Изобретение относится к молекулярной биологии и биотехнологии, а именно к способам диагностики нуклеиновых кислот в бесклеточных системах. Преимущественной областью использования является обнаружение специфических нуклеиновых кислот в анализируемом материале.
Известен ряд способов диагностики на основе экспоненциального размножения нуклеиновых кислот в бесклеточных системах, таких, как размножение РНК вирусными РНК-зависимыми полимеразами (ВРП), размножение ДНК в полимеразной цепной реакции (ПЦР) и изотермическая трехферментная система самоподдерживающегося размножения (ЗСР). В отличие от линейного размножения, происходящего, например, при синтезе РНК с помощью ДНК-зависимой РНК полимеразы, число молекул нуклеиновой кислоты в реакции экспоненциального размножения растет как экспоненциальная функция времени, что позволяет быстро получить большое количество нуклеиновой кислоты из небольшого числа исходных матриц-мишеней. В настоящее время реакции экспоненциального размножения проводят в жидкой среде, содержащей компоненты бесклеточной ферментной системы, включающей реакционный буфер, соответствующие ферменты, нуклеотидные субстраты и, когда необходимо, полимеризационные затравки. В этом случае нуклеиновокислотные продукты, синтезируемые на отдельных матрицах, распространяются по всему реакционному объему.
В ВРП реакции экспоненциальный синтез достигается благодаря тому, что как матричная РНК, так и синтезированная РНК остаются однонитевыми и обе служат одинаково эффективными матрицами в последующих циклах репликации. Эта реакция не требует затравок. Вирусные РНК полимеразы, такие, как Qβ репликаза, демонстрируют узкую матричную специфичность, основанную на узнавании ферментом особых структур, которые присутствуют в специфических матричных РНК, но отсутствуют в других РНК. РНК, содержащие такие структуры, называют "реплицирующимися РНК". Другие РНК, не являющиеся реплицирующимися, могут быть размножены в ВРП реакции, если они встроены в цепочку реплицирующейся РНК [1]. Такие рекомбинантные реплицирующиеся РНК были использованы в целях диагностики [2].
ПЦР используют для внеклеточного размножения ДНК. Для осуществления этой реакции необходимы две олигонуклеотидные затравки, которые комплементарны участкам цепочек двунитевой ДНК, ограничивающим размножаемый фрагмент (мишень). Эти затравки отжигаются с ДНК и наращиваются во взаимно встречных направлениях ДНК полимеразой. В отличие от ВРП реакции, матричная и синтезируемая цепочки ДНК оказываются в дуплексе, который необходимо расплавить при повышенной температуре, чтобы разрешить последующие циклы репликации, в которых каждая из цепочек служит матрицей. Процесс многократно повторяется путем циклической смены температур, при которых происходит отжиг затравки и плавление дуплекса ДНК, что приводит к экспоненциальному размножению ДНК-мишени [3]. В настоящее время ПЦР проводят с использованием термоустойчивых ДНК-полимераз, которые выдерживают многократную смену температур без потери активности [4] . Необходимость циклической смены температур требует наличия специального оборудования и делает ПЦР на порядок медленнее ВРП реакции. В то же время любая ДНК-мишень может быть размножена в ПЦР при наличии пары специфических затравок, без надобности в изготовлении рекомбинантной молекулы.
Способ размножения нуклеиновых кислот в трехферментной системе самоподдерживающегося размножения (ЗСР) соединяет преимущества ПЦР и ВРП реакции. ЗСР основана на совместном действии трех ферментов: ДНК-зависимой РНК-полимеразы, обратной транскриптазы и РНКазы Н [5]. В качестве стартовой матрицы может служить как фрагмент двунитевой ДНК, несущий на каждом конце промотор какой-либо РНК-полимеразы, так и однонитевая РНК. РНК-полимераза (например, РНК-полимераза фага Т7) использует двунитевую ДНК для синтеза многочисленных однонитевых РНК-транскриптов с последовательности ДНК, лежащей вслед за промотором на каждой цепочке ДНК. Обратная транскриптаза (например, обратная транскриптаза вируса миелобластоза птиц) синтезирует двунитевые кДНК-копии РНК-транскриптов, используя затравки, комплементарные 3'-концам транскриптов и содержащие последовательность промотора РНК-полимеразы для восcтановления этой последовательности на каждом конце кДНК. РНКаза Н разрушает РНК-матрицу, вовлеченную в РНК/ДНК гетеродуплекс после синтеза первой цепи кДНК, тем самым позволяя синтезироваться второй цепи кДНК. Действие РНК-полимеразы и РНКазы Н приводит к образованию однонитевых матриц, что позволяет экспоненциально размножать нуклеиновые кислоты в отсутствие циклического изменения температуры. Скорость ЗСР реакции сравнима со скоростью ВРП реакции, причем подобно ПЦР использование ЗСР не требует создания рекомбинантых нуклеиновых кислот. Продуктом ЗСР реакции является смесь молекул двунитевых ДНК и однонитевых РНК.
Благодаря экспоненциальной природе описанных реакций размножения, каждая из них может теоретически быть использована для быстрого получения многочисленного потомства одиночных молекул нуклеиновых кислот. Это позволило бы создать абсолютный метод диагностики нуклеиновых кислот, поскольку позволило бы размножать до легко детектируемого уровня даже одиночные молекулы мишени или мишень-зависимых реплицирующихся матриц, предложенных Крамером и Лизарди [6]. Однако такая возможность не была до сих пор реализована из-за практических проблем.
Как было заявлено Левисоном и Спигельманом [7], им удалось размножить одиночные молекулы РНК, используя Qβ репликазную систему экспоненциального размножения РНК. Они наблюдали синтез РНК в примерно половине образцов после добавления РНК-матрицы, разбавленной до такой степени, чтобы в среднем получить 0,5 молекулы РНК на образец. Однако в этих экспериментах продукт синтеза не был идентифицирован, а вывод авторов был поставлен под сомнение наблюдением, что синтез РНК в Qβ репликазной системе может происходить даже в отсутствие добавленной матрицы [8]. Недавно было показано, что этот спонтанный синтез РНК вызывается реплицирующимися РНК, загрязняющими окружающее пространство [9]. Из-за этого "шума" со стороны РНКовых загрязнений не удается довести технику диагностики, использующую ВРП, до уровня детектирования одиночных молекул нуклеиновых кислот, так как в среднем образец содержит до 100 посторонних молекул реплицирующихся РНК. В настоящее время практически достижимый предел чувствительности ВРП-диагностики составляет 103 - 104 молекул мишени в образце [10]. Проблема загрязнений также присуща ПЦР и ЗСР реакции, хотя она и не столь серьезна, как в случае ВРП реакции, поскольку размножение нуклеиновых кислот находится под контролем олигонуклеотидных затравок, комплементарных мишени. Более существенной в этом случае является ограниченная специфичность затравок. Из-за наличия неправильного спаривания оснований и гетерогенности олигонуклеотидных затравок возможен отжиг затравки на чужеродной матрице, присутствующей в образце, который начинает заметно конкурировать с правильным отжигом при понижении доли специфических матриц в образце ниже определенного предела. По крайней мере 100 копий специфической матрицы должно присутствовать в образце для надежного запуска ПЦР [11]. Таким образом, ни один из существующих методов внеклеточного размножения нуклеиновых кислот не позволяет достичь диагностирования одиночных молекул нуклеиновых кислот.
В качестве прототипа настоящего изобретения может быть взят любой известный способ диагностики, использующий экспоненциальное размножение нуклеиновых кислот в жидкой среде, например размножение рекомбинантных РНК в ВРП реакции [2].
В отличие от известных способов диагностики на основе экспоненциального размножения нуклеиновых кислот, включая указанный прототип, настоящим изобретением предлагается осуществлять размножение нуклеиновых кислот в иммобилизованной среде. Возможно использование любых бесклеточных систем экспоненциального размножения нуклеиновых кислот, таких, как ВРП реакция, ПЦР или ЗСР реакция. В отличие от ранее использовавшихся способов размножения нуклеиновых кислот в жидких средах, потомство (клон) каждой одиночной молекулы нуклеиновой кислоты образует колонию молекул в ограниченной зоне вокруг родительской матрицы. Разные колонии занимают, как правило, разные зоны иммобилизованной среды, что позволяет наблюдать индивидуальные колонии по отдельности и обнаруживать в анализируемом образце одиночные молекулы специфических нуклеиновых кислот, несмотря на загрязнение образца посторонними матрицами и на неспецифический отжиг затравок на чужеродных матрицах.
На фиг. 1 схематически изображен рост колоний нуклеиновых кислот в иммобилизованной среде; на фиг. 2 - колонии РНК, выросшие в тонком слое иммобилизованной среды, содержащей компоненты Qβ репликазной системы; на фиг. 3, 4 - схемы образования мишень-зависимых реплицирующихся матриц.
Иммобилизованная среда для размножения нуклеиновых кислот включает жидкую фазу, заключенную в твердой основе. Твердая основа иммобилизованной среды может иметь разнообразную структуру, например пористую, волокнистую, сетчатую, капиллярную, слоистую, складчатую. Основным требованием к твердой основе является наличие развитых поверхностей, пронизывающих жидкую фазу, среднее расстояние между которыми обеспечивает ту или иную степень иммобилизации жидкости за счет ее трения о поверхность и структурирования воды вблизи поверхности. Предпочтительно использовать твердую основу со средним расстоянием между поверхностями (размером "пор") от 100 мкм (крупнопористая основа) до 5 нм (мелкопористая основа). Верхний предел размера пор должен быть меньше расстояния, на которое размножаемые нуклеиновые кислоты способны диффундировать за время инкубации (порядка 1 мм за час инкубации при комнатной температуре для нуклеиновых кислот длиной около 100 нуклеотидов, ср. фиг. 2А - 2Б). Нижний предел размера пор должен быть несколько больше линейных размеров нуклеиновых кислот и/или ферментов, чтобы обеспечить их подвижность, необходимую для протекания реакции размножения. В качестве твердой основы может служить трехмерная сеть, образуемая отдельными молекулами полимеров или их агрегатами; склеенные, спеченные или спрессованные водонерастворимые порошки, мелкие гранулы или кристаллы; губчатые материалы; плотно упакованные волокна, капилляры или пленки (например, полимерная пленка или металлическая фольга). Основными требованиями к материалу, из которого изготавливают твердую основу, является его химическая инертность в условиях инкубации, гидрофильность формируемой им поверхности, а также отсутствие физических воздействий на ферменты (например, их необратимая сорбция на поверхности), влекущих их инактивацию. Подходящими материалами для изготовления твердой основы являются различные органические или неорганические носители, используемые в биотехнологии для хроматографии и электрофореза биополимеров, для иммобилизации ферментов, а также для выращивания микроорганизмов, клеток и вирусов. Примерами таких носителей являются агароза, полиакриламид, желатин, альгинат, карагеннан, целлюлоза, силикагель, губчатый титан, оксиапатит, химически сшитые агароза, декстран и полиэтиленгликоль, а также их комбинации и производные.
Согласно изобретению иммобилизованная среда для размножения нуклеиновых кислот содержит также компоненты системы размножения, включающей бесклеточную ферментную систему, способную к экспоненциальному размножению нуклеиновых кислот. Примеры таких бесклеточных систем рассмотрены выше. Ферменты, входящие в состав бесклеточной системы, могут быть либо растворены в жидкой фазе, либо иммобилизованы на твердой основе. Реакционный буфер, размножаемые нуклеиновые кислоты, субстраты и (где необходимо) затравки вводят в жидкую фазу.
При размножении в иммобилизованной среде нуклеиновые кислоты образуют колонии, как схематически показано на фиг. 1. На верхней диаграмме представлен фрагмент 11 иммобилизованной среды, образуемой сетью твердой основы 12 и водным раствором 13, содержащим молекулы фермента 14, нуклеиновокислотные матрицы 15, а также субстраты и реакционный буфер. В результате инкубации иммобилизованной среды в течение определенного времени при температурном режиме, подходящем для размножения нуклеиновых кислот, образуются колонии нуклеиновых кислот 16 в зонах локализации родительских матриц.
Предпочтительно, чтобы твердая основа иммобилизованной среды, используемой для размножения, могла обратимо взаимодействовать с нуклеиновыми кислотами. Это позволяет существенно подавить диффузию молекул нуклеиновых кислот и получить колонии меньшего размера, с более четкими краями и большей концентрацией нуклеиновых кислот; иными словами, значительно увеличить разрешающую способность метода. Большинство гидрофильных полимеров способно к взаимодействию с нуклеиновыми кислотами благодаря образованию водородных связей. Эта способность может быть усилена путем химической модификации твердой основы положительно заряженными группами, способными к ионным взаимодействиям с фосфатными остатками нуклеиновых кислот, и/или умеренно гидрофобными группами, способными к взаимодействиям с пуриновыми или пиримидиновыми кольцами. Например, твердая основа может быть модифицирована слабокатионными группами, такими, как этиламиноэтильными или диэтиламиноэтильными, или интеркалирующими красителями, такими, как этидиум или пропидиум. Интеркалирующие красители способны к гидрофобным взаимодействиям со стекингованными основаниями нуклеиновых кислот и при этом несут положительный заряд.
Чтобы предотвратить распространение нуклеиновых кислот в иммобилизованной среде в процессе роста колоний, очень важно не допускать наличие неиммобилизованной жидкости. В частности, нельзя допускать конденсации воды на поверхности иммобилизованной среды. В то же время нельзя допускать и пересыхания иммобилизованной среды, что может приводить к инактивации ферментов. Пересыхание иммобилизованной среды можно предотвратить путем проведения инкубаций в плотно закрытой камере, кассете или запаянном пластиковом пакете, а также оборачиванием среды водонепроницаемой пленкой или наслаиванием минерального масла.
Предпочтительно, чтобы реакция размножения не начиналась до полной иммобилизации среды. Это условие легко выполнимо в случае ПЦР, так как в этом случае реакция запускается циклическим изменением температуры. В случае реакций изотермического размножения, таких, как ВРП или ЗСР реакция, это условие выполняется либо путем приготовления среды при или около 0oC, когда скорость реакции мала, либо путем помещения ферментов и субстратов в разные зоны иммобилизованной среды, после чего субстраты диффундируют в ферментную зону, либо путем использования защищенных субстратов (химически недоступных ферменту) с последующим удалением защиты и высвобождением нормального субстрата. Примером защищенного субстрата является светочувствительное производное АТР, в котором γ -фосфат модифицирован 1-(2-нитро)фенилэтильной группой [12] и которое может быть превращено в АТР фотолизом для запуска реакции размножения.
Предпочтительно использовать иммобилизованную среду, приготовленную в виде тонкого слоя. В этом случае растущие колонии располагаются в двух измерениях, что облегчает их наблюдение и анализ, а также позволяет изготавливать реплики, например, путем частичного переноса колоний на мембранный фильтр. Реплики можно использовать для анализа колоний, для их пересева на новую среду, а также хранить в течение длительного времени. Малая толщина слоя уменьшает температурные градиенты в поперечном направлении (что особенно важно в случае ПЦР) и облегчает диффузию субстратов в ферментный слой при использовании сэндвичевых сред с раздельными ферментным и субстратным слоями (см. ниже). Предпочтительно, чтобы толщина слоя не превышала размера колоний (который определяется, главным образом, скоростью линейной диффузии нуклеиновых кислот), то есть была в пределах нескольких миллиметров. Наиболее предпочтительно использовать очень тонкие слои, так как это повышает разрешающую способность метода. Нижний предел толщины слоя зависит от механической прочности твердой основы, от способности предотвратить пересыхание слоя при манипуляциях и в процессе реакции размножения, а также от чувствительности метода, используемого для анализа колоний. На практике удовлетворительные результаты получаются при толщине слоя от 1 мм до 50 мкм. Конечно, могут быть использованы более толстые (например, 10 мм) и более тонкие (до 1 мкм) слои. Если используют более толстые слои, то предпочтительно помещать ферменты и субстраты в разные слои, которые накладывают друг на друга, и/или вводить анализируемый образец на поверхность слоя или между слоями, чтобы ограничить реакцию размножения и/или экспрессии узкой зоной вблизи поверхностей слоев. Более тонкие слои предпочтительно иммобилизовывать на подложке из прочного материала, такой, как стеклянная, металлическая или пластиковая пластинка или пленка.
Выбор техники приготовления тонких слоев зависит от свойств твердой основы, от температурного режима реакции, от свойств ферментов, используемых для реакции (как, например, их способность переносить условия формирования полимерной основы), а также от того, используют ли однослойную или многослойную иммобилизованную среду.
Однослойные среды удобно использовать, когда все компоненты ферментной системы могут быть смешаны без того, чтобы началась реакция (как в случае ПЦР). Во многих случаях предпочтительно использовать многослойные ("сэндвичевые") среды, в которых разные слои приводятся в непосредственный контакт друг с другом в нужный момент. Например, ферменты и субстраты могут быть разнесены в разные слои и реакция запускаться путем соприкосновения слоев благодаря диффузии субстратов в ферментный слой. Использование сэндвичевых сред может быть предпочтительным и в том случае, если разносить ферменты и субстраты не требуется, как, например, в ПЦР. Например, вторым слоем может быть мембранный фильтр, используемый для анализа колоний, на который нуклеиновые кислоты переносятся одновременно с их синтезом.
Тонкие слои могут быть изготовлены разными способами, такими, как пропитывание сухой твердой основы растворами, содержащими ферменты и/или субстраты, или включение ферментов и/или субстратов в процессе гелеобразования или синтеза (полимеризации) твердой основы.
Гелеобразующие растворы, такие, как основанные на агарозе, желатине, карагенане, смешивают с ферментами и/или субстратами и заливают, например, в чашки Петри или между двумя ровными поверхностями. В последнем случае можно получить очень тонкие слои геля и избежать образование мениска. Ферменты и/или субстраты могут быть также включены в слой в процессе синтеза твердой основы, например в процессе полимеризации акриламида [13] или фотоиндуцируемой полимеризации этиленгликоля [14]. В тех случаях, когда формирование твердой основы происходит в условиях, слишком жестких для ферментов и/или субстратов, сначала приготавливают твердую основу, а затем пропитывают ее соответствующим раствором. Например, повысить термоустойчивость агарозного геля (чтобы его можно было использовать в ПЦР) можно путем сшивания агарозы эпихлоргидрином или 2,3-дибромпропанолом; термоустойчивый гель можно также получить путем сшивания эпихлоргидрином или N, N-метиленбисакриламидом декстрана [15]. Однако сшивание происходит в условиях, ведущих к инактивации ферментов и субстратов реакции размножения. Поэтому тонкий слой геля сначала формируют в отсутствие ферментов и субстратов и затем пропитывают раствором, содержащим ферменты и/или субстраты. Таким же образом могут быть приготовлены слои из геля на основе полиакриламида или смеси полиакриламида и агарозы. Волокнистые слои (например, основанные на целлюлозе или нейлоне) или пористые слои (например, основанные на силикагеле или губчатом титане) могут быть приготовлены просто пропитыванием соответствующих сухих листов, мембранных фильтров или пластинок раствором, содержащим компоненты системы размножения и/или экспрессии.
Образец, содержащий анализируемые нуклеиновые кислоты, вводят в жидкую фазу твердой среды путем включения в ферментный и/или субстратный раствор перед его иммобилизацией или путем нанесения на слой или между слоями.
Реакцию запускают помещением иммобилизованной среды в соответствующие условия: повышение температуры среды, циклическое изменение температуры, воздействие света, приведение в соприкосновение слоев, содержащих ферменты и субстраты. Среду инкубируют в течение промежутка времени, позволяющего продуктам синтеза накопиться до детектируемого уровня. Необходимо, чтобы в каждой колонии образовалось по крайней мере 106 копий нуклеиновой кислоты (то есть должно произойти по крайней мере 20 циклов репликации при условии, что в каждом цикле число матриц удваивается), что соответствует нижнему пределу чувствительности существующих методов обнаружения нуклеиновых кислот [16].
Предложенный способ позволяет обнаруживать одиночные молекулы специфической нуклеиновой кислоты-мишени, даже если образец содержит большое количество чужеродных нуклеиновых кислот, и может быть использован как чрезвычайно чувствительный способ диагностики в клинической и сельскохозяйственной практике и в экологическом мониторинге. Разнообразные образцы, такие, как пробы биологических жидкостей и тканей, а также пробы воды, пищи и воздуха, могут быть исследованы на присутствие определенных вирусов, бактерий и других микроорганизмов или их останков. Способ позволяет также диагностировать наследственные и онкологические заболевания путем обнаружения соответствующих генов и их транскриптов в высших организмах. Процедура диагностики включает: (а) получение образца, подозреваемого на содержание искомых нуклеиновых кислот (мишени); (б) экспонирование мишени путем соответствующей обработки образца, такой, как экстракция фенолом, или лизис детергентами и/или гуанидинизотиоцианатом; (в) удаление (или разбавление) литического агента, и предпочтительно любого материала, который может мешать анализу, такого, как клеточный дебрис или пыль; (г) размножение мишени, ее фрагмента или мишень-зависимой реплицирующейся матрицы для получения детектируемых колоний в иммобилизованной среде, содержащей подходящую бесклеточную ферментную систему экспоненциального размножения нуклеиновых кислот; (д) обнаружение соответствующих мишени колоний (позитивных колоний) по их способности связывать специфические зонды, например гибридизоваться с мечеными олигонуклеотидами. Даже если среди множества чужеродных колоний встретится одиночная позитивная колония, она будет надежно зарегистрирована этим способом. Возможно количественное определение мишени: с этой целью приготавливают серию разведений образца и подсчитывают число позитивных колоний, полученных для каждого разведения. Способ позволяет также проводить определение одновременно нескольких мишеней (например, нескольких патогенных вирусов). В этом случае стадию (д) проводят с использованием нескольких специфических зондов; каждый зонд испытывают отдельно, либо последовательно, либо параллельно (для чего используют реплики, полученные переносом колоний на мембранные фильтры): или же используют смесь зондов, если все они мечены по-разному.
Из-за чрезвычайной чувствительности способа важно исключить возможность загрязнения образца мишенями до стадии размножения. В связи с этим предпочтительно проводить стадии, предшествующие размножению, и последующие стадии (анализ колоний) в разных лабораториях. Предпочтительно выращивать колонии в тонких слоях, помещенных в герметичные кассеты, которые вскрывают только в аналитической лаборатории. Полезно также инактивировать размноженные нуклеиновые кислоты перед вскрытием кассеты. Для этого можно использовать, например, фотоактивируемые производные изопсоралена [17], которые вводят в ростовый слой перед размножением. Эти соединения не мешают размножению, пока не разрушены под действием ультрафиолета (300 - 400 нм). Продукты их распада ковалентно модифицируют пиримидиновые основания, тем самым полностью лишая нуклеиновые кислоты способности служить матрицами для последующего размножения. В то же время сохраняется способность нуклеиновых кислот к специфической гибридизации с нуклеотидными зондами [18]. Важно также постоянно проверять надежность процедуры с помощью контрольных образцов, таких, как не содержащие добавленную мишень (для проверки уровня загрязнений) и содержащие известное количество мишени (для проверки чувствительности процедуры).
Пример 1. Выращивание колоний РНК с помощью ВРП реакции. Этот пример иллюстрирует способ размножения РНК в тонком слое иммобилизованной среды на примере RQ-РНК, являющихся природными матрицами Qβ репликазы. Порошок низкотемпературной агарозы (ultra-low gelling temperature agarose type IX, Sigma Chemical Company) расплавляют нагреванием в автоклавированном буфере A (80 мМ Трис-HCl pH 7,8, 2 мМ MgCl2, 1 мМ ЭДТА, 20% глицерин), охлаждают до примерно 40oC и тщательно перемешивают на роторном встряхивателе с концентрированным раствором Qβ репликазы, очищенной как описано Блюменталем [19]. Конечные концентрации агарозы и Qβ репликазы - 2% и 35 мкг/мл соответственно. Затем приготавливают сэндвичевую среду, как описано ниже. Когда необходимо, разведения РНК-мишени вводят в субстратный слой или в промежуток между ферментным и субстратным слоями в виде небольшой аликвоты.
130 мкл раствора, содержащего фермент и агарозу, заливают между покровным стеклом, используемом в микроскопии, и большей по размеру пластиковой пластинкой, между которыми имеется зазор 0,4 мм. Покровное стекло осторожно снимают и на агарозу накладывают нейлоновый мембранный фильтр, пропитанный субстратным раствором (по 4 мМ ATP, GTP, CTP и UTP) и высушенный. После инкубации среды в течение 20-40 мин для размножения РНК мембранный фильтр снимают с агарозы и отмывают от невключившихся субстратов. Колонии идентифицируют с помощью гибридизации мембранного фильтра со специфическими зондами, как описано ниже.
РНК пришивают к мембранному фильтру под действием ультрафиолета и гибридизуют с радиоактивно-меченым зондом путем инкубации в запаянном пластиковом пакете между листами фильтровальной бумаги [20]. Температуру гибридизации определяют по известной формуле [21] и оптимизируют в предварительных экспериментах. После гибридизации мембранный фильтр отмывают от негибридизовавшегося зонда и делают радиоавтограф. Для повторной гибридизации того же мембранного фильтра с другим зондом первый зонд отмывают кипячением мембранного фильтра в 0.1% растворе Na-додецилсульфата. На фиг. 2А и 2Б показаны негативы радиоавтографов, полученных в результате последовательной гибридизации одного и того же мембранного фильтра с радиоактивно-мечеными зондами, специфичными к RQ87-3 РНК и RQ135-1 РНК соответственно. Смесь этих РНК была введена в субстратный слой в количестве около 100 копий каждой из них. Этот эксперимент демонстрирует, что разные колонии содержат разные виды РНК и что число колоний соответствует количеству добавленных матриц.
Пример 2. Выращивание колоний нуклеиновых кислот с помощью ЗСР реакции.
Раствор низкотемпературной агарозы в буфере смешивают с ферментами и нуклеиновой кислотой, отожженной с олигонуклеотидными затравками при 65oC, и заливают ферментный слой как описано в примере 1. В качестве мишени используют РНК, ДНК или их смесь. Используют реакционный буфер, ферменты, затравки и условия инкубации согласно рекомендациям Гуателли и др. [5]. На ферментный слой наслаивают нейлоновый мембранный фильтр, пропитанный субстратным раствором, содержащим по 4 мМ dATP, dGTP, dCTP, dTTP и по 16 мМ ATP, GTP, CTP, UTP в реакционном буфере. После инкубации колонии нуклеиновых кислот идентифицируют при помощи гибридизации мембранного фильтра с меченым зондом, комплементарным внутренней области мишени, как описано в примере 1.
Пример 3. Выращивание колоний ДНК с помощью ПЦР.
Все компоненты реакции, включая буфер, термоустойчивую ДНК-полимеразу, образец ДНК, затравки и субстраты, смешивают с дегазированным раствором мономеров (смесь акриламида и N,N-метиленбисакриламида) и катализаторами полимеризации (персульфат аммония и N,N,N',N'-тетраметилендиамин) и заливают тонкий слой геля. По завершении полимеризации на гель наслаивают нейлоновый мембранный фильтр, смоченный в реакционном буфере, оборачивают термостойкой пленкой и помещают гель на металлический термостатируемый столик, температура которого контролируется в требуемом для ПЦР диапазоне автоматическим устройством, включающим два [22] или три [23] водяных термостата и соединенным шлангами с термостатируемым столиком. Используют реакционные компоненты и условия ПЦР, рекомендованные Сэйкай и др. [4]. После 20 - 35 температурных циклов колонии ДНК обнаруживают, как описано выше. Если в качестве образца используют РНК, ее сначала превращают в кДНК [24].
Пример 4. Использование способа размножения нуклеиновых кислот в иммобилизованных средах для диагностики.
В этом примере описана процедура, основанная на использовании ВРП системы, так как она включает специальные стадии, связанные с мишень-зависимым синтезом реплицирующихся матриц. Процедуры, использующие ЗСР реакцию и ПЦР, отличаются от описанной главным образом тем, что в них не требуется синтеза реплицирующихся матриц, а специфическое размножение мишени или ее фрагмента достигается благодаря использованию по крайней мере двух мишень-специфичных олигонуклеотидных затравок.
Образец (например, пробу крови пациента, содержащую примерно 5•106 клеток) обрабатывают 5 М гуанидинизотиоцианатом, что приводит к лизису клеток, денатурации белков (включая нуклеазы), а также высвобождению из клеточного дебриса и денатурации РНК и ДНК [25]. После разбавления гуанидинизотиоцианата до концентрации 2,5 М молекулы мишени вылавливают на магнитный порошок, ковалентно модифицированный олиго(dT), с помощью зондов-ловушек, которые содержат участок, комплементарный молекуле мишени, и участок, состоящий из поли(dA), комплементарный олиго(dT) [26]. Магнитный порошок промывают, отделяя его от жидкости осаждением в магнитном поле, связанные молекулы мишени высвобождают в раствор путем нагревания в буфере с низкой концентрацией соли и проводят мишень-зависимый синтез реплицирующихся матриц [6] одним из способов, описанных ниже.
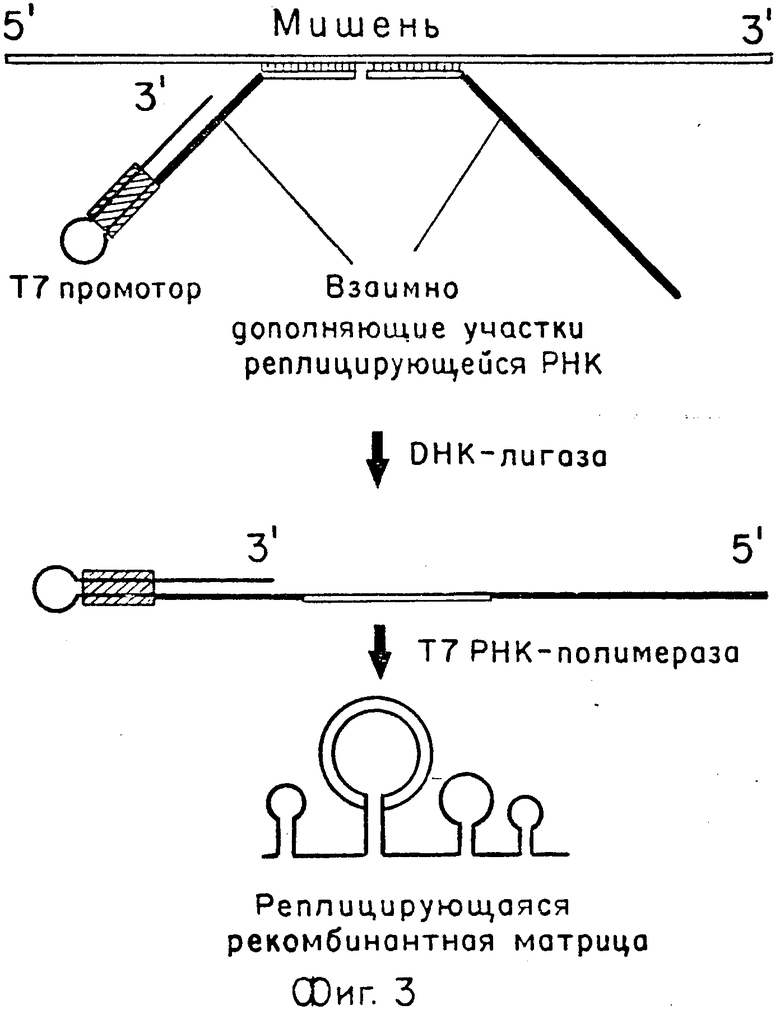
(а) Молекулы мишени гибридизуют с частями бинарного зонда, которые затем лигируют с помощью ДНК-лигазы для получения полного ДНК- зонда (фиг. 3). Части бинарного зонда представляют собой две однонитевые молекулы ДНК, которые содержат на лигируемых концах нуклеотидные последовательности, комплементарные двум соседним участкам молекулы мишени, и также включают в себя последовательности, кодирующие взаимно дополняющие части реплицирующейся РНК. На 3' конце одной из молекул находится двунитевой промотор Т7 РНК-полимеразы, предназначенный для транскрипции лигированного зонда. Части бинарного зонда могут быть лигированы только в том случае, если они одновременно гибридизованы с соседними участками одной и той же молекулы мишени, и только в этом случае может быть получен транскрипт, способный к репликации в ВРП реакции.
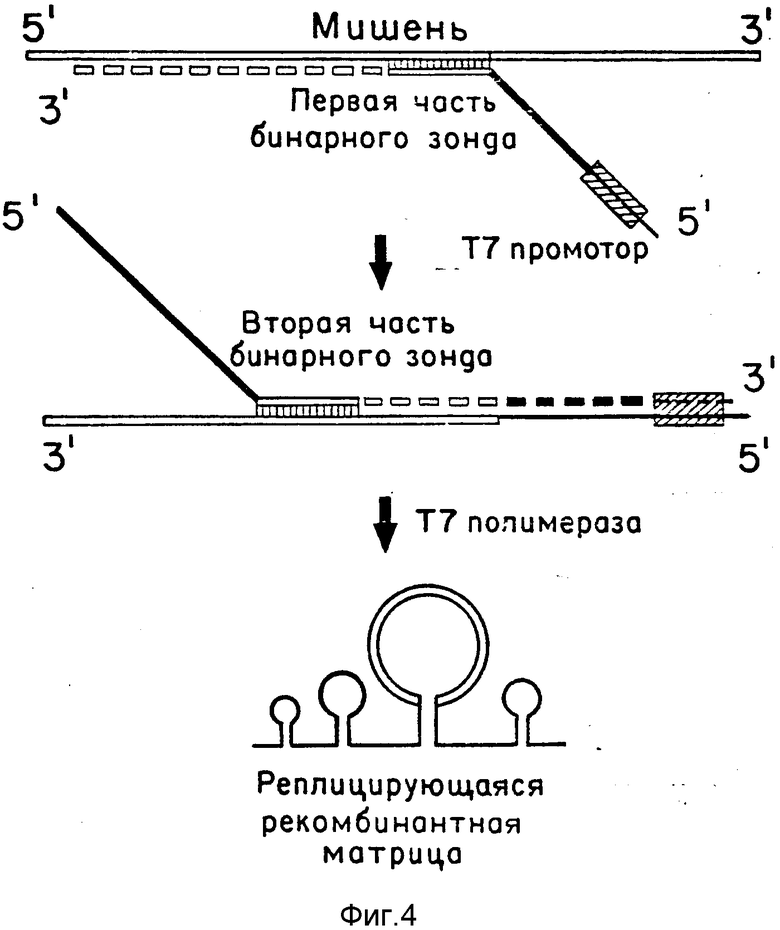
(б) Второй способ похож на первый, с той разницей, что полный бинарный зонд образуется в результате мишень-зависимой элонгации его частей (обозначено пунктирными линиями), которые здесь выступают в качестве затравок ДНК-полимеразной реакции (фиг. 4). Одна из частей зонда, несущая промотор Т7 РНК-полимеразы, гибридизуется с молекулой мишени и удлиняется с помощью ДНК-полимеразы на мишени как на матрице (или с помощью обратной транскриптазы, если в качестве мишени выступает РНК), как показано на верхней диаграмме. После расплавления полученного дуплекса удлиненная первая часть зонда выступает в качестве матрицы для удлинения второй части зонда (во встречном направлении), в результате чего образуется полный ДНК-зонд, содержащий функциональный двунитевой Т7 промотор (средняя диаграмма). Как и в первом случае, образование полного зонда возможно только при наличии в образце мишени.
В обоих способах после получения полного ДНК-зонда приготавливают серию разведений, которые вводят вместе с субстратами в нейлоновый мембранный фильтр, как описано в примере 1, или между мембранным фильтром и агарозным слоем. Помимо ВРП, агарозный слой содержит Т7 РНК-полимеразу [27], которая осуществляет транскрипцию молекул ДНК- зонда в тех местах, где они оказались в процессе приготовления иммобилизованной среды. Транскрипция приводит к образованию молекул реплицирующейся рекомбинантной РНК, несущей в средней части копию участка мишени (обозначена двойными линиями на нижних диаграммах фиг. 3 и 4). РНК-транскрипты размножают с помощью ВРП с образованием колоний РНК. Позитивные колонии идентифицируют и отличают от колоний, не несущих копии участка мишени, путем гибридизации с меченым олигонуклеотидным зондом, который комплементарен этому участку, как описано в примере 1.
Литература:
1. Miele, A. A., Mills, D.R. and Kramer, F.R. (1983). Autocatalytic Replication of a Recombinant RNA. J. Mol. Biol. 171, 281-295.
2. Lizardi, P.M., Guerra, C.E., Lomeli, H., Tussie-Luna, l. and Kramer, F. R. (1988). Exponential Amplification of Recombinant- RNA Hybridization Probes. Bio/Technology 6, 1197-1202 (прототип).
3. Saiki, R.K., Scharf, S., Faloona, F., Mullis, K. B., Horn, G. T., Erlich, H. A. and Arnheim, N. (1985). Enzymatic Amplification of β -Globin Genomic Sequence and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia. Science 230, 1350-1354.
4. Saiki, R. K., Gelfand, D. H., Stoffel, S., Scharf, S. J., Higuchi, R. , Horn, G. T., Mullis, K. B. and Erlich, H. A. (1988). Primer-directed Enzymatic Amplification of DNA with a Thermostable DNA Polymerase. Science 239, 487-491.
5. Guatelli, J.C., Whitfield, K.M., Kwoh, D.Y., Barringer, K.J., Richman, D.D. and Gingeras, T. R. (1990). Isothermal, in vitro Amplification of Nucleic Acids by a Multienzyme Reaction Modeled after Retroviral Replication. Proc. Nail. Acad. Sci. U.S.A. 87, 1874-1878.
6. Kramer, F. R. and Lizardi, P. M. (1989). Replicatable RNA Reporters. Nature 339, 401-402.
7. Levisohn, R. and Spiegelman, S. (1968). The Cloning of a Self-replicating RNA Molecule. Proc. Natl. Acad. Sci. U.S.A. 60, 866-872.
8. Sumper, M. and Luce, R. (1975). Evidence for de novo Production of Self-replicating and Environmentally Adapted RNA Structures by Bacteriophage Qβ Replicase. Proc. Natl. Acad. Sci. U.S.A. 72, 162-166.
9. Chetverin, A. B., Chetverina, H. V. and Munishkin, A. V. (1991). On the Nature of Spontaneous RNA Synthesis by Qβ Replicase. J. Mol. Biol. 222, 3-9.
10. Lomeli, H., Tayagi, S., Pritchard, C. G., Lizardi, P. M. and Kramer, F. R. (1989). Quantitative Assays Based on the Use of Replicatable Hybridization Probes. Clin. Chem. 35, 1826-1831.
11. Myers, T. W. and Gelfand, D. H. (1991). Reverse Transcription and DNA Amplification by a Thermus thermophilus DNA Polymerase. Biochemistry 30, 7661-7666.
12. Kaplan, J. H. , Forbush, B., III and Hoffman, J. F. (1978). Rapid Photolytic Release of Adenosine 5'-Triphosphate from a Protected Analogue: Utilization by the Na:K Pump of Human Red Blood Cell Ghosts. Biochemistry 17, 1929-1935.
13. O'Driscoll, K. F. (1976). Techniques of Enzyme Entrapment in Gels. Methods Enzymol. 44, 169-183.
14. Nojima, S. and Yamada, T. (1987). Large-scale Production of Photo-cross-linkable Resin-immobilized Yeast and Its Application to Industrial Ethanol Production. Methods Enzymol. 136, 380-394.
15. Osterman, L. A. (1986). Methods of Protein and Nucleic Acid Research, Vol. 3. Springer-Verlag, New York.
16. Landegren, U. , Kaiser, R., Caskey, C. T. and Hood, L. (1988). DNA Diagnostics - Molecular Techniques and Automation. Science 242, 229-237.
17. Cimino, G. D., Metchette, K. C., Tessman, J. W., Hearst, J. E. and Isaacs, S. T. (1991). Post-PCR Sterilization: a Method to Control Carryover Contamination for the Polymerase Chain Reaction. Nucleic Acids Res. 19, 99-107.
18. Isaacs, S. T., Tessman, J. W., Metchette, K. C., Hearst, J. E. and Cimino, G. D. (1991) Post-PCR Sterilization: Development and Application to an HIV-I Diagnostic Assay. Nucleic Acids Res. 19, 109- 116.
19. Blumenthal, T. (1979). Qβ RNA Replicase and Protein Synthesis Elongation Factors EF-Tu and EF-Ts. Methods Enzymol. 60, 628-638.
20. Jones, R. W. and Jones, M. J. (1992). Simplified Filter Paper Sandwich Blot Provides Rapid, Background-free Northern Blots.
BioTechniques 12, 685-688.
21. Bodkin, D. K. and Knudson, D. L. (1985). Sequence Relatedness of Palyam Virus Genes to Cognates of the Palyam Serogroup Viruses by RNA-RNA Blot Hybridization. Virology 143, 55-62.
22. Weier, H. U. and Gray, J. W. (1988). A Programmable System to Perform the Polymerase Chain Reaction. DNA 7, 44-47.
23. Torgensen, H. , Blaas, D. and Skern, T. (1989). Low Cost Apparatus for Primer-directed DNA Amplification Using Thermus aquaticus-DNA Polymerase. Analyt. Biochem. 176, 33-35.
24. Sambrook, J. , Fritsch, E. F. and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual, 2nd edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y.
25. Pellegrino, M. G., Lewin, M. Meyer, W. A., III, Lanciotti, R. S., Bhaduri-Hauck, L., Volsky, D. J., Sakai, K., Folks, T. M. and Gillespie, D. (1987). A Sensitive Solution Hybridization Technique for Detecting RNA in Cells: Application to HIV in Blood Cells. BioTechniques 5, 452-459.
26. Morrissey, D. V. , Lombardo, M., Eldredge, J. K., Kearney, K. R., Grody, E. P. and Collins, M. L. (1989). Nucleic Acid Hybridization Assays Employing dA-tailed Capture Probes, I. Multiple Capture Methods. Anal. Biochem. 181, 345-359.
27. Milligan, J. F., Groebe, D. R., Witherell, G. W. and Uhlenbeck, O. C. (1987). Oligoribonucleotide Synthesis Using T7 RNA Polymerase and Synthetic DNA Templates. Nucleic Acids Res. 15, 8783-8798.

Использование: биотехнология, молекулярная биология. Сущность изобретения: диагностику нуклеиновых кислот осуществляют путем получения исследуемого образца, его обработкой, размножением нуклеиновых кислот в среде, содержащей бесклеточную ферментную систему и субстраты, необходимые для экспоненциального размножения нуклеиновых кислот, контактированием с компонентами среды, далее жидкую фазу среды иммобилизуют заключением ее в твердую основу, обладающую развитой поверхностью со средним размером пор от 100 мкм до 5 нм, проводят инкубацию в условиях, обеспечивающих экспоненциальное размножение нуклеиновых кислот и обнаружение в образце исследуемых нуклеиновых кислот 3 ил .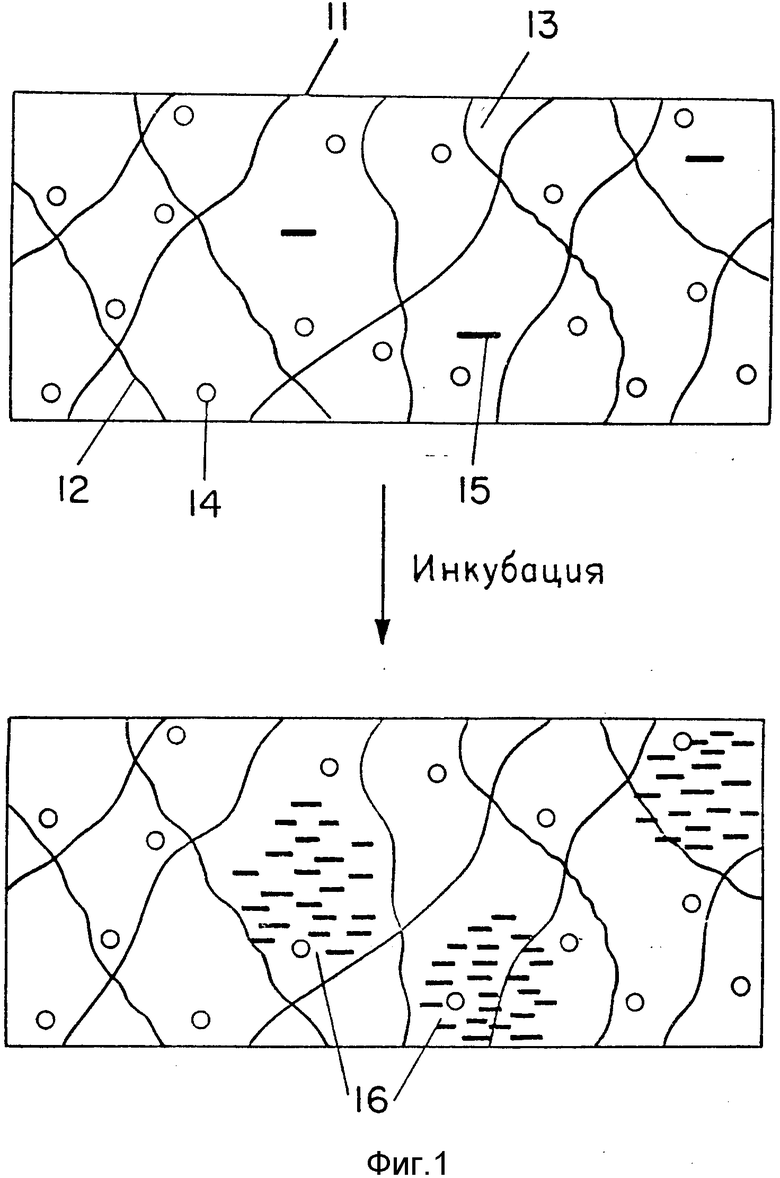
| Bio/Technology, N 6, 1988, с.1197-1202. |
