Изобретение относится к новым производным бис(фенил)этана, обладающим дифференцированной фармакологической, в частности противогиперпролиферирующей и противоопухолевой активностью, и фармакологическим композициям на их основе.
Известны производные салициловой кислоты на основе бис(фенил)этана, обладающие противовоспалительной активностью (патент N США 3657430, кл. A 61 K 27/00, 1972).
Задачей изобретения является создание новых производных бис(фенил)этана, обладающих высокой фармакологической активностью, позволяющей создать фармакологические композиции на их основе.
Поставленная задача достигается данными производными бис(фенил)этана общей формулы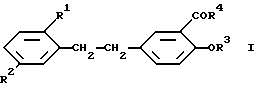
где
R1 и R2 каждый независимо представляет алкокси;
R3 - атом водорода или ацил;
R4 - алкоксигруппа в свободной форме в тех случаях, когда такие формы существуют, в форме соли, называемых здесь и далее "соединения изобретения".
Алкил, как часть такого заместителя как алкоксильная группа, предпочтительно содержит 1 - 4 атомов углерода, а особенно предпочтителен метил или этил.
Ацил предпочтительно представляет остаток карбоновой кислоты, в частности алкил-, арилалкил- или арилкарбоновой кислоты, причем арил предпочтительно представлен фенилом, а алкиленовая часть ацила, включая карбонильную группу, предпочтительно представляет группу, содержащую 1 - 5 атомов углерода. Предпочтительной ацильной группировкой служит ацетил.
В предпочтительной группе соединений изобретения R1 и R2 каждый независимо представляет алкоксильную группу, содержащую 1 - 4 атомов углерода, в частности метоксигруппу, а R3 и R4 имеют значения, определенные выше.
В другой подгруппе R1 и R2 каждый независимо представляет алкоксильную группу, содержащую 1 - 4 атомов углерода, R3 - атом водорода или алкилкарбонильная группа, в сумме содержащая 2 - 5 атомов углерода, и R4 - алкоксил, содержащий 1 - 4 атомов углерода (соединение Is); R1 и R2 особенно являются независимо метоксилом, этоксилом, R4 предпочтительно является метоксилом, этоксилом.
Предпочтительными соединениями являются соединения общей формулы I, где R1 и R2 представляют собой метоксигруппы и R3 и R4 являются соответственно либо H и OCH2CH3 или COCH3 и OCH3, или H и OCH2OCH2-CH3, или H и OCH3CH2CH2CH3 или H и OCH(CH3)2, или R1 и R2 представляют OCH2CH3, R3 - H, а R4 - OCH3; наиболее предпочтителен метиловый эфир 5-[2-(2,5-диматоксифенил)-этил]-2-гидроксибензойной кислоты.
Соединения по изобретению получают путем
а) восстановления соединения формулы II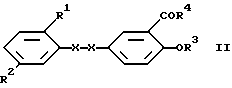
в которой
- X-X- представляет винилен или этинилен, путем обработки водородом в присутствии катализатора гидрирования или
соединения формулы I можно получить также тем, что соединение формулы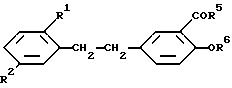
где R1 и R2 имеют указанные значения; R5 - гидрокси или алкоксигруппа, R6 - водород или ацильная группа,
подвергают этерификации или возможно переэтерификации или ацилированию обработкой соответственно спиртом в присутствии серной кислоты или уксусным ангидридом в среде пиридина. При этом функциональные группы могут быть защищены и защитные группы могут быть удалены после завершения реакции, полученное соединение формулы I выделяют либо в свободной форме, либо в виде фармакологически приемлемых солей.
Способ по изобретению может быть осуществлен традиционным образом.
Вариант а) способа обычно осуществляют путем гидрогенирования. Предпочтительно использовать в таком варианте водород вместе с катализаторами гидрирования, такими как палладий, платина или рений, более предпочтительно использовать палладий, нанесенный на активированный уголь.
Вариант б) способа также осуществляют традиционным путем. По крайней мере один из R5 и R6 является гидроксилом или водородом соответственно. Реакцию трансэтерификации также как этерификации предпочтительно осуществляют при взаимодействии со спиртом, соответствующим эфирной группировке, которую нужно получить в присутствии сильной кислоты, такой как серная кислота.
Удаление защитной группы также проводят традиционным образом. Функциональной группой, которая может быть защищена подходящим образом, например, служит гидроксильная группа, защищенная, например, триалкилсилилом. Удаление триалкилсилила может быть осуществлено путем обработки фтористоводородной кислотой в таком растворителе, как ацетонитрил.
Целевые соединения по изобретению могут быть выделены из реакционной смеси и очищены согласно известным методикам, например путем хроматографической очистки.
Исходные материалы также могут быть получены традиционным способом.
Соединения формулы II, в которой -X-X- представляет винилен, могут быть получены при взаимодействии соответствующего соединения формулы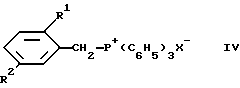
в которой X- - анион, предпочтительно бромид, с соответствующим соединением формулы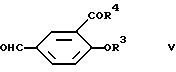
в условиях, характерных для реакции типа Виттига (Хорнера) Эммонса обработкой фосфоросодержащего компонента основанием, таким как алкиллитий, гидрид щелочного металла или амид щелочного металла, например амид натрия, диизопропиламид лития, или алкоголят щелочного металла, в температурном интервале от примерно -70 до примерно 100oC и одновременном или последовательном превращении с карбонильным компонентом при температурах от примерно -70 до примерно 120oC, предпочтительно от примерно -60 до примерно 60oC, в подходящих растворителях, таких как, например, тетрагидрофуран, толуол или диметилсульфоксид.
Соединения формулы II, в которой -X-X- - этилен, могут быть получены при взаимодействии соответствующего соединения формулы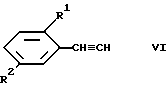
с соответствующим соединением формулы
в которой
Y представляет галоген, предпочтительно иод, следуя стандартным процедурам реакции Гека, а именно взаимодействию галогенолефинов с ацетиленами.
Соединения формулы III, в которой R6 - водород, а также соединения формулы III, в которой R5 представляет гидроксил, могут быть получены путем аналогично описанному выше или путем деацилирования или дезалкилирования соответствующих соединений формулы I, в которых R3 - ацил и/или R4 - алкоксил.
Исходные вещества известны или могут быть получены в соответствии с известными методиками или аналогично известным методикам, или согласно методам, описанным в примерах, так что их получение специально не описывается в рамках изобретения.
В следующих примерах, приводимых в качестве иллюстрации, все температуры приведены в градусах Цельсия; все спектры ЯМР - это 1H-ЯМР (CDCl3).
Пример 1. 5-[2-(2,5-Диметоксифенил)этил]-2-оксибензойной кислоты метиловый эфир.
Формула I: R1, R2, R4 - OCH3; R3 - H.
Вариант а) способа получения.
60 мг 5-[(2,5-диметоксифенил)этинил]-2-гидроксибензойной кислоты метилового эфира (формула II: -X-X- - этинилен) или 60 мг E/X смеси 5-{2-(2,5-диметоксифенил)этенил]-2-гидроксибензойной кислоты метилового эфира (формула II: -X-X- - винилен) растворили в 10 мл этилацетата. После добавления 10 мг 10 % Pd/C смесь перемешивали в атмосфере водорода в течение ночи, профильтровали через Целите-CeliteR и упарили до сухости при пониженном давлении. После хроматографической очистки получили соединение, название которого дано в заголовке (бесцветные кристаллы; т.пл. 65oC после хроматографической очистки; т.пл. 67oC после перекристаллизации из этанола).
Исходные материалы получают следующим образом.
В атмосфере аргона растворили 240 мг 2,5-диметоксифенилацетилена (формула VI) и 493 мг метил-5-иодосалицилата (формула VII) в 20 мл сухого и свободного от кислорода бензола. Смесь обработали 85 мг тетракис(тетрафенилфосфино)палладия, 20 мг иодида меди (I) и 450 мг триэтиламина, перемешивали при комнатной температуре в течение ночи и затем вылили в 100 мл водного pH 7 буфера. После экстрагирования этилацетатом и очистки путем хроматографирования на силикагеле получили метиловый эфир 5-[2-(2,5-диметоксифенил)этинил] -2-гидроксибензойной кислоты в виде бесцветных кристаллов с т.пл. 105-108oC. К суспензии 1 г 2,5-диметоксибензилтрифенилфосфоний бромида (формула IV) в 15 мл абсолютизированного тетрагидрофурана добавили при 40oC 6,6 ммоль литийдиизопропилбромида в тетрагидрофуран/н-гексане. После перемешивания в течение 1 ч смесь охладили до -70oC и обработали медленно 372 мг 3-метоксикарбонил-4-гидроксибензальдегида (формула V), растворенного в 5 мл абсолютизированного тетрагидрофурана. Смесь перемешивали в течение 1 ч при -70oC и в течение 2 ч при комнатной температуре, а затем вылили в водный раствор хлорида аммония. После экстрагирования этилацетатом и упаривания получена смесь E- и Z-изомеров метилового эфира 5-[2-(2,5-диметоксифенил)этенил] -2-гидроксибензойной кислоты, которая может быть подвергнута хроматографированию на силикагеле с использованием в качестве элюента смеси н-гексан/этилацетат 9:1. Первая фракция содержит Z-изомер (бесцветные кристаллы, т.пл. 78-80oC), вслед за ней получают E-изомер, т.пл. 80-82oC.
Пример 2. Этиловый эфир 5-[2-(2,5-диметоксифенил)этил]-2-гидроксибензойной кислоты.
Формула I: R1, R2 - OCH3; R3 - H; R4 - OCH2CH3.
Вариант б) способа получения.
В 10 мл сухого этанола растворили 250 мг 5-[2-(2,5-диметоксифенил)этил] -2-гидроксибензойной кислоты или 250 мг 5-[2-(2,5-диметоксифенил)этил]-2-гидроксибензойной кислоты метилового эфира (формула III: R5 - OH или OCH3; R6 -H) и обработали 0,2 мл концентрированной серной кислоты. Смесь нагревали с обратным холодильником в течение 48 ч и вылили в 150 мл воды. После экстрагирования этилацетатом и очистки путем хроматографирования на силикагеле получили соединение, название которого дано в заголовке (бесцветное масло).
ЯМР: 10,69 (с., 1H); 7,67 (д., J=2,2 Гц, 1H); 7,29 (дд., J=2,2 + 8,5 Гц, 1H); 6,91 (д., J=8,5 Гц, 1H); 6,66-6,83 (м., 3H); 4,42 (кв.д., J=7 Гц, 2H); 3,79 (с., 3H); 3,75 (с., 3H); 2,76-2,92 (м., 4H); 1,44 (тр., J=7 Гц, 3H).
Пример 3. Метиловый эфир 5-[2-(2,5-диметоксифенил)этил]-2-ацетоксибензойной кислоты.
Формула I: R1, R2, R4 - OCH3; R3 - COCH3.
Вариант б) способа получения.
В 2,5 мл уксусного ангидрида растворили 150 мг 5-[2-(2,5-диметоксифенил)этил] -2-гидроксибензойной кислоты метилового эфира и обработали 45 мг пиридина. Смесь перемешивали в течение ночи при комнатной температуре, вылили в 100 мл воды и проэкстрагировали этилацетатом. Объединенные органические экстракты энергично промыли водным раствором 0,1 M HCl, водным раствором бикарбоната натрия и водой. После упаривания растворителя получили озаглавленное соединение в виде бесцветного масла.
ЯМР: 7,85 (д., J=2 Гц, 1H); 7,34 (дд., J=2+8 Гц, 1H); 6,99 (д., J=8 Гц, 1H); 6,66-6,80 (м., 3H); 3,87 (с., 3H); 3,76 (с., 3H); 2,89 (с., 4H); 2,34 (с., 3H).
Следующие соединения (формула I) получены аналогичным способом (см. таблицу).
Соединения формулы I в свободной форме или в тех случаях, когда такие формы существуют в виде фармацевтических приемлемых солей, обладают преимущественными химиотерапевтическими свойствами. Их прописывают в качестве фармацевтических препаратов.
В частности, они обладают противогиперпролиферирующим, проотивовоспалительным и противоопухолевым действием. Ниже по тексту употребляются следующие сокращения:
CHO-KI - линия клеток, известная как "яичник китайского хомяка-KI",
DMEM - модифицированная по способу Дульбекко среда Игла (Gibco),
EGF - эпидермальный фактор роста,
FCS - фетальная телячья сыворотка,
HaCaT - линия клеток, известная под названием "кальциевая температура взрослой особи",
PPMI- - среда 1640 мемориального института Roswell Park 1640,
TGF α - трансформирующий фактор роста α .
Противогиперпролиферирующая, противовоспалительная активность и противоопухолевая активность могут быть определены следующим образом.
1. Ингибирование пролиферации в кератиноцитах человека линии клеток HaCaT (или клеточной линии CHO-KI).
HaCaT клетки, трансформированные спонтанно, TGF α- и EGF-рецептором позитивной неопухолевогенной линией клеток кератиноцита человека с сохранными в высокой степени характеристиками дифференциации фенотипа нормальных кератиноцитов (Boukamp et al., J. Cell. Biol. 106, 761-771 (1988), культивировали в среде DMEM, подпитываемой 2,2 г/л NaHCO3, 0,11 г/л пирувата натрия, 15 мМ Hepes, 5% FCS, пенициллином (100 М.Е./мл), стрептомицином (100 мг/мл) и глутамином )до достижения концентрации в конечном состоянии 4 мМ). CHO-KI клетки культивировали в приведенной среде с добавлением 40 мг/мл пролина. Для анализа пролиферации клетки разделили путем трипсинизации (клетки разделили, обработав их трипсином); суспендировали в свежей среде и высеяли в 96-луночные микротирационные планшеты в количестве 2000-4000 клеток (0,2 мл)/лунка. Через 24 ч среду заменили свежей средой, содержащей дифференцированные концентрации тестируемого соединения. Через 3-4 дня инкубирования измерили степень клеточной пролиферации методом колориметрического анализа, используя сульфородамин B (Skehan et al., J. Natl. Candcer Inst. 82, 1107-1112 (1990)). Результаты представляют как среднее ± стандартное отклонение трех измерений.
В приведенном опыте ИК50 соединений, ингибирующих пролиферацию HaCaT клеток, варьируются в пределах от 0,03 до примерно 3 мМ.
Противоопухолевая активность может быть определена следующим образом.
2. Ингибирование пролиферации опухолевых клеток.
Линии опухолевых клеток, например, K-562, L1210, Hela, SK-BR-3, MDAMB-468, MCF-7 или MDAMB-231 (все могут быть получены из Американской коллекции клеточных культур, MD 20852, USA) выращивали в среде RPMI-1640, подпитываемой 10% инактивированной теплом (56oC/30 мин) FCS и антибиотиками (1 х раствор GIBCO) пенициллин-стрептомицина). По достижении экспоненциального роста линий опухолевых клеток, выращиваемых в суспензии (K-562 и L1210), или 60-90% степени слияния вязких липких линий клеток (Hela, SK-BR-3, MDAMB-468, MCF-7 и MDAMB-231) клетки собирают (вязкие клетки трипсинизируют), суспендируют в свежеприготовленной ростовой среде и высевают в 96-луночные планшеты в концентрации, варьирующейся в пределах 1000 - 5000 клеток/лунка. Клетки выращивают в течение 2-4 дней в конечном объеме 0,2 мл/лунка при 37oC в увлажняемом инкубаторе, уравновешенном 5% CO2 в присутствии дифференцированных концентраций тестируемого соединения. Степень пролиферации клеток измеряют методом колориметрического анализа, используя MTT (Alley et al., Cancer Res. 48, 589-601 (1988)) для клеток, выращиваемых в суспензии, или сульфородамин B для вязких клеток.
В приведенном выше опыте значения ИК50 для соединений, ингибирующих пролиферацию четырех клеточных линий, упомянутых выше, варьируются в пределах от примерно 0,019 до примерно 3 мМ.
3. Влияние на рост опухолей человека, ксенотрансплантированных голым мышам.
Mia Pa Ca-2 - это линия клеток опухоли поджелудочной железы человека; A431 - это линия клеток, выделенных из эпидермальной карциномы наружных половых органов женщины. Эти обе клеточные линии можно получить в ATCC (ATCC - коллекция культур США). Эти культуры выращивались без антибиотиков и противогрибковых средств. Голые мыши - самки линии Balb/C массой 20 - 23 г содержали в группах по 5 животных в каждой со свободным доступом к питьевой воде и диете, лишенной патогенов, распространяемых грызунами. Опухоли у мышей инициировали путем инъекции подкожно культивированных клеток опухолевых клеточных линий в количестве 107 клеток. Как только опухоли достигали приблизительно 1 см в диаметре, их иссекали, разрезали на маленькие кусочки примерно по 3 мм и трансплантировали под кожу в обе боковые стороны живота голых мышей. Через одну и две недели после трансплантации измеряли размер опухолей, используя штанген-циркуль. Животные с растущими опухолями разделили на контрольные группы и группы для лечения, причем в обеих группах мыши имели идентичные значения размеров опухоли. Тестируемые соединения вводили перорально. Одинаковые растворы лекарственного препарата использовали для двух последовательных курсов лечения. Контрольные животные получали только наполнитель. С недельным интервалом проводили определение объемов опухоли и записывали как значение объема опухоли на одно животное в мм3. Результаты оценивали, используя статистические программы RS(1)BBN Software Products Corp.), а также тесты, включающие т-тест Стьюдента и тест Mann-Whitney.
В приведенном опыте соединения, вводимые в количестве 30 - 100 мг/кг перорально или 3-10 мг/кг внутривенно, ингибируют рост A431 опухолей (A431 - эпидермальные опухоли человека, сверхэкспрессирующие EGF рецептором) в значительной степени (P < 0,05 или < 0,01) в течение всего периода лечения. По окончании эксперимента (4 недели) объем опухоли у мышей, обработанных лекарством, составляют примерно 10 - 50% объема опухоли контрольной группы животных. Рост MiaPaCa опухолей (опухоли поджелудочной железы человека, экспрессирующие нормальные количества EGF рецептора) ингибируется аналогичным образом: через три недели лечения объем опухолей у мышей, получавших лекарственный препарат, значительно меньше объема опухоли у контрольной группы животных.
Противогиперпролиферирующая/противовоспалительная активность соединений при их местном нанесении может быть определена следующим образом.
4. Ингибирование у мыши аллергического контактного дерматита, индуцированного оксазолоном.
Чувствительность индуцируют однократным нанесением 23% оксазолона (10 мл) на кожу брюшной полости мышей (в группе - 8 животных). Гиперчувствительность, приводящую к опуханию наружного уха, выявляют вторичным нанесением 2% оксазолона на одно наружное ухо каждого животного через 8 дней.
Ингибирование припухлости наружного уха достигается путем двукратного нанесения тестируемого соединения местно (за 30 мин до и через 30 мин после выявления симптомов реакции). Эффективность определяли как разность веса наружного уха животных, обработанных лекарственным препаратом и обработанных только наполнителем (пропиленгликоль/ацетон 7:3) соответственно, и выражали как % набухания к случаю, когда наносили только один наполнитель.
В приведенном опыте примерно 20-60% ингибирования получали при дозе тестируемого соединения, равной 1,2%.
Соединения формулы I в свободном виде или, если такие формы существуют, в виде фармакологически приемлемых солей, таким образом, показаны для использования в качестве противогиперпролиферирующих/противовоспалительных и противоопухолевых средств при лечении гиперпролиферирующих/воспалительных заболеваний и рака, а также для суппрессии неопластических заболеваний, например воспалительных/гиперпролиферирующих кожных заболеваний, рака кожи, а также кожных и системных проявлений иммунологически опосредованных заболеваний и автоиммунных заболеваний, таких как псориаз, атопические дерматиты, контактные дерматиты и родственные экзематозные дерматиты, дерматиты себорейной природы, красный плоский лишай, пузырчатка, булезная пузырчатка, булезный эпидермолиз, крапивница, ангиоздема, васкулиты, эритемы, кожные эозинофильные лейкоциты, волчанка и очаговое облысение.
В этих случаях используемые дозы могут варьироваться, конечно, в зависимости, например, от конкретного используемого соединения, способа введения и требуемого лечения. Однако, как правило, удовлетворительные результаты получают, когда соединения вводят ежедневно в количестве примерно 0,1 - 10 мг/кг массы тела животного внутривенно или примерно 0,5 - 100 мг/кг перорально при дробном приеме препарата 2-4 раза в день. Для большинства крупных млекопитающих общая ежедневная доза составляет примерно 7 - 2000 мг удобно вводимых, например, при дробной дозировке до четырех раз в день или в пролонгированных формах выпуска. Однократная дозировочная форма составляет, например, примерно 1,75 - 1000 мг соединений в смеси с по крайней мере одним жидким или твердым фармацевтически приемлемым носителем или разбавителем.
Соединения могут быть введены способом, аналогичным известным стандартам, используемым в таких случаях. Соединения могут быть смешаны с традиционными химотерапевтически приемлемыми носителями и разбавителями, а также возможно с другими добавками, и вводиться они могут, например, перорально, в виде таких выпускных форм, как таблетки и капсулы.
В альтернативном варианте соединения можно использовать в формах, предназначенных для местного введения, таких, например, как мази или кремы, для парентерального введения типа внутривенного. Концентрации активного вещества будут, конечно, изменяться в зависимости, например, от используемого соединения, необходимого лечения и природы выпускной формы. Однако, как правило, удовлетворительные результаты получают, например, в формах, предназначенных для местного нанесения, при концентрациях активного субстрата примерно 0,05 - 5%, в частности примерно 0,1 - 1% по массе.
Фармацевтические композиции, содержащие соединение формулы I в свободной форме или, если такие формы существуют, в виде фармацевтически приемлемых солей вместе с по крайней мере одним фармацевтически приемлемым носителем или разбавителем также составляют часть изобретения.
Изобретение также содержит соединения формулы I в свободной форме или, если таковые имеются, в форме фармацевтически приемлемых солей для использования в качестве фармацевтического препарата(ов) в особенности при лечении гиперпролиферирующих/воспалительных заболеваний и рака, например карциномы поджелудочной железы или молочной железы.
Далее изобретение содержит способ лечения гиперпролиферирующих/воспалительных заболеваний и рака, который включает назначение терапевтически эффективного количества соединения формулы I в свободной форме или, если таковые существуют, в форме фармацевтически приемлемых солей пациенту, нуждающемуся в такого рода лечении.
Предпочтительным соединением является соединение примера 1, т.е. 5-[2-(2,5-диметоксифенил)этил] -2-гидроксибензойной кислоты метиловый эфир. Было, например, установлено, что в приведенном опыте 1 у этого соединения значения ИК50 равны 0,045 мМ, в то время как EGF-рецептор негативной клеточной линии CHO-KI ингибируется при ИК50, превышающих примерно 0,2 мМ.
В приведенном опыте 2 это соединение ингибирует три из четырех клеточных опухолевых линий молочной железы, а именно SK-BR-3, MDAMB-468 и MCF-7, при значениях ИК50, находящихся в пределах 20 - 50 нМ, в то время как клетки опухолевых MDAMB-231 молочной железы и опухолевые клетки K-562, L1210 и Hela, не относящиеся к молочной железе, ингибируются при ИК50, варьирующей в пределах 200 - 470 нМ, что свидетельствует о селективном ингибировании этим соединением некоторых опухолей молочной железы, в то время как колхицин ингибирует все опухолевые клетки, не отличаясь избирательностью, при ИК50 в интервале 5 - 20 нМ.
В приведенном опыте 3 с опухолями A431 при дозе соединения 30 мг/кг, вводимой перорально три раза в неделю, объем опухоли у мышей, получавших лекарственный препарат, составляет только 56,3% опухоли контрольной группы животных к концу эксперимента (4 недели), а при дозе 3 мг/кг, вводимой внутривенно, - примерно 40% от размера опухоли у контрольной группы животных. В случае опухолей панкреатической (поджелудочной железы) MiaPaCa размер опухоли у мышей, которых лечили, давая 30 мг/кг перорально соединения примера 1 в течение 2 недель, составлял примерно 46% размера опухоли у контрольной группы животных; в аналогичном эксперименте ингибирование цисплатиной, вводимой в стандартном, сопоставимом количестве 10 мг/кг внутрибрюшинно каждый третий день (цисплатину нельзя вводить обычно пероральным путем), достигло значимой величины только через 4 недели лечения (размер опухоли достиг 40% размера опухоли контрольной группы животных), хотя величина объема опухоли в начале лечения цисплатиной составляла только примерно половину размера опухоли, которую лечили соединением примера 1.
В приведенном опыте 4 соединение примера 1 показывает 46% ингибирование припухлости при концентрации 0,4%.
Таким образом, установлено, что в указанном антираковом применении соединение примера 1 может быть введено более крупным млекопитающим, например человеку, введение может осуществляться способами и в дозировках, аналогичных тем, которые традиционно используют при введении цисплатины. Эта сильная противоопухолевая активность наблюдается при дозах, не подавляющих иммунный ответ и гематопоэз, а опухолевые клетки, экспрессирующие фенотип, устойчивый к множеству лекарств, также чувствительны к соединению, как и их родительские копии.
Кроме того, изобретение включает способ получения лекарственного препарата, который включает смешение соединения формулы I в свободной форме или, если таковые существуют, в виде фармацевтически приемлемой формы вместе с по крайней мере одним фармацевтически приемлемым носителем или разбавителем.


Изобретение относится к соединениям формулы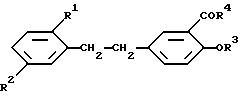
в которой R1 и R2 каждый независимо представляет алкоксильную группу, содержащую 1 - 4 атомов углерода, R3 - H или алкилкарбонильная группа, содержащая 2 - 5 атомов углерода, R4 - алкоксильная группа, содержащая 1 - 4 атомов углерода, в свободной форме, а также, если таковые существуют, в виде соли. Соединения обладают потенциальной противогиперпролиферирующей/противовоспалительной и противораковой активностью. 2 с. и 2 з.п. ф-лы. 1 табл.
\ \ \1 1. Производные бис-(фенил)этана общей формулы I \\\6 $$$ \\\1 где R<M^>1<D> и R<M^>2<D> - каждый независимо алкоксильная группа, содержащая от 1 до 4 атомов углерода; \\\4 R<M^>3<D> - атом водорода или алкилкарбонильная группа, в которой содержится суммарно от 2 до 5 атомов углерода; \\\4 R<M^ >4<D> - алкоксильная группа, содержащая 1 - 4 атома углерода, \\\1 в свободной форме или в форме солей, если они образуются. \\\2 2. Соединение по п. 1, отличающееся тем, что оно представляет метиловый эфир 5-[2-(2,5-диметоксифенил)-этил] -2-гидроксибензойной кислоты. \\\2 3. Соединение по п.1, отличающееся тем, что либо R<M^>1<D> и R<M^>2<D> представляют метоксигруппы и R<M^>3<D> и R<M^>4<D> представляют соответственно либо H и OCH<Mv>2<D>CH<Mv>3<D> или COCH<Mv>3<D> и OCH<Mv>3<D> или H и OCH<Mv>2<D>CH<Mv>2<D>CH<Mv>3<D>, или H и OCH<Mv>2<D>CH<Mv>2<D>CH<Mv>2<D>CH<Mv>3<D>, или H и OCH(CH<Mv>3<D>)<Mv>2<D>, или R<M^>1<D> и R<M^>2<D> представляют OCH<Mv>2<D>CH<Mv>3<D>, R<M^>3<D> - H, а R<M^>4<D> - OCH<Mv>3<D>. \\\2 4. Фармацевтическая композиция, проявляющая противоопухолевое, противогиперпролиферирующее и противовоспалительное действие, содержащая в качестве активного начала эффективное количество соединения формулы I, охарактеризованного в п.1, или его фармацевтически приемлемой соли и целевые добавки.
| US, патент, 3657430 , кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| EP, патент, 497740, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Journal of Natural Products, 52, N p.1252 - 1257. | |||
