Предметом настоящего изобретения являются определенные замещенные соединения бензамида и способы их получения, которые являются новыми, способ борьбы с выпреванием растений, в частности зерновых культур, с использованием соединений и фунгицидные составы, предназначенные для осуществления этого способа.
Выпревание представляет серьезную проблему при выращивании зерновых культур, в частности пшеницы и ячменя. Это заболевание вызывается почвенным грибом Gaeumannomyces graminis (Gg). Этот гриб поражает корни растения и прорастает в корневые ткани, вызывая черную корневую гниль. Развитие гриба в корнях и нижней части стебля не позволяет растению получать достаточное количество воды и/или питательных веществ из почвы, в результате проявляется в плохом развитии растения, а в особо серьезных случаях заболевания образуется "белый (седой) колос" без зерен или с небольшим числом неполноценных зерен, что ведет к снижению урожайности. Гриб Gaeumannomyces поражает также и другие зерновые культуры, например рис и овес, а также травяной покров.
До настоящего времени основным способом борьбы с потерями зерновых культур вследствие заражения почвенным грибом Gg была замена зерновых культур посевами других культур, устойчивых к воздействию гриба Gg. Однако в тех районах, где основными зерновыми культурами являются злаки, чередование культур является нежелательной практикой, поэтому здесь более необходимо эффективное средство борьбы с этим заболеванием.
Целью настоящего изобретения является создание соединений, обеспечивающих превосходное и неожиданное подавление развития гриба Gg в почве, благодаря чему сокращаются потери зерновых культур. Другой целью настоящего изобретения является создание эффективного и неожиданного способа борьбы с выпреванием растений. Еще одной целью этого изобретения является создание фунгицидных составов, которые можно использовать для превосходного подавления этого заболевания.
В международной заявке PCT/US 92/08663 раскрывается большое число соединений, предназначенных для борьбы с выпреванием. В объем настоящего изобретения входят соединения, которые отличаются особенно неожиданной и высокой эффективностью в отношении данного заболевания.
Ссылочные материалы, имеющие отношение к способам по настоящему изобретению, представлены в журналах Synth. Commun, 14, 621 (1984) и Synthesis 303 (апрель 1978 г.).
В качестве других противопоставленных материалов можно привести статью Гайда Т. и Звержака А. "Phase-Transfer- Catalysed-N-Alkylation of Carboxamides and Sulfonamides, " опубликованную в журнале Synthesis, стр. 1005-7, декабрь 1981 г., и статью Абико А. и др. "KMnO, Revisited: Oxidation of Aldehides to Carboxilic Acids in the tert-Butyl Alcohol-Aqueous NaH2PO4 System, " опубликованную в журнале Tetrahedron Letters, т. 27, N 38, 4637-4540, 1986 г. Кроме того, можно указать рефераты в Дервенте N 87-203436/29, 89- 013361/02, 90-213193/28, 91-061915/09, 93-062565/08 и 93- 096743/12. \\2 Настоящим изобретением предлагается соединение формулы (I)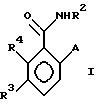
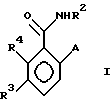
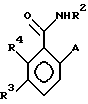
в которой R2 - этил, изопропил, пропил или аллил;
A - N(CH3)1-nHnR5 или OR6, где n означает 0 или 1, R5 представляет (CH3)m (CH3CH2)3-mС, 1-метил-1-циклопентил, 1- метил-1-циклогексил или 2, З-диметил-2-бутил, где m означает 0, 1, 2 или 3, и R6 независимо от других элементов представляет R5 или 2,3,3-триметил-2-бутил;
R3 представляет H или независимо R4; и
R4 представляет галоген или CH3,
при условии, что когда A представляет N(CH3)1-nHn R5, R3-Н и R5 - 1-метил-1-циклогексил или (CH3)m(CH3CH2)3-mС, где m означает 0 или 3, или если R3 - галоген и R2 - (CH3)m(CH3CH2)3-mC, где m означает 3, тогда R2 не может быть этилом;
и при условии, что когда A представляет OR6, тогда m является целым числом, равным или меньше 2, а если r3 - H или галоген и R2 - этил или изопропил, то R6 является (CH3)m(CH3CH2)3-mC, где m равно 1;
или его агрономически приемлемая соль.
Настоящим изобретением предлагается способ борьбы с заболеванием, вызываемым грибом Gaeumannomyces y растений, который включает обработку семян или почвы фунгицидно- эффективным количеством фунгицида формулы I.
Этим изобретением предлагаются также фунгицидные составы, содержащие фунгицидно-эффективное количество соединения формулы I и сельскохозяйственно-приемлемый носитель, пригодный для осуществления указанного способа.
Предпочтительным вариантом осуществления настоящего изобретения является соединение формулы I, в котором A представляет N(CH3)1-nHnC(CH3)m(CH2CH3)3-m, где n означает 0 или 1 и m равняется 1, 2 или 3, R2 - этил, пропил или аллил, R3 - метил и R4 - хлор, а также состав и способ его применения.
Другим предпочтительным вариантом осуществления настоящего изобретения является соединение формулы I, в которой A представляет OC(CH3)m(CH2CH3)3-m, где m равняется 1 или 2, или A представляет OC(CH3)2CH(CH3)2, R2 - аллил, R3 - H или CH3 и R4 - хлор, а также состав и способ его применения.
Еще одним предпочтительным вариантом осуществления настоящего изобретения является получение N-этил-2-[(1,1-диэтил) амино]-6-хлорбензамида, N-этил-2-[(1,1,2-триметилпропил) амино] -6-хлорбензамида, N-пропил-2-[(1,1-диметилпропил) амино] -6-хлорбензамида или N-аллил-2-[(1,1- диметилэтил)амино]-6-хлорбензамида.
В настоящее время установлено, что соединения формулы I, которые являются весьма активными при выполнении in vitro анализов, также демонстрируют очень хорошие результаты при выполнении in vivo анализов с учетом изменчивости почвы и в зависимости от используемых заместителей, сообщающих соединению повышенную гидрофильность.
Настоящим изобретением предусматривается также способ получения соединения формулы I, в котором A представляет OR6, а R6 имеет указанные выше значения, который включает:
стадию I) взаимодействия в растворителе соединения формулы (III)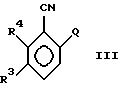
в котором Q - фтор или хлор, а R3 и R4 имеют указанные выше значения,
с соединением MA, где M представляет Li, Na или K, или с эквивалентом соединения MA, полученным in situ кипячением с обратным холодильником лития, натрия или кальция в избытке соединения AH, где A имеет указанные выше значения;
с образованием соединения формулы (IV)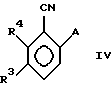
в которой A, R3 и R4 - имеют указанные выше значения;
стадию 2a), включающую нагрев соединения формулы IV с NaOH, H2O2, H2O и этанолом; или предпочтительно
стадию 2b) кипячением с обратным холодильником соединения формулы IV вместе с КОН в таком растворителе, как спирт или гликоль и предпочтительно трет-бутанол и еще предпочтительнее третичный амиловый спирт;
с образованием соединения формулы (V)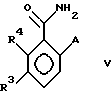
в которой A, R3 и R4 имеют указанные выше значения;
и стадию 3) обработки соединения формулы V соединением R2X, в котором X - хлор, бром, иод или -OSO2(OR2) в присутствии основания, или
стадию 3) обработки соединения формулы V соединением R2X, в котором R2 и X имеют указанные выше значения, в присутствии основания и агента межбазового переноса, с образованием вышеуказанного соединения формулы I, в которой A представляет OR6, а R6 имеет указанные выше значения;
стадию 2) взаимодействия вышеуказанного соединения формулы IV
а) с соединением HA1 (изобутил) в таком растворителе, как толуол или метиленхлорид, а затем
b) с соединением KMnO4 в спиртовом растворителе, таком как этанол или трет-бутанол, KH2PO4, H2O, при показателе pH примерно от 5 до 9, с образованием соединения формулы (VI)
в которой A, R3 и R4 имеют вышеуказанные значения, и стадию 3') обработки соединения формулы VI
а) тионилхлоридом или (COCl)2 в присутствии пиридина или диметилформамида в апротонном растворителе таком, как ацетонитрил, метиленхлорид или дихлорэтан;
b) а затем соединением H2NR2, в котором R2 имеет указанные значения;
с образованием вышеуказанного соединения формулы I, в которой A представляет OR6, a R6 имеет указанные значения.
Настоящим изобретением предусматриваются также новые соединения формул IV, V и VI.
Хотя вышеуказанные способы можно осуществлять в виде непрерывного процесса, то есть от получения соединения формулы III до соединения формулы I, каждая из стадий может быть также выполнена отдельно обычным образом. Например, соединение формулы VI можно подвергнуть обработке с образованием соединения формулы I, соединение формулы V можно подвергнуть обработке с образованием соединения формулы VI и соединение формулы IV можно подвергнуть обработке с образованием соединения формулы V.
Используемый в этом описании изобретения термин "галоген" означает радикал, выбираемый из хлора, брома, фтора и иода.
С заболеваниями, вызываемыми грибом Gg, в том числе и выпреванием, можно бороться разными способами, используя соответствующие химические средства. Эти средства можно вносить непосредственно в почву, зараженную грибом Gg, например, во время посева вместе с семенами. Альтернативно, эти средства можно вносить в почву после посева или появления всходов. Однако предпочтительно наносить такое средство на семена в виде покрытия до посева. Этот метод обычно применяется в отношении многих зерновых культур при использовании фунгицидов для борьбы с разными фитопатогенными грибами.
Составы по настоящему изобретению содержат эффективное в фунгицидном отношении количество одного или нескольких описанных выше соединений и одного или нескольких адъювантов. Активный ингредиент может присутствовать в таких составах в количестве от 0,01 до 95 вес.%. Этот состав может включать и другие фунгициды, в результате чего достигается более широкий спектр фунгицидного действия. Выбор фунгицидов зависит от зерновой культуры и заболеваний, имеющих распространение для нее в данном районе культивирования.
Фунгицидные составы по настоящему изобретению, включая концентраты, которые требуют разбавления перед применением, могут содержать по крайней мере один активный ингредиент и адъювант в жидкой или твердой форме. Эти составы получают путем смешивания активного ингредиента с адъювантом, включающим разбавители, наполнители, носители или улучшители, с образованием составов в виде тонкоизмельченных твердых веществ, гранул, растворов, дисперсий или эмульсий. Считается, что активный ингредиент можно использовать с адъювантом в виде тонкоизмельченного твердого вещества, с жидкостью органического происхождения, водой, смачивающим средством, эмульгатором или любым приемлемым сочетанием этих веществ.
Пригодные смачивающие средства включают алкилбензол и алкилнафталинсульфонаты, сульфатированные жирные спирты, амины или амиды кислот, кислые длинноцепочечные эфиры изотионата натрия, сложные эфиры сульфосукцината натрия, сульфатированные или сульфированные сложные эфиры жирной кислоты, нефтяные сульфонаты, сульфонированные растительные масла, двутретичные ацетиленгликоли, полиоксиэтиленовые производные алкилфенолов (в частности, изооктилфенол и нонилфенол) и полиоксиэтиленовые производные сложных эфиров одноосновных высших жирных кислот и ангидридов гексита (например, сорбит). Предпочтительными диспергаторами являются метилцеллюлоза, поливиниловый спирт, лигнинсульфонаты натрия, полимерные алкилнафталинсульфонаты, нафталинсульфонаты натрия и полиметиленбиснафталинсульфонат. Кроме того, для получения стабильных эмульсий можно использовать такие стабилизаторы, как силикат магния и алюминия и ксантановая камедь.
Другие составы представляют собой порошкообразные концентраты, содержащие от 0,1 до 60 вес.% активного ингредиента в приемлемом наполнителе, необязательно включающем другие адъюванты с целью улучшения потребительских свойств фунгицида, например, графит. Эти порошки можно разбавлять до концентраций в пределах 0,1 - 10 вес.%.
Концентраты могут также представлять собой водные эмульсии, получаемые путем перемешивания неводного раствора водонерастворимого активного ингредиента и эмульгатора с водой до образования однородной смеси в виде стабильной эмульсии тонкоизмельченных частиц Кроме того, можно получить водные суспензии путем измельчения смеси нерастворимого в воде активного ингредиента и смачивающих веществ, с образованием суспензии, отличающейся чрезвычайно мелким размером частиц, благодаря чему при разбавлении наносимое покрытие очень равномерно. Приемлемые концентраты этих составов содержат 0,1- 60%, предпочтительно 5-50 вес.% активного ингредиента.
Концентраты могут представлять собой растворы активного ингредиента в приемлемых растворителях вместе с поверхностно-активным веществом. Приемлемыми растворителями для активных ингредиентов по настоящему изобретению, предназначенных для обработки семян, являются пропиленгликоль, фурфуриловый спирт, другие спирты или гликоли и другие растворители, которые не оказывают существенного отрицательного действия на всхожесть семян. Если активный ингредиент предполагается вносить в почву, то можно использовать такие растворители, как N, N- диметилформамид, диметилсульфоксид, N-метилпирролидон, углеводород и несмешивающиеся с водой простые эфиры, сложные эфиры или кетоны.
Рассматриваемые здесь концентраты обычно содержат от 1,0 до 95 ч. (предпочтительно 5 - 60 ч. ) активного ингредиента, от 0,25 до 50 ч. (предпочтительно 1 - 25 ч.) поверхностно-активного вещества и при необходимости от 4 до 94 ч. растворителя, причем все части определяются по весу от общего веса концентрата.
Для внесения в почву во время посева можно использовать гранулированный состав. Гранулы - это физически стабильные составы в виде частиц, содержащие по крайней мере один активный ингредиент, адгезированный на или распределенный в основной массе инертного, тонкоизмельченного наполнителя в виде частиц. Для облегчения отделения активного ингредиента от частиц носителя в состав добавляется поверхностно-активное вещество, подобное указанным выше, или, например, пропиленгликоль. Примерами приемлемых минеральных наполнителей в виде частиц могут служить природные глины, пирофиллиты, иллит и вермикулит. Предпочтительными наполнителями являются пористые и абсорбирующие частицы, в частности предварительно измельченные или просеянные измельченные частицы аттапульгита или терморасширяющегося вермикулира в виде частиц и тонкоизмельченные глины, такие как каолин, гидратированный аттапульгит или бентонитовые глины. Эти наполнители напыляют на активный ингредиент или смешивают с ним с образованием фунгицидных гранул.
Гранулированные составы по настоящему изобретению могут содержать от 0,1 до 30 вес.ч. активного ингредиента на 100 вес.ч. глины и от 0 до 5 вес. ч. поверхностно-активного вещества на 100 вес. ч. измельченной глины.
Способ по настоящему изобретению может быть осуществлен путем смешивания состава, содержащего активный ингредиент с семенами перед посевом в количестве от 0,01 до 50 г на 1 кг семян, предпочтительно от 0,1 до 5 г на 1 кг и еще предпочтительнее от 0,2 до 2 г на 1 кг семян. В случае внесения в почву эти соединения можно использовать в количестве от 10 до 1000 г на 1 га, предпочтительно от 50 до 500 г на 1 га. В случае легких почв или большого количества осадков или того и другого вместе может потребоваться увеличение нормы внесения фунгицидов.
Соединения по настоящему изобретению можно получить в соответствии с хорошо известными способами. Следующие примеры служат для иллюстрации таких способов и никоим образом не ограничивают это изобретение.
Если нет специального указания, проценты представляют проценты по массе (вес. /вес. ), Температуры плавления и кипения не скорректированы. Тонкослойную хроматографию выполняли при элюировании смесями этилацетата и гексанов в разных концентрациях. Тетрагидрофуран и эфирные растворители отгоняли из смеси металлический натрий бензофенон непосредственно перед использованием. N, N, N, N'-(Тетраметил) этилендиамин отгоняли перед применением из гидрида кальция. Все другие реагенты были приобретены в фирме Aldrich или Lancaster и использовались без очистки. В каждом примере приводятся установленные физические свойства.
Используемые в тексте сокращения имеют следующие значения:
n-BuLi - н-бутиллитий
s-BuLi - втор-бутиллитий
t-BuLi - трет-бутиллитий
ДМФ - диметилформамид
DVSO - диметилсульфоксид
THF - тетрагидрофуран
TMEDA - N,N,N,N-(тетраметил) этилендиамин
eq - эквивалент (ы)
aq - водный
sat - насыщенный
min - минуты
h - часы
MeI - метилиодид
TLC - тонкослойная хроматография
HPLC - ВДЖХ
RC - радиальная хроматография
RT - ГЖХ комнатная температура
m.p. - температура плавления
Общие методики
Как правило, способ по настоящему изобретению осуществляется в соответствии с любой из следующих схем реакций.
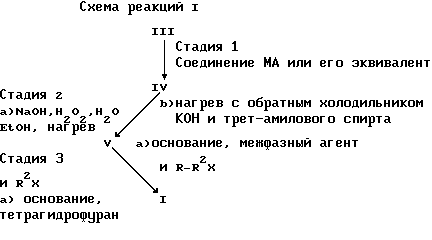
Цифрами I, III, IV и V обозначены соединения описанных выше формул с учетом того, что соединение формулы I имеет указанные выше значения, но A представляет OR6, a R6 MA, и R2 имеет указанные выше значения.
Предпочтительным вариантом осуществления способа, представленного в виде схемы реакций I по настоящему изобретению, является непрерывный процесс, включающий стадии 1, 2 и 3, в результате чего может быть достигнут 61% выход.
Другим предпочтительным вариантом осуществления способа, представленного в виде схемы реакций I по настоящему изобретению, является непрерывный процесс, включающий стадии 1 и 2.
Еще одним предпочтительным вариантом осуществления способа, представленного в виде схемы реакции I по настоящему изобретению, является непрерывный процесс, включающий стадии 2 и 3.
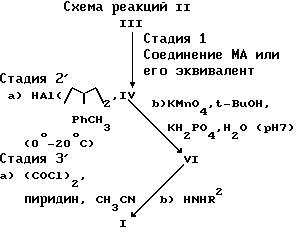
Цифрами I, III, IV и VI обозначены соединения описанных формул как они раскрыты в описании, включая ограничение, что соединение формулы I имеет указанные выше значения, где A представляет OR6, R6, MA и H2NR2, где R2 имеет указанные выше значения. Процесс, соответствующий стадии 1 в схемах реакций I и II, можно выполнять в таких растворителях, как тетрагидрофуран, диоксан, диметилформамид, диметилсульфоксид, AH, 1,2-диметоксиэтан или другие полярные апротонные растворители.
Исходные вещества
2-фтор-5,5-дихлорбензальдегид
1,3 М раствор втор-бутиллития в циклогексане (244 мл, 317 ммолей) добавляли к раствору N,N,N,N-(тетраметил) этилендиамина (34 г, 293 ммоля) в тетрагидрофуране (250 мл), охлажденному смесью сухого льда и ацетона, поддерживая внутреннюю температуру реакции <-70oC Полученную реакционную смесь охлаждали и помещали при температуре ≤ -90oC в ванну с простым эфиром и жидким азотом при одновременном добавлении по каплям 1,2-дихлор-4-фторбензола (40 г, 244 ммоля) в тетрагидрофурана (100 мл). Полученную реакционную смесь перемешивали в течение 1 часа при охлаждении смесью сухого льда и ацетона, после чего ее вводили с помощью канюли в охлажденный смесью простого эфира и жидкого азота и механически перемешиваемый раствор диметилформамида (89,1 г, 1,22 моля) в тетрагидрофуране (125 мл). Полученную смесь нагревали до -45oC и распределяли между разбавленным водным раствором HCl и простым эфиром. Органический раствор промывали водным раствором NaHCO3, сушили (MgSO4) и концентрировали с образованием 44,38 г 2-фтор-5,6- дихлорбензальдегида в виде масла.
2-фтор-5-метил-6-хлорбензальдегид
1,3М раствор втор-бутиллития в циклогексане (75 мл, 98 ммолей) добавляли к раствору N, N,N,N-(тетраметил) этилендиамина (9,67 г, 83 ммоля) в тетрагидрофуране (90 мл), охлажденному смесью сухого льда и ацетона, при сохранении внутренней температуры реакции < -70oC. Реакционную смесь охлаждали и помещали при температуре ≤ -80oC в ванну с простым эфиром и жидким азотом при одновременном добавлении по каплям раствора 2- хлор-4-фтортолуола (10 г, 69 ммолей) в тетрагидрофуране (10 мл). Полученную реакционную смесь перемешивали в течение 1 часа, охлаждая смесью сухого льда и ацетона, после чего ее вводили с помощью канюли в охлажденный смесью простого эфира и жидкого азота и механически перемешиваемый раствор диметилформамида (25,1 г, 345 ммолей) в тетрагидрофуране. Полученную смесь нагревали до -30oC и распределяли между разбавленным водным раствором HCl и простым эфиром. Органический раствор промывали водным раствором NaHCO3, сушили (MgSO4), концентрировали и растирали с гексанами, что позволило получить 2-фтор-5-метил-6-хлорбензальдегид в виде твердого кристаллического вещества.
2-фтор-5-метил-6-хлорбензонитрил
Хлористоводородный гидроксиламин (1,1 ч.) добавляли к раствору 2-фтор-5-метил-6-хлорбензальдегида в пиридине и полученную смесь перемешивали в течение 15 мин при комнатной температуре. Затем добавляли уксусный ангидрид (1,3 ч. ) и полученную смесь перемешивали в течение ночи при комнатной температуре, в результате чего происходила полная дегидратация оксима с образованием нитрила. Большую часть пиридина удаляли путем концентрирования в вакууме, после чего остаток распределяли между простым эфиром и водой. Органическую фазу промывали рассолом, сушили (MgSO4), фильтровали через силикагель, концентрировали и тритурировали в гексане с образованием 2-фтор-5-метил-6-хлорбензонитрила в виде светло- желтых кристаллов.
Способ A
NaN3 (2 ч. ) осторожно добавляли к 1,5 М раствору необязательно 5-замещенного 2-хлор-6-фторбензальдегида (1 ч.) в диметилсульфоксиде, после чего полученную смесь медленно нагревали до температуры 75oC в течение 2 ч. Затем температуру реакции повышали до 100oC и следили за завершением образования антранила в течение 3 ч с помощью анализа H1-ЯМР ароматической области. Темный раствор распределяли между водой и простым эфиром, а затем фильтровали через цеолит с целью фракционирования эмульсии. Водную фазу экстрагировали дополнительным количеством простого эфира, после чего соединенные органические экстракты промывали водой, сушили (MgSO4), концентрировали и перегоняли в аппарате Кюгельрора с образованием антранилов в виде светло-желтых твердых веществ, выход которых составлял 40-85%.
Смесь одного из этих антранилов (1 ч.) с трет-алканолом (от 1,1 до 1,2 ч. ) нагревали до растворения, а затем охлаждали в бане со льдом и водой при одновременном добавлении 70% перхлорной кислоты или 60% гексафторфосфорной кислоты (2 ч.) с такой скоростью, которая позволяла поддерживать внутреннюю температуру реакции ≤ 35oC. После окончания добавления реакционную смесь перемешивали при охлаждении смесью льда и воды в течение 30 мин до образования осадка. Этот осадок суспендировали в простом эфире, соли собирали фильтрованием, промывали сухим простым эфиром и сушили в условиях вакуума с образованием прехлората или гексафторфосфата N-трет-алкилантранила в виде бледно-желтых твердых веществ с высоким выходом.
Эту соль N-трет-алкилантранила (1 ч.) порциями добавляли к охлажденному смесью льда и воды 1,5 М раствору Et3N (3 ч.) в CH2Cl2. Полученный янтарный раствор перемешивали при комнатной температуре в течение 30 мин, затем концентрировали до небольшого объема, разбавляя сухим простым эфиром, и фильтровали с целью удаления солей. Этот раствор концентрировали с образованием масла, затем растворяли в гексанах и декантировали для удаления нерастворимого вещества. В результате концентрирования гексанового раствора был получен требуемый β-лактам в виде золотистого масла с высоким выходом.
Способ B
β-лактам, полученный в соответствии со способом A (1 ч.), необязательно растворенный в небольшом объеме органического растворителя, по каплям добавляли к охлажденному смесью льда и воды раствору первичного амина (от 5 до 10 ч. ) в CH2Cl2. Полученную смесь перемешивали в течение 0,5-1,0 ч, затем либо концентрировали и перекристаллизовывали из гексанов, либо распределяли между водой и органическим растворителем. Полученный экстракцией органический раствор сушили (MgSO4), концентрировали, суспендировали в гексанах и фильтровали с образованием N-алкил-2-трет-алкиламинобензамида в виде твердого вещества.
Способ C
β-лактам, полученный в соответствии со способом A (1 ч.), нагревали с обратным холодильником в метаноле (35 ч.) в течение 1 ч, затем концентрировали с образованием сложного метилового эфира в виде масла. 0,2 М раствор сложного метилового эфира в диметилформамиде соединяли с карбонатом калия (2 ч.) и йодистым метилом (5 ч.), затем нагревали в течение ночи при температуре 80oC в запаянной трубке. Этот раствор разбавляли простым эфиром, промывали водой, сушили (MgSO4) и концентрировали с образованием N-метилированного сложного эфира в виде темного масла.
К смеси бутиллития в гексанах (1 ч.) добавляли алкиламин (1,5 части) при температуре -78oC, в результате чего был получен гексановый раствор R-литиоалкиламина. Раствор N-литиоалкиламина (6 ч.) добавляли к полученному выше охлажденному до -78oC 1 М раствору N-метилированного сложного эфира (1 ч.) в тетрагидрофуране. Полученную реакционную смесь оставляли на ночь при температуре 0oC, затем разбавляли простым эфиром, промывали водой, сушили (MgSO4), концентрировали и очищали хроматографией с образованием требуемого N-алкилированного бензамида в виде твердого вещества.
Способ D
80%-ную масляную дисперсию гидрида натрия (1,2 ч.) добавляли к раствору 2-хлор-6-фторбензонитрила (1 ч.) и трет-алканола (1,2 ч.) в сухом 1,4-диоксане. Эту смесь кипятили с обратным холодильником в течение 16-20 ч, а затем распределяли между простым эфиром и водой. Органический слой промывали рассолом, сушили (MgSO4), фильтровали через силикагель и концентрировали. Этот неочищенный 2-трет-алкокси-6-хлорбензонитрил растворяли в трет-амиловом спирте и добавляли достаточное количество гранул гидроксида калия для сохранения насыщенного состояния при нагревании с обратным холодильником. Эту смесь нагревали с обратным холодильником в течение 2 ч, затем концентрировали в условиях вакуума, а остаток распределяли между простым эфиром и водой. Органический слой промывали рассолом, сушили (MgSO4) и фильтровали через силикагель. Затем фильтрат концентрировали, а остаток растирали с гексаном, что позволило получить требуемый 2-алкокси-6-хлорбензамид, который перекристаллизовывали из гексанов.
Способ E
35%-ную масляную дисперсию гидрида калия (1 ч.) добавляли к раствору трет-алканола в сухом 1,2- диметоксиэтане. Эту смесь недолго нагревали с обратным холодильником до полного образования алкоксида калия, затем охлаждали до комнатной температуры и добавляли 0,9 ч. 2-хлор-6-фторбензонитрила или 2-хлор-3-метил-6-фторбензонитрила. Полученную смесь перемешивали в течение 20 мин и распределяли между простым эфиром и водой. Органический слой промывали рассолом, сушили (MgSO4), фильтровали через силикагель и концентрировали. Остаток пропускали через 1 см слой силикагеля, производя элюирование сначала гексанами для удаления минерального масла, а затем смесью этилацетата и гексанов с соотношением 1:3, что позволило получить требуемый бензонитрил. Очищенный 2-трет-алкоксибензонитрил растворяли в трет-амиловом спирте и добавляли достаточное количество гранул гидроксида калия для сохранения насыщенного состояния во время нагревания с обратным холодильником. Эту смесь кипятили с обратным холодильником в течение 2 ч, затем концентрировали в условиях вакуума, а остаток распределяли между простым эфиром и водой. Органический слой промывали раствором, сушили (MgSO4) и фильтровали через силикагель. Затем этом фильтрат концентрировали, а остаток растирали с гексанами, что позволило получить 2-трет-алкокси-6-хлорбензамид или 2-трет-алкокси-5- метил-6-хлорбензамид, который перекристаллизовывали из гексанов
Способ F
N-хлорсукцинимид (2,4 ч. ) добавляли к раствору первичного бензамида (полученного в соответствии со способом D или E) в сухом ацетонитриле и полученную смесь кипятили с обратным холодильником в течение 1 ч. Эту смесь распределяли между простым эфиром и водным раствором Na2S2O3, после чего органический слой сначала промывали 10%-ным раствором NaOH, а затем рассолом, сушили (MgSO4), фильтровали через силикагель и концентрировали с образованием неочищенного 5-хлорбензамида.
Способ G
К раствору первичного бензамида, полученного в соответствии со способом D, E или F (1 ч.) в сухом тетрагидрофуране добавляли твердый бис (триметилсилил) амид лития (1,1 ч.) или 1,0 М раствор бис (триметилсилил) амида натрия в тетрагидрофуране (1,2 ч.). После перемешивания этой смеси в течение 5 мин добавляли соответствующий алкилгалогенид (2 ч.) и нагревали реакционную смесь с обратным холодильником в течение 3 ч. Эту смесь распределяли между простым эфиром и водой, а органический слой промывали рассолом, сушили (MgSO4), фильтровали через силикагель и концентрировали с образованием неочищенного N-алкилбензамида, который очищали посредством перекристаллизации или хроматографии.
Способ H
К раствору первичного бензамида, полученного в соответствии со способом D, E или F (1 ч.) и межфазного катализатора, бисульфата тетрабутиламмония (0,02 ч.) в толуоле добавляли такой же объем 50%-ного раствора NaOH и соответствующего алкилгалогенида (2,2 ч.), после чего эту смесь нагревали с обратным холодильником в течение 45 мин. Эту смесь распределяли между простым эфиром и водой, а органический слой промывали рассолом, сушили (MgSO4), фильтровали через силикагель и концентрировали с образованием неочищенного N-алкилбензамида, который очищали посредством перекристаллизации или хроматографии.
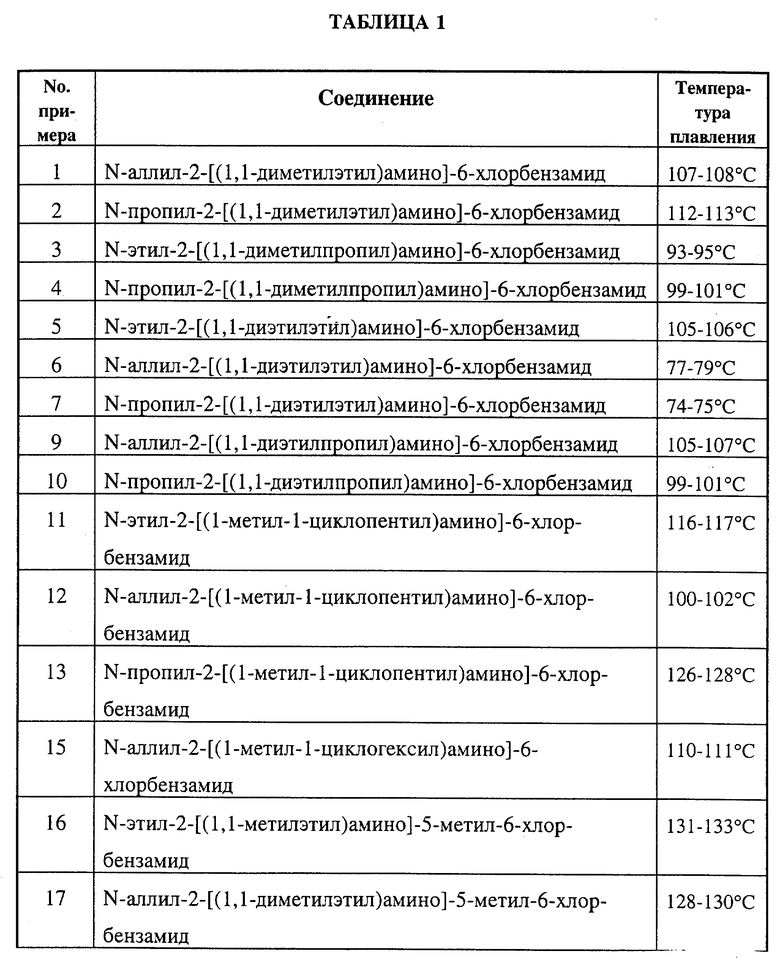
С помощью способов A и B были получены соединения, указанные в табл. 1.
С помощью способов A и C были получены соединения, указанные в табл. 2.
С помощью способа G и H были получены соединения, указанные в табл. 3.
Способы, используемые в настоящем изобретении, могут включать варианты, известные в этой области. Следующие примеры иллюстрируют некоторые типичные методы применения этих способов; они служат только для целей иллюстрации и никоим образом не ограничивают объем данного изобретения
Пример получения соединений по схеме реакции II
К раствору 2-фтор-6-хлорбензонитрила (10,35 г, 66,5 ммоля) в 1,2-диметоксиэтане (50 мл) при температуре 0oC добавляли третбутоксид калия (9,06 г, 80,7 ммоля). Эту смесь оставляли для медленного нагревания до комнатной температуры в течение 3 ч. Реакционную смесь выливали в воду и экстрагировали простым эфиром (3 раза). Органические слои промывали рассолом, сушили (MgSO4) и концентрировали с образованием 13,52 г (97%) 2-[(1,1-диметилэтил)окси]-6-хлорбензонитрила в виде бледно-желтого масла.
К раствору 2-[(1,1-диметилэтил)окси]-6- хлорбензонитрила (9,22 г, 44,0 ммолей) в толуоле (100 мл), охлажденному в ледяной бане, добавляли гидрид диизобутилалюминия (1 М раствор в гексане, 48,4 мл, 48,4 ммоля), поддерживая температуру ниже 10oC. Реакционную смесь перемешивали в течение 1 часа и выливали в смесь 10% водного раствора уксусной кислоты (100 мл) и льда. Эту смесь фильтровали через цеолит, слои разделяли и водный слой экстрагировали простым эфиром (2 раза). Соединенные органические слои промывали насыщенным раствором бикарбоната натрия и рассолом, сушили (MgSO4) и концентрировали с образованием 9,07 г (97%) 2-[(1,1-диметилэтил)окси]-6- хлорбензальдегида в виде желтого масла. К раствору 2-[(1,1- диметилэтил) окси]-6-хлорбензальдегида (8,40 г, 40,0 ммолей) в трет-бутаноле (200 мл) добавляли 1,25 М раствор KH2PO4 (pH 7, 200 мл) и 0,4 М водный раствор перманганата калия (200 мл, 80 ммолей). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и быстро охлаждали путем добавления насыщенного водного раствора сульфита натрия (200 мл) Коричневую суспензию подкисляли 2 н. раствором HCl при охлаждении льдом до полного растворения MnO2 (pH 4). Реакционную смесь экстрагировали этилацетатом (3 раза), органические слои промывали рассолом, сушили (MgSO4) и концентрировали до образования белого твердого вещества. Неочищенный продукт перекристаллизовывали из гексанов при температуре 0oC с образованием 7,14 г (78%) 2-[(1,1- диметилэтил)окси]-6-хлорбензойной кислоты в виде белых кристаллов: температура плавления 117 - 119oC.
2-[(1,1-диметилэтил)окси] -6-хлорбензойную кислоту (68,6 мг, 0,3 ммоля) вводили в пробирку емкостью 3,7 мл (1 драхм) с перегородкой и стержнем для микроперемешивания под слоем азота. Через микролитровые шприцы в эту пробирку в указанной последовательности вводили сухой ацетонитрил (300 микролитров), сухой пиридин (24 микролитра, 0,3 ммоля) и оксалилхлорид (26 микролитра, 0,3 ммоля). Однородный раствор желтого цвета перемешивали в течение 30 мин с образованием раствора 2-[(1,1-диметилэтил)окси]-6-хлорбензоилхлорида.
Хлористоводородный этиламин (16,3 мг, 0,20 ммоля) вводили в пробирку емкостью 3,7 мл (1 драхм) со стержнем для микроперемешивания. С помощью микролитровых шприцев в эту пробирку в указанной последовательности вводили воду (100 микролитров), ацетонитрил (300 микролитров) и триэтиламин (200 микролитров). В результате этого был получен светлый и бесцветный раствор, в который добавляли раствор 2-[(1, 1- диметилэтил) окси]-6-хлорбензоилхлорида (0,30 ммоля). Желтоватую реакционную смесь перемешивали в течение 30 мин.
Затем добавляли глутатион (50 мг, 0,16 ммоля) и реакционную смесь перемешивали еще 30 мин. Добавляли простой диэтиловый эфир (1,5 мл) и экстрагировали смесь водой (1 мл), 2,5 н. раствором NaOH (1 мл) и рассолом (1 мл) с целью удаления всех побочных продуктов. Это достигалось путем интенсивного перемешивания реакционной смеси при каждой промывке в течение нескольких минут с последующим удалением промывочного раствора 500 микролитровым шприцем. Простой эфир сушили над сульфатом натрия, фильтровали через пипетку одноразового применения, содержащую в качестве фильтра кусочек AccuWipe и концентрировали с образованием 44 мг чистого N-этил-2-[(1,1- диметилэтил)окси]-6-хлорбензамида в виде белого кристаллического твердого вещества с 86% выходом.
Пример выполнения стадии 3 схемы реакций 1 - межфазное моноалкилирование - получение соединения по примеру 49
Смесь 2-[(1,1-диэтил)окси]-6-хлорбензамида (1,00 г, 3,9 ммоля) и бисульфата тетрабутиламмония (0,13 г, 0,4 ммоля) в 50%-ном водном растворе гидроксида натрия (25 мл) и толуола (15 мл) нагревали до температуры 100oC и в течение 30 мин добавляли н- пропилбромид (0,43 мл, 4,7 ммоля) в толуоле (10 мл). Реакционную смесь нагревали в течение 1 ч, охлаждали и выливали в воду. Эту смесь экстрагировали простым эфиром, органические слои промывали рассолом, сушили (MgSO4) и концентрировали до образования белого твердого вещества. Неочищенный продукт очищали посредством радиальной хроматографии, что позволило получить 0,78 г (67%) N-пропил-2-[(1,1-диэтилэтил)окси]-6- хлорбензамида в виде белого твердого вещества: температура плавления 100-102oC.
Пример непрерывного процесса по схеме реакций 1
К суспензии трет-бутоксида калия (56,0 г, 0,5 моля) в трет-бутаноле (200 мл) при комнатной температуре добавляли 2-фторбензонитрил (12,1 г, 0,1 моля). Реакционную смесь нагревали до температуры кипения с обратным холодильником в течение 30 мин и добавляли воду (3, б мл, 0,2 моля). После этого реакционную смесь кипятили с обратным холодильником в течение 2 ч и на протяжении 40 мин порциями добавляли йодистый этил (40 мл, 0,5 моля), продолжая нагревать с обратным холодильником. После охлаждения и перемешивания в течение ночи при комнатной температуре реакционную смесь анализировали с помощью газовой хроматографии и масс-спектрометрии. Результаты этих анализов показали наличие N-этил-2-[(1,1-диметилэтил)окси]бензамида, выход которого составил 61%. Таким образом, при создании соответствующих условий соединения по настоящему изобретению можно получить с помощью одного непрерывного процесса.
Непрерывный процесс выполнения стадий 1 и 2 (b) схемы реакций 1 с целью получения 2-хлор-6-(1-этил-1-метил-пропокси) бензамида
К 100 г (980 ммолей) 3-метил-3-пентанола, нагретого до 120oC, добавляли небольшими порциями 15,0 г (385 ммолей) калия. После растворения всего калия эту смесь оставляли для охлаждения до 65oC и добавляли к ней раствор 50,0 г (322 ммоля) 2-хлор-6-фторбенэонитрила в 100 мл толуола. Эту смесь охлаждали в ледяной бане, производя добавление в течение 10 мин. Реакционную смесь продолжали перемешивать при комнатной температуре еще 15 мин, после чего эту смесь нагревали до 90oC. На этом этапе в реакционной смеси не оставалось исходное вещества. К смеси добавляли 50 г гранул гидроксида калия и 200 мл трет-амилового спирта. Эту смесь нагревали до температуры кипения с обратным холодильником в течение 45 мин и оставляли на ночь для охлаждения до комнатной температуры. Растворитель удаляли в условиях вакуума, а остаток распределяли между простым эфиром и водой. Органический слой промывали рассолом, сушили (MgSO4) и фильтровали через силикагель. Фильтрат выпаривали в условиях вакуума, а остаток перекристаллизовывали из гексана, что позволило получить 70,0 г (851) белых кристаллов.
Биологические испытания
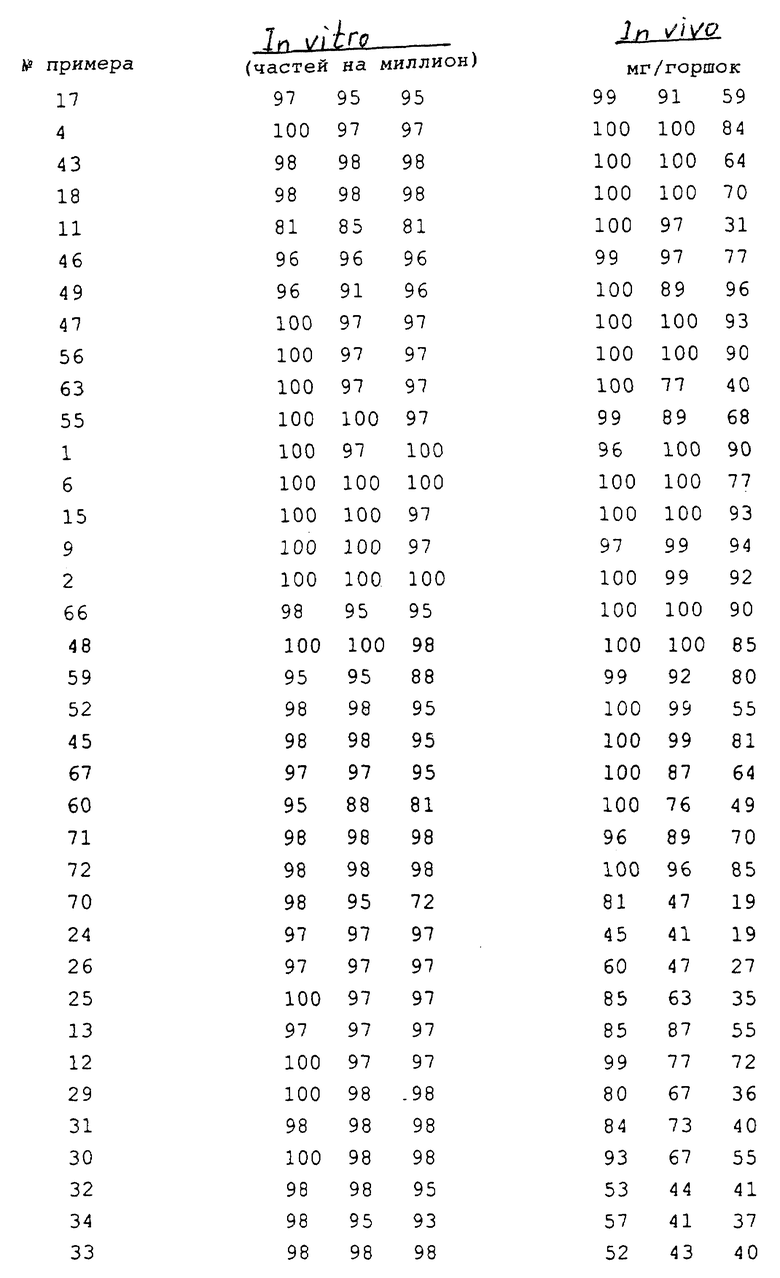
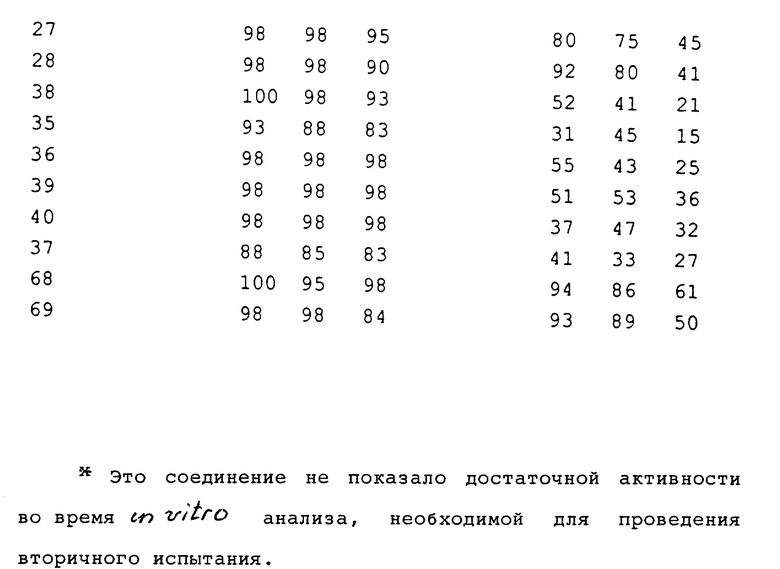
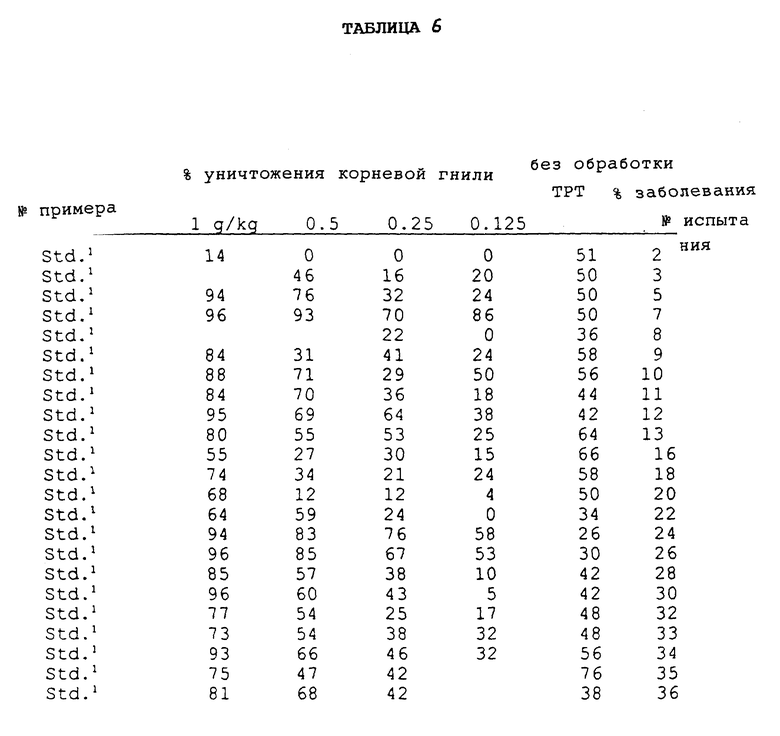
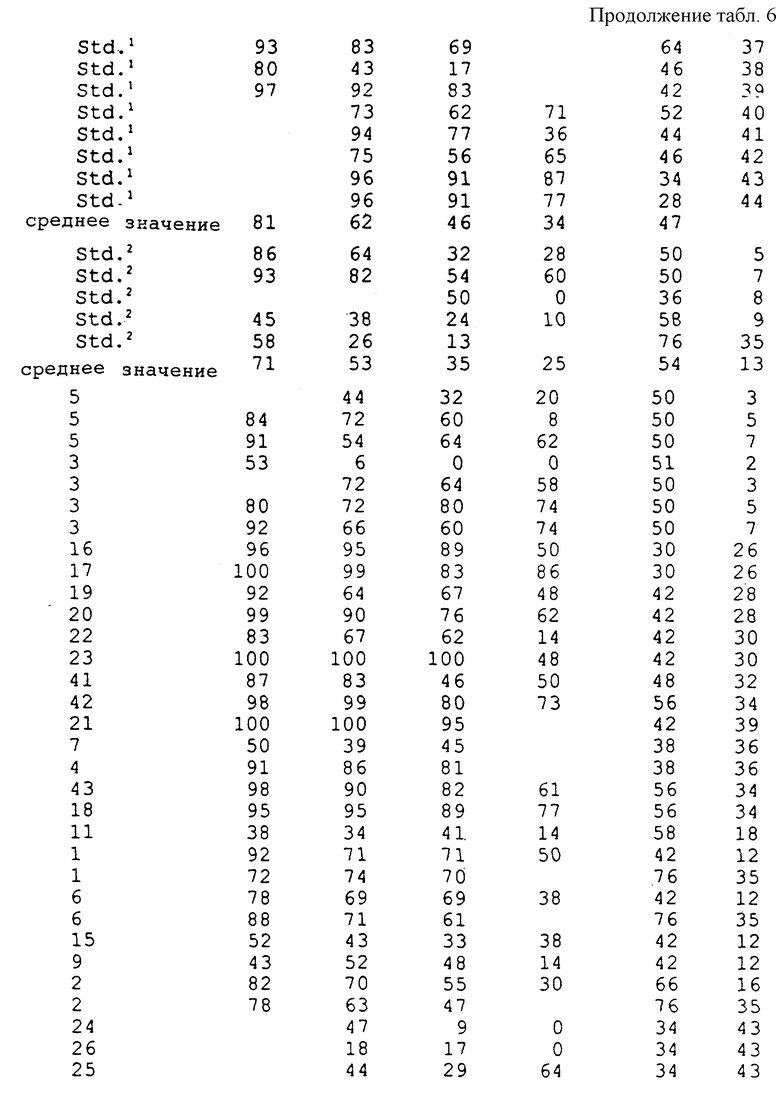
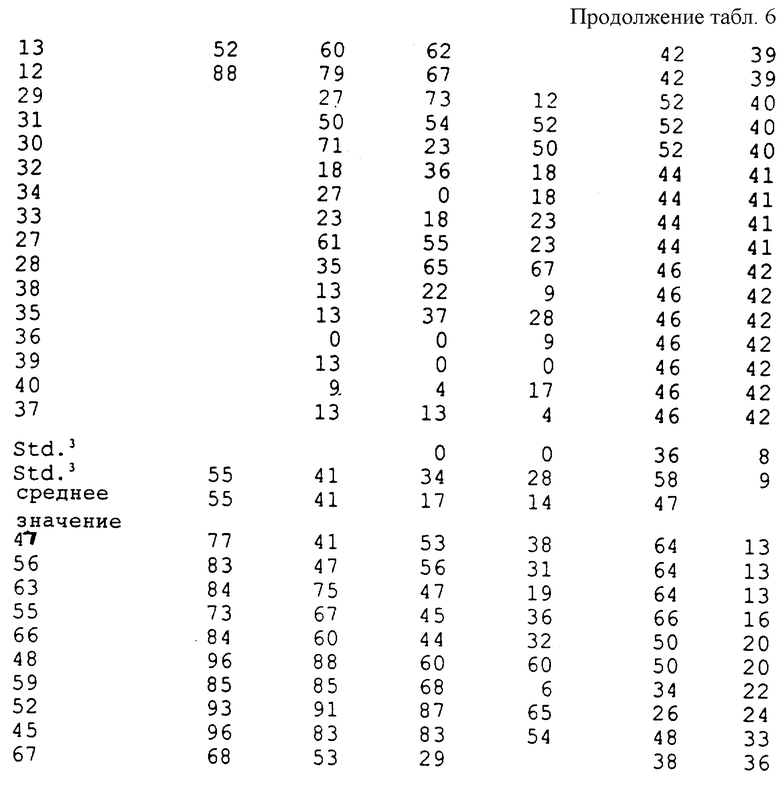
Соединения, полученные в соответствии с вышеуказанными примерами, испытывали в отношении эффективности уничтожения гриба Ggt с использованием одного или обоих приведенных ниже методов испытания. Полученные результаты представлены в таблицах.
In vitro анализ
Испытуемые соединения (0,25 мл соответствующего маточного раствора в ацетоне) помещали на 25 мл агара с минимальной питательной средой [агар получали путем автоклавирования раствора 17,5 г бульона Czapec Dox (Difco), 7,5 г очищенного агара или бактоагара (Difco) и 500 мл дистиллированной и деионизированной воды с последующим добавлением 50 мкл 1 мг/мл хлористоводородного тиамина и 50 мкл 1 мг/мл биотина в 5% этаноле/ и готовили чашки.
Чашки инокулировали, помещая в каждую из них в виде треугольника три 4 мм пробки с грибом Gaeumannomyces graminiss var. tritici (Ggt), выращенным на описанном агаре с минимальной питательной средой. Эти чашки инкубировали в темноте при температуре 19-20oC в течение 4-5 дней. Рост гриба определяли по увеличению диаметра мицелия. Полученные результаты выражены в виде процентного значения ингибирования, высчитываемого следующим образом [1-[(мм роста на обработанной чашке - 4) / (мм роста на контрольной чашке - 4)]] •100.
Обработка семян и почвы при выполнении in vivo анализа
Соединения испытывали в отношении уничтожения гриба Ggt при использовании пшеницы сортов "Bergenen" или "Anza", которую выращивали в 8 см (3 дюйма) квадратных горшках, содержащих почву, зараженную грибом Ggt. Заражение производили путем смешивания почвы с инокулятом, полученным в результате выращивания гриба Ggt чашках с картофельным агаром с декстрозой, характеризующимся 1/4 мощности (4,875 г картофельного агара с декстрозой, 5,0 г бактоагара, 500 мл дистиллированной и деионизированной воды), и использования пробок, взятых из чашек для заражения стерильных зерен овса (400 см3 цельных зерен овса, 350 мл деионизированной воды с обработанной в автоклаве). После инкубирования в течение одного месяца при комнатной температуре зерна овса сушили и смешивали с почвой в объемном отношении, равном 4%. На поверхность почвы в каждый горшок помещали четыре семени пшеницы. Испытуемые соединения готовили в виде раствора в ацетоне и воде с объемным отношением 1:9, который содержал 0,18% Tween® 20, что соответствовало норме обработки в количестве 0,5 и/или 0,1 мг активного ингредиента на горшок, при этом в каждый горшок вводили по 3 мл испытуемого раствора. Для каждой нормы обработки и контрольных образцов использовали пять горшков, которые содержали необработанные, инокулированные и неинокулированные образцы. После высушивания в течение одного часа семена покрывали дополнительным количеством соответствующей почвы и слоем вермикулита. Эти горшки помещали в камеру для выращивания и ежедневно поливали. Через четыре недели в каждом горшке определяли признаки заболевания путем осмотра первичных корешков у каждого растения под препаровальной лупой. Оценку производили по пятибальной шкале, имеющей следующие значения:
0 = отсутствие гифов или повреждения
1 = наличие небольшого количества гифов и небольших повреждений на < 10% корневой системы
2 = наличие гифов и небольших повреждений на 10-25% корневой системы
3 = наличие гифов и повреждений на 25-50% корневой системы
4 = наличие гифов и многочисленных крупных, сливающихся повреждений на > 50% корневой системы
5 = корневая система и стебель полностью поражены и имеют множество гифов
Из каждой серии, включающей пять репликатов, самую высокую или низкую оценку отбрасывали для гарантии получения наиболее репрезентативных оценок при вычислении среднего значения путем усреднения остальных оценок. Среднюю оценку затем сравнивали с оценкой, полученной для необработанного контрольного образца, и определяли степень ингибирования заболевания в процентах.
Результаты этих in vitro и in vitro анализов представлены в табл. 4. Если в результате вычислений получена оценка "0" или ниже по сравнению с необработанным контрольным образцом, то такой результат обозначается буквой "N", контрольного образца.
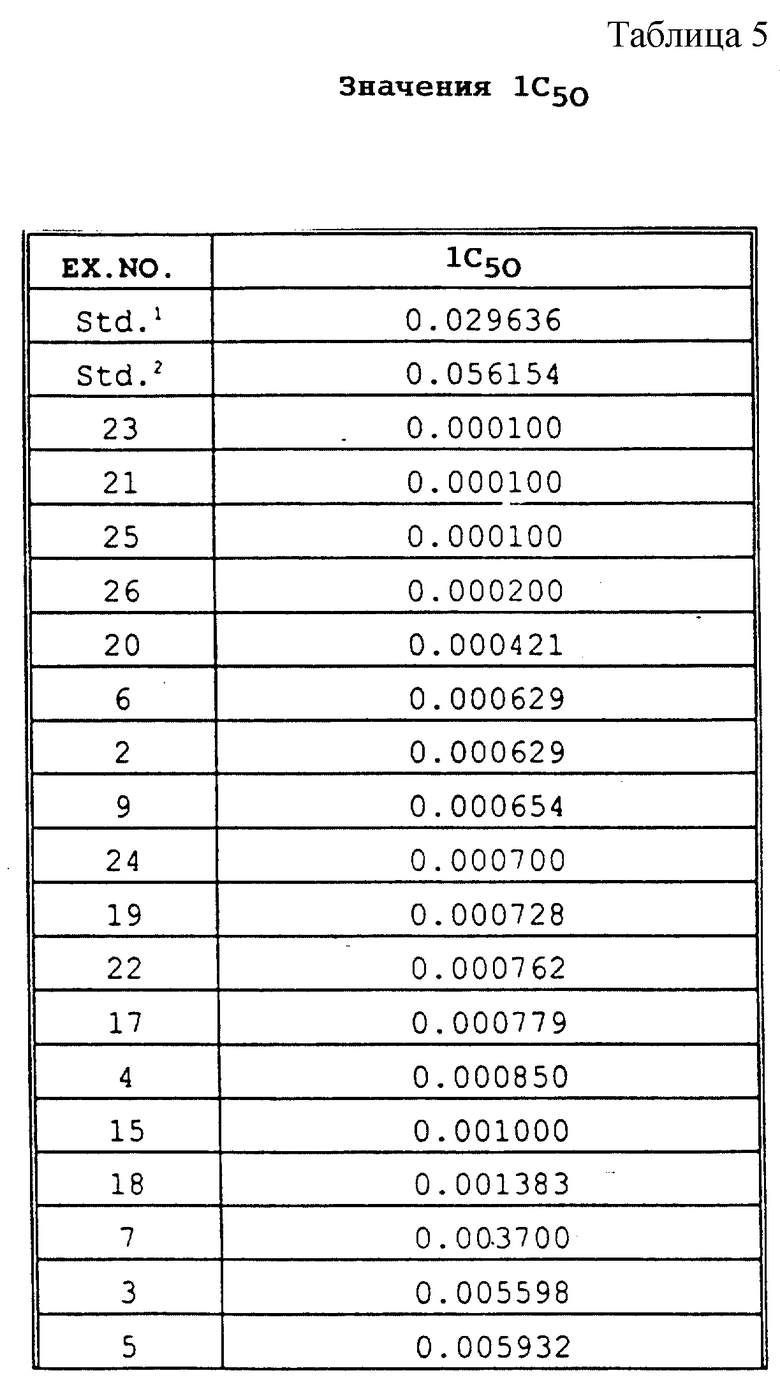
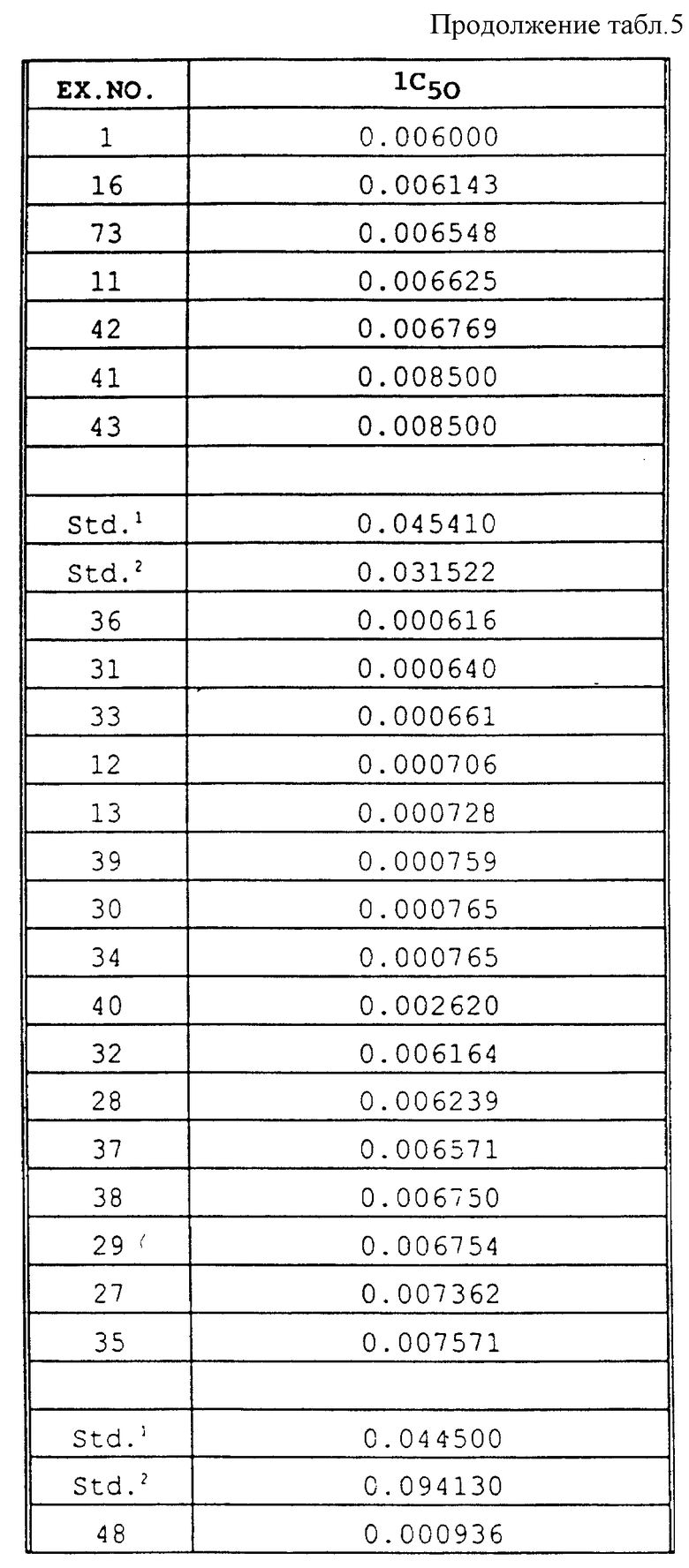
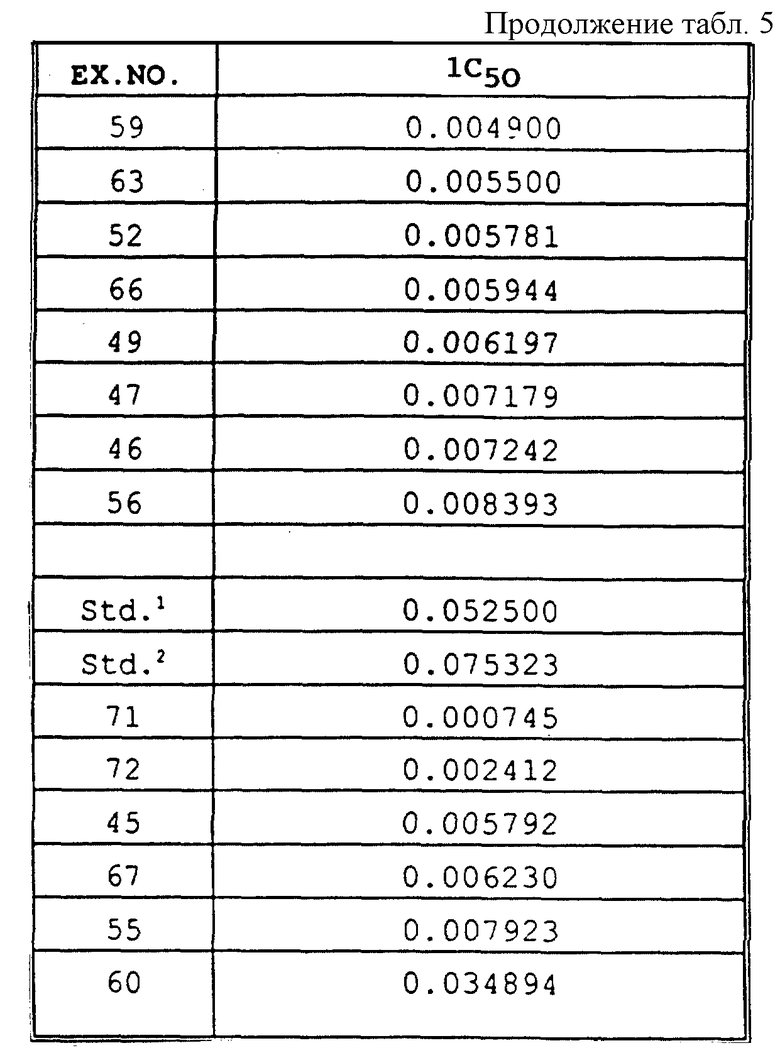
Для определения значений IC50 in vitro анализ выполняли в отношении каждого соединения, используемого в концентрациях 1, 0,1, 0,01, 0,001 и 0,0001 части на миллион. Величину ингибирования в процентном выражении высчитывали для каждой концентрации по уравнению, приведенному в разделе "Биологические испытания", посвященном in vitro анализам. При использовании двух показателей (концентрация, % ингибирования), которые обеспечивают 50% ингибирование роста гриба, концентрацию для 50% ингибирования рассчитывали с помощью следующего уравнения: IC50=[(50-I2)C1 + (I1-50)C2]/(I1 - I2), где C1 = 10C2. Результаты in vitro анализа представлены в табл. 5.
Обработка семян при выполнении in vivo анализа. Соединения испытывали в отношении уничтожения гриба Ggt с использованием пшеницы сортов "Bergen" или "Anza", которую выращивали в 15 см круглых горшках, заполненных почвой (равное количество трех компонентов, представляющих собой смесь "Metro-mix", песок и илистый суглинок, которую подвергали стерилизации паром). Семена обрабатывали раствором соединения по настоящему изобретению при норме 10000 частей на миллион маточного раствора в ацетоне. 20 мг соединения в 2 мл раствора обрабатывали 10 г семян при каждой из 4 норм обработки. При использовании 10000 частей на миллион маточного раствора для каждого соединения были получены следующие серии разбавлений:
Грамм активного ингредиента/100 кг состава
1. 100 1 мл маточного раствора
2. 50 1 мл маточного раствора + 1 мл ацетона
3. 25 1 мл соединения N 2 + 1 мл ацетона
4. 12,5 1 мл соединения N 3+1 мл ацетона (слив 1 мл или дальнейшее использование)
5. 6,25 1 мл соединения N 4+1 мл ацетона (слив 1 мл)
(5 - необязательная обработка, использовавшаяся не во всех испытаниях)
Примечание: в каждой пробирке находился 1 мл раствора, предназначенный для обработки 10 г семян. Готовили 10 г пакетики семян пшеницы (сорт "Bergen"), по одному для каждой обработки.
Сосуд, предназначенный для обработки семян, дважды промывали 3 мл ацетона. Затем 1 мл раствора взбалтывали так, чтобы он покрыл основание сосуда. В сосуд помещали 10 г семян и закрывали его крышкой, после чего этот сосуд взбалтывали и встряхивали до тех пор, пока на семена не наносилось равномерное покрытие. Через 30 с крышку удаляли, а встряхивание продолжали. Через 1 мин сосуд оставляли для высыхания. После высыхания семена снова высыпали в конверт с целью дальнейшего высевания в горшки или хранения до такого посева. Семена высевали следующим образом.
Анализ на ингибирование выпревания в теплице с использованием больших горшков:
15 см (6-дюймовые) горшки до края заполняли вышеуказанной почвенной смесью.
Метод посева:
а) Обработанные семена помещали на поверхность почвы (заполняющей горшки до края) при норме 8 семян на один горшок на расстоянии 5-7,5 см друг от друга. Для каждой обработки производили посев в 5 горшков (репликантов).
b) Отмеряли 15 мл инокулированного овса (примерно 4 г) и равномерно распределяли его по поверхности почвы в каждом горшке.
с) Почву, семена и инокулят покрывали 180 мл почвенной смеси (так, как описывалось выше). 150 мл химический стакан, наполненный до края, содержит примерно 180 мл.
d) Сначала каждый из подготовленных горшков несколько раз слегка поливали, чтобы смочить почву без вымывания семян.
е) В холодные зимние месяцы подготовленные горшки оставляли в теплице при температуре 16 - 18oC при минимальном дополнительном освещении. В более теплые месяцы подготовленные горшки помещали в камеру для выращивания при температуре 17oC на 3-4 недели с целью развития заболевания, а затем переносили в теплицу и оставляли там до полного созревания. Через 7-10 недель пшеницу срезали, промывали и оценивали состояние корней.
f) Площадь поражения корней выражали в процентах с присвоением следующих значений 1, 5, 10, 20, 30, 40, 50, 60, 80 или 100%.
Каждому горшку с растениями присваивали одну оценку.
Результаты 8-недельной обработки семян при выполнении in vivo анализа в почве представлены в табл. 6.
Полевые испытания
Соединения по примерам 1-73 соединяли с разными адъювантами, носителями и другими добавками и смешивали с семенами пшеницы и ячменя при норме от 0,01 до 50 г активного ингредиента на 1 кг семян, в результате чего обеспечивалось ингибирование развития гриба Gg на предварительно зараженных полях по сравнению с контрольными полями, засеянными необработанными семенами.
Примеры составов
Концентрат суспензии: - Вес. %
Соединение N 17 - 48.900
Блок-сополимер полиоксипропилена и полиоксиэтилена - 2,550
Лигнинсульфонат натрия - 2,040
10%-ная эмульсия диметилполисилоксана - 1,020
1%-ный раствор ксантановой камеди - 0,990
Вода - 43,250
Эмульгируемый концентрат: - Вес. %
Соединение N 19 - 13,5
Этоксилировнный сорбит (20EO) - 5,0
Ароматизатор C9 - 81,5
Смачиваемый порошок: - Вес. %
Соединение N 20 - 75,0
Лигнинсульфонат натрия - 3,0
N-метил-N-олеилтаурат натрия - 1,0
Каолинитовая глина - 11,0
Гранулированный состав: - Вес. %
Соединение N 21 - 1,0
Пропиленгликоль - 5,0
Монтмориллонит (24/48 меш) - 94,0
Порошок: - Вес. %
Соединение N 15 - 50,0
Графит - 10,0
Каолинитовая глина - 40,0
Из вышесказанного видно, что настоящее изобретение во всех отношениях соответствует поставленным целям и позволяет достичь очевидных преимуществ.
Вполне понятно, что возможны отличительные особенности и дополнительные комбинации, которые могут использоваться без ссылки на другие отличительные особенности и дополнительные комбинации. Все они подразумеваются и входят в объем формулы изобретения.
Поскольку существует много возможных вариантов осуществления настоящего изобретения, вариант, представленный в описании, следует рассматривать как иллюстрацию, но не как-либо ограничивающую объем изобретения.
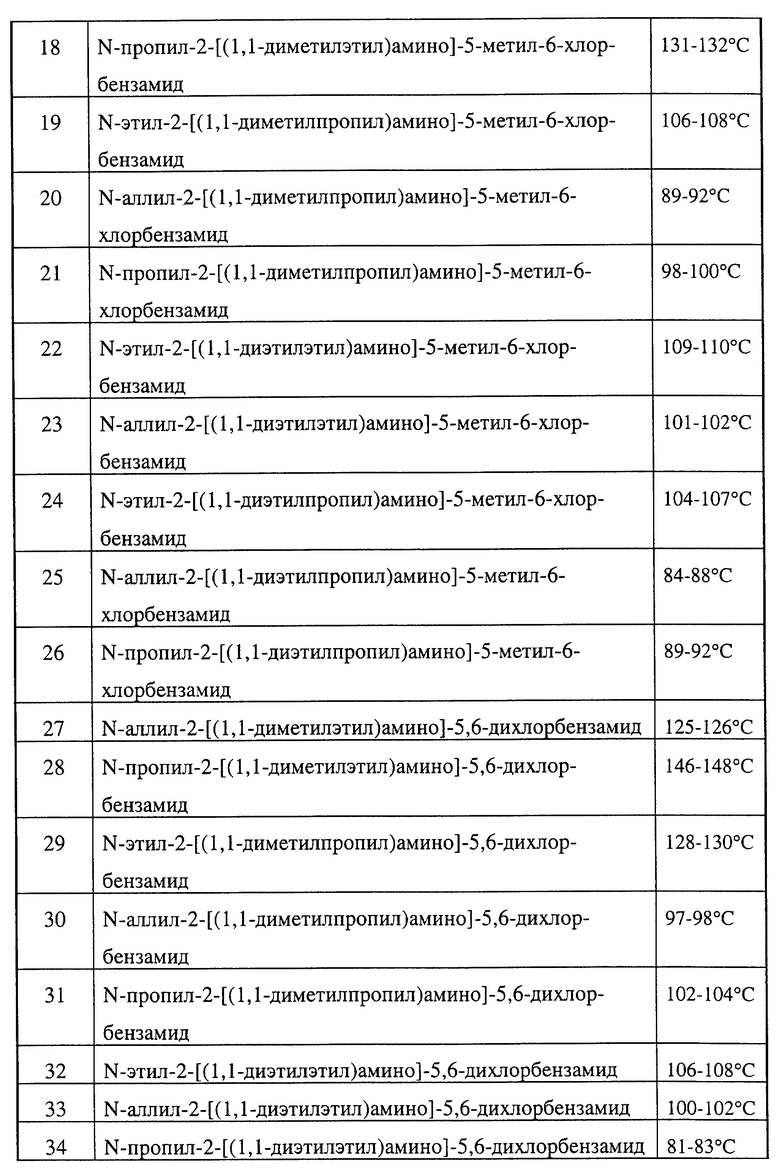
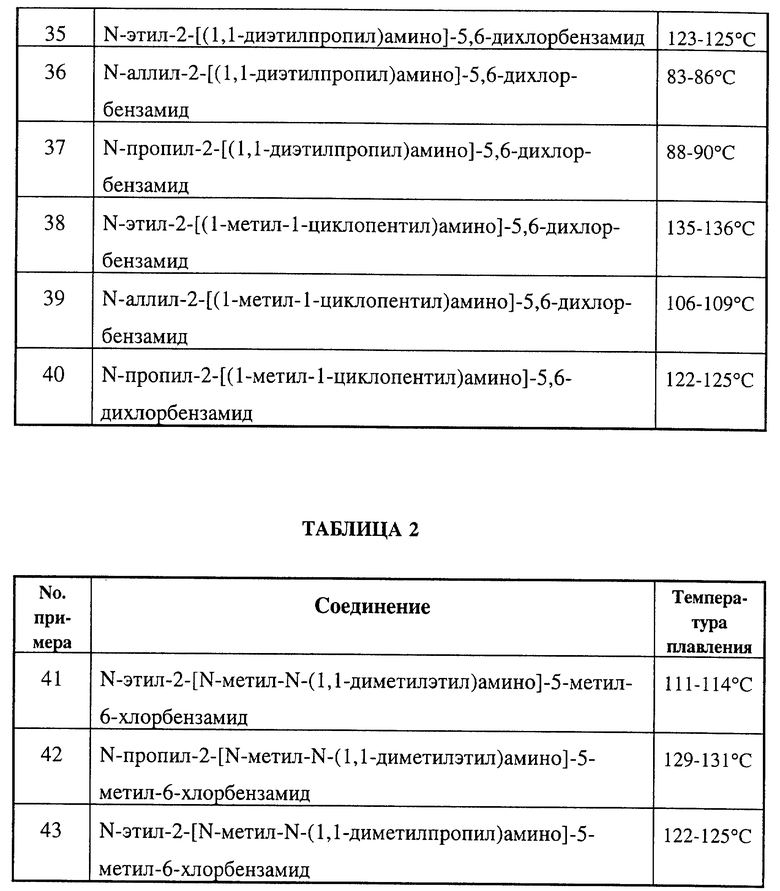
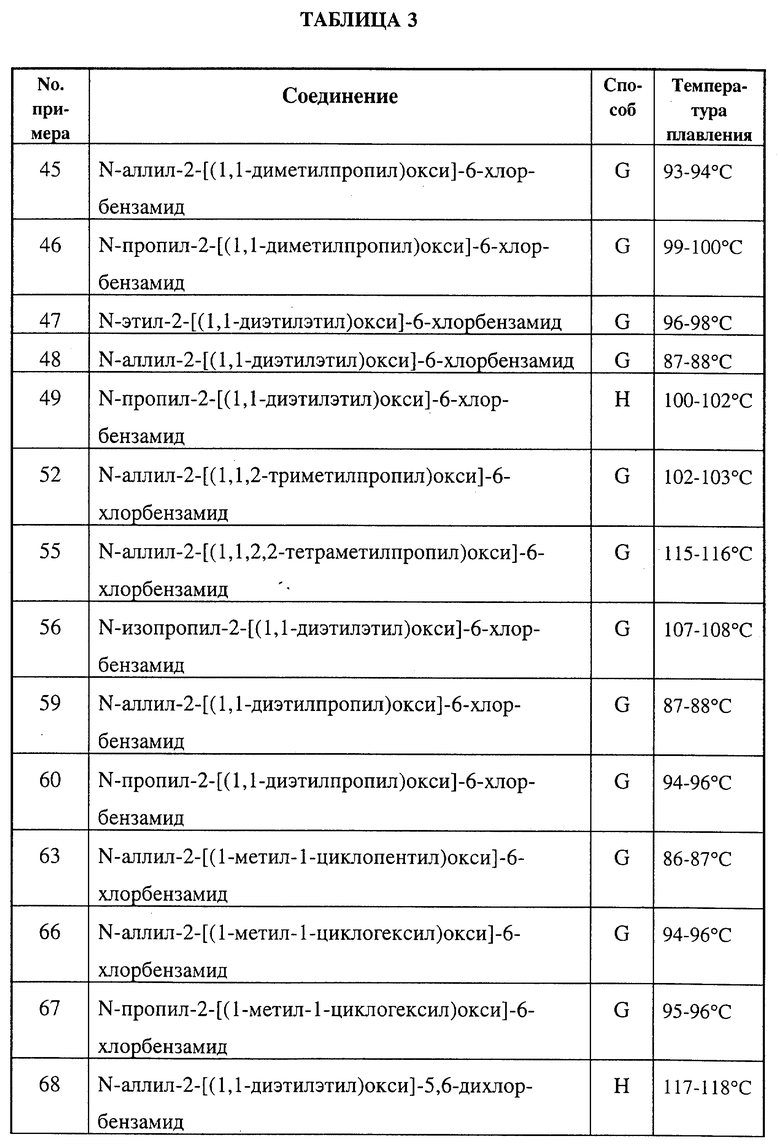

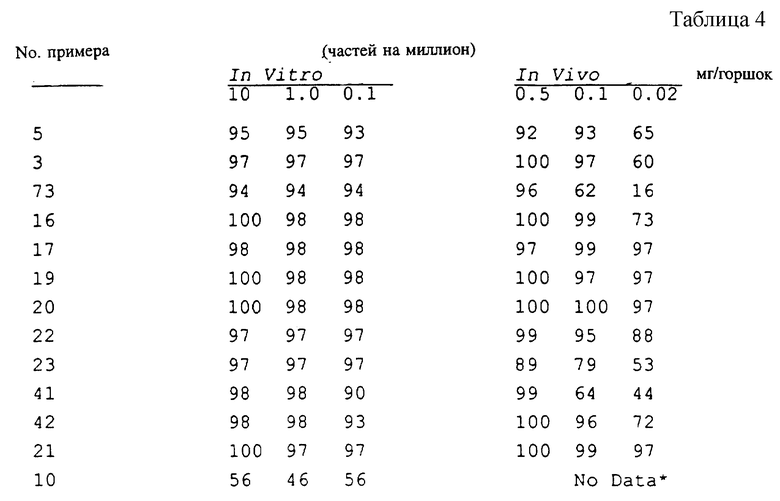
Замещенные бензамиды формулы I, где R2 - этил, изопропил или аллил; А - N(CH3)1-nHnR5 или OR6, n означает 0 или 1, R5 -(СН3)m(СН3СН2)3-mС, 1-метил-1-циклопентил, 1-метил-1-циклогексил или 2,3-диметил-2-бутил, m означает 0, 1, 2 или 3, a R6 независимо представляет R5 или 2,3,3-триметил-2-бутил (другие обозначения см. в п.1.формулы изобретения). Соединения пригодны для борьбы с заболеваниями, вызывающими выпревание растений. 8 c. и 8 з.п. ф-лы, 6 табл.
в которой R2 - этил, изопропил, пропил или аллил;
А - N(СН3)1-nHnR5 или ОR6, где n означает 0 или 1; R5 представляет (СН3)m(СН3СН2)3-mC, 1-метил-1-циклопентил, 1-метил-1-циклогексил или 2,3-диметил-2-бутил, где m означает 0,1,2 или 3, а R6 независимо представляет R5 или 2,3,3-триметил-2-бутил;
R3 представляет Н или независимо R4;
R4 представляет галоген или СН3;
при условии, что когда А является N(СН3)1-nHnR5, где n означает 0 или 1, R3 - Н и R5 -1-метил-1-циклогексил или (СН3)m(СН2СН3)3-mC, где m означает 0 или 3, или, если R3 - галоген и R5 является (СН3)m(СН2СН3)3-mC, где m означает 3, тогда R2 не может быть этилом;
а также при условии, что, когда А представляет ОR6, то m является целым числом, равным или меньше 2, и если R3 - Н или галоген, а R2 - этил или изопропил, тогда R6 является (СН3)m(СН3СН2)3-mC, где m означает 1;
или его сельскохозяйственно-приемлемая соль.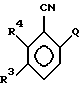
где Q представляет фтор или хлор;
R3 - водород, либо R3 и R4 независимо представляют собой галоген или СН3,
с соединением формулы МА, где М является Li, Nа или К, в растворителе или с эквивалентом соединения МА, полученным in situ кипячением с обратным холодильником лития, натрия или калия в избытке АН, где А имеет указанные выше значения, с получением соединения формулы IV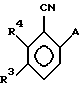
где А, R3 и R4 имеют указанные выше значения,
а затем нагревание полученного соединения формулы IV с NаОН, Н2О2, Н2О и этанолом, либо кипячение соединения формулы IV с обратным холодильником в растворителе с КОН, с получением соединения формулы V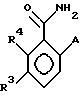
в которой А, R3 и R4 имеют указанные выше значения,
и обработку полученного соединения формулы V соединением формулы
R2Х,
где Х - хлор, иод или - OSО2(ОR2),
в присутствии основания, либо в присутствии основания, содержащего гидроксигруппу и катализатор межфазового переноса, с получением соединения формулы I, где А представляет ОR6, R6 имеет указанные выше значения.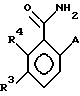
где А - ОR6, а R6 - независимо является R5 или 2, 3, 3-триметил-2-бутилом,
R3 - водород или независимо R4;
R4 - галоген или СН3;
соединением формулы
R2Х,
в которой R2 - этил;
Х представляет хлор, бром, иод или OSО2(ОR2),
в присутствии основания, или в присутствии основания и катализатора межфазового переноса с получением соединения формулы I по п.1, где А представляет ОR6, а R6 имеет указанные выше значения.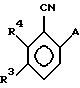
где А, R3 и R4 имеют указанные выше значения,
NаОН, Н2О2, Н2О и этанолом либо кипячением с обратным холодильником с КОН в трет-бутаноле или третичном амиловом спирте.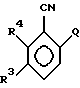
где Q - фтор или хлор;
R3 и R4 имеют указанные выше значения,
соединением формулы МА в растворителе, где М представляет Li, Nа или К, либо эквивалентом соединения МА, полученным in situ путем кипячения с обратным холодильником лития, натрия или калия в избытке АН, где А имеет указанные выше значения, с получением соединения формулы IV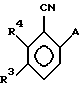
которое затем обрабатывают соединением HAl (изобутил)2 в растворителе, таком, как толуол или метиленхлорид, с последующим взаимодействием с КМnO4 в спиртовом растворителе, таком, как этанол или трет-бутанол, КН2РО4, Н2О при рН примерно от 5 до 9, с получением соединения формулы VI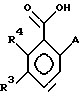
где А, R3 и R4 имеют указанные выше значения,
и обработку полученного соединения формулы VI тионилхлоридом или COCl2 в присутствии пиридина и диметилформамида в апротонном растворителе, таком, как ацетонитрил, метиленхлорид или дихлорэтан, а затем Н2NR2 с получением вышеуказанного соединения формулы I, где А представляет ОR6 и R6 имеет указанные выше значения.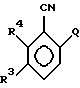
где Q - фтор или хлор;
R3 и R4 имеют указанные выше значения;
с соединением формулы МА в растворителе, где М является Li, Nа или К, или с эквивалентом соединения МА, полученным in situ кипячением с обратным холодильником лития, натрия или калия в избытке AH, где А имеет указанные выше значения.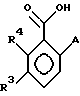
соединением COCl2 или тионилхлоридом в присутствии пиридина и диметилформамида в апротонном растворителе, а затем соединением Н2NR2.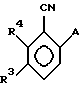
где А, R3 и R4 - определены выше,
Hal (изобутил)2, в растворителе, таком, как толуол или метиленхлорид, а затем КМnО4 в спиртовом растворителе, таком, как этанол или трет-бутанол, КН2РО4, Н2О при рН примерно от 5 до 9, с получением соединения формулы VI.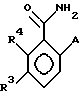
где А, R3 и R4, как определено в п.8.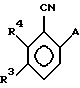
где А, R3 и R4, как определено в п.9.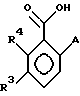
где А, R3 и R4, как определено в п.10 и 13.
| Устройство для пневмоподачи сыпучих материалов | 1975 |
|
SU538231A1 |
| Способ получения производных бензамида или их солей | 1976 |
|
SU645557A3 |
| Ароматические диаминобензанилиды в качестве мономеров для фенилированных полиамидов и полиамидхиназолонов | 1976 |
|
SU749826A1 |
| Вибратор для погружения или извлечения свай | 1973 |
|
SU487205A1 |
| US 4957533 A, 1990 | |||
| ФУНГИЦИД | 0 |
|
SU379067A1 |

