Изобретение касается новых 2,3,4,5-тетрагидро-1H-3-бензазепинов и их фармацевтических кислотно-аддитивных солей, способов из получения, фармацевтических композиций, содержащих их и их использования для лечения некоторых расстройств центральной нервной системы, например, психозов, боли или страдания, депрессии, нарушений сна, дискинезий, болезни Паркинсона, ударов (параличей).
В течение последнего десятилетия проводились интенсивные фармакологические исследования, касающиеся бензазепинов. Фармакологические свойства бензазепинов в значительной степени зависят от заместителей. Известны различные замещенные бензазепины, проявляющие нейтролептическое, антиагрессивное действие, действие, влияющее на болезнь Паркинсона и на сосудистую систему.
В европейском патенте N 0.200.455 (Ново Индастри А/С) описаны 2, 3,4,5-тетрагидро-1H-3-бензазепины с гетероциклической или орто-сконденсированной гетероциклической кольцевой системой в положении 5. Заявлено, что эти соединения оказывают антипсихозное и антидепрессивное воздействие.
В настоящее время обнаружено, что 5' - или 6' - замещенные или 5', 6' - ди замещенные (2,3-дигидробензофуран-7-ил)-2,3,4,5-тетрагидро-1H-3-бензазепиновые соединения обладают сильными антидопаминергическими свойствами, что делает полезными при психофармацевтических применениях.
Неожиданно оказалось, что соединения данного изобретения проявляют удивительно высокую антидопаминергическую активность при оральном приеме по сравнению с известными соединениями.
Согласно изобретению представляются 2,3,4,5-тетрагидро-1H-3-бензазепины общей формулы (I)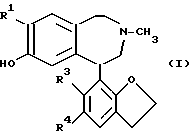 ,
,
где
R1 - Cl или Br;
R3 представляет собой Cl или Br;
R4 - галоген, NO2 или NH2; и их фармацевтически приемлемые кислотно-аддитивные соли.
Конкретными соединениями формулы (I) являются:
(+) 8-хлор-5-[5-бром-2,3-дигидробензафуран-7-ил] -7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин;
(+) 8-хлор-5-[2,3-дигидро-5-йодбензофуран-7-ил] -7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин;
(+) 8-хлор-5-[5,6-дихлор-2,3-дигидробензофуран-7-ил] -7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин;
(+) 8-хлор-5-[5-хлор-2,3-дигидробензофуран-7-ил] -7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин;
8-хлор-5-[5-нитро-2,3-дигидробензофуран-7-ил] -7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин;
8-хлор-5-[5-амино-2,3-дигидробензофуран-7-ил] -7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин;
8-хлор-5-[5-амино-2,6-дигидробензофуран-7-ил] -7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин.
Соединение формулы (I) могут быть представлены в виде смеси энантиомеров, которую можно разделить на индивидуальные чистые энантиомеры. Данное разделение может удобно осуществляться путем фракционной кристаллизации и различных растворителей солей соединений формулы (I) с оптически активными кислотами или другими способами, известными из литературы, например, с помощью хиральной колоночной хроматографии. Таким образом, изобретение включает все изомеры или в разделенном виде, или в виде смеси.
Особенно ценными воплощениями изобретения являются нетоксичные, фармацевтически приемлемые кислотно-аддитивные соли бензазепинов формулы (I). Такие соли включают соли, получаемые из неорганических и органических кислот, таких как соляная, бромистоводородная, серная, фосфорная, метансульфоновая, уксусная, молочная, малеиновая, фталевая и винная кислоты.
Эти соли могут быть получены способами, известными квалифицированными специалистами.
Изобретение касается также соединений формулы (I), в которой R3 или R4 представляют радиоактивные изотопы йода или брома, такие как применяемые в клинической практике: 1221, 1231, US1, 1311, 77Br, 82Br и 76Br.
Обнаружено, что эти соединения полезны в качестве визуализующих агентов в однофотонной эмиссионной компьютерной томографии (SPECT) или в позитронной эмиссионной томографии (PET).
Помимо этого изобретение представляет фармацевтические композиции, включающие соединения изобретения. Содержание активных соединений в дозированных рецептурах предпочтительно находится в интервале от 0,1 до примерно 1000 мг на дозу для орального применения. Обычная дозировка для достижения антипсихозного воздействия варьирует примерно между 0,5 и 10 мг/кг в день и делится на две или три дозы, применяемые орально.
Соединения изобретения могут быть получены различными способами. Эти способы включают:
а) галоидирование соединения формулы (II)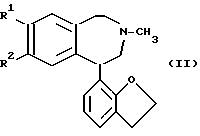 ,
,
где
R1 имеет значения, определенные выше;
R2 - O-C1-4 - алкил или O-CO-C1-4 - алкил, с образованием соединения общей формулы (III)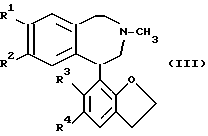 ,
,
где
R1 и R2 имеет значения, определенные выше;
R3 - галоген или H;
R4 - галоген;
и деацилирование или деалкилирование соединения формулы (III) с образованием соединения формулы (I), где R1, R3 и R4 имеют значения, как определены выше;
b) нитрование соединения формулы (II) ,
,
определенной в п. а) с образованием соединения общей формулы (III)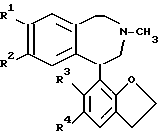 ,
,
где
R1 и R2 имеют значения в п. a);
R3 - H;
R4 - NO2,
и деацилирование или деалкилирование соединения формулы (III) с образованием соединения формулы (I), где R1, R3 и R4 являются такими, как определены выше;
или
с) восстановление или каталитическое гидрирование соединения формулы (IV) ,
,
где
R1 имеет значения, определенные выше;
R2 - O-C1-4- алкил или O-CO-C1-4 - алкил;
R3 - H,
в соединение формулы (III) ,
,
где
R1, R2 и R3 имеют значения как определены выше:
R4 - NH2,
и деацилирование или деалкилирование соединения формулы (III) с образованием соединения формулы (I), где R1, R3 и R4 имеют значения, определенные выше.
Исходные материалы, используемые в синтезе соединений формулы (I), известны, например, из EP 0.200.455.
Соединения изобретения полезны, благодаря их фармакологической активности. В частности эти соединения активны при испытаниях, свидетельствующих об антипсихозном воздействии. Так, соединения формулы (I) испытывались на их связывание с D1-рецептором допамина в гомогенатах полосатого тела крыс с использованием описанной методики (Life Scitnce vol.37, стр. 1971 (1985) P, Andersen et al.); результаты представлены в табл. 1 IC50 показывает сродство исследованных соединений к D1-рецептору допамина.
Ранее упомянутая высокая оральная антидопаминергическая активность соединений изобретения по сравнению с известными соединениями без заявляемых 5' - или 6' - заместителей (EP 200.455) может быть проиллюстрирована вычислением соотношения между способностью ингибировать стереотипизированное поведение мышей при приеме внутрь после метилфенидата (Acta Pharmacol. Tox col. 31, 1972, 488) и ингибированием связывания 3H- CH 23390 ин витро (мера антагонизма D1 - рецептора). Это дает следующие соотношения (табл. 2).
Соединение изобретения вместе с общепринятым вспомогательным средством (адъювантом) носителем или разбавителем, если необходимо в виде фармацевтически приемлемой кислотно-аддитивной соли этого соединения, может находиться в форме фармацевтической композиции и ее единичных доз и в этой форме может применяться в виде твердых веществ таких, как таблетки или капсулы, заполненные твердым веществом, или в жидком виде, например, в виде растворов, суспензий, эмульсий, эликсиров или капсул, заполненных ими; все - для орального приема, в форме свечей для ректального применения или в форме стерильных растворов для инъекций для парентерального использования включая подкожные. Подобные фармацевтические композиции и их формы единичных доз могут включать общепринятые ингредиенты в обычных пропорциях с дополнительными активными соединениями или компонентами или без них, и такие формы единичных доз могут содержать любое подходящее эффективное количество активного ингредиента, облегчающее заболевание центральной нервной системы, соразмерное с назначенным интервалом дневной дозы, которую необходимо принять. Таблетки, содержащие 0,1 - 1000 мг активного ингредиента, или более конкретно 0,5 - 10 мг активного ингредиента на таблетку, соответственно являются подходящим примером формы единичной дозы.
Так, соединения данного изобретения могут использоваться в рецептурах фармацевтических препаратов, например, для орального или парентерального назначения млекопитающим, включая людей, в соответствии с общепринятыми методами галеновой фармации.
Общепринятые эксципиенты представляют такие фармацевтически приемлемые органические или неорганические носители, пригодные для орального или парентерального применения, которые не вступают во вредные взаимодействия с активным соединением.
Примерами подобных носителей являются вода, растворы солей, спирты, полиэтиленгликоли, полигидроксиэтоксилированное касторовое масло, сироп, арахисовое масло, оливковое масло, желатин, лактоза, терраальба, сахароза, агар, пектин, акация (аравийская камедь), амилоза (крахмал), стеарат магния, тальк, кремниевая кислота, стеариновая кислота, моноглицериды и диглицериды жирных кислот, сложные эфиры пентаэрилидон.
Фармацевтические препараты могут стерилизоваться и смешиваться при желании со вспомогательными агентами, такими как смазывающие, предохраняющие агенты, стабилизаторы, смачивающие агенты, эмульгаторы, соли для воздействия на осмотическое давление, буферы и/или красители и другие, не вступающие во вредные взаимодействия с активным соединением.
Для парентерального применения особенно подходящими являются инъецируемые растворы или суспензии, предпочтительно водные растворы с активным соединением, растворенным в полигидроксилированном касторовом масле.
Удобной формой единичной дозировки являются ампулы. Для орального приема особенно подходят таблетки, драже или капсулы с носителем или связующим в виде талька и/или углевода или аналогичных веществ, причем предпочтительно носителем является лактоза и/или кукурузный и/или картофельный крахмал.
Когда можно применять подслащенный носитель, то могут использоваться сироп, эликсир или аналогичные вещества. В общем в более широких интервалах соединения изобретения выпускаются в форме единичных доз, включающих 0,05 - 100 мг на дозу в фармацевтически приемлемом носителе.
Типичная таблетка, которая может быть получена с помощью общепринятых приемов таблетирования, содержит, мг:
Активное соединение - 1,0
Лактоза - 67,8 мг Фарм. Евр.
АвицелR - 31,4
АмберлитR IPP 88 - 1,0
Стеарат магния - 0,25 мг Фарм. Евр.
Следующие примеры иллюстрируют получение новых соединений данного изобретения.
Пример 1. (+)8-Хлор-5-(5-бром-2,3-дигидробензофуран-7-ил)-7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин.
a) (+)8-Хлор-5-(2,3-дигидробензофуран-7-ил)-7- метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин (1,0 г; 2,9 моль) растворяли в уксусной кислоте (10 мл). К раствору при перемешивании добавляли бром (0,20 мл; 4,0 моль) в уксусной кислоте (5 мл) в течение 2 ч. Образовывался осадок. Белое твердое вещество отфильтровывали и промыли диэтиловым эфиром.
Выход (+)8-Хлор-5-(5-бром-2,3-дигидробензофуран-7-ил)-7-метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин. HBr в виде белого кристаллического порошка составлял 1,1 г (75%).
ЯМР (200 Мгц 1H-хим. сдвига в м.д.) свободного основания. С Cl3 в качестве растворителя, ТМС в качестве внутреннего стандарта.
(δ, м.д.): 2,42 (с., 3H); 2,40 - 2,55 (м., 1H); 2,95 (м., 5H);3,28 (т., 2H); 3,70 (с., 3H); 4,43 (д., 1H); 4,58 (т., 2H); 6,38 (с., 1H); 6,98 (д., 1H); 7,18 (с., 1H); 7,26 (д., 1H).
b) (+)8-Хлор-5-(5-бром-2,3-дигидробензофуран-7-ил)-7-метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин (3,9 г; 9,2 ммоль) растворяли в дихлорметане (50 мл). Раствор охлаждали на ледяной бане. К нему при перемешивании добавляли раствор BBr3 концентрации 10 об.% в дихлорметане (14 мл; 14,8 ммоль). Реакционную смесь перемешивали в течение 2 ч, затем нагревали до комнатной температуры. Смесь добавляли к метанолу и концентрировали при пониженном давлении. Добавляли метанол (50 мл), кипятили смесь с обратным холодильником в течение 2 ч, потом перемешивали при комнатной температуре всю ночь. Концентрировали смесь до густого маслянистого вещества. Добавляли метанол (15 мл) и затем при перемешивании добавляли NaOH (0,5 М) до образования осадка. Смесь перемешивали в течение 1 ч, далее охлаждали на ледяной бане. Коричневое твердое вещество отфильтровывали и промывали водой.
Выход соединения, указанного в заголовке, в виде кристаллического коричневатого порошка составил 3,2 г (85%).
ЯМР (200 Мгц 1H-хим. сдвига в м.д.) свободного основания. С Cl3 в качестве растворителя, ТМС в качестве внутреннего стандарта.
(δ, м. д.): 2,25 (т., 1H); 2,35 (с., 3H); 2,90 (м., 5H); 3,22 (т., 2H); 4,35 (д. , 1H); 4,51 (т., 2H); 6,30 (с., 1H); 6,96 (д., 1H); 7,10 (с., 1H); 7,22 (д., 1H).
Пример 2. (+)8-Хлор-5-(2,3-дигидро-5-йодбензофуран-7-ил)-7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин.
a) Йод (4,2 г; 16,3 ммоль) и 32% надуксусную кислоту (10,3 мл, 49 ммоль) перемешивали в уксусной кислоте (50 мл) в течение 15 мин при комнатной температуре. К смеси в течение 1 ч добавляли (+) 8-хлор-5-(2,3-дигидробензофуран-7-ил)-7-метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин (5,6 г; 16,3 ммоль) в уксусной кислоте (50 мл). Смесь перемешивали еще в течение 3 ч. Обрабатывали смесь тиосульфатом натрия и упаривали при пониженном давлении до образования коричневого твердого порошка. Добавляли дихлорметан и органическую фазу промывали водой, водным раствором NaOH, водой и затем сушили над безводным сульфатом магния. Экстракт отфильтровывали и концентрировали до образования коричневого твердого вещества. Продукт очищали с помощью колоночной хроматографии (силикагель: CH2Cl2 - MeOH).
Выход (+)8-Хлор-5-(2,3-дигидро-5-йодбензофуран-7-ил)-7-метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепина в виде кристаллического коричневатого порошка составил 4,8 г (63%).
ЯМР (200 Мгц 1H-хим. сдвига в м.д.) свободного основания. С Cl3 в качестве растворителя, ТМС в качестве внутреннего стандарта.
(δ, м. д.): 2,32 (м., 1H); 2,36 (с., 3H); 2,92 (м., 5H); 3,22 (т., 2H); 4,70 (с., 3H); 4,37 (дд., 1H); 4,52 (т., 2H); 6,35 (с., 1H); 7,13 (широк. с. , 2H); 7,42 (д., 1H).
b) (+)8-Хлор-5-(2,3-дигидро-5-йодбензофуран-7-ил)-метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин (300 мг; 0,65 ммоль) растворителя в дихлорметане (15 мл). К раствору при перемешивании в течение 1 ч добавляли раствор 10% BBr3 в дихлорметане (2,0 мл; 2,1 ммоль). Смесь перемешивали при комнатной температуре еще в течение 15 мин, разбавляли метанолом (25 мл) и концентрировали при пониженном давлении. Добавляли метанол (20 мл) и при перемешивании добавляли NaOH (0,5 М) до pH 7. Смесь перемешивали в течение 1 ч и добавляли воду до образования осадка. Твердое вещество отфильтровывали и промывали водой.
Выход указанного в заголовке соединения составил 207 мг (70%).
ЯМР (200 Мгц 1H-хим. сдвига в м.д.) свободного основания. С Cl3 в качестве растворителя, ТМС в качестве внутреннего стандарта.
(δ, м. д.): 2,28 (т., 1H); 2,38 (с., 3H); 2,90 (м., 5H); 3,22 (т., 2H); 4,37 (д. , 1H); 4,52 (т., 2H); 6,30 (с., 1H); 7,10 (с., 1H); 7,18 (д., 1H) 7,40 (д., 1H).
Пример 3. (+) 8-Хлор-5-(5,6-дихлор-2,3-дигидробензофуран-7-ил)-7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин.
1,65 (0,005 моль) (+) 8-Хлор-5-(2,3-дигидробензофуран-7-ил)-7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепина растворяли в 2,5 мл уксусного ангидрида и оставляли при комнатной температуре на 1 ч. Чистый раствор концентрировали в вакууме и вновь перемешивали с ледяной уксусной кислотой. Затем остаток вновь растворяли в 10 мл уксусной кислоты и к раствору при перемешивании медленно добавляли при комнатной температуре 7,2 мл (0,0055 ммоль) 0,77 - молярного раствора хлора в ледяной уксусной кислоте. Через 1 ч добавляли еще 7,1 мл раствора хлора. Реакционную смесь перемешивали еще в течение 1 ч. Затем добавляли 20 мл этанола и 1 мл концентрированной HCl и кипятили смесь с обратным холодильником в течение 1 ч. После охлаждения раствор разбавляли 20 мл этанола и доводили pH до 8 осторожным добавлением 10%-ного раствора Na2CO3.
Сырой продукт охлаждался спонтанно и собирался с помощью фильтрования. С помощью колоночной хроматографии (неподвижная фаза: C18 двуокись кремния; элюент: сульфат аммония/ацетонитрил 70 : 30; pH 3,3) получали чистое соединение, указанное в заголовке. Т.пл. 224 - 225oC.
1H-ЯМР в CDCl3 (δ, м.д.): 2,23 (т., 1H); 2,40 (с., 3H); 2,73 (дд., 1H); 2,80 - 3,37 (м., 6H); 4,57 (т., 2H); 4,93 (д., 1H); 6,17 (широк. с., 1H); 7,07 ( с., 1H); 7,25 (с., 2H).
Пример 4. (+)8-Хлор-5-(5-хлор-2,3-дигидробензофуран-7-ил)-7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин.
Синтез и обработку осуществляли по методике, описанной в примере 3, с одним отличием: для хлорирования брали 1,1 эквивалент хлора.
Сырой продукт очищали хроматографически, как описано выше, и получали свободное основание в виде кристаллического порошка т. пл. 182 - 186oC.
1H-ЯМР в CDCl3 (δ, м.д.): 2,40 (с., 3H); 2,30 - 3,20 (м., 8H); 3,90 (дд. , 1H); 4,15 (т., 2H); 5,96 (с., 1H); 6,40 (д., 1H); 6,65 (с., 1H); 6,70 (д., 1H).
Пример 5. 8-Хлор-5-(5-нитро-2,3-дигидробензофуран-7-ил)-7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин.
a) 1,50 г (0,00386 моль) 8-Хлор-5-(2,3-дигидробензофуран-7-ил)-7-метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепина растворяли в 20 мл уксусной кислоты. Добавляли 2,80 г (0,0116 моль) тригидрата натрия меди и перемешивали смесь при комнатной температуре в течение 18 ч. Растворитель упаривали в вакууме, остаток обрабатывали водным раствором аммиака и экстрагировали дихлорметаном. Промывали раствор водой и рассолом, концентрировали в вакууме и растворяли остаток в эфире. Нерастворимые примеси отделяли фильтрованием и получали 8-Хлор-5-(5-нитро-2,3-дигидробензофуран-7-ил)-7-метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин в форме гидрохлорида путем добавления избытка раствора HCl в эфире и выделения осадка фильтрацией. Белые кристаллы с т.пл. 251 - 254oC.
1H-ЯМР свободного основания в CDCl3 (δ, м.д.): 2,37 (с., 3H); 2,40 - 3,25 (м. , 6H); 3,34 (т., 2H); 3,70 (с., 3H); 4,45 (д., 1H); 4,73 (т., 2H); 6,36 (с., 1H); 7,20 (с., 1H); 7,86 (д., 1H); 8,03 (д., 1H).
b) 0,220 г (0,00052 моль) 8-хлор-5-(5-нитро-2,3-дигидробензофуран-7-ил)-7-метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепина гидрохлорида растворяли в 10 мл сухого дихлорметана и охлаждали в ледяной бане. К раствору при перемешивании добавляли 5 мл 1 М раствора трихлорида бора в гексане. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали ее в течение 4,5 ч. Затем раствор охлаждали на ледяной бане и проводили гидролиз, осторожно добавляя 15 мл метанола. Образовавшийся кристаллический гидрохлорид соединения, указанного в заголовке, выделяли фильтрацией и сушили. Розоватый кристаллический порошок. Т.пл. 265 - 270oC с разложением.
1H-ЯМР в d6-DMCO (δ, м.д.): 2,80 (д., 3H); 2,85 - 3,85 (м., 8H); 4,75 (т. , 2H); 4,90 (д., 1H); 6,22 (с., 1H); 7,27 (с., 1H); 8,02 (д., 1H); 8,25 (д., 1H); 9,95 (с., 1H); 11,30 (широк.с., 1H).
Пример 6. 8-Хлор-5-(5-амин-2,3-дигидробензофуран-7-ил)-7-гидрокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин.
a) Смесь 2,50 г (0,011 моль) сульфида натрия и 0,60 г (0,011 моль) хлорида алюминия в 10 мл н-пропанола нагревали при перемешивании до температуры кипения с обратным холодильником и по каплям добавляли раствор 0,44 г (0,0011 моль) 8-Хлор-5-(5-нитро-2,3-дигидробензофуран- 7-ил)-7-метокси-3-метил-2,3,4,5-тетрагидро-1H-3-бензазепин; продолжали кипячение с обратным холодильником в течение 16 ч. Растворитель удаляли в вакууме; остаток вновь растворяли в дихлорметане. Промывали раствор 0,1 н. NaOH, водой и рассолом. Упаривание приводило к образованию 8-хлор-5-(5-амино-2,3-дигидробензофуран-7-ил)-7-метокси-3-метил-2,3,4,5-тетрагидро- 1H-3-бензазепина в виде пены.
1H-ЯМР в CDCl3 (δ, м.д.): 2,37 (с., 3H, +м, 1H); 2,75 - 3,30 (м., 7H); 3,35 (широк. с., 2H); 3,68 (с., 3H); 4,33 (т., 1H); 4,46 (т., 2H); 6,21 (д., 1H); 6,43 (с., 1H); 6,53 (д., 1H); 7,12 (с., 1H).
b) 2,8 мл (0,0056 моль) 2 М раствора трихлорида бора в дихлорметане добавляли при перемешивании к охлажденному до ледяной температуры раствору 200 мг (0,00056 моль) продукт стадии a) в 10 мл дихлорметана. Реакционную смесь перемешивали в течение 8 ч при комнатной температуре и затем концентрировали в вакууме. Остаток вновь растворяли в эфире, промывали насыщенным раствором NaHCO3, водой и рассолом и сушили над Na2O4. Осаждение раствором HCl в эфире приводило к образованию указанного в заголовке соединения в виде гидрохлорида. Т.пл. 241 - 244oC.
1H-ЯМР в d6-DMCO (δ, м.д.): 2,80 (д., 3H); 3,00 (м., 2H); 3,31 (т., 2H); 3,35 - 3,67 (м., 4H); 4,60 (т., 2H); 4,77 (д., 1H); 6,23 (с., 1H); 6,98 (с., 1H); 7,26 (с. , 1H); 7,33 (с., 1H); 10,00 (с., 1H); 10,25 (широк. с., 2H); 10,00 (с., 1H).

Описываются 2,3,4,5-тетрагидро-1Н-3-бензазепины общей формулы (I), в которой R1 - Cl или Br; R3 - водород или галоген, R4 - галоген, NO2 или NH2. Способы их получения, а также фармацевтическая композиция на их основе, обладающая антидонаминергической активностью, например для связывания D1 рецептора при лечении заболевания нервной системы, а также способ связывания D1 рецептора 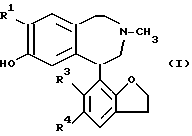 6 с. и 6 з.п.ф-лы, 2 табл.
6 с. и 6 з.п.ф-лы, 2 табл.

в которой R1 - хлор или бром;
R3 - водород или галоген;
R4 - галоген, нитро- или аминогруппа,
и их фармацевтически приемлемые кислотно-аддитивные соли.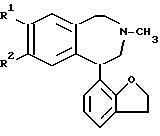
в которой R1 имеет указанные значения;
R2 - O-C1 - C4-алкил или O-CO-C1 - C4-алкил, подвергают галоидированию и полученное таким образом соединение общей формулы III
в которой R1 - R4 имеют указанные значения,
подвергают деацилированию или деалкилированию с получением соединения формулы I, определенного выше.
в которой R1 - R4 имеют указанные значения;
которое подвергают деацилированию или деалкилированию с получением соединения формулы I, определенного выше.
в которой R1 и R3 имеют указанные значения;
R2 - O-C1 - C4-алкил или O-CO-C1 - C4-алкил,
подвергают каталитическому гидрированию с получением соединения общей формулы III
в которой R1, R2 и R3 имеют указанные значения;
R4 - NH2,
которое подвергают деацилированию или деалкилированию с получением соединения формулы I, определенного выше.
| EP, 0200455 А2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, 0383247 А1, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| SU, 1149877 А, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

