ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области молекулярной биологии и биотехнологии, а именно к способам молекулярной диагностики, основанным на размножении нуклеиновых кислот в бесклеточных системах и на обнаружении продуктов размножения с помощью флуоресценции.
УРОВЕНЬ ТЕХНИКИ
В настоящее время, наибольшие чувствительность и надежность обнаружения нуклеиновых кислот достижимы с помощью метода молекулярных колоний (ММК) - способа размножения отдельных молекул нуклеиновых кислот в иммобилизованных средах, таких как гель. В таком формате копии каждой размножаемой молекулы концентрируются вокруг родительской матрицы в виде сферической колонии. Если размножение осуществляют в слое геля, то молекулярные колонии образуют двумерный рисунок, напоминающий колонии бактерий, выросших на поверхности питательного агара.
ММК раскрыт Четвериным и Четвериной в Патентах РФ №2048522, 2114175 и 2114915 [1-3] и в Патентах США №5616478, 5958698 и 6001568 [4-6]. В указанных патентах рассмотрены различные способы распределения нуклеиновых кислот в иммобилизованной среде, введения в среду компонентов системы размножения и собственно размножения нуклеиновых кислот с образованием молекулярных колоний. Рассмотрены разнообразные способы упаковок иммобилизованных сред в виде гранул, слоев или волокон, а также в виде тонких слоев. Указывается, что в качестве матрикса иммобилизованной среды могут быть использованы различные материалы, такие как агароза, полиакриламид, нейлон, желатин, альгинат, каррагенан, целлюлоза, силикагель, титановая губка, поперечно-сшитые агароза, декстран или полиэтиленгликоль. В качестве системы размножения может быть использована любая ферментная система экспоненциального размножения нуклеиновых кислот in vitro, среди которых можно выделить праймер-независимые (такие как Qβ-репликаза) и праймер-зависимые системы, в том числе как требующие циклических изменений температуры (ПЦР - полимеразная цепная реакция [7]), так и изотермические системы, такие как 3SR (Self-Sustained Sequence Replication [8]) и SDA (Strand Displacement Amplification [9]). Система размножения может быть введена в иммобилизованную среду либо при ее приготовлении, либо (особенно в случаях, когда условия приготовления среды слишком суровы для лабильных компонентов системы) путем пропитывания уже готовой иммобилизованной среды. Помимо рассмотренных в указанных патентах, в ММК могут быть использованы другие праймер-зависимые системы экспоненциального размножения, такие как NASBA (nucleic acid sequence-based amplification [10]), RCA (rolling circle amplification [11, 12] и LAMP (loop-mediated isothermal DNA amplification [13]).
Термин "система размножения" означает полный набор реагентов и иных компонентов, необходимых для синтеза нуклеиновых кислот на определенного вида матрицах (мишенях), в том числе один или более ферментов, включающих по крайней мере одну полимеразу нуклеиновых кислот; субстраты полимеризации (рибо- и/или 2'-дезоксирибонуклеозидтрифосфаты); реакционный буфер; а также мишень-специфичные олигонуклеотидные праймеры, если синтез нуклеиновых кислот осуществляют с помощью праймер-зависимой полимеразы.
Экспоненциальным называют такой способ размножения, когда число молекул нуклеиновых кислот (N) нарастает в соответствии с формулой N=ax(а>1), где x - число осуществленных циклов копирования матриц, а а - коэффициент увеличения количества матриц в одном цикле. Если размножение происходит по способу дупликации матриц (как, например, в случае ПЦР), то а=2 (в идеальном случае; на практике а несколько меньше). Экспоненциальное размножение возможно до тех пор, пока полимераза остается в молярном избытке по отношению к матрице.
Из различных способов размножения лучше всего известным и шире всего используемым является ПЦР. ПЦР-версия ММК, также называемая методом "полоний" [14], была модифицирована путем ковалентной иммобилизации по крайней мере одного из праймеров на матриксе среды; как следствие, одна из комплементарных цепей размноженной ДНК также оказывается ковалентно иммобилизованной [15].
По ряду параметров ММК превосходит способы, основанные на размножении нуклеиновых кислот в растворах. Благодаря обнаружению молекул в виде счетных колоний, ММК делает анализ цифровым способом и позволяет производить прямой подсчет молекул. Благодаря пространственному разделению размножаемых молекул, ММК значительно снижает конкуренцию между мишенями при одновременном определении нескольких мишеней, а также помехи со стороны неспецифического синтеза, вызванного ошибочной гибридизацией праймеров с посторонними нуклеиновыми кислотами. На примере ПЦР-версии ММК было продемонстрировано отсутствие помех со стороны другой одновременно размножаемой мишени, даже если ее количество было в миллион раз выше, и было показано, что единственная молекула ДНК вируса гепатита Б может быть надежно обнаружена в 100 мкл цельной крови, содержащих в триллион раз большее количество ДНК человека [16]. Для сравнения, нижний предел обнаружения с помощью жидкостной ПЦР другой ДНК-мишени, вируса натуральной оспы, составляет около 500 молекул, как установлено в "слепых" экспериментах по ПЦР в реальном времени, осуществленных с использованием амплификатора Smart Cycler (Cepheid, США) и набора TaqMan (Roche Molecular Systems, США) [17].
Однако существующие способы обнаружения молекулярных колоний имеют существенные недостатки, которые препятствуют использованию ММК в качестве инструмента диагностики. Способы, использующие окрашивание колоний флуоресцентными интеркалирующими красителями, такими как этидиум бромид [18, 19] или "SYBR Green I" [14], или включение радиоактивно меченых нуклеотидов [19] являются неспецифическими: они выявляют колонии, образованные при размножении как мишени (анализируемой ДНК или РНК), так и посторонних нуклеиновых кислот. Специфические способы основаны на гибридизации колоний с мишень-специфичными олигонуклеотидными зондами. В работах [19, 20] раскрыт способ, включающий перенос материала колоний на нейлоновую мембрану, его иммобилизацию на мембране и последующую гибридизацию с радиоактивно-мечеными зондами. Недостатками способа являются как необходимость использования радиоактивных материалов (32P, 33P или 35S, которые к тому же имеют короткий период полураспада), так и длительность процедуры получения радиоавтографа, часто более суток. Способ, раскрытый в работе [21], включает гибридизацию колоний in situ (в геле, где они были выращены) с флуоресцентными зондами или с немечеными олигонуклеотидными зондами, которые затем метят путем концевого включения флуоресцентных нуклеотидов. Благодаря использованию флуоресцентных меток отпадает необходимость использования радиоактивных материалов и время получения изображения сокращается до нескольких минут, однако осуществление гибридизации в геле существенно усложняет и удорожает процедуру, так как размноженные нуклеиновые кислоты необходимо ковалентно иммобилизовать на матриксе геля (для чего иммобилизуют используемые для размножения олигонуклеотидные праймеры), а также необходимо удалять из геля одну из комплементарных цепей размноженной мишени (для чего используют расшиваемый гель и после размножения осуществляют частичную расшивку с целью облегчения диффузии нуклеиновых кислот). Кроме того, при гибридизации в геле мишени оказываются менее доступными зондам и удаление несвязанных зондов из геля происходит медленнее, чем при гибридизации на мембране.
Решением проблемы могло бы стать осуществление гибридизации с флуоресцентными зондами на мембранах, однако имеющиеся в литературе сведения указывают на невозможность обнаружения молекулярных колоний таким способом из-за слишком низкого содержания нуклеиновых кислот в колонии и слишком высокого уровня фоновой флуоресценции мембран. Так, молекулярные колонии, образующиеся при осуществлении ПЦР в геле, содержат до 10 молекул ДНК [14, 16], что эквивалентно ≈10-16 моль или 0,1 фемтомоль. Было неоднократно заявлено, что такие количества невозможно обнаружить посредством гибридизации флуоресцентных зондов с иммобилизованными на нейлоновой мембране нуклеиновыми кислотами из-за высокой собственной флуоресценции нейлона [22-26]. Следует отметить, что именно нейлоновые мембраны наиболее широко используют для гибридизации нуклеиновых кислот [24].
Для преодоления этой проблемы было предложено несколько способов. Одним из них является замена нейлоновых мембран на мембраны из материалов с более низким уровнем собственной флуоресценции, таких как полипропилен, полиэтилен или политетрафторэтилен [22]. Однако Вейсс с соавторами заявили, что чувствительность детекции с использованием таких мембран в 100 раз ниже, чем требуется для обнаружения десятых долей фемтомоля ДНК, из-за неэффективной иммобилизации мишени [23]. Вместо этого они предложили использовать нейлоновую мембрану, но вместо флуоресцентных зондов - биотинилированные зонды, взаимодействующие с конъюгатом стрептавидина и щелочной фосфатазы, и субстрат, который под действием фосфатазы превращается во флуоресцирующий продукт, адсорбирующийся на мембране. Таким образом, в месте гибридизации каждой молекулы зонда образуется множество флуоресцирующих молекул, то есть происходит многократное усиление сигнала. В работе [24] предложен аналогичный способ, отличающийся тем, что вместо флуоресценции используют явление хемилюминесценции. Такое сопряжение с ферментативной реакцией позволяет увеличить чувствительность детекции до 0,01-0,1 фемтомоль мишени [23, 24], что достаточно для обнаружения молекулярных колоний, однако многократно увеличивает сложность и стоимость метода диагностики. То же самое можно сказать и о способе, использующем биотинилированный зонд и конъюгат стрептавидина с фикоэритрином [25] - белком, интенсивность флуоресценции которого в 30-100 раз выше, чем интенсивность флуоресценции низкомолекулярных флуорофоров, используемых при изготовлении флуоресцентных зондов [27].
Для снижения собственной флуоресценции нейлоновой мембраны авторы патента [26] предлагают внедрять в нее частицы сажи или пигментов, способные гасить флуоресценцию мембраны. Однако остается неясным, позволяет ли это увеличить чувствительность детекции нуклеиновых кислот (авторы не приводят каких-либо данных на этот счет), так как те же частицы могут аналогичным образом гасить флуоресценцию связанного с мембраной зонда.
Помимо указанных проблем, существует еще одна: длительность процедуры гибридизации олигонуклеотидных зондов с нуклеиновыми кислотами, иммобилизованными на мембране, которая занимает от 4-5 ч [23, 24] до 16 ч [22, 25], что многократно превышает время, необходимое для осуществления ПЦР в геле (около 1 ч, см. пример 3).
В качестве прототипа данного изобретения может быть рассмотрен способ обнаружения молекулярных колоний посредством гибридизации с радиоактивно-меченым зондом на нейлоновой мембране [19], включающий перенос материала колоний на нейлоновую мембрану, его иммобилизацию на мембране и последующую гибридизацию с радиоактивно-мечеными зондами. Недостатками способа являются необходимость использования радиоактивных материалов и длительность процедуры получения радиоавтографа.
Задача данного изобретения состоит в разработке способа детекции молекулярных колоний, который существенно упрощает и ускоряет процедуру анализа, а также делает процедуру анализа более безопасной и дешевой.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Поставленные задачи решаются тем, что в способе обнаружения и определения количества нуклеиновых кислот, который включает (а) приготовление образца, содержащего, по крайней мере, один вид определяемой нуклеиновой кислоты (мишени), (б) размножение мишени в виде молекулярных колоний посредством осуществления реакции размножения в иммобилизованной среде, содержащей бесклеточную ферментную систему экспоненциального размножения нуклеиновых кислот, (в) перенос содержимого молекулярных колоний на мембрану, (г) иммобилизацию перенесенных нуклеиновых кислот на мембране, (д) обнаружение нуклеиновых кислот на мембране посредством гибридизации с зондом, комплементарным последовательности мишени, используют флуоресцентно-меченый зонд и после гибридизации получают двумерное флуоресцентное изображение мембраны в режиме, обеспечивающем регистрацию флуоресценции зонда.
К другому аспекту изобретения относится то, что на этапе размножения используют праймер-зависимую ферментную систему экспоненциального размножения нуклеиновых кислот, включающую мишень-специфичные праймеры, а в качестве указанной праймер-зависимой системы используют либо систему полимеразной цепной реакции, причем для осуществления размножения осуществляют циклические изменения температуры иммобилизованной среды, обеспечивающие протекание этой реакции, либо используют изотермическую ферментную систему экспоненциального размножения, которую выбирают из группы систем, включающей 3SR, SDA, NASBA, RCA и LAMP.
К следующему аспекту изобретения относится то, что в качестве мишени используют ДНК или РНК, которую превращают в ДНК посредством обратной транскрипции, причем каждую из мишеней определяют, гибридизуя мембрану либо последовательно с соответствующими мишень-специфичными флуоресцентными зондами, либо одновременно со смесью соответствующих мишень-специфичных флуоресцентных зондов, спектральные характеристики которых различаются достаточно, чтобы обеспечить избирательную регистрацию флуоресценции каждого из них, и после гибридизации получают двумерные флуоресцентные изображения мембраны в режиме, обеспечивающем избирательную регистрацию флуоресценции каждого из зондов.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
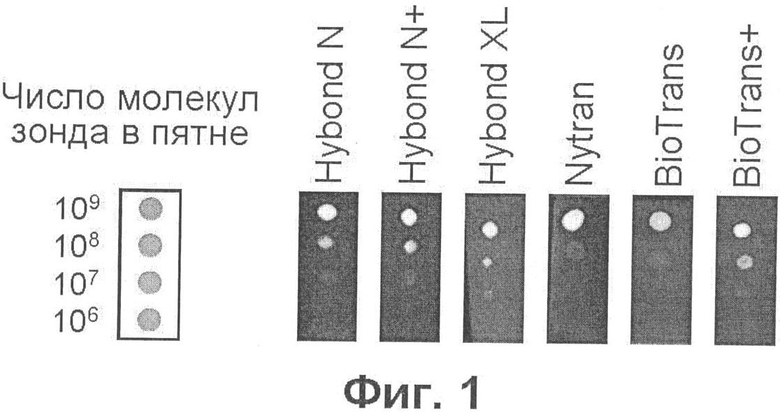
Фиг.1 демонстрирует чувствительность детекции флуоресцентного зонда, меченного флуорофором Су5, на нейлоновых мембранах разного типа и разных производителей.
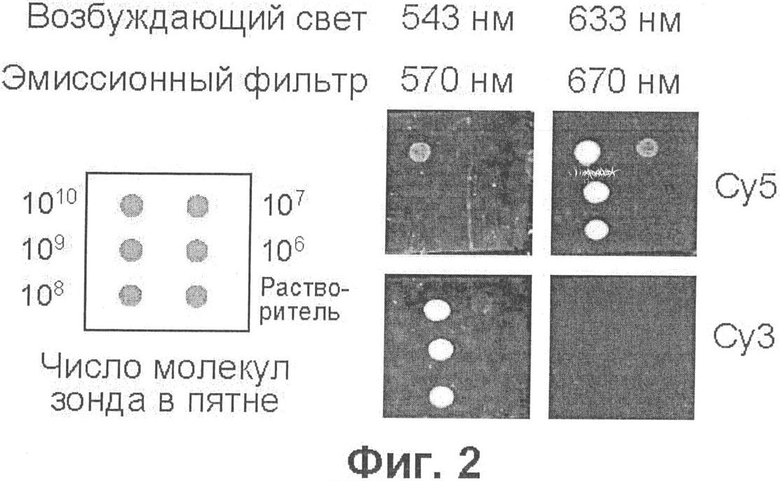
Фиг.2 демонстрирует чувствительность детекции зондов, меченных флуорофорами Су3 и Су5, на нейлоновой мембране Hybond N+.
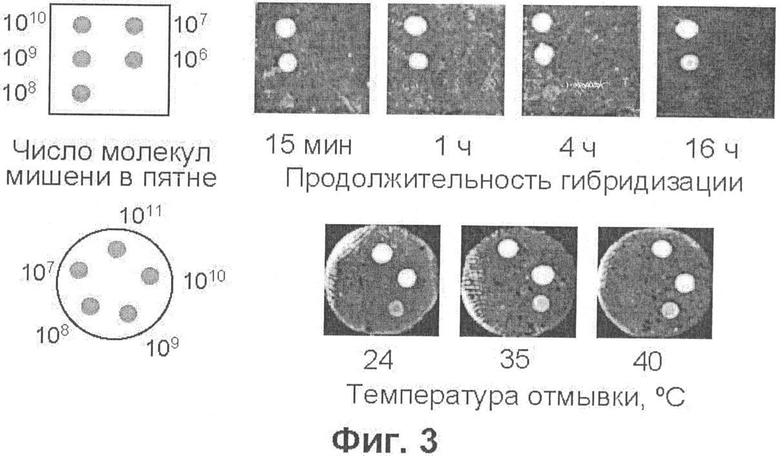
Фиг.3 демонстрирует детекцию известных количеств ДНК-мишени, нанесенной на нейлоновую мембрану Hybond N+, посредством гибридизации с флуоресцентным зондом, меченным флуорофором Су5, а также зависимость результата от времени гибридизации и от температуры отмывки несвязанного зонда.
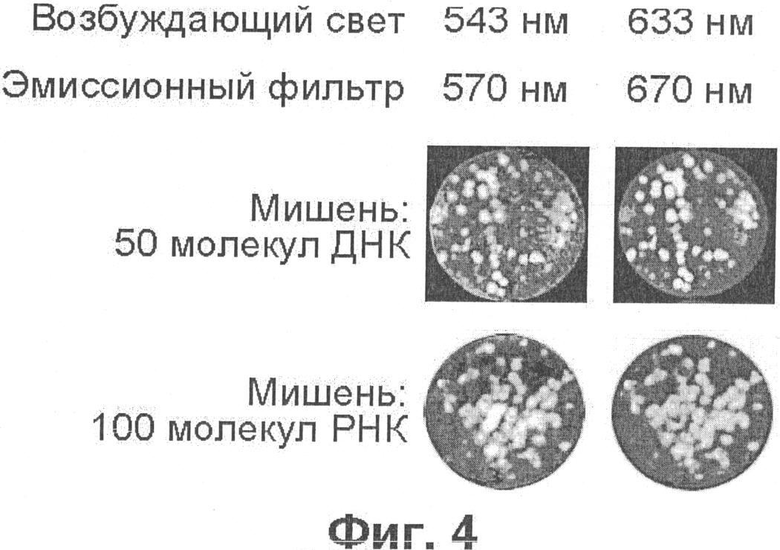
Фиг.4 демонстрирует обнаружение молекулярных колоний при помощи гибридизации на нейлоновой мембране Hybond N+с олигонуклеотидными зондами, меченными разными флуорофорами.
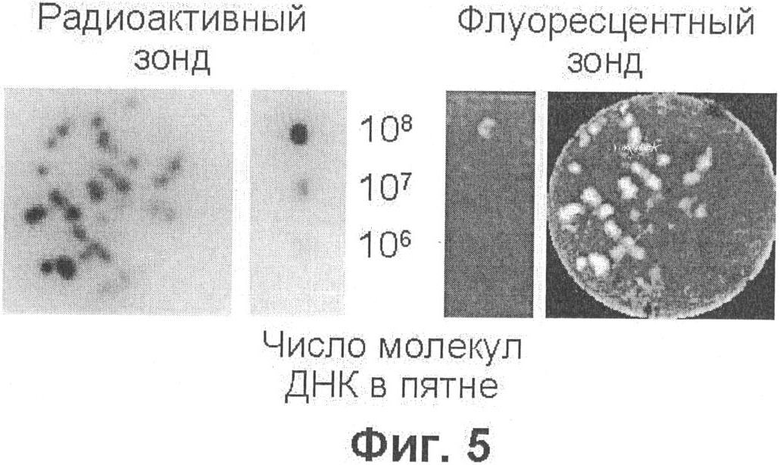
Фиг.5 демонстрирует результаты гибридизации молекулярных колоний и стандартов количества ДНК с радиоактивным и флуоресцентным зондами.
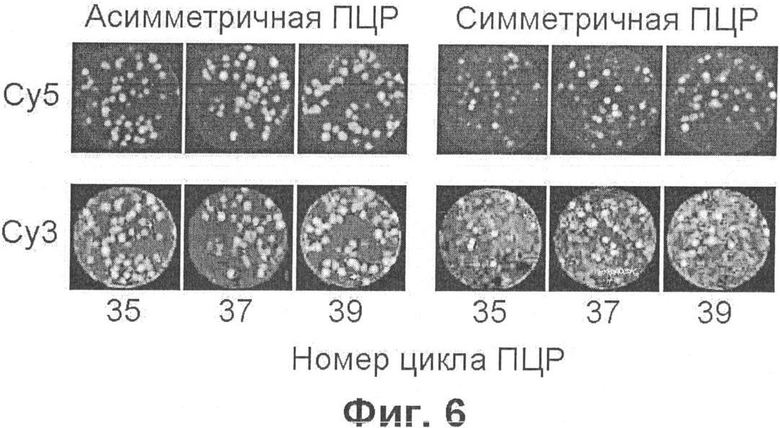
Фиг.6 демонстрирует обнаружение колоний ДНК, полученных в результате асимметричной и симметричной ПЦР, при помощи гибридизации с олигонуклеотидными зондами, меченными разными флуорофорами.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение основано на наблюдении, что молекулярные колонии, полученные в результате осуществления ПЦР в геле, могут быть обнаружены при помощи процедуры, включающей перенос материала колоний на мембрану, его иммобилизацию на мембране и гибридизацию с флуоресцентными олигонуклеотидными зондами. Показано, что сигнал, производимый гибридизованными зондами, достаточно велик, чтобы его можно было отличить от фоновой флуоресценции мембраны. Одна и та же мембрана может быть гибридизована одновременно с несколькими зондами, имеющими разную нуклеотидную последовательность и меченными разными флуоресцентными группами. При этом колонии, гибридизующиеся с каждым из зондов, можно наблюдать отдельно от других колоний посредством регистрации специфического излучения данного флуорофора, характеризующегося определенной длиной волны, при его возбуждении подходящим источником света. Также показано, что продолжительность процедуры гибридизации может быть существенно сокращена по сравнению с известными способами.
Как указано выше, нейлоновые мембраны наиболее широко используют для переноса и иммобилизации нуклеиновых кислот.Это связано с тем, что нейлоновые мембраны относительно дешевы и безопасны в производстве; они могут связывать нуклеиновые кислоты с высокой емкостью, на них легко ковалентно иммобилизовать нуклеиновые кислоты (например, путем обработки низкими дозами ультрафиолетовой радиации); они характеризуются низким уровнем сорбции олигонуклеотидных зондов и, следовательно, низким неспецифическим фоном гибридизации, а также высокой механической прочностью по сравнению с другими типами мембран, что позволяет многократно проводить повторные гибридизации. Поэтому нейлоновые мембраны прекрасно зарекомендовали себя при гибридизации с радиоактивными зондами.
Однако нейлоновые мембраны практически не используют для гибридизации нуклеиновых кислот с флуоресцентными зондами, особенно когда ожидаемое количество мишени (анализируемой нуклеиновой кислоты) относительно мало, поскольку считается, что нейлоновые мембраны обладают высоким уровнем собственной флуоресценции в широком диапазоне ультрафиолетового и видимого света, которая ухудшает соотношение сигнал/фон [22-26]. В частности, было заявлено, что количества специфической ДНК меньшие, чем 1 фемтомоль, можно обнаружить только при многократном усилении флуоресценции с использованием ферментных систем [23]. Отсюда, в частности, следовало, что выращенные с помощью ПЦР колонии ДНК не могут быть обнаружены путем гибридизации с простыми флуоресцентными зондами, так как одна колония содержит лишь около 108 молекул, то есть ≈0,1 фемтомоль ДНК [14, 16]. Более того, так как эффективность переноса ДНК на мембрану составляет около 10% [28], на месте каждой колонии на мембране может оказаться лишь 10 молекул, то есть ≈0,01 фемтомоль (10 аттомоль) ДНК, что близко к пределу чувствительности, а следовательно, не гарантирует надежную детекцию колоний даже при использовании ферментных систем [23, 24].
Поскольку не удалось найти в литературе точных данных о чувствительности детекции нуклеиновых кислот на мембранных фильтрах с помощью флуоресцентных зондов, проведено собственное исследование, результаты которого приведены ниже.
С целью определения нижнего предела чувствительности детекции, разные количества олигонуклеотидного зонда, меченного цианиновым красителем Су5 (флуоресцирующим в красной области), нанесли в виде точек на нейлоновые мембраны разного типа и разных производителей. Флуоресцентные изображения мембран (фиг.1) получены на сканере микрочипов в оптимальных условиях, как описано в примере 1. Результаты, представленные на фиг.1, показывают, что чувствительность детекции практически не зависит от производителя и что максимальная чувствительность (107 молекул) наблюдается при использовании мембран, несущих положительный заряд [Hybond N+, Hybond XL и BioTrans(+)]. По-видимому, более высокая чувствительность детекции на положительно заряженных мембранах является следствием того, то в этом случае образец (олигонуклеотидный зонд) меньше расплывается при нанесении на мембрану. Фиг.2 демонстрирует, что почти такая же чувствительность наблюдается при детекции зонда, меченного другим цианиновым красителем (Су3), флуоресцирующим в желто-зеленой области, а также что, используя подходящие значения длины волны возбуждающего и регистрируемого света, можно избирательно наблюдать флуоресценцию каждого из этих красителей.
Установленная в вышеприведенном эксперименте чувствительность детекции флуоресцентного зонда на нейлоновой мембране оказалась неожиданно высокой в свете литературных данных, а минимальное детектируемое количество зонда оказалась приблизительно равным количеству ДНК, переносимой из одной колонии на мембрану. Тем не менее, очевидно, что такая чувствительность не достаточна для обнаружения молекулярных колоний, так как эффективность гибридизации зонда, тем более на мембране (а не в растворе) и тем более с продуктом ПЦР (двутяжной ДНК), должна быть существенно ниже 100%. Действительно, прямые эксперименты показывают, что посредством гибридизации с флуоресцентным зондом не удается обнаружить количества специфической ДНК меньшие, чем 108 молекул (фиг.3 и пример 2).
Поэтому совершенно неожиданным стало наблюдение, что молекулярные колонии, образованные химерным геном AML1-ETO и соответствующей мРНК, являющимися маркерами одного из видов лейкоза, можно обнаружить посредством гибридизации на нейлоновой мембране с олигонуклеотидными зондами, меченными как Су5, так и Су3 (фиг.4; пример 3). Судя по тому, что число колоний совпадает, в пределах статистического разброса, с числом молекул ДНК, введенных в гель до начала ПЦР (верхний ряд на фиг.4), можно заключить, что выявляются все колонии ДНК. Молекулы РНК выявляются с эффективностью около 50% (нижний ряд на фиг.4), что совпадает с результатом, полученным при использовании вирусных мишеней и радиоактивно-меченых зондов [16].
Полученный результат можно было бы объяснить тем, что в опубликованных работах [14, 16] содержание ДНК в молекулярных колониях было занижено, либо тем, что молекулярные колонии, полученные в данном эксперименте с использованием этой конкретной мишени и конкретных праймеров, содержат необычно много ДНК. Чтобы проверить эти возможности, был проведен следующий эксперимент. Молекулярные колонии, полученные с использованием той же мишени и тех же праймеров, были сначала гибридизованы на нейлоновой мембране с радиоактивным 32Р-меченым зондом, как описано в примере 4 (фиг.5, слева). Из сравнения с гибридизацией того же зонда со стандартами количества ДНК-мишени (106, 107 и 108 молекул), нанесенными на такую же мембрану в виде миниатюрных аликвот, следует, что на месте каждой колонии на мембране оказывается в среднем ≈107 молекул ДНК, что, учитывая эффективность переноса [28], согласуется с предыдущими сообщениями [14, 16]. Затем мембраны с колониями и стандартами ДНК перегибридизовали с Су5-меченым зондом. Оказалось, что флуоресцентный зонд выявляет все молекулярные колонии, которые выявил радиоактивный зонд, причем даже те, из которых на мембрану попало порядка 106 молекул ДНК (фиг.5, справа). В то же время, из всех стандартов флуоресцентный зонд выявляет только пятно, содержащее максимальное количество мишени (10 молекул). Получается, что ДНК, перенесенная на мембрану из колоний, "светится" на 1-2 порядка ярче, чем ДНК, прямо нанесенная на мембрану.
Итак, полученные результаты указывают на то, что посредством гибридизации с флуоресцентными зондами на нейлоновой мембране можно обнаружить <107 молекул ДНК молекулярных колоний, то есть значительно меньшее количество, чем можно было ожидать, исходя из литературных данных и из результатов экспериментов, приведенных на фиг.1-3. Можно предположить что, в отличие от радиоактивного зонда, который одинаково хорошо "чувствует" ДНК колоний и стандартов, на чувствительность флуоресцентного зонда влияют некие параметры (например, соотношение сигнал/фон и квантовый выход), которые при детекции ДНК колоний оказываются иными и более благоприятными, чем при детекции ДНК, прямо нанесенной на мембрану. Какова бы ни была природа наблюдаемых эффектов, полученные результаты демонстрируют, что для детекции молекулярных колоний можно применять обычные флуоресцентные зонды вместо более дорогостоящих и менее стабильных систем, использующих флуоресцирующие белки или ферментативные способы усиления сигнала. Фактически, усиление сигнала достигается именно благодаря размножению нуклеиновых кислот в виде молекулярных колоний. Более того, установленная здесь чувствительность флуоресцентных зондов такова, что ее достаточно для детекции молекулярных колоний, полученных с помощью любых иных праймер-зависимых систем экспоненциального размножения нуклеиновых кислот, в том числе 3SR [8], SDA [9], NASBA [10], RCA [11, 12] и LAMP [13], так как во всех этих случаях коэффициент размножения (число копий, которые могут быть сделаны с одной молекулы мишени) превышает 107.
Очевидно, что для предложенного способа обнаружения молекулярных колоний вместо олигонуклеотидов в качестве гибридизационных зондов могут быть использованы аналоги олигонуклеотидов, в которых природный сахарофосфатный остов заменен на неприродный полимер, например, содержащий модифицированные фосфодиэфирные связи, неприродный сахарный компонент, химические аналоги фосфодиэфирной связи, полностью измененный остов, остов с 2'-5'-межнуклеотидными связями или составленный из α-нуклеотидных аномеров. Многочисленные примеры такого рода представлены в Патенте США №6329144 [29]. Примеры неприродных олигомеров, которые способны к правильному Уотсон-Криковскому спариванию с комплементарным участком нуклеиновой кислоты-мишени, включают, но не исчерпываются такими химическими аналогами, как фосфоротиоатные, метилфосфонатные, боранфосфонатные производные нуклеиновых кислот [30, 31], пептидные нуклеиновые кислоты, имеющие полиамидный остов [32, 33], и морфолиновые олигонуклеотиды [34, 35].
Очевидно также, что для предложенного способа обнаружения молекулярных колоний вместо нейлоновых мембран можно использовать другие мембраны, способность которых ковалентно связывать нуклеиновые кислоты лучше или не существенно хуже, а уровень собственной флуоресценции ниже или не существенно выше, чем нейлоновых мембран.
Фиг.3 демонстрирует, что мембрану можно гибридизовать со смесью флуоресцентных зондов и получать информацию отдельно о гибридизации каждого из зондов при условии, что спектральные характеристики зондов различаются достаточно, чтобы обеспечить избирательную регистрацию флуоресценции каждого из них. В данном случае, зонды мечены флуорофорами Су3 и Су5, возможность избирательной регистрации флуоресценции которых демонстрируется фиг.2. Фиг.3 демонстрирует возможность использования разных флуоресцентных зондов для обнаружения разных частей химерной молекулы, являющейся маркером лейкоза: один из зондов специфичен к AML-части и мечен красным флуорофором Су5, а другой специфичен к ЕТО-части и мечен желто-зеленым флуорофором Су3. Гибридизация обоих зондов с одной и той же колонией свидетельствует о том, что данная колония образована химерными молекулами. Это повышает специфичность диагностики, так как позволяет отсечь колонии, образованные нехимерными молекулами, содержащимися в нормальных лейкоцитах. Таким образом, для определения одной мишени может быть полезно использовать два или более двух флуоресцентных зондов, нуклеотидные последовательности которых различаются. Разные зонды комплементарны разным участкам мишени или разным вариантам одного участка мишени. Гибридизацию одновременно с несколькими флуоресцентными зондами, различающимися как нуклеотидными последовательностями, так и флуорофорами, можно использовать и в других целях, например, для раздельного определения сразу нескольких мишеней либо для определения вариантов последовательности одной мишени, например, для определения так называемого SNP (single nucleotide polymorphism, однонуклеотидного полиморфизма). Предпочтительно гибридизовать мембрану смесью зондов, спектральные характеристики которых различаются достаточно, чтобы обеспечить избирательную регистрацию флуоресценции каждого из них, для того чтобы после гибридизации получать двумерные флуоресцентные изображения мембраны в режиме, обеспечивающем избирательную регистрацию флуоресценции каждого из зондов.
Проведенные эксперименты показали, что процедура гибридизации может быть существенно ускорена и упрощена по сравнению с существующими методами. Оказалось, что время гибридизации может быть сокращено, по крайней мере, до 15 мин; увеличение продолжительности гибридизации до 4 ч (как рекомендовано в работах [23, 24]) или до 16 ч (как рекомендовано в работах [22, 25]) не приводит к увеличению флуоресцентного сигнала (фиг.3, верхний ряд). Также, отмывку негибридизованного зонда можно осуществлять при комнатной температуре; при этом уровень фоновой флуоресценции оказывается не выше, чем когда отмывку осуществляют при 40°С (фиг.3, нижний ряд).
Наконец, фиг.6 демонстрирует результат, позволяющий еще более упростить способ обнаружения молекулярных колоний. Если при осуществлении ПЦР в гель ввести больше того праймера, который используется при синтезе одной из цепей ДНК, то в каждой молекулярной колонии образуется молярный избыток этих цепей; иными словами, имеет место так называемая "асимметричная" ПЦР. Образующиеся в избытке цепи остаются в однотяжном состоянии, что позволяет осуществлять их гибридизацию с комплементарным флуоресцентным зондом, минуя стадию денатурации ДНК. Видно, что сигнал, получаемый при этом от молекулярных колоний (фиг.6, слева), не меньше, чем в случае обычной (симметричной) ПЦР, после которой ДНК подвергают щелочной денатурации перед ее иммобилизацией на мембране (фиг.6, справа).
Ниже приведены примеры, которые включают, но не ограничивают объем рассматриваемого изобретения.
Пример 1. Определение чувствительности детекции флуоресцентных зондов на нейлоновых мембранах.
Олигодезоксинуклеотиды Cy5-AML (последовательность SEQ ID NO:1 AATCACAGTGGATGGGCCCCGAGAA) и Су3-ЕТО (последовательность SEQ ID NO:2 ACCGTACTGAGAAGCACTCCACAAT) были синтезированы ЗАО "Синтол", Россия. Здесь Су3 (максимум возбуждения при 548 нм, максимум эмиссии при 562 нм) и Су5 (максимум возбуждения при 646 нм, максимум эмиссии при 664 нм) - цианиновые красители (торговая марка фирмы Amersham Biosciences Trading GmbH, в настоящее время подразделение GE Healthcare, Австрия); флуорофоры ковалентно связаны с олигонуклеотидами через ножку PO4(CH2)6NH2 (http://www.syntol.ru). Указанные олигонуклеотиды последовательно разводили в буфере А, содержащем 10 мМ трис-HCl (рН 9,0), 0,1 мМ ЭДТА (этилендиаминтетраацетат), 0.1% полиэтиленгликоль 6000 ("for synthesis"; арт. №807491; Merck, Германия) и 10 нг/мкл поли(А) (полиадениловой кислоты) (кат.№Р 9403; Sigma Chemical Company, США), до получения требуемой концентрации, и наносили в виде миниатюрных аликвот (0,2-0,5 мкл) на кусочки нейлоновых мембран следующих марок: Hybond N (кат. №RPN303N), Hybond N+(кат. №RPN303B) и Hybond XL (кат. №RPN303S) фирмы Amersham Biosciences; Nytran-N (NY 12 N, кат. №414420) фирмы Schleicher & Schull (Германия); BioTrans (кат. №810305) и BioTrans(+) (кат.№810204) фирмы ICN, в настоящее время МР Biomedicals (США). После высыхания пятен мембраны наклеивали на предметные стекла для микроскопии с помощью двусторонней липкой ленты и сканировали при разрешении 50 мкм с помощью конфокального сканера микрочипов ScanArray Express (PerkinElmer, США), используя зеленый (543 нм) лазер и эмиссионный фильтр 570 нм для регистрации флуоресценции Су3, или красный (633 нм) лазер и эмиссионный фильтр 670 нм для регистрации флуоресценции Су5. Результаты представлены на фиг.1 и 2 (расположение пятен, содержащих указанные количества зондов, показано на схемах слева).
Пример 2. Детекция ДНК посредством гибридизации на нейлоновой мембране с флуоресцентными зондами.
В качестве мишени использовали плазмиду (любезно предоставленную М. В. Фалалеевой, Институт белка РАН), несущую фрагмент кДНК химерной мРНК AML1-ЕТО, ассоциированной с острым миелоидным лейкозом, вызванным хромосомной транслокацией t (8; 21)(q22; q22) [36], вовлекшей гены AML1 (GeneBank accession number D43969) и ETO (GenBank accession number D14289). Плазмида была получена следующим образом. Участок мРНК AML1-ETO был размножен посредством обратной транскрипции с последующей ПЦР, с использованием в качестве матрицы суммарного препарата РНК, выделенного из лейкоцитов больного данным видом лейкоза, а также 5'-фосфорилированных праймеров AML1-A (последовательность SEQ ID NO:3 CTACCGCAGCCATGAAGAACC) и ЕТО-В (последовательность SEQ ID NO:4 AGAGGAAGGCCCATTGCTGAA), описанных в работе [37]. Продукт размножения длиной 395 п.о. лигировали в вектор pTZ19R (GeneBank accession number L08957), предварительно обработанный эндонуклеазой рестрикции Smal и дефосфорилированный. Клонированную вставку проверили прямым секвенированием. Полученная последовательность SEQ ID NO:5
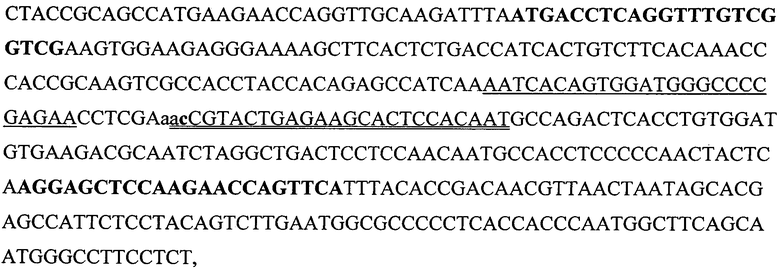
имела точечную замену по сравнению с опубликованной [38]: t→с, сразу после динуклеотида "аа", представляющего собой стыковочные нуклеотиды генов AML1 и ЕТО. Участок, подчеркнутый одинарной линией, идентичен последовательности зонда Cy5-AML, а участок, подчеркнутый двойной линией, идентичен последовательности зонда Су3-ЕТО; соответственно зонды способны гибридизоваться с комплементарной цепью ДНК.
Плазмиду выделяли по методу [39] и подвергали обработке эндонуклеазой рестрикции MspI для ее деградации на фрагменты с целью приближения ее свойств к свойствам ПЦР-продуктов. Полученный препарат последовательно разводили в буфере А (пример 1) до получения требуемой концентрации и наносили в виде миниатюрных аликвот (0,2-0,5 мкл) на кусочки нейлоновой мембраны Hybond N+. Концентрацию исходного препарата плазмиды определяли спектрофотометрически, полагая, что 1 OD260 единица равна 50 мкг/мл двутяжной ДНК [40], и принимая во внимание размер плазмиды и число Авогадро (6,02×1023) для пересчета измеренного количества ДНК в число молекул.
После высыхания пятен мембрану подвергали процедуре гибридизации, приспособленной для выявления молекулярных колоний. Мембрану промывали 80% этанолом в течение 5 мин при 4°С, высушивали в течение 15 мин при комнатной температуре и выдерживали при 80°С в течение 1,5 мин на фильтровальной бумаге №3 (Whatman, США), смоченной раствором, содержащим 0,5 М NaOH и 10 мМ ЭДТА, с целью денатурации ДНК. Денатурированную ДНК иммобилизовали на мембране (без высушивания) с помощью ультрафиолетового излучения в приборе Stratalinker (Stratagene, США) при энергии 160 мДж/см2, после чего мембрану промывали в буфере Б {1-кратный буфер SSPE [10 мМ фосфат натрия (рН 7,7), 180 мМ NaCl, 1 мМ ЭДТА], содержащий 0,5% SDS (додецилсульфат натрия)}, в течение 10 мин при комнатной температуре, а затем в буфере В (4-кратный SSPE, содержащий 1% SDS), в течение 5 мин при 50°С. Промытую мембрану гибридизовали при 50°С в запаянном полиэтиленовом пакете в буфере В, содержащем по 10 нг зондов Cy5-AML и Су3-ЕТО, из расчета 50 мкл буфера на 1 см2 мембраны, в течение 15 мин, 1 ч, 4 ч или 16 ч, после чего отмывали от негибридизованного зонда в буфере Б (5 мл на мембрану) дважды в течение 10 мин при комнатной температуре и третий раз при комнатной температуре (24°С), 35°С или 40°С. Мембрану высушивали и сканировали, как описано в примере 1. В первых экспериментах буферы Б и В также содержали 20% формамид. Однако, поскольку присутствие формамида не влияло на результаты гибридизации, в дальнейшем от его использования отказались. Результаты экспериментов представлены на фиг.3 (показаны изображения, полученные при регистрации флуоресценции Су5; расположение пятен, содержащих указанные количества плазмиды, показано на схемах слева).
Пример 3. Детекция молекулярных колоний посредством гибридизации на нейлоновой мембране с флуоресцентными зондами.
Молекулярные колонии выращивали в полиакриламидном геле, который приготавливали в лунках (диаметром 14 мм, глубиной 0,4 мм), высверленных в предметных стеклах толщиной 1 мм в мастерской Института белка РАН (Пущино, Московская область, РФ). Стекла обезжиривали путем вымачивания в 0,5 М NaOH в течение ночи, тщательно промывали и высушивали, после чего внутреннюю поверхность лунок обрабатывали с помощью реагента PlusOne™ Bind-Silane A 174 (кат. №17-1330-01; Amersham Biosciences) путем вливания в каждую лунку 80 мкл разбавленного реагента (0,4% в водном растворе 2% уксусной кислоты и 80% этанола). После испарения растворителя (около 1 ч при комнатной температуре) стекла промывали водой, затем отмывали этанолом (20 мл на стекло) в течение 15 мин при помешивании, снова промывали и высушивали. В каждую лунку вливали 65 мкл дегазированного раствора, содержащего 8% акриламид, 0,12% N,N'-метиленбисакриламид и 0,04% персульфат аммония, непосредственно перед использованием смешанного с TEMED (N,N,N',N'-tetramethylethylenediamine) до конечной концентрации 0,5%. В процессе заполнения лунки закрывали путем надвигания другого предметного стекла, предварительно обработанного реагентом PlusOne™ Repel-Silane ES (кат. №17-1330-01; Amersham Biosciences) по инструкции производителя. После инкубации в течение 40 мин при комнатной температуре и затем в течение 1 ч при 4°С, верхнее стекло снимали, нижнее стекло (содержащее гели) трижды кипятили в течение 20 мин в 40 мл/стекло деионизованной воды и высушивали при комнатной температуре в течение ночи.
Перед экспериментом гели восстанавливали до их первоначального объема путем пропитывания в полной ПЦР-смеси. С этой целью, вокруг каждой лунки наносили кольцо из 2-3 мкл минерального масла (для ИК-спектроскопии; кат. № 69808; Fluka Chemie, Швейцария), лунку наполовину закрывали покровным стеклом, под которое вливали 60-65 мкл ПЦР-смеси, одновременно надвигая покровное стекло так, чтобы заполнить лунку без пузырьков воздуха. Затем каждую лунку заклеивали поверх покровного стекла кусочком липкой фольги для ПЦР (кат. № 0030 127.471; Eppendorf AG, Германия) и до начала ПЦР инкубировали при 4°С в течение 1,5 ч с тем, чтобы гель полностью поглотил жидкость.
ПЦР-смесь имела следующий состав: 50 мМ трис-HCl (рН 8,7 при 20°С), 2,5 мМ MgCĺ2, 0,2 мМ каждого из dATP, dGTP, dTTP и dCTP, 0,2 мкМ каждого из праймеров AML1-C (последовательность SEQ ID NO:6 ATGACCTCAGGTTTGTCGGTCG) и ЕТО-D (последовательность SEQ ID NO:7 TGAACTGGTTCTTGGAGCTCCT), синтезированных ЗАО "Синтол", 1 мг/мл бычий сывороточный альбумин (БСА) (фракция V; кат.№735094; Roche Diagnostics, Германия), 1,2 нг/мкл Taq ДНК-полимеразы (приготовленной как описано [16]), а также ≈50 молекул ДНК-матрицы (плазмиды, описанной в примере 2) или продукт обратной транскрипции ≈100 молекул РНК-матрицы (РНК, полученной посредством транскрипции указанной плазмиды с помощью РНК-полимеразы фага Т7 [41]), осуществленной при 50°С в течение 60 мин в 10 мкл реакционной смеси следующего состава: 0,5 мкМ праймер ETO-D, 5 нг/мкл поли(А), 0,2 мМ каждого из dATP, dGTP, dTTP и dCTP, реакционный буфер для обратной транскрипции (Amersham Biosciences) и 50 единиц обратной транскриптазы M-MuLV (кат. № E70456Y, Amersham Biosciences).
Использованные в данном примере праймеры рекомендованы Европейской комиссией BIOMED-1 по международной стандартизации и клиническим испытаниям методов исследования минимальной остаточной болезни при острых лейкозах [37]. Участки гибридизации праймеров показаны жирным шрифтом в последовательности мишени (последовательность SEQ ID NO:5, пример 2), а задаваемая праймерами длина ПЦР-продукта составляла 260 пар оснований.
ПЦР осуществляли в амплификаторе UNO-Thermoblock™ (Biometra biomedizinische Analytik GmbH, Германия), оборудованном нагревательным блоком "in situ" с ровной поверхностью (кат. № 050-203) в следующем режиме: три первых ПЦР-цикла включали расплавление при 94°С в течение 15 с, отжиг при 58°С в течение 15 с и элонгацию при 72°С в течение 40 с. В последующих 40 циклах время расплавления составляло 6 с, а время элонгации в последнем цикле составляло 5 минут.
По окончании ПЦР материал колоний переносили на нейлоновую мембрану Hybond N+(Amersham Biosciences) посредством блоттинга (промокания) следующим образом. На гель накладывали мембрану диаметром 14 мм, один слой фильтровальной бумаги №3 (Whatman, США) диаметром 14 мм и резиновую прокладку, а сверху придавливали грузом (≈500 г на гель). После 20 минут блоттинга при комнатной температуре мембраны подвергали процедуре гибридизации, описанной в примере 2, а затем сканировали, как описано в примере 1. Результат приведен на фиг.4.
Пример 4. Детекция молекулярных колоний посредством гибридизации с радиоактивным зондом.
Молекулярные колонии выращивали как описано в примере 3 и, после переноса ДНК на нейлоновую мембрану и иммобилизации с помощью ультрафиолетового излучения, как описано в примере 2, подвергали следующей процедуре гибридизации.
Мембраны промывали в буфере Б в течение 10 мин при комнатной температуре, а затем в буфере Г (4-кратный SSPE, содержащий 1% SDS и 50% формамид), в течение 1 ч при 60°С. Промытые мембраны гибридизовали в течение ночи (18 часов) при 60°С в буфере Г, содержащем 32P-меченый радиоактивный зонд (≈1.000.000 распадов в 1 мин на 1 мл буфера), синтезированный посредством транскрипции описанной в примере 2 плазмиды с помощью РНК-полимеразы фага Т7 в присутствии [α-32P] [19, 20]. Гибридизацию осуществляли в запаянном полиэтиленовом пакете, помещая мембраны между листами фильтровальной бумаги, смоченными гибридизационным раствором из расчета 40 мкл/см2. После гибридизации мембраны промывали буфером Б (5 мл на мембрану) дважды в течение 15 мин при комнатной температуре и третий раз в течение 15 мин при 60°С. Мембраны экспонировали в течение ночи на экран MS и получали их изображение, сканируя экран на фосфоримиджере Cyclone™ (Packard Instrument, США).
Для перегибридизации с флуоресцентным зондом радиоактивный зонд отмывали в денатурирующем растворе (0,5 М NaOH и 10 мМ ЭДТА), затем мембраны промывали в 1×SSPE и гибридизовали с зондом Cy5-AML, как описано в примере 2, минуя стадию денатурации и пришивки. Мембрану высушивали и сканировали, как описано в примере 1, в режиме регистрации флуоресценции Су5.
Пример 5. Детекция молекулярных колоний, полученных с помощью асимметричной ПЦР.
Асимметричную ПЦР в геле проводили аналогично симметричной ПЦР (пример 3) с той разницей, что концентрация праймера AML1-C (последовательность SEQ ID NO:6) была снижена до 0,06 мкМ и время элонгации в последнем цикле не удлиняли. После осуществления указанного числа циклов (фиг.6) материал колоний переносили на нейлоновые мембраны, как описано в примере 3, и подвергали процедуре гибридизации, описанной в примере 2, минуя стадию денатурации ДНК: после промывания 80% спиртом и высушивания, мембраны сразу подвергали ультрафиолетовому облучению. После гибридизации в течение 15 мин и отмывке при комнатной температуре (пример 2), мембраны высушивали и сканировали, как описано в примере 1.
Параллельный эксперимент по симметричной ПЦР осуществляли так же, с той разницей, что использовали 0,2 мкМ праймер AML1-C и ДНК перед иммобилизацией денатурировали.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Настоящее изобретение направлено на усовершенствование способов идентификации и определения количества нуклеиновых кислот, обладающих заданной нуклеотидной последовательностью. Использование ММК вместо жидкостных систем размножения нуклеиновых кислот позволяет существенно повысить чувствительность, точность и надежность анализа. Использование флуоресцентных меток вместо радиоактивных позволяет существенно снизить стоимость и сократить время, а также повысить безопасность процедуры анализа. Проведение гибридизации на мембранном фильтре позволяет упростить процедуру по сравнению с гибридизацией в геле; в частности, это позволяет исключить необходимость ковалентной иммобилизации гибридизуемой цепи нуклеиновой кислоты на матриксе геля. Изобретение может быть использовано во многих областях фундаментальной биологии, биотехнологии и медицины, например, с целью сверхчувствительного обнаружения вирусов и микроорганизмов, ранней диагностики рака и мониторинга минимальной остаточной болезни, а также обнаружения следов генетически модифицированных материалов в пищевых продуктах.
Список литературы
1. Четверин А.Б., Четверина Е.В. (1995). Способ размножения нуклеиновых кислот, способ их экспрессии и среда для их осуществления. Патент РФ №2048522.
2. Четверин А.Б., Четверина Е. В. (1998). Способ клонирования нуклеиновых кислот. Патент РФ №2114175.
3. Четверин А.Б., Четверина Е.В. (1998). Способ диагностики нуклеиновых кислот. Патент РФ №2114915.
4. Chetverin А.В., Chetverina H.V. (1997). Method for amplification of nucleic acids in solid media. US Patent 5616478.
5. Chetverin А.В., Chetverina H.V. (1999). Method for amplification of nucleic acids in solid media and its application for nucleic acid cloning and diagnostics. US Patent 5958698.
6. Chetverin А.В., Chetverina H.V. (1999). Solid medium for amplification and expression of nucleic acids as colonies. US Patent 6001568.
7. Saiki R.K. et al. (1988). Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239, 487-491.
8. Guatelli J.C. et al. (1990). Isothermal in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication. Proc. Natl. Acad. Sci. USA 87, 1874-1878.
9. Walker G.T., Little M.C., Nadeau J.G., Shank D.D. (1992). Isothermal in vitro amplification of DNA by a restriction enzyme/DNA polymerase system. Proc. Natl. Acad. Sci. USA 89, 392-396.
10. Compton, J. (1991). Nucleic acid sequence-based amplification. Nature 350, 91-92.
11. Fire A., Xu S.-Q. (1995). Rolling replication of short DNA circles. Proc. Natl. Acad. Sci. USA 92, 4641-4645.
12. Lizardi P.M. (2002). Molecular cloning using rolling circle amplification. U.S. Patent Application 20020048761.
13. Notomi Т. et al. (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63.
14. Mitra R.D., Church G.M. (1999). In situ localized amplification and contact replication of many individual DNA molecules. Nucleic Acids Res. 27, e34.
15. Church G.M., Mitra R. (2002). Replica amplification of nucleic acid arrays. US Patent 6485944.
16. Chetverina H.V., Samatov T.R., Ugarov V.I., Chetverin A.B. (2002). Molecular colony diagnostics: detection and quantitation of viral nucleic acids by in-gel PCR. BioTechniques 33, 150-156.
17. LeDuc J.W. et al. (2002). Smallpox Research Activities: U.S. Interagency Collaboration, 2001. Emerg. Infect. Dis. 8, 743-745.
18. Chetverin A.B., Chetverina H.V., Munishkin A.V. (1991). On the nature of spontaneous RNA synthesis by QP replicase. J. Mol. BioL, 222, 3-9.
19. Chetverina H.V., Chetverin A.B. (1993). Cloning of RNA molecules in vitro. Nucleic Acids Res. 21, 2349-2353 (прототип).
20. Chetverin A.B., Chetverina H.V., Demidenko A.A., Ugarov V.I. (1997). Nonhomologous RNA recombination in a cell-free system: evidence for a transesterification mechanism guided by secondary structure. Cell 88, 503-513.
21. Mitra R.D. et al. (2003). Digital genotyping and haplotyping with polymerase colonies. Proc. Natl. Acad. Sci. USA, 100, 5926-5931.
22. Caldwell K.D., Chu T.-J., Pitt W.G. (1992). DNA sequencing using fluorescence background electroblotting membrane. US Patent 5112736.
23. Weiss R.B., Kimball A.W., Gesteland R.F., Ferguson P.M., Dunn D.M., Di Sera L.J., Cherry J.L. (1995). Automated hybridization/imaging device for fluorescent multiplex DNA sequencing. US Patent 5470710.
24. Guerasimova A., Ivanov I., Lehrach H. (1999). A method of one-step enzyme labelling of short oligonucleotide probes for filter hybridisation. Nucleic Acids Res. 27, 703-705.
25. Guerasimova A. et al. (2001). New tools for oligonucleotide fingerprinting. Biotechniques 31, 490-495.
26. Andreoli R., Amin M., Meyering M., Chesterson R., Ostreicher E. (2004) Low fluorescence nylon/glass composites for micro-analytical diagnostic applications. US Patent 6734012.
27. Haugland R.P. (2002). Nucleic acid detections and genomics technology. In: Handbook of Fluorescent Probes and Fluorescent Products, 9th Edn., Molecular Probes, Inc., Chapter 6.
28. Samatov T.R., Chetverina H.V., Chetverin A.B. (2005). Expressible molecular colonies. Nucleic Acids Res. 33, e145.
29. Kubista M., Svanvik N. (2001). Probe for analysis of target nucleic acids. US Patent 6329144.
30. Eckstein F., Ed. (1991). Oligonucleotides and Analogues: a Practical Approach. IRL Press, Oxford.
31. Agrawal S., Ed. (1993). Protocols for Oligonucleotides and Analogs: Synthesis and Properties. Humana Press, Totowa, New Jersey.
32. Nielsen P.E., Egholm M., Berg R.H., Buchardt O. (1991). Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 254, 1497-1500.
33. Nielsen P.E., Egholm M., Eds. (1999). Peptide Nucleic Acids: Protocols and Applications. Horizon Scientific Press, Wymondham, UK.
34. Summerton J.E., Weller D.D., Stirchak E. (1992). Uncharged polynucleotide-binding polymers. US Patent 5142047.
35. Summerton J.E., Weller D.D. (1993). Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages. US Patent 5185444.
36. Viehmann S. et al. (2003). Monitoring of minimal residual disease (MRD) by real-time quantitative reverse transcription PCR (RQ-RT-PCR) in childhood acute myeloid leukemia with AML1/ETO rearrangement. Leukemia 17,1130-1136.
37. Van Dongen J.J.M. et al. (1999). Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: Investigation of minimal residual disease in acute leukemia. Leukemia 13, 1901-1928.
38. Miyoshi H. et al. (1993). The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J. 12, 2715-2721.
39. Ugarov V.I., Samatov T.R., Chetverina H.V., Chetverin A.B. (1999). Plasmid purification using hot Mg2+ treatment and no RNase. BioTechniques 26, 194-198.
40. Sambrook J., Russell D.W. (2001). Molecular cloning. Edn. 3, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
41. Gurevich V.V., Pokrovskaya I.D., Obukhova T.A., Zozulya S.A. (1991). Preparative in vitro mRNA synthesis using SP6 and T7 RNA polymerases. Anal. Biochem. 195, 207-213.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕСКОНТАКТНЫЕ СПОСОБЫ ОБНАРУЖЕНИЯ МОЛЕКУЛЯРНЫХ КОЛОНИЙ, НАБОРЫ РЕАГЕНТОВ И УСТРОЙСТВО ДЛЯ ИХ ОСУЩЕСТВЛЕНИЯ | 2006 |
|
RU2394915C2 |
| СПОСОБ КЛОНИРОВАНИЯ НУКЛЕИНОВЫХ КИСЛОТ | 1993 |
|
RU2114175C1 |
| СПОСОБ ДИАГНОСТИКИ НУКЛЕИНОВЫХ КИСЛОТ | 1993 |
|
RU2114915C1 |
| СПОСОБ АМПЛИФИКАЦИИ СПЕЦИФИЧНЫХ ФРАГМЕНТОВ НУКЛЕИНОВЫХ КИСЛОТ С ПОМОЩЬЮ РЕКУРРЕНТНОЙ ЦЕПНОЙ РЕАКЦИИ | 2007 |
|
RU2414510C2 |
| ОБНАРУЖЕНИЕ МИШЕНИ TSG-ПРАЙМЕРОМ | 2010 |
|
RU2551321C2 |
| СПОСОБ ГОМОГЕННОЙ ДЕТЕКЦИИ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО ПРОДУКТА ОДНОЦЕПОЧЕЧНОЙ АМПЛИФИКАЦИИ | 2005 |
|
RU2460804C2 |
| СПОСОБ АМПЛИФИКАЦИИ И ДЕТЕКЦИИ НУКЛЕОТИДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ В РЕАКЦИОННОЙ СМЕСИ И ИСПОЛЬЗУЕМЫЙ В НЕМ НАБОР | 2009 |
|
RU2523589C2 |
| НУКЛЕИНОВО-КИСЛОТНЫЙ ЗОНД | 2014 |
|
RU2668154C2 |
| Способ выявления структурных перестроек генома опухолевых клеток (химерных генов), определяющих выбор терапии и прогноз при острых лейкозах у детей, с использованием ОТ-ПЦР и последующей гибридизацией с олигонуклеотидным биологическим микрочипом (биочипом) | 2016 |
|
RU2639513C1 |
| СПОСОБ РАЗМНОЖЕНИЯ НУКЛЕИНОВЫХ КИСЛОТ, СПОСОБ ИХ ЭКСПРЕССИИ И СРЕДА ДЛЯ ИХ ОСУЩЕСТВЛЕНИЯ | 1992 |
|
RU2048522C1 |
Изобретение относится к области молекулярной биологии и биотехнологии и может быть использовано в молекулярной диагностике. Способ включает размножение анализируемой нуклеиновой кислоты в виде молекулярных колоний, перенос полученных колоний на мембрану с последующей иммобилизацией и обнаружением колоний нуклеиновых кислот на мембране. Обнаружение колоний нуклеиновых кислот на мембране осуществляют посредством гибридизации с флуоресцентно меченными зондами и последующего получения двумерного изображения мембраны в режиме, обеспечивающем регистрацию флуоресценции хотя бы одного использованного зонда. Применение изобретения позволяет обнаружить специфические молекулы РНК и ДНК в биологическом материале по более совершенной технологии. 26 з.п. ф-лы, 6 ил.
| Nucleic Acids Res | |||
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| СПОСОБ ДИАГНОСТИКИ НУКЛЕИНОВЫХ КИСЛОТ | 1993 |
|
RU2114915C1 |
| СПОСОБ РАЗМНОЖЕНИЯ НУКЛЕИНОВЫХ КИСЛОТ, СПОСОБ ИХ ЭКСПРЕССИИ И СРЕДА ДЛЯ ИХ ОСУЩЕСТВЛЕНИЯ | 1992 |
|
RU2048522C1 |
| СПОСОБ КЛОНИРОВАНИЯ НУКЛЕИНОВЫХ КИСЛОТ | 1993 |
|
RU2114175C1 |
| KR 1020060015668 A, 17.02.2006 | |||
| US 6696246, 24.02.2004 | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Methods Mol Med | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| BMC Biotechnol | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |

