Данное изобретение относится к усовершенствованному способу получения водорастворимых сульфированных диспергаторов.
Более точно, данное изобретение относится к способу получения водорастворимых сульфированных диспергаторов посредством взаимодействия ароматических фракций с SO3.
В патентной литературе приводятся методы, которые используются для получения водорастворимых сульфированных диспергаторов посредством взаимодействия ароматических фракций с SO3. Взаимодействие вышеуказанных ароматических фракций с трехокисью серы в особых температурных условиях вызывает помимо обычного образования сульфонатов окислительную олигомеризацию.
Реакция получения сульфированных диспергаторов посредством взаимодействия ароматических фракций с SO3 является очень сложной. Вероятно, она состоит из первой стадии, на которой происходит преимущественное сульфирование субстратов, и последующей стадии, в процессе которой преобладает реакция окислительной олигомеризации, которой подвержены ароматические соединения, в особенности те, в молекуле которых содержатся два или более кольца, и в результате которой образуются структуры с увеличенным молекулярным весом и выделяется SO2. Можно предположить, что во время этой стадии реакция протекает следующим образом:
2HR-SO3H+SO3_→ H3OS-R-R-SO3H+SO2+H2O (1)
В заявке GB-A-2159536 описан вышеуказанный процесс, проведенный при использовании в качестве ароматической фракции коксовального дегтя или его фракции и в качестве растворителя - хлорированного соединения. Недостатком данного процесса является то, что в любом случае хлорированный растворитель подвергается хотя бы минимальному разложению, загрязняя таким образом диспергатор и создавая проблемы с восстановлением и повторным использованием растворителя.
В заявке EP-A-379749 описан метод, в котором фракции тяжелого жидкого топлива из парофазного крекинга обрабатывают SO3, с использованием SO2 в качестве растворителя.
По сравнению с процессом, описанным в заявке GB-A-2159536, метод, приведенный в заявке EP-A-379749, проявляет несомненное преимущество, исключая явление разложения растворителя и давая, таким образом, возможность легко использовать тот же растворитель в рецикле.
Неожиданно этот процесс обнаружил некоторые недостатки, происходящие, кроме всего, из трудностей, которые встречаются при попытке добиться полного удаления SO2 и из необходимости управления под давлением: фактически необходима стадия нагревания до максимальной температуры приблизительно 120oC. Неполное удаление SO2 является причиной увеличения расхода NaOH на стадии нейтрализации с последующим увеличением содержания сульфита натрия в конечном продукте.
В European Application Public N 580194 описан процесс, в котором в качестве ароматической фракции используется инден или его смесь с упомянутыми выше ароматическими фракциями промышленного происхождения.
В любом случае в соответствии со всеми этими процессами реакция протекает в присутствии растворителя, который используется главным образом для того, чтобы снимать теплоту реакции, которая выделяется в очень большом количестве в продолжении стадии сульфонирования.
Авторы данного изобретения в настоящее время разработали упрощенный процесс, в котором отсутствуют вышеуказанные недостатки, путем проведения стадии окислительной олигомеризации в отсутствие растворителей.
В соответствии с этим данное изобретение относится к методу получения водорастворимых сульфированных диспергаторов инициированием взаимодействия трехокиси серы с ароматическими фракциями в весовом соотношении, заключенном в пределах от 0,8:1 до 1,5:1, отличающимся тем, что указанная реакция осуществляется посредством последовательности следующих стадий:
/1/ преобладающее сульфирование ароматических фракций в присутствии SO2 при температуре, значение которой заключено в пределах от 5 до 45oC, с преимущественным образованием продукта сульфирования;
/2/ удаление SO2 и нагрев твердого продукта сульфирования до температуры, значения которой заключены в пределах от 70 до 110oC, и выдерживание твердого материала при вышеуказанной температуре в течение времени, значение которого заключено в пределах от 10 до 200 мин,
причем обе вышеуказанные стадии осуществляют в условиях, когда твердые сульфонаты разрушаются и перемещаются.
В соответствии с предпочтительным воплощением данного изобретения весовое соотношение "трехокись серы: ароматические фракции" заключено в пределах от 0,85:1 до 1,3:1.
Под термином "ароматическая фракция" в данном изобретении подразумевают углеводородные композиции, которые представляют собой преимущественно ароматические соединения и получены из различных источников, например из процесса пирролиза каменного угля, из парофазного крекинга лигроина или газойля или их смеси с получением легких олефинов, из процесса получения смазочных масел. Термин "ароматические фракции" включает также инден и вышеуказанные фракции, смешанные с инденом.
В любом случае минимальное содержание ароматических соединений в вышеуказанных ароматических фракциях должно составлять по весу 70%, предпочтительно заключаться от 80% до 100% по весу, что определяется методом колоночной хроматографии в соответствии с ASTM 2549; причем ароматические виды вышеуказанных фракций составляют по крайней мере на 75% ароматические соединения и алкилароматические соединения, имеющие два или более конденсированных циклов.
Следующие требования здесь имеют место:
/1/ тяжелое жидкое топливо из парофазного крекинга, например высококипящий жидкий остаток крекинга лигроина и/или газойля, приводящего к получению легких олефинов, в частности этилена /см. Ulmann's Encyclopedia of Industrial Chemistry", Vol. A. 10, p. 47/;
/2/ коксовальный деготь - бипродукт процесса пиролиза каменного угля /см."Ulmann's Encyclopedia of Industrial Chemistry", Vol. A 7, р. 275/;
/3/ так называемые "ароматические экстракты" - бипродукты процесса получения смазочных масел;
/4/ инден один или в смеси с вышеуказанными фракциями - как определено в European Application Public N 580194.
"Ароматические фракции", определенные выше, также включают дистилляты или остатки из процессов переработки /например, дистилляции или экстракции/, перечисленных в четырех вышеприведенных пунктах, а также любые композиции из любого источника при условии, что они удовлетворяют требованиям содержания ароматических веществ и их типа, как определено выше.
В соответствии с предпочтительным воплощением ароматической фракцией является тяжелое жидкое топливо из процесса парофазного крекинга.
Реакция окислительного сульфирования /это выражение используется для обозначения общей реакции, включающей сульфирование и окислительную олигомеризацию/ протекает в соответствующем реакторе для обеспечения давления в присутствии в качестве растворителя двуокиси серы. Давление, которое должен выдерживать реактор, является по существу функцией давления паров двуокиси серы. Ввиду того, что на практике SO2 присутствует только на первой стадии реакции, давление никогда не превосходит приблизительно 5 бар. Это приводит к значительному упрощению процесса по сравнению с методом, описанным в заявке EP-A-379749, в котором давление может достигать значения 30 - 35 бар. Возможность проведения процесса при более низком давлении представляет собой значительное преимущество, особенно в отношении материалов для изготовления реактора и изолирования реактора.
SO2 может быть одинаково использован как для разбавления ароматической фракции или SO3, так и для разбавления их обоих. Благодаря легкости, с которой SO3 стремится к затвердеванию при даже минимальном отклонении температуры, может быть предпочтительным, чтобы SO3 разбавлялся по крайней мере порцией SO2. Соотношение SO2 и реагентов не является определяющим, вследствие дешевизны весовое соотношение SO2: реагенты /где под реагентами подразумевают SO3 и ароматическую фракцию/, заключенное в пределах от 0,5 до 4, является предпочтительно используемым.
При использовании вышеуказанных отношений SO2 к реагентам значительное количество тепла, выделяемого в процессе реакции сульфирования, где термин "сульфирование" означает простое замещение атома водорода ароматического субстрата фрагментом SO3H, должно быть удалено. Указанное удаление тепла может быть эффективно проведено посредством испарения SO2 и/или охлаждением реактора извне. Давление, которое должен выдерживать реактор, в котором проводится сульфирование, в значительной степени определяется давлением паров SO2 при температуре, которая поддерживается в реакторе.
Процесс в соответствии с данным изобретением может быть оформлен либо как периодический, либо как непрерывный.
В том случае, когда процесс протекает периодически, стадия сульфонирования может быть осуществлена посредством постепенной подачи SO3, предпочтительно разбавленного SO2, в реактор, который содержит ароматическую фракцию, предпочтительно также разбавленную в SO2. В продолжении этой стадии температура должна поддерживаться в пределах от 5 до 45oC, предпочтительно от 10 до 40oC. В этих условиях и при данной концентрации реагентов образующийся сульфированный продукт плохо растворим в реакционной среде и, следовательно, стремится перейти в твердую форму.
Когда эта стадия реакции, в которой, как оказывается, преобладает простое сульфирование ароматических субстратов, завершена, начинается вторая стадия процесса, которая состоит из нагрева сульфированного продукта до температуры, которая заключена в области от 70 до 110oC, предпочтительно от 80 до 100oC. В продолжении этой стадии /открытием верхнего клапана в верхней части реактора/ осуществляется удаление SO2. Эта операция может быть проведена при таком же давлении, которое имело место на стадии сульфирования, либо при атмосферном давлении, либо при пониженном давлении. Удаление SO2 может быть осуществлено выделением его при температуре, близкой к температуре сульфирования, и последующим нагревом растворителя, свободного от твердого сульфоната, или SO2 может быть удален одновременно с нагревом в продолжении второй стадии реакции. Удаленный из реактора таким образом SO2 может быть конденсирован и использован в рецикле для последующей реакции.
Очевидно, что отсутствие растворителя в процессе этой второй стадии реакции не создает компромисса для окислительной олигомеризации.
Установленное время протекания этой стадии реакции обычно заключено в пределах от 10 до 200 мин, предпочтительно от 20 до 100, как функция молекулярного веса, который желают получить посредством удаления SO2.
В конце процесса, описанного выше, продукт реакции теперь в виде сухого, рыхлого порошка может быть высвобожден из реактора в твердой форме и затем нейтрализован, или в виде водного раствора основания, предпочтительно гидроокиси натрия, в котором сульфонат в солевой форме непосредственно растворим, и который может быть добавлен в реактор.
Для того, чтобы указанный процесс протекал успешно, важно, чтобы обе стадии - сульфирование и окислительная олигомеризация протекали в присутствии соответствующих средств для измельчения и перемешивания твердых материалов. Фактически в отсутствии таких устройств во время первой стадии сульфонат занял бы весь объем реактора, налипая на стенки и все неподвижные части реактора, создавая тем самым проблемы при необходимости удаления твердого сульфоната для проведения последовательной второй стадии процесса.
Что касается второй стадии окислительной олигомеризации, только эффективное измельчение твердого продукта делает возможным контролирование необходимой температуры и предотвращение термического разложения сульфированного продукта, вызываемого локальным перегревом, и дает возможность легко удалить SO2. Кроме того, поддержка перемешивания твердого продукта является необходимой для удаления его из реактора.
Для этой цели полезными являются реакторы, которые состоят из горизонтальной цилиндрической камеры, снабженной термостатирующей рубашкой и аксиально расположенным валом, обеспечивающим благодаря лопастям перемешивание материала и, в случае необходимости, направление продукта реакции к месту разгрузки реактора.
Указанный вал может быть сконструирован в соответствии с задачей термостатирования, и цилиндрическая емкость реактора может быть оснащена фиксированным контр-ножом для очистки движущихся лопастей.
Когда процесс осуществляется периодическим способом, в продолжении стадии сульфирования реактор охлаждают, а в течение последующей стадии удаления растворителя и окислительной олигомеризации реактор нагревают.
Кроме того, при периодическом исполнении процесс в соответствии с данным изобретением может быть осуществлен в двух реакторах. В первом реакторе протекает стадия сульфирования, а затем твердый сульфированный продукт после предварительного удаления SO2 непосредственно поступает во второй реактор /уже при действующих условиях температуры и давления/, внутри которого вторая стадия окислительной олигомеризации имеет место.
При осуществлении процесса в виде непрерывного должны быть использованы два реактора в виде каскада, например один охлажденный реактор, в котором протекает реакция сульфирования, и один нагретый реактор, в котором протекает реакция окислительной олигомеризации. Помимо обеспечения измельчения и перемешивания твердого продукта оба реактора должны быть способны обеспечивать передвижение продукта от входного устройства к выходному устройству, сохраняя таким образом условия подачи и удаления.
Помимо вышеуказанных преимуществ /пониженное потребление NaOH, почти полное отсутствие сульфита натрия в конечном продукте, более низкое давление процесса/ процесс в соответствии с данным изобретением дает возможность использовать SO2 в качестве растворителя и SO2, полученный в процессе окислительной олигомеризации, возвращать обратно с почти количественным выходом.
Сульфированная форма сульфоната является хорошо растворимой в воде и используется в качестве диспергатора для концентрированных смесей угля и в качестве агента для обеспечения супертекучести бетонов.
Диспергатор может быть также использован в различных областях в виде концентрированного водного раствора или в твердой форме после удаления воды.
В тех случаях, когда сульфонат, полученный в соответствии с методом данного изобретения, используется в качестве диспергатора для СЦВ, его эффективное количество заключено в пределах от 0,2 до 1,0% веса относительно к общему весу пульпы, предпочтительно от 0,4 до 0,8%.
В тех случаях, когда сульфонат используется в качестве супертекучего агента для бетона, его эффективное количество заключено в пределах от 0,4 до 1,5% по весу относительно веса цемента, предпочтительно от 0,5 до 1,0%. При приготовлении бетона сульфонаты, полученные согласно методу данного изобретения, могут быть использованы, если это необходимо, вместе с антипенным агентом.
Для лучшей иллюстрации данного изобретения приведены следующие примеры.
Примеры 1 - 4
Используют реактор Дискотерм /зарегистрированная торговая марка фирмы List AG/ объемом 1,5 л, который состоит главным образом из горизонтальной цилиндрической емкости, оснащенной аксиальным перемешивающим устройством. Такая мешалка оснащена срезающими ножами, которые расположены по всей длине. Кроме того, на поверхности цилиндрического реактора расположены соответствующие ножи крючкообразной формы, которые сохраняют срезающие ножи чистыми. На указанном валу расположен второй ряд скребков, которые расположены между соседними лопастями, и которые, в свою очередь, сохраняют чистыми срезающие ножи крючкообразной формы.
Вышеуказанное перемешивающее устройство способно передвигать и измельчать твердый материал, образованный в процессе реакции. Реактор, кроме того, оснащен охлаждающей и/или нагревающей рубашкой, помещенной в кожух, внутри которой может циркулировать вода.
Проведены четыре опыта, в которых используемым ароматическим субстратом является тяжелое жидкое топливо из парофазного крекинга /ЖТПК/.
Пример 1
221 г ЖТПК, разбавленного 510 г SO2, взаимодействуют с 224 г SO3, разбавленной 440 г SO2 /общее соотношение SO2:SO3:ЖТПК равно 4,3:1:1/. Сульфирование проводят при температуре 27 - 30oC подачей раствора SO3 в течение 57 мин.
Систему выдерживают при температуре приблизительно 30oC в течение 10 мин, затем нагревают при перемешивании и открытом верхнем клапане /время нагрева до приблизительно 75oC равно 53 мин/, удаляя таким образом SO2. Реакционную смесь выдерживают при температуре 75 - 89oC в течение приблизительно 80 мин, реактор очищают азотом и выдерживают при уменьшенном давлении в течение приблизительно 15 мин.
В продолжении всего времени реакции давление составляет менее 5,0 бар.
Реакционную смесь нейтрализуют добавлением в реактор 95 г NaOH, растворенного в 700 мл воды, до достижения pH значения 8,9.
Для аналитических целей водный раствор сушат вымораживанием с получением 512,2 г твердого вещества, которое содержит 9,9% остаточной воды, 11,75% Na2SO4, <0,5% Na2SO3, 78,4% активного вещества.
Специфический расход, относящийся к 1 кг 100% диспергатора, составляет 550,4 г ЖТПК, 557,9 г SO3 и 236,6 г NaOH.
Пример 2
333,3 г ЖТПК, разбавленного 510 г SO2, реагируют с 269 г SO3, разбавленной 560 г SO2 /общее соотношение SO2:SO3: ЖТПК равно 3,21:0,81:1/.
Сульфирование проводят при температуре 26 -30oC подачей раствора в SO3 в течение 39 мин.
Систему выдерживают при температуре приблизительно 30oC в течение 10 мин, затем нагревают при перемешивании и открытом верхнем клапане /время нагрева до приблизительно 77oC равно 100 мин/, реакционную смесь выдерживают при температуре 77 - 82oC в течение приблизительно 30 мин и реактор выдерживают под уменьшенным давлением в течение приблизительно 15 мин.
В продолжении всей реакции давление составляет менее 5,0 бар.
Смесь нейтрализуют добавлением в реактор 107,5 г NaOH, растворенными в 800 мл воды, до достижения pH значения 9,8.
Для аналитических целей водный раствор сушат вымораживанием с получением 669,6 г твердого вещества, которое содержит 4,3% остаточной воды, 7,75% Na2SO4, < 0,5% Na2SO3, 88,0% активного вещества.
Специфический расход, отнесенный на 1 кг 100% диспергатора, составляет 566,0 г ЖТПК, 457,1 г SO3 и 182,5 г NaOH.
Пример 3
264,4 г ЖТПК, разбавленного 415 г SO2, реагируют с 354,4 г SO3, разбавленными 565 г SO2 /общее соотношение SO2:SO3:ЖТПК равно 3,71:1,34:1/.
Сульфирование проводят при температуре 22-29oC подачей раствора SO3 в течение 43 мин.
Систему выдерживают при температуре приблизительно 29oC в течение 10 мин, затем нагревают при перемешивании и открытом верхнем клапане /время нагрева до приблизительно 77oC равно 97 мин/. Реакционную смесь выдерживают при температуре 77 - 84oC в течение приблизительно 15 мин, последние следы SO2 удаляют под уменьшенным давлением /приблизительно 30 мин/.
Максимальное давление в продолжении всего времени реакции; 5,2 бара.
Реакционную смесь нейтрализуют добавлением в реактор 133,1 г NaOH, растворенных в 900 мл воды, до достижения pH значения 8,5.
Для аналитических целей водный раствор сушат вымораживанием с получением 712,3 г твердого вещества, содержащего 7,1% остаточной воды, 14,2% Na2SO4, < 0,5% Na2SO3, 78,7% активного вещества.
Специфический расход, отнесенный к 1 кг 100% диспергатора, составляет 471,1 г ЖТПК, 632,3 г SO3 и 237,5 г NaOH.
Пример 4
300,1 г ЖТПК, разбавленного 650 г SO2, реагируют с 291 г SO3, разбавленной 420 г SO2 /общее соотношение SO2:SO3:ЖТПК равно 3,56:0,97:1/.
Сульфирование проводят при температуре 25 - 29oC подачей раствора SO3 в течение 50 мин.
Систему выдерживают при температуре приблизительно 29oC в течение 10 мин, затем начинают вытеснение SO2, открыв верхний клапан /60 мин при приблизительно 27 - 29oC/; затем реакционную смесь выдерживают под вакуумом в течение приблизительно 30 мин, удаляя таким образом последние следы SO2.
Нагревание начинают под вакуумом /25 мбар/ с повышением температуры от 29 до 80oC в течение 40 мин; затем реакционную смесь выдерживают при 80 - 82oC в течение приблизительно 30 мин, сохраняя уменьшенное давление.
Максимальное давление /4,3 бара/ наблюдают в продолжение первой стадии сульфирования.
Реакционную смесь нейтрализуют добавлением в реактор 110,6 г NaOH, растворенного в 830 мл воды, до достижения значения pH, равного 9,0.
Для аналитических целей водный раствор сушат вымораживанием с получением 627 г твердого вещества, содержащего 6,2% остаточной воды, 10% Na2SO4, < 0,5% Na2SO3, 83,8% активного вещества.
Специфический расход, отнесенный на 1 кг 100% диспергатора, составляет 570,4 г ЖТПК, 553,1 г SO3 и 210,2 г NaOH.
Сравнительные примеры 5 - 7
Сравнительные примеры выполнены в вертикальном автоклаве емкостью 22 л, снабженном магнитной мешалкой с турбиной диаметром 180 мм с четырьмя лопастями, расположенными под углом 45oC. В качестве ароматической фракции используют то же самое тяжелое жидкое топливо из процесса крекинга парофазного крекинга, как и в примерах 1 - 4.
Сравнительный пример 5
4375 г ЖТПК, разбавленного 4900 г SO2, сульфируют 3500 г SO3, разбавленной 1540 г SO2 /общее соотношение по весу SO2:SO3:ЖТПК равно 1,47:0,80:1/ при температуре, значение которой заключено в пределах от 21 до 37oC, в течение 72 мин. После выдерживания в течение 10 мин при 37oC температуру реакционной массы повышают с 37oC до 80oC в течение 58 мин, затем выдерживают 30 мин при 80 - 87oC, удаляют SO2, что требует 40 мин, и очищают реактор азотом. Общее время реакции составляет 260 мин.
Максимальное давление во время реакции составляет 21 бар.
Реакционную смесь нейтрализуют 1871,4 г NaOH, растворенных в 15,5 л воды.
Получают 9075 г твердого продукта, который содержит 4,68% воды, 7,5% Na2SO4, 8,8% Na2SO3 и 79,0% активного вещества.
Специфический расход, отнесенный к 1 кг 100% диспергатора, составляет: 610,2 г ЖТПК, 488,2 г SO3, 261,0 г NaOH. В примере 2, в котором используют ту же величину весового соотношения для SO3: ЖТПК, равную 0,8:1, полученный специфический расход значительно ниже /566,0 г ЖТПК, 457,1 г SO3 и 182,5 г NaOH/.
Сравнительный пример 6
4002 г ЖТПК, разбавленного 4790 г SO2, сульфируют 3940 г SO3, разбавленной 1130 г SO2 /общее соотношение по весу SO2:SO3:ЖТПК равно 1,48:0,98:1/ при температуре, значение которой заключено в пределах от 17 до 41oC, в течение 52 мин. После выдерживания смеси в течение 10 мин при температуре 41oC температуру повышают от 41 до 80oC в течение 57 мин, выдерживают 30 мин при 89 - 91oC, удаляют SO2, что требует 60 мин, и очищают реактор азотом. Общее время реакции составляет 265 мин.
Максимальное давление во время реакции составляет 22,5 бар.
Реакционную смесь нейтрализуют 2198 г NaOH, растворенного в 16 л воды.
Получают 8032 г твердого продукта, который содержит 5,97% воды, 11,1% Na2SO4, 10,9% Na2SO3 и 72,03% активного вещества.
Специфический расход, отнесенный к 1 кг 100% диспергатора, составляет: 691,7 г ЖТПК, 681,0 г SO3, 379,9 г NaOH.
В примерах 1 и 4, в которых используют то же весовое соотношение для SO3: ЖТПК, равное 1:1, полученный специфический расход значительно ниже /в примере 1: ЖТПК = 550,4 г, SO3 = 557,9 г, NaOH = 236,6 г; в примере 4: ЖТПК = 570,4 г, SO3 = 553,1 г, NaOH = 210,2 г.
Сравнительный пример 7
3035 г ЖТПК, разбавленного 3240 г SO2, сульфируют 3930 г SO3, разбавленной 1250 г SO2 /общее соотношение по весу SO2:SO3:ЖТПК равно 1,48:1,29: 1/, при температуре, значение которой заключено в пределах от 15 до 38oC, в течение 62 мин. После выдерживания в течение 10 мин при температуре 38oC температуру повышают от 38 до 100oC в течение 40 мин, затем выдерживают смесь при 100 - 110oC в течение 30 мин, удаляют SO2, что требует 58 мин, и очищают реактор азотом. Общее время реакции составляет 285 мин.
Максимальное давление во время реакции составляет 35 бар.
Реакционную смесь нейтрализуют 2075 г NaOH, растворенного в 16,0 л воды.
Получают 7714,3 г твердого продукта, который содержит 5,01% воды, 17,5% Na2SO4, 4,6% Na2SO3 и 72,89% активного вещества.
Специфический расход, отнесенный на 1000 г 100% диспергатора, составляет 539,8 г ЖТПК, 698,9 г SO3, 369,1 г NaOH.
В примере 3, где использовалось то же соотношение для SO:ЖТПК, равное 1,30: 1, специфический расход значительно ниже (471,5 г ЖТПК, 632,3 г SO3 и 237,5 г NaOH).
Изучение текучих свойств на смеси уголь/вода (УВС)
Уголь подвергают предварительному сухому измельчению для получения гранул с максимальным размером 3 мм. Затем получают смесь указанного предварительно измельченного угля, воды и органического сульфоната, полученного, как показано, в примерах 1 - 7. Органический сульфонат присутствует в каждой смеси в качестве активного вещества в количестве 1% по весу.
Эти смеси подвергают мокрому измельчению с использованием следующих дробильных устройств:
шары весом 3,2 кг из стали марки A1S1 диаметром 31 мм;
шары весом 4,8 кг из стали марки A1S1 диаметром 25,40 мм;
шары весом 3,2 кг из стали марки A1S1 диаметром 12,70 мм;
шары весом 4,8 кг из стали марки A1S1 диаметром 9,53 мм и мельницы с внутренним размером 240 мм х 203 мм.
1,8 кг каждой смеси измельчают со скоростью вращения барабана, равной 70 об/мин. Измельчение проводят в течение 2 ч до тех пор, пока средний размер частиц не уменьшится до приблизительно 7 - 8 мкм.
Затем окончательное измельчение проводят в стержневой мельнице с внутренними размерами, такими же "как у микронной коллоидной мельницы. В эту мельницу дозируют предварительно измельченный уголь (65% по весу в шламе составляет уголь) и предварительно измельченный в микронной мельнице уголь (содержащий остальные 35% угля) до получения общего веса, равного 1,4 кг.
Получающаяся концентрация добавки составляет 0,5% от веса конечной суспензии.
Измельчение проводят в течение приблизительно 10 мин, уменьшая средний радиус частиц до приблизительно 16 мкм, верхний предел размера до 25 мкм (максимально 1 - 2% имеют размер 25 мкм).
Далее конечный продукт, полученный из стержневой мельницы, перемешивают при температуре приблизительно 20oC для оптимизации его реологических характеристик. Указанное перемешивание осуществляют в лабораторных условиях при скорости вращения лопастей 450 об./мин.
Значение вязкости суспензий определяют с использованием ротационного вискозиметра Contraves Rheomat 115 с DIN 145. Реологические свойства суспензий изучают измерением кажущейся вязкости при 10 с-1.
Явление распада суспензии под действием перекачки насосом (или изучение динамической стабильности) моделируют перемешиванием 200 г суспензии, термостатированной при 20oC, при скорости перемешивания 1000 об./мин с лабораторной моделью миксера марки IKF- WERK KW20DZM, оснащенного мешалкой со стальными коррозионно-устойчивыми лопастями. Конечной целью опыта разложения является предположение значения усилия на срез при 10 с-1, которая является приблизительно удвоенной относительно начальной величины. Время, необходимое для достижения этой величины, оценивается как индекс сопротивления перекачки насосом или динамической стабильности суспензии. Должно быть очевидно то, что высокая скорость перемешивания (1000 об./мин) выбирается только для ускорения процесса разложения, поэтому полученные таким образом результаты должны рассматриваться только для сравнения и не могут представлять характеристику суспензии в процессе перекачки насосом.
Гранулометрическое распределение угля измеряют с использованием лазерного дифракционного анализатора размера частиц.
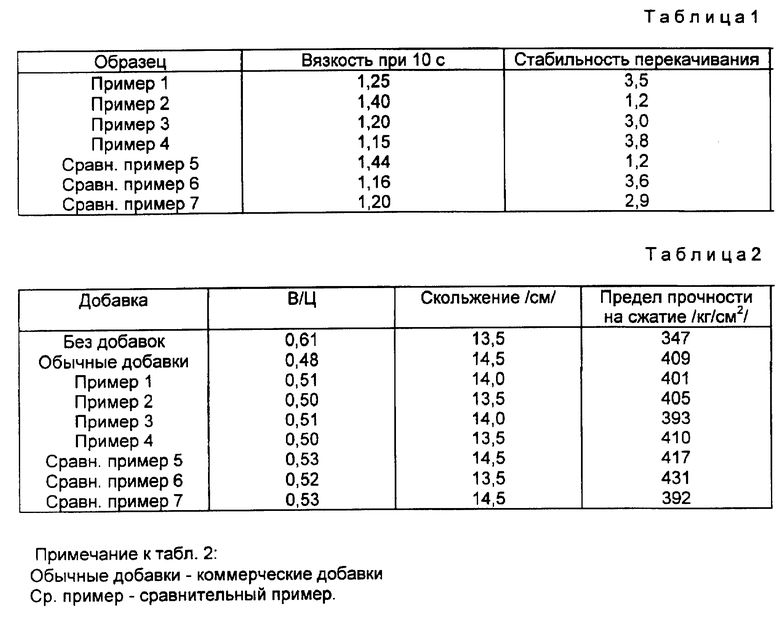
В табл. 1 представлены характеристики суспензий, смешанных с сульфированными продуктами, описанными в примерах 1 - 4 и в сравнительных примерах 5 - 7, при использовании угля Polish, имеющего следующие характеристики (измерено на сухом угле): летучих веществ 30,5%, золы 9,87% и твердого угля (вычитанием) 59,98%. Все эти величины - весовые проценты.
Значения вязкости и стабильности к перекачке насосом являются относительными величинами, сравненными с характеристиками, которые были получены при использовании (при прочих равных условиях) коммерческих добавок, принадлежащих к тому же классу натрийнафталенсульфонатформальдегидных конденсатов.
Что касается вязкости суспензий, величина меньше 1 для соотношения показывает более высокую текучесть продукта, соответствующего данному изобретению, в сравнении с коммерческим продуктом.
Большее, чем 1, значение стабильности перекачки насосом показывает преимущество продуктов, соответствующих данному изобретению, в сравнении с коммерческим продуктом, взятым в качестве эталона.
Данные, представленные в табл. 1, показывают, что продукты, соответствующие данному изобретению, полученные при более низком давлении и с более низким потреблением реагентов, но в присутствии SO2, проявляют характеристики в концентрированной смеси угля с водой, приблизительно равные характеристикам продуктов, полученных в обычном реакторе высокого давления.
Оценка сверхтекучих свойств
Опыты на бетоне
Добавки из примеров 1 - 7 данного изобретения изучили на соответствие как характеристикам текучести свежеприготовленного бетона, так и механическим свойствам бетонной конструкции.
Для каждого приготовления бетона используют следующую композицию, в которой процентное количество сверхтекучего агента и пеногасителя отнесено к цементу:
Песок 0 - 3 мм - 22,4 кг
Галька 6 - 25 мм - 22,4 кг
Портландцемент 425 - 10 кг
Добавка для достижения сверхтекучести - 0,7%
Подавитель пены - 0,04%
Вода: - Для достижения необходимого скольжения
Скольжение измеряют на свежеприготовленном бетоне, а кубики из бетона получают при использовании металлических кубических форм со стороной, равной 150 мм, расположенных на вибрационном столе в соответствии со стандартами.
После 24-часового времени отверждения кубики удаляют из форм и подвергают старению в воде в течение 28 дней. У кубиков, полученных таким образом, измеряют предел прочности на сжатие, выраженный в кг/см2. Для сравнения приведены данные, которые относятся к бетону без добавок и к бетону, смешанному для придания высокотекучих свойств с промышленными агентами класса натрийнафталенсульфонатформальдегидных конденсатов. Данные приведены в табл. 2, в которой отношение В/Ц представляет собой весовое соотношение вода: цемент.
Данные табл. 2 показывают, что продукты, полученные методом, соответствующим данному изобретению, проявляют высокотекучие свойства части, которые приблизительно эквивалентны свойствам сверхтекучести, которыми обладают коммерческие продукты и эти продукты, полученные в обычном реакторе высокого давления.
Для получения водорастворимых сульфированных диспергаторов проводят взаимодействие триоксида серы с ароматическими фракциями при массовом соотношении 0,8-1,5:1 и температуре 5 - 45oC в присутствии диоксида серы. После удаления диоксида серы продукт сульфирования нагревают до температуры 70 - 110oC и выдерживают при этой температуре от 10 до 200 мин. Стадии взаимодействия, удаления диоксида серы, нагревания и выдерживания проводят в условиях, обеспечивающих измельчение и принудительное поддерживание движения твердого продукта сульфирования. 6 з.п. ф-лы, 2 табл.
| GB, заявка, 2159536, кл | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
