Настоящее изобретение относится к терапевтически активным гетероциклическим соединениям, а также способу их получения, фармацевтическим композициям, включающим данные соединения, и кроме того, способу лечения с применением указанных соединений.
В частности, настоящее изобретение относится к производным [1,2,4]триазол[4,3-а]хиноксалинона, которые полезны для лечения любого показания, вызванного гиперактивностью возбуждающих аминокислот.
Различные родственные соединения известны из предшествующего уровня техники.
Так, в патенте EP-A-0040401, между прочим, описывают семейство триазолхиноксалин-4-онов, замещенных по триазольному кольцу, например, алкильной, ацильной или карбалкоксильной группой. Эти соединения заявлены как обладающие соответствующей антигипертензивной активностью.
В патенте США N 5153196 раскрыты некоторые антагонисты рецепторов возбуждающих аминокислот и способы их применения. Эти соединения, между прочим, соответствуют триазолхиноксалинонам, имеющим один заместитель, такой как H, алкил, ароматический или CF3 в кольце триазола.
Кроме того, в опубликованной международной заявке N WO 93/20077 раскрыты конденсированные производные хиноксалинона, произвольно замещенные в кольце триазола низшим алкилом, который может быть замещен моно- или ди (низший алкил) амином.
L-глутаминовая кислота, L-аспарагиновая кислота и ряд других близкородственных аминокислот обладают способностью активировать нейроны центральной нервной системы (ЦНС). Биохимические, электрофизиологические и фармакологические исследования подтвердили это и продемонстрировали, что кислые аминокислоты являются медиаторами для большого числа возбуждающих нейронов ЦНС млекопитающих.
Воздействие глутаминовой кислоты, опосредующей передачу нервного возбуждения, считали важным методом при лечении неврологических и психических заболеваний. Так, известные антагонисты возбуждающих аминокислот проявляют сильные анксиолитические (Stephens et al., Psychopharmacology 90, 143-147, 1985), противосудорожные свойства (Croucher et al., Science 216, 899-901, 1982) и свойства мышечного релаксанта (Turski et al., Neurosci Lett. 53, 321-326, 1985).
Предполагалось, что внеклеточное накопление возбуждающих аминокислот, с последующей чрезмерной стимуляцией нейронов, может объяснять деградацию нервных клеток, наблюдаемую при неврологических расстройствах, таких как амиотрофический латеральный (боковой) склероз, паркинсонизм, болезнь Альцгеймера, болезнь Гентингтона, эпилепсия, а также умственная неполноценность и недостаточная двигательная активность, возникающие после состояний ишемии мозга, аноксии и гипогликемии или травмы головы и спинного мозга (McGeer et al. , Nature 263, 517-519, 1976; Simon et al., Science 226, 850-852, 1984; Wieloch, Science 230, 681-683, 1985; Faden et al., Science 244, 798-800, 1989, Turski et al., Nature 349, 414-418, 1991).
Другими возможными показаниями являются психоз, мышечная ригидность, рвота и аналгезия.
Возбуждающие аминокислоты осуществляют свое действие через специфические рецепторы, локализованные постсинаптически или пресинаптически. Такие рецепторы в настоящее время удачно разделили на три группы на основании электрофизиологических и нейрохимических данных: 1 группа представлена рецепторами NMDA (N-метил-D-аспартат), 2 группа представлена AMPA-рецепторами и 3 группа представлена каинатными рецепторами. L-глутаминовая кислота и L-аспарагиновая кислота, вероятно, активируют все вышеуказанные типы рецепторов возбуждающих аминокислот и, вероятно также, другие типы рецепторов.
Вышеприведенная классификация возбуждающих аминокислотных рецепторов на NMDA, AMPA и каинатные рецепторы изначально основаны на следующих электрофизиологических и нейрохимических данных.
1) N-метил-D-аспарагиновые (NMDA) рецепторы проявляют высокую избирательность к возбуждению NMDA. Иботеновая кислота, L-гомоцистеиновая кислота, D-глутаминовая кислота и транс-2,3-пиперидиндикарбоновая кислота (транс-2,3-PDA) сильно влияют на небольшую агонистическую активность этих рецепторов. Наиболее сильными и избирательными антагонистами являются D-изомеры 2-амино-5-фосфонокарбоновых кислот, например, 2-амино-5-фосфоновалериановая кислота и 3-[(±)-2-карбокси-пиперазин-4-ил] -пропил-1-фосфоновая кислота (CPP), а D-изомеры длинноцепочечных 2-аминодикарбоновых кислот (например, D-2-аминоадипиновая кислота) и длинноцепочечных диаминодикарбоновых кислот (например, диаминопимелиновая кислота) проявляют умеренную антагонистическую активность. Синаптические ответы, индуцированные NMDA, интенсивно изучали в ЦНС млекопитающих, особенно в спинном мозге (J.Davies et al., J.Physiol. 297, 621-635, 1979) и было показано, что эти ответы сильно ингибировались ионами Mg2+.
2) AMPA-рецепторы избирательно активируются AMPA (2-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислотой), другими сильными агонистами, представленными хискваленовой кислотой и L-глутаминовой кислотой. Диэтиловый эфир глутаминовой кислоты (GDEE) представляет собой селективный, но очень слабый антагонист этого рецептора. AMPA-рецепторы относительно нечувствительны к ионам Mg2+.
Долгое время полагали, что высвобождение глутамата играет главную роль в гибели нейронов при церебральной ишемии (Benveniste, Н. Et al., J.Neurochem. 43, 1369-1374, 1984). Хорошо известно, что приток ионов Ca2+, вызванный NMDA-рецептором, является важным механизмом при ишемической утрате нервных клеток. Рецептор NMDA, не присоединяющий ионофор, становится непроницаемым для кальция. Однако возбуждение через коллатерали Шеффера осуществляется в области СА1 не рецепторами NMDA, и этот факт является важным для хода событий в постишемический период. Недавние исследования показали, что селективные AMPA-антагонисты обладают нейрозащитным действием при глобальной ишемии у песчанки даже при их даче спустя несколько часов после реперфузии (Sheardown et al., Science 247, 571-574, 1990).
Следовательно, антагонисты AMPA пригодны для лечения церебральной ишемии.
3) Каинат-рецепторы. Ответы возбуждения на каиновую кислоту относительно нечувствительны к антагонистам NMDA и GDEE и поэтому предположили, что каиновая кислота активирует третий подкласс рецепторов кислых аминокислот. Некоторые лактонизированные производные каиновой кислоты представляют собой селективные антагонисты (O. Goldberg et al., Neurosci. Lett. 23, 187-191, 1981) и дипептид 3-глутамилглицин также проявляет некоторую селективность в отношении каинатных рецепторов. Ионы Ca2+, но не Mg2+, являются сильным ингибитором связывания каиновой кислоты.
Сродство вещества по одному или большему числу разных типов рецепторов возбуждающих аминокислот исследовали в простых экспериментах по связыванию. В сущности способ включает в себя инкубацию отдельно взятого лиганда с введенной радиоактивной меткой и отдельного специфического вещества, чтобы исследовать гомогенат мозга, содержащий рецептор. Измерение заполнения рецептора осуществляли путем определения связывания радиоактивной метки с гомогенатом и вычитания неспецифического связывания.
Связывание рецептора AMPA исследовали, применяя 3H-AMPA в качестве радиолиганда.
Влияние аналогов глутаминовой кислоты на вторичные эффекты взаимодействия глутаматного рецептора можно исследовать ин витро, благодаря использованию феномена распространяющейся депрессии в сетчатке цыпленка. Такие эксперименты позволяют получать сведения об эффективности (агонист/антагонист) испытываемых веществ. Их необходимо рассматривать в сопоставлении с результатами исследований связывания, в которых получали данные только о сродстве соединений к рецептору.
Нами установлено, что соединения настоящего изобретения обладают сродством с рецепторами AMPA и представляют собой антагонисты, в соответствии с этим типом рецептора, что делает их полезными для лечения любого из многочисленных показаний, вызванных гиперактивностью возбуждающих аминокислот, разрушающих, главным образом, нейроны, как это наблюдается при амиотрофическом боковом склерозе, хорее Генингтона, болезни Паркинсона, эпилепсии и старческом слабоумии или психических и двигательных дисфункциях, обнаруживающихся после состояний ишемии мозга, кислородной недостаточности, гипогликемии и травме головного и спинного мозга. Другими возможными показаниями являются психоз, мышечная ригидность, рвота, острое и хроническое воспалительное заболевание и аналгезия.
Соединения настоящего изобретения представлены общей формулой I.
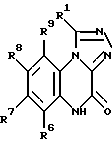
в которой R1 представляет собой POX'X'' или прямой или разветвленный C1-6-алкил, замещенный COX' или POX'X'', где X' и X'' независимо представляют гидрокси- или C1-6-алкокси-группу, а
R6, R7, R8 и R9 независимо представляют собой водород; C1-6-алкил; галоген; NH2; NO2, CN; CF3; SO2NY'Y''; COZ', в которой Z' представляет собой NY'Y'' или C1-6-алкил, а Y' и Y'' независимо представляют водород или C1-6-алкил; триазолил; имидазолил; пиперидин; пиперазинил; морфолино; тиоморфолино, кольца которых необязательно замещены одним или более фенилом или C1-6-алкилом, причем фенил необязательно замещен C1-6-алкокси-группой, и их фармацевтически приемлемые соли.
Термин "C1-6-алкил", как он используется здесь, относится к прямой или разветвленной насыщенной углеводородной цепи, имеющей 1-6 углеродных атомов, таких как метил, этил, п-пропил, изопропил, п-бутил, 2-бутил, третбутил, 3-пентил, неопентил или п-гексил.
Термин "C1-6-алкокси", как он используется здесь, сам по себе или в сочетании, относится к моновалентному заместителю, включающему в себя C1-6-алкильную группу, связанную через эфирный кислород, имеющему свою свободную валентную связь при эфирном кислороде, например, метокси, этокси, пропокси, изопропокси, циклопропилметокси, бутокси, пентокси.
Термин "галоген", как он используется здесь, означает фтор, хлор, бром и иод.
В предпочтительном варианте осуществления настоящего изобретения R1 представляет C1-6-алкил, замещенный группой COX' или POX'X''.
В другом предпочтительном варианте осуществления настоящего изобретения R6, R7, R8 и R9 независимо представляют водород, хлор; NO2, NC; CF3; пиперидино; морфолино; тиоморфолино; пиперазинил; пиперазинил, замещенный на метил, фенил или метоксифенил; триазолил, замещенный на метил; имидазолил, двузамещенный на метил, этил, фенил.
Еще в одном предпочтительном варианте осуществления настоящего изобретения R6 и R9 представляют водород.
Предпочтительными соединениями настоящего изобретения являются: 1-(этокси-гидрокси-фосфорилметил)-8-(4-метил-2-фенил- 1H-имидазол-1-ил)-7-трифторметил[2,4-1]триазоло[4,3-а] хиноксалин-4(5H)-он;
8-(4-метил-2-фенил-1H-имидазол-1-ил)-1-фосфонометил-7- трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он;
1-(этокси-гидрокси-фосфорилметил)-8-(2-этил-4- метил-1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-a] хиноксалин-4(5H)-он;
8-(2-этил-4-метил-1H-имидазол-1-ил)-1- фосфонометил-7-трифторметил[1,2,4]триазоло[4,3-а] хиноксалин-4(5H)-он;
8-морфолино-1-фосфонометил-7-трифторметил [1,2,4]триазоло[4,3-а]хиноксалин-4(5H)-он;
8-морфолино-1-(1-фосфоноэтил)-7-трифторметил[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
8-пиперидино-1-фосфонометил-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
1-(2-этоксикарбонилэтил)-8-морфолино-7-трифторметил[1,2,4] триазоло [4,3-a]хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-8-морфолино-7-трифторметил[1,2,4] триазоло [4,3-а] хиноксалин-4(5H)-он;
1-(этокси-гидрокси-фосфорилметил)-8-(1H-имидазол-1-ил)- 7-трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он;
8-(1H-имидазол-1-ил)-1-(фосфонометил-7-трифторметил [1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
1-(этокси-гидрокси-фосфорилметил)-8-(2-изопропил-1H-имидазол-1-ил)-7- трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он;
8-(2-изопропил-1H-имидазол-1-ил)-1-фосфонометил-7- трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он.
Другими предпочтительными соединениями настоящего изобретения являются:
8-(2,4-диметил-1H-имидазол-1-ил)-1-фосфонометил-7- трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-циано-8-(2,4-диметил-1H-имидазол-1-ил)-1-фосфонометил [1,2,4] триазоло[4,3-а]хиноксалин-4(5H)-он;
8-(2,4-диметил-1H-имидазол-1-ил)-7-нитро-1-фосфонометил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-циано-8-(2-этил-4-метил-1H-имидазол- 1-ил)-1-фосфонометил[1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он;
7-циано-8-морфолино-1-фосфонометил[1,2,4] триазоло [4,3-a] хиноксалин-4(5H)-он;
8-морфолино-7-нитро-1-фосфонометил[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
7-циано-1-фосфонометил-8-тиоморфолино[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
1-фосфонометил-8-тиоморфолино-7- трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-циано-1-фосфонометил-8-пиперидино[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
1-фосфонометил-8-(пиперазин-1-ил)-7- трифторметил[1,2,4]триазоло[4,3-a] хиноксалин-4(5H)-он;
7-циано-1-фосфонометил-8-(пиперазин-1-ил)[1,2,4]триазоло[4,3-a] хиноксалин-4(5H)-он;
8-(4-пиперазин-1-ил)-1-фосфонометил-7- трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-циано-8-(4-фенилпиперазин-1-ил)-1-фосфонометил[1,2,4] триазоло [4,3-a] хиноксалин-4(5H)-он;
8-(4-(3-метоксифенил)пиперазин-1-ил)-1-фосфонометил-7- трифторметил[1,2,4]триазоло[4,3-а]хиноксалин-4(5H)-он;
8-(4-(4-метоксифенил)пиперазин-1-ил)-1-фосфонометил-7- трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он;
8-(2,4-диметил-1H-имидазол-1-ил)-1-фосфоноэтил-7- трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-хлоро-8-(2,4-диметил-1H-имидазол-1-ил)-1-фосфонометил[1,2,4] триазоло[4,3-а]хиноксалин-4(5H)-он;
8-(3,5-диметил-1,2,4-триазол-1-ил)-1-фосфонометил-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
8-(4-метилпиперазин-1-ил)-1-фосфонометил-7-трифторметил [1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-8-(2,4-диметил-1H- имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-a] хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-8-(2-этил-4-метил-1H- имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-a] хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-7-циано-8-(2,4- диметил-1H-имидазол-1-ил[1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-8-(4-фенилпиперазин-1-ил)-7- трифторметил[1,2,4]триазоло[4,3-a)хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-8-(2,4-диметил-1H-имидазол-1-ил)-7-нитро [1,2,4]триазоло[4,3-а]хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-7-циано-8-морфолино[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-8-морфолино-7-нитро [1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-8-(4-метилпиперазин-1-ил)-7-трифторметил [1,2,4] триазоло[4,3-а]хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-7-хлоро-8-(2,4-диметил-1H-имидазол-1-ил) [1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-8-(4-(4- метоксифенил)пиперазин-1-ил)-7-трифторметил[1,2,4]триазоло [4,3-а]хиноксалин-4(5H)-он;
1-(2-карбоксиэтил)-8-пиперидино-7-трифторметил[1,2,4]триазоло [4,3-a]хиноксалин-4(5H)-он,
1-фосфонометил-8-(1H-1,2,4-триазол-1-ил)-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
8-(4-метил-1H-имидазол-1-ил)1-фосфонометил-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
8-(2-метил-1H-имидазол-1-ил)-1-фосфонометил-7- трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он;
8-(2-фенил-1H-имидазол-1-ил)-1-фосфонометил-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-циано-8-(1H-имидазол-1-ил)-1-фосфонометил[1,2,4] триазоло[4,3-а] хиноксалин-4(5H)-он;
8-(1H-имидазол-1-ил)-1-(1-фосфоноэтил)- 7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
8-(2-этил-1H-имидазол-1-ил)-1-фосфонометил-7-трифторметил[1,2,4] триазоло[4,3-а]хиноксалин-4(5H)-он;
1-фосфонометил-8-(2-н-пропил-1H-имидазол-1-ил)-7-трифторметил[1,2,4] триазоло[4,3-а]хиноксалин-4(5H)-он;
8-(1H-имидазол-1-ил)-7-нитро-1-фосфонометил[1,2,4] триазоло[4,3-а] хиноксалин-4(5H)-он;
8-(1H-имидазол-1-ил)-7-нитро-1-(1-фосфоноэтил)[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
8-(2-метил-1H-имидазол-1-ил)-7-нитро-1-фосфонометил[1,2,4] триазоло [4,3-а]хиноксалин-4(5H)-он;
7-циано-8-(2-метил-1H-имидазол-1-ил)-1- фосфонометил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-циано-8-(2-фенил-1H-имидазол-1-ил)-фосфонометил[1,2,4]триазоло [4,3-а] хиноксалин-4(5H)-он;
8-(4-фенил-1H-имидазол-1-ил)-1-фосфонометил-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-циано-8-(2-изопропил-1H-имидазол-1-ил)-1-фосфонометил [1,2,4] триазоло[4,3-а]хиноксалин-4(5H)-он;
7-циано-8-(2-этил-1H-имидазол-1-ил)-1-фосфонометил[1,2,4] триазоло [4,3-a]хиноксалин-4(5H)-он;
7-циано-8-(1H-имидазол-1-ил)-1-(1- фосфоноэтил)[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
7-(1H-имидазол-1-ил)-1-фосфонометил-8-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-(2-метил-1H-имидазол-1-ил)-1-фосфонометил-8-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
8-циано-7-(1H-имидазол-1-ил)-1-фосфонометил[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
7-(1H-имидазол-1-ил)-8-нитро-1-фосфонометил[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
1-фосфонометил-7-(1H-1,2,4-триазол-1-ил)-8-трифторметил[1,2,4] триазоло[4,3-а]хиноксалин-4(5H)-он;
7-(1H-имидазол-1-ил)-1-(1-фосфоноэтил)-8-трифторметил [1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
7-циано-8-(4-метил-1H-имидазол-1-ил)-1-фосфонометил[1,2,4] триазоло[4,3-а]хиноксалин-4(5H)-он;
7-циано-8-(2-н-пропил-1H-имидазол-1-ил)-1-фосфонометил[1,2,4] триазоло [4,3-a]хиноксалин-4(5H)-он;
7-циано-8-(4-фенил-1H-имидазол-1-ил)-1-фосфонометил[1,2,4] триазоло [4,3-a]хиноксалин-4(5H)-он;
7-циано-1-фосфонометил-8-(1H-1,2,4-триазол-1-ил)[1,2,4] триазоло [4,3-a] хиноксалин-4(5H)-он;
7-нитро-1-фосфонометил-8-(1H-1,2,4-триазол-1-ил)[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он;
7-хлоро-8-(1H-имидазол-1-ил)-1-фосфонометил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он;
8-хлоро-7-(1H-имидазол-1-ил)-1-фосфонометил[1,2,4] триазоло [4,3-a]хиноксалин-4(5H)-он.
Данные соединения настоящего изобретения могут быть представлены в различных таутомерных формах. Поэтому настоящее изобретение включает в себя все такие таутомерные формы.
Следующий вариант осуществления настоящего изобретения представляет фармацевтически приемлемые соли, производные [1,2,4]триазоло[4,3-а]хиноксалинона формулы I. Такие соли включают в себя соли, полученные из неорганических и органических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, уксусная кислота, серная кислота, азотная кислота, щавелевая кислота, фумаровая кислота, винная кислота и др. Другие соли включают в себя соли щелочных металлов, такие как натриевые или калиевые соли; соли щелочноземельных металлов, такие как кальциевые или магниевые соли; и аммонийные соли.
Кроме того, в соответствии с другой целью настоящее изобретение относится к соединению общей формулы (I) или его фармацевтически приемлемой соли для применения в качестве лекарственного средства, предпочтительно для применения в качестве лекарственного средства для лечения показания, связанного с гиперактивностью возбуждающих нейромедиаторов, в частности, рецепторов AMPA.
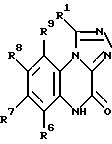
Настоящее изобретение относится также к способу получения вышеуказанных соединений. Представленные соединения формулы I получают: а) алкилированием соединения, имеющего формулу II: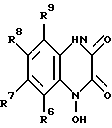 (II)
(II)
в которой R6, R7, R8 и R9 имеют значения, определенные выше, бензилгалогенидом с образованием соединения формулы III: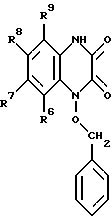 (III)
(III)
в которой R6, R7, R8 и R9 имеют значения, определенные выше, и галогенированием соединения с образованием соединения формулы IV: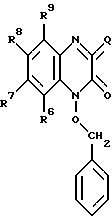 (IV)
(IV)
в которой R6, R7, R8 и R9 имеют значения, определенные выше, a Q представляет Br, Cl или I; взаимодействием данного соединения с гидразином с образованием соединения формулы V: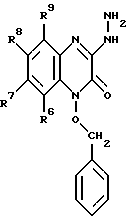 (V)
(V)
в которой R6, R7, R8 и R9 имеют значения, определенные выше, и ацилированием данного соединения ацилхлоридом общей формулы VI:
R1-COCl, (VI)
в которой R1 имеет значение, как определили выше, для соединения общей формулы I, в которой X' и X'' представляют C1-6-алкокси, с образованием соединения формулы VII: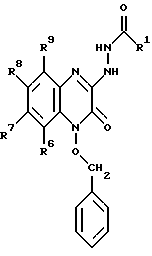 (VII)
(VII)
в которой R1, R6, R7, R8 и R9 имеют значения, определенные выше, с последующим гидрогенолизом данного соединения с образованием соединения формулы VIII: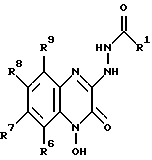 (VIII)
(VIII)
в которой R1, R6, R7, R8 и R9 имеют значения, определенные выше, а последующей термической циклизацией и одновременной дезоксигенизацией с образованием соединения формулы I, в которой X' и X'' независимо представляют гидрокси- или C1-6-алкокси-группу; или
b) взаимодействием соединения, имеющего формулу IX: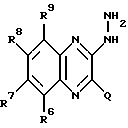 (IX)
(IX)
в которой R6, R7, R8 и R9 имеют значения, определенные выше, а Q представляет собой Br, Cl или I, с соединением общей формулы VI:
R1-COCl, (VI)
в которой R1 имеет значение, как показано выше для соединения общей формула I, в которой X' и X'' представляют C1-6-алкокси-группу, с образованием соединения формулы XI: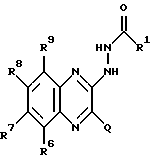 (XI)
(XI)
в которой R1, R6, R7, R8 и R9 имеют значения, определенные выше, а Q представляет собой Br, Cl или I, и затем либо циклизацией с последующим гидролизом, либо одновременно циклизацией и гидролизом с образованием соединения формулы I, в которой X' и X'' независимо представляют гидрокси- или C1-6-алкокси-группу; или
с) замещением соединения формулы XII: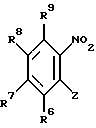 (XII)
(XII)
в которой R6, R7, R8 и R9 имеют значения, определенные выше, а Z представляет собой галоген, либо C1-6-алкокси-группу, моно-, ди- или триметокси-замещенную бензиламином, с образованием соединения формулы XIII: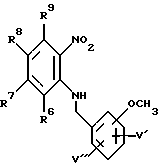 (XIII)
(XIII)
в которой R6, R7, R8 и R9 имеют значения, определенные выше, а V' и V'' независимо представляют водород или метокси-группу, и взаимодействием данного соединения с этилоксалилхлоридом с образованием соединения формулы XIV: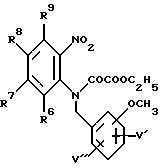 (XIV)
(XIV)
в которой R6, R7, R8 и R9 имеют значения, определенные выше, а V' и V'' независимо представляют водород или метокси-группу, и затем либо гидрированием, с образованием промежуточного циклизованного N-гидрокси-соединения и последующим дезоксигенированием, либо циклизацией гидрированием с образованием соединения формулы XV: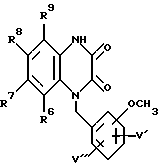 (XV)
(XV)
в которой R6, R7, R8 и R9 имеют значения, определенные выше, а V' и V'' независимо представляют водород или метокси-группу, с последующим галогенированием полученного соединения формулы XV, взаимодействием полученного таким образом соединения с гидразином с последующим ацилированием ацилхлоридом общей формулы VI, как показано выше, и затем циклизацией с образованием соединения формулы XVI: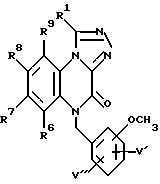 (XVI)
(XVI)
в которой R1, R6, R7, R8 и R9 имеют значения, определенные выше, а V' и V'' независимо представляют водород или метокси-группу, и гидролизом с образованием соединения формулы I, в которой X' и X'' независимо представляют водород или C1-6-алкокси-группу, или
d) гидролизом соединения формулы I, в которой X' и X'' представляют C1-6-алкокси-группу, водным основанием с образованием соединения формулы I, в которой X' представляет гидрокси-группу, а X'' представляет C1-6-алкоксигруппу, или
е) взаимодействием соединения формулы I, в которой X' представляет гидрокси-группу или C1-6-алкокси-группу, а X'' представляет C1-6-алкоксигруппу, с галотриметилсиланом с образованием соединения формулы I, в которой X' и X'' представляют гидрокси-группу.
Фармацевтически приемлемые соли можно получить в соответствии со стандартными методиками, в результате обработки соединения формулы I соответствующими кислотами или основаниями.
Исходные материалы, получение которых здесь не представили, являются либо известными соединениями (например, из международной заявки N PCT-DK94/00170), либо соединениями, которые можно получить по аналогии с получением известных соединений, либо по аналогии с известными способами.
Фармакологические свойства соединений настоящего изобретения можно проиллюстрировать путем определения их способности к вытеснению радиоактивно меченной 2-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (AMPA) из рецепторов типа AMPA. Антагонистические свойства данных соединений показаны по их способности противодействовать хискваленовой кислоте, стимулирующей распространяющуюся депрессию в сетчатке цыпленка.
Активность замещения соединениями можно показать в результате определения значения IC50, которое отражает концентрацию соединения (мкМ), которая вызывает 50% замещение специфически связанной 3H-AMPA.
Этот антагонизм измеряли путем определения значения IC50, отражающего концентрацию, при которой получают максимальное 50% ингибирование хискваленовой кислоты, стимулирующей распространяющуюся депрессию в сетчатке цыпленка.
Связывание 3H-AMPA (Опыт 1)
500 мкл оттаянного гомогената церебральных кортикальных мембран крысы в трис-HCl (30 мМ), CaCl2 (2,5 мМ) и KSCN (100 мМ) рН 7,1 инкубировали при 0oC в течение 30 мин с 25 мкл 3H-AMPA (конечная концентрация 5 нМ), испытуемым соединением и буфером. Неспецифическое связывание определяли путем инкубирования с L-глутаминовой кислотой (конечная концентрация 600 мкМ). Реакцию связывания прекращали добавлением 5 мл ледяного буфера с последующей фильтрацией через фильтры из стекловолокна Whatman GF/C и промывкой 2х5 мл ледяным буфером. Связавшуюся радиоактивную метку измеряли на сцинтилляционном счетчике. IC50 определяли с помощью анализа Hill'а не менее чем в четырех концентрациях испытуемого соединения.
Распространяющаяся депрессия (Опыт 2)
Цыплят (в возрасте 3-10 дней) обескровливали, глаза, энуклеировали и рассекали вдоль экваториальной плоскости. После удаления передней камеры и стекловидного тела, заднюю камеру каждого глаза помещали в небольшую чашку Петри, содержащую физиологический солевой раствор (P.S.S.) следующего состава (в мМ): NaCl (100), KCl (6,0), CaCl2 (1,0), MgSO4 (1,0), NaHCO3 (30), NaH2PO4 (1,0), глюкоза (20).
Раствор насыщали O2 до 100% и держали при температуре 26oC.
Вначале глаза инкубировали в обычном P.S.S. в течение 15-30 мин, а затем переносили в P.S.S., содержащий хисквалат (1 мкг/мл). В этом "стимулирующем растворе" S. D. S. начинались спонтанно, обычно от края сетчатки, и легко наблюдалась невооруженным глазом. Измеряли время, необходимое для запуска S. D. в каждом глазу.
После последующих 15 мин инкубации в обычном P.S.S. глаза переносили в обычный P. S.S., содержащий испытуемое соединение, и инкубировали в течение 15 мин. Затем глаза переносили в "стимулирующий раствор", содержащий ту же концентрацию испытуемого соединения. Вновь измеряли время, необходимое для запуска S. D. в каждом глазу. Затем глаза возвращали в обычный P.S.S. и спустя 15 мин снова измеряли время, необходимое для запуска S.D. с тем, чтобы определить степень восстановления после воздействия соответствующего препарата.
Увеличение времени, необходимого для запуска S. D. , по сравнению с контролем более чем на 30 с считали 100% ингибированием S.D. Поэтому действие лекарственного средства выражали как процентную долю максимального ответа, полученную для данной дозы. Опытное значение расценивали поэтому как концентрацию испытуемого вещества, которое вызывает максимальное 50% ингибирование (IC50).
Результаты опытов, полученные при тестировании некоторых соединений настоящего изобретения, показаны в нижеследующей таблице 1.
Полученные фармацевтические препараты, включающие в себя соединения настоящего изобретения, можно вводить людям или животным пероральным, ректальным или парентеральным способом.
Эффективное количество активного соединения или фармацевтически приемлемой его соли можно определить в соответствии с обычными показателями, такими как происхождение и серьезность состояния и вес млекопитающего, нуждающегося в лечении.
Традиционные эксипиенты представляют собой такие фармацевтически приемлемые органические или неорганические вещества, в качестве носителей, которые пригодны для парентерального применения или через кишечник, которые являются нейтральными по отношению к активным соединениям.
Примерами таких носителей являются вода, растворы солей, спирты, полиэтиленгликоли, полигидроксиэтоксилированное касторовое масло, желатин, лактоза, амилоза, магния стеарат, тальк, кремниевая кислота, моноглицериды и диглицериды жирных кислот, пентаэритритные эфиры жирных кислот, гидроксиметилцеллюлоза и поливинилпирролидон.
При необходимости фармацевтические препараты можно стерилизовать и смешивать с дополнительными агентами, такими как увлажнители, консерванты, стабилизаторы, смачивающие агенты, эмульгаторы, соли, влияющие на осмотическое давление, буферы и/или красители и им подобные, которые при взаимодействии с активными соединениями не разрушают их.
Инъецируемые растворы или суспензии, предпочтительно водные растворы активного соединения, растворенные в полигидроксилированном касторовом масле, особенно подходят для парентерального введения.
Ампулы традиционно представляют собой единичные дозированные формы.
Таблетки, драже или капсулы, содержащие тальк и/или носитель или связывающее вещество, или им подобные, особенно пригодны для перорального введения. Носитель предпочтительно представляет собой лактозу и/или кукурузный крахмал, и/или картофельный крахмал.
Сироп, эликсир или им подобные можно использовать в случаях, когда используют или необходимо использовать подслащенный наполнитель.
Вообще, соединения настоящего изобретения приготовляются в виде единичной дозы, включающей в себя по 0,5-1000 мг активного ингредиента в фармацевтически приемлемом носителе, или вместе с ним, в расчете на единичную дозу.
В соответствии с настоящим изобретением, при введении пациентам, например - людям, в качестве лекарственного средства, доза соединения по изобретению составляет 1-500 мг/день, например около 50-100 мг, в расчете на дозу.
Обычная таблетка, которую можно изготовить по традиционной технологии таблетирования, содержит:
Ядро:
Активное соединение (в виде свободного соединения или его соли) - 100 мг
Коллоидная двуокись кремния (Aerosil®) - 1,5 мг
Целлюлоза, микрокрист (Avicel®) - 70 мг
Модифицированная целлюлозная камедь (Ac-Di-Sol®) - 7,5 мг
Магния стеарат - 1 мг
Оболочка
НРМС - приблиз. 9 мг
*Mywacett® 9-40Т - приблиз. 0,9 мг
* Ацилированный моноглицерид, используемый в качестве пластификатора для оболочечного пленочного покрытия.
Свободные соединения настоящего изобретения, которые образуют соли щелочных металлов или щелочноземельных металлов, можно использовать в такой солевой форме. Такие соли щелочных металлов или щелочноземельных металлов обыкновенно образуются путем взаимодействия соединения с эквивалентным количеством или избытком избранного щелочного металла или щелочноземельного металла в виде гидроокиси, часто и соответственно в результате смешивания в присутствии нейтрального растворителя, из которого соль можно осадить или извлечь другим традиционным способом, например выпариванием. Введение соединения настоящего изобретения часто является предпочтительным в виде фармацевтически приемлемых водорастворимой соли щелочного металла или щелочноземельного металла пероральным, ректальным или парентеральным путем в форме фармацевтического препарата, в котором он присутствует вместе с фармацевтически приемлемой жидкостью или твердым носителем или разбавителем.
Соединения настоящего изобретения, вместе с обычным адъювантом, носителем или разбавителем можно представить в форме фармацевтических препаратов и его единичных доз, и в такой форме можно использовать в твердом виде, например в виде таблеток или заполненных капсул, или в жидком виде, как например, в виде растворов, суспензий, эмульсий, эликсиров или наполненных ими капсул, - все для перорального применения, в форме суппозиториев для ректального введения; или в форме стерильных растворов для инъекций при парентеральном применении (в том числе подкожном). Такой фармацевтический препарат и его единичные дозированные формы могут включать в себя обычные ингредиенты в обычных пропорциях, с дополнительными активными соединениями или их составными частями или без них, и такие единичные дозированные формы могут содержать любое подходящее эффективное количество активного ингредиента, антагонистичного AMPA, соответствующего назначенной ежедневной, в пределах колебаний, применяемой дозе. Таблетки, содержащие 1-500 мг активного ингредиента, или более точно - 10-200 мг на таблетку, соответствуют подходящей единичной дозированной форме.
Благодаря высокому уровню антагонистической активности, по отношению к AMPA, и низкой токсичности, что создает наиболее благоприятный терапевтический индекс, соединения настоящего изобретения можно ввести субъекту, например в организм животного, нуждающемуся в таком лечении, ликвидации, облегчении или уменьшении интенсивности показания, чувствительного к изменению состояния рецептора AMPA, например склероза, паркинсонизма, болезни Альцгеймера, болезни Гентингтона, эпилепсии; дефектах, вызванных ишемией, аноксии, гипогликемии, травм головного и спинного мозга, психоза, мышечной ригидности, рвоты и аналгезии, зачастую предпочтительно в форме щелочного металла или соли щелочноземельного металла, параллельно, одновременно или вместе с фармацевтически приемлемым носителем или разбавителем, особенно и предпочтительно в форме его фармацевтического препарата, пероральным, или ректальным, или парентеральным (включая подкожный) путем, в эффективном количестве.
Подходящая дозировка колеблется в пределах 1-500 мг ежедневно, предпочтительно 10-200 мг ежедневно, и особенно - 50-100 мг ежедневно, в зависимости от точности соблюдения режима введения, формы введения, показания, против которого предприняли введение, субъекта, подвергающегося введению, и веса тела этого субъекта, а также предпочтений и опыта врача или ветеринара, осуществляющего введение.
Такой способ лечения можно описать как лечение показания, обусловленного или связанного с гиперактивностью возбуждающих нейромедиаторов, и особенно рецепторов AMPA у субъектов, в нем нуждающихся, которое включает стадию введения указанному субъекту неврологически эффективного количества соединения настоящего изобретения, противодействующего AMPA или фармацевтически приемлемой его соли.
Кроме того, настоящее изобретение относится к использованию соединения настоящего изобретения для изготовления лекарственного средства для лечения показания, обусловленного или связанного с гиперактивностью возбуждающих нейромедиаторов, и особенно AMPA-рецепторов, у субъекта, в нем нуждающегося.
Далее настоящее изобретение более конкретно поясняется со ссылками на нижеследующие примеры.
Пример 1
1-(Этокси-гидрокси-фосфорилметил)-8-(4-метил-2- фенил-1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-а] хиноксалин-4(5H)-он
а. 1-Бензилокси-3-хлоро-6-(4-метил-2- фенил-1H-имидазол-1-ил)-7-трифторметилхиноксалин-2(1H)-он, гидрохлорид
Раствор 20% фосгена в толуоле (18,2 мл, 35 ммолей) по каплям добавляли к перемешиваемому раствору 1-бензилокси-6-(4-метил-2- фенил-1H-имидазол-1-ил)-7-трифторметилхиноксалин-2,3 (1H,4H)-диона (8,8 г, 17,5 ммолей) в 100 мл безводного N, N-диметилформамида при 0oC. Смесь перемешивали при комнатной температуре в течение ночи, а твердый осадок отделяли фильтрованием и промывали эфиром, получая 8,0 г (84%) вышеназванного соединения.
1H-ЯМР (DMSO-d6): δ 2,42 (s, 3H), 5,35 (s, 2H), 7,30-7,61 (m, 10H), 7,62 (s, 1H), 7,75 (s, 1H), 8,48 (s, 1H).
b. 1-Бензилокси-3-гидразино-6-(4-метил-2-фенил-1H-имидазол-1-ил) -7-трифторметилхиноксалин-2(1H)-он
Смесь 1-бензил-3-хлоро-6-(4-метил-2-фенил-1H-имидазол-1- ил)-7-трифторметилхиноксалин-2(1H)-она, гидрохлорида (2,0 г, 3,6 ммоля) и гидратированного гидразина (0,74 мл, 15 ммоля) в 40 мл дихлорметана перемешивали при 0oC в течение 1 ч и выпаривали досуха под вакуумом. Остаток растирали с водой, получая 1,59 г (87%) вышеназванного соединения. Т.пл. 127-130oC.
1H-ЯМР (DMSO-d6): δ 2,21 (s, 1H), 5,34 (s, 2H), 7,01 (s, 1H), 7,18 (s, 1H), 7,27 (s, 5H), 7,37-7,45 (m, 3H), 7,48 (s, 1H), 7,51-7,60 (m, 2H).
c. 1-Бензилокси-3-[2-[(диэтоксифосфорил) ацетил] гидpaзино] - 6-(4-метил-2-фенил-1H-имидазол-1-ил)-7- трифторметилхиноксалин-2(1H)-он
Раствор (диэтоксифосфорил)ацетилхлорида (0,67 г, 3,1 ммоля) в 20 мл безводного тетрагидрофурана по каплям добавляли в перемешиваемый раствор 1-бензилокси-3-гидразино-6-(4-метил-2- фенил-1H-имидазол-1-(-ил)-7-трифторметилхиноксалин-2(1H)-она (1,52 г, 3,0 ммоля) и безводного триэтиламина (0,43 мл, 3,1 ммоля) в 50 мл безводного тетрагидрофурана.
Смесь перемешивали в течение ночи при комнатной температуре и затем выпаривали досуха под вакуумом. Остаток растирали с водой, получая 1,8 г (88%) вышеназванного соединения. Т.пл. > 90oC с разложением.
1H-ЯМР (DMSO-d6): δ 1,10-1,21 (m, 6H), 2,20 (s, 3H), 2,99 (d, 2H), 4,01 (пятикр, 4H), 5,40 (s, 2H), 7,04 (s, 1H), 7,24 (s, 6H), 7,37-7,46 (m, 3H), 7,50-7,62 (m, 3H), 10,26 (s, 1H), 10.38 (s, 1H).
d. 3-[2-[(Диэтоксифосфорил)ацетил]гидразино]-1-гидрокси- 6-(4-метил-2-фенил-1H-имидазол-1-ил)-7-трифторметилхиноксалин-2 (1H)-он
Суспензию из 1-бензилокси-3-[2-[(диэтоксифосфорил)ацетил] гидразино]-6-(4-метил-2-фенил-1H-имидазол-1-ил)-7- трифторметилхиноксалин-2(1H)-она (1,8 г, 2,6 ммоля) и 50 мг 5% палладия на угле в 50 мл этанола гидрировали при комнатной температуре и атмосферном давлении в течение 9 ч. Катализатор отделяли фильтрацией, фильтрат выпаривали досуха под вакуумом и конечный остаток растирали с эфиром, получая 1,51 г (97%) вышеназванного соединения. Т.пл. > 177oC с разложением.
1H-ЯМР (DMSO-d6): δ 1,10-1,22 (m, 6H), 2,23 (s, 3H), 2,98 (d, 2H), 4,00 (квинтет, 4H), 7,12 (s, 1H), 7,18-7,30 (m, 5H), 7,31 (s, 1H), 7,92 (s, 1H), 10,24 (s, 2H), 12,52 (br.s, 1H).
e. 1-(Этокси-гидрокси-фосфорилметил)-8-(4-метил-2-фенил- 1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-а] хиноксалин-4(5H)-он
Раствор 3-[2-[(диэтоксифосфорил)ацетил] гидразино]-1- гидрокси-6-(4-метил-2-фенил-1H-имидазол-1-ил)-7-трифторметил- хиноксалин-2(1H)-она (1,5 г, 2,5 ммоля) и трифенилфосфина (1,3 г, 5 ммолей) в 50 мл ледяной уксусной кислоты перемешивали в течение ночи при 120oC.
Охлажденную смесь фильтровали, а отделенный продукт промывали эфиром, получая 0,64 г (48%) вышеназванного соединения. Т.пл. 303-308oC.
1H-ЯМР (DMSO-d6): δ 1,10 (t, 3H), 2,25 (s, 3H), 3,87 (квинтет, 2H), 3,97 (d, 2H), 7,12 (s, 1H), 7,14-7,45 (m, 5H), 7,72 (s, 1H), 8,62 (s, 1H), 12,4 (s, 1H).
Пример 2
8-(4-Метил-2-фенил-1H-имидазол -1 -ил)-1- фосфонометил-7-трифторметил[1,2,4]триазоло[4,3-а]хиноксалин- 4(5H)-он
Бромтриметилсилан (1 мл, 7 ммолей) по каплям добавляли к перемешиваемому раствору 1-(этокси-гидрокси-фосфорилметил)-8-(4- метил-2-фенил-1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло [4,3-а]хиноксалин-4(5H)-она (500 мг, 0,94 ммоля) в 20 мл безводного N,N-диметилформамида.
Раствор перемешивали при комнатной температуре в течение ночи и выпаривали досуха под вакуумом. Остаток растирали с 10 мл воды, и твердый осадок отделяли фильтрацией. После промывки небольшим количеством воды и этанола получали 0,45 г (95%) чистого вышеназванного соединения. Т.пл. 321-325oC.
1H-ЯМР (DMSO-d6): δ 2,35 (s, 3H), 3,93 (d, 2H), 7,22-7,52 (m, 6H), 7,74 (s, 1H), 8,79 (s, 1H), 12,4 (s, 1H).
Пример 3
1-(Этокси-гидрокси-фосфорилметил)-8-(2-этил- 4- метил-1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-а] хиноксалин-4(5H)-он
Названное соединение получили из 1-бензилокси-6-(2-этил-4- метил-1H-имидазол-1-ил)-7-трифторметилхиноксалин-2,3 (1H,4H)-диона способом, аналогичным способу, описанному в примере 1, за тем исключением, что конечный продукт (теоретически 10,8 ммоля) обработали следующим образом. К охлажденному раствору добавили 100 мл дихлорметана и 100 мл эфира. Твердый осадок отделили фильтрованием и экстрагировали кипящей водой (2 х 100 мл). Эту водную фазу сконцентрировали до примерно 100 мл и охладили в бане со льдом. Результирующий осадок отделили фильтрованием и высушили, получив 0,90 г (17%) вышеназванного соединения.
1H-ЯМР (CF3COOD): δ 1,38 (t, 3H), 1,45 (t, 3H), 2,51 (s, 3H), 2,72-3,10 (m, 2H), 4,31 (квинтет, 1H), 4,58 (dd, 2H (частично обмениваемый)), 7,22 (s, 1H), 8,32 (s, 1H), 9,00 (s, 1H).
Пример 4
8-(2-Этил-4-метил-1H-имидазол-1-ил)-1-фосфонометил-7- трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он
Названное соединение получили из 1-(этокси- гидроксифосфорилметил)-8-(2-этил-4-метил-1H-имидазол-1-ил)-7- трифторметил[1,2,4.]триазоло[4,3-а]хиноксалин-4(5H)-она (870 мг, 1,8 ммоля) способом, аналогичным способу, приведенному в примере 2. Получили: 710 мг (86%). Т.пл. > 300oC.
1H-ЯМР (DMSO-d6): δ 1,10 (t, 3H), 2,20 (s, 3H), 2,27-2,77 (m, 2H), 3,62-3,95 (m, 2H), 7,07 (s, 1H), 7,85 (s, 1H), 8,55 (s, 1H); MS (FAB): m/e 457 (MH+).
(C17H16N6F3O4P • 1/2H2O)
Расчет: C 43,48 H 3,68 N 18,06
Обнаружено: C 44,07 H 3,56 N 18,02
Пример 5
8-Морфолино-1-фосфонометил-7-трифторметил[1,2,4] триазоло [4,3-а]хиноксалин-4(5H)-он
a. 1-(Этокси-гидрокси-фосфорилметил)-8-морфолино-7-трифторметил [1,2,4] триазоло[4,3-a]хиноксалин-4(5Н)-он
Названное соединение получили из 1-бензилокси-6-морфолино-7- трифторметилхиноксалин-2,3 (1H,4H)-диона способом, аналогичным способу, описанному в примере 1, за тем исключением, что конечный продукт выделили следующим образом. Полученную смесь выпарили досуха под вакуумом, а остаток поместили в смесь из 200 мл дихлорметана и 50 мл хлороформа. Этот результирующий раствор экстрагировали водой (6х100 мл) и водный раствор выпаривали досуха при пониженном давлении путем азеотропной перегонки с 1-пропанолом, чтобы получить неочищенный продукт, который использовали без дополнительной очистки на следующей стадии.
b. 8-Морфолино-1-фосфонометил-7-трифторметил[1,2,4]триазоло [4,3-a]хиноксалин-4(5H)-он
Названное соединение получили из сырого 1-(этокси- гидрокси-фосфорилметил)-8-морфолино-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-она способом, аналогичным способу, описанному в примере 2. Т.пл. > 300oC с разложением (этанол).
1H-ЯМР (DMSO-d6): δ 2,9-3,03 (m, 4H), 3,66-3,78 (m, 4H), 3,98 (d, 2H), 7,68 (s, 1H), 8,39 (s, 1H), 12,18 (s, 1H).
Пример 6
8-Морфолино-1-(1-фосфоноэтил)-7-трифторметил[1,2,4]триазоло [4,3-а]хиноксалин-4(5H)-он
а. 1-(1-(Этокси-гидрокси-фосфорил)этил)-8-морфолино-7- трифторметил[1,2,4]-триазоло[4,3-а]хиноксалин-4(5H)-он
Названное соединение получили из 1-бензилокси-6-морфолино-7-трифторметилхиноксалин-2,3 (1H,4H)-диона способом, аналогичным способу, описанному в примере 5, за тем исключением, что 2-(диэтоксифосфорил)пропионилхлорид использовали вместо (диэтоксифосфорил)ацетилхлорида. Полученный сырой продукт использовали без дальнейшей очистки на следующей стадии.
b. 8-Морфолино-1-(1-фосфоноэтил)-7-трифторметил[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он
Названное соединение получили из неочищенного 1-(1-(этоксигидрокси-фосфорил)этил)- 8-морфолино-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-она способом, аналогичным способу, описанному в примере 2, за тем исключением, что полученный продукт очищали с помощью колоночной хроматографии. Т.пл. > 300oC с разложением.
1H-ЯМР (DMSO-d6): δ 1,73 (dd, 3H), 2,87-3,02 (m, 4H), 3,68-3,78 (m, 4H), 4,11-4,38 (m, 1H), 7,68 (s, 1H), 8,38 (s, 1H), 12,18 (s, 1H).
Пример 7
8-Пиперидино-1-фосфонометил-7-трифторметил [1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он
а. 1-(Этокси-гидрокси-фосфорилметил)-8-пиперидино-7-трифторметил [1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он
Данное названное соединение получили из 1-бензилокси-6- пиперидино-7-трифторметилхиноксалин-2,3 (1H, 4H)-диона способом, аналогичным способу, описанному в примере 3, за тем исключением, что полученный сырой продукт использовали без дальнейшей очистки на следующей стадии.
b. 8-Пиперидино-1-фосфонометил- 7-трифторметил[1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он
Данное названное соединение получили из неочищенного 1-(этоксигидроксифосфорилметил)- 8-пиперидино-7-трифторметил [1,2,4]триазоло [4,3-а] хиноксалин-4(5H)-она способом, аналогичным способу, описанному в примере 2.
1H-ЯМР (DMSO-d6): δ 1,48-1,72 (m, 6H), 2,83-2,98, (m, 4H), 3,90 (d, 2H), 7,65 (s, 1H), 8,32 (s, 1H), 12,12 (s, 1H)
Пример 8
1-(2-Этоксикарбонилэтил)-8-морфолино-7- трифторметил[1,2,4] триазоло[4,3-а]хиноксалин-4(5H)-он
Данное названное соединение получили из 1-бензилокси-6- морфолино-7-трифторметилхиноксалин-2,3(1H,4H)-диона и этилсукцинилхлорида способом, аналогичным способу, описанному в примере 1, за тем исключением, что данный конечный продукт наработали следующим образом. Полученную охлажденную смесь выпаривали досуха под вакуумом и подвергали выделению очисткой с помощью флэш-хроматографии последовательно дихлорметаном и этилацетатом. Растирание с эфиром позволило получить чистый продукт. Т. пл. 204-210oC.
1H-ЯМР (DMSO-d6): δ 1,22 (t, 3H), 2,88-3,00 (m, 4H), 3,02 (t, 2H), 3,61-3,82 (m, 6H), 4,12 (g, 2H), 7,71 (s, 1H), 7,99 (s, 1H), 12,2 (br.s, 1H).
Пример 9
1-(2-Карбоксиэтил)-8-морфолино-7-трифторметил [1,2,4] триазоло[4,3-a] хиноксалин-4(5H)-он
Суспензию 1-(2-этоксикарбонилэтил)-8-морфолино-7- трифторметил[1,2,4] триазоло[4,3-а] хиноксалин-4(5H)-она (365 мг, 0,83 ммоля) в 10 мл 2 н гидроокиси калия перемешивали при комнатной температуре в течение 3 ч.
Полученный результирующий раствор фильтровали и фильтрат подкисляли 4 М хлористоводородной кислотой до получения осадка. Полученный продукт отделяли фильтрованием, промывали водой и высушивали до получения вышеназванного соединения. Т. пл. 170-176oC.
1H-ЯМР (DMSO-d6): δ 2,88-3,02 (m, 6H), 3,67 (t, 2H), 3,68-3,80 (m, 4H), 7,71 (s, 1H), 7,99 (s, 1H), 12,26 (br.s, 1H).
Пример 10
1-(Этокси-гидрокси-фосфорилметил)-8-(1H-имидазол-1-ил) -7-трифторметил[1,2,4]триазоло[4,3-а]хиноксалин-4(5H)-он
Стадия а. 1-Бензилокси-3-[2-[(диэтоксифосфорил)ацетил] гидpaзинo]-6-(1H-имидазол-1-ил)-7-трифторметилхиноксалин-2(1H)-он
Раствор (диэтоксифосфорил) ацетилхлорида (2,4 г, 11 ммолей) в 20 мл безводного тетрагидрофурана добавляли по каплям к перемешиваемому раствору 1-бензилокси-3-гидразино-6-(1H-имидазол- 1-ил)-7-трифторметилхиноксалин-2(1H)-она (4,16 г, 10 ммолей) и безводного триэтиламина (1,53 мл, 11 ммолей) в 150 мл безводного тетрагидрофурана.
Смесь перемешивали в течение 2 ч при комнатной температуре и выпаривали досуха под вакуумом. Остаток суспендировали в 100 мл воды и pH доводили примерно до 7,0, насыщенным водным раствором натрия кислого углекислого.
Сырой продукт отделяли фильтрацией и перекристаллизовывали из этилацетата/эфира с получением 4,7 г (79%) вышеназванного соединения. Т.пл.: 159-161oC.
1H-ЯМР (DMSO-d6): δ 1,23 (t, 6H), 3,04 (d, 2H), 4,06 (квинтет, 4H), 5,39 (s, 2H), 7,08 (s, 1H), 7,34-7,68 (m, 8H), 7,81 (s, 1H), 10,33 (br.s. 2H, обмениваемый).
Стадия b. 3-[2-[(Диэтоксифосфорил)ацетил] гидразино]-1-гидрокси-6-(1H- имидазол-1-ил)-7-трифторметилхиноксалин-2(1H)-он
Суспензию 1-бензилокси-3-[2-[(диэтоксифосфорил)ацетил] гидразино]-6-(1H-имидазол-1-ил)-7-трифторметилхиноксалин-2(1H)- она (3,86 г, 6,5 ммолей) и 500 мг 10% палладия на угле в 250 мл этанола гидрогенизировали при атмосферном давлении и при комнатной температуре в течение 3 ч. Катализатор удаляли фильтрацией и промывали небольшими порциями этанола. Объединенный фильтрат и промывки выпаривали досуха под вакуумом, и сухой остаток растирали в порошок с эфиром, получая 2,80 г (86%) вышеназванного соединения.
1H-ЯМР (DMSO-d6): δ 1,23 (t, 6H), 3,03 (d, 2H), 4,08 (пятикратн. 4H), 7,07 (s, 1H), 7,40 (s, 1H), 7,41 (s, 1H), 7,83 (s, 1H), 7,90 (s, 1H), 10,1-10,3 (2H), 12,5 (очень br.s., 1H).
Стадия c. 1-(Этокси-гидрокси-фосфорилметил)-8-(1H-имидазол-1-ил)-7- трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4(5H)-он
Раствор 3-[2-[(диэтоксифосфорил)ацетил] гидразино]-1- гидрокси-6-(1H-имидазол-1-ил)- 7-трифторметилхиноксалин-2(1H)-она (2,52 г, 5 ммолей) и трифенилфосфина (2,62 г, 10 ммолей) в 100 мл ледяной уксусной кислоты перемешивали при 120oC в течение 23 ч. К охлажденной смеси добавили 150 мл эфира.
Твердый осадок отделяли фильтрованием и промывали эфиром и ацетоном, получая 1,09 г (43%) вышеназванного соединения. Т.пл. 324-328oC.
1H-ЯМР (DMSO-d6): δ 1,10 (t, 3H), 1,91 (s, 3H), 3,88 (квинтет, 2H), 4,05 (d, 2H), 7,21 (s, 1H), 7,50 (s, 1H), 7,82 (s, 1H), 8,06 (s, 1H), 8,50 (s, 1H), 12,49 (s, 1H).
Пример 11
8-(1H-Имидазол-1-ил)-1-фосфонометил- 7-трифторметил[1,2,4] триазоло [4,3-a]хиноксалин-4(5H)-он
Бромтриметилсилан (2 мл, 14 ммоля) по каплям добавляли к перемешиваемому раствору 1-(этокси-гидрокси-фосфорилметил)- 8-(1H- имидазол-1-ил)-7-трифторметил [1,2,4] триазоло[4,3-a] -она (450 мг) в 20 мл безводного N,N-диметилформамида. Раствор перемешивали при комнатной температуре в течение 3 дней, добавляли 25 мл воды и выпаривали досуха под вакуумом. Маслянистый остаток растирали с небольшим количеством воды, и полученный осадок отделяли фильтрацией и промывали небольшими порциями холодной воды, этанолом и эфиром, получая 210 мг вышеназванного соединения. Т.пл. 349-350oC.
1H-ЯМР (DMSO-d6): δ 3,98 (d, 2H), 7,20 (s, 1H), 7,48 (s, 1H), 7,83 (s, 1H), 8,05 (s, 1H), 8,50 (s, 1H), 12,4 (br.s, 1H).
(C14H10N6F3O4P • 1,5H2O)
Расчет: C 38,11; H 2,97; N 19,05.
Обнаружено: C 38,21; H 3,02; N 18,75.
Пример 12
1-(Этокси-гидрокси-фосфорилметил)-8-(2- изопропил-1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-а] хиноксалин-4(5H)-он
Стадия а. 1-Бензилокси-3-хлор-6-(2-изопропил-1H- имидазол-1-ил)-7-фторметилхиноксалин-2(1H)-он
Раствор 20% фосгена в толуоле (18 мл, 34,7 ммолей) добавляли по каплям к перемешиваемому раствору 1-бензилокси-6-(2-изопропил- 1H-имидазол-1-ил)-7-трифторметилхиноксалин-2,3(1H,4H)-диона (5,04 г, 11,3 ммоля) в 50 мл безводного N, N-диметилформамида при 0oC. Смесь перемешивали при 25oC в течение ночи, а твердый осадок отделяли фильтрацией и промывали эфиром, получая 4,81 г (85%) вышеназванного соединения в виде гидрохлорида.
1H-ЯМР (DMSO-d6): δ 1,33 (искаженная t, 6H), 2,65-2,80 (m, 1H), 5,87 (s, 2H), 7,40-7,67 (m, 5H), 7,77 (s, 1H), 7,92 (s, 1H), 7,96 (s, 1H), 8,62 (s, 1H).
Стадия b. 1-Бензилокси-3-гидразино-6-(2-изопропил-1H-имидазол-1- ил)-7-трифторметилхиноксалин-2(1H)-он
Смесь 1-бензилокси-3-хлор-6-(2-изопропил-1H-имидазол-1-ил) -7-трифторметилхиноксалин-2(1H)-он гидрохлорида (4,8 г, 9,6 ммолей) и гидроокиси гидразина (2,0 мл, 41 ммоль) в 100 мл дихлорметана перемешивали при 0oC в течение 2 ч. Смесь выпаривали досуха под вакуумом и остаток растирали с водой, получая 4,1 г (93%) вышеназванного соединения.
1H-ЯМР (DMSO-d6): δ 1,10 (d, 6H), 2,42-2,65 (m, 1H), 5,32 (s, 2H), 6,91 (s, 1H), 7,07 (s, 1H), 7,28 (s, 1H), 7,37-7,47 (m, 3H), 7,50 (s, 1H), 7,52-7,60 (m, 2H).
Стадия c. 1-Бензилокси-3-[2-[(диэтоксифосфорил)ацетил]гидразино]-6-(2- изопропил-1H-имидазол-1-ил)-7-трифторметилхиноксалин-2(1H)-он
Раствор (диэтоксифосфорил)ацетилхлорида (1,93 г, 9 ммолей) в 25 мл безводного тетрагидрофурана по каплям добавляли к перемешиваемому раствору 1-бензилокси-3-гидразино-6-(2-изопропил- 1H-имидазол-1-ил)-7-трифторметилхиноксалин-2(1H)-она (4,0 г, 8,7 ммоля) и безводного триэтиламина (1,25 мл, 9 ммолей) в 75 мл безводного тетрагидрофурана.
Смесь перемешивали в течение ночи при комнатной температуре и выпаривали досуха. Осадок вносили в 500 мл воды и фильтровали. pH фильтрата доводили до 8,0 насыщенным водным раствором натрия кислого углекислого и экстрагировали этилацетатом (5 х 50 мл). Объединенный органический экстракт обезвоживали (безводным сульфатом натрия), фильтровали и выпаривали досуха под вакуумом, получая 4,3 г (78%) сырого вышеназванного соединения.
1H-ЯМР (DMSO-d6): δ 1,25 (t, 12H), 2,63-2,77 (m, 1H), 3,03 (d, 2H), 4,07 (квин. , 4H), 5,40 (s, 2H), 7,38-7,48 (m, 3H), 7,55-7,63 (m, 2H), 7,68 (s, 1H), 7,79 (s, 2H), 7,87 (s, 1H), 10,4 (br.s, 1H), 10,5 (br.s, 1H).
Стадия d. 3-[2-[(Диэтоксифосфорил)ацетил]гидразино] -1-гидрокси-6-(2-изопропил-1H-имидазол-1-ил)-7-трифторметил- хиноксалин-2(1H)-он
Суспензию 1-бензилокси-3-[2-[(диэтоксифосфорил)ацетил] гидразино]-6-(2-изопропил-1H-имидазол-1-ил)-7-трифторметил хиноксалин-2(1H)-она (4,3 г, 6,6 ммоля) и 100 мг 5% палладия на угле в 100 мл этанола гидрогенизировали в течение 2 ч. Катализатор удаляли фильтрацией и промывали этанолом. Объединенный фильтрат выпаривали досуха под вакуумом и остаток растирали с эфиром, получая 3,0 г (83%) вышеназванного соединения. Т.пл. > 190oC с разложением.
1H-ЯМР (DMSO-d6): δ ≈ 1,09 (два d, 6H), 1,22 (t, 6H), 2,46-2,69 (m, 1H), 3,02 (d, 2H), 4,05 (квинтет, 4H), 6,94 (s, 1H), 7,15 (s, 1H), 7,38 (s, 1H), 7,94 (s, 1H).
Стадия e. 1-(Этокси-гидрокси-фосфорилметил)-8-(2-изопропил- 1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-a] хиноксалин-4(5H)-он
Раствор 3-[2-[(диэтоксифосфорил)ацетил] гидразино]-1-гидрокси-6-(2-изопропил-1H-имидазол-1-ил)-7- трифторметилхиноксалин-2(1H)-она (3,0 г, 5,5 ммолей) и трифенилфосфина (2,9 г, 11 ммолей) в 50 мл ледяной уксусной кислоты перемешивали при 120oC в течение 23 ч. Смесь охлаждали при комнатной температуре и твердый осадок отделяли фильтрацией, промывали эфиром, получая 1,84 г вышеназванного соединения. Т.пл. 335-338oC.
1H-ЯМР (DMSO-d6): δ 1,68 (t, 3H), 1,82 (искаженная t, 6H), 3,37-3,57 (m, 1H), 4,62 (квинт. 2H), 4,78-5,00 (m, 2H), 7,88 (s, 2H), 8,69 (s, 1H), 9,35 (s, 1H).
Пример 13
8-(2-Изопропил-1H-имидазол-1-ил)-1-фосфонометил- 7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4(5H)-он
Бромтриметилсилан (2,5 мл, 17,5 ммолей) по каплям добавляли в перемешиваемый раствор 1-(этокси-гидрокси-фосфорилметил)-8- (2-изопропил-1H-имидазол-1-ил)-7-трифторметил [1,2,4]триазоло[4,3-а]хиноксалин-4(5H)-она (1,5 г, 2,7 ммоля) в 10 мл безводного N,N-диметилформамида.
Раствор перемешивали при комнатной температуре в течение 3 дней и выпаривали досуха под вакуумом. Маслянистый остаток растирали с 20 мл воды, полученный твердый осадок отделяли фильтрованием и промывали водой. Сырой продукт обрабатывали 50 мл 1М (pH 7,4) буфером однозамещенного калийфосфата, получаемую калиевую соль отфильтровывали, затем растворяли в 50 мл воды, обесцвечивали углем, фильтровали и, наконец, переосаждали с концентрированной соляной кислотой. Полученный продукт отделяли фильтрованием, промывали водой и высушивали, получая 0,76 г (54%) вышеназванного соединения. Т.пл. > 325oC.
1H-ЯМР (DMSO-d6): δ 1,07 (d, 3H), 1,19 (d, 3H), 2,79 (квинтет, 1H), 3,57-4,01 (m, 2H), 7,04 (s, 1H), 7,17 (s, 1H), 7,88 (s, 1H), 8,48 (s, 1H).

Предложены производные [1,2,4]триазоло[4,3-а]хиноксалинона общей формулы (I), в которой R1 представляет прямой или разветвленный С1-6-алкил, замещенный COX или POX'X'', где X' и X'' независимо представляют гидрокси- или алкоксигруппу, R6 и R9 представляют водород, R7 представляет CF3 и R8 представляет имидазолил, пиперидино или морфолино, кольца которых необязательно замещены одним или двумя заместителями, выбранными из фенила и C1-6-алкила, или их фармацевтически приемлемые соли. Соединения (I) обладают антагонистической активностью по отношению к АМРА и пригодны для лечения показаний, обусловленных гиперактивностью возбуждающих нейромедиаторов. Предложены также способ получения соединений (I), фармацевтическая композиция на их основе и способ ингибирования рецепторов АМРА. 4 с. и 4 з.п.ф-лы, 1 табл.
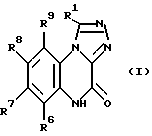
в которой R1 представляет прямой или разветвленный C1-6-алкил, замещенный COX' или POX' X'', где X' и X'' независимо представляют гидрокси- или C1-6-алкоксигруппу;
R6 и R9 представляют водород;
R7 представляет CF3;
R8 представляет имидазолил, пиперидино или морфолино, кольца которых необязательно замещены одним или двумя заместителями, выбранными из фенила и C1-6-алкила,
или их фармацевтически приемлемые соли.
1-(этоксигидроксифосфорилметил)-8-(4-метил-2-фенил-1H-имидазол-1-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4-(5H)-он;
8-(4-метил-2-фенил-1H-имидазол-1-ил)-1-фосфонометил-7-трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4-(5H)-он;
1-(этоксигидроксифосфорилметил)-8-(2-этил-4-метил-1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4-(5H)-он;
8-(2-этил-4-метил-1H-имидазол-1-ил)-1-фосфонометил-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4-(5H)-он;
8-морфолино-1-фосфонометил-7-трифторметил[1,2,4]триазоло [4,3-a]хиноксалин-4-(5H)-он;
8-морфолино-1-(1-фосфоноэтил)-7-трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4-(5H)-он;
8-пиперидино-1-фосфонометил-7-трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4-(5Н)-он;
1-(2-этоксикарбонилэтил)-8-морфолино-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4-(5H)-он;
1-(2-карбоксиэтил)-8-морфолино-7-трифторметил[1,2,4] триазоло [4,3-a]хиноксалин-4-(5H)-он;
1-(этоксигидроксифосфорилметил)-8-(1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4-(5H)-он;
8-(1H-имидазол-1-ил)-1-фосфонометил-7-трифторметил[1,2,4]триазоло[4,3-a] хиноксалин-4-(5H)-он;
1-(этоксигидроксифосфорилметил)-8-(2-изопропил-1H-имидазол-1-ил)-7-трифторметил[1,2,4]триазоло[4,3-a]хиноксалин-4-(5H)-он;
8-(2-изопропил-1H-имидазол-1-ил)-1-фосфонометил-7-трифторметил[1,2,4] триазоло[4,3-a]хиноксалин-4-(5H)-он.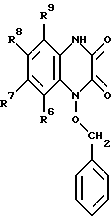
в которой R6, R7, R8 и R9 имеют значения, определенные в п.1,
с получением соединения формулы IV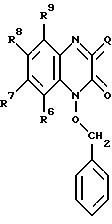
в которой R6, R7, R8 и R9 имеют значения, определенные выше;
Q представляет собой Br, Cl, J,
и взаимодействие данного соединения с гидразином с образованием соединения формулы V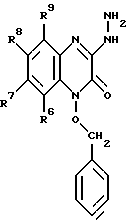
в которой R6, R7, R8 и R9 имеют значения, определенные выше,
и ацилирование соединения формулы V ацилхлоридом, общей формулы VI
R1 - COCl
в которой R1 представляет прямой или разветвленный C1 - C6-алкил, замещенный COX1 или POX1X'', где X' и X'', представляют C1-6-алкокси,
с образованием соединения формулы VII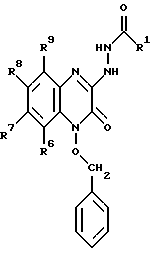
в которой R1, R6, R7, R8 и R9 имеют значения, определенные выше,
и гидрогенолиз данного соединения с образованием соединения формулы VIII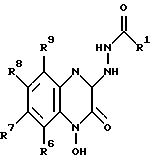
в которой R1, R6, R7, R8 и R9 имеют значения, определенные выше,
и последующую температурную циклизацию и одновременное дезоксигенирование с образованием соединения формулы I, в которой X' и X'' независимо представляют гидрокси - или C1-6-алкоксигруппу,
и в случае необходимости осуществляют гидролиз соединения формулы I, в которой X' и X'' представляют C1-6-алкоксигруппу, водным основанием с образованием соединения формулы I, в которой X' представляет гидроксигруппу, а X'' представляет C1-6-алкоксигруппу, и/или взаимодействие соединения формулы I, в которой X' представляет гидрокси- или C1-6-алкоксигруппу, а X'' представляет C1-6-алкоксигруппу, с галотриметилсиланом с образованием соединения формулы I, в которой X' и X'' представляют гидроксигруппу.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Зажим для канатных подвесных дорог | 1933 |
|
SU40401A1 |
| US 5153196 A, 06.10.92 | |||
| Способ получения 5-пиперидино-7- @ N-(н-пентил)-N-( @ -оксиэтил)-амино @ -S-триазоло (1,5- @ ) пиримидина | 1985 |
|
SU1468423A3 |
| СПОСОБ ПОЛУЧЕНИЯ СУХОЙ СМЕСИ ДЛЯ ДЕТСКОГО ПИТАНИЯ | 2003 |
|
RU2260353C2 |
| Способ получения производных оксотиазолидина или их солей | 1987 |
|
SU1493107A3 |
