продукт растворяли в 100 мл ДМФ и добавляли 17,87 г (0,262 моля) имидазола, охлаждали до 0oC и затем добавляли 31,50 г (0,21 моля) хлористого трет.- бутилдиметилсилила. Полученную смесь перемешивали в течение часа при 0oC и затем нагревали до комнатной температуры. Через 20 часов реакцию прекращали с помощью 10 мл метанола и реакционную смесь концентрировали до половины объема. Добавляли 100 мл забуференной воды с pH 7 и водный слой экстрагировали EtOAc (4 х 100 мл), объединенные органические слои промывали 10% HCl (2 х 50 мл), водой (3 х 75 мл) и рассолом (1 х 50 мл), сушили над MgSO4 и концентрировали с получением целевого соединения. Полученный материал непосредственно использовали на следующей стадии.
Стадия 9: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(трет. -бутилдиметилсилилокси)-5-(1-(4-(1,1- диметилэтоксикарбониламино)))-2(S)-N-(трет.-бутилкарбоксамидо) -пиперазинил)-пентанамида.
К 27,0 г (0,0446 моля) сырого материала со стадии 6, растворенного в 180 мл ДМФ и охлажденного до 0oC, добавляли 8,98 г (0,0468 моля) ЕДС, 6,32 г (0,0468 моля) HOBt и 7,31 г (0,049 моля) аминогидроксииндана. Добавляли триэтиламин (6,52 мл, 0,0468 моля) и реакционную смесь перемешивали в течение 2 часов при 0oC, при комнатной температуре в течение 16 часов, и реакцию прекращали разбавлением 500 мл EtOAc. Органический слой промывали 10% HCl (2 х 100 мл), насыщенным NaHCO3 (1 х 100 мл), водой (3 х 150 мл), рассолом (1 х 75 мл), сушили над MgSO4 и концентрировали с получением целевого соединения в виде белой пены.
1H-ЯМР (400 МГц, CDCl3) δ : 7.4-7.17 (мультиплет, 9H), 6.51 (широкий синглет, 1H), 5.79 (широкий синглет, 1H), 5.23 (мультиплет, 1H), 4.23 (широкий синглет, 1H), 4.06 (мультиплет, 1Н), 3.96-3.84 (мультиплет, 2H), 3.07-2.78 (мультиплет, 8Н), 3.65 (двойной дублет, J= 9.6 и 4.1 Гц, 1H), 2.56-2.44 (мультиплет, 2H), 2.29 (двойной дублет, J=12.0 и 4.5 Гц, 1H), 2.17-2.09 (мультиплет, 1H), 1.79 (широкий синглет, 1Н), 1.44 (синглет, 9H), 1.35 (синглет, 9H), 1.10 (синглет, 2H), 0.84 (синглет, 9H), 0.12 (синглет, 3H), 0.08 синглет, 3H).
Стадия 10: Получение N-(2(R)-гидрокси-1(S)-инданил)- 2(R)-фенилметил-4(S)-(гидрокси)-5-(1-(4-(1,1-диметил- этоксикарбониламино)))-2(S)-N-(трет. -бутилкарбоксамидо) пиперазинил)-пентанамида.
К 32,20 г (0,0437 моля) N-(2(R)-гидрокси-1(S)-инданил)-2- (R)-фенилметил-4(S)-(трет. -бутилдиметилсилилокси)-5-(1- (4-(1,1-диметилэтоксикарбониламино))-2(S)-N-(трет. -бутилкарбоксамидо)- пиперазинил))-пентанамида добавляли 437 мл (0,437 моля) фтористого тетрабутиламмония (1,0 М раствор в ТГФ, Алдрич). Реакционную смесь перемешивали в течение 18 часов и затем концентрировали до объема в 200 мл и разбавляли 700 мл EtOAc. Полученный раствор промывали водой (2 х 100 мл), рассолом (1 х 50 мл) и водные слои подвергали обратной экстракции EtOAc (2 х 200 мл). Объединенные органические слои сушили над MgSO4 и концентрировали до состояния масла. В результате очистки методом хроматографии мгновенного испарения (колонка 120х150 мм, градиентное элюирование смесью CH2Cl2:CHCl3, насыщенной смесью NH3:метанол, с повышением количества метанола в ряду 1%, 1,5%, 2%) получали целевое соединение в виде белой пены.
Спектр 1H-ЯМР (400 МГц, CDCl3) δ : 7.31-7.11 (мультиплет, 9H), 6.41 (широкий синглет, 1H), 6.23 (дублет, J=8.6 Гц, 1H), 5.25 (двойной дублет, J= 8.6 и 4.7 Гц, 1H), 4.21 (мультиплет, 1H), 3.83-3.82 (мультиплет, 2H), 3.78-3.61 (мультиплет, 2H), 3.22-3.19 (мультиплет, 2Н), 3.03-2.78 (мультиплет, 8H), 2.62-2.58 (мультиплет, 1H), 2.41-2.35 (мультиплет, 2H), 2.04-2.02 (мультиплет, 1H), 1.57-1.50 (мультиплет, 1H), 1.45 (синглет, 9H), 1.32 (синглет, 9H).
Стадия 11: Получение N-(2(R)-гидрокси-1(S)-инданил)- 2(R)-фенилметил-4(S)-(гидрокси)-5-(1-(2(S)-N-(трет. - бутилкарбоксамидо)-пиперазинил) пентанамида. Соединение 14.
К 21,15 г (0,034 ммоля) N-(2(R)-гидрокси-1(S)-инданил)-2 (R)-фенилметил-4(S)-(гидрокси)-5-(1-(4-(1,1-диметилэтокси- карбониламино))-2(S)-N-(трет.-бутилкарбоксамидо)- пиперазинил))-пентанамида, растворенного в 350 мл хлористого метилена и охлажденного до 0oC, добавляли 22,43 мл (0,204 моля) 2,6-лутидина и затем 32,85 мл (0,170 моля) триметилсилилтрифлата в течение 5 минут. Через 0,5 часа реакцию прекращали с помощью 10% HCl (80 мл) и полученную смесь перемешивали в течение 0,5 часа. К полученной смеси добавляли 100 мл насыщенного NaHCO3 и затем твердый NaHCO3 до pH 8. Затем водный слой экстрагировали EtOAc (4 х 100 мл) и объединенные органические слои промывали водой (1 х 50 мл), рассолом (1 х 75 мл), сушили над MgSO4 и концентрировали. Остаток очищали методом колонной хроматографии (120х150 мм, градиентное элюирование смесью CH2Cl2:CHCl3, насыщенной смесью NH3:MeOH, медленно повышая концентрацию метанола в ряду 2%, 3%, 4%, 5%, 6% и до 10%). В результате получали, целевой продукт в виде белой пены.
Спектр 1H-ЯМР (400 МГц, CDCl3) δ: 7.53 (синглет, 1H), 7.29-7.09 (мультиплет, 9H), 6.52 (дублет, J=1.83 Гц, 1H), 5.24 (двойной дублет, J=1.82 и 4.9 Гц, 1H), 4.23 (двойной дублет, J= 4.7 и 4.03 Гц, 1H), 4.25-4.00 (широкий синглет, 1H), 3.83-3.81 (мультиплет, 1H), 3.03-2.88 (мультиплет, 4H), 2.82-2.73 (мультиплет, 7H), 2.50- 1.60 (широкий синглет, 2H), 2.45 (дублет, J= 6.2 Гц, 2Н), 2.32-2.29 (мультиплет, 1H), 1.98 (мультиплет, 1H), 1.51 (мультиплет, 1H), 1.33 (синглет, 9H).
Пример 2. Получение N-(2(R)-гидрокси-1(R)-инданил)- 2(R)-фенилметил-4(R)-гидрокси-5-(1-(4-(3- фуро[2,3-b] пиридилметил)-2(R)-N-(трет.-бутилкарбоксамидо) - пиперазинил))-пентанамида
Стадия 1: Получение (фуро[2,3-b]-бипиридин-2,5- дикарбоновой кислоты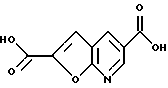
К раствору известного (Snyder H. R., Ebetino F.F., J. Het. Chem., 1, 202-205 (1966) диэтил-(фуро-[2,3-b] пиридин-2,5-дикарбоксилата (1,22 г, 4,923 ммоля) в 10 мл 95%-го этанола добавляли раствор гидроксида калия (0,66 г, 11,81 ммоля) в 10 мл воды. Реакционную смесь нагревали в течение 3 часов до 80oC, охлаждали до комнатной температуры и фильтровали. Двухкалиевую соль растворяли в воде и подкисляли 10% HCl до pH 2. Осадок отфильтровывали и сушили в вакууме с получением 850 мг белого твердого вещества.
Спектр 1H-ЯМР (400 МГц, (CD3)2SO) δ : 8.98 (дублет, J=2.2 Гц), 8.76 (дублет, J=2.2 Гц), 7.69 (синглет, 1H), 4.25 (широкий синглет, 3H).
Стадия 2: Получение фуро[2,3-b]пиридин-5-карбоновой кислоты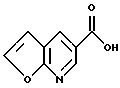
К суспензии фуро[2,3-b]пиридин-2,5-дикарбоновой кислоты (0,36 г, 1,484 ммоля) в 3 мл хинолина, в атмосфере Ar, добавляли порошкообразную медь (180 мг, 2,82 ммоля) и реакционную смесь нагревали в течение 1,5 часов до 210oC. Реакционную смесь охлаждали до комнатной температуры и разбавляли 50 мл хлористого метилена и фильтровали через целит. Органический слой экстрагировали насыщенным раствором (2 х 40 мл) Na2CO3 подкисляли до pH 3 с помощью 3 N HCl и фильтровали с получением 80 мг желтовато-коричневого твердого вещества. Водный слой экстрагировали смесью эфир/метанол (85/15) (3х50 мл) и промывали рассолом (1 х 10 мл), сушили над MgSO4 фильтровали и концентрировали с получением еще 35 мг продукта.
Спектр 1H-ЯМР (400 МГц, CD3OD) δ 8.89 (синглет, 1H), 8.67 (дублет, J=2.0 Гц, 1H), 7.97 (дублет, J=2.5 Гц, 1Н), 7.01 (дублет, J=2.4 Гц, 1H).
Стадия 3: Получение метил-фуро-[2,3-b]пиридин-5- карбоксилата.
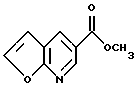
К фуро-[2,3-b] пиридин-5-карбоновой кислоте (3,0 г, 18,40 ммоля), растворенной в 40 мл метанола, добавляли 160 мл хлороформа и затем медленно добавляли триметилсилилдиазометан (42 мл, 10% раствор в гексенах). Через 0,5 часа добавляли 4 капли ледяной уксусной кислоты и реакционную смесь концентрировали. В результате получали 3,20 г белого твердого вещества.
Спектр 1H-ЯМР (400 МГц, CDCl3) δ/ : 9.02 (дублет, J=2.0 Гц, 1H), 8.60 (дублет, J=2.0 Гц, 1H), 7.9 (дублет, J= 2.5 Гц, 1H), 6.87 (дублет, J=2.5 Гц, 1H), 3.98 (синглет, 3H).
Стадия 4: Получение 5-гидроксиметил-фуро[2.3-b]пиридина.
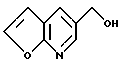
В высушенную на пламени 500 мл круглодонную колбу загружали метил-фуро-[2,3-b] пиридин-5-карбоксилат (3,20 г, 18,08 ммоля), растворенный в ТГФ и охлажденный до 0oC. К полученному раствору добавляли диизобутилалюминий гидрид (46 мл 46,1 ммоля, 1 М раствор в гексанах) в течение 10 минут и охлаждающую баню снимали. Через 4 часа реакционную смесь охлаждали до 0oC и медленно тушили реакцию с помощью солей Рошель (100 мл). Еще через 18 часов слои разделяли и водный слой экстрагировали этилацетатом (4 х 40 мл). Объединенные органические слои промывали рассолом (1 х 20 мл), сушили над MgSO4, фильтровали и концентрировали. Остаток очищали методом хроматографии мгновенного испарения (колонка 40х150 мм, градиент элюирования CH2Cl2:CHCl3, насыщенный смесью NH3:MeOH, в соотношении 60-39-1,0 (1000 мл), 60:38:2 (1000 мл), 60-37-3 (1000 мл), 60-36-4 (1000 мл). В результате получали 2,16 г белого твердого вещества.
Спектр 1H-ЯМР (400 МГц, CDCl3) δ : 8.19 (дублет, J=2.0 Гц, 1H), 7.92 (дублет, J= 2.0 Гц, 1H), 7.64 (дублет, J= 2.5 Гц, 1H), 6.69 (дублет, J=2.4 Гц, 1H), 4.78 (дублет, J=3.8 Гц, 2H), 4.69 (широкий синглет, 1Н).
Стадия 5: Получение гидрохлорида 3-хлорметил-фуро[2,3-b] пиридина.
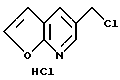
К раствору 5-гидроксиметил-фуро[2,3-b] пиридина, растворенного в 9 мл хлористого метилена, охлажденного до 0oC, добавляли хлористый тионил (4,23 мл, 57,99 ммоля). Ледяную баню снимали и через 1 час реакционную смесь концентрировали с получением 2,86 г белого твердого вещества.
Спектр 1H-ЯМР (400 МГц, CDCl3) δ: 8.40 (дублет, J=2.0 Гц, 1H), 8.13 Дублет, J= 2.2 Гц,1E), 7.80 (дублет, J=2.4 Гц, 1Н), 6.86 (дублет, J=2.4 Гц, 1H), 4.74 (синглет, 2H).
Стадия 6: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(1-(4-(3-фуро[2,3-b] пиридилметил) - 2(S)-N'-(трет.-бутилкарбоксамидо)-пиперазинил))- пентанамида.
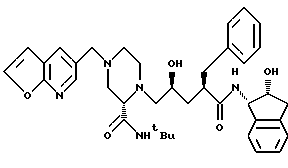
К раствору N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил- 4(S)-гидрокси-5(-2(S)-N'-(трет.-бутилкарбоксамидо)-пиперазинил- пентанамида (6,50 г, 12,48 ммоля), растворенному в 12 мл диметилформамида, в атмосфере аргона, добавляли 3-хлорметилфуро [2.3-b]пиридин гидрохлорид (2,80 г, 13,72 ммоля) и триэтиламин (5,21 мл, 38.44 ммоля). Через 18 часов реакционную смесь разбавляли 400 мл этилацетата и промывали насыщенным NaHCO3 (1 x 25 мл), водой (5 х 20 мл) и рассолом (1 х 25 мл).
Полученный раствор сушили над MgSO4, фильтровали и концентрировали до состояния масла. Остаток очищали методом хроматографии мгновенного испарения (колонка 60х150 мл, градиентное элюирование смесью CH2Cl2:CH2Cl2, насыщенной смесью NH3:MeOH в соотношении 60:39:1 (1000 мл), 60:38:2 (1000 мл), 60:37:3 (1500 мл), 60:36:4 (1500 мл). Полученную в результате пену обрабатывали этилацетатом и желаемый продукт фильтровали и сушили в течение ночи в высоком вакууме при 65oC с получением 5,30 г белого кристаллического твердого вещества. Смешанные фракции с хроматографической колонки могут быть объединены и подвергнуты повторной очистке с получением дополнительного количества продукта, т.пл. 183,5-184,5oC.
Спектр 1H-ЯМР (400 МГц, CDCl3) δ : 8.25 (дублет, J=2.2 Гц, 1H), 7.85 (дублет, J= 2.0 Гц, 1H), 7.5 (синглет, 1Н), 7.73 (дублет, J=2.4 Гц, 1H), 7.32-7.10 (мультиплет, 9H),6.75 (дублет, J=2.4 Гц, 1H), 5.95 (дублет, J=8.6 Гц, 1H), 5.27 (двойной дублет, J=8.5 и 4.8 Гц, 1H), 4.27-4.26 (мультиплет, 1H), 4.12 (широкий синглет, 1H), 3.89-3.83 (мультиплет, 1H), 3.51 (синглет, 2H), 3.29 (двойной дублет, J=17.5 и 4.0 Гц, 1H), 3.16 (двойной дублет, J= 3.36 и 3.48 Гц, 1H), 3.15 (двойной дублет, J=6.6 и 5.1 Гц, 1H), 2.94-2.50 (мультиплет, 11H), 2.36-2.34 (мультиплет, 1Н), 1.66 (синглет, 1H), 1.62-1.47 (мультиплет, 1H), 1.35 (синглет, 9H).
Элементный анализ: вычислено для C38H47N5O5
C 69.81, H 7.25. N 10.71;
Найдено: C 69.46, H 7.22, N 10.69.
Пример 3.
Используя практически ту же методику, что описана в примере 2, но проводя реакцию между N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)-гидрокси-5-(1-(2(S)-N'-(трет. бутилкарбоксамидо)пиперазинил))-пентанамидом, используемым в этом примере (соединение (1), см. ниже), и алкилирующим агентом (11), указанным ниже, вместо алкилирующего агента, используемого на стадии 6, получали следующие продукты формулы (III), см. в конце описания.
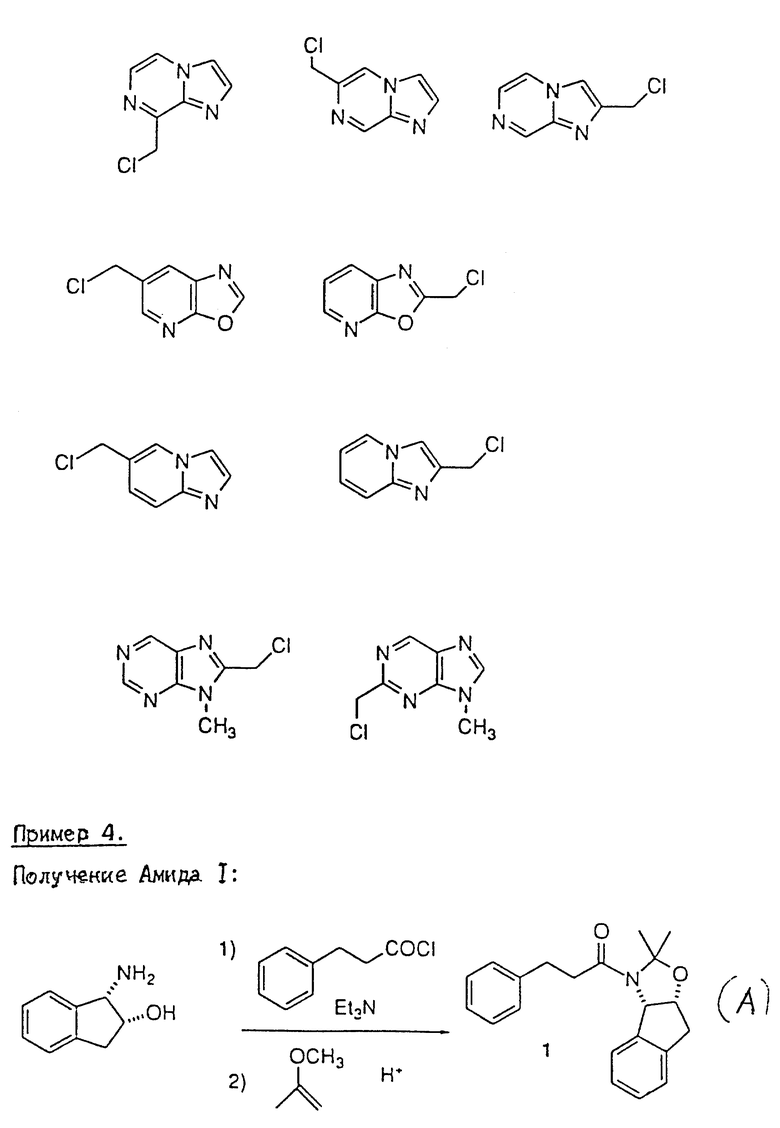
Пример 4. Получение амида I (см. в конце описания).
Раствор (-)-цис-1-аминоиндан-2-ола (884 г, 5,93 моля) в 17,8 л сухого ТГФ (KF= 55 мг/мл) (KF - обозначает титрование воды по Карлу Фишеру) и триэтиламин (868 мл, 6,22 моля) в 50 л круглодонной колбе, снабженной карманом для термопары, механической мешалкой, устройством для ввода азота и барботером, охлаждали до 15oC. Затем в течение 75 минут добавляли 3-фенилпропилхлорид (1000 г, 5,93 моля), поддерживая температуру внутри колбы в интервале 14-24oC с помощью охлаждающей бани, содержащей смесь льда с водой. После завершения добавления смеси давали стареть при 18-20oC в течение 30 минут и с помощью анализа методом HPLC проверяли исчезновение (2)-цис-1-аминоиндан-2-ола.
За ходом реакции следили методом хроматографии высокого разрешения: 25 см колонка Дюпон C8-RX, 60-40 ацетонитрил/10 мМ (KH2PO4/K2HPO4), 1,0 мл/мин, вводимый объем = 20 мл, детекция = 200 нм, приготовление образца: разбавление в 500 раз. Примерные времена удерживания: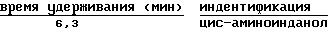
Реакционную смесь обрабатывали п-толуолсульфонатом пиридиния (241 г, 0,96 моля, 0,16 эквивалентов) и перемешивали в течение 10 минут (pH смеси после разбавления 1 мл образца равным объемом воды составляло 4,3- 4,6). Затем добавляли 2-метоксипропен (1,27 л, 13,24 моля, 2,2 эквивалента) и реакционную смесь нагревали до 38-40oC в течение 2 часов. Реакционную смесь охлаждали до 20oC и распределяли между этилацетатом (12 л) и 5% водным раствором NaHCO3 (10 л). Смесь перемешивали и слои разделялись. Этилацетатный экстракт промывали 5% водным раствором NaHCO3 (10 л) и водой (4 л). Этилацетатный экстракт сушили дистилляцией при атмосферном давлении, и растворитель меняли на циклогексан (общий объем ~ 30 л). После окончания дистилляции и концентрирования (объем экстракции этилацетатом 20% об.) горячий циклогексановый раствор медленно охлаждали до 25oC с кристаллизацией продукта реакции. Полученную в результате суспензию дополнительно охлаждали до 10oC и давали ей стареть. Продукт реакции выделяли фильтрацией и влажный осадок на фильтре промывали холодным (10oC) циклогексаном (2 х 800 мл). Промытый осадок на фильтре сушили в вакууме (26" Н; примерно 66 мм рт.ст.) при 40oC с получением 1,65 кг ацетонида 1 (86,4%, 98% площади, согласно HPLC).
Спектр 1H-ЯМР (300.13 МГц, CDCl3 основной ротамер) δ : 7.36-7.14 (мультиплет, 9H), 5.03 (дублет, J=4.4 Гц, 1Н), 4.66 мультиплет, 1H), 3.15 (мультиплет, 2H), 3.06 (широкий синглет, 2H), 2.97 (мультиплет, 2H): 1.62 (синглет, 3H), 1.37 (синглет, 3H);
13C-ЯМР (75,5 Мгц, CDCl3 основной ротамер) δc : 168.8, 140.9, 140.8, 140.6,128.6, 128.5, 128.4,127.1, 126.3, 125.8, 124.1, 96.5, 78.6, 65.9, 38.4, 36.2, 31.9, 26.5, 24.1.
Элементный анализ: вычислено для C21H23NO2
C 78.47, H 7.21, N 4.36;
Найдено; С 78.65, H 7.24, N 4.40.
Пример 5. Получение эпоксида 3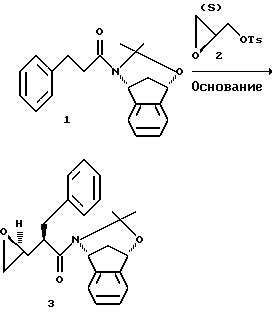
Раствор ацетонида 1 (1000 г, 3,11 моля) и 2(S)-глицидилтозилата 2 (853 г, 3,74 моля, 1,2 эквивалента) в 15,6 л ТГФ (KF=22 мг/мл) в 50 л четырехгорлой круглодонной колбе, снабженной термопарой, механической мешалкой, капельной воронкой и адаптером для ввода азота, трижды дегазировали, создавая вакуум и осуществляя продувку азотом, и полученную смесь охлаждали до -56oC. Затем в течение 2 часов добавляли гексаметилдисилазид лития (LiN/(CH3)3 Si/2) (2,6 л, 1,38 моля, 1,15 эквивалентов), поддерживая при добавлении температуру внутри колбы в интервале -50 - -45oC. Затем реакционную смесь перемешивали в течение 1 часа при -45 - -40oC и давали ей нагреваться до -25oC в течение 1 часа. Смесь перемешивали в течение 4 часов при -25 - -22oC (или до тех пор, пока исходный ацетонид не занимал 3,0% площади).
За ходом реакции следили с помощью анализа методом HPLC: колонка Зорбакс Оксид кремния, размерами 25 см х 4,6 нм, 20% этилацетата в гексане, 2,0 мл/мин, вводимый объем = 20 мл, детекция = 254 нм, приготовление образца = 100-кратное разбавление. Примерные времена удерживания:
Время удерживания (мин) - Идентификация
5,5 - амид 1
6,5 - глицидилтозилат 2
13,5 - эпоксид 3
Реакционную смесь обрабатывали деионизированной водой (6,7 л) при -15oC и обрабатывали этилацетатом (10 л). Смесь перемешивали и разделяли слои. Этилацетатный экстракт промывали смесью 1% водного раствора NaHCO3 (5 л) и насыщенным раствором NaCl (0,5 л). Этилацетатный экстракт (28,3 л) концентрировали вакуумной дистилляцией (28" Hg; 71,1 мм рт.ст.) и добавляли дополнительное количество этилацетата для замены растворителя на этилацетат (конечный объем = 11,7 л). В этилацетатном концентрате проводили замену растворителя на MeOH для кристаллизации продукта и систему концентрировали до конечного объема в 3,2 л. Оставшийся этилацетатный растворитель удаляли путем загрузки 10 л метанола и сбора 10 л дистиллата. Полученную в результате суспензию перемешивали в течение 1 часа при 22oC, затем охлаждали до 5oC и осуществляли старение в течение 0,5 часа. Продукт выделяли фильтрацией и влажный осадок на фильтре промывали холодным метанолом (2 х 250 мл). Промытый осадок на фильтре сушили в вакууме (26" Hg; примерно 66 мм рт.ст.) при 25oC с получением 727 г эпоксида 3 (61,2%, 98,7% площади основного эпоксида, согласно HPLC).
13C ЯМР (300 МГц, CDCl3) δ : 171.1, 140.6, 140.5, 139.6, 129.6, 128.8, 128.2, 127.2, 126.8, 125.6, 124.1, 96.8, 79.2, 65.8, 50.0, 48.0, 44.8, 39.2, 37.4, 36.2, 26.6, 24.1
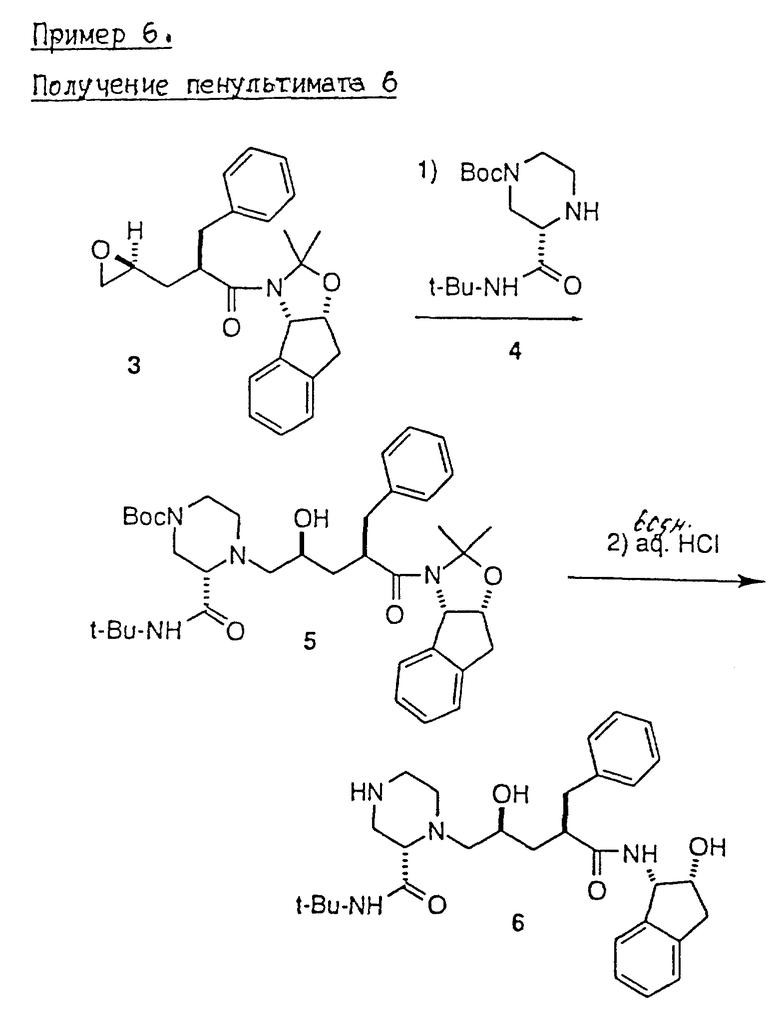
Пример 6. Получение пенультимата 6 (см. в конце описания).
Суспензию 2(S)-трет. -бутилкарбоксамидо-4-N-Вос-пиперазина 4 (1950. г, 6,83 моля, > 99,5%ее) (ee = энантиомерный избыток) и эпоксида 3 (2456 г, смесь в соотношении 97,5: 2,5 эпоксидов 4 S/R, 6,51 моля) в изопропаноле (2-пропанол, 18,6 л), помещенную в 72-литровую круглодонную колбу с четырьмя входными отверстиями, снабженную механической мешалкой, конденсатором флегмы, паровой баней, покрытой тефлоном термопарой и входом для азота, нагревали до начала дефлегмации (внутренняя температура 84-85oC). Через 40 минут образовывался гомогенный раствор. Смесь нагревали при дефлегмации в течение 28 часов.
Внутренняя температура в ходе дефлегмации составляла 84-85oC. За ходом реакции следили анализом с помощью HPLC: 25 см колонка Дюпон С8-РХ, соотношение ацетонитрил/10 мМ (KH2PO4/K2HPO4 60:40, 1,0 мл/мин, детекция = 220 нм, приготовление образца: 2 мкл, реакционную смесь разбавляли до объема 1 мл ацетонитрилом. Примерные времена удерживания:
Время удерживания (мин) - Отнесение
4,8 - пиперазин 4
8,9 - эпоксид 3
15,2 - продукт сочетания 5
Через 28 часов оставшийся эпоксид 3 и продукт сочетания 5 (в соответствии с данными HPLC) составили 1,5% площади и 91-93% площади соответственно. Смесь охлаждали до 0-5oC и добавляли 20,9 л 6 NHCl, поддерживая температуру на значении ниже 15oC. После завершения добавления смесь нагревали до 22oC. В этот момент наблюдалось выделение газа (изобутилен). Смеси давали стареть при 20-22oC в течение 6 часов.
За ходом реакции следили методом HPLC: используя те же условия, что были описаны выше. Примерные времена удерживания были следующими:
Время удерживания (мин) - Отнесение
7,0 - цис-аминоинданол
11,9 - пенультимат 6
15,1 - продукт сочетания 5
Смесь охлаждали до 0 С и медленно добавляли 7,5 л 50% раствора NaOH с целью установления pH смеси на значении pH 11.6, поддерживая температуру на значении ниже 25oC в ходе добавления. Смесь распределяли между этилацетатом (40 л) и водой (3 л). Смесь перемешивали и разделяли слои. Органическую фазу (60 л) концентрировали при пониженном давлении (29" Hg; 73,7 мм рт.ст.) и растворитель заменяли на ДМФ и полученную смесь концентрировали до конечного объема 10,5 л (KF = 1,8 мг/мл). Согласно данным HPLC анализа выход соединения 6 в этилацетате составил 86,5%. Соединение 6 в ДМФ непосредственно использовали на следующей стадии без дополнительной очистки. Для выделенного соединения 6:
13C ЯМР (75,4 МГц, CDCl3) δ : 175.2, 170.5, 140.8, 140.5, 139.9, 129.1, 128.5, 127.9, 126.8, 126.5, 125.2, 124.2, 73.0, 66.0, 64.8, 62.2, 57.5, 49.5, 47.9, 46.4, 45.3, 39.6, 39.3, 38.2, 28.9
Пример 7. Пиразин-2-трет.-бутилкарбоксамид 9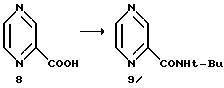
2-Пиразинкарбоновая кислота (8) - 3,35 кг (27 ммолей)
Хлористый оксалил - 3,46 кг (27,2 моля)
Трет.-бутиламин (KF = 460 мкг/мл) - 9,36 л (89 молей)
EtOAc (KF = 56 мкг/мл) - 27 л
ДМФ - 120 мл
1-Пропанол - 30 л
Карбоновую кислоту 8 суспендировали в 27 л EtOAc и 120 мл ДМФ в 72-литровой трехгорлой колбе, снабженной механической мешалкой, в атмосфере N2 и суспензию охлаждали до 2oC. Хлористый оксалил вводили в систему, поддерживая температуру в интервале 5-8oC.
Добавление завершали за 5 часов. В ходе экзотермического добавления выделялись CO и CO2. Происходило образование HCl, который, в основном, оставался в растворе. В системе присутствовал осадок, вероятно представляющий собой HCl-соль хлорангидрида пиразиновой кислоты. Анализ на образование хлорангидрида кислоты проводили путем тушения безводного образца реакционной смеси трет.-бутиламином. После завершения реакции оставалось менее 0,7% кислоты 8.
Анализ на полноту образования хлорангидрида кислоты имеет важное значение, поскольку неполное протекание реакции приводит к образованию примесей бис-трет.-бутилоксамида.
За ходом реакции можно следить методом HPLC: колонка Дюпон Зорбакс RXC8 размерами 25 см, скорость потока 1 мл/мин и детекция при 250 нм; линейный градиент с 98% 0,1% водного раствора H3PO4 и 2% CH3CN до 5% водного раствора H3PO4 и 50% CH3CN в течение 30 минут. Времена удерживания: кислоты 8 = 10,7 мин, амида 9 = 28,1 мин.
Реакционной смеси давали стареть в течение 1 часа при 5oC. Полученную в результате суспензию охлаждали до 0oC, добавляли трет.-бутиламин с такой скоростью, чтобы поддерживать температуру внутри реактора ниже 20oC.
Добавление требует 6 часов, если реакция была сильно экзотермичной. Небольшую часть образовавшегося гидрохлорида трет.- бутиламмония выводили из реакционной системы в виде пушистого белого твердого вещества.
Смеси давали стареть еще в течение 30 минут при 18oC. Осадившиеся аммониевые соли удаляли фильтрацией. Осадок на фильтре промывали 12 л EtOAc. Объединенные органические фазы промывали 6 л 3% NaHCO3 и 2х2л насыщенного водного раствора NaCl. Органическую фазу обрабатывали 200 г углерода Дарко G 60 и фильтровали через Золка Флок и осадок на фильтре промывали 4 л EtOAc.
Обработка углеродом эффективно удаляет пурпурный цвет продукта.
Раствор соединения 9 в EtOAc концентрировали при давлении 10 миллибар до 25% исходного объема. Добавляли 30 л 1-пропанола и дистилляцию продолжали до конечного объема 20 л.
К этому моменту количество EtOAc составляло, величину ниже предела детекции методом 1H-ЯМР (< 1%). Внутренняя температура такой замены растворителя составляла величину < 30oC. Раствор 1-пропанол/EtOAc соединения 3 был устойчив до начала дефлегмации при атмосферном давлении в течение нескольких дней.
Выпаривание аликвоты давало желтовато-коричневое твердое вещество, т.пл. 87-88oC.
13C ЯМР (75 МГц, CDCl3, ч/млн): 161.8, 146.8, 154.0, 143.8, 142.1, 51.0, 28.5
Пример 8. рац-2-трет.-бутилкарбоксамид пиперазин 10.
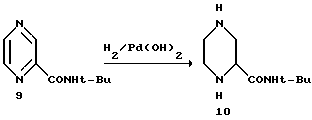
Материалы:
Пиразин-2-трет. бутилкарбоксамид 9 2,4 кг (23,4 моля), в растворе 1-пропанола 12 л 20% Pd(OH)2/C 16% вес., вода 144 г.
Раствор пиразин-2-трет.-бутилкарбоксамид/1-пропанол помещали в автоклав емкостью 5 галлонов (18,3 л). Катализатор добавляли в систему и смесь гидрировали при 65oC и давлении H2 40 фунт/дюйм2 (3 атм).
Через 24 часа реакционная смесь поглощала теоретическое количество водорода и метод ГХ показал наличие < 1% соединения 9. Смесь охлаждали, продували N2 и катализатор удаляли фильтрацией через Золка Флок. Катализатор промывали 2 л теплого 1-пропанола.
Было установлено, что использование теплого 1-пропанола в ходе промывки осадка на фильтре улучшает фильтрацию и уменьшает потери продукта из осадка на фильтре.
За ходом реакции следили методом ГХ: колонка Мегабор длиной 30 м, программирование температуры в интервале 100-160oC со скоростью 10oC/мин, выдержка 5 минут, затем подъем температуры до 250oC со скоростью 10oC/мин, времена удерживания: соединение 9 = 7,0 минут, соединение 10 = 9,4 минут. За ходом реакции можно также следить методом ТСХ, используя систему EtOAc/MeOH (50:50) в качестве растворителя и нингидрина в качестве проявляющего агента.
Выпаривание аликвоты показало, что выход в ходе амидирования и гидрирования составил 88% и что концентрация соединения 10 составила 133 г/л.
В результате выпаривания аликвоты получали соединение 10 в виде белого твердого вещества, с т.пл. 150-151oC;
13С ЯМР (75 МГц, D2О, ч/млн): 173.5, 59.8, 52.0, 48.7, 45.0, 44.8, 28.7
Пример 9. (S)Соль (S)-2-трет.-бутилкарбоксамидпиперазин бис(S)камфоросульфокислоты 11.
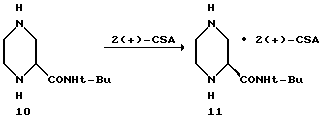
Материалы:
рац-2-трет. -бутилкарбоксамид-пиперазин 10 в растворе 1-пропанола - 4,10 кг (22,12 моля) в 25,5 кг растворителя
(S)-(+)-10-камфоросульфокислота - 10,0 кг (43,2 моля)
1-пропанол - 12 л
Ацетонитрил - 39 л
Вода - 2,4 л
Раствор амина 10 в 1-пропаноле загружали в 100 л колбу, соединенную с центрифугой (концентратором) периодического действия. Раствор концентрировали при давлении 10 миллибар и при температуре < 25oC до объема около 12 л.
К этому моменту продукт, осаждался из раствора, но возвращался обратно в раствор при нагревании смеси до 50oC.
Анализ гомогенной аликвоты показал, что концентрация соединения 10 составляла 341 г/л. Концентрацию определяли методом HPLC: колонка Дюпон Зорбакс PXC8 длиной 25 см, скорость подачи 1,5 мл/мин, детекция при длине волны 210 нм, изократный (98/2) CH3CN/0,1% водного раствора H3PO4. Время удерживания соединения 10: 2,5 минуты.
Добавляли ацетонитрил (39 л) и воду (2,4 л) с образованием прозрачного, слегка коричневатого раствора.
Определение содержания воды KF титрованием и соотношения CH3CN/1-пропанол интегрированием данных 1H-ЯМР показало, что соотношение CH3CN /1-пропанол/H2O составляет 26/8/1,6. Концентрация в растворе составила 72,2 г/л.
(S)-10-камфоросульфокислоту загружали в течение 30 минут 4-мя порциями при 20oC. Перед добавлением CSA температура повышалась до 40oC. Через несколько минут образовывался вязкий белый осадок. Белую суспензию нагревали до 76oC для растворения всех твердых веществ, затем слегка коричневатый раствор в течение 8 часов охлаждали до 21oC.
Продукт осаждался при 62oC. Продукт фильтровали без старения при 21oC и осадок на фильтре промывали 5 л смеси растворителей CH3CN /1-пропанол/H2O в соотношении 26/8/1,6. Полученное твердое вещество сушили при 35oC в вакуумной печи с подачей N2 с образованием 5,6 кг (39%) соединения 11 в виде белого кристаллического твердого вещества, т.пл. 288-290oC (при разложении).
[α]
13C ЯМР (75 МГц, D2O, ч/млн): 222.0, 164.0, 59.3, 54.9, 53.3, 49.0, 48.1, 43.6, 43.5, 43.1, 40.6, 40.4, 28.5, 27.2, 25.4, 19.9, 19.8
Значение "ee" материала составляло 95% согласно следующему хиральному HPLC анализу: аликвоту соединения 11 (33 мг) суспендировали в 4 мл EtOH и 1 мл Et3N. Добавляли Boc2О (11 мг) и реакционной смеси давали стареть в течение 1 часа. Растворитель полностью удаляли in vacuo и остаток растворяли, примерно в 1 мл EtOAc, и фильтровали с помощью пипетки Пастера через SiO2 с использованием в качестве элюента EtOAc. Выпаренные фракции продукта повторно растворяли в гексанах в количестве 1 мг/мл. Энантиомеры разделяли на колонке Дайсел Хираселл AS с помощью растворительной системы гексан/IPA (97: 3) при скорости потока 1 мл/мин, проводя детекцию при 228 нм. Времена удерживания: S антипод = 7,4 минут, R = 9,7 минут.
Пример 10. (S)-2-трет.-Бутилкарбоксамид-4-трет.- бутоксикарбонил-пиперазин 4 из соли 11.
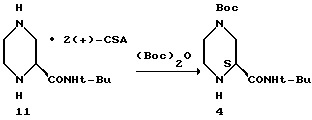
Материалы;
(S)-2-трет. -бутилкарбоксамид-пиперазиновая Бис(S)- (+)-CSA соль 11, 95% ее - 5,54 кг (8,53 моля)
Ди-трет.-бутилдикарбонат - 1,86 кг (8,53 моля)
Et3N - 5,95 л (42,6 моля)
EtOHПунктилиус, крепость 200 - 55 л
EtOAc - 2 л
К (S)-CSA соли 11 в трехгорлой колбе емкостью 110 л, снабженной капельной воронкой, в атмосфере N2 добавляли EtOH, после чего при 25oC добавляли триэтиламин. Твердое вещество легко растворялось при добавлении Et3N. Boc2O растворяли в EtOAc и загружали в капельную воронку. Раствор Boc2O в EtOAc добавляли с такой скоростью, чтобы поддерживать температуру ниже 25oC. Добавление продолжали в течение 3 часов. Реакционной смеси давали стареть в течение 1 часа после завершения добавления раствора Boc2O.
За ходом реакции можно следить методом HPLС: колонка Дюпон Зорбакс РХС8 при скорости подачи 1 мл/мин, детекция при. 228 нм, изократическая смесь (50/50) CH3CN/0,1 М KH2PO4, при установлении pH 6,8 с помощью NaOH. Время удерживания соединения 4 = 7,2 минут. Хиральный анализ проводили с использованием той же системы, что и на предыдущей стадии. За ходом реакции можно также следить с использованием метода ТСХ, применяя в качестве растворителя 100% EtOAc (Rf= 0,7).
Затем раствор концентрировали до объема примерно 10 л при внутренней температуре < 20oC в центрифуге (концентраторе) периодического действия, в вакууме с давлением 10 миллибар. Замену растворителя завершали добавлением в 20 л EtOAc и реконцентрированием до объема 10 л. Реакционную смесь промывали в экстрактор с помощью 60 л EtOAc. Органическую фазу промывали 16 л 5% водного раствора Na2CO3, 2 х 10 л деионизированной воды и 2 х 6 л насыщенного водного раствора хлористого натрия. Объединенные водные промывные жидкости подвергали обратной экстракции 20 л EtOAc и органическую фазу промывали 2 х 3 л воды и 2 х4 л насыщенного водного раствора хлористого натрия. Объединенные EtOAc экстракты концентрировали в вакууме при давлении 10 миллибар, при температуре внутри реакционного сосуда > 20oC в центрифуге периодического действия емкостью 100 л, до объема около 8 л. Замену растворителя на циклогексан осуществляли медленным добавлением примерно 20 л циклогексана и повторным концентрированием до объема 8 л. К суспензии добавляли 5 л циклогексана и 280 мл EtOAc, и смесь нагревали до дефлегмации, когда все содержимое переходило в раствор. Раствор охлаждали и добавляли при 58oC затравку (10 г). Суспензию охлаждали до 22oC в течение 4 часов и продукт выделяли фильтрацией через 1 час старения при 22oC. Осадок на фильтре промывали 1,8 л циклогексана и сушили в вакуумной печи при 35oC с подачей N2, в результате чего получали 1,87 кг (77%, > 99,9% площади, согласно данным HPLC R-изомер присутствовал в количестве ниже уровня детекции) соединения 4 в виде слегка желтовато-коричневого порошка.
[α]
13C ЯМР (75 МГц, CDCl3 ч/млн): 170.1, 154,5, 79.8, 58.7, 50.6; 46.6, 43.6, 43.4, 28.6, 38.3.
Хотя в настоящем описании излагаются принципы настоящего изобретения, причем примеры представлены в целях иллюстрации, следует иметь в виду, что практическая реализация изобретения охватывает все обычные изменения, адаптации или модификации, входящие в сферу следующей формулы изобретения и эквивалентов пунктов формулы изобретения.
Приложение 1. Получение соединения п. 13 (в уточненной формуле п.7) (называемого далее как соединение 13).
Первая стадия - Получение бензофуран-2-карбинола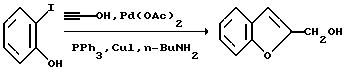
2-Иодфенол (500 мг, 2,27 ммоль), пропаргиловый спирт (265 мкл, 4,54 ммоль), Pd(OAc)2 (5,1 мг, 0,03 ммоль), трифенилфосфин (12 мг, 0,046 ммоль), н-бутиламин (450 мкл, 4,5 ммоль) и CuI (8,6 мг, 0,045 ммоль) объединяют в 4,5 мл ТТФ и смесь нагревают при 40oC в атмосфере N2 в течение 36 ч. Смесь охлаждают до комнатной температуры и растворители удаляют в вакууме и остаток очищают SiO2 колоночной хроматографией на 100 г силикагеля, элюируя 20% этилацетатом в гексанах. 2-(Гидроксиметил) бензофуран получают путем концентрирования фракций, содержащих продукт. [Kudu, N. G.; Pal, М.; Nahanty, J.S.; Dasgupta, F.K.; JCS Chem. Com. 1992, 41].
Вторая стадия - Получение бензофуран-2-хлорметила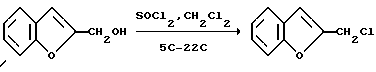
Бензофуран-2-карбинол (10,38 г, 68,4 ммоль) растворяют в метиленхлориде (100 мл) и охлаждают до 5oC. Через 5 минут загружают тионилхлорид (5,49 мл, 75,2 ммоль) и реакционную смесь выдерживают при 5oC в течение 30 минут и нагревают до 22oC. Реакционную смесь выдерживают при 22oC в течение 4 часов. Метиленхлоридную загрузку промывают ДИ водой (4 х 60 мл) и фильтруют через силикагель. Раствор концентрируют в вакууме, чтобы получить твердое вещество при охлаждении. Сырое твердое вещество растворяют в гексанах(120 мл) и обрабатывают Darco G-60 (1,0 г). Суспензию фильтруют и раствор концентрируют в вакууме, чтобы получить соединение бензофуран-2-хлорметил в виде твердого вещества.
Третья стадия - Алкилирование бензофуран-2-хлорметилом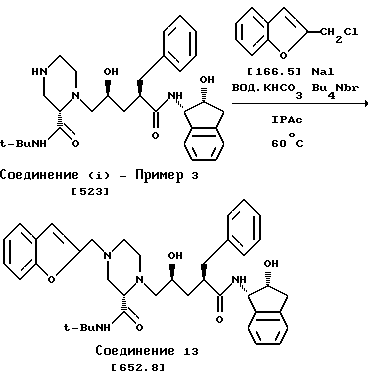
Выделенное предпоследнее твердое вещество (13,1 г, 25 ммоль) объединяют с IPAc (60 мл), водой (20 мл), KHCO3 (4,25 г, 42,5 ммоль), иодидом натрия (1,88 г, 12,5 ммоль) и бромидом тетрабутиламмония (600 мг, 1,86 ммоль) и смесь нагревают до 45oC под атмосферой азота. Добавляют 2- (хлорметил)бензофуран (4,6 г, 27,5 ммоль) и полученную смесь нагревают до 59-61oC в течение 5 ч. Смеси позволяют остыть до комнатной температуры и разбавляют IPAc (100 мл) и отделяют водный слой. Органический слой промывают 3 х 50 мл водой, затем 50 мл солевого раствора и сушат (MgSO4) и фильтрат концентрируют в вакууме и промывают 100 мл IPAc и концентрируют в атмосферных условиях до 80 мл, охлаждают до 25oC, вводят затравку и выдерживают при перемешивании 2 ч. Твердое вещество отфильтровывают и промывают холодным IPAc (2 х 15 мл), чтобы получить Соединение 13 (температура плавления = 152-153,5oC).
Приложение 2. Биологические данные для соединения 13.
Анализ на ингибирование экспрессированной микроорганизмами ВИЧ протеазы
Исследования ингибирования реакции протеазы, экспрессированной в Eschericia coli, с пептидной структурой [Val-Ser-Gln-Asn-(бета- нафтил)Ala-Pro-Ile-Val, 0,5 мг/мл, во время инициирования реакции проводят в 50 мМ Na ацетате, pH 5,5, при 30o С в течение 1 часа. Различные концентрации ингибитора в 1,0 мкл ДМСО добавляют к 25 мкл пептидного раствора в воде. Реакцию инициируют добавлением 15 мкл 0,33 нМ протеазы (0,11 нг) в растворе 0,133 М Na ацетата pH 5,5 и 0,1% альбумина бычьей сыворотки. Реакцию гасят 160 мкл 5%-ной фосфорной кислоты. Продукты реакции разделяют путем ВЭЖХ (VYDAC широкая пора 5 см C-18 обратная фаза, градиент ацетонитрила, 0,1% фосфорная кислота). Степень ингибирования реакции определяют по высотам пиков продуктов. ВЭЖХ продуктов, независимо синтезированных, подтверждает соответствие качественным стандартам анализа и подтверждает состав продукта. Соединение 13 показывает величины IC50 около 0,17 нМ.
Анализ распространения в клетках.
Ингибирование распространения ВИЧ в культуре клеток измеряют согласно Nunberg et al. , J. Virol. , 65, 4887 (1991). В этом анализе Т-лимфоидные клетки МТ-4 инфицируют ВИЧ-1 (дикий тип, кроме иначе указанного), используя заранее определенный инокулят, и культуры инкубируют в течение 24 часов. При этом времени выявляют ≤ 1% положительных клеток путем непрямой иммунофлуоресценции. Затем клетки экстенсивно промывают и распределяют в 96-луночные планшеты для культивирирования. Последовательно двукратно разбавленный ингибитор добавляют в лунки и культивирование продолжают в течение 3 дополнительных дней. За 4 дня после инфицирования 100% клеток в контрольных культурах оказываются зараженными. Накопление ВИЧ-1 р24 непосредственно соотносят с распространением вируса. Ингибирующую концентрацию для клеточной культуры определяют как концентрацию ингибитора в наномолях/литр, которая уменьшает распространение инфекции по меньшей мере на 95% или CIC95. CIC95 для Соединения 13 - 25 нМ.
Приложение 3. Фармацевтические композиции, содержащие соединение 13.
Первая стадия - Образование соли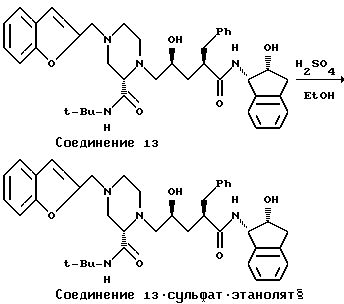
Свободное основание Соединения 13 (25 г, 38,3 ммоль) растворяют в абсолютном этаноле (150 мл) при 22oC. Эту порцию фильтруют через 5 мкм фильтр и фильтр промывают абсолютным спиртом (50 мл). Раствор серная кислота/этанол готовят при < 5oC путем введения концентрированной серной кислоты (3,91 г, 38,3 ммоль) в охлажденный раствор < 5oC) абсолютного этанола (50 мл) с такой скоростью, чтобы температура оставалась < 5oC. Часть кислотного раствора (10 мл, 20 об.%) загружают в раствор порции Соединения 13 при 22oC. В этот момент в порцию Соединения 13 необязательно может быть внесена затравка. Соединение 13*сульфат*этанолят (500 мг), при 22oC. Идеально вносить затравку в порцию Соединения 13, когда затравка помогает пересыщению во время кристаллизации. Суспензию выдерживают при 20-25oC в течение 30 минут. Остальной раствор кислоты загружают в порцию через канюлю свыше 60 минут. Во время добавления температура порции остается 20-25oC (примечание: раствор кислоты держат при < 5oC).
Окончательную суспензию порции выдерживают при 20-25oC в течение 60 минут и фильтруют. Осадок на фильтре промывают абсолютным этанолом (2 х 25 мл) и сушат в вакууме (635 мм Hg, 20oC) в течение 18 часов в токе азота, чтобы получить моноэтанолят сульфата Соединения 13. Моноэтанолят сульфата характеризуется кривой дифференциальной сканирующей калориметрии (DSC) при скорости нагревания 10oC/мин в открытом сосуде под текущим азотом, обнаруживающей эндотерму с экстраполированной начальной температурой около 190oC, пиковой температурой около 193oC и ассоциированным теплом около 120 Дж/г. На основе результатов TG и TG-FTIR эндотерма является следствием сочетания потери этанола и плавления с разложением. Порошковая рентгенограмма характеризуется d-интервалами 11,72, 5,56, 5,20, 5,00, 4,60, 4,50, 4,40, 4,26, 4,17, 4,08, 3,90, 3,81, 3,69, 3,24 и 3,33 Ангстрем.
Вторая стадия - Препарат А
100 мг сульфатной соли Соединения 13 с Первой стадии смешивают с достаточно тонко измельченной лактозой, чтобы получить общее количество 580-590 мг для заполнения капсулы из твердого геля О-вида.
Третья стадия - Препарат В.
Оральную суспензию готовят путем суспендирования 400 мг сульфатной соли Соединения 13 в 20 мл воды.
Приложение 4. Характеристики соединений из примера 3.
Температуры плавлений (mp) типичных соединений: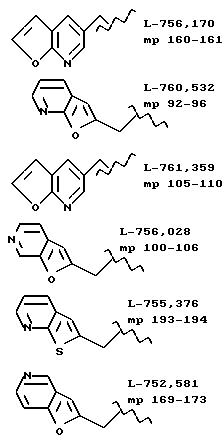






Описываются новые соединения формулы (I) или фармацевтически приемлемая соль такого соединения, в которой  представляет собой устойчивый 8-10 членный бициклический гетероцикл, каждое из колец которого может быть насыщенным или ненасыщенным, и указанный гетероцикл состоит из углеродных атомов и 1-3 гетероатомов, выбранных из группы, состоящей из N, S или O, причем указанный гетероцикл может быть незамещенным или замещенным галогеном или низшим C1-4 алкилом, при условии, что
представляет собой устойчивый 8-10 членный бициклический гетероцикл, каждое из колец которого может быть насыщенным или ненасыщенным, и указанный гетероцикл состоит из углеродных атомов и 1-3 гетероатомов, выбранных из группы, состоящей из N, S или O, причем указанный гетероцикл может быть незамещенным или замещенным галогеном или низшим C1-4 алкилом, при условии, что  не является группами (А). Соединения являются ингибиторами протеазы, закодированной вирусом иммунодефицита человека. Описываются также фармацевтическая композиция на основе соединений формулы (I) и способ ингибирования HIV протеазы. 3 с. и 11 з.п. ф-лы.
не является группами (А). Соединения являются ингибиторами протеазы, закодированной вирусом иммунодефицита человека. Описываются также фармацевтическая композиция на основе соединений формулы (I) и способ ингибирования HIV протеазы. 3 с. и 11 з.п. ф-лы.
 -
-
или фармацевтически приемлемая соль такого соединения,
где  представляет собой устойчивый 8-10-членный бициклический гетероцикл, каждое из колец которого может быть насыщенным или ненасыщенньм и указанный гетероцикл состоит из углеродных атомов и 1-3 гетероатомов, выбранных из группы, состоящей из N, S или O, причем указанный гетероцикл может быть незамещенным или замещенным галогеном или низшим C1-4 алкилом;
представляет собой устойчивый 8-10-членный бициклический гетероцикл, каждое из колец которого может быть насыщенным или ненасыщенньм и указанный гетероцикл состоит из углеродных атомов и 1-3 гетероатомов, выбранных из группы, состоящей из N, S или O, причем указанный гетероцикл может быть незамещенным или замещенным галогеном или низшим C1-4 алкилом;
при условии, что 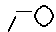 не является группами;
не является группами;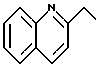
или 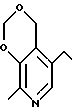
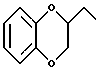
2. Соединение по п.1, в котором 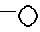 представляет собой устойчивый 8-10-членный бициклический гетероцикл, любое кольцо которого может быть насыщенным или ненасыщенным и указанный гетероцикл, состоит из углеродных атомов и 2 гетероатомов, выбранных из группы, состоящей из N или O, причем гетероатомы присутствуют в различных кольцах, или фармацевтически приемлемая соль такого соединения.
представляет собой устойчивый 8-10-членный бициклический гетероцикл, любое кольцо которого может быть насыщенным или ненасыщенным и указанный гетероцикл, состоит из углеродных атомов и 2 гетероатомов, выбранных из группы, состоящей из N или O, причем гетероатомы присутствуют в различных кольцах, или фармацевтически приемлемая соль такого соединения.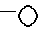 представляет собой:
представляет собой:
Х представляет собой O или S,
или фармацевтически приемлемая соль такого соединения.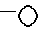 ограничено фрагментом
ограничено фрагментом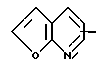
или фармацевтически приемлемая соль такого соединения.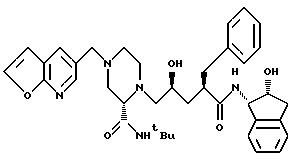
называемое N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)-гидрокси-5-(1-(4-(3-фуро[2,3-b] пиридилметил)-2(S)-N'-(трет-бутилкарбоксамидо)пиперазинил))пентанамид, или фармацевтически приемлемая соль такого соединения.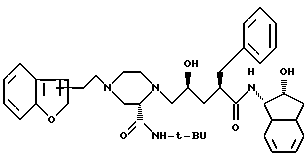
или его фармацевтически приемлемая соль.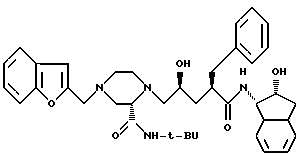
или его фармацевтически приемлемая соль.
| Способ получения производных бензодиоксола | 1973 |
|
SU468412A3 |
| Устройство для возведения двоичных чисел в степень | 1974 |
|
SU541168A1 |
| РАЗЪЕМНЫЙ БЕСКОНТАКТНЫЙ ИНДУКТОР | 1971 |
|
SU432695A1 |
| УСТРОЙСТВО ДЛЯ ПРОЯВЛЕНИЯ ФОРМАТНЫХ ФОТОМАТЕРИАЛОВ__В__П^Т БФОНЛ | 1972 |
|
SU434365A1 |
| Terry A | |||
| Lyll, Journal of Medicinal Chem., 1991, 34, № 3, с | |||
| Прибор для выделения минерального масла из смеси его с водой | 1920 |
|
SU1228A1 |
| Noel A | |||
| Roberts et al, Science, 1990, 248, № 20, с | |||
| Зажим для канатной тяги | 1919 |
|
SU358A1 |
