Изобретение относится к эстроген агонистам и антагонистам и их фармацевтическому применению.
Ценность эстрогенов природного происхождения и синтетических композиций, демонстрирующих "эстрогенную" активность, состоит в их медицинском и терапевтическом применении. Традиционный перечень терапевтических применений эстрогенов как таковых или в комбинации с другими активными агентами включает: пероральная контрацепция; ослабление симптомов менопаузы; профилактика угрожающего или привычного выкидыша; ослабление дисменореи; ослабление дисфункционального маточного кровотечения; вспомогательное средство в овариальном развитии; лечение угрей; уменьшение избыточного роста волос на теле женин (гирсутизм); профилактика сердечно-сосудистого заболевания; лечение остеопороза; лечение рака предстательной железы и подавление постнатальной лактации [Goodman and Gilman, The Prarmacological Basis of Therapeutics (Seventh Edition) Macmillan Publishing Company, 1985, стр. 1421-1423]. Таким образом существует повышенный интерес к открытию вновь синтезированных композиций и к новым применениям ранее известных соединений, которые являются очевидно эстрогенными, т.е. способными имитировать действие эстрогена в эстроген чувствительной ткани.
С точки зрения фармаколога, заинтересованного в разработке новых лекарственных средств, полезных для лечения человека и специфических патологических состояний, наиболее важно обеспечить соединения с некоторой доказуемой эстроген-подобной функцией, но лишенные пролиферативных побочных эффектов. В подтверждение этого последнего заключения, остеопороз, заболевание в котором кость становится значительно более ломкой, существенно снижается при применении полностью активных эстрогенов; однако, ввиду признанного возрастающего риска карциномы матки у пациенток, постоянно подвергающихся обработке активными эстрогенами, клинически не целесообразно лечить остеопороз интактной женщины полностью активными эстрогенами по причине увеличения длительности менструации. Т.о. эстроген агонисты представляют основной интерес и центральный аспект проблемы.
Остеопороз является системным заболеванием скелета, характеризующимся низкой костной массой и разрушением костной ткани, с последующим увеличением ломкости костей и подверженностью переломам. В США этим заболеванием поражено более 25 млн. человек и каждый год оно является причиной более 1.3 млн. переломов, включая 500,000 переломов позвоночника, 250,000 переломов тазобедренного сустава и 240,000 переломов запястья ежегодно. Это обходится государству более чем в 10 миллиардов долларов. Наиболее серьезными являются переломы тазобедренного сустава с 5-20% смертности пациентов за год, а свыше 50% оставшихся в живых становятся нетрудоспосбными.
Пожилые люди наиболее подвержены остеопорозу, и поэтому можно ожидать, что проблема значительно возрастет с увеличением возраста населения. Предполагается трехкратное увеличение распространенности переломов в течение ближайших 60 лет, и изучение заболеваний дает возможность предположить, что в 2050 году во всем мире будет 4,5 млн. случаев перелома тазобедренного сустава. Женщины подвергаются большему риску заболевания, чем мужчины. Женщины испытывают резкое ускорение разрежения кости в течение пяти следующих за менопаузой лет. Другие увеличивающие риск факторы включают курение, злоупотребление алкоголем, сидячий образ жизни и недостаточное потребление кальция.
Эстроген выбирается в качестве лекарственного средства для предотвращения остеопороза или после менопаузального разрежения кости у женщин; это единственный курс лечения, который определенно снижает переломы. Однако эстроген стимулирует матку и связан с возрастанием риска внутриматочной карциномы. Хотя предполагается, что риск образования внутриматочной карциномы снижается одновременным применением прогестогена, все же при применении эстрогена существует опасность возможного увеличения случаев рака молочной железы.
Black и др. в EP 0605193А1 сообщают, что эстроген, в частности, при пероральном введении снижает уровни плазмы LDL и повышает уровни благотворных альфа-липопротеинов высокой плотности (HDL's). Однако продолжительная терапия с использованием эстрогена влечет за собой разнообразные нарушения, включая увеличение риска возникновения маточной карциномы и, возможно, рака молочной железы, заставляющие многих женщин избегать этого лечения. Недавно предложены терапевтические схемы, которые стараются уменьшить риск появления карцином, такие как введение комбинаций прогестогена и эстрогена, приводящие к тому, что пациентки испытывают нежелательное кровотечение. Кроме того, комбинация прогестерона с эстрогеном, по-видимому, притупляет воздействие эстрогена, понижающее холестерин сыворотки. Значительные нежелательные воздействия, связанные с терапией с использованием эстрогена подтверждают необходимость развития альтернативной терапии для (гипер)холестеринемии, оказывающей заданное воздействие на LDL сыворотки, но не вызывающей нежелательные эффекты. Существует потребность в улучшенных эстроген агонистах, оказывающих избирательные воздействия на различные ткани организма. Тамоксифен, 1-(4- β диметиламиноэтоксифенил)-1,2-дифенил-бут-1-ен, является антиэстрогеном, оказывающим временное облегчающее воздействие на рак молочной железы, но, по сообщениям, проявляет эстрогенную активность в матке.
Gill-Sharma и др. J. Reproducition and Fertility (1993) 99, 395, раскрывают, что тамоксифен при 200 и 400 мг/кг/день понижает массу "testes" и вторичных половых органов у самцов крыс.
Недавно поступило сообщение (Osteoporosis Conference Scrip N 1812/13 Апрель 16/20, 1993, стр. 29), что ралоксифен, 6-гидрокси-2(4-гидроксифенил)-3-[4-(2-пиперидинэтокси)бензоил] бензо[b] тиофен, имитирует благоприятное воздействие эстрогена на кости и липиды, но в отличии от эстрогена обладает минимальным стимулирующим воздействием на матку (Breast Cancer Res. Treat. 10(1). 1997 p. 31-36, Jordan. V.C. и др.).
Neubauer и др. The Рrostate 23: 245 (1993) сообщают, что обработка ралоксифеном самцов крыс приводит к регрессии вентральной простаты.
Ралоксифен и родственные соединения описываются как антиандрогенные вещества, которые эффективны в лечении рака молочной железы и предстательной железы. См. Патент США 4,418,068 и Charles D. Jones, и др., J. Med. Chem. 1984, 27, 1057-1066.
Jones и др., в патенте США 4,133,814 описывают производные 2-фенил-3-ароил-бензотиофена и 2-фенил-3-ароилбензотиофен-1-окислов, которые полезны в качестве противозачатоных средств, а также в качестве средства, подавляющего рост опухолей молочной железы.
Lednicer и др. , J.Med. Chem., 12, 881 (1969) описали эстроген антагонисты структуры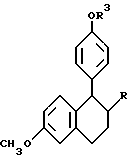
в которой R2 представляет фенил или циклофенил и R3 обозначает H, 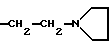 или -CH2CHOHCH2OH.
или -CH2CHOHCH2OH.
Bencze и др. , J.Med. Chem., 10, 138 (1967) получили серии тетрагидронафталинов, предназначенных для достижения разделения эстрогенной, противозачаточной и гипохолестеринемической активностей.
Эти структуры имеют общую формулу (I)
в которой R1 обозначает H или OCH3; R2 обозначает H, OH, OCH3, OPO(OC2H5)2, OCH2CH2 N(C2H5)2, OCH2COOH или OCH(CH3) COOH.
Патент США N 3,234,090 относится к соединениям, обладающим эстрогенным и противогрибковым свойствам, формулы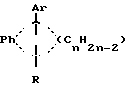
в которой Rh обозначает 1,2-фенилен радикал, Ar представляет моноциклическую карбоциклическую арил группу, замещенную третичным низшим-амино алкил-окси, в которой третичная амино группа отделена от окси как минимум двумя атомами углерода, R обозначает водород, алифатический радикал, карбоксициклическй арил радикал, карбоксициклический арил-алифатический радикал, гетероциклический арил радикал или гетероциклический арил алифатический радикал, группа формулы -(CnH2n-2)-обозначает неразветвленный алкилен радикал, имеющий от трех до пяти углеродных атомов и несущей группы Ar и R, их солям, N-окислам, солям N-окислов или четвертичным аммониевым соединениям, а также к способу получения таких соединений.
Патент США N 3,277,106 относится к неполным сложным эфирам многоатомных спиртов с эстрогенным, гипохолестеринемическим и противозачаточным действиями, имеющим формулы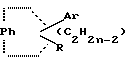
в которой Rh обозначает 1,2-фенилен радикала, Ar представляет моноциклический арилрадикал, замещенный по крайней мере одной низшей амино аклилоксигруппой, в которой атом азота отделен от атома кислорода по крайней мере двумя атомами углерода, R обозначает арил радикал, радикал -(CnH2n-2)- обозначает низший алкилен, образующий с Ph шести- или семичленный цикл, два углеродных атома, входящие в состав кольца, несут группы Ar и R, их солям, N-окислам, солям N-окислов и четвертичным аммониевым соединениям.
Lednicer и др. , в J.Med. Chem 10 78(1967) и патенте США N 3,274,213 приводят соединения формулы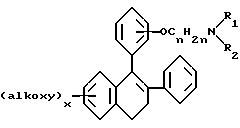
в которой R1 и R2 выбраны из класса, состоящего из низшего алкила и низшего алкила, связанных вместе таким образом, что образуется 5-7 членный насыщенный гетероциклический радикал.
Краткое описание изобретения
Изобретение предлагает соединения формулы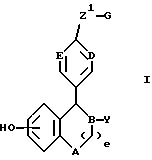
в которой: A выбирают из CH2 и NR;
B, D и E независимо выбирают из CH и N;
Y обозначает
(a) фенил, необязательно замещенный 1-3 заместителями, независимо выбранными из R4;
(b) нафтил, необязательно замещенный 1-3 заместителями, независимо выбранными из R4;
(c) C3-C8 циклоалкил, необязательно замещенный 1-2 заместителями, независимо выбранными из R4;
(d) C3-C8 циклоалкенил, необязательно замещенный 1-2 заместителями, независимо выбранными из R4;
(e) пятичленный гетероцикл, содержащий до двух гетероатомов, выбранных из группы, состоящей из -O-, -NR2- и -S(O)n-, необязательно замещенный 1-3 заместителями, независимо выбранными из R4;
(f) шестичленный гетероцикл содержащий до двух гетероатомов, выбранных из группы, состоящей из -O-, -NR2- и -S(O)n-, необязательно замещенный 1-3 заместителями, независимо выбранными из R4; или
(g) бициклическая кольцевая система, содержащая пяти или шестичленный гетероцикл, сконденсированный с фенильным кольцом, где указанный гетероцикл содержит до двух гетероатомов, выбранных из группы, включающей -O-, -NR2- и -S(O)n-, необязательно замещенный 1-3 заместителями, независимо выбранными из R4;
Z1 обозначает
(a) -(CH2)p W (CH2)q-;
(b) -O(CH2)p CR5R6-;
(c) -O(CH2)pW(CH2)q;
(d) -OCHR2CHR3-; или
(e) -SCHR2CHR3-;
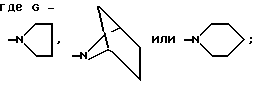
G обозначает
(a) -NR7R8;
(b)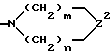
в котором n равно 0,1 или 2; m равно 1,2 или 3; Z2 обозначает -NH-, -O-, -S- или -CH2-; необязательно сконденсированный по смежным углеродным атомам с одним или двумя фенильными циклами и необязательно независимо замещенный по углероду одним - тремя заместителями и необязательно независимо по азоту - химически пригодным заместителем, выбранным из R4; или
(C) бициклический амин, содержащий от пяти до двенадцати атомов углерода, либо связанный мостиковой связью, либо сконденсированный и необязательно замещенный 1-3 заместителями, независимо выбранными из R4; или
Z1 и G в комбинации могут представлять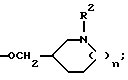
W обозначает
(a) -CH2--
(b) -CH=CH-;
(c) -O-;
(d) -NR2-;
(e) -S(O)n-;
(f) 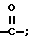
(g) -CR2(OH)-;
(h) -CONR2-;
(i) -NR2CO-;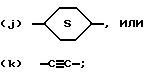
R представляет водород или C1-C6-алкил;
R2 и R3 независимо обозначают
(a) водород; или
(b) C1-C4-алкил;
R4 представляет
(a) водород;
(b) галоген;
(c) C1-C6-алкил;
(d) C1-C4-алкокси,
(e) C1-C4-ацилокси,
(f) C1-C4-алкилтио,
(g) C1-C4-алкисульфонил,
(h) C1-C4-алкилсульфонил,
(i) гидрокси-(C1-C4)-алкил,
(j) арил-(C1-C4)-алкил,
(k) -CO2H,
(l) -CN,
(m) -CONHOR,
(n) -SO2NHR,
(o) -NH2,
(p) C1-C4-алкиламино,
(q) C1-C4-диалкиламино,
(r) -NHSO2R,
(s) -NO2,
(t) -арил или
(u) -OH.
R5 и R6 представляет независимо C1-C8-алкил или вместе образуют C3-C10-карбоцикл,
R7 и R8 обозначают независимо
(a) фенил,
(b) C3-C10 карбоцикл, насыщенный или ненасыщенный,
(c) C3-C10 гетероцикл, содержащий до двух гетероатомов, выбранных из -O-, -N- и -S-,
(d) H,
(e) C1-C6-алкил или
(f) образуют 3-8-членный азот, содержащий цикл с R5 или R6,
R7 и R8 в форме либо линейной, либо циклической могут необязательно содержать до трех заместителей, независимо выбранных из: C1-C6-алкил, галоген, алкокси, гидрокси и карбокси;
цикл, образованный R7 и R8, может необязательно быть конденсированным с фенильным кольцом;
e равно 0, 1 или 2;
m равно 1, 2 или 3;
n равно 0, 1 или 2;
p равно 0, 1, 2 или 3;
q равно 0, 1, 2, или 3;
и их оптические и геометрические изомеры, а также их нетоксичные фармакологически приемлемые соли кислотного присоединения, N-окиси и четвертичные аммониевые соли.
Предпочтительными соединениями изобретения являются соединения формулы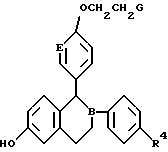
R4 представляет H, OH, F или Cl;
B и E независимо выбирают из CH и N.
Наиболее предпочтительными соединениями являются:
Цис-6-(4-фтор-фенил)-5-[4-(2-пиперидин-1-ил-этокси)-фенил] -5,6,7,8- тетрагидронафталин-2-ол;
(-)-Цис-6-фенил-5-[4-(2-пирролидин-1-ил-этокси)-фенил] -5,6,7,8- тетрагидронафталин-2-ол;
Цис-6-фенил-5-[4-(2-пирролидин-1-ил-этокси)-фенил] -5,6,7,8- тетрагидро-нафталин-2-ол;
Цис-1-[6'-пирролидиноэтокси-3'-пиридил] -2-фенил-6-гидрокси-1,2, 3,4-тетрагидронафталин;
1-(4'-Пирролидиноэтоксифенил)-2-(4''-фторфенил)-6-гидрокси-1,2, 3,4-тетрагидроизохинолин;
Цис-6-(4-гидроксифенил)-5-[4-(2-пиперидин-1-ил-этокси)-фенил] -5,6, 7,8-тетрагидронафталин-2-ол и
1-(4'-Пирролидинолэтоксифенил)-2-фенил-6-гидрокси-1,2,3,4-тетрагидроизохинолин.
С другой стороны, данное изобретение предлагает способы лечения или профилактики болезненных состояний, включающих рак молочной железы, остеопороз, внутриматочное или сердечно-сосудистое заболевание и (гипер)холестеринемию у мужских и женских особей млекопитающих и доброкачественную гипертрофию предстательной железы и карциномы предстательной железы у мужских особей млекопитающих, которые включают введение указанному млекопитающему количества соединения формулы I и желательно предпочтительного соединения из соединений формулы I, указанных выше, которое эффективно в лечении или профилактике указанного состояния.
С другой стороны, данное изобретение предлагает способ лечения или предупреждения ожирения у млекопитающих, который включает введение указанному млекопитающему количества соединения формулы I и желательно предпочтительного соединения формулы I, указанного выше, эффективного в лечение или профилактике ожирения.
Дальнейший аспект данного изобретения составляет фармацевтическая композиция для лечения или предупреждения рака молочной железы, остеопороза, ожирения, сердечно-сосудистого заболевания, (гипер)холестеринемии, внутриматочного заболевания и заболевания предстательной железы, включающая соединение формулы I и фармацевтически приемлемый носитель.
Другой аспект данного изобретения составляют промежуточные соединения, полезные для получения соединений формулы I. Это 1-{2-[4-(6-метокси-3,4-дигидронафталин-1-ил)фенокси] этил} пирролидин и 1-{2-[4-(2-бром-6-метокси-3,4-дигидронафталин-1-ил)фенокси]этил}пирролидин.
Подробное описание изобретения
Термины C1-C3 хлоралкил и C1-C3 фторалкил включают метил, этил, пропил и изопропил, замещенный до некоторой заданной степени атомами хлора или фтора, от одного атома до полного замещения. Термин C5-C7 циклоалкил включает циклопентил, циклогексил и циклогептил.
Галоид означает хлор, бром, иод и фтор. Арил (Ar) включает фенил и нафтил, необязательно замещенные одним-тремя заместителями, выбранными из R4, как определено выше. DTT означает дитиотреитол. DMSO означает диметилсульфоксид. EDTA - этилендиаминтетрауксусная кислота.
Эстроген агонисты определяются здесь как химические соединения, способные связываться по эстроген рецепторным участкам в ткани млекопитающих и имитирующие действия эстрогена в одной или более тканях.
Эстроген антагонисты определяются здесь как химические соединения, способные связываться по эстроген рецепторным участкам в ткани млекопитающих и блокирующие действия эстрогена в одной или более тканях.
Каждому специалисту понятно, что некоторые перечисленные в данном изобретении соединения будут химически несовместимы одно с другим или с гетероатомами в соединениях, и следует избегать этих несовместимостей при выборе соединений данного изобретения. Аналогично, некоторые функциональные группы (в ходе синтезов) могут нуждаться в защитных группах, известных каждому специалисту-химику.
Каждому специалисту-химику понятно, что некоторые соединения данного изобретения содержат атомы, которые могут давать отдельные оптические и геометрические конфигурации. Все такие изомеры включены в данное изобретение; предпочтительны левовращающие изомеры в цис конфигурации. Аналогично, химику очевидно, что из некоторых соединений данного изобретения могут быть получены разнообразные фармакологически приемлемые сложные эфиры и соли. Все такие эфиры и соли включены в данное изобретение.
Используемое в данном описании понятие заболевания простаты означает доброкачественную гиперплазию простаты или рак предстательной железы.
Лекарственные средства для лечения заболеваний простаты, рака молочной железы, ожирения, сердечно-сосудистого заболевания, (гипер) холестеринемии и остеопороза в данном изобретении включают в качестве активного ингредиента соединение формулы I или его соль, или сложный эфир. Фармацевтически приемлемыми солями соединений формулы I являются обычно используемые нетоксичные соли, такие как соли с органическими кислотами (например, муравьиная, уксусная, трифторуксусная, лимонная, малеиновая, щавелевая кислоты, метансульфокислота, бензолсульфокислота и толуолсульфокислота), неорганическими кислотами (например, соляная, бромистоводородная, серная или фосфорная кислоты) и аминокислотами (например, аспарагиновая или глутаминовая кислоты). Такие соли могут быть получены известными каждому специалисту-химику способами.
Лекарственные средства для лечения заболеваний простаты, рака молочной железы, ожирения, сердечно-сосудистого заболевания, (гипер) холестеринемии и остеопороза данного изобретения могут вводиться животным, включая человека, перорально или парентерально в обычно используемых для препаратов формах, таких как капсулы, микрокапсулы, таблетки, гранулы, порошки, лепешки, пилюли, суппозитории, инъекции, суспензии и сиропы.
Лекарственные средства данного изобретения для лечения заболеваний простаты, рака молочной железы, ожирения, сердечно-сосудистого заболевания, (гипер)холестеринемии и остеопороза могут быть получены обычными способами с применением обычных органических или неорганических добавок, таких как наполнитель (например, сахароза, крахмал, маннитол, сорбитол, лактоза, глюкоза, целлюлоза, тальк, фосфат кальция или карбонат кальция), связывающие вещества (например, целлюлоза, метилцеллюлоза, гидроксиметилцеллюлоза, полипропилпирролидин, поливинилпирролидон, желатин, гуммиарабик, полиэтиленгликоль, сахароза или крахмал), разрыхлитель (например, крахмал, карбоксиметилцеллюлоза, гидроксипропилкрахмал, мало замещенная гидроксипропилцеллюлоза, бикарбонат натрия, фосфат кальция или цитрат кальция), смазывающее вещество (например, стеарат магния, маловязкая безводная кремневая кислота, тальк или лаурилсульфат натрия), корригент (например, лимонная кислота, ментол, гликокол или апельсиновый порошок), консервант (например, бензоат натрия, бисульфит натрия, метилпарабен или пропилпарабен), стабилизатор (например, лимонная кислота, цитрат натрия или уксусная кислота), суспендирующее вещество (например, метилцеллюлоза, поливинилпирролидон или стеарат алюминия), диспергирующее вещество (например, гидроксипропилметилцеллюлоза), растворитель (например, вода) и восковая основа (например, масло какао, белый вазелин или полиэтиленгликоль). Количество активного ингредиента в медицинской композиции должно быть на таком уровне, чтобы оказывать желаемый терапевтический эффект; например, приблизительно от 0,1 до 50 мг в стандартной дозе как для перорального, так и для парентерального введения.
Активный ингредиент обычно может вводиться пациенту от одного до четырех раз в день со стандартной дозой от 0,1 до 50 мг, но данная доза может быть соответственно изменена в зависимости от возраста, веса тела и медицинского состояния пациента и типа введения. Предпочтительная доза для человека составляет от 0,25 до 25 мг. Предпочтительно введение одной дозы в день.
Соединения изобретения легко получить по приведенным ниже реакционным схемам.
Определенные соединения формулы I можно получить из ненасыщенного промежуточного соединения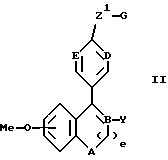
путем гидрирования с благородным металлом в качестве катализатора в реакционно инертном растворителе. Давление и температура не являются решающими и гидрирование обычно завершается за несколько часов при комнатной температуре и при давлении водорода 20-80 фунт/кв.дюйм (1,4-26,7 кг/см2).
Продукт гидрирования выделяют, очищают при желании и простую эфирную группу расщепляют с кислотным катализатором в реакционно инертном растворителе при температуре между 0 и 100oC в зависимости от используемого кислотного катализатора. Найдено, что в этой реакции эффективны: бромистый водород при повышенных температурах, трибромид бора и хлористый алюминий при температурах от 0oC до температуры окружающей среды.
Продукт формулы I выделяют и очищают стандартными способами.
Промежуточные продукты формулы II, в которых A представляет CH2, и B, D и E обозначают CH, описаны в патенте США 3,274,213; J.Med. Chem. 10, 18 (1967); J. Med. Chem. 10, 138 (1967); и J. Med. Chem. 12, 881(1969), каждый из которых представлен здесь в качестве ссылки. Они могут также быть получены по описанным ниже методикам.
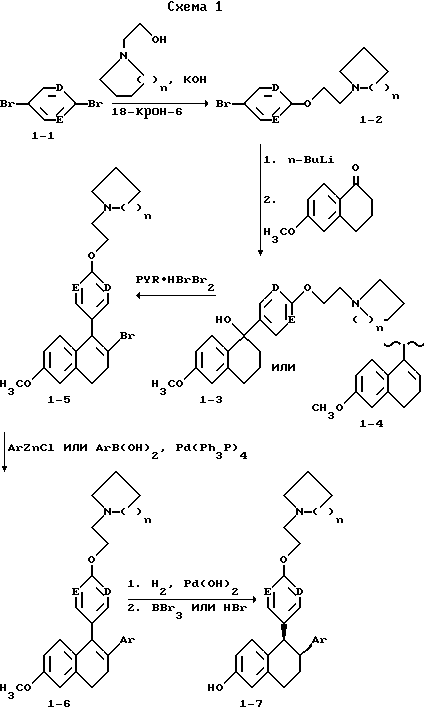
Получение соединений формулы I, в которых e=1, A=CH2, Z1=OCH2CH2, G= циклоалкиламин, B= CH, показано в схеме 1. Соединения 1-2, в которых D и E представляют CH, получают алкилированием 4-бромфенола с соответствующим N-хлорэтиламином, используя карбонат кальция в качестве основания в полярном апротонном растворителе, таком как диметилформамид, при повышенных температурах. Предпочтительна температура в 100oC. Соединение 1-2, в которых D или E или оба обозначают N, синтезируют, используя реакцию нуклеофильного замещения, выполненного на дибромидах (I-1) с использованием гидроксиэтилциклоалкиламинов в условиях фазового переноса, что дает бромамины (I-2). Synthesis, 77, 573(1980). Последующий металлогалоидного обмена с использованием н-бутиллития или металлического магния бромаминов (I-2), дающий соответствующие литиевые или магниевые реагенты, позволяющие проводить реакцию при низких температурах, предпочтительно в присутствии цезий хлорида (в отсутствии цезий хлорида реакция также протекает) с 6-метокси-1-тетралоном с получением карбинолов (I-3) или стиролов (I-4) после обработки кислотой. Обработка карбинолов (I-3) или стиролов (I-4) бромирующим агентом, таким как пиридиний бромид, дает бромстиролы (I-5). Арил или гетероарил хлориды цинка или арил или гетероарил борные кислоты реагируют с бромидами (I-5) в присутствии металлического палладия в качестве катализатора, такого как тетракис трифенил фосфин палладий (0), с образованием диарил стиролов (I-6). [Pure & Applied Chem. 63, 419, (1991) и Bull. Chem. Soc. Jpn. 61, 3008-3010, (1988)] . Для получения предпочтительных соединений в этой реакции используют замещенные фенил хлориды цинка или замещенные фенилборных кислот. Арил хлориды цинка получают гашением соответствующего литиевого реагента безводным хлористым цинком. Арил борные кислоты, которые не выпускаются промышленностью, получают гашением соответствующего арил литиевого реагента триалкил боратом, предпочтительно триметил или триизопропил боратом, с последующей обработкой водной кислотой. Acta Chemica Sсan. 47, 221-230 (1993). Литиевые реагенты, не выпускаемые промышленностью, получают путем металло-галоидного обмена соответствующего бромида или галогенида с н-бутил или т-бутиллитием. Или же иначе, литиевый реагент получают путем облегченной гетероатомной литизации, как описано в Organic Reactions, Volumer 27, Chapter I. Каталитическое гидрирование (I-6) в присутствии гидроокиси палладия на активированном угле, например, дает соответствующие дигидрометокси промежуточные продукты, которые впоследствии диметилируют, используя трибромид бора при 0oC в метилен хлориде или 48% бромистый водород в уксусной кислоте при 80-100oC, что дает целевые структуры (I-7). Эти соединения рацемические и могут быть разделены на энантиомеры жидкостной хроматографией высокого давления с применением колонки с хиральной неподвижной фазой, такой как колонки Chiracel OD.
Иначе оптическое разделение может быть выполнено перекристаллизацией диастереомеромерных солей, образованных с оптически чистыми кислотами, такими как 1,1'-динафтил-2,2'-диил кислый фосфат (см. пример 8).
Цис соединения (I-7) могут быть изомеризованы в транс соединения обработкой основаниями (см. пример 2).
Когда D и/или E является азотом, промежуточные соединения (формула II) и соединения формулы I могут быть получены из соответствующих дигалоидпиридинов или пиримидинов, как показано в схеме 1 и как подробно описано для 6-фенил-5-[6-(2-пирролидин-1-ил-этокси)пиридин-3-ил] -5,6,7,8- тетрагидронафталин-2-ола в примере 6.
Простой метиловый эфир соединения формулы I, в котором e=1, A=CH2, Z1= OCH2CH2, G= пирролидин, D, E, B=CH, Y=Ph, удобно получать на первой стадии гидрирования нафоксидина (Upjohn & Co. , 700 Portage Road, Kalamazoo, M1 49001) в реакционно инертном растворителе в присутствии благородного металла в качестве катализатора. Давление и температура не имеют решающего значения; реакция обычно проходит в этаноле при комнатной температуре приблизительно за 20 часов при 50 фунт/кв.дюйм (10,5 кг/см2).
Второй стадией является расщепление метокси группы, которое выполняется обычно при комнатной температуре с кислотным катализатором, таким как трибромид бора в реакционно инертном растворителе или при 80-100oC с бромистым водородом в уксусной кислоте. Продукт затем выделяют обычными способами и превращают при желании в кислую соль.
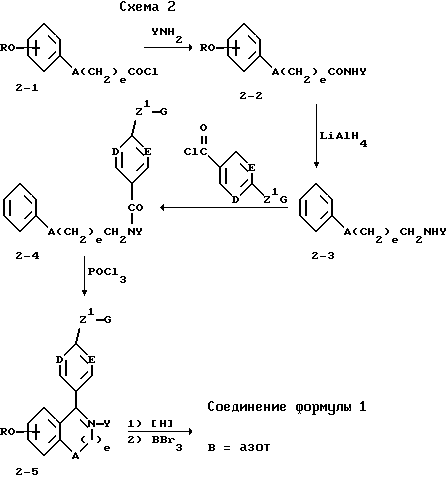
Соединения формулы I, в которых B обозначает азот, получают способами, показанными в схеме 2 и примерах 3-5 и 10-12.
Синтез соединений формулы I, в которых B=N, приведен в схеме 2. Арил хлорангидриды (2-1) при обработке первичными аминами дают арил вторичные амиды (2-2), которые восстанавливают алюмогидридом лития в эфирных растворителях, получая вторичные амины (2-3). Последующее ацилирование (2-3) ароил хлорангидридами приводит к третичным амидам (2-4), которые циклизуют в горячем оксихлориде фосфора, получая соли дигидро изохинолина (2-5). Восстановление боргидридом натрия до алкокситетрагидроизохинолинов и последующее деметилирование в метиленхлориде дают требуемые структуры.
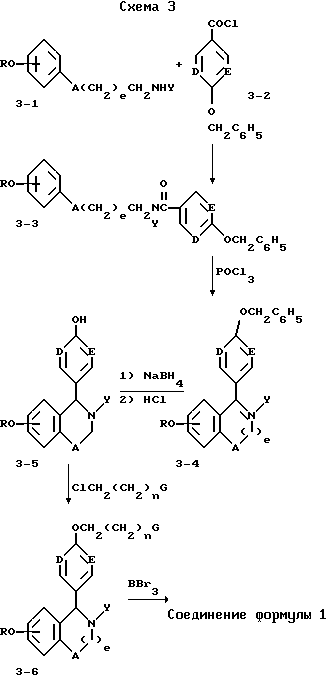
Синтез соединений формулы I, в которых B=N, также описан ниже на схеме 3. Вторичные амины (3-1) при ацилировании бензилоксиароилхлоридами (3-2) дают третичные амиды (3-3), которые при циклизации с горячим оксихлоридом фосфора образуют соли дигидро изохинолина (3-4). Восстановление боргидридом натрия (3-4) с последующим дебензилированием с водной соляной кислотой дают изохинолины (3-5), которые алкилируют соответствующими функциональными хлоридами и деметилируют трибромидом бора, получая заданные целевые структуры.
Соединения данного изобретения являются ценными эстроген агонистами и фармацевтическими средствами или промежуточными продуктами для них. Те соединения, которые являются эстроген агонистами, полезны для пероральной контрацепции; для ослабления симптома менопаузы; предупреждения угрожающего или привычного выкидыша; ослабления дисменореи; ослабления дисфункционального маточного кровотечения; как вспомогательное средство в овариальном развитии; при лечении угрей; для уменьшения избыточного роста волос на теле женщин (гирсутизм); для профилактики и лечения сердечно-сосудистого заболевания; для профилактики и лечения атеросклероза; профилактики и лечения остеопороза; лечения доброкачественной простатической гиперплазии и карциномы предстательной железы; ожирения и подавления пост-натальной лактации. Эти соединения оказывают также благотворное воздействие на уровни липидов плазмы и сами по себе полезны в лечении и профилактике (гипер)-холестеринемии.
Хотя соединения данного изобретения являются эстроген агонистами в кости, они служат также антиэстрогенами в ткани молочной железы и как таковые полезны в лечении и профилактике рака молочной железы.
Предупреждение и борьба с эндометриозом.
Протокол хирургически индуцируемого эндометриоза идентичен протоколу, описанному Jones, Acta Endoerinol (Copenh) 106:282-8. Используют взрослые особи Charles River Spraque-Dawley CD® самок крыс (200-240 г). Косой вентральный надрез делают через кожу и мускулатуру стенки тела. Вырезают сегмент правой трубы матки, мышечную оболочку матки отделяют от эндометрия и сегмент режут продольно. 5х5 мм разрез эндометрия с эпителиальной выстилкой, соединенной со стенкой тела, сшивают по его четырем углам с мышцей, используя полиэфирную нить (Ethiflex, 7-0®). Критерием жизнеспособного трансплантата является аккумуляция жидкости, аналогичная аккумуляциии, которая происходит в результате стимуляции эстрогеном.
Спустя три недели после трансплантации эндометриальной ткани (+3 недели) животными производят лапаротомию, объем эксплантата (длина х ширина х высота) в мм измеряют циркулем и начинают обработку лекарственными препаратами. Животным впрыскивают подкожно в течение 3 недель от 10 до 1000 мг/кг/день соединения формулы I. В качестве контрольных животных используют с эндометриальным трансплантатом, вводя подкожно в течение 3 недель кукурузное масло по 0,1 мл/день. По окончании 3 недельного периода обработки (+6 недель), животным производят лапоротомию и определяют объем эксплантата. Спустя 8 недель после прекращения обработки лекарственными средствами (+14 недель) животных забивают; эксплантат измеряют вновь. Статистический анализ объема эксплантата производят путем анализа отклонений.
Воздействие на вес простаты
Самцам Sprague-Dawley крыс, в возрасте трех месяцев, подкожно инъекцитируют одним из: растворитель (10% этанол в воде), эстрадиол (30 мг/кг), тестостерон (1 мг/кг) или соединение формулы I - ежедневно в течение 14 дней (n= 6/группа). Спустя 14 дней животных забивают, простату удаляют и определяют вес влажной простаты. Определяют средний вес и статическую значимость (p < 0,05) по сравнению с группой, обработанной растворителем, используя t-тест Student's.
Соединения формулы I существенно (P < 0,05) снижают вес простаты по сравнению с растворителем. Тестостерон не оказывает никакого действия, хотя экстрадион при концентрации 30 мкг/кг также значительно уменьшает вес простаты.
Минеральная плотность костей
Минеральная плотность кости, степень минерального содержания кости, отвечают за более чем 80% прочности кости. Снижение минеральной плотности кости с возрастом и/или заболеванием делает ее более склонной к переломам. Степень минерального содержания кости точно измеряют у людей и животных путем абсорбциометрии с помощью двойного рентгеновского луча (DEXA), так что могут быть определены такие малые как 1% изменения. Использован утилизированный DEXA для оценки изменений в минеральной плотности костей, обусловленной дефицитом эстрогена, вызванным овариэктомией (хирургическое удаление яичников) и обработкой растворителем, эстрадиолом (E2), кеоксифеном (ралоксифеном) или другими агонистами эстрогена. Целью этих изучений является оценка способности соединений данного изобретения предупреждать вызываемое дефицитом эстрогена разрежение кости, измеренное посредством DEXA.
Самок (S-D) крыс в возрасте 4-6 месяцев подвергают овариэктомии или симулированному хирургическому вмешательству и дают оправиться от наркоза. Крыс обрабатывают путем подкожной инъекции или перорально через зонд различными дозами (10-1000 мг/кг/день, например) соединения формулы I ежедневно в течение 28 дней. Все соединения взвешивают и растворяют в 10% этаноле в стерильном физиологическом растворе. Спустя 28 дней крыс забивают и бедра удаляют и очищают от мяса. Бедренную кость помещают на Hologic QDR1000W (Hologic, Inc. Waltham, MA) и определяют минеральную плотность кости в периферической части бедренной кости в месте, расположенном на 1-2 см от конца бедренной кости, используя software высокого разрешения, поставляемый Hologic. Минеральную плотность кости определяют путем деления костного минерального содержания на площадь дистальной бедренной кости. Каждая группа состоит по крайней мере из 6 животных. Получают среднюю минеральную плотность кости для каждого животного и определяют t-тестом статистические разности (p<0,05) для группы, обработанной растворителем, овариэктомией и симулированным хирургическим вмешательством.
In vitro (в лабораторном сосуде) испытание эстроген-рецепторного связывания
Для определения способности соединений настоящего изобретения к эстроген-рецепторному связыванию используют in vitro испытание эстроген-рецепторного связывания, которое определяет способность соединений настоящего изобретения к замещению [3H]-эстрадиола из человеческого эстроген-рецептора, полученного рекомбинантными способами в дрожжах. В данном испытании использованы следующие вещества: (I) буферный раствор для испытания, TD - 0.3 (содержащий 10 нМ Трис, pH 7,6, 0,3 М хлористого калия и 5 мМ DTT, pH 7,6); (2) используют [3H]-эстрадиол радиолиганд, полученный от New England Nuclear; (3) в качестве холодного лиганда использован эстрадиол, полученный от Sigma; (4) рекомбинантный человеческий эстроген рецептор, hER.
Раствор подлежащего испытанию соединения получают в TD-0,3 с 4% DMCO и 16% этанола. Насыщенный тритием эстрадиол растворяют в TD-0.3 так, чтобы конечная концентрация в опыте составляла 5 нМ. hER также растворяют в TD-0.3 так, чтобы 4-10 мг общего белка было в каждой ячейке для испытания. На используемых пластинах для микротитрования в каждый инкубат добавляют 50 (мкл) холодного эстрадиола (неспецифическое связывание) или раствор соединения, 20 (мкл) насыщенного тритием эстрадиола и 30 (мкл) растворов hER. Каждая пластина содержит тройное повторение общего связывания и различных концентраций соединения. Пластины инкубируют в течение ночи при 4oC. Затем реакцию связывания прерывают добавлением и перемешиванием со 100 мл 3% гидроксилапатита в 10 мМ трис, pH 7,6 и инкубируют 15 минут при 4oC. Смеси центрифугируют и осадок промывают четыре раза 1% Triton x 100 в 10 мМ Трис, pH 7,6. Осадки гидроксилапатита суспендируют в Ecoscint A и определяют радиоактивность, используя сцинциграфию. Определяют среднее значение из трех полученных точек (подсчет в минуту, cpm's). Специфическое связывание рассчитывают путем вычитания неспецифических cpm's (определенных как подсчет остатка после разделения реакционной смеси, содержащей рекомбинантный рецептор радиолиганд и избыток немеченного лиганда) из cpm's общего связывания (определенных как подсчет остатка после разделения реакционной смеси, содержащей только рекомбинантный рецептор, радиолиганд). Активность соединения оценивают, определяя значение IC50 (концентрация соединения, необходимая для ингибирования 50% от общего специфического связывания насыщенного тритием эстрадиола). Определяют специфическое связывание в присутствии различных концентраций соединения и рассчитывают как процент специфического связывания от общего специфического связывания радиолиганда. Данные представляют графически как процент ингибирования соединением (линейная шкала) в зависимости от концентрации соединения (log шкала).
Влияние на общие уровни холестерина
Влияние соединений настоящего изобретения на плазменные уровни общего холестерина определяют следующим образом. Образцы крови отбирают путем сердечной пункции у анестезированных самок (S-D) крыс 4-6 месячного возраста, которым билатерально произведена овариэктомия и обработанных соединением (10-1000 мг/кг/день, например, подкожно или перорально в течение 28 дней или растворителем в течение того же времени), или произведена симулированная операция. Кровь помещают в пробирки, содержащие 30 мкл ЭДТА (10 мкл ЭДТА/1 мл крови). После центрифугирования при 2500 об/мин в течение 10 минут при 20oC плазму собирают и сохраняют стандартный образец при -20oC. Общий холестерин анализируют, используя стандартный набор ферментативного определения из Sigma Diagnostics (методика N 352).
Воздействие на ожирение
Sprague-Dawley самкам крыс в возрасте 10 месяцев, весящим приблизительно 450 г, производят симулированную операцию (симуляцию) или овариэктомию (OVX) и обрабатывают перорально растворителем, 17α этинилэстрадиолом при 30 мг/кг/день или соединением формулы I при 10-1000 мг/кг/день в течение 8 недель. В каждой подгруппе используют 6-7 крыс. В последний день изучения определяют состав тела всех крыс, используя рентгеновский абсорбциометр двойной мощности (Hologic QDR-1000/W), снабженный сканирующим software для всего тела, который показывает соотношение массы жирового тела и массы обезжиренного тела.
Снижение массы жирового тела показывает, что эстроген агонисты формулы I полезны для предупреждения и лечения ожирения.
Специалисту в области фармации ясно, что физиологически активные соединения, обладающие доступными гидроксильными группами, вводятся в форме фармацевтически приемлемых сложных эфиров. Литература, описывающая подобные соединения, такие как эстрадиол, дает большое число примеров таких эфиров. Соединения данного изобретения не являются исключением в этом отношении и могут быть эффективно введены в виде сложных эфиров, образованных по гидрокси группам, именно так, как может предполагать каждый специалист в фармацевтической химии. Хотя механизм пока еще не исследован, предполагается, что эфиры метаболически расщепляются в теле и что фактическим лекарственным средством, вводимым в такой форме, является само гидрокси соединение. Как известно в фармацевтической химии, возможно регулирование скорости выделения или длительности воздействия соединения путем соответствующего выбора эфирных групп.
Определенные сложноэфирные группы являются предпочтительными в качестве составных частей соединений данного изобретения. Соединения формулы I могут содержать сложноэфирные группы в различных частях, как определено выше, и эти группы обозначения как -COOR9. R9 представляет C1-C14-алкил, C1-C3-хлоралкил, C1-C3 фторалкил, C5-C7 циклоалкил, C1-C4 алкокси, фенил, или фенил моно-, или дизамещенный C1-C4 алкилом, C1-C4 алкокси, гидрокси, нитро, хлором, фтором или три(хлор или фтор)метилом.
Фармацевтически приемлемые соли кислотного присоединения соединений данного изобретения могут быть образованы как самими соединениями, так и любыми их сложными эфирами, и включают фармацевтически приемлемые соли, которые часто используются в фармацевтической химии. Например, соли могут быть образованы с неорганическими или органическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, сульфокислотами, включающими такие соединения, как нафталинсульфо-, метансульфо- и толуолсульфокислоты, серной кислотой, азотной кислотой, фосфорной кислотой, винной кислотой, пиросерной кислотой, метанфосфорной кислотой, янтарной кислотой, муравьиной кислотой, фталевой кислотой, молочной кислотой и тому подобными, наиболее предпочтительно с хлористоводородной кислотой, лимонной кислотой, бензойной кислотой, малеиновой кислотой, уксусной кислотой и пропионовой кислотой. Обычно желательно вводить соединение данного изобретения в форме соли кислотного присоединения, как это принято для введения фармацевтических препаратов, имеющих основную группу, такую как пирролидиновое кольцо.
Соединения данного изобретения, как обсуждалось выше, очень часто вводятся в форме солей кислотного присоединения. Эти соли удобно получать, как обычно в органической химии, реакцией соединения данного изобретения с подходящей кислотой, такой как описано выше. Соли быстро образуются с высоким выходом при умеренных температурах, и часто их получают просто отделением соединения от соответствующей кислоты промывкой в качестве конечной стадии синтеза. Солеобразующую кислоту растворяют в подходящем органическом растворителе или водном органическом растворителе, таком как алканол, кетон или сложный эфир. С другой стороны, если соединение данного изобретения желательно получить в форме свободного основания, его отделяют от основания конечной стадией промывки, в соответствии с общепринятой практикой. Предпочтительный способ получения хлористоводородных солей состоит в растворении свободного основания в подходящем растворителе и тщательном высушивании раствора, например, над молекулярными ситами, перед барботированием через него газообразного хлористого водорода.
Доза соединения данного изобретения, требующаяся для введения человеку, изменяется в довольно широких пределах и определяется лечащим врачом. Следует отметить, что может быть необходимо подбирать дозу соединения, при введении его в форме соли, такой как лаурат, в которой солеобразующая составляющая имеет значительный молекулярный вес. Обычный интервал эффективных доз введения соединений составляет приблизительно от 0,05 мг/день до 50 мг/день. Предпочтительный интервал доз составляет приблизительно от 0.25 мг/день до 25 мг/день. Конечно, часто практикуется введение суточной нормы соединения порциями, в различное время дня. Однако в любом приведенном случае, количество вводимого соединения будет зависеть от таких факторов, как растворимость активного компонента, используемый состав и способ введения.
Способ введения соединений данного изобретения не имеет решающего значения. Известно, что соединения абсорбируются из пищеварительного тракта, и поэтому обычно желательно вводить соединение перорально из соображений удобства. Однако соединения могут быть в равной степени эффективно введены подкожно или в виде суппозиториев путем абсорбции через прямую кишку, если это желательно в данной ситуации.
Соединения настоящего изобретения обычно вводятся в виде фармацевтических композиций, что является еще одним воплощением изобретения. Могут быть использованы все обычные типы композиций, включая таблетки, жевательные таблетки, капсулы, растворы, парентеральные растворы, лепешки, суппозитории и суспензии. Составы готовят так, чтобы они содержали суточную дозу или нужную часть суточной дозы, в стандартной дозе, которая может представлять отдельную таблетку, или капсулу, или подходящий объем жидкости.
Любые соединения могут быть легко составлены в виде таблеток, капсул и тому подобного; желательно готовить растворы водорастворимых солей, таких как хлористоводородная соль.
Обычно все составы готовят по общепринятым в фармацевтической химии методикам.
Капсулы получают смешением соединения с подходящими растворителем и заполнением капсул соответствующим количеством смеси. Обычные разбавители включают инертные порошкообразные вещества, такие как крахмал различных видов, порошкообразная целлюлоза, особенно кристаллическая или микрокристаллическая целлюлоза, сахара, такие как фруктоза, маннитол и сахароза, мука различных видов и тому подобные пригодные в пищу порошки.
Таблетки получают путем непосредственного прессования, путем влажного или сухого гранулирования. Их составы обычно включают растворители, связывающие, смазывающие вещества и разрыхлители наряду с самими соединениями. Типичные разбавители включают, например, различные типы крахмала, лактозу, маннитол, каолин, фосфат или сульфат кальция, неорганические соли, такие как хлористый натрий и порошкообразный сахар. Полезны также порошкообразные производные целлюлозы. Типичные связывающие вещества, входящие в таблетку, включают, например, такие как крахмал, желатин и сахара, такие как лактоза, фруктоза, глюкоза и тому подобные. Используются также подходящие натуральные и синтетические смолы, включающие сок акации, альтинаты, метилцеллюлозу, поливинилпирролидин и тому подобные. Полиэтиленгликоль, этилцеллюлоза и парафины могут служить связывающими веществами.
Смазывающие вещества необходимы в составе таблеток для препятствия слипанию таблеток и перфораций в штампе. Смазывающее средство выбирают из таких скользких твердых веществ, как тальк, стеарат магния или кальция, стеариновая кислота и гидрированные растительные масла.
Разрыхлителями для таблеток служат вещества, которые разбухают при смачивании, разрушая таблетку и высвобождая соединение. Они включают крахмалы, глины, целлюлозу, альгины и смолы. Более предпочтительно кукурузный и картофельный крахмал, метилцеллюлоза, агар, бентонит, древесная целлюлоза, порошкообразная природная губка, катион-обменные смолы, альгиновая кислота, кизельгуровая смола, цитрусовая мякоть и карбоксиметилцеллюлоза, например, могут быть использованы наравне с лаурил сульфатом натрия.
Таблетки часто покрывают сахаром в качестве вкусовой добавки и изолирующего слоя или пленкообразующими защитными веществами для модификации способности таблетки к растворению. Соединения могут также быть составлены в виде жевательных таблеток путем использования в их составе разнообразных приятных на вкус веществ, таких как маннитол, что в настоящее время хорошо технически отработано.
Когда желательно вводить соединение в виде суппозитория, могут быть использованы стандартные основы. Масло какао является традиционной основой суппозитория, которая может быть модифицирована добавкой парафинов, чтобы слегка повысить его температуру плавления. Широко используются смешиваемые с водой основы суппозиториев, в частности, включающие полиэтиленгликоли различных молекулярных весов.
Действие соединений может быть замедлено или отсрочено применением соответствующего состава. Например, может быть получен и включен в таблетки или в капсулы слабо растворимый шарик соединения. Методика может быть улучшена изготовлением гранул с несколькими различными скоростями растворения и заполнением капсул смесью шариков. Таблетки или капсулы могут быть покрыты пленкой, которая может препятствовать растворению в течение заданного времени. Даже парентеральные препараты могут быть изготовлены длительно действующими путем растворения или суспендирования соединения в масле или эмульгирующих растворителях, которые позволяют ему диспергировать, только медленно, в сыворотку.
Следующие примеры служат для иллюстрации, но не ограничения изобретения, которое определено формулой изобретения.
В приведенных в экспериментальной части примерах ПМР-спектров приняты следующие обозначения:
S - синглет,
m - мультиплет,
t - триплет,
d - дублет,
dd - двойной дублет,
q - квартет,
b - уширенный сигнал
Примеры
Пример 1
Цис-6-фенил-5-[4-(2-пирролидин-1-илэтокси)фенил] -5,6,7,8- тетрагидронафталин-2-ол
Стадия A
Цис-1-{2-[4-(6-метокси-2-фенил-1,2,3,4-тетрагидронафталин-1-ил) фенокси] этил}пирролидин.
Раствор 1-{ 2-[4-(6-метокси-2-фенил-3,4-дигидронафталин-1-ил)- фенокси] этил}пирролидин гидрохлорида (нафоксиден гидрохлорид) (1,0 г, 2, 16 ммоль) в 20 мл абсолютного этанола, содержащего 1,0 г гидроокиси палладия на углероде, гидрируют при 50 фунт/кв.дюйм (10,5 кг/см2) при 20oC в течение 19 часов. Фильтрация и упаривание дают 863 мг (93%) цис-1-{2-[4-(6-метокси-2-фенил-1,2,3,4-тетрагидронафталин-1-ил)фенокси] этил} - пирролидина: 1H-ЯМР (CDCl3): δ 3.50-3.80 (m, 3H), 3,85 (S, 3H), 4.20-4.40 (m, 3H), 6.80-7.00 (m, 3H); MS 428 (P+ + 1).
Стадия B
К раствору 400 мг (0.94 ммоль) продукта стадии A в 25 мл метиленхлорида при 0oC добавляют по каплям при перемешивании 4,7 мл (4,7 ммоль) 1,0 М раствора трибромида бора в метиленхлориде. После 3 часов при комнатной температуре реакционную смесь выливают в 100 мл быстро перемешиваемого насыщенного водного раствора бикарбоната натрия. Органический слой отделяют, сушат над сульфатом натрия, фильтруют и концентрируют, получая 287 мг (74%) указанного в заглавии вещества в виде свободного основания. ПМР (CDCl3): δ 3.55 (dd, 1H), 4.00 (t, 2H), 4.21 (d, 1H), 6.35 (ABq, 4H).
Соответствующую хлористоводородную соль получают обработкой раствора основания избытком 4 н. HCl в диоксане с последующим упариванием досуха и растиранием с диэтиловым эфиром (MS:415[P++1]).
Ниже описан альтернативный способ, используемый для получения соединения примера 1.
Стадия А
1-{2-[4-(6-Метокси-3,4-дигидронафталин-1-ил)фенокси]этил}пирролидин
Смесь безводного CeCl3 (138 г, 560 ммоль) и ТГФ (500 мл) энергично перемешивают в течение 2 часов в сосуде для разделения жидкостей, раствор 1-[2-(4-бромфенокси)этил] пирролидина (100 г, 370 ммоль) в ТГФ (1000 мл) охлаждают до -78oC и медленно добавляют в течение 20 минут н-бутиллитий (н-Buli) (2,6 М в гексане, 169 мл, 440 ммоль). Через 15 минут раствор добавляют к суспензии CeCl3, охлажденной до -78oC, через стеклянную трубку и реакционную смесь перемешивают 2 часа при -78oC. Через трубку к арилцериевому реагенту при -78oC добавляют раствор 6-метокси-1-тетралона (65, 2 г, 370 ммоль) в ТГФ (1000 мл). Реакционной смеси дают медленно нагреться до комнатной температуры и перемешивают в общей сложности 16 часов. Смесь фильтруют через слой целита. Фильтрат концентрируют в вакууме и добавляют 3 н. HCl (500 мл) и Et2O (500 мл). После перемешивания в течение 15 минут слои разделяют. Водный слой дополнительно промывают Et2O (2 раза). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют, получая 6-метокси-1-тетралон (22 г). Водный слой подщелачивают до pH 12 с помощью 5 н. NaOH и 15% водный (MH4)2CO3 (1000 мл). Водную смесь экстрагируют CH2Cl2 (2 раза). Органический раствор сушат (MgSO4), фильтруют и концентрируют, получая коричневое масло. Примеси отгоняют (110-140oC @ 0.2 мм Hg), получая продукт (74 г, 57%).
ПМР (250 MHz, CDCl3): δ 7,27 (d, J = 8.7 Hz, 2H), 6.92-6.99 (m, 3H), 6.78 (d, J = 2.6 Hz, 1H), 6.65 (dd, J = 8.6, 2.6 Hz, 1H), 5.92 (t, J = 4.7 Hz, 1H), 4.15 (t, J = 6.0 Hz, 2H), 3.80 (s, 3H), 2.94 (t, J = 6.0 Hz, 2H), 2.81 (t, J = 7.6 Hz, 2H), 2.66 (m, 2H), 2.37 (m, 2H), 1.84 (m, 4H).
Стадия B
1-{ 2-[4-(2-Бром-6-метокси-3,4-дигидронафталин-1-ил)фенокси] этил}пирролидин
Пербромид пиридиний бромид (21,22 г, 60,55 ммоль) добавляют порциями к раствору 1-{2-[4-(6-метокси-3,4-дигидронафталин-1-)фенокси]этил}пирролидина (23 г, 72 ммоль) в ТГФ (700 мл). Реакционную смесь перемешивают 60 часов. Осадок фильтруют через слой целита с добавлением ТГФ. Не совсем белый твердый продукт растворяют в CH2Cl2 и MeOH отфильтровывают через целит. Органический раствор промывают 0.5 н. HCl и далее насыщенным N2HCO3 (водн.). Органический раствор сушат (MgSO4), фильтруют и концентрируют, получая коричневый твердый продукт (21,5 г, 83%).
ПМР (250 MHz, CDCl3): δ 7.14 (d, J = 8.7 Hz, 2H), 6.97 (d, J = 8.8 Hz, 2H), 6.71 (d, J = 2.2 Hz, 1H), 6.55 (m, 2H), 4.17 (t, J = 6.0 Hz, 2H), 3.77 (s, 3H), 2.96 (m, 4H), 2.66 (m, 4H), 1.85 (m, 4H).
Стадия C
1-{ 2-[4-(6-Метокси-2-фенил-3,4-дигидронафталин-1-ил)фенокси]этил}пирролидин гидрохлорид (Нафоксиден гидрохлорид)
К смеси 1-{ 2-[4-(2-бром-6-метокси-3,4-дигидронафталин-1-ил)-фенокси] этил} пирролидина (19 г, 44 ммоль), фенилборной кислоты (7,0 г, 57 ммоль) и тетракис(трифенилфосфоний)палладиния (1,75 г, 1,51 ммоль) в ТГФ (300 мл) добавляют Na2CO3 (13 г, 123 ммоль) в H2O (100 мл). Реакционную смесь нагревают при температуре кипения с обратным холодильником 18 часов. Слои разделяют и органический слой промывают H2O и впоследствии раствором соли. Органический раствор сушат (MgSO4), фильтруют и концентрируют, получая 17,96 г коричневого твердого продукта. Остаток растворяют в смеси 1:1 CH2Cl2 и EtOAc (250 мл) и добавляют 1 н. HCl в Et2O (100 мл).
После перемешивания в течение 2 часов продукт оставляют кристаллизоваться из раствора и путем фильтрации собирают 11 г вещества. Концентрирование маточного раствора до половины его объема дает дополнительно 7.3 г продукта.
Стадия D
Цис-1-{2-[4-(6-метокси-2-фенил-1,2,3,4-тетрагидронафталин 1-ил)-фенокси] этил}пирролидин
1-{ 2-[4-(6-Метокси-2-фенил-3,4-дигидронафталин-1-ил)фенокси] этил} пирролидин гидрохлорид (нафоксиден гидрохлорид) (75 г, 162 ммоль) растворяют в 1000 мл EtOH и 300 мл MeOH. Добавляют сухой Pd (OH) на углероде и смесь гидрируют на вибраторе Парра при 50oC и 50 фунт/кв. (10,5 кг/см2) дюйм в течение 68 часов. Катализатор отфильтровывают с помощью целита и растворители удаляют в вакууме. Образовавшийся твердый белый продукт растворяют в CH2Cl2 и раствор промывают насыщенным NaHCO3 (водн.). Органический раствор сушат, (MgSO4), фильтруют и концентрируют, получая не совсем белый твердый продукт (62.6 г, 90%).
Стадия E
Цис-6-фенил-5-[4-(2-пирролидин-1-ил-этокси)фенил] 5,6,7,8-тетрагидронафталин- 2-ол
Смесь цис-1-{ 2-[4-(6-метокси-2-фенил-1,2,3,4-тетрагидронафталин-1-ил)фенокси] этил} пирролидина (12 г, 28 ммоль), уксусной кислоты (75 мл) и 48% HB (75 мл) нагревают при 100oC 15 часов. Раствор охлаждают и образовавшийся белый осадок собирают фильтрацией. Гидробромидную соль (9.6 г, 69%) растворяют в CHCl3/MeOH и перемешивают с насыщен. NaHCO3 (водн.). Слои разделяют и водный слой экстрагируют в дальнейшем CHCl3МeOH. Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют, получая продукт в виде не совсем белой пены.
ПМР (250 MH, CDCl3): δ 7.04 (m, 3H), 6.74 (m, 2H), 6.63 (d, J = 8.3 Hz, 2H), 6.50 (m, 3H), 6.28 (d, J = 8.6 Hz, 2H), 4.14 (d, J = 4.9 Hz, 1H), 3.94 (t, J = 5.3 Hz, 2H), 3.24 (dd, J = 12.5, 4.1 Hz, 1H), 2.95 (m, 4H), 2.78 (m, 4H), 2.14 (m, 1H), 1.88 (m, 4H), 1.68 (m, 1H).
Пример 2
Транс-6-фенил-5-[4-(2-пирролидин-1-ил-этокси)фенил] -5,6,7,8-тетрагидронафталин- 2-ол
Стадия A
К раствору цис-1-{2-[4-(6-метокси-2-фенил-1,2,3,4-тетрагидронафталин-1-ил)фенокси] этил} пирролидина (500 мг, 1,17 ммоль) в 10 мл диметилсульфоксида при 10oC медленно добавляют 4,7 мл (11,7 ммоль) 2,5 М н-бутиллития в гексане. Реакционной смеси дают нагреться до 20oC и перемешивают 19 часов. После добавления воды и экстракции диэтиловым эфиром органические слои объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха, получая 363 мг (73%) транс-6-метоксидигидронафталина.
ПМР (CDCl3): δ 3.45 (m, 2H), 3.82 (s, 3H), 4.06 (d, 1H), 4.45 (m, 2H), 6.80 (d, 2H).
Стадия B
Используя бор трибромидный способ снятия защиты, описанный в примере 1, стадии B, 363 мг (0.85 ммоль) продукта стадии A превращают в 240 мг (68%) указанного в заглавии соединения.
ПМР (CDCl3): δ 4.02 (d, 1H), 4.45 (m. 2H), 7.00 (d, 2H).
Соответствующую хлористоводородную соль получают, как описано в стадии B примера 1 (MS 414 P+ + 1).
Пример 3
1-(4'-Пирролидиноэтоксифенил)-2-(4''-гидроксифенил)-6-гидрокси- 1,2,3,4-тетрагидроизохинолин гидрохлорид
Стадия A
3-Метоксифенилацета-4'-метоксианилид.
Раствор 20,0 г (0,120 моль) 3-метоксифенилуксусной кислоты и 40 мл (65,3 г, 0,549 моль) тионилхлорида в 100 мл бензола нагревают при температуре кипения с обратным холодильником в течение 2 часов и упаривают досуха, что дает соответствующий хлорангидрид (предположительно 0,120 моль). Хлорангидрид перемешивают в 50 мл диэтилового эфира и добавляют к смеси 4-метоксианилина в 100 мл диэтилового эфира при 0oC. После перемешивания при 20oC в течение ночи суспензию фильтруют, получая твердый продукт, который промывают водой, 5,5% водн. HCl, водой и диэтиловым эфиром. Последующее высушивание над P2O5 в вакууме в течение 4 часов дает 19,7 г (60%) указанного в заглавии вещества в виде белого твердого продукта.
ПМР (CDCl3): δ 3.70 (s, 2H), 3.77 (s, 3H), 3.81 (s, 3H).
Стадия B
N-(4-Метоксифенил)-2'-(3''-метокси фенетиламин)гидрохлорид
Суспензию 19,6 г (0,072 моль) продукта стадии A и 6,04 г (0,159 моль) алюмогидрида лития в 130 мл диэтилового эфира и 75 мл диоксана нагревают при 35oC в течение 48 часов. Добавляют избыток декагидрата сульфита натрия и смесь фильтруют и промывают 5% водн. HCl. Органический слой сушат над безводным сульфатом магния и концентрируют, получая 10,84 г указанного вещества в виде - соли (51%).
ПМР (CDCl3): δ 3.15 (m, 2H), 3.42 (m, 2H), 3.71 (s, 3H), 3.74 (s, 3H).
Стадия C
N-2-(3'-Метоксифенетил)-4''-бензилоксибен-4-'''-метоксианилид
К суспензии 4,83 г (0,164 моль) продукта стадии B и 2,12 г (0,0164 моль) диизопропилэтиламина в 50 мл диэтилового эфира добавляют 0,013 моль 4-бензилоксибензоил хлорида (полученного из 3,00 г (0,013 моль) соответствующей бензойной кислоты и 7,13 г (0,059 моль) тионилхлорида в 35 мл бензола) в 50 мл диэтилового эфира при 20oC и реакционную смесь перемешивают в течение ночи. После декантации с осадка эфирный раствор промывают 5% водн. HCl, водой, раствором соли, сушат над сульфатом магния, фильтруют и упаривают досуха, получая 5,58 г указанного в заглавии вещества (73%).
ПМР (CDCl3): δ 3.00 (m, 2H), 3.75 (m, 9H), 4.05 (m, 2H).
Стадия D
1-(4'-Бензилоксифенил)-2-(4''-метоксифенил)-6-метокси-3,4-дигидроизохинолиний хлорид
Раствор 1,04 г (2,22 ммоль) продукта стадии C в 5 мл оксихлорида фосфора нагревают при 100oC в течение 2.5 часов. Реакционную смесь упаривают досуха и распределяют в смеси этилацетат/вода. Этилацетатный слой сушат над безводным сульфатом магния и концентрируют, получая 1,03 г указанного в заглавии соединения в виде масла (96%).
ПМР (CDCl3): δ 3.46 (t, 2H), 3.80 (s, 3H), 4.00 (s, 3H), 4.55 (t, 2H).
Стадия E
1-(4'-Бензилоксифенил)-2-(4''-метоксифенил)-6-метокси-1,2,3,4- тетрагидроизохинолин
К продукту стадии (1,00 г, 2,07 ммоль) в 10 мл метанола добавляют 200 мг (5,28 ммоль) боргидрида натрия. После перемешивания в течение 19 часов при 25oC осадок собирают и сушат в вакууме, получая 611 мг (66%) указанного в заглавии соединения в виде пены.
ПМР (CDCl3): δ 2.95 (m, 2H), 3.50 (m, 2H), 3.71 (s, 3H), 3.78 (s, 3H), 5.09 (s, 1H).
Стадия F
1-(4'-Гидроксифенил)-2-(4''-метоксифенил)-6-метокси-1,2,3,4- тетрагидроизохинолин гидрохлорид
Раствор 611 мг (1,35 ммоль) продукта стадии E в 6 мл конц. водн. HCl и 6 мл диоксина нагревают при 90oC в течение 5 часов. Диоксан удаляют в вакууме и водный слой разбавляют водой. Указанное в заглавии соединение выделяют (155 мг, 29%) в виде выпавшей в осадок хлористоводородной соли.
ПМР (CDCl3): δ 3.72 (s, 3H), 3.76 (s, 3H), 5.94 (s, 1H).
Стадия G
1-(4'-Пирролидиноэтоксифенил)-2-(4''-метокси-фенил)-6-метокси-1, 2,3,4-тетрагидроизохинолин
К суспензии 152 мг (0,382 ммоль) продукта стадии в 5 мл диоксана и 1 мл ДМФ добавляют 152,8 мг (3,82 ммоль) 60% дисперсии гидрида натрия в растительном масле. После перемешивания при 45oC в течение 0,5 часа, 65 мг (0,382 ммоль) 2-хлорэтилпирролидин гидрохлорида медленно добавляют порциями и перемешивают 3 часа при 45oC. После добавления воды реакционную смесь экстрагируют этилацетатом. Этилацетатный слой сушат над безводным сульфатом магния и концентрируют, получая 203 мг сырого продукта, который хроматографируют на силикагеле смесью хлороформ/метанол (99:1), получая 78 мг (45%) указанного в заглавии вещества.
ПРМ (CDCl3): δ 2.85 (m, 2H), 3.72 (s, 3H), 3.79 (s, 3H), 4.00 (t, 2H), 5.50 (s, 1H).
Стадия H
1-(4'-Пирролидиноэтоксифенил)-2-(4''-гидроксифенил)-6-гидрокси-1,2,3,4- тетрагидроизохинолин гидрохлорид
К раствору 0,75 мг (0,164 ммоль) продукта стадии G в 5 мл метиленхлорида при 0oC добавляют по каплям 0,82 мл (0,82 ммоль) 1,0 М трибромида бора в метиленхлориде. После перемешивания при 0oC в течение 0,5 часа, реакции проводят при 20oC в течение 2 часов. Реакционную смесь выливают в охлажденный льдом насыщенный раствор бикарбоната натрия. Супернатант отфильтровывают от смолы, которую растворяют в метаноле, сушат над сульфатом магния, фильтруют и упаривают досуха, получая 53 мг (75%) указанного в заглавии соединения в виде пены.
ПМР (CD3ОD): δ 4.02 (m, 2H), 5.50 (s, 1H), 6.50-7.00 (m, 11H).
Хлористоводородную соль получают аналогичным способом в виде белого твердого продукта:
MS 431 (P++1).
Пример 4
1-(6'-Пирролидиноэтокси-3'-пиридил)-2-(4''-гидроксифенил)-6-гидрокси-1, 2,3,4-тетрагидроизохинолин гидрохлорид
Стадия A
1-(6'-Хлор-3'-пиридил)-2-(4''-метоксифенил)-6-метокси-1,2,3,4- тетрагидроизохинолин
Используя методику, описанную для примера 3, стадии C, заменяя 4-бензилоксибензоил хлорид на 6-хлорникотиноил.
Стадия B
1-(6'-Пирролидиноэтокси-3'пиридил)-2-(4''-метоксифенил)-6-метокси-1,2, 3,4-тетрагидроизохинолин
Продукт стадии A (500 мг, 1,31 ммоль) суспендируют в 10 мл толуола и обрабатывают 364 мг (5,52 ммоль) гидроокиси калия, 346 мг (1,31 ммоль) 18-краун-6 и 318 мг (2,76 ммоль) 1-(2-гидрокси-этил)пирролидина. После нагревания при 80oC в течение 18 часов реакционную смесь охлаждают, разбавляют водой и экстрагируют этилацетатом. Объединенные органические экстракты промывают раствором соли, сушат над сульфатом магния, фильтруют и концентрируют досуха, получая 575 мг пены. Хроматография на силикагеле с использование 97,5% смеси хлороформ/метанол (9:1) и 2,5% конц. NH4OH дает 127 мг (21%) указанного в заглавии соединения.
ПМР (CDCl3): δ 2.50 (m, 4H), 2.90 (m, 4H), 3.42 (m, 2H), 3,72 (s, 3H), 3.79 (s, 3H), 4.39 (t, 2H), 5.05 (s, 1H).
Стадия C
С продукта стадии B снимают защиту согласно методике примера 1 и превращают в хлористоводородную соль обычным способом, получая указанное в заглавии соединение.
ПМР (CDCl3): δ 2.55 (m, 2H), 5.45 (s, 1H), MS (P++1) 432.
Пример 5
1-(4-Азабициклогептаноэтоксифенил)-2-(4''-гидроксифенил)-6- гидрокси-1,2,3,4-тетрагидроизохинолин гидрохлорид
Используя методику примера 3, заменяя на стадии C 4-бензилокси бензойную кислоту на 4-(2'-азабицикло[2.2.1]-гептаноэтокси)бензойную кислоту и последующим применением стадий D, E и H, получают указанное в заглавии соединение в виде белого твердого продукта.
ПМР (CDCl3): δ 2.95 (m, 3H), 3.90 (s, 1H), 4.15 (t, 3H), 5.42 (s, 1H):
MS 457 (P+ + 1).
Пример 6
(-)-цис-6-Фенил-5-[6-(2-пирролидин-1-ил-этокси-)пиридин-3-ил] -5,6,7,8- тетрагидронафталин-2-ол
Стадия A
5-Бром-2-(2-пирролидин-1-илэтокси)пиридин
Раствор 2,5-дибромпиридина (15,0 г, 63,3 ммоль), порошкообразного KOH (6,39 г, 114 ммоль), 1-(2-гидроксиэтил)пирролидина (14,58 г, 126,6 ммоль) и 18-краун-6 (300 мг, 1,14 ммоль) в сухом толуоле (100 мл) нагревают при 70oC в течение 1 часа. Раствор охлаждают до комнатной температуры и добавляют воду и EtOAc. Органический слой промывают водой и раствором соли. Раствор сушат (MgSO4), фильтруют и концентрируют в вакууме. Молекулярная перегонка (153oC/ 0,1 мм Hg) дает указанное в заглавии соединение в виде бесцветного масла, кристаллизующегося при охлаждении (14,9 г, 87%).
ПМР (250 MHz, CDCl3): δ 8.15 (d, J = 2.4 Hz, 1H), 7.65 (dd, J = 2.4, 8.4 Hz, 1H), 6.67 (d, J = 8,4 Hz, 1H), 4.38 (t, J = 5.8 Hz, 2H), 2.84 (t, J = 5.8 Hz, 2H), 2.62 (m, 4H), 1.82 (m, 4H).
Стадия B
6-Метокси-1-[6-(2-пирролидин-1-илэтокси)пиридин-3-ил] -1,2,3,4- тетрагидронафталин-1-ол
К раствору 5-бром-2-(2-пирролидин-1-илэтокси)пиридин (7,0 г, 26 ммоль) в сухом ТГФ (50 мл) при -78oC добавляют по каплям H-BuLi (2,5 М в гексане, 12,4 мл, 31,0 ммоль). Через 30 минут добавляют 6-метокси-1-тетралон (4,55 г, 25,8 ммоль) в сухом ТГФ. После перемешивания в течение 15 минут при -78oC реакционной смеси дают нагреться до комнатной температуры. Через 30 мин реакционную смесь выливают в водн. NaHCO3 (насыщ.). Водный слой экстрагируют EtOAc(2x). Объединенные органические растворы сушат (MgSO4), фильтруют и концентрируют. Флэш-хроматография (CHCl3:MeOH, 95:5) дает целевое соединение (4,23 г, 44%) в виде белого твердого вещества.
ПМР (250 MHz, CDCl3): δ 8.07 (d, J = 2.5 Hz, 1H), 7,49 (dd, J = 2.5, 8.7 Hz, 1H), 7.00 (d, J = 8.5 Hz, 1H), 6.73 (m, 3H), 4.45 (t, J = 5.7 Hz, 2H), 3.79 (s, 3H), 2.92 (t, J = 5.7 Hz, 2H), 2.76 (m, 2H), 2.67 (m, 4H), 2.11 (s, 1H), 2.08 (m, 3H), 1.82 (m. 5H).
Стадия C
5-(2-Бром-6-метокси-3,4-дигидронафталин-1-ил)-2-(2-пирролидин-1- илэтокси)пиридин
Пиридиний бромид пербромид (3,5 г, 12,2 ммоль) добавляют к раствору 6-метокси-1-6-(2-пирролидин-1-илэтокси)пиридин-3-ил-1,2,3,4- тетрагидронафталин-1-ола (3,3 г, 8,9 ммоль) в CH2Cl2 (50 мл). Реакционную смесь перемешивают 18 часов и добавляют водный NaHCO3 (насыщ.). Водный слой экстрагируют CH2Cl2 и объединенный органический раствор промывают водой и раствором соли. Органический раствор сушат (MgSO4), фильтруют и концентрируют. Флэш-хроматография (CHCl3):MeOH, 95:5) дает заданный винил бромид (2,65 г, 70%).
ПМР (250 MHz, CDCl3): δ 8.0 (d, J = 2.4 Hz, 1H), 7.41 (dd, J = 2.4, 8.4 Hz, 1H), 6.83 (d, J = 8.4 Hz, 1H), 6.69 (m, 1H), 6.55 (m, 2H), 4.92 (t, J = 5.8 Hz, 2H), 3.76 (s, 3H), 2.94 (m, 6H), 2.64 (m, 4H), 1.82 (m, 4H).
Стадия D
5-(6-Метокси-2-фенил-3,4-дигидронафталин-1-ил)-2-(2-пирролидин-1 илэтокси)пиридин
Фениллитий (1,8 М в смеси циклогексан/диэтиловый эфир, 3,8 мл, 7,0 ммоль) медленно добавляют к хлористому цинку (0,5 М в ТГФ, 14 мл, 7,0 ммоль) при 0oC. После перемешивания в течение 15 минут добавляют 5-(2-бром-6-метокси-3,4-дигидронафталин-1-ил)-2-(2-пирролидин-1-илэтокси) пиридин (1,0 г, 2,3 ммоль) в сухом ТГФ (20 мл) и впоследствии Pd(PPh3)4 (200 мг, 0,173 ммоль). Реакционную смесь нагревают до комнатной температуры и затем кипятят с обратным холодильником 4 часа. Реакционную смесь выливают в водный раствор NH4Cl (насыщ.). Водный слой промывают CHCl3 (2x) и объединенные органические растворы промывают водой и затем раствором соли. Органический раствор сушат (MgSO4), фильтруют и концентрируют в вакууме. Флэш-хроматография (CHCl3:MeOH, 95:5) дает указанное в заглавии соединение (680 мг, 68%).
ПМР (250 MHz, CDCl3): δ 7.78 (d, J = 2.1 Hz, 1H), 7.27 (m, 1H), 7.07 (m, 5H), 6.68 (m, 4H), 4.40 (t, J = 5.8 Hz, 2H), 3.80 (s, 3H), 2.88 (m, 6H), 2.71 (m, 4H), 1.81 (m, 4H).
Стадия E
цис-5-(6-Метокси-2-фенил-1,2,3,4-тетрагидронафталин-1-ил)-(2-пирролидин- 1-илэтокси)пиридин
Pd(OH)2 (20%, 77 мг) сушат в вакууме пламени и добавляют к раствору 5-(6-метокси-2-фенил-3,4-дигидронафталин-1-ил)-2-(2-пирролидин-1- илэтокси)пиридина (286,4 мг, 0,6714 ммоль) в уксусной кислоте (50 мл). Смесь гидрируют в вибраторе Парра при 50 фунт/кв.дюйм (10,5 кг/см2) и при 50oC в течение 16 часов. Катализатор отфильтровывают с помощью целита и уксусную кислоту удаляют в вакууме. (ПМР показал, что реакция не завершена и остаток вновь выдерживали в реакционных условиях (50 фунт/кв.дюйм (10,5 кг/см2) и 60oC дополнительно 6 часов). Катализатор удаляют фильтрацией через целит и растворитель отгоняют в вакууме. Заданный продукт (207 мг, 72%) получают радиальной хроматографией (градиент растворителя от CH2Cl2 до 10% MeOH в CH2Cl2).
ПМР (250 MHz, CDCl3): δ 7.19 (m, 4H), 6.84 (m, 3H), 6.75 (d, J = 2.4 Hz, 1H), 6.68 (d, J = 2.4, 8.4 Hz, 1H), 6.59 (dd, J = 2.4, 8.4 Hz, 1H), 6.40 (d, J = 8.4 Hz, 1H), 4.35 (t, J = 5.7 Hz, 2H), 4.21 (d, J = 4.8 Hz, 1H), 3.82 (s, 3H), 3.38 (m, 1H), 3.06 (m, 2H), 2.90 (t, J = 5.7 Hz, 2H), 2.69 (m, 4H), 2.11 (m, 2H), 1.84 (m, 4H).
Стадия F
Цис-6-фенил-5-[6-(2-пирролидин-1-илэтокси)пиридин-3-ил] -5,6,7,8- тетрагидронафталин-2-ол
К раствору цис-5-(6-метокси-2-фенил-1,2,3,4-тетрагидронафталин-1-ил)-2- (2-пирролидин-1-илэтокси)пиридина (69,6 мг, 0,162 ммоль) в сухом CH2Cl2 (3 мл) при 0oC добавляют AlCl3 (110 мг, 0,825 ммоль) и впоследствии избыток EtSH (400 мкл). Через 0,5 часов реакционную смесь нагревают до комнатной температуры и добавляют дополнительно AlCl3 (130 мг). Спустя 0,5 часов осторожно добавляют водный NaHCO3 (насыщ.) и водный слой экстрагируют CH2Cl2/MeOH (3x). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют. Радиальная хроматография (градиент растворителя, от CH2Cl2 до 15% MeOH в CH2Cl2) дает соединение со снятой защитой (64,6 мг, 96%) в виде не совсем белого твердого вещества.
ПМР (250 MHz, CDCl3): δ 7.18 (m, 3H), 6.96 (d, J = 2.4 Hz, 1H), 6.82 (m, 2H), 6.70 (d, J = 2.4 Hz, 1H), 6.67 (d, J = 8.4 Hz, 1H), 6.62 (dd, J = 2.4, 8.5 Hz, 1H), 6.52 (dd, J = 2.4, 8.4 Hz, 1H), 5.80 (d, J = 8.5 Hz, 1H), 4.45 (m, 2H), 4.18 (d, J = 4.8 Hz, 1H), 3.40 (m, 1H), 3.04 (m, 3H), 2.75 (m, 6H), 2.11 (m, 1H), 1.88 (m, 4H).
Два энантиомера разделяют хроматографически на 5 см внутр. диаметр х 5 см Chiracel OD колонке, используя 5% этанол/95% гептан с 0,05% диэтиламина.
Энантиомер 1: - Rt = 17,96 мин, [α]d = +242(c=1, MeOH);
Энантиомер 2: - R7 = 25,21 мин, [α]d = -295(c=1, MeOH).
Пример 7
Цис-6-(4-фтор-фенил-)-5-[4-(2-пиперидин-1-илэтокси)-фенил] -5,6,7,8- тетрагидронафталин-2-ол
Стадия A
К 1 г 1-[4'-пиперидино этокси-фенил]-2-[4''фторфенил]-6-метокси-3,4-дигидронафталина (который может быть получен, как в примере 1, но заменой фенилборной кислоты в стадии C на 4-фторфенилборную кислоту) в 35 мл уксусной кислоты добавляют гидроокись палладия на углероде (20%, 1Г) (высушен пламенем в вакууме). Смесь гидрируют на вибраторе Парра при 50oC и 50 фунт/кв (10,5 кг/см2) дюйм в течение 4 часов. Фильтрация через Целит и концентрация дает 1,2 г сырого реакционного продукта, который используют в следующей стадии без дополнительной очистки.
ПМР (250 MHz, CDCl3): δ 1.91 (m), 3.1 (m), 3.25 (m), 3.8 (s, 3H), 4.2 (d, 1H), 4.25 (bd), 6.35 (d, 2H), 6.5 (d, 2H), 6.65 (m), 6.75 (m), 6.8-6.88 (m). m/z 460 (M+1).
Стадия B
Раствор цис-1-[4'-пиперидиноэтокси фенил] -2-[4''-фтор-фенил]-6-метокси-1,2,3,4-тетрагидро нафталин-1-ил]фенокси}-этил)-пиперидина (540 мг, 1,17 ммоль) в безводном CH2Cl2 охлаждают до 0oC и впоследствии добавляют BBr3 5,8 мл (IM раствор в CH2Cl2), 5.88 ммоль по каплям. Реакционной смеси дают нагреться до комнатной температуры и перемешивают еще один час. После завершения реакции реакционную смесь вновь охлаждают до 0oC и осторожно добавляют водный NaHCO3. Водный слой экстрагируют CH2Cl2 (3x). Органический слой сушат над MgSO4, фильтруют и концентрируют. Сырой продукт подвергают круговой хроматографии (растворитель 4: 1 диэтиловый эфир/гексан, 1% триэтиламин), получая незащищенный продукт. HCl-соль продукта получают с IM HCl/диэтил. эфир раствором с последующим растиранием со смесью EtOAc /ТГФ, что дает 126 мг продукта.
ПМР (250 MHz, CDCl3): δ 6.80 (m, 4H), 6.63 (m, 4H), 6.50 (dd, 1H), 6.40 (d, 2H), 4.22 (dd, 3H), 3.72 (m, 2H), 3.48 (dd, 2H), 3.0 (bm, 2H), 1.83 (m, 9H). m/z 446 (M+1).
Пример 8
(-)Цис-6-фенил-5-[4-(2-пирролидин-1-ил-этокси)фенил] -5,6,7,8- тетрагидронафталин-2-ол
Рацемическое соединение примера 1 (3 г) подвергают энантиомерному разделению на 5 см х 5 см Chiracell OD колонке, используя 99,95% (5% EtOH/95% гептан)/ 0,05% диэтиламин в качестве элемента, что дает 1 г быстро вымываемого (+) энантиомера и 1 г медленно вымываемого (-) энантиомера, которые проявляются на ПМР, MS и ТСХ как рацемат. Или же иначе, для разделения может быть применена методика кристаллизации с использованием R-binap фосфорной кислоты. В 20 мл метанола и 20 мл метиленхлорида добавляют 7.6 г (0,0184 моль) продукта примера 1 и 6,4 г (0,0184 моль) R-(-)-1,1'-бинафтил-2,2'-диил кислого фосфата. После завешения растворения, упаривание растворителя с последующим растиранием с диэтиловым эфиром дает 14,2 г рацемической соли. Этот твердый продукт суспендируют в 500 мл диоксана и 25 мл метанола и полученную смесь нагревают до растворения исходного твердого продукта. При выдерживании в течение 1 часа образуется белый осадок (6.8 г), который собирают и ВДЖХ которого (использование приведенных выше условий) показывает приблизительно 73% чистоту энантиомера. Этот продукт суспендируют в 250 мл абсолютного этанола и нагревают до получения раствора, после этого раствор оставляют стоять при комнатной температуру в течение ночи.
Собранные кристаллы промывают охлажденным этанолом и затем диэтиловым эфиром, получая 3,1 г 98% энантиомерно чистой соли; вторичной обработкой получают также 588 мг. Распределение между 1:2 метанол/метилен хлорид и 1% водной гидроокисью натрия дает соответствующее основание, которое превращают в хлористоводородную соль (HCl в диоксане). Перекристаллизация из смеси ацетонитрил/метиленхлорид дает левовращающий предпочтительный энантиомерный гидрохлорид соответствующий примеру 1.
[α]d - 330.6 (C=0.05, CH2Cl2]; Т.пл. 260-263oC.
Пример 9
Цис-6-(4'-гидроксифенил)-5-[4-(2-пиперидин-1-илэтокси)фенил] -5,6,7,8- тетрагдиронафталин-2-ол
Указанное в заглавии соединение получают, следуя методике, описанной в способе примера 1.
ПМР (CDCl3): δ 3.12 (m, 1H), 3.90 (m, 2H); 4.15 (d, 1H); 6.15-6.72 (m, 11H);
FAB MS (M+1) 430.
Пример 10
1-(4'-Пиперидиноэтоксифенил)-2-(4''-гидроксифенил)-6-гидрокси-1, 2,3,4-тетрагидроизохинолин гидрохлорид
Используя методику примера 3, описанную в стадии G, заменяя N-2-хлорэтилпирролидин гидрохлорид на N-2-хлорэтилпиперидин гидрохлорид, получают соединение, указанное в заглавии.
ПМР (CDCl3): δ 2.65 (m, 2H), 2.75 (m, 2H); 5.45 (s, 1); 6.50-7.00 (m, 11H).
FAB MS (M+1) 445
Пример 11
1-(4'-Пирролидиноэтоксоксифенил)-2-(4''-фторфенил)-6-гидрокси-1,2,3,4- тетрагидроизохинолин гидрохлорид
Указанное в заглавии соединение получают, используя методику примера 3, описанную в стадии A, заменяя 4-метоксианилин на 4-фторанилин.
ПМР (CDCl3): δ 2.12 (m, 2H); 3.65 (m, 2H); 4.45 (m, 2H); 6.10 (s, 1H); 7.5 (m, 2H);
FAB MS (M + 1) 433.
Пример 12
1-(4'-Пирролидиноэтоксифенил)-2-фенил-6-гидрокси-1,2,3,4- тетрагидроизохинолин гидрохлорид
Указанное в заглавии соединение получают, используя методику примера 3, описанную в стадии A, заменяя 4-метоксианилин анилином.
ПМР (CDCl3): δ 1.70 (m, 4H); 2.70 (m, 2H); 4.00 (m, 2H); 5.70 (s, 1H); 6.60-7.25 (m, 12H);
FAB MS (M+1) 415.
Пример 13. Данные, проведенного in vivo анализа связывания эстроген-рецептор (по методике, описанной на стр. 28)
Соединение - IC50 (нм)
N примера
1 - 11,3
2 - 9,75
3 - 37,5
4 - 16,0
5 - 37,2
6 - 7,0
8 - 11,1
12 - 14,8
Пример 14
Испытание эффективности соединения, полученного по примеру 1, в отношении потери костной ткани, уровня холестерина и увеличения веса тела.
Используют 60 самок крыс (Sprague - Dawley) в возрасте 5 месяцев, весом приблизительно 280 г (Charles River Laboratory, Wilmington, mA).
Крысы аклиматизировались в условиях вивария (24oC, 12 часов - свет, 12 часов - темнота) в течение 2 недель. Все крысы имели свободный доступ к воде и пище, содержащей 0,97% кальция, 0,85% фосфора и 1,05 IU/2 витамина D3.
Десять крыс оперировали и им перорально вводили растворитель (10% этанола и 90% физиологического раствора, 1 мл/день); оставшихся крыс овариектомизировали и вводили перорально растворитель: 17 β- эстрадиол (E2, 30 мг/кг, ежедневно подкожной инъекцией), или соединение примера 1 (5, 10 или 20 мг/кг, ежедневно перорально) в течение 4 недель.
Всем крысам подкожно вводили 10 мг/кг кальцеина (флюорисцентная метка для кости) за 12 и 2 дня до умерщвления для того, чтобы исследовать динамические изменения костной ткани.
После 4-х недельной обработки крыс умерщвляли и определяли динамические изменения костной ткани, увеличение веса тела и содержание холестерина в крови.
Исследования показали, что введение соединения примера 1 овариектомизированным крысам в количестве приблизительно 10 мг/кг в день перорально предотвращает потерю костной массы, значительно снижает уровень холестерина в крови и значительно снижает вес тела по сравнению с контрольными овариектомизированными животными.
Изобретение относится к новым соединениям формулы I, где В и Е независимо выбраны из СН или N; R4 - водород, галоген, гидрокси; G представляет собой соединения формулы II (a, b, c) и их оптические и геометрические изомеры; и нетоксичные фармацевтически приемлемые кислотно-аддитивные соли. Описываются также фармацевтическая композиция и способ лечения с использованием соединения формулы I, обладающего эстроген антагонистической /агонистической активностью. 4 c. и 17 з.п.ф-лы.
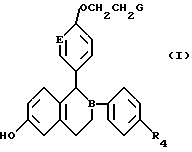

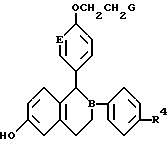
где B и E независимо выбраны из CH или N;
R4 представляет собой (а) водород, (b) галоген, (с) гидрокси;
G представляет собой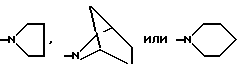
и их оптические и геометрические изомеры,
и нетоксичные фармацевтически приемлемые кислотно-аддитивные соли.
| EP 0313295 A1, 1989 | |||
| Горелка пламенной печи | 1974 |
|
SU566069A2 |
| Гаситель энергии потока воды | 1976 |
|
SU586229A1 |
| 0 |
|
SU401623A1 | |
| Поворотный переключатель | 1974 |
|
SU506072A1 |
| Способ получения производных изохинолина или их солей | 1974 |
|
SU528035A3 |
| Способ получения карбостирильных производных или их фармацевтически приемлемых солей | 1982 |
|
SU1215621A3 |
| Способ получения производных изохинолина или их фармацевтически пригодных аддитивных кислых солей | 1983 |
|
SU1207393A3 |
| Станок для шлифования шаров | 1956 |
|
SU105210A1 |
| Сметнев А.С | |||
| и др | |||
| Внутренние болезни | |||
| - М.: Медицина, 1981, с.62-202 | |||
| Малая медицинская энциклопедия | |||
| - М.: Советская энциклопедия, 1991, т.2, т.4. | |||
