Изобретение относится к фармацевтической промышленности и предназначено для профилактики и лечения заболеваний вирусной природы, таких как гепатиты, энцефалиты, хламидиозы, герпес, а также ВИЧ-обусловленных иммунодефицитных состояний.
В связи с широким распространением вирусных инфекций, необходимостью проведения длительных курсов лечения, а также острой потребностью в профилактике вирусных инфекций целесообразно создание пероральной лекарственной формы.
Известны пероральные лекарственные формы на основе замещенных акриданонов, а именно 10-карбоксиметил-2-хлор-9-акриданона, предназначенных для лечения вирусных заболеваний (пат. США N 3681360, C 07 D 37/20, заявл. 09.04.71, опубл. 01.08.72).
Прототип имеет следующий состав:
в качестве активного вещества - 10-карбоксиметил-2-хлор-9-акриданон структурной формулы (I)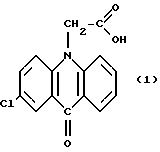
в количестве 42-79%;
традиционные связующие добавки: крахмал, масло какао, стеарат кальция или магния, лактоза, желатин, дизамещенный фосфат кальция, тальк в количестве суммарно 58-21%.
Активное вещество смешивается с неактивными связующими добавками и полученный гранулят прессуется в виде таблеток, содержащих по 250 или 500 мг активного вещества.
Пероральное лекарственное средство, описанное выше, практически не проявляет биологической активности, вследствие низкой биодоступности.
Заявителем было экспериментально установлено, что при попадании таблетки в желудок происходит высвобождение активного вещества в виде акридонуксусной кислоты, которая не всасывается в желудке и выводится с содержимым кишечника в неизменном виде, таким образом, упомянутое выше лекарственное средство не обладает биодоступностью.
Для создания биодоступного перорального лекарственного средства, обладающего высокой лечебной эффективностью, активное вещество должно находиться в таблетке в виде фармакологически приемлемой соли акридонуксусной кислоты, а распадаться такая таблетка должна, минуя желудок, в кишечнике.
При введении таблетки, содержащей активное вещество в виде фармакологически приемлемой соли в желудок, и в случае ее распадения в желудке, где pH желудочного сока составляет 1-2, произойдет выделение активного вещества в виде нерастворимой акридонуксусной кислоты, которая, как было описано выше, не является биодоступной и не оказывает лечебного противовирусного эффекта.
В случае распадения таблетки в кишечнике, где, как известно, щелочная среда, активное вещество выделится в виде соли, которая обладает биодоступностью, хорошо всасывается и оказывает высокое биологическое действие.
Введение в кишечник активного вещества в виде фармакологически приемлемой соли приводит к быстрому всасыванию вещества и проявлению высокого лечебного эффекта.
При введении в кишечник активного вещества в виде акридонуксусной кислоты, а не ее фармакологической соли, экспериментально установлена низкая (в 8-10 раз меньше) лечебная эффективность препарата по сравнению с заявляемым лекарственным средством. Это связано с недостаточно высоким значением pH кишечника для образования соли из акридонуксусной кислоты и ограниченным временем пребывания таблетки в организме, за которое препарат не успевает всосаться и оказать высокий лечебный эффект.
Задачей изобретения является создание перорального препарата, обладающего высокой биодоступностью и проявляющего высокую противовирусную эффективность.
Поставленная задача решается тем, что заявляется два варианта перорального лекарственного средства. Первый вариант лекарственного средства представляет собой таблетированное лекарственное средство для взрослых, содержащее активное вещество и целевые добавки, при этом в качестве активного вещества в него включена соль 10-карбоксиметил-9-акриданона структурной формулы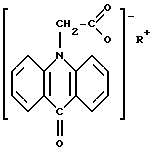
где
R = NH
а на ядро таблетки дополнительно нанесено защитное кислотоустойчивое покрытие, выбранное из группы: шеллак, ацетилфтолилцеллюлоза при следующих соотношениях компонентов, в мас. ч.:
Указанная соль 10-карбоксиметил-9-акриданона - 269-900
Целевые добавки - 50-225
Шеллак или ацетилфталилцеллюлоза - 35-50
Второй вариант лекарственного средства представляет собой гранулы, предназначенные для детей, содержащие активное вещество и целевые добавки, нанесенные на инертную частицу, при этом в качестве активного вещества в него включена соль 10-карбоксиметил-9-акридона структурной формулы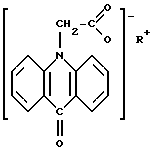
где
R = NH
а на гранулу дополнительно нанесено защитное кислотоустойчивое покрытие, выбранное из группы: шеллак, ацетилфтолилцеллюлоза при следующем соотношении компонентов, в мас. ч.:
Указанная соль 10-карбоксиметил-9-акриданона - 187-195
Инертная частица - 380-400
Целевые добавки - 10-15
Шеллак или ацетилфталилцеллюлоза - 40- 45
Обе заявляемые лекарственные формы направлены на решение одной и той же задачи - повышения биодоступности и увеличения противовирусной активности препарата и основаны на едином изобретательском замысле, заключающемся в том, что в композиции в качестве активного вещества используется 10-карбоксиметил-9-акриданон в виде фармакологически приемлемой соли, обеспечивающей быстрое проникновение активного вещества в органы и ткани организма. На таблетку или гранулу нанесено кислотоустойчивое защитное покрытие, способствующее высвобождению активного вещества в кишечнике.
Решение поставленной задачи иллюстрируется экспериментальными данными по структуре, способу получения и биологической активности активного вещества заявляемых лекарственных форм. Активное вещество заявляемых лекарственных форм относится к солям производных акриданонов, например, N-метилглюкаминовой соли акридонуксусной кислоты, общей формулы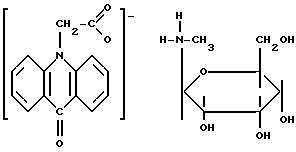
обладающим широким спектром биологической активности: интерфероногенной, противовирусной, в том числе антиВИЧ.
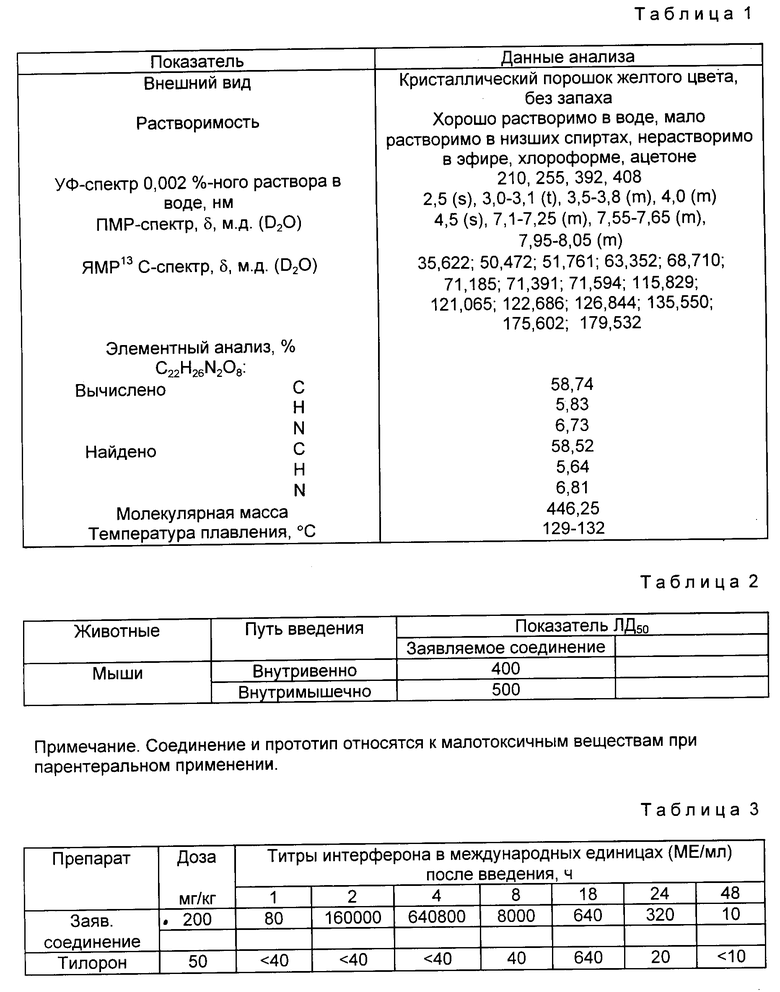
Пример 1. 99,6 г N-метил-N-(α, D-глюкопиранозиламина) /N-метилглюкамина/ растворяют в 200 мл дистиллированной воды и прибавляют порциями при перемешивании 125 г тщательно измельченной 2-(акридон-9-он-10-ил) уксусной кислоты, продолжают перемешивание при комнатной температуре до полного растворения и прибавляют 1000 мл этилового спирта. Выпавший осадок отфильтровывают, промывают на фильтре 100 мл этилового спирта и сушат при 60oC в течение 1 ч. В форме желтых кристаллов получают 224 г (100%) N-метил-N-(α, D-глюкопиранозил)-аммония-2-(акридон-9-он-10-ил) ацетата /соль N-метилглюкамина и акридонуксусной кислоты/, который после перекристаллизации из низших спиртов имеет т.пл. 129-131oC.
Элементный анализ C22H26N2O8:
Вычислено,%: C 58,74, H 5,83, N 6,73
Найдено,%: C 58,52, H 5,64, N 6,81.
Физико-химические свойства активного вещества заявляемых лекарственных форм представлены в табл. 1.
Мутагенным, терратогенным, эмбриотоксическим и аллергенным действием N-метил-N-(α, D-глюкопиранозил)-аммония-2-(акридон-9-он-10-ил) ацетат не обладает.
Изучение медико-биологических свойств N-метил-N-(α, D-глюкопиранозил)-аммония-2-(акридон-9-он-10-ил)ацетата проведено в эксперименте на животных, при этом выявлен широкий спектр биологической активности заявляемого вещества, сочетающийся с его низкой токсичностью, хорошей переносимостью и высоким индексом терапевтической эффективности.
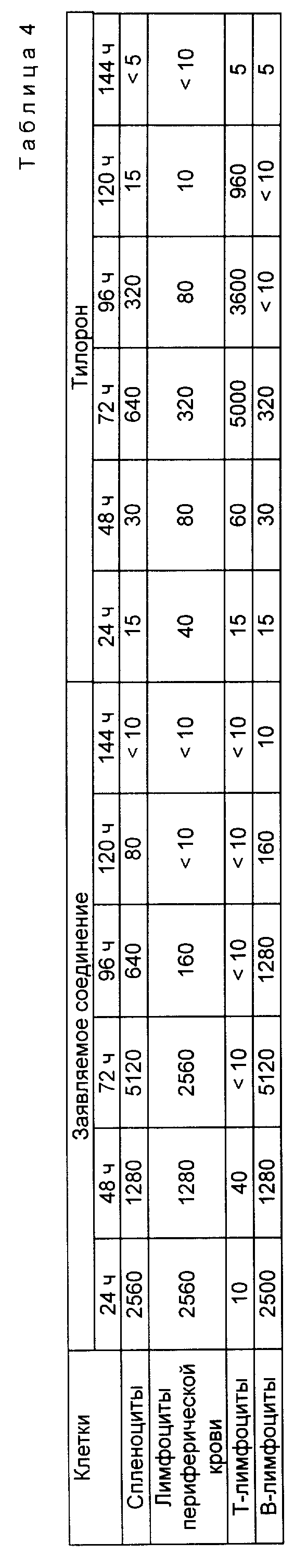
Опыт А. Острая токсичность активного вещества изучена на беспородных белых мышах линии ристал массой 18-20 г.
Препарат вводили внутривенно и внутримышечно в различных дозах от 2000 мг/кг и ниже с двухкратным шагом. Срок наблюдения 14 дней. Летальную дозу (ЛД50) рассчитывали по методу Кербера. Данные по изучению острой токсичности представлены в табл. 2.
Известно, что основой противовирусной активности любого химического соединения может служить выработка организмом универсального противовирусного белка - интерферона. Заявителем установлено, что этим свойством обладает и заявляемое соединение.
Опыт Б. Интерфероногенная активность.
Интерфероноиндуцирующую активность активного вещества изучали на мышах и в культуре клеток животного и человеческого происхождения.
Мышам линии CBA массой 10-12 г вводили однократно подкожно активное вещество в дозе 200 мг/кг и тилорон в дозе 50 мг/кг. Через определенные интервалы времени определяли титры интерферона в крови стандартным методом на гомологических клетках L-929, выращенных в 96-луночных планшетах в термостате с CO2. Тест-вирусом служил вирус энцефаломиокардита. Сравнительные данные интерфероноиндуцирующей активности заявляемого соединения и тилорона приведены в табл. 3.
Как видно из представленных данных, заявляемое соединение индуцирует продукцию раннего (2-8 ч) интерферона в высоких титрах. Пик продукции интерферона в этом случае превышает уровень интерферона в сыворотке мышей в ответ на введение тилорона в 1000 раз. Высокая интерфероноиндуцирующая активность заявляемого соединения подтверждена также на культуре клеток.
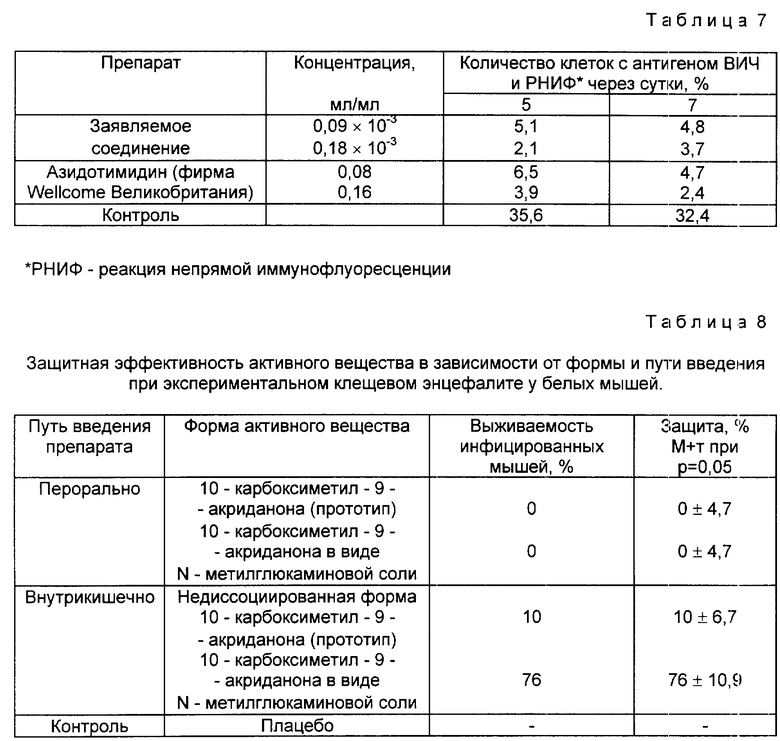
Из мышей линии CBA выделяли лимфоциты селезенки и лимфоциты периферической крови и индуцировали их заявляемым соединением и тилороном. Посадочная концентрация лимфоцитов составляла 5•106 кл/мл. Исследовали динамику накопления интерферона в культуральной жидкости лимфоцитов, выращенных на пластиковых панелях в присутствии CO2.
Титры интерферона в спленоцитах и клетках крови после введения заявляемого соединения и тилорона представлены в табл. 4.
Результаты, представленные в табл. 4, демонстрируют, что заявляемое соединение индуцирует существенно более высокие уровни интерферона в смешанной культуре лимфоцитов селезенки и периферической крови, чем тилорон.
Подтверждена интерфероногенная активность заявляемого соединения при действии в культуре клеток человека. С этой целью лимфоциты периферической крови человека получали путем разделения лейкоцитарной массы донорской крови. Посадочная концентрация лимфоцитов составляла 2•106 кл/мл. Клетки выращивали в 24-луночных пластиковых панелях. Индукцию лимфоцитов осуществляли заявляемым соединением и тилороном в концентрации 600 и 200 мг/мл соответственно. Интерферон в культуральной жидкости определяли титрованием на диплоидных человеческих клетках M-19. Тест-вирусом служил вирус везикулярного стоматита. Пробы интерферона нейтрализовали стандартными антисыворотками к α-9 и β-9 человеческого интерферона, контролем служили международные стандарты человеческого интерферона.
Данные индукции интерферона в лимфоцитах человека приведены в табл. 5.
Как следует из данных табл. 5, заявляемое соединение индуцирует продукцию интерферона в лимфоцитарных культурах периферической крови человека в 90 раз выше, чем тилорон.
Опыт В. Противовирусная активность заявляемого соединения.
1В. Установлена высокая активность N-метил-N-( α , D-глюкопиранозил)аммония-2-(акридон-9-он-10-ил) ацетата при экспериментальной инфекции мышей, зараженных вирусом клещевого энцефалита (КЭ).
В настоящее время для борьбы с этой инфекцией используется только инактивированная вакцина, обладающая весьма низкой эффективностью и требующая ежегодных прививок в связи с быстрым снижением резистентности к этой инфекции. Других средств защиты от КЭ практическая медицина не имеет.
Для установления эффективности заявляемого соединения в отношении КЭ использовали вирус КЭ штамм АБСЕТТАРОВ. Титр вируса при внутримозговом заражении составил 7,0-8,0 lg LD50, при подкожном заражении - 5,0 lg LD50. Эксперимент проводили на нелинейных белых мышах массой 12-14 г. Заражение животных осуществляли введением вируса КЭ подкожно. Заявляемое соединение растворяли в физрастворе и вводили опытной группе мышей подкожно. Контрольной группе мышей вводили подкожно физраствор. Наблюдение за экспериментальными животными проводилось в течение 21 дня. Критериями оценки эффективности заявляемого соединения служили выживаемость и средняя продолжительность жизни животных опытной группы по сравнению с контрольной. Результаты проведенного эксперимента представлены в табл. 6.
Представленные данные свидетельствуют о выраженном защитном действии препарата. Кроме того, специально проведенные исследования показали, что на 4 и 7 сутки после заражения вирус КЭ в мозгу мышей, получавших заявляемое соединение, не был обнаружен, в то время как в контрольной группе животных вирус репродуцировался в высоких титрах.
2В. Установлена активность заявляемого соединения в отношении вируса иммунодефицита человека (ВИЧ). В настоящее время известен единственный препарат, разрешенный для применения в медицинской практике как средство для лечения синдрома приобретенного иммунодефицита (СПИД). Это нуклеозидной природы азидотимидин (ретровир, зидовудин), существенными недостатками которого являются высокая токсичность, быстрое формирование устойчивости к нему штаммов вируса. Кроме того, препарат не обеспечивает выживаемости, а только пролонгирует продолжительность жизни больных СПИД.
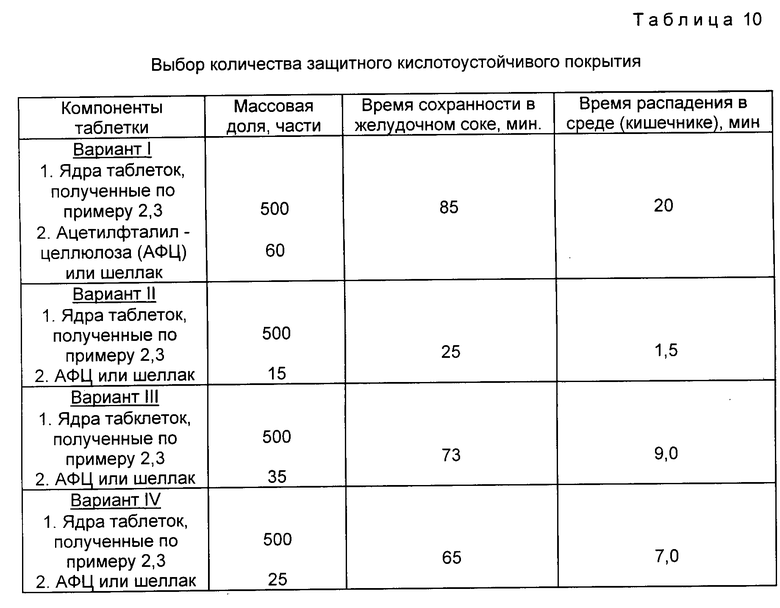
Для оценки эффективности заявляемого соединения на репродукцию ВИЧ использовали культуру клеток моноцитов линии И-937. Клетки выращивали в пробирках на среде RPMI-1640 с 20%-й фетальной телячьей сывороткой в конечной концентрации 0,5-0,7•106 кл/мл. Перед внесением препаратов в состав поддерживающей среды суспензию клеток инфицировали концентратом культуральной жидкости, питая полученные из клеток линии НТА-41, продуцирующих ВИЧ. Затем клетки инкубировали при 37oC в течение 3-4 суток с последующей сменой питательной среды. На 5 и 7 дни после инфицирования определяли наличие вирус-специфического антигена в реакции непрямой иммунофлуоресценции с использованием иммуноглобулинов к ВИЧ.
Результаты оценки ингибирующего действия заявляемого соединения на эспрессию антигенов ВИЧ в клетках моноцитов линии И-937, индуцирующих ВИЧ, в сравнении с азидотимидин представлены в табл. 7.
Таким образом, заявляемое соединение в концентрациях, почти в 1000 раз меньших, чем азидотимидин, оказывает равный эффект на экспрессию антигенов ВИЧ в культуре клеток моноцитов.
Аналогичными свойствами обладает аммониевая ( NH
Лечебная эффективность препарата изучалась на моделях клещевого энцефалита - заболевания вирусной природы у белых мышей. В эксперименте были задействованы три группы животных.
Первая группа - инфицированным белым мышам вводили перорально активное вещество в виде 10-карбоксиметил-9-акриданона, описанного в прототипе, и в виде N-метилглюкаминовой соли, приведенной в формуле изобретения.
Второй группе инфицированных животных те же соединения и в тех же дозах вводились непосредственно в кишечник, минуя желудок.
Третья группа инфицированных мышей лечения не получала (плацебо).
В результате эксперимента учитывали выживаемость инфицированных \\мышей и защитное действие препарата.
Как следует из табл. 8, при введении препарата внутрикишечно в виде соли эффективность лечения возрастает в 8-10 раз по сравнению с прототипом, что свидетельствует о высокой биодоступности лекарственного средства. В связи с тем, что заявляемое лекарственное средство является пероральным, необходимо нанесение защитного кислотоустойчивого покрытия, обеспечивающего доставку препарата и его высвобождение в кишечнике.
Медико-биологическими испытаниями установлено, что терапевтические лечебные дозы солей 10-карбоксиметил-9-акриданона составляют 269-900 мг на человека в сутки при однократном применении. В соответствии с этой дозой заявителем разработан оптимальный состав лекарственного средства в виде таблеток и гранул.
Ниже приведены экспериментальные данные по выбору оптимального состава ядра таблеток, табл. 9.
Экспериментальным путем устанавливалось соотношение активного вещества и связующих добавок. Критериями оценки выбираемых соотношений служили показатели биодоступности и качественные технологические характеристики ядра таблетки: прессуемость, прочность, гигроскопичность.
Как видно из табл. 9, оптимальными вариантами состава ядра таблетки являются варианты I и IV, обеспечивающие максимальную биодоступность активного вещества и высокие качественные технологические характеристики ядра таблетки.
Приведем способы получения ядра таблетки.
Пример 2. В стакане растворяют 550 мас.ч. аммониевой соли 10-карбоксиметил-9-акриданона (активного вещества) в 100 мас.ч. дистиллированной воды, 75 мас.ч. лактозы, 2 мас.ч. твина-80 и 13 мас.ч. стеарата кальция (связующие добавки). Полученную пасту высушивают при комнатной температуре в течение 18 ч в слое толщиной не более 0,5 см. Высушенный гранулит растирают, просеивают через сито и подают в смеситель таблеточной машины, где прессуют таблетки массой 640 мг. Каждая такая таблетка содержит 500 мг активного вещества.
Пример 3. В стакан вносят 45 мас.ч. дистиллированной воды, 445 мас.ч. активного вещества в виде N-метилглюкаминовой соли 10-карбоксиметил-9-акриданона, 13 мас.ч. твин-80 (связующая добавка). Полученную пасту тщательно перемешивают и высушивают, как в примере 2. В смеситель таблеточной машины загружают высушенную и просеянную пасту и 12 мас.ч. талька, после чего прессуют таблетки массой 500 мг. Каждая такая таблетка содержит 250 мг активного вещества.
Полученные таким образом таблетки являются полуфабрикатом (ядром) и покрываются сверху кислотоустойчивым покрытием, обеспечивающим защиту таблетки от распадения в желудочном соке в течение не менее одного часа (прохождение через желудок) и быстрый распад в кишечнике в течение 5-10 мин. Эти свойства лекарственной формы зависят от толщины слоя кислотоустойчивого покрытия, которое установлено нами экспериментально и регулируется количеством подаваемого в аппарат соответствующего ингредиента.
В табл. 10 приведено обоснование выбора количества защитного кислотоустойчивого покрытия, наносимого на ядро таблетки.
Критериями отбора служили два показателя:
время сохранности таблетки в желудочном соке;
время распадения таблетки в щелочной среде (кишечнике).
Как следует из табл. 10, сохранность таблеток в желудочном соке в течение 60-73 мин и быстрая распадаемость их в щелочных средах, имитирующих среду кишечника, обеспечивается в вариантах III и IV, когда количество защитной кислотоустойчивой оболочки составляет 35-50 мас.ч., что составляет 5-7% от массы ядер таблеток.
При увеличении количества защитного покрытия (вариант I) имеет место длительное время распадения таблетки в кишечнике, что приводит к снижению эффективности препарата.
При уменьшении количества защитного покрытия (вариант II) происходит распадение таблетки в желудке и, как было показано выше, лекарственное средство полностью утрачивает свою эффективность.
Приведем пример нанесения защитного кислотоустойчивого покрытия на ядро таблетки: 350 мас. ч. ядер таблеток, полученных по способу, описанному в примерах 2 и 3, помещают в установку псевдоожиженного слоя, куда распыляют 20 мас. ч. АФЦ или шеллака в виде 1% раствора в подходящем растворителе (например, спирте, хлороформе, ацетоне). При этом поверхность ядер таблеток равномерно покрывается защитной кислотоустойчивой оболочкой. После подачи всего количества АФЦ осуществляют подачу воздуха для удаления растворителя и закрепления защитной оболочки на поверхности ядра таблетки еще в течение не менее 1,5-2 ч.
Приведем пример осуществления изобретения на основе различных композиций.
I композиция
Активное вещество в виде аммониевой соли 10-карбоксиметил-9-акриданона - 269 мг
Целевые добавки - 231 мг
Шеллак или ацетилфталилцеллюлоза - 35 мг
Итого: - 535 мг
II композиция
Активное вещество в виде N-метилглюкаминовой соли 10-карбоксиметил-9-акриданона - 445 мг
Целевые добавки - 55 мг
Шеллак или ацетилфталилцеллюлоза - 35 мг
Итого: - 535 мг
III композиция
Активное вещество в виде аммониевой соли 10-карбоксиметил-9-акриданона - 538 мг
Целевые добавки - 102 мг
Ацетилфталилцеллюлоза - 45 мг
Итого: - 685 мг
IV композиция
Активное вещество в виде N-метилглюкаминовой соли 10-карбоксиметил-9-акриданона - 890 мг
Целевые добавки - 55 мг
Ацетилфталилцеллюлоза - 47 мг
Итого: - 992 мг
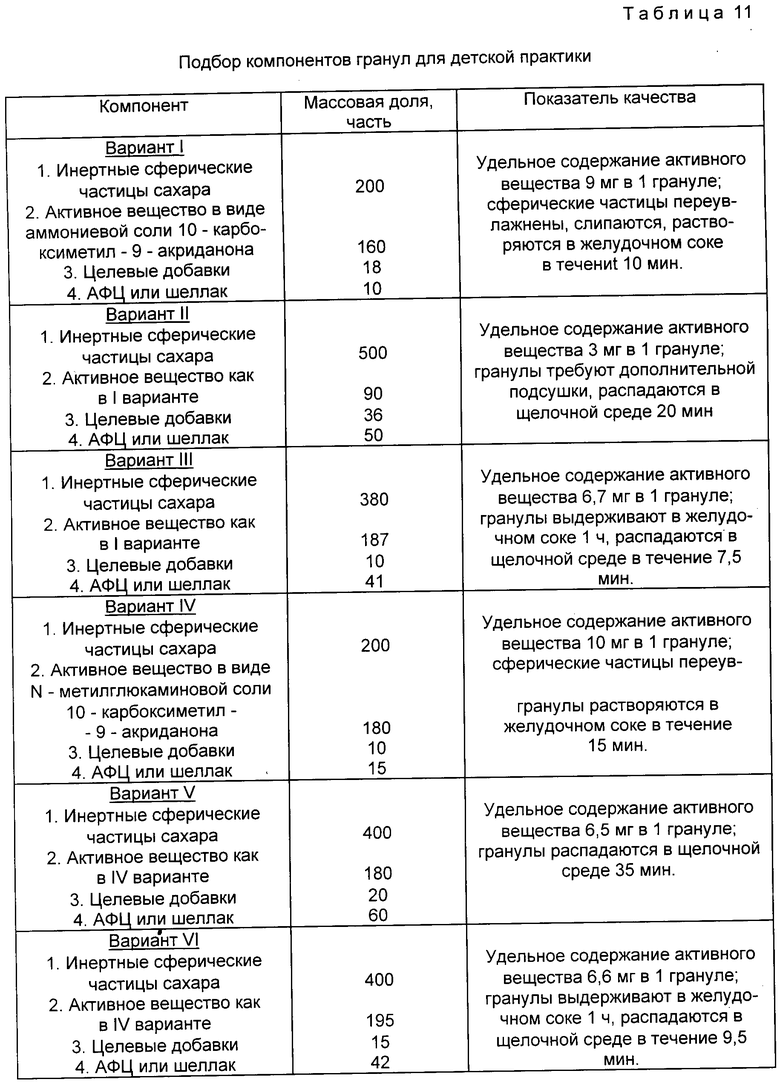
Учитывая возможность применения соли 10-карбоксиметил-9-акриданона в качестве противовирусного средства в детской практике и невозможность дробления таблеток, покрытых защитной кислотоустойчивой оболочкой, была разработана аналогичная пероральная лекарственная форма в виде гранул для педиатрии.
Как указывалось выше, экспериментально установлена эффективная лечебная доза соли 10-карбоксиметил-9-акриданона 269-900 мг на человека в сутки, что составляет в пересчете на 1 кг массы тела 3,8 - 12,8 мг. Для легкости дозировки количества гранул на один прием для детей заявители выбрали содержание активного вещества в одной грануле 6-7 мг, соответствующее суточной потребности на 1 кг массы тела ребенка.
При получении гранулы сохранены все основные принципы создания эффективного перорального лекарственного средства, а именно:
для обеспечения максимальной биодоступности 10-карбоксиметил-9-акриданон вводится в виде фармакологически приемлемой соли;
каждая гранула покрывается защитной кислотоустойчивой оболочкой, способствующей высвобождению активного вещества в кишечнике.
В качестве основы для гранул использованы стандартно получаемые инертные сферические частицы сахара, откалиброванные на сите с диаметром пор 2-3 мм.
Экспериментально установлены оптимальные соотношения компонентов для получения гранул. При этом основными критериями являлись: удельное содержание активного вещества в одной грануле, которое должно составлять 6-7 мг, технологические показатели качества гранулята (влажность, слипаемость), время сохранности гранул в желудочном соке. Данные эксперимента приведены в табл. 11.
Как следует из табл. 11, оптимальными вариантами для получения гранул являются соотношения компонентов, приведенные в варианте III и IV. Количество защитного кислотоустойчивого покрытия составляет 40-45 мас. ч., что соответствует 5-7% от массы гранул, так же, как для описанной выше таблетки. Превышение этого содержания защитного кислотоустойчивого покрытия в грануле приводит к увеличению времени распадения гранулы в кишечнике (варианты II, IV, V табл. 11), что отрицательно сказывается на эффективности препарата.
Приведем пример получения гранул перорального лекарственного средства для педиатрии.
390 мас.ч, инертных сферических частиц сахара, откалиброванных на сите с диаметром пор 2-3 мм, помещают в аппарат псевдоожиженного слоя, в который распыляют водный раствор, состоящий из смеси 10 мас.ч. метилцеллюлозы (связующие добавки) и 180 мас.ч, активного вещества. После распыления указанного раствора слой полученных гранул подсушивают в течение 1,5-2 ч, затем продолжают нанесения раствора 40 мас.ч. АФЦ или шеллака в виде 5% раствора в ацетоне или другом легко летучем растворе (хлороформ, спирт), после чего продувают воздух в течение 1,5-2 ч для закрепления защитного слоя и полного удаления растворителя.
Таким образом, заявляемые пероральные лекарственные средства являются высокоэффективными препаратами для профилактики и лечения широкого спектра заболеваний вирусной этиологии у взрослых и детей.



Изобретение относится к фармацевтической промышленности. Твердая дозированная форма для перорального применения обладает противовирусным действием. Дозированная форма может быть выполнена в виде таблетки или в виде гранулы. Форма содержит в качестве активного вещества аммониевую или N- метилглюкаминовую соль 10-карбоксиметил-9-акриданона и целевые добавки. Дозированная форма покрыта защитным кислотоустойчивым покрытием из шеллака или ацетил-фталилцеллюлозы. Таблетки или гранулы предназначены для профилак- тики и лечения заболеваний вирусной природы. 2 с.п. ф-лы, 11 табл.
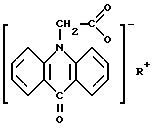
где R - NH
с нанесенным защитным кислотоустойчивым покрытием из шеллака или ацетилфталилцеллюлозы, при этом компоненты взяты в следующем соотношении, мас. ч.:
Указанная соль 10-карбоксиметил-9-акриданона - 269 - 900
Целевые добавки - 50 - 225
Шеллак или ацетилфталилцеллюлоза - 35 - 50
2. Твердая дозированная форма для перорального применения, обладающая противовирусной активностью, содержащая в качестве активного вещества фармацевтически приемлемую соль 10-карбоксиметил-9-акриданона и целевые добавки, отличающаяся тем, что она выполнена в виде гранулы с защитным кислотоустойчивым покрытием из шеллака или ацетилфталилцеллюлозы, содержащей фармацевтически приемлемую соль 10-карбоксиметил-9-акриданона общей формулы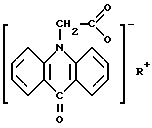
где R - NH
нанесенную вместе с целевыми добавками на инертную частицу, при этом компоненты взяты в следующем соотношении, мас.ч.:
Указанная соль 10-карбоксиметил-9-акриданона - 187 - 195
Инертная частица - 380 - 400
Целевые добавки - 10 - 15
Шеллак или ацетилфталилцеллюлоза - 40 - 45щ
| US 3681360 A (Hoffmann-la Roche Inc.), 01.08.72, C 07 D 37/20. |

