Данное изобретение относится у новым фторалкенильным соединениям, их сельскохозяйственно приемлемым солям, композициям на их основе для борьбы с заражением растений нематодами и способы борьбы и системного подавления заражения растений нематодами.
Фторированные алкены давно известны как средство борьбы с насекомыми и нематодами при внесении их в почву. Патенты США NN 3510503, 3654333 и 3780050 раскрывают такие соединения. Позднее в патенте США N 4952580 были описаны полигалоидалкены, пригодные в качестве нематоцидов, некоторые из которых обладают спускающейся вниз системой активности, так как могут до некоторой степени устранять заражение нематодой корневой системы при обработке листвы растений. Большинство соединений, описанных в этих патентах, являются неполярными соединениями, что является желательной характеристикой для пестицидов, вносимых в почву, обеспечивая более длительный период действия, но гораздо менее эффективны при обработке листвы, для достижения системного действия. Патент США N 4950666 раскрывает некоторые полученные соединения, производимые дифторалкенилалканов, пригодные в качестве инсектицидов и нематоцидов системного действия. Однако, остается потребность в средствах борьбы с нематодами, насекомыми и акаридами, которые имеют улучшенную общую подвижность и, желательно, низкий уровень эффективного применения.
Согласно настоящему изобретению предлагаются фторалкенильные соединения общей формулы I: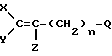
где
X и Y - фтор;
Z - водород или фтор;
n = 1, 3, 5, 7, 9 или 11;
Q - представляет собой CH2NHR6, CH2NO2, CH2N=CHR2, CH2N=C=O, CH2N+R3R4R5W- или (C=O)-RII при условии, что когда Q - (C=O)-RII каждый из X, Y и Z - фтор, а n = 1;
W- - анион минеральной или органической кислоты;
R2 - фенил, необязательно замещенный по крайней мере одной группой, выбранной из гидрооксила, нитро, галогена, ди(C1-C4)алкиламино; или пиразолил
R3, R4, R5 - водород или взятые вместе с атомом азота группы Q образуют пирролидинил или тетраазотрицикло-[3,3,1,1-(суперскрипт-3-суперскрипт-7)] декан;
R6 - представляет собой водород;(C1-C6) алкил, необязательно замещенный амино или трет-бутоксикарбониламиногруппой; (C1-C5)алкил-COOH, необязательно замещенный карбоксильный группой; (C1-C5)алкил-CONH2; (C1C5)алкил-COO(C1C4)алкил; (C1-C5)алкил-COO(C1C4)алкилфенил; (C1-C6)алкенил, замещенный галогеном; дигидро-3-оксопиразолидинил, тиенил и фенил, необязательно замещенный карбоксилом или (C1-C4)алкоксикарбонилом; (C=O)R7, где R7 представляет собой (C=O)R14; (C1-C6)алкил, необязательно замещенный по крайней мере одной группой, выбранной из ряда: галоген, аминогруппа или (C3-C6)циклоалкил, третбутоксикарбоналаминогруппа, гидрокси, фенил, (C1-C4)алкилтиогруппа; (C1-C5), алкил-COOH или его эфиры, необязательно замещенные аминогруппой; (C1-C5)алкилCONH2, необязательно замещенный аминогруппой; (C1-C6)алкенил, замещенный галогеном; или N-содержащую группу, которая взятая вместе с карбоксамидом, представляет собой остаток мочевины, необязательно замещенный (C1-C4)алкилом, возможно замещенным гидроксилом; или
R6 взятый вместе с азотом группы Q, представляет собой гидразин, гуанидин, трифторметилфенилсульфонамид;
R11 представляет собой галоген, NHOH; OR12; SR12; NR12'', где R12 - водород; (C1-C8)алкил, необязательно замещенный аминогруппой или фенилом; фенил, необязательно замещенный нитрогруппой;
R12 - водород, (C1-C8)алкил, необязательно замещенный аминогруппой или фенилом; фенил, необязательно замещенный карбоксилом;
R12'' - водород, (C1-C6)алкил;
R13 - водород; (C2-C4)алкенилCOOH; (C1-C6)алкил, необязательно замещенный по крайней мере одной группой, выбранной из (C1-C4)алкоксикарбонила, амина, фенила; (C2-C6)алкилCOOH, необязательно замещенный карбоксилом или фенилом; (C1-C6)алкиламин; замещенный карбоксилом или (C1-C4)алкоксикарбонилом; фенил, необязательно замещенный карбоксилом; или R12'' и R13 вместе с атомом азота группы NR12''R13 образуют остаток аминокислоты природного белка, выбранной из ряда: глицил, валил, аланил, аспарагил, фенилаланил, пролил, лизил, морфолиногруппу, пирролидинил или пиразолонил, возможно замещенный карбоксилом;
R14 представляет собой OH, (C1-C6)алкокси, NH2 или NHNH2, или их сельскохозяйственно приемлемые соли, при условии, что когда n=1, а X, Y и Z означают фтор, Q не является CH2NH2 или ее минеральной солью.
Предпочтительны фторалкенильные соединения, где R2 представляет собой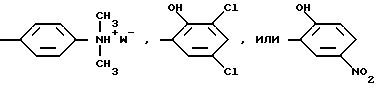
либо фторалкенильные соединения, где n= 1, X и Y каждый представляет собой фтор;
в том числе фторалкенильное соединение, где Z представляет собой водород, а Q представляет собой CH2NH2 или CH2NH3 +W-, или Z представляет собой фтор,
например, фторакенильное соединение, где Z - фтор, Q представляет собой CH2NHR6 и R6 представляет собой амид аминокислоты Q,
или R6 представляет собой амид основной аминокислоты Q, где основная аминокислота предпочтительно является метионином.
Предпочтительны соединения формулы I, где W- представляет собой хлорид, иодид, бромид, оксалат, сульфат, ацетат, цитрат или 3,4,4-трифтор-3-бутеноат.
Среди предпочтительных соединений можно назвать фторалкенильные соединения, представляющие собой 4,4-дифтор-3-бутен-1-амин или его сельскохозяйственно приемлемые соли; 3,4,4-трифтор-3-бутеновую кислоту или его сельскохозяйственно приемлемые соли; 3,4,4-трифтор-3-бутен-1-амина 3,4,4-трифтор-3-бутеноат; 2-(3,4,4-трифтор-1-оксо-3-бутенил)гидразид 3,4-трифтор-3-бутеновой кислоты.
Настоящее изобретение также относится к композиции для борьбы с заражением растений нематодами, включающей активный ингредиент и целевые добавки. В качестве активного ингредиента композиция содержит соединение формулы I в эффективном количестве. Предпочтительно в качестве активного ингредиента она содержит N-(3,4,4-трифтор-1-оксо-3-бутен-ил)глицин или 3,4,4-трифторбутеновую кислоту или их соли.
Объектом настоящего изобретения является также способ борьбы с заражением растений нематодами путем обработки локуса растений активным соединением, в качестве которого используют соединение формулы I в эффективном количестве.
Изобретение относится также к способам системного подавления заражения растений нематодами путем обработки растений активным соединением. В качестве активного соединения используют соединение формулы:
где:
n = 1, 3, 5, 7, 9 или 11; Q1 представляет собой CH2NHR6, (C=O)-R11 или CH2NH3W-; каждый из X, Y и Z представляет собой водород или фтор или по крайней мере один из X и Y представляет собой фтор, при условии, что когда Q1 представляет собой (C-O)-R11, Z представляе6т собой фтор и n = 1;
R6 имеет вышеуказанные значения;
R11 представляет собой галоген, NHOH; OR12'; SR12; NR12''R13, где R12 - водород; (C1-C8)алкил, необязательно замещенный аминогруппой или фенилом; фенил, необязательно замещенный нитрогруппой и имеет полярность, обеспечивающую проникновение соединения во флоэму листа без снижения их нематоциидной активности; R12' - (C1-C8)алкил, необязательно замещенный аминогруппой или фенилом, фенил, необязательно замещенный карбоксилом R12'' - водород, (C1-C6)алкил; R13 - водород; (C2-C4)алкенилCOOH; (C1-C6)алкил, необязательно замещенный по крайней мере одной группой, выбранной из (C1-C4)алкоксикарбонила, амина, фенила; (C2-C6)алкилCOOH, необязательно замещенный карбоксилом или фенилом; (C1-C6)алкиламин, замещенный карбоксилом или (C1-C4)алкоксикарбонилом; фенил, необязательно замещенный карбоксилом; или R12'' или R13 вместе с атомом азота группы NR12''R13 образуют остаток аминокислоты природного белка, выбранной из ряда: глицил, валил, аланил, аспарагил, фенилаланил, пролил, лизил, морфолиногруппу, пирролидинил или пиразолонил, возможно замещенный карбоксилом;
W- - анион минеральной или органической кислоты, или их сельскохозяйственно приемлемые соли в эффективном количестве.
Предпочтительно в качестве соединения использовать соединение, у которого n равно 1, X и Y каждый представляет собой фтор, в частности, соединение у которого Z представляет собой фтор, и у которого Q1 представляет собой CH2N+H3W- и W- является сельскохозяйственно приемлемым анионом.
Предпочтительно использовать соединение, у которого W- представляет собой хлорид, иодид, бромид, оксалат, сульфат, фосфат, ацетат, цитрат или 3,4,4-трифтор-3-бутеноат, либо соединения, где Z - фтор, а Q1 представляет собой COOH или его соль; или Q1 представляет собой CH2NHR6, а R6 представляет собой остаток 3,4,4-трифтор-3-бутен-1-амина или его соли или группу, которая может трансформироваться в него после применения на растении или внутри растения, или R6 представляет собой амид основной аминокислоты Q1, в частности, используют соединение, у которого R6 представляет собой остаток метионина.
Другим способом согласно настоящему изобретению является способ системного подавления заражения растений нематодами путем обработки растения активным соединением, в качестве которого используют N-(3,4,4-трифтор-1-оксо-3-бутен-ил)глицина или 3,4,4-трифторбутеновую кислоту или их сельскохозяйственно приемлемые соли в эффективном количестве.
В настоящее описание также включены способы получения 3,4,4-трифтор-3-бутен-1-амина и новых промежуточных соединений - 3,4,4-трифтор-3-бутен-1-тозилата и 3,4,4-трифтор-3-бутен-1-мезилата.
Подробное описание изобретения
Новые соединения настоящего изобретения пригодны для борьбы с заражением растения нематодами, насекомыми и акаридами. Многие из этих соединений, будучи полярными, являются высокоэффективными для системной борьбы, то есть, когда они применяются для обработки листвы или стеблей растения, или способны двигаться по флоэме и ксилеме растения и обеспечить борьбу с нематодами, насекомыми и акаридами на других частях растения. Полагают, что механизм этого воздействия представляет собой скорее отпугивающее или отвращающее от поедания действие, чем действие, причиняющее вред. Другие, в частности, неполярные соединения являются эффективными только тогда, когда вносятся в почву непосредственно. Некоторые соединения могут обеспечить оба типа воздействия.
Предлагаемые способы общего воздействия на нематоды, насекомых и акарид используют способные продвигаться по флоэме соединения настоящего изобретения, или такие соединения, которые имеют ди- или трифторалкенильную группу, замещенную CH2NHR, или трифторалкенильную группу, несущую заместитель (C=O)-R, обладающие достаточной полярностью, чтобы обеспечить флоэмную подвижность без устранения противонематодной активности фторалкеновой частицы. Существует несколько различных теорий, касающихся флоэмной подвижности, в соответствии с которыми соединения, обладающие полярностью, должны быть достаточно подвижными во флоэме, чтобы распространиться вниз по растению. Полагают, что полярность молекулы как целое должна быть достаточной для молекулы, чтобы удержаться ей во флоэме, но не настолько сильной, чтобы молекула вовсе не могла выйти.
Для эффективной и системной борьбы с нематодами или другими вредителями при обработке надземных частей растения, соединения должны быть способными проникать сквозь кожицу листвы или стебля растения, проникать во флоэму и оставаться там достаточно долго, чтобы быть транспортированными по растению и двигаться до необработанных участков, включая корни. Здесь они могут просочиться или иным путем вступить в контакт с вредителями в такой степени, чтобы уничтожить или отпугнуть вредителей, и вред, который вредители нанесут растению, уменьшается или устраняется. Во время стадий транспортировки от отработанных листьев или стеблей через растение, соединение может подвергаться химическим реакциям, таким как гидролиз, или биологическим реакциям, таким как ферментативное воздействие. Кроме того, могут быть предложены соединения, которые при попадании на растение, до проникновения в растение, могут подвергаться реакциям, которые дадут в результате соединение, которое способно легко поглощаться, перемещаться и быть эффективным для предотвращения вредного действия вредителя. Примерами таких соединений являются соединения, содержащие защитные группы, неустойчивые к действию УФ-излучения, которые при выдержке в условиях естественного освещения, претерпевают реакцию, и в результате образуются активные и подвижные соединения. Другим примером являются силилированные производные аминов.
Следовательно, то, что попадает на листву или стебель растения, может не быть соединением, которое в действительности транспортируется или которое фактически воздействует на вредителей. Таким образом, способы настоящего изобретения предусматривают применение соединений, которые могут быть изменены химическими и биологическими реакциями, чтобы приобрести полярность, подходящую для общего воздействия.
Способы настоящего изобретения включают обработку растения, предпочтительно его листвы 3,4,4-трифтор-3-бутен-1-амином или его солями. Кроме того, обработку можно вести композицией, содержащей водный раствор 3,4,4-трифтор-3-бутен-1-амина, или его солей в агрономически приемлемом носителе.
В описанных выше способах для борьбы с нематодами предпочтительно, чтобы n был равен 1, а X, Y и Z все представляли бы собой фтор, и предпочтительно, когда Q1 представляет собой CH2NH3 +W-, то есть соль 3,4,4-трифтор-3-бутен-1-амина. Когда Q1 представляет собой CH2NHR, предпочтительно, чтобы R являлся α -аминокислотой, присоединенной через пептидную (амидную) связь, предпочтительнее - аминокислотой белка. Когда Q1 представляет собой (C=O)-R, предпочтительно, чтобы R представлял собой гидроксил, и соединение, таким образом, будет представлять собой 3,4,4-трифтор-3-бутеновую кислоту или ее соль, включая соль, образованную с 3,4,4-трифтор-3-бутен-1-амином, то есть 3,4,4-трифтор-3-бутен-1-амин3,4,4-трифтор-3-бутеноат.
W- может представлять собой любой агрономически приемлемый анион. К числу таких анионов относятся, но не ограничиваются ими, хлорид, иодид, бромид, оксалат, сульфат, фосфат, цитрат, ацетат или фторалкенкарбоксилат, например, F2C=CFCH2COO-.
Кроме соединений, особо описанных выше, все агрономически приемлемые соли соединений входят в объем изобретения. Например, соединение настоящего изобретения, имеющее свободную аминогруппу, может также существовать в протонированной форме, имеющей различные ассоциированные с ней анионы, например, - но пример не является ограничительным - хлорид, бромид, иодид, фосфат, оксалат, сульфат, цитрат и ацетат. Кроме того, противоионом может быть фторалкенкарбоксилатный ион, такой как F2C=CFCH2COO-. Соединение настоящего изобретения, содержащее карбоксильную или гидроксильную группу, может существовать в виде соли, имеющей различные ассоциированные с ней катионы, например, - но пример не является ограничительным - щелочноземельных металлов, таких как натрий, кальций и калий, магния, или ионы четвертичного аммония, такие как аммоний, моно-, ди- или триалкиламмоний, например, изопропиламмоний, или пиридиний. Кроме того, катионом может быть ион фторалкениламмония, такой как F2C=CFCH2C-H2NH3+. Все такие соединения и другие соединения, имеющие подобные характеристики, будучи агрономически приемлемыми, охватываются настоящим изобретением.
Используемые здесь термины "галоид", "галогенид" или "галоген" означают фтор, хлор, бром или иод, или их производные.
Термин "алкил" означает группы с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода. Термин "низший алкил" означает такую группу, которая содержит от 1 до 4 атомов углерода. Термин "алифатический" означает насыщенные или ненасыщенные, с прямыми или разветвленными цепями группы, содержащие от 1 до 10 атомов углерода.
Термин "алкокси" означает низшую алкильную группу, присоединенную через атом кислорода. Термин "алкилтио-" означает, что низшая алкильная группа связана через атом серы. Термин "алкоксикарбонил" означает сложный низший алкиловый эфир карбоксильной группы.
Термин "алифатический амин" означает алифатическую группу, в которой по крайней мере атом водорода замещен -NH2. Термин "алифатическая карбоновая кислота, ее сложные эфиры, сложные тиоэфиры и амиды" означает алифатическую группу, в которой по крайней мере один атом углерода входит в состав карбокисльной группы -COOH, или ее сложного (низший алкил) - эфира, сложного (низший алкил) - триоэфира или амида.
Термин "ароматическая группа" или "арил" означает фенил, необязательно замещенный по крайней мере одной группой, выбираемой среди гидроксила, алкокси, галоидо-, нитро-, амино-, тио- и алкилтиогруппы, карбоксила, алкоксикарбонила и фенила. Термин "ароматическая группа" может быть использован также для гетероциклических остатков таких, как например, тиадиазол, пиридин, тиазол, изотиазол, оксазол, пиразол, триазол, бензотиазол, тиофен, фуран и тому подобное, и все они также могут быть необязательно замещенными.
Фраза "амид аминокислоты Q" (или Q1)" означает, что R6 (или R, соответственно) представляет собой аминокислоту, связанную через пептидную (амидную) связь с N или CH2NHR6. Аминокислота может быть природной, то есть аминокислотой белка, или аминокислотой искусственного происхождения. Аминогруппа аминокислоты может быть заместителем у любого атома углерода в группе, например, в положении α,β или γ относительно карбонила.
Термин "алкил- или арилгидразин" означает гидразинную группу, замещенную низшим алкилом или фенильной группой, которые, в свою очередь, могут быть необязательно замещенными.
Синтез соединений
Соединения вышеупомянутой формулы, в которых X, Y и Z представляют собой фтор, и Q представляет собой производное -CH2N-, вообще получают, получая в первую очередь нужный трифторалкениламин, который, когда n=1, представляет собой 3,4,4-трифтор-3-бутен-1-амин. Один из способов получения этого соединения описан в примере 6 патента США 4952580, указанного выше. Настоящее изобретение предлагает улучшенный и новый способ изготовления такого соединения, 4-Бром-1,1,2-трифтор-1-бутен, который доступен коммерчески, может быть непосредственно превращен прямым взаимодействием с тозилатной солью, например, с тозолатом серебра, мезилатной солью или другой, отдающей сульфогруппу. Образованные в результате промежуточные соединения, например, 3,4,4-трифтор-3-бутен-1-тозилат или 3,4,4-трифтор-3-бутен-1-мезилат, являются новыми соединениями. За этой стадией следует конверсия до производного фталимида с использованием соли фталимида, такой как калиевая соль. Реакция может быть успешно осуществлена без выделения промежуточного тозилата или мезилата. Фталимид затем превращают в нужный амин взаимодействием с гидразином. Этот новый способ получения 3,4,4-трифтор-3-бутен-1-амина имеет то преимущество, что дает улучшенный выход по сравнению с методом синтеза, о котором сообщалось прежде, что частично связано с отсутствием выделения бромистого водорода в процессе конверсии соединения брома. Таким образом, N-(3,4,4-трифтор-3-бутенил)-фталимид получают более чем с 80%-ным выходом, и превращают в нужный амин с выходом свыше 85%. Преимуществом является также то, что, когда в качестве реагента с отщепляющейся группой выбирают тозилат серебра, ионы серебра затем могут быть восстановлены и тозилат серебра регенерирован.
Алкениламины с более длинной цепью могут быть получены несколькими путями. Например, чтобы получить соединение настоящего изобретения, в котором n=3, этиленоксид можно ввести в реакцию с 4-бром-1,1,2-трифтор-1-бутеном известными способами для получения 6-гидрокси-1,1,2-трифтор-1-гексана. Это соединение затем может быть превращено в соль амина, используя тозилхлорид и фталимид, как это описано выше для бутена. По другому способу 1,1,2-трихлор-1,2,2-трифторэтан (Г113) может быть введен в реакцию с алкеном или алкином, содержащими на конце бром, в окислительно-восстановительных условиях, как описано в Tetrahedron Letters 31, pp. 1307 - 1308, 1990. Продукт реакции затем дехлорируют с Zn, чтобы получить трифторалкенбромид, который может быть превращен в нужный амин, как описано выше для бутена.
Дифторалкениламины могут быть получены двумя различными способами, в зависимости от положения атомов фтора.
Чтобы получить дифторсоединение на конце, такое как 4,4-дифтор-3-бутен-1-амин-гидрохлорид, может быть использован следующий путь: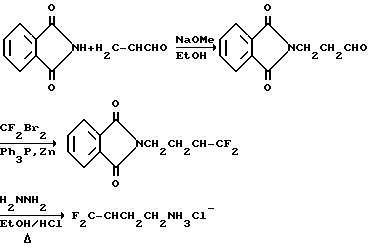
Другие дифторсоединения, то есть соединения, в которых один из X и Y является H, а Z представляет собой F, получают по следующему пути: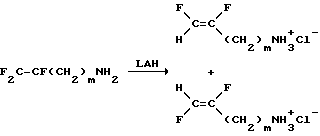
где
m = 2, 4, 6, 8, 10 или 11; LAH означает алюмогидрид лития. Изомерные формы E и Z могут быть выделены отгонкой из смеси продуктов, полученных в этой реакции.
Подобным образом, монофторсоединения настоящего изобретения получают двумя различными путями, в зависимости от того, будет ли фтор на конце или в середине молекулы, как это показано на следующих схемах:
Чтобы получить монофторсоединение, в котором фтор находится в середине молекулы, можно использовать следующую схему реакций: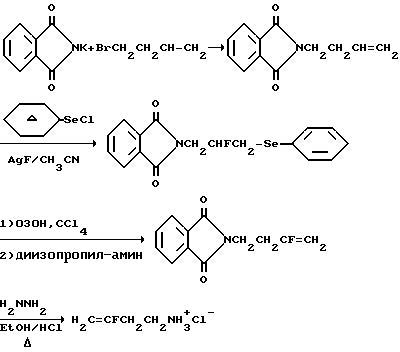
Многие другие соединения настоящего изобретения, в которых Q представляет собой CH2NHR6, затем легко получают при взаимодействии выбранного реагента с соответствующим фторалкениламином или его солью способами, известными специалистам. Например, чтобы получить амидопроизводные, в которых R6 представляет собой -(C=O)-производное, соответствующую кислоту вводят во взаимодействие с соответствующим фторированным алкениламином обычными приемами. Кислота может находиться в форме галоидангидрида или ангидрида для наиболее эффективной реакции с амином. Например, когда R6 представляет собой (C=O)-CF3, используют ангидрид трифторуксусной кислоты. Когда нужно получить производное сукцинаминовой кислоты, может быть использован янтарный ангидрид, м когда нужно получить производное щавелевой кислоты, может быть использован хлорангидрид кислоты.
Когда R6 представляет собой α -аминокислоту, связанную пептидной (амидной) связью с атомом азота группы CH2NHR6, то есть, когда R6, взятый вместе с атомом азота, представляет собой амид α -аминокислоты, для образования пептидной (амидной) связи могут быть использованы типичные пептидные или амидные связывающие реагенты, такие как карбонилдиимидазол или DCC. В случае замещенных кислотных групп, любая функциональная группа, которая может воздействовать или испытывать воздействие со стороны групп, образующих пептидную (амидную) связь, должна быть надежно защищена. Например, функциональная аминогруппа будет защищаться трет.-БОК-группой. Кислотные или спиртовые группы могут быть замещены образованием сложных или, соответственно, простых бензиловых или трет.-бутиловых эфиров. Тиогруппа (сульфгидрильная группа) может быть защищена тритильной группой. Затем осуществляют отщепление защитных групп, используя известные способы, чтобы получить нужное соединение.
Когда R6 представляет собой алифатическую карбоновую кислоту, существует два пути синтеза. Нужный фторалкениламин может быть введен во взаимодействие с соответствующим электрофильным реагентом, например, галоидалкилкарбоксилатом. Полученный в результате сложный эфир может быть использован или гидролизован до свободной кислоты, которая затем известными способами может быть переведена в ее производные - соли, амиды и тиоэфиры. По другому способу бромфторалкен может быть введен во взаимодействие с соответствующей аминокислотой.
Когда R6 представляет собой низшую алифатическую группу, выбранный фторалкениламин вводят во взаимодействие с соответствующим электрофильным агентом, например, галоидалкилом, таким как иодистый метил. Предпочтительно, чтобы амин присутствовал в избытке по отношению к галоидалкилу для того, чтобы свести к минимуму дальнейшее замещение амина.
Когда R6 представляет собой (C1-C12)алкиламин, в качестве исходного вещества обычно вместо амина используют бромид или тозилат, и его вводят во взаимодействие с избытком нужного амина, одна аминогруппа которого защищена. Дальнейшее отщепление защитной группы известными способами будет давать в результате нужный продукт.
Когда R6, взятый вместе с азотом Q или Q1 группы CH2NHR6 (или RH2NHR), представляет собой гуанидин, соответствующий фторалкениламин в виде соли, как описано выше, вводят во взаимодействие с цианамидом.
Когда R6, взятый вместе с азотом Q или Q1 группы CH2NHR6 (или CH2NHR), представляет собой алкил- или арилсульфонамид, необязательно замещенный, соответствующий фторалкениламин вводят во взаимодействие с соответствующим сульфонилхлоридом, например, с тозилхлоридом.
Когда R6, взятый вместе с азотом Q или Q1 группы CH2NHR6 (или CH2NHR), представляет собой мочевину, карбамат, тиокарбамат или гидразин, выбранный фторалкениламин сначала превращают в изоцианат (Q представляет собой CH2N=C= O). Этот изоцианат получают взаимодействием амина с дифенилкарбамоилхлоридом и вслед за этим при высокой температуре получают изоцианат, в соответствии с представленной ниже схемой
Из азоцианата могут быть получены другие соединения при взаимодействии с аммиаком или аминами - для получения мочевин, со спиртами - для получения карбаматов, тиоспиртами - для получения тиокарбаматов, или гидразинами - для получения семикарбазидов. В последнем случае гидразин должен быть замещен, например, трет. -БОК-группой, и затем защитная группа должна быть удалена, чтобы получить семикарбазид.
Соединения, в которых Q представляет собой CH2N=CH-R2, также получают из соответствующего фторалкениламина, например, 3,4,4-трифтор-3бутен-амина. Его вводят в реакцию, используя известные методы, с соответствующим ароматическим альдегидом, предпочтительно с бензальдегидом, например, с п-(N,N-диметиламино)-бензальдегидом или с 2-гидрокси-5-нитробензальдегидом.
Соединения, в которых Q представляет собой CH2N+R3R4R5W-, иногда могут быть получены из фторалкениламина, например, 3,4,4-трифтор-3-бутен-1-амина. Аминогруппу кватернизируют известными способами, например, избытком галоидалкила, например, иодистого метила. Таким образом может быть получено соединение 3,4,4-трифтор-3-бутенилтриметиламмонийиодид.
Для других соединений, в которых Q представляет собой CH2N+R3R4R5W-, может быть использован фторалкенилбромид. Например, когда R3, R4 и R5, взятые вместе с атомом азота, образуют циклическую группу четвертичного аммония, соответствующий циклический амин, например, гексаметилентетрамин, вводят во взаимодействие с фторалкенилбромидом, чтобы получить нужное четвертичное аммониевое соединение. Когда один из R3, R4 и R5 представляет собой гидроксильную группу, соединение получают взаимодействием фторалкенилбромида с избытком O-триметилсилилгидроксиламина, чтобы получить фторалкенилгидроксиламин, защищенный O-триметилсилилом, который затем гидролизуют метанолом, и вслед за этим обрабатывают кислотой, чтобы получить соль нужного гидроксиламина.
Соединение, в котором Q представляет собой CH2NO2 , может быть получено известными способами из фтоалкенилбромида и нитрита серебра.
Соединения настоящего изобретения, которые представляют собой трифторалкенилкарбоновые кислоты и их производные, то есть, когда Q представляет собой (C=O)-RII (X, Y и Z являются F) подобны некоторым соединениям, раскрытым в патенте США 4950666, но не могут быть получены по методике реакции Виттига. Их получают совершенно другим путем, путем превращения 4-бром-1,1,2-трифтор-1-бутена в спирте через промежуточный сложный эфир. Например, для получения фенилацетата трифторбутена используют фенилуксусную кислоту, затем эфир гидролизуют и получают трифторбутенол, и затем окисляют до кислоты, как показано на схеме
Кислоты с более длинной цепью, в которых n = 3, 5 и т.д., могут быть получены таким же образом из галогенидов с более длинной цепью, полученных, как описано выше. Производные этих кислот, например, соли, сложные эфиры, амиды и т.д., могут быть легко получены способами, известными специалистам.
Подробности таких реакций даются в следующих примерах конкретных синтезов, которые приводятся для иллюстрации, и не означают каких-либо ограничений.
Пример синтеза 1
Получение 3,4,4-трифтор-3-бутен-1-амина и его солей
a) К 25 г (0,0896 моль) тозилата серебра в 100 мл ацетонитрила при комнатной температуре и медленном перемешивании добавляют 13,2 г (0,07 моль) 4-бром-1,1,2-трифтор-1-бутена. Реакционную смесь, защищенную от света, затем нагревают и перемешивают в течение ночи при температуре кипения с обратным холодильником. После охлаждения осадок отфильтровывают, и растворитель удаляют из фильтрата при пониженном давлении. К остатку добавляют 10 мл этилацетата и затем промывают 3 раза водой и сушат над сульфатом магния. К этому этилацетатному раствору 3,4,4-трифтор-3- бутетолизата добавляют 25 мл ДМФА и 16,7 г (0,09 моль) фталимида калия. Эту реакционную смесь перемешивают при температуре кипения с обратным холодильником в течение 24 часов. После охлаждения осадок отфильтровывают и промывают этилацетатом, который объединяют с фильтратом, промывают один раз водой, один раз 5%-ным раствором NaOH, и затем 3 раза водой, и, наконец, сушат на сульфатом магния. Растворитель удаляют при пониженном давлении и получают 14,45 г N-(3,4,4-трифтор-3-бутенил)фталимида в виде твердого вещества рыжевато-коричневого цвета.
b) К 13,91 г (0,054 моль) продукта стадии a), растворенного в 100 мл этанола, добавляют 1,76 г (0,054 моль) безводного гидразина. Реакционную смесь затем перемешивают при температуре кипения с обратным холодильником в течение 3 часов. Затем медленно, через обратный холодильник, добавляют 40 мл конц. HCl и смесь перемешивают при температуре кипения с обратным холодильником в течение еще 2 часов. После охлаждения реакционную смесь разбавляют водой, осадок фильтруют и промывают дополнительным количеством воды, которую соединяют с фильтратом. Объединенные фильтраты промывают эфиром, и эфирный слой отбрасывают. Водный слой охлаждают в ледяной бане, и затем добавляют 50% NaOH до тех пор, пока раствор не станет щелочным. Водный слой затем дважды экстрагируют хлороформом и объединенные хлороформные экстракты, содержащие нужный амин, сушат над сульфатом магния.
c) В хлороформенный раствор стадии b), чтобы выделить хлористоводородную соль нужного амина, барботируют избыток газообразного HCl. После перемешивания в течение 15 мин хлороформ удаляют при пониженном давлении и получают 7,4 г гидрохлорида 3,4,4-трифтор-3-бутен-1-амина в виде белого твердого вещества, т. пл. 191 - 193oC. Это соединение I, используемое в биологических испытаниях, описанных ниже.
d) Другие соли амина могут быть получены подобным способом, или они могут быть получены из хлористоводородной соли методом, широко известным в технике. Например, хлористоводорожная соль может быть нейтрализована вновь до свободного амина. Избыток свободного амина затем добавляют к различным кислотам, растворенным в метаноле, и получают нужные соли.
Пример синтеза 2
Получение гидрохлорида N-(3,4,4-трифтор-3-бутенил_глицина
(соединение 9)
a) К суспензии 55 г (0,34 моль) соединения I, полученного так, как описано выше, в 600 мл тетрагидрофурана (ТГФ) добавляют 34,4 г (0,34 моль) триэтиламина. Затем при комнатной температуре добавляют по каплям 16,1 г (0,105 моль) метилбромацетата и реакционную смесь перемешивают при комнатной температуре в течение 4 часов. Осадок отфильтровывают и растворитель удаляют из фильтрата при пониженном давлении. К остатку добавляют эфир, и полученную смесь перемешивают в течение 20 минут. Дополнительно образовавшийся осадок отфильтровывают, и в эфирный фильтрат барботируют избыток безводного газообразного HCl. Образовавшийся осадок отфильтровывают, промывают эфиром и сушат при пониженном давлении, получают 23,4 г метилового эфира N-(3,4,4-трифтор-3-бутенил)-глицина-гидрохлорида, соединение 8, в виде белого твердого вещества, выход 96%.
b) Соединение 8 - 19 г (0,081 моль) - кипятят с обратным холодильником в 90 мл 6N HCl. После удаления растворителя при пониженном давлении остаток перемешивают в течение 2 часов в эфире. Осадок отфильтровывают, промывают эфиром и сушат при пониженном давлении, получают 16,25 г названного в заголовке соединения в виде белого твердого вещества, выход 92%, т.пл. 186 - 188oC.
Пример синтеза 3
Получение гидрохлорида N-(3,4,4-трифтор-3-бутенил)валина
(соединение 10)
a) К раствору 8,88 г (0,053 моль) гидрохлорида метилового эфира DL-валина в 75 мл ДМФА в атмосфере азота добавляют 7,39 мл (0,053 моль) триэтиламина. В образовавшуюся суспензию добавляют по каплям 4 г (0,021 моль) 4-бром-1,1,2-трифтор-1-бутена, и реакционную смесь перемешивают в течение 7 дней. Растворитель удаляют при пониженном давлении. К остатку добавляют эфир, и осадок отфильтровывают. Эфир их фильтрата удаляют при пониженном давлении. Остаток хроматографируют методом высокоэффективной жидкостной хроматографии (7% этилацетата в гексане), и выделяют 0,33 г нужного амина, растворяют в эфире и обрабатывают избытком газообразного HCl. Образовавшийся осадок отфильтровывают и сушат, получают 0,29 г гидрохлорида метилового эфира N-(3,4,4-трифтор-3-бутенил)-валина, соединение 49.
b) В Течение ночи при температуре кипения с обратным холодильником нагревают 0,47 г (0,0017 моль) соединения 49 в 10 мл ^N HCl. Реакционную смесь охлаждают и растворитель удаляют при пониженном давлении. Остаток суспендируют в эфире и продукт отфильтровывают, получают 0,43 г названного в заголовке соединения в виде твердого вещества белого цвета, выход 98%.
Пример синтеза 4
Синтез N,N'-бис-(3,4,4-трифтор-3-бутенил)этандиамида
(соединение 21)
К 4 г (0,0248 моль) соединения I и 3,72 г (0,037 моль) триэтиламина в 30 мл ТГФ медленно добавляют 0,79 г (0,0062 моль) оксалилхлорида. Все это перемешивают в течение ночи при комнатной температуре. Осадок отфильтровывают, и растворитель из фильтрата удаляют при пониженном давлении. Остаток суспендируют в воде в течение 1 часа. Осадок отфильтровывают, промывают водой и сушат. Сырой продукт перекристаллизовывают из этилацетата и получают 0,46 г названного в заголовке соединения в виде твердого рыжевато-коричневого цвета, выход 24%, т.пл. 139 - 141oC.
Пример синтеза 5
Синтез 4-оксо-4-[(3,4,4-трифтор-3-бутенил)амино]бутеновой кислоты
(соединение)
Смешивают 0,8 г (0,005 моль) соединения I с (0,005 моль) янтарного ангидрида в 30 мл ТГФ. К этой смеси добавляют 0,015 моль триэтиламина при перемешивании. Смесь перемешивают при комнатной температуре 48 часов. Растворитель удаляют испарением, и остаток растворяют в воде и подкисляют конц. HCl. Нужный продукт отфильтровывают и перекристаллизовывают из смеси этилацетата и циклогексана, получают 0,7 г названного в заголовке соединения в виде иглоподобных кристаллов, т.пл. 64 - 66oC.
Пример синтеза 6
Синтез 4-нитро-2-[[(3,4,4-трифтор-3-бутенил)имино]метил]фенола
(соединение 23)
К 0,005 молям 5-нитросалицилальдегида в 20 мл этанола добавляют 0,005 молей NaOH в виде 10%-ного водного раствора. К этому раствору добавляют 0,005 молей соединения I в 20 мл этанола. Полученную смесь перемешивают в течение 3 часов при комнатной температуре и затем в течение 5 часов при 50oC. Пока реакционная смесь горячая, добавляют немного воды. По охлаждении образуются кристаллы, выход 0,6 г названного в заголовке соединения в виде иглоподобных кристаллов желтого цвета, т.пл. 96 - 98oC.
Пример синтеза 7
Синтез бромида N-(3,4,4-трифтор-3-бутенил)гексаметилентетрамина
(соединение 26)
Нагревают 10 г (0,053 моль) 4-бром-1,1,2-трифтор-1-бутена, 3,7 г (0,026 моль) гексаметилентетрамина и 27 мл хлороформа при температуре кипения с обратным холодильником в течение 6,5 часов. Осадок отфильтровывают из горячей реакционной смеси и промывают 50 мл горячего хлороформа. Продукт сушат в вакууме и получают 1,1 г названного в заголовке соединения в виде твердого вещества белого цвета, выход 13%.
Пример синтеза 8
Синтез гидрохлорида 3,4,4-трифтор-N-гидрокси-3-бутен-1-амина
(соединение 27)
К 15 г О-ТМС-гидроксиламина в высушенной пламенем трубке высокого давления, продутой азотом, добавляют 7,5 г (0,027 моль) 3,4,4-трифтор-3-бутентозилата. Трубку закупоривают и реакционную смесь перемешивают в течение ночи при 75oC. После декантирования жидкости с осадка жидкость перегоняют и собирают продукт, который кипит при 29oC при 1 мм рт.ст., и получают 0,8 г названного в заголовке соединения, защищенного О-ТМС. Его перемешивают в метаноле в 5 мл в течение ночи. Затем в реакционную смесь барботируют избыток газообразного HCl. Растворитель удаляют при пониженном давлении, продукт сушат, и получают 0,6 г названного в заголовке соединения в виде вязкого желтого масла.
Пример синтеза 9
Синтез 2-(3,4,4-трифтор-3-бутенил)гидразида бензойной кислоты
(соединение 28)
К 20 г (0,147 моль) бензоилгидразина и 3,7 г (0,037 моль) триэтиламина в 60 мл ДМФА добавляют 7 г (0,037 моль) 4-бром-1,1,2-трифтор-1-бутена, и смесь перемешивают в течение 2 дней при комнатной температуре. Твердые вещества отфильтровывают и растворитель удаляют из фильтрата при пониженном давлении. К остатку добавляют эфир и суспендируют в течение 1 часа. Осадки отфильтровывают. Эфир из фильтрата удаляют при пониженном давлении. Остаток хроматографируют методом высокоэффективной жидкостной хроматографии (40% этилацетата в гексане), получают 1,35 г названного в заголовке соединения в виде светло-желтого твердого вещества, выход 15%, т.пл. 74 - 76oC.
Пример синтеза 10
Синтез гидрохлорида N-(3,4,4-трифтор-3-бутенил)гидразина
(соединение 29)
Кипятят с обратным холодильником в течение ночи 1 г (0,0041 моль) соединения 28, полученного так, как описано в примере 9, в 10 мл 6N HCl. После охлаждения образуется осадок побочного продукта. Водный слой промывают 4 раза эфиром. Воду удаляют при пониженном давлении и получают 0,6 г названного в заголовке соединения в виде липкого коричневого вещества, выход 83%.
Пример синтеза 11
Синтез гидрохлорида N-(3,4,4-трифтор-3-бутенил)гуанидина
(соединение 30)
Кипятят 3 г (0,0186 моль) соединения I и 0,78 г (0,0186 моль) цианамида с обратным холодильником в 25 мл абсолютного этанола в течение 4 дней. Растворитель удаляют при пониженном давлении и остаток высушивают при пониженном давлении, получают 3,66 г названного в заголовке соединения в виде янтарного вязкого масла, выход 97%.
Пример синтеза 12
Синтез 3,4,4-трифтор-3-бутенилнитрата
(соединение 31)
К 4 г (0,026 моль) нитрата серебра в 30 мл CH3CN добавляют по каплям 2,6 г (0,14 моль) 4-бром-1,1,2-трифтор-1-бутена. Все это перемешивают в течение ночи в темноте при комнатной температуре. Осадок отфильтровывают и растворитель удаляют при пониженном давлении. Воду добавляют к остатку и продукт экстрагируют эфиром. Эфир промывают 3 раза водой, сушат над сульфатом магния и удаляют при пониженном давлении, получают 250 мг сырого вещества, которое перегоняют и получают названное в заголовке соединение, т.кип. 25 - 28oC при 0,85 торр.
Пример синтеза 13
Синтез N-(3,4,4-трифтор-3-бутенил)мочевины
(соединение 32)
К 2 г (0,0124 моль) соединения I и 2,5 г (0,024 моль) триэтиламина в 14 мл ТГФ добавляют 1,5 г (0,0124 моль) ТМС-изоцианата. Смесь перемешивают в течение ночи при комнатной температуре и затем 2 часа при 70oC. После охлаждения осадок отфильтровывают и растворитель удаляют при пониженном давлении. Полученное в результате твердое вещество суспендируют в эфире. Эфир декантируют с нерастворившегося вещества и удаляют при пониженном давлении. К остатку добавляют метанол, перемешивают в течение ночи при комнатной температуре и затем удаляют при пониженном давлении. Остаток суспендируют в смеси эфира с петролейным эфиром (1:1), твердое вещество отфильтровывают и сушат при пониженном давлении, получают 0,6 г названного в заголовке соединения, выход 30%, т.пл. 86 - 88oC.
Пример синтеза 14
Синтез N-(п-трифторметилбензолсульфонил)-3,4,4-трифтор-3-бутен-1-амина
(соединение 33)
Соединение I (0,01 моль) смешивают со 100 мл метиленхлорида и добавляют к п-трифторметилбензолсульфонилхлориду (0.01 моль) в 20 мл метиленхлорида. Смесь охлаждают и перемешивают, затем добавляют 0,023 молей триэтиламина. После перемешивания в течение 4 часов при комнатной температуре добавляют 100 мл воды и два слоя разделяют. Метиленхлоридный слой промывают раствором NaHCO3 в воде, и сушат над сульфатом магния. Растворитель упаривают и белый остаток перекристаллизовывают из этилацетата и циклогексана, получают 2,6 названного в заголовке соединения в виде белых кристаллов, т.пл. 68 - 70oC.
Пример синтеза 15
Синтез гидрохлорида 2-амино-N-(3,4,4-трифтор-2-бутенил)-пропанамида
(соединение 35)
a) К 3,8 г (0,02 моль) N-трет.-ВОК-L-аланина в 40 мл безводного ТГФ добавляют 3,2 г (0,02 моль) карбонилдиимидазола с выделением газа. Через 1 час добавляют 3,6 г 3,4,4-трифтор-3-бутен-1-амина, полученного так, как в примере 1, и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют при пониженном давлении. К остатку добавляют воду и продукт реакции экстрагируют эфиром. Эфир промывают 3 раза водой, сушат над сульфатом магния и удаляют при пониженном давлении, получают 4,31 защищенного трет. -ВОК-группой аналога названного в заголовке соединения (соединение 34) в виде белого твердого вещества, выход 73%, т.пл. 78 - 79oC.
b) Растворяют в эфире 1,5 г (0,0051 моль) соединения 34 и в полученный раствор барбортируют избыток газообразного HCl. Реакционную смесь перемешивают в течение 5 часов при комнатной температуре. Полученный в результате белый осадок отфильтровывают и сушат при пониженном давлении, получают 1,1 г названного в заголовке соединения в виде твердого вещества белого цвета, выход 98%, т.пл. 164 - 166oC.
Пример синтеза 16
Синтез 3,4,4-трифтор-3-бутеновой кислоты (соединение 44) и ее солей
a) К 50 г (0,37 моль) фенилуксусной кислоты и 55,9 г (0,37 моль) 1,8-диазабицикло[5,4,о]ундец-7-ена (ДВУ) в 400 мл CH3CN добавляют 98 г (0,52 моль) 4-бром-1,1,2-трифтор-1-бутена. Полученную смесь перемешивают при температуре кипения с обратным холодильником в течение 2 дней. После охлаждения растворитель удаляют при пониженном давлении. К остатку добавляют воду и продукт экстрагируют эфиром. Эфирный экстракт промывают 2 раза 5% NaOH, 2 раза водой, сушат над сульфатом магния и эфир удаляют при пониженном давлении; получают 40,87 г трифторбутенильного эфира фенилуксусной кислоты. Этот эфир добавляют к 7,2 г (0,18 моль) NaOH, растворенного в 70 мл воды. Все это энергично перемешивают в течение ночи при комнатной температуре. В реакционную смесь добавляют эфир чтобы проэкстрагировать продукт реакции. Отделенный эфирный слой сушат над сульфатом магния и перегоняют. Собирают продукт, получая 15,78 г 4-гидрокси-1,1,2-трифтор-1-бутена в виде светлой жидкости, т.кип. 120oC при 760 мм рт.ст.
b) К 84,27 г (0.843 моль) триоксида хрома в 500 мл уксусной кислоты и 75 мл воды добавляют по каплям 26,43 г (0,21 моль) спирта, полученного в a), поддерживая температуру ниже 10oC. После завершения добавления реакционную смесь перемешивают при 5oC в течение 2 часов, а затем при комнатной температуре. Смесь затем разбавляют 1 л воды и дважды экстрагируют эфиром. Объединенные эфирные слои промывают 3 раза водой, сушат над сульфатом магния и затем растворитель удаляют при пониженном давлении. Остаток перегоняют коротким путем при 1 мм рт.ст., и собирают фракцию, кипящую при 55oC. Этот дистиллат растворяют в эфире и затем дважды экстрагируют насыщенным NaHCO3. Объединенные слои NaHCO3 промывают 3 раза эфиром и затем подкисляют конц. HCl. Затем продукт экстрагируют эфиром. Эфирный экстракт промывают 3 раза водой, сушат над сульфатом магния и эфир удаляют при пониженном давлении; получают 8,22 г названного в заголовке соединения в виде светлой жидкости.
c) Может быть получено соединение 44, и затем нейтрализовано известными способами до любой агрономически приемлемой соли. Это включает и соль трифторбутенамина, такого как 3,4,4-трифтор-3-бутен-1-амин, полученного в примере 1.
Пример синтеза 17
Синтез 3,4,4-трифтор-3-бутен-1-амида
(соединение 45)
К 1 г (0,0071 моль) соединения 44, полученного так, как в примере 16, растворенного в 20 мл безводного ТГФ, добавляют 1,15 г (0,0071 моль) карбонилдиимидазола. После перемешивания в течение 20 минут в реакционную смесь барботируют избыток безводного газообразного NH3 и смесь перемешивают в течение ночи при комнатной температуре. Затем растворитель удаляют при пониженном давлении. К остатку добавляют этилацетат и полученную смесь экстрагируют 2 раза 1-% HCl, сушат над сульфатом магния и растворитель удаляют при пониженном давлении. Сырой продукт затем сублимируют (50 - 55oC при 1 мм рт. ст. ) и получают 0,39 г названного в заголовке соединения в виде твердого вещества белого цвета.
Пример синтеза 18
Синтез 2,2,2-трифтор-N-(3,4,4-трифтор-3-бутенил)ацетамида
(соединение 50)
К 1,07 г (0,0066 моль) соединения I и 0,67 г (0,0066 моль) триэтиламина в 10 мл ТГФ добавляют 1,7 г (0,008 моль) трифторуксусного ангидрида и смесь перемешивают в течение ночи при комнатной температуре. Твердые вещества отфильтровывают и растворитель удаляют при пониженном давлении. К остатку добавляют воду и продукт экстрагируют эфиром. Добавляют к эфирному экстракту 0,5 мл триэтиламина, промывают его 4 раза водой, сушат над сульфатом магния и удаляют эфир при пониженном давлении. Продукт отгоняют из остатка, получают 0,5 г названного в заголовке соединения в виде светлой жидкости; выход 35;.
Пример синтеза 19
Синтез 1,1,2-трифтор-4-изоцианат-1-бутена
(соединение 72)
a) Смешивают безводный пиридин и 0,033 моля соединения 1, и охлаждают до 0oC. Добавляют 0,022 моля дифенилкарбамоилхлорида и реакционную смесь перемешивают в течение ночи при комнатной температуре в атмосфере азота. Образовавшуюся суспензию выливают в ледяную воду, осадок отфильтровывают и промывают водой. Осадок растворяют в растворе эфира в этилацетате, сушат над сульфатом магния и концентрируют. Полученный в результате осадок перекристаллизовывают из горячей смеси этилацетата с гексаном и получают 4,57 г N,N-дифенил-N'-(3,4,4-трифтор-3-бутенил)мочевины; выход 65%; т.пл. 116 - 177oC.
b) Нагревают в атмосфере азота 0,016 молей мочевины стадии a) до тех пор, пока не перестанут выделяться пары, образовавшиеся при пиролизе. Собирают названное в заголовке соединение, так как оно отгоняется, получают 1,67 г соединения в виде светлого масла.
Пример синтеза 20
Синтез 4-метил-N-[[(3,4,4-трифьло-3-бутенил)амино]карбонил]бензол-сульфонамида (соединение 75)
При комнатной температуре перемешивают 0,01 моль соединения I в 40 мл ТГФ, добавляя в это время 0,01 моль п-толуолсульфонилизоцианата. Осуществляют охлаждение и добавляют 0,01 моль триэтиламина. Смесь перемешивают при комнатной температуре в течение 48 часов и растворитель удаляют при пониженном давлении. Остаток растворяют в метиленхлориде и промывают 3 раза 50 мл воды. Раствор сушат над сульфатом магния и растворитель упаривают. Остаток перекристаллизовывают из этанола и получают 1,1 г названного в заголовке соединения в виде белых кристаллов, т.пл. 134 - 136oC.
Пример синтеза 21
Синтез гидрохлорида 4-амино-1,1-дифтор-1-бутена (соединение 46)
a)Нагревают до 48oC 100 г (0,68 моль) фталимида в 250 мл этанола и 0,08 г метоксида натрия, и добавляют по каплям 50,8 г (0,91 моль) акролеина в 40 мл этанола. Смесь перемешивают в течение ночи и растворитель удаляют при пониженном давлении. Продукт перекристаллизовывают из метиленхлорида и сушат в вакууме. Получают 150 г белых кристаллов.
b) Расплавляют 51,63 г трифенилфосфина и растворяют в 100 мл сухого диметилацетамида. Раствор охлаждают до -5oC и добавляют по каплям 41,34 г (0,197 моль) дибромдифторметана. Добавляют 20,0 г (0,0984 моль) продукта стадии a), растворенного в 70 мл метиленхлорида, и вслед за этим добавляют 12,88 г Zn-катализатора. Смесь перемешивают в течение 2 часов и фильтруют. Фильтрат разделяют, используя 200 мл метиленхлорида в 200 мл воды. Органический слой дважды промывают водой и экстрагируют 100 мл 5%-ного раствора гидроксида натрия, 100 мл 10% HCl и 200 мл воды. Растворитель удаляют в вакууме, и получают N-(4,4-дифтор-3-бутенил)фталимид.
c) Смешивают 51,0 г (0,215 моль) продукта стадии b) с 250 мл этанола и 24,11 г гидразина. После перемешивания и нагревания при температуре кипения с обратным холодильником в течение 45 минут к смеси добавляют 71,38 г HCl и 70 мл воды, и продолжают кипячение с обратным холодильником в течение 30 минут. Растворитель удаляют при пониженном давлении и оставшуюся смесь распределяют между 300 мл воды и 200 мл метиленхлорида. Поднимают величину pH водного слоя до 12 водным 50%-ным раствором гидроксида натрия, и смесь дважды экстрагируют метиленхлоридом (200 мл). Объединенные метиленхлоридные растворы добавляют в 100 мл 6N HCl и растворитель удаляют при пониженном давлении. Сырую соль амина добавляют к 30 г гидроксида натрия (пластинки) и отгоняют свободный амин. Амин добавляют к 50 мл 6N HCl и воду удаляют при пониженном давлении, получают 13,31 названного в заголовке соединения, соединение 46, в виде белых кристаллов.
Из соединения 46 могут быть получены дифтораналоги производных трифторбутенамина, полученных в предыдущих примерах, например, гидрохлорид метилового эфира N-(4,4-дифтор-3-бутенил)глицина (соединение 76).
Пример синтеза 22
Синтез 5,6,6-трифтор-5-гексеновой кислоты (соединение 104)
a) В 1-литровую колбу в атмосфере азота помещают 5,5 г (0,226 моль) магниевой стружки и 250 мл безводного эфира. Добавляют по каплям 40 г (0,212 моль) 4-бром-1,1,2-трифторбутена до тех пор, пока последует энергичное обратное возвращение флегмы. Оставшуюся часть затем добавляют по каплям с такой скоростью, чтобы поддерживать спокойный процесс возвращения флегмы. После завершения добавления, реакционную смесь еще перемешивают в течение 30 минут. Реакционную смесь затем охлаждают до температуры между -30oC и -50oC с помощью смеси сухого льда с ацетоном. Вслед за этим добавляют 4,04 г CuI и затем 8,5 мл (0,017 моль) конденсированного оксида этилена. Эту смесь оставляют стоять при температуре от -30oC до -10oC в течение 20 минут и затем нагревают до комнатной температуры. Начинается кипение и реакционную смесь охлаждают на ледяной бане, а затем выдерживают при комнатной температуре в течение 3 дней. Затем медленно добавляют 200 мл 10% HCl, а вслед за этим 35 мл конц. HCl. После перемешивания в течение 2 часов осадок отфильтровывают. Отделившийся эфирный слой промывают 1 раз водой, 1 раз насыщенным NaHCO3, снова 1 раз водой, сушат над MgSO4 и удаляют растворитель в вакууме. Остаток дважды перегоняют. Собирают 4,8 г фракции, кипящей при 58 - 59oC при 2 мм рт.ст.
b) К 3 г (0,019 моль) соединения, полученного на стадии a), в 48 мл ацетона добавляют 15 мл реактива Джона (Fieser and Fieser, vol. I., p. 142) по каплям, и во время добавки поддерживают температуру приблизительно на уровне 20oC с помощью ледяной бани. После завершения добавления реакционную смесь перемешивают еще полчаса при комнатной температуре. Отделяют хроматные соли, пропуская реакционную смесь через силикагель. После промывания ацетоном, ацетон удаляют в вакууме. Остаток разбавляют водой и продукт экстрагируют эфиром. Эфир промывают 3 раза водой, сушат над MgSO4, и эфир удаляют в вакууме. Сырой продукт растворяют в эфире и экстрагируют насыщенным NaHCO3. Слой NaHCO3 промывают эфиром, затем подкисляют конц. HCl. нужный продукт переходит в эфир. Эфирный экстракт промывают один раз водой, сушат над MgSO4 и эфир удаляют в вакууме; получают 1 г продукта в виде светлой жидкости.
Пример синтеза 23
Синтез 3,4,4-трифтор-N-гидрокси-3-бутенамида (соединение 105)
К 1,0 г (0,0071 моль) соединения 44 в 10 мл безводного ТГФ добавляют 1,16 г (0,0071 моль) карбонилдиимидазола. Через 20 минут добавляют 0,75 г (0,0071 моль) О-ТМС-гидроксиламина (Aldrich) к реакционной смеси. Все это перемешивают при комнатной температуре в течение 2 дней. Затем растворитель удаляют в вакууме. Остаток растворяют в этилацетате и промывают 2 раза минимальным количеством 10% HCl. Этилацетатный раствор сушат над MgSO4 и растворитель удаляют в вакууме. Остаток затем смешивают с метанолом в течение 2 часов. Метанол удаляют в вакууме. Остаток сублимируют (при 80oC при 102 мм рт.ст.). Сублимированное твердое вещество 3 раза смешивают со смесью эфира с петролейным эфиром (растворитель декантируют каждый раз), затем перекристаллизовывают из смеси 30% этилацетата с гексаном, получают 100 мг продукта в виде белого твердого вещества, т.пл. 99 - 100oC.
Пример синтеза 24
Синтез (2,2-диметил-1,3-диоксолан-4-ил)метилового эфира 3,4,4-трифтор-3-бутеновой кислоты (соединение 106)
К 1,0 г (0,0071 моль) соединения 44 в 10 мл безводного ТГФ добавляют 1,16 г (0,0071 моль) карбонилдиимидазола. Через 20 минут добавляют 0,94 г (0,0071 моль) 2,2-диметил-1,3-диоксолан-4-илметанола и реакционную смесь перемешивают при комнатной температуре в течение 3 дней. Растворитель удаляют в вакууме. Остаток растворяют в петролейном эфире. Петролейный эфир промывают 4 раза водой, сушат над MgSO4 и удаляют растворитель в вакууме; получают 0,44 г продукта в виде светлой жидкости.
Пример синтеза 25
Синтез метилового эфира 4-[(3,4,4-трифтор-3-бутенил)амино)] бензойной кислоты (соединение 107)
Осторожно нагревают 4,1 г (0,027 моль) метил-4-аминобензоата и 2 г (0,00714 моль) 3,4,4-трифтор-3-бутентозилата при 130oC в течение 4 часов. Полученный в результате продукт растворяют (после охлаждения) в этилацетате. Осадок отфильтровывают, этилацетат промывают один раз водой, сушат над MgSO4 и удаляют в вакууме. Сырой продукт хроматографируют методом высокоэффективной жидкостной хроматографии (15% этилацетата в гексане); получают 1,48 г продукта в виде светлой жидкости, которую превращают в твердое вещество, т. пл. 47 - 49oC.
Пример синтеза 26
Синтез 4-[3,4,4-трифтор-3-бутенил)амино]бензойной кислоты
(соединение 109)
При комнатной температуре в течение ночи перемешивают 1 г (0,00386 моль) соединения 107 и 0,15 г (0,00386 моль) NaOH в 10 мл воды и 10 мл этанола. Растворитель удаляют в вакууме. К остатку добавляют воду и промывают один раз эфиром. Водный слой затем подкисляют конц. HCl. Образовавшийся осадок отфильтровывают, промывают водой и сушат; получают 0,33 г нужного продукта в виде твердого белого вещества, т.пл. 149 - 151oC.
Пример синтеза 27
Синтез моногидрохлорида (2-[3,4,4-трифтор-3-ьутенил)амино] ацетамида (соединение 108)
К 15 г (0,093 моль) соединения I 9,6 г (0,093 моль) триэтиламина и 180 мл ТГФ медленно добавляют 3,9 г (0,028 моль) бромацетамида, и перемешивают в течение ночи при комнатной температуре. Осадок отфильтровывают и растворитель удаляют в вакууме из фильтрата. Остаток дважды смешивают со смесью эфира с метиленхлоридом и каждый раз осадок отфильтровывают. Затем в раствор барботируют избыток газообразного HCl, полученный в результате осадок отфильтровывают, промывают эфиром и сушат в вакууме. Продукт растворяют в воде и водный раствор дважды промывают эфиром. Воду удаляют в вакууме и полученный сырой продукт перекристаллизовывают из этанола, получают 2,39 г продукта в виде твердого белого вещества, т.пл. 198 - 200oC.
Пример синтеза 28
Синтез 7,8,8-трифтор-7-октеновой кислоты
(соединение 84)
Раствор 4-броммасляной кислоты (8,35 г, 0,05 моль) в безводном ТГФ (100 мл) обрабатывают, добавляя по каплям, магнийметилхлоридом (0,051 моль, 17 мл 3М раствора в ТГФ) при -25oC и перемешивании в течение 15 минут. Раствор дополнительно перемешивают в течение 15 минут при 0oC и обрабатывают дилитийтетрахлоркупратом (0,002 моль, 20 мл 0,1М раствора в ТГФ), и вслед за этим - 3,4,4-трифтор-3-бутенилмагнийбромидом (0,0565 моль, полученного отдельно из 3,4,4-трифтор-3-бутенилбромида и магниевой стружки в ТГФ). Смесь перемешивают при 0oC в течение 2 часов и при комнатной температуре в течение ночи. Раствор затем выливают в 400 мл эфира и 150 мл 10; водной серной кислоты. Эфирный слой экстрагируют 10% NaOH (2 х 50 мл). Водный слой промывают эфиром, подкисляют конц. HCl и экстрагируют эфиром (3 х 100 мл). Эфирный экстракт сушат, концентрируют и остаток перегоняют под вакуумом; получают 4,1 г нужного продукта в виде бесцветной жидкости, выход 42;, т.кип. 125 - 127oC (10 торр).
Пример синтеза 29
Синтез фенилметилового эфира 3,4,4-трифтор-3-бутеновой кислоты
(соединение 113)
Раствор 3,4,4-трифтор-3-бутенилхлорида (2,8 г 0,0176 моль) и бензилового спирта (0,9 г 0,0083 моль) в 20 мл дихлорметана нагревают при температуре кипения с обратным холодильником в течение 40 часов. Раствор охлаждают до комнатной температуры, разбавляют дихлорметаном (15 мл), промывают последовательно 5; раствором бикарбоната натрия, водой и молевым раствором, и сушат. Выпаривание растворителя дает 1,75 г аналитически чистого продукта в виде бледно-желтого масла, выход 92%.
Пример синтеза 30
Синтез 4-нитрофенилового эфира 3,4,4-трифтор-3-бутеновой кислоты
(соединение 117)
Раствор 4-нитрофенола) 1,10 г, 0,0079 моль) и 3,4,4-трифтор-3-бутеноилхлорида (1,85 г, 0,0116 моль) в сухом эфире (15 мл) обрабатывают, добавляя по каплям, триэтиламином (1,01 г, 0,01 моль) при -78oC и перемешивании. Смесь перемешивают при -78oC в течение 10 минут и оставляют достигать комнатной температуры. Реакционную смесь разбавляют эфиром (20 мл), смешивают с 15 мл 2N HCl. Органический слой последовательно промывают водой, 5; бикорбонатом натрия, солевым раствором и сушат. Темно-коричневый остаток, полученный после выпаривания растворителя, очищают, пропуская через короткую колонку с силикагелем, и получают 1,9 г продукта в виде коричневого твердого вещества; выход 91%, т.пл. 58 - 62oC.
Пример синтеза 31
Синтез 2-(3,4,4-трифтор-1-оксо-3-бутенил)гидразида 3,4,4-трифтор-3-бутеновой кислоты (соединение 120)
Раствор 3,4,4-трифтор-3-бутеноилхлорида (2,4 г, 0,0151 моль) в сухом эфире (20 мл) обрабатывают, добавляя по каплям, безводным гидразином (0,48 г, 0,15 моль) при перемешивании при -78oC. Смесь оставляют нагреваться до комнатной температуры, белый осадок отфильтровывают и растворяют в этилацетате. Этилацетатный раствор промывают 5% бикарбонатом натрия и сушат. Выпаривание растворителя дает 1,1 г нужного продукта в виде белого твердого вещества; выход 47;, т.пл. 191 - 193oC.
Пример синтеза 32
Синтез S-октилового эфира 3,4,4-трифтор-3-бутентиоловой кислоты
(соединение 121)
Смесь 3,4,4-трифтор-3-бутеноилхлорида (1,7 г, 0,0107 моль) и 1-октантиола (0,72 г, 0,0049 моль) нагревают при 70oC в течение 12 часов. Сырой продукт очищают, пропуская через колонку с силикагелем, и получают 1,2 г нужного продукта в виде светло-желтого масла; выход 91%.
Пример синтеза 33
Синтез гидрохлорида S-2-аминоэтилового эфира 3,4,4-трифтор-3-бутентиоловой кислоты (соединение 127)
Смесь 3,4,4-трифтор-3-бутенилхлорида (2,5 г, 0,00158 моль) и 2-аминоэтантиода-гидрохлорида (1,14 г, 0,010 моль) медленно нагревают до температуры кипения с обратным холодильником в течение 15 минут. Смесь охлаждают до комнатной температуры, обрабатывают сухим эфиром и фильтруют. Продукт перекристаллизовывают из абсолютного этанола и получают 0,8 г названного в заголовке соединения в виде не совсем белого твердого вещества, выход 34%, т.пл. 95 - 115oC.
Пример синтеза 34
Синтез моногидрохлорида 2-аминоэтилового эфира 3,4,4-трифтор-3-бутеновой кислоты (соединение 130)
Раствор 3,4,4-трифтор-3-бутеноилхлорида (1,97 г, 0,0124 моль) и N-третю-БОК-аминоэтанола (1,61 г, 0,010 моль) в сухом эфире (20 мл) обрабатывают триэтиламином (1,25 г, 0,0124 моль) при -78oC и при перемешивании. Смесь перемешивают в течение 30 минут и оставляют достигать комнатной температуры, и затем выливают в 20 мл воды. Эфирный слой последовательно промывают 5; бикарбонатом натрия и солевым раствором, и сушат. Эфирный раствор насыщают сухим HCl (газообразным) и перемешивают при комнатной температуре в течение 30 минут. Осадок отфильтровывают и сушат; получают 1,6 г названного в заголовке соединения в виде белого твердого вещества, выход 77%, т.пл. 94 - 100oC.
Пример синтеза 35
Синтез метилового эфира [(3,4,4-трифтор-3-бутенил)амино]-оксоуксусной кислоты (соединение 142)
Раствор 3,4,4-трифтор-3-бутен-1-амина (12,5 г, 0,1 моль) и триэтиламина (10,1 г, 0,1 моль) в сухом эфире (200 мл) обрабатывают, добавляя по каплям, оксалилхлоридом (12,3 г, 0,1 моль, в 30 мл эфира) при 0oC и при перемешивании. Смесь перемешивают при комнатной температуре в течение 30 минут и обрабатывают 30 мл воды. Органический слой последовательно промывают 2N HCl водой, 5% бикарбонатом натрия и солевым раствором, и сушат. Остаток, полученный после выпаривания растворителя, очищают перегонкой и получают 16,42 г нужного продукта в виде белого твердого вещества; выход 78;, т.пл. 33 - 34oC.
Пример синтеза 36
Синтез N-(3,4,4-трифтор-3-бутенил)этандиамида
(соединение 143)
Раствор соединения 142 (5,0 г, 0,0237 моль) в метаноле (50 мл) насыщают сухим газообразным аммиаком при комнатной температуре. Осадок отфильтровывают, промывают метанолом и сушат; получают 3,16 г названного в заголовке соединения в виде белого твердого вещества, выход 68;, т.пл. 190 - 220oC.
Пример 37
Синтез гидразида N-[(3,4,4-трифтор-3-бутенил)амино]оксоуксусной кислоты (соединение 144)
Раствор соединения 142 (4,22 г, 0,02 моль) в абсолютном этаноле (50 мл) обрабатывают моногидратом гидразина (1,6 г, 0,032 моль) при перемешивании. Осадок отфильтровывают, промывают этанолом и сушат; получают 2,54 г названного в заголовке соединения в виде белого твердого вещества, выход 60;, т.пл. 145 - 200oC.
Пример синтеза 38
Синтез [(3,4,4-трифтор-3-бутенил)амино]оксоуксусной кислоты (соединение 145)
Раствор соединения 142 (3,16 г, 0,015 моль) в метаноле (25 мл) обрабатывают раствором NaOH (0,8 г, 0,02 моль) в воде (5 мл). Раствор перемешивают при комнатной температуре в течение 30 минут и концентрируют. Остаток растворяют в воде (20 мл) и экстрагируют дихлорметаном (2 х 30 мл). Водный слой подкисляют конц. HCl и экстрагируют этилацетатом. Органический слой сушат и упаривают; получают 0,75 г названного в заголовке соединения в виде белого твердого вещества, выход 25;, т.пл. 95 - 100oC.
Пример синтеза 39
Синтез N-бутил-3,4,4-трифтор-3-бутенамида
(соединение 103)
К смеси воды (15 мл), дихлорметана (15 мл) и н-бутиламина (2,34 г, 0,032 моль) добавляют 2,5 г (0,0158 моль) 3,4,4-трифтор-3-бутеноилхлорида при 0oC и при перемешивании. После перемешивания в течение 30 минут органический слой последовательно промывают 2N HCl, водой 5% бикарбонатом натрия и солевым раствором, и сушат. Выпаривание растворителя дает 2,85 г названного в заголовке соединения в виде белого твердого вещества; выход 92;, т.пл. 34 - 36oC.
Пример синтеза 40
Синтез 2-[(3,4,4-трифтор-1-оксо-3-бутенил)амино]бензойной кислоты
(соединение 128)
К смеси 3-аминобензойной кислоты (1,37 г, 0,01 моль), бикарбоната натрия (0,84 г, 0,01 моль), воды (20 мл) и дихлорметана (20 мл) добавляют 2,2 г (0,0139 моль) 3,4,4-трифтор-3-бутеноилхлорида при 0oC и при перемешивании. После перемешивания в течение 15 минут при комнатной температуре смесь разбавляют этилацетатом (100 мл) и водой (50 мл). Органический слой промывают 2N HCl и солевым раствором, и сушат. Остаток, полученный после выпаривания растворителя, порошкуют с сухим эфиром и фильтруют; получают 1,8 названного в заголовке соединения в виде светло-розового твердого вещества, выход 69;, т.пл. 252 - 255oC.
Пример синтеза 41
Синтез 2,4-дигидро-4-[[(3,4,4-трифтор-3-бутенил)амино] метилен] -3Н-пиразол-3-она (соединение 131)
Раствор 3,4,4-трифтор-3-бутен-1-амина (2,25 г, 0,018 моль) и 4,5-дигидро-5-оксо-1Н-пиразол-4-карбоксальдегида (1,12 г, 0,01 моль) в абсолютном этаноле (30 мл) нагревают при температуре кипения с обратным холодильником в течение 15 минут. Раствор концентрируют и остаток перекристаллизовывают из смеси эфира с дихлорметанам; получают 1,49 г названного в заголовке соединения в виде желтого твердого вещества, выход 68;, т.пл. 126 - 130oC.
Пример синтеза 42
Синтез 3,4,4-трифтор-3-бутеноилхлорида (соединение 101)
К 46,7 г (0,334 моля) свежеперегнанного 3,4,4-трифтор-3-бутеновой кислоты (соединение 44) в 100 мл дихлорметана, содержащего 2 капли диметилформамида, при 0oC добавляют 31 мл (355 моль) оксалилхлорида в течение 5 минут. Смесь перемешивают при 0oC и оставляют на ночь нагреваться до температуры окружающей среды. Смесь быстро перегоняют на фракции при нормальном давлении через 20-см колонку Вигре, снабженную короткой дистилляционной насадкой. Чистый хлорангидрид получают в виде бесцветной жидкости, 31,8 г (выход 60%), т. кип. 90 - 97oC (температура масляной бани 130oC). При хранении при комнатной температуре происходит медленное разложение.
Другие галогенангидриды, например 3,4,4-трифтор-3-бутонилбромид, могут быть получены из соединения 101, или подобным способом из соединения 44.
Как очевидно для специалистов в этой области техники, другие активные кислотные соединения, такие как симметричные или несимметричные ангидриды или имидазолкарбонилы, могут быть получены из кислот или галогенангидридов настоящего изобретения, и могут быть пригодны для способов борьбы с вредителями, которые здесь описаны.
Пример синтеза 43
Синтез 1,1-диметилэтилового эфира N-(3,4,4-трифтор-1-оксо-3-бутенил)глицина (соединение 90)
К 5,6 г (66,7 ммоль) бикарбоната натрия, суспендированного в 40 мл воды, при 0oC добавляют 20 мл дихлорметана и вслед за этим добавляют 5,6 г (33.4 ммоль) гидрохлорида тет.-бутилглицината. К смеси в несколько приемов в течение 5 минут добавляют 5,25 г (33,2 ммоль) 3,4,4-трифтор-3-бутеноилхлорида (соединение 101). Смесь перемешивают в течение 30 минут при 0oC и фазы разделяют. Водную фазу экстрагируют дихлорметаном и органические фазы объединяют, разбавляют эфиром и промывают насыщенным водным раствором хлорида натрия. Раствор сушат над сульфатом магния и концентрируют. Остаток перегоняют, используя трубку с шаровым расширением, при 90 - 95oC (0,1 мм рт.ст. ), и получают 6,85 г (82;) бесцветного кристаллического твердого вещества, т.пл. 52 - 54oC.
Пример синтеза 44
Синтез N-(3,4,4-трифтор-1-оксо-3-бутенил)глицина
(соединение 91)
К 7,7 г (30,4 ммоль) 1,1-диметилэтилового эфира N-(3,4,4-трифтор-1-оксо-3-бутенил)глицина (соединение 90) добавляют 8 мл трифторуксусной кислоты. Раствор перемешивают при температуре окружающей среды в течение 24 часов и концентрируют. Остаток перекристаллизовывают из смеси этилацетата и эфира, получают 1,7 г бесцветных кристаллов, т.пл. 115 - 116oC. Маточную жидкость концентрируют и из смеси этилацетата с эфиром выкристаллизовывают дополнительное количество вещества, и общее количество составляет 4,4 г (73%).
Пример синтеза 45
Синтез гидрохлорида N-(2-аминоэтил)-3,4,4-трифтор-3-бутенамида
(соединение 96)
a) К 3,5 г (21,9 ммоль) 1,1-диметилэтилового эфира (2-аминоэтил)карбаминовой кислоты (полученной, как описано в Krapcho, A.P. Kuell, C. S. Synthetic Communications 1990, 20, 2559 - 2564) в 40 мл дихлорметана при 0oC добавляют 15 мл воды и 2,02 г (24 ммоль) бикарбоната натрия. Смесь перемешивают в течение 5 минут и в течение 2 минут добавляют 3,46 г (21,9 ммоль) 3,4,4-трифтор-3-бутеноилхлорида (соединение 101). В процессе добавления образуется белый осадок. Смеси позволяют нагреваться до температуры окружающей среды и для растворения продукта добавляют 150 мл дихлорметана. Фазы разделяют и органическую фазу смешивают с безводным сульфатом натрия, фильтруют и концентрируют. Остаток перекристаллизовывают из этилацетата и получают 4,25 г (69%) белого твердого вещества, т.пл. 123 - 124oC.
b) К 2,45 г (8,69 ммоль) карбамата, полученного на стадии a), в 15 мл метанола при температуре окружающей среды добавляют раствор HCl в метаноле, приготовленный добавлением 0,68 мл (6,5 ммоль) ацетилхлорида к 5 мл метанола при энергичном перемешивании. Смесь перемешивают при температуре окружающей среды в течение 3 часов и концентрируют. Перекристаллизация остатка из смеси 2-пропанола с этилацетатом дает 200 мг соли в виде порошка белого цвета, т. пл. 138 - 146oC. Вторая порция дает чистую хлористоводородную соль в виде бесцветных пластинок, т.пл. 145 - 147oC. Выход в двух порциях составляет 470 мг (33%).
Пример синтеза 46
Синтез моногидрохлорида 7,8,8-трифтор-7-октен-1-амина
(соединение 59)
a) В 1-литровую 4х-горловую колбу загружают магниевую стружку (15,4 г, 0,635 моль) и безводный ТГФ (100 мл) в атмосфере N2. Добавляют несколько кристаллов иода и смесь нагревают до тех пор, пока не исчезнет окраска иода. Добавляют по каплям раствор 1-бром-3,4,4-трифтор-3-бутена (100 г, 0,529 моль) в безводном ТГФ (500 мл) с такой скоростью, чтобы ТГФ спокойно кипел и стекал в смесь. После завершения добавления (около 1 часа) раствор нагревают при температуре кипения с обратным холодильником в течение 30 минут. Раствор охлаждают до 30oC и переносят в капельную воронку под давлением N2, используя трубку, и оставляя в колбе непрореагировавшие магниевые стружки.
b) В 3-литровую 4х-горловую колбу загружают 1,4-дибромбутан (114,4 г, 0,530 моль, 63,3 мл), дилитийтетрахлоркупрат (80 мл 0,1М раствора в ТГФ, 8,0 ммоль) и безводный ТГФ (250 мл). Смесь охлаждают до 5oC и обрабатывают, добавляя по каплям, реактивом Гриньяра, полученным на стадии a), при перемешивании при 5 - 10oC. Добавление завершают за 30 минут. Смесь затем перемешивают при 5 - 10oC в течение 3 часов и в течение ночи при комнатной температуре. Реакционную смесь разбавляют эфиром (1200 мл), охлаждают на бане с ледяной водой и медленно обрабатывают 5% серной кислотой (500 мл). Эфирный слой последовательно промывают 5% серной кислотой (300 мл), водой (200 мл), насыщенным NaHCO3 (200 мл), солевым раствором (200 мл) и сушат MgSO4. Растворитель выпаривают и остаток перегоняют при пониженном давлении; получают 100,2 г сырого продукта в виде бесцветного масла, т.пл. 85 - 100oC при 35 мм рт.ст. Эту фракцию растворяют в диметилсульфоксиде (ДМСО) (400 мл, безводный) и обрабатывают BaN3 (88.4 г, 1,36 ммоль) при перемешивании при комнатной температуре. Через несколько минут образуется толстый слой белого кристаллического вещества, добавляют дополнительные 20 мл ДМСО и энергично перемешивают в течение 1 часа. Затем смесь обрабатывают 600 мл воды и экстрагируют эфиром (2 х 600 мл). Эфирный экстракт промывают водой (3 х 300 мл), солевым раствором (300 мл) и сушат при MgSO4. Выпаривание растворителя дает 77.0 г светло-желтого масла. Это масло растворяют в 400 мл ДМСО и обрабатывают одной порцией трифенилфосфина (225 г, 0,858 моль) при перемешивании. Реакционную смесь охлаждают на водно-ледяной бане до тех пор, пока не перестанет выделяться тепло, и перемешивают при комнатной температуре в течение 5 часов. Затем добавляют 750 мл конц. гидроксида аммония и смесь перемешивают в течение ночи. Смесь разбавляют эфиром (1500 мл) и осадок отфильтровывают. Фильтрат промывают водой и эфирный слой экстрагируют 10; HCl. Водный слой затем снова экстрагируют дихлорметаном и концентрируют при пониженном давлении. Остаток обрабатывают 60% толуола в абсолютном этаноле и выпаривают для удаления следов воды. Остаток (10,1 г) представляет собой бледно-желтое твердое вещество, выход 8,7%
Пример синтеза 47
Синтез моногидрохлорида 5,6,6-трифтор-5-гексен-1-амина (соединение 74)
a) К магниевой стружке (26,7 г, 1,7 мль) в сухом эфире (500 мл) добавляют несколько кристаллов иода. Смесь нагревают до тех пор, пока не закипит эфир, и добавляют 4-бром-1,1,2-трифтор-1-бутен (189 г, 1,0 моль) в сухом эфире (400 мл) по каплям и при перемешивании, и с такой скоростью, чтобы эфир кипел медленно. Добавление занимает 1,5 часа. После завершения добавления, добавляют 200 мл сухого эфира, и смесь нагревают при температуре кипения с обратным холодильником в течение 30 минут. Смесь охлаждают до - 30oC и добавляют CuI (19,0 г, 0,1 моль). Этиленоксид, сконденсированный в капельной воронке, с помощью смеси сухого льда и ацетона (55 г, 1,25 моль), добавляют по каплям и перемешивании при -30oC приблизительно в течение 30 минут. Затем смесь оставляют достигать комнатной температуры на ночь. Затем смесь охлаждают до 0oC, медленно обрабатывают 500 мл 10% HCl и 150 мл конц. HCl, и фильтруют. Слои разделяют и водный слой экстрагируют эфиром. Объединенные эфирные экстракты промывают насыщенным NaHCO3, вслед за этим - солевым раствором, и сушат над MgSO4. После тщательного упаривания растворителя получают 195 г масла красного цвета. Перегонка при пониженном давлении дает 155 г бесцветного масла, т. кип. 90 - 100oC при 80 - 75 мм рт.ст. Продукт затем обрабатывают 1,5 л гексана и промывают 4 раза водой. Гексановый слой сушат над безводным Na2SO4 и концентрируют, получают 100 г светлого масла, которое перегоняют при пониженном давлении, и получают 95 г 5,6,6-трифтор-5-гексанола в виде светлого масла, выход 61,7%.
b) Раствор мезилхлорида (46,5 мл, 600 ммоль) в дихлорметане (250 мл) добавляют по каплям к раствору спирта, полученного на стадии a) (77,0 г, 500 ммоль) и триэтиламина (104,5 мл, 750 ммоль) в дихлорметане (750 мл) при -25oC при перемешивании. Перемешивание продолжают в течение 30 минут при -20oC. Затем смесь обрабатывают 10% HCl (300 мл) и органический слой промывают 10% HCl (200 мл), а затем насыщенным NaHCO3 (200 мл), солевым раствором (200 мл) и сушат над MgSO4. Выпаривание растворителя дает 116 г желтого масла. Его растворяют в ДМСО (400 мл) и вводят во взаимодействие с NaN3 (65 г, 1000 ммоль) при комнатной температуре в течение ночи. Затем в реакционную смесь при охлаждении добавляют трифенилфосфин (157,4 г, 600 ммоль). Смесь перемешивают при комнатной температуре в течение 5 часов и добавляют конц. гидроксид аммония. Смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь затем разбавляют эфиром (1500 мл) и экстрагируют водой. Осадок отфильтровывают и эфирный слой промывают водой и экстрагируют 10% HCl. Водный слой снова экстрагируют 3 раза дихлорметаном, и концентрируют в роторном испарителе при пониженном давлении; получают нужный продукт в виде светло-желтого твердого вещества, 74,6 г, выход 78,7%.
Используя способы и исходные вещества, описанные выше и проиллюстрированные в предшествующих примерах, получают и другие соединения. Ниже приведена таблица соединений, которые могут быть получены как указано выше.
КОМПОЗИЦИИ
Для обычного применения соединения формулы I, как правило, используют в свободном виде, без смешения или разбавления, а обычно используются в соответствующим образом составленной композиции, совместимой со способом применения, и содержащей эффективное количество соединения. Соединения настоящего изобретения, подобно большинству сельскохозяйственных препаратов, могут быть смешаны с приемлемыми для сельского хозяйства поверхностно-активными веществами и носителями, обычно применяемыми для облегчения диспергирования активных ингредиентов, с учетом признанного факта, что состав и способ применения могут влиять на активность вещества. Предлагаемые соединения могут применяться, например, в виде жидкостей для растения, дустов или гранул, на пространствах, на которых требуется провести борьбу с вредителями, причем тип применения изменяется в зависимости от вредителя и окружающей среды. Так, соединения настоящего изобретения могут входить в состав гранул большого размера, дустов, смачивающихся порошков, эмульгируемых концентратов, растворов и тому подобное.
Гранулы могут содержать пористые и непористые частицы, такие как частицы аттапульгитной глины или песка, например, которые служат в качестве носителей для предлагаемых соединений. Гранульные частицы являются относительно большими, диаметром, как правило, 400 - 2500 мкм. Частицы либо пропитывают соединением изобретения из раствора, либо покрываются соединением, причем иногда используют адгезив. Гранулы обычно содержат 0,05 - 10%, предпочтительно 0,5 - 5%, активного ингредиента.
Дусты представляют собой смеси соединений с тонко измельченными твердыми веществами, такими как тальк, аттапульгитная глина, кизельгур, пирофиллит, мед, диатомовая земля, фосфаты кальция, карбонаты кальция и магния, сера, различная мука, и другие органические и неорганические твердые вещества, которые действуют как носители для соединения. Эти твердые тонко измельченные вещества имеют средний размер частицы менее 50 мкм. Типичная дустовая композиция содержит 1 часть соединения и 99 частей талька.
Соединения настоящего изобретения могут быть изготовлены в виде жидких концентратов путем растворения или эмульгирования в подходящих жидкостях, и в виде твердых концентратов путем смешивания с тальком, глинами и другими известными твердыми носителями, применяемыми в химических препаратах для сельского хозяйства. Концентраты представляют собой композиции, содержащие 5 - 50% активного соединения и 95 - 50% инертного материала, который включает поверхностно-активные диспергаторы, эмульгаторы и смачивателя, но в порядке эксперимента могут быть использованы и более высокие концентрации активного ингредиента. При практическом применении в виде жидкостей для пульверизации концентраты разбавляют водой или другими жидкостями, а при использовании в виде дустов их разбавляют дополнительным количеством твердого носителя.
Типичный состав 50% смачивающегося порошка будет содержать 50% (вес/вес) активного ингредиента, 22% аттарульгитного разбавителя, 22% каолинового разбавителя и 6% натриевых солей сульфированного сульфатного лигнина как эмульгатора.
Типичные носители для твердых концентратов (также называемых смачивающимися порошками) включают фуллерову землю, глины, кремнеземы и другие сильные абсорбенты, легко смачивающиеся неорганическими разбавителями. Подходящая композиция твердого концентрата может содержать по 1,5 части лигносульфоната натрия и лаурилсульфата натрия, в качестве смачивателей, 25 частей активного соединения и 72 части аттапульгитной глины.
Производственные концентраты пригодны для перевозки низкоплавких продуктов настоящего изобретения. Такие концентраты получают, расплавляя низкоплавкие твердые продукты вместе с 1% или большим количеством растворителя, чтобы получить концентрат, который не затвердевает при охлаждении до точки замерзания чистого продукта или до более низкой температуры.
Подходящие жидкие концентраты включают эмульгируемые концентраты, которые представляют собой гомогенные жидкие или пастообразные композиции, легко диспергируемые в воде или в других жидких носителях. Они могут полностью состоять из активного соединения и жидкого или твердого эмульгатора, или они могут также содержать жидкий носитель, такой как ксилол, тяжелые ароматические фракции лигроина, изофорон и другие относительно нелетучие органические растворители. При применении эти концентраты диспергируют в воде или других жидких носителях, и применяют обычным образом в виде жидкостей для пульверизации на пространствах, которые подлежат обработке.
Типичный состав эмульгируемого концентрата (50 г на литр) будет состоять из 5,9% (вес/вес) соединения изобретения, эмульгаторов - 1,80% смеси кальциевой соли додецилбензосульфоната и неионного продукта конденсации 6-молярного этиленоксида с нонилфенолом, 2,7% смеси кальциевой соли додецилбензолсульфоната и неионного продукта конденсации 30-молярного этиленоксида с нонилфенолом, 1,50% неионной пасты простого эфира полиалкиленгликоля, и 88,10% очищенного ксилола в качестве растворителя.
Типичные поверхностно-активные смачиватели, диспергаторы и эмульгаторы, используемые в композициях для сельского хозяйства, включают, например, алкил- и арил-сульфонаты и сульфаты и их натриевые соли, алкиламидосульфонаты, включая жирные метилтауриды, алкиларильные простые полиэфиры спиртов, сульфированные высшие спирты, поливиниловые спирты, полиэтиленоксиды, сульфированные растительные и животные масла, сульфированные нефтяные масляные дистилляты, эфиры жирных кислот и многоатомных спиртов и продукты присоединения этиленоксида к таким эфирам, и аддукты длинноцепных меркаптанов и этиленоксида. Поверхностно-активное вещество обычно содержит 1 - 15 вес.% активного ингредиента.
Другие подходящие составы включают простые растворы активного ингредиента в растворителе, в котором он полностью растворяется в нужной концентрации, таких как вода, ацетон или другие органические растворители. Предпочтительная композиция для обработки листвы представляет собой водный раствор, содержащий обычно глицерин и такое поверхностно-активное вещество как Tween® 20, и как правило, 1% глицерина и 0,1% Tween® 20.
Композиции могут быть составлены и применяться с подходящими ингредиентами, обладающими пестицидной активностью, включая инсектициды, акарициды, фунгициды, регуляторы роста, гербициды, удобрения и т.д.
СПОСОБЫ ПРИМЕНЕНИЯ
Настоящее изобретение предлагает способы борьбы с различными вредителями, которые наносят вред урожаю сельскохозяйственных культур, а именно, нематодами, насекомыми и клещами. С заражением растения любыми из этих вредителей можно бороться, применяя для обработки частей растения эффективное количество любого из соединений настоящего изобретения. Обработка может быть выполнена различными способами, включая обработку соединением или композицией, его содержащей, почвы до или после посева или появления всходов, и семян или частей семян до или при посеве, и листвы, стеблей или стволов растения. Обработка может быть осуществлена в период роста не однажды, с перерывом между обработками в один или более число дней. Соответствующий режим обработки будет зависеть от жизненного цикла вредителя, с которым проводится борьба, температуры окружающей среды и уровня влажности. Кроме того, на режим обработки могут влиять возраст или размер растения.
Способы настоящего изобретения могут применяться для различных культур. К таким культурам относятся, но не ограничиваются ими, плодовые и овощные культуры, такие как картофель, сладкий картофель, морковь, томаты, виноград, персики, цитрусовые, бананы, кукуруза и культурная соя, табак и хлопчатник.
Соединения формулы I настоящего изобретения могут быть использованы для борьбы с любыми фитопатгенными нематодами. К насекомым, с которыми можно бороться способами настоящего изобретения, относятся, но не ограничиваются ими, лиственные вредители, такие как тля персиковая, и вредители, зарождающиеся в почве, такие как южная длинноусая блошка. Акариды, с которыми можно бороться способами настоящего изобретения, включают, но не ограничиваясь ими, клещиков паутинных двупятнистых. Испытания, проводимые с несколькими соединениями настоящего изобретения, показывают недостаточный уровень борьбы с совкой или колорадским жуком.
ПРИМЕРЫ ИСПЫТАНИЙ
A. Испытания с томатной и соевой нематодой
Соединения настоящего изобретения формулы I, полученные так, как описано выше, испытывают на эффективность действия против нематоды корневого нароста (Meloidogyne incognita). Соединения испытывают, обрабатывая листву и смачивая почву. Результаты четырех различных способов испытаний представлены в таблице A. Способы испытаний описаны далее.
СПОСОБ 1. Обработка листвы растений томата
Растение томата с "Rutgers" выращивают по одному растению в горшке размером 2,5х18,8 см. Через 19 дней после посадки обе стороны листьев опрыскивают испытуемым соединением при концентрациях, указанных в таблице A. Испытуемые соединения готовят, растворяя сначала каждое в воде или ацетоне, в соответствии с назначением, и смешивают с водой, содержащей 0,05% Tween® 20 и 1,0% глицерина. Для каждой группы испытаний опрыскивают четыре растения 29 мл раствора. Растения оставляют высыхать, прежде чем их помещают в камеры для выращивания, в которых поддерживается температура 25 - 28oC. Здесь их орошают, но недостаточно, чтобы предотвратить стекание капель с листьев на почву. Через 2 - 3 дня после химической обработки на корни и почву наносят пипеткой суспензию личинок 2,4500 на горшок. Через 3 недели после инокуляции количество повреждений на промытых листьях сравнивают с количеством повреждений на контрольных листьях, обработанных водой. Результаты выражают как процент устранения заболевания.
СПОСОБ 2. Обработка листвы растений томата
Осуществляют стадии способа 1, за исключением того, что (1) растения томата высаживают в горшки размером 7,5х6,3 см и (2) инокуляцию осуществляют, нанося пипеткой приблизительно 8000 яиц в 5 мл на почву в каждый горшок на 20 день после посадки. На следующий день каждое растение опрыскивают 4 мл раствора для обработки. Степень повреждения определяют через 3 недели после инокуляции. Используют шкалу оценки от 0 до 3, причем 0 соответствует обширному повреждению и 3 означает отсутствие повреждений.
СПОСОБ 3. Обработка почвы под семена сои
По два семени культурной сои CV"Williams" помещают в каждый горшок размером 2,5 см2 и инокулируют приблизительно 8000 яиц. Добавляют испытуемые соединения по 1 мг на горшок в 2 мл раствора, приготовленного так, как описано в способе 1. Для каждого уровня обработки используют один горшок. Семена покрывают вермикулитом и слегка смачивают. Растения ограниченно орошают один раз в день в течение 4 недель. Затем оценивают состояние промытых корней, как в способе 2.
СПОСОБ 4. Обработка почвы под растения томата
Следуют стадиям способа 2, за исключением того, что на следующий день после инокуляции почву обрабатывают, нанося пипеткой 4 мл раствора для обработки на почву в каждом горшке. Повреждения оценивают через 3 недели после инокуляции, как в способу 2.
СПОСОБ 5. Обработка почвы и листвы растений томата
Выполняют стадии способа 2 и 4, за исключением того, что растения томатов выращивают в горшках размером 5,0х5,0 см2, и инокуляцию осуществляют, нанося пипеткой приблизительно 7000 яиц в 4 мл на почву в каждом горшке через 20 дней после посадки. На следующий день после инокуляции обрабатывают почву, нанося пипеткой 2 мл раствора для обработки в каждый горшок на поверхность почвы, и опрыскивают листья 1,5 мл раствора для обработки. Повреждения оценивают через 3 недели после инокуляции, как в способе 2.
B. Испытания с цистой соевой нематоды
Соединение I испытывают на эффективность в борьбе с цистой соевой нематоды (Heterodera glycines) при обработке листвы. Песчаную почву заражают приблизительно 100 цист соевой нематоды на 1 см3 почвы. Приблизительно по 350 см3 зараженной почвы помещают в каждый горшок с основанием 25,8 см2. Три семени сои, Glycine max = вариации Williams-82, помещают на поверхность зараженной почвы и закрывают слоем незаряженной песчаной почвы.
Обработку листвы начинают, когда растение достигнет стадии развития одного листа. Обработку осуществляют как единичную, опрыскивания этот один лист, или проводят ряд последовательных опрыскиваний - одного листа плюс через 7 дней, или одного листа плюс через 7 дней, плюс через 14 дней. Испытуемое соединение растворяют в воде 1% глицерина и 0,1% Tween® 20. При каждой обработке поверхность почвы закрывают, чтобы предотвратить контакт вещества с почвой. Обработку проводят до точки роста, используя дозы, указанные ниже.
Окончательную оценку заболевания проводят приблизительно через 5 недель после посева, подсчитывая число цист на каждой корневой системе. Устранение заболевания определяют как процент уменьшения среднего числа цист на корнях обработанных растений по сравнению с инокулированными, обработанными контрольной композицией, состоящей из воды с глицерином и Tween® 20 .
C. Испытания с насекомыми и акаридами
Соединения I настоящего изобретения испытывают на эффективность в борьбе с клещиком паутинным двупятнистым, Tetranychus urticae или TSSM, персиковой тлей, Myzus persicae или GPA, и южный длинноусой блошкой, Diabrotica undecimpunctata или CPW, при обработке листвы.
В начале испытаний по определению действия против тлей трехнедельные растения китайской капусты (Brassica chinensis) заражают взрослыми насекомыми персиковой тли с кусочков листа культурного растения. Заново инфицированные растения оставляют в камерах для выращивания до тех пор, пока популяции насекомых не вырастут более, чем до сорока насекомых на листе. Для испытаний выбирают растения, которые являются наиболее однородно зараженными. Соответствующие количества испытуемых материалов растворяют непосредственно в водных составах, содержащих 1% глицерина и 0,1% Tween® 20. Растения опрыскивают до точки роста, нанося раствор как на верхнюю, так и на нижнюю поверхность листьев. Каждым испытуемым раствором опрыскивают по 5 растений. Опрысканные растения перемещают под колпак, где их держат до тех пор, пока они не высохнут. Растения снова переносят в камеры для выращивания на 5 дней, прежде чем определить окончательный результат действия.
Для испытаний с двупятнистым паутинным клещиком используют сою (Glycine max) на стадии одного листа, или хлопчатник (Gossypium hirsutum), у которого полностью раскрылись семядоли. С культурных растений, зараженных взрослыми клещиками и их яйцеклетками, удаляют листовые диски, изготовленные с помощью сверла для кормовых пробок # 6, помещают их на верхнюю поверхность первого листа растений сои или на верхнюю поверхность семядольных листьев растений хлопчатника. После того, как клещики расползутся и заразят все растение (6 - 10 дней), листовые диски удаляют. Наиболее однородно зараженные растения отбирают для испытаний и опрыскивают испытуемыми растворами, как описано выше, используя 5 растений на каждую обработку. Через 5 дней после обработки проверяют все листья и считают взрослых клещиков на нижней стороне листьев. Действие вычисляют как процент снижения среднего числа клещиков на обработанных растениях по сравнению с обработанными водным составом.
Испытания с южной длинноусой блошкой проводят, используя семена кукурузы (Zea mays), которые высевают по 4 в каждый из 7,5-см горшков, и выращенных в теплице до высоты растения приблизительно в 15 см. В поверхности почвы около корней каждого растения процарапывают небольшие отверстия, и в отверстия помещают 10 личинок длинноусой блошки первой стадии развития. На другой день после заражения листья растений кукурузы опрыскивают до точки роста испытуемыми растворами такого же состава, как указано выше. Через 7 дней после опрыскивания личинки извлекают из почвы, пропитывая горшки 10М раствором MgSO4. Усредняют число личинок для каждого вида обработки и вычисляют процент устранения.
D. Обработка семян картофеля
Соединения I настоящего изобретения испытывают на эффективность в борьбе с нематодой коричневого нароста (Meliodogine incgnita) при обработке кусков картофельных семян. Соединения также испытывают, обрабатывая листву, - для сравнения.
Картофельные клубни разрезают таким образом, чтобы на каждом куске оставалось по крайней мере 2 глазка. Для каждого уровня обработки кусочки клубней взвешивают и помещают в смесь соединения и достаточного количества измельченной глины, чтобы выдержать кусочки при определенной дозе обработки при постоянном объеме обработки. Кусочки перемешивают до тех пор, пока не покроются поверхности, и высаживают по одному кусочку в 3,8 л (1-галлонный) горшок. Каждый горшок инокулируют 50000 яиц.
При обработке листвы каждое растение опрыскивают раствором соединения I в воде с 1% глицерина и 0,05% Tween® 20 . Опрыскивание выполняют таким образом, чтобы в результате распределить соединение по поверхности всех листьев. Каждое растение опрыскивают 4 раза с интервалом в 2 недели, и инокулируют либо при посадке, либо за 3 дня до обработки. Результаты представлены в таблице D.

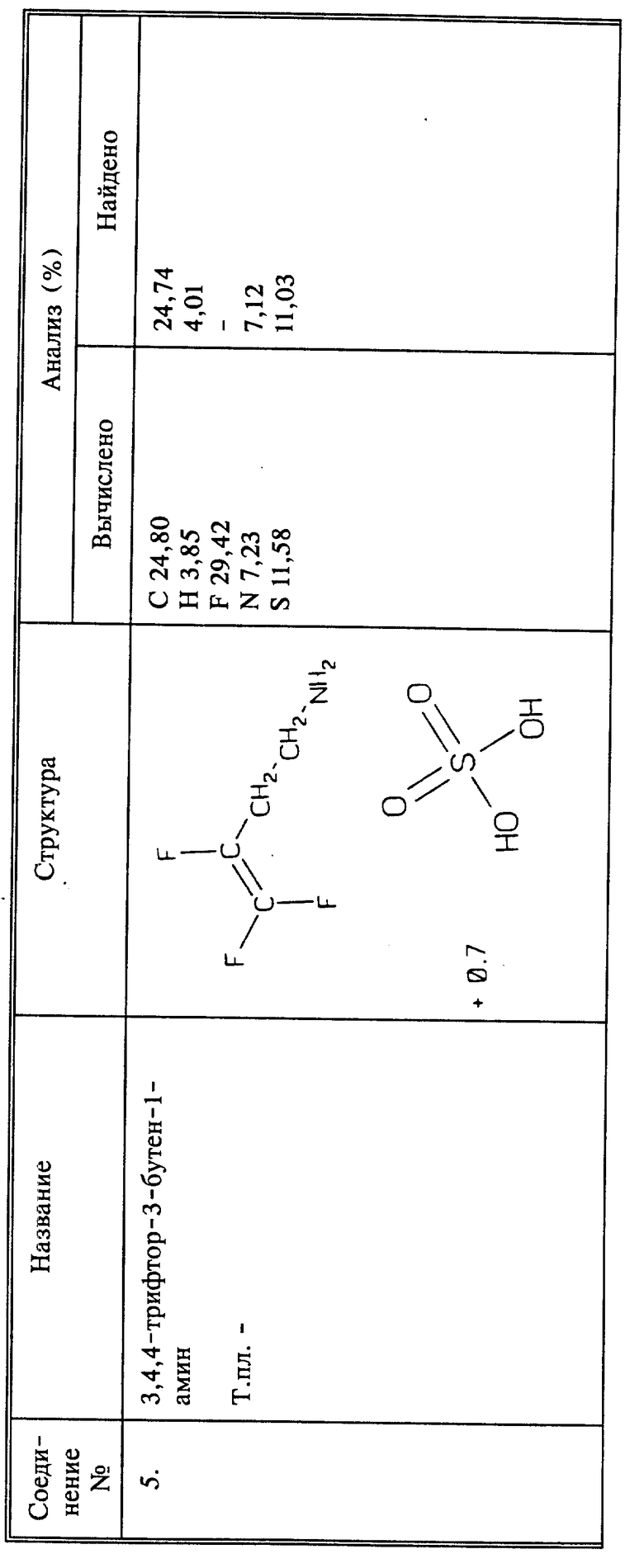
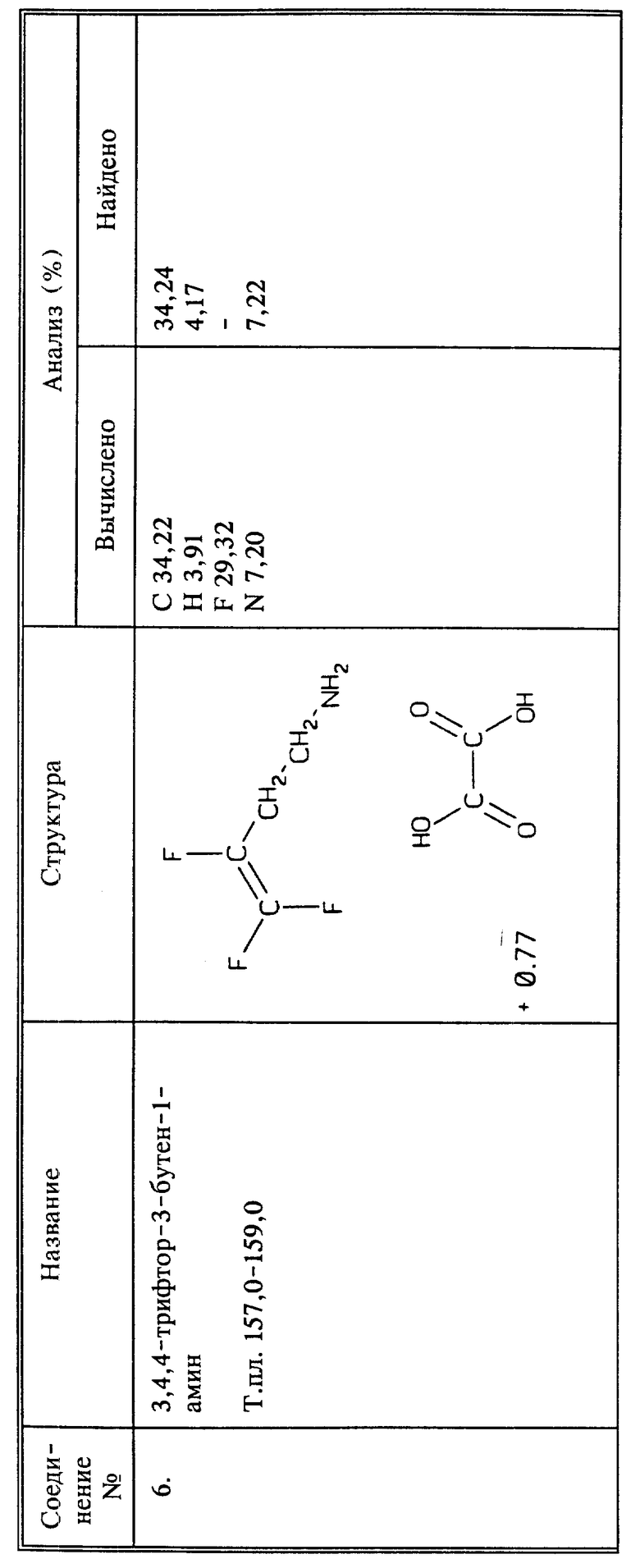
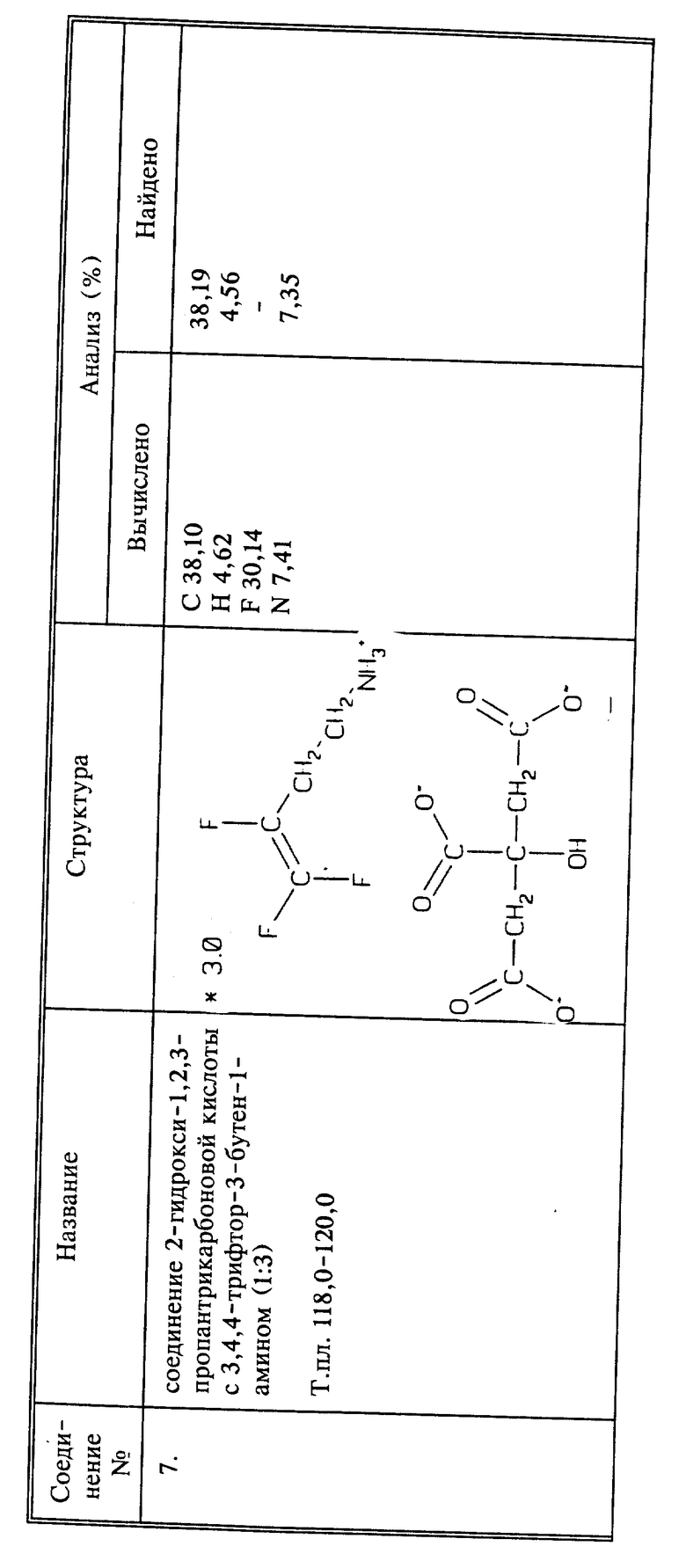
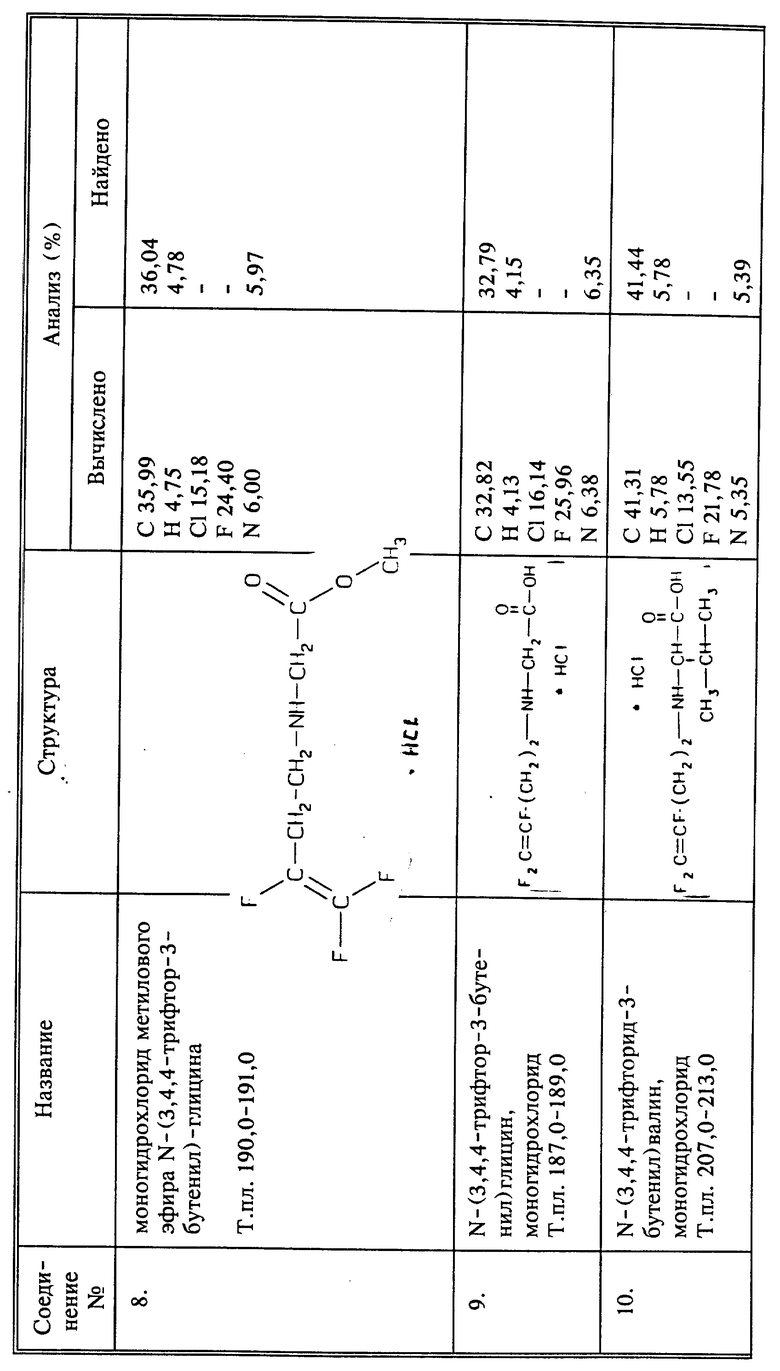

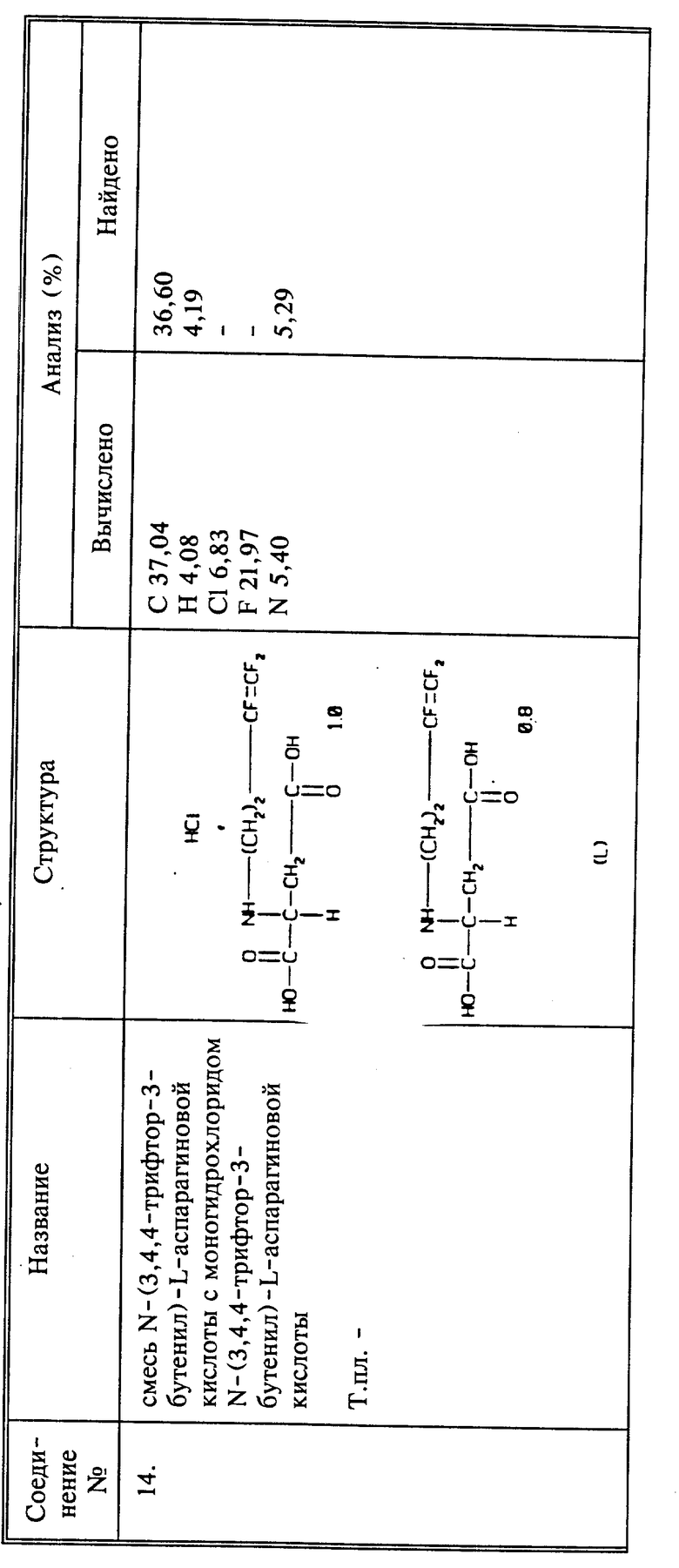
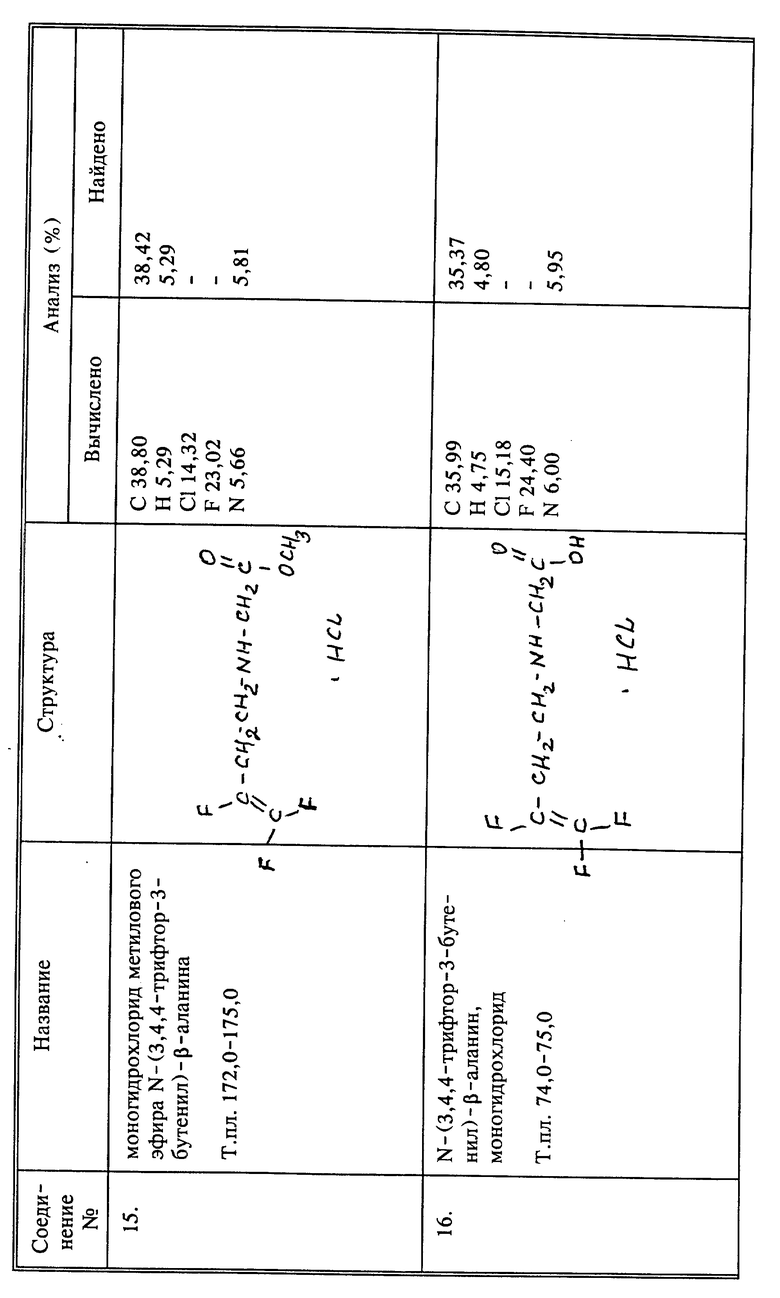
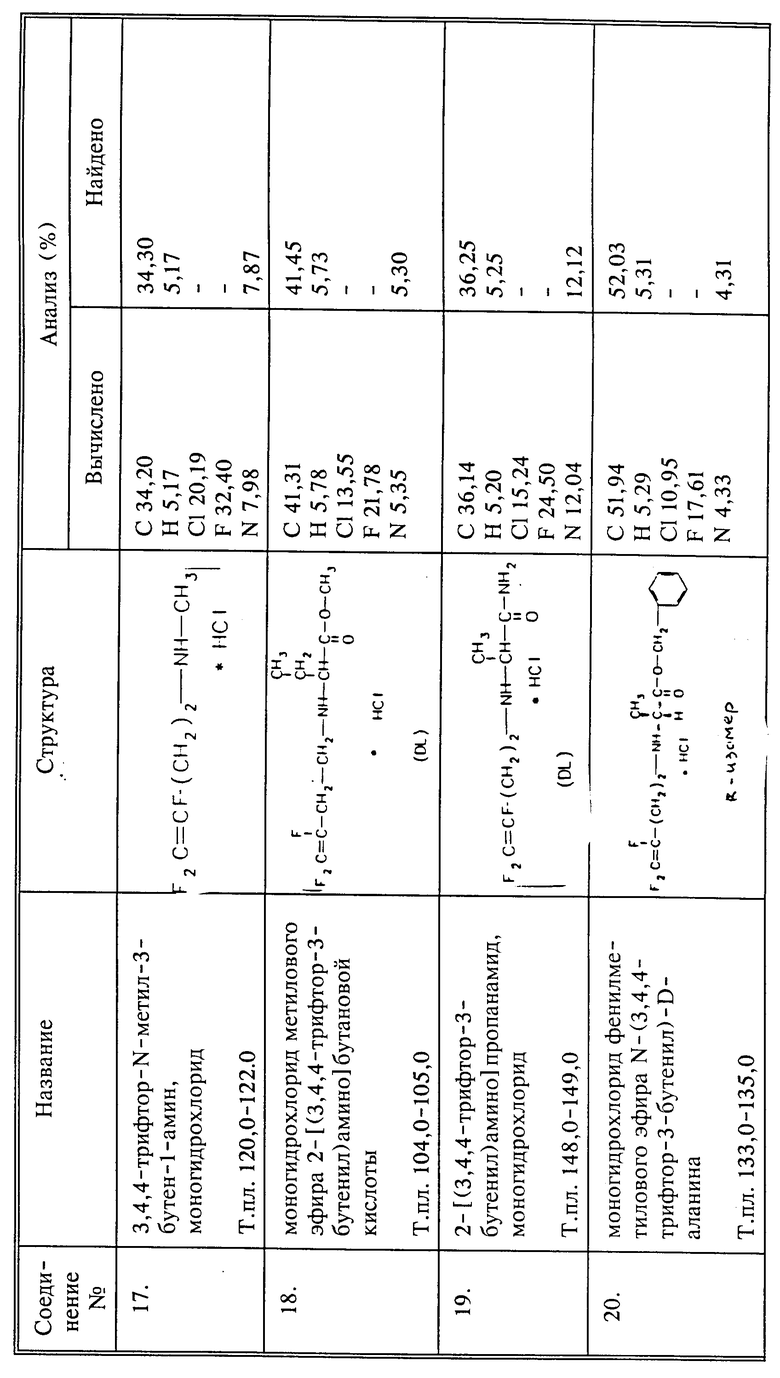
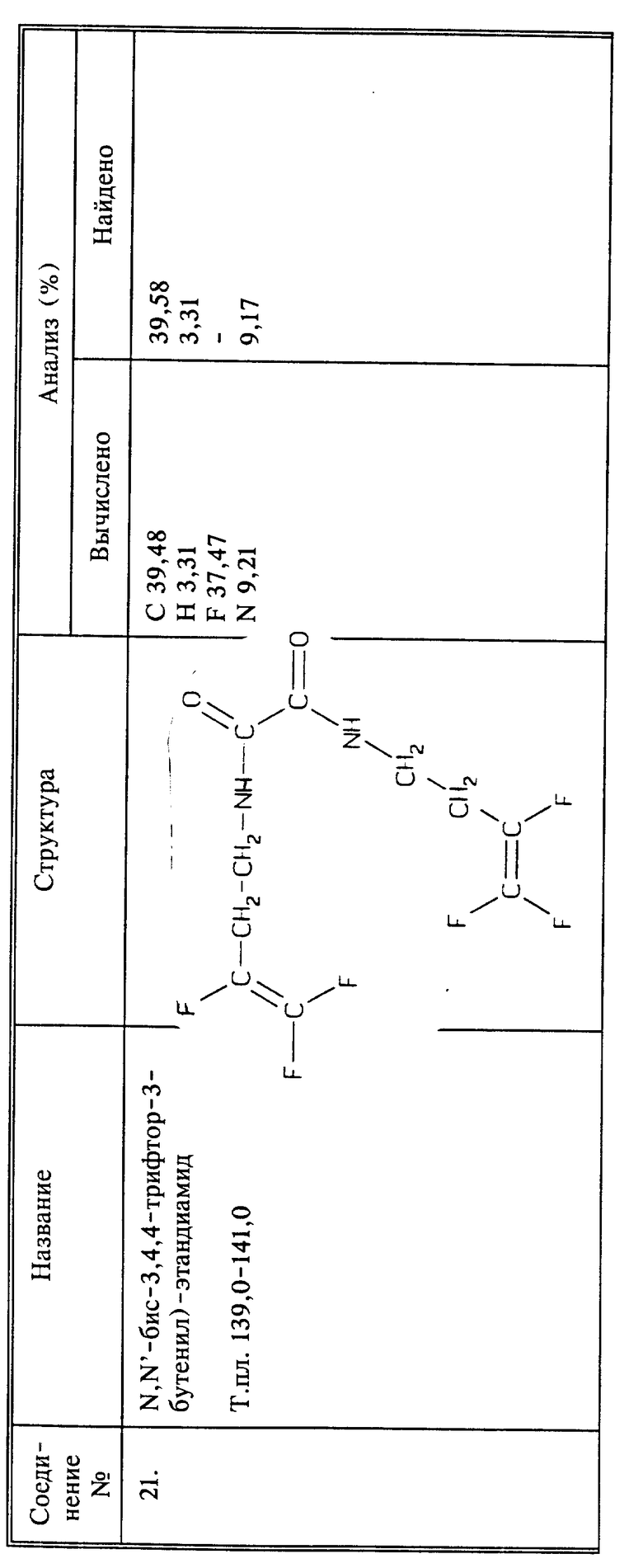
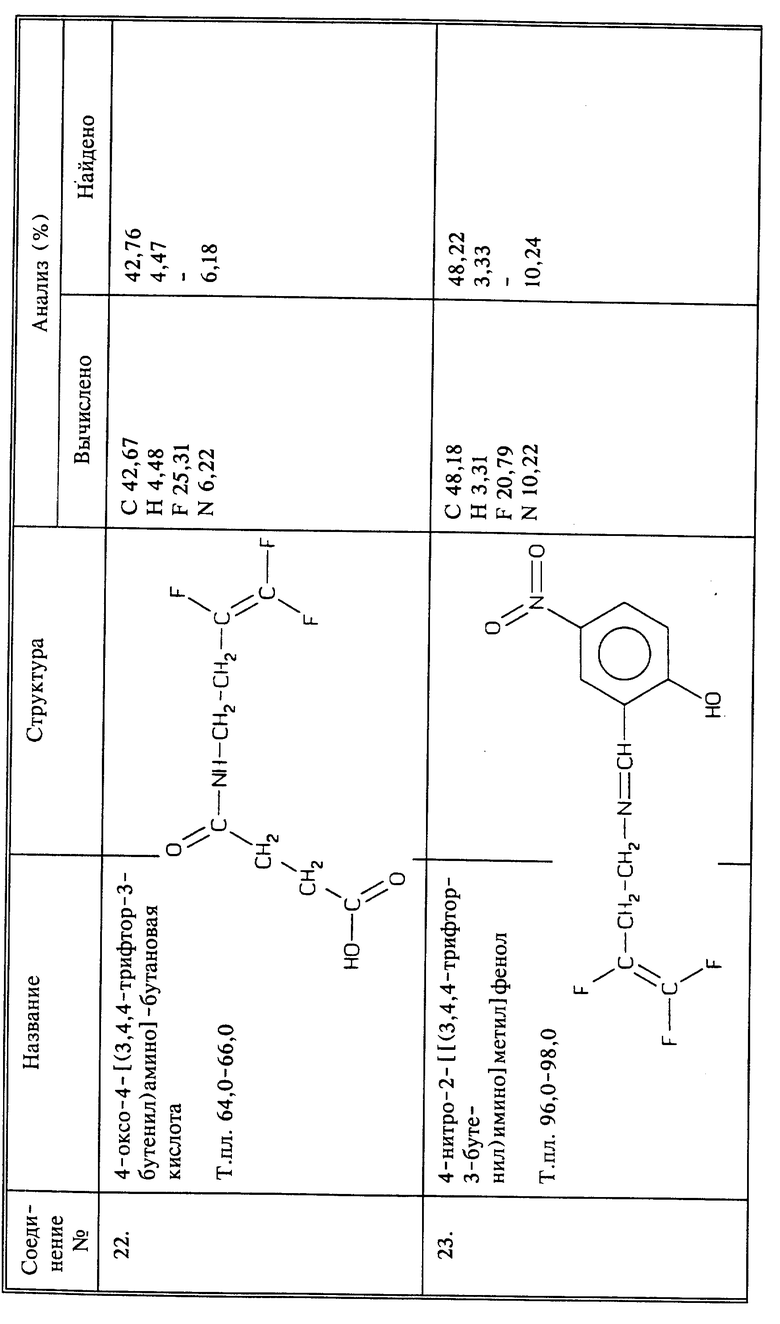
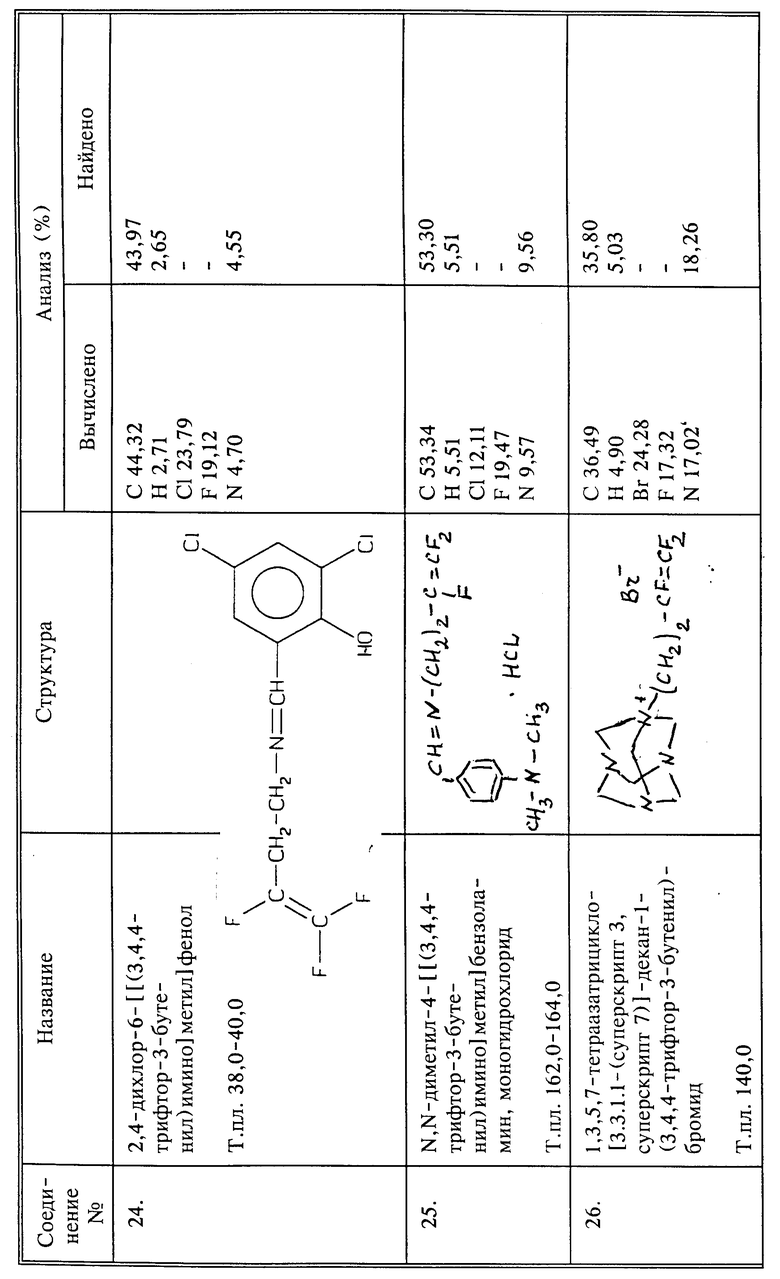
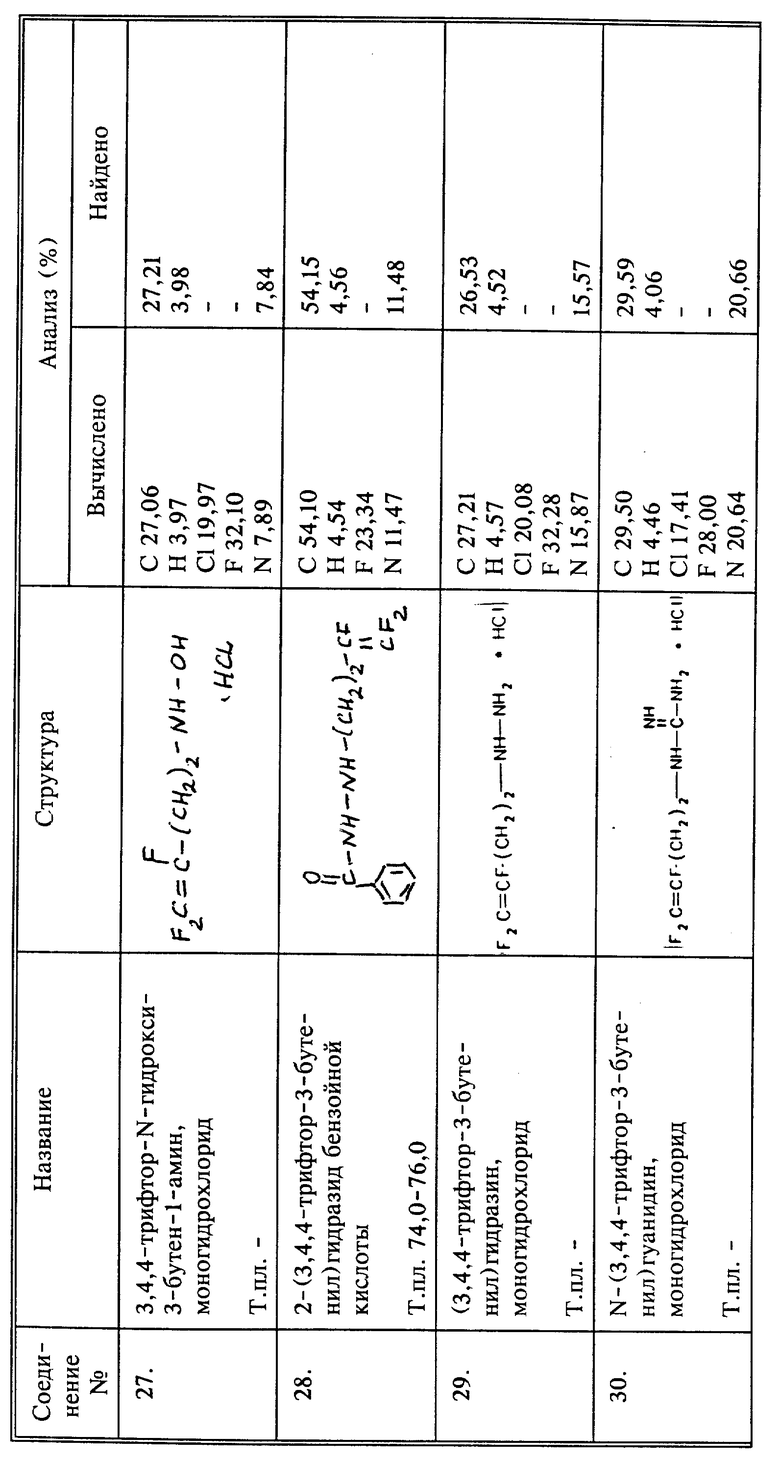
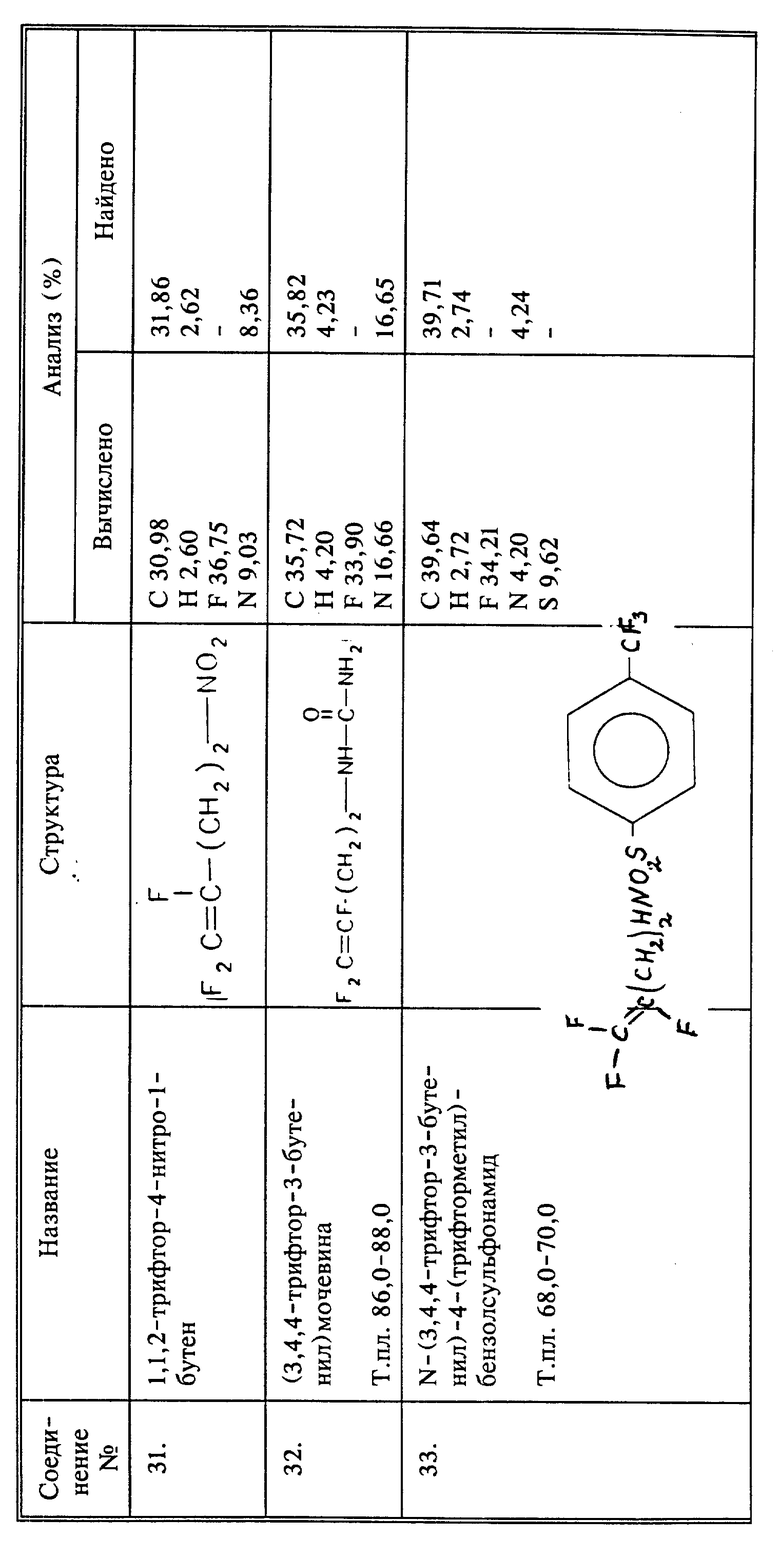
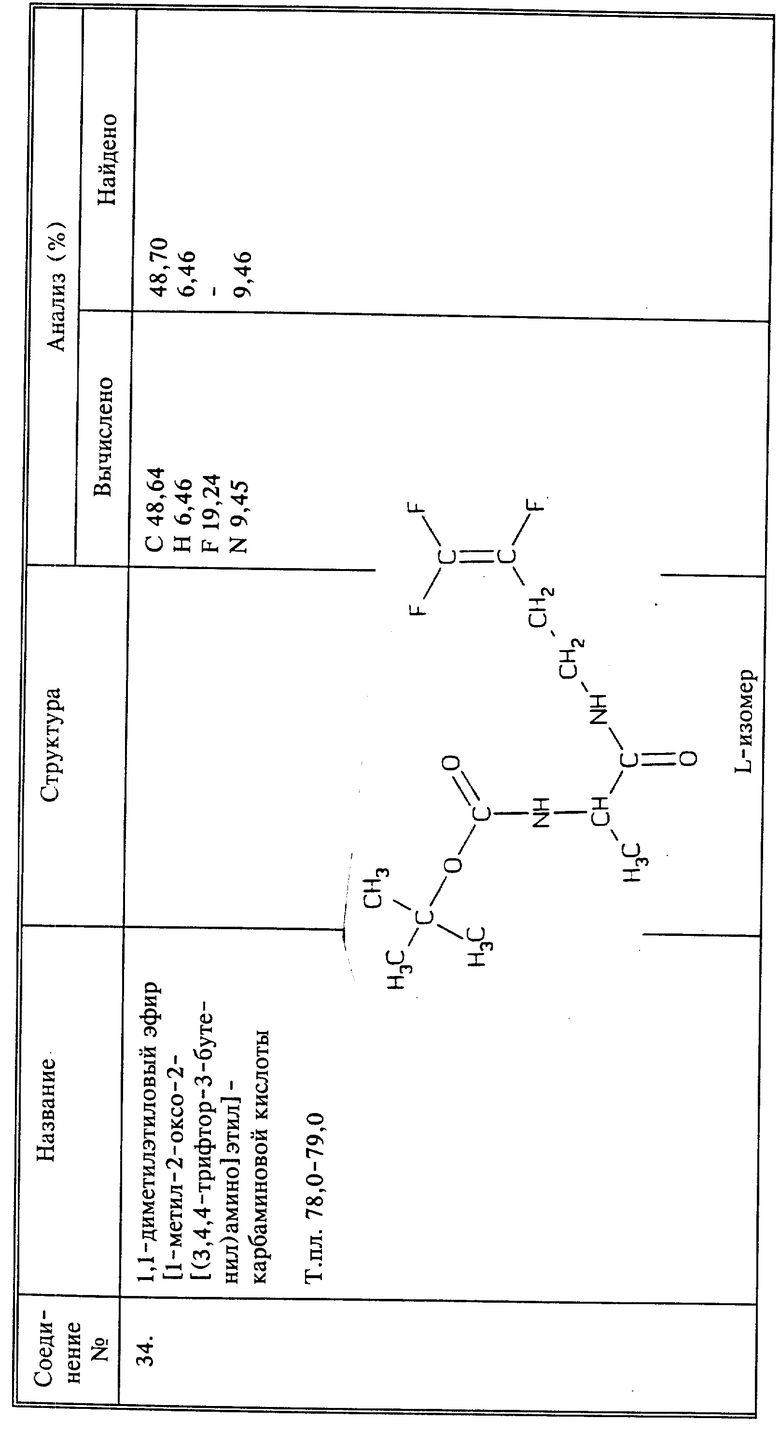
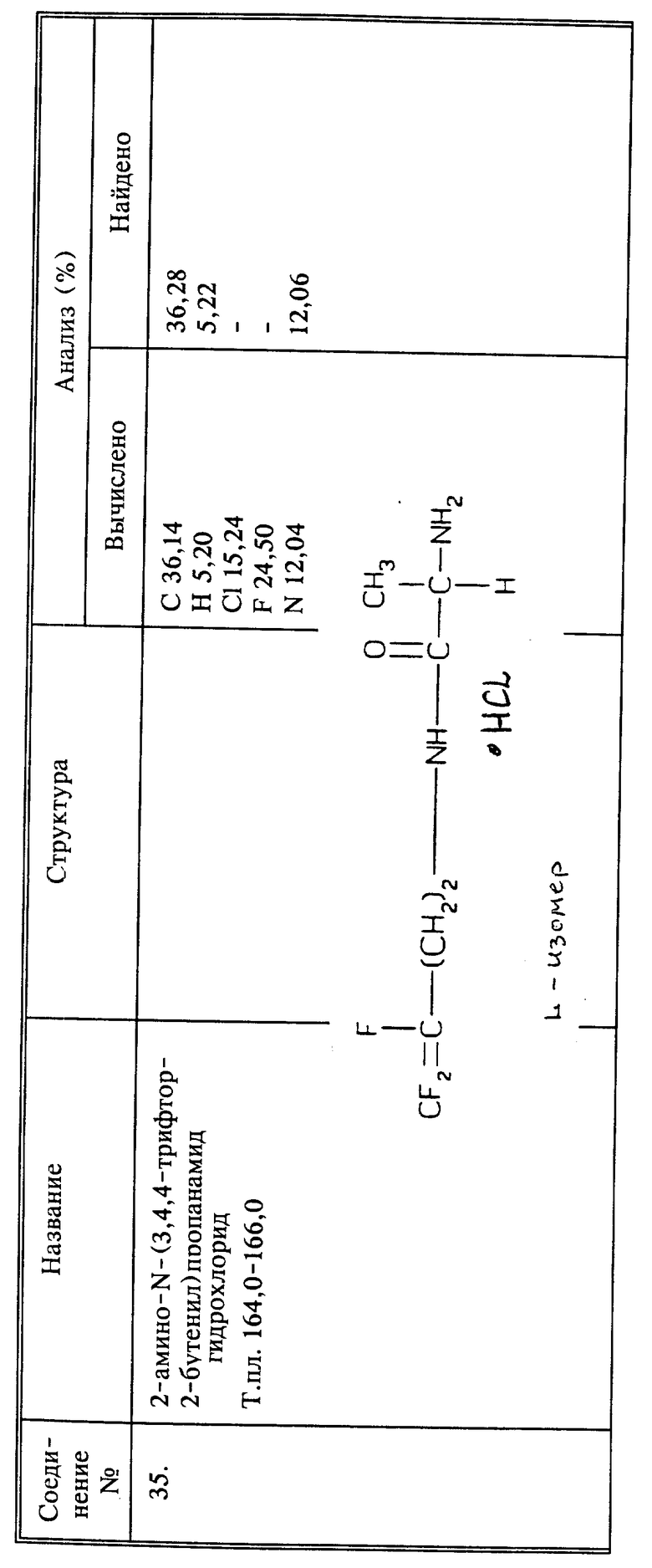
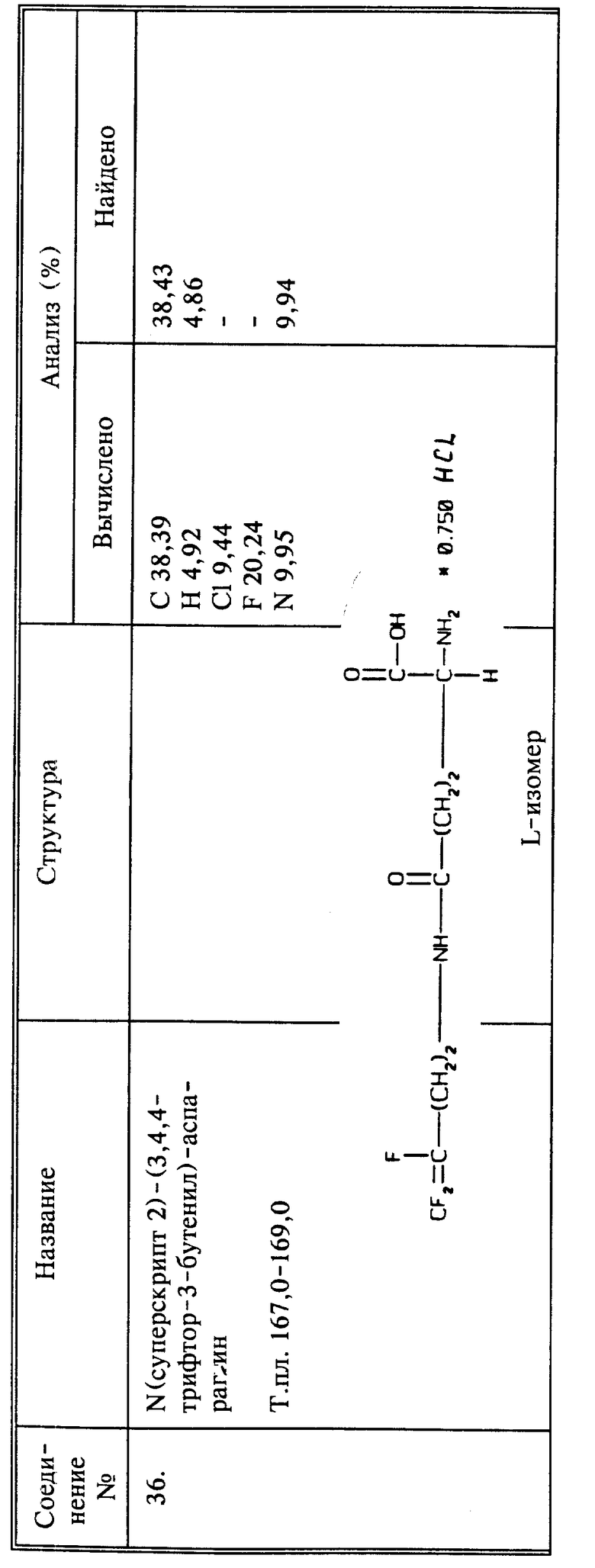

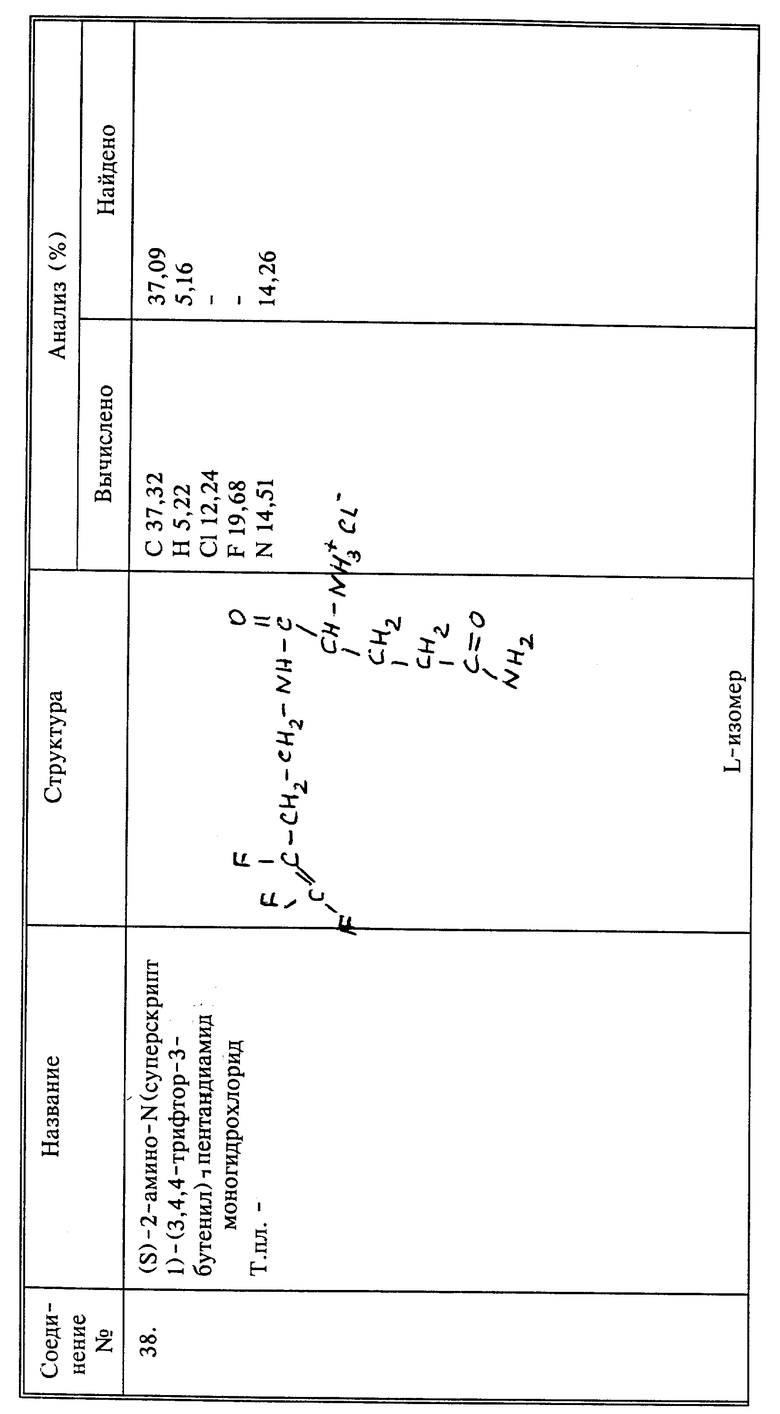
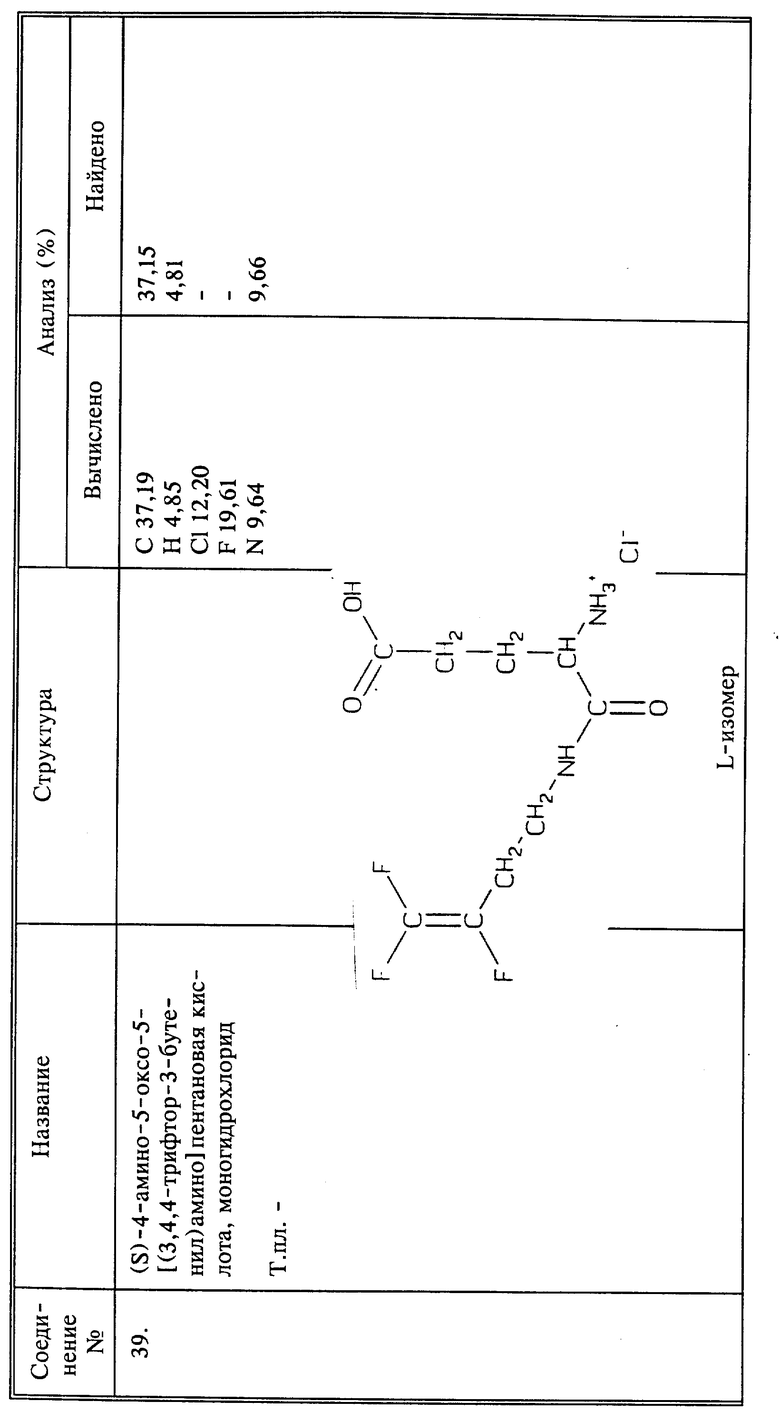


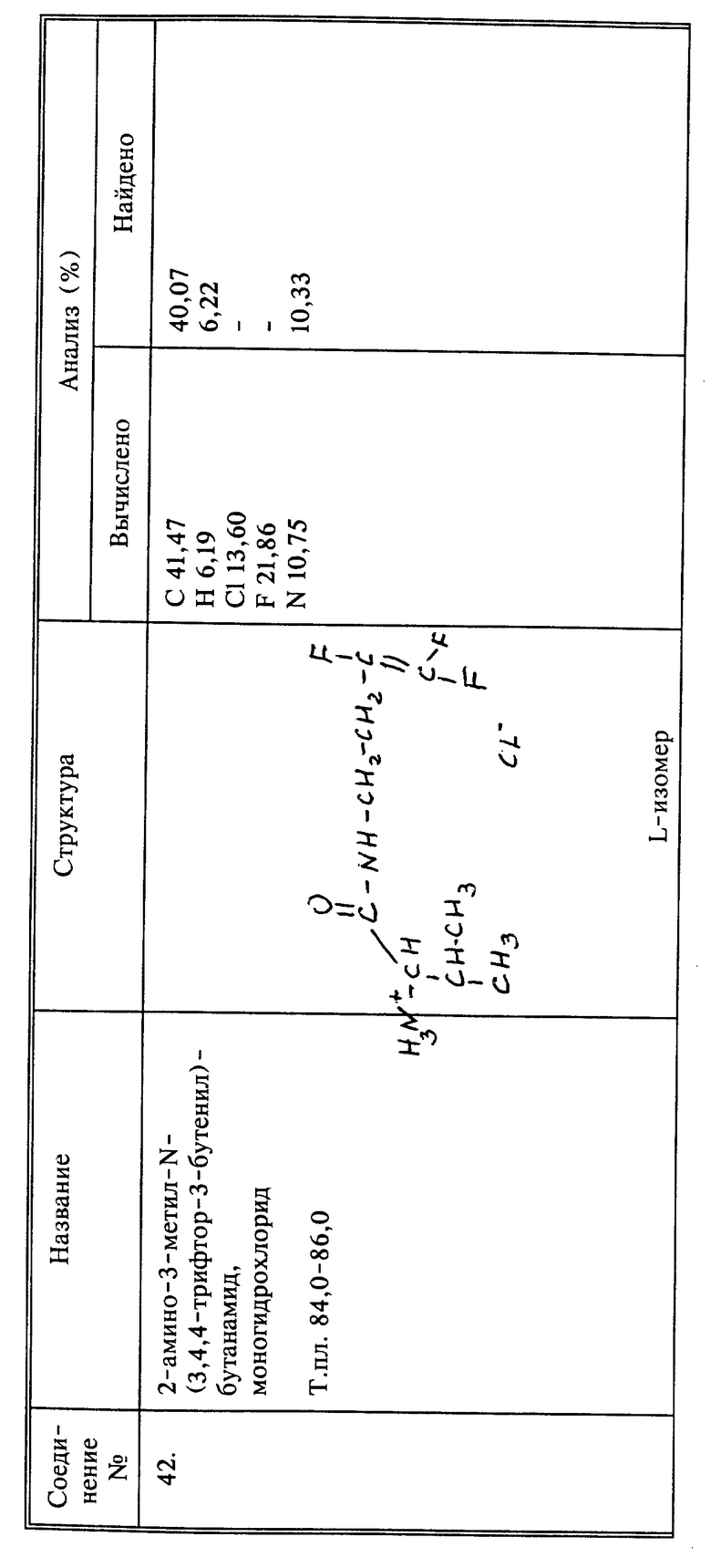
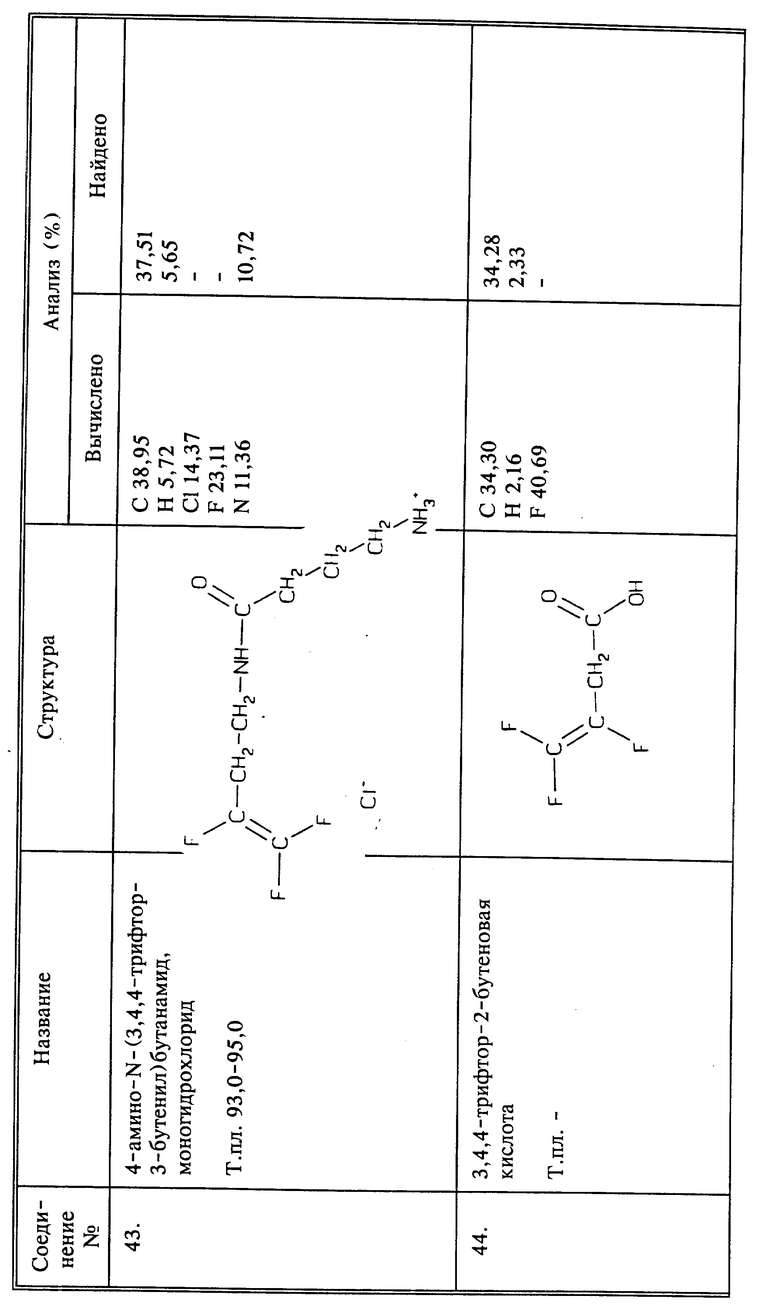

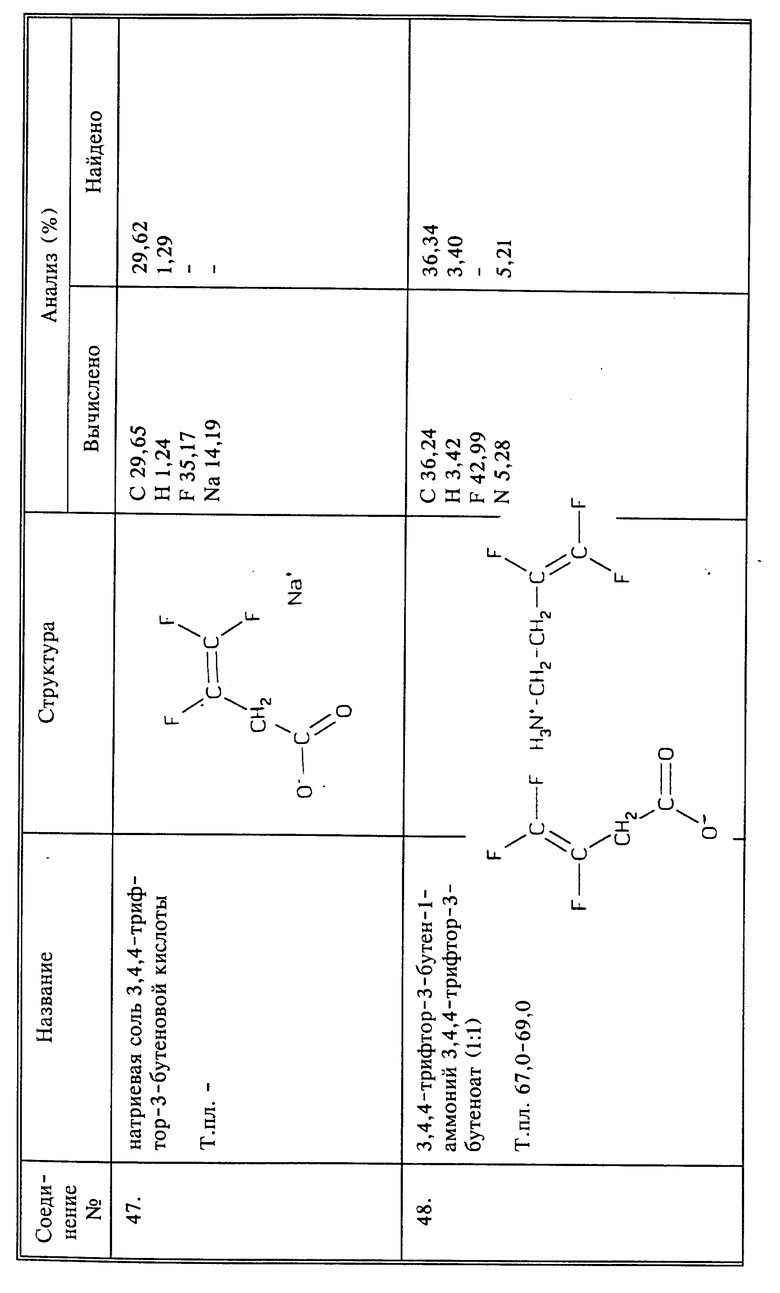
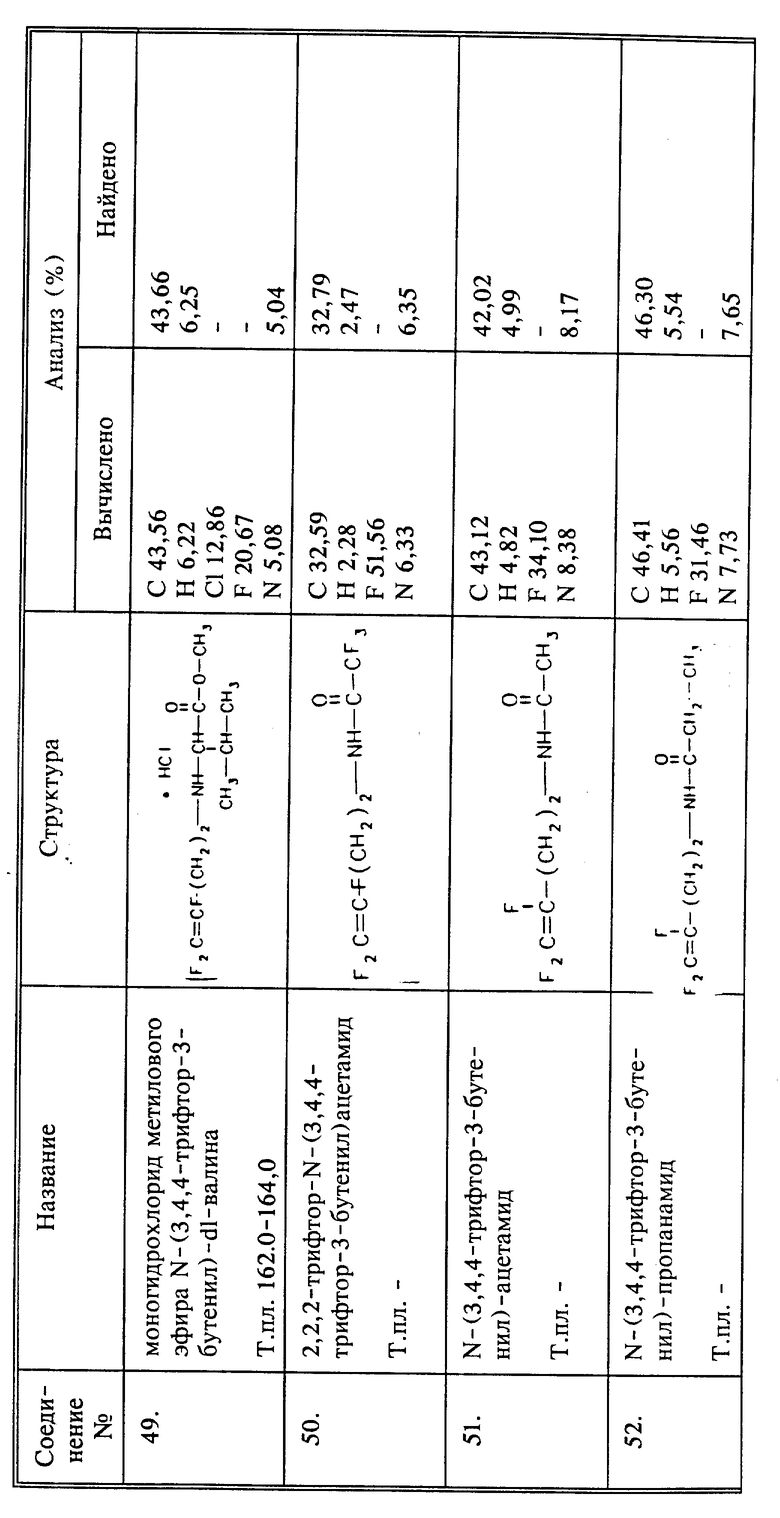
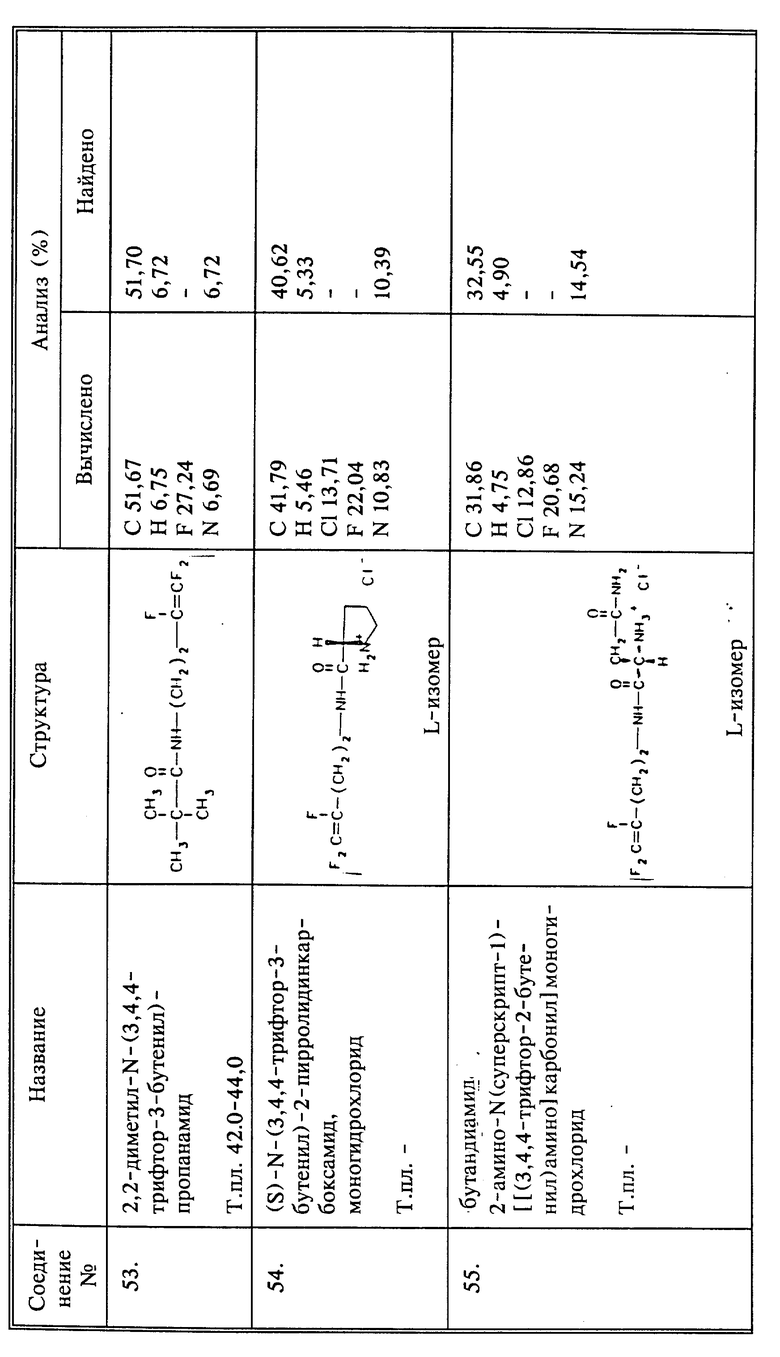
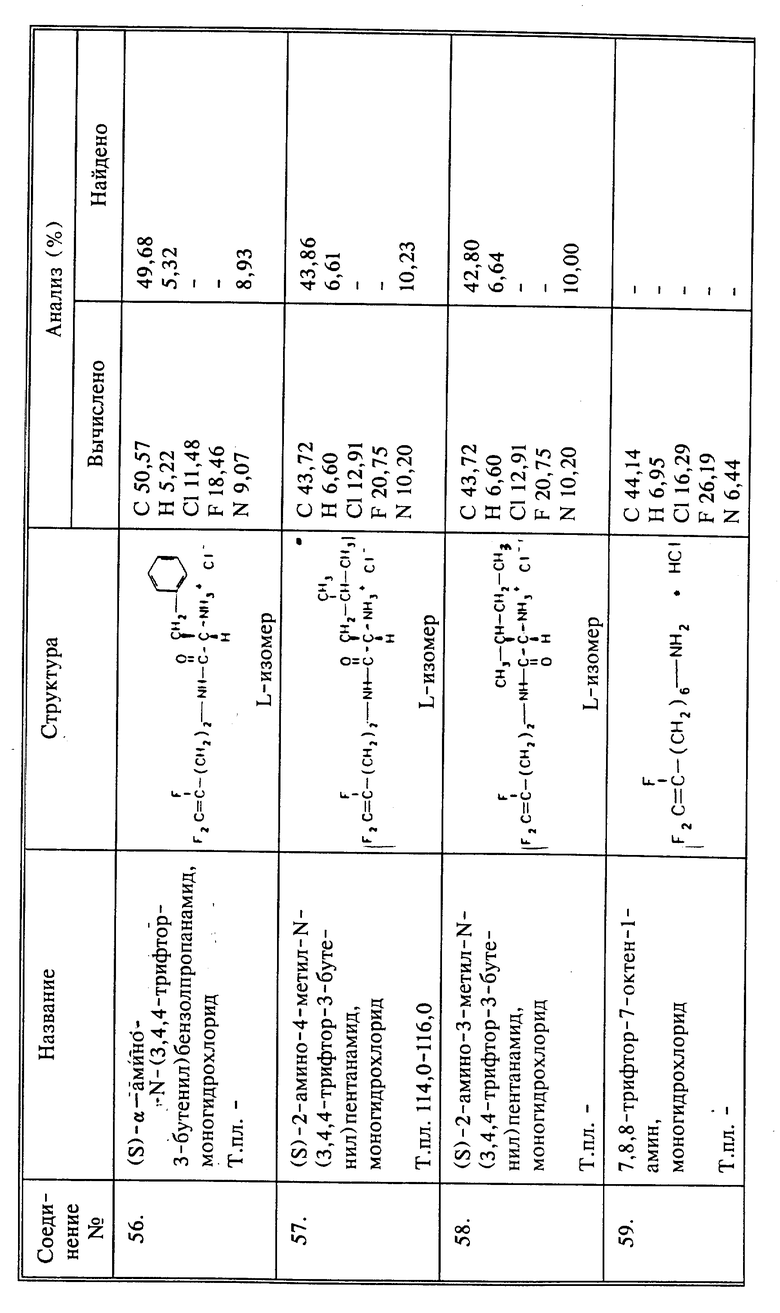
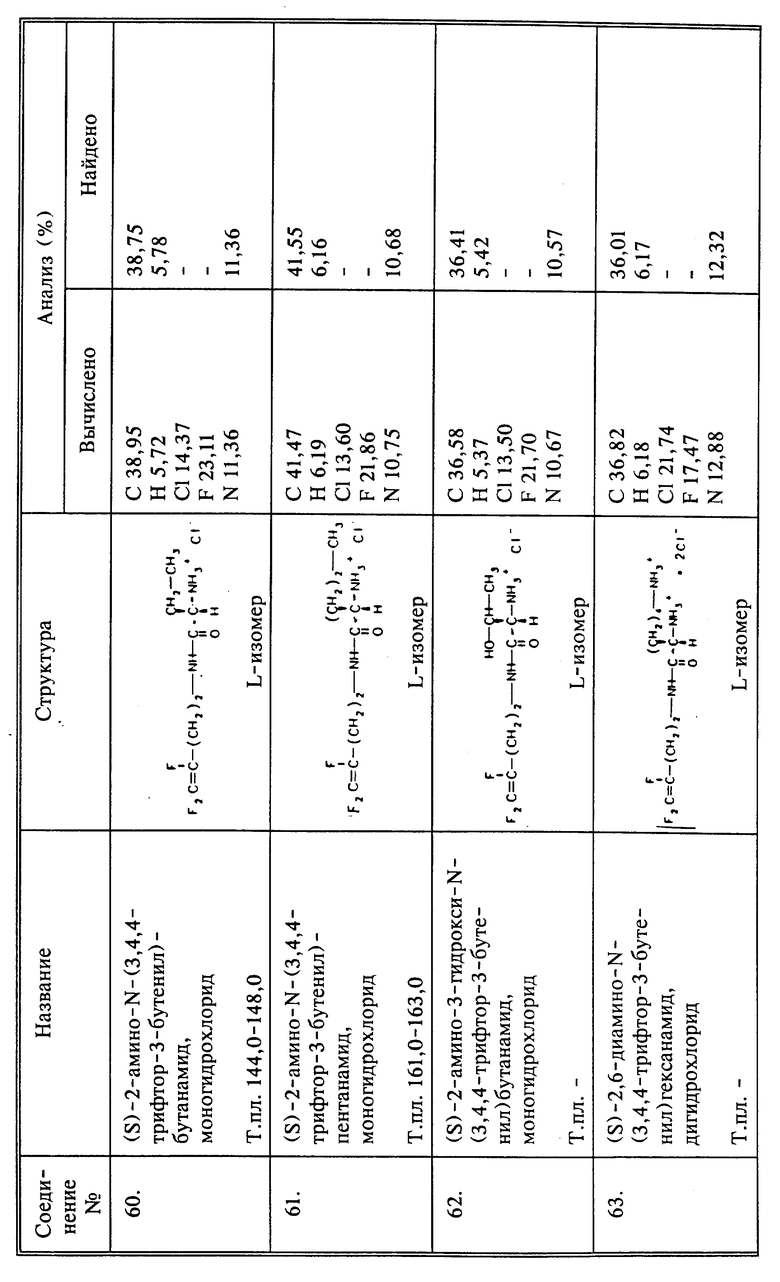
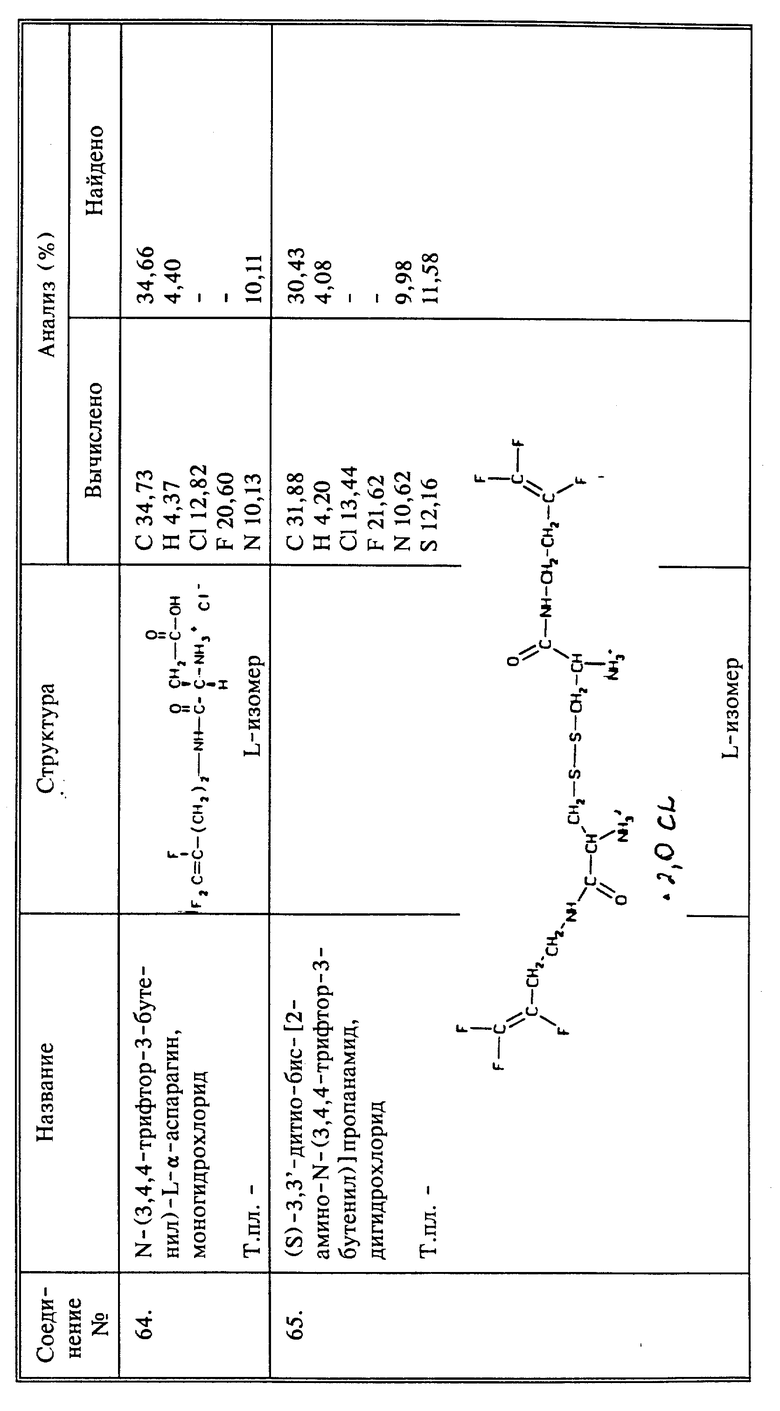
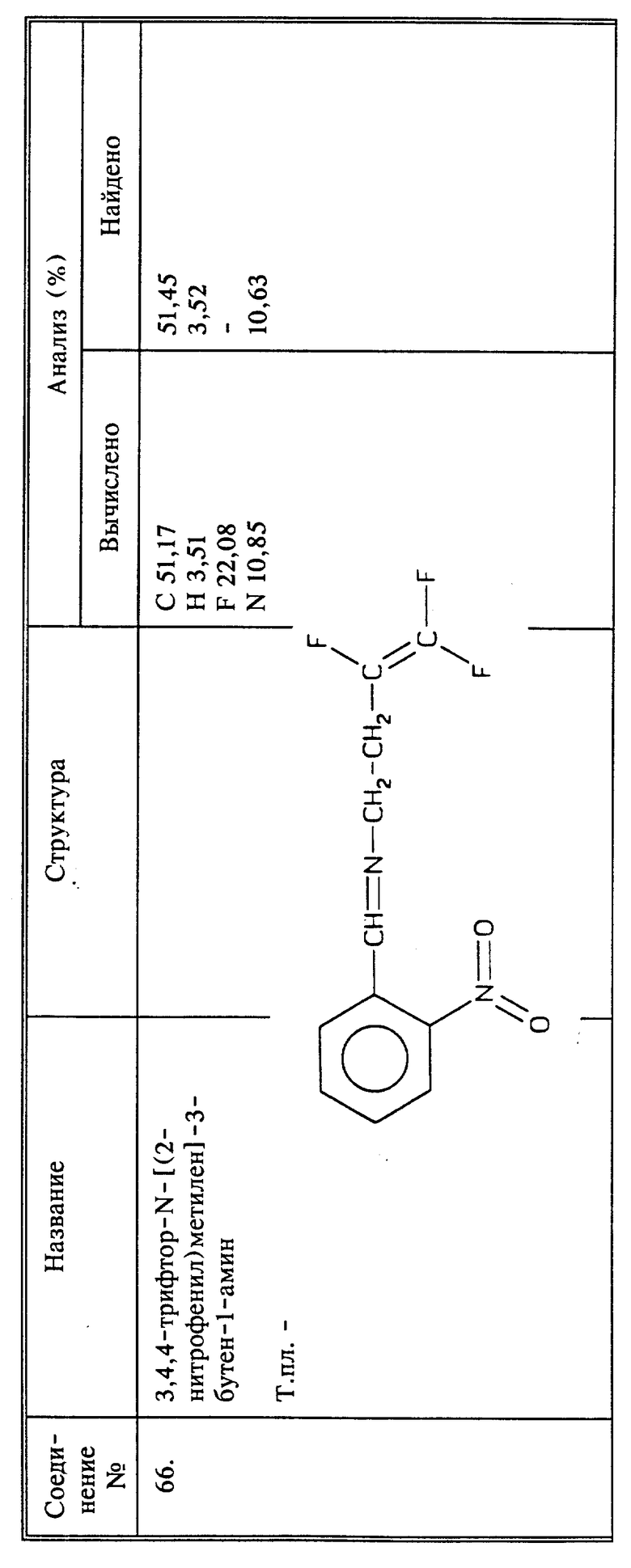
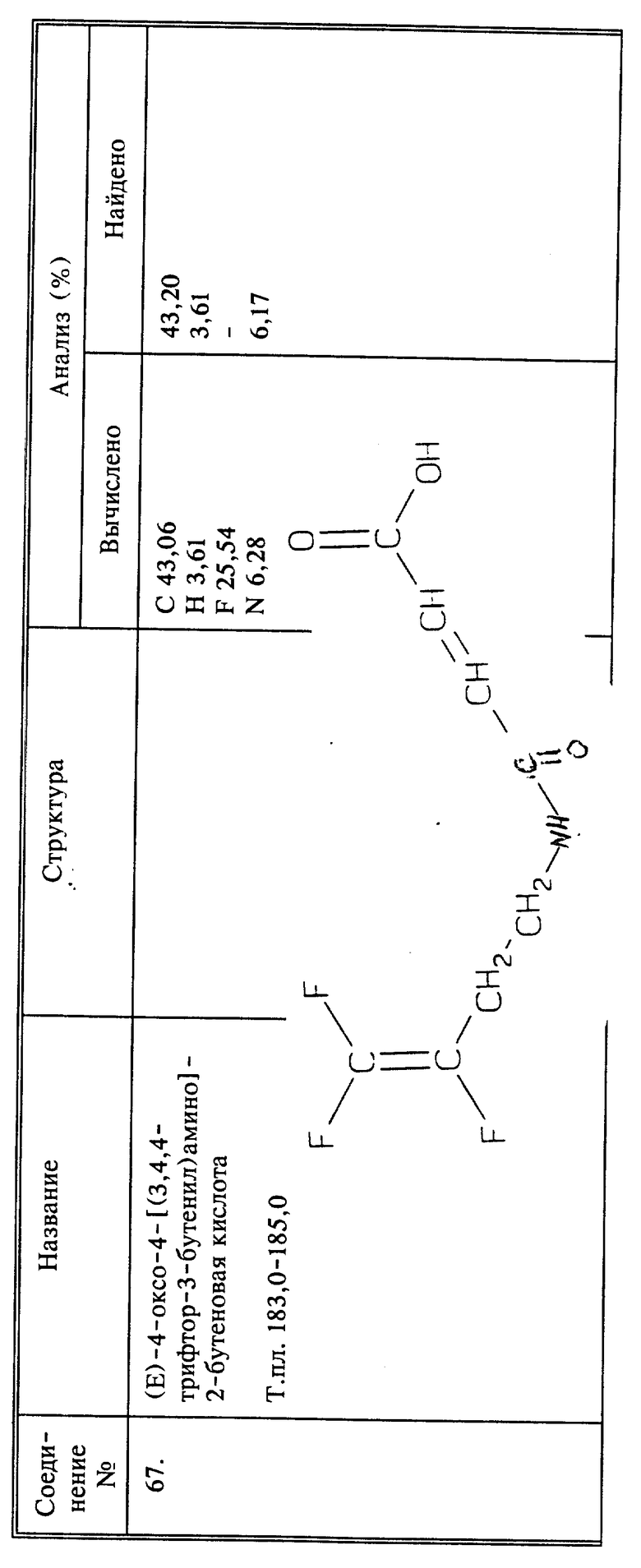
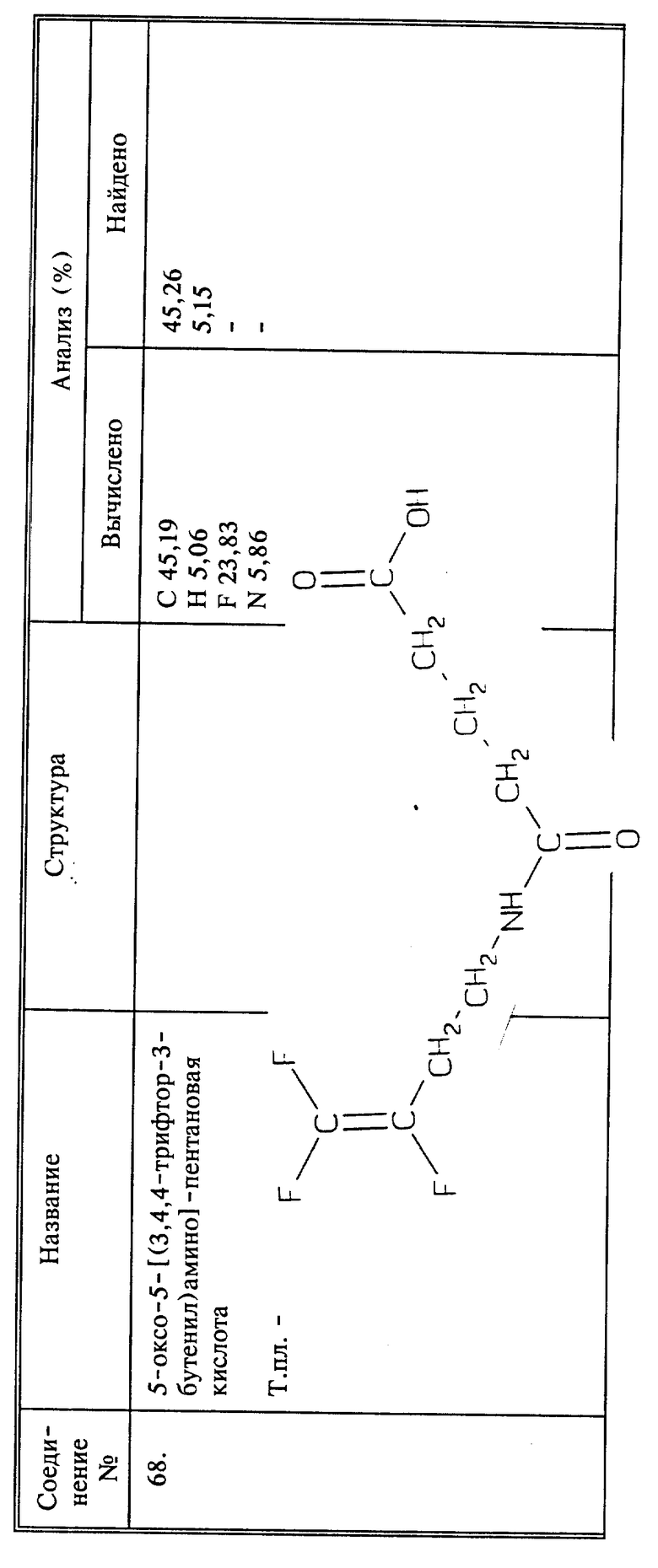
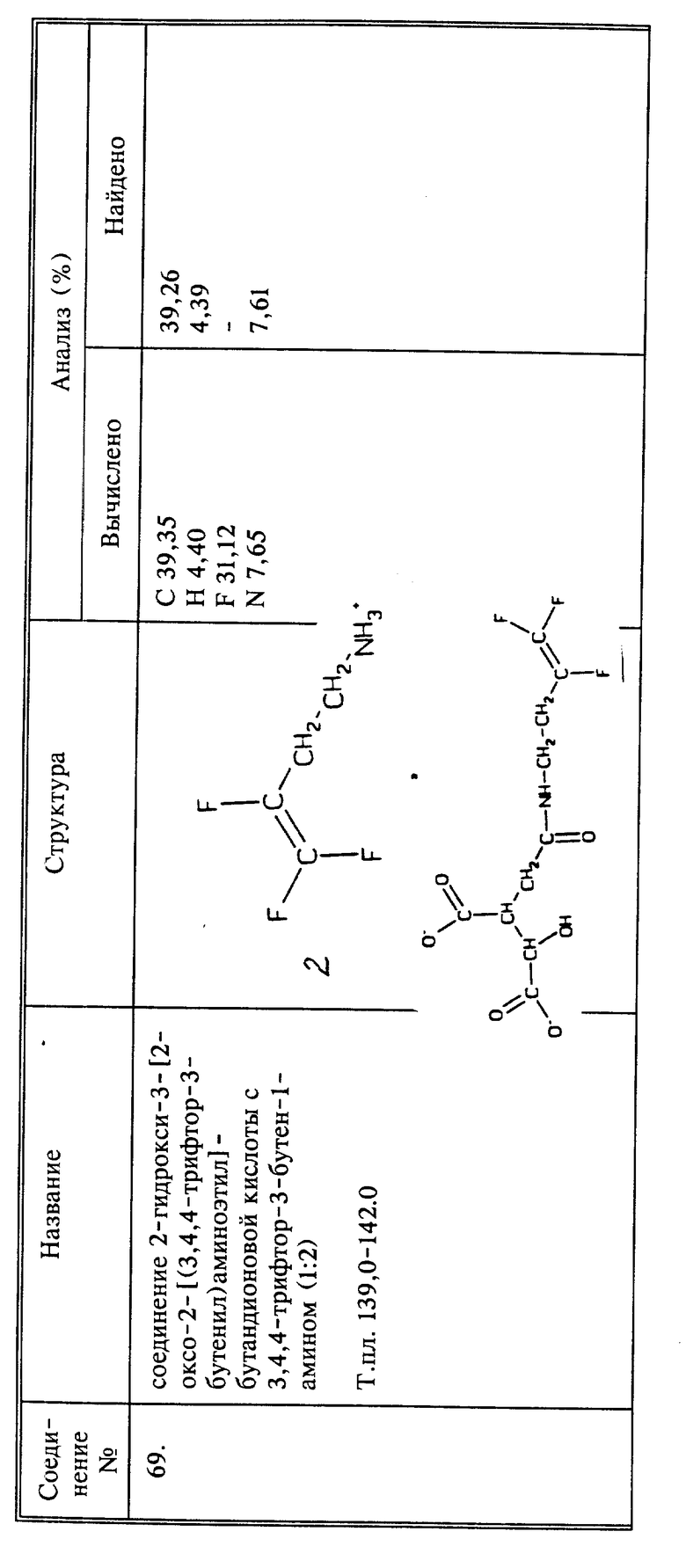
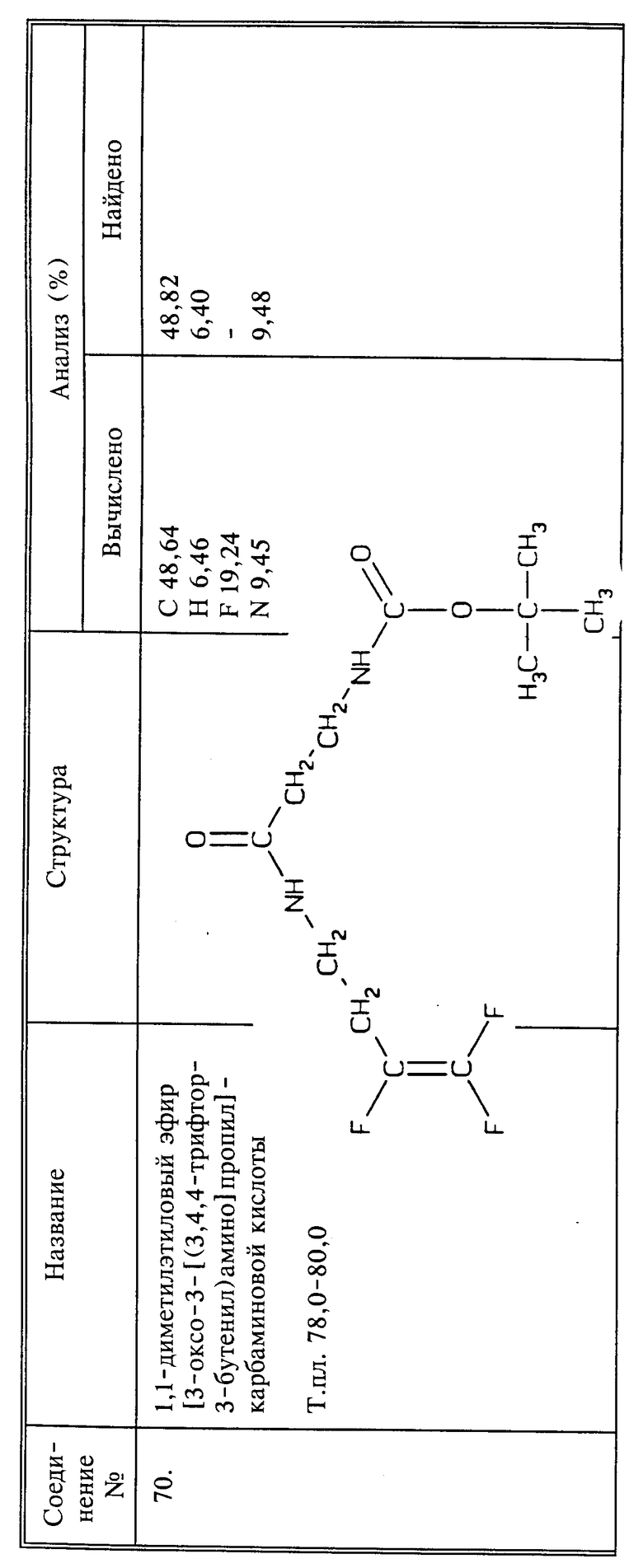

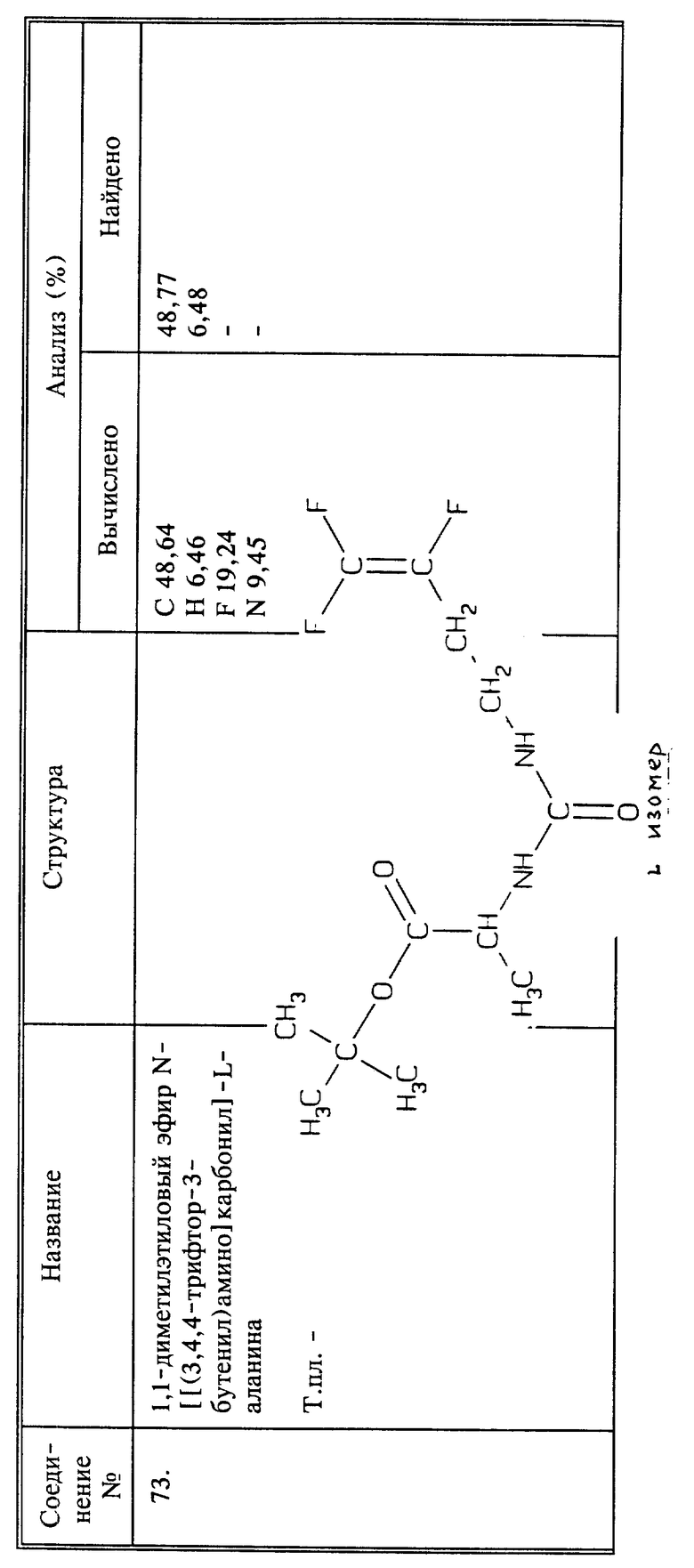
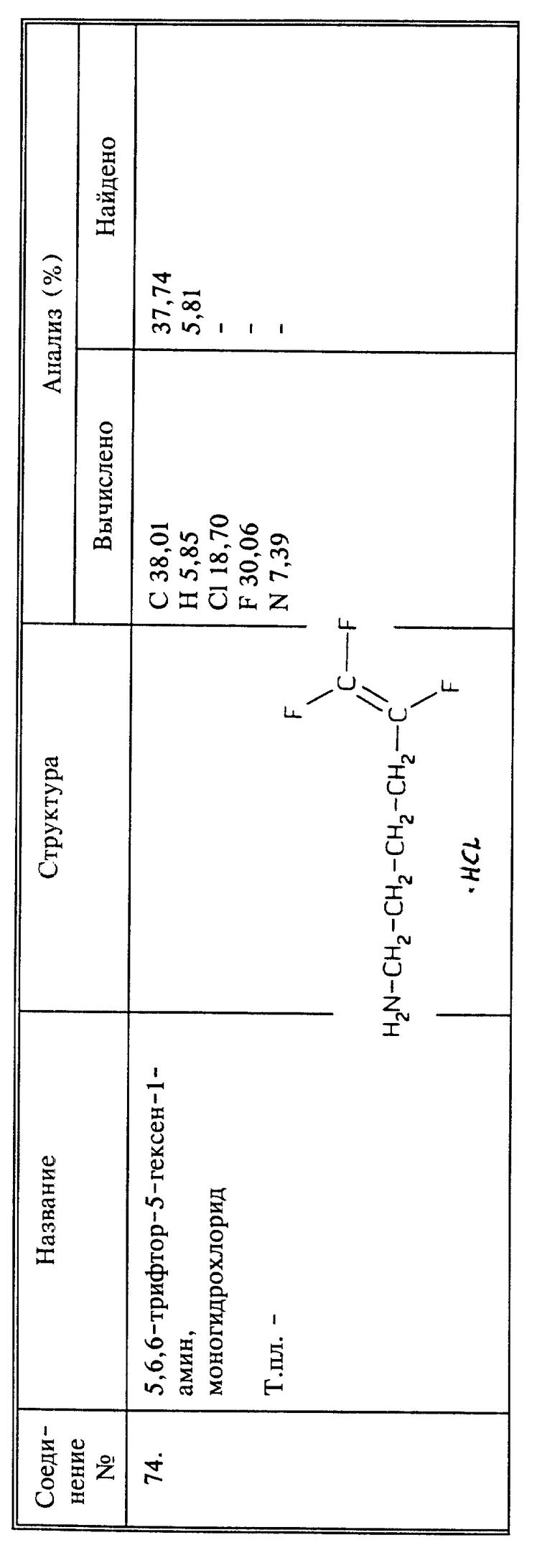
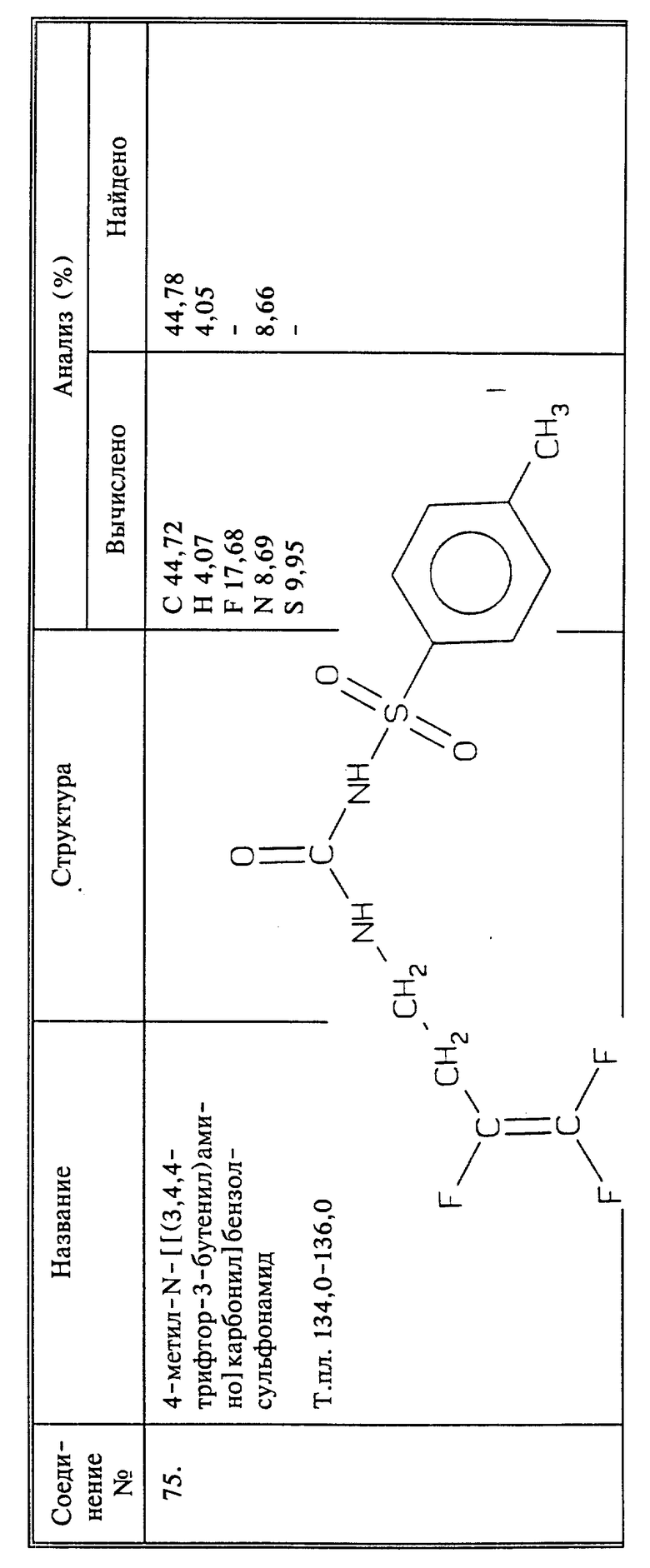

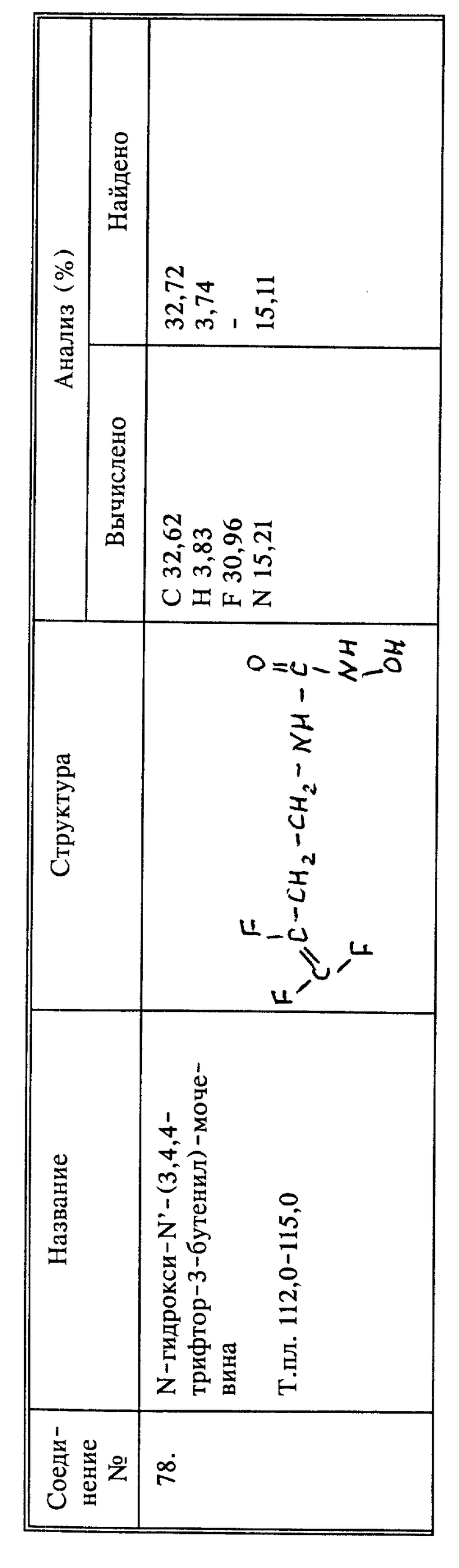
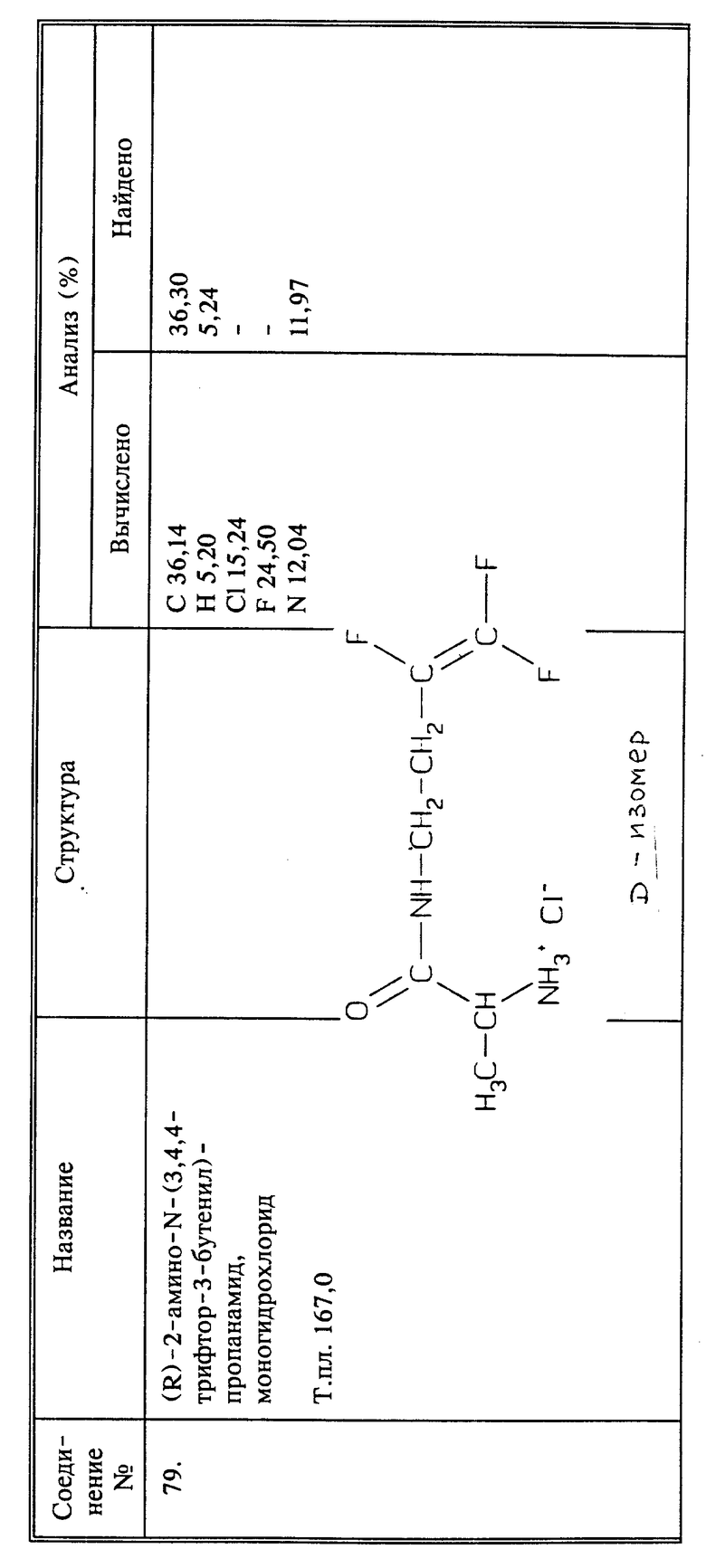
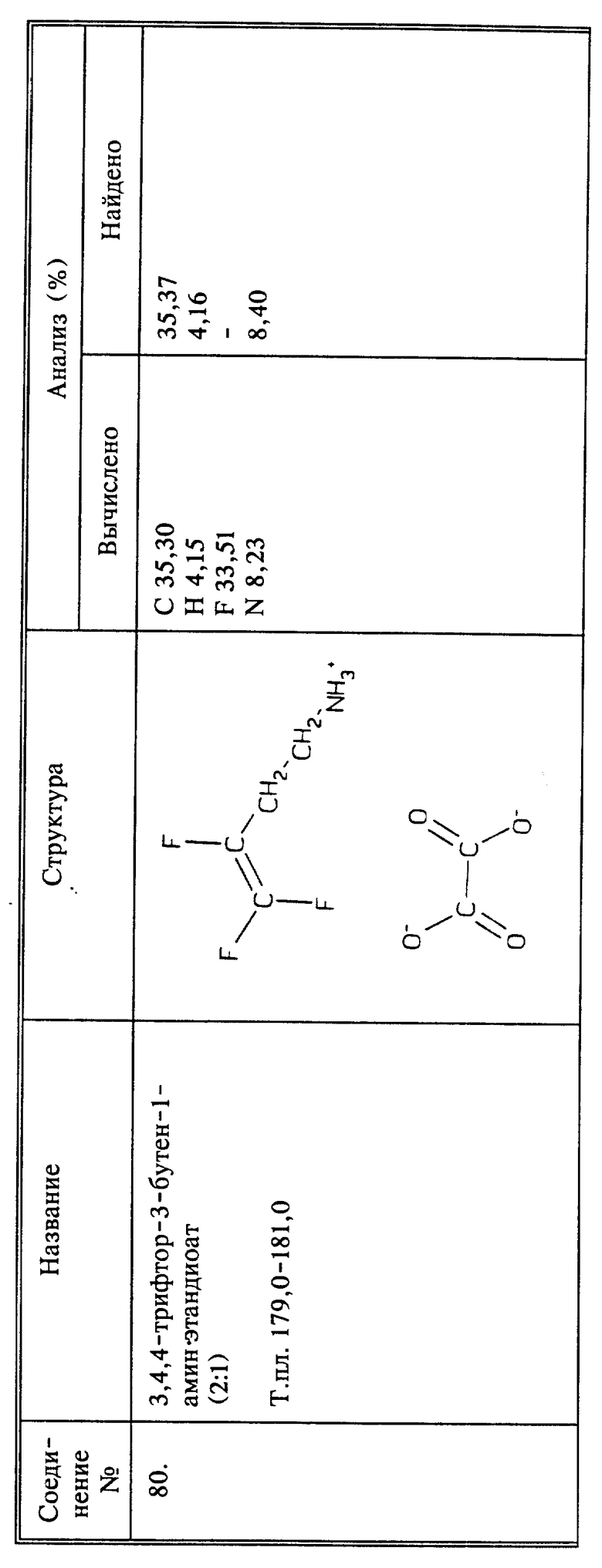

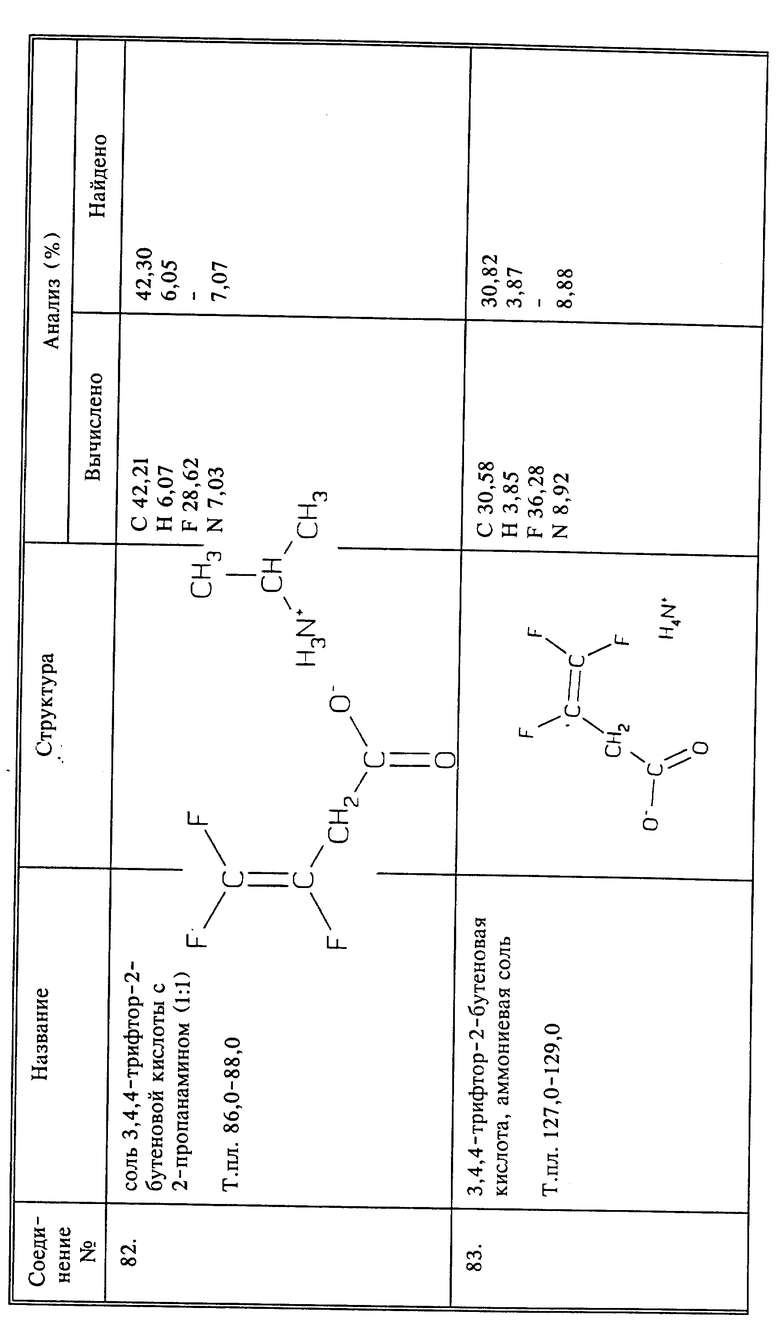
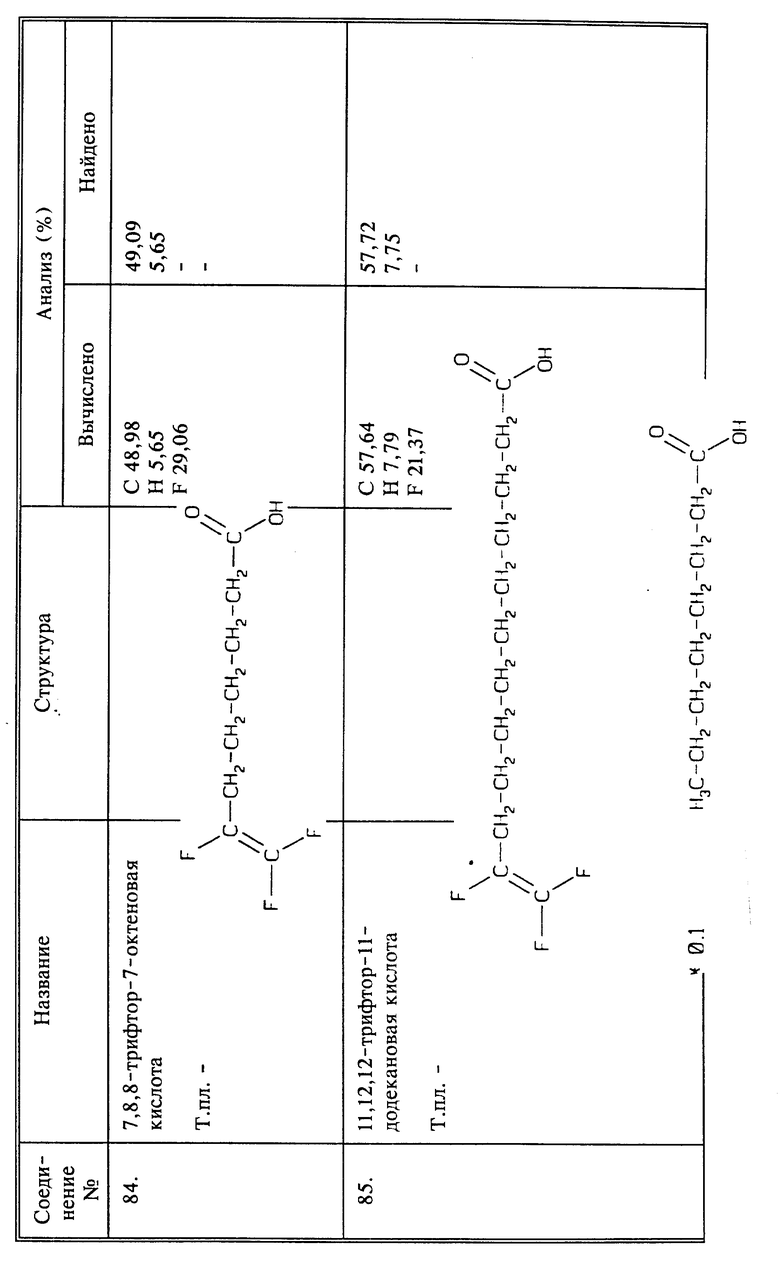

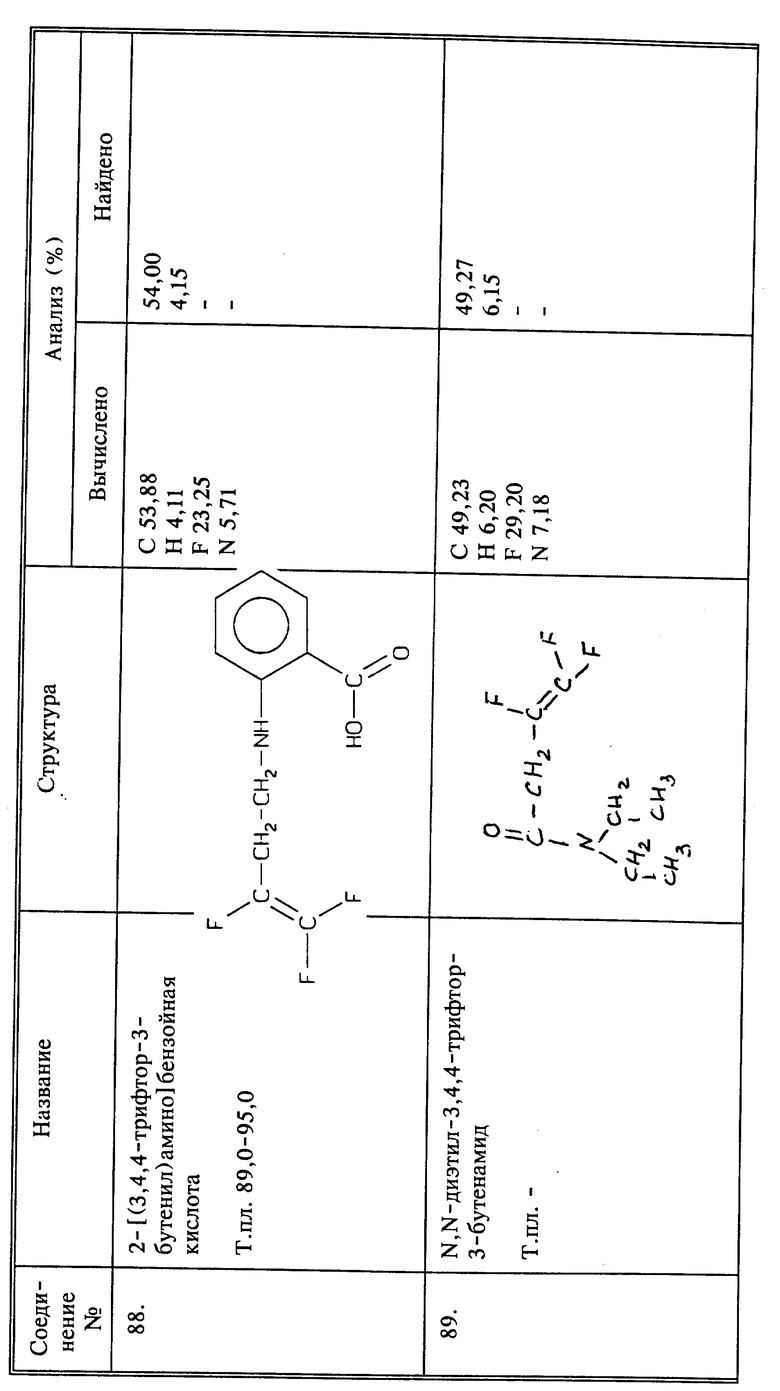
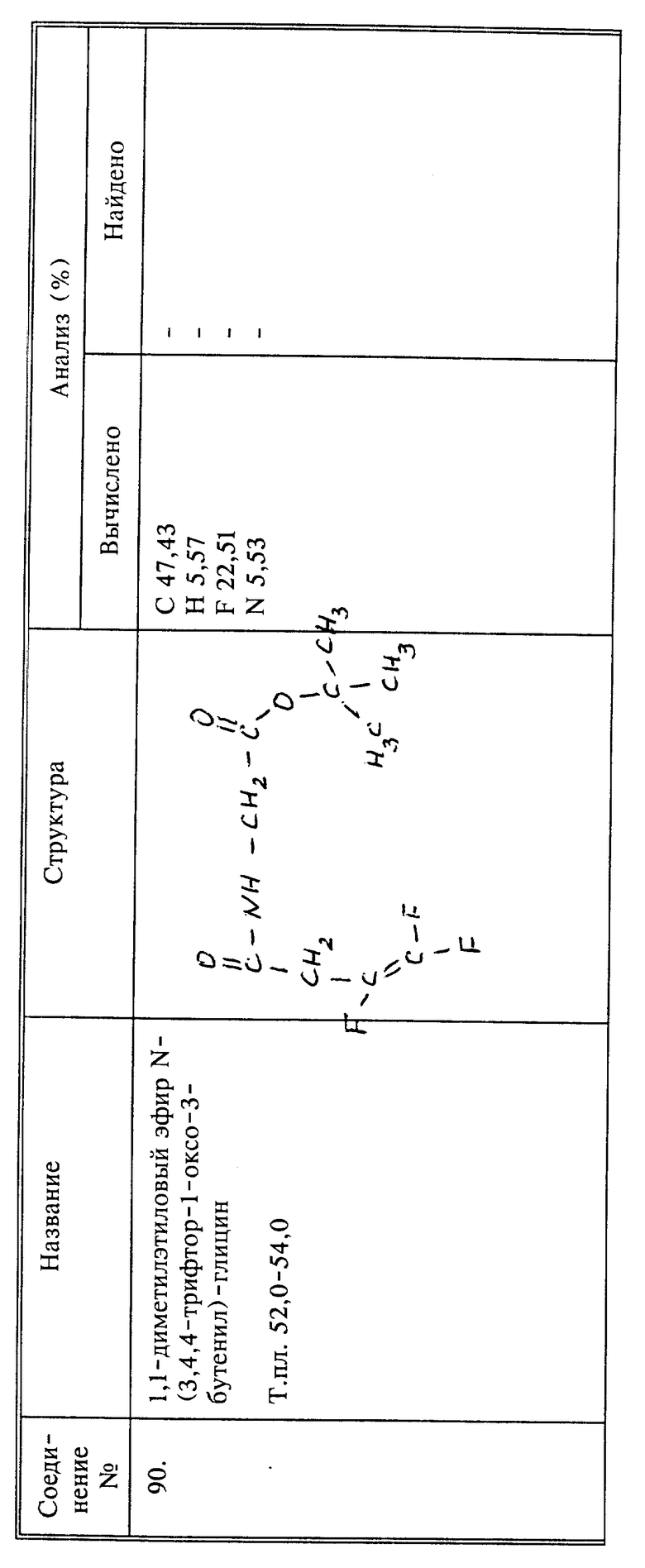
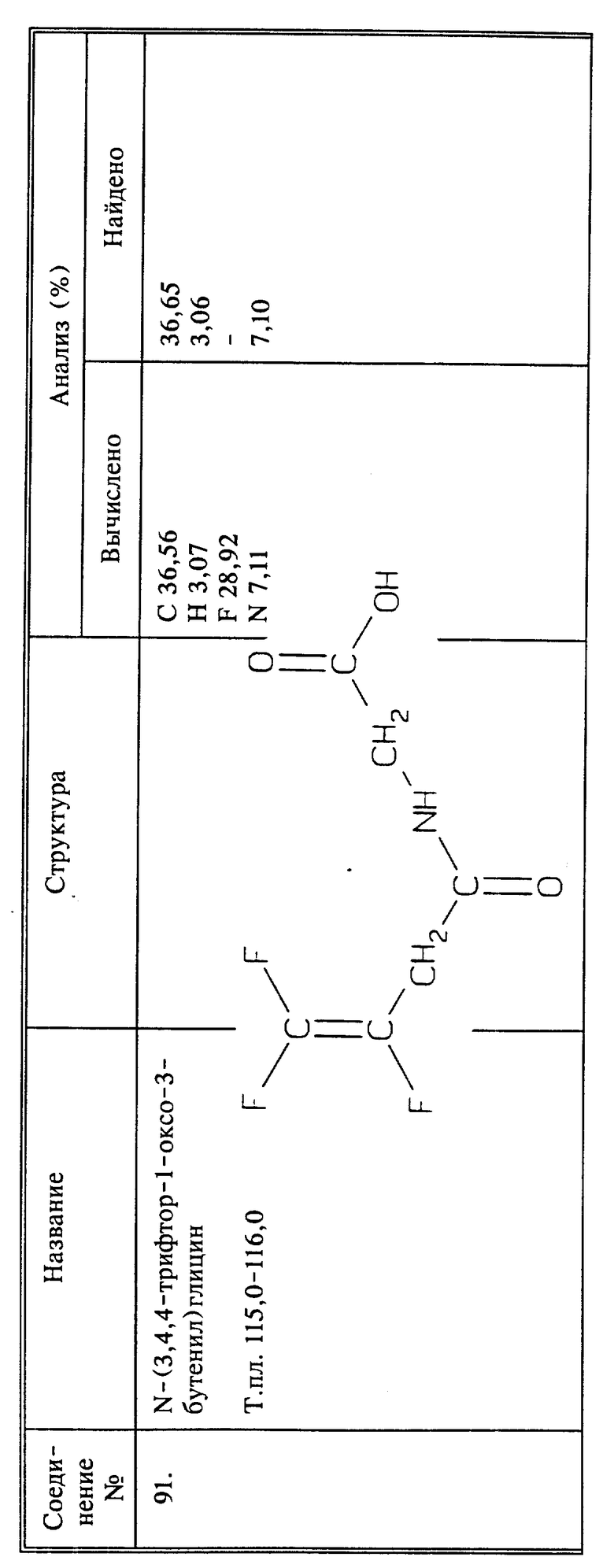
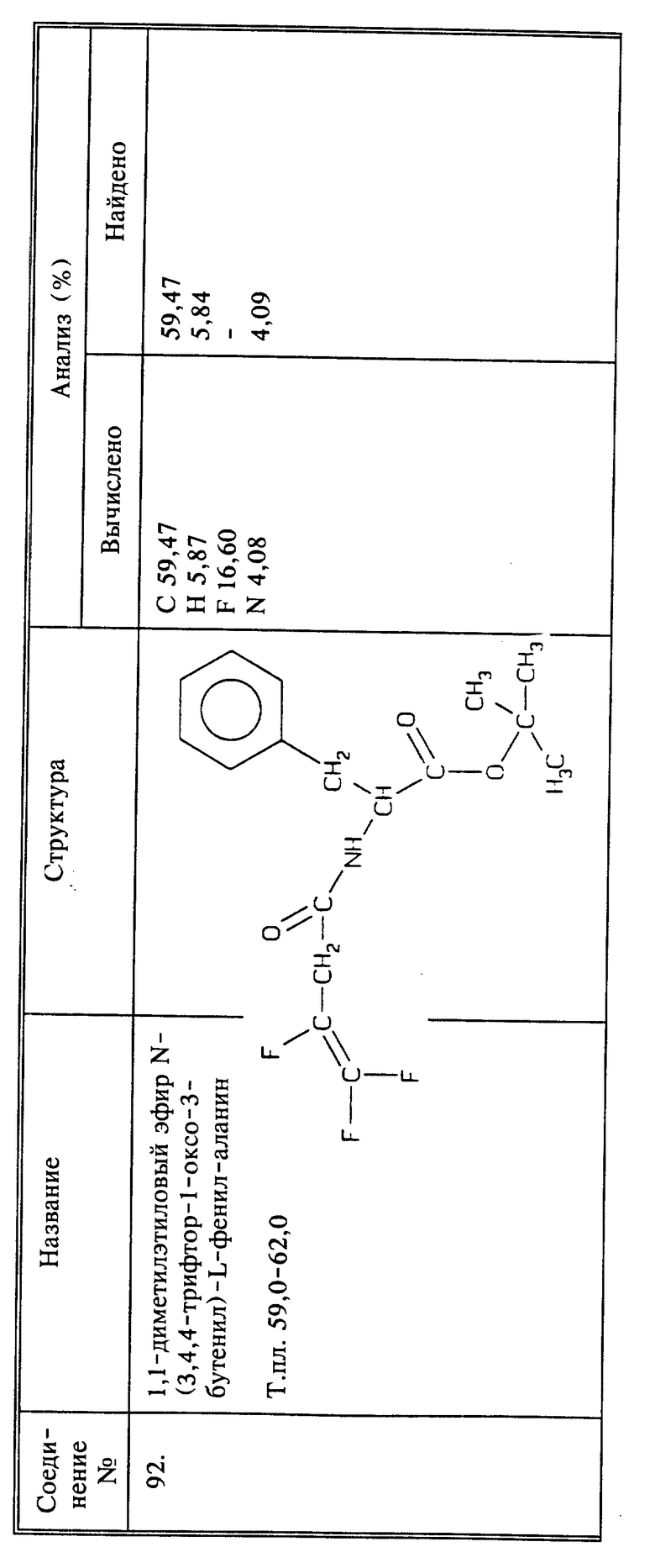
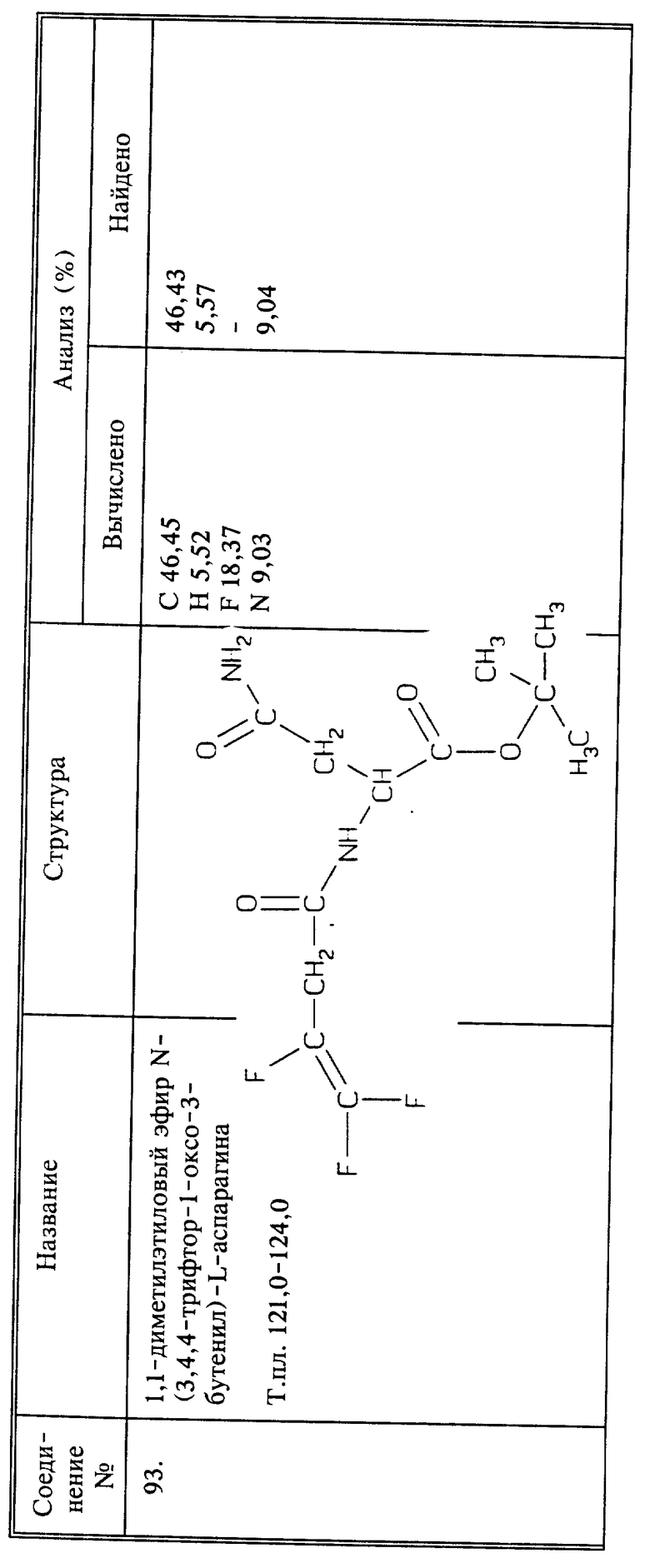
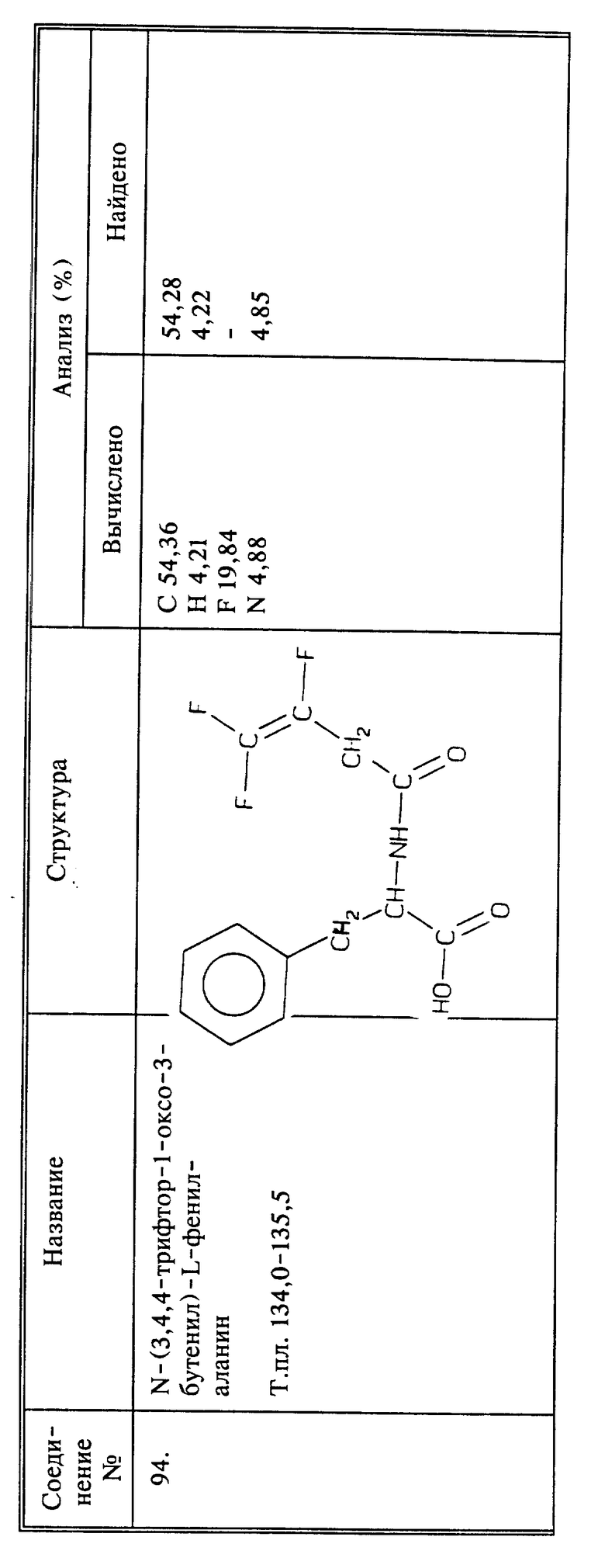
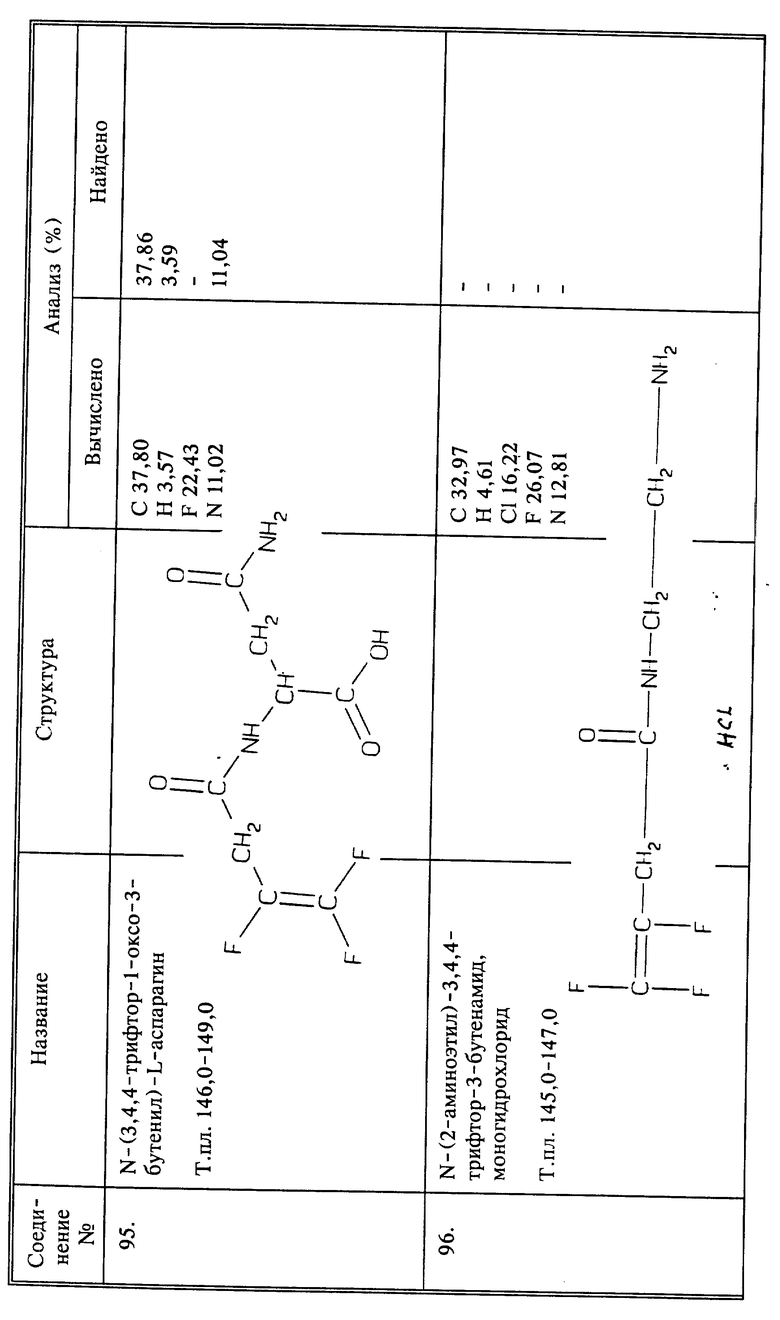
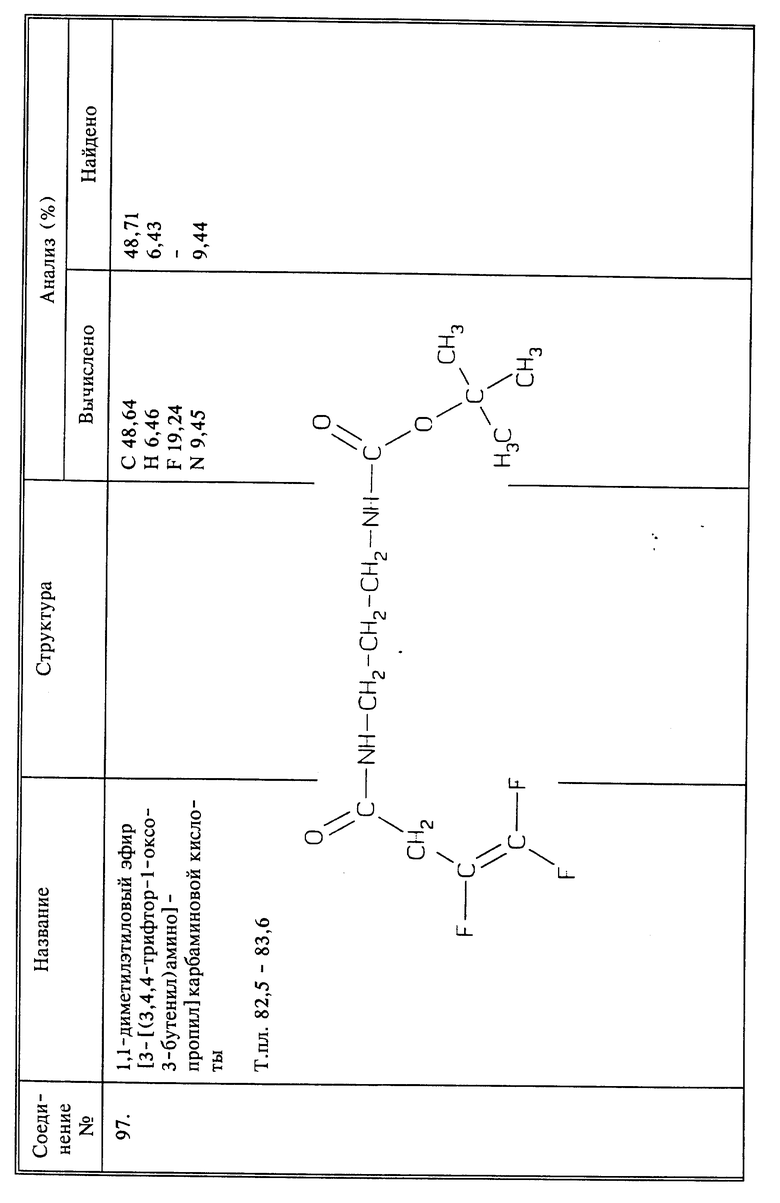
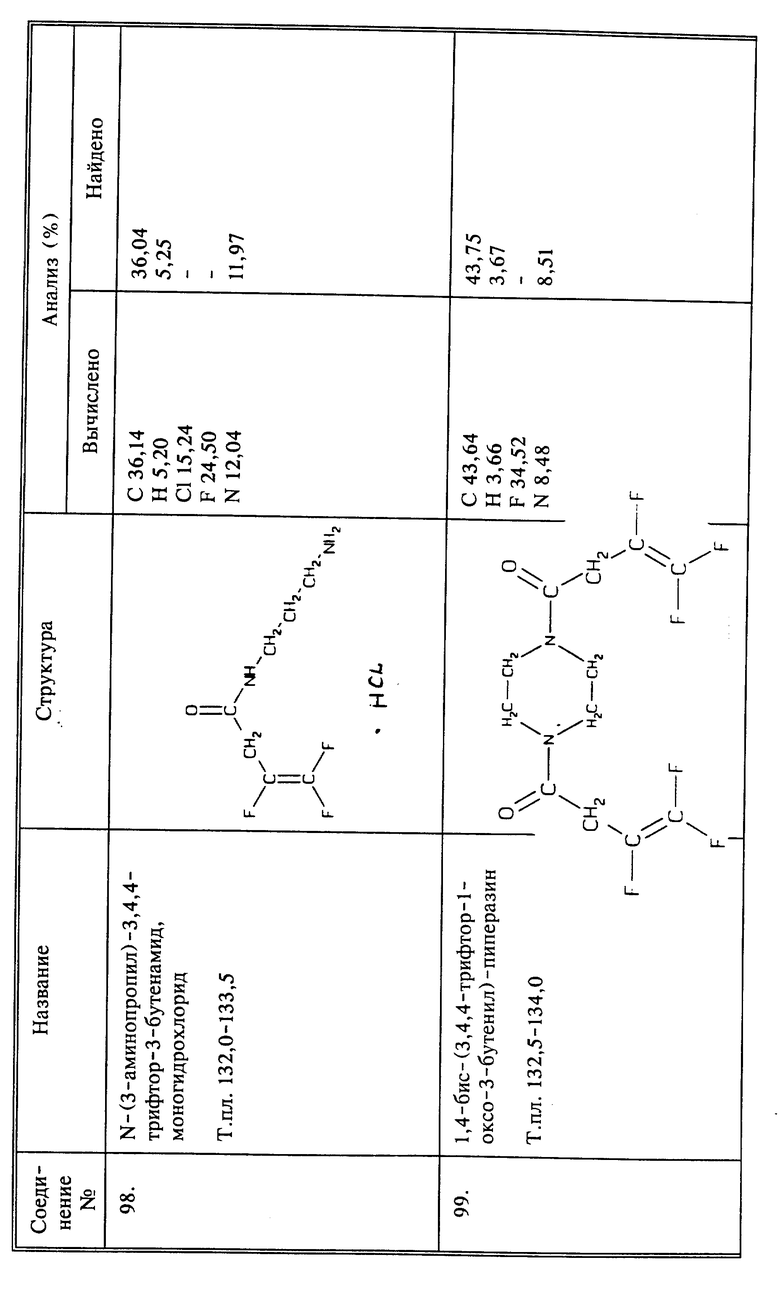
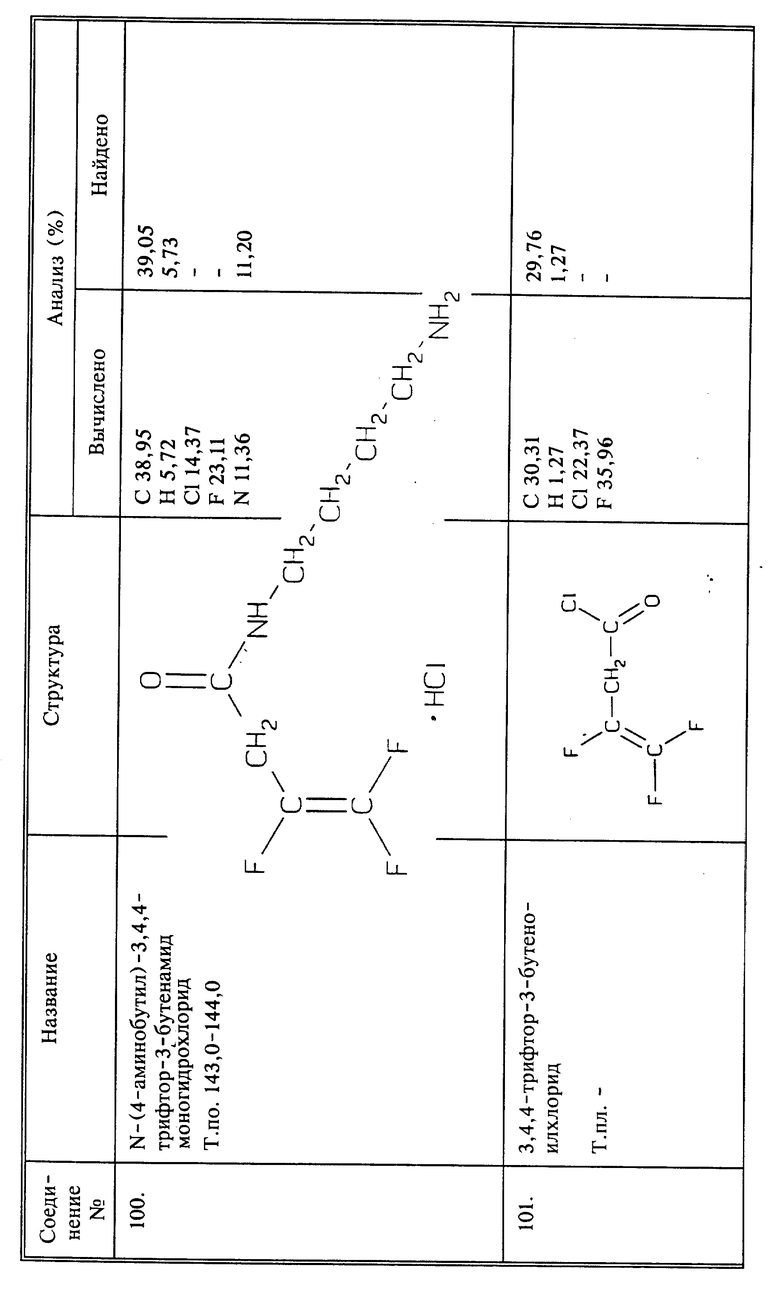
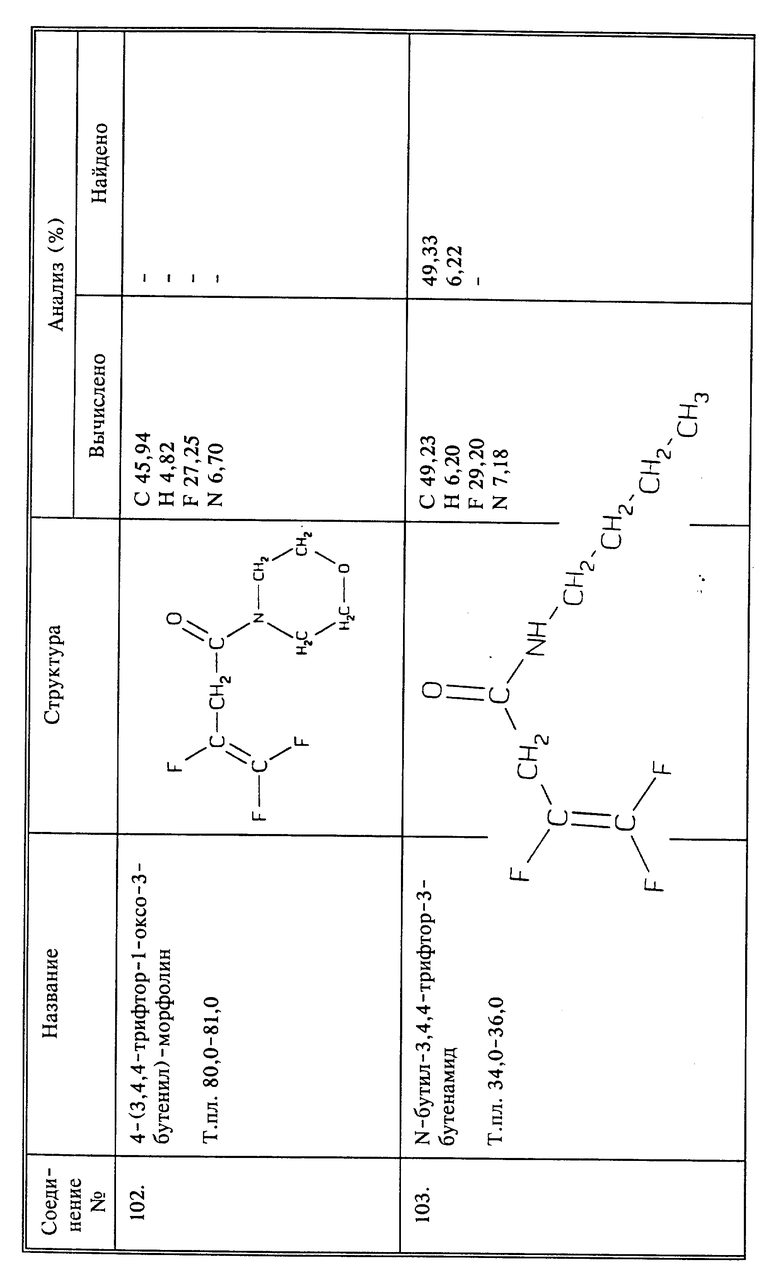
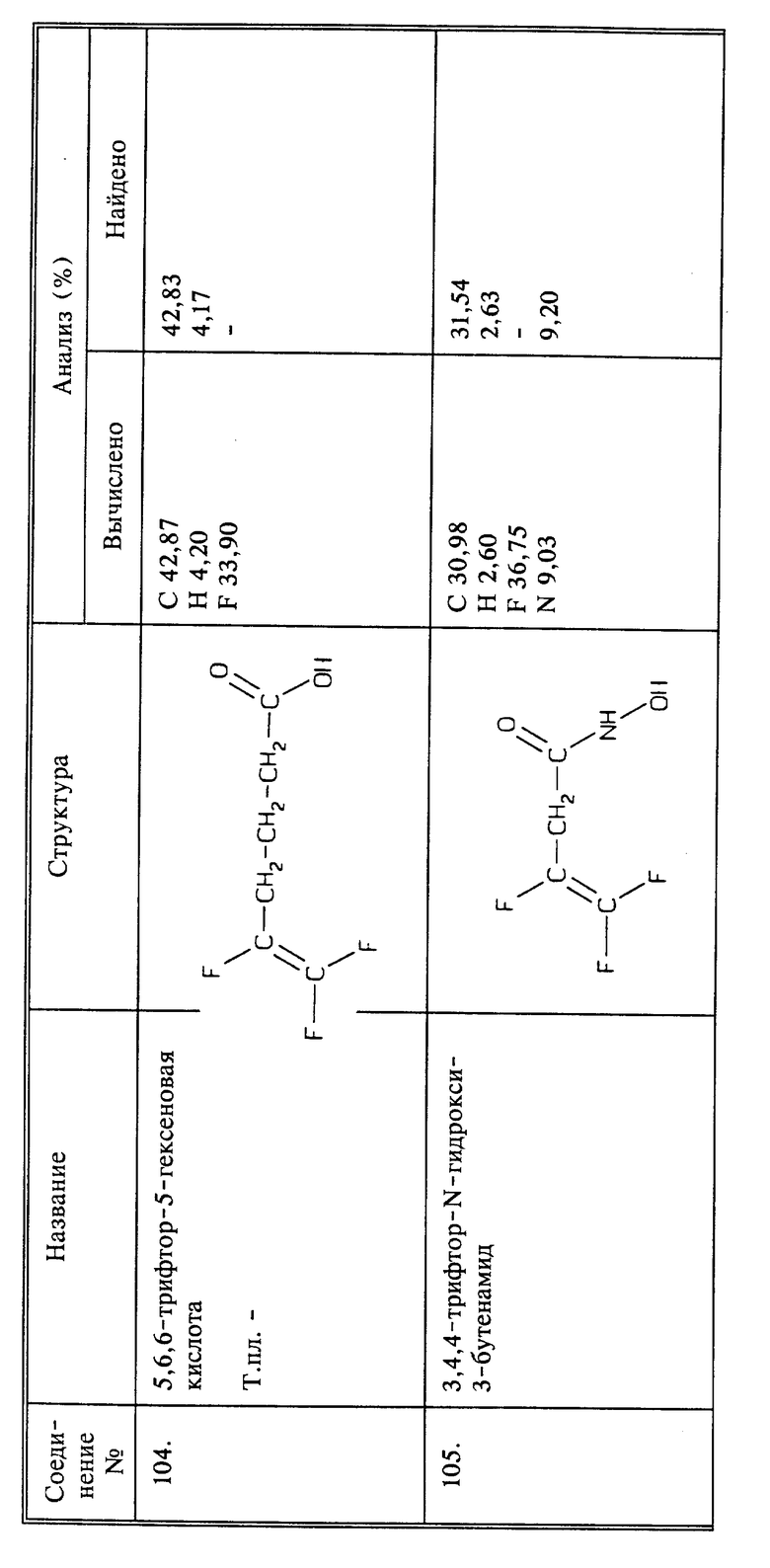
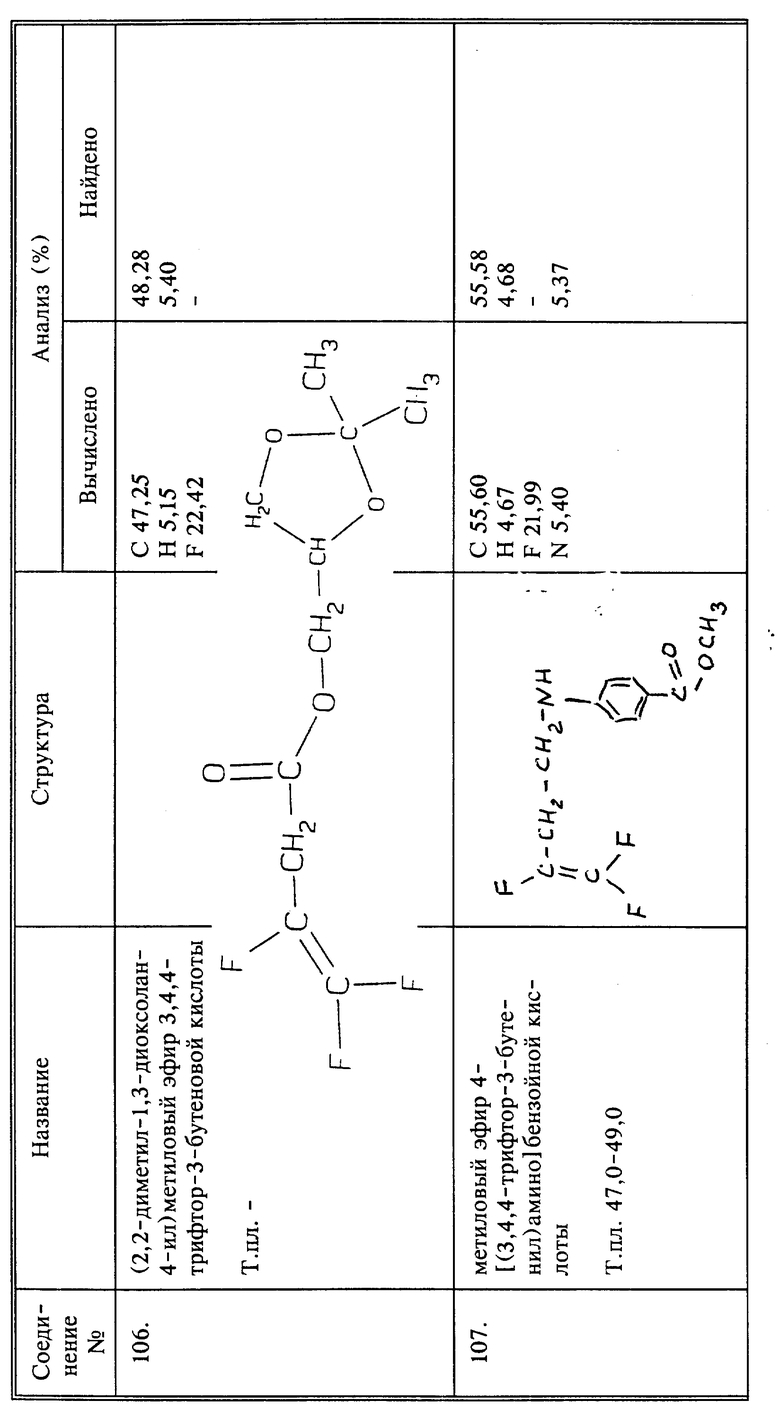
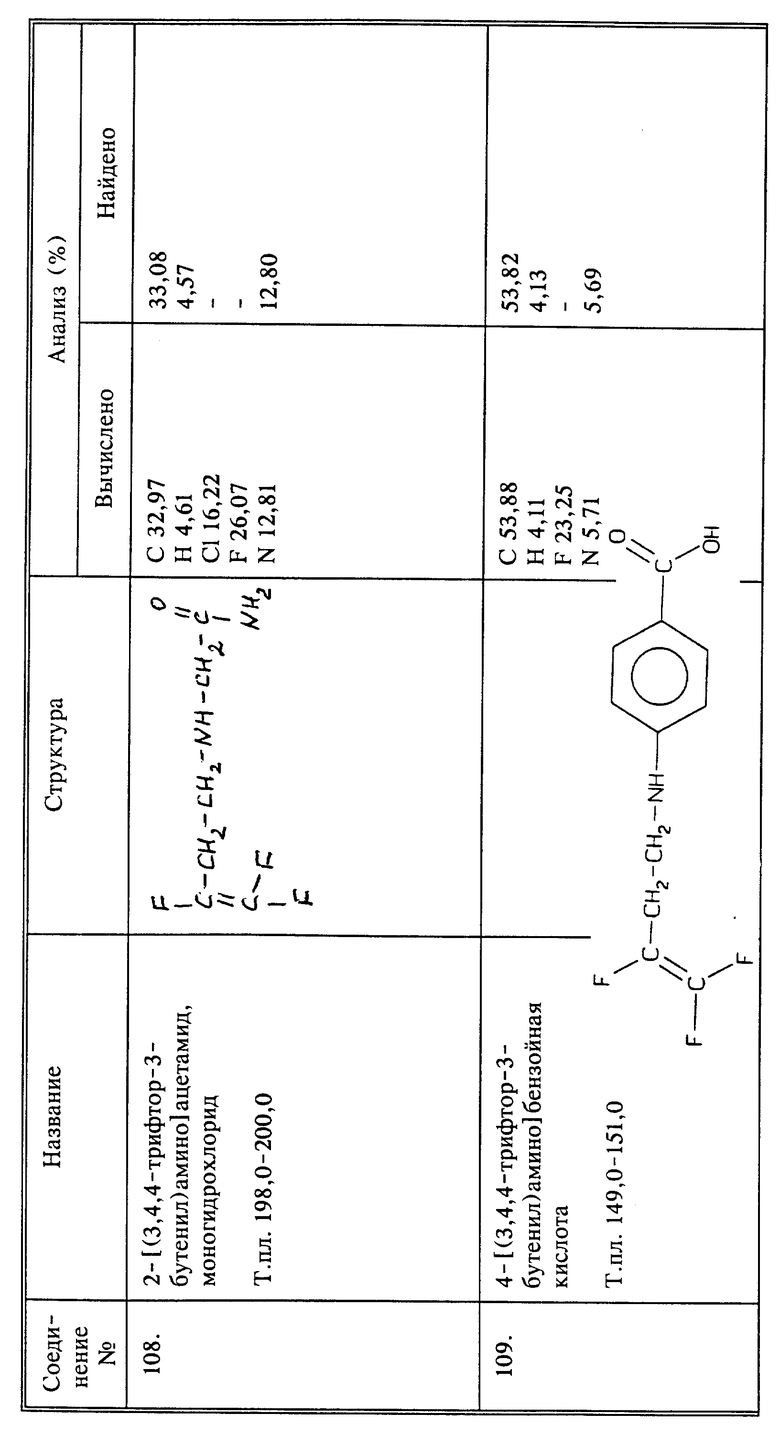
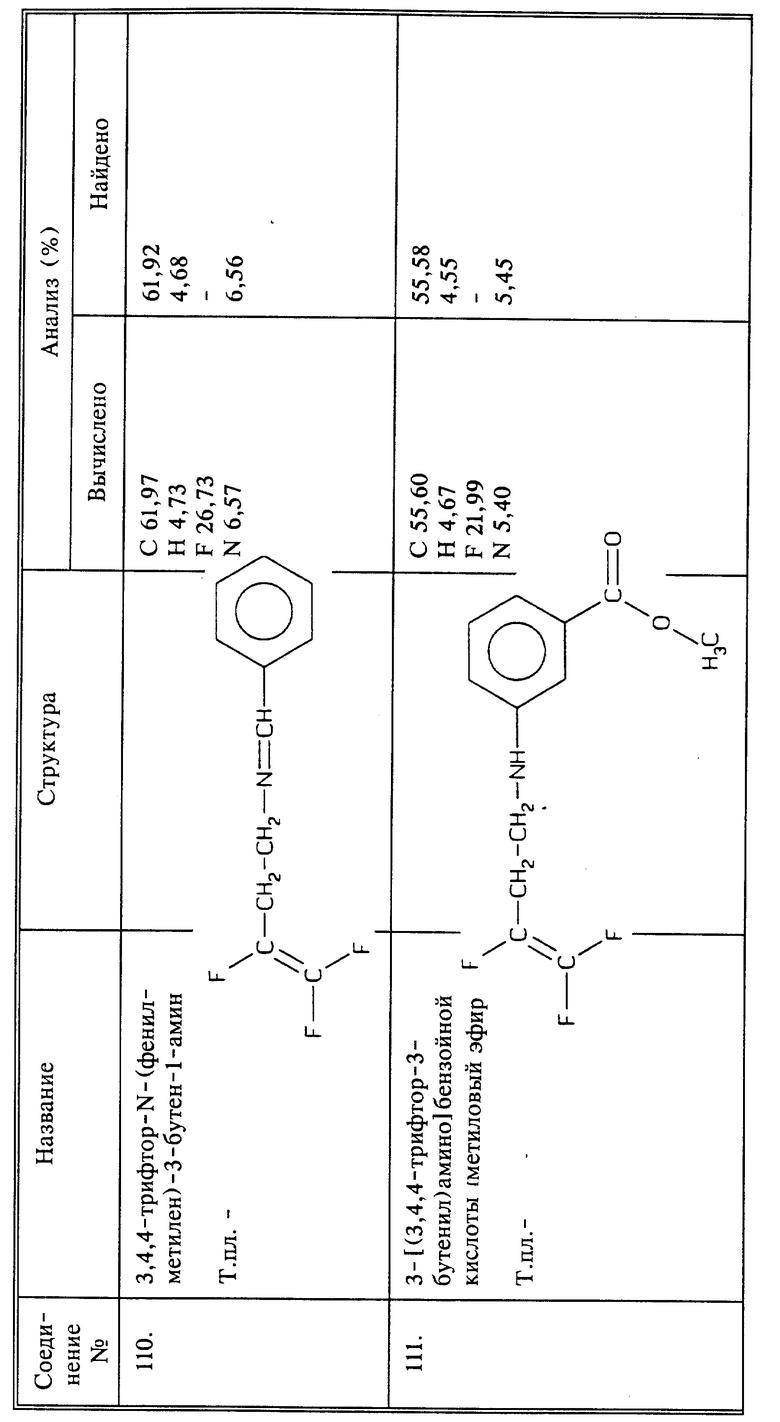
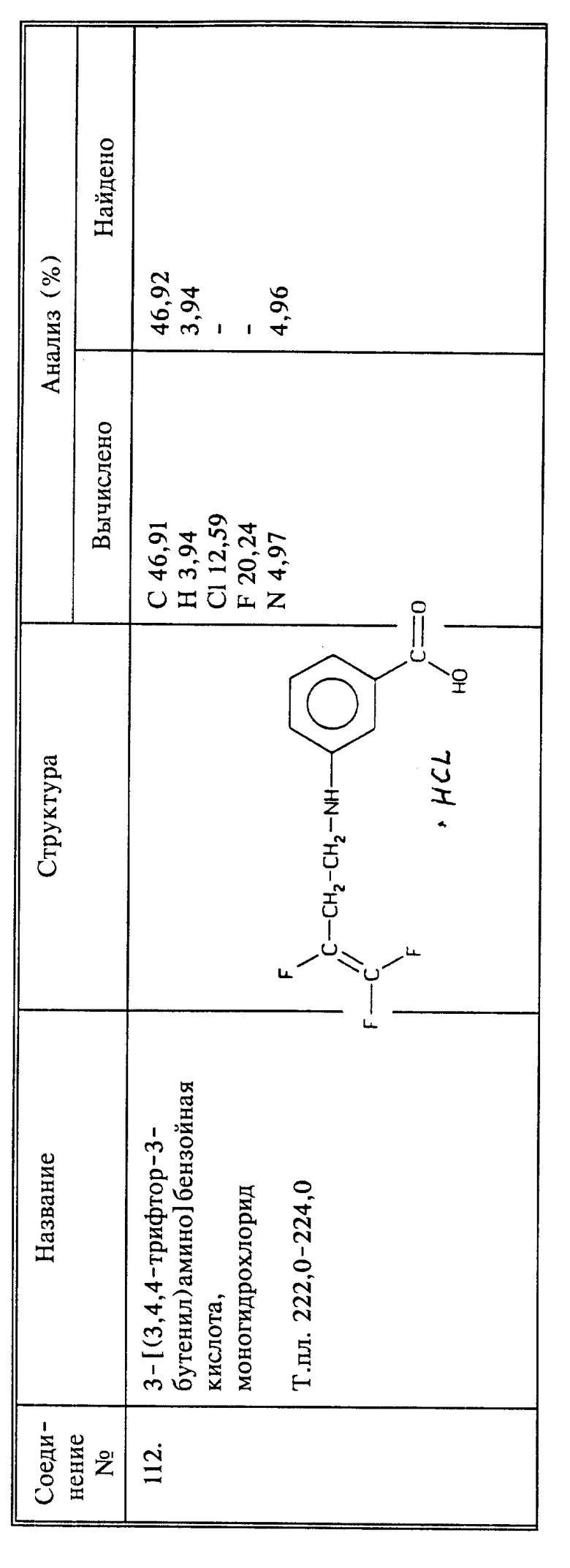

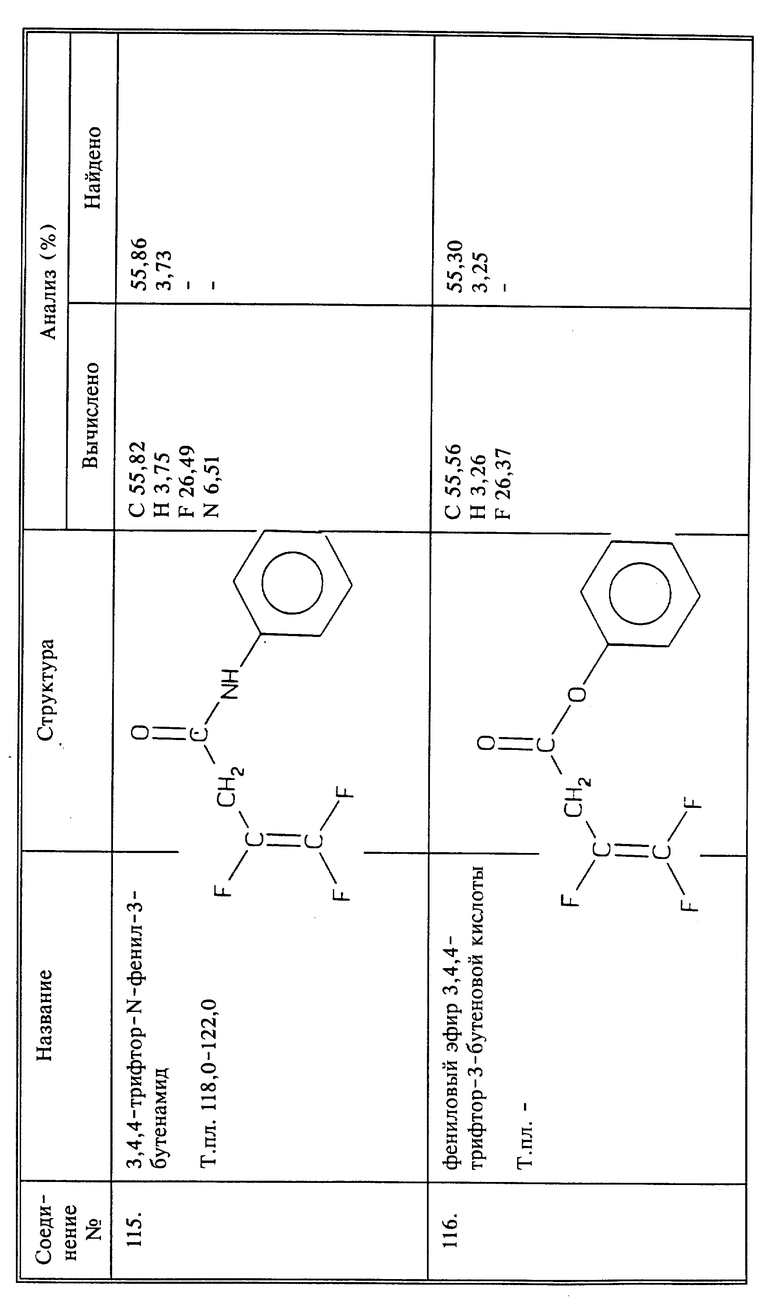

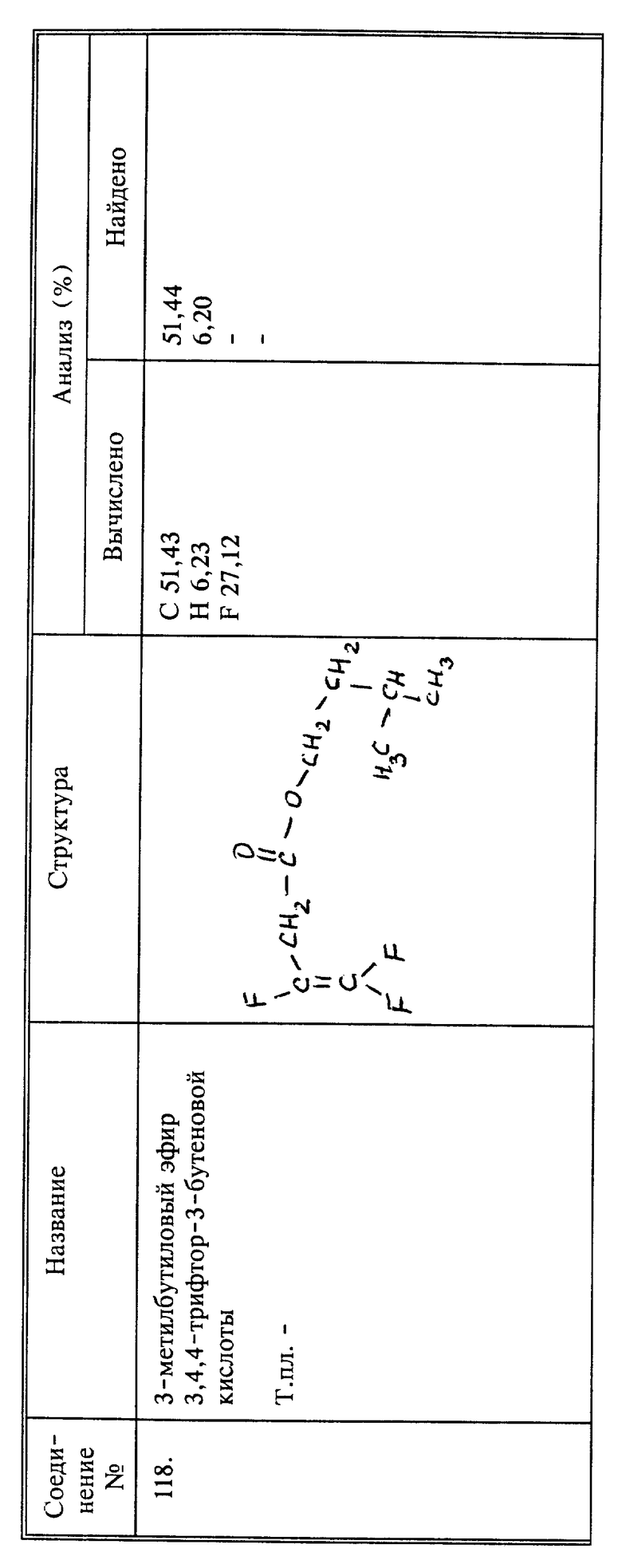
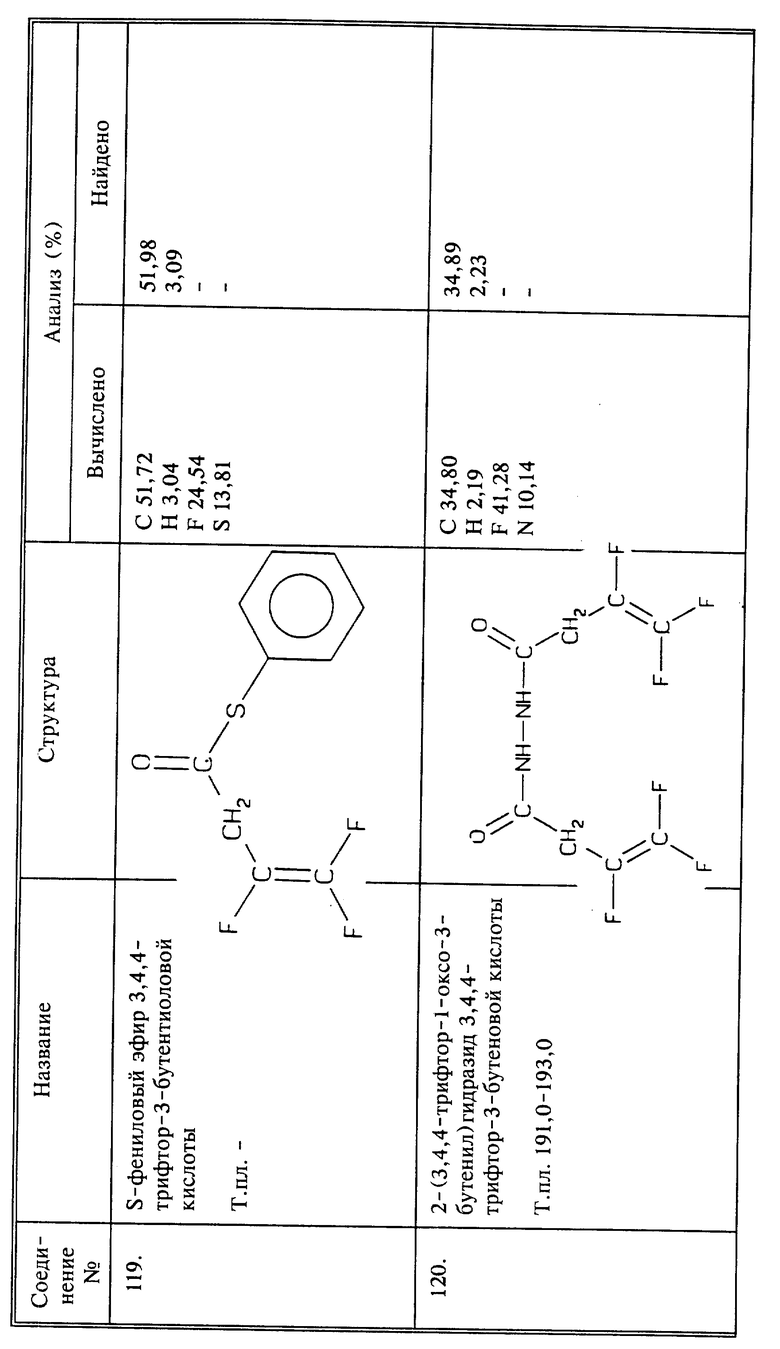
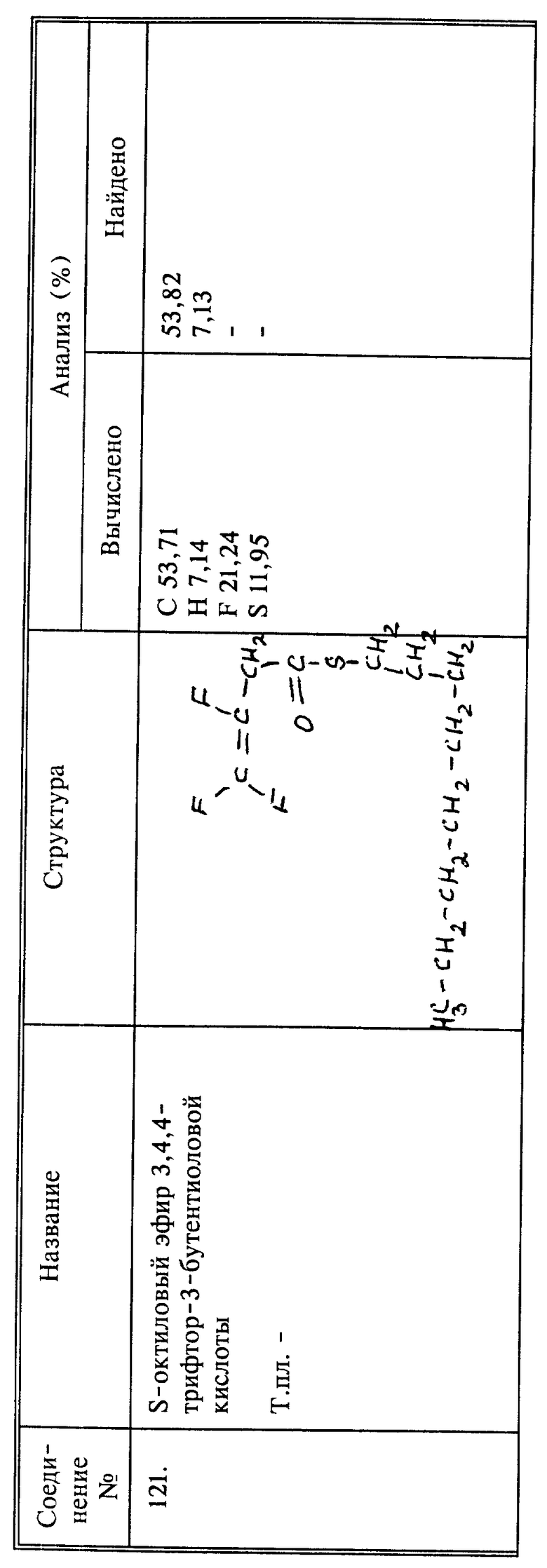

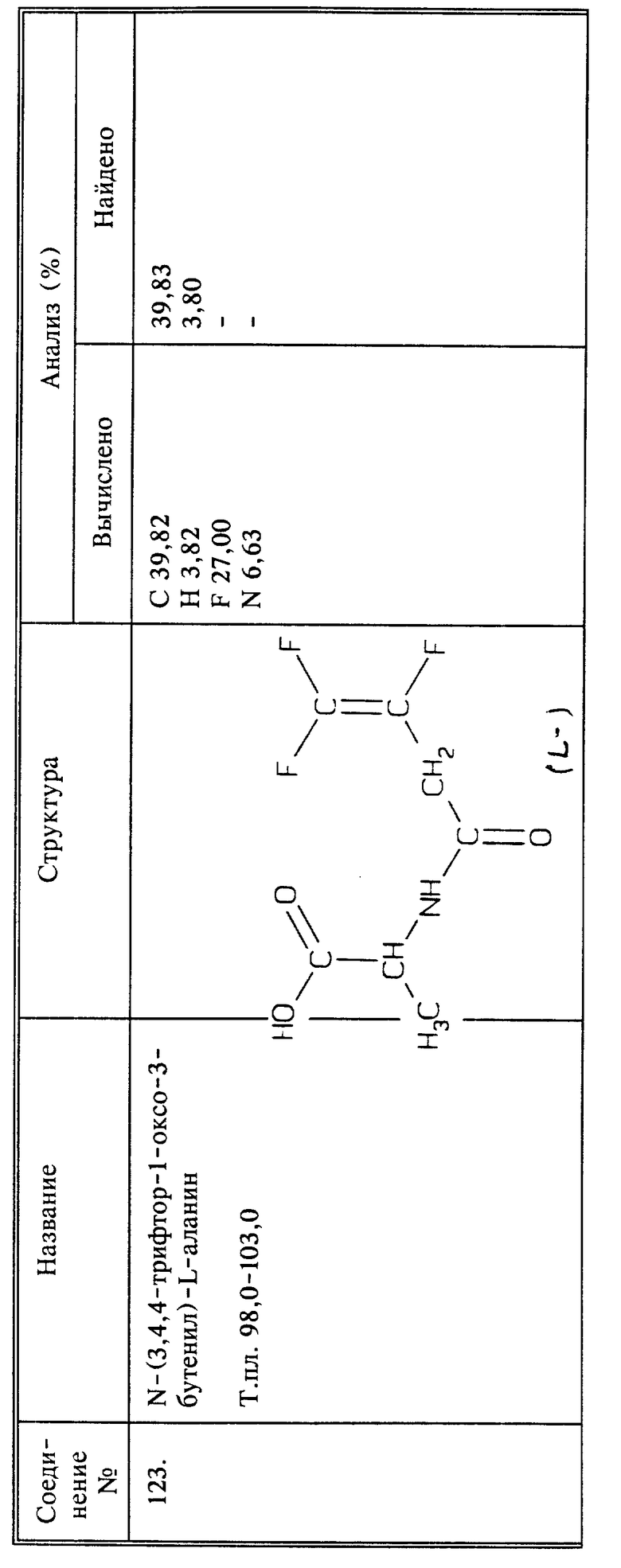

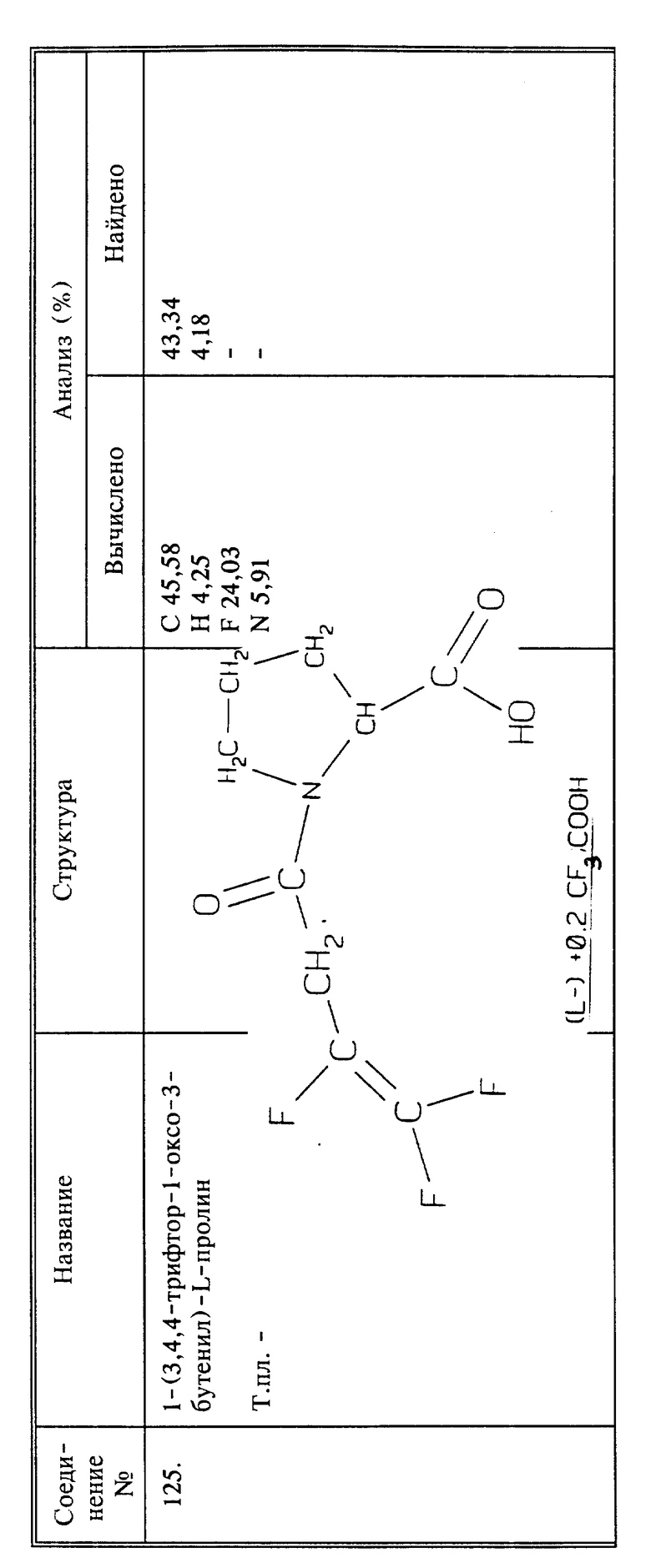
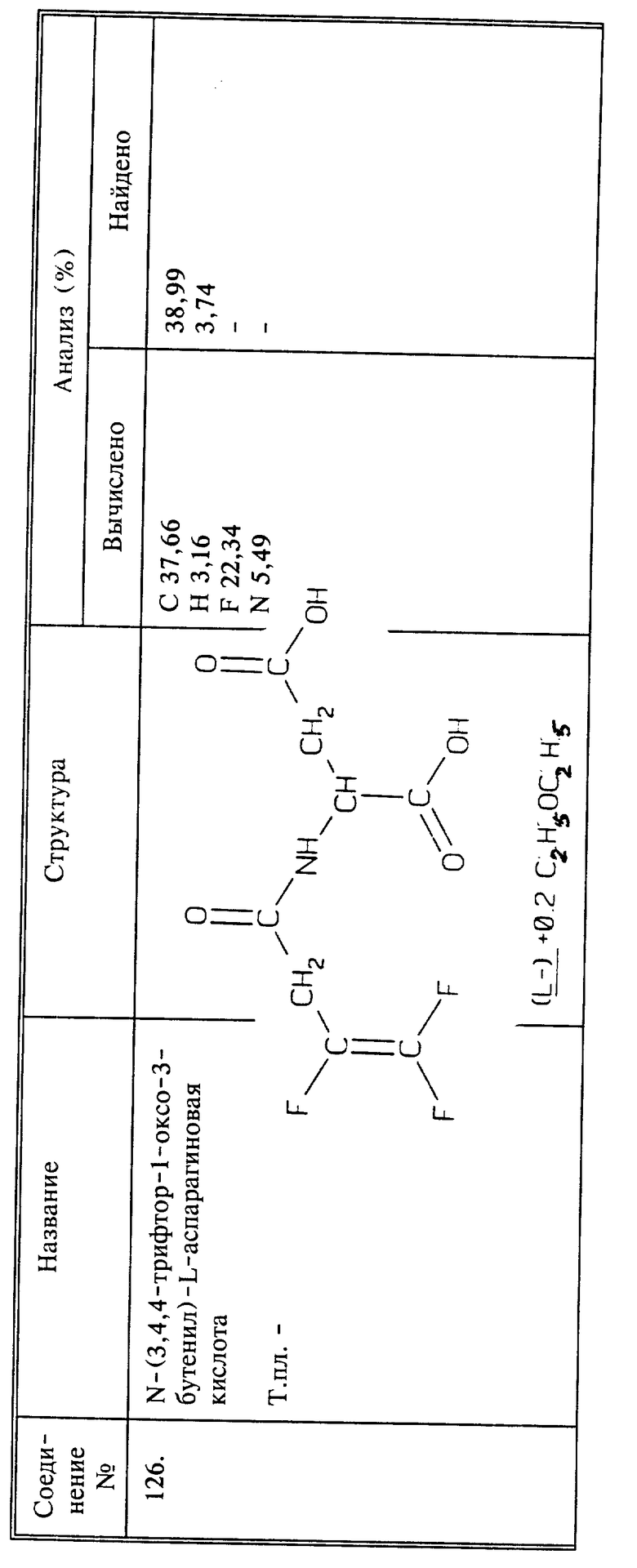
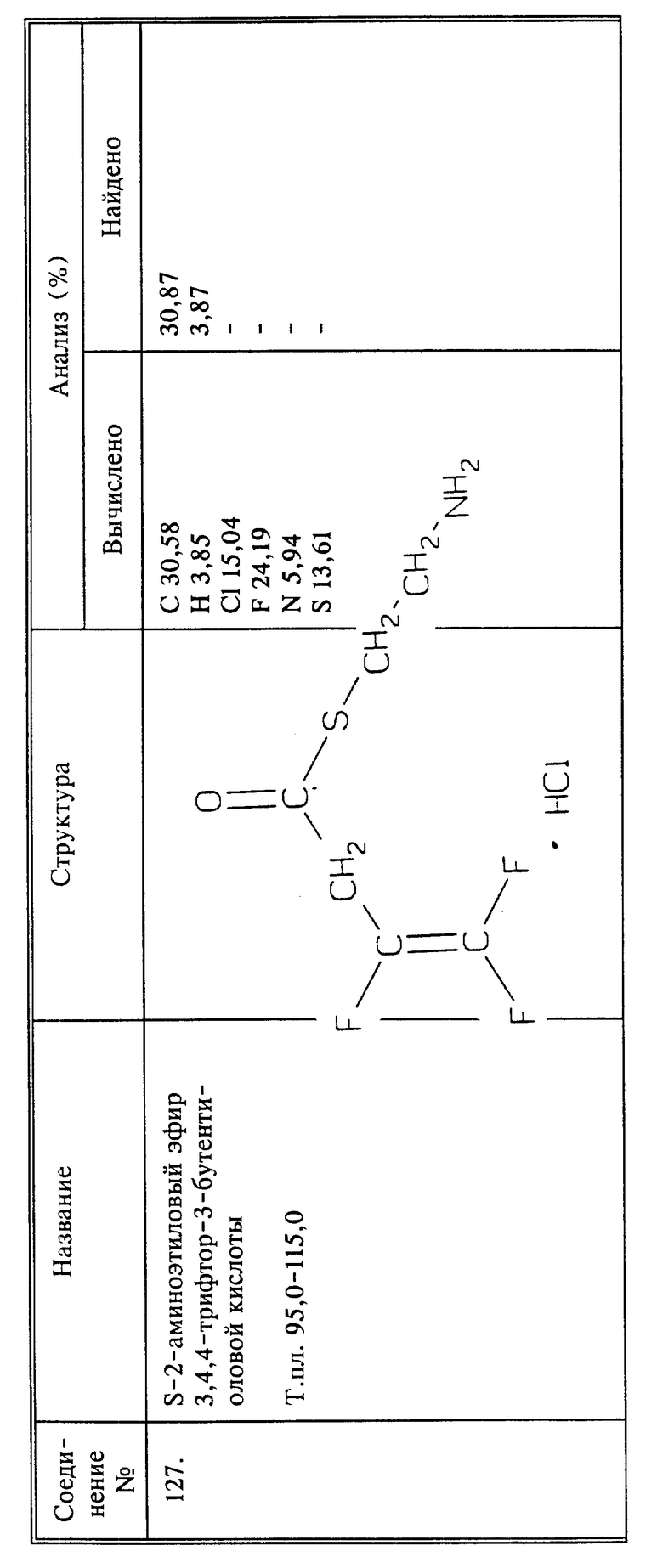
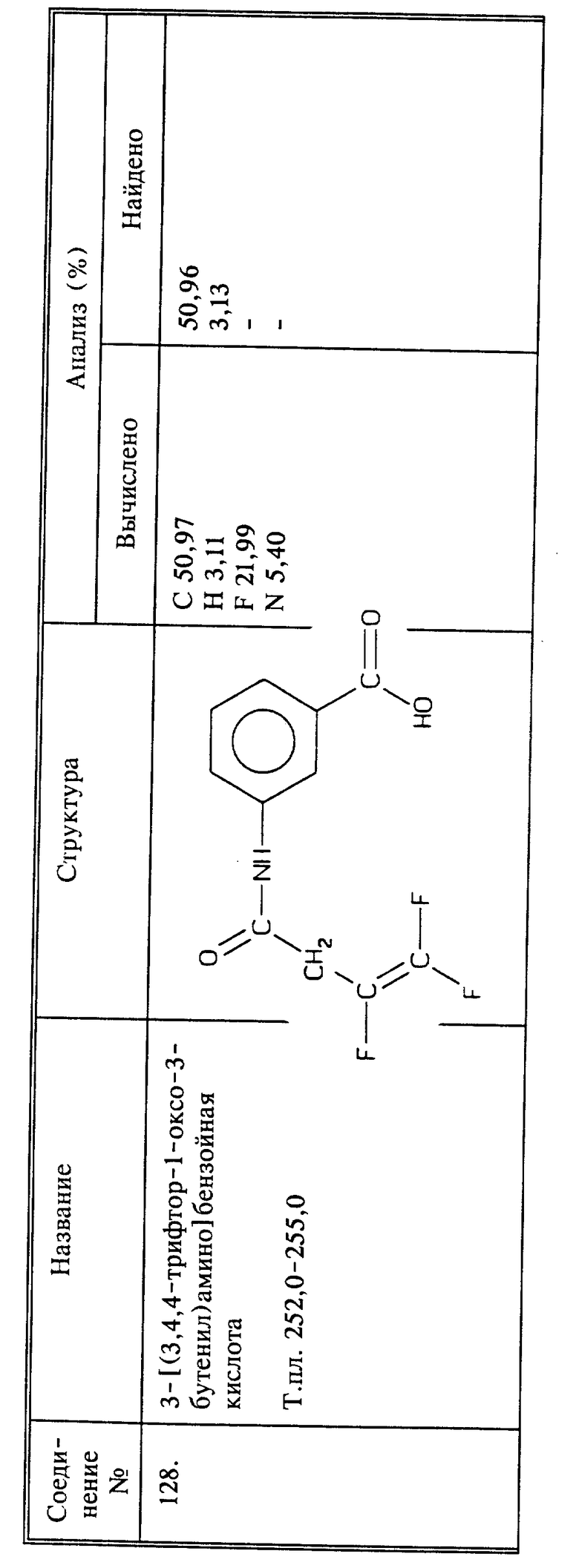
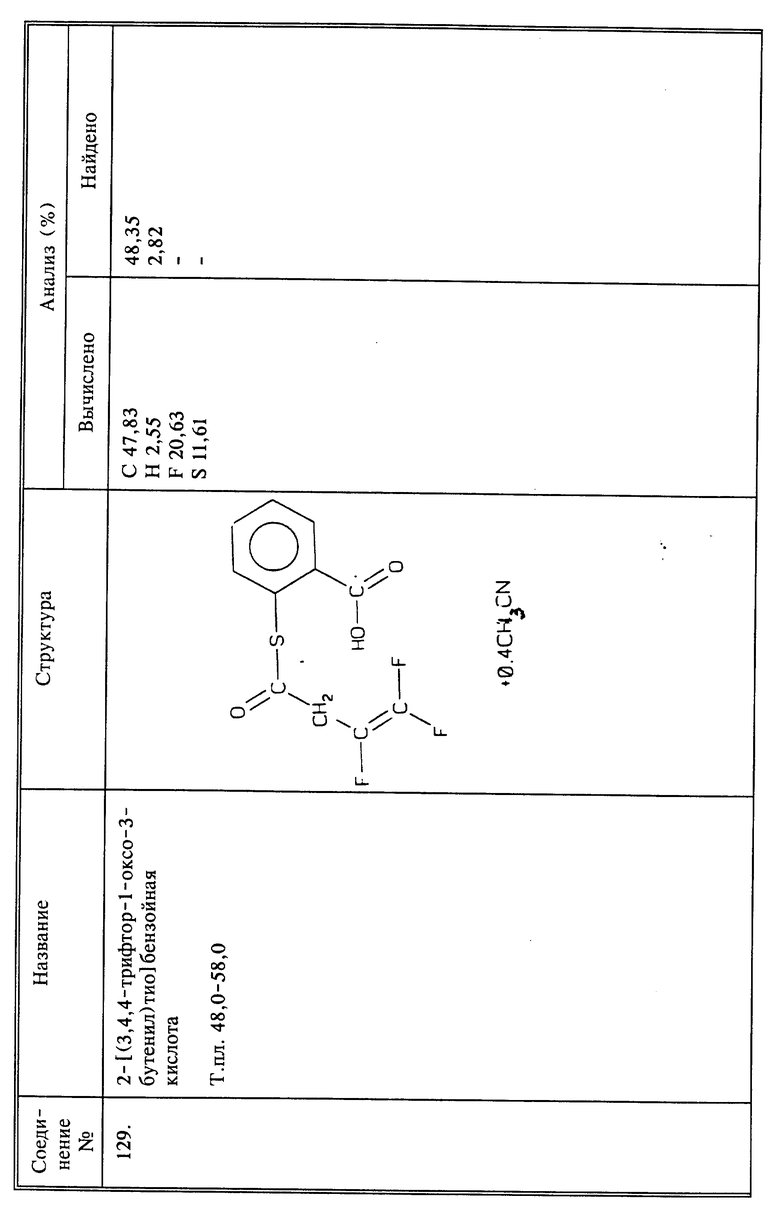
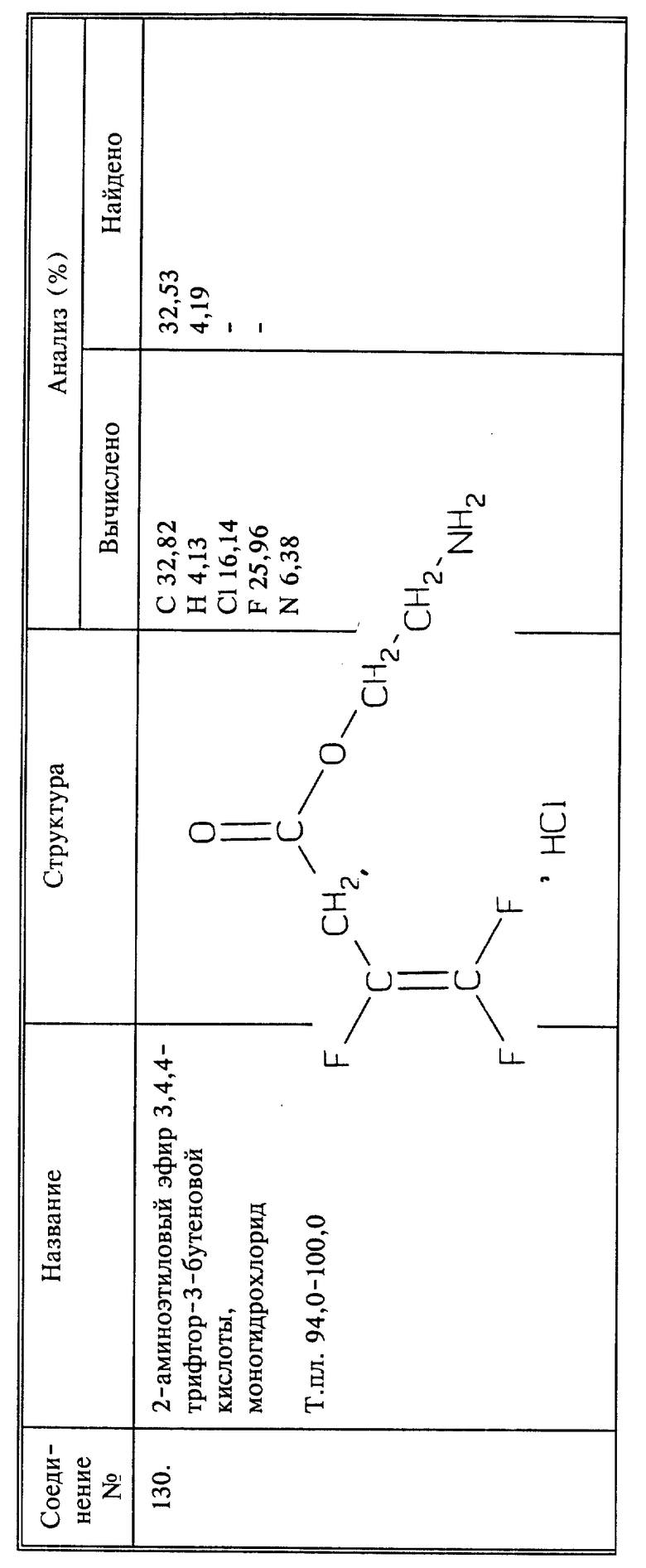
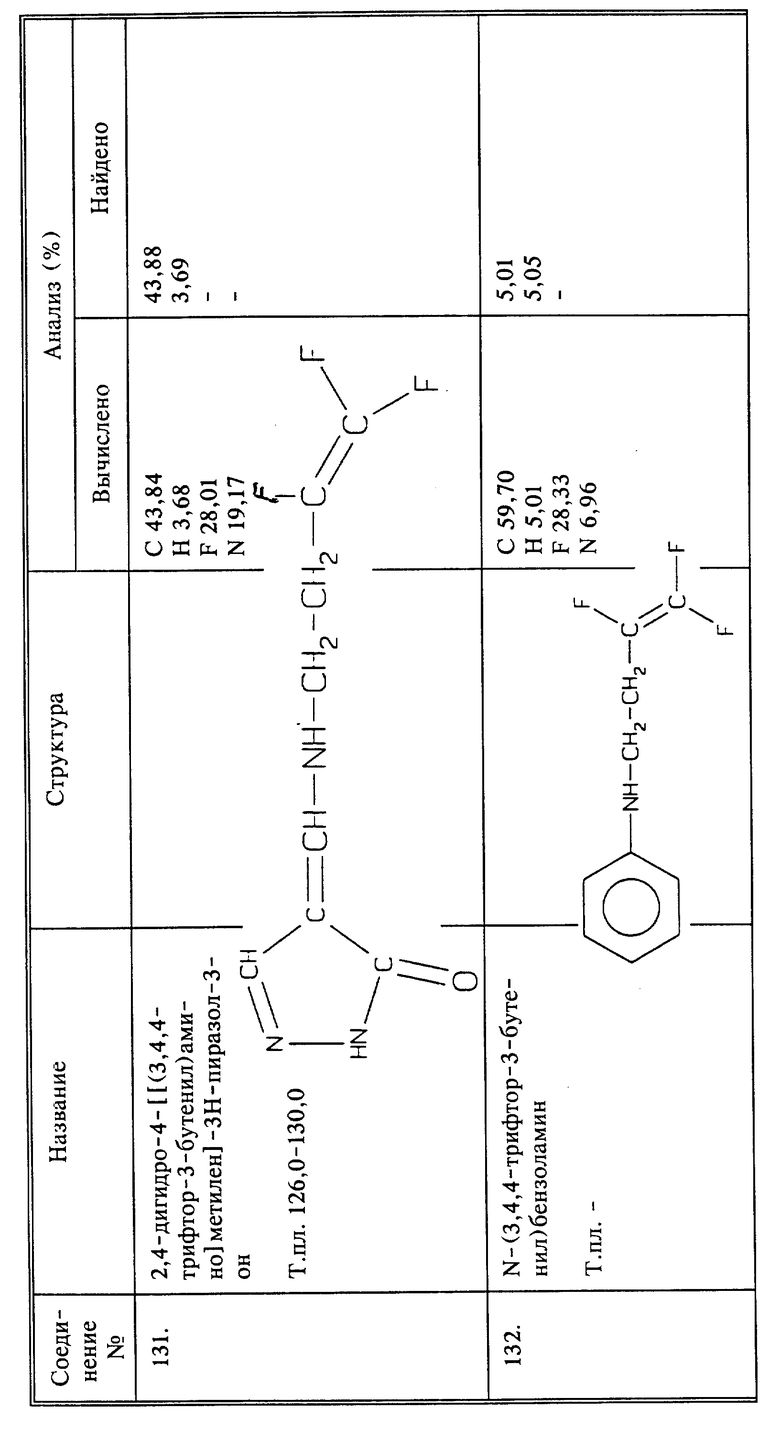
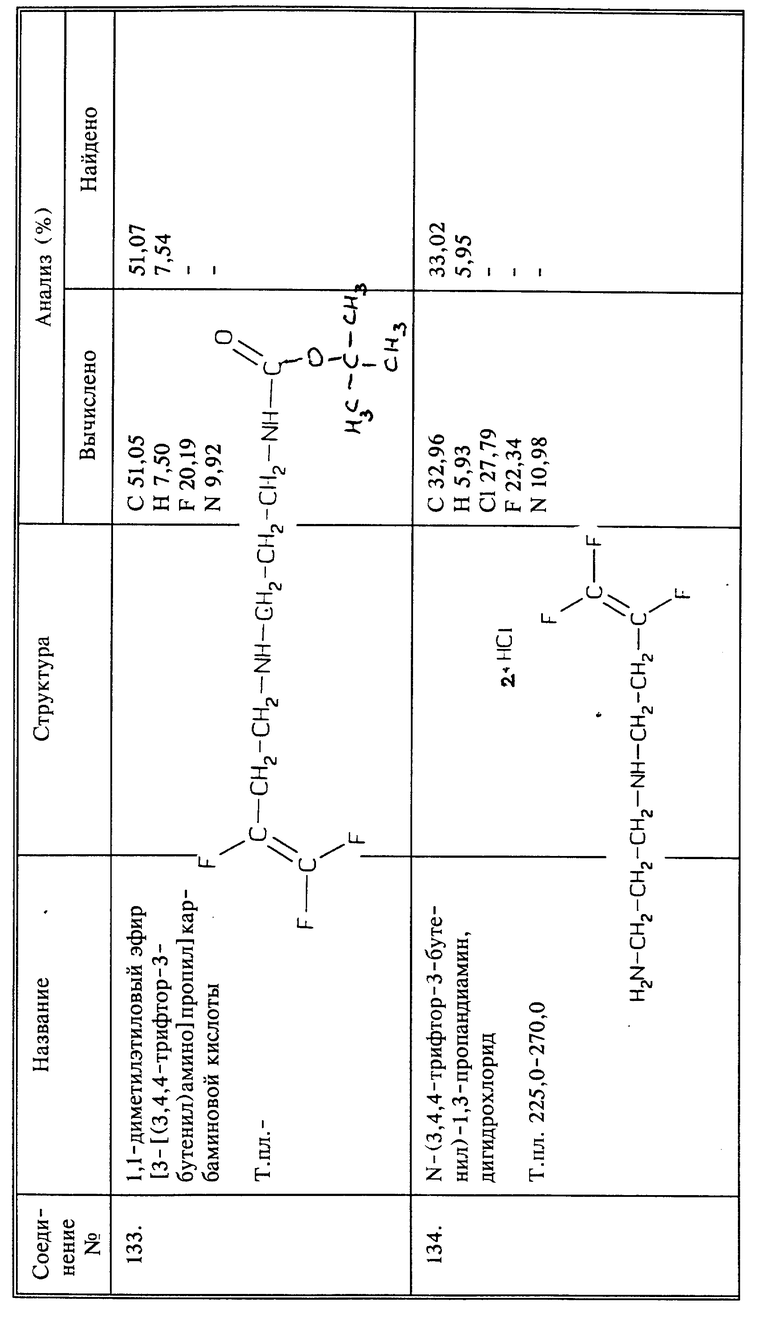
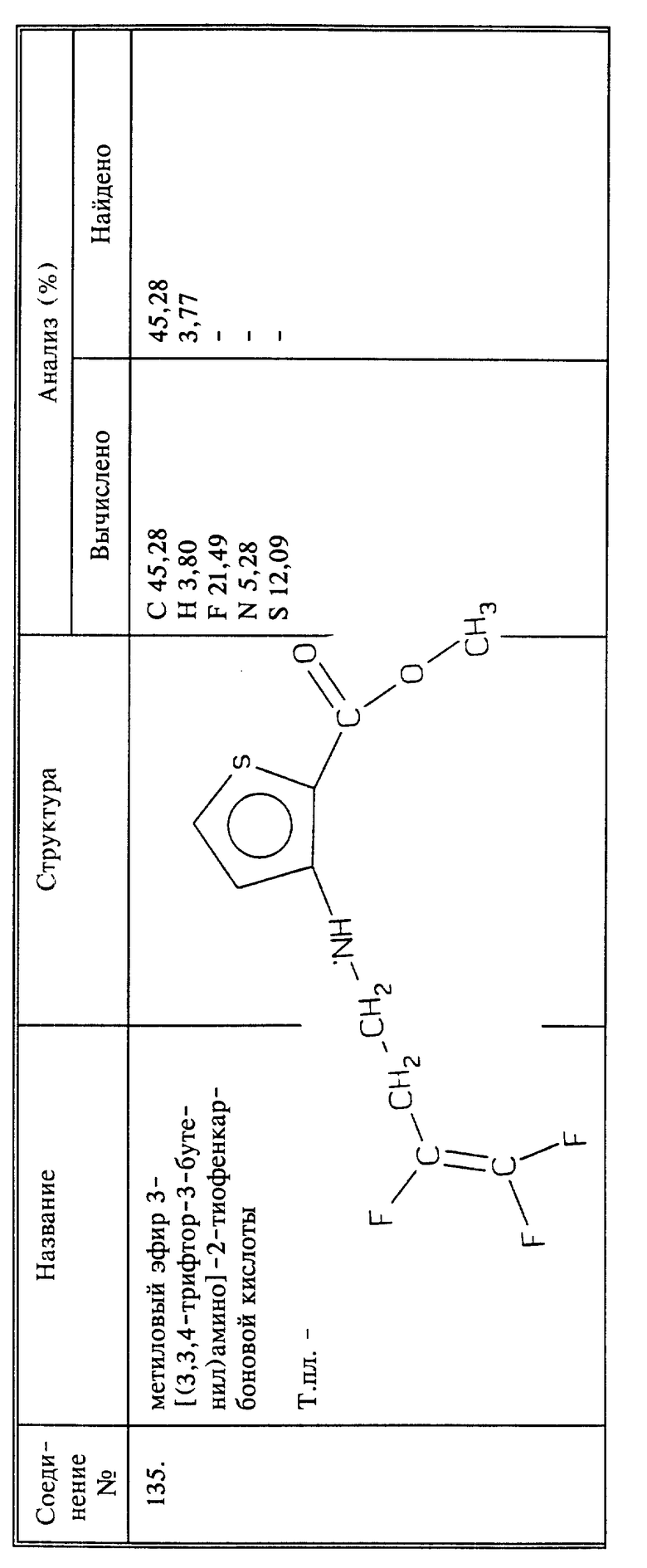
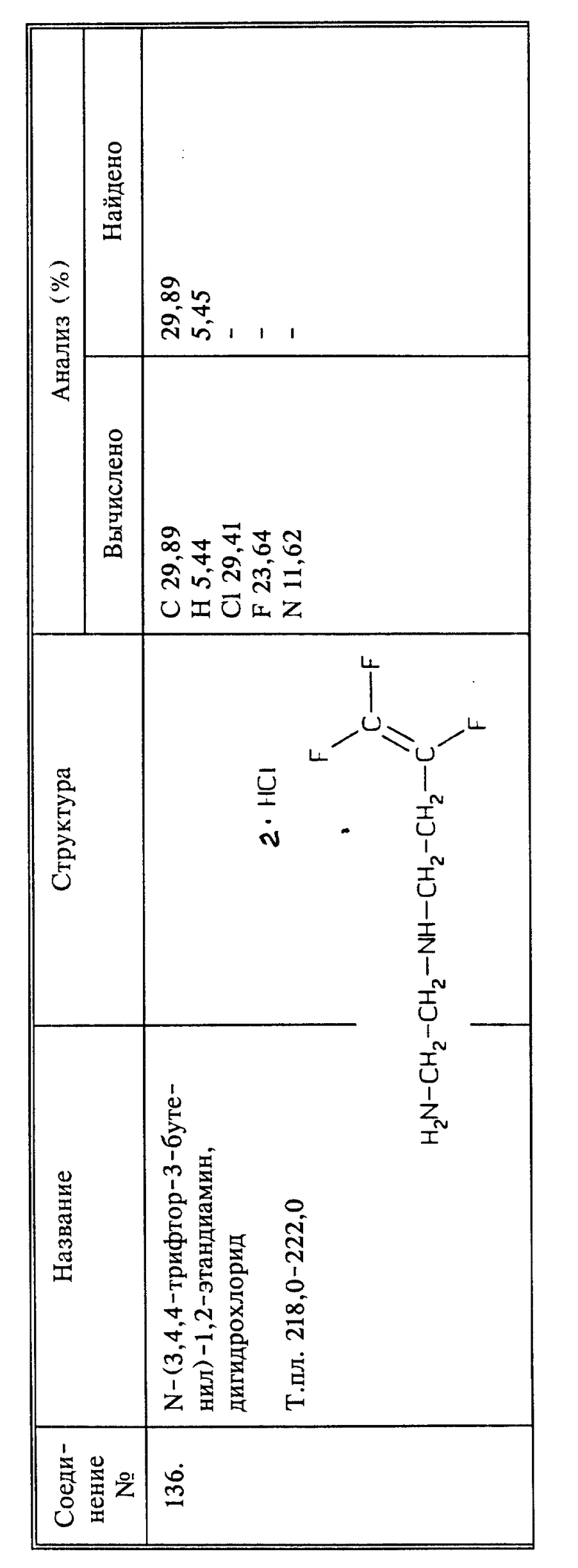
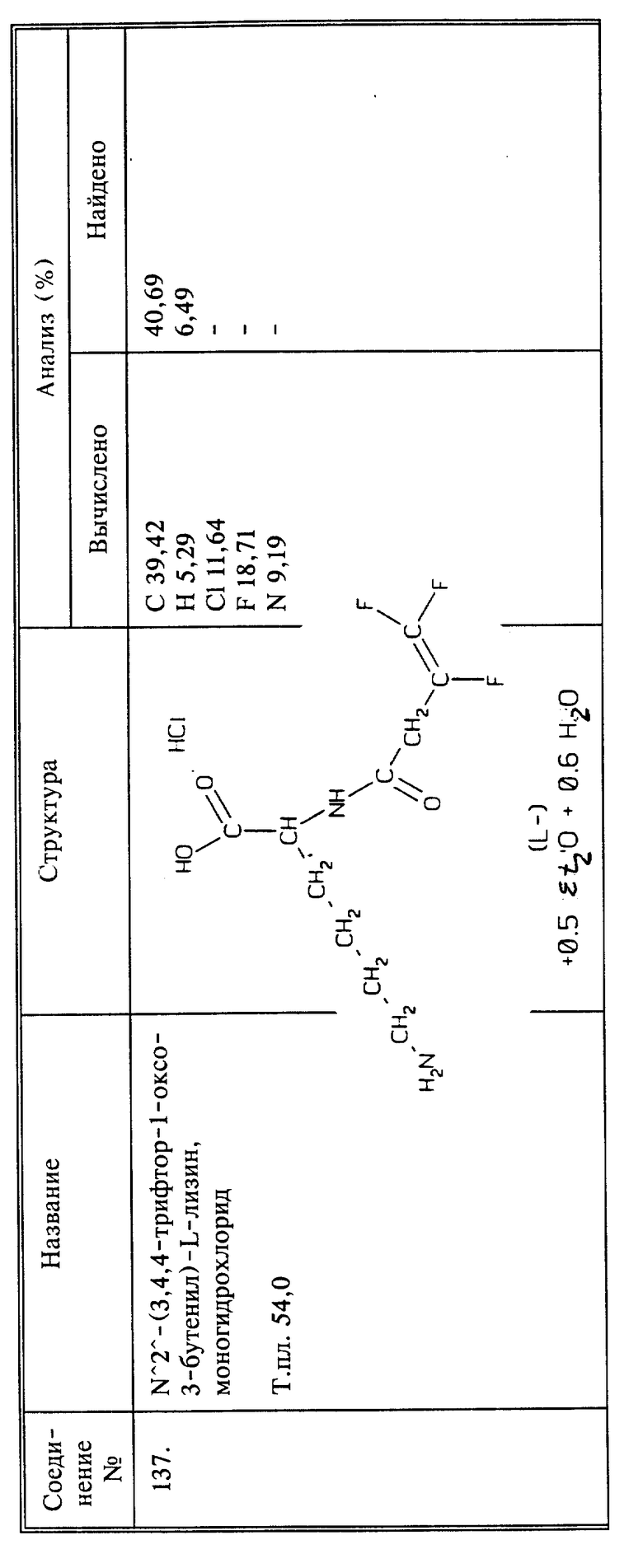
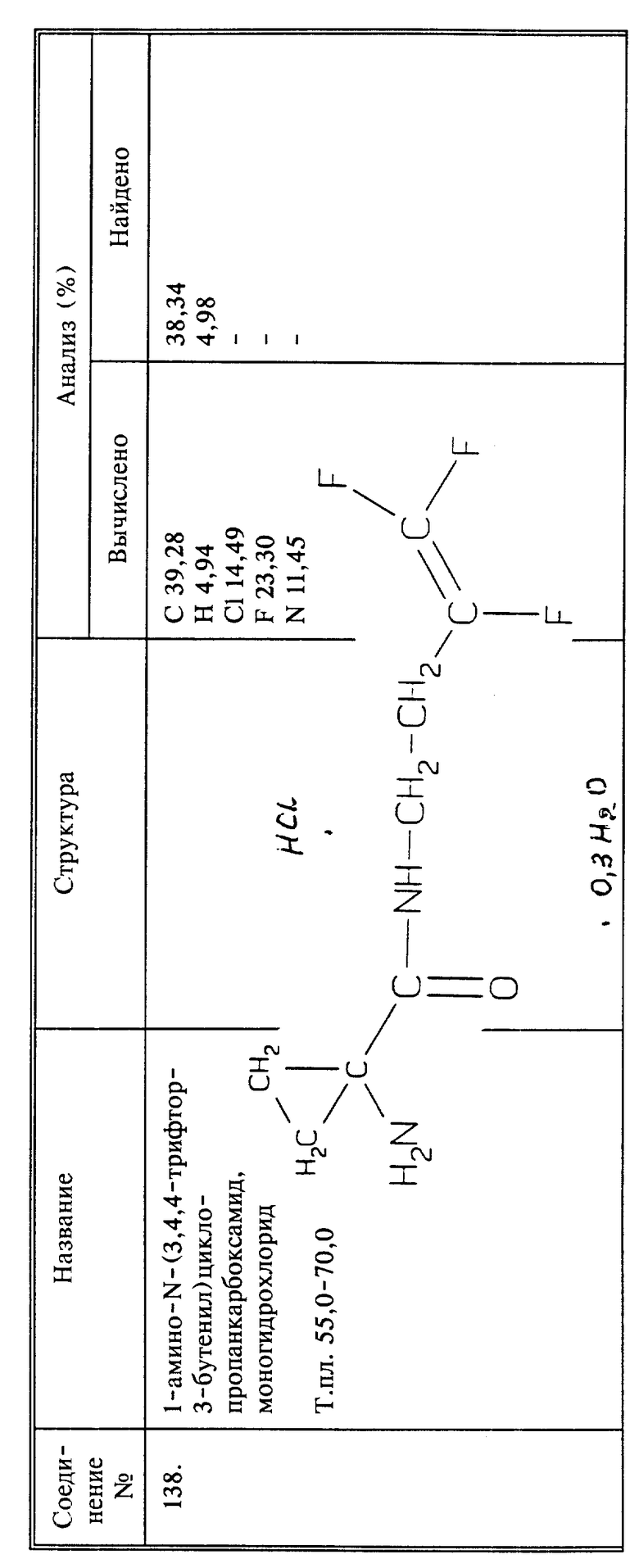
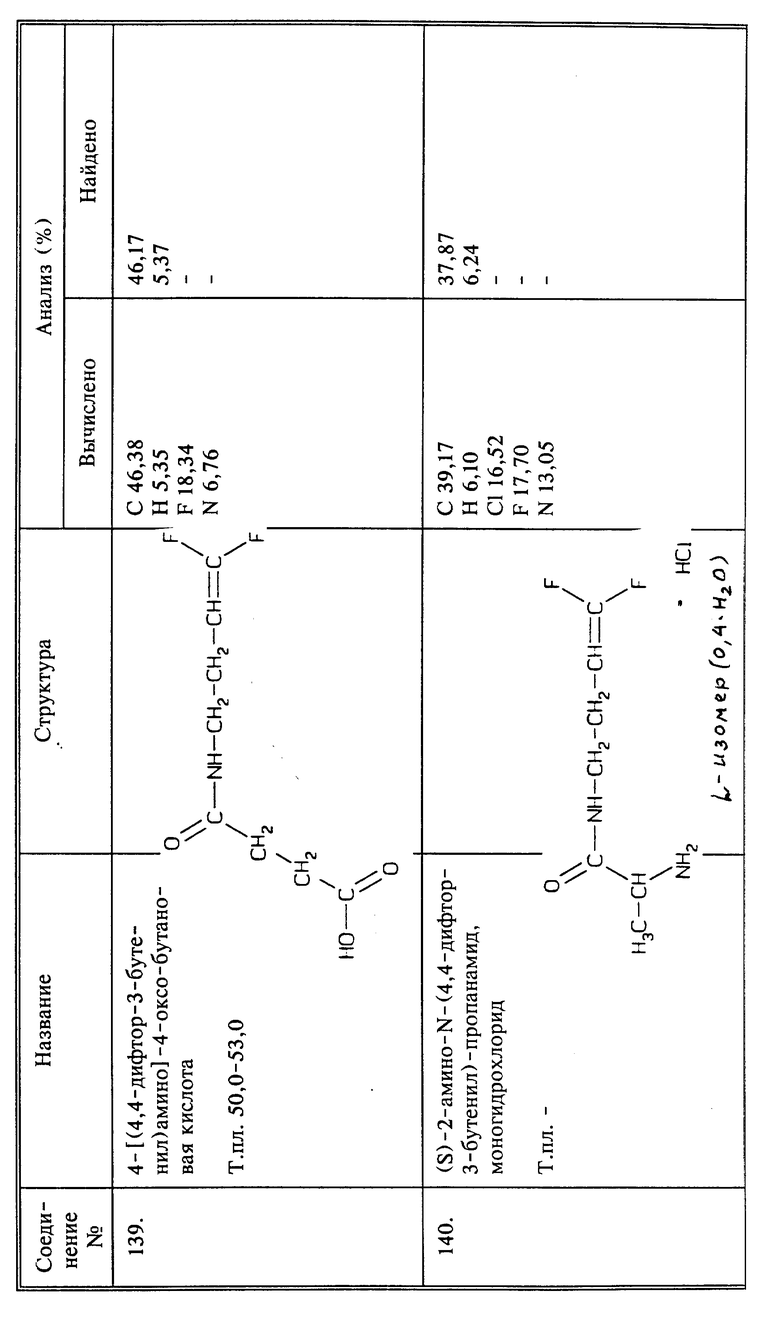
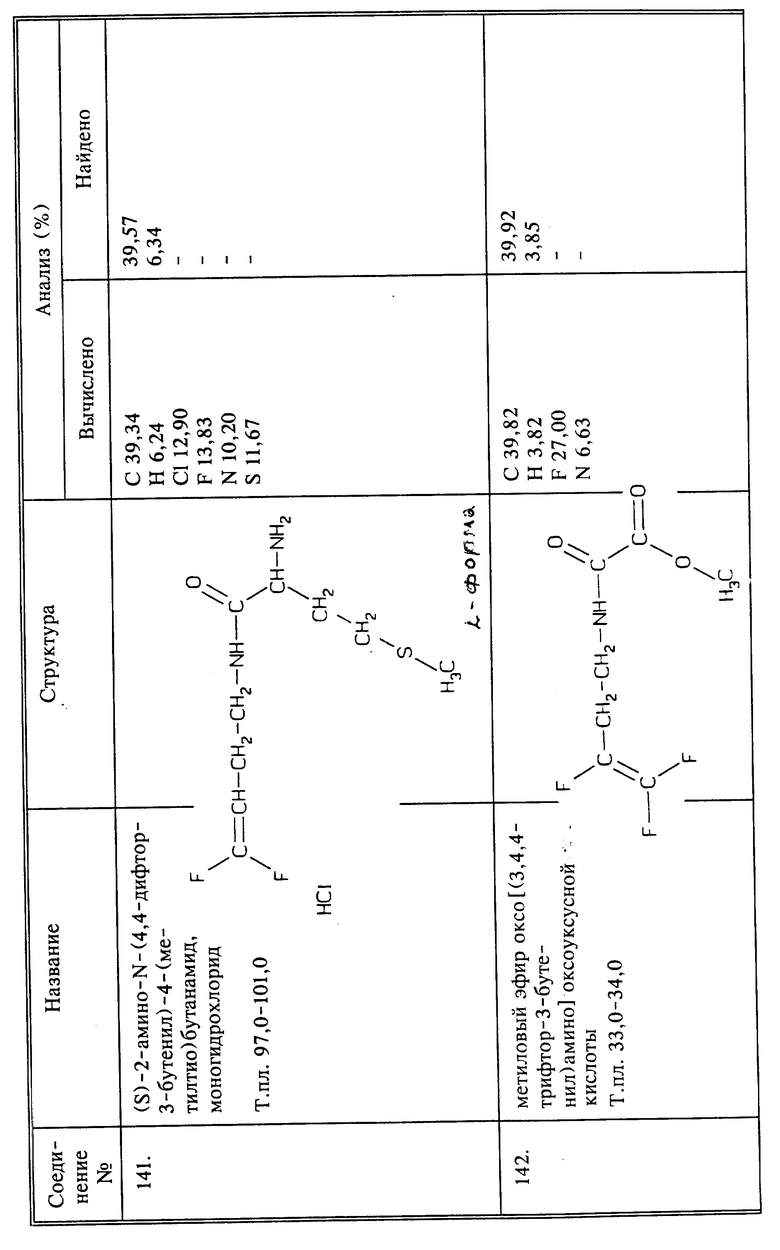
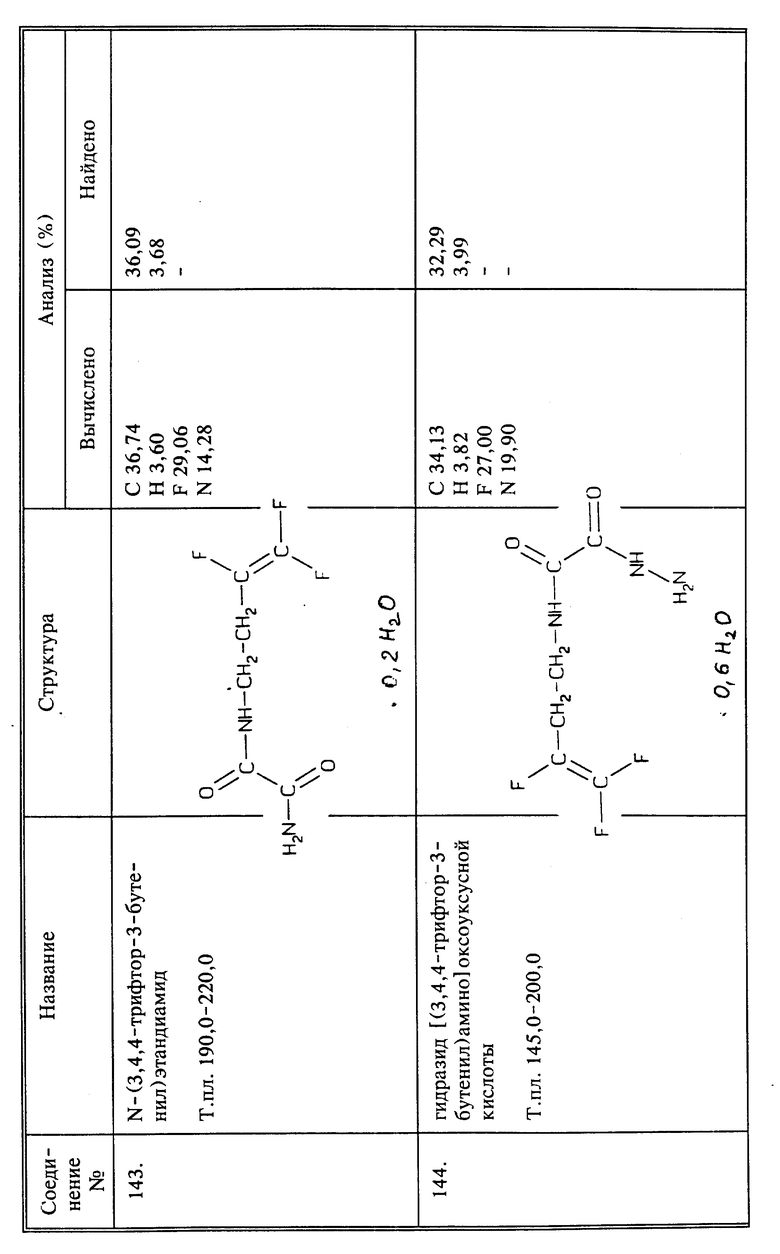
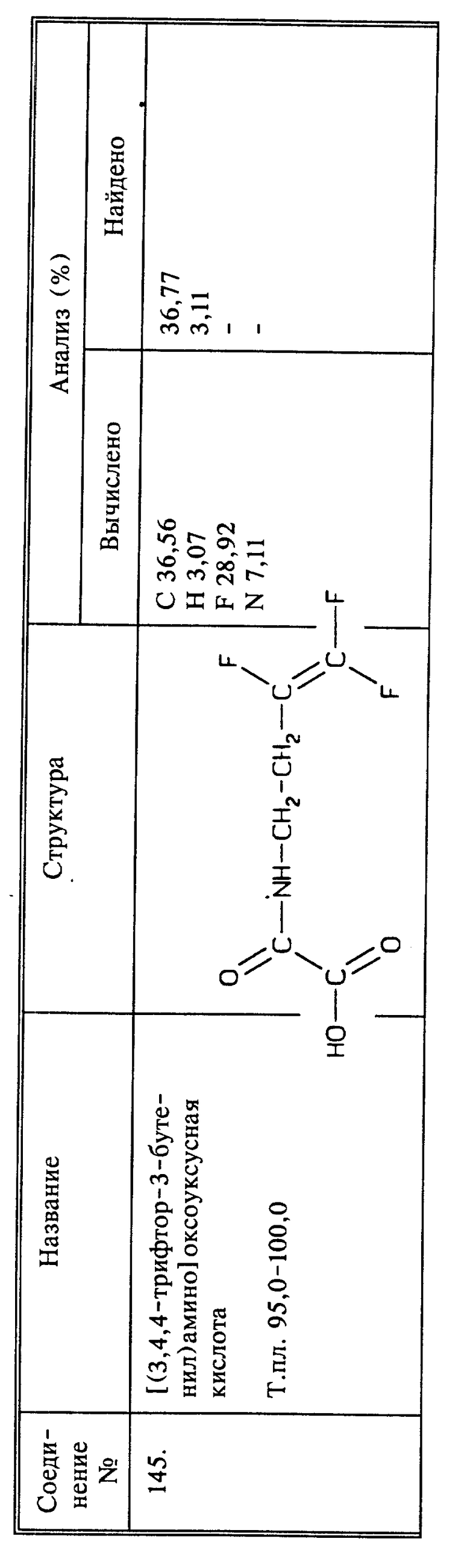
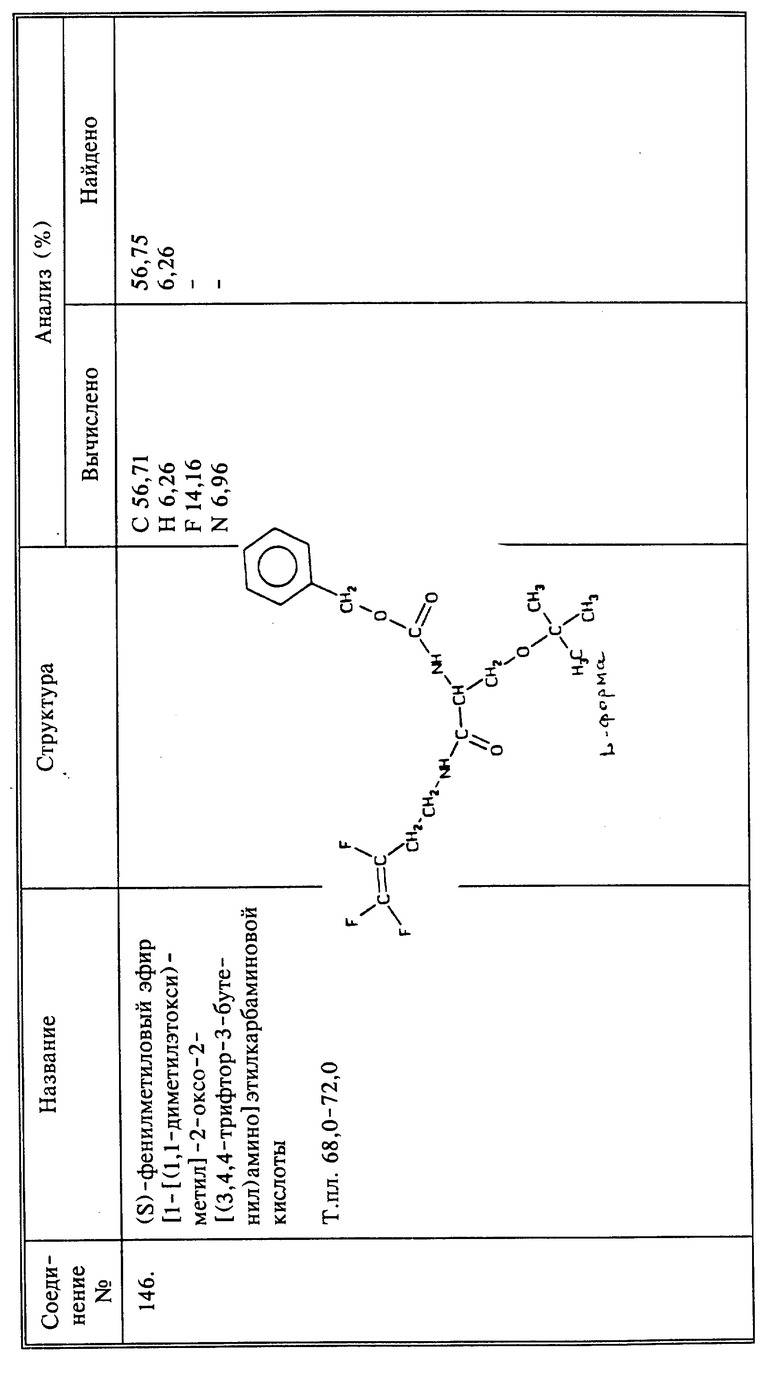
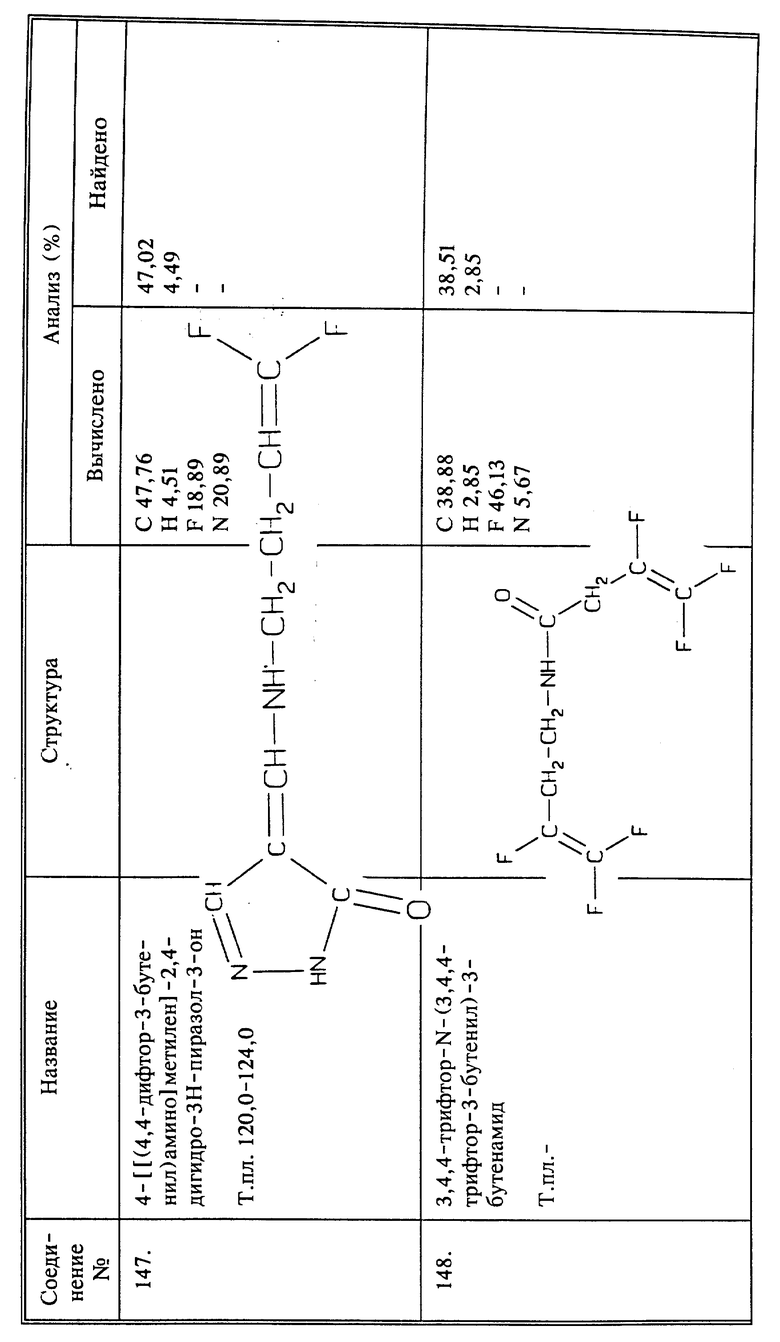

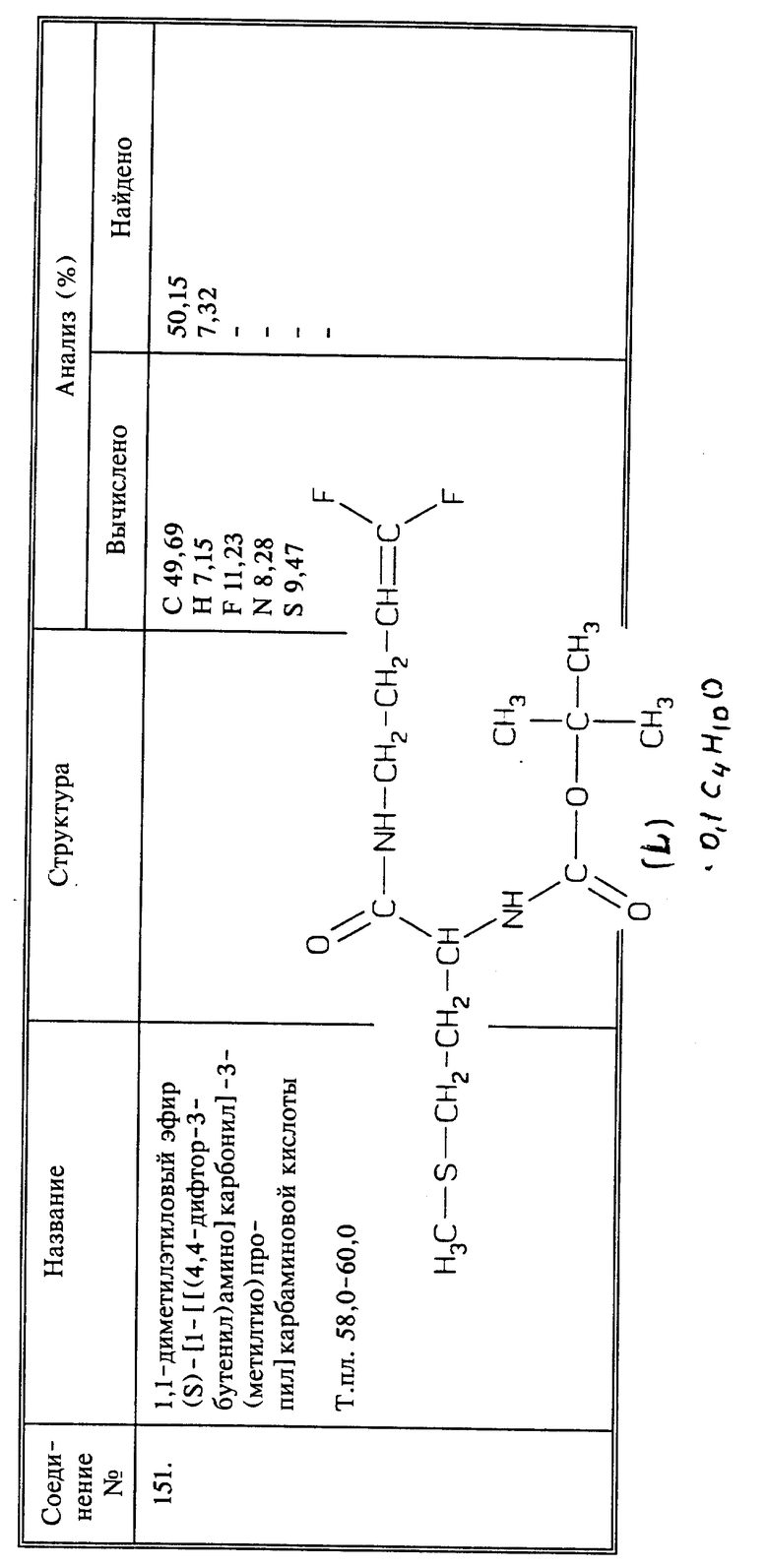
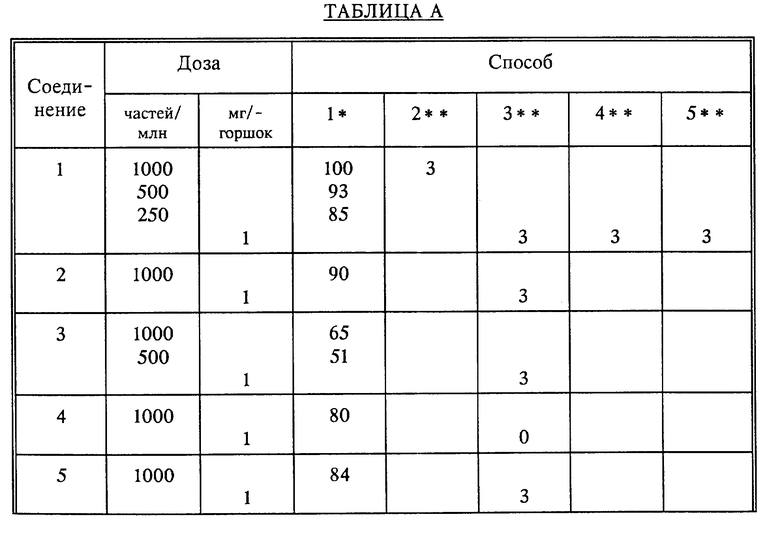
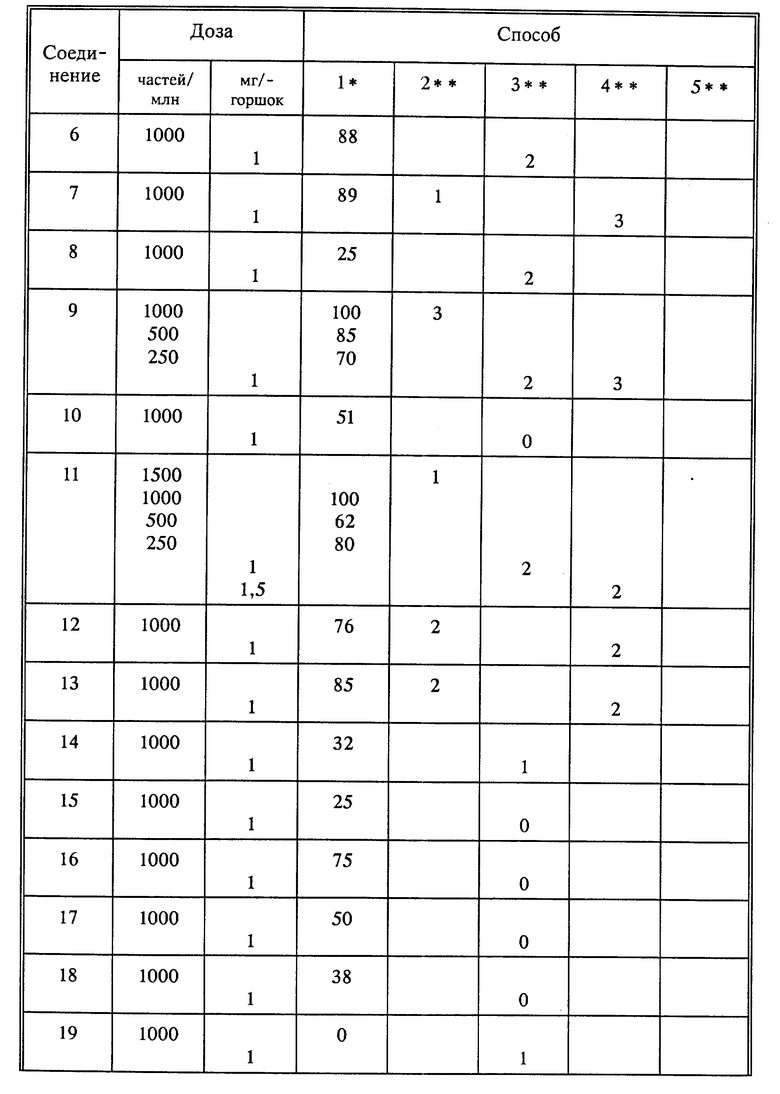
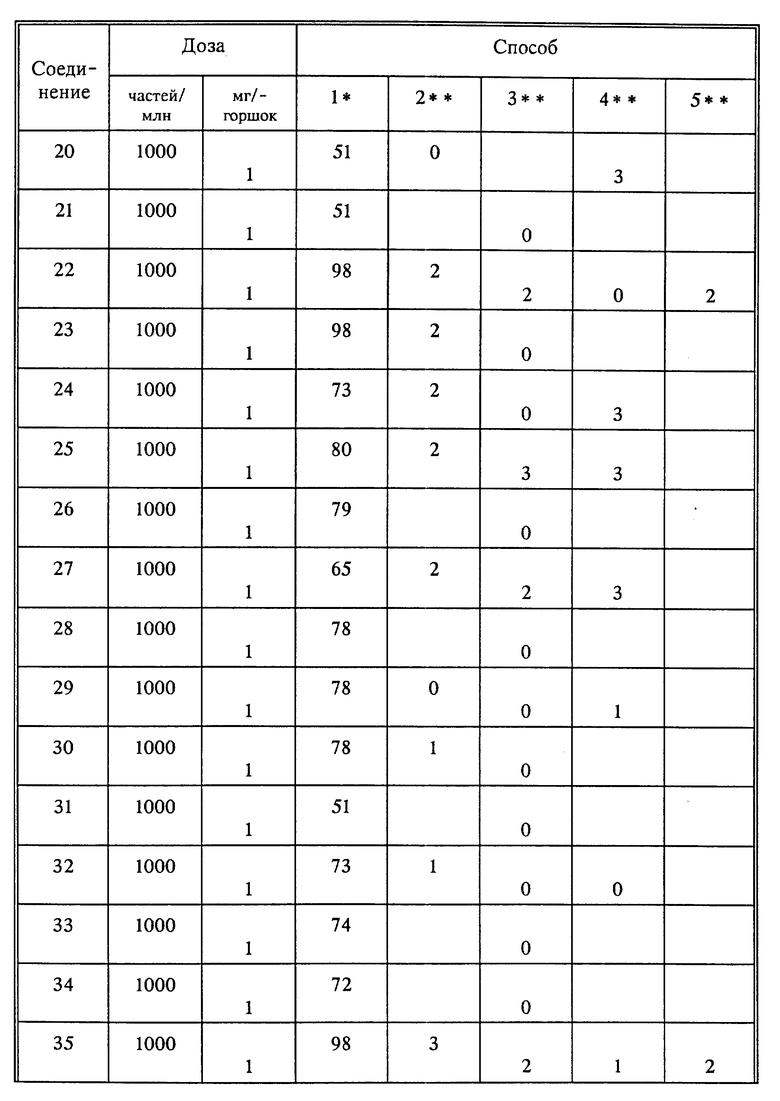
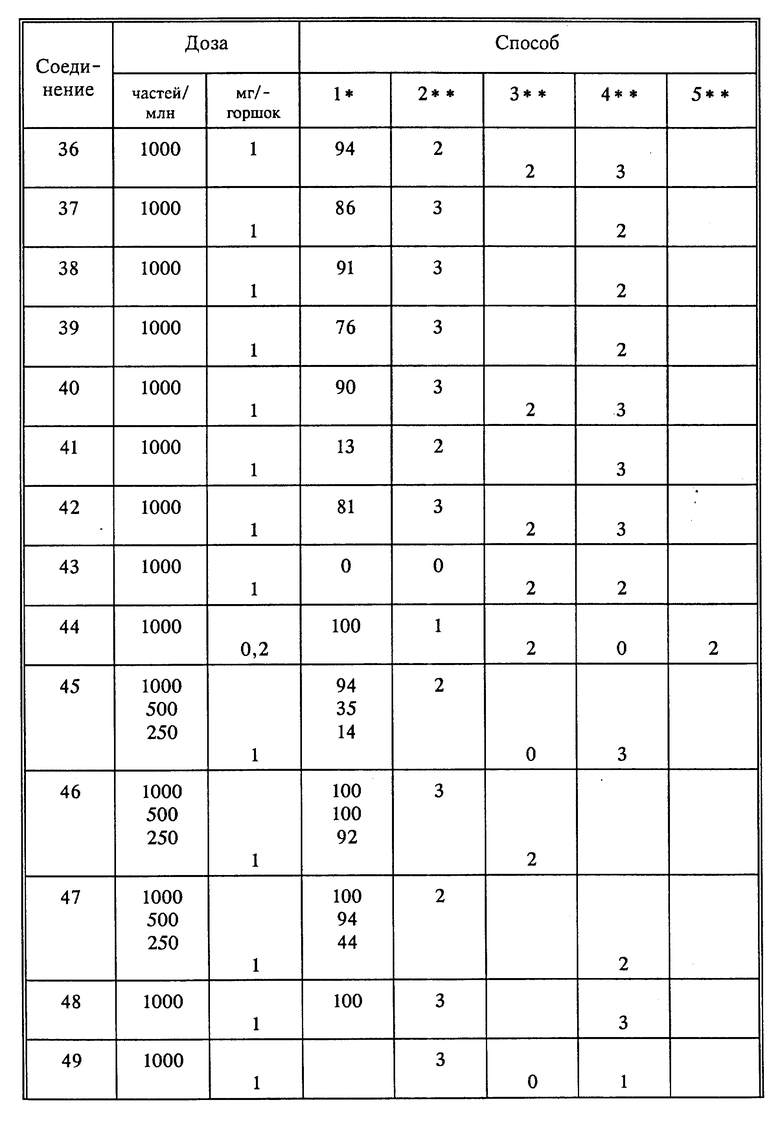
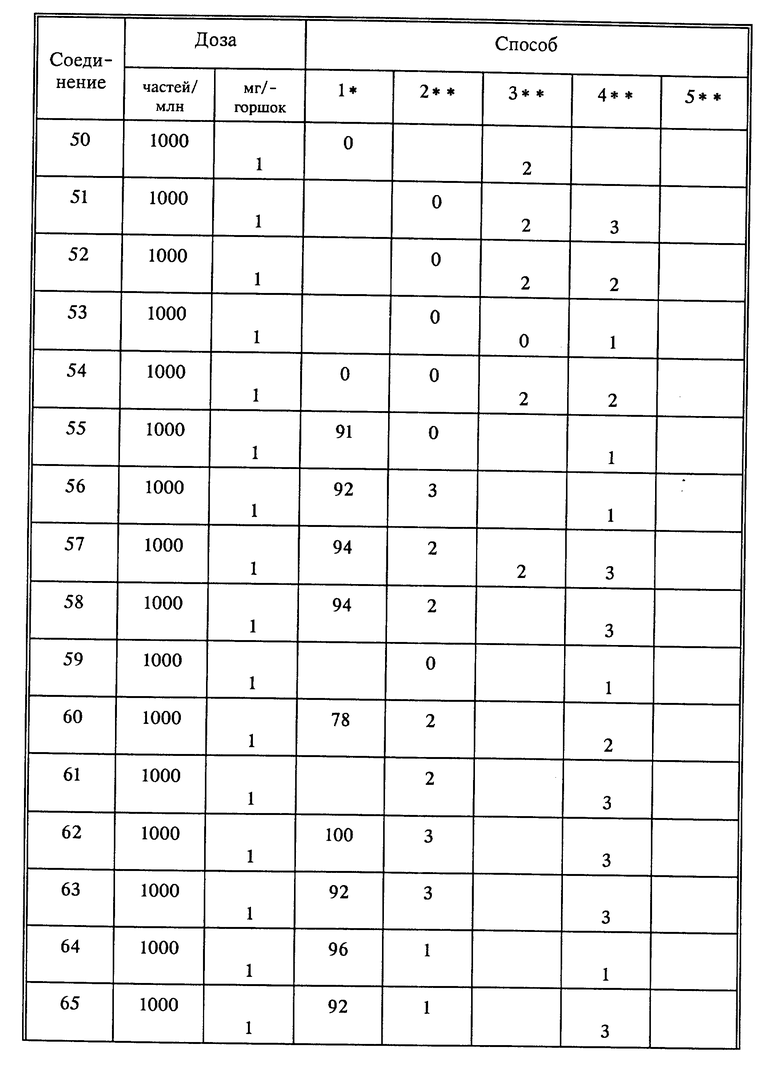
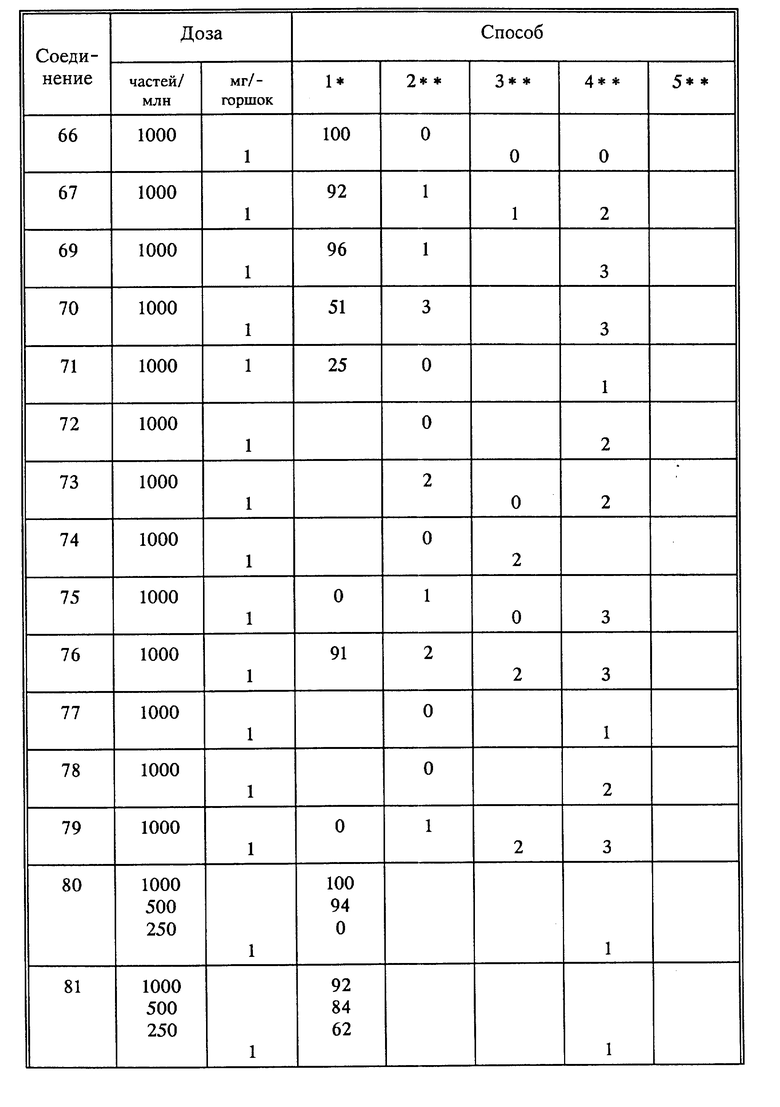
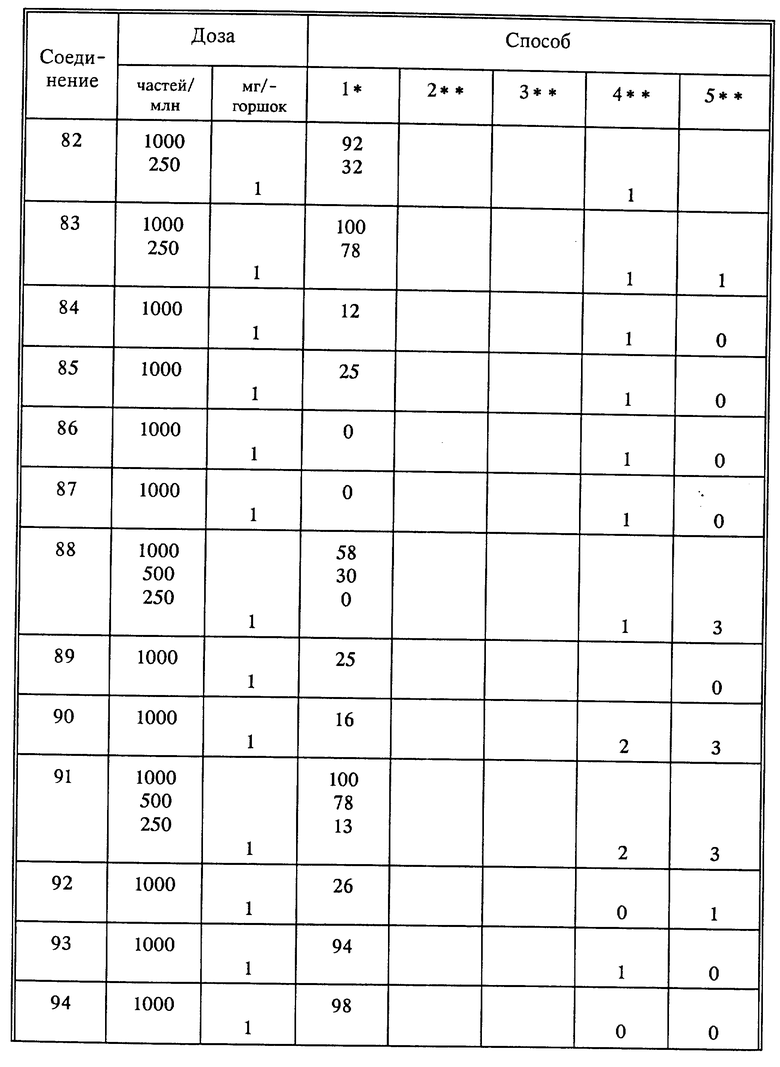

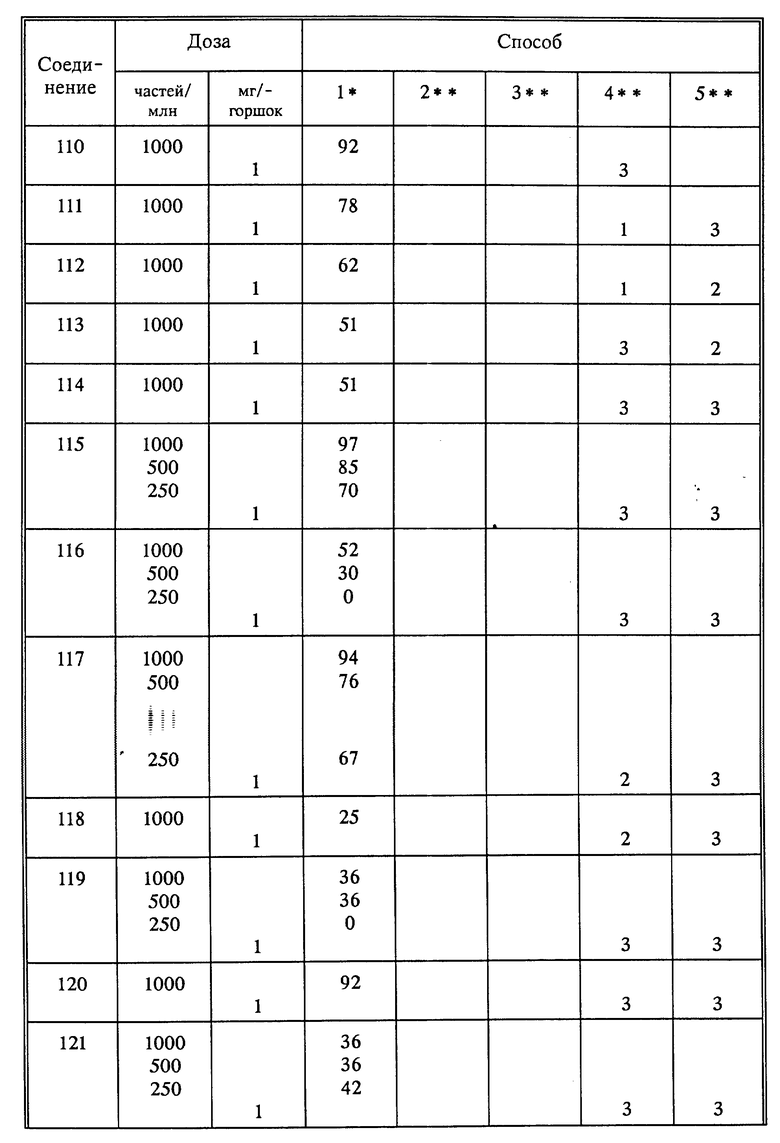
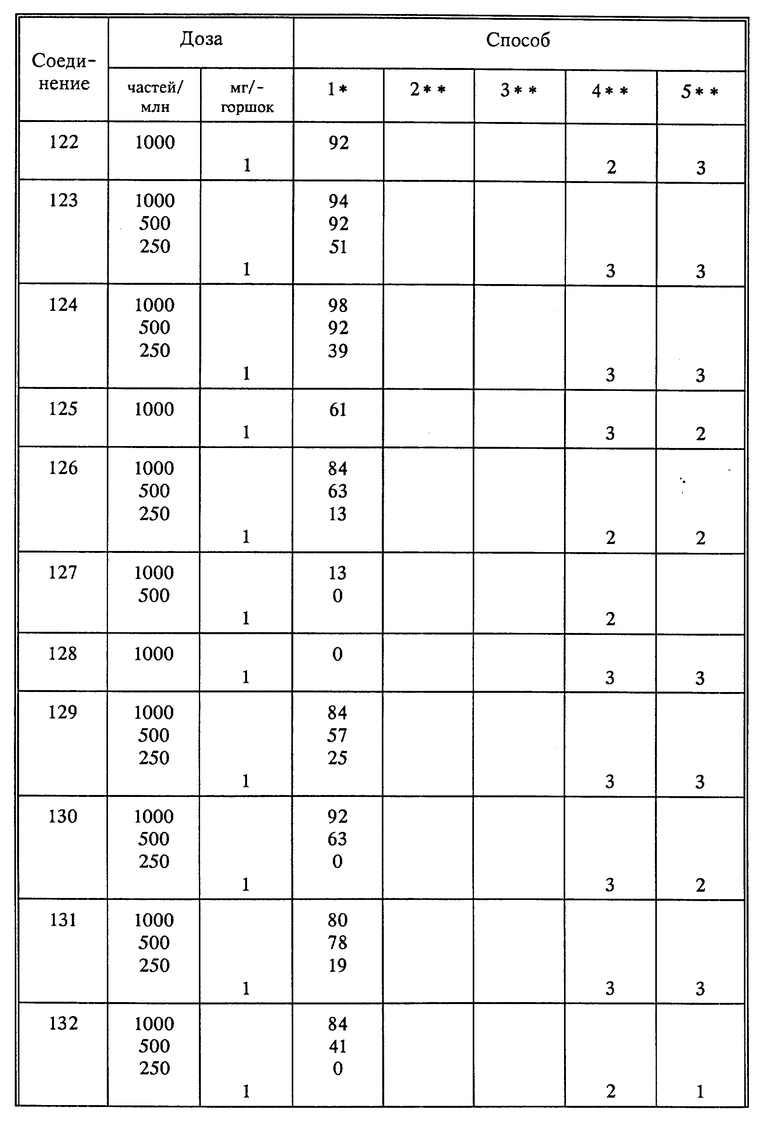
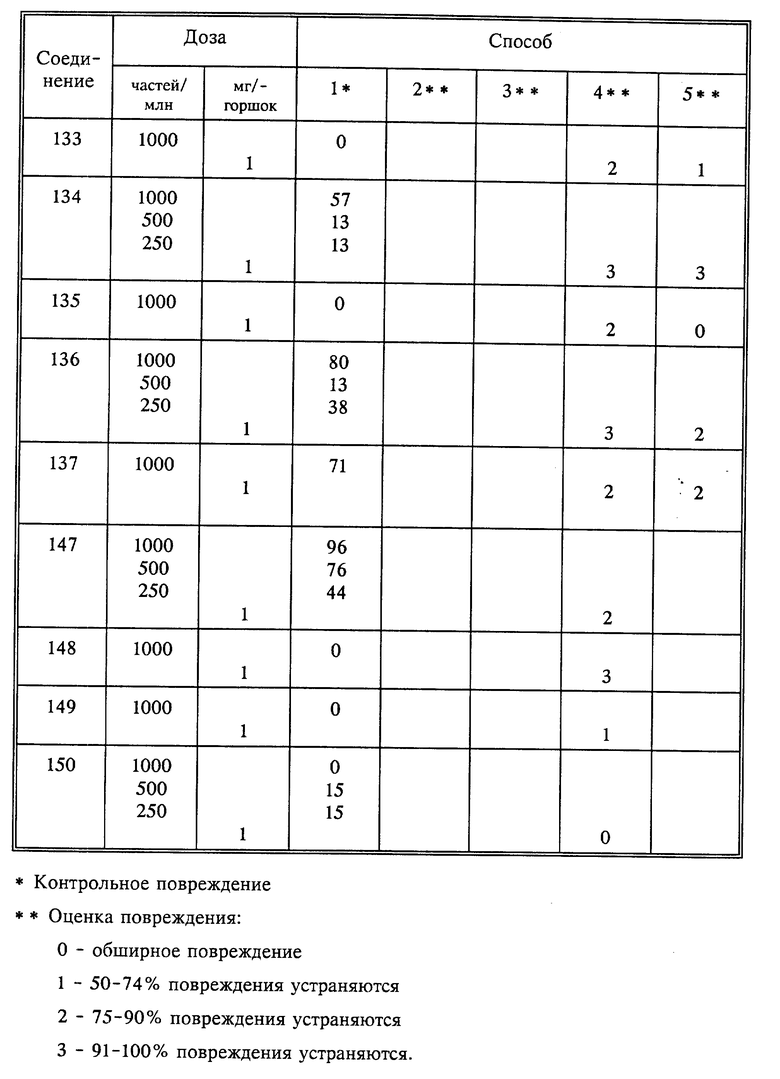

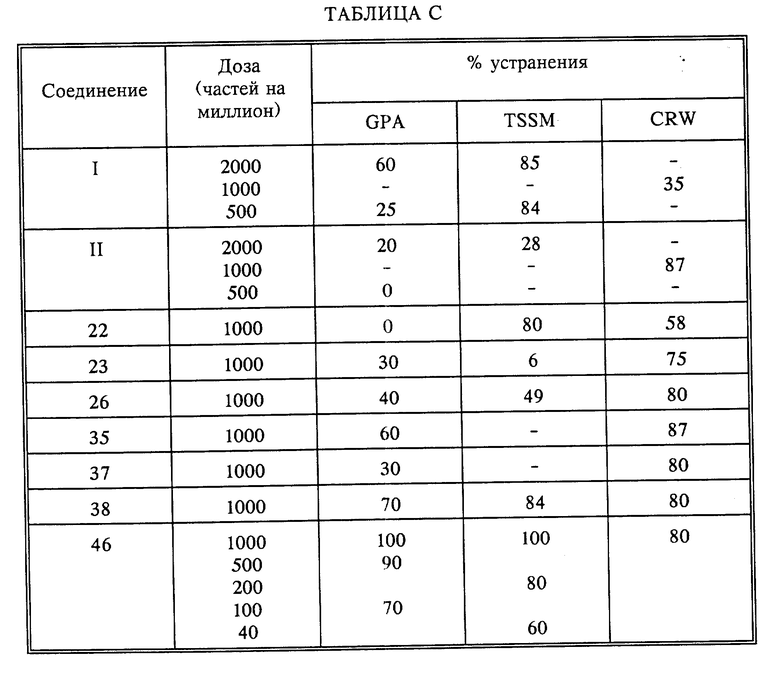
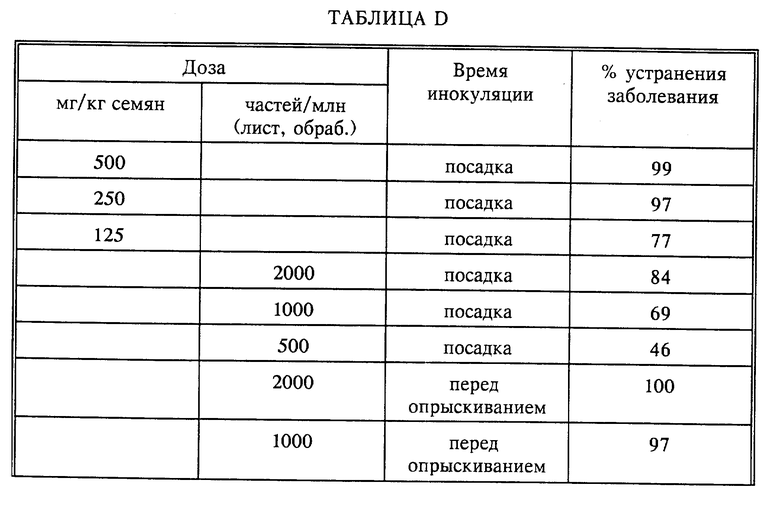

Изобретение относится к фторалкенильным соединениям общей формулы (X)(Y)C=C(Z)-(CH2)n-Q(I), где X и Y-фтор; Z - водород, фтор; n = 1, 3, 5, 7, 9, 11; Q-CH2NHR6, СH2NO2, CHN=CHR2,CY2N=C=O, CH2N+R3R4R5W-, (C=O)-R11, при условии, что если X и/или Y - фтор, а Q-(C=O)-R11, каждый X, Y, Z - фтор, а n = 1, W-анион минеральной или органической кислоты; R2- возможно замещенный фенил, R3, R4, R5-водород и др., R6-водород, (C1-C6)-алкил и др., R11-галоген, NHOH и др., и к их сельскохозяйственно приемлемым солям. Изобретение относится также к композиции для борьбы с заражением растений нематодами, содержащей соединение формулы I, к способу борьбы с заражением растений нематодами, который включает обработку локуса растения соединением формулы I, а также к способу системного подавления заражения растений нематодами путем обработки растений соединением формулы I. 4 с. и 23 з.п. ф-лы, 5 табл.
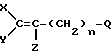
где X и Y - фтор;
Z - водород или фтор;
n = 1, 3, 5, 7, 9 или 11;
Q - CH2NHR6, CH2NO2, CH2N = CHR2, CH2N = C = O, CH2N+R3R4 R5W- или (C = O) - R11, при условии, что когда Q - (C = O) - R11, Z - фтор, а n = 1, W- - анион минеральной или органической кислоты;
R2 - фенил, необязательно замещенный по крайней мере одной группой, выбранной из гидроксила, нитро, галогена, ди(C1 - C4)алкиламино, или пиразолил;
R3 - R5 - водород или взятые вместо с атомом азота группы Q образуют пирролидинил или тетраазотрицикло-[3,3,1,1-(суперскрипт-3-суперскрипт-7)]декан;
R6 - водород, (C1 - C8)алкил, необязательно замещенный амино- или трет-бутоксикарбониламиногруппой, (C1 - C5)алкил-COOH, необязательно замещенный карбоксильной группой, (C1 - C5)алкил-CONH2, (C1 - C5)алкил-COO(C1 - C4)-алкил, (C1 - C5)алкил-COO(C1 - C4)алкилфенил, (C1 - C6)алкенил, замещенный галогеном, дигидро-3-оксопиразолидинил, тиенил и фенил, необязательно замещенный карбоксилом или (C1 - C4)алкоксикарбонилом, (C = O)R7, где R7 - (C = O)R14, (C1 - C6)алкил, необязательно замещенный по крайней мере одной группой, выбранной из ряда галоген, аминогруппа или (C1 - C6)циклоалкил, трет-бутоксикарбониламиногруппа, гидроксил, фенил, (C1 - C4)алкилтиогруппа, (C1 - C5)алкил-COOH или его эфиры, необязательно замещенные аминогруппой, (C1 - C5)алкил-CONH2, необязательно замещенный аминогруппой, (C1 - C6)алкенил, замещенный галогеном, или N-содержащая группа, которая, взятая вместе с карбоксамидом, представляет собой остаток мочевины, необязательно замещенная (C1 - C4)-алкилом, возможно замещенным гидроксилом, или R6 взятый вместе с азотом группы Q представляет собой гидразин, гуанидин, трифторметилфенилсульфонамид;
R11 - галоген, NHOH, OR12, SR12′,NR12″R13, где R12 - водород, (C1 - C8)алкил, необязательно замещенный аминогруппой или фенилом, фенил, необязательно замещенный нитрогруппой;
R12′- (C1 - C8)алкил, необязательно замещенный аминогруппой или фенилом, фенил, необязательно замещенный карбоксилом;
R12″- - водород, (C2 - C6)алкил;
R13 - водород, (C2 - C4)алкенилCOOH, (C1 - C6)алкил, необязательно замещенный по крайней мере одной группой, выбранной из (C1 - C4)алкоксикарбонила, амина, фенила, (C2 - C6) алкилCOOH необязательно замещенный карбоксилом или фенилом, (C1 - C6)алкиламин, замещенный карбоксилом или (C1 - C4)алкоксикарбонилом, фенил, необязательно замещенный карбоксилом;
или R12" и R13 вместе с атомом азота группы NR12″R13 образуют остаток аминокислоты природного белка, выбранный из ряда глицил, валил, аланил, аспарагил, фенил аланил, пролил, лизил, а также морфолиногруппу, пирролидинил или пиразолонил, возможно замещенный карбоксилом;
R14 - OH, (C1 - C6)алкокси, NH2 или NHNH2,
или их сельскохозяйственно приемлемые соли, при условии, что когда n = 1, а X, Y и Z - фтор, Q не является CH2NH2, или ее минеральной солью.
3. Соединение по п.1, отличающееся тем, что n = 1, X и Y - фтор.
5. Соединение по п.3, отличающееся тем, что Z - фтор.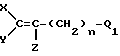
где n = 1, 3, 5, 7, 9 или 11;
Q1 - CH2NHR6, (C = O) - R11 или CH2N+H3W-;
X, Y и Z каждый - водород или фтор, или по крайней мере один из X и Y - фтор, при условии, что когда Q1 - (C = O) - R11, Z - фтор и n = 1;
R6 - водород, (C1 - C8)алкил, необязательно замещенный амино- или трет-бутоксикарбониламиногруппой, (C1 - C5)алкил-COOH, необязательно замещенный карбоксильной группой; (C1 - C5)алкил-CONH2, (C1 - C5)алкил-COO(C1 - C4)алкил, (C1 - C5)-алкил-COO(C1 - C4)алкилфенил, (C1 - C6)алкенил, замещенный галогеном, дигидро-3-оксопиразолидинил, тиенил и фенил, необязательно замещенный карбоксилом или (C1 - C4)алкоксикарбонилом, (C = O)R7, где R7 - (C = O)R14, (C1 - C6)алкил, необязательно замещенный по крайней мере одной группой, выбранной из ряда, галоген, аминогруппа или (C1 - C6)-циклоалкил, трет-бутоксикарбониламиногруппа, гидроксил, фенил, (C1 - C4)алкилтиогруппа, (C1 - C5)алкил-COOH или его эфиры, необязательно замещенные аминогруппой, (C1 - C5)алкил CONH2, необязательно замещенный аминогруппой, (C1 - C6)алкенил, замещенный галогеном, или N-содержащая группа, которая, взятая вместе с карбоксамидом, представляет собой остаток мочевины, необязательно замещенная (C1 - C4)алкилом, возможно замещенным гидроксилом, или R6, взятый вместе с азотом группы Q1 представляет собой гидразин, гуанидин, трифторметилфенилсульфонамид;
R11 - галоген, NHOH1, OR12, SR12′,NR12″R13, где R12 - водород, (C1 - C8)алкил, необязательно замещенный аминогруппой или фенилом, фенил, необязательно замещенный нитрогруппой, и имеет полярность, обеспечивающую проникновение соединения во флоэму листа без снижения их нематоцидной активности;
R12′-(C1 - C8)алкил, необязательно замещенный аминогруппой или фенилом, фенил, необязательно замещенный карбоксилом;
R12″-водород, (C1 - C6)алкил;
R13 - водород, (C1 - C4)алкенилCOOH, (C1 - C6)алкил, необязательно замещенный по крайней мере одной группой, выбранной из (C1 - C4)алкоксикарбонила, амина, фенила, (C2 - C6)алкилCOOH, необязательно замещенный карбоксилом или фенилом, (C1 - C6)алкиламин, замещенный карбоксилом или (C1 - C4)алкоксикарбонилом, фенил, необязательно замещенный карбоксилом, или
R12″и R13 вместе с атомом азота группы NR12″R13 образуют остаток аминокислоты природного белка, выбранной из ряда глицил, валил, аланил, аспарагил, фенилаланил, пролил, лизил, либо морфолиногруппу, пирролидинил или пиразолонил, возможно замещенный карбоксилом;
W- - анион минеральной или органической кислоты,
или их сельскохозяйственно приемлемые соли в эффективном количестве.
Приоритет по пунктам:
01.03.91 по пп.1, 2 - 12, 14, 16, 18 - 25;
03.02.92 по пп.1, 13, 15, 17, 26, 27.
| US, патент, 4952580, C 07 D 285/13, 1990 | |||
| EP, патент, 0432861, C 07 C 69/618, 1991. |
