Настоящее изобретение относится к новым антрациклиновым гликозидам, которые обладают противоопухолевой активностью, к способам их получения и к содержащим их фармацевтическим композициям.
В настоящем изобретении предложены антрациклиновые гликозиды, родственные даунорубицину и доксорубицину, в которых 3'-аминогруппа сахарного остатка замкнута в азиридиновое кольцо, и необязательно гидроксигруппа у C-4' сахара может быть защищена в форме сульфоната. В настоящем изобретении предложены также их растворимые в воде производные и фармацевтически приемлемые соли присоединения кислот.
В настоящем изобретении предложено соединение, которое является антрациклиновым гликозидом формулы 1 или 2: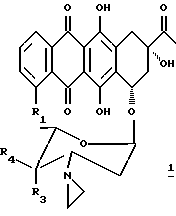
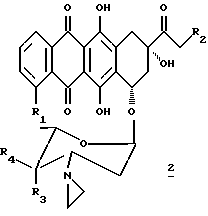
где R1 представляет водород или метоксигруппу; R2 представляет водород, гидроксигруппу или представляет ацилоксиостаток формулы 3:
-O-COR5
где R5 представляет линейный или разветвленный C1-C8 алкил, моно- или бициклический арил, предпочтительно фенил, или гетеро- моно- или бициклическое кольцо, предпочтительно пиридил, причем каждая из групп может быть необязательно замещена (a) аминогруппой -NR6R7, где R6 и R7 независимо представляют водород или C1-C4 алкил, или (b) карбоксигруппой; R3 и R4 оба представляют водород, или один из R3 и R4 представляет водород, а другой представляет гидроксигруппу или группу формулы -OSO2R8, где R8 может быть линейной или разветвленной алкильной группой, содержащей от 1 до 6 атомов углерода, например, от 1 до 4 атомов углерода; R8 может представлять, в частности, метил, этил, н-пропил или изопропил.
В другом варианте R8 может представлять арильную группу, такую, как фенил, незамещенный или замещенный 1 - 3 заместителями, каждый из которых может быть независимо линейной или разветвленной алкильной или алкоксигруппой, содержащей от 1 до 6 атомов углерода, например 1 - 3 атома углерода, атомом галоида или нитрогруппой. Примеры атомов галоида включают фтор, хлор, бром и йод, предпочтительно, фтор или хлор, более предпочтительно хлор.
В настоящем изобретении арильная группа представляет моноциклический или бициклический ароматический углеводород, содержащий от 6 до 10 атомов углерода, например, фенил или нафтил. Гетероциклическое кольцо представляет 5- или 6-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее, по крайней мере, один гетероатом, выбранный из O, S и N, которое необязательно конденсировано со второй 5- или 6-членной, насыщенной и ненасыщенной гетероциклической группой.
Примеры насыщенных и ненасыщенных гетероциклических колец включают пиразолильное, имидазолильное, пиридильное, пиразильное, пиримидильное, пиридазинильное, морфолино, тилморфолино, фурильное и тиенильное кольца.
Предпочтительно, чтобы R2 представлял гидрокси- или O-никотинил, R3 представлял гидрокси- или -OSO2R8, где R8 представлял бы C1-C4 алкил, а R4 представлял бы водород.
Примеры соединений настоящего изобретения включают:
(1a) 3'-диамино-3'-[1-азиридинил]-4-O-метансульфонил-даунорубицин (R1 = OCH, R4 = H, R3 = OSO2CH3)
(1b) 4-деметокси-3'-деамино-3'[1-азиридинил] -4'-O-метансульфонил-даунорубицин (R1 = R4 = H, R3 = OSO2CH3)
(1c) 3'-деамино-3'[1-азиридинил] -даунорубицин (R1 = OCH3, R4 = H, R3 = OH)
(1d) 4-деметокси-3'-деамино-3'-[1-азиридинил]-даунорубицин (R1 = R4 = H, R = OH)
(2a) 3'-деамино-3'-[1-азиридинил] -4'-O-метансульфонил-14-никотинат- даунорубицин (R1 = OCH3, R2 = O-никотинил, R4 = H, R3 = OSO2CH3)
(2b) 3'-деамино-3'-[1-азиридинил]-14-никотинат-доксорубицин (R1 = OCH3, R2 = O-никотиноил, R4 = H, R3 = OH)
(2c) 3'-деамино-3'-[1-азиридинил]-4'-O-метансульфонил- доксорубицин (R1 = OCH3, R2 = OH, R4 = H, R3 = OSO2CH3)
(2d) 4-деметокси-3'-деамино-3'-[1-азиридинил] - 4'-O-метансульфонил-доксорубицин (R1 = R4 = H, R2 = OH, R3 = OSO2CH3)3
(2e) 3'-деамино-3'-[1-азиридинил]-доксорубицин (R1 = OCH3, R4 = H, R2 = R3 = OH)
(2f) 4-деметокси-3'-деамино-3'-[1-азиридинил]-доксорубицин (R1 = R4 = H, R2 = R3 = OH)
(2g) 3'-деамино-3'-[1-азиридинил] -4'-иододоксорубицин (R1 = OCH3, R2 = OH, R4 = H, R3 = 1)
(2h) 3'-деамино-3'-[1-азиридинил]-4'-деоксидоксорубицин (R1 = OCH3, R2 = OH, R3 = R4 = H)
и такие их фармацевтически приемлемые соли, как соли хлористоводородной кислоты (соляной кислоты).
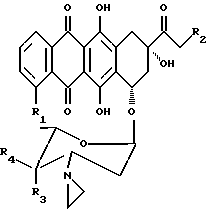
Далее, в настоящем изобретении предложен способ получения азиридиноантрациклинового гликозида формулы 1 или 2, как указано ранее, или его фармацевтически приемлемой соли, который включает:
(a) превращение антрациклина общей формулы 4: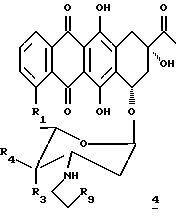
где R1, R3 и R4 имеют указанные ранее значения, а R9 представляет сульфонатную группу или атом галоида, предпочтительно атом хлора, в антрациклин формулы 1, причем соединение формулы 4, предпочтительно растворяют в безводном органическом растворителе в присутствии безводной соли щелочного металла и мягкого основания; и, при желании,
(b) гидролиз производного формулы 5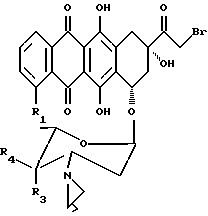
где R1, R3, R4 имеют указанные ранее значения (которое можно получить из соединения формулы 1 по способу, описанному в патенте США N 3803124) до получения азиридино-антрациклинового производного формулы 2, где R2 представляет гидроксильную группу; и, при желании,
(c) осуществление взаимодействия соединения формулы 5, как определено ранее, с солью производного формулы 3'
X+-OCOR5
где R5 имеет указанные ранее значения, при условии, что R5 не представляет остатка, содержащего первичную аминогруппу, aX+ представляет ион, предпочтительно ион натрия или калия, и, при желании, превращение полученного таким образом соединения формулы 2 в его фармацевтически приемлемую соль; или
(d) осуществление взаимодействия соединения формулы 5, как определено ранее, с солью производного формулы 3', где R5 представляет первичную аминогруппу, защищенную чувствительной к кислоте защитной группой, с последующим удалением защитной группы, и, при желании, превращение полученного таким образом соединения формулы 2 в его фармацевтически приемлемую соль.
В настоящем изобретении предложен другой способ получения азиридиноантрациклинового гликозида формулы 2, как определено ранее, или его фармацевтически приемлемой соли, который включает:
(a) обработку атрациклина общей формулы 6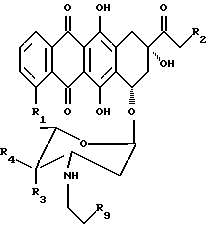
где R1, R2, R3, R4 и R9 имеют указанные ранее значения (такие соединения были раскрыты также в WO 93/01201), силикагелем, и, при желании, превращение полученного таким образом соединения формулы 2, в его фармацевтически приемлемую соль.
Следует отметить, что антрациклины формулы 4 или 6 также могут образовывать азиридиновое кольцо, если их обработать силикагелем. Для такой обработки можно использовать мягкие условия, что позволяет получать соединения формулы 2, исходя из чувствительных к основаниям сложноэфирных производных, таких, как те, которые имеют формулу 6.
В соответствии со способом настоящего изобретения предпочтительные условия реакции получения азиридиноантрациклинов формулы 1 включают растворение соединения формулы 4, как было определено ранее, в безводном органическом растворителе, таком как безводный метиленхлорид, в присутствии безводной щелочной соли металла, например безводного карбоната или бикарбоната натрия или калия, при перемешивании, при температуре от 0 до 30oC, предпочтительно при комнатной температуре, и в течение от 15 минут до 2 часов, предпочтительно около 30 минут.
В другом способе соединения формулы 4 растворяют в смеси таких органических растворителей, как сухой метиленхлорид и метанол, в соотношении от 1: 1 до 1:3 по объему, затем этот раствор обрабатывают силикагелем, предпочтительно 230 - 400 мешей, при перемешивании при температуре от 0 до 30oC, предпочтительно при комнатной температуре, в течение промежутка времени от 15 минут до 2 часов, предпочтительно около 30 минут.
В аналогичном процессе условия реакции для превращения соединений формулы 6, как определено ранее, в азиридиноатрациклины формулы 2, предпочтительно включают растворение соединений формулы 6 в безводном органическом растворителе, например в сухом метиленхлориде и метаноле, и обработку полученного раствора силикагелем, предпочтительно, 230 - 400 мешей, при перемешивании при температуре от 0 до 30oC, предпочтительно, при комнатной температуре в течение промежутка времени от 15 минут до 2 часов, предпочтительно в течение около 30 минут.
Такой полярный растворитель, как метанол, в способе обработки силикагелем используют для удаления антрациклина из силикагеля.
В другом способе получения азиридиноантрациклинового гликозида формулы 2 или его фармацевтически приемлемой соли, где R2 представляет группу формулы 3, в которой R5 не представляет остатка, содержащего первичную аминогруппу, предпочтительные условия реакции включают осуществление взаимодействия соединения формулы 5 с производным соли кислоты формулы 3', как было определено ранее, в безводном полярном растворителе, предпочтительно ацетоне или диметилформамиде, при температуре от 20 до 60oC, предпочтительно при комнатной температуре, в течение от 4 до 15 часов, предпочтительно от 5 до 12 часов.
Условия реакции для получения азиридиноантрациклинового гликозида общей формулы 2, где R2 представляет группу формулы 3, в которой R5 представляет первичную аминогруппу, включают осуществление взаимодействия соединений формулы 5, как было определено ранее, с производным соли кислоты формулы 3', в которой аминогруппа защищена группой, чувствительной к кислоте, например аминогруппа, защищена Шиффовым основанием, в полярном растворителе, таком как ацетон или диметилформамид, при температуре от 20 до 60oC, предпочтительно при комнатной температуре, в течение от 4 до 15 часов, предпочтительно от 5 до 12 часов, затем полученное N-защищенное-сложноэфирное производное деблокируют, растворяя его, например, в метиленхлориде, и добавляя дистиллированную воду и водную соляную кислоту, предпочтительно примерно такой же объем воды, что и метиленхлорида, а соляную кислоту в количестве, соответствующем примерно трем эквивалентам 0,1 н HCl. Полученную смесь интенсивно перемешивают при температуре от 0 до 20oC, предпочтительно, около 15oC, в течение промежутка времени от 30 минут до 2 часов, предпочтительно от 45 до 90 минут, выделяют и водную фазу сушат вымораживанием до получения растворимой соли соляной кислоты C-15 сложноэфирного производного формулы 2. Предпочтительно защищать первичную аминогруппу группой метилендифенила.
В следующем аспекте в настоящем изобретении предложены фармацевтические композиции, содержащие антрациклиновый гликозид формулы 1 или 2, или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым разбавителем или носителем.
Можно использовать обычные носители разбавители. Композиции можно выполнять и вводить обычными способами.
Подходящие способы введения включают парэнтеральное введение. Для парэнтерального введения жидкую композицию можно приготовить, используя активное соединение и стерильный разбавитель или носитель, в котором активное соединение либо может раствориться, либо в нем получают его суспензию. Парэнтеральную композицию можно приготовить в форме стерильного твердого вещества, которое необходимо снова растворить перед употреблением в таком подходящем носителе, как физиологический раствор, стерильная вода или другие стерильные носители.
Соединения настоящего изобретения пригодны для терапевтического лечения организмов человека и животных. Они полезны как противоопухолевые агенты, особенно для лечения лейкемии или аденокарциномы толстой кишки. Терапевтически эффективное количество соединения вводят пациенту, у которого имеется опухоль, для улучшения состояния пациента. Можно вводить такое количество соединения, которого достаточно для ингибирования роста опухоли.
Вводимые дозы можно оценить, зная интервалы доз для доксорубицина и даунорубицина, модифицированные относительно активности, демонстрируемой соединениями настоящего изобретения в тестах против опухолей ин витро и ин виво. Обычно подходящие дозы попадают в интервал от 1 до 200 мг/м2 площади поверхности тела, предпочтительно от 1 до 100 мг/м2, в зависимости от характера и степени серьезности заболевания, которое нужно лечить, и от общего состояния пациента.
Нижеследующие примеры иллюстрируют изобретение.
Пример 1
Получение 3'-деамино-3'-[1-азиридинил]-4'-O-метансульфонилдаунорубицина
(R1=OCH3, R4=H, R3=OSO2CH3)
3'-N-(2-хлорэтил)-4'-O-метансульфонилдаунорубицин (4a, R1=OCH3, R4=H, R3=OSO2CH3, R9=Cl) (0,33 г, 0,05 ммоля), полученный по способу WO/93/012001, растворяют в смеси 10 мл безводного метиленхлорида и 20 мл метанола, и перемешивают с силикагелем (Мегск, 200-400 мешей, 2 г) при комнатной температуре в течение 30 минут. Полученный раствор фильтруют, концентрируют досуха и сырой материал обрабатывают с помощью флешхроматографии на колонке с силикагелем, используя в качестве элюента смесь метиленхлорида и метанола (95:5 по объему), до получения указанного в заглавии соединения 1а (выход 0,22 г).
ТСХ на пластинах Kieselgel F254 (Мегск) с использованием в качестве элюента системы метиленхлорид:метанол (98:2 по объему) дает Rf = 0,65. Масс-спектр с полевой десорбцией: м/z [M+] 631.
2H-ЯМР (400 МгГц, CDCl) δ : 1,16, 1,25 (м, 2H, водороды азиридина); 1,36 (д, J = 6,5 Гц, 3H, CH3-5'); 1,52 (м, 1H, H-3'); 1,73 (v, 2H, водороды азиридина); 1,80 (м, 1H, H-2'экв); 2,09 (м, 1H, H-2'акс.); 2,12 (м, 1H, H-8 акс); 2,31 (м, 1H, H-8экв); 2,39 (с, 3H, COCH3); 2,98 (д, J = 19,2 Гц, 1H, H-10 акс.); 3,21 (дд, J = 1,7, 19,2 Гц, 1H, H-10 экв); 3,22 (с, 3H, CH3SO2); 4,09 (кв, J = 6,4 Гц, 1H, H-5'); 4,10 (с, 3H, ОCH3); 4,44 (с, 1H, ОH-9); 4,75 (с, 1H, H-4'); 5,28 (м, 1H, H-7); 5,55 (д, J = 3,4 Гц, 1H, H-1'); 7,41 (д, J = 8,1 Гц, 1H, H-3); 7,80 (дд, J = 7,7, 8,1 Гц, 1H-H-2); 8,05 (д, J = 7,7 Гц, 1H, H-1); 13,30 (с, 1H, OH-11); 14,00 (с, 1H, OH-6).
Пример 2
Получение 4 -деметокси-3'-деамино-3'-[1-азиридинил] -4'-O-метансульфонилдаунорубицина
(1b: R1=R4=H, R3=OSO2CH3)
4-деметокси-N-(2-гидроксиэтил)даунорубицин (4b: R1-R4=H, R3=OSO2CH3, R9= OH, 0,3 г, 0,5 ммоля) растворяют в смеси метиленхлорида (10 мл) и метанола (5 мл), и встряхивают при комнатной температуре с 3 г силикагеля в течение 30 минут. Затем органический раствор отфильтровывают, а растворитель удаляют при пониженном давлении. Остаток обрабатывают с помощью флеш-хроматографии на колонке с силикагелем, используя в качестве системы элюента смесь метиленхлорида и метанола (95:5 по объему), до получения указанного в заглавии соединения 1b (0,18 г). ТСХ на пластинах Kieselgel F254 (Мегск) с использованием в качестве элюирующей системы метиленхлорида и метанола (20:1 по объему) дает Rf=0,42.
Масс-спектр с полевой десорбцией: m/z (M+) 601.
Пример 3
Получение 3'-деамино-3'-[1-азиридинил] -4'-O-метансульфонил-14-никотинат-доксорубицина.
(2a: R1=OCH3, R2=O-никотиноил, R4=H, R3=OSO2CH3)
3'-деамино-3'-[1-азиридинил] -4-O-метан-сульфонилдаунорубицин (1a, 0,63 г, 1 ммоль), полученный по способу примера 1, растворяют в смеси 6 мл безводного метанола и 13 мл диоксана, добавляют 0,5 мл этилортоформата и затем полученную смесь обрабатывают раствором брома (1 г) в 5 мл безводного метиленхлорида при 10oC в течение 1,5 часов. Затем реакционную смесь осаждают смесью этилового эфира (100 мл) и петролейного эфира (50 мл). Осадок собирают и снова растворяют в смеси ацетона (15 мл) и 0,25 н. водного бромистого водорода (15 мл). Полученную смесь выдерживают при 30oC в течение 20 часов, затем экстрагируют H-бутанолом (50 мл). Органический растворитель удаляют при пониженном давлении, а остаток, растворенный в сухом ацетоне (200 мл), обрабатывают никотинатом калия (2 г) при кипении с обратным холодильником в течение часа. Растворитель удаляют при пониженном давлении, а сырой материал обрабатывают хроматографически на колонке с силикагелем, используя в качестве элюирующей системы смесь метиленхлорида и метанола (95:5 по объему) до получения указанного в заглавии соединения 2a (0,35 г). Т.плавления 148-149oC (с разложением).
ТСХ на пластинах Kieselgel F254 (Мегск), с использованием в качестве элюирующей системы метиленхлорида и метанола (10:1 по объему) дает Rf=0,37.
Масс-спектр с полевой десорбцией: m/z (M+) 752
Пример 4
Получение 3'-деамино-3'-[1-азиридинил]-4'-O-метансульфонилдоксорубицина
(2c: R1=OCH3, R2=OH, R4=H, R3=OSO2CH3)
3'-N-(2-хлорэтил)-4'-метансульфонилдоксорубицин (6a: R1-OCH3, R2=OH, R9= Cl, R3=OSO2CH3, R4=H), полученный по способу патента Великобритании 9114549, превращают в указанное в заглавии соединение 2c по способу примера 1. ТСХ на пластинах Kieselgel F254 (Мегск) с использованием в качестве элюирующей системы метиленхлорида и ацетона (8:2 по объему) дает Rf=0,35
Масс-спектр с полевой десорбцией: m/z (M+) 647
Пример 5
Получение 3'-деамино-3'-[1-азиридинил]-4'-иододоксорубицина
(2g: R1=OCH3, R2=OH, R4=H, R3=1)
3'-N-(2-хлорэтил)-4'-иододоксорубицин (6b: R1=OCH3, R2=OH, R9=Cl, R3=1, R4= H), полученный по способу патента Великобритании 9114549, превращают в указанное в заглавии соединении 2g по способу примера 1. ТСХ на пластинах Kieselgel F254 (Мегск) с использованием в качестве элюирующей системы метиленхлорида и ацетона (9:1 по объему), дает Rf=0,45.
Масс-спектр с полевой десорбцией, m/z (M+) 679
Пример 6
Получение 3'-деамино-3'-[1-азиридинил]-4'-деоксидоксорубицина
(2h: R1=OCH3, R2=OH, R3=R4=H).
3'-N-(2-хлорэтил)-4'-деоксидоксорубицин (6c: R1=OCH3, R2=OH, R9=Cl, R3= R4= H), полученный по способу патента Великобритании 9114549, превращают в указанное в заглавии соединение 2h по способу примера 1. ТСХ на пластинах Kieselgel F245 (Мегск), с использованием в качестве элюирующей системы метиленхлорида и ацетона (20:1 по объему), дает Rf = 0,33.
Масс-спектр с полевой десорбцией: m/z (M+) 553.
Биологическая активность
3'-деамино-3'-[1-азиридинил] -4'-O-метансульфонилдаунорубицин (1a), 4-деметокси-3'-деамино-3'-[1-азиридинил]-4'-O-метансульфонилдаунорубицин (1b),
3'-деамино-3'-[1-азиридинил] -4'-O-метансульфонил-14-никотинатдоксорубицин (2a) и
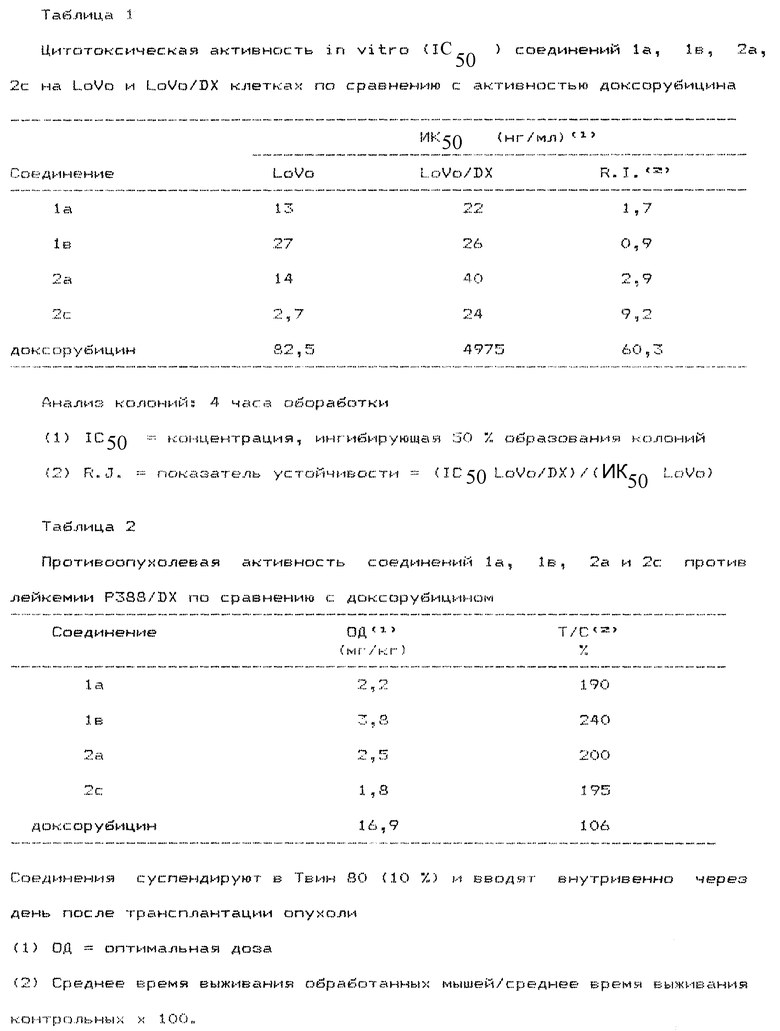
3-'деамино-3'-[1-азиридинил] -4'-O-метансульфонилдоксорубицин (2c), были тестированы ин витро на двух человеческих клеточных линиях, LoVo (аденокарцинома ободочной кишки человека) и LoVo/DX (аденокарцинома, устойчивая к доксорубицину), в сравнении с доксорубицином.
Цитотоксическая активность приводится в виде ИК50, т.е. концентрация, ингибирующая 50% образования колоний, рассчитываемая на основании кривых концентрация-реакция. Показателем устойчивости R.J. является отношение ИК50 для устойчивых клеток к ИК50 для чувствительных клеток. Соединения 1a, 1b, 2a и 2c демонстрируют высокую активность против обеих клеточных линий и низкий показатель устойчивости (таблица 1).
Соединения 1a, 1b, 2a и 2c оценивали также ин виво против P388 мышиной лейкимии, устойчивой к доксорубицину (105 клеток/мышь трансплантируют внутривенно мышам BD2F1) в сравнении с доксорубицином.
Соединения 1a, 1b, 2a и 2c демонстрируют также поразительно более высокую активность, чем доксорубицин (таблица 2).
Животные
Для оценки противоопухолевой активности использовались инбредные DBA/2 и C57BI/6, гибридные C57BL/6 x DBA/2 (B6D2F1) и BALB/c х DBA/2 (CD2F1) взрослые самки мыши. Животным было по 2-3 месяца, их вес составлял 20-22 г, содержались в обычных лабораторных условиях. В экспериментах с ксенотрансплантатом опухоли человека использовали швейцарских мышей Nu/Nu в возрасте 4-6 недель, весом 20-25 г, которых содержали в клетках с подстилкой из фильтровальной бумаги; корм и подстилку стерилизовали, а воду подкисляли. Колонию мышей подвергали обычному тестированию на отсутствие антител к ряду патогенов, включая гепатит мышей, вирус Сендай и Mycoplasma pulmonis. Всех животных поставляли Charles River (Calco, Como, Italy) и Harlan Nossan (Correzzana, Milano, Italy).
Лейкозы
Сублинию P388/DX Johnson поддерживали посредством еженедельных и/п пассажей на мышах BD2F1 в дозе 106 клеток на мышь. В эксперименте мышам той же линии инъецировали по 105 клеток в/в.
Лейкоз мышей L1210 (первоначально полученный из NCl) и сублинии, резистентные к L-PAM (L1210/L-PAM, первоначально полученный из NCl), к cDDP (L1210/cDDF, первоначально полученный из NCl), и к DX (L1210/DX) поддерживали посредством еженедельных и/п пассажей на мышах DBA/2N в дозе 106 клеток на мышь.
Животных с резистентными опухолями лечили следующим образом:
- мышам L1210/L-PAM вводили и/п 7,5 мг/кг L-PAM через 48 часов после трансплантации опухоли
- мышам L1210/cDDP вводили и/п 5 мг/кг цисплатина через 96 часов после трансплантации опухоли
- мышам L1210/DX вводили и/п 6 мг/кг DX через 48 часов после трансплантации опухоли.
В эксперименте мышам CD1F1 в/в инъецировали по 105 клеток.
Солидные опухоли человека
Карциному молочной железы MX1 (первоначально полученную из Национального института рака [NCI], NHI, Bethesda, MD), карциномы яичника A2780 (любезно представленные д-ром Ozols, Национальный институт рака, NHI, Bethesda, MD) и H207 (первоначально полученную из каталога ATCC), и мелкоклеточную карциному легких N592 трансплантировали п/к бестимусным мышам, используя 15-20 мг опухолевой взвеси. Для экспериментов опухоли вырезали и их фрагменты имплантировали п/к в область левого бока.
Введение лекарственных препаратов
Все лекарственные растворы приготавливали непосредственно перед употреблением. Растворы вводили в/в в объеме 10 мг/кг веса тела. Схемы лечения представлены в каждой таблице (3, 4).
Оценка противоопухолевой активности и токсичности
В экспериментах с моделями лейкозов активность лекарственных препаратов определяли путем сравнения медианы срока выживания (MCB) в экспериментальной группе с тем же показателем в контрольной группе, и результаты, выраженные как УПЖ%, были следующими:
Количество выживших в течение длительного времени (ВДВ) относится к мышам, которые выжили в течение периода времени более 60 дней со дня имплантации опухоли.
В экспериментах с солидными опухолями активность оценивалась как ингибирование роста опухоли через неделю после последнего введения лекарственного препарата. Рост опухоли оценивали с помощью кронциркуля: фиксировали два диаметра и рассчитывали вес опухоли по следующей формуле: d2x D/2 (где d= меньшему диаметру, D = большему диаметру). Ингибирование роста опухоли (ИРО%) рассчитывали согласно уравнению:
В экспериментах с солидными опухолями человека излеченными считались мыши, не имевшие опухоли спустя 30 дней после ее имплантации.
Токсичность оценивалась на основании макроскопических находок при вскрытии и потери веса.
Фармацевтически приемлемая композиция
Для приготовления соединений для инъекций применяли следующую процедуру.
Соединение 1b (2 мг) растворяли в 1 мл смеси кремофора и этанола 65:5 (по объему) при встряхивании, затем добавляли физиологический раствор (4 мл) и раствор встряхивали в течение 2 минут.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТРАЦИКЛИНОВЫЙ ГЛИКОЗИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2118328C1 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРСВЯЗАННЫЕ АНТРАЦИКЛИНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2145965C1 |
| СВЯЗУЮЩИЙ АГЕНТ ДЛЯ БИОАКТИВНЫХ ПРЕПАРАТОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2116087C1 |
| ПРОИЗВОДНЫЕ АЗА-АНТРАЦИКЛИНОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159245C2 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНОНА И ИХ ИСПОЛЬЗОВАНИЕ ПРИ АМИЛОИДОЗЕ | 1995 |
|
RU2167661C2 |
| АНТРАЦИКЛИН ГЛИКОЗИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2081878C1 |
| 8-ФТОРАНТРАЦИКЛИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1995 |
|
RU2146261C1 |
| АНТРАЦИКЛИНОВЫЕ ГЛИКОЗИДЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ДИИОДОПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1991 |
|
RU2100366C1 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНА | 1995 |
|
RU2159619C2 |
| АНТРАЦИКЛИНОВЫЕ ДИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1994 |
|
RU2144540C1 |



Описываются новые производные 3'-азиридино-антрациклина общей формулы 1, где R1 представляет водород или метоксигруппу; R2 представляет водород, гидроксигруппу или представляет ацилоксиостаток формулы (3): -O-COR5, где R5 представляет собой пиридил; R3 и R4 оба представляют водород или один из R3 и R4 представляет водород, а другой представляет гидроксигруппу, иод или группу формулы -OSO2CH2; или их фармацевтически приемлемые соли, проявляющие противоопухолевую активность. Описывается также способ их получения и фармацевтическая композиция на основе соединений формулы 1. 4 с. и 4 з.п.ф-лы, 4 табл. 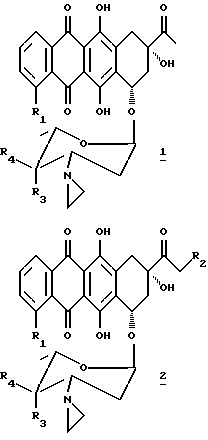
где R1 представляет собой водород или метоксигруппу;
R2 представляет водород, гидроксигруппу или представляет ацилоксиостаток формулы 3
-О-СОR5,
где R5 представляет пиридил;
R3 и R4 оба представляют водород или один из R3 и R4 представляет водород, а другой представляет гидроксигруппу, иод или группу формулы -OSO2СН3,
или их фармацевтически приемлемые соли.
где R1, R2, R3, R4 имеют значения, указанные в п.1;
R9 - гидрокси или атом галогена,
растворенного в безводном полярном органическом растворителе, который представляет собой сухой метиленхлорил, метанол или их смесь в объемном соотношении 1:1 - 1:3, при перемешивании при температуре 0 - 30oC от 15 мин до 2 ч силикагелем, и, при желании, превращение полученного таким образом антрациклинового гликозида формулы 2 в его фармацевтически приемлемую соль.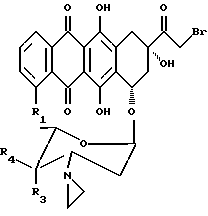
где R1, R3 и R4 имеют значения, указанные в п.1,
с производным соли формулы 3'
Х+-ОСОR5,
где R5 имеет значения, указанные в п.1.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ получения производных морфолинилдоксорубицина | 1984 |
|
SU1450749A3 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 4789665 А, 06.12.88. | |||

