Область техники
Настоящее изобретение относится к гербицидным соединениям, в общем определенными родовыми признаками в названии изобретения, к содержащим их составам (композициям) и способам получения указанных соединений.
Предпосылки создания изобретения
В литературе известны различные соединения производные замещенного 3-арил- и 5-арилпиразола. Такие соединения имеют различную полезность, например применяются к качестве химических промежуточных соединений, фармацевтических средств и гербицидов.
Среди замещенных 3-арил-5-(галоид)алкилпаразолов и 5-арил-3-(галоид)алкилпаразолов в данной области техники известны соединения, имеющие различные заместители-радикалы в арильно и/или пиразольном фрагментах соединения, например алкил, карбоксил, алкоксикарбонил, формил, фенил и фенил, замещенный различными группами, такими как алкил, галоид- или нитрогруппы и т.д. Например, известны соединения этого типа, в которых арильная часть представляет собой замещенный или незамещенный фенильный радикал, в котором заместителями являются алкил, циклоалкил, трифторметил и т.д., а радикал пиразола замещен в разных положениях у атомов азота или углерода алкилом, галогеном, алкоксилом, гетероциклами, S(O)nR-группами, в которых n есть 0-2, а R может быть различными радикалами, такими как радикалы, замещенные в арильной или пиразольной части.
Соединения вышеуказанного типа, имеющие полезность как гербициды, обычно требуют норм расхода в 5 или 10 или более килограмм на гектар для достижения адекватных результатов в борьбе с сорняками. В соответствии с этим целью настоящего изобретения является предложение нового класса соединений типа арилпиразола, имеющих уникально высокую единичную фитотоксическую активность против спектра сорняков, включающего узколиственные и широколиственные сорняки, при высокой степени безопасности для многих видов культур, в особенности мелкозернистых хлебов и/или пропашных культур, таких как пшеница, ячмень, кукуруза, соя-бобы, земляные орехи и т.д.
Описываемые здесь 1-(гало)алкил-3-(замещенный)арил-4-гало-5-галоалкпиразолы и 1-(гало)-5-(замещенный)5-арил-4-гало-3-галоалкилпиразолы являются новыми.
Краткое изложение сущности изобретения
Настоящее изобретение относится к гербицидно активным соединениям, составам, содержащим эти соединения, способам получения этих соединений и способам применения их в качестве гербицидов.
Гербицидные соединения согласно настоящему изобретению включают соединения, характеризуемые структурой формулы I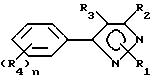
и их сельскохозяйственно-приемлемые соли и гидраты, в которых
R1 представляет собой независимо C1-8-алкил, C3-8-циклоалкил, циклоалкенил, циклоалкилалкил или циклоалкенилалкил; C2-8-алкенил или алкинил, бензил, в которых вышеуказанные члены могут быть произвольно замещены галогеном, амино, нитро, циано, гидрокси, алкокси, алкилтио,
CYR8, -CR9, YR10 или NR11R12,
R2 представляет собой C1-5-галоалкил,
R3 представляет собой галоген
R4 представляет собой R1-член, тиоалкил, алкоксиалкил или полиалкоксиалкил, карбамил, галоген, амино, нитро, циано, гидрокси, C1-10-гетероцикл, содержащий 1-4 O, S(O)m и/или NR18-гетероатомов, C6-12-арил аралкил или алкарил, 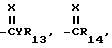 YR15 или NR16R17-группу. Любые две R4-группы могут быть объединены посредством насыщенной и/или ненасыщенной углеродной, -(C=X)- и/или гетеро O, S(O)m и/или NR18-связи с образованием циклического кольца, имеющего до 9 членов в кольце, и которое может быть замещено любым из R4-членов,
YR15 или NR16R17-группу. Любые две R4-группы могут быть объединены посредством насыщенной и/или ненасыщенной углеродной, -(C=X)- и/или гетеро O, S(O)m и/или NR18-связи с образованием циклического кольца, имеющего до 9 членов в кольце, и которое может быть замещено любым из R4-членов,
X есть O, S(O)m, NR19 или CR20R21,
Y есть O, S(O)m или NR22,
R8-22представляет собой водород или один из R4-членов
m есть 0-2 и
n есть 1-5.
Предпочтительным подклассом замещенных арилпиразольных соединений в настоящем изобретении являются соединения соответствующие формуле II: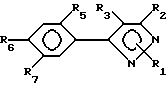
и их сельскохозяйственно-приемлемые соли и гидраты, в которых:
R1 представляет собой C1-15-алкил, алкилтио, алкоксиалкил, C2-4-алкенил, бензил, которые возможно замещены галогеном, амино, нитро, циано, гидрокси или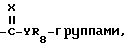
R2, R3, X, Y и R8 имеют значения, указанные для формулы I,
R5 представляет собой галоген или водород,
R6 и R7 имеют значения, указанные для R4-члена в формуле I.
Особо предпочтительными соединениями настоящего изобретения являются соединения согласно формуле III: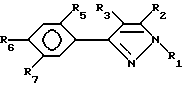
и их сельскохозяйственно-приемлемые соли и гидраты, в которых:
R1 представляет собой C1-5-алкил
R2, R3 и R5 имеют значения, указанные выше,
R6 представляет собой галоген, нитро, циано, YR10,
R7 представляет собой водород или R4-член и
R6 и R7 объединены посредством насыщенной и/или ненасыщенной углеродной, -(C= X)- и/или гетеро O, S(O)m и/или NR18-связи с образованием циклического кольца, имеющего до 9 членов кольца, и которое может быть замещено любым из R4-членов, при условии, что когда указанная связь содержит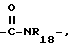 указанное циклическое кольцо имеет по меньшей мере шесть членов кольца, и
указанное циклическое кольцо имеет по меньшей мере шесть членов кольца, и
X, Y, R18 и m имеют вышеуказанные значения.
Еще более предпочтительны соединения в соответствии с формулой III и их сельскохозяйственно-приемлемые соли и гидраты, в которых:
R1 представляет собой метил,
R2 представляет собой CF3, CF2Cl или CF2H,
R3 представляет собой хлор или бром,
R5 представляет собой фтор,
R6 представляет собой хлор,
R7 представляет собой пропаргилокси, аллилокси, полиалкокси, OCH(R23)COR24, где R23 есть водород, метил или этил, а R24 есть YR10 или NR11R12,
R6 и R7 объединены посредством -OCH2(C=O)N(R18)-связи с образованием конденсированного шестичленного кольца и
Y, R10-R12 и R18 имеют вышеуказанное значение.
Предпочтительные конкретные соединения в соответствии с настоящим изобретением включают следующие:
4-хлор-3-(4-хлор-2-фтор-5-пропаргилоксифенил)-1-метил-5- (трифторметил)-1H-пиразол,
2-(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)- 4-фторфенокси)пропановая кислота, этиловый эфир,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4- фторфенокси)уксусная кислота, 1-метилэтиловый эфир,
4-хлор-3-(4-хлор-2-фтор-5-(метоксиметокси)фенил)-1-метил-5- (трифторметил)-1H-пиразол,
4-хлор-3-(4-хлор-2-фтор-5-(метоксиэтокси)фенил)-1-метил-5- (трифторметил)-1H-пиразол,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)- 4-фторфенокси)уксусная кислота, 1,1-диметилэтиловый эфир,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)- 3-фторфенокси)уксусная кислота,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)- 4-фторбензойная кислота, 2-этокси-1-метил-2-оксоэтиловый эфир,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)- 4-фторбензойная кислота, 2-метокси-1-метил-2-оксоэтиловый эфир,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)- 4-фторбензойная кислота, этиловый эфир,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)- 4-фторбензойная кислота, 1-метилэтиловый эфир и
6-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-7-фтор- 4-(2-пропинил)-2H-1,4-бензоксазин-3-(4H)-он.
Хотя все вышеприведенные соединения проявили высокую эффективность при использовании их на большом количестве культур, испытания, проведенные на настоящее время, показывают, что наибольший интерес представляют соединения NN 135, 137, 261, 282 и 446. Эти соединения коллективно обеспечивают прекрасную защиту от таких устойчивых широколистных сорняков, как щирица, дурнишник, канатник Теофраста и конопля (hemp sesbania) в различных культурах, таких как кукуруза, соя-бобы и орехи и в лесном хозяйстве против деревьев и вьющихся растений. Другие из соединений согласно настоящему изобретению проявляют прекрасные гербицидные свойства против сорняков в других культурах, таких как пшеница и ячмень.
Некоторых из соединений согласно настоящему изобретению могут иметь более одного возможного стереоизомера, и эти стереоизомеры могут разливаться в их гербицидной эффективности. Показанные структуры показаны с намерением включать все возможные стереоизомеры.
Вышеприведенные соединения допускают различный режим их применения, например довсходовая и/или послевсходовая обработка, поверхностное нанесение, предпосевное внедрение и т.д.
Другой аспект настоящего изобретения относится к способам получения соединений согласно формулам I - III и их предшественников, промежуточных и/или исходных соединений. Эти аспекты будут более подробно обсуждены ниже.
Другие аспекты настоящего изобретения относятся к гербицидным составам, содержащим соединения формулы I - III и к способам применения этих составов в качестве средств уничтожения нежелательных сорняков.
Далее, к области настоящего изобретения относится то, что замещенные арилпиразольные соединения согласно формулам I - III могут быть включены в рецептуру составов, содержащих другие гербицидные соединения в качестве согербицидов, например ацетамиды, особенно ацетанилиды, тиокарбаматы, мочевины, сульфонилмочевины, сульфонамиды, имидазолиноны, бензойная кислота и ее производные, дифениловые эфиры, соли глифосатов и т.д.
В такие гербицидные рецептуры могут быть включены другие добавки как желательные и подходящие, например антидоты (добавки, повышающие безопасность) для гербицидов, средства предупреждения болезней растений, такие как фунгициды, инсектициды, нематоциды и другие пестициды.
Используемые здесь термины "алкил", "алкенил", "алкинил", взятые отдельно или в составной форме, например галоидалкил, галоидалкенил, алкокси, алкоксиалкил и т. д. охватывают как линейные, так и соединения с разветвленной цепью. Предпочтительные представители алкилов - это низшие алкилы, имеющие от 1 до 4 углеродных атомов, а предпочтительными представителями алкенилов и алкинилов являются таковые, имеющие от 2 до 4 углеродных атомов.
Термин галоидалкил (галоалкил) употреблен для обозначения алкильных радикалов, замещенных одним или более чем одним атомом галогена (хлор, бром, иод или фтор), предпочтительными членами этого класса являются те соединения, которые имеют от 1 до 4 углеродных атомов, в особенности галоидметильные радикалы, например трифторметил. В полигалоидалкилах галогены могут быть одинаковыми или смешанными.
Не исчерпывающий перечень представителей алкильных, алкенильных, алкинильных, циклоалкильных, циклоалкилалкильных, циклоалкенильных и циклоалкенилалкильных радикалов включает:
метил, этил, изомерные пропилы, бутилы, пентилы, гексилы, гептилы, октилы, нонилы, децилы и т.д.; винил, аллил, кротил, металлил, изомерные бутенилы, пентенилы, гексенилы, гептенилы, октенилы; этинилы, изомерные пропинилы, бутинилы, пентинилы, гексинилы и т.д.; алкокси-, полиалкокси-, алкоксиалкил- и полиалкоксиалкильные аналоги вышеприведенных алкильных групп, например метокси, этокси, пропоксилы, бутоксилы, петоксилы и гексоксилы и соответствующие полиалкоксилы и алкоксиалкилы, например метоксиметокси, метоксиэтокси, этоксиметокси, этоксиэтокси, метоксиметил, метоксиэтил, этоксиметил, этоксиэтил, пропоксиметил, изопропоксиметил, бутоксиметил, изобутоксиметил, трет.-бутоксиметил, пентоксиметил, гексоксиметил, и т.д., циклопропил, циклобутил, циклопентил, циклогексил, циклопентил, циклопропилметил, циклобутилметил, циклопентилметил и т.д.; изомерные циклопентены, циклогексены и циклогептены, имеющие моно- или ди-ненасыщенность; представители арильных, аралкильных и алкарильных групп включают фенил, изомерные толилы и ксилилы, бензил, нафтил и т.д.
Представители моно-, ди- и три-галоидалкилов включают: хлорметил, хлорэтил, бромметил, бромэтил, иодметил, иодэтил, хлорпропил, бромпропил, иодпропил, 1,1-дихлорметил, 1,1-дибромметил, 1,1-дихлорпропил, 1,2-дибромпропил, 2,3-дибромпропил, 1-хлор-2-бромэтил, 2-хлор-3-бромпропил, трифторметил, трихлорметил, и т.д.
Представители гетероциклических соединений включают: алкилтиодиазолил, пиперидил, пиперидилалкил, диоксоланилалкил, тиазолил, алкилтиазолил, бензотиазолил, галоидбензотиазолил, фурил, алкилзамещенный фурил, фурилалкил, пиридил, алкилпиридил, алкилоксазолил, тетрагидрофурилалкил, 3-циантиенил, тиенилалкил, алкилзамещенный тиенил, 4,5-полиалкилен-тиенил, пиперидинил, алкилпиперидинил, пиридил, ди- или тетрагидропиридинил, алкилтетрагидроморфолил, алкилморфолил, азабициклононил, диазабициклоалканил, бензоалкилпирролидинил, оксазолидинил, пергидрооксазолидинил, алкилоксазолидил, фурилоксазолидинил, тиенилоксазолидинил, пиридилоксазолидинил, пиримидинилоксазолидинил, бензооксазолидинил, C3-7-спироциклоалкилоксазолидинил, алкиламиноалкенил, алкилиденимин, пирролидинил, пиперидонил, пергидроазепинил, пергидроазоцинил, пиразолил, дигидропиразолил, пиперазинил, пергидро-1,4-диазепинил, хинолинил, изохинолинил, ди-, тетра- и пергидрохинолил- или -изохинолил, индолил и ди- и пергидроиндолил и указанные гетероциклические члены, замещенные радикалами, такими как радикалы, определенные в формулах I - III.
Под "сельскохозяйственно-приемлемыми солями" соединений, определенных вышеуказанными формулами, имеется в виду соль или соли, которые легко ионизируются в водной среде, образуя катион или анион указанных соединений и соответствующий солевой анион и катион, причем соли не уменьшают гербицидных свойств данного гербицида и позволяют приготовлять различные смеси, например гербицидно-антидотные композиции без ненужных проблем смешения, суспензии, стабильности, применения оборудования по нанесению, упаковки и т.д.
Под "гербицидно эффективным" имеется в виду количество гербицида, требуемое для существенного повреждения или разрушения значительной части обработанных нежелательных растений или сорняков. Хотя это и не твердое правило, но с коммерческой точки зрения желательно уничтожение 80 - 85 или более процентов сорняков, однако коммерчески значительное подавление роста сорняков может быть достигнуто и при значительно более низких уровнях, в частности с некоторыми очень вредными, стойкими к гербицидам растениями.
Подробное описание изобретения
Ниже описываются соединения в соответствии с настоящим изобретением, получаемые в ряде способов.
В широком аспекте предпочтительный общий способ получения соединений формул I - III лучше всего может быть рассмотрен по отдельным стадиям способа, требующимся для получения необходимых промежуточных продуктов, непосредственных предшественников и конечных продуктов вышеприведенных формул. Продукты "способа I" являются промежуточными продуктами, необходимыми для "способов II - XVI". Продукты в соответствии с формулами I - III получаются либо одним способом "II - XVI" либо любой комбинацией "способов II - XVI". Следует ясно понимать, что рассматриваются различные модификации, очевидные для специалиста в данной области. Конкретные варианты описываются ниже в примерах 1 - 42.
В последовательности стадий процесса, описываемых ниже, различные символы, определяющие радикалы-заместители, например R1 - R24, X, Y и т.д. имеют те же значения, что и для соединений формулы I - III, если не указано иное.
Способ I.
Этот способ описывает приготовление важных промежуточных соединений формулы B или изомерных смесей их, полезных в общем процессе получения формул I - III.
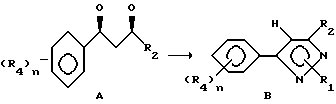
Способ получения соединения формулы B исходит из соединений формулы A. Соединения формулы A готовятся известными средствами из замещенных ацетофенонов, которые также известны из уровня техники. Имеется в виду, что структура, представляемая формулой A, воплощает все возможные таутомерные формы или смеси ее. Соединения формулы A могут быть приготовлены в любом безводном растворителе или смеси растворителей; предпочтительными растворителями являются эфир, спирты, диметилсульфоксид, толуол, бензол, и т.д., реакцией замещенного ацетофенона в присутствии эфира с сильным основанием, таким как алкоксид щелочного металла, амид щелочного металла или гидрид щелочного металла, причем предпочтительны алкоксиды щелочного металла, такие как метоксид натрия. Температура реакции находится в пределах от -100oC до 200oC, предпочтительно от -78oC до 50oC. Период реакции может быть выбран в пределах от нескольких минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. По окончании реакции соединение формулы A выделяют разбавлением реакционной смеси водой, возможно с последующим подкислением водного слоя или, альтернативно, разбавлением реакционной смеси водным раствором кислоты. Затем продукт выделяется методом кристаллизации или экстракции растворителем. Если необходимо, продукт очищается стандартными методами. Циклизация этого промежуточного соединения с образованием соединений формулы B может быть приведена в любом подходящем растворителе действием гидразина или замещенных гидразинов, предпочтительно алкилгидразинами. Температура реакции в пределах от -78oC до 200oC, предпочтительно от 10oC до 120oC. Период реакции может быть выбран в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. Продукт выделяют после окончания реакции путем фильтрации и/или концентрирования реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
В случае добавления гидразина к соединениям формулы A получающийся пиразол формулы C может быть обработан алкилирующим агентом для получения соединений формулы B. В этом случае продукты формулы B могут быть получены обработкой вышеупомянутых соединений алкилирующим агентом, таким как метилиодид, аллилбромид, диметилсульфат и т.д. Предпочтительными растворителями являются толуол, диметилсульфоксид, ацетон, диметилформамид, диоксан, и т.д. Реакция может быть проведена с основанием или без него. В случае применения основания можно применять карбонаты щелочного металла или металла, такие как карбонат натрия или гидроксид натрия. Температура реакции находится в пределах от -78oC до 200oC, предпочтительно 10oC - 120oC. Время реакции может быть выбрано в пределах от немногих минут до нескольких недель в зависимости от количества реагентов, температуры реакции и т.д. Продукт выделяют после окончания реакции фильтрацией и/или концентрированием реакционной смеси. При необходимости продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
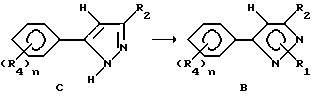
Соединения, представленные формулой C, могут существовать в двух возможных таутомерных формах, либо как 5-арилпиралол либо как 3-арилпиразол. Имеется в виду, что 5-арилпиразол, изображенный формулой C, включает обе возможные таутомерные формы. Таблица 1 показывает типичные примеры соединений формулы C.
Во всех таблицах в данном описании точки кипения и точки плавления измерены в градусах Цельсия (oC) и, если не указано иное, приведенные показатели преломления относятся к 25oC.
2-фтор-4-хлор-5-метоксиацетофенон, используемый для получения соединения NN 3, 4 и 9 по вышеприведенному способу, получали из 2-хлор-4-фторфенола методами, известными из уровня техники (C.A. Buehler and D.E. Pearson, Survey of Organic Synthesis, pages 285 - 382, Wiley - Interscience, New York, 1970). Обработка 2-хлор-4-фторанизола четыреххлористым титаном и дихлорметилметилэфиром при комнатной температуре дает 2-фтор-4-хлор-5-метоксибензальдегид. 2-фтор-4-хлор-5-метоксибензальдегид превращали в 2-фтор-4-хлор-5-метоксиацетофенон обработкой метилом Гриньяра с последующим окислением с использованием стандартных методов, известных из уровня техники.
Вышеупомянутый 2-фтор-4-хлор-5-метоксиацетофенон и его аналог - предшественник 2-фтор-4-хлор-5-метоксибензальдегид и способы получения их составляют предмет изобретения других лиц (Брус С. Хэмпер и Киндрик Л. Лешински), которое использовано здесь правопреемником.
Таблицы 2 и 3 показывают типичные примеры соединений, полученных по способу 1.
Способ II
В данном описании способа один класс продуктов в соответствии с формулой I, в которой R3 представляет собой галоген, получали галогенированием соответствующего соединения формулы B. В данном способе R1 может иметь вышеуказанное значение и включать еще гидроген.
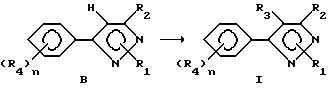
Любой инертный растворитель может быть использован в этой реакции, который заметно не препятствует протеканию реакции или реакция может проводиться без растворителя. Такие растворители включают, но не ограничены ими, органические кислоты, неорганические кислоты, углеводороды, галогенированные углеводороды, ароматические углеводороды, эфиры и сульфиды, сульфоксиды или сульфоны. Галогенирующие агенты, пригодные для вышеозначенных реакций, включают бром, хлор, N-бромсукцинамид, N-хлорсукцинамид, сульфурилхлорид и т. д. С некоторыми галогенирующими агентами предпочтительно использовать органическую перекись или свет в качестве катализатора. Количество галогенирующего агента может варьировать от менее чем один молярный эквивалент до избытка. Температура реакции лежит в пределах от -100oC до 200oC, предпочтительно от 10oC до 100oC. Время реакции может быть выбрано в пределах от немногих минут до нескольких недель в зависимости от количества реагентов, температуры реакции и т.д. По окончании реакции продукт выделяют разбавлением реакционной смеси водой и затем продукт выделяют методом кристаллизации или экстракцией растворителем. Если необходимо, продукт очищают стандартными методами.
Способ III
Этот раздел описывает способ получения соединений в соответствии с формулой D (соединение формулы I, в котором один из R4-остатков является нитрогруппой) исходя из соединений формулы I.
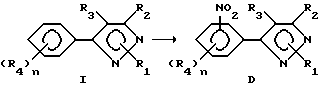
Нитрующие агенты, как то концентрированная азотная кислота, дымящая серная кислота, алкилнитралы и ацетилнитраты, пригодны для этой реакции. Могут быть использованы растворители, как то: минеральные кислоты, органические кислоты, органические растворители, как то: уксусный ангидрид или метиленхлорид и вода или смеси этих растворителей. Нитрирующий агент может быть использован в эквимолярных количествах или в избытке, Температура реакции находится в пределах от -100oC до 200oC, предпочтительно от -10oC до 100oC. Время реакции может быть выбрано в пределах от немногих минут до нескольких дней в зависимости от количества реагентов, температуры реакции и т.д. По окончании реакции продукт выделяют разбавлением реакционной смеси водой и продукт выделяют такими методами, как кристаллизация или экстракция растворителем. Если необходимо, продукт очищают стандартными методами.
Способ IV
В описании этого способа один класс продуктов в соответствии с формулой F (разновидность соединений формулы II) получали замещением Z-радикала соответствующего соединения формулы E, в которой Z является любой подходящей покидающей группой вышеуказанных R4-членов.
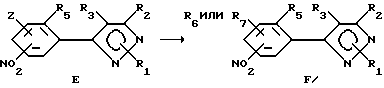
Образование продуктов формулы E может быть достигнуто обработкой соединений формулы D алкоксидом, тиоалкоксидом, амином и т.д. или спиртом, меркаптаном, амином и т.д. в присутствии основания в любом подходящем растворителе или смеси растворителей. Предпочтительные растворители - диметилсульфоксид, ацетон, диметилформамид, диоксан, вода и т.д. или смеси растворителей, включая двухфазные смеси (такие как вода и метиленхлорид или другой органический растворитель). Основанием может быть органическое основание (такое как триалкиламин или другой органический амин) или неорганическое основание (карбонат щелочного металла, как то: карбонат калия или натрия или гидроокись щелочного металла, такая как гидроокись натрия). В случае двух несмешивающихся жидких фаз желательно добавлять катализатор фазового переноса, такой как галоидный бензилтриалкиламмоний или другую аммониевую соль. Температура реакции находится в пределах от -100oC до 200oC, предпочтительно от -10oC до 100oC. Время реакции может быть выбрано в пределах от немногих минут до нескольких недель в зависимости от количества реагентов, температуры реакции и т.д. Продукт выделяют после окончания реакции путем фильтрации и/или концентрации реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
Способ V
При описании этого способа соединения формулы I получаются из соединений формулы G (соединения формулы I, в которой один из R4-членов является нитроостатком).
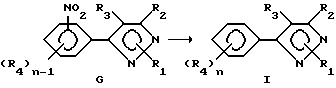
А. На первой стадии этого двухстадийного способа соединения в соответствии с формулой G восстанавливаются с получением производного в соответствии с формулой I, где один из R4-радикалов является аминогруппой. Восстановители, пригодные в кислой среде, включают, но не ограничены этим, металлы, такие как железо, цинк или олово. Растворитель для реакции может включать либо органические, либо неорганические кислоты, такие как уксусная кислота или хлористоводородная кислота, и может быть использован в виде концентрированных растворов кислот или разбавленных водных растворов. Температура реакции находится в пределах от 0oC до 200oC, предпочтительно от 10oC до 120oC. Время реакции может быть выбрано в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д.
По окончании реакции продукт выделяют разбавлением реакционной смеси водой и продукт выделяют такими методами, как кристаллизация или экстракция растворителем. Если необходимо, продукт очищают стандартными методами.
Альтернативно соединения формулы G могут быть восстановлены каталитическим гидрированием. Для каталитического гидрирования, которое может проводиться при атмосферном или повышенном давлении, подходящие катализаторы включают никель Ренея, палладированный уголь, палладиевую чернь, палладий на любом подходящем носителе, окись палладия, платину, платиновую чернь и т.д. Растворители включают любой инертный растворитель, который заметно не препятствует реакции, включая спирты, эфиры и т.п. Продукт выделяют фильтрацией и концентрированием реакционной смеси. Если необходимо, продукт очищается стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
Б. Аминовый радикал продукта стадии А может быть превращен в целый ряд функциональных групп, например галоген (предпочтительно), цианогруппу, гидроксил и т.д. В случае конверсии радикала амина в галоген раствор или суспензия продукта стадии А обрабатывается медными солями, включая галогениды меди (II), галогениды меди (I) или другие медные соли и их смеси, и алкилнитритом или органическим нитратом, таким как т-бутилнитрит. В этой реакции может быть применен любой подходящий растворитель, хотя предпочтительны безводные растворители, такие как безводный ацетонитрил. Температура реакции находится в пределах от 0oC до 200oC, предпочтительно от 10oC до 100oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количества реагентов, температуры реакции и т.д. Продукт выделяют после окончания реакции фильтрацией и/или концентрированием реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
Операции альтернативного способа по превращению радикаламина в различные функциональные группы, в том числе упомянутые в предыдущем абзаце, включают использование различных обычных процедур, например реакции Сандмауера, Меервайна и т.д., в которых применяются в качестве промежуточных продуктов соли диазония.
Способ VI
При описании этого способа соединения согласно формуле I, в которой один из R4-членов является YH, готовят из соединений формулы I, в которой один из R4-членов представляет собой YR15, а R15 не является водородом.
Реакция может проводиться как в растворе или суспензии в любом подходящем растворителе, так и без растворителя. Может применяться кислота Льюиса, такая как (но этот перечень не ограничен) BBr3, AlCl3 и т.д., или неорганические или органические кислоты, такие как хлористоводородная, серная, бромистоводородная, уксусная кислоты и т.д. Альтернативно могут быть использованы нуклеофильные реагенты деалкилирования, в том числе триметилсилилиодид, цианидные соли, меркаптидные соли, галогениды щелочных металлов и т. д. Температура реакции находится в пределах от 0oC до 200oC, предпочтительно от 10oC до 100oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количества реагентов, температуры реакции и т.д. Продукт выделяют после окончания реакции фильтрацией и/или концентрированием реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
Способ VII
При описании этого способа соединения в соответствии с формулой I (которая включает соединения формул II и III), в которой один из R4-членов представляет собой YR15, а R15 не является водородом, готовят из соединений в соответствии с формулой I, в которой один из R4-членов представляет собой YH или NR16R17.
В представительных вариантах этого способа образование вышеуказанных продуктов может быть осуществлено обработкой исходных материалов алкилирующими агентом, таким как алкилгалоид или алкилсульфонат, например метилиодид, аллилбромид, пропаргилбромид, метиловый эфир фенилсульфоновой кислоты и т.д. , или ацилирующим агентом. Реакция может быть проведена в любом подходящем растворителе или смеси растворителей, с катализатором или без него, в присутствии или отсутствии основания. Предпочтительными растворителями являются диметилсульфоксид, ацетон, диметилформамид, диоксан и т.д. или смеси растворителей, включая двухфазные смеси (такие как вода и метиленхлорид или другой органический растворитель). В случае двух несмешивающихся жидких фаз желательно добавлять катализатор фазового переноса, такой как бензилтриалкиламмонийгалоид или другую аммониевую соль. Основанием может быть органическое основание (такое как триалкиламин или другой органический амин) или неорганическое основание (карбонат щелочного металла или щелочной металл, такой как карбонат натрия или калия или гидроокись натрия). Температура реакции находится в пределах от 0oC до 200oC, предпочтительно от 10oC до 100oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количества реагентов, температуры реакции и т.д. Продукт выделяют после окончания реакции фильтрацией и/или концентрированием реакционной смеси. При необходимости продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
Способ VIII
Этот способ описывает получение соединений формулы K (формулы II, в которой R7 представляет собой YCH2-p(R25)p-COYR27) из соответствующих соединений формулы. В радикалах R25-27 члены R4 имеют вышеуказанное значение; Y - члены независимо имеют вышеуказанное значение и p представляет собой целое число от 0 до 2.
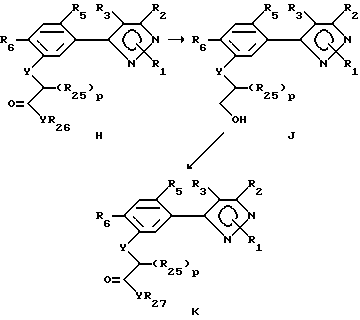
А. На первой стадии этого двухстадийного способа соединения формулы Н превращают в соединения формулы J путем гидролиза радикала YR26. Реакция может проводиться в любом подходящем растворителе или смеси растворителей, с катализатором или без катализатора, в присутствии основания или кислоты. Предпочтительными растворителями являются вода, спирты, диоксан, диметилсульфоксид, уксусная кислота, ацетон, диметилформамид и т.д. В случае щелочного гидролиза предпочтительны неорганические основания, такие как гидроксиды щелочных металлов. В случае кислотного гидролиза могут применяться неорганические кислоты, такие как концентрированная хлористоводородная кислота или серная кислота, органические кислоты или смеси таких кислот. Температура реакции находится в пределах от примерно 0oC до 200oC, предпочтительно от 10oC до 100oC. Период реакции может быть выбран от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т. д. По окончании реакции продукт выделяют разбавлением реакционной смеси водой и/или обработкой раствора кислотой (в случае щелочного гидролиза) и продукт затем выделяют таким методом, как кристаллизация или экстракция. Если необходимо, продукт очищают стандартными методами.
Б. Последняя стадия этого способа включает преобразование соединений формулы J в соединения формулы K с помощью любого из целого ряда стандартных методов получения производных карбоновых кислот. Эта стадия процесса представляет собой этерификацию или реакцию образования амида. Этерификация может быть проведена с использованием избытка спирта в соответствии с целевым эфиром в присутствии минеральной кислоты (например, серной кислоты). Производные амида могут быть получены обработкой соединений формулы K желательным аминоэфиром - без растворителя или в подходящем растворителе. Реакции этерификации или амидообразования могут также быть проведены в присутствии инертного растворителя и дегидратирующего агента.
Альтернативно продукт стадии А может быть превращен в галоидангидрид или ангидрид кислоты и обработан спиртом или амином. Приготовление галоидангидрида кислоты осуществляется в присутствии галогенирующего агента, такого как тионилхлорид, пятихлористый фосфор, оксалилхлорид и т.д. (этот перечень не является исчерпывающим) с растворителями или без растворителя. Может быть использован любой инертный растворитель, не вступающий в реакцию. С целью ускорения этой реакции может быть добавлено каталитическое количество аминного основания, такого как триэтиламин, пиридин или диметилформамид и т.д. Температура реакции находиться в пределах от -20oC до точки кипения используемого растворителя. Продолжительность реакции варьирует от нескольких минут до 48 часов в зависимости от количеств используемых реагентов и температуры реакции. По окончании реакции избыток галогенирующего агента и растворителя (растворителей) удаляют из продукта реакции выпариванием или дистилляцией. Полученный галоидангидрид кислоты может быть подвергнут действию амина или спирта непосредственно или очищен обычными средствами.
Галоидангидрид кислоты обрабатывают спиртом или амином, получая соединение формулы К. Реакция может проводиться в отсутствии растворителя, в присутствии инертного растворителя или смесей растворителей, включая двухфазные смеси (такие как вода и метиленхлорид или другой органический растворитель). С целью ускорения этой реакции может быть прибавлено основание, такое как триэтиламин, пиридин, гидроокись щелочного металла и/или каталитическое количество катализатора фазового переноса, такого как хлористый бензилтриалкиламмоний или другая аммониевая соль. Температура реакции находится в пределах от -20oC до точки кипения применяемого растворителя. Продолжительность реакции варьируют от нескольких минут до 48 часов в зависимости от количеств применяемых реагентов и температуры реакции. Продукт выделяют после окончания реакции путем фильтрации и/или концентрирования реакционной смеси. При необходимости продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
Соединения, требуемые в качестве исходных материалов для способов IX-XI, получаются путем вышеприведенных способов II-VIII.
Способ IX
При описании этого способа соединения в соответствии с формулой N получаются из соединений в соответствии с формулой M (соединения формулы II, где R6 представляет собой YCH2-q(R28)qCOOR29, R7-нитро-радикал, Y имеет вышеуказанное значение, q целое число от 0 до 2, а радикалы R28-30 определены выше для указанных R4-членов), как описывается ниже.
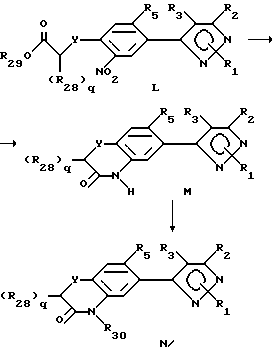
A. На первой стадии этого двухстадийного процесса соединения в соответствии с формулой L превращают в соединения формулы M восстановлением нитро-радикала в радикал амина и последующей циклизацией. Выбором условий реакции можно получить либо нециклизованный амин (соединения формулы L, где нитрорадикал замещается радикалом амина), либо циклизованный продукт. Как правило, условия реакции выбираются таким образом, чтобы циклизованный продукт получался непосредственно. Альтернативно нециклизованный амин может быть выделен стандартным методом и циклизован с получением соединения формулы M в отдельной стадии с использованием стандартных условий. Восстановители, применяемые в кислой среде, включают (но не ограничивают этим перечнем) металлы, такие как железо, цинк или олово. Растворитель, используемый в реакции, может включать органические или неорганические кислоты, такие как уксусная или хлористоводородная кислота, и может быть использован либо как концентрированный кислый раствор или разбавленные водные растворы. Температура реакции находится в пределах от 0oC до 200oC, предпочтительно от 10oC до 120oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д.
По окончании реакции продукт выделяют разбавлением реакционной смеси водой и выделяют таким методом, как кристаллизация и экстракция растворителем. Если необходимо, продукт очищают стандартными методами.
Альтернативно соединения формулы L могут быть восстановлены каталитическим гидрированием. Катализаторы, подходящие для каталитического гидрирования, которое может быть осуществлено при нормальном или повышенном давлении, включают никель Ренея, палладий на угле, палладиевую чернь, палладий на любом подходящем носителе и т.д. Растворы включают любой инертный растворитель, который заметно не препятствует реакции, включая спирты, эфиры и т.д. Выбором условий реакции можно получить либо нециклизованный амин (соединения формулы M, в которой нитро-радикал замещен аминовым радикалом), или циклизованный продукт. Как правило, условия реакции выбирают такими, чтобы непосредственно получался циклизованный продукт. С другой стороны, нециклизованный амин может быть выделен стандартными методами и циклизован с получением соединений формулы M в отдельной стадии с использованием стандартных условий. Продукт после окончания реакции выделяют фильтрацией и концентрированием реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, хроматография на колонках и т. д.
B. В этой стадии продукт со стадии A превращают в соединения формулы N. Образование продуктов, определенных выше, может проводиться путем обработки соединений формулы M алкилирующим агентом, таким как алкилгалоид или алкилсульфонат, например метилиодид, аллилбромид, пропаргилбромид, метиловый эфир фенилсульфокислоты и т.д., или ацилирующим агентом. Реакция может быть осуществлена в подходящем растворителе или смеси растворителей, с катализатором или без катализатора, в присутствии или отсутствии основания. Предпочтительными растворителями являются диметилсульфоксид, ацетон, диметилформамид, диоксан и т. д. или смеси растворителей, включая двухфазные смеси (такие как вода и метиленхлорил или другой органический растворитель). В случае двух несмешивающихся жидких фаз желательно добавлять катализатор фазового переноса, такой как бензилтриалкиламмонийгалоид или другая аммониевая соль. Основанием может быть органическое основание (такое как триалкиламин или другой органический амин) или неорганическое основание, такое как карбонат или гидроокись калия или натрия. Температура реакции находится в пределах от 0oC до 200oC, предпочтительно от 10oC до 120oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количества реагентов, температуры реакции и т.д. Продукт после окончания реакции выделяют фильтрацией и/или концентрированием реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, хроматография на колонках и т.д.
Способ X
При описании этого способа соединения в соответствии с формулой Q, в которой R33 не является водородом, получают из соединений согласно формуле O (соединения формулы II, в которой R6 представляет собой нитро-радикал, R7 YCH2-r(R31)r)-COOR32, Y имеет вышеуказанное значение, r представляет собой целое число от 0 до 2, а радикалы R31-33 определены ранее для R4-членов.
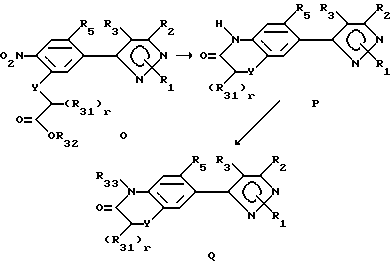
A. На первой стадии этого двухстадийного способа соединения в соответствии с формулой O превращают в соединения формулы P восстановлением нитрорадикала в радикал амина и последующей циклизацией. Выбором условий реакции можно получать либо нециклизованный амин (соединения формулы H, в которой нитрорадикал замещен аминовым радикалом), либо циклизованный продукт. Как правило, условия реакции выбираются так, чтобы получать непосредственно циклизованный продукт. Впрочем, нециклизованный амин может быть выделен стандартными методами и циклизован с получением соединений формулы P в отдельной стадии с использованием стандартных условий. Восстановители, подходящие в кислой среде, включают (не ограничены этим перечнем) металлы, такие как железо, цинк или олово. Растворитель, применяемый в реакции, может включать либо органические, либо неорганические кислоты, такие как уксусная или хлористоводородная кислота, и может быть применен в виде концентрированных кислых растворов или разбавленных водных растворов. Температура реакции находится в пределах от 0oC до 200oC, предпочтительно от 10oC до 120oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количества реагентов, температуры реакции и т.д.
По окончании реакции продукт отделяют разбавлением реакционной смеси водой и затем выделяют такими методами, как кристаллизация или экстракция растворителем. Если необходимо, продукт очищают стандартными методами.
Альтернативно соединения формулы O могут быть восстановлены каталитическим гидрированием. Катализаторы, подходящие для каталитического гидрирования, которое может проводиться при нормальном или повышенном давлении, включают никель Ренея, палладированный уголь, палладиевую чернь, палладий на любом подходящем носителе, окись палладия, платину, платиновую чернь и т.д. Растворители включают любой инертный растворитель, заметно не препятствующий реакции, включая спирты, эфиры и т.д. Выбором условий реакции можно получать либо нециклизованный амин (соединения формулы O, в которой нитрорадикал замещен аминовым радикалом) либо циклизованный продукт. Как правило, условия реакции подбираются так, чтобы непосредственно получать циклизованный продукт. Впрочем, нециклизованный амин может быть выделен стандартными методами и циклизован, используя стандартные условия. Продукт изолируют по окончании реакции путем фильтрации или концентрированием реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, хроматография на колонках и т.д.
Б. На этой стадии продукт со стадии A превращают в соединения формулы Q, в которой R33 не является водородом. Образование продуктов, определенных выше, может быть проведено обработкой соединений формулы P алкилирующим агентом, таким как алкилгалоид или алкилсульфонат, например метилиодид, алкилбромид, пропаргилбромид, метиловый эфир фенилсульфоновой кислоты и т.д. , или ацилирующим агентом. Реакция может быть проведена в любом подходящем растворителе или смеси растворителей, с катализатором или без катализатора, в присутствии или отсутствии основания. Предпочтительными растворителями являются диметилсульфоксид, ацетон, диметилформамид, диоксан и т.д. Основанием может быть органическое основание (такое как триалкиламин или другой органический амин) или неорганическое основание, такое как карбонат или гидроксид калия или натрия. Температура реакции находится в пределах от 0oC до 200oC, предпочтительно от 10oC до 120oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. Продукт выделяют после окончания реакции фильтрацией и/или концентрированием реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, хроматография на колонках и т.д.
Способ XI.
Этот раздел описывает способ получения соединений в соответствии с формулой S из соединений формулы R (соединения формулы II, в которой R6 является аминовым радикалом, R7 представляет собой YC(R34)SCCR35, Y имеет вышеуказанное значение, s есть целое число от 0 до 2, а радикалы R34-36 являются ранее определенными R4-членами).
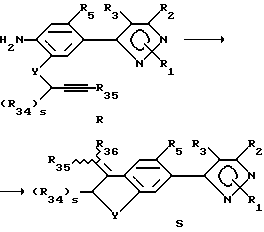
Способ получения соединений формулы S исходит из соединений формулы R. В этой реакции может применяться любой подходящий растворитель, хотя предпочтительны безводные растворители, такие как ацетонитрил. Раствор или суспензию соединения формулы R обрабатывают медными солями, включая галогениды меди (II), галогениды меди (I), смеси галогенидов меди (II и I) и других медных солей и их смесей, и алкилнитратом или органическим нитритом, таким как т-бутилнитрит. Температура реакции находится в пределах от 0oC до 200oC, предпочтительно 10oC до 100oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. Продукт выделяют после окончания реакции фильтрацией и/или концентрированием реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
Способ XII.
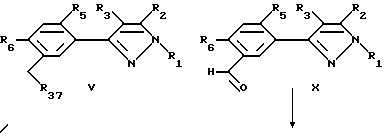
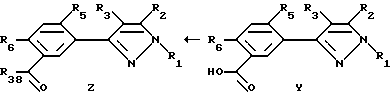
Этот способ описывает получение соединений формулы U, V, W, X, Y или Z (соединения формулы II, в которой заместитель R7 представляет собой алкил, замещенный алкил, галоидалкил, карбоксальдегид, карбоновую кислоту или производное карбоновой кислоты, такое как CXYR8 или CXR9) из соединений формулы T. Радикалы R37 и R38 имеют то же значение, которое было ранее определено для R4-членов, а X1 и X2 являются галогенами. Схемы процесса показаны ниже.
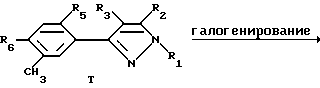
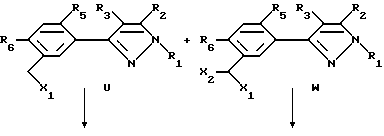
В первой стадии этого процесса соединения формулы T превращаются либо в соединения формулы U или W, либо в смесь этих продуктов. Любой инертный растворитель может быть использован в этой реакции, который не препятствует заметно протеканию реакции. Такие растворители включают, но не исчерпываются этим перечнем, органические кислоты, неорганические кислоты, углеводороды, галогенированные углеводороды, ароматические углеводороды, эфиры, сульфоксиды или сульфоны. Галогенирующие агенты, подходящие для вышеуказанных реакций, включают бром, хлор, N-бромсукцинамид, N-хлорсукцинамид, сульфурилхлорид и т.д. С некоторыми галогенирующими агентами желательно использовать органическую перекись или свет в качестве катализатора. Количество галогенирующего агента может варьировать от менее чем один моль до избытка. Температура реакции находится в пределах от -78oC до 200oC, предпочтительно от 10oC до 120oC. Продолжительность реакции может быть выбрана от немногих минут до нескольких недель в зависимости от количества реагентов, температуры реакции и т.д. По окончании реакции продукт или продукты выделяют, разбавляя реакционную смесь водой и выделяя продукт (продукты) таким методом, как кристаллизация или экстракция растворителями. Если необходимо, продукт (продукты) очищают стандартными методами.
Соединения формулы U могут быть превращены в соединения формулы V замещения радикала галогена X1 подходящим нуклеофилом. Образование продуктов формулы C может происходить в результате обработки соединений формулы U анионом алкоксида, тиоалкоксида, цианида, амина, алкила или арила и т.д. или спиртом, меркаптаном, амином и т.д. в присутствии основания в любом подходящем растворителе или смеси растворителя. Предпочтительными растворителями являются диметил-сульфоксид, ацетон, диметилформамид, диоксан, вода и т.д. или смесь растворителей, включая двухфазные смеси (такие как вода и метиленхлорид или другой органический растворитель). Основанием может быть органическое основание (такое как триалкиламин или другой органический амин) или неорганическое основание (карбонат щелочного металла, такой как карбонат калия или карбонат натрия или гидроокись щелочного металла, такая как гидроокись натрия). В случае двух несмешивающихся жидких фаз желательно добавлять катализатор фазового переноса, такой как бензилтриалкиламмонийгалоид или другая аммониевая соль. Температура реакции находится в пределах от -78oC до 200oC, предпочтительно от 10oC до 120oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. Продукт отделяют после окончания реакции фильтрацией и/или концентрированием реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
Образование продуктов формулы X может происходить в результате кислотного гидролиза соединений формулы W. Для осуществления кислотного гидролиза соединения формулы W подвергают действию избытка минеральной кислоты, такой как хлористоводородная или серная кислоты, причем предпочтителен избыток серной кислоты. Температура реакции находится в пределах от 0oC до точки кипения инертного растворителя, предпочтительно от 10oC до 100oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т. д. По окончании реакции продукт или продукты отделяются разбавлением реакционной смеси водой и выделяются, например, методами кристаллизации или экстракции растворителем. Если необходимо, продукт (продукты) очищают стандартными методами.
Соединения формулы Y получаются окислением соединений формулы X. В этой реакции можно пользоваться любым инертным растворителем, включая углеводороды, ароматические углеводороды, пиридин и его производные, воду и т.д. Применяемые окислители включают, но не ограничиваются этим перечнем, перманганат калия или дихромат калия. Температура реакции находится в пределах от -50oC до точки кипения инертного растворителя, предпочтительно от 10oC до 100oC. продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. По окончании реакции продукт или продукты отделяют разбавлением реакционной смеси водой и выделением таким методом, как кристаллизация или экстракция растворителем. Если необходимо, продукт (продукты) очищают стандартными методами.
Последняя стадия этого процесса включает преобразование соединений формулы Y в соединения формулы Z посредством любого из целого ряда стандартных способов получения производных карбоновых кислот. Эта стадия процесса является этерификацией или реакцией амидообразования. Это может быть осуществлено непосредственно из соединений формулы Y или через соль щелочного металла соединения формулы Y. Этерификацию можно проводить, используя избыток спирта, соответствующего целевому эфиру, в присутствии минеральной кислоты (например, серной кислоты). Производные амида могут быть получены обработкой соединения формулы Y желаемым аминоэфиром без растворителя или в проходящем растворителе. Реакции этерификации или амидообразования могут быть также проведены в присутствии инертного растворителя и дегидратирующего агента.
Альтернативно соединения формулы Y могут быть превращены в галоидангидрид или ангидрид кислоты и обработаны спиртом или амином. Приготовление галоидангидрида кислоты осуществляется в присутствии галогенирующего агента, такого как тионилхлорид, пятихлористый фосфор, оксалилхлорид и т.д. (этот перечень не исчерпывающий) в присутствии инертного растворителя или без растворителя. Может быть использован любой инертный растворитель, который не препятствует реакции. Может быть добавлено каталитическое количество аминового основания, такого как триэтиламин, пиридин или диметилформамид или тому подобное, для ускорения этой реакции. Температура реакции находится в пределах от -20oC до точки кипения использованного растворителя. Продолжительность реакции находится в пределах от нескольких минут до 48 часов в зависимости от количеств применяемых реагентов и температуры реакции. По окончании реакции избыточный галогенирующий агент и растворитель (растворители) удаляют из продукта реакции выпариванием или дистилляцией. Галоидангидрид кислоты обрабатывают спиртом или амином, получая соединение формулы Z. Реакция может быть проведена в отсутствии растворителя, в присутствии инертного растворителя или со смесью растворителей, включающей двухфазные смеси (такие как вода и метиленхлорид или другой органический растворитель). С целью ускорения этой реакции может быть добавлено основание, такое как триэтиламин, пиридин, щелочной металл, и/или каталитическое количество катализатора фазового переноса, такого как бензилтриалкиламмонийгалоид или другая аммониевая соль. Температура реакции находится в пределах от -20oC до точки кипения применяемого растворителя. Продолжительность реакции составляет от нескольких минут до 48 часов в зависимости от количеств применяемых реагентов и температуры реакции. Продукт выделяют после окончания реакции фильтрацией и/или концентрацией реакционной смеси. Если необходимо, продукт очищают стандартными методами, такими как экстракция, кристаллизация, колоночная хроматография и т.д.
Способ XIII
Этот раздел описывает способ получения соединений в соответствии с формулой I, в которой один из R4-остатков является тиоловой группой (формула AA), исходя из соединений в соответствии с формулой I.
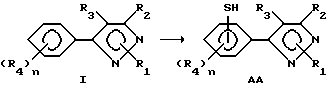
В этом способе желаемые продукты получают путем приготовления производного галоидсульфонила и последующего восстановления с получением соединений AA. Может быть использован любой растворитель, который не препятствует течению реакции, такой как галогенированные углеводороды, эфиры, алкилнитрилы, минеральные кислоты, и т.д. Для образования производных хлорсульфонила предпочтителен избыток хлорсульфоновой кислоты как в качестве реагента, так и в качестве растворителя. Температура реакции находится в пределах от 25oC до точки кипения применяемого растворителя. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. После окончания реакции продукт или продукты отделяются разбавлением реакционной смеси водой, и продукты затем выделяются таким методом, как кристаллизация или экстракция растворителем. Если необходимо, продукт (продукты) очищаются стандартными методами.
Восстановление производного галоидсульфонила может проводиться в органических или неорганических кислотах, таких как уксусная кислота или хлористоводородная кислота или смесях указанных кислот в инертных растворителях. Восстановители, подходящие в кислой среде, включают, но не ограничены этим перечнем, металлы, такие как железо, цинк или олово. Температура реакции находится в пределах от 0oC до 150oC, предпочтительно от 10oC до 150oC. Продолжительность реакции может быть выбрана в пределах от немногих минут до несколько недель в зависимости от количеств реагентов, температуры реакции и т.д.
После окончания реакции продукт отделяют разбавлением реакционной смеси водой и продукт затем выделяют таким методом как кристаллизация или экстракция растворителем. Если необходимо, продукт очищают стандартными методами.
Способ XIV.
Этот раздел описывает способ получения соединений согласно формуле I, в которой один из R4-остатков представляет собой циклический (тио)кетальный или (тио)ацетальный радикал (формула CC), исходя из соединений в соответствии с формулой BB.
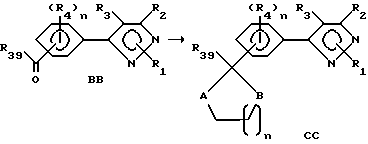
R39 представляет собой водород или ранее определенный R4-член, A и B представляют собой независимо O или S, а n является целым числом от 0 до 2. В этом способе желаемые соединения формулы CC готовятся из соединений формулы BB превращением карбионильной группы в циклическую (тио)ацетальную или (тио)кетальную группу. Альдегидная или кетонная группа соединения формулы BB обрабатывается диолом, дитиолом или гидроксиотилом. Может быть использован любой растворитель, который не препятствует прогрессу реакции, такой как галогенированные углеводороды, ароматические углеводороды, эфиры, алкилнитрилы, минеральные кислоты и т.д. Альтернативно реакция может быть проведена в отсутствии растворителя. Как правило, реакция может проводиться в присутствии кислоты, такой как минеральные кислоты, органические кислоты и т. д. Температура реакции находится в пределах от 25oC до точки кипения применяемого растворителя. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. После окончания реакции продукт или продукты выделяют концентрированием реакционной смеси и очищают таким методом, как кристаллизация или экстракция растворителем. Если необходимо, продукт (продукты) подвергают дальнейшей очистке стандартными методами.
Способ XV.
Этот раздел описывает способ получения соединений согласно формуле DD, исходя из соединений согласно формуле BB.
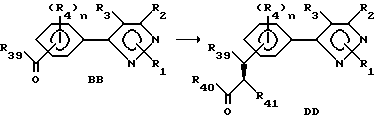
R39-41 представляют собой прежде определенные R4-члены. Соединения формулы DD получают конверсией кетонной или альдегидной группы соединений формулы BB в алкеногруппу. Преобразование может быть осуществлено обработкой соединения формулы BB реактивом типа реактива Виттига, таким как алкилиденфосфоран, илиды, полученные из солей фосфония или эфиров фосфония, алкилиденсульфураны и т.д. Подходящие растворители включают, но не ограничиваются этим перечнем, ароматические углеводороды, спирты, алканы, эфиры, галогенированные углеводороды и т.д. Температура реакции находится в пределах от -50oC до точки кипения применяемого растворителя. Продолжительность реакции может быть выбрана от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. После окончания реакции продукт или продукты выделяют путем концентрирования реакционной смеси и продукт (продукты) затем очищают методом кристаллизации или экстракции растворителем. Если необходимо, продукт (продукты) подвергают дальнейшей очистке стандартными методами.
Способ XVI.
Этот раздел описывает способ получения соединений в соответствии с формулой EE, исходя из соединений согласно формуле BB.
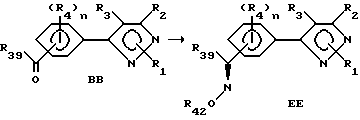
R39 и R42 представляют собой прежде определенные R4-члены. В этой стадии процесса соединения формулы EE, которые имеют оксимный заместитель в качестве одного из фенильных радикалов, получают из соединений формулы BB. Кетонный или альдегидный заместитель соединения формулы BB может быть превращен в оксим любым из двух методов. Исходный альдегид или кетон формулы BB может быть обработан O-замещенным оксимом с получением оксима формулы EE. Из этого соединения в дальнейшем могут быть получены производные стандартными методами, известными специалисту. Примеры этого подхода включают, но не ограничиваются этим, обработку альдегида или кетона (аминокси) уксусной кислоты или другой 2-(аминоокси)карбоновой кислотой и конверсию получающейся карбоновой кислоты в любое из целого ряда производных карбоновой кислоты, как то: амиды, сложные эфиры, сложные тиоэфиры и т.д. Альтернативно оксим может быть получен обработкой соединений формулы BB гидроксиламином или гидроксиламиновыми солями. Получающийся оксим может быть алкилирован с получением производных путем обработки алкилирующим агентом, таким как галоидалкилы, алкилсульфонаты и т.д. Подходящими для вышеуказанных реакций включают, но не ограничены этим перечнем, ароматические углеводороды, алканы, эфиры, спирты, галогенированные углеводороды и т.д. Температура реакции находится в пределах от -50oC до точки кипения применяемого растворителя. Реакция может быть проведена с основанием или без основания. В случаях применения основания им может быть ацетат натрия, карбонаты щелочных металлов, такой как карбонат натрия, или гидроокиси щелочного металла, такие как гидроокись натрия. Продолжительность реакции может быть выбрана в пределах от немногих минут до нескольких недель в зависимости от количеств реагентов, температуры реакции и т.д. По окончании реакции продукт или продукты выделяют концентрированием реакционной смеси, и продукт (продукты) очищают такими методами, как кристаллизация или экстракция растворителем. Если необходимо, продукт (продукты) подвергают дальнейшей очистке.
Нижеследующие примеры 1 - 42 описывают конкретные варианты осуществления способа получения представителей соединений в соответствии с настоящим изобретением.
Примеры 1 - 4 описывают конкретные воплощения способа I.
Пример 1.
Этот пример описывает получение 3-(2,5-дифторфенил)-1-метил-5- (трифторметил)-1H-пиразола (соединение N 40) и 5-(2,5- дифторфенил)-1-метил-3-(трифторметил)-1H-пиразола (соединение N 20).
A. 28,5 г 2,5-дифторацетофенона и 26 г этилового эфира трифторуксусной кислоты перемешивали в 400 мл безводного эфира и охлаждали в ледяной бане. Затем прибавляли в течение более 5 минут 42 мл 25%-ного (вес.) раствора метоксида натрия в метаноле. После перемешивания в течение 1 часа при комнатной температуре реакционную смесь экстрагировали водой, воду подкисляли и экстрагировали метиленхлоридом, получая 42 г 1-(2,5-дифторфенил)-3-(трифторметил)-пропан-1,3-дион.
B. 34,5 г 1-(2,5-дифторфенил)-3-трифторметил)-пропан-1,3-дион растворяли в 250 мл уксусной кислоты и медленно добавляли к раствору 9,5 мл метилгидразина. Смесь нагревали при 100oC в течение 5 минут, затем охлаждали и разбавляли водой. Раствор эфира промывали водой и раствором карбоната калия, затем сушили сульфатом магния, фильтровали и концентрировали. Остаток хроматографировали, получая 9,5 г 3-(2,5-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола.
Аналитич.
рассчитано для C11H7N2F5: C 50,39%, H 2,69%, N 10,68%
найдено: C 50,48%, H 2,72%, N 10,64%.
и 21,11 г 5-(2,5-дифторфенил)-1-(метил-3-(трифторметил)-1H- пиразол (т. пл. 38 - 39oC).
Аналитич.
рассчитано для C11H7N2F5: C 50,39%, H 2,69%, N 10,68%,
найдено: C 50,63%, H 2,65%, N 10,40%.
Пример 2.
Этот пример описывает получение 5-(2,4-дифторфенил)-3-(трифторметил)-1H-пиразола (соединение N 6).
A. К раствору 40,0 г (0,256 моля) 2',4'-дифторацетофенона (имеющегося в продаже) в 400 мл диэтилового эфира при 0oC прибавляли (0,405 моля) этилового эфира трифторуксусной кислоты. При 5oC добавляли в течение более 15 минут 80 мл 25%-ного (вес.) раствора метоксида натрия в метаноле (0,37 моля). Реакционную смесь перемешивали в течение ночи при 25oC. Смесь выливали на 300 мл ледяной воды и добавляли 21,3 мл (0,37 моля) уксусной кислоты. Органический слой дважды промывали водой, сушили над безводным MgSO4 и концентрировали в вакууме, получая 62,85 г (97%) 4-(2,4-дифторфенил)-1,1,1-трифтор-4-гидрокси-3-бутен-2-она в виде желтого масла. 1H-ЯМР (CDCl3) ч/млн: 6,61 (с, 1H), 6,87 (м, 1H), 6,97 (м, 1H), 7,97 (м, 1H).
Аналитич.
рассчитано для C10H5F5O2: C 47,64%, H 2,00%
найдено: C 47,70, H 1,96%.
B. При 24oC 15,0 г (0,06 моля) продукта стадии A растворяли в 50 мл ледяной уксусной кислоты и обрабатывали 2 мл (0,064 моля) безводного гидразина, добавляемого в течение более 5 минут. Реакционную смесь нагревали до 95oC в течение 30 минут. Реакционную смесь охлаждали и выливали в 300 мл ледяной воды. Суспензию отфильтровывали, и осадок на фильтре промывали водой и сушили воздухом, получая 13,86 г (94%) 5-(2,4-дифторфенил)-3-(трифторметил)-1H-пиразола в виде твердого вещества белого цвета, т.пл. 157 - 158oC.
Аналитич.
рассчитано для C10H5F5N2: C 48,40, H 2,03, N 11,29
найдено: C 48,38, H 2,03, N 11,32.
Пример 3.
Этот пример описывает получение 3-(2,4-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола (соединение N 42) и 5-(2,4-дифторфенил)-1-метил-3-(трифторметил)-1H-пиразола (соединение N 21.
Суспензию 13,6 г (0,055 моля) продукта стадии Б, 7,7 г (0,056 моля) K2CO3 и 3,7 мл (0,06 моля) метилиодида в 150 мл ацетона перемешивали в течение ночи при 25oC. Раствор разбавляли 300 миллилитрами холодной воды и экстрагировали трижды этилацетатом. Этилацетатные вытяжки промывали рассолом, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически, используя 5%-ный этилацетат в гексане в качестве элюента, получая 78,3 г (58%) 3-(2,4-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола в виде твердого вещества белого цвета, т.пл. 51oC.
Аналитич.
рассчитано для C11H7F5N2: C 50,39, H 2,69, N 10,68
найдено: C 50,36, H 2,70, N 10,70.
С помощью хроматографии, описанной в вышеприведенном способе приготовления, получали вторую фракцию, которую собирали, концентрировали и остаток кристаллизовали, получая 4,0 г (28% выход) 5-(2,4-дифторфенил)-1-метил-3-(трифторметил)-1H-пиразола в виде твердого вещества белого цвета, т. пл. 37 - 38oC.
Аналитич. рассчитано для C11H7F5N2: C 50,39, H 2,69, N 10,68
найдено: C 50,40, H 2,67, N 10,67.
Пример 4.
Этот пример описывает приготовление 3-(2,5-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола (соединение N 40).
Раствор 8,5 г (34 ммоля) сухого 5-(2,5-дифторфенил)-1H-3- (трифторметил)-1H-пиразола в 100 мл безводного толуола нагревали с обратным холодильником в аппарате, оснащенном ловушкой Дина-Старка, и обрабатывали 3,25 мл диметилсульфата. Смесь нагревали с обратным холодильником 5 часов, давали остыть и промывали 10%-ным (вес./об.) водным NaOH. Органическую фазу сушили MgSO4 и концентрировали, получая 7,74 г (86,2%) прозрачного почти бесцветного масла, nD 1,4925 (25oC).
Аналитич.
рассчитано для C11H7N2F5: C 50,39, H 2,69, N 10,68%
найдено: C 50,48, H 2,72, N 10,64%.
Примеры 5 - 7 описывают конкретные воплощения способа 11.
Пример 5.
Этот пример описывает приготовление 4-хлор-5-(2,5-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола (соединение N 361).
При 25oC 5,24 г (0,02 моля) 3-(2,5-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола растворяли в 40 мл ледяной уксусной кислоты и в течение более 1 часа барботировали 2,1 г (0,03) моля хлоргаза. Реакционную смесь перемешивали 2 часа. Реакционный раствор выливали в 200 мл ледяной воды и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaHCO3, рассолом и сушили над безводным MgSO4 и десорбировали в вакууме. Остаток очищали хроматографически с использованием 3%-ного раствора этилацетата в гексане в качестве элюента, получая 5,87 г (99%) 4-хлор-5-(2,5-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола в виде легкого желтого масла, n
Аналитич. рассчитано для C11H6Cl1F5N2: C 44,56, H 2,04, N 9,44%, Cl 11,95.
найдено: C 44,53, H 2,00, N 9,44, Cl 11,94%.
Пример 6.
Этот пример описывает получение 4-хлор-3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)- 1H-пиразола (соединение N 389).
К 5,00 г 3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)- 1H-пиразола, растворенным в 50 мл уксусной кислоты, прибавляли 15 мл сульфурилхлорида. Смесь мягко орошали 2 мл порциями сульфурилхлорида, добавляемыми каждые 15 минут. После 6 часов смесь охлаждали, затем разбавляли водой и экстрагировали эфиром. Эфир промывали трижды водой, сушили безводным сульфатом магния, фильтровали и концентрировали. Остаток хроматографировали, получая количественный выход 4-хлор-3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)-1H- пиразола.
Аналитически рассчитано для C11 H5N3O2Cl1F5: C 38,67, H 1,48, N 12,30%
найдено: C 38,73, H 1,48, N 12,34%.
Пример 7.
Этот пример описывает получение 4-хлор-3-(4-хлор-2-фтор-5- метоксифенил)-1-(1-метилэтил)-5-(трифторметил)-1H-пиразола (соединение N 489).
К раствору 1,6 г 3-(4-хлор-2-фтор-5-метоксифенил)-1-(1- метилэтил)-5-(трифторметил)-1H-пиразола в 20 мл диметилформамида прибавляли 2,0 г N-хлорсукцинимида. Раствор нагревали до 80oC в течение 2 часов, давали остыть и выливали в ледяную воду. Водную смесь экстрагировали трижды метиленхлоридом, объединенные органические вытяжки промывали водой, сушили MgSO4 и концентрировали, получая сырое масло. Масло очищали хроматографией и перегоняли из колбы в колбу, получая 1,54 г 4-хлор-3-(4-хлор-2-фтор-5-метоксифенил)-1-(1-метилэтил)-5- (трифторметил)-1H-пиразола в виде желтого масла, nD 1,5192 (24oC).
Аналитически рассчитано для C14H12N2O1F4Cl1: C 45,31, H 3,26, N 7,55%
найдено: C 45,19, H 3,27, N 7,49%.
Примеры 8 - 10 описывают конкретные воплощения способа III.
Пример 8.
Этот способ описывает получение 3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)-1H-пиразола (соединение N 388).
К охлажденному льдом раствору 50 мл дымящей азотной кислоты (90%) медленно прибавляли 8,29 г 3-(2,5-дифторфенил)-1-метил-5- (трифторметил)-1H-пиразола. После добавления смеси давали нагреться до комнатной температуры и затем осторожно нагревали до 52oC. Нагревание вели 2,5 часа, затем охлаждали и выливали на лед. Образовавшуюся смесь экстрагировали эфиром, который затем промывали дважды водой, сушили безводным сульфатом магния, фильтровали и растворитель удаляли путем концентрирования под вакуумом. Остаток очищали, используя комбинацию хроматографии и кристаллизации, получая 5,62 г 3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)-1H-пиразола, т.пл. 80 - 81oC.
Аналитически рассчитано для C11H6N3O2F5: C 43,01, H 1,97, N 13,68%
найдено: C 42,99, H 1,97, N 13,68%.
Пример 9.
Этот пример описывает получение 4-бром-3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)-1H-пиразола (соединение N 396).
При 15oC 9,5 г (0,03 моля) 4-бром-3-(2,5-дифторфенил)-1- метил-5-(трифторметил)-1H-пиразола медленно добавляли к 100 мл дымящей азотной кислоты. Реакцию нагревали до 28oC в течение периода 20 минут. Реакционную смесь перемешивали при 30oC в течение 4 часов. Смесь выливали в 500 мл льда. После перемешивания в течение 1 часа суспензию экстрагировали трижды метиленхлоридом. Метиленхлоридные вытяжки промывали водой, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически, используя 10%-ный раствор этилацетата в гексане в качестве элюента, и получали 5,84 г (55%) 4-бром-3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)-1H- пиразола в виде белого твердого вещества, т.пл. 45,5oC.
Аналитически рассчитано для C11H5Br1F5N3O2: C 34,22, H 1,31, N 10,88%
найдено: C 34,25, H 1,38, N 10,76%/
Пример 10.
Этот пример описывает получение 4-хлор-3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)-1H-пиразола (соединение N 389).
Раствор 5,9 г 4-хлор-5-(2,5-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола в 6 мл концентрированной H2SO4 охлаждали до 15oC и подвергали действию по каплям раствора 1,8 г 70% HNO3 в 2 мл концентрированной H2SO4. Реакционную смесь перемешивали при 30oC в течение 5 часов и затем обрабатывали дополнительно 1,8 г 70%-ной HNO3. После перемешивания в течение ночи при комнатной температуре смесь выливали в 250 мл ледяной воды и экстрагировали метиленхлоридом. Метиленхлоридные вытяжки трижды промывали насыщенным водным NaHCO3, дважды водой, сушили MgSO4 и концентрировали в вакууме. Полученный в результате материал хроматографировали через кремневый ангидрид, используя 10%-ный раствор этилацетата в гексане в качестве элюента, получая 3,93 г (58%) 4-хлор-3-(2,5-дифтор-4-нитрофенил)-1-метил-5- (трифторметил)-1H-пиразола.
Аналитически рассчитано для C11H5N3O2Cl1F5: C 38,67, H 1,48, N 12,30%
найдено: C 38,73, H 1,48, N 12,34%.
Примеры 11-15 описывают конкретные воплощения способа IV.
Пример 11.
Этот пример описывает приготовление 4-хлор-3-(2-фтор-5-метокси- 4-нитрофенил)-1-метил-5-(трифторметил)-1-пиразола (соединение N 390).
5,04 г 4-хлор-3-(2,5-дифтор-4-нитрофенил)-1-метил-5- (трифторметил)-1H-пиразола растворяли в безводном эфире и раствор охлаждали на ледяной бане, затем прибавляли 3,7 мл 25%-ного (вес.) раствора метоксида натрия в метаноле. После добавления ледяную баню удаляли и смесь перемешивали в течение 30 минут при комнатной температуре. Затем раствор экстрагировали 4 раза водой, сушили над безводным сульфатом магния, фильтровали и концентрировали. Остаток хроматографировали, получая 4,63 г 4-хлор-3-(2-фтор-5-метокси-4-нитрофенил)-1-метил-5-(трифторметил)- 1H-пиразола, т. пл. 115 - 116oC.
Аналитически рассчитано для C12H8N3O3Cl1F4: C 40,75, H 2,28, N 11,88%
найдено: C 40,84, H 2,24, N 11,88%.
Пример 12.
Этот пример описывает получение 4-хлор-3-(2-фтор-4-метокси-5- нитрофенил)-1-метил-5-(трифторметил)-1H-пиразола (соединение N 387).
При 35oC 13,7 г (0,04 моля) 4-хлор-3-(2,4-дифтор-5-нитрофенил)-1-метил-5-(трифторметил)-1H- пиразола, 5,5 г (0,04 моля) K2CO3 и 100 мл метанола перемешивали в течение 1 часа. Реакционную смесь охлаждали, разбавляли 100 мл холодной воды и экстрагировали четыре раза этилацетатом. Этилацетатные вытяжки промывали рассолом, сушили над безводным MgSO4 и десорбировали в вакууме. Остаток очищали хроматографически с использованием 25%-ного раствора этилацетата в гексане в качестве элюента, получая 13,0 г (90%) 4-хлор-3-(2-фтор-4-метокси-5-нитрофенил)-1-метил-5-(трифторметил)- 1H-пиразола в виде белого твердого вещества, т.пл. 116oC.
Аналитически рассчитано для C12H8Cl1F4N3O3: C 40,75, H 2,28, N 11,88%
найдено: C 40,74, H 2,34, N 11,90%.
Пример 13.
Этот пример описывает получение (5-(4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил)-4-фтор-2-нитрофенил)тиоуксусной кислоты, этиловый эфир (соединение N 393).
При 25oC 1,5 г (4,5 ммоля) 4-хлор-3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)-1H- пиразола, 0,69 г (5,0 ммоля) K2CO3, 0,55 мл (5,0 ммоля) этилового эфира меркаптоуксусной кислоты и 0,05 г (0,5 ммоля) CuF2 были суспендированы в 15 мл 1-метил-2-пирролидинона. Реакционную смесь перемешивали при 28oC в течение 24 часов. Смесь охлаждали, разбавляли 100 мл холодной воды и экстрагировали четыре раза этилацетатом. Этилацетатные вытяжки промывали рассолом, сушили над безводным MgSO4 и десорбировали в вакууме. Остаток очищали хроматографически, с использованием 10%-ного раствора диэтилового эфира и 15%-ного раствора метиленхлорида в гексане в качестве элюента, получая 0,86 г (43%) (5-(4-хлор-1-метил-5-(трифторметил)-1H- пиразол-3-ил)-4-фтор-2-нитрофенил)тиоуксусную кислоту, этиловый эфир в виде твердого вещества желтого цвета, т. пл. 79oC.
Аналитически рассчитано для C15H12Cl1F4N3O4S1: C 40,78, H 2,74, N 9,51, S 7,26%.
найдено: C 40,89, H 2,69, N 9,61, S 7,31%.
Пример 14.
Этот пример описывает приготовление 5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фтор-N- метил-2-нитро-N-пропилбензоламин (соединение N 402).
При 25oC 6,83 г (0,02 моля) 4-хлор-3-(2,5-дифтор-4-нитрофенил)-1-метил-5-(трифторметил)-1H- пиразола, 4,1 г (0,03 моля) K2CO3, 3,1 мл (0,03 моля) N-метил-N-пропиламина и каталитическое количество CuF2 суспендировали в 50 мл 1-метил-2-пирролидинона. Реакционную смесь перемешивали при 35oC в течение 2 часов. Смесь охлаждали, разбавляли 100 мл холодной воды и экстрагировали четыре раза этилацетатом. Этилацетатные вытяжки промывали рассолом, сушили над безводным MgSO4 и десорбировали в вакууме. Остаток очищали хроматографически с использованием 15%-ного раствора этилацетата в гексане в качестве элюента, получая 6,8 г (86%) 5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фтор-N- метил-2-нитро-N-пропилбензоламина в виде оранжевого масла, n
Аналитически рассчитано для C15H15Cl1F4N4O2: C 45,64, H 3,83, N 14,19
найдено: C 45,52, H 3,87, N 14,32%.
Пример 15.
Этот пример описывает приготовление (4-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-5-фтор-2- нитрофенокси)уксусной кислоты, бутилового эфира (соединение N 498).
Раствор 3,4 г (0,01 моля) 4-хлор-3-(2,4-дифтор-5-нитрофенил)-1-метил-5-(трифторметил)-1H- пиразола и 1,4 мл (0,011 моля) бутилгликолата в 25 мл безводного ТГФ охлаждали до 0oC. Поддерживая температуру ниже 5oC, порциями добавляли 0,33 г (0,11 моля) NaH. Как только добавление было закончено, реакционной смеси давали нагреться до 25oC. После 3 часов смесь осторожно закаляли водой и экстрагировали этилацетатом. Этилацетатные вытяжки промывали рассолом, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически 20%-ным этилацетат/гексаном, получая 3,25 г (72%) (4-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-5-фтор-2- нитрофенокси)уксусной кислоты, бутилового эфира в виде легкого желтого масла, т.пл. 65oC.
Аналитически рассчитано для C17H16Cl1F4N3O5:
C 45,00, H 3,55, N 9,26%
найдено: C 44,97, H 3,56, N 9,29%
Примеры 16-19 описывают конкретные воплощения способа V.
Пример 16.
Этот пример описывает получение 4-хлор-3-(4-хлор-2-фтор-5-метоксифенил)-1-метил-5-(трифторметил)- 1H-пиразола (соединение N 312).
A. К раствору 4,05 г 4-хлор-3-(2-фтор-5-метокси-4-нитрофенил)-1-метил-5-(трифторметил)- 1H-пиразола в 50 мл уксусной кислоты прибавляют 1,39 г (0,0249 моля) железного порошка. Реакционную смесь нагревали близко к орошению в течение 2 часов, обрабатывали 1,39 граммами железного порошка и нагревали близко к орошению еще один час. После охлаждения, концентрирования и хроматографии выделяли 3,54 г 4-хлор-3-(4-амино-2-фтор-5-метоксифенил)-1-метил-5-(трифторметил)- 1H-пиразола.
Б. 3,064 г 4-хлор-3-(4-амино-2-фтор-5-метоксифенил)-1-метил-5-(трифторметил)-1H-пиразола растворяли в 50 мл безводного ацетонитрила и прибавляли 1,90 г безводного хлорида меди (II). Затем по каплям добавляли в течение 10 минут 1,93 мл т-бутилнитрита (техн. 90%), растворенного в 10 мл безводного ацетонитрила, еще 20 минут перемешивали и затем концентрировали. Остаток собирали в этилацетат, экстрагировали трижды 10%-ной соляной кислотой, сушили безводным сульфатом магния, фильтровали, концентрировали и хроматографировали, получая 2,10 г 4-хлор-3-(4-хлор-2-фтор-5-метоксифенил)-1-метил-5-(трифторметил)-1H-пиразола, т.пл. 70-71oC.
Аналитически рассчитано для C12H8N2O1Cl1F4:
C 42,01, H 2,35, N 8,16%
найдено: C 42,15, H 2,34, N 8,18%.
Пример 17.
Этот пример описывает получение 2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фтор- N-метил-N-пропилбензоламина (соединение N 166).
A. Раствор 5,2 г (0,013 моля) 5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фтор- N-метил-2-нитро-N-пропилбензоламина в 100 мл уксусной кислоты нагревали до 80oC в атмосфере азота. Тепло и азот удаляли и прибавляли в течение 5 мин. тремя порциями 2,2 г (0,039 моля) железного порошка. Раствор перемешивали при 80oC еще 30 мин. Раствор охлаждали через Celite®. Фильтрат разбавляли 100 мл воды и экстрагировали трижды этилацетатом. Этилацетатные вытяжки промывали насыщенным раствором NaHCO3, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически с использованием 30%-ного раствора этилацетата в гексане в качестве элюента, получая 3,85 г (80%) 5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фтор- N-метил-N-пропил-1,2-бензендиамина в виде легкого желтого масла, n
Аналитически рассчитано для C15H17Cl1F4N4:
C 49,39, H 4,70, N 15,36%.
найдено: C 49,40, H 4,64, N 15,16%
Б. Все оборудование сушили пламенем в атмосфере азота. Раствор 3,35 г (9,2 моля) продукта стадии A в 60 мл ацетонитрила при 25oC подвергали действию 0,9 г (9,2 ммоля) CuCl и 1,8 г (13,3 ммоля) CuCl2. В течение 5 минут прибавляли раствор 2,2 мл (18,4 ммоля) 90%-ного т-бутилнитрита. Через 2 часа смесь десорбировали при 28oC в вакууме. Остаток от реакции собирали в этилацетате и промывали трижды 10%-ным раствором HCl, дважды рассолом и сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически с использованием 20%-ного раствора этилацетата в гексане в качестве элюента, получая 2,45 г (70%) 2-хлор-5-(4-хор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фтор- N-метил-N-пропилбензоламина в виде прозрачного бесцветного масла,
n
Аналитически рассчитано для C15H15Cl2F4N3:
C 46,89, H 3,94, N 10,94%
найдено: C 46,84, H 3,83, N 10,93%.
Пример 18.
Этот пример описывает приготовление 4-бром-3-(4-хлор-2-фтор-5-метоксифенил)-1-метил-5-(трифторметил)-1H-пиразола (соединение N 313).
A. Раствор 3,16 г (7,9 ммоля) 4-бром-3-(2-фтор-5-метокси-4-нитрофенил)-1-метил-5-(трифторметил)-1H-пиразола в 59 мл уксусной кислоты нагревали до 80oC в атмосфере азота. Тепло и азот удаляли и прибавляли в течение 5 мин тремя порциями 1,76 г (31,6 ммоля) железного порошка. Раствор перемешивали при 80oC в течение еще 30 мин. Раствор охлаждали и фильтровали через Celite®. Фильтрат разбавляли 100 мл воды и экстрагировали трижды диэтиловым эфиром. Эфирные вытяжки промывали рассолом, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически с использованием 40%-ного раствора этилацетата в гексане в качестве элюента, получая 2,4 г (83%) 4-(4-бром-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-5-фтор-2- метокси-бензенамина в виде твердого вещества белого цвета, т.пл. 85 - 86oC.
Аналитически рассчитано для C12H10Br1F4N3O1:
C 39,15, H 2,74, N 11,41%
найдено: C 39,13, H 2,74, N 11,40%.
Б. Все оборудование сушили пламенем в атмосфере азота. Раствор 6,6 г (0,0179 моля) продукта стадии A в 100 мл акрилонитрила охлаждали до 5oC. Прибавляли 1,8 г (0,018 моля) CuCl и 3,7 г (0,027 моля) CuCl2 при 5oC. Добавляли раствор 4,8 мл (0,036 моля) 90%-ного т-бутилнитрита в 15 мл ацетонитрила в течение 15 минут и затем давали нагреться до 28oC. Через 2 часа при 28oC реакционную смесь десорбировали под вакуумом. Остаток от реакции собирали в диэтиловом эфире и трижды промывали 10%-ным раствором HCl, дважды рассолом и сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически с использованием 20%-ного раствора этилацетата в гексане в качестве элюента, получая 6,3 г (91%) 4-бром-3-(4-хлор-2-фтор-5-метоксифенил)-1-метил-5-(трифторметил)- 1H-пиразола в виде твердого белого тела, т.пл. 85-86oC.
Аналитически рассчитано для C12H8Br1Cl1F4N2O1:
C 37,19, H 2,08, N 7,23%
найдено: C 37,23, H 2,08, N 7,24%.
Пример 19.
Этот пример описывает приготовление 4-хлор-3-(5-хлор-2,4-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола (соединение N 354).
A. Раствор 3,4 г (0,01 моля) 4-хлор-3-(2,4-дифторфенил-5-нитрофенил)-1-метил-5-(трифторметил)-1H-пиразола в 50 мл уксусной кислоты нагревали до 80oC в атмосфере азота. Тепло и азот удаляли и прибавляли в течение 5 мин тремя порциями 1,7 г (0,03 моля) железного порошка. Раствор перемешивали при 80oC еще 30 мин. Раствор охлаждали и фильтровали через Celite®. Фильтрат разбавляли 100 мл воды и экстрагировали трижды этилацетатом. Этилацетатные вытяжки промывали насыщенным раствором NaHCO3, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически с использованием 35%-ного этилацетата в гексане в качестве элюента, получая 2,46 г (79%) 5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-2,4- дифторбензоламина в виде твердого белого вещества, т.пл. 82oC.
Аналитически рассчитано для C11H7Cl1F5N3:
C 42,40, H 2,26, N 13,48%
найдено: C 42,40, H 2,26, N 13,49%.
Б. Все оборудование сушили пламенем в атмосфере азота. Раствор 2,0 г (6,4 ммоля) продукта стадии A в 50 мл ацетонитрила при 25oC обрабатывали 0,63 г (6,4 ммоля) CuCl и 1,2 г (9,4 ммоля) CuCl2. В течение 5 минут добавляли раствор 1,74 мл (5,0 ммоля) 90%-ного т-бутилнитрита. Через 4 часа при 28oC реакционную смесь десорбировали в вакууме. Остаток от реакции собирали в этилацетате и промывали трижды 10%-ным раствором HCl, дважды рассолом и сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически с использованием 10%-ного раствора этилацетата в гексане в качестве элюента, получая 1,63 г (78%) 4-хлор-3-(5-хлор-2,4-дифторфенил)-1-метил-5-(трифторметил)-1H-пиразола в виде белого твердого вещества, т.пл. 50-51oC.
Аналитически рассчитано для C11H5Cl2F5N2:
C 39,91, H 1,52, N 8,46%
найдено: C 39,89, H 1,52, N 8,39%.
Примеры 20-21 описывают конкретные воплощения способа VI.
Пример 20.
Этот способ описывает приготовление 4-хлор-3-(4-хлор-2-фтор-5-гидроксифенил)-1-метил-5-(гидроксифенил)- 1-метил-5-(трифторметил)-1H-пиразола (соединение N 325).
1,39 г 4-хлор-3-(4-хлор-2-фтор-5-метоксифенил)-1-метил-5-(трифторметил)- 1H-пиразола растворяли в 80 мл безводного метиленхлорида и затем охлаждали на сухой ледяной/ацетоновой бане и прибавляли 0,14 мл трибромида бора. После того как ей давали нагреться до комнатной температуры, в смесь добавляли еще 0,28 мл трибромида бора. Еще добавляли 1,0 мл трибромида бора и перемешивали при комнатной температуре в течение 6 часов. После перемешивания добавляли 30-50 мл охлажденной ледяной воды и смесь перемешивали в течение 10 минут. Органическую фазу экстрагировали водой, сушили безводным сульфатом магния, фильтровали и концентрировали, получая 1,258 г 4-хлор-3-(4-хлор-2-фтор-5-гидроксифенил)-1-метил-5-(трифторметил)- 1H-пиразола, т.пл. 123,0 - 126,0oC.
Аналитически рассчитано для C11H6N2O1Cl2F4:
C 40,15, H 1,84 N 8,51%
найдено: C 40,08, H 1,87, N 8,48.
Пример 21.
Этот пример описывает получение 4-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-5-фтор-2-нитрофенола (соединение N 429).
Раствор 1,4 г (4 ммоля) 4-хлор-3-(2-фтор-4-метокси-5-нитрофенил)-1-метил-5-(трифторметил)-1H-пиразола в 20 мл метиленхлорида охлаждали до 0oC. Затем медленно в течение 10 минут прибавляли 5,0 мл 1М раствора BBr3 (4,9 ммоля) в метиленхлориде. Раствор перемешивали в течение ночи при комнатной температуре. Раствор промывали дважды водой, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток перекристаллизовывали из гексана, получая 0,7 г (54%) 4-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-5-фтор-2- нитрофенол в виде твердого вещества бежевого цвета, т.пл. 89-90oC. Аналитически рассчитывали для C11H6Cl1F4N3O3:
C 38,90, H 1,78, N 12,37%
Найдено: C 38,93, H 1,78, N 12,16%
Примеры 22-24 описывают конкретные воплощения способа VII.
Пример 22.
Этот пример описывает приготовление 4-хлор-3-(4-хлор-2-фтор-5-пропаргилоксифенил)-1-метил-5-(трифторметил)-1H- пиразола (соединение N 261).
1,01 г 4-хлор-3-(4-хлор-2-фтор-5-гидркосифенил)-1-метил-5-(трифторметил)-1H-пиразола, 0,44 г безводного карбоната калия и 0,5 мл пропаргилбромида (80% по весу в толуоле) растворяли в 20-30 мл безводного ДМФ. Смесь нагревали при 65oC в течение 90 минут. После охлаждения смесь разбавляли водой и затем экстрагировали трижды эфиром. Объединенные эфирные вытяжки дважды промывали водой, сушили безводным сульфатом магния, фильтровали, концентрировали и хроматографировали, получая 1,05 г 4-хлор-3-(4-хлор-2-фтор-5-пропаргилоксифенил)-1-метил-5-(трифторметил)-1H -пиразола, т. пл. 89,5-91,0oC.
Аналитически рассчитано для C14H8N2O1Cl2F4:
C 45,80, H 2,20, N 7,63%
найдено: C 45,93, H 2,21, N 7,61%.
Пример 23.
Этот пример описывает приготовление (4-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-5-фтор-2-нитрофенокси) уксусной кислоты, этилового эфира (соединение N 386).
При 25oC 6,11 г (0,018 моля) 4-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-5-фтор-2-нитрофенола, 2,5 г (0,019 моля) K2CO3 и 2,0 мл (0,019 моля) этилового эфира бромуксусной кислоты суспендировали в 100 мл ацетона. Реакционную смесь перемешивали при 40oC в течение 4 часов. Смесь охлаждали, разбавляли 100 мл холодной воды и экстрагировали четыре раза этилацетатом. Этилацетатные вытяжки промывали рассолом, сушили над безводным MgSO4 и десорбировали в вакууме. Остаток перекристаллизовывали из метилциклогексана, получая 7,5 г (99%) 4-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-5-фтор-2-нитрофенокси) уксусной кислоты, этиловый эфир, в виде легкого желтого масла, т.пл. 95-96oC.
Аналитически рассчитано для C15H12Cl1F4N3O5:
C 42,32, H 2,84, N 9,87%
найдено: C 42,30, H 2,83, N 9,85%
Пример 24.
Этот пример описывает получение (2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторфенокси) уксусной кислоты, этилового эфира (соединение N 290).
При 25oC 13,16 г (0,04 моля) 4-хлор-3-(4-хлор-2-фтор-5-гидроксифенил)-1-метил-5-(трифторметил)-1H-пиразола, 6,1 г (0,44 моля) K2CO3 и 4,8 мл (0,44 моля) этилового эфира бромуксусной кислоты суспендировали в 25 мл ацетона. Реакционную смесь перемешивали при 25oC в течение 16 часов. Реакционный раствор выливали в 150 мл ледяной воды, фильтровали, промывали водой и сушили воздухом. Остаток перекристаллизовывали из гексана, получая 16,6 г (100%) (2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторфенокси)-уксусной кислоты, этиловый эфир в виде твердого вещества белого цвета, т.пл. 130-131oC.
Аналитически рассчитано для C15H12Cl2F4N2O3:
C 43,40, H 2,91, N 6,75%
найдено: C 43,54, H 2,91, N 6,77%.
Примеры 25 и 26 описывают конкретные воплощения способа VIII.
Пример 25.
Этот пример описывает получения 2-(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4- фторфенокси)-N-метил-пропанамида (соединение N 237).
А. К суспензии 1,4 г (3,3 ммоля) 2-(2-хлор-5-(4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил)-4-фторфенокси)-пропановой кислоты, этиловый эфир, в 50 мл воды и 30 мл 1,4-диоксана добавляли 1,3 мл (3,3 ммоля) 10%-ного раствора NaOH. Через 30 минут раствор охлаждали и устанавливали pH на значении 3 с помощью концентрированной HCl. Реакционную смесь экстрагировали диэтиловым эфиром. Раствор эфира промывали водой, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток перекристаллизовывали из метилциклогексана, получая 1,3 г (100%) 2-(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторфенокси)- пропановую кислоту в виде твердого вещества белого цвета, т.пл. 150-151oC.
Аналитически рассчитано для C14H10Cl2F4N2O3:
C 41,92, H 2,51, N 6,98%
найдено: C 41,96, H 2,48, N 7,00%.
Б. К раствору 0,8 г (2,0 ммоля) продукта стадии А в 100 мл метиленхлорида прибавляли 0,5 мл (6,0 ммоля) оксалилхлорида в течение 5 минут, вызывая выделение газа. Когда выделение газа закончилось, добавляли одну каплю ДМФ и раствор перемешивали до тех пор, пока не прекращалось выделение газа. Раствор десорбировали до сухости в вакууме. Остаток растворяли в 10 мл ТГФ и прибавляли к раствору 5 мл 40% водного метиламина и 10 мл ТГФ при 0oC в течение 5 минут. Реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Раствор разбавляли 100 мл холодной воды, экстрагировали этилацетатом. Этилацетат промывали рассолом, сушили над безводным MgSO4 и концентрировали в вакууме. Твердое вещество перекристаллизовывали из метилциклогексана, получая 0,83 г (99%) 2-(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторфенокси) -N-метилпропанамид в виде твердого вещества белого цвета, т.пл. 134,5 - 135,5oC.
Аналитически рассчитано для C15H13Cl2F4N3O2:
C 43,50, H 3,16, N 10,16%
найдено: C 43,70, H 3,16, N 10,20%.
Пример 26.
Этот пример описывает получение 2-(2-хлор-5-(4-хлор- 1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторфенокси)пропановой кислоты, 3-метилбутилового эфира (соединение N 288).
К раствору 1,9 г (5,0 ммоля) 2-(хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторфенокси)- пропановой кислоты в 50 мл метиленхлорида прибавляли 1,3 мл (15,0 ммоля) оксалилхлорида в течение 5 минут, вызывая выделение газа. Когда выделение газа прекращалось, добавляли одну каплю ДМФ и раствор перемешивали до тех пор, пока не прекращалось выделение газа. Раствор десорбировали до сухости в вакууме. Хлорангидрид кислоты растворяли в 40 мл 3-метил-1-бутанола и нагревали до образования флегмы в течение одного часа. Реакционную смесь охлаждали, разбавляли 100 мл холодной воды и экстрагировали этилацетатом. Этилацетат промывали рассолом, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически с использованием 25%-ного раствора этилацетата в гексане в качестве элюента, получая 2,17 г (95%) 2-(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4- фторфенокси)-пропановой кислоты, 3-метилбутилового эфира, в виде твердого вещества белого цвета, т.пл. 128oC.
Аналитически рассчитано для C18H18Cl2F4N2O3:
C 47,28, H 3,97, N 6,13%
найдено: C 47,32, H 3,95, N 6,17%.
Пример 27.
Этот пример описывает получение 2H-1,4-бензоксазин-3 (4H)-она, 6-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-7-фтор-4-(2-пропинил)-2H-1,4-бензоксазин-3 (4H)-она (соединение N 446) и является конкретным воплощением способа IX.
А. Раствор 4,5 г (0,0106 моля) (4-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-5-фтор-2-нитрофенокси)- уксусной кислоты, этилового эфира, в 75 мл уксусной кислоты нагревали до 80oC в атмосфере азота. Тепло и азот удаляли и прибавляли в течение 5 минут тремя порциями 1,8 г (0,033 моля) железного порошка. Раствор перемешивали при 80oC еще 3 часа. Раствор охлаждали и фильтровали через Celite®. Фильтрат разбавляли 100 мл воды и экстрагировали трижды этилацетатом. Этилацетатные вытяжки промывали насыщенным раствором NaHCO3, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток перекристаллизовывали из метилциклогексан/этилацетата, получая 2,95 г (80%) 6-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-7-фтор-2H-1,4-бензоксазин -3(4H)-она в виде твердого вещества белого цвета, т.пл. 207oC.
Аналитически рассчитано для C13H8Cl1F4N3O2:
C 44,65 H 2,31 N 12,02%
найдено: C 44,66, H 2,31, N 11,97.
Б. При 25oC 3,0 г (8,6 ммоля) продукта стадии А, 1,22 г (6,0 ммоля) K2CO3 и 0,79 мл (8,8 ммоля) 80%-го пропаргилбромида суспендировали в 50 мл ацетона. Реакционную смесь перемешивали пи 40oC в течение 6 часов. Реакцию охлаждали, разбавляли 100 мл холодной воды и экстрагировали четыре раза этилацетатом. Этилацетатные вытяжки промывали рассолом, сушили над безводным MgSO4 и десорбировали в вакууме. Остаток перекристаллизовывали из метилциклогексана, получая 2,97 г (89%) 2H-1,4-бензоксазин-3(4H)-она, 6-(4-хлор- 1-метил-5-(трифторметил)1H-пиразол-3-ил)-7-фтор-4-(2-пропинил)-2H-1,4 -бензоксазин-3(4H)-она в виде твердого вещества бежевого цвета, т.пл. 142-143oC.
Аналитически рассчитано для C16H10Cl1F4N3O2:
C 49,57, H 2,60, N 10,84%
найдено: C 49,58, H 2,62, N 10,85%
Пример 28.
Этот пример описывает получения 7-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-6-фтор-4-(2-пропинил)- 2H-1,4-бензоксазин-3(4H)-она (соединение N 479) и является конкретным воплощением способа Х.
А. Раствор 2,3 г (5,4 ммоля) (5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фтор-2-нитрофенокси) уксусной кислоты, этилового эфира, в 50 мл уксусной кислоты нагревали до 80oC в атмосфере азота. Тепло и азот убирали и в течение 5 минут тремя порциями прибавляли 0,9 г (16,2 ммоля) железного порошка. Раствор перемешивали при 80oC еще 50 минут. Раствор охлаждали и фильтровали через Celite®. Фильтрат разбавляли 100 мл воды и экстрагировали трижды этилацетатом. Этилацетатные вытяжки промывали насыщенным раствором NaHCO3, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток перекристаллизовывали из метилциклогексан/этилацетата, получая 0,96 г (50%) 7-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-6-фтор-2H-1,4-бензоксазин- 3(4H)-она в виде белого твердого вещества, т.пл. 242oC.
Аналитически рассчитано для C13H8Cl1F4N3O2:
C 44,65, H 2,31, N 12,02%
найдено C 44,61, H 2,27, N 11,99%.
Б. При 25oC 2,7 г (7,7 ммоля) продукта стадии А, 1,1 г (8,0 ммоля) K2CO3 и 0,9 мл (8,0 ммоля) 80%-ного пропаргилбромида суспендировали в 25 мл ДМСО. Смесь перемешивали при 45oC в течение 10 часов. Смесь охлаждали, разбавляли 100 мл холодной воды и экстрагировали четыре раза этилацетатом. Этилацетатные вытяжки промывали рассолом, сушили над безводным MgSO4 и десорбировали в вакууме. Остаток очищали хроматографически с использованием метиленхлорида в качестве элюента, получая 2,7 г (90%) 7-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол- 3-ил)-6-фтор-4-(2-пропинил)-2H-1,4-бензоксазин-3(4H)-она в виде белого твердого вещества, т.пл. 184oC.
Аналитически рассчитано для C16H10Cl1F4N3O2:
C 49,57, H 2,60, N 10,84%
найдено: C 49,48, H 2,56, N 10,95%.
Пример 29.
Этот пример описывает получение цис- и транс-4-хлор-3- (3-хлорметилен)-5-фтор-2,3-дигидро-6-бензофуранил)-2-метил-5- (трифторметил)-1H-пиразола (соединение NN 481 и 482) и является конкретным воплощением способа XI.
Все оборудование сушили пламенем под азотом. Раствор 2,0 г (5,75 ммоля) 4-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол- 3-ил)-5-фтор-2-(2-пропилокси)бензоламина в 100 мл ацетонитрила при 25oC обрабатывали 0,6 г (5,75 ммоля) CuCl и 0,8 г (5,75 ммоля) CuCl2. В течение 5 минут добавляли 1,1 мл (8,6 ммоля) 90%-ного т-бутилнитрита. Через 6 часов при 28oC реакционную смесь десорбировали в вакууме. Остаток от реакции собирали в этилацетате и промывали трижды 10%-ным раствором HСl, дважды рассолом и сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически с использованием 20%-ного раствора этилацетата в гексане в качестве элюента, получая 0,73 г (35%) цис-4-хлор-3-(3-(хлор- метилен)-5-фтор-2,3-дигидро-6-бензофуранил)-1-метил-5- (трифторметил)-1H-пиразола в виде белого твердого вещества, т.пл. 140,5 - 142,5oC.
Аналитически рассчитано для C14H8Cl2F4N2O1:
C 45,80, H 2,20, N 7,63, Cl 19,31%
найдено: C 45,64, H 2,22, N 7,60, Cl 19,29.
Хроматография, описанная выше, давала вторую фракцию после основного компонента. Эту фракцию собирали, десорбировали и остаток кристаллизовали из гексана, получая 0,68 г (32%) транс-4-хлор-3- (3-хлорметилен)-5-фтор-2,3-дигидро-6-бензофуранил)-1-метил-5- (трифторметил)-1H-пиразола в виде твердого вещества цвета беж, т.пл. 132 - 135oC.
Аналитически рассчитано для C14H8Cl2F4N2O1:
C 45,80, H 2,20, N 7,63, Cl 19,31%
найдено: C 45,71, H 2,23, N 7,63, Cl 19,28%.
Примеры 30 - 37 описывают конкретные воплощения способа XII.
Пример 30.
Этот пример описывает получение 3-[5-(бромметил)-4-хлор-2- фторфенил]-4-хлор-1-метил-5-(трифторметил)-1H-пиразола (соединение N 108).
Суспензию 3-[5-метил-4-хлор-2-фторфенил]-4-хлор-1-метил- 5-(трифторметил)-1H-пиразола (25 г, 76,4 ммоля) и N-бромсукцинимида (13,6 г, 76,4 ммоля) в 100 мл четыреххлористого углерода в 500-миллилитровой круглодонной колбе, оснащенной магнитной мешалкой, обрабатывали каталитическим количеством бензоила. Температуру поднимали для образования флегмы в течение одного часа. Реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали, получая 31,5 г белого твердого вещества. Материалы дважды перекристаллизовывали из гексана, получая 15,3 г (49%) 3-[5-(бромметил)-4-хлор-2-фторфенил]-4-хлор- 1-метил-5-(трифторметил)-1H-пиразола в виде белого твердого вещества, т.пл. 112 - 114oC.
Аналитически рассчитано для C12H7N2F4Cl2Br1:
C 35,50, H 1,74, N 6,90%
найдено: C 35,57, H 1,76, N 6,88%.
Пример 31.
Этот пример описывает получение (((2-хлор-5-(4-хлор-1-метил- 5-(трифторметил)-1H-пиразол-3-ил)-4-фторпропенил)метил)тио)уксусной кислоты, этилового эфира (соединение N 123).
Смесь 1,62 г (4,0 ммоля) 3-[5-(бромметил)-4-хлор-2-фторфенил]- 4-хлор-1-метил-5-(трифторметил)-1H-пиразола, 0,44 мл этилового эфира меркаптоуксусной кислоты и 0,55 г K2CO3 суспендировали в 25 мл ацетона. Реакционную смесь оставляли перемешиваться на ночь при комнатной температуре. После разбавления 100 мл холодной воды смесь экстрагировали этилацетатом, органические вытяжки промывали водой, сушили MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически, получая 1,7 г (96%) (((2-хлор-5-(4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил)-4-фторфенил)метил)тио)уксусной кислоты, этилового эфира в виде твердого вещества белого цвета, т.пл. 63oC.
Аналитически рассчитано для C16H14Cl2F4N2O2S1:
C 43,16, H 3,17, N 6,29%
найдено: C 43,16, H 3,16, N 6,27%.
Пример 32.
Этот пример описывает получение 2-хлор-5-[4-хлор-1-метил- 5-(трифторметил)-1H-пиразол-3-ил]-4-фторбензометанола (соединение N 122).
К раствору 7,1 г (0,0175 ммоля) 3-[5-(бромметил)-4-хлор- 2-фторфенил]-4-хлор-1-метил-5-(трифторметил)-1H-пиразола в 20 мл ДМФ прибавляли 1,5 г (0,018 ммоля) ацетата натрия. Смесь выливали в 100 мл холодной воды и твердое отфильтрованное вещество сушили. Продукт перекристаллизовывали из этанол/воды и получали 6,0 г (90%) 2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4- фторбензолметанол, ацетата (эфир), т.пл. 90oC. Ацетат растворяли в 10 мл 1,4-диоксана и 10 мл воды и добавляли 6,3 мл (0,0158 моля) 10%-го раствора NaOH. Через 30 минут раствор нейтрализовали концентрированной HCl, фильтровали и твердое вещество сушили. Твердое вещество перекристаллизовывали из этанол/воды, получая 5,4 г (99%) 2-хлор-5-[4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил] -4- фторбензолметанола в виде твердого вещества белого цвета, т.пл. 103oC.
Аналитически рассчитано для C12H8N2O1F4Cl2:
C 42,01, H 2,35, N 8,16%
найдено: C 41,88, H 2,34, N 8,09%.
Пример 33.
Этот пример описывает получение ((2-хлор-5-(4-хлор-1-метил- 5-(трифторметил)-1H-пиразол-3-ил)-4-фторфенил)метокси)-уксусной кислоты, 1-метилэтилэфира (соединение N 119).
При 25oC 1,7 г (5,0 ммоля) 2-хлор-5-[4-хлор-1-метил- 5-(трифторметил)-1H-пиразол-3-ил]-4-фторбензолметанола, 0,8 г (5,5 ммоля) K2CO3 и 0,7 мл (5,5 ммоля) изопропилового эфира бромуксусной кислоты суспендировали в 15 мл ДМСО. Смесь перемешивали всю ночь при 45oC. Смесь охлаждали, разбавляли 100 мл холодной воды и экстрагировали четыре раза этилацетатом. Этилацетатные вытяжки промывали рассолом, сушили над безводным MgSO4 и десорбировали в вакууме. Остаток очищали хроматографически с использованием 10%-ного раствора этилацетата в гексане в качестве элюента, получая 0,9 г (41%) ((2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол- 3-ил)-4-фторфенил)метокси)уксусной кислоты, 1-метилэтилового эфира в виде твердого вещества белого цвета, т.пл. 55oC.
Аналитически рассчитано для C17H16Cl2F4N2O3:
C 46,07, H 3,64, N 6,32%
найдено: C 46,21, H 3,69, N 6,11%.
Пример 34.
Этот пример описывает получение 4-хлор-3-[4-хлор-5- (дибромметил)-2-фторфенил]-1-метил-5-(трифторметил)-1H-пиразола (соединение N 132).
В 250 миллилитровой круглодонной колбе, оснащенной магнитной мешалкой, готовили суспензию 3-[5-метил-4-хлор-2-фторфенил] -4- хлор-1-метил-5-(трифторметил)-1H-пиразола (8,18 г, 25 ммоля) и N-бромсукцинимида (8,9 г, 50,0 ммоля) в 50 мл четыреххлористого углерода. Прибавляли каталитическое количество перекиси бензоила и поднимали температуру до образования флегмы и поддерживали ее в течение 3,5 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали. Остаток очищали хроматографически, получая 10,6 г (85%) 4-хлор-3-[4-хлор-5- (дибромметил)-2-фторфенил] -1-метил-5-(трифторметил)-1H-пиразола в виде твердого вещества белого цвета, т. пл. 89 - 92oC.
Аналитически рассчитано для C12H6N2F4Cl2Br2:
C 29,72, H 1,25, N 5,78%
найдено: C 29,72, H 1,25, N 5,78%.
Пример 35.
Этот пример описывает получение 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил]-4-фторбензальдегида (соединение N 133).
В 100 миллилитровой круглодонной колбе, оснащенной магнитной мешалкой, 4-хлор-3-[4-хлор-5-(дибромметил)-2-фторфенил] -1-метил-5- (трифторметил)-1H-пиразол (5,0 г, 10,0 ммоля) перемешивали в течение 30 минут в 20 мл серной кислоты. Получающийся прозрачный желтый раствор оставляли стоять при комнатной температуре на 10 дней, перемешивали короткое время для удаления цвета и выливали на 200 мл льда/воды. Водную смесь экстрагировали эфиром и органический слой сушили MgSO4, фильтровали и концентрировали, получая 3,15 г белого цвета твердого вещества, которое перекристаллизовывали из холодных гексанов, получая 2,5 г (71%) 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил] -4-фторбензальдегида в виде твердого вещества белого цвета, т. пл. 70 - 72oC.
Аналитически рассчитано для C12H6N2O1F4Cl2:
C 42,26, H 1,77, N 8,21%
найдено: C 42,22, H 1,78, N 8,24%.
Пример 36.
Этот пример описывает получение 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил]-4-фторбензойной кислоты (соединение N 149).
К раствору 2-хлор-5-[4-хлор-1-метил-5-(трифторметил)-1H-пиразол- 3-ил] -4-фторбензальдегида (4,5 г, 13,2 ммоля) в 14 мл ацетона прибавляли 13 мл (26 ммоля) реактива Джоунса. Раствор перемешивали при температуре окружающей среды в течение 2 часов и выливали в 400 мл воды. Получающееся твердое вещество отфильтровывали и сушили на воздухе, получая 4,5 г (96%) 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил]-4-фторбензойой кислоты в виде твердого вещества белого цвета. Пробу для анализа перекристаллизовывали из эфира/гексанов, т. пл. 179 - 181oC.
Аналитически рассчитано для C13H6N2O2F4Cl2:
C 40,36, H 1,69, N 7,84%
найдено: C 40,49, H 1,74, N 7,77%.
Пример 37.
Этот пример описывает получение 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил] -4-фторбензойной кислоты, 1-метилэтилового эфира (соединение N 135).
К раствору 4,3 г (0,012 моля) 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил] -4-фторбензойной кислоты в 50 мл метиленхлорида прибавляли 3,1 мл (0,036 моля) оксалилхлорида, вызывая выделение газа. Когда это выделение прекращалось, добавляли одну каплю ДМФ и раствор перемешивали, пока не прекратится выделение газа. Раствор концентрировали в вакууме и получающийся остаток растворяли в 25 мл изопропанола и нагревали до 60oC в течение 1 часа. Раствор охлаждали, выливали в 200 мл холодной воды и твердое вещество отфильтровывали и сушили. Продукт перекристаллизовывали из этанола/воды, получая 1,69 г (70%) 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил] -4-фторбензойной кислоты, 1-метилэтилового эфира, в виде твердого вещества белого цвета, т. пл. 69oC.
Аналитически рассчитано для C15H12Cl2F4N2O2:
C 45,13, H 3,03, N 7,02%
найдено: C 45,14, H 3,04, N 7,03%.
Примеры 38 и 39 описывают конкретные воплощения способа XIII.
Пример 38.
Этот пример описывает получение 2-хлор-5-[4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил] -4-фторбензолсульфонилхлорида (соединение N 346).
Раствор 4-хлор-3-(4-хлор-2-фторфенил)-1-метил-5- (трифторметил)-1H-пиразола в 20 мл хлорсульфоновой кислоты нагревали при 120oC в масляной бане в течение четырех часов и давали остыть до комнатной температуры. Прибавляли метиленхлорид и раствор прибавляли по каплям к перемешиваемой смеси льда и воды (осторожно, крайне реакционноспособен). Слои разделяли, водный слой промывали метиленхлоридом. Объединенные органические слои сушили MgSO4, фильтровали и концентрировали и получающийся твердый остаток промывали очень малым количеством эфира и перекристаллизовывали из гексанов, получая 1,65 г (63%) 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил]-4-фторбензолсульфонилхлорид в виде твердого вещества белого цвета, т. пл. 116 - 117oC.
Аналитически рассчитано для C11H5N2O2S1F4Cl3:
C 32,10, H 1,22, N 6,81, Cl 25,84%
найдено: C 32,15, H 1,17, N 6,76, Cl 25,77%.
Пример 39.
Этот пример описывает получение 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил]-4-фтортиофенол (соединение N 343).
К раствору 12,8 г (0,031 моля) 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил]-4-фторбензолсульфонилхлорида в 100 мл уксусной кислоты прибавляют 40,7 г (0,62 моля) цинкового порошка. Суспензию перемешивали при 80oC в течение 4 часов, давали ей остыть и фильтровали через Celite®. Фильтрат выливали в 1,0 л воды, твердое вещество отфильтровывали и сушили. Твердое вещество перекристаллизовывали из этанол/воды, получая 10,2 г (95%) 2-хлор-5-[4-хлор-1-метил-5-(трифторметил)- 1H-пиразол-3-ил]-4-фтортиофенола в виде твердого вещества белого цвета, т. пл. 56 - 58oC.
Аналитически рассчитано для C11H6N2S1F4Cl2:
C 38,28, H 1,75, N 8,12%
найдено: C 38,29, H 2,02, N 8,12%.
Пример 40.
Этот пример описывает получение 4-хлор-3-(4-хлор-5- (1,3-диоксолан-2-ил)-2-фторфенил)-1-метил-5-(трифторметил)-1H- пиразола (соединение N 100) и является конкретным воплощением способа XIV.
В аппарате, оснащенном ловушкой Дина-Старка для азеотропного удаления воды, нагревали 2,4 г (7,0 ммоля) 2-хлор-5-[4-хлор-1-метил- 5-(трифторметил)-1H-пиразол-3-ил] -4-фторбензальдегида, 0,4 мл (7,7 ммоля) этиленгликоля и каталитическое количество п-толуолсульфоновой кислоты в 50 мл толуола до образования флегмы в течение 24 часов. Образующуюся смесь концентрировали и остаток сушили хроматографически, получая 1,65 г (61%) 4-хлор-3-(4-хлор-5-(1,3-диоксолан-2-ил)-2-фторфенил)-1-метил-5-(трифторметил)-1H- пиразола в виде прозрачного бесцветного масла, n
Аналитически рассчитано для C14H10Cl2F4N2O2:
C 43,66, H 2,62, N 7,27%
найдено: C 43,67, H 2,59, N 7,24%.
Пример 41
Этот пример описывает получение 3-(2-хлор-5-(4-хлор-1-метил- 5-(трифторметил)-1H-пиразол-3-ил)-4-фторфенил)пропановой кислоты, метилового эфира (соединение N 128) и является конкретным воплощением способа XV.
К раствору 2,3 г (6,8 ммоля) 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил] -4-фторбензальдегида в 25 мл метанола прибавляли 2,27 г (6,8 ммоля) метил(трифенилфосфоранилиден)ацетата, поддерживая температуру ниже 35oC. Реакционную смесь оставляли перемешиваться на 15 минут и разбавляли этилацетатом, промывали рассолом, сушили над безводным MgSO4 и концентрировали в вакууме. Остаток очищали хроматографически с использованием 20%-ного раствора этилацетата в гексане в качестве элюента, получая 2,0 г (74%) 3-(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)- 4-фторфенил)пропановой кислоты, метилового эфира в виде твердого вещества белого цвета, т. пл. 117oC.
Аналитически рассчитано для C15H10Cl2F4N2O2:
C 45,36, H 2,54, N 7,05%
найдено: C 45,41, H 2,59, N 7,03%.
Пример 42.
Этот пример описывает получение ((((2-хлор-5-(4-хлор-1-метил- 5-(трифторметил)-1H-пиразол-3-ил] -4-фторфенил)метилен)амино) окси)уксусной кислоты (соединение N 130) и является конкретным воплощением способа XVI.
Смесь 3,4 г (0,01 моля) 2-хлор-5-[4-хлор-1-метил-5- (трифторметил)-1H-пиразол-3-ил]-4-фторбензальдегида, 2,73 г (0,0125 моля) полугидрохлорида карбоксиметоксиламина и 1,03 г (0,0125 моля) ацетата натрия в 50 мл этанола нагревали до образования флегмы в течение 2 часов. Реакционную смесь оставляли охлаждаться, обрабатывали 150 мл воды, полученный осадок собирали и сушили. Продукт перекристаллизовывали из метилциклогексана с минимальным количеством этилацетата, получая 3,35 г (81%) ((((2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)- 4-фторфенил)метилен)амино)окси)уксусной кислоты в виде твердого вещества белого цвета, т. пл. 170oC.
Аналитически рассчитано для C14H19Cl2F4N3O3:
C 40,60, H 2,19, N 10,15%
найдено: C 40,54, H 2,28, N 10,17%.
Таблицы 4 - 6 показывают примеры соединений, полученных в соответствии со способами II - XVI. В таблице 4 перечислены примеры 1-метил-5-арилпиразольных соединений. В таблице 5 приведены примеры 1-метил-3-арилпиразолов. Таблица 6 содержит различные соединения, большинство из которых включает соединения, в которых R6 и R7 циклизованы, образуя конденсированные гетероциклические структуры.
В таблице 6 представлены различные соединения в соответствии с формулой I, структуры которых неудобны для указания в таблицах 4 и 5.
Предвсходовые испытания гербицида.
Как отмечалось выше, соединения согласно настоящему изобретению оказались неожиданно эффективными в качестве гербицидов.
Испытания на предвсходовую гербицидную активность проводились следующим образом:
верхний слой почвы помещали в алюминиевый лоток и уплотняли на глубину 0,95 - 1,27 см от верха лотка. Затем в почву высевалось заданное число семян каждого вида различных однодольных и двудольных однолетних растений и/или вегетативные побеги различных видов многолетних растений. Затем непосредственно на грядку с семенами наносили известное качество активного ингредиента, растворенного или суспендированного в органическом растворителе, например в ацетоне, или в воде в качестве носителя, и грядку затем накрывали слоем необработанной почвы, заполняя лоток вровень с его краями. После обработки лотки устанавливали на скамью в теплице, где их поливали снизу как требуется для обеспечения достаточной влаги для прорастания и роста.
Приблизительно через 10 -14 дней (обычно 11 дней) после высева и обработки лотки осматривали и результаты (% ингибирования) записывали.
Таблицы 7 и 7A содержат результаты испытаний предвсходовой гербицидной активности на сорняках соединений согласно настоящему изобретению. Гербицидная оценка, показанная в этих таблицах, представляет собой процент ингибирования каждого вида растений. Виды растений, рассматриваемые как сорняки, которые использовались в одной из серий испытаний, данные о которых показаны в таблицах, идентифицированы по буквенным рубрикам над графами в соответствии со следующим
Yens - Yellow nutsedge (желтая осока)
Anbg - мятлик однолетний (Annual bluegrass)
Sejg - саженец джонсоновой травы (гумай) - Seedling johnsongrass
Dobr - костер кровельный - Dawny Brome
Bygr - просо петушье - Barnyardgrass
Modl - вьюнок пурпурный (ипомеа) - Morningglory
Cobu - дурнушник - Coklebur
Vele - канатник Теофраста - Velvetleaf
Inmu - горчица индийская - Indian Mustard
Wibw - гречишка вьюнковая - Wild buckwheat
В таблицах ниже отмечено, что символ "C" представляет собой 100%-ное уничтожение сорняков, а символ "N" указывает, что соответствующий вид высевали, но по тем или иным причинам данные не получены.
Послевсходовые испытания гербицида
Послевсходовая гербицидная активность некоторых различных соединений согласно настоящему изобретению была продемонстрирована испытанием в теплице следующим образом. Верхний слой почвы помещали в алюминиевый лоток, имеющий отверстия в днище, и уплотняли до глубины 0,95 - 1,27 см от верха лотка. В почву вносили заданное количество семян каждого из различных видов однолетних двудольных и однодольных растений и/или вегетативные отростки многолетних видов растений и вдавливали их в поверхность почвы. Семена и/или вегетативные отростки покрывали почвой и выравнивали. Лотки затем помещали на скамью в теплице и поливали снизу, как требовалось. После того как растения достигали желаемого возраста (две или три недели), каждый лоток индивидуально перемещали в оросительную камеру и орошали с помощью распылителя, работающего при давлении распыления 170,3 кПа (10 фунтов на кв. дюйм) при отмеченных расходных нормах. Раствор для распыления содержал определенное количество смеси эмульгирующего агента, такое, что содержание эмульгатора в распылительном растворе или суспензии составляло около 0,4% по объему. Распылительный раствор или суспензия содержит достаточное количество испытуемого химиката, чтобы получить нормы расхода активного ингредиента, соответствующие таковым в таблицах 8 и 8A, при применении общего количества раствора или суспензии, эквивалентного 1870 л/га (200 галлон/акр). Лотки возвращали в теплицу и поливали как прежде, и приблизительно через 10 - 14 дней (обычно 11 дней) наблюдали повреждение растений в сравнении с контрольными, а в некоторых случаях наблюдали снова через 24 - 28 дней (обычно через 25 дней) после орошения. Послевсходовая гербицидная активность, показанная в этих таблицах, представляет собой процент ингибирования каждого вида растений.
Гербицидные составы согласно настоящему изобретению, включая концентраты, требующие разбавления перед применением, могут содержать по меньшей мере один активный ингредиент и одну добавку в жидкой или твердой форме. Составы готовят смешением активного ингредиента с добавкой, включающей разбавители, наполнители, носители и модифицирующие агенты для получения составов в форме тонко размельченных твердых частиц, гранул, шариков, растворов, дисперсий и эмульсий. Так, предполагается, что активный ингредиент мог бы быть использован с такими добавками, как тонко дисперсные твердые вещества, жидкости органического происхождения, вода, смачивающий агент, диспергатор, эмульгатор и любое подходящее их сочетание.
Подходящие смачивающие агенты включают алкилбензол- и алкилнафталинсульфонаты, сульфированные жирные спирты, амины или амиды кислот, длинноцепные кислотные эфиры изотионата натрия, эфиры сульфосукцината натрия, эфиры сульфированных или сульфонированных жирных кислот, сульфонаты нефти, сульфонированные растительные масла, дитрет-ацетиленовые гликоли, полиоксиэтиленовые производные алкилфенолов (в частности изооктилфенола и нонилфенола) и полиоксиэтиленовые производные моно-высших жирно-кислотных эфиров гекситангидридов (например, сорбитана). Предпочтительными диспергирующими агентами являются метилцеллюлоза, поливиниловый спирт, лигнинсульфонаты натрия, нафталинсульфонаты натрия, полимерные алкилнафталинсульфонаты, нафталинсульфонаты натрия и полиметилен-нафталинсульфонат. Смачивающими порошками являются вододиспергируемые составы, содержащие один или несколько активных ингредиентов, инертный твердый наполнитель-расширитель и один или более смачивающих и диспергирующих агентов. Инертными твердыми наполнителями-расширителями являются обычно минеральное происхождение, как-то природные глины, диатомовая земля и синтетические минералы, производные кремнезема и т.п. Примеры таких расширителей включают каолиниты, аттапульгитная глина и синтетический силикат магния. Составы смачивающих порошков согласно настоящему изобретению обычно содержат от 0,5 до 60 частей (предпочтительно от 5 до 20 частей) активного ингредиента, от 0,25 до 25 частей (предпочтительно от 1 до 15 частей) смачивающего агента, от 0,25 до 25 частей (предпочтительно от 1,0 до 15 частей) диспергатора и от 5 до 95 частей (предпочтительно 5 - 50 частей) инертного твердого наполнителя-расширителя, все части суть весовые части от общего состава. Где требуется, примерно от 1,0 до 2,0 частей твердого инертного расширителя, может быть заменено ингибитором коррозии или противовспенивающим агентом или обоими.
Другие рецептуры включают дустовые концентраты, содержащие от 0,1 до 60% по весу активного ингредиента на подходящем расширителе; эти дусты могут быть разбавлены для применения в концентрациях в пределах от 0,1 до 10% по весу.
Водные суспензии или эмульсии могут быть получены путем перемешивания неводного раствора водонерастворимого активного ингредиента и эмульгатора с водой до получения однородного состояния и последующего гомогенизирования с получением устойчивой эмульсии весьма тонко распределенных частиц. Получающаяся концентрированная водная суспензия характеризуется ее крайне малыми размерами частиц, так что после разбавления и распыления достигается весьма равномерное покрытие. Подходящие концентрации таких рецептур составляют от 0,1 до 60%, предпочтительно от 5 до 50% по весу, активного ингредиента, причем верхний предел определяется пределом растворимости активного ингредиента в растворителе. Концентраты - это обычно растворы активного ингредиента в несмешивающихся с водой или частично несмешивающихся с водой растворителях вместе с поверхностно-активным веществом. Подходящие растворители для активного ингредиента согласно настоящему изобретению включают диметилформамид, диметилсульфоксид, метилпирролидон, углеводороды и не смешивающиеся с водой эфиры, сложные эфиры или кетоны. Однако могут быть приготовлены другие жидкие концентраты высокой крепости растворением активного ингредиента в растворителе с последующим разбавлением, например керосином, до концентраций распыления.
Составы концентратов, описываемые здесь, обычно содержат от 0,1 до 95 частей (предпочтительно 5 - 60 частей) активного ингредиента, от 0,25 до 50 частей (предпочтительно от 1 - 25 частей) поверхностно-активного вещества и, где требуется, от 5 до 94 частей растворителя, все части суть весовые части от общего веса эмульгированного масла.
Гранулы являются физически устойчивыми дисперсными композициями, содержащими активный ингредиент, сцепленный с основной матрицей инертного тонкодисперсного наполнителя-расширителя или распределенный через нее. Для облегчения выщелачивания активного ингредиента из дисперсного наполнителя-расширителя в составе может присутствовать поверхностно-активное вещество. Природные глины, пирофиллиты, иллит и вермикулит являются примерами классов применимых дисперсных минеральных расширителей. Предпочтительными расширителями являются пористые адсорбирующие предварительно формованные частицы, такие как предварительно гранулированный и просеянный аттапульгит или термически расширенный дисперсный вермикулит и тонкодисперсные глины, такие как каолиновые глины, гидратированный аттапульгит или бентонитные глины. Эти расширители распыляются на активный ингредиент или смешиваются с ним для образования гербицидных гранул.
Гранулированные составы согласно настоящему изобретению могут содержать от 0,1 до примерно 30 частей по весу активного ингредиента на 100 частей по весу глины и от 0 до 5 частей по весу поверхностно-активного вещества на 100 частей дисперсной глины.
Составы согласно настоящему изобретению могут также содержать другие добавки, например удобрения, другие гербициды, другие пестициды, добавки, повышающие безопасность применяемых составов и т.п., используемые как таковые или в комбинации с вышеописанными добавками. Химикаты, которые могут использоваться в сочетании с активными ингредиентами настоящего изобретения, включают, например, триазины, мочевину, сульфонилмочевину, карбаматы, ацетаамиды, ацетанилиды, урацилы, производные уксусной кислоты или фенола, тиокарбаматы, триазолы, азолопиримидины, бензойную кислоту и ее производные, нитрилы, бифениловые эфиры, нитробензолы и т.п., как-то:
Гетероциклические производные азота/серы
2-хлор-4-этиламино-6-изопропиламино-S-триазин
2-хлор-4,6-бис(изопропиламино)-S-триазин
2-хлор-4,6-бис(этиламино)-S-триазин
3-изопропил-1H-2,1,3-бензотиадиазин-4-(3H)-он 2,2-диоксид
3-амино-1,2,4-триазол
6,7-дигидродипиридо (1,2-2',1'-c)-пиразидииниевая соль
5-бром-3-изопропил-6-метилурацил
1,1'-диметил-4,4'-бипиридин
2-(4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил)-3- хинолинкарбоновая кислота
Изопропиламиновая соль 2-(4-изопропил-4-метил-5-оксо-2-имидазолин-2-ил)никотиновой кислоты
Метиловый эфир 6-(4-изопропил-4-метил-5-оксо-2-имидазолин-2- ил)-м-толиловой кислоты и метиловый эфир 2-(4-изопропил-4-метил-5- оксо-2-имидазолин-2-ил)-п-толиловой кислоты
Мочевина/сульфомочевина
N-(4-хлорфенокси)фенил-N,N-диметилмочевина
N,N-диметил-N'-(3-хлор-4-метилфенил)мочевина
3-(3,4-дихлорфенил)-1,1-диметилмочевина
1,3-диметил-3-(2-бензотиазолил)мочевина
3-(п-хлорфенил)-1,1-диметилмочевина
1-бутил-3-(3,4-дихлорфенил)-1-метилмочевина
2-хлор-N/(4-метокси-6-метил-1,3,5-триазин-2-ил)аминокарбонил/-бензолсульфонамид
N-(2-метоксикарбонилфенил сульфонил)-N'-(4,6-бисдифторметоксипиримидин-2-ил)мочевина
Метиловый эфир 2-(((((4,3-диметил-2-пиримидинил)амино)карбонил) амино)сульфонил)бензойной кислоты
Этиловый эфир 1-/метил-2-(((((4,6-диметил-2-пиримидинил) амино)карбонил)амино)сульфонил)/бензойной кислоты
Метил-2 ((4,6-диметокси-пиримидин-2-ил)аминокарбонил)амино- сульфонилметил)бензойной кислоты
Метиловый эфир 2-(((((4-метокси-6-метил-1,3,5-триазин- 2-ил)-амино)карбонил)амино)сульфонил)бензойной кислоты
Карбаматы/тиокарбаматы
2-хлораллиловый эфир диэтилдитиокарбаминовой кислоты
S-(4-хлорбензил)N,N-диэтилтиокарбамат
Изопропиловый эфир N-(3-хлорфенил)карбаминовой кислоты
S-2,3-дихлораллиловый эфир N,N-диизопропилтиокарбаминовой кислоты
S-N,N-дипропилтиокарбамат
S-пропиловый эфир N,N-дипропилтиокарбаминовой кислоты
S-2,3,3-трихлораллил-N,N-диизопропилтиокарбамата
Ацетамиды/ацетанилиды/анилины/амиды
2-хлор-N,N-диаллилацетамид
N,N-диметил-2,2-дифенилацетамид
N-(2,4-диметилтиен-3-ил)-N-(1-метоксипроп-2-ил)-2-хлорацетамид
N-(1H-пиразол-1-илметил-N-(2,4-диметилтиенил-3-ил)-2-хлорацетамид
N-(1-пиразол-1-илметил)-N-(4,6-диметоксипиримидин-5-ил)-2-хлорацетамид
N-(2,4-диметил-5-///Трифторметил)сульфонил/амино/фенил/-ацетамид
N-изопропил-2-хлорацетанилид
N-изопропил-1-(3,5,5-триметилциклогексен-1-ил)-2-хлорацетамид
2',6'-диэтил-N-(бутоксиметил)-2-хлорацетанилид
2',6'- диэтил-N-(2-н-пропоксиэтил)-2-хлорацетанилид
2',6'-диметил-N-(1-пиразол-1-илметил)-2-хлорацетанилид
2',6'-диэтил-N-метоксиметил-2-хлорацетанилид
2'-метил-6'-этил-N-(2-метоксимпроп-2-ил)-2-хлорацетанилид
2'-метил-6'-этил-N-(этоксиметил)-2-хлорацетанилид
α,α,α- трифтор-2,6-динитро-N,N-дипропил-п-толуидин
N-(1,1-диметилпропинил)-3,5-дихлорбензамид
Кислоты/сложные эфиры/спирты
2,2-дихлорпропионовая кислота
2-метил-4-хлорфеноксиуксусная кислота
2,4-дихлорфеноксиуксусная кислота
Метил-2-/4-(2,4-дихлорфенокси)фенокси/пропионат
3-амино-2,5-дихлорбензойная кислота
2-метокси-3,6-дихлорбензойная кислота
2,3,6-трихлорфенилуксусная кислота
N-1-нафтилполуамид фталевой кислоты
5-/2-хлор-4-(трифторметил)фенокси/-2-нитробензоат натрия
4,6-динитро-о-втор.-бутилфенол
N-(фосфонметил)глицин и его соли
Бутиловый эфир (R)-2-/4-/(5-трифторметил)-2-пиридинил/окси/ фенокси/пропановой кислоты
Простые эфиры
2,4-дихлорфенол-4-нитрофениловый эфир
2-хлор -δ,δ,δ- трифтор-п-толил-3-этокси-4-нитродифениловый эфир
5-(2-хлор-4-трифторметилфенокси)-N-метилсульфонил-2-нитробензамид
1'-(карбоэтокси)этиловый эфир 5-/2-хлор-4-(трифторметил)-фенокси/-2-нитробензойной кислоты
Разное
2,6-дихлорбензонитрил
Кислая натриевая соль метансернокислоты
Натриевая соль метанмышьяковой кислоты
2-(2-хлорфенил)метил-4-, 4-диметил-3-изоксазолидинон
7-оксабицикло-(2.2.1)-гептан, 1-метил-4-(1-метилэтил)-2-(2- метилфенилметокси)-, экзо-глифосат и его соли.
Удобрения, которые могут использоваться в сочетании с активными ингредиентами, включают, например, нитрат аммония, мочевину, поташ и суперфосфат. Другие полезные добавки включают материалы, в которых укореняются и растут растительные организмы, такие как компост, навоз, гумус, песок и т.п.
Гербицидные рецептуры описанных выше типов, рассматриваемые как находящиеся в сфере настоящего изобретения, показаны ниже в нескольких иллюстративных примерах.
1. Эмульгируемые концентраты - Весовые проценты
А. Соединение N 308 - 11,00
Свободная кислота комплексного органического фосфата или ароматического или алифатического гидрофобного основания (например, GAFAC RE-610, зарегистрированная торговая марка GAF Corp. - 5,59
Блок-сополимер полиоксиэтилен/полиоксипропилена с бутанолом (например, тергитол XH, зарегистрированная торговая марка Union Carbide Corp.) - 1,11
Фенол - 5,34
Монохлорбензол - 76,96 - 100,00
Б. Соединение N 261 - 25,0
Свободная кислота комплексного органического фосфата ароматического или алифатического гидрофобного основания (например, GAFAC RE-610) - 5,00
Блок-сополимер полиоксиэтилена/полиоксипропилена с бутанолом (например, тергитол XH) - 1,60
Циклогексанон - 4,75
Монохлорбензол - 63,65 - 100,0
В. Соединение N 291 - 12,0
Свободная кислота комплексного органического фосфата или ароматическое или алифатическое гидрофобное основание (например, GAFAC RE-610, зарегистрированная торговая марка GAF Corp.) - 6,00
Блок-сополимер полиоксиэтилена/полиоксипропилена с бутанолом (например, тергитол ХН, зарегистрированная торговая марка Union Carbide Corp.) - 1,5
Циклогексанон - 5,5
Монохлорбензол - 75,0 - 100,0
Г. Соединение N 229 - 20,0
Свободная кислота комплексного органического фосфата ароматического или алифатического гидрофобного основания (например, GAFAC RE-610) - 5,00
Блок-сополимер полиоксиэтилена/полиоксипропилена с бутанолом (например, тергитол XH) - 2,0
Циклогексанон - 5,0
Монохлорбензол - 68,0 - 100,0
Д. Соединение N 312 - 11,0
Свободная кислота комплексного органического фосфата или ароматическое или алифатическое гидрофобное основание (например, GAFAC RE-610, зарегистрированная торговая марка GAFАС Corp.) - 5,59
Блок-сополимер полиоксиэтилена/полиоксипропилена с бутанолом (например, тергитол XH, зарегистрированная торговая марка Union Carbide Corp.) - 1,11
Циклогексанон - 5,34
Монохлорбензол - 76,96 - 100,0
Е. Соединение N 282 - 25,0
Свободная кислота комплексного органического фосфата ароматического или алифатического гидрофобного основания (например, GAFAC RE-610) - 5,00
Блок-сополимер полиоксиэтилена/полиоксипропилена с бутанолом (например, тергитол XH) - 1,60
Циклогексанон - 4,75
Монохлорбензол - 63,65 - 100,0
II. Текучие рецептуры - Весовые проценты
А. Соединение N 261 - 25,0
Метилцеллюлоза - 0,3
Аэрогель кремнезема - 1,5
Лигносульфонат натрия - 3,5
N-метил-N-олеилтаурат натрия - 1,0
Вода - 67,7 - 100,00
Б. Соединение N 270 - 45,0
Метилцеллюлоза - 0,3
Аэрогель кремнезема - 1,5
Лигносульфонат натрия - 3,5
N-метил-N-олеилтаурат натрия - 1,0
Вода - 47,7 - 100,00
В. Соединение N 294 - 30,0
Метилцеллюлоза - 0,3
Аэрогель кремнезема - 1,5
Лигносульфонат натрия - 3,5
N-метил-N-олеилтаурат натрия - 3,0
Вода - 62,0 - 100,00
Г. Соединение N 135 - 23,0
Метилцеллюлоза - 0,5
Аэрогель кремнезема - 2,0
Лигносульфонат натрия - 3,5
N-метил-N-олеилтаурат натрия - 2,0
Вода - 69,00 - 100,00
Д. Соединение N 148 - 45,0
Метилцеллюлоза - 0,3
Аэрогель кремнезема - 1,3
Лигносульфонат натрия - 3,5
N-метил-N-олеилтаурат натрия - 1,0
Вода - 47,7 - 100,00
III Смачивающиеся порошки - Весовые проценты
А. Соединение N 261 - 25,0
Лигносульфонат натрия - 3,0
N-метил-N-олеил-таурат натрия - 1,0
Аморфный кремнезем (синтетический) - 71,0
Б. Соединение N 312 - 45,0
Диоктилсульфосукцинат натрия - 1,25
Лигносульфонат кальция - 1,75
Аморфный кремнезем (синтетический) - 52,0 - 100,00
В. Соединение N 237 - 10,0
Лигносульфонат натрия - 3,0
N-метил-N-олеил-таурат натрия - 1,0
Каолинитовая глина - 86,0 - 100,00
Г. Соединение N 463 - 30,0
Лигносульфонат натрия - 3,0
N-метил-N-олеил-таурат натрия - 1,0
Каолин - 56,0
Аморфный кремнезем (синтетический) - 10,0 - 100,0
Д. Соединение N 446 - 75,0
Диоктилсульфосукцинат натрия - 1,25
Лигносульфонат кальция - 1,75
Каолин - 12,0
Аморфный кремнезем синтетический - 10,0 - 100,00
Е. Соединение N 482 - 15,0
Лигносульфонат натрия - 3,0
N-метил-N-олеил-таурат натрия - 1,0
Аморфный кремнезем, синтетический - 10,0
Каолинитовая глина - 71,0 - 100,00
IV. Гранулы
А. Соединение N 74 - 15,0
Диипропиленгликоль - 5,0
Гранулированный аттапульгит (20/40 меш) - 80,0 - 100,00
Б. Соединение N 390 - 15,0
Диипропиленгликоль - 5,0
Диатомная земля (20/40) - 80,0 - 100,00
В. Соединение N 399 - 1,0
Этиленгликоль - 5,0
Метиленовый синий - 0,1
Пирофиллит - 93,9 - 100,00
Г. Соединение N 393 - 5,0
Этиленгликоль - 5,0
Пирофиллит (20/40) - 90,00 - 100,00
Д. Соединение N 312 - 15,0
Пропиленгликоль - 5,0
Гранулированный аттапульгит (20/40 меш) - 80,00 - 100,00
Е. Соединение N 324 - 25,00
Диатомная земля (20/40 меш) - 75,00 - 100,00
Ж. Соединение N 261 - 5,0
Этиленгликоль - 5,0
Метиленовый синий - 0,5
Пирофиллит - 94,5 - 100,0
З. Соединение N 262 - 10,0
Пропиленгликоль - 5,0
Пирофиллит (20/40) - 85,0 - 100,0
V. Суспензионные концентраты - Весовые проценты
А. Соединение N 262 - 16,0
Нонилфенолэтоксилат 9,5 моля EO Стерокс NJ эн-джей - 13,8
Лигносульфонат натрия (реакс 88В) - 12,2
Вода - 58,0 - 100,0
Б. Соединение N 446 - 32,5
Калиевая соль конденсата нафталинсульфоната с формальдегидом (DAXAD IIKLS) - 9,0
Нонилфенолэтоксилат 10 моль EО (Айджипал си-оу-660) - 9,0
Вода - 49,5 - 100,0
В. Соединение N 76 - 10,0
Диоксилсульфосукцинат натрия, аэрозоль ОТВ - 11,0
Касторовое масло +36 этиленоксид (Флоумоу 3 джи) - 11,00
Вода - 70,0 - 100,0
Е. Соединение N 261 - 15,0
Нонилфенолэтоксилат 9,5 моля EO Стерокс эн-джей
,0
Лигносульфонат натрия (Реакс 88В) - 5,0
Вода - 79,00 - 100,00
Д. Соединение N 290 - 30,0
Калиевая соль конденсата нафталинсульфоната с формальдегидом (DAXAD II KLS) - 4,0
Нонилфенолэтоксилат 10 молей EO (Айджипал си-оу-660) - 2,0
Вода - 64,0 - 100,0
Е. Соединение N 135 - 18,0
Нонилфенолэтоксилат 9,5 моля EO Стерокс эн-джей - 14,0
Лигносульфонат натрия (Реакс 88B) - 12,0
Вода - 56,0 - 100,0
Ж. Соединение N 148 - 34,0
Калиевая соль конденсата нафталинсульфоната с формальдегидом (DAXAD aag) - 8,0
Нонилфенолэтоксилат 10 моля EО (Айджипал си-оу-660) - 10,0
Вода - 48,0 - 100,0
З. Соединение N 482 - 14,0
Диоктилсульфосукцинат натрия, аэрозоль ОТВ - 3,0
Касторовое масло + 36 этиленоксид (Флоумоу 3 Джи) - 3,0
Вода - 80,0 - 100,0
VI. Жидкие концентраты - Весовые проценты
А. Соединение N 76 - 20,0
Ксилол - 80,0 - 100,0
Б. Соединение N 229 - 10,0
Ксилол - 90,0 - 100,0
В. Соединение N 217 - 25,0
Ксилол - 75,0 - 100,0
Г. Соединение N 482 - 15,0
Ксилол - 85,0
100,0
VII. Микрокапсулы - Весовые проценты
А. Соединение N 135, капсулированное в оболочку из полимочевины - 4,5
Реэкс C-21 - 1,5
NaCl - 5,0
Вода - 89,0 - 100,0
Б. Соединение N 137, капсулированное в оболочку из полимочевины - 20,0
Реэкс 88В - 2,0
NaNO3 - 10,0
Ксилол - 30,0
Вода - 38,0 - 100,0
В. Соединение N 138, капсулированное в оболочку из полимочевины - 4,8
Реэкс 88В - 1,2
NaNO3 - 5,0
Керосин - 20,0
Вода - 69,0 - 100,0
Г. Соединение N 148, капсулированное в мочевино-формальдегидную полимерную оболочку - 50,0
Реэкс C-21 - 1,5
NaCl - 8,5
Петролейное масло (ароматик 200) - 20,0
Вода - 20,0 - 100,0
Е. Соединение N 229, капсулированное в тиомочевино-формальдегидную полимерную оболочку - 30,0
Реэкс C-21 - 2,0
NaCl - 8,0
Ксилол - 30,0
Вода - 30,0 - 100,0
Ж. Соединение N 261, капсулированное в полимочевинную оболочку - 7,5
Реэкс 88В - 1,5
NaCl - 8,0
Аромэтик 200 - 30,0
Вода - 53,0 - 100,0
З. Соединение N 308, капсулированное в оболочку из меламин-формальдегидного сополимера - 9,0
Реэкс 88B - 2,0
NaNO3 - 10,0
Керосин - 40,0
Вода - 39,0 - 100,0
И. Соединение N 446, капсулированное в полимерную мочевино-формальдегидную оболочку - 15,0
Реэкс 88B - 10,0
NaNO3 - 8,0
Ксилол - 42,0
Вода - 25,0 - 100,0
К. Соединение N 312, капсулированное в полимочевинную оболочку - 22,0
Реэкс 88B - 2,0
NaCl - 8,0
Ксилол - 35,0
Вода - 33,0 - 100,0
При осуществлении настоящего изобретения эффективные количества соединений согласно настоящему изобретению вносят в почву, содержащую семена или вегетативные ростки, или внедряют их любым подходящим способом. Внесение жидких или твердых дисперсных составов в почву может быть осуществлено обычными способами, например с помощью механического опыливателя, стрельчатых и ручных разбрызгивателей и аэрозольных распылителей. Составы могут вноситься также с самолетов в виде пыли или аэрозоля благодаря эффективности их при малых дозах. Точное количество активного ингредиента, которое подлежит применению, зависит от различных факторов, включая вид растения, стадию его развития, тип и характеристику почвы, количество осадков в виде дождя и применяемых конкретных соединений. При избирательном довсходовом применении или при применении в почву дозировка составляет обычно от 0,02 до 11,2 кг/га, предпочтительно от 0,1 до 5,60 кг/га. В некоторых случаях могут потребоваться более высокие и более низкие нормы расхода. Специалист легко может определить из данной спецификации, включая вышеприведенные примеры, оптимальную норму расхода в каждом отдельном случае.
Термин "почва" употреблен в его наиболее широком смысле, включающем все обычные "почвы", как они определяются в Webster's New International Dictionary, второе издание, несокращенное (1961). Таким образом, термин относится к любому веществу или любой среде, в которой растительность может пустить корни и произрастать, и включает не только землю, но и также компост, навоз, мусор, гумус, суглинок, ил, грязь, глину, песок и т.п., приспособленные для поддержки роста растений.
Хотя настоящее изобретение описано на конкретных примерах, детали этих воплощений не могут рассматриваться как ограничения. Различные эквиваленты, изменения и модификации могут предприниматься, не выходя за рамки сущности и объема настоящего изобретения, и такие эквивалентные воплощения понимаются как часть настоящего изобретения.
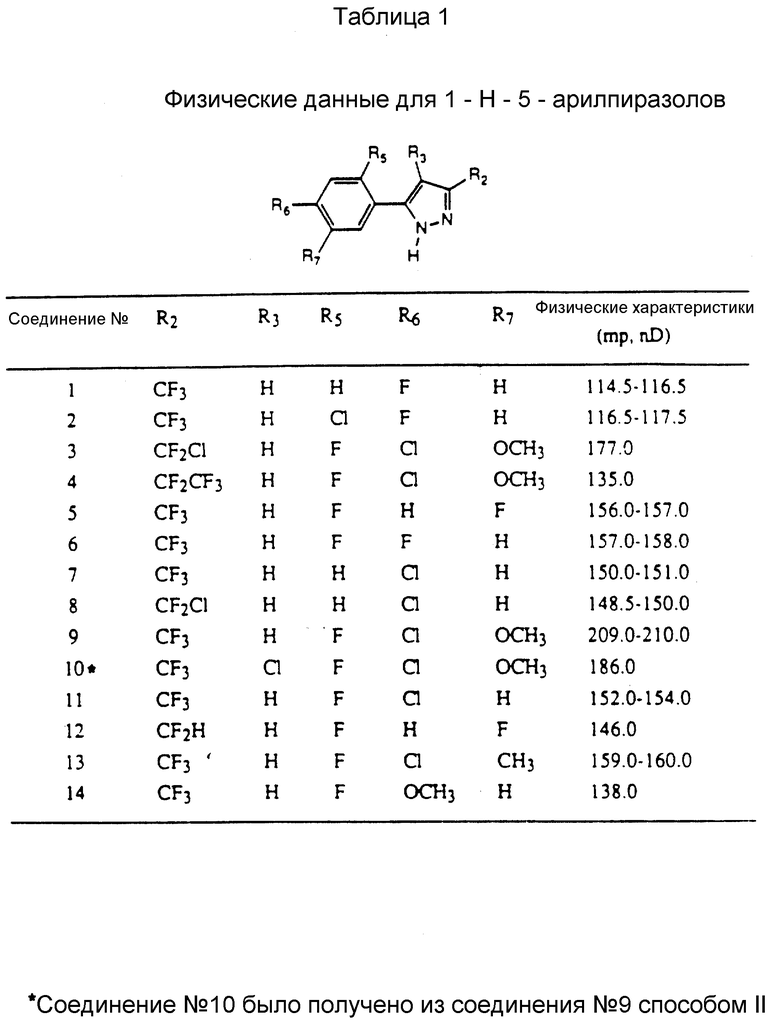
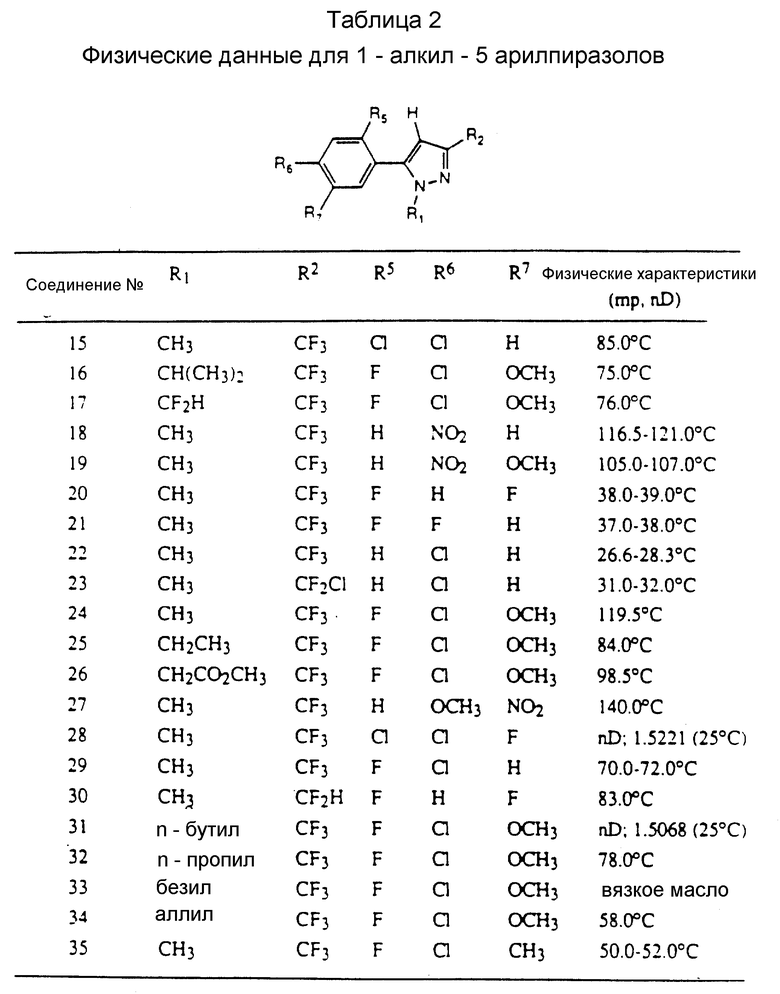
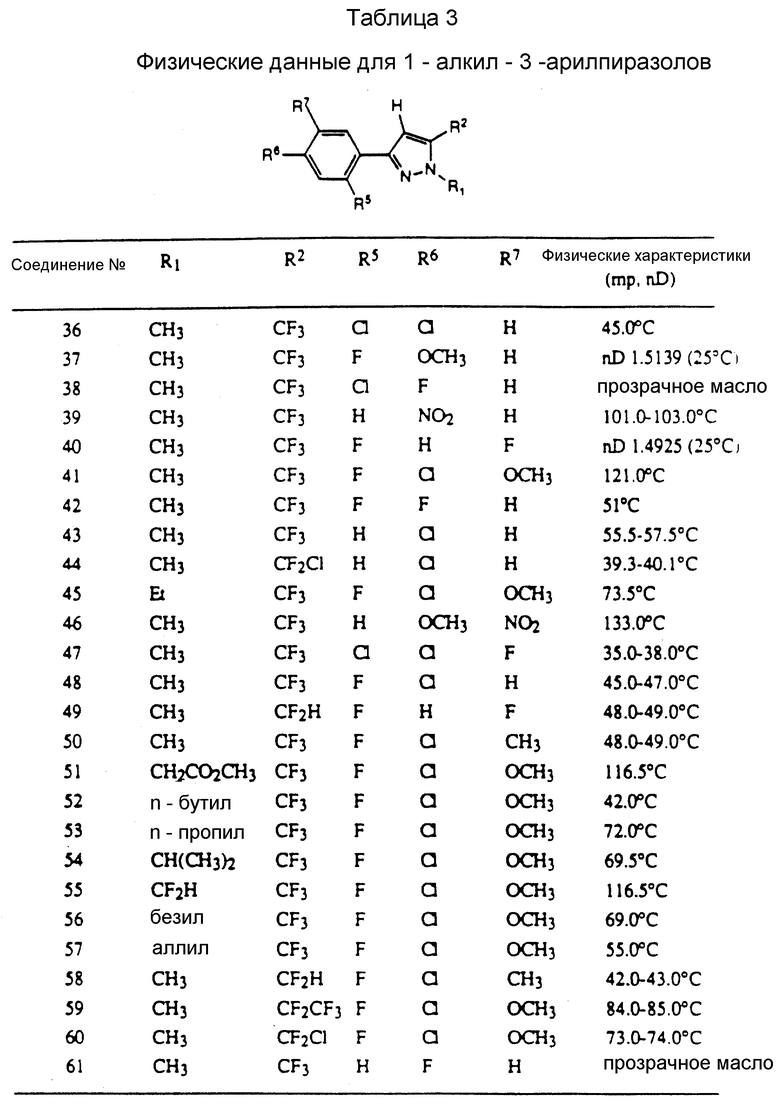
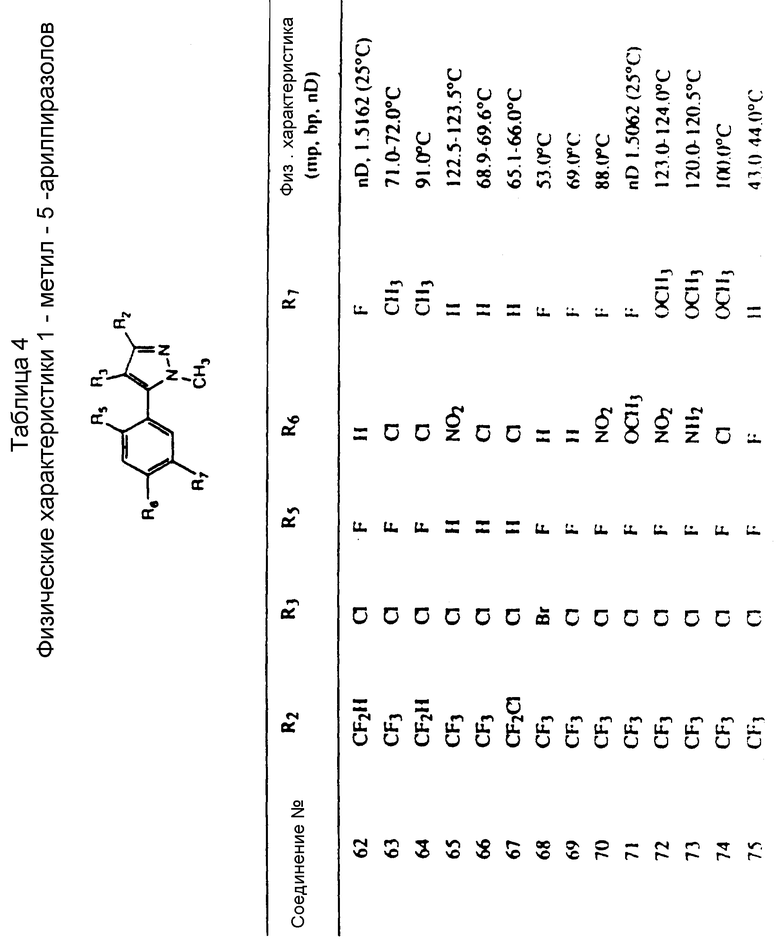
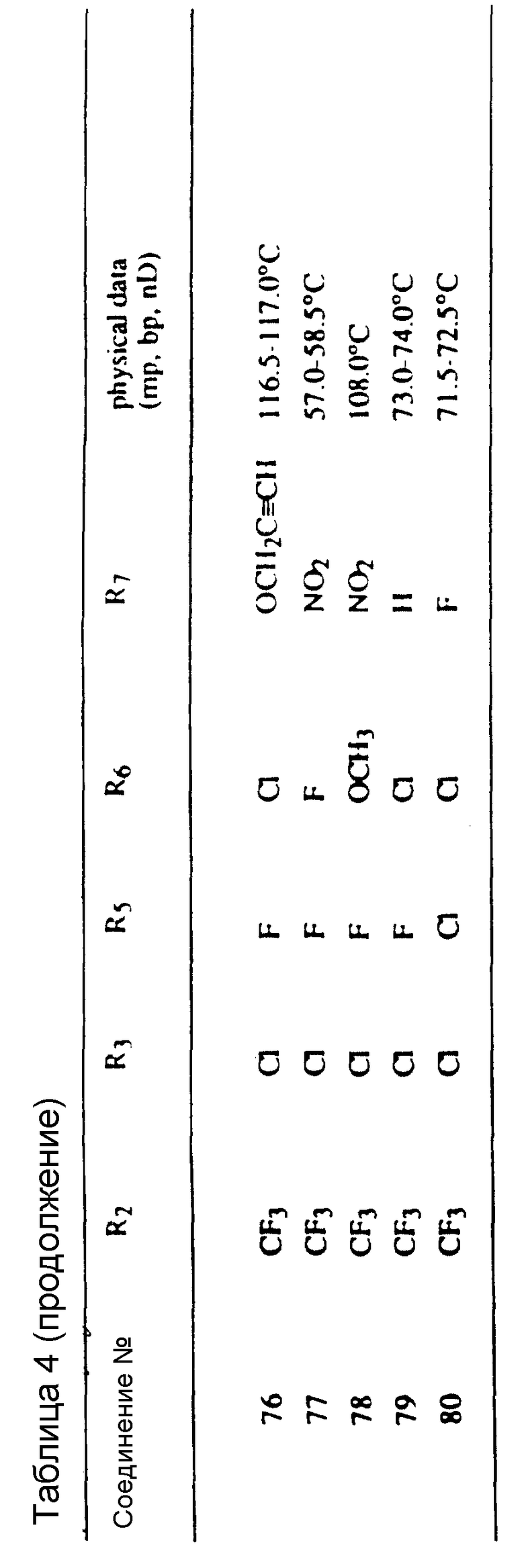
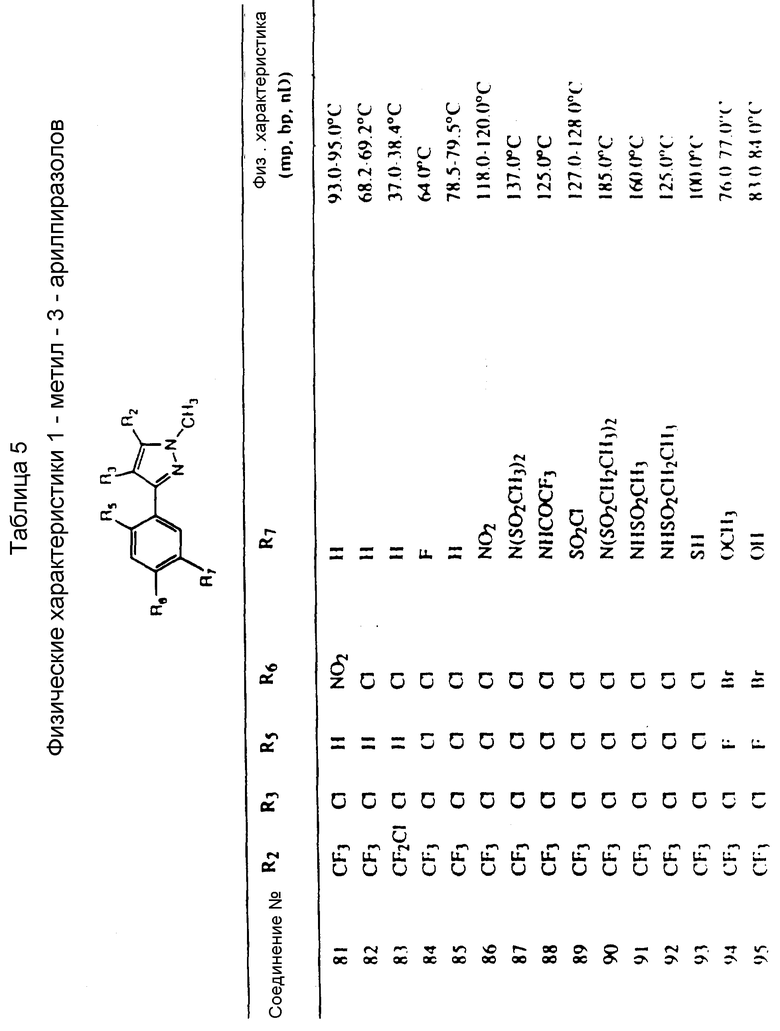
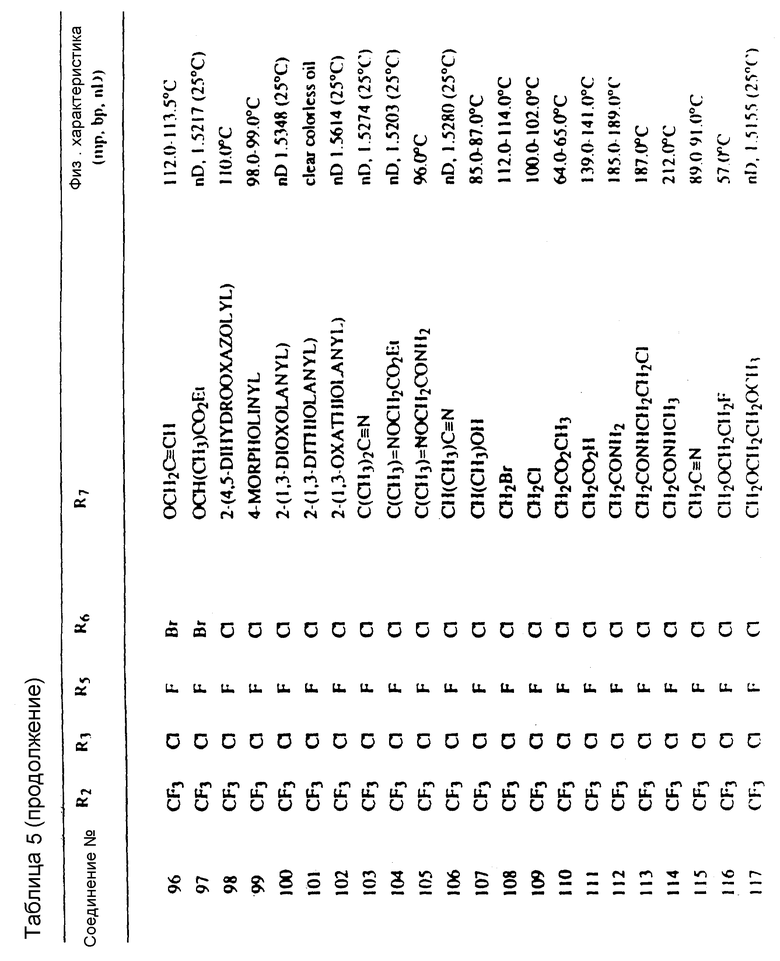
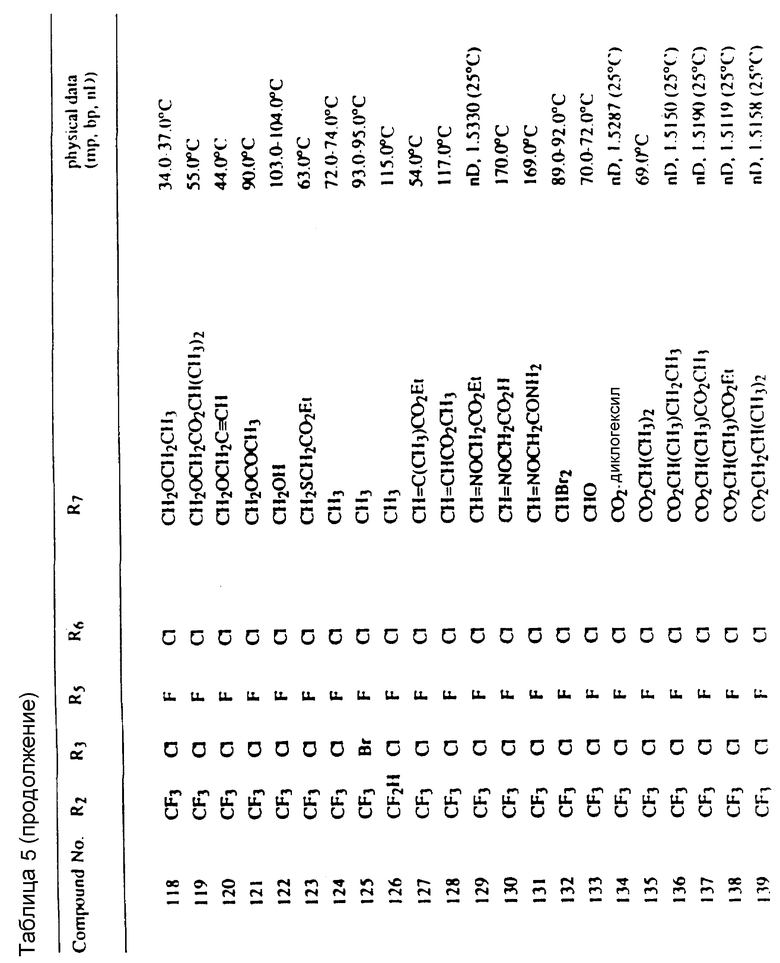
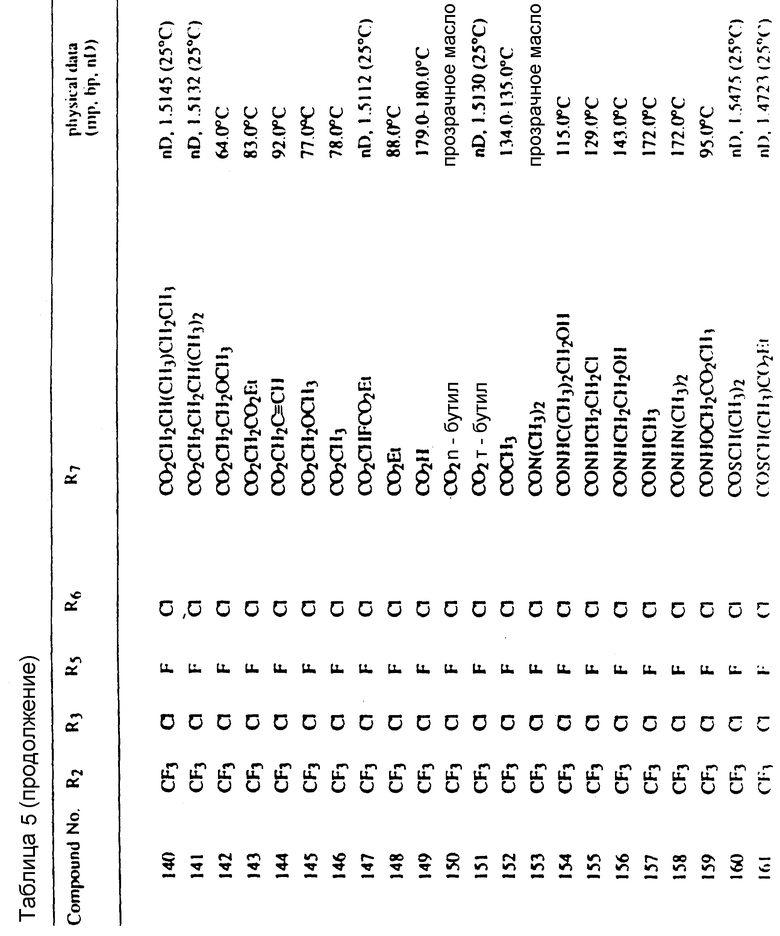
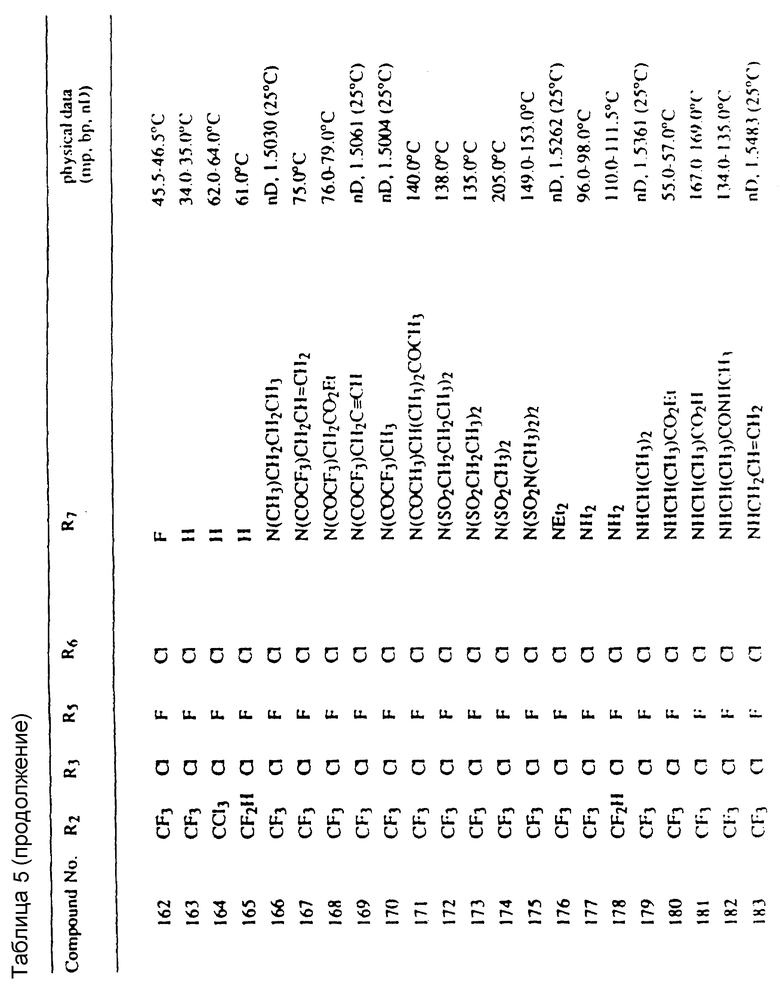
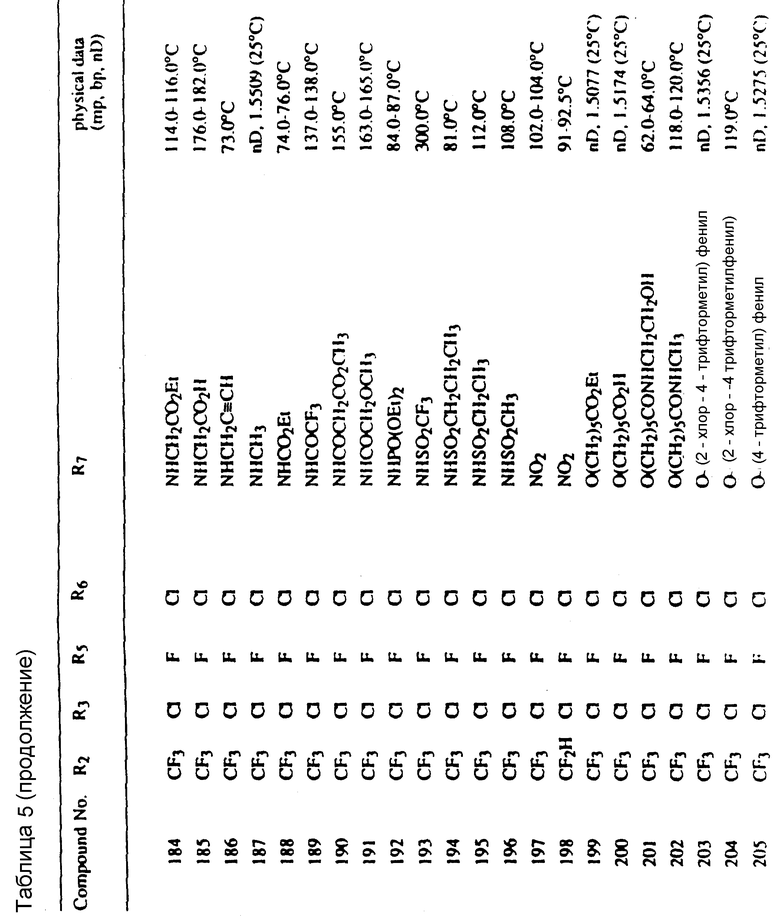
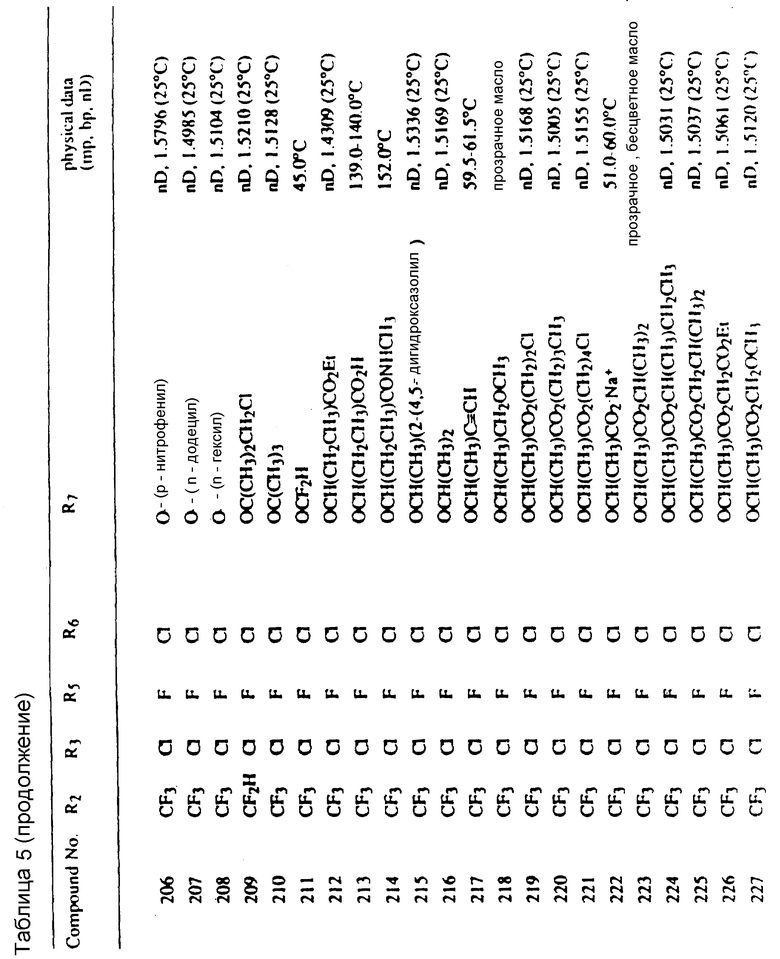
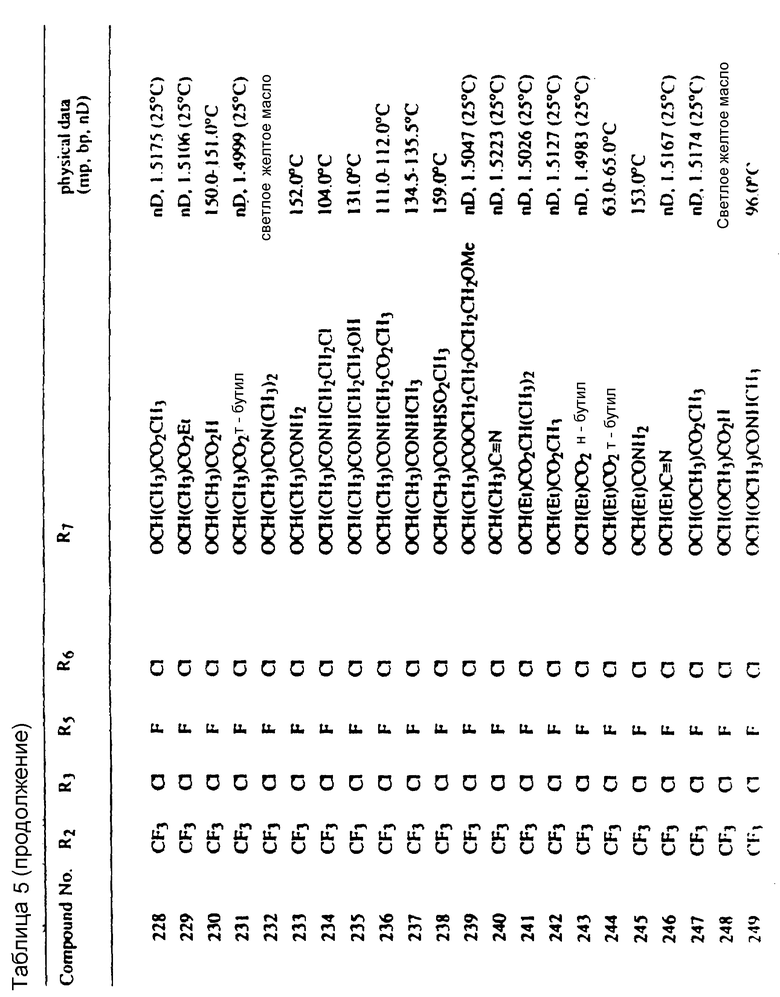
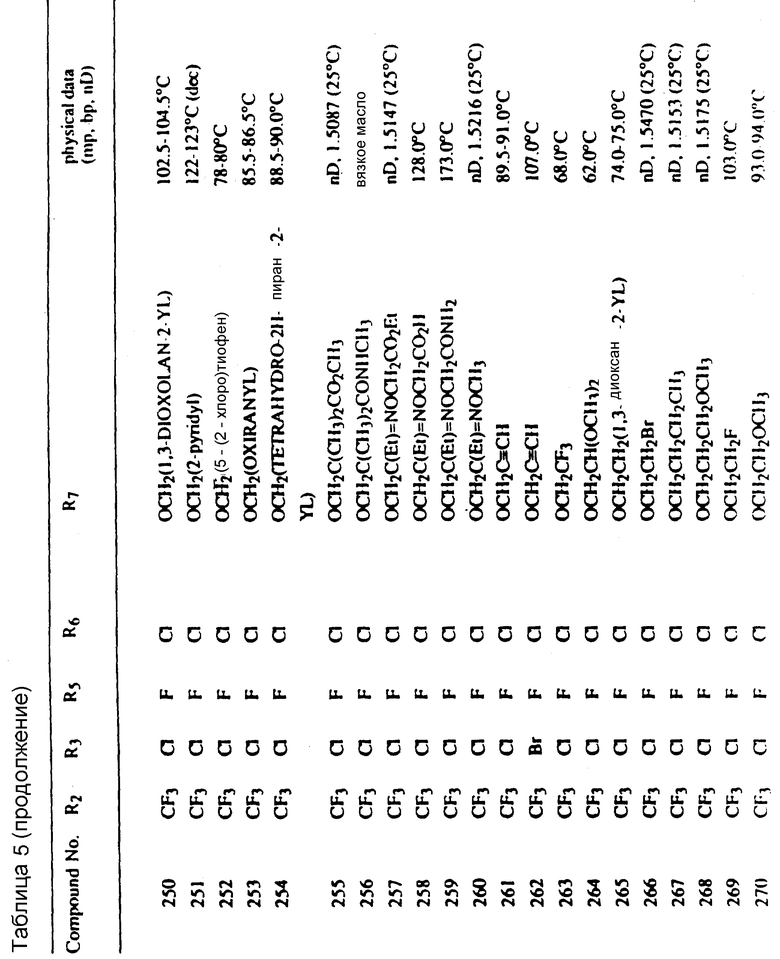
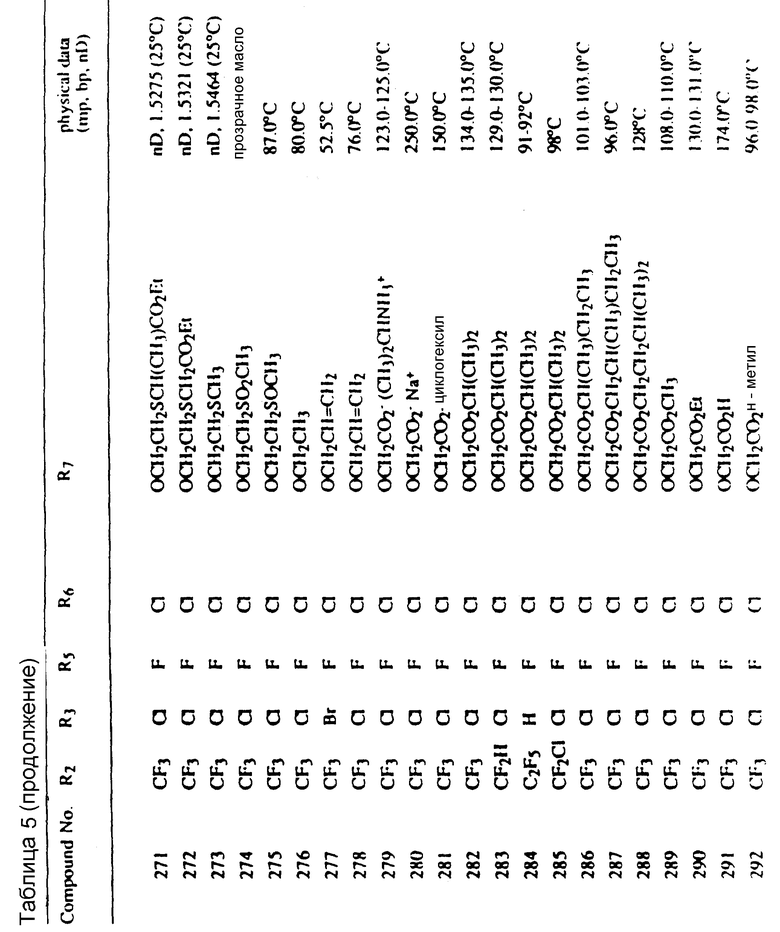
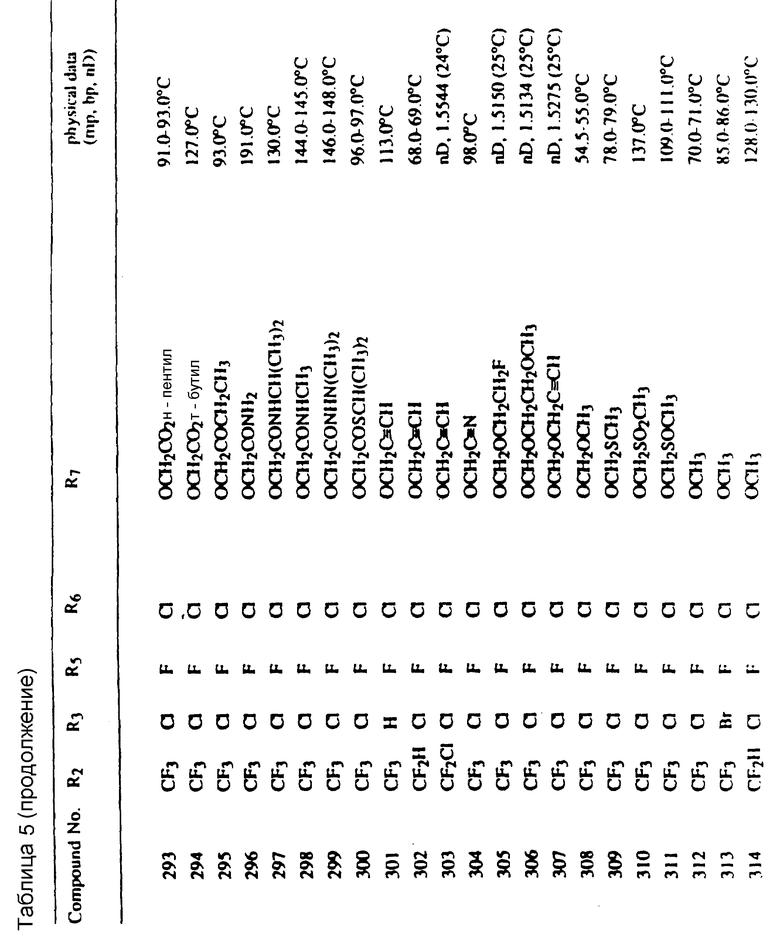
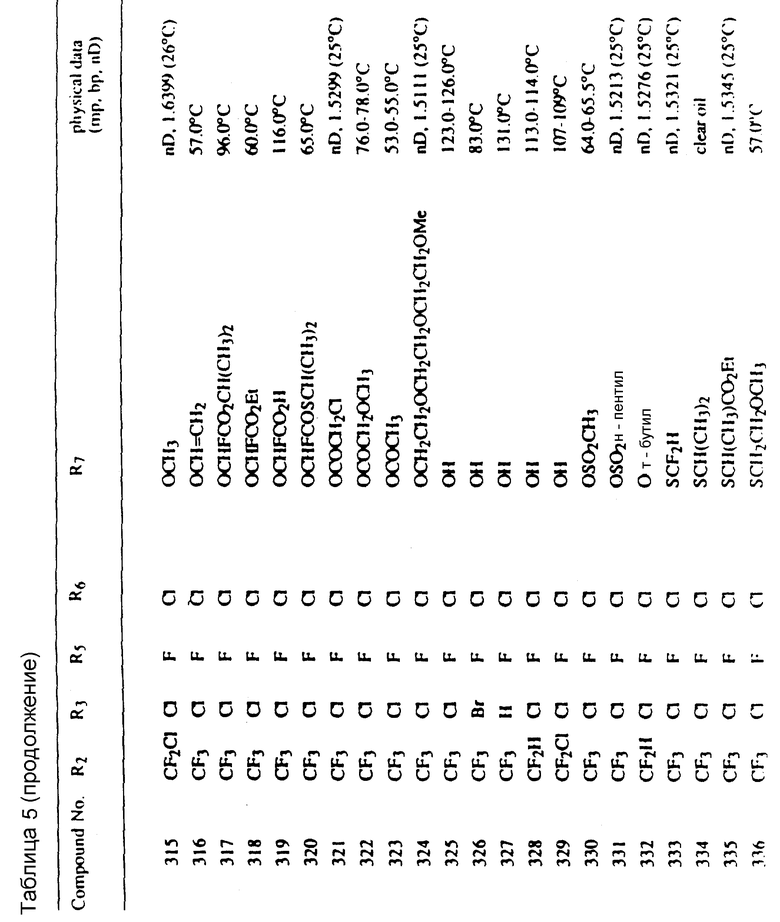
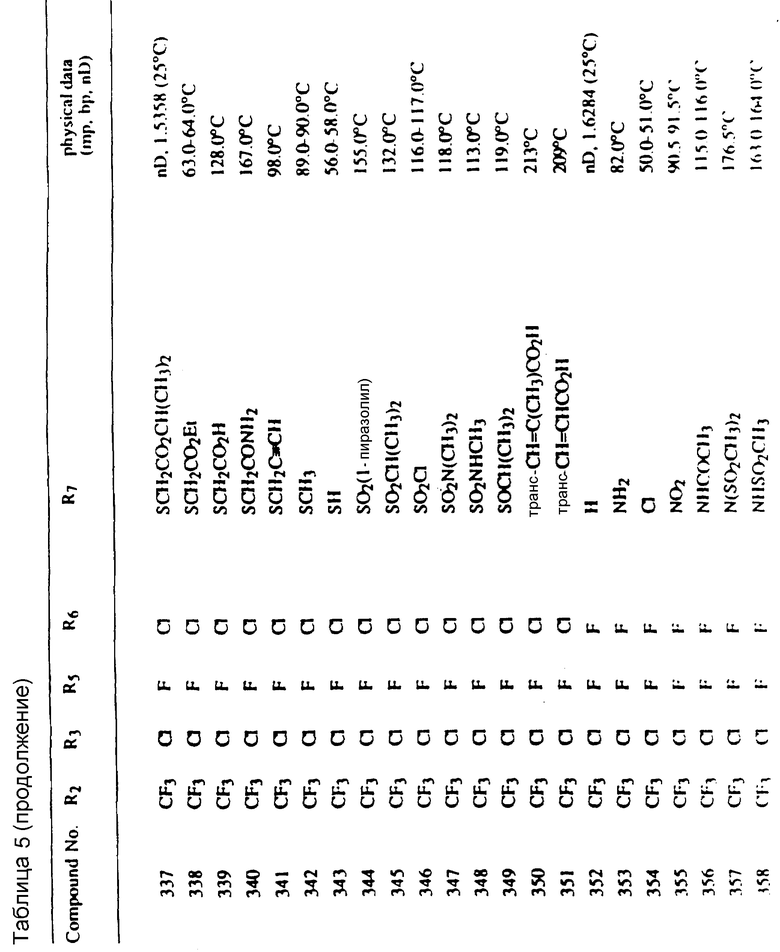
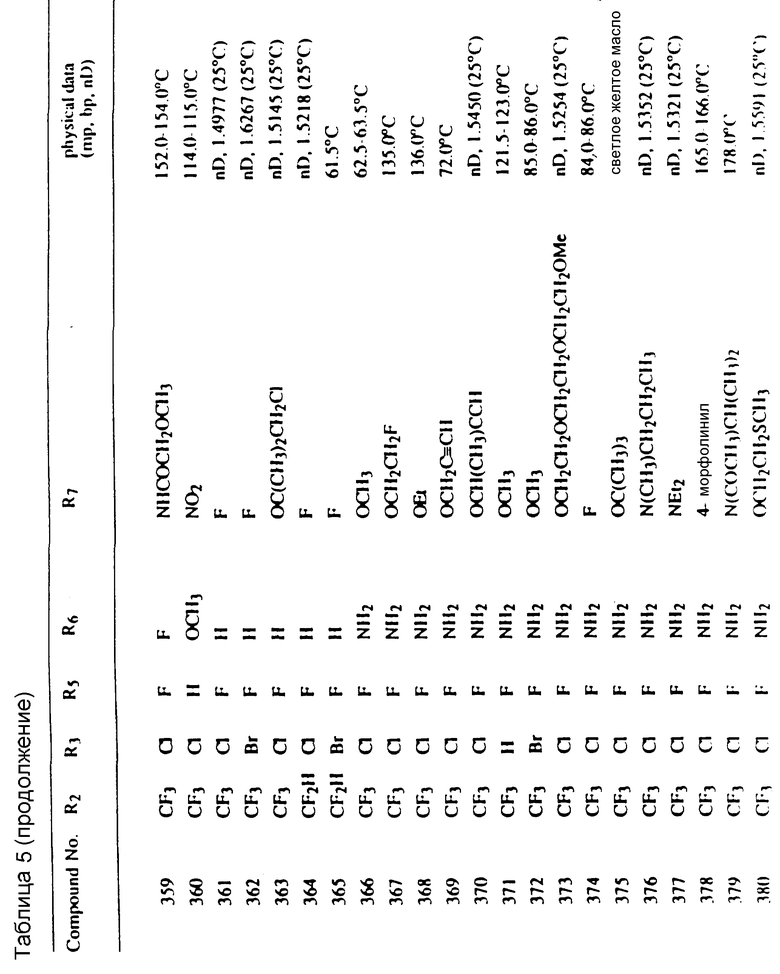
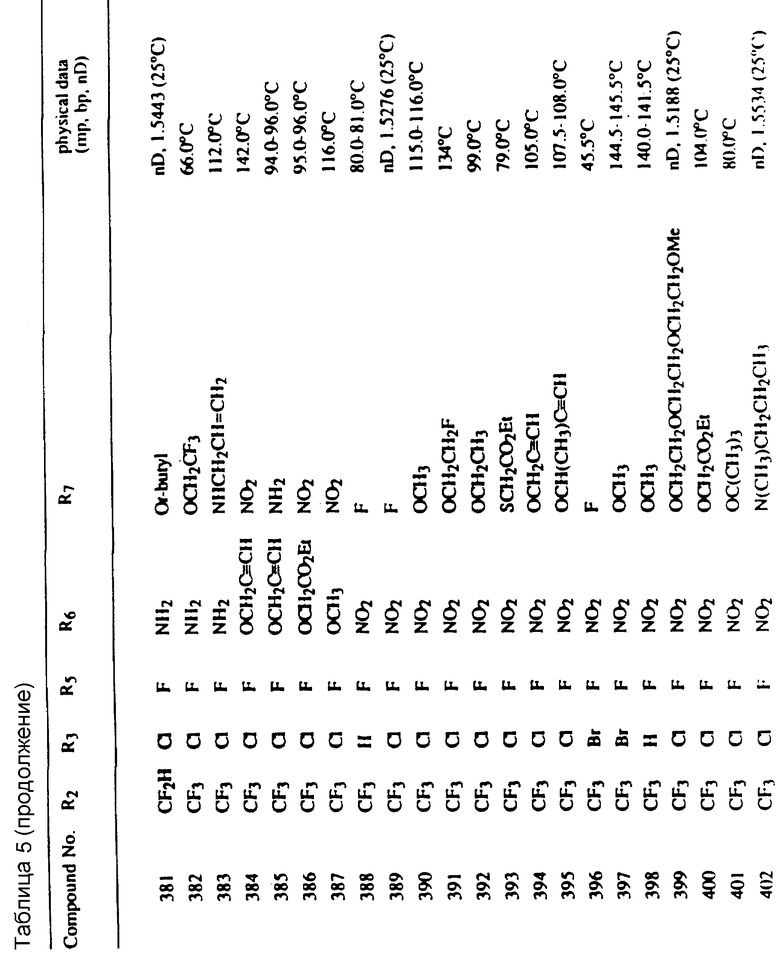
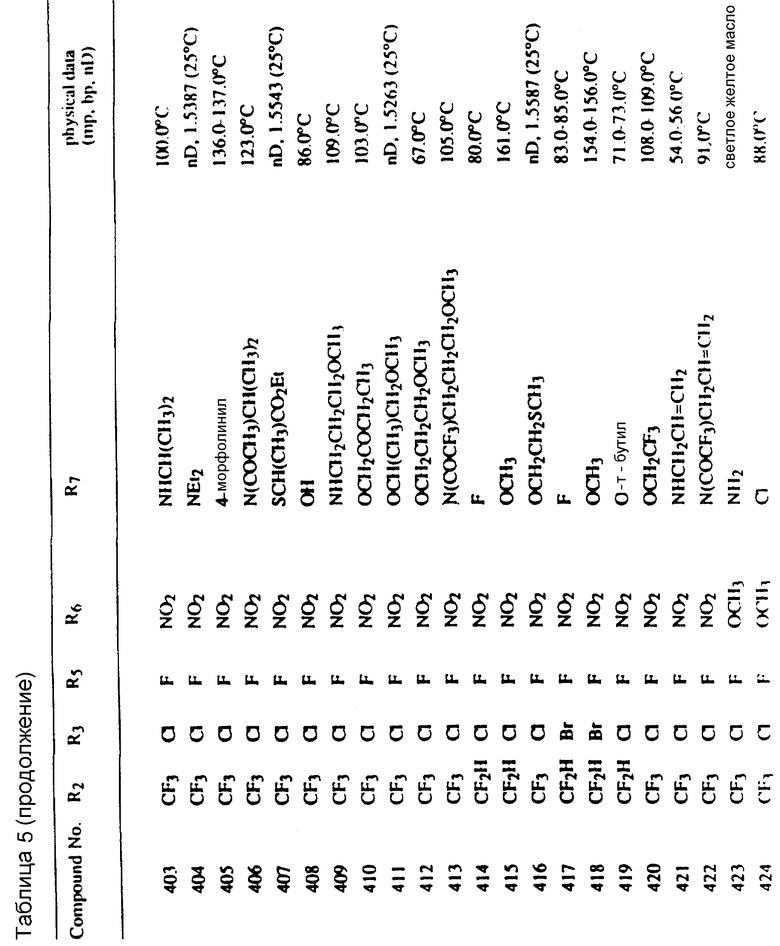

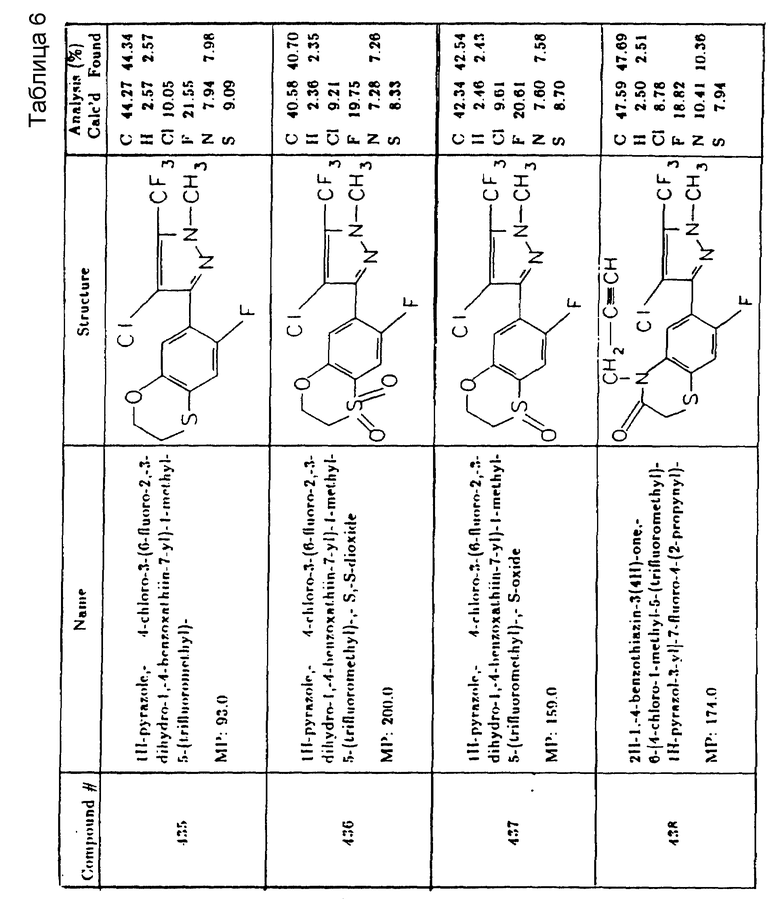
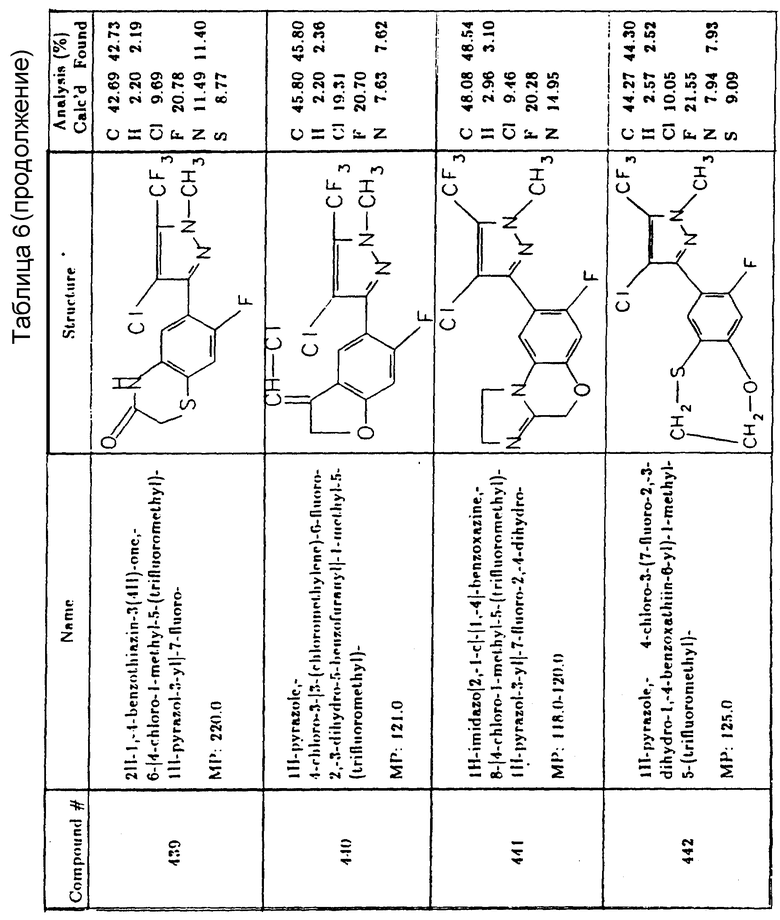
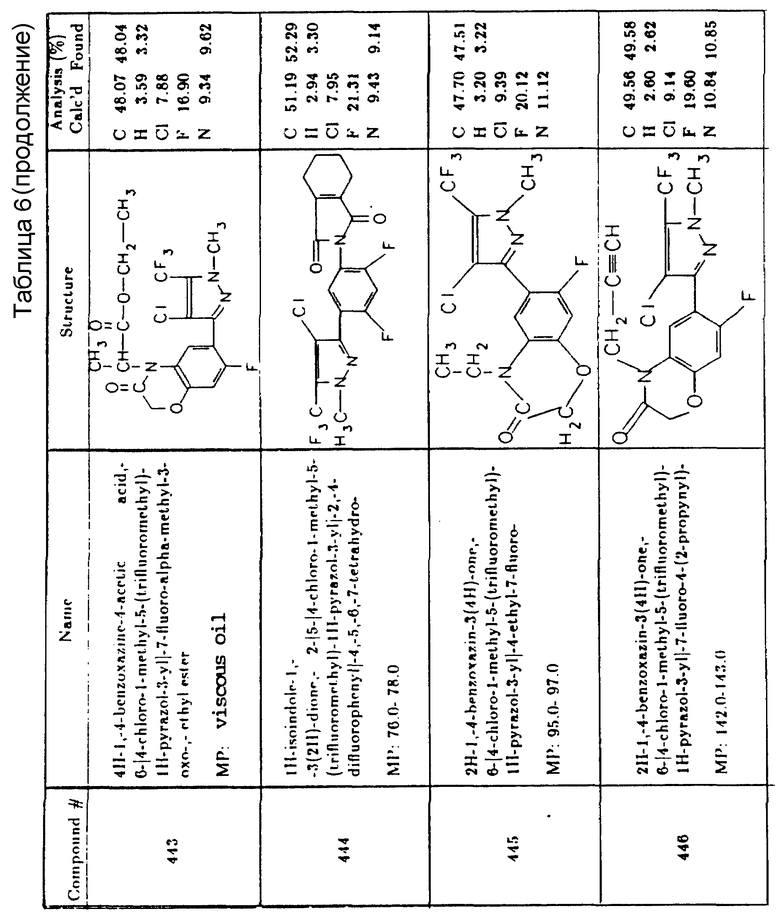
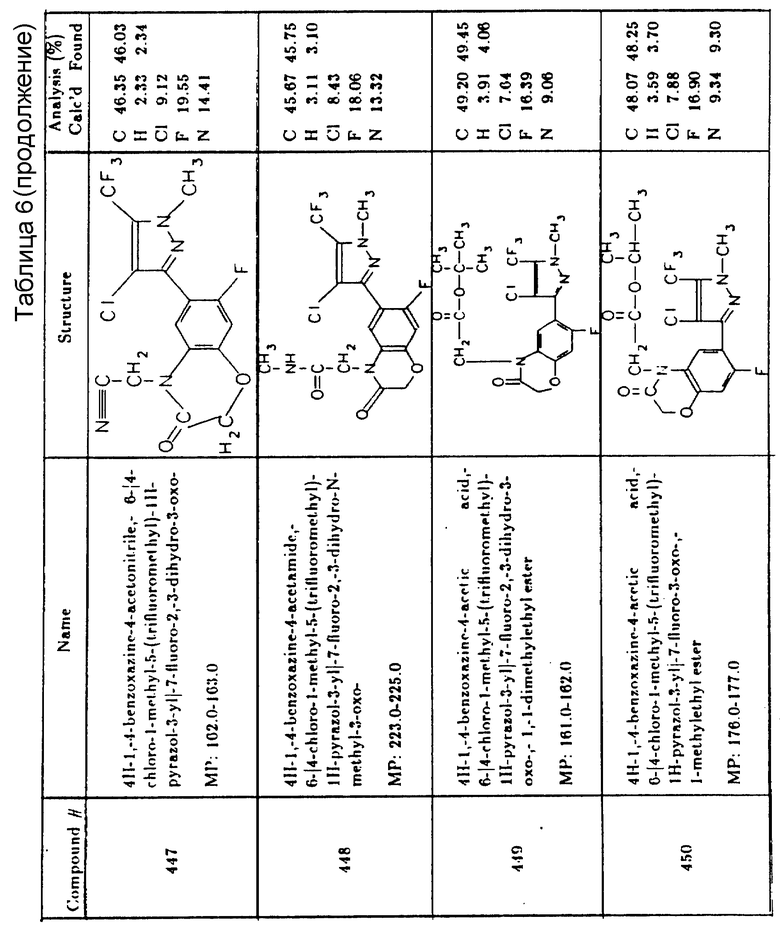
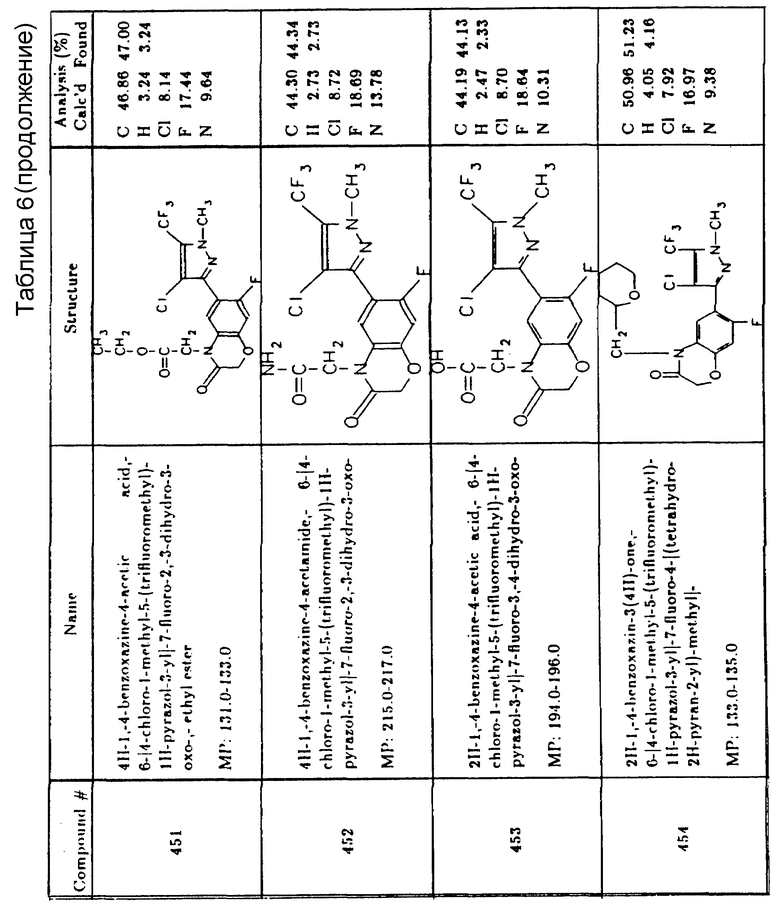
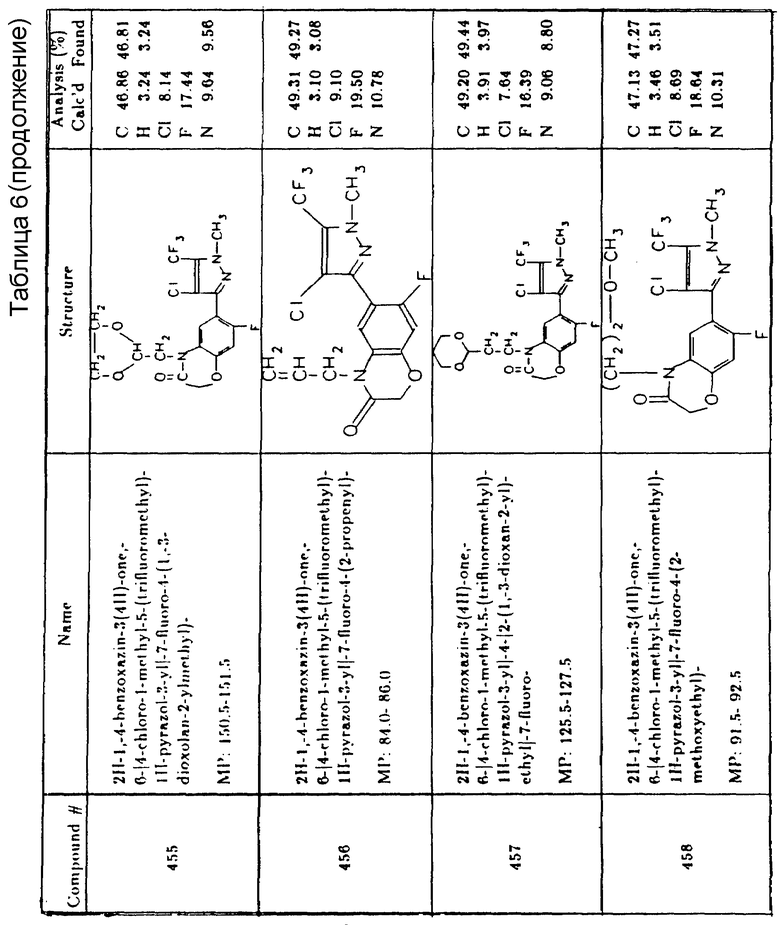
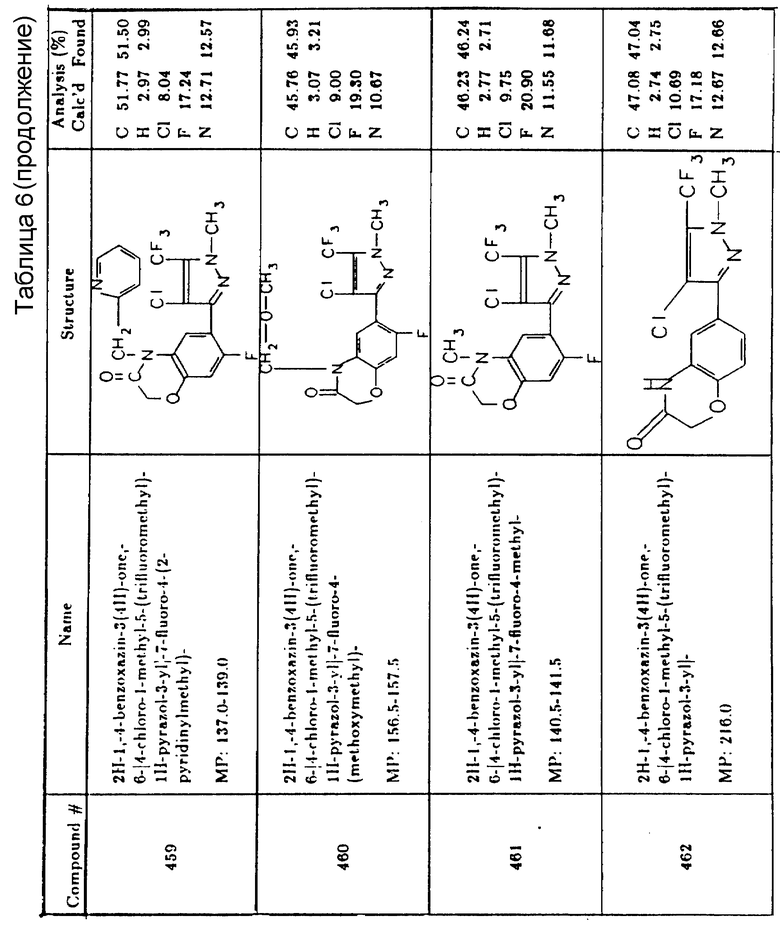
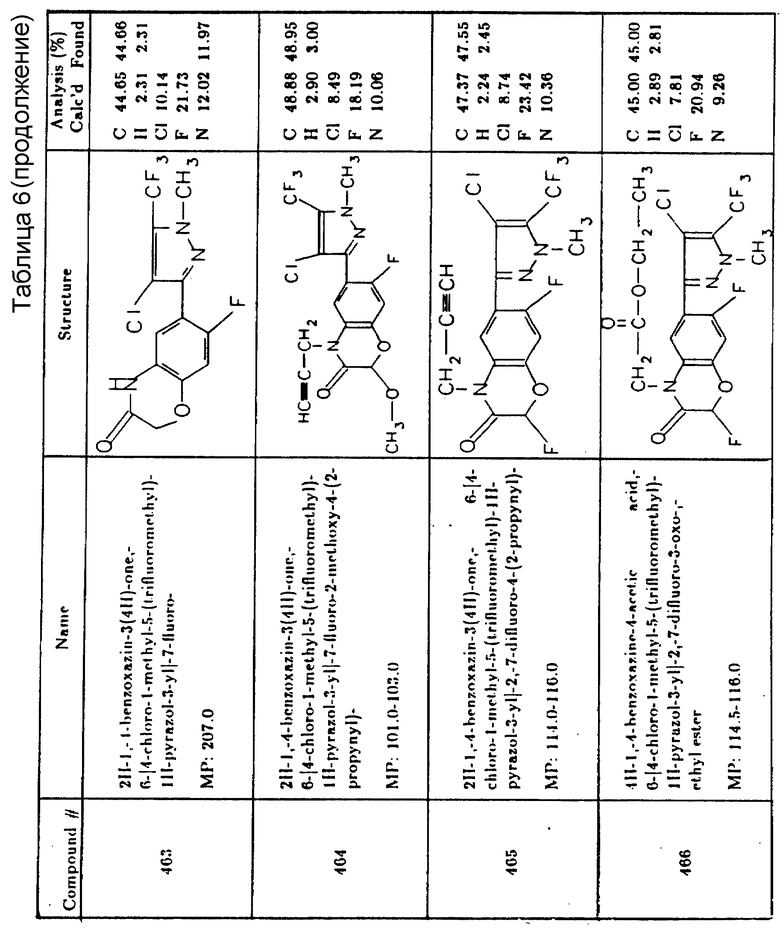
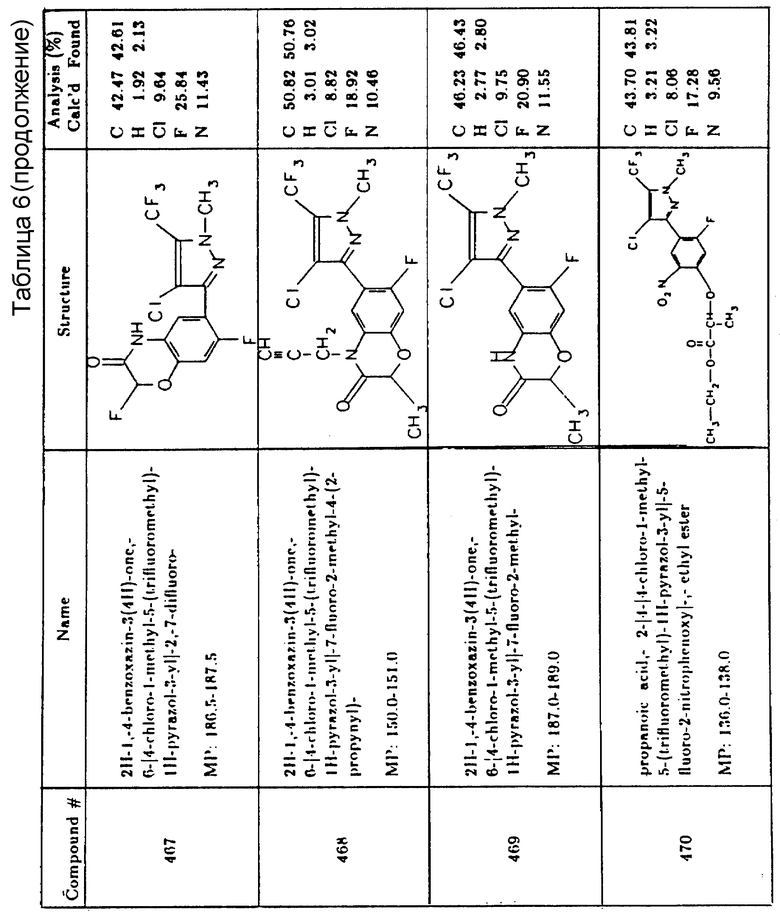
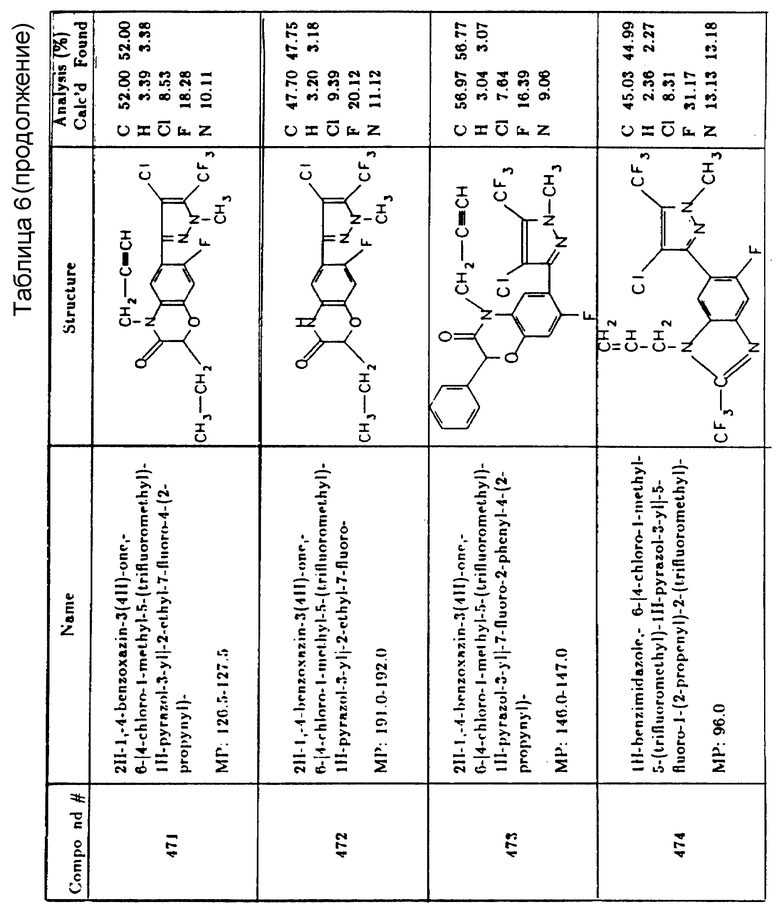
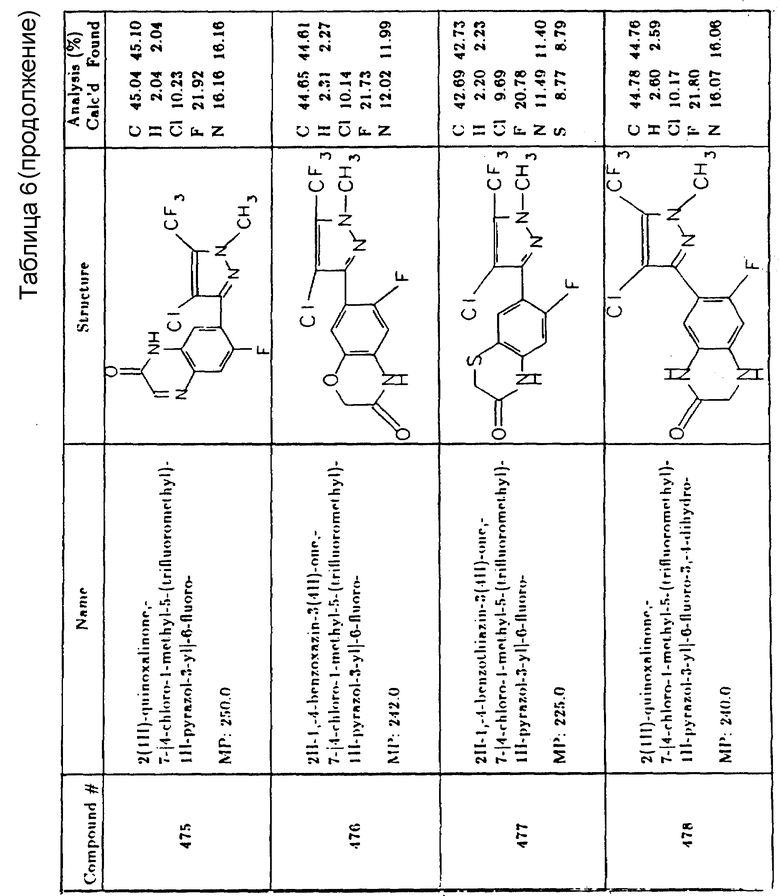
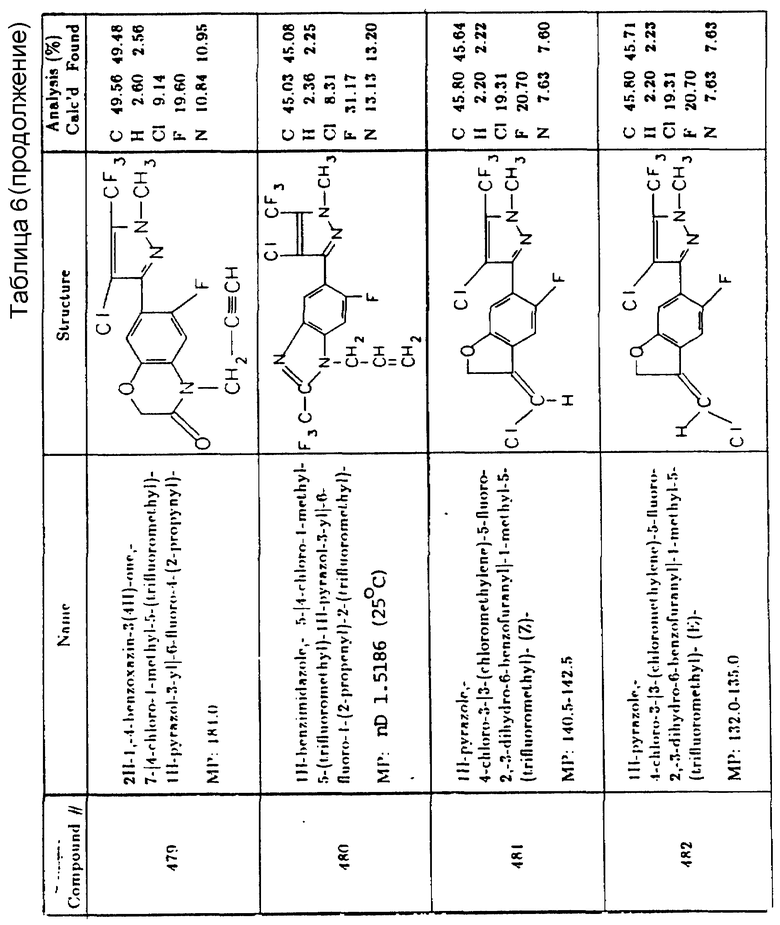
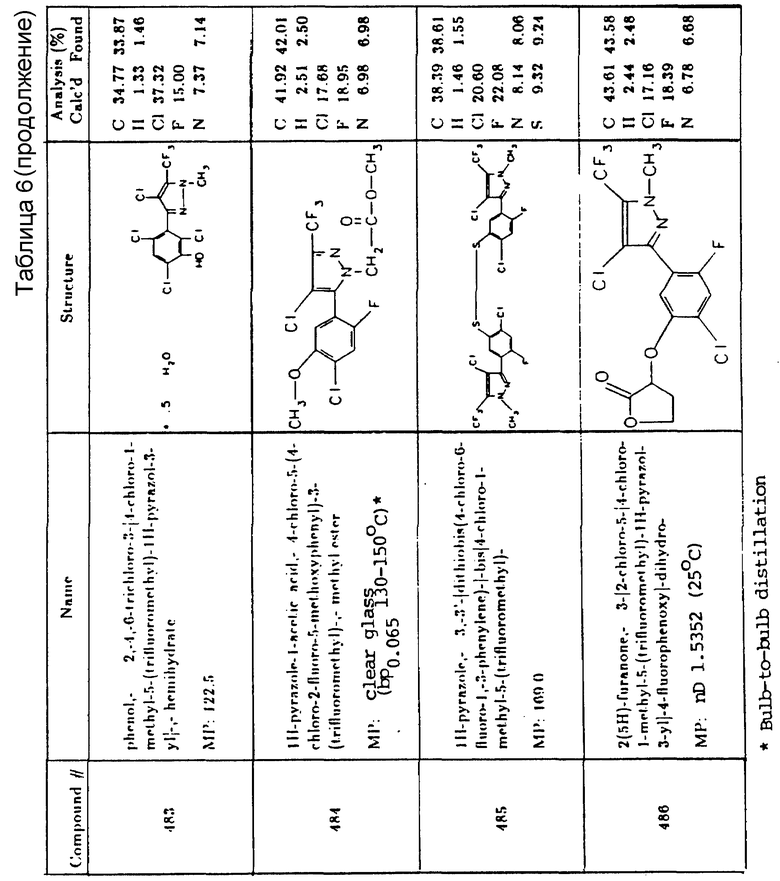
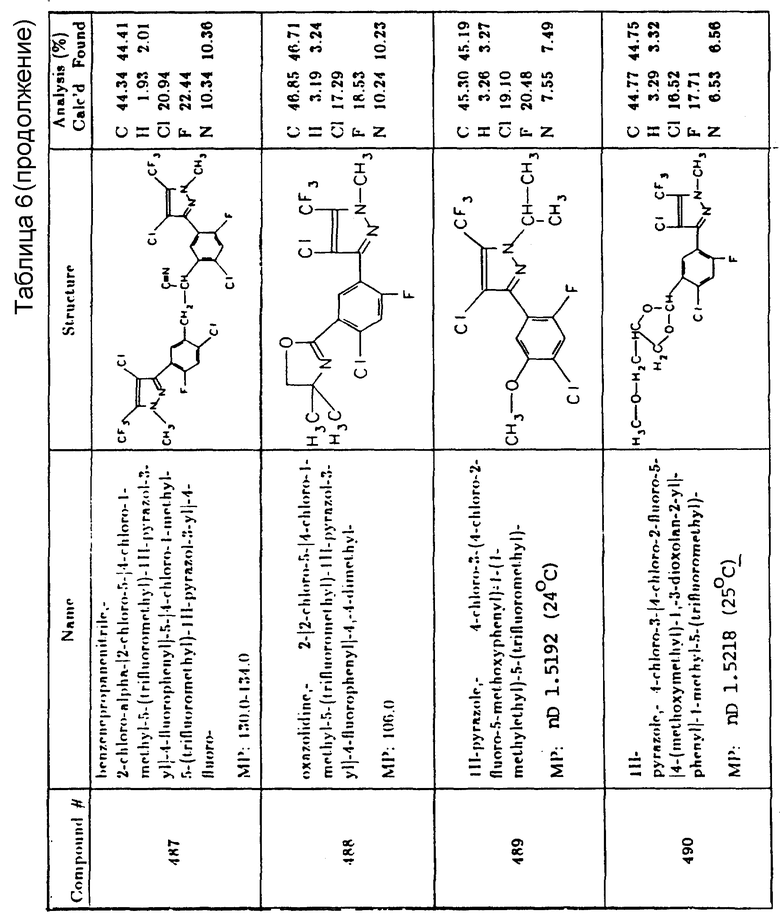
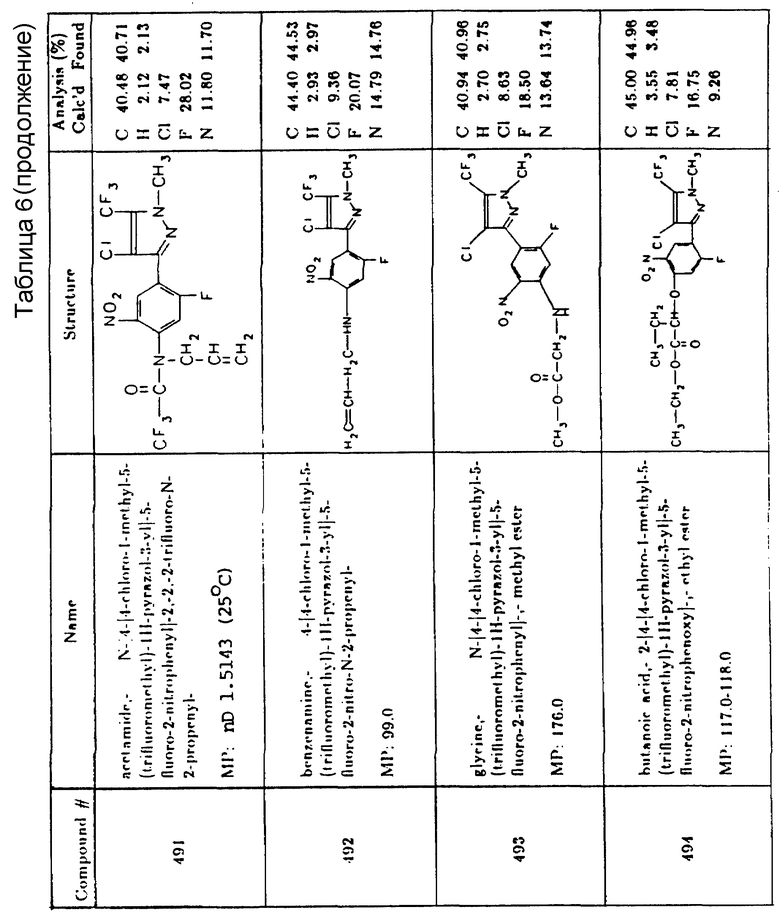
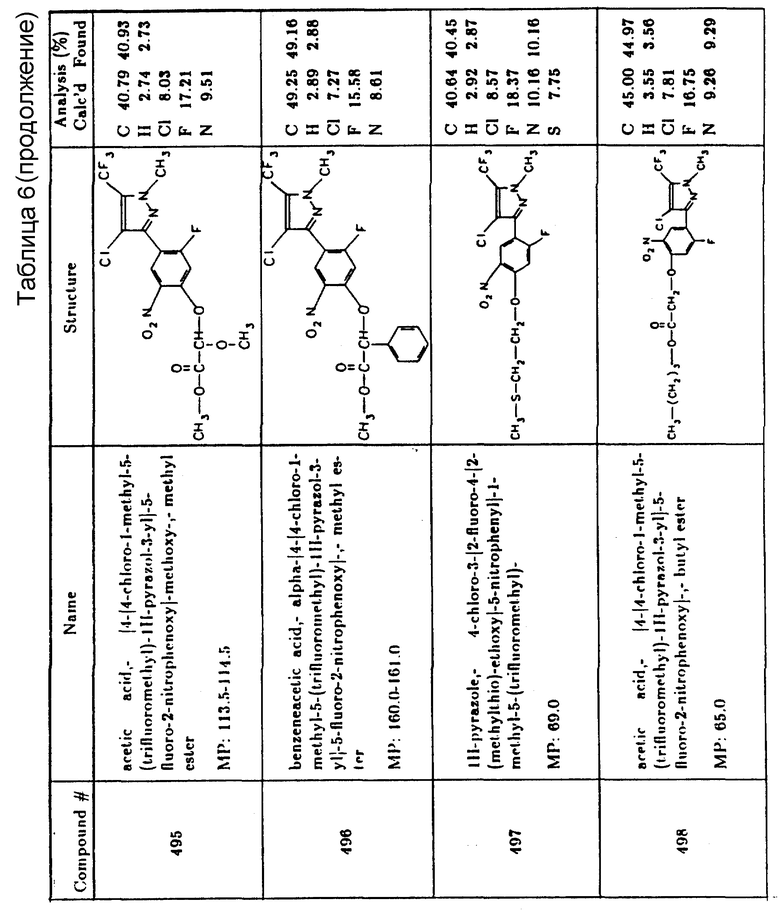
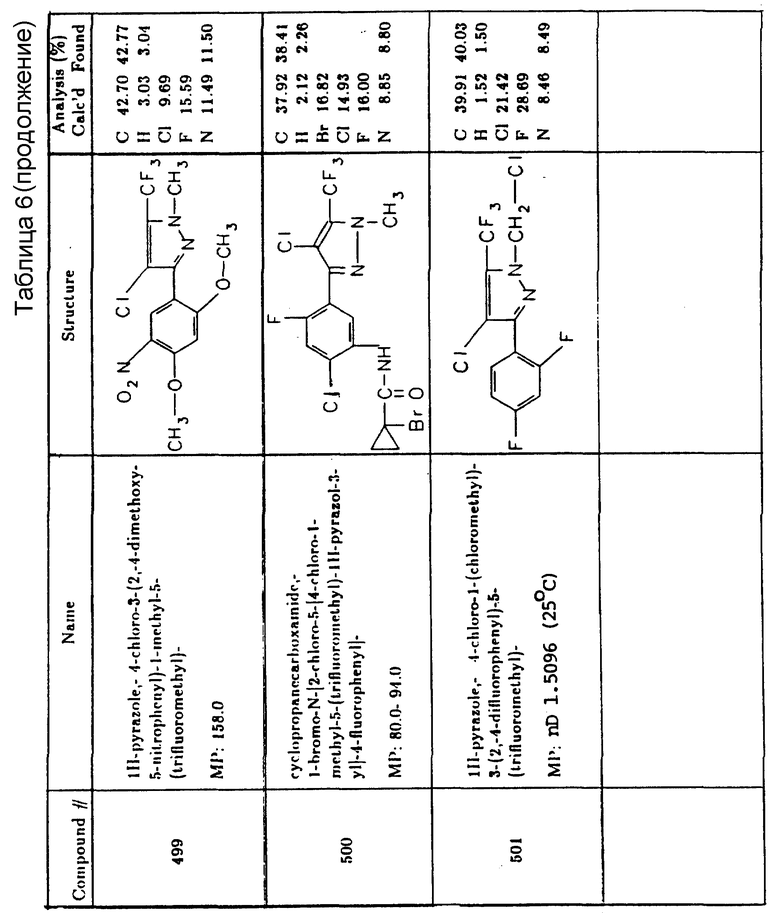











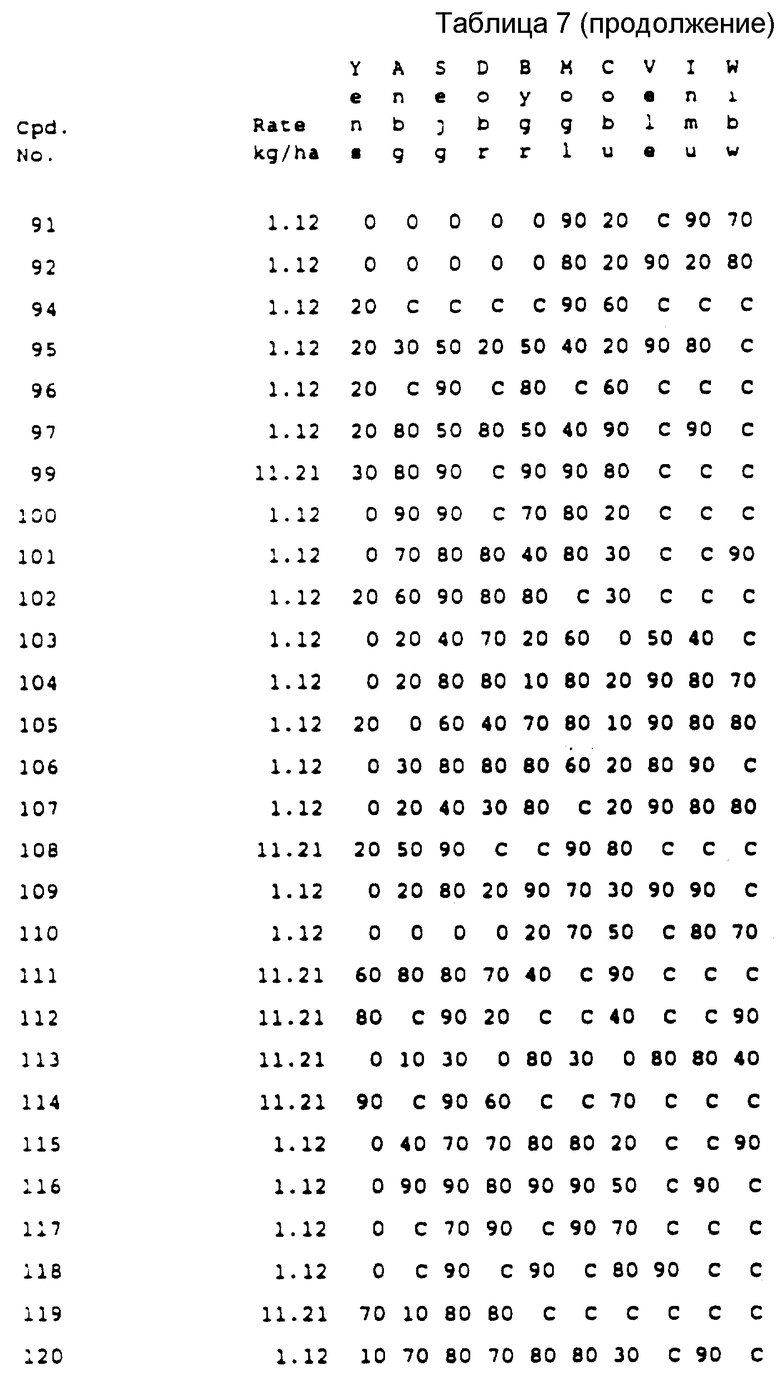








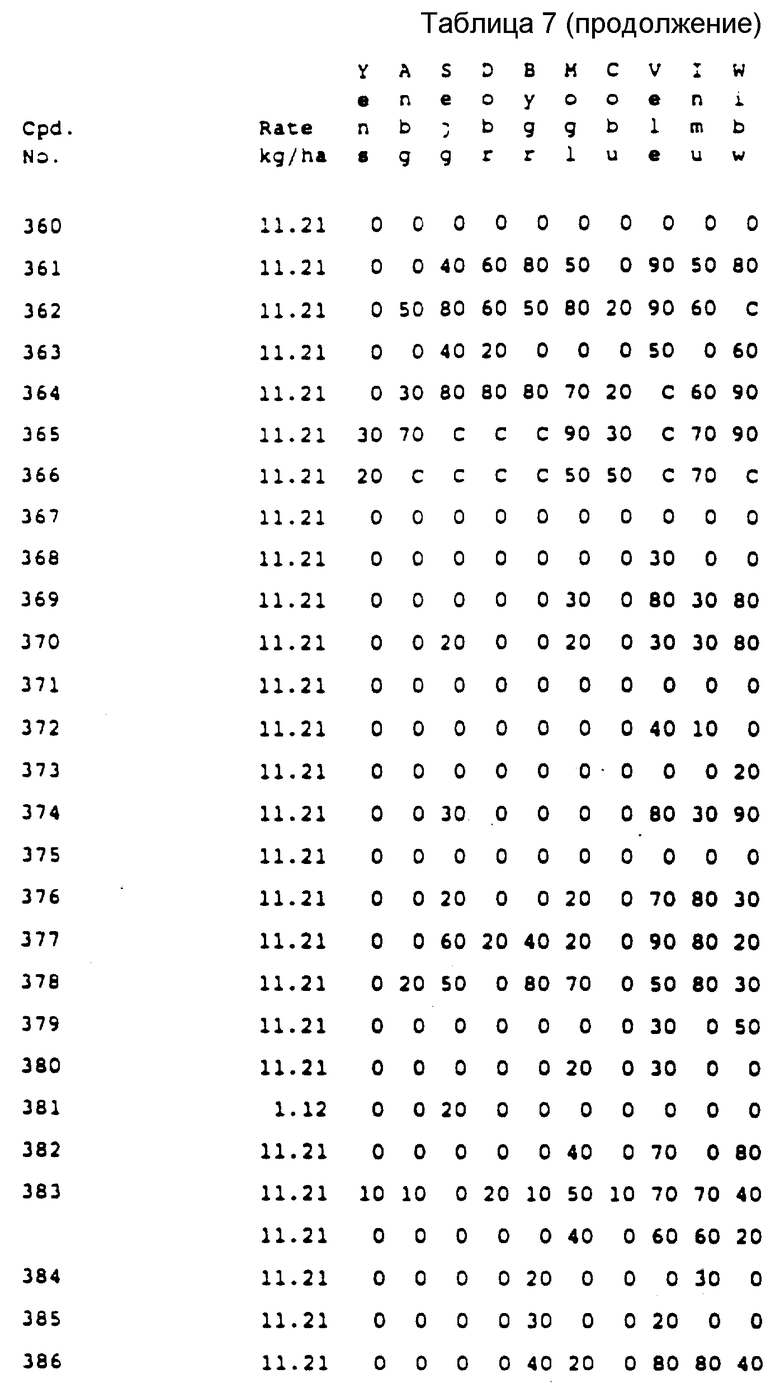

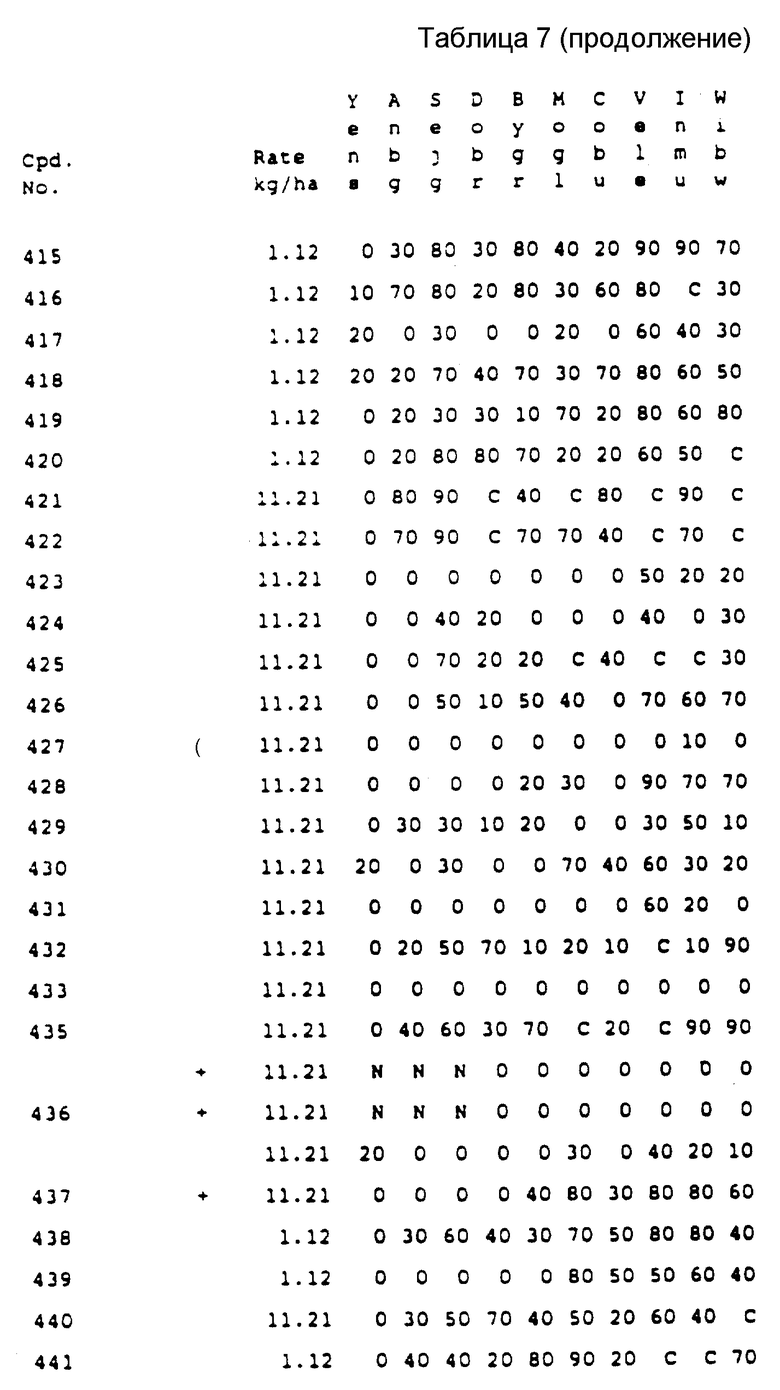


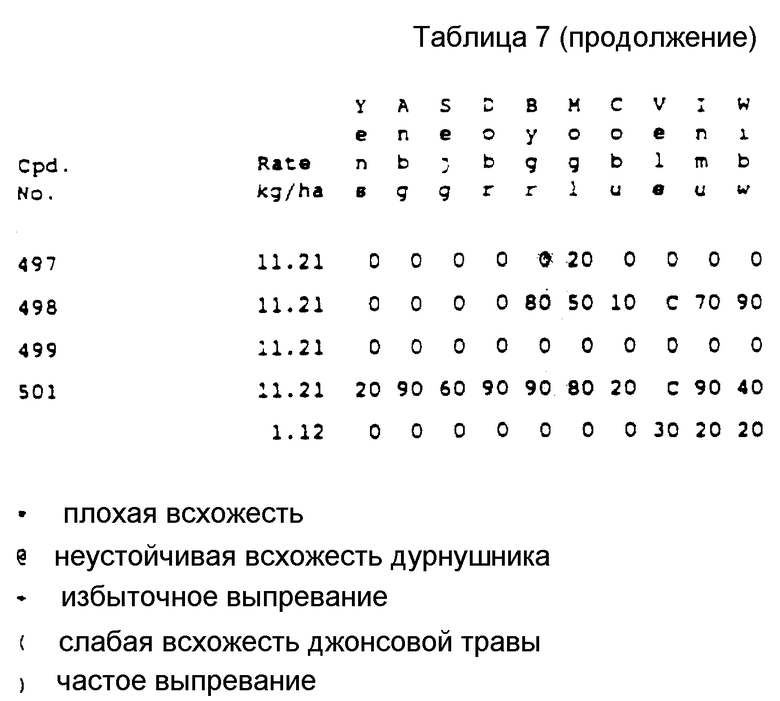
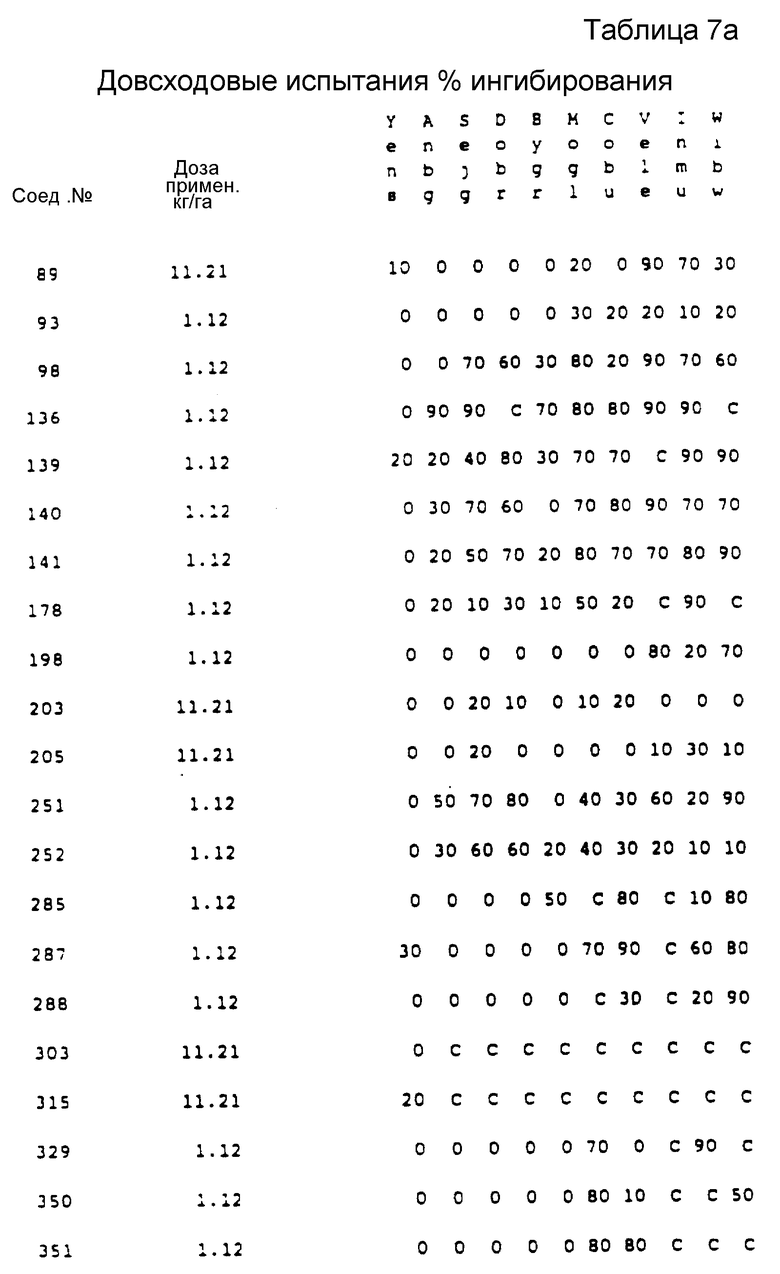











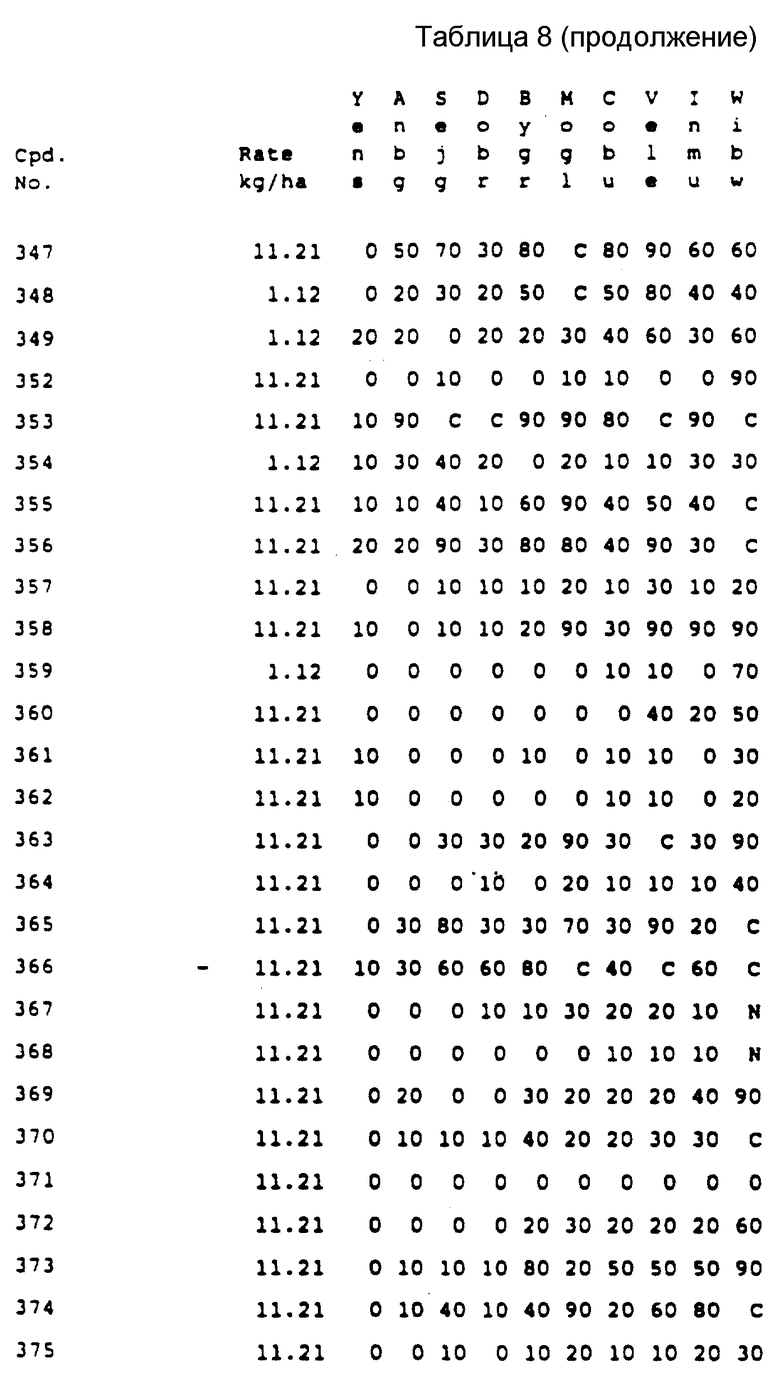


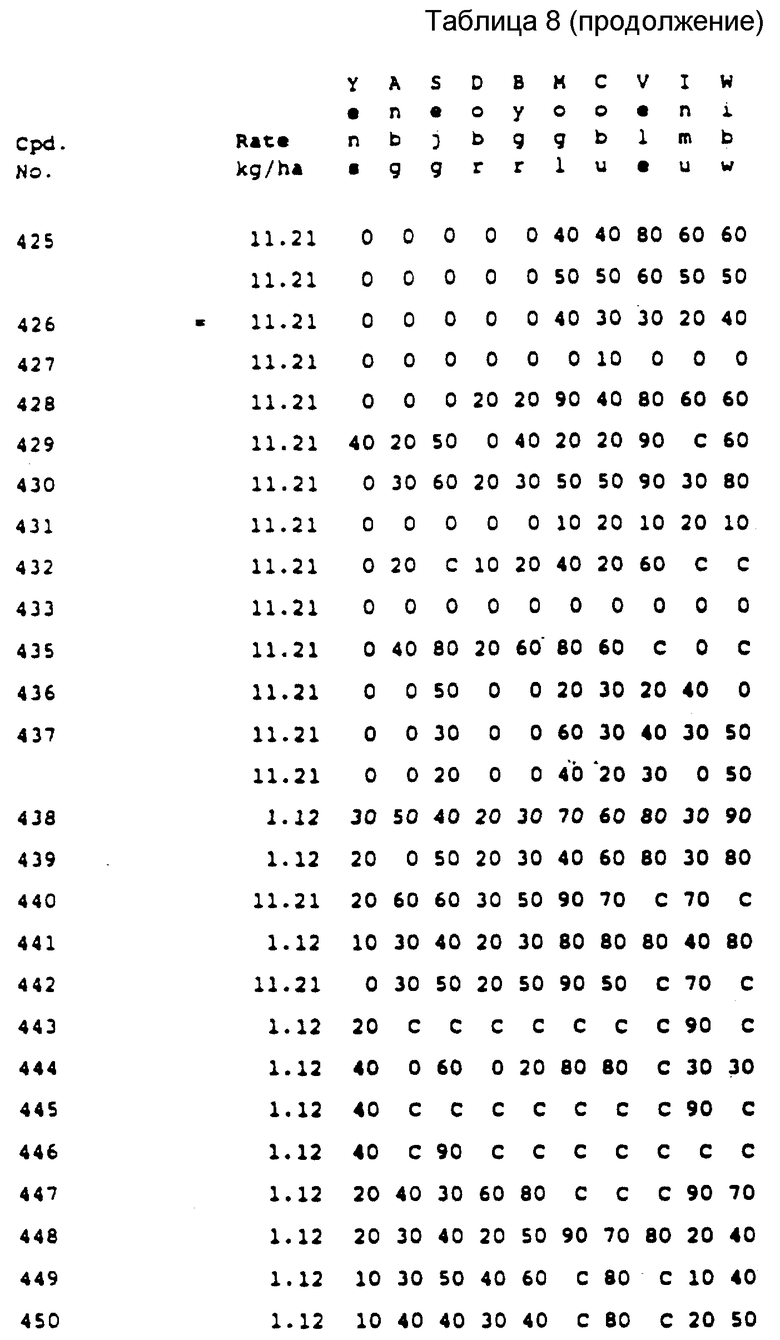

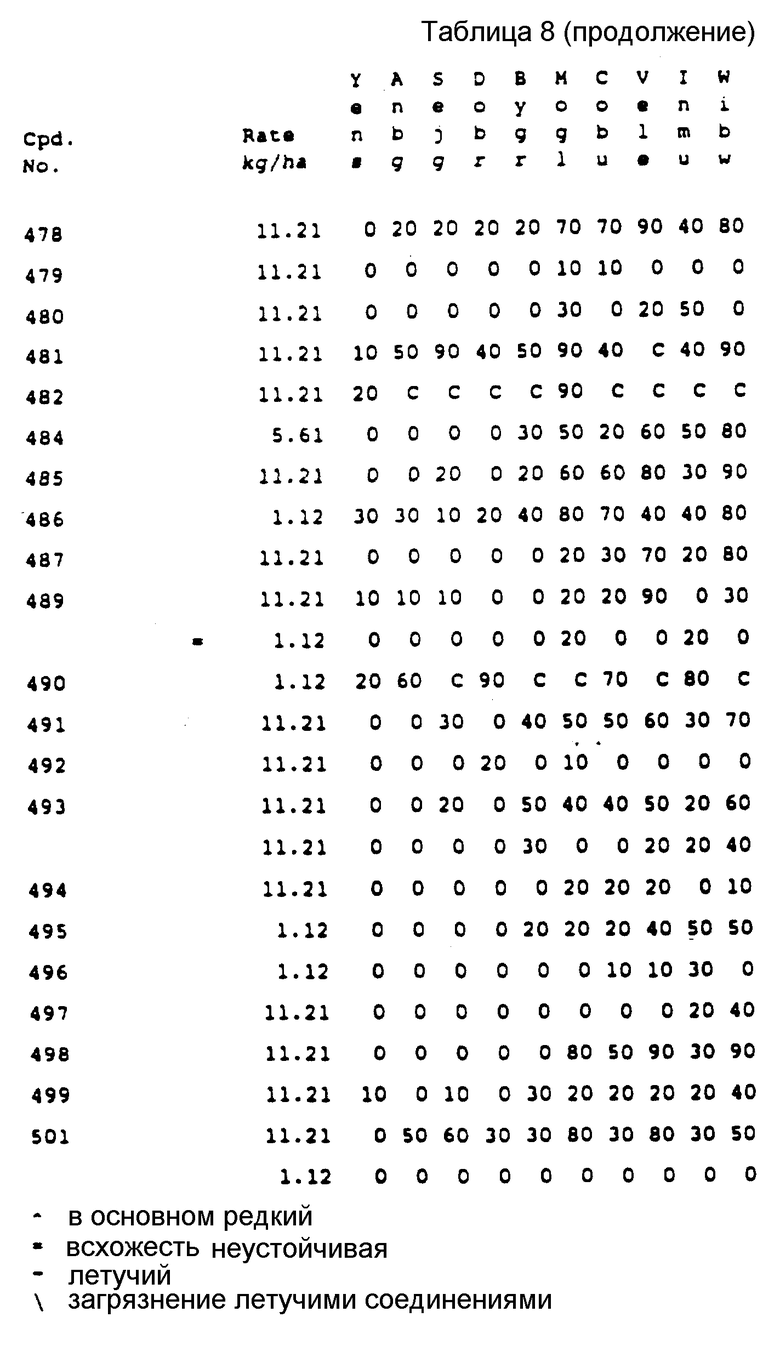


Описываются новые производные пиразола формулы I, где значения R1 - R4 указаны в п.1 формулы изобретения, которые могут найти применение в борьбе с нежелательными растениями. Описывается способ получения вышеуказанных соединений, гербицидная композиция и способ борьбы с использованием соединений фломулы I. 7 с. и 31 з. п. ф-лы, 8 табл.
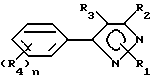
и их сельскохозяйственно-приемлемые соли и гидраты, где
R1-С1-3-алкил, галоидметил, или (С1-С3)-алкоксикарбонилметил;
R2-С1-С3-галоидалкил;
R3 - галоген;
R4 - независимо друг от друга представляют С1-3-алкил, (С2-4-алкенил)-R19, (С1-3)алкокси(С1-3)алкил, (С1-3)алкокси (С1-3)алкокси(С1-3)алкил, С1-3-галоидалкил, галоген, нитрогруппа, цианогруппа, циклические заместители, такие, как дигидрооксазолил, морфолинил, диоксоланил, дитиоланил, оксатиоланил, изоиндол-3-дион, которые могут быть незамещены или необязательно замещены заместителями R15 и R16,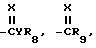
YR10, NR11R12, (C1-3-алкил)-R19, -C(CH3)=NOCH2R19, либо две группы R4 связаны с образованием кольца, которое в комбинации с фенильным кольцом дает замещенную или незамещенную бициклическую группу, которую выбирают из 1,4-бензоксатиенила, бензоксатиазинона, бензофуранила, имидазобензоксазина, бензоксазина, бензоксазинона, бензимидазола или хиноксалинона, каждая из которых может быть замещена в гетероциклическом кольце одной или двумя группами R22;
Х - независио O или S;
Y - O или S(O)m;
m - 0-2;
n - от 1 до 4;
R8 - водород, С3-7 - циклоалкил, С1-5-алкил, С2-5-алкенил, С2-5-алкинил, (С1-4-алкил)-R19 или (С1-4-галоидалкил)-R19;
R9 - водород, С1-6-алкил или NR17R18 или NHOR20;
R10 - водород, галоген, C1-10-алкил, С1-4-галоидалкил, С2-4-алкенил, С2-4-алкинил, (С1-5-алкил)-R19, (С1-4-галоидалкил)-R19, (С=О)-R16, пиразолил, NR11R12 или фенил, необязательно замещенный одним или двумя заместителями R15 и R16
R11 и R12 независимо представляют водород, С1-4-алкил, С2-4-алкенил, С2-4-алкинил, -(С=Х)-YR13, -(С=Х)-R14, -РO(R14)2, (С1-3-алкил)-R19, YR20;
R13 - H, С1-3-алкил, С1-3-галоидалкил, С1-3-алкоксильная группа, С2-4-алкоксиалкил, С1-3-алкоксикарбонилметил;
R14 - Н, С1-3-алкил, С1-3-галоидалкил, С1-3-алкоксильная группа, С2-4-алкоксиалкил, С1-3-алкоксикарбонилметил;
R15 и R16 независимо представляют водород, галоген, нитрогруппу, С1-3-алкил, С1-3-галоидалкил, С2-4-алкоксиалкил;
R17 и R18 независимо представляют водород, С1-4-алкил, С1-4-галоидалкил, С1-4-гидроксиалкил;
R19 - водород, нитрил, ацетил, COYR20, CONHR20, YR20, С(С1-3-алкил)NOCH2R23, 1,3-диоксолан-2-ил, 2-пиридил, 2-хлортиофен, (С1-3алкил)-дигидрооксазолил, (С1-3алкил)-оксиранил, (С1-3алкил)-тетрагидро-2Н-пиранил, (С1-3алкил)-диоксоланил;
R20 - С1-5-алкил, С1-4-галидалкил, С2-8-алкоксиалкил, С1-4-гидроксиалкил, С3-6-циклоалкил, С1-3-алкоксикарбонилметил, С1-3-алкоксикарбонилэтил, N(CH3)2, SO2CH3;
R22 - С1-4-алкил, (С1-4-алкил) - R23, С2-4-алкоксиалкил, С3-8-циклоалкоксиалкил, С2-5-алкенил, С3-5-алкинил, пиридинилметил, фенил;
R23 - СООН, СОNН2, СОNH(С1-3-алкил), СОО(С1-3-алкил), тетрагидрофуранил, 1,3-диоксанил или 1,3-диоксоланил.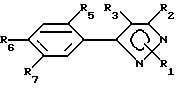
и их сельскохозяйственно-приемлемые соли и гидраты, где
R1, R2 и R3 определены для формулы I;
R5 - галоген или водород;
R6 и R7 имеют значения, определенные для R4 формулы I.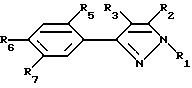
и их приемлемые в сельском хозяйстве соли и гидраты, где
R1 - С1-3-алкил,
R2, R3 и R5 имеют значения, определенные в п.2;
R6 - галоген, нитрогруппа, циано группа или YR10;
R7 - COOR8;
R8 определен в п.1;
Y представляет собой 0.
4-хлор-3-(4-хлор-2-фтор-5-пропаргилоксифенил)-1-метил-5-(трифторметил)-1Н-пиразола,
2-(2-хлор-5-(4-хлор-1-метил-5(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)пропановой кислоты, этилового эфира,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)уксусной кислоты, 1-метилэтилового эфира,
4-хлор-3(4-хлор-2-фтор-5-(метоксиметокси)фенил-1-метил-5-(трифторметил)-1Н-пиразола,
4-хлор-3-(4-хлор-2-фтор-5-(метоксиэтокси)фенил)-1-метил-5-(трифторметил)-1Н-пиразола,
(2-хлор-5-(4-хлор-1-метил-5(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)уксусной кислоты, 1,1-диметилэтилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)уксусной кислоты,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторбензойной кислоты, 2-этокси-1-метил-2-оксоэтилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторбензойной кислоты, 2-метокси-1-метил-2-оксоэтилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторбензойной кислоты, этилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторбензойной кислоты, 1-метилэтилового эфира и
6-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-7-фтор-4-(2-пропинил)-2Н-1,4-бензоксазин-3-(4Н)-она.
4-хлор-3-(4-хлор-2-фтор-5-пропаргилоксифенил)-1-метил-5-(трифторметил)-1Н-пиразола,
2-(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)пропионовой кислоты, этилового эфира,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)уксусной кислоты, 1-метилэтилового эфира,
4-хлор-3-(4-хлор-2-фтор-5-(метоксиметокси)фенил)-1-метил-5-(трифторметил)-1Н-пиразола,
4-хлор-3-(4-хлор-2-фтор-5-(метоксиэтокси)фенил-1-метил-5-(трифторметил)-1H-пиразола,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)уксусной кислоты, 1,1-диметилэтилового эфира,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)уксусной кислоты,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторбензойной кислоты, 2-этокси-1-метил-2-оксоэтилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторбензойной кислоты, 2-метокси-1-метил-2-оксоэтилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторбензойной кислоты, этилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторбензойной кислоты, 1-метилэтилового эфира и
6-(4-хлор-1-метил-5-(трифторметил)-2Н-пиразол-3-ил)-7-фтор-4-(2-пропинил)-2Н-1,4-бензоксазин-3-(4Н)-она.
4-хлор-3-(4-хлор-2-фтор-5-пропаргилоксифенил)-1-метил-5-(трифторметил)-1H-пиразола,
2-(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)пропановой кислоты, этилового эфира,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)уксусной кислоты, 1-метилэтилового эфира,
4-хлор-3-(4-хлор-2-фтор-5-(метоксиметокси)фенил)-1-метил-5-(трифторметил)-1Н-пиразола,
4-хлор-3-(4-хлор-2-фтор-5-(метоксиэтокси)фенил)-1-метил-5-(трифторметил)-1Н-пиразола,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторфенокси)уксусной кислоты, 1,1-диметилэтилового эфира,
(2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси)уксусной кислоты,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторбензойной кислоты, 2-этокси-1-метил-2-оксоэтилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пираэол-3-ил)-4-фторбензойной кислоты, 2-метокси-1-метил-2-оксоэтилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторбензойной кислоты, этилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1H-пиразол-3-ил)-4-фторбензойной кислоты, 1-метилэтилового эфира и
6-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-7-фтор-4-(2-пропинил)-2Н-1,4-бензоксазин-3-(4Н)-она.
4-хлор-3-(4-хлор-2-фтор-5-пропаргилоксифенил)-1-метил-5-(трифторметил)-1Н-пиразола,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторфенокси уксусной кислоты, 1-метилэтилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторбензойной кислоты, 2-метокси-1-метил-2-оксоэтилового эфира,
2-хлор-5-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-4-фторбензойной кислоты, 1-метилэтилового эфира и
6-(4-хлор-1-метил-5-(трифторметил)-1Н-пиразол-3-ил)-7-фтор-4-(2-пропинил)-2Н-1,4-бензоксазин-3-(4H)-она.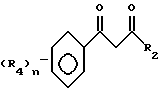
где R2, R4 и n определены в п.1,
с замещенным или незамещенным гидразином R1NHNH2, где R1, R2 и R4 определены в п. 1, с получением соединения формулы В в случае замещенного гидразина: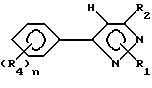
или соединения формулы С в случае незамещенного гидразина при условии, что полученное соединение формулы С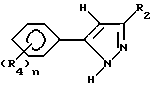
где R2, R4 и n определены в п.1,
подвергают взаимодействию с алкилирующим агентом для получения соединения В, которое затем дополнительно обрабатывают галогенирующим агентом для получения соединения формулы I.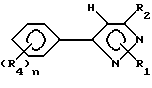
отличающийся тем, что осуществляют взаимодействие соединения формулы А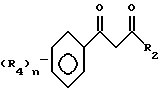
с замещенным или незамещенным гидразином при условии, что когда гидразин не замещен, получающееся соединение формулы С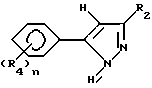
подвергают взаимодействию с алкилирующим агентом и в котором в вышеприведенных формулах R1, R2, R4 и n имеют ранее указанное значение.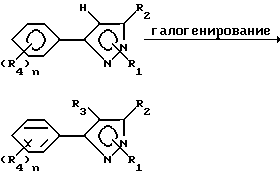
34. Способ по п.33, отличающийся тем, что получают указанные соединения формулы I, которые являются соединениями формулы III по п.3, где R1-R7 определены в п.1, при условии, что R6 также может быть водородом.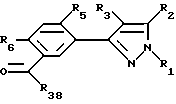
где R1-3, R5, R6 определены в пп. 1 и 2, а R38 представляет собой NR17R18, YR8, отличающийся тем, что производные пиразола формулы V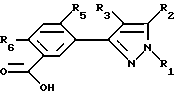
в которой R1-3, R5 и R6 имеют указанные значения, подвергают эстерификации для получения Z, где R38-YR8, или амидированию для получения Z, где R38-NR17R18.
Приоритет по пунктам:
18.10.90 - пп.1-37;
25.09.91 - пп.1-37 (разновидности радикалов).
| Способ получения производных @ -фенилпиразола | 1983 |
|
SU1309909A3 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНОПОЛИХЛОРПИКОЛИПОВ | 0 |
|
SU290904A1 |
| СПОСОБ СВАРКИ ДЕТАЛЕЙ КОСВЕННОЙ ДУГОЙ | 0 |
|
SU289879A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭПОКСИСОЕДИНЕНИЙ | 0 |
|
SU269806A1 |
