Изобретение относится к синтезу новых биологически активных соединений группы пирролидина, в частности к новым производным пирролидина, к способу их получения в виде солей или энантиомеров, к способу разделения энантиомеров с помощью хиральной фазы, и к использованию новых соединений в фармацевтических композициях.
Новые соединения отвечают общей формуле I
где либо
R - метилен-, этилен-радикал, SO, SO2, CHOH или сера, R1 - пиридил-радикал, при необходимости замещенный одним или несколькими алкил-, фурилрадикалами, при необходимости замещенными одним или несколькими алкил-, тиэнилрадикалами, при необходимости замещенными одним или несколькими алкил-, хинолилрадикалами, при необходимости замещенными одним или несколькими алкил-, нафтилрадикалами, при необходимости замещенными одним или несколькими алкил-, индолилрадикалами, при необходимости замещенными одним или несколькими алкил- или фенилрадикалами, при необходимости замещенными одним или несколькими заместителями, выбранными из атомов галогена и алкил-, алкокси-, гидрокси-, нитро-, амино-, моноалкиламино-, диалкиламино-, алкоксикарбонил-, -CO-NR7R8-, -NH-CO-CH3-, трифторметил- или трифторметокси-радикалами, а R5 - водород;
либо
R - метиленрадикал, а R1 - водород, а R5 - фенилрадикал;
либо
R - CHR6, R1 и R5 - каждый, водород;
R2 - алкоксикарбонил-, циклоалкилоксикарбонил-, циклоалкилалкилоксикарбонил-, -CONR9R10- или фенил-радикал, при необходимости замещенный одним или несколькими заместителями, выбранными из алкил, алкокси- или гидрокси-радикалов;
R3 - фенилрадикал (при необходимости замещенный одним или несколькими заместителями, выбранными из атомов галогена и алкил-, алкокси-, или алкилтиорадикалов), нафтил-, индолил-, хинолил- или фениламинорадикал, фенильное ядро которого при необходимости замещено одним или несколькими заместителями, выбранными из атомов галогена или алкил-, алкокси-, алкилтио-, трифторметил-, карбокси-, алкоксикарбонил-, гидрокси-, нитро-, амино-, ацил-, циано-, сульфамоил-, карбамоил-, гидроксииминоалкил-, алкоксииминоалкил-, гидроксииминокрбонил-, алкоксииминокарбонил-, тетразолил-5-, тетразолил-5 алкил-, трифтор-, метилсульфонамидо-, алкилсульфинил-, моно- или полигидроксиалкил-, сульфо-, -алк-O-CO-алк-, -алк-COOX-, -O-алк-COOX-, -CH=CH-COOX-, -CO-COOX-, -алк- SO3H-, в форме соли, -CH=CH-алк'-, -C(=NOH)-COOX-, -S-алк-COOX, -O-CH2-алк'-COOX-, -CX=N-O-алк-COOX, -алк-N(OH)-CO-алк-радикал или диметил-2,2 диоксо-4,6 диоксан-1,3-ил-5;
R4 - водород или алкилрадикал;
R6 - фенилрадикал;
R7 - водород или алкил-, фенилалкил- или фенилрадикал, при необходимости замещенный одним или несколькими заместителями, выбранными из атомов галогена и алкил-, алкокси- и алкилтиорадикалов;
R8 - алкил-, фенилалкил- или фенилрадикал, при необходимости замещенный одним или несколькими заместителями, выбранными из атомов галогена и алкил-, алкокси- и алкилтиорадикалов;
или же
R7 и R8 образуют с атомом азота, с которым они связаны, один насыщенный или ненасыщенный моно- или полицилический гетероцикл, содержащий 4 - 9 атомов углерода или один или несколько гетероатомов (O, N) и при необходимости замещенный одним или несколькими радикалами алкила;
R9 - водород или алкил-, циклоалкилалкил-, циклоалкил-, фенилалкил- или фенилрадикал, при необходимости замещенный одним или несколькими заместителями, выбранными из атомов галогена или алкил-, алкокси- или алкилтиорадикалов;
R10 - алкил-, циклоалкилалкил-, циклоалкил-, фенилалкил- или фенилрадикал, при необходимости замещенный одним или несколькими заместителями, выбранными из атомов галогена или алкил-, алкокси- или алкилтиорадикалов;
или же
R9 и R10 образуют с атомом азота, с которым они связаны, один насыщенный или ненасыщенный моно- или полицилический гетероцикл, содержащий 4 - 9 атомов углерода или один или несколько гетероатомов (O, N, S) и, при необходимости замещенный одним или несколькими радикалами алкила;
X - водород, алкил- или фенилалкил-радикал;
алк - алкил- или алкилен-радикал;
алк' - гидроксиалкил-, гидроксиалкилен-, алкоксиалкил- или алкоксиалкилен-радикал.
В предыдущих и нижеследующих определениях, если не оговорено иного, алкил-, алкилен- и алкоксирадикал и участки алкила, алкилена и алкокси, содержат 1 - 4 атома углерода с прямой или разветвленной цепью, радикалы и участки алкила содержат 2 - 4 атома углерода, а радикалы и участки циклоалкила содержат 3 - 6 атомов углерода.
Когда R7 и R8 образуют с атомом азота, с которым они связаны, гетероцикл, последний предпочтительно является пиперидино-циклом, при необходимости замещенным одним или несколькими радикалами алкила или тетрагидро-1,2,3,4 хинолеиновым циклом.
Когда R9 и R10 образуют с атомом азота, с которым они связаны, гетероцикл, последний предпочтительно является пиперидино-, 1-пергидроазепинил-, 1,2,3,6-тетрагидро 1-пиридил, 1,2,3,4-тетрагидро 1-хинолил-, 1-пирролидинил-, 1,2,3,4-тетрагидро 2-изоквинолил-, тиоморфолино- или 1-индолил-циклами, которые могут при необходимости быть замещены по крайней мере одним алкильным радикалом.
Соединения формулы I, содержащие один или несколько асимметричных центров, имеют форму изомеров. Рацематы и энантиомеры этих соединений также являются частью изобретения.
Соединения формулы I, где R обозначает метилен-, этилен-, CHOH, CHR6-радикал или серу, а R3 - фениламино-радикал, где фенильное ядро при необходимости замещено, могут приготавливаться путем воздействия активного производного карбаминовой кислоты, полученного при необходимости "ин ситу" путем воздействия активного производного угольной кислоты, выбранного из N, N'-димидазолкарбонила, фосгена, дифосгена и хлорфомиата п-нитрофенила, на производное формулы II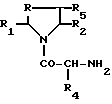
где
R - метилен-, этилен-, CHOH-, CHR6-радикал или сера, а R1, R2, R3, R4, R5 и R6 имеют те же обозначения, что и в формуле I, на анилин, фенильное ядро которого при необходимости замещено одним или несколькими заместителями, выбранными из атомов галогена и алкил-, алкокси-, алкилтио-, трифторметил-, карбокси-, алкоксикарбонил-, гидрокси-, нитро-, амино-, ацил-, циано-, сульфамоил-, карбамоил-, гидроксииминоалкил-, алкоксииминоалкил-, гидроксиаминокарбонил-, алкоксиамино-карбонил, 5-тетразолил-, 5-тетразолилалкил-, трифторметилсульфонамидо-, алкилсульфинил-, моно- или полигидроксиалкил-, сульфо-, -алк-O-CO-алк, -алк-COOX-, -алк-O-алк-, -алк'-COOX-, -O-алк-COOX-, -CH= CH-COOX, -CO-COOX-, -алк-SO3H-, -CH=CH-алк'-, -C(=NOH)-COOX, -S-алк-COOX, -O-CH2-алк'-COOX-, -CX=N-O-алк-COOX- или -алк-N(OH)-CO-алк радикал или диметил-2,2 диоксо-4,6 диоксан-1,3-ил-5.
Эта реакция осуществляется, как правило, в инертном растворителе, таком как тетрагидрофуран, диметилформамид, хлорсодеражщий растворитель (например, хлороформ, дихлоро-1,2 этан), при температуре в пределах от 20oC до температуры кипения растворителя.
Активное производное карбаминовой кислоты может быть получено при тех же условиях среды и температуры.
Производные формулы II могут быть получены путем снятия защиты производного формулы III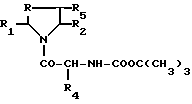
где
R - метилен, этилен-, CHOH-, -CHR6-радикал или сера, а R1, R2, R4, R5 и R6 имеют те же обозначения, что и в формуле I.
Снятие защиты осуществляется предпочтительно посредством иодотриметилсилана в инертном растворителе, таком как хлорсодержащий растворитель (например, хлороформ, 1,2-дихлорэтан) при 15 - 40oC.
Производные формулы (III) могут быть получены путем воздействия производного формулы IV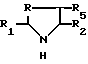
где
R - метилен-, этилен-, CHOH-, CHR6-радикал или сера, а R1, R2, R5 и R6 имеют те же обозначения, что и в формуле I, на кислоту формулы V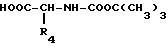
где
R4 определен как в формуле I.
Эта реакция осуществляется в инертном растворителе, таком как ацетонитрил, тетрагидрофуран, или хлорсодержащий растворитель, в присутствии сгустителя, используемого в химии пептидов, такого как карбодиимид (например, N, N'-дициклогексил-карбодиимид) или алкил-хлорформиат, при 10 - 40oC.
Производные формулы V могут быть получены обычными методами защиты аминокислот.
Производные формулы V могут приготавливаться, применяя или адаптируя методы, описанные в литературе, или описанными выше методами.
Производные формулы V, где R2 обозначает алкоксикарбонил-, циклоалкилоксикарбонил- или циклоалкилалкилоксикарбонил-радикал, могут быть получены путем этерификации кислоты формулы VI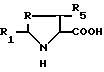
где
R - метилен-, этилен-, CHOH-, CHR6-радикал или атом серы, а 1, R5 и R6 имеют те же обозначения, что и в формуле I.
Этерификация осуществляется, как правило, с помощью спирта R13OH, где R13 обозначает алкил-, циклоалкил- или циклоалкилалкил-радикал, в кислотной среде при температуре кипения реакционной смеси. Для соединения формулы IV, где R2 обозначает трет-бутоксикарбонильный радикал, проводят реакцию изобутилена с продуктом формы VI в инертном растворителе, таком как хлорсодержащий растворитель, в присутствии кислоты, например, серной кислоты, при температуре приблизительно 20oC.
Производные формулы VI, где R означает метилрадикал, а R1 и R5 определены как в формуле I, могут быть получены, применяя или адаптируя известный метод (H. Gerchon et coll., J. Org. Chem., 26, 2347, 1961).
Производные формулы VI, где R означает CHR6, а R1, R5 и R6 определены как в формуле I, могут быть получены, используя или адаптируя известный метод (J. K. Thottathil et coll., Tetrahedron Letters, 26, 151, 1986 и D.R. Kronenthal et coll., Tetrahedron Letters, 31, 1241, 1990).
Производные формулы VI, где R означает метилен-радикал, R1 - водород, а R5 - фенилрадикал, могут быть получены, используя или адаптируя известный метод (Y.N. Belokon et coll., J. Chem. Soc. Perkin Trans, 1, 2075, 1988 и J. Rivier et G.R. Marshall, Peptides, Chemistry, Structure and bilogy, Proceedings of the Eleventh American Peptide Symposium, July. 9-14, 1989, 9-14, 1989 - La Jolla California USA-ESCOM Leiden, 1990).
Производные формулы VI, где R обозначает серу, R1 определен как в формуле I, а R5 водород, могут быть получены реакцией производного формулы VII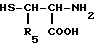
где
R5 - водород, с альдегидом формулы VIII
R1 - CHO
где
R1 имеет те же значения, что и в формуле I
Эта реакция осуществляется предпочтительно в спирте при температуре кипения реакционной среды.
Производные формулы VI, где R обозначает этилен-радикал, R1 определен как в формуле I, а R5 - водород, могут быть получены путем восстановления производных формулы IX
где
R1 имеет те же значения, что и в формуле I.
Реакция восстановления осуществляется, как правило, с помощью водорода в инертном растворителе, таком как спирт, в присутствии катализатора, такого как окись платины, при 20 - 100oC, при необходимости под давлением, или с помощью боргидрида натрия и карбоната калия в водно-спиртовой среде (предпочтительно, этанол) при 0 - 20oC.
Производные формулы IX могут быть получены путем реакции алкил-ацетамидомалоната с производным формулы X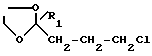
где
R1 имеет те же значения, что и в формуле I,
с последующим гидролизом, декарбоксилированием и дегидратацией полученного продукта путем нагревания в водной соляной кислоте, причем реакция алкил-ацетамидомалоната с продуктом формулы X осуществляется в спирте в присутствии основания, такого как алкоголят щелочного металла, при температуре кипения растворителя.
Производные формулы X могут быть получены, используя или адаптируя известный метод (M.T> Vills et coll., J. Org. Chem., 45 (12), 2495, 1980).
Производные формулы IV также могут быть получены путем снятия защиты производного формулы VI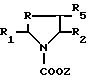
где
R - метилен-, этилен-, CHOH-, CHR6-радикал или атом серы;
R1, R2, R5 и R6 имеют те же обозначения, что и в формуле I;
X - алкил- и, предпочтительно, трет.бутил-радикал, при этом, если R2 означает трет-бутоксикарбонильный радикал, Z не может быть метилом или этилом.
Эта реакция осуществляется в инертном растворителе, таком как хлорсодержащий растворитель, с помощью иодотриметилсилана при температуре от 15oC до температуры кипения реакционной среды.
Производные формулы XI, где R означает метилен-радикал, R1 - фенил-, при необходимости замещенный 2-тиенил-, при необходимости замещенный 2-фурил- или, при необходимости замещенный 3-индолил-радикал, R2 - алкоксикарбонил-, циклоалкилоксикарбонил-, циклоалкилалкилоксикарбонил-радикалы, а R5 - водород, могут быть получены путем реакции производного формулы VII
R1H
где
R1 - фенил-, при необходимости замещенный 2-тиэнил-, при необходимости замещенный 2-фурил- или, при необходимости замещенный 3-индолил-радикал, с производным формулы VIII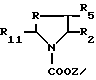
где
R - метилен-радикал;
R2 и R5 имеют вышеуказанные значения;
R11 - алкокси-радикал, содержащий 1 или 2 атома углерода;
Z - алкильный радикал.
Эта реакция осуществляется, как правило, в присутствии сильной кислоты, такой как п-толуолсульфоновая кислота, или кислоты Lewis, такой как трихлорид алюминия, при необходимости в инертном растворителе, таком как ароматический растворитель, при температуре от 20oC до температуры кипения реакционной среды.
Производные формулы XIII могут быть получены, используя или адаптируя известный метод (T. Shono et coll., J. Am. Chem. Soc., 104, 6697, 1982).
Производные формулы XI, где R2 означает алкоксикарбонил-, циклоалкилоксикарбонил-, циклоалкилалкилоксикарбонил-радикал, Z - трет.бутил-радикал, могут быть получены путем этерификации кислоты формулы XIV
где
R - метилен-, этилен-, CHOH-, CHR6-радикал или сера;
R1, R5 и R6 имеют те же обозначения, что и в формуле I.
Этерификация осуществляется в условиях, описанных ранее для этерификации кислот формулы VI, или с помощью спирта в присутствии хлорида тозила в пиридине.
Кислоты формулы XIV могут быть получены путем реакции дитрет-бутилкарбоната с кислотой формулы VI.
Эта реакция осуществляется в инертном растворителе, таком как вода, диоксан или смесь этих растворителей, в присутствии карбоната щелочного металла, при температуре приблизительно 20oC.
Производные формулы XI, где R2 означает остаток -CONR9R10, а Z - трет. бутил-радикал, могут быть получены реакцией кислоты формулы XIV или активного производного этой кислоты на амин формулы XV
HNR9R10
где
R9 и R10 имеют те же значения, что и в формуле I.
При использовании кислоты действуют в присутствии агента конденсации, используемого в химии пептидов, такого как карбодиимид (например, N,N'-дициклогексилкарбодиимид) или N,N'-диимидазол карбонил, в инертном растворителе, таком как эфир (например, тетрагидрофуран, диоксан), амид (диметилформамид) или хлорсодержащий растворитель (например, метиленхлорид, 1,2-дихлорэтан, хлороформ) при температуре от 0oC и до температуры флегмы реакционного раствора.
При использовании активного производного кислоты можно провести реакцию ангидрида, смешанного ангидрида или эфира (который можно выбрать из активированных или неактивированных сложных эфиров кислоты).
Действуют либо в органической среде, при необходимости в присутствии акцептора кислоты, такого как азотсодержащее органическое основание (например, триалкиламин, пиридин, 1,8-диаза бицикло /5.4.0/ 7-ундецен или 1,5-диаза бицикло /4.3.0/ 5-ненен), в растворителе, таком как указан выше, или смеси этих растворителей при температуре от 0oC до температуры флегмы реакционной смеси, либо в гидроорганической бифазной среде в присутствии щелочного или щелочноземельного основания (едкий натр, гидроокись натрия), или карбоната или бикарбоната щелочного или щелочноземельного металла при 0-40oC.
Производные формулы IV, где R означает метилен-радикал, R1 определен как в общей формуле I, за исключением радикалов или заместителей, которые могут видоизменяться при восстановлении (например, хинолил-радикал или нитро-заместитель), R2 - фенил-радикал, при необходимости замещенный одним или несколькими радикалами, выбранными из алкильных, алкокси- и гидрокси-радикалов, а R5 - водород, могут быть получены, используя или адаптируя известные методы (C.G. Overberger et coll., J. Amer. Chem. Soc., 91 887, 1969). Этот метод использует восстановление пирролов, которые могут быть получены, используя или адаптируя известные методы (Synthesis, 613, 1991; Tetrahedron Letters, 4407-4410, 1986).
Производные формулы IV, где R означает метилен-радикал, R1 - фенил-радикал, при необходимости замещенный одним или несколькими радикалами, выбранными из алкильных, алкокси- и гидрокси-радикалов, R2 - фенил-радикал, при необходимости выбранными из алкильных, алкокси- и гидрокси-радикалов, R5 - водород, могут быть получены реакцией этилена и производного формулы XVI
R1-CH=N-CH2-R2
где
R1 и R22 имеют обозначения, указанные ранее.
Этилен может быть образован ин ситу путем разложения тетрагидрофурана в присутствии основания, такого как бутиллитий, при 0-25oC. Можно добавить этилен в присутствии диизопропиламидида лития в тетрагидрофуран при температуре приблизительно 20oC.
Производные формулы XVI могут быть получены путем воздействия альдегида формулы VIII, где R1 имеет значения, указанные выше, на амин формулы XVII
R2-CH2-NH2
где
R2 имеет значения, указанные выше.
Эта реакция осуществляется, обычно, в инертном растворителе, таком как углеводород (например, бензол, толуол), хлорсодержащий растворитель (например, дихлорметан, хлороформ), при необходимости в присутствии п-толуолсульфоновой кислоты, при температуре кипения реакционной смеси.
Соединения формулы IV, где R означает метилен-радикал или CHOH, R1 - алкоксикарбонил-, циклоалкоксикарбонил-, циклоалкилалкилоксикарбонил- или фенил-радикал, при необходимости замещенный одним или несколькими заместителями, выбранными из алкил-, алкокси- или гидрокси-радикалов, R5 - водород, могут быть получены восстановлением производного формулы XVIII
где
R, R1 и R2 имеют значения, указанные выше.
Эта реакция осуществляется предпочтительно с помощью водорода в присутствии катализатора, такого как окись платины, в инертном растворителе, таком как этанол, при температуре приблизительно 20oC, или с помощью боргидрида натрия и карбоната калия в водно-спиртовой смеси (предпочтительно, этанол), при 0 - 20oC.
Производные формулы XVIII могут быть получены, используя или адаптируя известные методы (A. Mkairi et J. Hamelin, Tetrahedron Letters, 28 1397, 1987; A. Vander Werf et R. M. Kellogg, Tetrahedron Letters, 32 3727, 1991; E. Kato et coll., Chemm. Pharm. Bull, 33 4836, 1985; J. Ackermann et coll., Helv. Chim. Acta, 73 122, 1990).
Производные формулы XVIII могут быть также получены путем снятия защиты и дегидратации производного формул XIX и XX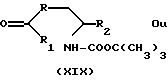
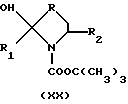
где
R, R1 и R2 имеют значения, указанные выше,
или смеси этих производных.
Реакции снятия защиты и дегидратации осуществляются, как правило, с помощью трифторуксусной кислоты или иодотриметилсилана в инертном растворителе, таком как хлорсодержащей растворитель (например, дихлорметан), при температуре приблизительно 20oC.
Производные формулы XIX и XX могут быть также получены путем воздействия производного формулы XXI
R1 - M
где
R1 имеет значения, указанные выше;
а
R1 - M означает магнийорганическое, литийорганическое производное или купрат, на карбонилсодержащее производное формулы XXII
где
R и R2 имеют те же значения, что выше.
Эта реакция осуществляется в инертном растворителе, таком как тетрагидрофуран, при (-78) - 20oC.
Производные формулы XXII могут быть получены, используя или адаптируя известные методы (J. Ackermann et coll., Helv. Chim. Acta, 73, 122, 1990; T. Ohta, Chem. Lett., 2091, 1987 или T. Ohta et coll., Tetrahedron Letters, 29 329, 1988). Предпочтительно, проводят реакцию дикарбоната дитрет-бутила с производным формулы XXIII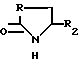
где
R и R2 имеют те же значения, что и ранее.
Эта реакция осуществляется, как правило, в присутствии триэтиламина и 4-диметиламинопиридина в хлорсодержащем растворителе, таком как дихлорметан, при температуре приблизительно 20oC.
Производные формулы XXII могут быть получены, используя или адаптируя известные методы (T. Kolasa et coll., J. Org. Chem., 55, 1711, 1990; A. L. Johnson et coll., J. Med. Chem., 28, 1596, 1985 и B. Rigo et coll., J. Het. Chie. 25, 49, 1988; R. W. Rosenmund и P. Engels, Arch. Parm., 284 16, 1951; C. F. Koelsch и C. H. Stratton, J. Am. Chem. Soc., 66 1883, 1944; S. Sidequist, ArK. Kemi. Mimeral., Geil., 26 1, 1948; J. Sinnreich и D. Elad, Tetrahedron Letters, 24 4509, 1968; G. R. Brown et coll., J. Chem. Soc. Chem. Commun., 1973, 1984).
Производные формулы IV, где R обозначает метилен-радикал, R1 - пиридил-, хинолил-, нафтил- или фенилрадикал, при необходимости замещенный одним или несколькими заместителями, выбранными из атомов галогена и алкил-, алкокси-, гидрокси-, амино-, моноалкиламино-, диалкиламино-, алкокси-карбонил-радикал, -CO-NH7R8 или -NH-CO-CH3, а R2 - алкоксикарбонил-, циклоалкилоксикарбонил- или циклоалкилалкилоксикарбонил-радикал, а R5 - водород, также могут быть получены путем восстановления производного формулы XXIV
Эта реакция осуществляется, как правило, с помощью амальгаммы ртуть-натрий в присутствии дигидрофосфата натрия или гидрофосфата натрия в растворителе, таком как спирт (например, метанол), тетрагидрофуран, вода или смесь этих растворителей, при (-10) - 40oC, или с помощью магния в инертном растворителе, таком как спирт (например, метанол), при температуре от 20oC и температурой кипения реакционной среды.
Производные формулы XXIV могут быть получены путем реакции производного формулы XVI, где R1 и R2 имеют те же значения, что и выше, и фенилвинилсульфона.
Эта реакция осуществляется, как правило, в присутствии соли металла, такой как бромид лития или ацетат серебра, и триалкиламина, такого как триэтиламин, в инертном растворителе, таком как ацетонитрил, при температуре приблизительно 20oC.
Производные формулы IV, где R означает серу, R1 определен как в формуле I, R2 - фенил-радикал, а R5 - водород, могут быть получены путем реакции производного формулы VIII и 2-амино-2-фенилэтантиола, фенильное ядро которого, при необходимости замещено одним или несколькими заместителями, выбранными из алкил-, алкокси-, гидрокси-радикалов.
Эта реакция осуществляется, как правило, в инертном растворителе, таком как спирт, при температуре кипения реакционной среды.
2-амино-2-фенилэтантиолы, фенильное ядро которых, при необходимости замещено, могут быть приготовлены, используя или адаптируя известный метод (патент JP 197447), где используются 2-амино-2-фенилэтанолы, которые приготавливаются, используя или адаптируя известные методы (Z.L. Kis и J. Morly, EP 258191, J. Pless, CH 590 820, S. Miyamoto et coll., EP 432661, J. Suzuki et coll., EP 345775).
Производные формулы IV, где R2 означает фенил, при необходимости замещенный одним или несколькими заместителями, выбранными из алкил-, алкокси- или гидрокси-радикалов, R - метилен-радикал, R1 - водород, а R5 - фенил-радикал, могут быть получены, используя или адаптируя известные методы (W.H. Pearson et coll., J.Am. Chem. Soc., 1141329, 1992; O. Tsuge et cool., Bull. Chem. Soc. Japan, 592537, 1986).
Эти производные также могут быть приготовлены путем восстановления пирролов и соответствующих пирролинов, используя и адаптируя известные методы (C.G. Overberger et coll., J. Am. Chem. Soc., 91687, 1969).
Эти перролы и пирролины могут быть получены, используя или адаптируя известные методы (M. Onno et coll., Tetrahedron Letters, 325093, 1991; S.C. Cherkofsky US 4267184, S.C. Cherkofsky u G.A. Boswell Jr., EP 25884, O. Tsuge et coll., Bull. Chem. Soc. Japan, 591809, 1986).
Производные формулы IV, где R означает этилен-радикал, R2 - фенил, при необходимости замещенный одним или несколькими заместителями, выбранными из алкил-, алкокси- или гидрокси-радикалов, R5 - водород, а R1 имеет те же значения, что и в формуле I, могут быть получены, используя или адаптируя известные методы (C.G. Overberger et coll., J. Am. Chem. Soc., 796430, 1957; J. Thesing et H. Meyer, Ann. 60946, 1957; D.Y. Jackson u P.G. Schultz, J.Am. Chem. Soc. , 1132319, 1991; C.G. Overberger U L.P. Herin, J. Org. Chem., 272423, 1962).
Некоторые из этих методов используют реакцию восстановления пиперидинов, которые также могут быть получены, используя или адаптируя известные методы (H. Quast u B. Miller, Chem. Ber., 1163931, 1983; R. Weil u N. Collignon, C. Rend. Acad. Ser. C., 275299, 1972 и Bull. Soc. Chim. Fr., 258, 1974).
Производные формулы IV, где R2 означает фенил, при необходимости замещенный одним или несколькими заместителями, выбранными из алкил-, алкокси- или гидрокси-радикалов, R - CHR6, R1 и R5 - каждый, водород, R6 - фенильный радикал, могут быть получены, используя или адаптируя известные методы (M.C. Koezel, J. Am. Chem. Soc., 692271, 1947; W.H. Pearson et coll., J. Am. Chem. Soc. , 1141329, 1992; O. Tsuge et coll., Bull. Soc. Japan, 592537, 1986; M. Carriou et coll., Can. J. Chem., 612359, 1983; E. Brewer et, D. Melumad, J. Org. Chem., 373949, 1972).
В некоторых из этих методов применяются реакции восстановления пирролилов и пирролинов, которые могут быть получены, используя или адаптируя известные методы (C. F. H. Allen et, C.V. Wilson, Org. Synth. Coll. Vol., 111358, 1955; W. Savey et D.J. Tivey, J. Chem. Soc., 2276, 1958; W. Chen et coll., Chin. Chem. Lett, 2439, 1991; S.M. Blomm et P.P Gorcia, US, 3883555 и US, 3691161).
Производные формулы III, где R2 означает алкоксикарбонил-, циклоалкоксикарбонил- или циклоалкилалкилоксикрабонил-радикал, также могут быть получены путем этерификации кислоты формулы XXV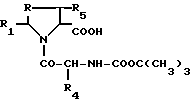
где
R, R1, R4, R5 и R6 имеют те же значения, что в и формуле I.
Эта реакции осуществляется предпочтительно при тех же условиях, что описаны ранее для этерификации соединений формулы V.
Кислоты формулы XXV могут быть получены путем гидролиза метиловых или этиловых сложных эфиров, соответствующих формуле III.
Гидролиз осуществляется, как правило, в инертном растворителе, таком как вода, диоксан или смесь этих растворителей, с помощью основания, такого как гидроокись щелочного металла (едкий натр, гидроокись калия), при температуре приблизительно 20oC.
Производные формулы III, где R означает метилен-радикал, R1 - пиридил-, хинолил-, нафтил- или фенил-радикал, при необходимости замещенный одним или несколькими заместителями, выбранными из атомов галогена или алкил-, алкокси-, гидрокси-, амино-, моноалкиламино-, диалкиламино-, алкокси-карбонил-радикалов, -CO-NR7R8 или -NH-CO-CH3, а R2 - алкоксикарбонил-, циклоалкилоксикарбонил- или циклоалкилалкилоксикарбонил-радикал, а R5 - водород, также могут быть получены путем восстановления производного формулы XXVI
где
Z1 - трет-бутоксикарбонил-радикал или радикал CO-CH(R4)-NH-COOC (CH3)3;
R1 и R2 имеют те же значения, что и ранее;
R4 имеет те же значения, что и в формуле I.
Эта реакция осуществляется при тех же условиях, что описаны ранее для реакции восстановления производных формулы XXIV.
Производные формулы XXVI могут быть получены путем воздействия кислоты формулы V или ди-карбоната ди-трет.бутила, XXIV.
Эти реакции осуществляются в инертном растворителе, таком как ацетонитрил, тетрагидрофуран или хлорсодержащий растворитель, в присутствии агента конденсации, используемого в химии перидов, такого как карбодиимид (например, N,N'-дициклогексил-карбодиимид) или хлортоформиата алкила, при 10 - 40oC.
Анилины при необходимости замещенные коммерчески доступны или могут быть получены, используя или адаптируя известные методы (R. Schroeter, Methoden der organischen Chemie, Houben Weil, Band XI/I, p. 360; G.J. Esselen et coll. , J. Am. Chem. Soc., 36, 322, 1914; G.Adriant et coll., Bull. Soc. Chim. Fr. 1511, 1970; W.A. Jacobs et coll., J. Am. Chem. Soc., 39, 2438, 1917 и J. Am. Chem. Soc., 39, 1438, 1917).
Соединения формулы I, в которой R означает радикал метилен, этилен, CHOH, CHR6 или серу, R3 - радикал фениламино, фенильное ядро которого может быть замещено одним или несколькими заместителями, выбранными из атомов галогена и радикалов алкил, алкокси, алкилтио, трифторметил, нитро, ацил, циано, сульфамоил, алкоксикарбонил, алк-O-алк, трифторметилсульфонамидо, алк-SO3H, радикалов в форме соли, алк-COOX или алк'-COOX, где X не является водородом, могут быть получены взаимодействием производного формулы II с фенилизоцианатом, фенильное ядро которого может быть замещено одним или несколькими заместителями, выбранными из атомов галогена и радикалов алкил, алкокси, алкилтио, трифторметил, нитро, ацил, циано, сульфамоил, алкоксикарбонил, алк-O-алк, трифторметилсульфонамидо или алк-SO3H, в форме соли.
Эту реакцию осуществляют обычно в среде инертного растворителя, такого как тетрагдирофуран, диметилформамида, хлорсодержащего растворителя (хлороформ, 1,2-дихлорэтан), ароматического растворителя (бензол, толуол) при температуре от 10oC до температуры кипения растворителя.
Фенилизоцианаты можно найти в продаже или могут быть получены адаптацией или применением известных методов (Р. Рихтер и др. The chemistry of Cyanate and their thio derivatives, S. Patal, часть 2, Wiley New Jork, 1977).
Соединения формулы I, где R означает метилен-, этилен-, CHOH-, CHR6-радикал или сера, а R3 - радикал фениламино, фенильное ядро которого замещено карбокси-, алк-COOH-, -O-алк-COOH-, -алк'-COOH-, -CH=CH-COOH-, -CO-COOH-, -S-алк-COOH-, -C(=NOH)-COOH, OCH2алк'COOH- или CX=N-O-алк-COOH, а R1, R2, R5 и R6 определены как в формуле I, также могут быть получены реакцией гидролиза или, в случае необходимости, реакцией гидрогенолиза соответствующих сложных эфиров.
При использовании сложных эфиров алкила или фенилалкила проводить гидролиз лучше с помощью основания, такого как едкий натр, гидроокись калия или гидроксид лития, в инертном растворителе, таком как тетрагидрофуран, диоксан, вода или смесь этих растворителей, при 20 - 40oC. При использовании сложного эфира триметилсилилэтила, лучше работать в инертном растворителе, таком как тетрагидрофуран, с помощью фторида, такого как фторид тетрабутиламмония, при 10 - 40oC. При использовании сложных эфиров фенилалкила также лучше осуществлять гидрогенолиз с помощью водорода или формиата аммония в присутствии катализатора, такого как палладий на угле в растворителе, таком как метанол или этилацетат.
Эфиры триметилсилилэтила могут быть получены, используя или адаптируя метод, описанный в примерах.
Соединения формулы I, где R обозначает метилен-, этилен-, CHOH-, CHR6-радикал или серу, а R3 - радикал фениламино, фенильное ядро которого при необходимости замещено гидроксииминоалкил- или алкоксииминоалкил-радикалом, R1, R2, R5 и R6 определены как формуле I могут также быть получены реакцией соответствующего алкилированного производного и производного формулы XXVII
H2N - OR12
где
R12 - водород или алкил-радикал.
Эта реакция проводится, как правило, в инертном растворителе, таком как спирт (например, метанол, этанол), вода или смесь этих растворителей, при температуре кипения растворителя и при необходимости в присутствии основания, такого как пиридин.
Соединения формулы I, где R обозначает метилен-, этилен-, CHOH-, CHR6-радикал или серу, а R3 - радикал фениламино, фенильное ядро которого, при необходимости замещено несколькими заместителями, выбранными из атомов галогена и алкил-, алкокси-, алкилтио-, трифторметил-, нитро-, ацил-, циано-, сульфамоил-, алкоксикарбонил-, трифторметилсульфонамидо-, -алк-O-алк, -алк-COOX- или -алк'-COOX-радикалов, где X не является атомом водорода, а R1, R2, R5 и R6 определены как в формуле I, могут также быть получены реакцией производного формулы IV на кислоту формулы XXVIII
где
R3 имеет вышеуказанные значения или активное производное этой кислоты, а R4 имеет те же значения, что и в формуле I.
Эта реакция проводится предпочтительно в присутствии агента конденсации, используемого в химии пептидов, такого как карбодиимид, в растворителе, таком как ацетонитрил, тетрагидрофуран или хлорсодержащий растворитель, или с помощью тионилхлорида в дихлорметане при температуре от 10oC до температуры кипения растворителя.
Кислоты формулы XXVIII могут быть получены, используя или адаптируя известный метод (J. R. Johnson et coll., J. Am. Chem. Soc., 69, 2370, 1947), или соединения, где R3 обозначает фениламинорадикал, при необходимости замещенный путем реакции фенилизоцианата, фенильное ядро которого, при необходимости замещено одним или несколькими заместителями, выбранными из атомов галогена и алкил-, алкокси-, алкилтио-, трифторметил-, нитро- ацил-, циано-, сульфамоил-, алкоксикарбонил-, -алк-O-алк, -алк-COOX- или -алк'-COOX-радикалов, где X не является водородом, или трифторметилсульфонамино, на производное формулы XXIX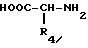
где
R4 имеет те же значения, что и в формуле I.
Эта реакция осуществляется, как правило, в водном растворителе в присутствии основания, такого как бикарбонат щелочного металла или в водном диоксане, при температуре около 20oC.
Соединения формулы I, где R обозначает SO или SO2, R1, R2 и R5 определены, как в общей формуле I, могут быть получены путем окисления соответствующих соединений формулы I, где R обозначает серу, при том другие радикалы и другие заместители выбирают так, чтобы они были нечувствительны к условиям реакции.
Эта реакция окисления проводится, как правило, с помощью оксонаR (пероксимоносульфат калия), выпускаемого "Aldrich" в спирте, таком как метанол или смесь метанол-вода, при температуре около 25oC.
Для осуществления способов изобретения, описанных выше, иногда бывает необходимым вводить группы защиты амино-, гидрокси-, карбокси-функций во избежание вторичных реакции. Амино-функции могут, например, быть блокированы в форме карбаматов трет.-бутила или метила, затем регенерированы с помощью иодотриметилсилана или карбаматов бензила, затем регенерированы гидрогенизацией после осуществления способа, описанного в изобретении. Гидроксифункции могут, например, быть блокированы в виде бензоата, затем регенерированы гидролизом в щелочной среде после осуществления способа, описанного в изобретении.
Энантиомеры соединений формулы I, содержащие по крайней мере один асимметричный центр, могут быть получены путем разделения рацематов, например, путем хроматографии на хиральной колонке или путем синтеза из хиральных предшественников.
В качестве хиральной фазы используют предпочтительно фазу, хиральный селектор которой, являющийся предпочтительно 33,5-динитро-бензоил-D-фенилглицином, удален от двуокиси кремния с помощью аминоалканоильного звена, содержащего 3 - 14 атомов углерода, фиксированного на амино-функциях силикоаминопропила и чьи свободные силанол-функции блокированы триалкилсилил-радикалами.
Эта хиральная фаза, которая является также объектом изобретения, может быть определена следующей структурой XXX:
где
R' - одинаковы или различны;
R'' - одинаковы или различны, алкил-радикалы, содержащие 01 - 10 атомов углерода;
G1 - электро-акцепторная группа;
n = 3 - 13.
Предпочтительно один из символов R' обозначает алкилрадикал, содержащий 7 - 10 атомов углерода, а два других обозначают алкил-радикал, содержащий 1 - 2 атома углерода, и предпочтительно метил-радикал, символы R'' идентичны и обозначают метил- или этил-радикал; G1 обозначает бензоил-радикал, при необходимости замещенный предпочтительно одним или несколькими радикалами нитро-, такими как 3,5-динитро-бензоил-радикал, а n = 10.
Новая хиральная фаза, описанная в изобретении, может быть получена реакцией силико-аминопропила ангидрида аминоалканоильной кислоты, содержащей 3 - 14 атомов углерода, амино-функции которой защищены защитной группой, такой как трет.бутоксикарбонил-радикал, с последующей блокировкой части силанол-функций с помощью Si(R')3-радикалов, определенных выше, затем, после удаления защитных групп амино-функици, осуществляют амидирование с помощью D-фенилглицина, амино-функция которого защищена группой электро-акцеторной G1, определенной выше, и наконец, осуществляют блокировку остаточных силанол-функций с помощью Si(R'')3-радикалов, определенных выше.
Как правило, реакция ангидрида защищенной аминоалканоильной кислоты с силико-аминопропилом, проводится в безводном органическом растворителе, таком как диметилформамид, при температуре около 20oC.
Блокировка силанол-функций с помощью Si(R)3-групп, определенных выше, осуществляется путем реакции галогентриалкилсилана с силико-аминопропилом, привитом с помощью аминоалканоильных остатков, в органическом растворителе, таком как метиленхлорид, в присутствии агента основного характера, такого как пиридин.
Удаление защитных групп аминоалканоильных остатков обычно осуществляется, если защитной группой является трет. бутоксикарбонильный радикал, реакцией трифторуксусной кислоты в органическом растворителе, таком как метиленхлорид.
Амидирование с помощью D-фенилглицина с защищенной аминной функцией осуществляется в присутствии агента конденсации, такого как N-этоксикарбонил-2-этокси-1,2-дигидрохинолин, в безводном органическом растворителе, таком как диметилформамид.
Блокировка остаточных силанол-функций Si(R'')3-радикалами, определенными выше, осуществляется, как правило, с помощью триалкилсилилимидазола в органическом растворителе, таком как метиленхлорид.
Соединения формулы I могут быть очищены известными и обычно применяемыми методами, например, кристаллизацией, хроматографией или экстрагированием.
Соединения формулы I могут быть при необходимости быть превращены в аддитивные соли с помощью неорганической или органической кислоты путем реакции этой кислоты в органическом растворителе, таком как спирт, кетон, эфир или хлорсодержащий растворитель.
Соединения формулы I, содержащие карбокси-, сульфо- или алк-SO3H-функции, также могут быть превращены в соли металлов или аддитивные соли с азотсодержащими основаниями по известным методикам. Эти соли могут быть получены реакцией основания металла (например, щелочного или щелочноземельного), аммиака, амина или соли органической кислоты с соединением формулы I в растворителе. Образующуюся соль отделяют обычными методами.
Эти соли составляют часть изобретения.
В качестве примеров фармацевтически приемлемых солей можно привести аддитивные соли с неорганическими или органическими кислотами (такие как ацетат, пропионат, сукцинат, бензоат, фумарат, малеат, оксалат, метансульфонат, изетионат, теофиллинацетат, салицилат, метил-бис- β- оксинафтоат, хлоргидрат, сульфат, нитрат и фосфат), соли с щелочными металлами (натрий, калий, литий), или с щелочноземельными металлами (кальций, магий), соль аммония, соли азотсодержащих оснований (этаноламин, триметиламин, метиламин, бензиламин, N-бензил- β- фенэтиламин, холин, аргинин, лейцин, лизин, N-метил глюкамин).
Соединения формулы I имеют интересные с фармакологической точки зрения свойства. Эти соединения обладают высоким сродством с рецепторами холецистокинина (ХЦК) и гастрина и, таким образом, используются при лечении или профилактике расстройств, связанных с ХЦП и гастрином на уровне центральной нервной системы и желудочно-кишечного тракта.
Таким образом, эти соединения могут применяться при лечении и профилактике психозов, состояний беспокойства, болезни Паркинсона, замедленной дискинезии, синдрома раздраженной ободочной кишки, острого панкреатита, язв, расстройства моторики кишечника, некоторых видов новообразований (опухолей), восприимчивых к ХЦК, а также для регуляции аппетита.
Эти соединения обладают эффектом потенциализации анальгетической активности наркотических и ненаркотических медицинских препаратов. Кроме того, они сами обладают анальгетическим действием.
Кроме того, соединения, имеющие высокое сродство с рецепторами ХЦК, модифицируют возможности запоминания. В следствие этого эти соединения могут эффективно применяться при расстройствах памяти.
Сродство соединений формулы I к рецепторам ХЦК было определено по методике, опирающейся на методике A. Saito et coll., (J. Neuro. Chem., 7, 483 - 490, 1981), на уровне головного мозга и поджелудочной железы.
Результатов этих тестов показали, что CI50 соединений формулы I, как правило, ниже или равно 1000 нМ.
Кроме того, известно, что продукты, которые "распознают" центральные рецепторы ХЦК, имеют те же специфические свойства по отношению к рецепторам гастрина в желудочно-кишечном тракте (Bock et coll., J. Med. Chem., 32, 16-23, 1989; Reyfeld et coll., Am. J. Physiol., 240, G255-266, 1981; Beinfeld et coll., Neuropeptides, 3, 411-427, 1983).
Соединения формулы I обладают слабой токсичностью. DL50, как правило, выше 40 мг/кг при подкожном введении в опытах на мышах.
Особый интерес вызывают соединения формулы I, где R обозначает метилен-радикал, серу или SO-радикал, R1 - фенил-радикал, при необходимости замещенный, R2 - фенил- или алкоксикарбонил-радикал, R4 и R5 - водород, R3 - радикал фениламино, фенильное ядро которого замещено карбокси-, -алк-COOH, -S-алк-COOH, гидроксиалкил-, алк'-COOH или алк SO
Особый интерес вызывают следующие соединения:
1-(2-(3-(3-(1-гидрокси-этил-(RS)фенил)уреидо)ацетил)5-фенил пролинат трет-бутила-(2RS, 5SR);
2-(3-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил (2S,5R))2-оксо-этил)уреидо)фенил)пропионовая кислота (форма B);
(3-3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо)фенилтио) уксусная-(2RS, 5SR) кислота;
3-(3-((4-трет-бутоксикарбонил-2-(2-фтор-3-тиазолидинил) 2-оксо-этил)уреидо)фенилуксусная-(2R, 4R) кислота;
2-(3-(3-(2-(2-трет-бутоксикарбонил-2-(2-фтор-фенил)-3- тиазолидинил-(2R, 4R))-2-оксо-этил)уренидо)фенил) пропионовая- (2R, 4R) кислота (форма B);
1-(3-(3-(2-(4-трет-бутоксикарбонил-2-фенил-3-тиазолидинил (2R, 4R)2-оксо-этил)уреидо)фенил)этансульфонат калия - (RS), смесь форм A и B;
1-(3-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил- (2S, 5R))2-оксо-этил)уреидо)фенил)этансульфонат калия (RS);
3-(3-(2-(2-третбутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо)1-фенил-метансульфонат калия-(2S, 5R);
3-(3-(2-(2-трет-бутоксикарбонил 5-фенил 1-пирролидинил)2- оксо-этил)уреидо) бензойная- (2S, 5R) кислота;
3-(3-(2-(2-трет-бутоксикарбонил-5)2-фтор-фенил)1- пирролидинил 2-оксо-этил)уреидо) бензойная-(2RS, 5SR) кислота
3-(3-(2-(2,5-дифенил 1-пирролидинил) 2-оксо-этил)уреидо бензойная-(цис) кислота;
3-(2-(2-(2-гидрокси-фенил)-5-фенил-1-пирролидинил 2- оксо-уреидо) фенилуксусная- (2RS, 5SR) кислота;
3-(3-(2-(4-трет-бутоксикарбонил-2-фенил-3-тиазолидинил) 2-оксо-этил)уреидо) фенилуксусная- (2R, 4R) кислота;
3-(3-(2-(4-трет-бутоксикарбонил 2-фенил 3-тиазолидинил) 2-оксо-этил)уреидо) бензойная-(2R, 4R)кислота;
2-(3-(3-(2-(4-трет-бутоксикарбонил 2-(2-фтор-фенил)-1- оксид 3-тиазолидинил-(1RS, 2R, 4R)) 2-оксо-этил)уреидо)фенил) пропионовая кислота (форма A);
3-(3-(4-трет-бутоксикарбонил 2-(2,3-дифтор-фенил) 3-тиазолидинил) 2-оксо-этил)уреидо)фенилуксусная-(2R, 4R) кислота;
1-(2-(3-(3-(1-гидроксиимино-этил) фенил-(Е))уреидо) ацетил)5-фенил пролинат трет-бутила-(RS, 5SR).
Пример 1. К суспензии 3,1 г трет.бутил-5-фенил-пролината (2RS, 5SR) и 2,6 г 2-(3(3-метил-фенил-уреидо)уксусной кислоты в 100 см3 безводного 1,2-дихлорэтана, нагретой до температуры флегмы, добавляют медленно 0,9 см3 хлористого сульфинила. Реакционную смесь снова нагревают с обратным холодильником в течение 15 мин, затем охлаждают до 50oC и нейтрализуют до pH 7 - 8 добавлением 10%-ного водного раствора кислого карбоната натрия. Органическую фазу промывают 3 раза 50 см3 воды, высушивают над сульфатом магния, фильтруют и концентрируют досуха при пониженном давлении. Полученный продукт очищают путем хроматографии на двуокиси кремния (элюент: дихлорметан-метанол (98/2 объемов)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и температуре приблизительно 45oC. После рекристаллизации в ацетонитриле получают 1 г 1 (2-(3-(3-метилфенил)уреидо)ацетил)-5-фенил-трет. бутил пролината (2RS, 5SR), плавящегося при 156oC.
А) 2-(3-(3-Метилфенил)уреидо) уксусная кислота может быть получена следующим образом: к раствору 30 г глицина и 53 г кислого карбоната натрия в 600 см3 воды добавляют в течение 15 мин 53,2 г 3-метилфенилизоцианата. Реакционную смесь перемешивают в течение 4 ч при температуре приблизительно 25oC, затем промывают 200 см3этилацетата и подкисляют до pH 1 с помощью 200 см3 водного раствора соляной кислоты 4N. Нерастворимый продукт отделяют путем фильтрования, промывают три раза 80 см3 воды и высушивают на воздухе. Получают 72 г 2-(3-(3-метилфенил)уреидо) уксусной кислоты с температурой плавления 208oC.
B) Трет-бутил-5-фенилпролинат (2RS, 5SR) может быть получен следующим образом: суспензию 45 г хлоргидрата 5-фенилпролина-(2RS, 5SR) в 500 см3 безводного хлороформа перемешивают и охлаждают до температуры приблизительно 5oC. Прикапывают 5,5 см3 концентрированной серной кислоты, затем реакционную смесь насыщают изобутеном в течение 2 ч, перемешивая и поддерживая температуру при 5oC. После нагревания до температуры приблизительно 20oC перемешивание осуществляют в течение 20 ч. Затем реакционная смесь доводится до pH 8 с помощью 4N водного раствора едкого натра. Органическую фазу декантируют, промывают 3 раза 100 см3 воды, сушат над сульфатом магния и концентрируют досуха при пониженном давлении и температуре приблизительно 40oC. Таким образом, получают 40 г трет.бутил-5-фенилпролината-(2RS, 5SR) в виде оранжевого масла, используемого при последующем синтезе.
Хлоргидрат 5-фенилпролина-(2RS, 5SR) может быть получен известным методам (H.GERSHON и A.SCALA, J. Org. Chem., 26, 2347-50, 1961).
Пример 2. К раствору 2 г 1-(2-аминоацетил) 5-фенил-трет.бутилпролината (2RS, 5SR) в 20 см3 безводного тетрагидрофурана добавляют 0,9 см3 3-метоксифенилизоцианата. Реакционную смесь перемешивают в течение 20 ч при температуре приблизительно 25oC. После выпаривания растворителя при пониженном давлении и температуре приблизительно 45oC полученный исходный продукт очищают путем хроматографии на двуокиси кремния (элюент: дихлорметан-метанол (95/5 об.)). Фракции, содержащие целевой продукт, объединяют, затем концентрируют досуха при пониженном давлении. После рекристаллизации в ацетонитриле получают 1,9 г 1-(2-(3-(3-метоксифенил)уреидо)ацетил) 5-фенил- трет.бутилпролината (2RS, 5SR), плавящегося при 174oC.
A) 1-(2-Амино-ацетил) 5-фенил-трет.бутилпролинат (2RS, 5SR) может быть получен следующим образом: к раствору 16 г 1-(2-трет.бутоксикарбониламино-ацетил) 5-фенил-трет.бутилпролината (2SR, 5SR) в 150 см3 хлороформа прикапывают 5,6 см3 иодтриметилсилана при температуре приблизительно 25oC. Реакционную смесь перемешивают в течение 20 ч при этой температуре, затем концентрируют досуха при пониженном давлении и температуре приблизительно 45oC. Полученный исходный продукт очищают путем хроматографии на двуокиси кремния (элюент: дихлорметан-метанол (90/10 об.)). Фракции, содержащие целевой продукт, объединяют, затем концентрируют досуха при пониженном давлении. Получают 10 г 1-(2-аминоацетил) 5-фенил-трет.бутилпропината (2RS, 5SR) в виде аморфного белого продукта, используемого при последующем синтезе.
ЯМР протона (250 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре: 1,5 (s, 9H, C(CH3)3), 1,8-2,4 (bm, 4H, CH2-CH2), 2,7, 3,25, 3,45 и 3,6 (bd, 2H, AB, CH2CO2), 4,3 (bt, 1H, CH), 5,05 (bm, 1H, CH), 7,2-7,8 (m, 5H, ароматические).
Б) 1-(2-Трет-бутоксикарбониламино-ацетил) 5-фенил-трет.бутокси пролинат (2RS, 5SR) может быть приготовлен следующим образом: к раствору 11,5 г 5-фенил-трет. бутокси пролината (2RS, 5SR) и 8,2 г 2-трет-бутоксикарбониламиноуксусной кислоты в 150 см3 безводного ацетонитрила, выдерживаемому при температуре приблизительно 0oC, добавляют за 30 мин раствор 9,6 г N,N'-дициклогексилкарбодиимида в 50 см3 безводного ацетонитрила. Реакционную смесь перемешивают в течение 16 ч при температуре приблизительно 25oC, затем нерастворимый продукт отделяют фильтрованием и промывают три раза в 30 см3 дихлорметана. Фильтрат концентрируют досуха при пониженном давлении и температуре приблизительно 45oC. После рекристаллизации в пентане получают 16 г 1-(2-трет-бутоксикарбониламино-ацетил) 5-фенил-трет.бутокси пролината (2RS, 5SR), плавящегося при +112oC.
Пример 3. К раствору 1,8 г N,N'-диимидазол-карбонила в 50 см3 1,2-дихлорэтана безводного, добавляют медленно раствор 3,1 г 1-(2-амино-ацетил) 5-фенил-трет.бутил-пролината (2RS, 5SR) в 50 см3 1,2-дихлорэтана безводного. Реакционную смесь перемешивают в течение 1 ч при температуре приблизительно 25oC, затем добавляют 1,4 г 1-(3-амино-фенил) этанола (RS). Реакционную смесь нагревают с обратным холодильником при перемешивании в течение 4 ч. После охлаждения смесь промывают 3 раза по 50 см3 воды, органическую фазу высушивают над сульфатом магния, а растворитель выпаривают досуха при пониженном давлении и температуре 45oC. Полученный маслянистый остаток очищают путем хроматографии на двуокиси кремния (элюент: дихлорметан-метанол (95/5 об. )), а фракции, содержащие целевой продукт, объединяют, затем концентрируют досуха при пониженном давлении, после рекристаллизации в ацетонитриле получают 1,8 г 1-/2-(3-(3-(1-гидроксиэтил-(RS))фенил)уреидо)ацетил) 5-фенил-трет.бутил пролината (2RS,5SR), плавящегося при 160oC.
Пример 4. По способу, описанному в примере 3, но исходя из 1,8 г N,N'-диимидазол-карбонила, 3,1 г 1-(2-амино-ацетил) 5-фенил-трет.бутил-пролината (2RS, 5SR), растворенного в 100 см3 1,2-дихлорэтана безводного, и из 1,3 см3 3-метилтио-анилина получают после рекристаллизации в ацетонитриле 1,8 г 1-(2-(3-(3-метилтио-фенил)уреидо)ацетил) 5-фенил-трет. бутил пролината (2RS, 5SR), плавящегося при 163oC.
Пример 5. По способу, описанному в примере 3, но исходя из 1,8 г N,N'-диимидазолкарбонила, 3,1 г 1-(-2-амино-ацетил) 5-фенил-трет.бутил-пролината (2RS, 5SR), растворенного в 100 см3 безводного 1,2-дихлорэтана, и из 1,25 г 3-амино-фенилметанола получают после рекристаллизации в ацетонитриле 1,8 г 1-(2-(3-гидроксиметил-фенил)уреидо)ацетил)-5-фенил-трет. бутил- пролината (2RS, 5SR), плавящегося при 163oC.
Пример 6. По способу, описанному в примере 3, но исходя из 1,8 г N,N'-диимидазол-карбонила, 3,1 г 1-(-2-амино-ацетил)-5-фенил- трет.бутил пролината (2RS, 5SR), растворенного в 100 см3 1,2-дихлорэтана безводного, и из 1,5 г 3-амино-ацетофенона получают после рекристаллизации в смеси циклогексан-метанол (9/1 объемов) 1,1 г 1-(2-(3-(3-ацетил-фенил)уреидо)ацетил) 5-фенил-трет.бутил-пролината (2RS, 5SR), плавящегося при 122oC.
Пример 7. К раствору 3г 1-(2-(3-(3-ацетил-фенил)уреидо)ацетил) 5-фенил-трет.бутил-пролината (2RS, 5SR) в 12 см3 метанола и 6 см3 пиридина добавляют 0,5 г хлоргидрата гидроксиламина, растворенного в 6 см3 воды. Реакционную смесь нагревают с обратным холодильником в течение 2 ч. После выпаривания растворителей при пониженном давлении и температуре приблизительно 45oC, остаток извлекают с помощью 100 см3 этилацетата, и органическую фазу промывают 3 раза в 50 см3 воды, высушивают над сульфатом магния, фильтруют и концентрируют досуха при пониженном давлении. Полученный сырой продукт очищают путем хроматографии на двуокиси кремния (элюент: дихлорметан-метанол (95/5 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и температуре приблизительно 40oC. После рекристаллизации в ацетонитриле получают 1,1 г 1-(2-(-3(3-(1-гидроксимино-этилфенил)уреидо)ацетил) 5-фенил-трет. бутил-пролината (2RS, 5SR), плавящегося при 118 oC.
Пример 8. По способу, описанному в примере 3, но исходя из 12,5 г N, N'-диимидазол-карбонила, 21,3 г 1-(2-амино-ацетил) 5-фенил-трет.бутил-пролината (2RS, 5SR), растворенного в 400 см3 1,2-безводного дихлорэтана, и из 10,5 см3 3-амино-этилбензоата получают 24,8 г 1-(2-(3-(3-этоксикарбонил- фенил)уреидо)ацетил)-5-фенил-трет. бутил-пролината (2RS, 5SR) в виде воздушной массы белого цвета.
Протонный ЯМР (250 МГц, DNSO D6, δ в ppm), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, общее описание подходит для других продуктов серии: 1,35 (t, 3H, CH3 этила), 1,50 (bs, 9H, (CH3)3), 1,90-2,5 (m, 4H, H в 3 и 4 пирролидина), 3,9 и 3,5 (ABX, 2H, CH2N), 4,3 (q, 2H, CH2O), 4,5 (vbdd, 1H, H в 2 пирролидина), 5,15 (dd, 1H, H в 5 пирролидина), 6,2 (bdd, 1H, NH), 7,2-7,6 (m, 8H, ароматические), 8 (bs, 1H, H в 2 фенилмочевины), 8,7 (bs, 1H, NH).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3375, 3150, 3090, 3065, 3030, 2980, 2930, 2870, 1720, 1635, 1610, 1596, 1555, 1490, 1450, 1430, 1390, 1365, 1300, 1285, 1235, 1180, 1150, 1105, 1030, 860, 840, 755, 700, 685.
Пример 9. К раствору 8 г 1-(2-(3-(3-этоксикарбонил-фенил)уреидо)ацетил 5-фенил-трет. бутил-пролината (2RS, 5SR) в 120 см3 метанола добавляют 0,9 г гидроокиси калия, растворенного в 60 см3 дистиллированной воды. Реакционную смесь перемешивают в течение 3 ч при температуре приблизительно 25oC, затем концентрируют до 50 см3 при пониженном давлении. Полученный раствор разбавляют 30 см3 воды, промывают 2 раза 50 см3 этилацетата, подкисляют до pH 2 с помощью водного раствора 4N соляной кислоты и экстрагируют 3 раза по 100 см3 дихлорметана. Органические фазы объединяют, промывают 2 раза в 50 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и температуре приблизительно 40oC. Полученный исходный продукт очищают путем хроматографии на двуокиси кремния (элюент: дихлорметан-метанол (90/10 об.). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. После рекристаллизации в этилацетате получают 4,5 г 3(3-(2-(2-трет-бутоксикарбонил 5-фенил 1-пирролидинил) 2-оксоэтил) уреидо) бензойной кислоты (2RS, 5SR), плавящейся при 236oC.
Пример 10. По способу, описанному в примере 3, но исходя из 1,8 г N, N'-диимидазолкарбонила, 3,1 г 1-(2-аминоацетил) 5-фенилтрет.бутил-пролината (2RS, 5SR), растворенного в 100 см3 безводного 1,2-дихлорэтана, и из 1,4 г 2-(3-амино-фенил)этанола получают после рекристаллизации в ацетонитриле 1,5 г 1-(2-(3-(3-(2-гидроксиэтил)фенил)уреидо)ацетил) 5-фенил-трет. бутил-пролината (2RS, 5SR), плавящегося при 162oC.
A) 2-(3-Аминофенил)этанол может быть получен следующим образом: к раствору 15 г 2-(3-нитро-фенил)этанола в 250 см3 этанола добавляют 0,75 г 5%-ного палладия на угле. Суспензию перемешивают в течение 2 ч при температуре приблизительно 25oC в атмосфере водорода (130 кПа). Катализатор отделяют путем фильтрования, а фильтрат концентрируют досуха при пониженном давлении и 45oC. Таким образом, получают 12 г 2-(3-амино-фенил)этанола в виде масла оранжевого цвета, используемого при последующем синтезе.
Пример 11. По способу, аналогичному описанному в примере 3, но исходя из 3,6 г N,N'-диимидазол-карбонила, 6,2 г 1-(2-амино-ацетил) 5-фенил-трет.бутил-пролината (2RS, 5SR), растворенного в 150 см3 безводного 1,2-дихлорэтана, и из 3,9 г 3-амино-этилциннамата -(E) получают 4,8 г 3-(3-(2-(2-трет. бутоксикарбонил-5-фенил-1-пирролидинил- (2 RS, 5SR)) 2-оксо-этил)уреидо)этилциннамата -(E).
Протонный ЯМР (250 МГц, DMCO D6, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 120oC: 1,3 (t, 3H, CH3), 1,5 (bs, 9H, (CH3)3), 4,25 (q, 2H, CH2O), 6,4 (d, 1H, J = 15, CH=транс.), 7,10-7,70 (m, 10H, ароматические и CH=транс).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3375, 3150, 3070, 3035, 2980, 2940, 2880, 1740, 1700, 1640, 1610, 1590, 1560, 1495, 1490, 1455, 1430, 1395, 1370, 1310, 1270, 1220, 1180, 1160, 1040, 990, 860, 840, 790, 760, 705, 685.
3-амино-этил-циннамат (E) может быть получен по известному методу (заявка на патент NL 7416449, С.А. 84, 58882q).
Пример 12. По способу, описанному в примере 9, но исходя из 3,7 г 3-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил- (2 RS, 5SR)) 2-оксо-этил)уреидо)-этилциннамата (E), растворенного в 60 см3 метанола, и 0,4 г гидроокиси калия, растворенного в 20 см3 воды, после обработки получают 1 г 3-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил)- (2 RS, 5SR)) 2-оксо-этил)уреидо)коричной (E) кислоты.
Протонный ЯМР (250 МГц, DMCO D6 δ ч/млн, J в Гц), 2 ротамеров при комнатной температуре, описание преобладающего ротамера, общее описание пригодно для других продуктов серии: 1,5 (bs, 9H, (CH3)3), 1,9 и 2,2 (2 m, 4H, H в 3 и 4 на пирролидине), 3,2 и 3,9 (ABX, 2H, CH2N), 4,35 (dd, 1H, H в 2 пирролидина), 5,20 (dd, 1H H в 5 пирролидина), 6,30 (dd, 1H, NH), 6,4 (bd, 1H, J = 15, CH = транс.), 7,1-7,7 (m, 10H, ароматические и CH = транс.), 8,7 (s, 1H, NH).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3380, 3075, 3030, 3700-2250 с максимумом при 2745, 2980, 2935, 2875, 1735, 1705, 1695, 1640, 1610, 1590, 1560, 1495, 1450, 1430, 1395, 1370, 1315, 1250, 1225, 1155, 985, 910, 890, 860, 840, 790, 760, 705, 685.
Пример 13. По способу, описанному в примере 3, но исходя из 3,6 г N, N'-диимидазол-карбонила, 6,2 г 1-(2-амино-ацетил) 5-фенил-трет.бутил пролината (2RS, 5SR), растворенного в 125 см3 безводного 1,2-дихлорэтана, и из 3,9 г 3-(3-амино-фенил) этилпропионата получают после кристаллизации в смеси пентан-изопропанол (60/40 об. ) 5,3 г 3-(3-(2-(2-трет-бутоксикарбонил -5-фенил 1-пирролидинил)2-оксоэтил)уреидо)-3-фенилэтил пропионата (2RS, 5SR), плавящегося при 96oC.
3-(3-Амино-фенил)этилпропионат может быть получен по способу, аналогичному описанному в примере 10, A, но исходя из 16,8 г 3-нитро-этилциннамата (E), растворенного в 500 см3 этанола, и 0,9 г 5% палладия на угле. Таким образом, получают 14,2 г 3-(3-амино-фенил)этил-пропионата в виде масла, используемого при последующем синтезе.
3-Нитро-этилциннамат (E) может быть получен следующим образом: в раствор 31 г 3-нитро-коричной-(E) кислоты в 300 см3 этанола добавляют 5 см3 чистой серной кислоты. Реакционную смесь перемешивают с обратным холодильником в течение 3 ч. После охлаждения и добавления 59 см3 воды раствор концентрируют до объема приблизительно 60 см3 при пониженном давлении и при 49oC. Добавляют 250 см3 этилацетата, затем органическую фазу промывают последовательно 2 раза по 100 см3 водного раствора едкого натра 2N, 2 раза по 100 см3 воды, затем высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 32 г 3-нитро-этилциннамата (E), плавящегося при 70oC.
3-Нитро-коричная-(E) кислота может быть получена следующим образом: смесь 30,2 г 3-нитро-бензальдегида, 2,8 г малоновой кислоты, 15,8 г пиридина и 0,15 см3 пиперидина нагревают с обратным холодильником в течение 1 ч. После охлаждения добавляют 50 см3 воды, а нерастворимый продукт отделяют фильтрованием, промывают 3 раза в 50 см3 воды и высушивают на воздухе. Таким образом, получают 31 г 3-нитро-коричной-(E) кислоты, плавящейся при 205oC.
Пример 14. По способу, описанному в примере 9, но исходя из 3,9 г 3-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксоэтил)уреидо)3-фенил-этилпропионата (2RS, 5SR), растворенного в 60 см3 метанола, и из 0,45 г гидроокиси калия, растворенного в 20 см3 воды, после обработки получают 2,1 г 3-(3-(2-(2-третбутоксикарбонил-5- фенил-1-пирроллидинил) 2-оксо-этил)уреидо) 3-фенил-пропионовой-(2RS, 5SR)кислоты.
Протонный ЯМР (250 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC: 1,5 (bs, 9H, (CH3)3), 2,5 (t, 2H, CH2), 2,8 (t, 2H, CH2), 6,8-7,60) (m, 9H, ароматические).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3380, 3700-2250 с максимумом 2625, 3160, 3060, 3030, 2980, 2930, 2880, 1735, 1610, 1595, 1560, 1495, 1450, 1440, 1395, 1370, 1310, 1225, 1155, 905, 890, 865, 840, 790, 760, 705.
Пример 15. По способу, аналогичному описанному в примере 3, но исходя из 2,9 г N, N'-карбонил-диимидазол, 5 г 1-(2-амино-ацетил) 5-фенил-трет.бутил пролината (2RS, 5SR), растворенного в 100 см3 безводного 1,2-дихлор-этана, и из 3,2 г 3-амино-феноксиэтил пропионата получают 4,9 г 3-(3-(2-(2-трет- бутоксикарбонил-5-фенил-1-пирролидинил)2-оксо-этил)уреидо) феноксиэтилацетата (2RS, 5SR).
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, описание преобладающего ротамера: 1,15 (bt, 3H, CH3 этила), 1,50 (bs, 9H, (CH3)3), 4,2 (q, 2H, CH2 этила), 4,5 (bs, 2H, OCH2CO), 6,4 (bd, 1H, H в 6 фенилмочевины), 6,8-7,75 (m, 8H, ароматические).
Инфракрасный спектр (КBr), характерные полосы в см-1: 3375, 3150, 3090, 3060, 3030, 2980, 2930, 2875, 1758, 1735, 1700, 1638, 1600, 1550, 1495, 1450, 1430, 1390, 1365, 1295, 1220, 1190, 1155, 1085, 1030, 860, 840, 760, 700, 690.
3-Амино-феноксиэтилацетат может быть получен как описано в примере 10, A, но исходя из 18 г 3-нитро-феноксиэтилацетата, растворенного в 250 см3 этанола, и 0,2 г 5% палладия на угле. Таким образом, получают 15 г 3-амино-феноксиэтилацетата в виде масла, используемого при последующем синтезе.
3-Нитро-феноксиэтилацетат может быть получен следующим образом: в раствор 13,9 г 3-нитро-фенола в 125 см3 безводного диметилформамида добавляют в течение 20 мин 4,8 г масляной суспензии (50 мас.%) гидрида натрия. Полученную смесь перемешивают при температуре приблизительно 25oC в течение 30 мин, затем добавляют в течение 10 мин 10,8 см3 этилхлорацетата. Реакционную смесь перемешивают в течение 20 ч при температуре приблизительно 20oC, затем выливают в 400 см3 воды и экстрагируют 3 раза 200 см3 этилацетата. Объединенные органические фазы высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и температуре 35oC. Получают 18 г 3-нитро-феноксиэтилацетата в виде масла оранжевого цвета, используемого при последующем синтезе.
Пример 16. По способу, аналогичному описанному в примере 9, но исходя из 3,7 г 3-(2-(2-трет. бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо)феноксиэтилацетата (2RS, 5SR), растворенного в 80 см3 метанола, и из 0,4 г гидроокиси калия, растворенного в 40 см3 воды. После обработки и рекристаллизации в изопропилацетате получают 1,4 г 3-(3-(2-(2-трет-бутоксикарбонил-5- фенил-1-пирролидинил) 2-оксо-этил)уреидо)феноксиуксусной-(2RS, 5SR) кислоты, плавящейся при 192oC.
Пример 17. По способу, описанному в примере 3, но исходя из 3,6 г N, N'-диимидазол-карбонила, 6,2 г 1-(2-амино-ацетил)-5-фенил-трет.бутил пролината (2RS, 5SR), растворенного в 150 см3 безводного 1,2-дихлорэтана, и из 4,2 г (3-аминофенилтио) этилацетата получают 4,9 г 3-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил)2-оксо-этил)уреидо) фенилтио) этил ацетата (2RS,5SR).
Протонный ЯРМ (300 МГц, DMCO D6, δ в г/млн), 2 ротамера при комнатной температуре, описание преобладающего ротамера: 1,2 (t, 3H, CH3 этила), 1,5 (bs, 9H, (CH3)3), 3,8 (bs, 2H, CH2), 4,2 (q, 2H, CH2O этила), 6,9-7,7 (m, 9H, ароматические).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3365, 3130, 3085, 3065, 3030, 2980, 2930, 2875, 1735, 1700. 1635, 1585, 1545, 1480, 1498, 1425, 1450, 1395, 1365, 1305, 1295, 1275 1220, 1150, 1030, 885, 865, 840, 780, 760, 700, 690.
(3-Аминофенилтио)этилацетат может быть получен следующим образом: в раствор 12,5 г 3-амино-тиофенола в 200 см3 этанола добавляют в течение 5 мин 16,7 г этилбромацетата. Смесь перемешивают при температуре приблизительно 20oC в течение 3, затем концентрируют досуха при пониженном давлении и при 40oC. Полученный продукт растворяют в 100 см3 этилацетата и промывают в 100 см3 водного раствора едкого натра 1N. Органическую фазу отделяют, промывают 2 раза в 50 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении. Полученный продукт очищают путем хроматографии на двуокиси кремния (элюэнт: этилацетат-циклогексан (70/30 об.)). Фракции, содержащие целевой продукт объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 13 г (3-аминофенилтио) этилацетата в виде масла, используемого при последующем синтезе.
Пример 18. По способу, описанному в примере 9, но исходя из 4 г 3-(3-2-трет-бутоксикарбонил-5-фенил-1-пирролидинил)2-оксоэтил)уреидо) фенилтиоэтилацетата (2RS, 5SR), растворенного в 80 см3 метанола, и из 0,45 г гидроокиси калия, растворенного в 40 см3 воды, после обработки и кристаллизации в смеси окись изопропилацетат изопропила (50/50 об.), получают 2 г 3-(3-(2-(2-третбутоксикарбонил-5-фенил-1-пирролидинил)2-оксоэтил)уреидо) фенилтиоуксусной-(2RS, 5SR) кислоты, плавящейся при 136oC.
Пример 19. По способу, аналогичному описанному в примере 3, но исходя из 3,6 г N,N'-диимидазол-карбонила, 6,2 г 1-(2-аминоацетил) 5-фенил-трет.бутилпролината (2RS, 5SR), растворенного в 150 см3 безводного 1,2-дихлорэтана и 3,7 г 5-амино-этил-салицилата, получают 3,1 г 5-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил)2-оксоэтил)уреидо) этилсалицилата (2RS, 5SR), плавящегося при 150oC.
5-Амино-этилсалицилат может быть приготовлен способом, аналогичным описанному в примере 10, A, но исходя из 10 г 5-нитроэтилсалицилата, растворенного в 200 см3 этанола, и из 0,5 г 5%-ного палладия на угле. Получают 8,5 г 5-амино-этилсалицилата в виде оранжевого масла, используемого в следующих синтезах.
5-Нитро-этилсалицилат может быть получен следующим методом: к раствору 10 г 5-нитросалициловой кислоты в 250 см3 этанола добавляют 3 см3 концентрированной серной кислоты. Реакционную смесь перемешивают с обратным холодильником в течение 70 ч. После охлаждения и добавления 50 см3 воды, раствор концентрируют приблизительно до объема 60 см3 при пониженном давлении и при 40oC. Добавляют 250 см3 этилацетата, затем органическую фазу промывают 2 раза в 100 см3 дистиллированной воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: дихлорметан). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 10 г 5-нитро-этилсалицилата, плавящийся при 96oC.
Пример 20. По способу, описанному в примере 9, но исходя из 4,7 г 5-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил)2-оксоэтил)уреидо) этилсалицилата (2RS, 5SR), растворенного в 80 см3 метанола, и 1,04 г гидроокиси калия, растворенного в 40 см3 воды, после обработки получают 2,3 г 5-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил)2-оксоэтил)уреидо) салициловой (2RS, 5SR) кислоты, плавящейся при 190oC.
Пример 21. По способу, аналогичному описанному в примере 2, но исходя из 6,2 г 1-(2 амино-ацетил)5-фенил-трет.бутилпролината (2RS, 5SR), растворенного в 150 см3 безводного тетрагидрофурана, и из 4,2 г 3-изоцианат фенилметилацетата получают 6 г 3-(3-(2-(2-трет- бутоксикарбонил-5-фенил-1-пирролидинил)2-оксоэтил)уреидо)фенилметилацетата (2SR, 5SR).
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротомера при комнатной температуре, коалесценция линий при 120oC: 1,5 (s, 9H, (CH3)3), 3,6 (s, 2H, CH2CO), 3,65 (s, 3H, OCH3), 6,8-7,7 (m, 9H, ароматические).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3365, 3155, 3110, 3090, 3060, 3030, 2975, 2950, 2930, 2875, 1738, 1700, 1650, 1610, 1595, 1560, 1495, 1435, 1395, 1365, 1315, 1250, 1220, 1155, 1-15, 905, 890, 860, 845, 780, 760, 700.
3-Изоцианатфенилметилацетат приготавливают следующим образом: в суспензию 1 г угля и 6 см3 дифосгена в 70 см3 толуола добавляют при температуре приблизительно -20oC и в атмосфере аргона 8,25 г 3-аминофенилметилацетат, растворенного в 100 см3 толуола. Реакционную смесь перемешивают и выдерживают при -20oC в течение 15 мин, затем после нагревания до температуры приблизительно 20oC нагревают с обратным холодильником в течение 2 ч. 30 мин. Смесь дегазируют путем барботирования аргона в течение 30 мин, фильтруют на целите, промывают в 50 см3 дихлорметана и концентрируют при пониженном давлении и температуре приблизительно 50oC. Таким образом, получают 9,30 г 3-изоцианатфенилметилацетата в виде жидкости желтого цвета, содержащейся в атмосфере аргона и используемой при последующем синтезе.
3-Аминофенилметилацетат может быть получен по методу, аналогичному описанному в примере 10, A, но исходя из 37,1 г 3-нитрофенилметилацетата, растворенного в 550 см3 метанола, и 2 г палладия на угле. Таким образом, получают 28,2 г 3-аминофенилметилацетата в виде жидкости темножелтого цвета, используемой при последующем синтезе.
3-Нитрофенилметилацетат может быть получен по известному методу (SEGERS и A.BRUYLANTS, Bull.Soc.Chim.Belg.64, 87, 1955).
Пример 22. По способу, аналогичному описанному в примере 9, но исходя из 4,9 г 3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксоэтил)уреидо)фенилметилацетата (2SR, 5SR), растворенного в 80 см3 метанола, и 0,56 г гидроокиси калия, растворенной в 40 см3 воды, после обработки получают 1 г 3-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил)2-оксоэтил)уреидо) фенилуксусной (2RS, 5SR) кислоты.
Протонный ЯРМ (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC: 1,5 (bs, 9H,(CH3)3), 3,5 (s, 2H, CH2CO), 6,8-7,7 (m, 9H, ароматические).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3380, 3700-2250 с максимумом 2625, 3155, 3110, 3090, 3030, 2975, 2930, 2880, 1735, 1635, 1610, 1595, 1560, 1495, 1450, 1395, 1365, 1310, 1225, 1155, 905, 890, 980, 840, 780, 760, 705.
Пример 23. По способу, описанному в примере 1, но исходя из 2,2 г 5-фенилэтилпролината (2RS, 5SR), 2,1 г 2-(3-(3-метилфенил)уреидо) уксусной кислоты в суспензии в 50 см3 безводного 1,2-дихлорэтана и из 0,72 см3 сульфинилхлорида получают после рекристаллизации в ацетонитриле 1,2 г 1-(2-(3-(3-метилфенил)уренидо)ацетил)5-фенилэтилпролината (2RS, 5SR), плавящегося при 115oC.
5-фенилэтилпролинат (2RS, 5SR) может быть получен по известному методу (F. Leonard, патент GB 997097, C.A. 62P, 9109e, 1965).
Пример 24. По способу, аналогичному описанному в примере 3, но исходя из 4,1 г N,N'-диимидазол-карбонила, 7 г 1-(2-аминоацетил)-5-фенилэтилпролината (2RS, 5SR), растворенного в 135 см3 безводного 1,2-дихлорэтана, и из 4,1 г 3-аминоэтилбензоата получают 1,5 г 1-(2-(3-этоксикарбонилфенил)уреидо)ацетил) 5-фенилэтилпролината 2RS, 5SR), плавящегося при 136oC.
1-(2-Аминоацетил)-5-фенилэтилпролината (2RS, 5SR) может быть получен по способу, аналогичному описанному в примере 2, A, но исходя из 11,1 г 1-(2-трет-бутоксикарбониламиноацетил) 5-фенилэтилпролината (2RS, 5SR) и из 4,3 см3 иодтриметилсилана, растворенного в 150 см3 безводного хлороформа. Таким образом, получают 7 г 1-(2-аминоацетил)-5-фенилэтилпролината (2RS, 5SR), используемого при последующем синтезе.
1-(2-Трет. бутоксикарбониламиноацетил)-5-фенилэтилпролинат (2RS, 5SR) может быть получен как описано в примере 2,B, но исходя из раствора, содержащего 7 г 5-фенилэтилпролината (2RS, 5SR), 5,6 г 2-трет.бутоксикарбониламиноуксусной кислоты и 6,6 г N,N'-дициклогексилкарбодимида в 65 см3 безводного ацетонитрила. Таким образом, получают 11,1 г 1-(2-трет.бутоксикаробниламино-ацетил)-5-фенилэтилпролината (2RS, 5SR) в виде масла оранжевого цвета, используемого при последующем синтезе.
Пример 25. По способу, описанному в примере 1, но на основе 2 г 5-фенилциклопропилметилпролината (2RS, 5SR), 1,7 г 2-(3-(3-метилфенил)уреидо)уксусной кислоты в суспензии в 50 см3 безводного 1,2-дихлорэтана и 0,6 см3 сульфинилхлорида получают после перекристаллизации в ацетонитриле 1,1 г 1-(2-(3-(3-метилфенил)уреидо)ацетил)- 5-фенилциклопропилметилпролината (2RS, 5RS), плавящегося при 130oC.
А) 5-Фенилциклопропилметилпролината (2RS, 5RS) может быть получен следующим способом: в раствор 3,5 г 1-(трет.бутоксикарбонил) 5-фенилциклопропилметилпролината (2RS, 5RS) в 50 см3 безводного хлороформа прикапывают 1,5 см3 иодотриметилсилана. Реакционную смесь перемешивают в течение 20 ч при температуре приблизительно 25oC, затем концентрируют досуха при пониженном давлении и температуре 45oC. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: дихлорметанметанол (97/3 об.)). Фракции, содержащие целевой продукт, объединяют, затем концентрируют досуха при пониженном давлении. Таким образом, получают 2 г 5-фенилциклопропилметилпролината (2RS, 5RS) в виде масла оранжевого цвета, используемого при последующем синтезе.
Б) 1-(трет. бутоксикарбонил)-5-фенилциклопропилметилпролинат (2RS, 5RS) может быть получен следующим образом: в раствор 7 г 1-(трет-бутоксикарбонил)-5-фенилпролина-(2RS, 5RS) и 4,6 г паратолуолсульфонилхлорида в 403 безводного пиридина прикапывают, перемешивают, 1,8 г циклопропилметанола при температуре приблизительно 0oC. После возвращения к температуре 20oC перемешивание продолжают в течение 20 ч, затем экстрагируют 3 раза в 100 см3 этилацетата. Объединенные органические фазы промывают 2 раза в 100 см3, 2 раза в 100 см3 водного раствора 1N соляной кислоты, 2 раза в 100 см3 водного раствора 1N едкого натра, затем 3 раза в100 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и температуре приблизительно 45oC. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: дихлорметан-метанол (97/3 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и температуре приблизительно 45oC. Таким образом, получают 3,6 г 1-(трет.бутоксикарбонил)-5-фенилциклопропилметилпролината (2RS, 5SR) в виде масла оранжевого цвета, используемого при последующем синтезе.
В 1-(трет. бутоксикарбонил)-5-фенилпролин-(2RS, 5SR) может быть получен следующим образом: в раствор 22,8 г хлоргидрата 5-фенилпролина (2RS, 5SR) и 22 г карбоната натрия в 160 см3 воды прикапывают, перемешивая, 21,8 г бикарбоната дитрет-бутила, растворенного в 120 см3 диоксана. Реакционную смесь перемешивают 20 ч при температуре приблизительно 20oC, затем образующийся осадок отфильтровывают. Фильтрат промывают 2 раза 100 см3 этилацетата и подкисляют до pH водным раствором 4N соляной кислоты. Водную кислую фазу экстрагируют 3 раза 150 см3 дихлорметана. Объединенные органические экстракты промывают 2 раза в 50 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и температуре приблизительно 40oC. После рекристаллизации в ацетонитриле получают 24 г 1-(трет.бутоксикарбонил) 5-фенилпролина-(2RS, 5SR), плавящегося при 170oC.
Пример 26. По способу, описанному в примере 1, но исходя из 3 г 5-фенилизопропилпролината (2RS, 5SR), 2,7 г 2-/3-(3-метилфенил)уреидо/уксусной кислоты в суспензии в 75 см3 безводного 1,2-дихлорэтана и из 1 см3 сульфинилхлорида получают после очистки 1,2 г 1-(2-(3-(3-метилфенил)уреидо)ацетил)-5-фенилизопропилпролината (2RS, 5SR).
Протонный ЯМР (200 МГц, CDCl3) δ в ч/млн), 2 ротамера при комнатной температуре, описание преобладающего ротамера: 1,1 (d, 6H, (CH3)2), 1,7 - 2,5 (m, 4H, H в 3 и 4 пирролидина), 2,15 (s, 3H, CH3), 3 и 4,1 (2bd, 2H, CH2N), 4,4 (bt, 1H, H в 2 пирролидина), 4,9 (m, 2H, CH изопропила и H в 5 в пирролидине), 6,6 - 7,5 (m, 9H, ароматические).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3365, 3150, 3060, 3030, 2980, 2935, 2875, 1738, 1700, 1645, 1615, 1595, 1560, 1495, 1450, 1430, 1375, 1305, 1295, 1280, 1210, 1190, 1145, 1120, 915, 890, 860, 780, 755, 705.
5-Фенилизопропилпролината (2RS, 5SR) может быть получен следующим образом, аналогичным описанному в примере 25, A, но исходя из 5 г 1-(трет.бутоксикарбонил)-5-фенилизопропилпролината (2RS, 5SR) и 2,4 см3 иодтриметилсилана в растворе в 50 см3 безводного хлороформа. Таким образом, получают 3 г 5-фенилизопропилпролината (2RS, 5SR) в виде масла желтого цвета, используемого при последующем синтезе.
1-(Трет. бутоксикарбонил)-5-фенилизопропилпролинат (2RS, 5SR) может быть получен как описано в примере 25, Б, но исходя из 5,85 г 1-(трет.бутоксикарбонил)-5-фенилпролина-(2RS, 5SR), 3,85 г паратолуолсульфонилхлорида и 1,6 г 2-пропанола в 30 см3 безводного пиридина. После обработки, получают 5 г 1-(трет. бутоксикарбонил)-5-фенилизопропилпролината (2RS, 5SR) в виде масла желтого цвета, используемого при последующем синтезе.
Пример 27. Энантиомеры 1-(2-(3-(3-метилфенил)уреидо)ацетил) 5-фенил-трет. бутилпролината (2RS, 5SR) разделяют высокоскоростной жидкой хроматографией на хиральной фазе типа PIRCKLE, используя 400 г (Д)-N-3,5-динитробензоилфенилглицина, привитого на силикоаминопропиле в качестве стационарной фазы, загруженной в колонку длиной 200 мм и диаметром 80 мм, используя в качестве подвижной фазы смесь гексан-2 пропанол-метиленхлорид (87/7, 5/7,5). Исходя из 1 г рацемата получают 0,48 г 1-(2-(3-(3-метилфенил)уреидо)ацетил)-5-фенилтрет. бутилпролината (2S, 5R), плавящегося при 79oC, /α/
Носитель может быть приготовлен следующим образом:
В 6-литровой трехгорлой колбе суспензируют 600 г силикоаминопропила (  -10 мкм - NH2, Macherey - Nagel) в 2 л диметилформамида. Добавляют 95 г ангидрида N-трет-бутоксикарбониламино-11-ундекановой кислоты и перемешивают реакционную смесь в течение 18 ч при температуре около 20oC. Двуокись кремния отфильтровывают, промывают последовательно 2 раза в 1500 см3 дихлорметана, затем 2 раза в 1500 см3 диметилформамида. Промытую двуокись кремния снова суспензируют в 2 л диметилформамида и добавляют 95 г ангидрида N-трет-бутоксикарбониламино-11-ундекановой кислоты, затем перемешивают в течение 18 ч при температуре около 20oC. Двуокись кремния отфильтровывают, промывают последовательно два раза в 600 см3 дихлорметана, два раза в 600 см3 тетрагидрофурана, два раза в 600 см3 метанола и два раза в 600 см3 диэтилового эфира, затем высушивают при пониженном давлении и температуре около 20oC. Таким образом, получают 610 г двуокиси кремния, имеющей название "BOC-C11 - C3-двуокись кремния", в виде белого порошка, структура которого подтверждена инфракрасным спектром и элементарный анализ которого: C% = 8,8, H% = 1,7, N% = 1,2.
-10 мкм - NH2, Macherey - Nagel) в 2 л диметилформамида. Добавляют 95 г ангидрида N-трет-бутоксикарбониламино-11-ундекановой кислоты и перемешивают реакционную смесь в течение 18 ч при температуре около 20oC. Двуокись кремния отфильтровывают, промывают последовательно 2 раза в 1500 см3 дихлорметана, затем 2 раза в 1500 см3 диметилформамида. Промытую двуокись кремния снова суспензируют в 2 л диметилформамида и добавляют 95 г ангидрида N-трет-бутоксикарбониламино-11-ундекановой кислоты, затем перемешивают в течение 18 ч при температуре около 20oC. Двуокись кремния отфильтровывают, промывают последовательно два раза в 600 см3 дихлорметана, два раза в 600 см3 тетрагидрофурана, два раза в 600 см3 метанола и два раза в 600 см3 диэтилового эфира, затем высушивают при пониженном давлении и температуре около 20oC. Таким образом, получают 610 г двуокиси кремния, имеющей название "BOC-C11 - C3-двуокись кремния", в виде белого порошка, структура которого подтверждена инфракрасным спектром и элементарный анализ которого: C% = 8,8, H% = 1,7, N% = 1,2.
В 6-литровой трехгорлой колбе суспензируют 607 г двуокиси кремния "BOC-C11 - C3-двуокись кремния" в 2 л дихлорметана и 68 см3 пиридина. Прикапывают 530 см3 диметилоктилхлорсилана и перемешивают реакционную смесь в течение 16 ч при температуре около 20oC. Полученное твердое вещество отфильтровывают и промывают последовательно два раза в 1 л дихлорметана, два раза в 1 л метанола, 2 раза в 1 л тетрагидрофурана, два раза в 1 л дихлорметана и два раза в 1 л диэтилового эфира, затем высушивают при пониженном давлении и температуре около 20oC. Таким образом, получают 712 г двуокиси кремния, имеющей название "ВОС-C11-C3-двуокись кремния-O-Si(CH3)2 (CH2)7CH3", в виде белого порошка, структура которого подвержена инфракрасным спектром и элементарный анализ которого показывает: C% = 12,1, H% = 2,4, N% = 1,0.
В 6-литровой трехгорлой колбе суспензируют 711 г двуокиси кремния "ВОС-C11-C3-двуокись кремния-O-Si(CH3)2 (CH2)7CH3" в 2200 см3 6% (в объемном выражении) раствора трифтоуксусной кислоты в дихлорметане. Реакционную смесь перемешивают в течение 5 ч при температуре около 20oC. Двуокись кремния отфильтровывают и промывают последовательно два раза в 1 л дихлорметана, два раза в 1 л смеси дихлорметан-диизопропилэтиламин (70/30 об.), в 1 л дихлорметана, два раза в 1 л тетрагдирофурана, два раза в 1 л метанола и два раза в 1 л диэтилового эфира, затем высушивают при пониженном давлении и температуре около 50oC. Промытую и высушенную двуокись кремния снова суспензируют в 2 л 6% (в объемном выражении) раствор трифторуксусной кислоты в дихлорметане. Реакционную смесь перемешивают в течение 16 ч при температуре около 20oC. Двуокись кремния отфильтровывают и промывают последовательно два раза 1,5 л дихлорметана, два раза в 1 л смеси дихлорметан-диизопропилэтиламин (70/30 об. ), в 1,5 л дихлорметана, два раза в 2 л тетрагидрофурана, два раза в 2 л метанола и два раза в 2 л диэтилового эфира, затем высушивают при пониженном давлении и температуре около 50oC. Таким образом, получают 607 г двуокиси кремния имеющей название "C11-C3-двуокись кремния-O-Si(CH3)2(CH2)7CH3", в виде белого порошка, структура которого подтверждена инфракрасным спектром и элементарный анализ которого дает: C% = 8,8, H% = 1,7, N% = 1,3.
В 4-литровой трехгорлой колбе суспензируют 400 г двуокиси кремния "C11-C3-двуокись кремния-O-Si(CH3)2(CH2)7CH3" в 1800 см3 диметилформамида. Добавляют 42 г 3,5-динитробензоил-Д-фенилглицина и 30 г 2-этокси-1-этоксикарбонил-1,2-дигидрохинолеина и реакционную смесь перемешивают в течение 16 ч при температуре около 20oC. Двуокись кремния отфильтровывают и промывают последовательно два раза в 1 л дихлорметана, два раза в 1 л тетрагидрофурана, два раза в 1 л метанола и два раза в 1 л диэтила эфира. Двуокись кремния, промытая таким образом, снова суспензируется в 2 л диметилформамида и добавляют 30 г 2-этокси-1-этоксикрабонил-1,2-дигидрохинолеина и 42 г 3,5-динитробензоил-Д-фенилглицина, затем реакционную смесь перемешивают в течение 5 ч при температуре около 20oC. Двуокись кремния отфильтровывают, промывают последовательно два раза в 1 л диметилформамида, два раза в 1 л дихлорметана, два раза в 1 л тетрагидрофурана, два раза в 1 л метанола и два раза в 1 л диэтилового эфира, затем высушивают при пониженном давлении при температуре около 140oC. Таким образом, получают 434 г двуокиси кремния, имеющей название "DNB-Д-Phg- C11-C3-двуокись кремния-O-Si(CH3)2 (CH2)7CH3", в виде белого порошка, структура которого подтверждена инфракрасным спектром и элементарный анализ которого дает: C% = 12,3, H% = 1,8, N% = 2,1.
В 4-литровой трехгорлой колбе суспензируют 434 г двуокиси кремния "DNB-Д-Phg-C11-C3-двуокись кремния-O-Si(CH3)2(CH2)7CH3" в 1,3 л дихлорметана и добавляют 100 см3 диметилоктилметоксилилана, затем реакционную смесь перемешивают в течение 54 ч при температуре около 20oC. Двуокись кремния отфильтровывают, промывают последовательно два раза в 1 л дихлорметана, два раза в 1 л метанола, два раза в 1 л тетрагидрофурана и два раза в 1 л дихлорметана, затем высушивают при пониженном давлении при температуре около 140oC. Таким образом, получают 425 г двуокиси кремния, имеющей название "DNB-D-Phg-C11-C3-двуокись кремния-OSi(CH3) 2(CH2)7CH3 реоктилированная", в виде белого порошка, структура которого подтверждена инфракрасным спектром и элементарный анализ которого дает: C% = 12,7, H% = 1,9, N% = 2,0.
В 4-литровой трехгорлой колбе суспензируют 425 г двуокиси кремния "DNB-D-Phg-C11-C3-двуокись кремния-OSi(CH3)2(CH2)7CH3 реоктилированной" в 1,3 л дихлорметана. Прикапывают 545 см3 триметилсилилимидазола и реакционную смесь перемешивают в течение 15 ч при температуре около 20oC. Полученный твердый продукт отделяют фильтрацией и промывают последовательно два раза в 1 л тетрагидрофурана, два раза в 1 метанола, два раза в 1 л ацетона и два раза в 1 л дихлорметана, затем высушивают при пониженном давлении и температуре около 20oC. Таким образом, получают 431 г двуокиси кремния, имеющей название "DNB-D-Phg-C11-C3-двуокиси кремния-/OSi(CH3)2(CH2)7CH3/- /O-Si(CH3)3/", в виде белого порошка, структура которого подтверждена инфракрасным спектром и элементарный анализ которого дает: C% = 13,7, H% = 2,2, N% = 2,0.
Ангидрид N-трет. бутоксикарбониламино-11-ундекановой кислоты может быть получен следующим образом: в раствор 30,1 г N-трет-бутоксикарбониламино-11 ундекановой кислоты в 480 см3 этилацетата, поддерживая температуру около 5oC, добавляют в течение 10 мин раствор 10,63 г N,N'-дициклогексилкарбодиимида в 120 см3 этилацетата. Реакционную смесь перемешивают в течение 1 ч при температуре около 5oC, затем в течение 16 ч при температуре около 20oC. Выпавший осадок отфильтровывают и промывают в 30 см3 этилацетата. Фильтрат концентрируют при пониженном давлении и при 30oC. Полученный твердый продукт высушивают в вакууме при температуре около 30oC. Таким образом, получают 31 г ангидрида N-трет-бутоксикарбониламино-11 ундекановой кислоты, плавящегося при 58oC.
N-трет-бутоксикарбониламино-11-ундекановая кислота может быть получена по известному методу (I.T. Sparrow, J. Org. Chem., 41, 1350, 1976).
Пример 28. В раствор 0,08 г 1-(2-амино-ацетил)-5-фенилтрет.бутилпролината (2S, 5R), в 10 см3 безводного тетрагидрофурана, добавляют 0,05 см3 изоцианата 3-метилфенила. Реакционную смесь перемешивают в течение 3 ч при температуре около 25oC, затем растворитель выпаривают при пониженном давлении при 45oC. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: дихлорметан-метанол (98/2 об.). Фракции, содержащие целевой продукт объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 0,04 г 1-(2-(3-(3-метилфенил)уреидо)ацетил) 5-фенилтрет. бутилпропилата-(2S, 5R), аналитические данные которого соответствуют аналитическим данным правовращающегося энантиомера, полученного хроматографией на стационарной хиральной фазе рацемата, /α/
1-(2-Аминоацетил)-5-фенилтрет. бутилпролинат (2S,5R) может быть получен следующим образом: в раствор 0,08 г 1-(2-трет.бутоксикарбониламиноацетил)-5-фенил-2-пирролидинкарбоновой -(2S,5R) кислоты в 20 см3 хлороформа, охлажденный до 5oC, добавляют 0,1 см3 концентрированной серной кислоты. Реакционную среду насыщают изобутиленом в течение 2 ч, перемешивая и поддерживая температуру при 5oC. После повышения температуры до приблизительно 20oC, продолжают перемешивание в течение 20 ч. Раствор доводят до pH 8, прибавляя 10%-ный водный раствор бикарбоната натрия, а водную фазу отделяют и затем экстрагируют 3 раза с помощью 30 см3 хлороформа. Объединенные органические фазы промывают 2 раза в 10 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 0,08 г 1-(2-аминоацетил)-5-фенилтрет.бутилпролината (2S,5R) в виде желтого цвета, используемого при последующем синтезе.
1-(2-Трет. бутоксикарбониламиноацетил) 5-фенил-2- пирролидинкарбоновая-(2S, 5R) кислота может быть получена следующим образом: в охлажденный до 5oC раствор 0,16 г 1-(2-трет.бутоксикарбониламиноацетил) 5-фенилметилпролината (2S, 5R) в 15 см3 смеси диоксан-вода (70/30 об.) добавляют 0,5 см3 водного раствора едкого натра 1N. После повышения температуры до приблизительно 20oC продолжают перемешивание в течение 12 ч, затем добавляют снова 0,5 см3 водного раствора 1 N едкого натра. После 3 ч перемешивания при температуре около 20oC реакционную смесь промывают 2 раза в 10 см3 эфира, затем доводят pH приблизительно до 2, добавляя 20 см3 водного раствора 0,1N соляной кислоты. Реакционную смесь экстрагируют 3 раза с помощью 40 см3 этилацетата. Объединенные органические фазы высушиваются над сульфатом магния и концентрируются досуха при пониженном давлении и при 35oC. Таким образом, получают 0,09 г 1-(2-трет.бутоксикарбониламиноацетил)-5-фенил-2-пирролидинкарбоновой (2S,5R) кислоты в виде бесцветной смолы, используемой при дальнейшем синтезе.
1-(2-Трет.бутоксикарбониламиноацетил)-5-фенилметилпролинат (2S,5R) может быть получен следующим образом: в раствор 0,1 г 5-фенилметилпролината-(2S, 5R) и 0,1 г 2-трет.бутоксикарбониламиноуксусной кислоты в 10 см3 безводного ацетонитрила, выдержанный при температуре около 0oC, добавляют сразу 0,12 г N, N'-дициклогексилкарбодиимида. Реакционную смесь перемешивают в течение 16 ч при температуре около 25oC, затем нерастворимый продукт отфильтровывают и промывают три раза в 5 см3 дихлорметана. Фильтрат концентрируют досуха при пониженном давлении и при 40oC. Полученный маслянистый остаток очищают хроматографией на двуокиси кремния (элюент: этилацетатциклогексан (30/70 об.). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 0,16 г 1-(2-трет.бутоксикарбониламиноацетил)-5- фенилметилпролината(2S, 5R) в виде бесцветного масла, используемого при последующем синтезе.
5-Фенилметилпролинат (2S,5R) может быть получен следующим образом: к раствору 0,6 г 1-метоксикарбонил-5-фенилметилпролината (2S,5R) в 15 см3 хлороформа прикапывают 0,45 см3 иодтриметилсилана при температуре около 25oC. Смесь нагревают с обратным холодильником в течение 18 ч. После охлаждения до температуры около 25oC добавляют 20 см2 воды. Водную фазу отделяют, затем экстрагируют 2 раза 20 см2 хлороформа. Органические фазы объединяют, промывают 2 раза в 10 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Полученную смесь двух диастереизомеров очищают хроматографией на двуокиси кремния (элюент: этилацетатциклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом получают 0,12 г 5-фенилметилпролината (2S,5R) в виде масла, используемого при последующем синтезе.
1-Метоксикарбонил-5-фенилметилпролинат (2S,5R) может быть получен следующим образом: в раствор 5,1 г 5-метокси-1-метоксикарбонилметилпролината (2S, 5RS) в 400 см3 бензола, охлажденный до температуры около 5oC, добавляют 3,1 г трихлорида алюминия. Реакционный раствор перемешивают в течение 12 ч при температуре около 25oC, затем добавляют 50 см3 насыщенного водного раствора бикарбоната натрия. Гидроокись алюминия отфильтровывают и промывают 2 раза в 20 см3 метиленхлорида. Объединенные органические фазы промывают 2 раза в 50 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетатциклогексан (20/80 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 0,6 г 1-метоксикарбонил-5-фенилметилпролината-(2S, 5RS) в виде масла светло-желтого цвета, смесь (40/60 вес.) изомеров (2S,5R) и (2S,5S), использующуюся при последующем синтезе.
5-Метокси-1-метоксикарбонилметилпролинат (2S,5RS) может быть получен по известному методу (T. Shono et coll., J. Am. Chem. Soc., 104, 6697, 1982).
Пример 29. Энантиомеры 3-(3-(2-(2-трет.бутоксикарбонил-5-фенил- 1-пирролидинил)2-оксо-этил)уреидо)бензойной-((2RS, 5SR) кислоты отделяют высокоскоростной жидкой хроматографией на хиральной фазе типа PIRCKLE, используя 400 г (D)-N-3,5-динитробензоилфенилглицина, привитого на силикоаминопропиле, в качестве стационарной фазы хроматографической колонки длиной 220 мм и диаметром 70 мм, а в качестве подвижной фазы используют смесь гептан-2-пропанолтрифторуксусная кислота (80/20/0,05 об.). Исходя из 3,5 г рацемата получают:
0,72 г 3-(3-(2-(2-трет.бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксоэтил)уреидо)бензойной-((2S,5R) кислоты, плавящейся при 204oC, /α/
0,54 г 3-(3-(2-(2-трет.бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксоэтил)уреидо)бензойной-((2SR,5RS) кислоты, плавящейся при 240oC, /α/
Пример 30. По способу, описанному в примере 2, но исходя из 2 г 1-(2-аминоацетил)-2,5-дифенил-пирролидина-(цис) и 0,9 см3 изоцианата 3-метилфенила в растворе в 25 см3 безводного тетрагидрофурана получают после перекристаллизации в 2-пропаноле 1,4 г 1-/2-(2,5-дифенил-1-пирролидинил)2-оксоэтил/-3-(3-метилфенил) мочевины-((2RS,5SR), плавящейся при 168oC.
1-(2-Аминоацетил)-2,5-дифенилпирролидин-(цис) может быть получен по способу, аналогичному описанному в примере 2, А, но исходя из 6,6 г 1-(2-трет. бутоксикарбониламиноацетил)-2,5-дифенилпирролидина-(цис) и 2,5 см3 иодтриметилсилана, растворенного в 70 см3 безводного хлороформа. Таким образом получают 4,8 г 1-(2-аминоацетил)-2,5-дифенилпирролидина-(цис) в виде масла оранжевого цвета, используемого при последующем синтезе.
Протонный ЯМР (300 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре: 1,8-2,4 (bm, 4H, CH2-CH2), 2,7 и 3,3 (bd, 2H, AB, CH2CO2, 5,0 (bm, 1H, CH), 5,2 (bm, 1H, CHN), 7,2-7,5 (m, 10H, ароматические).
Масса (электронный удар, 70 эВ, m/z), 280 (M+), 263 (M+-NH2), 222 (M+-COCH2NH2), 91 (C6H5CH
1-(2-Трет.бутоксикарбониламиноацетил)-2,5-дифенилпирролидин- (цис) может быть получен, как описано в примере 2, Б, но на основе раствора 6,6 г 2,5-дифенилпирролидина (смесь изомеров цис-транс), 5,2 г 2-(трет.бутоксикарбониламино)уксусной кислоты и 6,1 г N,N'-дициклогексилкарбодиимида в 50 см3 безводного ацетонитрила. Получают после отделения хроматографией на двуокиси кремния (элюент: дихлорметан-метанол (98/2 об.)) 3,6 г 1-(2-трет.бутоксикарбониламиноацетил)-2,5-дифенилпирролидин-(цис) в виде масла оранжевого цвета, используемого при последующем синтезе.
2,5-Дифенилпирролидин (смесь изомеров цис-транс) может быть получена по известному методу (C.G. Overberger, M.Valentine et J.-P. Anselme, J. Amer. Chem. Soc., 91, 687-94, 1969).
Пример 31. По способу, описанному в примере 9, но исходя из 3 г 3-(3-(2-(2,5-дифенил-1-пирролидинил)-2-оксо-этил)уреидо)этилбензоата -(цис), растворенного в 50 см3 метанола, и 0,7 г гидроокиси калия, растворенного в 20 см3 воды, после обработки и рекристаллизации в этаноле получают 2 г 3-(3-(2-(2,5-дифенил-1-пирролидинил)-2-оксо-этил)уреидо)-бензойной- (цис) кислоты, плавящейся при 255oC.
3-(3-(2-(2,5-Дифенил-1-пирролидинил)-2-оксо-этил)уреидо)- этилбензоат-(цис) может быть получен способ, аналогичным описанному в примере 3, но исходя из 3,4 г 1-(2-аминоацетил)-2,5-дифенил- пирролидина)-(цис), 2,2 г N, N'-диимидазолкарбонила, растворенного в 75 см3 безводного 1,2-дихлорэтана и 2 г 3-аминоэтиллбензоата. После очистки получают 3 г 3-(3-(2-(2,5-дифенил-1-пирролидинил)-2-оксо-этил)уреидо)этилбензоата (цис) в виде воздушной массы белого цвета, используемого при последующем синтезе.
Пример 32. По способу, аналогичному описанному в примере 2, но исходя из 1,8 г 1-(2-аминоацетил)-6-фенил-2-пиперидин- трет.бутилкарбоксилата (2RS, 5SR), растворенного в 30 см3 безводного тетрагидрофурана, добавляют 0,7 см3 изоцианата 3-метилфенила. Получают после рекристаллизации в окиси диизопропила 0,7 г 1-(2-(3-(3-метилфенил)уреидо)ацетил)-6-фенил-2-пиперидин- трет. бутилкарбоксилата (2RS,5SR), плавящегося при 123oC.
1-(2-Аминоацетил)-6-фенил-2-пиперидинтрет. бутилкарбоксилат (2RS, 5SR) может быть получен по способу, аналогичному описанному в примере 2, А, но исходя из 5,9 г 1-(2-трет. бутоксикарбониламиноацетил)-6-фенил-2-пиперидинтрет. бутилкарбоксилата (2RS,5SR) и 2 см3 иодтриметилсилана, растворенного в 50 см3 безводного хлороформа. Таким образом, получают 1,8 г 1-(2-аминоацетил)-6-фенил-2-пиперидин- трет.бутилкарбоксилат (2RS,5SR) в виде масла оранжевого цвета, используемого при последующем синтезе.
Масса (электронный удар, 70 эВ, m/z), 318 (M+), 159 (M+-COCH2NH2 и -CO2 трет.бутил).
1-(2-Трет. бутоксикарбониламиноацетил)-6-фенил-2-пиперидин- трет.бутилкарбоксилат-(2RS,5SR) может быть получен, как описано в примере 2, Б, но исходя из раствора 5,1 г 6-фенил-2-пиперидин-трет.бутилкарбоксилата-(2RS,5SR), 3,4 г 2-(трет.бутоксикарбониламино) уксусной кислоты и 4 г N,N'-дициклогексилкарбодиимида, растворенного в 90 см3 безводного ацетонитрила. Получают 5,9 г 1-(2-трет. бутоксикарбониламиноацетил)6-фенил-2-пиперидин- трет.бутилкарбоксилата (2RS,5SR) в виде масла оранжевого цвета, используемого при последующем синтезе.
6-Фенил-2-пиперидинтрет. бутилкарбоксилат-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 1, Б, но исходя из 14 г хлоргидрата 6-фенил-2-пиперидинкарбоновой-(2RS, 5SR) кислоты, в суспензии в смеси 5 см3 концентрированной серной кислоты в 250 см3 безводного хлороформа. Смесь насыщают изобутиленом. После обработки получают 5,2 г 6-фенил-2-пиперидинкарбоксилата трет-бутила-(2RS, 5SR), плавящегося при 68oC.
Хлоргидрат 6-фенил-2-пиперидинкарбоновой-(2RS, 5SR) кислоты может быть получен следующим образом: 14 г хлоргидрата 6-фенил-2,3,4,5-тетрагидро-2-пиридинкарбоновой кислоты, 0,7 г окиси платины в суспензии в 200 см3 этанола перемешивают в течение 3 ч при температуре около 20oC в атмосфере водорода (130 кПа). Катализатор отфильтровывают и фильтрат концентрируют досуха при пониженном давлении и при 45oC. После перекристаллизации в ацетонитриле получают 14 г хлоргидрата 6-фенил-2-пиперидинкарбоновой-(2RS, 5SR) кислоты, плавящейся при 184oC.
Хлоргидрат 6-фенил-2,3,4,5-тетрагидро-2-пиридинкарбоновой кислоты может быть получен следующим образом: в раствор этилата натрия, полученного из 4,1 г натрия в 180 см3 безводного этанола, добавляют 38,3 г этил-ацетамидомалоната. После 1 ч перемешивания при температуре около 20oC выливают 40 г 2-(3-хлорпропил)-2-фенил-1,3-диоксолана и 2,1 г иодида калия. Реакционную смесь нагревают с обратным холодильником в течение 24 ч, затем концентрируют досуха при пониженном давлении и температуре около 45oC. Пастообразный остаток извлекают с помощью 400 см3 дихлорметана, промывают 3 раза в 100 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: дихлорметан). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 25 г масла оранжевого цвета, которое суспензируют в 102 см3 воды и 102 см3 12 N соляной кислоты. Реакционную смесь нагревают с обратным холодильником, перемешивая, в течение 5 ч. После охлаждения кислый раствор промывают 3 раза в 100 см3 диэтилового эфира, затем концентрируют досуха при пониженном давлении и температуре около 50oC. После перекристаллизации в ацетонитриле получают 14 г хлоргидрата 6-фенил-2,3,4,5-тетрагидро-2-пиридинкарбоновой кислоты, плавящейся при 146oC.
2-(3-Хлорпропил)-2-фенил-1,3-диоксолан может быть получен известным способом (M. T. WILLS, J.E. WILLS, L. Von DOLLEN, B.L. BUTLER et J. PORTER, J, Org. Chem., 45(12), 2495, 1980).
Пример 33. В суспензию 0,5 г 5-(2-фурил) трет.бутилпролината (2S, 5R) и 0,48 г 2/3-(3-метилфенил)уреидо/уксусной кислоты в 50 см3 безводной смеси ацетонитрил/-1,2-дихлорэтан (80/20 об.), поддерживаемой при температуре около 0oC, добавляют за 10 мин раствор 0,48 г N,N'-дициклогексилкарбодиимида в 10 см3 безводного ацетонитрила. Реакционную смесь перемешивают в течение 16 ч при температуре около 25oC, затем нерастворимый продукт отфильтровывают и промывают три раза в 5 см3 дихлорметана. Фильтрат концентрируют досуха при пониженном давлении и при 40oC. Масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (40/60 об.)). Фракции, содержащие целевой продукт, объединяют, затем концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают после перекристаллизации в смеси этилацетатциклогексан (50/50 об.) 0,15 г 5-(2-фурил)-1- (2-(3-(3-метилфенил)уреидо)ацетил)трет. бутилпролината (2S, 5R), плавящегося при 144oC, /α/
Протонный ЯМР (250 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция при 120oC, описание при 120oC: 1,50 (bs, 9H, (CH3)3), 2,10 (bm, 2H, CH2), 2,30 (bm, 2H, CH2), 2,30 (s, 3H, CH3), 3,8 и 4,0 (ABX, 2H, CH2N), 4,45 (bt, 1H, NCHCOO), 5,20 (bdd, 1H, NCHC), 6,15 (bt, 1H, NH), 6,40 (bdd, 1H, C4H4O в позиции 4), 6,50 (bd, 1H, C4H4O в позиции 3), 6,70 (bd, 1H, N-C6H4C в позиции 4), 7,15 - 7,25 (m, 3H, ароматические), 7,50 (bs, 1H, C4H4O в позиции 5), 8,40 (bs, 1H, NH).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3365, 2975, 2930, 2850, 1735, 1640, 1615, 1595, 1560, 1490, 1390, 1365, 1150, 780, 745, 695.
5-(2-Фурил)трет. бутилпролинат-(2S, 5R) может быть получен следующим образом: в раствор 3,8 г 1-трет.бутоксикарбонил-5-(2-фурил)трет.бутилпролината (2S, 5RS) в 50 см3 хлороформа прикапывают 1,77 см3 иодтриметилсилана при температуре приблизительно 25oC. Перемешивание производят в течение 30 мин, затем добавляют 20 см3 воды. Водную фазу отделяют, затем экстрагируют 2 раза 20 см3 хлороформа. Органические фазы объединяют, промывают последовательно в 30 см3 насыщенного водного раствора кислого карбоната натрия и два раза в 30 см3 воды, затем высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Полученную смесь обоих диастереоизомеров отделяют хроматографией на двуокиси кремния (элюент: этилацетатциклогексан (20/80 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 0,50 г 5-(2-фурил)трет.бутилпролината-(2S, 5R) в виде масла желто-оранжевого цвета, используемого при последующих реакциях синтеза.
1-Трет-бутоксикарбонил-5-(2-фурил)трет. бутил пролинат (2S, 5RS) может быть получен следующим образом: в раствор 9,6 г 1-трет.бутоксикарбонил-5-метокситрет. бутил пролината (2S, 5RS) в 100 см3 фурана добавляют 0,42 г паратолуолсульфоновой кислоты. Реакционную смесь перемешивают в течение 24 ч при температуре около 25oC, затем добавляют 1 г кислого карбоната натрия. Гетерогенную среду концентрируют досуха при пониженном давлении и температуре около 30oC. Полученный остаток извлекают 250 см3 этилацетата и промывают 2 раза 50 см3 воды. Органическую фазу высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 30oC. Полученный масляный остаток очищается хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (5/95 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при 35oC. Таким образом, получают 3,8 г 1-трет-бутоксикарбонил-5-(2-фурил)трет. бутилпролината (2S, 5RS), плавящегося при 70oC, смесь изомеров (30/70 по весу) (2S, 5R) и (2S, 5S) используется при последующих реакциях синтеза.
1-Трет-бутоксикарбонил-5-метокситрет. бутилпролинат (2S, 5RS) может быть получен по известному методу (T. SHONO et coll., J. Am. Chem. Soc., 104, 6697, 1982), но исходя из 1-трет-бутоксикарбонил трет-бутил пролината (2S).
1-Трет. бутоксикарбонил трет. бутилпролинат (2S) может быть получен по известному методу (S. YOSHIFUJI et coll. , Chem. Pharm. Bull., 34, 3873, 1986).
Пример 34. В раствор 2,0 г 3-(2-аминоацетил)-2-фенил-4-тиазолидин трет. бутилкарбоксилата (2R, 4R) в 25 см3 безводного тетрагидрофурана, добавляют 0,8 см3 изоцианата 3-метилфенила. Реакционную смесь перемешивают в течение 12 ч при температуре около 25oC, затем разбавляют 100 см3 этилацетата. Органическую фазу промывают 2 раза в 40 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: метиленхлорид-метанол (98/2 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают после встряхивания в гептане 0,5 г 3-(2-(3-(3-метил-фенил)уреидо)ацетил) 2-фенил-4-тиазолидинтрет. бутилкарбоксилата (2R, 4R) в виде аморфного белого порошка, /α/
Протонный ЯМР (200 МГц, CDCl3, δ в ч/млн), 2 ротамера при комнатной температуре, описание преобладающего ротамера при 25oC: 1,45 (bs, 9H, (CH3)3), 2,2 (s, 3H, CH3), 3,2 (A2X, 2H, CH2S), 3,2 и 4,3 (ABX, 2H, CH2N), 4,9 (t, 1H, CHN), 6,1 (bdd, 1H, NH), 6,2 (s, 1H, SCHN), 6,8-7,7 (m, 9H, ароматические).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3375, 2975, 2930, 1735, 1650, 1610, 1595, 1490, 1455, 1370, 1150, 780, 730, 695.
А) 3-(2-Аминоацетил)-2-фенил-4-тиазолидинтрет.бутилкарбоксилат (2R, 4R) может быть получен следующим образом: в раствор 2,5 г 3-(2-трет.бутоксикарбониламиноацетил)-2-фенил-4- тиазолидинтрет.бутоксикарбоксилата (2R, 4R) в 15 см3 хлороформа прикапывают 0,9 см3 иодтриметилсилана при температуре около 25oC. Реакционную смесь перемешивают в течение 1 ч при температуре около 25oC, затем добавляют 15 см3 воды. Водную фазу отделяют, затем экстрагируют 2 раза 10 см3 хлороформа. Органические фазы объединяют, промывают последовательно в 20 см3 воды, 20 см3 насыщенного водного раствора кислого карбоната натрия и 20 см3 воды, затем высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 2 г 3-(2-амино-ацетил)-2-фенил- 4-тиазолидин-трет.бутилкарбоксилата (2R, 4R) в виде масла светло-желтого цвета, используемого при последующих реакциях синтеза.
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре: 1,5 (s, 9H, C(CH3)3), 2,9-4,4 (vbm, 4H, 2CH2), 4,8-5,4 (2m, 1H, NCHCO), 6,2-6,55 (2s, 1H, NCHS), 7,2-7,8 (m, 5H, ароматические), 8,3 (vbs, 1H, NH
Б) 3-(2-Трет.бутоксикарбониламиноацетил)-2-фенил-4-тиазолидин- трет.бутилкарбоксилат (2R, 4R) может быть получен следующим образом: в раствор 37,4 г 2-фенил-4-тиазолидин-трет.бутилкарбоксилата (2RS, 4R) и 24,7 г 2-трет.бутоксикарбониламиноуксусной кислоты в 150 см3 безводного ацетонитрила, поддерживая температуру около 0oC, добавляют за 30 мин раствор 29,1 г N,N'-дициклогексилкарбодиимида в 80 см3 безводного ацетонитрила. Реакционную смесь перемешивают в течение 16 ч при температуре около 25oC, затем нерастворимый продукт отфильтровывают и промывают три раза в 20 см3 ацетонитрила. Фильтрат концентрируют досуха при пониженном давлении и при 45oC. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 39,6 г 3-(2-трет.бутоксикарбониламиноацетил)-2-фенил-4- тиазолидинтрет.бутилкарбоксилата (2R, 4R) в виде вязкого масла желтого цвета, которое используется при последующих реакциях синтеза, /α/
В) 2-Фенил-4-тиазолидинтрет.бутилкарбоксилат (2RS, 4R) может быть получен следующим образом: в суспензию 53,7 г 2-фенил-4-тиазолидинкарбоновой-(2RS, 4R) кислоты в 590 см3 хлороформа, охлажденную до температуры около 5oC, прикапывают 15 см3 концентрированной серной кислоты. Реакционную среду насыщают изобутиленом в течение 5 ч, перемешивая и поддерживая температуру при 5oC. После повышения температуры до приблизительно 20oC, перемешивание продолжают в течение 20 ч. Раствор доводят до pH 8, добавляя насыщенный водный раствор кислого карбоната натрия, затем водную фазу отделяют и экстрагируют 2 раза 300 см3 хлороформа. Объединенные органические фазы промывают 2 раза в 300 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (20/80 об. )). Фракции, содержащие целевой продукт, объединяют, затем концентрируют досуха при пониженном давлении. Таким образом, получают 42,8 г 2-фенил-4-тиазолидинтрет. бутилкарбоксилата (2RS, 4R) в виде масла светло-желтого цвета, смесь (40/60 вес. ) изомеров (2R, 4R) и (2S, 4R) используется при последующих реакциях синтеза.
Г) 2-фенил-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена следующим образом: в суспензию 2,4 г L-цистеина в 35 см3 этанола прикапывают при температуре около 50oC 2,3 см3 бензальдегида. Реакционную среду нагревают с обратным холодильником в течение 2 ч. После охлаждения до температуры около 25oC нерастворимый продукт отфильтровывают и промывают последовательно в 20 см3 этанола и 20 см3 диэтилового эфира. Таким образом, получают 3,7 г 2-фенил-4-тиазолидинкарбоновой- (2RS, 4R) кислоты, плавящейся при 190oC, которая используется при последующих реакциях синтеза.
Пример 35. Работают аналогично примеру 34, но исходя из 0,3 г 3-(2-аминоацетил)-2-фенил-4-триазолидинтрет. бутилкарбоксилата (2S, 4S) и из 0,12 см3 3-метилфенилизоцианата. Получают после встряхивания в циклогексане 0,16 г 3(2-(3(3-метилфенил)уреидо)ацетил)- 2-фенил-4-триазолидинтрет.бутилкарбоксилата (2S, 4S) в виде аморфного бледно-желтого порошка, /α/
ЯМР протона (250 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция полос при 120oC; общее описание действительно к другим продуктам группы тиазолидина (при 120oC): 1,50 (bc, 9H, (CH3)3), 2,3 (s, 3H, CH3), 3,25 и 3,5 (ABX, 2H, CH2S), 3,7 и 4,05 (ABX, 2H, CH2N), 5,0 (bdd, 1H, CHN), 6,2 (bds, 1H, NH), 6,4 (bs, 1H, SCHN), 6,7 (bd, 1H, N-C6H4-C в положении 4 или 6), 7,05-7,2 (m, 3H, N-C6H4-C в положении 2, 4 и 5), 7,35 (m, 3H, C6H5), 7,65 (bd, 2H, C6H5), 8,35 (bs, 1H, NH).
Спектр ИК (KBr), характеристические полосы в см-1: 3380, 2990, 2940, 2860, 1745, 1650, 1615, 1600, 1560, 1495, 1460, 1375, 1155, 785, 735, 700).
3-(2-Аминоацетат)-2-фенил-4-триазолидин-трет. бутилкарбоксилат (2S, 4S) может быть получен как в примере 34, A, но исходя из 0,53 г 3-(2-трет.бутоксикарбониламиноацетил)-2-фенил-4-тиазолидин- трет.бутилкарбоксилата (2S, 4S) и из 0,18 см3 иодтриметилсилана. Получают 0,31 г 3-(2-аминоацетил)-2-фенил-4-тиазолидин- трет. бутилкарбоксилата (2S, 4S) в форме бледно-желтого масла, используемого при последующих синтезах.
3-(2-Трет.бутоксикарбониламиноацетил)-2-фенил-4-тиазолидин- трет.бутилкарбоксилат (2S, 4S) может быть получен, как описано в примере 34, Б, но исходя из 0,75 г 2-фенил-4-тиазолидинтрет. бутилкарбоксилата (2RS, 4S), 0,53 г 2-трет-бутоксикарбониламиноуксусной кислоты и 0,62 г N,N'-дициклогексилкарбодиимида. Таким образом, получают 0,56 г 3-(2-трет-бутоксикарбониламиноацетил)-2-фенил-4-тиазолидин трет. бутилкарбоксилата-(2S, 4S) в виде аморфного порошка бежевого цвета, который используется при последующих реакциях синтеза, /α/
2-Фенил-4-тиазолидинтрет. бутилкарбоксилат (2RS, 4S) может быть получен, как описано в примере 34, В, но исходя из 4,5 г 2-фенил-4-тиазолидинкарбоновой-(2RS, 4S) кислоты, 1,3 см3 концентрированной серной кислоты и избытка изобутилена. Таким образом, получают 1,5 г 2-фенил-4-тиазолидинитрет.бутил карбоксилата (2RS, 4S) в виде масла светло-желтого цвета, смесь (50/50 вес.) изомеров (2R, 4S) и (2S, 4S) используют при последующих реакциях синтеза.
2-Фенил-4-тиазолидинкарбоновая-(2RS, 4S) кислота может быть получена, как в примере 34, Г, но исходя из 3,0 г Д-цистеина и 2,85 см3 бензальдегида. Таким образом, получают 4,5 г 2-фенил-4-тиазолидинкарбоновой- (2RS, 4S) кислоты, плавящейся при 190oC, которая используется при последующих реакциях синтеза.
Пример 36. В раствор 1,1 г N,N'-диимидазол-карбонила в 30 см3 хлороформа добавляют раствор 0,97 г 3-аминоэтилбензоата в 10 см3 хлороформа. Реакционную смесь перемешивают в течение 6 ч при температуре около 25oC, затем добавляют 1,3 г 3-(2-аминоацетишл)-2-фенил-4- тиазолидинтрет.бутилкарбоксилата (2R, 4R), растворенного в 10 см3 хлороформа. Реакционную смесь перемешивают в течение 24 ч при температуре около 25oC, затем промывают последовательно в 50 см3 воды, 40 см3 водного раствора, 0,5 N соляной кислоты и 50 см3 воды. Органическую фазу отделяют, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (35/65 об. )), а фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают после встряхивания в гептане 1,0 г 3-(2-(3-(3-этоксикарбонилфенил)уреидо)ацетил)-2-фенил-4-тиазолидинтрет. бутилкарбоксилата (2R, 4R) в виде аморфного белого порошка, плавящегося при 85oC, /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 1,3 (s, 3H, CH3), 4,3 (q, 2H, CH2O), 7,3-7,7 (m, 8H ароматические), 8,0 (bs, 1H, N-C6H4-C в позиции 2).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3375, 2980, 2930, 1720, 1655, 1610, 1600, 1560, 1460, 1370, 1240, 1155, 760,735, 700, 690.
Пример 37. В раствор 0,6 г N,N'-диимидазол-карбонила в 15 см3 хлороформа добавляют раствор 1,0 г 3-(2-аминоацетил) 2-фенил-4- тиазолидинтрет.бутилкарбоксилата-(2R, 4R) в 10 см3 хлороформа. Реакционную смесь перемешивают в течение 2 ч при температуре около 25oC, затем добавляют 0,44 г 2-(3-аминофенил)-этанола, растворенного в 5 см3 хлороформа. Реакционную смесь перемешивают в течение 12 ч при температуре около 25oC, затем концентрируют досуха при пониженном давлении и при 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат), а фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают после перекристаллизации в ацетонитриле 0,18 г 3(-2(3-(3-(2-гидроксиэтил) фенил)уреидо)ацетил)-2-фенил-4-тиазолидинтрет.бутилкарбоксилата- (2R, 4R) в виде белых кристаллов, плавящихся при 164oC, /α/
Протонный ЯМР (300 МГц, DMCO D6, δ/ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 2,7-3,60 (2t, 4H, CH2CH2O), 6,8 (6d, 1H, N-C6H4-C в позиции 4 или 6), 7,05-7,20 (m, 3H, N-C6H4-C в позиции 2,5 и 6), 7,3 (m, 3H, C6H5), 7,6 (d, 2H, C6H5).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3320, 2975, 2930, 2880, 2850, 1740, 1660, 1610, 1590, 1560, 1510, 1480, 1450, 1365, 1150, 1060, 790, 730, 695).
Пример 38. В раствор 1,3 г N,N'-диимидазол-карбонила в 15 см3 безводного тетрагидрофурана добавляют за 2 ч раствор 2,6 г (3-(2-аминоацетил) 2-фенил-4-тиазолидинтрет. бутилкарбоксилата (2R, 4R) в 15 см безводного тетрагидрофурана. Реакционную смесь перемешивают при температуре около 25oC в течение 12 ч, затем концентрируют досуха при пониженном давлении и 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат). Раствор 2,8 г 3-/2-(1-имидазолилкарбоксамидо)ацетил/2-фенил-4-тиазолидинтрет. бутилкарбоксилата -(2R, 4R), полученного таким образом, и 2,7 г 3-аминофенилметилацетата в 100 см3 толуола нагревают с обратным холодильником в течение 4 ч. После охлаждения при температуре около 25oC, реакционную смесь промывают последовательно в 50 см3 воды, 50 см3 водного раствора 1N соляной кислоты и 2 раза в 50 см3 воды. Органическую фазу отделяют, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (50/50 об.)). Фракции, содержащие целевой продукт, объединяют, затем концентрируют досуха при пониженном давлении и 30oC. Таким образом, получают после взбалтывания в гептане, 1,9 г 3-(3-(2-(4-трет. бутоксикарбонил-2-фенил-3-тиазолидинил) 2-оксо- этил)уреидо)фенилметилацетата-(2R, 4R) в виде порошка светло-бежевого цвета, плавящегося при 69oC, /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC 3,7 (2S, 5H, CH2CO2CH3), 6,8 (d, 1H, N-C6H4-C в позиции 4 или 6), 7,1-7,7 (m, 8H, ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3370, 2980, 2950, 2930, 1740, 1650, 1610, 1595, 1560, 1495, 1370, 1240, 1155, 780, 735, 700.
Пример 39. В раствор 1,2 г 3-(3-(2-(4-трет-бутоксикарбонил-2- фенил-3-тиазолидинил)2-оксо-этил)уреидо) фенилметилацетата-(2R, 4R) в 25 см3 смеси вода-тетрагидрофуран (30/70 об.) добавляют при температуре около 5oC 0,1 г гидроокиси лития. Реакционную смесь перемешивают в течение 12 ч при температуре около 25oC, затем концентрируют до объема 5 см3 при пониженном давлении и 40oC. Полученный раствор разбавляют 50 см3 воды, промывают 2 раза в 30 см3 этилового эфира, затем подкисляют до pH 2 с помощью 2,8 см3 водного раствора 1N соляной кислоты. Нерастворимый продукт отфильтровывают, промывают 3 раза в 10 см3 воды, затем высушивают на воздухе. Таким образом, получают 0,7 г 3-(3-(2-(4-трет-бутоксикарбонил-2-фенил-3- тиазолидинил) 2-оксо-этил)уреидо) фенилуксусной-(2R, 4R) кислоты, плавящейся при 140oC, /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн.), 2 ротамера при комнатной температуре, коалесценция полос при 120oC, характеристические химические сдвиги при 120oC: 3,60 (bs, 2H, CH2CO2H), 6,8 (bd, 1H, N-C6H4-C в позиции 6), 7,15 - 7,80 (m, 8H ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3385, 2985, 2945, 2625, 1735, 1650, 1615, 1600, 1560, 1500, 1375, 1240, 1155, 785, 735, 705.
Пример 40. По способу, описанному в примере 33, но исходя из 3,5 г 2-(2-фторфенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 2,3 г 2-(3-(3-метилфенил)уреидо)уксусной кислоты и 2,3 г N,N'-дициклогексилкарбодиимида. Таким образом, получают после взбалтывания в гептане 1,05 г 3-(2-(3-(3-метилфенил)уреидо)ацетил)-2-(2-фторфенил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в виде белого твердого вещества, плавящегося при 107oC, /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC: 2,2 (bs, 3H, CH3), 6,7 (bd, 1H, N-C6H4-C в позиции 4 или 6), 7,1 - 7,40 (m, 6H ароматические), 7,9 (bm, 1H, C6H4F в позиции 3).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3370, 2975, 2925, 1735, 1645, 1615, 1560, 1490, 1460, 1370, 1150, 775, 760, 690.
2-(2-Фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилат-(2RS, 4R) может быть получен, как описано в примере 34, B, но исходя из 5,7 г 2-(2-фторфенил)-4-тиазолидинкарбоновой-(2RS, 4H) кислоты, растворенной в 60 см3 хлороформа, из 1,5 см3 концентрированной серной кислоты и избытка изобутилена. Таким образом, получают 5,8 г 2-(2-фтор-фенил)-4-тиазолидинтрет.бутилкарбоксилата-(2RS, 4R) в виде масла желтого цвета, смесь (50/50 вес) изомеров (2R, 4R) и (2S, 4R), которое используется при последующих реакциях синтеза.
2-(2-Фторфенил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как описано в примере 34, Г, но исходя из 21,2 г L-цистеина и 19,2 см3 2-фтор-бензальдегида. Таким образом, получают 28,2 г 2-(2-фторфенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 147oC, которая используется при последующих реакциях синтеза.
Пример 41. В раствор 4,2 г 3-(3-(2-(4-трет.бутоксикарбонил-2- (3-метоксифенил)-3-тиазолидинил) 2-оксо-этил)уреидо)-2- триметилсилилэтилбензоата (2R, 4R) в 80 см3 тетрагидрофурана добавляют при температуре около 25oC 13,7 см3 раствора 1M фторида тетрабутиламмония в тетрагидрофуране. Реакционную смесь перемешивают в течение 12 ч при температуре около 25oC, затем подкисляют с помощью 15 см3 водного раствора 1N серной кислоты. После экстрагирования 3 раза 50 см3 этилацетата, объединенные органические фазы промывают в 60 см3 воды, затем экстрагируют 2 раза 20 см3 водного раствора 1N едкого натра. Водные фазы отделяют, промывают 2 раза в 40 см3 диэтилового эфира, подкисляют до pH 4 водным раствором 4N серной кислоты и экстрагируют 3 раза 50 см3 диэтилового эфира. Органические фазы объединяют, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 30oC. Полученное твердое вещество растворяют в 10 см3 водного раствора 0,5N едкого натра. Полученный раствор фильтруют и подкисляют добавлением 5 см3 водного раствора 1N соляной кислоты. Нерастворимый продукт отфильтровывают 2 раза в 5 см3 воды и высушивают на воздухе. Таким образом, получают 1,48 г 3-(3-2-(4-трет. бутоксикарбонил-2-(3-метоксифенил)-3-тиазолидинил) -2-оксо-этил)уреидо) бензойной-(2R, 4R) кислоты, плавящейся при 125oC, /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 3,8 (s, 3H, OCH3) 6,85 (bd, 1H, O-C6H4-C в позиции 4), 7,2 - 7,6 (m, 6H ароматические), 8,0 (bs, 2H, N-C6H4- в позиции 2).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2830, 2600, 1715, 1690, 1655, 1600, 1555, 1490, 1370, 1230, 1150, 1050, 755, 690, 680.
A) 3-(3-(2-(4-Трет.бутоксикарбонил-2-(3-метоксифенил)-3-тиазолидинил)-2- оксо-этил)уреидо)-2-триметилсилилэтилбензоат -(2R, 4R) может быть получен следующим образом: к раствору 3,5 г 2-(3-метоксифенил)-4-тиазолидинтрет.бутилкарбоксилата (2RS, 4R) и 4,0 г 2-(3-(3-(2-триметилсилилэтоксикарбонил)фенил)уреидо) уксусной кислоты в 20 см3 безводного тетрагидрофурана, охлажденному до температуры около 5oC, добавляют за 10 мин. раствор 2,45 г N, N'-дициклогексилкарбодиимида в 10 см3 безводного тетрагидрофурана. Реакционную смесь перемешивают в течение 16 ч при температуре около 25oC. Нерастворимый продукт отфильтровывают и промывают 2 раза в 10 см3 тетрагидрофурана. Фильтрат концентрируют досуха при пониженном давлении и 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 4,85 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(3-(метоксифенил)-3-тиазолидинил) -2-оксо-этил)уреидо-2-триметилсилилэтилбензоата-(2R, 4R) в виде аморфного бежевого порошка, который используется при последующем синтезе. /α/
Б) 2-(3-метоксифенил)-4-тиазолидинтрет.бутилкарбоксилат (2RS, 4R) может быть получен по способу, аналогичному описанному в примере 34, B, но исходя из 14,0 г 2-(3-метоксифенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, 3,0 см3 концентрированной серной кислоты и избытка изобутилена. Полученный сырой продукт очищается хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом получают 11 г 2-(3-метоксифенил)-4-тиазолидинтрет.бутилкарбоксилата (2RS, 4R) в виде масла желтого цвета, смесь (40/60 вес.) изомеров (2R, 4R) и (2S, 4R), которую используют при последующем синтезе.
В) 2-(3-Метоксифенил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена как описано в примере 34, Г, но исходя из 21,2 г L-цистеина и 23 см3 3-метоксибензальдегида. Таким образом, получают 38, г 2-(3-метоксифенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 171oС, которая используется при последующем синтезе.
Г) 2-(3-(3-(2-Триметилсилилэтоксикарбонил(фенил)уреидо)уксусная кислота может быть получена следующим образом: в раствор 24,7 г 3-(3-этоксикарбонилметилуреидо)-2-триметилсилилэтилбензоата в 500 см3 тетрагидрофурана добавляют раствор 4,45 г гидроокиси калия в 160 см3. Реакционную смесь перемешивают в течение 12 ч при температуре около 25oC, затем концентрируют до 150 см3 при пониженном давлении и при 40oC. Полученный раствор разбавляют 200 см3 воды, промывают 2 раза в 50 см3 этилацетата, подкисляют с помощью 68 см3 водного раствора 1N соляной кислоты и экстрагируют 3 раза 100 см3 диэтилового эфира. Органические фазы объединяют, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении. Получают после кристаллизации в окиси диизопропила 17,2 г 2-(3-(3-(2-триметилсилилэтоксикарбонил)фенил)уреидо) уксусной кислоты, плавящейся при 173oC.
Д) 3-(3-Этоксикарбонилметилуреидо)-2-триметилсилилэтилбензоат может быть получен следующим образом: в раствор 29,5 г 2-(1-имидазолилкарбоксамидо)-этилацетата и 36,2 г 3-амино-2-триметилсилилэтилбензоата в 1000 см3 толуола перемешивают с обратным холодильником в течение 3 ч. После охлаждения до температуры около 25oC реакционную смесь промывают 3 раза в 50 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (40/60 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 24,7 г 3-(3-этоксикарбонилметилуреидо)-2-триметилсилилэтилбензоата в виде вязкого масла желтого цвета, используемого при последующем синтезе.
E) 2-(1-Имидазолилкарбоксамидо)-этилацетат может быть получен следующим образом: в раствор 50,0 г N,N'-диимидазол-карбонила и 43 см3 триэтиламина в 800 см3 безводного тетрагидрофурана добавляют в течение 2 ч 42,3 г хлоргидрата 2-амино-этилацетата. Смесь перемешивают в течение 24 ч при температуре около 25oC, затем нерастворимый продукт отфильтровывают и промывают 2 раза в 50 см3 тетрагидрофурана. Фильтрат концентрируют досуха при пониженном давлении и 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 36,7 г 2-(1-имидазолилкарбоксамидо)-этилацетата, плавящегося при +103oC.
Ж) 3-Амино-2-триметилсилилэтилбензоат может быть получен следующим образом: в раствор 42,7 г 3-нитро-2-триметилсилил-этилбензоата в 600 см3 этанола добавляют 2,1 г 5%-ного палладия на саже. Суспензию перемешивают в течение 2 ч при температуре около 25oC в атмосфере водорода (100 кПа). Затем катализатор отфильтровывают, а фильтр концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 36,2 г 3-амино-2-триметилсилил-этилбензоата в виде масла, используемого при последующем синтезе.
З) 3-Нитро-2-триметилсилил-этилбензоат может быть получен следующим образом: в раствор 66,0 г 3-нитро-бензоилхлорида в 1000 см3 1,2 дихлорэтана, охлажденный до 5oC, добавляют раствор 41,8 г 2-триметилсилил-этанола в 200 см3 1,2-дихлорэтана и 51 см3 триэтиламина. Реакционную смесь перемешивают в течение 12 ч при температуре около 25oC, затем нерастворимый продукт отфильтровывают и промывают 2 раза в 50 см3 1,2-дихлорэтана. Фильтрат концентрируют досуха при пониженном давлении и 40oC. Полученный масляный остаток очищается хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 76,9 г 3-нитро-2-триметилсилил-этилбензоата в виде масла, используемого при последующем синтезе.
Пример 42. По способу, описанному в примере 41, но исходя из 2,3 г 3-(3-(2-(4-трет-бутоксикарбонил-2-(2-фтор-фенил)-3- тиазолидинил)2-оксо-этил)уреидо)2-триметилсилил-этилбензоата- (2R, 4R) и 7,6 см3 1М раствора фторида тетрабутиламмония. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-метанол (90/10 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают после встряхивания в окиси диизопропила: 0,4 г 3-(3-(2-(4-трет-бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил)-1- оксо-этил)уреидо)бензойной-(2R, 4R)-кислоты в виде аморфного твердого тела, /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 7,2-7,6 (m, 6H ароматические), 7,9 (bt, 1H, F-C6H4- в позиции 3, JHF = 9 Гц), 8,0 (bs, 1H, N-C6H4 в положении 2).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2980, 2930, 2600, 1735, 1690, 1655: 1610, 1590, 1555, 1490, 1455, 1370, 1230, 1150, 760, 680.
3-(3-(2-(4-Трет-бутоксикарбонил-2-(2-фтор-фенил)-3- тиазолидинил)-2-оксо-этил)уреидо)2-триметилсилил-этилбензоат-(2R, 4R) может быть получен по способу, аналогичному, описанному в примере 41, А, но исходя из 3,35 г 2-(2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 4,0 г 2-(3-(3-(2-триметилсилил-этоксикарбонил)фенил)уреидо)уксусной кислоты и 2,45 г N, N'-дициклогексилкарбодиида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 2,4 г 3-(3-(2-(4-трет-бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил)-2- оксоэтил)уреидо-2-триметилсилил-этилбензоата-(2R, 4R) в виде аморфного твердого вещества бежевого цвета, используемого при последующем синтезе, /α/
Пример 43. По способу, описанному в примере 41, но исходя из 1,6 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(3-фтор-фенил)-3-тиазолидинил)-2- оксо-этил)уреидо-2-триметилсилил-этилбензоата-(2R, 4R) и 5,3 см3 1М раствора фторидатетрабутиламмония. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат метанол (90/10 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают после встряхивания в окиси диизопропила, 0,22 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(3-фтор-фенил)-3-тиазолидинил)-2- оксо-этил)уреидо) бензойной (2R, 4R) кислоты в виде аморфного твердого вещества, /α/
Протонный ЯМР (250 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, характеристические химические сдвиги при 25oC: 1,5 (bs, 9H, (CH3)3), 7,1-7,6 (m, 7H ароматические), 8,0 (bs, 1H, N-C6H4- в позиции 2).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2600, 1715, 1695, 1650, 1610, 1590, 1560, 1490, 1450, 1370, 1240, 1150, 780, 755, 685.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(3-фтор-фенил)-3- тиазолидинил)-2-оксо-этил)уреидо)2-триметилсилил-этилбензоат (2R, 4R) может быть получен способом, аналогичным описанному в примере 41, А, но исходя из 1,68 г 2-(3-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 2,0 г 2-(3-(3-(2-триметилсилил-этоксикарбонил)фенил)уреидо)уксусной кислоты и 1,23 г N, N'-дициклогексилкабодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,6 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(3-фтор-фенил)-3- тиазолидинил)-2-оксо-этил)-уреидо)2-триметилсилил-этилбензоата-(2R, 4R) в виде аморфного твердого вещества бежевого цвета, используемого при последующем синтезе, /α/
2-(3-Фтор-фенил)-4-тиазолидинтрет.бутилкарбоксилата-(2RS, 4R) может быть получен способом, аналогичным описанному в примере 34, В, но исходя из 11,4 г 2-(3-фтор-фенил)-4-тиазолидинбутилкарбоновой-(2RS, 4R) кислоты, растворенной в 200 см3 хлороформа, 3,0 см3 концентрированной серной кислоты и избытка изобутилена. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 10,3 г 2-(3-фтор-фенил)-4-тиазолидинтрет.бутилкарбоксилата-(2RS, 4R) в виде масла желтого цвета, смесь (30/70 вес.) изомеров (2R, 4R), и (2S, 4R), которое используется при последующем синтезе.
2-(3-Фтор-фенил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как описано в примере 34, Г, но исходя из 21,2 г L-цистеина и 20 см3 3-фтор-бензальдегида. Таким образом, получают 30,2 г 2-(3-фтор-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 178oC, используемой при последующем синтезе.
Пример 44. По способу, описанному в примере 41, но исходя из 2,0 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(4-фтор-фенил)-3-тиазолидинил)2-оксо-этил) уреидо)-2-триметилсилил-этилбензоата (2R, 4R) и 6,6 см3 1М раствора фторида тетрабутиламмония. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают после встряхивания в оксиде диизопропила 0,74 г 3-(2-(2-(4-трет.бутоксикарбонил-2-(4-фтор-фенил)-3-тиазолидинил)-1-оксо-этил) уреидо) бензойной-(2R, 4R) кислоты, плавящейся при 154oC, /α/
Протонный ЯМР (250 МГц, ДМСО Д6, δ в ч/мин), 2 ротамера при комнатной температуре, характеристические химические сдвиги при 25oC: 1,5 (bs, 9H, (CH3)3), 7,1-7,8 (m, 7H ароматические), 8,0 (bs, 1H, N-C6H4 - в позиции 2).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2985, 2945, 2625 ,1695, 1650, 1610, 1560, 1510, 1490, 1375, 1235, 1155, 800, 760, 685.
3-(3-(2-(4-Трет.бутоксикарбонил-2-(4-фтор-фенил)-3-тиазолидинил)- 2-оксо-этил)уреидо)-2-триметилсилил-этилбензоат (2R, 4R) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 1,68 г 2-(4-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 2,0 г 2-(3-(3-(2-триметилсилил-этоксикарбонил) фенил) уреидо) уксусной кислоты и 1,23 г N, N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 2,0 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(4-фтор-фенил)-3-тиазолидинил)-2-оксоэтил) уреидо-2-триметилсилил-этилбензоата (2R, 4R) в виде аморфного твердого вещества бежевого цвета, используемого при последующем синтезе, /α/
2-(4-Фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилат-(2RS, 4R) может быть получен способом, аналогичным описанному в примере 34, B, но исходя из 11,4 г 2(4-фтор-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, 3,0 см3 концентрированной серной кислоты и избытка изобутилена. Полученный сырой продукт очищают хроматографией на силикагеле (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 10,0 г 2-(4-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R) в виде масла желтого цвета, смесь (40/60 вес.) изомеров (2R, 4R) и (2S, 4R), которую используют при последующем синтезе.
2-(4-Фтор-фенил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как описано в примере 34, Г, но исходя из 21,2, г L-цистеина и 20 см3 4-фтор-бензальдегида. Таким образом, получают 34,3 г 2-(4-фтор-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 186oC, используемой при последующем синтезе.
Пример 45. По способу, описанному в примере 41, но исходя из 2,0 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(2-хлор-фенил)-3-тиазолидинил) 2-оксо-этил)уреидо)-2-триметилсилил-этилбензоата-(2R, 4R) и 6,5 см3 1M раствора фторида тетрабутиламмония. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-метанол (90/10 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают после встряхивания в оксиде диизопропила, 0,65 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(2-хлор-фенил)-3-тиазолидинил) 2-оксо-этил) уреидо) бензойной-(2R, 4R) кислоты в виде аморфного твердого тела, /α/
Протонный ЯМР (200 МГц, ДМСО Д6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 7,3-7,6 (m, 7H ароматические), 7,95 (bs, 1H, N-C6H4- в позиции 2).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3390, 2985, 2940, 2600, 1735, 1660, 1610, 1595, 1560, 1375, 1240, 1155, 755, 685.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(2-хлор-фенил)-3- тиазолидинил) 2-оксо-этил) уреидо)-2-триметилсилил-этилбензоат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 1,6 г 2-(2-хлор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 2,0 г 2-(3-(3-(2-триметилсилил-этоксикарбонил)-фенил)уреидо) уксусной кислоты и 1,23 г N, N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 2,0 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(2-хлор-фенил)-3-тиазолидинил)-2- оксоэтил)уреидо)-2-триметилсилил-этилбензоата-(2R, 4R) в виде аморфного твердого вещества бежевого цвета, используемого при последующем синтезе, /α/
2-(2-Хлор-фенил)-4-тиазолидинтрет. бутилкарбоксилат-(2RS, 4R) может быть получен способом, аналогичным описанному в примере 34, B, но исходя из 12,2 г 2-(2-хлор-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, 3,0 см3 концентрированной серной кислоты и избытка изобутилена. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 11,0 г 2-(2-хлор-фенил)-4-тиазолидинтрет. бутилкарбоксилата (2RS, 4R) в виде масла желтого цвета, смесь (40/60 вес.) изомеров (2R, 4R) и (2S, 4R), которую используют при последующем синтезе.
2-(2-Хлор-фенил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 21,2 г L-цистеина и 25,8 г 2-хлор-бензальдегида. Таким образом, 21,9 г 2-(2-хор-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 144oC, используемой при последующем синтезе.
Пример 46. По способу, описанному в примере 41, но исходя из 4,0 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(3-гидрокси-фенил)-3- тиазолидинил)-2-оксо-этил)уреидо)-2-триметилсилил-этилбензоата (2R, 4R) и 11,8 см3 1М раствора хлорида тетрабутиламмония. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Твердый остаток растворяют в 8,0 см3 водного раствора едкого натрия 0,5 N. Раствор отфильтровывают и подкисляют с помощью 4,1 см3 водного раствора 1N соляной кислоты. Нерастворимый продукт отфильтровывают, промывают 2 раза в 5 см3 воды и высушивают на воздухе. Таким образом, получают 0,65 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(3-гидроксифенил)-3-тиазолидинил)-2- оксо-этил)уреидо)бензойной (2R, 4R) кислоты, плавящейся при 150oC. /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC: 6,8 (bd, 1H, O-C6H4-C в позиции 4), 7,0-7,6 (m, 6H ароматические), 8,0 (bs, 1H, N-C6H4-в позиции 2).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2625, 1700, 1650, 1590, 1560, 1490, 1370, 1235, 1150, 785, 760, 690, 680.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(3-гидрокси-фенил)-3- тиазолидинил) 2-оксо-этил)уреидо)-2-триметилсилил-этилбензоат- (2R, 4R) может быть получен способом, аналогичным описанному в примере 41, А, но исходя из 1,66 г 2-(3-гидрокси-фенил)-4- тиазолидинтрет.бутилкарбоксилата-(2RS, 4R), 2,0 г 2-(3-(3-(2-триметилсилил-этоксикарбонил)фенил)уреидо) уксусной кислоты и 1,33 г N, N'-дициклогексилкарбодиимида. Таким образом, получают 4,0 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(3-гидрокси- фенил)-3-триазолидинил)-2-оксо-этил)уреидо-2-триметилсилил- этилбензоата (2R, 4R) в виде аморфного порошка желтого цвета, используемого при последующем синтезе.
2-(3-Гидрокси-фенил)-4-тизолидинтрет. бутилкарбоксилат-(2RS, 4R) может быть получен способом, аналогичным описанному в примере 34, B, но исходя из 11,3 г 2-(3-гидрокси-фенил)-4-тиазолидинкарбоновой- (2RS, 4R) кислоты, растворенной в 200 см3 хлороформа, 3,0 см3 концентрированной серной кислоты и избытка изобутилена. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат/циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 4,5 г 2-(3-гидрокси-фенил)-4-тиазолидинтрет. бутилкарбоксилата (2RS, 4R) в виде масла желтого цвета, смесь (50/50 вес.) изомеров (2R, 4R) и (2S, 4R), которую используют при последующем синтезе.
2-(3-Гидрокси-фенил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 21,2 г L-цистеина и 22,8 г 3-гидроксибензальдегида. Таким образом получают 37,5 г 2-(3-гидрокси-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 207oC, используемой при последующем синтезе.
Пример 47. По способу, описанному в примере 41, но исходя из 1,3 г 3-(3-(2-(4-трет. бутоксикарбонил-2-фенил-3-тиазолидинил) 2-оксо-этил)уреидо)-2-триметилсилил-этилбензоата (2R, 4R) и 4,5 см3 молярного раствора фторида тетрабутиламмония. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-метанол (95/5 об.)). Фракции, содержащие целевой продукт объединяют и концентрируют досуха при пониженном давлении и 40oC. Твердый остаток растворяют в 4,0 см3 0,5 N водного раствора едкого натра. Раствор отфильтровывают и подкисляют с помощью 2,1 см3 1N водного раствора соляной кислоты. Нерастворимый продукт отфильтровывают, промывают 2 раза в 5 см3 воды и высушивают на воздухе. Таким образом, получают 0,33 г 3-(3-(2-(4- трет. бутоксикарбонил-2-фенил-3-тиазолидинил) 2-оксо-этил)уреидо) бензойной-(2R, 4R) кислоты, плавящейся при 134oC. /α/
Протонный ЯМР (250 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 7,3-7,7 (m, 8H ароматические), 8,0 (bs, 1H, N-C6H4-C в позиции 2).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2980, 2930, 2600, 1690, 1650, 1610, 1590, 1555, 1490, 1370, 1235, 1150, 760, 730, 700, 680.
3-(3-(2-(4-Трет. бутоксикарбонил-2-фенил-3-тиазолидинил) 2-оксоэтил)уреидо)-2-триметилсилил-этилбензоат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 38, но исходя из 1,7 г 3-(2-(1-имидазолил-карбоксамидо)ацетил)-2-фенил-4- тиазолидинтрет.бутилкарбоксилата-(2R, 4R) и 12 г 3-амино-2- триметилсилил-этилбензоат. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,3 г 3-(3-(2-(4-трет. бутоксикарбонил-2-фенил-3-тиазолидинил) 2-оксо-этил)уреидо) 2-триметилсилил-этилбензоата-(2R, 4R) в виде масла оранжевого цвета, используемого при последующем синтезе.
Пример 48. По способу, описанному в примере 41, но исходя из 4,0 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(4-диметиламино-фенил)-3- тиазолидинил)-2-оксо-этил)уреидо)-2-триметилсилил-этилбензоата (2R, 4R) и 4,2 см3 1М раствора фторида тетрабутиламмония. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-метанол (90/10 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, после встряхивания в окиси диизопропила получают 0,35 г 2-(3-(2-(4-трет. бутоксикарбонил-2-(4-диметиламин-фенил)-3- тиазолидинил (2-оксо-этил)уреидо) бензойной-(2R, 4R) в виде аморфного твердого продукта, /α/
Протонный ЯМР (200 МГц, DMCO D6,δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 6,7 (bd, 2H, (Me)2) N-C6H4 в позиции 3 и 5), 7,3 - 7,6 (m, 5H, ароматические), 8,0 (bs, 1H, N-C6H4 в позиции 2).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2805, 2600, 1715, 1690, 1650, 1610, 1555, 1520, 1485, 1365, 1230, 1150, 815, 760, 685.
А) 3-(3-(2-(4-Трет. бутоксикарбонил-2-(4-диметиламин-фенил)- 3-тиазолидинил)-2-оксо-этил)уреидо)-2-триметилсилил-этилбензоат (2R, 4R) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 1,66 г 2-(4-диметиламинофенил)-4- тиазолидинтрет.бутилкарбоксилата (2RS, 4R), 2,0 г 2-(3-(3-(2-триметилсилилэтоксикарбонил)фенил)уреидо)уксусной кислоты и 1,23 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,3 г 3-(3-(2-(4-трет.бутоксикарбонил-2- (4-диметиламин-фенил)-3-тиазолидинил)-2-оксо-этил)уреидо)-2- триметилсилил-этилбензоата-(2R, 4R) в виде аморфного твердого вещества бежевого цвета, используемого при последующем синтезе, /α/
Б) 2-(4-диметиламинофенил)-4-тиазолидинтрет.бутилкарбоксилат- (2RS, 4R) может быть получен следующим образом: в раствор 8,1 г 3-трет.бутоксикарбонил-2-(4-диметиламинофенил)-4- тиазолидинтрет.бутилкарбоксилата (2RS, 4R) в 100 см3 хлороформа прикапывают 2,95 см3 иодтриметилсилана при температуре около 25oC. Реакционную смесь перемешивают в течение 18 ч при этой температуре, затем добавляют 50 см3 воды. Органическую фазу отделяют, промывают 2 раза в 30 см3 насыщенного водного раствора кислого карбоната натрия и 2 раза в 30 см3 воды, затем экстрагируют 3 раза с помощью 30 см3 водяного раствора соляной кислоты 1N. Водные фазы объединяют, промывают 2 раза в 20 см3 дихлорметана, доводят до pH 7 водным раствором едкого натрия 4N и экстрагируют 3 раза с помощью 30 см3 дихлорметана. Органические фазы объединяют и высушивают над сульфатом магния и концентрируют досуха при небольшом давлении и при 40oC. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: окись диизопропила). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 2,3 г 2-(4-диметиламинофенил)-4-тиазолидин- трет. бутилкарбоксилата-(2RS, 4R), плавящийся при 142oC, смесь (50/50 вес.) изомеров (2R, 4R) и (2S, 4R), используемую при последующем синтезе.
В) 3-трет.бутоксикарбонил-2-(4-диметиламинофенил)-4- тиазолидинтрет.бутилкарбоксилат (2RS, 4R) может быть получен следующим образом: в раствор 14,8 г 3-трет. бутоксикарбонил-2- (4-диметиламинофенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты и 16,0 г паратолуолсульфонилхлорида в 70 см3 безводного пиридина, охлажденный до температуры приблизительно 0oC, вводят 3,1 г трет. бутанола. После возврата к температуре 20oC перемешивают еще в течение 20 ч, затем смесь выливают в 200 см3 воды и экстрагируют 3 раза 100 см3 этилацетата. Объединенные органические фазы промывают последовательно 2 раза в 100 см3 воды, 2 раза в 100 см3 водного раствора едкого натрия 1N и 2 раза в 100 см3 воды, затем высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 40oC. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: окись диизопропила). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при температуре около 45oC. Таким образом, получают 8,1 г 3-трет. бутоксикарбонил-2- (4-диметиламинофенил)-4-тиазолидинтрет.бутил карбоксилата (2RS, 4R) в виде масла оранжевого цвета, которое используется при последующем синтезе.
Г) 2-Трет. бутоксикарбонил-2-(4-диметиламинофенил)-4- тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена следующим образом: в раствор 12,6 г 2-(4-диметиламинофенил)-4- тиазолидинкарбоновой-(2RS, 4R) кислоты, 5,25 г карбоната натрия в 100 см3 воды прикапывают, перемешивая, 10,9 г дикарбоната дитрет. бутила, растворенного в 50 см3 диоксана. Реакционную смесь перемешивают в течение 20 ч при температуре около 20oC, затем образовавшийся осадок отфильтровывают. Фильтрат промывают 2 раза в 100 см3 этилацетата, подкисляют до pH 4 водным раствором соляной кислоты 4N, затем экстрагируют 3 раза 100 см3 этилацетата. Объединяют органические фазы промывают 2 раза в 50 см3, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают после кристаллизации в окиси диизопропила, 14,8 г 3-трет.бутоксикарбонил-2-(4-диметиламинофенил)-4-тиазолидинкарбоновой- (2RS, 4R) кислоты, плавящейся при 175oC.
2-(4-Диметиламинофенил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 21,2 г L-цистеина и 27,5 г 4-диметиламинобензальдегида. Таким образом, получают 41,5 г 2-(4-диметиламинофенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 184oC, используемой при последующем синтезе.
Пример 49. По способу, описанному в примере 41, но исходя из 0,95 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(3-пиридил)-3- тиазолидинил)-2-оксо-этил)уреидо)-2-триметилсилил-этилбензоата- (2R, 4R) и 3,2 см3 1М раствора фторида тетрабутиламмония. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают, после встряхивания в окиси диизопропила, 0,36 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(3-пиридил)-3-тиазолидинил)-2- оксо-этил)уреидо)бензойной-(2R, 4R) кислоты, плавящейся при 147oC, /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линии при 120oC, характеристические химические сдвиги при 120o: 7,3 - 7,6 (m, 5H ароматические), 8,0 (bs, 1H, N-C6H4-C в позиции 2), 8,5 (bs, 1H, H в позиции 6 пиридила).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3385, 2975, 2930, 2600, 2500, 1730, 1660, 1610, 1590, 1560, 1485, 1370, 1240, 1150, 760, 710, 685.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(3-пиридил)-3-тиазолидинил)- 2-оксо-этил)уреидо)-2-триметилсилил-этилбензоат-(2R, 4R) может быть получен способом, аналогичным описанному в примере, 41, A, но исходя из 2,0 г 2-(3-пиридил)-4-тиазолидинтрет.бутилкарбоксилата-(2RS, 4R), 2,5 г 2-(3-(3-(2-триметилсилил-этоксикарбонил)фенил)уреидо) уксусной кислоты и 1,8 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (40/60 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,0 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(3-пиридил)-3-тиазолидинил)- 2-оксо-этил)уреидо)-2-триметилсилил-этилбензоата (2R, 4R) в виде масла желтого цвета, используемого при последующем синтезе.
2-(3-Пиридил)-4-тиазолидинтрет.бутилкарбоксилат (2RS, 4R) может быть получен способом, аналогичным описанному в примере 43, Б, но исходя из 3,4 г 3-трет. бутоксикарбонил-2-(3-пиридил)-4- тиазолидинтрет. бутилкарбоксилата (2RS, 4R) и 1,7 см3 иодтриметилсилана. Таким образом, получают 2,1 г 2-(3-пиридил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R) в виде масла желтого цвета, используемого при последующем синтезе.
3-Трет. бутоксикарбонил-2-(3-пиридил)-4- тиазолидинтрет.бутилкарбоксилат (2RS, 4R) может быть получен способом, аналогичным описанному в примере 48, В, но исходя из 9,0 г 3-трет.бутоксикарбонил-2-(3-пиридил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, 5,7 г паратолуолсульфонилхлорида и 2,2 г трет-бутанола. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (50/50 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,25 г 3-трет.бутоксикарбонил-2-(3-пиридил)-4-тиазолидинтрет. бутилкарбоксилата (2RS, 4R) в виде масла оранжевого цвета, используемого при последующем синтезе.
3-Трет. бутоксикарбонил-2-(3-пиридил)-4-тиазолидинкарбоновая- (2RS, 4R) кислота может быть получена способом, аналогичным описанному в примере 48, Г, но исходя из 26,5 г 2-(3-пиридил)-4- тиазолидинкарбоновой-(2RS, 4R) кислоты, 130 см3 1N раствора едкого натра и 28,4 г дикарбоната дитрет.бутила. Таким образом, получают 31,6 г 3-трет.бутоксикарбонил-2-(3-пиридил)-4- тиазолидинкарбоновой-(2RS, 4R) кислоты в виде аморфного порошка желтого цвета, используемого при последующем синтезе.
2-(3-Пиридил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 25,0 г L-цистеина и 22 см3 никотинальдегида. Таким образом, получают 26,6 г 2-(3-пиридил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 149oC, используемой при последующем синтезе.
Пример 50. По способу, описанному в примере 39, но исходя из 2,5 г 3-(3-(3-(2-(4-трет. бутоксикарбонил-2-фенил-3-тиазолидинил)-2- оксо-этил)уреидо)фенил)этилпропионата-(2R, 4R) и 0,2 г гидроокиси лития. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-метанол (90/10 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 0,25 г 3-(3-(3-(2- (4-трет.бутоксикарбонил-2-фенил-3-тиазолидинил)-2- оксо-этил)уреидо)фенил)этилпропионовой-(2R, 4R) кислоты в виде аморфного твердого вещества бежевого цвета, /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, характеристические химические сдвиги при 25oC: 1,5 (bs, 9H, (CH3)3), 2,5 (bt, 2H, CH2), 2,8 (bt, 2H, CH2), 6,8 (bd, 1H, N-C6H4-C в позиции 4), 7,1 - 7,5 (m, 7H, ароматические) 7,7 (bd, 2H, ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2620, 1730, 1645, 1610, 1590, 1555, 1490, 1370, 1235, 1150, 785, 730, 695.
3-(3-(3-(2-(4-Трет.бутоксикарбонил-2-фенил-3-тиазолидинил)- 2-оксо-этил)уреидо)фенил)этилпропионат-(2R, 4R) может быть получен способом, аналогичным описанному в примере, 38, но исходя из 3,75 г 3-(2-(1-имидазолил-карбоксамидо)ацетил) 2-фенил-4- тиазолидинтрет.бутилкарбоксилата-(2R, 4R) и 3,5 г 3-(3-амино-фенил)этилпропионата. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об. )). Фракции, содержащие целевой продукт, объединяют, затем концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 2,5 г 3-(3-(2-(4-трет.бутоксикарбонил-2-)- фенил-3-тиазолидинил)-2-оксо-этил)уреидо)фенил)этилпропионата (2R, 4R) в виде масла, используемого при последующем синтезе.
Пример 51. По способу, описанному в примере 39, но исходя из 2,4 г 3-(3-(2-(4-трет.бутоксикарбонил-2-фенил-3-тиазолидинил)-2-оксо- этил)уреидо)феноксиэтилацетата-(2R, 4R) и 0,2 г гидрокиси лития. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают, после встряхивания в окиси диизопропила, 1,3 г 3-(3-(2-(4-трет.бутоксикарбонил-2-фенил-3-тиазолидинил) -2-оксо-этил)уреидо)феноксиуксусной -(2R, 4R) кислоты в виде аморфного твердого вещества, /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 4,5 (bs, 2H, OCH2CO2), 6,5 (ddd, 1H, N-C6H4-C в позиции 4), 6,9 (d, 1H ароматический), 7,1-7,7 (m, 7H ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2630, 2550, 1735, 1640, 1600, 1555, 1490, 1370, 1240, 1150, 770, 730, 695.
3-(3-(2-(4-Трет. бутоксикарбонил-2-фенил-3-тиазолидинил)- 2-оксоэтил)уреидо)феноксиэтилацетат (2R, 4R) может быть получен способом, аналогичным описанному в примере 38, но исходя из 4,4 г 3-(2-(1-имидазолил-карбоксамидо)ацетил)2-фенил- 4-тиазолидинтрет.бутилкарбоксилата (2R, 4R) и 4,1 г 3-аминофеноксиэтил ацетата. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (35/65 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 2,5 г 3-(3-(2-(4-трет.бутоксикарбонил-2-фенил- 3-тиазолидинил)2-оксо-этил)уреидо)феноксиэтилацетата-(2R, 4R) в виде аморфной пудры бежевого цвета, используемой при последующем синтезе.
Пример 52. В раствор 2,5 г 3-(2-(3-(3-ацетил-фенил)уреидо)ацетил)2- фенил-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в 10 см3 метанола и 5 см3 пиридина добавляют 0,4 г хлоргидрата гидроксиамина, растворенного в 5 см3 воды. Реакционную смесь перемешивают с обратным холодильником в течение 2 ч. После выпаривания растворителей при пониженном давлении и при 45oC, остаток извлекают с помощью 100 см3 этилацетата. Органическую фазу промывают 3 раза в 50 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат/циклогексан (50/50 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Получают, после встряхивания в окиси диизопропила, 0,5 г 3-(2-(3-(3-(1-гидроксиимидо-этил) фенил-(E)/уреидо)ацетил) 2-фенил 4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в виде аморфного твердого тела, /α/
Протонный ЯМР (300 МГц, DMCO D6, δ/ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 2,1 (s, 3H, CH3), 7,2-7,6 (m, 8H ароматические), 7,7 (bs, 1H, N-C6H4-C в позиции 2), 10,8 (bs, 1H, NOH). Эффект NOE был отмечен между метилом при 2,1 ч/млн и оксимом NOH при 10,8 ч/млн.
Инфракрасный спектр (KBr), характерные полосы в см-1: 3350, 2975, 2930, 1740, 1650, 1610, 1590, 1560, 1370, 1240, 1150, 1005, 790, 730, 695.
3-(2-(3-(3-Ацетил-фенил)уреидо)ацетил) 2-фенил-4-тиазолидинтрет.бутилкарбоксилат (2R, 4R) может быть получен способом, аналогичным описанному в примере 38, но исходя из 3,75 г 3-(2-(1-имидазолил-карбоксамидо)ацетил) 2-фенил-4-тиазолидинтрет. бутилкарбоксилата (2R, 4R) и 1,46 г 3-аминоацетофенона. Полученный масляный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (50/50 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 2,5 г 3-(2-(3-(3-ацетил-фенил)уреидо)ацетил) 2-фенил-4-тиазолидинтрет. бутил карбоксилата (2R, 4R) в виде аморфного порошка желтого цвета, используемого при последующем синтезе.
Пример 53. По способу, описанному в примере 38, но исходя из 2,3 г 3-(2-(1-имидазолил-карбоксамидо)ацетил) 2-фенил-4-тиазолидинтрет. бутилкарбоксилата (2R, 4R) и 4,7 г 3-амино-бензилсульфоната тетра-н-бутил-аммония. Полученный сырой продукт растворяют в 20 см3 ацетона и добавляют раствор 1,86 г нонафторбутансульфоната калия в 20 см3 ацетона. Реакционную среду перемешивают в течение 18 ч при температуре около 25oC, затем добавляют 80 см3 окиси диизопропила. Нерастворимый продукт отфильтровывают, промывают 2 раза в 3 см3 смеси ацетона и окиси диизопропила (30/70 об.). Таким образом, получают после встряхивания в ацетонитриле, 0,75 г 3-(3-(2-(4-трет.бутоксикарбонил-2-фенил-3-тиазолидинил) 2-оксо-этил)уредино)бензилсульфоната калия-(2R, 4R), плавящегося при 185oC, /α/
Протонный ЯМР (300 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 3,7 (AB, 2H, CH2SO3), 6,9 (bd, 1H, N-C6H4-C в позиции 6), 7,1-7,6 (m, 8H ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3400, 2980, 2945, 1740, 1660, 1615, 1600, 1560, 1500, 1375, 1220, 1200, 1160, 1050, 800, 735, 700.
3-Амино-бензилсульфонат тетра-н-бутил аммония может быть получен способом, аналогичным описанному в примере 41, Ж, но исходя из 11,6 г 3-нитробензилсульфоната тетра-н-бутиламмония и 0,3 г 5%-ного палладия на саже. Таким образом, получают 10,5 г 3-аминобензилсульфоната тетра-н-бутиламмония в виде масла, используемого при последующем синтезе.
3-Нитро-бензилсульфонат тетра-н-бутиламмония может быть получен следующим образом: к 800 см3 водного раствора гидрогенфосфата калия 0,5 М добавляют 6,9 г 3-нитробензилсульфоната натрия, затем 9,9 г кислого сульфоната тетра-н-бутиламмония. Смесь экстрагируют с помощью 500 см3 метиленхлорида. Органическую фазу высушивают над сульфатом магния и концентрируют досуха при небольшом давлении и 40oC. Таким образом, получают 13 г 3-нитробензилсульфоната тетра-н-бутиламмония в виде масла, используемого при последующем синтезе.
3-Нитро-бензилсульфонат натрия может быть получен по известному методу (PURGOTTI et MONTI, Gazz. Chim. Ital., 30, 11, 247).
Пример 54. Действуют по способу примера 39, но исходя из 1,5 г 2-(3-(3-(2-(4-трет. бутоксикарбонил-2-фенил-3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уредо)фенил)метилпропионата-(RS) и 0,11 г гидроокиси лития. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат). Фракции, содержащие целевой продукт, после встряхивания в окиси диизопропила, 0,25 г 2-(3-(3- (4-трет.бутоксикарбонил-2-фенил-3-тиазолидинил-(2R, 4R)2-оксо-этил) уреидо)фенил)пропионовой-(RS) кислоты в виде аморфного твердого тела.
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 1,4 (d, 3H, CH3), 3,5 (bm, 1H, Ph-CH-CO2), 6,9 (bd, 1H, N-C6H4-C в позиции 4), 7,1-7,4 (m, 6H ароматические), 7,7 (bd, 2H ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3385, 2975, 2935, 1735, 1650, 1610, 1560, 1495, 1455, 1370, 1240, 1155, 785, 730, 700.
2-(3-(3-(2-(4-Трет. бутоксикарбонил-2-фенил-3-тиазолидинил- (2R, 4R))-2-оксо-этил)уреидо)фенил)метилпропионат-(RS) может быть получен способом, аналогичным описанному в примере 38, но исходя из 3,75 г 3-(2-(1-имидазолил-карбоксамидо)ацетил)-2-фенил-4- тиазолидинтрет. бутилкарбоксилата-(2R, 4R) и 3,22 г 2-(3-аминофенил)метилпропионата (RS). Полученный масляный остаток очищают хроматографией на силикагеле (элюент: этилацетат), а фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 3,1 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-фенил-3-тиазолидинил- (2R, 4R) 2-оксо-этил)уреидо)фенилметилпропионата(RS) в виде аморфного порошка бежевого цвета, который используется при последующем синтезе.
2-(3-Амино-фенил)метилпропионат-(RS) может быть получен способом, аналогичным описанному в примере 41, Ж, но исходя из 4 г 3-(3-нитро-фенил)метилпропионата-(RS) и 0,3 г 5% палладия на саже. Таким образом, получают 3,3 г 2-(3-амино-фенил)метилпропионата-(RS) в виде масла, используемого при последующем синтезе.
3-(3-Нитро-фенил)метилпропионат-(RS) может быть получен следующим способом: в течение 3 ч барботируют газообразную соляную кислоту в растворе 5 г 2-(3-нитро-фенил)пропионитрила-(RS) в 40 см3 метанола. Полученную смесь перемешивают с обратным холодильником в течение 30 мин, нерастворимый продукт отфильтровывают. Фильтрат концентрируют досуха при пониженном давлении и 40oC. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: петролейный эфир-этилацетат (80/20 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 4 г 3-(3-нитро-фенил)-метилпропионата-(RS) в виде масла, используемого при последующем синтезе.
2-(3-Нитро-фенил)пропионитрил-(RS) может быть получен по известному способу (E. Felder et coll., J. Med. Chem., 13, 559, 1970).
Пример 55. По способу, описанному в примере 41, А, но исходя из 1,7 г 2-(4-фтор-фенил) 4-тиазолидин-трет.бутилкарбоксилата-(2R, 4R), растворенного в 20 см3 безводного ацетонитрила, 1,24 г N,N'-дициклогексилкарбодиимида и 1,3 г 2-(2-индолил-крабоксамидо)-уксусной кислоты в смеси 40 см3 безводного ацетонитрила и 40 см3 дихлорметана, после кристаллизации в окиси диизопропила, 1,7 г 2-(4-фтор-фенил)-3-(2-(2-индолил-карбоксамидо)ацетил/4- тиазолидинтрет.бутилкарбоксилата-(2R, 4R), плавящегося при 176oC.
2-(2-Индолил-карбоксамидо)уксусная кислота может быть получена по известному способу (J.R. Johnson et coll., J. Am. Chem. Soc., 69, 2370, 1947).
Пример 56. По способу, описанному в примере 1, но исходя из 1,2 г N-метил-N-фенил-5-фенил-пролинамида-(2RS, 4SR), 0,94 г 2-(3-(3-метил-фенил)уреидо)уксусной кислоты в суспензии с 25 см3 1,2-дихлор-этана и 0,33 см3 сульфинилхлорида, получают после рекристаллизации в ацетонитриле, 1 г N-метил-1-(3-(3-метил-фенил)уреидоацетил)-N-фенил-5-фенилпролинамида- (2RS, 4SR), плавящегося при 210oC.
N-метил-N-фенил-5-фенилпролинамид-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 25, А, но исходя из 2,2 г 1-(трет.бутоксикарбонил)-N-метил-N-фенил-5-фенилпролинамида- (2RS, 5SR) в 25 см3 безводного хлороформа и 0,85 см3 иодотриметилсилана. Получают 1,2 г N-метил-N-фенил-5-фенилпролинамида-(2RS, 5SR) в виде масла оранжевого цвета, используемого при последующем синтезе.
А) 1-(трет. бутоксикарбонил)-N-метил-N-фенил-5-фенилпролинамид- (2RS, 5SR) может быть получен следующим образом: в суспензию 2,9 г 1-(трет.бутоксикарбонил)-5-фенилпролинамид-(2RS, 5SR) и 1,1 см3 N-метиланилина в 25 см3 безводного ацетонитрила прикапывают при температуре около 0oC раствор 2,1 г N,N'-дициклогексилкарбодиимида в 15 см3 безводного ацетонитрила. Реакционную среду перемешивают в течение 30 мин при температуре около 0oC, затем в течение 20 ч при температуре около 20oC. Затем нерастворимый продукт отфильтровывают и промывают 2 раза в 20 см3 дихлорметана. Фильтрат концентрируют досуха при пониженном давлении и температуре около 40oC. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: дихлорметан-метанол (99/1 в растворе)). Фракции, содержащие целевой продукт, объединяют, затем концентрируют досуха при пониженном давлении. Таким образом, получают после кристаллизации в пентане 2,2 г 1-(трет.бутоксикарбонил)-N-метил-N-фенил-5- фенилпролинамина-(2RS, 5SR), плавящегося при 132oC.
Пример 57. По способу, описанному в примере 1, но исходя из 3 г 5-фенил-(1-, 2,3,4-тетрагидро-1-хинолин)-2-карбонилпирролидина (2RS, 5SR), 2 г 2-(3-(3-метил-фенил)уреидо)уксусной кислоты в суспензии с 100 см3 1,2-дихлорэтана и 0,7 см3 сульфинилхлорида, получают после перекристаллизации в ацетонитриле 1,5 г 1-(2-(3-(3-метил-фенил)уреидо)ацетил)-5-фенил-(1,2,3,4- тетрагидро-1-хинолил)-2-карбонилпирролидина-(2RS, 5SR), плавящегося при 158oC.
5-Фенил-(1,2,3,4-тетрагидро-1-хинолин)-2-карбонилпирролидин (2RS, 5SR) может быть получен способом, аналогичным описанному в примере 25, А, но исходя из 4,1 г 1-(трет.бутоксикарбонил)- 5-фенил-(1,2,3,4-тетрагидро-1-хинолин)-2-карбонилпирролидина (2RS, 5SR) в 50 см3 безводного хлороформа и 1,4 см3 иодотриметилсилана. Получают 3 г 5-фенил-(1,2,3,4-тетрагидро-1-хинолин)-2-карбонилпирролидина- (2RS, 5SR) в виде масла оранжевого цвета, используемого при последующем синтезе.
1-(Трет. бутоксикарбонил)-5-фенил-(1,2,3,4-тетрагидро-1- хинолил)-2-карбонилпирролидин-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 56, А, но исходя из 8,7 г 1-(трет.бутоксикарбонил)-5-фенилпролина-(2RS, 5SR), 3,8 см3 1,2,3,4-тетрагидрохинолеина в 75 см3 безводного ацетонитрила и 6,2 г N,N'-дициклогексилкарбодиимида в 45 см3 безводного ацетонитрила. Получают после кристаллизации в гексане 4,1 г 1-(трет.бутоксикарбонил)-5-фенил-(1,2,3,4-тетрагидро-1-хинолил) -2-карбонилпирролидина-(2RS, 5SR), плавящегося при 132oC.
Пример 58. По способу, описанному в примере 1, но исходя из 1,8 г (3,3-диметилпиперидино)-2-карбонил-5-фенилпирролидина-(2RS, 5SR), 1,3 г 2-(3-(3-метил-фенил)уреидо)уксусной кислоты в 75 см3 1,2-дихлорэтана и 0,5 см3 сульфинилхлорида, получают после перекристаллизации в ацетонитриле 0,7 г (3,3-диметилпиперидино)-2-карбонил-1-(2-(3-метил-фенил)уреидо) ацетил) 5-фенилпирролидина-(2RS, 5SR), плавящегося при 150oC.
(3,3-Диметилпиперидин)-2-карбонил-5-фенил-пирролидин- (2RS, 5SR) может быть получен по способу, аналогичному описанному в примере 25, А, но исходя из 5,0 г 1-(трет.бутоксикарбонил)-(3,3-диметилпиперидино)- -2-карбонил-5-фенилпирролидина-(2RS, 5SR) в 50 см3 безводного хлороформа и 1,9 см3 иодотриметилсилана. Получают кристаллизацией в окиси диизопропила 1,8 г (3,3-диметилпиперидино)-2-карбонил-5-фенилпирролидина- (2RS, 5SR), плавящегося при 163oC.
1-(Трет. бутоксикарбонил)-(3,3-диметилпиперидино)- -2-карбонил-5-фенилпирролидин-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 56, А, но исходя из 5,8 г 1-(трет.бутоксикарбонил)-5-фенилпролина-(2RS, 5SR), 2,6 см3 3,3-диметилпиперидина в 50 см3 безводного ацетонитрила и 4,1 г N,N'-дициклогексилкарбодиимида в 30 см3 безводного ацетонитрила. Получают после кристаллизации в гексане 4 г 1(трет.бутоксикарбонил)-(3,3-диметилпиперидино)-2-карбонил- 5-фенилпирролидина-(2RS, 5SR), плавящегося при 110oC.
Пример 59. По способу, описанному в примере 2, но исходя из 1,9 г 1-(2-амино-ацетил)-5-фенил-N-трет. бутил-пролинамида-(2RS, 5SR), растворенного в 20 см3 безводного тетрагидрофурана и 0,82 см3 изоцианата 3 метил-фенила, получают после перекристаллизации в ацетонитриле 0,7 г N-трет.бутил-1-(3-(3-метилфенил)уреидоацетил)-5-фенилпролинамида- (2SR, 5SR), плавящегося при 169oC.
1-(2-Амино-ацетил)-5-фенил-N-трет.бутил-пролинамид-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 2, A, но исходя из 2,7 г 1-(2-трет. бутоксикарбониламино-ацетил)-N-трет. -бутил-5-фенилпролинамида- (2RS, 5SR), растворенного в 50 см3 безводного хлороформа и 0,95 см3 иодтриметилсилана. Получают 1,9 г 1-(2-аминоацетил)-5-фенил-N-трет.бутил-пролинамида-(2RS, 5SR) в виде аморфного порошка, используемого при последующем синтезе.
Протонный ЯРМ (250 М Гц, DMCO D6, δ в ч/млн), 2 ротамера при комнатной температуре, 1,35 (s, 9H, C(CH3)3, 1,8-2,4 (bm, 4H, CH2-CH2), 2,7, 3,25 и 3,8 (d, 2H, AB, CH2CO2), 4,45 (bm, 1H, CHN), 5,1 (bm, 1H, CHN), 7,2-8 (m, 6H, ароматические и NH).
1-(2-Трет. бутоксикарбониламино-ацетил)N-трет-бутил-5- фенилпролинамид может быть получен способом, аналогичным описанному в примере 2, Б, но исходя из 1,8 г N-трет.бутил-5-фенилпролинамида-(2RS, 5SR), 1,28 г 2-трет.бутокси-карбониламиноуксусной кислоты в 50 см3 безводного ацетонитрила и 1,5 г N, N'-дициклогексилкарбодиимида в 25 см3 безводного ацетонитрила. Получают после кристаллизации в петролийном эфире 2,7 г 1-(2-трет.бутоксикарбониламиноацетил)-N-трет.бутил-5-фенилпролинамида, плавящегося при 117oC.
N-трет. бутил-5-фенилпролинамид-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 25, A, но исходя из 3,5 г 1-(трет.бутоксикарбонил)-N-трет. бутил-5-фенилпролинамида-(2RS, 5SR) в 50 см3 безводного хлороформа и 2,1 см3 иодтриметилсилана. Получают 1,8 г N-трет.бутил-5-фенилпролинамида-(2RS, 5SR) в виде масла желтого цвета, используемого при последующем синтезе.
1-(Трет.бутоксикарбонил)-N-трет.бутил-5-фенилпролинамид-(2RS, 5SR) может быть получен следующим образом: в раствор 2,9 г 1-трет.бутоксикарбонил-5-фенилпролина-(2RS, 5SR) в 50 см3 1,2-дихлорэтана добавляют при температуре около 20oC 1,8 г N,N'-диимидазолкарбонила. Реакционную среду перемешивают в течение 2 ч при температуре около 20oC, затем добавляют 1 см3 трет.бутиламина и нагревают с обратным холодильником в течение 5 ч, перемешивая. После охлаждения реакционную смесь разбавляют 100 см3 дихлорметана и промывают последовательно 2 раза в 50 см3 стандартного водного раствора соляной кислоты, затем 3 раза в 50 см3 воды. Органическую фазу высушивают над сульфатом магния, затем выпаривают досуха под пониженным давлением. Полученный остаток очищают хроматографией на двуокиси кремния (элюент: дихлор-метанол (97,5/2,5 об. )), а фракции, содержащие целевой продукт, объединяют и концентрируют досуха под пониженным давлением. Таким образом, получают 3 г 1-(трет.бутоксикарбонил)-N-трет. бутил-5-фенилпролинамида-(2RS, 5SR) в виде масла желтого цвета, используемого при последующем синтезе.
Пример 60. По способу, описанному в примере 9, но исходя из 10,6 г 3-(3-(2-(2,5-дифенил-1-пирролидинил) 2 оксо-этил)уреидо)фенил-метилацетата-(цис), растворенного в 150 см3 метанола и 1,7 г гидроокиси калия, растворенной в 30 см3 воды, и после обработки и перекристаллизации в этаноле получают 1,2 г 3-(3-(2-(2,5-дифенил-1-пирролидинил)2-оксо-этил)уреидо) фенилуксусной (цис) кислоты, плавящейся при 152oC.
3-(3-(2-(2,5-Дифенил-1-пирролидинил)2-оксо-этил)уреидо) фенилметилацетат-(цис) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 4,45 г 2,5-дифенилпирролидина-(цис), 5,32 г 2-(3-(3-метоксикарбонилметил-фенил)уреидо)-уксусной кислоты и 4,2 г N,N'-дициклогексилкарбодиимида в 40 см3 ацетонитрила. После обработки получают 10,6 г 3-(3-(2-(2,5-дифенил-1-пирролидинил) 2-оксо-этил)уреидо)фенилметилацетата-(цис) в виде масла, используемого при последующем синтезе.
2-(3-(3-Метоксикарбонилметил-фенил)уреидо) уксусная кислота может быть получена по способу, аналогичному описанному в примере 1, A, но исходя из 9,42 г глицина, 34, 69 г карбоната калия в 220 см3 воды и 24 г 3-изоцианат-фенилметилацетата, растворенного в 170 см3 1,4-диоксана. После обработки и перекристаллизации в этилацетате получают 46,85 г 2-(3-(3-метоксикарбонилметил-фенил)уреидо) уксусной кислоты, плавящейся при 136oC.
Пример 61. По способу, аналогичному описанному в примере 9, но исходя из 4,8 г 3-(3-(2-(2,5-дифенил-2-пирролидинил) 2-оксо-этил)уреидо)-фенил)этилпропионата-(цис), растворенного в 25 см3 метанола и 0,25 г гидроокиси калия, растворенной в 5 см3 воды и после обработки получают 0,9 г (3-(3-(3-(2-(2,5-дифенил-1-пирролидинил) 2(оксо-этил)уреидо)фенил)пропионовой-(цис) кислоты, плавящейся при 170oC.
3-(3-(3-(2-(2,5-Дифенил-1-пирролидинил)2-оксо-этил)уреидо)фенил) этилпропионат-(цис) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 0,68 г 2,5-дифенилпирролидина-(цис), 0,9 г 2-(3-(3-(2-этоксикарбонил-этил)уреидо)фенил)уксусной кислоты и 0,63 г N,N'-дициклогексилкарбодиимида в 6 см3 ацетонитрила. После обработки получают 1,8 г 3-(3-(3-(2,5-дифенил-1-пирролидинил)- 2-оксо-этил)уреидо)фенил)этилпропионата (цис) в виде масла, используемого при последующем синтезе.
2-(3-(3-(2-Этоксикарбонил-этил)уреидо)фенил)уксусная кислота может быть получена способом, аналогичным описанному в примере 1, A, но исходя из 0,44 г глицина, 1,63 г карбоната калия в 10 см3 воды и 1,3 г 3-(3-изоцианат-фенил) этилпропионата, растворенного в 8 см3 1,4-диоксана. После обработки получают 0,9 г 2-(3-(3-(2-этоксикарбонил-этил)уреидо)фенил) уксусной кислоты в виде аморфного тела, используемого при последующем синтезе.
3-(3-Изоцианат-фенил) этилпропионат может быть получен следующим способом: в суспензию 0,45 г угля в 3 г 3-(3-амино-фенил) этилпроионата, растворенного в 60 см3 толуола, добавляют в течение 30 мин при температуре около -20oC раствор 2,36 г карбоната бис-трихлорметила в 20 см3 толуола. Реакционную смесь нагревают до 110oC в течение 2 ч 30 мин, охлаждают до температуры около 20oC, фильтруют на целите и концентрируют при пониженном давлении. Таким образом, получают 3,5 г 3-(3-изоцианат-фенил)этилпропионата в виде масла, используемого при последующем синтезе.
Пример 62. По способу, описанному в примере 9, но исходя из 1,2 г 3-(3-(2-(2-(2-метокси-фенил) 5-фенил-1-пирролидинил) 2-оксо-этил)уреиодо)фенилметилацетата-(2RS, 5SR), растворенного в 20 см3 метанола и 0,19 г гидроокиси калия, растворенной в 4 см3 воды, после обработки и кристаллизации в диэтиловом эфире получают 0,6 г 3-(3-(2-(2-(2-метокси-фенил) 5-фенил-1-пирролидинил) 2-оксо-этил)уреиодо)фенилуксусной-(2RS, 5SR) кислоты, плавящейся при 140oC.
3-(3-(2-(2-Метокси-фенил) 5-фенил-1-пирролидинил) 2-оксоэтил)уреидо)фенилметилацетат (2RS, 5SR) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 0,57 г 2-(2-метокси-фенил) 5-фенилпирролидина-(2RS, 5SR), 0,6 г 2-(3-(3-метоксикарбонилметил-фенил)уреидо)уксусной кислоты и 0,46 г N,N'-дициклогексилкарбодиимида в 4,5 см3 ацетонитрила. После обработки получают 1,2 г 3-(3-(2-(2-(2-метокси-фенил 5-фенил-1-пирролидинил)-2-оксо-этил) уреидо)фенилметилацетата-(2RS, 5SR), в виде масла, используемого при последующем синтезе.
А) 2-(2-метокси-фенил) 5-фенилпирролидин-(2RS, 5SR) может быть получен следующим образом: в раствор 2,25 г N-бензил-о-метоксибензальдимина в 20 см3 тетрагидрофурана прикапывают, перемешивая, 12,8 см3 1,6M раствора бутиллития в гексане при температуре около -78oC. После повышения температуры до приблизительно 20oC продолжают перемешивать в течение 40 ч, затем реакционную смесь выливают в 100 см3 насыщенного водного раствора хлорида аммония, а продукт экстрагируют с помощью 50 см3, затем 2 раза 25 см3 оксида диизопропила. Органические фазы объединяют и высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и температуре около 35oC. Остаток растворяют в 25 см3 стандартного водного раствора соляной кислоты и раствор нагревают с обратным холодильником в течение 1 мин. После охлаждения до температуры приблизительно 20oC раствор промывают три раза в 25 см3 окиси диизопропила, затем pH доводят до 9 добавлением 1N водного раствора гидроокиси натрия, а продукт экстрагируют 3 раза 25 см3 окиси диизопропила. Органические экстракты объединяют, промывают два раза в 25 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и температуре около 35oC. Полученный продукт очищают хроматографией на двуокиси кремния (элюент: петролейный эфир-окись диизопропила (50/50 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и температуре около 45oC. Таким образом, получают 0,9 г 2-(2-метокси-фенил) 5-фенилпирролидина-(2RS, 5SR) в виде аморфного тела, используемого при последующем синтезе.
Б) N-бензил-о-метоксибензальдимин может быть получен следующим способом: раствор 60 см3 ортометоксибензальдегида, 55 см3 бензиламина и 50 мг паратолуолсульфокислоты в 150 см3 толуола нагревают в течение 3 ч с обратным холодильником, собирая образующуюся воду с помощью прибора Дин-Старка. После возврата к температуре приблизительно 20oC органическую фазу промывают в 100 см3 насыщенного водного раствора кислого карбоната натрия, затем в 100 см3 воды, высушивают над сульфатом магния и концентрируют при пониженном давлении. После перегонки получают 57,3 г N-бензил-о-метоксибензальдимина, отгоняемого при 155 - 159oC при 130 Па.
Пример 63. По способу, описанному в примере 9, но исходя из 0,46 г 3-(3-(2-(2-(3-гидрокси-фенил) 5-фенил-1-пирролидинил) 2-оксо-этил)уреидо) фенилметилацетата-(2RS, 5SR), растворенного в 10 см3 метанола, и 0,1 г гидрокиси калия, растворенной в 2 см3 воды, и после обработки и перекристаллизации в этаноле получают 0,2 г 3-(3-(2-(2-(3-гидрокси-фенил) 5-фенил-1-пирролидинил) 2-оксо-этил)уреидо) фенилуксусной-(2RS, 5SR) кислоты, плавящейся при 166oC.
3-(3-(2-(2-(3-Гидрокси-фенил) 5-фенил-1-пирролидинил) 2-оксоэтил)уреидо) фенилметилацетат-(2RS, 5SP) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 0,48 г 2-(3-гидрокси-фенил) 5-фенилпирролидина-(2RS, 5SR), 0,54 г 2-(3-(3-метоксикарбонилметил-фенил)уреидо)уксусной кислоты и 0,42 г N, N'-дициклогексилкарбодиимида в 5 см3 ацетонитрила. После обработки получают 0,.73 г 3-(3-(2-(2-(3-гидрокси-фенил) 5-фенил-1-пирролидинил)-2-оксо-этил)уреидо)фенилметилацетата-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
2-(3-Гидрокси-фенил) 5-фенилпирролидин-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 62, A но исходя из 4,22 г N-бензил-м-гидроксибензальдимина, растворенного с 40 см3 тетра-гидрофурана и 33,6 см3 2,5 M раствора бутиллития в гексане. После обработки получают 3,3 г 2-(3-гидрокси-фенил) 5-фенилпирролидин-(2RS, 5SR) в виде масла, используемого при последующем синтезе.
N-бензил-м-гидроксибензальдимин может быть получен способом, аналогичным описанному в примере 62, A, но исходя из 61,1 г 3-гидрокси-бензальдегида и 55 см3 бензиламина в 450 см3 толуола. После обработки получают 100,3 г N-бензил-м-гидроксибензальдимина, плавящегося при 151oC.
Пример 64. По способу, описанному в примере 9, но исходя из 4 г 4-(3-(2-(2,5-дифенил-1-пирролидинил) 2-оксо-этил)уреидо)фенилбензилацетата-(цис), растворенного в 65 см3 метанола, и 0,61 г гидроокиси калия, растворенной в 12 см3 воды, после обработки получают 1,8 г 4-(3-(2-(2,5-дифенил-1-пирролидинил) 2-оксо-этил)уреидо)фенилуксусной-(цис) кислоты, плавящейся при 221oC.
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 1,4 - 2,4 (m, 4H, CH2-CH2), 3,3 (s, 2H, CH2CO2), 3,2 - 3,8 (ABX, 2H, CH2NH), 5,1 (m, 2H, 2 CHN), 6,1 (t, 1H обменные, NH), 6,9 (d, 2H, ароматические в "пара"), 7,1 - 7,4 (m, 12H, ароматические), 8,6 (s, 1H, обменный, NH).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3375, 3095, 3070, 3035, 2980, 2880, 1735, 1700, 1630, 1610, 1515, 1450, 1560, 805, 760, 705).
4-(3-(2-(2,5-Дифенил-1-пирролидинил) 2-оксо-этил)уреидо)-фенилбензилацетат-(цис) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 1,7 г 2,5-дифенилпирролидина-(цис), 2,6 г 2-(3-(4-бензилоксикарбонилметил-фенил)уреидо)уксусной кислоты и 1,6 г N, N'-дициклогексилкарбодиимида в 15 см3 ацетонитрила. После обработки получают 4 г 4-(3-(2-(2,5-дифенил-1-пирролидинил) 2-оксо-этил)уреидо)фенилбензилацетата-(цис) в виде воздушной массы, используемой при последующем синтезе.
2-(3-(4-Бензилоксикарбонилметил-фенил)уреидо)уксусная кислота может быть получена способом, аналогичным описанному в примере 1, A, но исходя из 3,68 г глицина, 4,1 г кислого карбоната натрия в 150 см3 воды и 13 г 4-изоцианато-фенилбензилацетата, растворенного в 60 см3 1,4-диоксана. После обработки получают 5,2 г 2-(3-(4-бензилоксикарбонилметил-фенил)уреидо)-уксусной кислоты, плавящейся при 192oC.
А) 4-Изоцианатофенилбензилацетат может быть получен следующим способом: в суспензию 1 г угля в смеси 5,9 см3 хлорформиата трихлорметила и 50 см3 толуола добавляют в течение 15 мин при температуре около -25oC, раствор 11,8 г 4-аминофенилбензилацетата в 150 см3 толуолда. Реакционную смесь перемешивают при температуре около 25oC в течение 2 ч, затем нагревают до 110oC в течение 2 ч. После охлаждения до температуры приблизительно 25oC, реакционную среду дегазируют барботированием азота, отфильтровывают на бумажном фильтре и концентрируют при пониженном давлении и температуре около 52oC. Таким образом, получают 15,4 г 4-изоцианат-фенилбензил ацетата в виде масла, используемого при последующем синтезе.
4-Амин-фенилбензилацетат может быть получен по известному методу (E. ZURABY et coll, lzv Akad Nauk SSSR, Ser. Khim. (II) 2036, 1964).
Пример 65. По способу, описанному в примере 9, но исходя из 3,5 г 3-(3-(2-(2-(2-метил-фенил) 5-фенил-1-пирролидин 2-оксоэтил)уреидо) фенилметилацетата-(2RS, 5SR), растворенного в 60 см3 метанола, и 0,56 г гидроокиси калия, растворенной в 11 см3 воды, после обработки и перекристаллизации в этилацетате получают 2,2 г 3-(3-(2-(2-(2-метил-фенил) 5-фенил-1-пирролидинил) 2-оксо-этил)-уреидо)фенилуксусной кислоты-(2RS, 5SR), плавящейся при 133oC.
3-(3-(2-(2-(2-Метил-фенил) 5-фенил-1-пирролидинил) 2-оксо-этил) -уреидо)фенилметилацетат-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 41, А, но исходя из 1,6 г 2-(2-метил-фенил) 5-фенилпирролидина-(2RS, 5SR), 1,8 г 2-(3-(3-метоксикарбонилметил-фенил)уреидо)уксусной кислоты и 1,4 г N,N'-дициклогексилкарбодиимида в 15 см3 ацетонитрила. После обработки получают 3,5 г 3-(3-(2-(2-(2-метил-фенил) 5-фенил-1-пирролидин) 2-оксо-этил)уреидо)фенилметилацетата-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
2-(2-Метил-фенил) 5-фенилпирролидин-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 62, А, но исходя из 4,18 г N-бензил-о-метилбензальдимина, растворенного в 40 см3 тетрагидрофурана, и 25,6 см3 2,5M раствора бутиллития в гексане. После обработки получают 1,7 г 2-(2-метил-фенил) 5-фенилпирролидина-(2RS, 5SR) в виде масла, используемого при последующем синтезе.
N-Бензил-о-метилбензальдимин может быть получен по известной методике (A. PADVA et coll., J.Amer. Chem. Soc., 91 2653, 1969).
Пример 66. По способу, описанному в примере 9, но исходя из 1,8 г 3-(3-(2-(2-(3-гидрокси-фенил)-5-(3-метокси-фенил)-1-пиролидинил) 2-оксо-этил)уреидо)этилбензоата-(2RS, 5SR), растворенного в 30 см3 метанола, и 0,29 г гидроокиси калия, растворенной в 6 см3 воды, после обработки и кристаллизации в диэтиловом эфире получают 1 г 3-(3-(2-(2-(3-гидрокси-фенил)-5-(3-метокси-фенил) 1-пирролидинил 2-оксо-этил)уреидо)бензойной-(2R*, 5S*) кислоты, плавящейся при 172oC.
3-(3-(2-(2-(3-Гидрокси-фенил)-5-(3-метокси-фенил)-1-пирролидинил) 2-оксо-этил)уреидо)этилбензоат-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 41, А, но исходя из 1,3 г 2-(3-гидрокси-фенил)-5-(3-метокси-фенил)-пирролидина-(2RS, 5SR), 1,3 г 2-(3-(3-этоксикарбонил-фенил)уреидо) уксусной кислоты и 1 г N,N'-дициклогексилкарбодиимида в 11 см3 ацетонитрила. После обработки получают 1,8 г 3-(3-(2-(2-(3-гидрокси-фенил)-5-(3-метосифенил)-1-пирролидинил 2-оксо-этил)уреидо)этилбензоата-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
2-(3-Гидрокси-фенил)-5-(3-метокси-фенил)-пирролидин-(2RS, 5SR) может быть получен способом, аналогичном описанному в примере 62, А, но исходя из 7,2 г N-/(3-гидрокси-фенил)метилен/-(3-метокси-фенил)-метиламина в 60 см3 тетрагидрофурана и 50,4 см3 2,5M раствора бутиллития в гексане. После обработки получают 4,5 г 2-(3-гидрокси-фенил)-5-(3-метокси-фенил)пирролидина-(2RS, 5SR) в виде масла, используемого при последующем синтезе. N-/(3-гидрокси-фенил)метилен/-(3-метокси-фенил)метиламин может быть получен способом, аналогичным описанному в примере 62, Б, но исходя из 13,2 г 3-гидроксибензальдегида и 13,7 г м-метоксибензиламина в 90 см3 толуола после обработки получают 20,7 г N-/(3-гидрокси-фенил)метилен/- (3-метоксифенил)метиламина, плавящегося при 103oC.
2-/3-(3-Этоксикарбонил-фенил)уреидо/уксусуная кислота может быть получена, как описано в примере 1, А, но исходя из 9,85 г глицина, 11 г кислого карбоната натрия, растворенного в 150 см3 воды, и 25 г 3-изоцианат этилбензоата. После обработки получают 16 г 2-(3-(3-этоксикарбонил-фенил)уреидо)уксусной кислоты, плавящейся при 174oC.
Пример 67. По способу, описанному в примере 9, но исходя из 1,8 г 4-(3-(2-(2-(3-гидрокси-фенил)-5-фенил-1-пирролидинил)2-оксо-этил) уреидо)фенилбензилацетата-(2RS, 5SR), растворенного в 30 см3 метанола, и 0,27 гидроокиси калия, растворенной в 6 см3 воды, после обработки и кристаллизации в диэтиловом эфире получают 0,5 г 4-(3-(2-(2-(3-гидрокси-фенил)-5-фенил-1-пирролидинил) 2-оксо-этил)-уреидо)фенилуксусной-(2RS, 5SR) кислоты, плавящейся при 260oC.
4-(3-(2-(2-(3-Гидрокси-фенил)-5-фенил-1-пирролидинил)2-оксоэтил) уреидо)фенилбензилацетат-(2RS, 5SR) может быть получен способом, аналогичном описанному в примере 41, А, но исходя из 1,4 г 2-(3-гидрокси-фенил)-5-фенилпирролидина-(2RS, 5SR), 2,0 г 2-(3-(3-метоксикарбонилметил-фенил)уреидо)уксусной кислоты и 1,2 г N,N'-дициклогексилкарбодиимида в 11 см3 ацетонитрила. После обработки получают 1,8 г 4-(3-(2-(2-(3-гидрокси-фенил)-5-фенил-1-пирролидинил)-2- оксо-этил)уреидо)фенилбензилацетата-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
Пример 68. По способу, описанному в примере 9, но исходя из 1,3 г (4-(3-(2-(2,5-дифенил-1-пирролидинил)2-оксо-этил)уреидо)-фенилтио) трет. бутилацетата-(цис), растворенного в 20 см3 метанола, и 0,17 г гидроокиси калия, растворенной в 4 см3 воды, после обработки получают 0,5 г (4-(3-(2-(2,5-дифенил-1-пирролидинил)2-оксо-этил)уреидо)фенилтио) уксусной-(цис) кислоты, плавящейся при 200oC.
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 1,8-2,4 (m, 4H, CH2-CH2), 3,35 (s, 2H, CH2S), 3,3 и 3,85 (ABX, 2H, CH2N), 5,1 (m, 2H, 2 CHN), 6,5 (t, 1H обменный NH), 7,1-7,4 (m, 14H, ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3400, 3090, 3060, 3030, 2975, 2875, 1640, 1595, 1540, 1495, 1450, 1400, 825, 760, 700.
(4-(3-(2-(2,5-дифенил-1-пирролидинил)2-оксо-этил)уреидо) фенилтио)трет-бутилацетат-(цис) может быть получен способом аналогичным описанному в примере 41, А, но исходя из 0,46 г 2,5-дифенилпирролидина, 0,7 г 2-(3-(4-трет. бутоксикарбонилметилтио-фенил)уреидо)-уксусной кислоты и 0,42 г N,N'-дициклогексилкарбодиимида в 5 см3 ацетонирила. После обработки получают 1,3 г (4-(3-(2-(2,5-дифенил-1-пирролидинил)2- оксо-этил)уреидо)фенилтио)-трет.бутилацетата-(цис) в виде воздушной массы, используемой при последующем синтезе.
2-(3-(4-Трет.бутоксикарбонилметилтио-фенил)уреидо)уксусная кислота может быть получена способом, аналогичным описанному в примере 1, А, но исходя из 0,65 г глицина, 2,4 г карбоната калия в 15 см3 воды и 2,3 г (4-изоцианатфенилтио)трет-бутилацетата, растворенного в 12 см3 1,4-диоксана. После обработки получают 1,7 г 2-(3-(4-трет.бутоксикарбонилметилтио-фенил)уреидо)уксусной кислоты в виде воздушной массы, используемой при последующем синтезе.
(4-Изоцианат-фенилтио)трет.бутилацетат может быть получен способом, аналогичным описанному в примере 64, А, но исходя из 3,1 г (4-аминофенилтио)трет. бутилацетата в 117 см3 толуола, 0,26 угля и 1,56 см3 хлорформиата трихлорметила в 30 см3 толуола. После обработки получают 2,3 г (4-изоцианат-фенилтио)трет. бутилацетата в виде масла, используемого при последующем синтезе.
(4-Амино-фенилтио)трет. бутилацетат может быть получен способом, аналогичным описанному в примере 17 для получения (3-амино-фенилтио)этилацетата, но исходя из 5 г 4-амино-тиофенола и 6,4 см3 бромацетата трет.бутила в 80 см3 этанола. Таким образом, получают 7,45 г (4-амин-фенилтио)ацетата трет. бутила в виде масла, используемого при последующем синтезе.
Пример 69. По способу, описанному в примере 9, но исходя из 0,8 г 3-(3-(2-(2-(2-гидрокси-фенил) 5-фенил-1-пирролидинил)-2-оксо-этил(уреидо) этилбензоата-(2RS, 5SR), растворенного в 15 см3 метанола, и 0,27 г гидроокиси калия, растворенной в 3 см3 воды, после обработки и кристаллизации в окиси диизопропила получают 0,21 г 3-(3-(2-(2-(2-гидрокси-фенил) 5-фенил-1-пирролидинил) 2-оксо-этил)уреидо) бензойной-(2RS, 5R) кислоты, плавящейся при 160oC.
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 1,8-2,5 (2m, 4H, CH2-CH2), 3,3 и 4 (ABX, 2H, CH2N), 5,2 (m, 2H, 2CHN), 6,7-7,4 (m, 13H, ароматические), 6,8 (bt), 9(s), 9,8(bs), 12,8(vbs) (4H обменные, 1 OH, 1 CO2H и 2NH).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3400, 3070, 2980, 1695, 1630, 1560, 1490, 760, 700, 685.
3-(3-(2-(2-(2-Гидрокси-фенил) 5-фенил-1-пирролидинил) 2-оксоэтил)уреидо)этилбензоат-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 0,5 г 2-(2-гидрокси-фенил) 5-фенилпирролидина-(2RS, 5SR), 0,56 г 2-(3-(3-этоксикарбонил-фенил)уреидо)уксусной кислоты и 0,43 г N,N'-дициклогексилкарбодиимида в 7,5 см3 ацетонитрила. После обработки получают 0,85 г 3-(3-(2-(2-(2-гидрокси-фенил) 5 фенил-1-пирролидинил)-2-оксо-этил)уреидо) этилбензоата-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
2-(2-Гидрокси-фенил) 5-фенилпирролидин-(2RS, 5SR) может быть получен следующим способом: в раствор 8,6 см3 диизопропиламина в 60 см3 тетрагидрофурана добавляют за 10 мин, при температуре около -78oC, 24 см3 2,5 M раствора бутиллития. Затем реакционную среду перемешивают при температуре около 20oC в течение 15 мин, затем охлаждают до температуры приблизительно -70oC. Добавляют за 5 мин при температуре около -70oC раствор 4,2 г N-бензил-о-гидроксибензальдимина в 5 см3 тетрагидрофурана. Раствор перемешивают в течение 10 мин при температуре около -70oC, затем оставляют до повышения температуры до 20oC. Затем реакционную среду насыщают барботированием этилена в течение 40 ч, затем выливают в 150 см3 насыщенного водного раствора хлорида аммония, а продукт экстрагируют 100 см3, затем два раза 50 см3 диэтилового эфира. Объединенные органические фазы высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и температуре около 35oC. Остаток растворяют в 150 см3 нормального водного раствора соляной кислоты и раствор нагревают с обратным холодильником в течение 1 мин. После охлаждения до температуры около 20oC раствор промывали три раза в 100 см3 диэтилового эфира, затем доводят pH до 11 с помощью 4N водного раствора гидроокиси натрия и раствор экстрагируют три раза 75 см3 диэтилового эфира. Органические экстракты объединяют, промывают два раза в 50 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и температуре около 35oC. Полученный продукт очищают хроматографией на двуокиси кремния (элюент: дихлорметан, затем дихлорметан-метанол (95-5 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и температуре около 45oC. Таким образом, получают 0,5 г 2-(2-гидрокси-фенил) 5-фенилпирролидин-(2RS, 5SR) в виде масла, используемого при последующем синтезе.
N-бензил-о-гидроксибензальдимин может быть получен способом, аналогичным описанному в примере 62, Б, но исходя из 21,2 см3 2-гидроксибензальдегида и 22 см3 бензиламина в 180 см3 толуола. После обработки получают 38,2 г N-бензил-о-гидроксибензальдимина в виде твердого вещества, плавящегося ниже 50oC.
Пример 70. По способу, описанному в примере 9, но исходя из 1,4 г 3-(3-(2-(2-(2-гидрокси-фенил) 5-фенил-1-пирролидинил) 2-оксоэтил)уреидо)фенилметилацетата-(2RS, 5SR), растворенного в 30 см3 метанола, и 0,49 г гидроокиси калия, растворенной в 6 см3 воды, после обработки и кристаллизации в окиси диизопропила получают 0,45 г 3-(2-(-2-(2-(2-гидрокси-фенил) 5-фенил-1-пирролидин) 2-оксо-этил)-уреидо)фенилуксусной-(2RS, 5SR) кислоты, плавящейся при 165oC.
3-(3-(2-(2-(2-гидрокси-фенил) 5-фенил-1-пирролидинил) 2-оксоэтил(уреидо) фенилметилацетат-(2RS, 5SR) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 1,8 г 2-(3-гидрокси-фенил)-5-фенилпирролидина-(2RS, 5SR), 2г 2-(3-(3-метоксикарбонилметил-фенил)уреидо) уксусной кислоты и 1,55 г N,N'-дициклогексилкарбодиимида в 30 см3 ацетонитрила. После обработки получают 1,4 г 3-(3-(2-(2-(2-гидрокси-фенил) 5-фенил-1-пиррорлидинил)-2-оксо-этил)уреидо) фенилметилацетата-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
Пример 71. По способу, описанному в примере 41, но исходя из 0,2 г 3-(3-(2-(2-трет. бутоксикарбонил-5-(3-индилил)-1- пирролидинил)2-оксо-этил)уреидо)2-триметилсилил-этилбензоата-(2S, 5R) и 1,0 см3 1M раствора фторида тетрабутиламмония. Сырой продукт растворяют в 25 см3 этилацетата и экстрагируют два раза 5 см3 0,1N водного раствора гидроокиси натрия. Водные фазы доводят до pH 2, прибавляя 1N водный раствор соляной кислоты. Выпавший в осадок продукт отфильтровывают, промывают 2 раза в 5 см3 дистиллированной воды и высушивают на воздухе. Таким образом, получают 0,12 г 3-(3-(2-(2-трет. бутоксикарбонил-5-(3-индилил)-1-пирролидинил) 2-оксо-этил)уреидо) бензойной-(2S, 5R) кислоты в виде твердого вещества, плавящегося при 160oC, /α/
Протонный ЯМР (300 МГц, DMCO D6, δ в ч/млн, J в Гц), 1,5 (S, 9H, -C(CH3)3), 1,8-2,5 (m, 4H, -CH2-CH2-), 3,6 и 4,1 (2dd, J-17,5 и 5,1, 2H, N-CO-CH2N), 4,26 (dd, J=9 и 8, 1H, N-CH-COO-), 5,52 (d,J=8,5, 1H, N-CH-Ar), 6,38 (t, J= 5,1 H, -NH-CO-), 7,0-7,7 (m, 7H, ароматические), 7,68 (s, 1H, N-CH= ), 8(bs, 1H, в позиции 2 в NH-Ph-COO), 9,03 (массив, 1H, Ph-NH-CO-), 11,05 (массив, 1H, -NH-), эффект NOE был отмечен между N-CH= индолила и протоном в позиции 5 пирролидина).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2980, 2935, 2700-2250 (широкая полоса), 1720-1695 (широкая полоса), 1635, 1595, 1560, 1490, 1460, 1370, 1155, 745, 685.
3-(3-(2-(2-Трет. бутоксикарбонил-5-(3-индолил)-1-пирролидинил)- 2-оксо-этил) уреидо)2-триметилсилил-этилбензоат-(2S, 5R) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 0,5 г (5-(3-индолил)-2-пирролидинтрет. бутилкарбоксилата-(2S, 5RS), 0,59 г 2-(3-(3-(2-триметилсилилэтоксикарбонил)фенил)уреидо)уксусной кислоты и 0,36 г N,N'-дициклогексилкарбодиимида. После хроматографии на двуокиси кремния (элюент: этилацетат-циклогексан (40-60 об.)). Фракции, содержащие смесь диастереоизомеров, концентрируют досуха при пониженном давлении и при 40oC, полученный остаток очищают второй хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (с градиентом этилацетата)). Фракции, содержащие каждая два диастереоизомера, объединяют и концентрируют досуха при пониженном давлении и при 40oC. Таким образом, получают 0,20 г 3-(3-(2-(4-трет.бутоксикарбонил-5-(3-индолил)-1-пирролидинил) 2-оксо-этил)уреидо)2-триметилсилил-этилбензоата-(2S, 5R) (первый продукт элюирования) и 0,26 г 3-(3-(2-(4-трет. бутоксикарбонил-5-(3-индонил)-1-пирролидинил) 2-оксо-этил)уреиодо 2-триметилсилил-этилбензоата-(2S, 5S) (второй продукт элюирования).
5-(3-Индолил)-2-пирролидинтрет. бутилкарбоксилат-(2S, 5SR) может быть получен способом, аналогичным описанному в примере 48, Б, но исходя из 0,7 г 1-трет-бутоксикарбонил-5-(3-индолил)-2-пирролидинтрет. бутилкарбоксилата- (2S, 5R) и 0,25 см3 иодтриметилсилана. Таким образом, получают 0,50 г 5-(3-индолил)-5-пирролидинтрет. бутилкарбоксилата-(2S, 5RS) в виде масла, используемого при последующем синтезе.
1-Трет. бутоксикарбонил-5-(3-индолил)-2-пирролидинтрет. бутилкарбоксилат (2S, 5R) может быть получен следующим способом: в раствор 10,0 г 1-трет.бутоксикарбонил-5-метокси-2-пирролидинтрет. бутилкарбоксилата -(2S, 5RS) и 0,6 г паратолуолсульфоксилоты в 100 см3 безводного метиленхлорида, охлажденный до температуры около 5oC, добавляют раствор 3,5 г индола в 30 см3 метиленхлорида. В конце добавления температуру реакционной среды доводят до приблизительно 25oC, перемешивают в течение 2 ч при этой температуре, затем гидролизуют добавлением 40 см3 насыщенного водного раствора кислого карбоната натрия. Органическую фазу декантируют, водную фазу экстрагируют 2 раза 50 см3 метиленхлорида. Органические фазы объединяют, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и при 30oC. Полученный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (10/90 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 35oC. Таким образом, получают после взбалтывания в окиси диизопропила 1,2 г 1-трет.бутоксикарбонил-5-(3-индолил)-2-пирролидин-трет. бутилкарбоксилата-(2S, 5R) (первый продукт элюирования), плавящийся при 214oC, и 0,8 г I-трет.бутоксикарбонил-5-(3-индолил)-2- пирролидинтрет. бутилкарбоксилата-(2S, 5S) (второй продукт элюирования), плавящегося при 194oC.
Пример 72. По способу, описанному в примере 41, но исходя из 0,25 г 3-(3-(2-(2-трет. бутоксикарбонил-5-(3-индолил)-1-пирролидинил) 2-оксо-этил)уреидо) 2-триметилсилил-этилбензоата-(2S, 5S) и 1,5 см3 1M раствора фторида тетрабутиламмония. Сырой продукт растворяют в 25 см3 этилацетата и экстрагируют 2 раза 10 см3 0,1N водного раствора гидроокиси натрия. Водные фазы доводят до pH 2 добавляют 1N водного раствора соляной кислоты. Выпавший в осадок продукт отфильтровывают, промывают 2 раза в 5 см3 дистиллированной воды и высушивают на воздухе. Таким образом, получают 0,09 г 3-(3-(2-(2-трет. бутоксикарбонил-5-(3-индолил)-1-пирролидинил) 2-оксо-этил)уреидо) бензойной-(2S, 5S) кислоты в виде аморфного продукта, /α/
Протонный ЯМР (300 МГц, DMCO D6), δ в ч/млн), J в Гц), 1,44 (S, 9H, -C(CH3)3, 1,7 - 2,4 (m, 4H, -CH2-CH2-), 3,59 и 3,99 (2 dd, J = 18 и 6, 2H, N-CO-CH2-N), 4,55 (d, J = 9 Гц, 1H, N-CH-COO-), 5,53 (d, J = 8, 1H, N-CH-Ar), 6,3 (t, J = 6,1 Гц, 1H, -NH-CO-), 6,9 - 7,6 (m, 7H, ароматические), 7,15 (bs, 1H, N-CH=), 7,94 (s широкое, 1H в позиции 2 в NH-Ph -COO), 8,95 (массив, 1H, Ph -NH-CO), 11,02 (массив, 1H, -NH-), 12,75 (массив, 1H, COOH).
Были отмечены два эффекта NOE между N-CH= индола и протонами в позиции 2 и 5 пирролидина.
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3385, 2975, 2930, 2700-2250 (широкая полоса), 1730, 1630, 1640, 1560, 1490, 1460, 1395, 1370, 1155, 740, 685.
Пример 73. По способу, описанному в примере 38, но исходя из 4,84 г 3-(2-(1-имидазолил-карбоксамидо)ацетил) 2-фенил-4-тиазолидин-трет. бутилкарбоксилата-(2R, 4R) и 8,85 г 1-(3-аминофенил)-этансульфоната тетра-н-бутиламмония-(RS). В полученный сырой продукт в виде соли тетра-н-бутиламмония, растворенный в 20 см3 ацетона, добавляют раствор 2,5 г нонафторбутансульфоната калия в 15 см3 ацетона. После 2 ч перемешивания при температуре около 25oC добавляют 220 см3 окиси диизопропила. Нерастворимый продукт отфильтровывают, промывают 2 раза в 3 см3 смеси ацетона и окиси диизопропила (30/70 об. ) и очищают хроматографией на двуоксиси кремния (элюент: этилацетат-метанол (85/15)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают после встряхивания в ацетонитриле 0,40 г 1-(3-(3-(2-(4-трет.бутоксикарбонил-2-фенил-3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил) этансульфоната калия-(RS) (смесь форм A и B), плавящегося при 240oC, в виде твердого тела, /α/
Протонный ЯМР (200 МГц, DMCO D6 + несколько капель CD3COOD, δ в ч/млн, J в Гц) 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические сдвиги при 120oC: 1,51 (m, 12H, -C(CH3)3) и CH3), 3,28 и 3,45 (2 dd, J = 12,5 и 6, 2H, S -CH2-), 3,7 (m, 2H, -CH и 1H N-COCH2-N), 4,05 (d, J = 17, 1H, другой H -N-COCH2-N), 5,0 (t, J = 6, 1H, N-CH-COO), 6,4 (s, 1H, S -CH-N), 6,98 (d широкая, J = 8, 1H, в позиции 4 в CO-NH-Ph), 7,08 (t, J = 8, 1H, в позиции 5 в CO-NH-Ph), 7,2 - 7,5 (m, 5H, ароматические), 7,64 (bd, J = 8, 2H, ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3370, 2970, 2920, 2870, 1725, 1655, 1610, 1590, 1555, 1490, 1450, 1365, 1205, 1150, 1025, 725, 695).
1-(3-Аминофенил)этансульфонат тетра-н-бутиламмония-(RS) может быть получен способом, аналогичным описанному в примере 41, Ж, но исходя из 22,4 г 1-(3-нитро-фенил) этансульфоната тетра-н-бутиламмония-(RS) и 0,8 г 5% палладия на саже. Таким образом, получают 20,7 г 1-(3-амино-фенил) этансульфоната тетра-н-бутиламмония в виде масла, используемого при последующем синтезе.
1-(3-Нитро-фенил) этансульфонат тетра-н-бутиламмония-(RS) может быть получен следующим способом: в раствор 9,45 г сульфита натрия в 125 см3 дистиллированной воды добавляют 11,5 г 3-(1-бромэтил) нитробензола-(5). Реакционную среду нагревают в течение 3 ч при температуре около 70oC, охлаждают до температуры приблизительно 25oC, затем переливают в 1450 см3 0,5M водного раствора гидрогенфосфата калия. Затем добавляют 17,8 г кислого сульфата тетра-н-бутиламмония и экстрагируют смесь с помощью 300 см3 метиленхлорида. Органическую фазу промывают последовательно 3 раза в 300 см3 воды и 300 см3 насыщенного водного раствора хлорида натрия, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 22,4 г 1-(3-нитро-фенил) этансульфоната тетра-н-бутиламмония-(RS) в виде масла, используемого при последующем синтезе.
3-(1-Бром-этил) нитробензол-(RS) может быть получен по известному методу (E.FELDER et coll., J. Med. Cheh., 13, 559, 1970).
Пример 74. По способу, описанному в примере 38, но исходя из 2,42 г 3-(2-(1-имидазолил-карбоксамидо)ацетил/-2-фенил-4-тиазолидин- трет. бутилкарбоксилата-(2R, 4R) и 1,4 г 1-(3-амино-фенил)- этанола. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (75/25 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают после перекристаллизации в ацетонитриле 0,62 г 3-(2-(3-(3-(1-гидрокси-этил)фенил-(RS)/уреидо)ацетил)2-фенил-4- тиазолидинтрет. бутил-карбоксилата-(2R, 4R) (смесь форм A и B), плавящегося при 160oC /α/
Протонный ЯМР (200 МГц, плюс несколько капель CD3COOD, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характерные химические смещения при 120oC: 1,35 (d, J = 6,5, 3H, -CH3), 1,5 (s, 9H, -C(CH3)3), 3,29 и 3,45 (2dd, J = 12,5 и 6, 2H, S-CH2-), 3,72 (bd, 1H, N-COCH2-N), 4,06 (d, J = 17, 1H, другой H-N-COCH2-N), 4,68 (q, J = 6,5, 1H, -CH-O-), 5,0 (t, J = 6, 1H, N-CH-COO), 6,4 (s, 1H, S-CH-N), 6,93 (d широкая J = 8, 1H, в позиции 4 в CO-NH-Ph), 7,14 (t, J = 8, 1H, в позиции 5 в CO-NH-Ph), 7,22 (dd, J = 3 и 2,5, 1H, в позиции 2 в CO-NH-Ph), 7,2-7,5 (m, 4H, ароматические), 7,64 (bd, J = 8, 2H, ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3350, 2975, 2930, 1740, 1660, 1615, 1590, 1560, 1480, 1445, 1365, 1155, 1060, 730, 705, 700.
Пример 75. По способу, описанному в примере 34, но исходя из 2,0 г 3-(2-амино-ацетил)2-фенил-4-тиазолидинтрет.бутилкарбоксилата -(2R, 4R) и 1,6 г изоцианата 4-хлор-3-трифторметилфенила. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат/циклогексан (30/70 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают после встряхивания в 20 см3 гексана 0,7 г 3-(2-(3-(4-хлор-3-трифторметил-фенил)уреидо)-ацетил)2- фенил-4-тиазолидинтрет. бутилкарбоксилата (2R, 4R) в виде аморфного порошка, /α/
Протонный ЯМР (250 МГц, DMCO D6, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 130oC, характеристические химические сдвиги при 130oC: 1,53 (s, 9H, -C(CH3)3), 3,29 и 3,47 (2dd, J = 12,5 и 6,5, 2H, S-CH2-), 3,73 (массив, 1H, N-COCH2-N), 4,10 (dd, J = 17,5 и 4,1H, другой H в N-COCH2-N), 5,0 (массив, 1H, N-CH-COO), 6,33 (массив, 1H, -NHCO-), 6,4 (s, 1H, S-CH-N), 7,2-7,8 (m, 7H, ароматические), 7,97 (bs, 1H, в позиции 2 в CO-NH-Ph), 8,98 (массив, 1H, -CO-NH-Ph).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3375, 2980, 2930, 2875, 1740, 1640, 1595, 1550, 1485, 1455, 1370, 1325, 1175, 1150, 895, 730, 695).
Пример 76. По способу, описанному в примере 41, но исходя из 1,37 г 3-(3-(2-(4-трет. бутоксикарбонил(2-(2-метил-фенил)-3- тиазолидинил)2-оксо-этил)уреидо)-2-триметилсилил-этилбензоата-(2R, 4R) и 4,6 см3 1M раствора фторида тетрабутиламмония. Сырой продукт растворяют в 25 см3 насыщенного раствора кислого карбоната натрия и промывают в 25 см3 этилацетата. Водную фазу доводят до pH 2 добавлением водного раствора соляной кислоты 4N. Выпавший в осадок продукт отфильтровывают, промывают дистиллированной водой до нейтральности промывных вод и высушивают на воздухе. Таким образом, получают 0,89 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(2-метил-фенил)-3-тиазолидинил)2- оксо-этил)уреидо)бензойной-(2R, 4R) кислоты в виде твердого вещества, плавящегося при 152oC, /α/
Протонный ЯМР (200 МГц, DMCO D6 плюс несколько капель CD3COOD, δ ч/млн, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 110oC, характеристические химические сдвиги при 110oC: 1,56 (s, 9H, -C(CH3)3), 2,42 (s, 3H, Ar -CH3), 3,28 и 3,48 (2dd, J = 12 и 6,5, 2H, S-CH2-), 3,65 (массив, 1H, N-COCH2-N), 4,05 (d, J = 17,5, 1H, другой H в N-COCH2-N, 4,98 (t, J = 6,5, 1H, N-CH-COO), 6,24 (массив, 1H, -NH-CO), 6,46 (S, 1H, S-CH-N), 7,10-7,65 (m, 5H, ароматические), 7,33 (t, J = 8, 1H, в позиции: в CO-NHPh), 7,94 (массив, 1H, в позиции 6 в S-CH-Ph), 8,0 (dd, J = 2 и 3, 1H, в позиции 2 в CO-NH-Ph), 8,67 (массив, 1H, -CO-NH-Ph).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2980, 2935, 2700-2250 (широкая полоса), 1715-1695 (широкая полоса), 1655, 1610, 1595, 1560, 1490, 1370, 1155, 760, 745, 685.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(2-метил-фенил)-3-тиазолидинил 2-оксо-этил)уреидо)2-триметилсилил-этилбензоат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 41, А, но исходя из 2,1 г 2-(2-метил-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 2,5 г 2-(3-(3-(2-триметилсилилэтоксикарбонил)фенил)уреидо)уксусной кислоты и 1,6 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат/циклогексан (40/60 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,7 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(2-метил-фенил)-3-тиазолинил)-1- оксо-этил)уреидо)2-триметилсилил-этилбензоата-(2R, 4R) в виде аморфного твердого вещества, используемого при последующем синтезе.
2-(2-Метил-фенил)-4-тиазолидинтрет.бутилкарбоксилат-(2RS, 4R) может быть получен способом, аналогичным описанному в примере 34, В, но исходя из 10,0 г 2-(2-метил-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, растворенной в 130 см3 хлороформа, 3,0 см3 концентрированной серной кислоты и избытка изобутана. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (10/90 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 3,8 г 2-(2-метил-фенил)-4-тиазолидинтрет.бутилкарбоксилата-(2RS, 4R) в виде масла, используемого при последующем синтезе.
2-(2-Метил-фенил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 12,5 г L-цистеина и 12,0 см3 2-метил-бензальдегида. Таким образом, получают 18,4 г 2-(2-метил-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при последующем синтезе.
Пример 77. В раствор 0,95 3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил) 2-оксо-этил)уреидо)фенилметилацетата-(2R, 4R) в 6 см3 смеси вода-метанол (30/70 об.) добавляют при температуре около 25oC, 0,12 г гидроокиси калия. Реакционную смесь перемешивают в течение 3 ч при температуре около 25oC, затем концентрируют приблизительно на половину объема при пониженном давлении. Полученный раствор разбавляют 40 см3 воды, промывают 2 раза в 30 см3 этилового эфира, затем доводят до pH 2 добавлением 2,1 см3 1N водного раствора серной кислоты. Нерастворимый продукт отфильтровывают и очищают хроматографией на двуокиси кремния (элюент: этилацетатметанол (90/10 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Полученный таким образом аморфный продукт (0,26 г) растворяют в 1N водном растворе гидроокиси натрия. Этот раствор фильтруют, затем подкисляют добавлением 1N водного раствора серной кислоты. Выпавший в осадок продукт отфильтровывают, промывают 3 раза в 10 см3 воды, затем высушивают на воздухе. Таким образом, получают 0,26 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(2- фтор-фенил)-3-тиазолидинил)-2-оксо-этил)уреидо)фенилуксусной-(2R, 4R) кислоты, плавящейся при 110oC, /α/
Протонный ЯМР (200 МГц, DMCO D6 плюс несколько капель CD3COOD, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 1,55 (s, 9H, -C(CH3)3), 3,34 и 3,52 (2dd, J = 12,5 и 6,5, 2H, S-CH2-), 3,51 (S, 2H, Ar CH2COO-), 3,8 и 4,07 (2d, J = 17,5, 2H, N-COCH2-N), 5,02 (t, J = 6,5, 1H, N-CH-COO), 6,55 (s, 1H, S-CH-N), 6,85 (bd, J = 8, 1H, в позиции 4 в CO-NH-Ph), 7,05 - 7,4 (m, 6H ароматические), 7,93 (bt, J = 8,5, 1H, в позиции 6 в S-CH-Ph).
Инфракрасный спектр (KBr), характерные полосы в см-1: 3380, 2980, 2930, 2700 - 2250 (широкая полоса), 1735, 1650, 1615, 1595, 1560, 1490, 1460, 1370, 1230, 1155, 760, 705.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(2-фтор-фенил)-3- тиазолидинил)-2-оксо-этил)уреидо)фенилметилацетат-(2R, 4R) может быть получен способом, аналогичным описанному в примере, 41, A, но исходя из 1,46 г 2-(2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 1,37 г 2-(3-(3-(метоксикарбонилметил)фенил)уреидо)уксусной кислоты и 1,15 г N,N'-дициклогексилкарбодимиида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (50//50 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 0,95 г 3-(3-(2-(4-трет.бутоксикарбонил-2- (2-фтор-фенил)-3-тиазолидинил)-2-оксо-этил)уреидо)фенилметилацетата- (2R, 4R) в виде твердого аморфного продукта, используемого при последующем синтезе.
Пример 78. По способу, описанному в примере 77, но исходя из 0,76 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(2-метокси-фенил)-3- тиазолидинил)-2-оксо-этил)уреидо)фенилметилацетата-(2R, 4R) в 6 см3 смеси вода-метанол (30/70 об. ) и 0,09 г гидроокиси калия. Сырой продукт (0,45 г) растворяют в 7,5 см3 0,1 N водного раствора гидроокиси и натрия. Полученный раствор промывают 2 раза в 10 см3 диэтилового эфира, доводят до pH 2 добавлением 1N водного раствора серной кислоты, затем экстрагируют 2 раза с помощью 20 см3 этилацетата. Органические фазы объединяют, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают после встряхивания в диизопропиле, 0,25 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(2-метокси-фенил)-3- тиазолидинил)2-оксо-этил)уреидо)фенил-уксусной-(2S, 4R) кислоты в виде аморфного продукта, /α/
Протонный ЯМР (200 МГц, DMCO D6 плюс несколько капель CD3COOD, δ в ч/млн J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 1,55 (s, 9H, -C(CH3)3), 3,22 и 3,45 (2dd, J = 12,5 и 7, 2H, S-CH2-), 3,51 (s, 2H, ArCH2COO-), 3,65 (bd, 1H, N-COCH2-N), 3,93 (s, 3H, -OCH3), 4,05 (d, J = 17,5, 1H, другой H в N-COCH2-N), 4,91 (t, J = 7, 1H, N-CH-COO), 6,5 (s, 1H, N-CH-N), 6,85 (bd, J = 8, 1H, в позиции 4 в CO-NH-Ph), 6,9 - 7,4 (m, 6H, ароматические), 7,9 (bd, J = 8, 1H, в позиции 6 в S-CH-Ph).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3390, 2975, 2930, 2840, 2700 - 2250 (широкая полоса), 1735, 1645, 1610, 1600, 1595, 1560, 1370, 1245, 1155, 1025, 760, 705.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(2-метокси-фенил)-3- тиазолидинил)2-оксо-этил)уреидо)фенилметилацетат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 41, А, но исходя из 1,9 г 2-(2-метокси-фенил)-4-тиазолидинтретбутоксикарбоксилата- (2RS, 4R), 1,73 г 2-(3-(3-метоксикарбонилметио-фенил)уреидо)уксусной кислоты и 1,4 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (50/50 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,5 г 3-(3-(2- (4-трет.бутоксикарбонил-2-(2-метокси-фенил)-3-тиазолидинил)2-оксоэтил)уреидо)-фенилметилацетата-(2R, 4R) в виде аморфного твердого белого продукта, используемого при последующем синтезе.
2-(2-Метокси-фенил)-4-тиазолидинтрет. бутилкарбоксилат-(2RS, 4R) может быть получен способом, аналогичным описанному в примере 34, В, но исходя из 3,5 г 2-(2-метокси-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, растворенной в 50 см3 хлороформа, 1,0 см3 концентрированной серной кислоты и избытка изобутилена. Полученный исходный продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 2,0 г 2-(2-метокси-фенил)- 4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R) в виде масла, смесь изомеров (2R, 4R) и (2S, 4R), используемую при последующем синтезе.
2-(2-Метокси-фенил)-4-тиазолидинтрет. бутилкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 7,26 г L-цистеина и 8,9 г 2-метокси-бензальдегида. Таким образом, получают 3,7 г 2-(2-метокси-фенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 170oC, используемой при последующем синтезе.
Пример 79. По способу, аналогичному в примере 77, но исходя из 0,70 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(2-фтор-фенил)-3- тиазолидин-(2R, 4R)2-оксо-этил)уреидо)-метилманделата-(RS) в 7 см3 смеси вода-метанол (30/70 об.) и 0,08 г гидроокиси калия. Сырой продукт (0,45 г) растворяют в 8,5 см3 0,1N водного раствора гидроокиси натрия. Полученный раствор промывают 2 раза в 25 см3 этилацетата, фильтруют и подкисляют до pH 2 добавлением 1N водного раствора соляной кислоты. Выпавший в осадок продукт отфильтровывают, промывают 2 раза в 10 см3 воды и высушивают на воздухе. Таким образом, получают 0,15 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил)-3- тиазолидинил (2R, 4R)/2-оксо-этил)уреидо)мендальной-(RS) кислоты в виде белого продукта, плавящегося при 135oC.
Протонный ЯМР (200 МГц, DMCO D6 плюс несколько капель CD3COOD, δ в ч/млн J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 110oC, характеристические химические сдвиги при 110oC: 1,55 (s, 9H, -C(CH3)3), 3,32 и 3,53 (2dd, J = 12,5 и 6,5 2H, S-CH2-), 3,8 (bd, 1H, N-COOCH2-N), 4,08 (d, J = 17,5, 1H, другой H в N-COCH2-N), 5 (m и s, 2H в совокупности, соответственно N-CH-COO и Ar-CH-), 6,2 (массовое, 1H, -NHCO-), 6,54 (s, 1H, S-CH-N), 7,02 (bd, J = 7,5, 1H, в позиции 4 в CO-NH-Ph), 7,1 - 7,5 (m, 6H, ароматические) 7,94 (dd, J = 7,5 и 9, 1H, в позиции 6 в S-CH-Ph), 8,5 (массовое), -CONH-Ar).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3390, 2980, 2930, 2840, 2700 - 2250 (широкая полоса), 1735, 1650, 1610, 1560, 1490, 1455, 1370, 1230, 1150, 1060, 790, 760, 700.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(2-фтор-фенил)-3- тиазолидин-(2R, 4R)/2-оксо-этил)уреидо)-метилманделат-(2R, 4R) может быть получен по способу, аналогичному описанному в примере 38, но исходя из 3,2 г 3-(2-(1-имидазолил-карбоксамидо)ацетил-2- (2-фторфенил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) и 2,7 г 3-аминометилманделата-(RS). Сырой продукт очищают двумя последовательными хроматографиями на двуокиси кремния (элюент: метиленхлорид-метанол (95/5 об.)) для первой хроматографии, элюент: этилацетат для второй хроматографии). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 1,6 г 3-(3-(2-(4-трет. бутоксикарбонил-2- (2-фторфенил)-3-тиазолидинил-(2R, 4R)/2-оксо-этил)уреидо)метилманделат- (R, S) в виде воздушной массы, используемой для последующего синтеза.
3-(2-(1-Имидазолил-карбоксамида)ацетил)-2-(2-фтор-фенил)-4- тиазолидинтрет. бутилкарбоксилат (2R, 4R) может быть получен следующим способом: в раствор 7 г 3-(2-амино-ацетил)-2-(2-фтор-фенил) 4-тиазолидинтрет.бутилкарбоксилата-(2R, 4R) в 100 см3 безводного тетрагидрофурана, медленно добавляют при температуре около 25oC, раствор 4,8 г N,N'-диимидазол-карбонила в 50 см3 безводного тетрагидрофурана. Реакционную среду перемешивают в течение 12 ч при этой температуре, затем концентрируют досуха при пониженном давлении и 40oC. Полученный остаток растворяют в 200 см3 этилацетата и промывают 2 раза в 50 см3 воды. Органическую фазу высушивают над сульфатом магния и выпаривают досуха при пониженном давлении и 40oC. Таким образом, получают 8,7 г 3-(2-(1-имидазолил-карбоксамида)ацетил/-2- (2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата (2R, 4R) в виде масла, используемого при последующем синтезе.
3-(2-Амино-ацетил)-2-(2-фтор-фенил)-4- тиазолидинтрет. бутилкарбоксилат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 34, А, но исходя из 15,0 г 3-(2-трет.бутоксикарбониламиноацетил)-2-(2-фтор-фенил)-4- тиазолидинтрет. бутилкарбоксилата-(2R, 4R) и 5,34 г иодотриметилсилана. Таким образом, получают 10 г 3-(2-амино-ацетил)-2- (2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в виде масла, используемого при последующем синтезе.
3-(2-Трет. бутоксикарбониламино-ацетил)-2-(2-фтор-фенил)- 4-тиазолидинтрет. бутилкарбоксилат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 34, Б, но исходя из 25,0 г 2-(2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 15,5 г 2-трет.бутоксикарбониламино-уксусной кислоты и 18,2 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 25,0 г 3-(2-трет. бутоксикарбониламино-ацетил)-2-(2-фтор-фенил)- 4-тиазолидин-трет. бутилкарбоксилата-(2R, 4R) в виде масла, используемого при последующем синтезе.
3-Амино-метилманделат-(RS) может быть получен следующим образом: в раствор 15 г 3-нитрометилманделата-(RS) в 150 см3 этанола добавляют 0,5 г 5% палладия на угле. Суспензию перемешивают в течение 2 ч при температуре около 25oC в атмосфере водорода (100 кПа). Затем отфильтровывают катализатор, а фильтрат концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 13,1 г 3-аминометилманделата-(RS) в виде масла, используемого при последующем синтезе.
3-Нитрометилманделат (RS) может быть получен по известному методу (L.S. FOSDICK et J.C.CALANDRA, J.Am. Chem. Soc.,63,1101,1941).
Пример 80. По способу, аналогичному в примере 38, но исходя из 2,17 г 3-(2-(1-имидазолил-карбоксамидо)ацетил)-2-(2-фтор-фенил)- 4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) и 1,37 г 2-(3-аминофенил) этанола. Сырой продукт очищают двумя последовательными хроматографиями на двуокиси кремния (элюент: метиленхлорид-метанол (95/5 об.) для первой хроматографии, элюент: этилацетат - для второй хроматографии). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 0,5 г 3-(2-(3-(2-гидрокси-этил)фенил)уреидо) ацетил)-2-(2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в виде аморфного продукта, /α/
Протонный ЯМР (200 МГц, DMCO D6 плюс несколько капель CD3COOD, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 110oC, характеристические химические сдвига при 110oC, 1,55 (s, 9H, -C(CH3)3), 2,72 (t, J = 7, 2H, Ar CH2), 3,31 и 3,52 (2dd, J = 12 и 6, 2H, S-CH2), 3,67 (t, J = 7, 2H, -CH2O-), 4,1 (d, J = 17, 1H, другой H в N-COCH2-N), 3,8 (bd, 1H, в N-COCH2-N), 5,01 (t, J = 6, 1H, N-CH-COO), 6,2 (массив, 1H, - NHCO-), 6,54 (s, 1H, S -CH-N), 6,81 (bd, J = 7,5, 1H, в позиции 4 в CO-NH-Ph-), 7,05-7,45 (m, 6H, ароматические), 7,93 (bt, J = 8,5, 1H, в позиции 6 в S-CH-Ph), 8,36 массив, 1H, Ar NHCO-).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2875, 1740, 1655, 1610, 1590, 1560, 1490, 1460, 1370, 1230, 1150, 1050, 760, 700.
2-(3-Амино-фенил) этанол может быть получен по известному методу (B. CARNMALM et coll., Acta Pharm Suecica, 11,33,1974).
Пример 81. По способу, описанному в примере 41, но исходя из 1,37 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(2-хлор-6-фторфенил)-3- тиазолидинил)-2-оксо-этил)уреидо)2-триметилсилил-этилбензоата (2R, 4R) и 4,3 см3 1М раствора фторида тетрабутиламмония. Сырой продукт растворяют в 10 см3 0,5М водного раствора гидроокиси натрия и промывают 2 раза в 20 см3 этилацетата. Водную фазу доводят до pH 2 добавлением 1N водного раствора серной кислоты. Выпавший в осадок продукт отфильтровывают, промывают 2 раза в 5 см3 дистиллированной воды и высушивают на воздухе. Таким образом, получают 0,12 г 3-(3-(2-(4-трет. бутоксикарбонил-2- (2-хлор-6-фторфенил)-3-тиазолидинил)-2-оксо-этил)уреидо) бензойной-(2R, 4R) кислоты, плавящейся при 148oC /α/
Протонный ЯМР (250 МГц, DMCO D6, δ в ч/млн J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 1,5 (s, 9H, -C(CH3)3), 3,48 (dd, J = 12,5 и 6,5, 1H в S-CH2), 3,62 (dd, J = 12,5 и 5, 1H, другой H в N-CH2-) 3,96 (dd, J = 17,5 и 5, 1H, в N-COCH2-N), 4,18 (dd, J = 17,5 и 5,5, 1H, другой H в N-COCH2-N), 5,11 (dd, J = 6,5 и 5, N-CH-COO), 6,24 (массив, 1H, -NHCO-), 6,01 (S, 1H, S-CH-N), 7,05-7,65 (m, 6H, ароматические), 7,98 (bs, 1H, в позиции 2 в CO-NH-Ph), 8,66 (массив, 1H, Ar NHCO-).
Инфракрасный спектр (KBr), [характеристические полосы в см-1: 3380, 2980, 2930, 2700-2250 (широкая полоса), 1715, 1695, 1655, 1605, 1590, 1560, 1490, 1460, 1370, 1150, 785, 760, 680.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(2-хлор-6-фторфенил)-3- тиазолидинил) 2-оксо-этил)уреидо) 2-триметилсилил-этилбензоат (2R, 4R) может быть получен способом, аналогичным описанному в примере 34, но исходя из 1,40 г 3-(2-амино-ацетил)-2-(2-хлор-6-фторфенил)-4- тиазолидинтрет.бутилкарбоксилата-(2R, 4R) и 1,4 г 3-изоцианатбензоата 2-триметилсилил-этилена. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетатциклогексан (30/70 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,44 г 3-(3-(2-(4- трет.бутоксикарбонил-2-(2-хлор-6-фторфенил)-3-тиазолидил) 2-оксо-этил)уреидо)-2-триметилсилил-этилбензоата- (2R, 4R) в виде воздушной массы, используемого при последующем синтезе.
3-(2-Амино-ацетил)-2-(2-хлор-6-фторфенил)-4-тиазолидин- трет.бутилкарбоксилат-(2R, 4R) может быть получен способом, аналогичным описанному а примере 34, A, но исходя из 1,86 г 3-(2-трет.бутоксикарбониламино-ацетил)-2-(2-хлор-6-фторфенил)-4- тиазолидинтрет. бутилкарбоксилата-(2R, 4R) и 0,67 см3 иодотриметилсилана. Таким образом, получают 1,4 г 3-(2- амино-ацетил)-2-(2-хлор-6-фторфенил)-4- тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в виде пасты, используемой при последующем синтезе.
3-(2-Трет.бутоксикарбониламиноацетил)-2-(2-хлор-6-фторфенил) -4-тиазолидин-трет. бутилкарбоксилат (2R, 4R) может быть получен аналогично методу, описанному в примере 34, Б, но исходя из 2,2 г 2-(2-хлор-6-фторфенил)-4-тиазолидин-трет.бутилкарбоксилата (2RS, 4R), 1,22 г 2-трет.бутоксикарбониламиноуксусной кислоты и 1,45 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: метиленхлорид-метанол (98/2 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Получают 1,56 г 3(2-трет.бутоксикарбониламиноацетил)-2-(2-хлор-6-фторфенил)-4- тиазолидинтрет.бутилкарбоксалата (2R, 4R) в форме твердого вещества, используемого на последующих синтезах.
2-(2-Хлор-6-фторфенил)-4-тиазолидинтрет.бутилкарбоксилат (2RS, 4R) может быть получен способом, аналогичным описанному в примере 34, В, но исходя из 6,1 г 2-(2-хлор-6-фторфенил)-4-тиазолидинкарбоновой- (2R, 4R) кислоты, растворенной в 60 см3 хлороформа, 1,4 см3 концентрированной серной кислоты и избытка изобутилена. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (25/75 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 4,4 г 2-(2-хлор-6-фторфенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R) в виде масла, используемого при последующем синтезе.
2-(2-хлор-6-фторфенил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 11,4 г L-цистеина и 16,7 г 2-(2-хлор-6-фтор-фенил)-4-тиазолидинкарбоновой- (2RS, 4R) кислоты, плавящейся при 148oC, используемой при последующем синтезе.
3-Изоцианат-бензоат 2-триметилсилил-этила может быть получен, как в примере 21, но исходя из 2,37 г 3-амино-бензоат 2-триметилсилил-этила, 1,32 см3 хлорформиата трихлорметила и 0,21 г угля. Таким образом, получают 21,6 г 3-изоцианат-бензоат 2-триметилсилил-этила в виде масла, используемого при последующем синтезе.
Пример 82. По способу, описанному в примере 77, но исходя из 0,38 г 3-(3-(1-(4-трет. бутоксикарбонил-2-(2-фтор-метил)-3- тиазолидинил-(2R, 4R)) 1-оксо-2-пропил-(2S)уреидо)фенилметилацетата в 6 см3 смеси вода-метанол (30/70 об. ) и 0,05 г гидроокиси калия. Сырой продукт растворяют в 3,5 см3 0,1 N водного раствора гидроокиси натрия. Полученный раствор промывают 2 раза в 10 см3 этилацетата, фильтруют и доводят до pH 2 добавлением 1N водного раствора серной кислоты. Выпавший в осадок продукт отфильтровывают, промывают 2 раза в 5 см3 воды и высушивают на воздухе. Таким образом, получают 0,13 г 3-(3-(1-(4-трет.бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил- (2R, 4R))1-оксо-2-пропил-(2S))уреидо)фенилуксусной кислоты, плавящейся при 110oC, /α/
Протонный ЯМР (200 МГц, DMCO D6 δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 1,05 (d, J = 7, 3H, -CH3), 1,55 (s, 9H, -C(CH3)3), 3,25 (dd, J = 12,5 и 7,1H, в S-CH2): 3,52 (dd, J = 12,5 и 6, 1H, другой H в S-CH2-), 3,53 (s, 2H, ArCH2COO), 4,4 (m, 1H, N-COCH-N), 4,86 (dd, J = 7 и 6, N-CH-COO), 6,3 (d, J = 8, 1H, N-HCO-), 6,83 (s, 1H, S-CH-N), 6,89 (bd, J = 8, 1H: в позиции 4 в CO-NH-Ph), 7,10-7,5 (m, 6H, ароматические), 8,0 (bt, J = 8,5, 1H, в позиции 6 в S -CH-Ph), 8,29 (массив, 1H, Ar NHCO-).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2980, 2935, 2700-2250 (широкая полоса),1735, 1640, 1615, 1595, 1560, 1490, 1460, 1370, 1235, 1155, 760, 705.
3-(3-(1-(4-Трет. бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил- (2R, 4R))1-оксо-2-пропил-(2S))уреидо)фенилметилацетат может быть получен способом, аналогичным описанному в примере 34, но исходя из 0,85 г 3-(2-амино-пропионил-(2S))-2-(2-фтор-фенил)-4- тиазолидинтрет.бутилкарбоксилата(2R, 4R) и 0,53 г 3-изоцианат-фенилметилацетата. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетатциклогексан (30/70 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 0,38 г 3-(3-(1-(4-трет.бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил- (2R, 4R))1-оксо-2-пропил-(2S))уреидо)фенилметилацетата в виде масла, используемого при последующем синтезе.
3-(2-Амино-пропионил-(2S))-2-(2-фтор-фенил)-4-тиазолидин- трет. бутилкарбоксилат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 34, А, но исходя из 1,0 г 3-(2-трет.бутоксикарбониламино-(2S))-2-(2-фтор-фенил)-4- тиазолидинтрет.бутилкарбоксилат-(2R, 4R) и 0,39 см3 иодотриметилсилана. Таким образом, получают 0,85 г 3-(2-амино-пропионил-(2S))-2-(2-фтор-фенил)-4- тиазолидинтрет.бутилкарбоксилата-(2R, 4R) в виде масла, используемого в последующем синтезе.
3-(2-Трет.бутоксикарбониламино-(2S))-2-(2-фтор-фенил)-4- тиазолидинтрет. бутилкарбоксилат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 34, Б, но исходя из 2,0 г 2-(2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 1,36 г N-трет.бутоксикарбонил-L-аланин и 1,47 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (15/85 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 1,0 г 3-(2-трет.бутоксикарбониламино-пропионил-(2S))-2-(2-фтор-фенил)-4- тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в виде масла, используемого при последнем синтезе.
Пример 83. Действуют по способу, описанному в примере 77, но исходя из 0,48 г 3-(3-(1-(4-трет.бутоксикарбонил-2-(2-фтор-фенил) 3-тиазолидинил-(2R, 4R))1-оксо-2-пропил-(2R))уреидо)фенилметилацетата в 7 см3 смеси вода-метанол (30/70 об.) и 0,06 г гидроокиси калия. Полученный после фильтрации на двуокиси кремния продукт (элюент: этилацетат-метанол (90/10 об.)) растворяют в 7,0 см3 0,1N водного раствора гидроокиси натрия. Полученный раствор фильтруют и доводят до pH 2 добавлением 1N водного раствора серной кислоты. Выпавший в осадок продукт отфильтровывают, промывают 2 раза в 5 см3 воды и высушивают на воздухе. Таким образом, получают 0,25 г 3-(3-(1-(4-трет.бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил- (2R, 4R))1-оксо-2-пропил)уреидо-(2R)) фенилуксусной кислоты, плавящейся при 200oC, /α/
Протонный ЯМР (250 МГц, DMCO6, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 1,27 (d, J = 7,5, 3H, -CH3), 1,54 (s, 9H, -C(CH3)3, 3,40 и 3,56 (2dd, J = 12,5 и 6,5, 2H, S-CH2-), 3,50 (s, 2H, -CH2COO-), 4,50 (m, 1H, N-COCH-N), 5,45 (массовое, 1H, N-CH-COO), 6,3 (d, J = 8,5, 1H, -NHCO-), 6,50 (s, 1H, S-CH-N), 6,9 (bd, J = 8, 1H, в позиции 4 в CO-NH-Ph), 7,0-7,415 (m, 6H, ароматические), 7,9 (bt, J = 8,5, 1H, в позиции 6 в S-CH-Ph-), 8,3 (массив, 1H, ArNHCO-).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2700-2250 (широкая полоса), 1730, 1640, 1610, 1595, 1555, 1490, 1455, 1365, 1230, 1150, 755, 700.
(3-(3-(1-(4-Трет. бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил-(2R, 4R)) 1-оксо 2-пропил-(2R)) уреидо) фенилметил ацетат может быть получен способом, аналогичным описанному в примере 34, но исходя из 0,55 г 3-(2-амино-пропионил-(2R))-2-(2-фтор-фенил)-4-тиазолидинтрет.бутилкарбоксилата -(2R, 4R) и 0,37 г 3-изоцината фенилметилацетата. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (40/60 в об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 0,48 г 3-(3-(1-(4-трет. бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил-(2R, 4R)) 1-оксо-2-пропил-(2R))уреидо) фенилметилацетата в виде аморфного продукта, используемого при последующем синтезе.
3-(2-Амино-пропионил-(2R))-2-(2-фтор-фенил)-4-тиазолидин-трет. бутилкарбоксилат -(2R, 4R) может быть получен способом, аналогичным описанному в примере 34, A, но исходя из 0,8 г 3-(2-трет.бутоксикарбониламино-пропионил (2R))-2-(2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) и 0,30 см3 иодотриметилсилана. Таким образом, получают 0,55 г 3-(2-амино-пропионил-(2R)-2-(2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата -(2R, 4R) в виде масла, используемого при последующем синтезе.
3-(2-Трет. бутоксикарбониламино-пропионил-(2R))-2-(2-фторфенил)- 4-тиазолидин-трет. бутилкарбоксилат может быть получен способом, аналогичным описанному в примере 34, Б, но исходя из 2,0 г 2-(2-фтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата- (2RS, 4R), 1,36 г N-трет.бутоксикарбонил-Д-аланина и 1,47 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (15/85 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 1,0 г 3-(2-трет.бутоксикарбонил-амино-пропионил-(2R))-2-(2-фторфенил)-4 -тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в виде масла, используемого при последующем синтезе.
Пример 84. В растворе 0,35 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фторфенил)-3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенилбензил)пропионата (форма B) в 40 см3 этилацетата добавляют 0,1 г 10% палладия на угле. Суспензию перемешивают в течение 48 ч при температуре около 25oC в атмосфере водорода (100 кПа). Затем отфильтровывают катализатор, а фильтрат концентрируют досуха при пониженном давлении и 40oC. Полученный остаток растворяют в 10 см3 0,1 N водного раствора гидроокиси натрия и промывают два раза в 10 см3 диэтилового эфира. Водную фазу доводят до pH 2 добавлением 1N водного раствора серной кислоты. Выпавший в осадок продукт отфильтровывают, промывают 2 раза в 10 см3 воды и высушивают на воздухе. Таким образом, получают 0,12 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил) 3-тиазолидинил-(2R, 4R)) 2-оксоэтил)уреидо)фенил)пропионовой кислоты (форма B), плавящейся при 126oC /α/
Протонный ЯМР (200 МГц, ДМСО Д6 плюс несколько капель СД3СООД, δ в ч/м, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические сдвиги при 120oC: 1,4 (d, J = 7,5, 3H, -CH3), 1,54 (s, 9H, -C(CH3)3), 3,32 (dd, J = 12 и 6,5, 1H, S - CH2), 3,52 (dd, J = 12 и 7, 1H, другой H в S -CH2-), 3,62 (q, J = 7,5, 1H, Ar -CO-COOO), 3,78 (bd, 1H, в N-COCH2-N), 4,07 (d, J = 17, 1H, другой H в N -COCH2-N), 5,0 (dd, J = 7 и 6,5, 1H, N-CH-COO), 6,17 (массив, 1H, -NHCO-), 6,53 (s, 1H, S-CH-N), 6,86 (bd, J = 8, 1H, в позиции 4 в CO-NH-Ph-), 7,10-7,5 (m, 6H, ароматические), 7,92 (bt, J = 8,5, 1H, в позиции 6 в S-CH-Ph-), 8,43 (массив, 1H, ArNHCO-).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2650-2250 (широкая полоса), 1735 1650, 1610, 1595, 1560, 1490, 1455, 1370, 1230, 1150, 760, 700.
А) 2-(3-(3-(2-(4-Трет. бутоксикарбонил-2-(2-фтор-фенил)-3- диазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил)бензилпропионат (форма B) может быть получен следующим способом: аналогично описанному в примере 34, но исходя из 1,02 г 3-(2-аминоацетил-2-(2-фтор-фенил)-4-тиазолидинтрет.бутилкарбоксилата-(2R, 4R) и 0,89 г (2-(3-изоцианат-фенил) бензилпропионата (форма B). Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (50/50 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом получают 1,2 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил)бензилпропионата (форма B) в виде пасты желтого цвета, используемой при последующем синтезе.
Б) 2-(3-Изоционат-фенил)бензилпропионат (форма B) может быть получен, как в примере 21, но исходя из 2,85 г (+)-2-(3-аминофенил)бензилпропионата 1,48 см3 хлорформиата трихлорметила и 0,24 г угля. Таким образом, получают 3,1 г 2-(3-изоционатфенил)бензилпропионата (форма B) в виде масла оранжевого цвета, используемого при последующем синтезе.
В) (+)-2-(3-амино-фенил)бензилпропионат может быть получен следующим способом: в смесь 8,0 г (+)-2-(3-нитро-фенил)бензилпропионата в 35 см3 метанола и 300 см3 воды, добавляют 75 г хлорида аммония и 37,0 цинка в порошке. Реакционную среду нагревают с обратным холодильником в течение 1 ч, затем охлаждают до 0oC. Нерастворимые соли отфильтровывают, а фильтрат экстрагируют 3 раза по 200 см3 диэтилового эфира. Собранные органические фазы промывают последовательно в 100 см3 воды и 100 см3 насыщенного водного раствора хлорида натрия. Таким образом, получают, после высушивания над сульфатом магния и концентрации досуха при пониженном давлении и 40oC, 6,7 г (+)-2-(3-амино-фенил)бензилпропионата в виде масла желтого цвета, используемого при последующем синтезе.
Г) (+)-2-(3-нитро-фенил)бензилпропионат может быть получен следующим способом: в смесь, содержащую 9,75 г (+)-2-(3-нитро-фенил)пропионовой кислоты и 0,5 см3 диметилформамида в 100 см3 1,2-дихлорэтана, добавляют медленно 4,72 см3 оксалилдихлорида. Реакционную среду перемешивают в течение 3 ч при температуре около 25oC, затем добавляют в течение 12 ч при этой температуре, затем реакционную смесь промывают последовательно 2 раза в 200 см3 насыщенного водного раствора кислого карбоната натрия, в 100 см3 воды и 100 см3 насыщенного водного раствора хлорида натрия. Объединенные органические фазы высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 40oC. Полученный остаток очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 11,5 г (+)-2-(3-нитро-фенил)бензилпропината в виде масла желтого цвета, используемого при последующем синтезе.
Д) (+)-2-(3-нитро-фенил) пропионовая кислота может быть получена следующим способом: 21,5 г 2-(3-нитро-фенил)-N-(2-гидрокси-1-фенил-этил-(R))-пропионамида (форма B), растворенного в смеси 450 см3 диоксана, и 450 см3 4N водного раствора соляной кислоты, нагревают при температуре около 80oC в течение 5 ч, затем перемешивают в течение 12 ч при температуре около 25oC. Реакционную среду концентрируют наполовину выпариванием при пониженном давлении и 40oC, разбавляют добавлением 500 см3 воды и экстрагируют 2 раза по 500 см3 диэтилового эфира. Собранные органические фазы промывают последовательно 3 раза по 250 см3 воды и 250 см3 насыщенного водного раствора хлорида натрия. Таким образом, получают, после высушивания над сульфатом магния и концентрируют досуха при пониженном давлении и 40oC, 14 г (+)-2-(3-нитро-фенил)пропионовой кислоты (форма B) в виде сметанообразного продукта, используемого при последующем синтезе.
Е) 2-(3-Нитро-фенил)-N-(2-гидрокси-1-фенил-этил-(R))- пропионамид (форма B) может быть получен следующим способом: в смесь, содержащую 39,0 г 2-(3-нитро-фенил)пропионовой-(RS) кислоты и 0,5 см3 диметилформамида в 400 см3 1,2-дихлор-этана, добавляют медленно 17,2 см3 оксалилдихлорида. Реакционную среду перемешивают в течение 3 ч при температуре около 25oC, затем концентрируют досуха при пониженном давлении и 40oC. Полученный остаток растворяют в 150 см3 1,2-дихлорэтана. Полученный раствор хлорангидрида кислоты добавляют в раствор 27,4 г 2-фенил-гилицинола (2R), поддерживая температуру реакционной среды ниже 10oC. После добавления смесь перемешивают в течение 12 ч при температуре около 25oC, затем промывают последовательно 1 раз в 100 см2 воды, 500 см3 1N водного раствора соляной кислоты, два раза по 500 см3 воды и один раз в 500 см3 насыщенного водного раствора хлорида натрия. Собранные органические фазы высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 40oC. Полученные два диастереоизомера отделяют хроматографией на двуокиси кремния (элюент: метиленхлорид-этилацетат (70/30 об.)). Фракции, содержащие каждая два диастереоизомера, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 21,0 г 2-(3-нитро-фенил)-N-(2-гидрокси-1-фенил-этил-(R))-пропионамида (форма A) (первый продукт элюирования), плавящийся при 135oC, и 19,0 г 2-(3-нитро-фенил)-N-(2-гидрокси-фенил-этил-(R)/-пропионамида (форма B) (второй продукт элюирования), плавящегося при 150oC.
Пример 85. Действуют по способу, описанному в примере 84, но исходя из 3,8 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фторфенил) 3-тиазолидинил-(2R, 4R) 2-оксо-этил)уреидо)фенил)бензилпропионата (форма A) и 0,8 г 10% палладия на угле. Таким образом, получают 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил) 3-тиазолидинил-(2R, 4R) 20 оксо-этил)уреидо)фенил) пропионовой кислоты (форма A), плавящейся при 145oC. /α/
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, каолесценция линий при 120oC, характеристические химические сдвиги при 120oC:1,4 (d, J = 3, 3H, -CH3), 1,53 (s, 9H, -C(CH3)3), 3,3 и 3,48 (dd, J = 12,5 и 6, 2H, S -CH2), 3,6 (q, J = 7, 1H, Ar -CH-COO), 3,77 и 4,05 (2 dd, J = 17,5 и 5,5, 2H, N -COCH2-N), 5,0 (t, J = 6, 1H, N -CH-COO), 6,17 (t, J = 5,5 1H, -NHCO-), 6,53 (s, 1H, S -CH-N), 6,85 (bd, J = 7,5, 1H, в позиции 4 в CO-NH-Ph-), 7,05 - 7,45 (m, 6H, ароматические), 7,91 (dt, J = 8 и 1, 1H, в позиции 6 в S -CH-Ph-), 8,43 (массовое, 1H, Ar NHCO-).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 3100 - 300, 2975, 2930, 2875, 2750 - 2350 (широкая полоса), 1735, 1650, 1615, 1595, 1555, 1490, 1490, 1460, 1420, 1395, 1370, 1155, 760, 700.
2-(3-(3-(2-(4-Трет.бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил- (2R, 4R)) 2-оксо-этил)уреидо)фенил)бензилпропионат (форма A) может быть получен способом, описанным в примере 34, но исходя из 4,0 г 3-(2-амино-ацетил)-2-(2-фтор-фенил) 4-тиазолидинтрет.бутилкарбоксилат-(2R, 4R) и 4,0 г 2-(3-изоцианат -фенил)бензилпропионата (форма A). Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (40/60 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 3,8 г 2-(3-(3-(2-(4-трет. бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил -(2R, 4R)) 2-оксо-этил)уреидо)фенил)бензил пропионата (форма A) в виде аморфного продукта, используемого при последующем синтезе.
2-(3-Изоцианат-фенил)-бензилпропионат (форма A) может быть получен, как описано в примере 21, но исходя из 4,0 г (-)-2-(3-амино-фенил) бензилпропионата 2,1 см3 хлорформиата трихлорметила и 0,33 г угля. Таким образом, получают 4,7 г 2-(3-изоцианатфенил)-бензилпропионата (форма A) в виде масла оранжевого цвета, используемого при последующем синтезе.
(-)-2-(3-Амино-фенил)бензилпропионат может быть получен способом, описанным в примере 84, B, но на основе 5,3 г (-)-2-(3-нитро-фенил)бензилпропионата, 50 г хлорида аммония и 24,8 г цинка в порошке. Таким образом, получают 4,2 (-)-2-(3-аминофенил)бензилпропионата в виде желтого масла, используемого при последующем синтезе.
(-)-2-(3-Нитро-фенил)бензилпропионат может быть получен способом, описанным в примере 84, Г, но исходя из 4,45 г 2-(3-нитро-фенил)пропионовой (-)-кислоты, 0,3 см3 диметилформамида и 2,15 см3 оксалилдихлорида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 5,6 г (-)-2-(3-нитро-фенил)бензилпропионата в виде желтого масла, используемого при последующем синтезе.
2-(3-Нитро-фенил) пропионовая (-)-кислота может быть получена способом, аналогичным описанному в примере 84, Д, но исходя из 9,4 г 2-(3-нитро-фенил)-N-/2-гидрокси-1-фенил-этил-(R)/-пропионамида (форма A), растворенного в смеси 200 см3 диоксана и 200 см3 4N водного раствора соляной кислота. Таким образом, получают 5,85 г 2-(3-нитро-фенил) пропионовой (-)-кислоты в виде сметанообразного продукта, используемого при последующем синтезе.
Пример 86. Действуя по способу, аналогичному описанному в примере 77, но исходя из 0,68 г 3-(3-(2-(4-трет.бутоксикарбонил-2- (2,3-дифтор-фенил) 3-тиазолидинил) 2-оксо-этил(уреидо)фенил-метилацетата-(2R, 4R) в 6 см3 смеси вода-метанол (30/70 об.) и 0,08 г гидроокиси калия. Сырой продукт (0,45 г) растворяют в 7,5 см3 0,1N водного раствора гидроокиси натрия. Полученный раствор промывают 2 раза по 10 см3 диэтилового эфира доводят до pH 2 добавлением 1N водного раствора серной кислоты, затем экстрагируют 2 раза по 20 см3 этилацетата. Органические фазы объединяют, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 40oC. Полученный продукт хроматографией на двуокиси кремния (элюент: метиленхлорид-метанол (80/20 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом получают 0,05 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(2,3-дифтор-фенил) 3-тиазолидинил) 2-оксо-этил)уреидо)фенилметилуксусной-(2R, 4R) кислоты в виде аморфного продукта. /α/
Протонный ЯМР (200 МГц, DMCO D6 плюс несколько капель CD3COOD, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, коалесценция линий при 120oC, характеристические химические смещения при 120oC: 1,52 (s, 9H, -C(CH3)3), 3,31 и 3,51 (2dd, J = 12,5 и 6, 2H, S-CH2), 3,46 (s, 2H, Ar-CH2-COO), 3,82 (bd, 1H, в COCH2-N), 4,05 (d, J = 17, 1H, другой H в N-COCH2-N), 5,0 (t, J = 6, 1H, N-CO-COO), 6,15 (массив, 1H, -NHCO-), 6,5 (s, 1H, S-CH=N), 6,82 (bd, J = 7,5, 1H, в позиции 4 в CO-NH-Ph-), 7,05-7,45 (m, 5H, ароматические), 7,86 (bt, J = 8, 1H, в позиции 6 в S-CH-Ph-), 8,7 (массив, 1H, ArNHCO-).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 3100-3000, 2975, 2930, 2750-2350 (широкая полоса), 1735, 1655, 1615, 1595, 1560, 1490, 1405, 1370, 1150, 775, 745.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(2.3-дифтор-фенил)3- тиазолидинил)2-оксо-этил)уреидо)фенилметилацетат-(2R, 4R) может быть получен способом, описанным в примере 41, А, но исходя из 1,5 г 2-(2,3-дифтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R), 1,33 г 2-(3-(3-метоксикарбонилметил-фенил)уреидо)уксусной кислоты и 1,0 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния: (элюент: этилацетат-циклогексан (40/60 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 0,4 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(2,3-дифтор-фенил)3- тиазолидинил) 2-оксо-этил)уреидо)фенилметилацетата-(2R, 4R) в виде бесцветного масла, используемого при последующем синтезе.
2-(2,3-Дифтор-фенил)-4-тиазолидинтрет. бутилкарбоксилат (2RS, 4R) может быть получен способом, описанным в примере 34, В, но исходя из 14,3 г 2-(2,3-дифтор-фенил)4-тиазолидинкарбоновой-(2RS, 4R) кислоты, растворенной в 50 см3 хлороформа, 3,5 см3 концентрированной серной кислоты и избытка изобутилена. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (10/90 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 14,0 г 2-(2,3-дихлорфенил)4-тиазолидинтрет.бутилкарбоксилата-(2RS, 4R) в виде густого масла желтого цвета смесь изомеров (2R, 4R) и (2S, 4R), используемого при последующем синтезе.
2-(2,3-дифтор-фенил)4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 8,5 г L-цистеина и 10,1 г 2,3-дифтор-бензальдегида. Таким образом, получают 3,7 г 2-(2,3-дифтор-фенил)4-тиазолидинкарбоновой-(2RS, 4R)кислоты, плавящейся при 120oC, используемой при последующем синтезе.
Пример 87. По способу, описанному в примере 41, А, но исходя из 1,2 г 2,4-дифенилтиазолидина(2RS, 4R), 1,04 г 2-(3-(3-метилфенил)уреидо)уксусной кислоты и 1,03 г N,N'-дициклогексилкарбодиимида в 15 см3 ацетонитрила после обработки, получают 0,9 г 1-(2-(3-(3-метил-фенил)уреидо)ацетил)2,4-дифенилтиазолидина-(2RS, 4R) в виде воздушной массы.
Масса (химическая ионизация аммиаком, 70 эВ, m/z), 432 (M+).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3340, 3085, 3060, 3030, 2920, 2920, 1635, 1610, 1490, 1450, 1560, 780, 765, 695.
2,4-Дифенилтиазолидин-(2RS, 4R) может быть получен, как в примере 34, Г, но исходя из 1,9 г хлоргидрата 2-амино-2-фенил-этантиола-(R), 2,75 см3 тиэтиламина и 1,15 см3 бензальдегида в 25 см3 этанола. Таким образом получают 1,15 г 2,4-дифенил-тиазолидина-(2RS, 4R), плавящегося при 120oC.
2-Амино 2-фенил-этантиол-(R) может быть получен по известному методу (заявка на патент JP 57193447, C.A. 98, 17894r).
Пример 88. По способу, описанному в примере 9, но исходя из 1,35 г 3-(3-(2-(2-(2-фтор-фенил)-4-фенил-3-тиазолидинил) 2-оксоэтил)уреидо) фенилметилацетата-(2RS, 4R), растворенного в 25 см3 метанола, и 0,22 г гидроокиси калия, растворенной в 5 см3 воды, после обработки получают 0,45 г 3-(3-(2-(2-(2-фтор-фенил)-4-фенил-3-тиазолидинил)2-оксо-этил)уреидо) фенилуксусной-(2RS, 4R)кислоты, в виде воздушной массы.
Масса (химическая ионизация аммиаком, 70 эВ, m/z), 494 (M+), 343.
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3375, 3060, 3025, 2930, 2700-2250, 1710, 1640, 1610, 1485, 1455, 1560, 760, 700.
3-(3-(2-(2-(2-Фтор-фенил)-4-фенил-3-тиазолидинил)2-оксо-этил) уреидо)фенилметилацетат-(2RS, 4R) может быть получен, как описано в примере 41, А, но исходя из 1,3 г 2-2-фторфенил)4-фенил-тиазолидина-(2RS, 4R), 1,33 г 2-(3-(3-метоксикарбонилметил-фенил)уреидо)уксусной кислоты и 1,03 г N,N'-дициклогексилкарбодиимида в 15 см3 ацетонитрила. После обработки получают 1,5 г
3-(3-(-(2-(2-(2-фторфенил)4-фенил-3-тиазолидинил/2-оксо-этил) уреидо)фенилметилацетата-(2RS, 4R) в виде масла, используемого при последующем синтезе.
2-(2-фтор-фенил)4-фенил-3-тиазолидинил-(2RS, 4R) может быть получен как в примере 34, Г, но исходя из 1,9 г хлоргидрата 2-амино-2-фенил-этантиола-R), 2,75 см3 триэтиламина и 1,37 г 2-фтор-бензальдегида в 25 см3 этанола. Таким образом, получают 1,5 г 2-(2-фтор-фенил)4-фенил-3-тиазолидинил-(2RS, 4R) в виде масла, используемого при последующем синтезе.
Пример 89. Действуют по способу, описанному в примере 9, но исходя из 0,9 г 1-(2-(3-(3-этоксикрабонил-фенил)уреидо)ацетил)-5- (2-метилфенил)трет. бутилпролината-(2RS, 5SR), растворенного в 30 см3 метанола и 0,1 г гидроокиси калия, растворенной в 15 см3 воды. После обработки и кристаллизации в смеси окиси диизопропила-ацетатизопропила (9-1 объемов), получают 0,35 г 3-(3-(-(2-(2-трет. бутоксикарбонил 5-(2-метил-фенил)1-пирролидинил) 2-оксо-этил)уреидо)бензойной-(2RS, 5SR)кислоты, плавящейся при 235oC.
1-(2-(3-(3-Этоксикрабонил-фенил)уреидо)ацетил)-5- (2-метилфенил)трет.бутилпролината-(2RS, 5SR) может быть получен, как описано в примере 41, А, но исходя из 1,3 г 5-(2-метил-фенил)трет.бутилпролината-(2RS, 5SR), 1,32 г 2-(3-(3-этоксикарбонил-фенил)уреидо)уксусной кислоты и 1,03 г N,N'-дициклогексилкарбодиимида в 50 см3 тетрагидрофурана. После обработки получают 1,9 г 1-(2-(3-(3-этоксикарбонил-фенил)уреидо)ацетил)5-(2-метил-фенил) трет.бутилпролината-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
5-(2-Метил-фенил)трет. бутилпролината-(2RS, 5SR) может быть получен следующим образом: суспензию 3 г магния и растворе 4 г смеси двух эпимеров в положении 4 5-(2-метил-фенил)-4-фенилсульфонил-трет. бутилпролината-(2RS, 5SR) в 200 см3 метанола перемешивают в течение 3 ч при температуре около 20oC. Реакционную смесь затем выливают в 20 см3 нормального водного раствора соляной кислоты, затем экстрагируют три раза по 200 см3 дихлорметана. Органические экстракты объединяют, промывают два раза по 100 см3 насыщенного водного раствора хлорида натрия, высушивают над сульфатом магния и концентрируют при пониженном давлении. Остаток хроматографируют на двуокиси кремния (элюент: циклогексан-этилацетат (70/30 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении. Таким образом, получают 0,6 г 5-(2-метил-фенил)трет.бутилпролината-(2RS, 5SR) в виде масла, используемого при последующем синтезе.
А) 5-(2-метил-фенил)4-фенилсульфонилтрет.бутилпролинат-(2RS, 5SR) может быть получен следующим способом: в суспензию 5 г ацетата серебра в растворе 3,4 г фенилвинилсульфона и 4,7 N-(ортометилбензилиден)-трет.бутилгицината в 150 см3 ацетонитрила, прикапывают 2,8 см3 триэтиламина при температуре около 20oC. Реакционную смесь перемешивают в течение 2 ч при температуре около 20oC, затем выливают в 200 см3 насыщенного водного раствора хлорида аммония. Водную фазу фильтруют и экстрагируют три раза по 100 см3 дихлорметана. Органические экстракты объединяют, промывают в 100 см3 насыщенного водного раствора хлорида натрия, высушивают над сульфатом магния и концентрируют при пониженном давлении. Таким образом, получают после кристаллизации в диэтиловом эфире 5 г 5-(2-метил-фенил) 4-фенилсульфонил-трет.бутилпролината-(2RS, 5SR), (смесь двух эпимеров в 4), плавящегося при 180oC.
Б) N-(ортометилбензилиден)-трет. бутилглицинат может быть получен следующим способом: в суспензию 3 г молекулярного сита  в растворе 3,35 г хлоргидрата трет.бутилглицината в 2,4 см3 ортотолуальдегида и 50 см3 дихлорметана, прикапывают 2,8 см3 триэтиламина при температуре около 20oC. Реакционную смесь перемешивают в течение 20 ч при температуре около 20oC, фильтруют и концентрируют при пониженном давлении. Остаток извлекают 250 см3 диэтилового эфира, фильтруют и концентрируют при пониженном давлении. Таким образом, получают 4,7 г N-(ортометилбензилидена)-трет.-бутилглицината в виде масла, используемого при последующем синтезе.
в растворе 3,35 г хлоргидрата трет.бутилглицината в 2,4 см3 ортотолуальдегида и 50 см3 дихлорметана, прикапывают 2,8 см3 триэтиламина при температуре около 20oC. Реакционную смесь перемешивают в течение 20 ч при температуре около 20oC, фильтруют и концентрируют при пониженном давлении. Остаток извлекают 250 см3 диэтилового эфира, фильтруют и концентрируют при пониженном давлении. Таким образом, получают 4,7 г N-(ортометилбензилидена)-трет.-бутилглицината в виде масла, используемого при последующем синтезе.
Пример 90. Действуют по способу, описанному в примере 9, но исходя из 1,1 г 1-(2-(3-(3-этоксикарбонил-фенил)уреидо-)ацетил) 5-(2-фторфенил) трет-бутилпролината (2RS, 5SR), растворенного в 30 см3 метанола, и 0,12 г гидроокиси калия, растворенной в 15 см3 воды. После обработки и кристаллизации в окиси диизопропила получают 0,4 г 3-(3-(2-(2-трет.бутоксикарбонил 5-(2-фтор-фенил) 1-пирролидинил) 2-оксо-этил)уреидо)бензойной-(2RS, 5SR) кислоты, плавящейся при 180oC.
1-(2-(3-(3-этоксикарбонил-фенил)уреидо)ацетил)-5-(2фтор-фенил)- трет.бутилпролинат (2RS, 5SR) может быть получен, как описано в примере 41, A, но исходя из 0,8 г 5-(2-фтор-фенил)трет.бутилпролината (2RS, 5SR), 0,8 г 2-(3-(3-(этоксикарбонил-фенил)уреидо) уксусной кислоты и 0,62 г N, N'-дициклогексилкарбодиимида в 30 см3 тетрагидрофурана. После обработки, получают 1,5 г 1-(2-(3-(3-этоксикарбонилфенил)уреидо)ацетил)-5-(2-фтор-фенил) трет. бутилпролината (2RS, 5SR) в виде воздушной массы ,используемой при последующем синтезе.
А) 5-(2-Фтор-фенил)трет.бутилпролинат (2RS, 5SR) может быть получен следующим образом: суспензию 5,7 г двунатриевого кислого фосфата и 7,7 г амальгамы с 6% натрия (в ртути) в растворе 4,06 г смеси двух эпимеров в положении 4 5-(2-фтор-фенил)-4-фенилсульфонил- трет. бутилпролината-(2RS, 5RS) в 150 см3 метанола, перемешивают в течение 20 ч при температуре около 20oC. Затем реакционную смесь выливают в 200 см3 воды, а ртуть отделяют декантированием. Водную фазу экстрагируют три раза по 100 см3 этилацетата. Органические экстракты объединяют, промывают два раза по 100 см3 воды, высушивают над сульфатом магния и концентрируют при пониженном давлении. Остаток подвергают хроматографии на двуокиси кремния (элюент: циклогексан-этилацетат (70/30 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении. Таким образом, получают 0,4 г 5-(2-фтор-фенил) трет. бутилпролината (2RS, 5SR) в виде масла, используемого при последующем синтезе.
5-(2-Фтор-фенил)-4-фенилсульфонилтрет. бутилпролинат-(2RS, 5RS) может быть получен, как описано в примере 89, A, но исходя из 4,8 г N-(ортофторбензилиден)-трет. бутилглицината, 5 г ацетата серебра, 3,4 г фенилвинилсульфона и 2,8 см3 триэтиламина. После обработки и кристаллизации в диэтиловом эфире получают 8 г 5-(2-фтор-фенил)-4- фенилсульфонилтрет.бутилпролината (2RS, 5RS), смесь двух эпимеров в положении 4), плавящуюся при 200oC.
N-(ортофторбензилиден) трет. бутилглицинат может быть получен, как описано в примере 89, Б, но исходя из 2 см3 ортофторбензальдегида, 3,35 г хлоргидрата трет.бутилглицината, 2,8 см3 триэтиламина и 3 г молекулярного сита 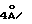 в 50 см3 дихлорметана. После обработки получают 4,8 г N-(ортофторбензилиден)-трет. бутилглицината в виде масла, используемого при последующем синтезе.
в 50 см3 дихлорметана. После обработки получают 4,8 г N-(ортофторбензилиден)-трет. бутилглицината в виде масла, используемого при последующем синтезе.
Пример 91. Действуют по способу, описанному в примере 2, но исходя из 1,12 г 1-(2-амино-ацетил)-5-фенил-трет.бутил-(2RS, 5SR) и 1,3 см3 изоцианата парахлорфенила в 50 см3 тетрагидрофурана. После обработки получают 1,2 г 1-(2-(3-(4-хлор-фенил)уреидо) ацетил)5-фенил-трет.бутилпролината-(2RS, 5SR) в виде аморфного продукта.
Протонный ЯМР (250 МГц, DMCO D6, δ в ч/млн) 1,50 (s, 9H, (CH3)3), 1,85 (m, 2H, CH2) 2,2 и 2,4 (2m, 2H, CH2), 3,25 и 3,85 (ABX, 2H, CH2), 4,30 (dd, 1H, CHN), 5,20 (dd, 1H, CHN), 6,3 (t, 1H заменяемый, NH), 7,2 - 7,6 (m, 9H, ароматические), 9 (s, 1H заменяемый, NH).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3370, 3065, 3030, 2980, 2930, 2875, 1735, 1555, 1490, 1450, 1445, 1365, 1150, 830, 760, 700.
Пример 92. Действуя по способу, аналогичному в примере 3, но исходя из 3,05 г 1-(2-амино-ацетил)-5-фенил-трет.бутилпролината (2RS, 5SR), 1,78 г N, N'-диимидазол-карбонила и 4,5 г 1-(3-аминофенил)этансульфоната тетрабутиламмония-(RS) в 100 см3 1,2-дихлорэтана. После обработки получают 0,6 г 1-(3-(3-(2-(2-трет.бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо)фенил)этансульфоната калия-(2RS, 5SR), смесь двух эпимеров в 1) в виде аморфного продукта.
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн), 1,6 (s + d, 12H, CCH3 и (CH3)3), 1,8-2,4 (m, 4H, 2CH2), 3,5 (m, 1H, CHSO3), 3,1 и 3,8 (ABX, 2H, CH2N) 4,2 (dd, 1H, CHN), 5,1 (dd, 1HCHN), 6,2 (t, 1H заменяемый, NH), 6,8-7,6 (m, 9H, ароматические), 8,7 (bs, 1H заменяемый, NH).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 3060, 3025, 2975, 2930, 2875, 1730, 1555, 1490, 1445, 1390, 1360, 1215, 1115, 1030, 790, 755, 700.
Пример 93.
Действуя по способу, аналогичному в примере 41, A, но исходя из 4,95 г 5-фенил-трет. бутилпролината (2RS, 5SR), 5,04 г 2-(3-(3-метоксикарбонил-фенил)уреидо) уксусной кислоты и 4,12 г N,N'-дициклогексилкарбодиимида в 75 см3 тетрагидрофурана. После обработки и кристаллизации в смеси оксида диизопропилацетата изопропила (90 - 10 об.) получают 1,2 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо)ацетил) 5-фенил-трет. бутилпролината (2RS, 5SR), плавящегося при 141oC.
2-(3-(3-Метоксикарбонил-фенил)уреидо)уксусная кислота может быть получена, как описано в примере 1, A, но исходя из 7,5 г глицина, 8,4 г кислого карбоната натрия в 150 см3 воды и 17,7 г 3-изоцианатметилбензоата. После обработки получают 14,5 г 2-(3-(3-метоксикарбонил-фенил)уреидо)уксусной кислоты, плавящейся при 215oC.
Пример 94. Действуют по способу, аналогичному в примере 9, но исходя из 2,6 г 1-(2-(3-(3-этоксикарбонил-фенил)уреидо)ацетил)-5- фенил-трет.бутилпролината-(2S, 5R) ,растворенного в 40 см3 метанола, и 0,33 г гидроокиси калия, растворенной в 20 см3 воды. После обработки получают 1,8 г 3-(3-(2-(2-трет. бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо бензойной-(2S, 5R) кислоты, аналитические данные которой соответствуют аналитическим данным правовращающегося энантиомера, полученного хроматографией на стационарной хиральной фазе рацемического продукта.
1-(2-(3-(3-Этоксикарбонил-фенил)уреидо)ацетил) 5-фенил-трет. бутилпролинат-(2S, 5R) может быть получен следующим способом: исходят из 6,6 г 1-(2-(3-(3-этоксикарбонил-фенил)уреидо) ацетил) 5-фенил-трет.бутилпролината-(2RS, 5SR), отделяют высокоскоростной жидкостной хроматографией на 400 г подложки, способ изготовления которой описан ниже, содержащейся в колонке длиной 26 см и диаметром 6 см, используя смесь гексан-этанол (70 - 30 об.) в качестве подвижной фазы с расходом 70 см3/мин, осуществляя последовательное элюирование: 2,9 г 1-(2-(3-(3-этоксикарбонил-фенил)уреидо)ацетил) 5-фенил-трет. бутилпролината-(2S, 5R) с вращательной способностью /α/
Пример 95. Действуют по способу, описанному в примере 9, но исходя из 3,5 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо)ацетил)- 5-фенил-трет. бутилпролината-(2S, 5R), растворенного в 60 см3 метанола, и 0,45 г гидроокиси калия, растворенной в 30 см3 воды. После обработки получают 2,0 г 3-(3-(2-(2-трет. бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо) бензойной-(2S, 5R) кислоты, аналитические данные которой соответствуют аналитическим данным правовращающегося энантиомера, полученного хроматографией на стационарной хиральной фазе рацемата.
1-(2-(3-(3-Метоксикарбонил-фенил)уреидо)ацетил)-5-фенил- трет. бутилпролинат-(2S, 5R) может быть получен, как описано в примере 41, A, но исходя из 0,1 г 5-фенил-трет.бутилпролината-(2S, 5R), 0,1 г 2-(3-(3-метоксикарбонил-фенил)уреидо)уксусной кислоты и 0,08 г N,N'-дициклогексилкарбодиимида в 5 см3 тетрагидрофурана. После обработки получают 0,1 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо)ацетил) 5-фенил-трет. бутилпролината (2S, 5R), аналитические данные которого соответствуют аналитическим данным правовращающегося энантиомера, полученного хроматографией на стационарной хиральной фазе рацемата.
5-Фенил-трет.бутилпролинат-(2S, 5R) может быть получен следующим способом: в эмульсию 0,83 г 5-фенил- Δ- -5-пирролин-2-трет.бутилкарбоксилата-(S) в смеси 0,5 см3 этанола и 1,5 см3 воды добавляют в течение 5 мин при температуре около 2oC раствор 0,14 г боргидрида натрия и 0,07 г карбоната калия в 0,8 см3 воды. Реакционную среду перемешивают в течение 20 ч при температуре около 20oC, затем добавляют раствор 0,14 г боргидрида натрия и 0,07 г карбоната калия в 0,8 см3 воды. Реакционную смесь снова перемешивают в течение 70 ч при температуре около 20oC, затем разбавляют 25 см3 воды и экстрагируют три раза по 20 см3 дихлорметана. Органические экстракты объединяют, промывают в 10 см3 воды, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении и 50oC. Остаток очищают хроматографией на двуокиси кремния (элюент: дихлорметан). Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении. Таким образом, получают 0,1 г 5-фенилпролината трет-бутила-(2S, 5R) в виде масла, используемого при последующем синтезе.
5-фенил- Δ-5-пирролин-2-трет. бутилкарбоксилат-(S) может быть получен следующим способом: в раствор 1,8 г 2-трет.бутоксикарбониламино-5-оксо-5-фенил-трет-бутил-(S)-пентаноата в 25 см3 дихлорметана добавляют при температуре около 20oC 2,3 см3 трифторуксусной кислоты. Реакционную смесь перемешивают в течение 6 ч при температуре около 20oC, затем добавляют 120 см3 насыщенного водного раствора кислого карбоната натрия. Органическую фазу отделяют декантированием, промывают в 20 см3 дистиллированной воды, высушивают над сульфатом магния и концентрируют при пониженном давлении. Таким образом, получают 0,9 г 5-фенил- Δ- -5-пирролин-2-трет.бутил-(S)-карбоксилата в виде масла, используемого при последующем синтезе.
2-Трет. бутоксикарбониламино-5-оксо-5-фенилтрет.бутил (S)пентаноат может быть получен следующим способом: в суспензию 0,72 г магния в 20 см3 тетрагидрофурана добавляют в течение 35 мин при 20 - 30oC раствор 2,8 см3 бромбензола в 60 см3 тетрагидрофурана. Реакционную среду снова перемешивают при температуре около 24oC в течение 145 мин, затем вводят в течение 20 мин в раствор 5,7 г 1-трет.бутоксикарбонил-5-оксопирролидин-2-трет.бутилкарбоксилата (S) в 80 см3 тетрагидрофурана, поддерживаемый при температуре около -75oC. Реакционную среду снова перемешивают в течение 3 ч при температуре около -70oC, затем нагревают до температуры около -15oC. Добавляют в течение 15 мин 100 см3 10%-ного водного раствора хлорида аммония. Водная фаза отделяется декантированием и экстрагируется три раза по 100 см3 диэтилового эфира. Органические фазы объединяют и промывают два раза по 25 см3 воды, высушивают над сульфатом магния и концентрируют при пониженном давлении и температуре около 50oC. Остаток очищают кристаллизацией в 20 см3 пентана. Таким образом, получают 2,5 г 2-трет.бутуксикарбониламино 5-оксо-5-фенил-трет. бутил-(S) пентаноата, плавящегося при 107oC. Этот продукт может находиться в виде 1-трет. бутоксикарбонил-5-гидрокси-5-фенил-пирролидин-2- трет-бутилкарбоксилата (2S, 5RS), плавящегося при 85oC.
1-Трет. бутоксикарбонил-5-оксо-пирролидин-2-трет. бутил-(S) карбоксилат может быть получен по известному методу (J. ACKERMANN et M. MATTHES, Helv. Chim. Acta, 73, 122 - 132, 1990).
1-(2-(3-(3-Метоксикарбонил-фенил)уреидо)ацетил)-5-фенил-трет. бутилпролинат (2S, 5R) может быть получен следующим способом: исходят из 7,8 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо)ацетил) 5-фенил-трет. бутилпролината (2RS, 5SR), отделяют высокоскоростной жидкой хроматографией на 400 г подложки, способ получения которой описан в предыдущем примере, находящейся в колонке длиной 26 см и диаметром 6 см, используя смесь гексан-этанол (70 - 30 об. ) в качестве подвижной фазы, с расходом 70 см3/мин последовательно элюируя 3,7 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо)ацетил)-5- фенил трет. бутилпролината-(2S, 5R) с вращательной способностью /α/
Пример 96. Действуют по способу, описанному в примере 9, но исходя из 2,6 г 1-(2-(3-(3-этоксикарбонил-фенил)уреидо)ацетил)- 5-фенил трет-бутилпролината-(2R, 5S), растворенного в 60 см3 метанола, и 0,3 г гидроокиси калия, растворенной в 40 см3 воды. После обработки получают 1,3 г 3-(3-(2-(2-трет.бутоксикарбонил 5-фенил-1-пирролидинил)2-оксо-этил)уреидо)бензойной-(2R, 5S) кислоты, аналитические данные которой соответствуют аналитическим данным левовращающегося энантиомера, полученного хроматографией на стационарной хиральной фазе рацемата.
Пример 97. Действуют по способу, описанному в примере 9, но исходя из 3,38 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо)ацетил) 5-фенил-трет. -бутилпролината-(2R, 5S), растворенного в 60 см3 метанола, и 0,45 г гидроокиси калия, растворенной в 20 см3 воды. После обработки получают 2,0 г 3-(3-(2-(2-трет.бутоксикарбонил-5- фенил-1-пирролидинил)2-оксо-этил)уреидо)бензойной-(2R, 5S) кислоты, аналитические данные которой соответствуют аналитическим данным левовращающегося энантиомера, полученного хроматографией на стационарной хиральной фазе рацемата.
Пример 98. Действуют, как в примере 9, но исходя из 1,9 г 3-(3-(2-(2-трет.бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо)фенилметилацетата (2S, 5R), растворенного в 60 см3 метанола, и 0,22 г гидроокиси калия, растворенной в 30 см3 воды. После обработки получают 0,65 г 3-(3-(2-(2-трет.бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо) фенилуксусной-(2S, 5R) кислоты, плавящейся при 112oC, /α/
3-(3-(2-(2-Трет. бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо) фенилметилацетат-(2S, 5R) может быть получен, как описано в примере 41, A, но исходя из 1,2 г 5-фенил-трет.бутилпролината-(2S, 5R), 1,29 г 2-(3-(3-метоксикарбонилметил-фенил) уреидо) уксусной кислоты и 1 г N,N'-дициклогексилкарбодиимида в 50 см3 ацетонитрила. После обработки получают 1,9 г 3-(3-(2-(2-трет. бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо) фенилметилацетата-(2S, 5R) в виде воздушной массы, используемой при последующем синтезе.
5-Фенилтрет. бутилпролинат-(2S, 5R) может быть получен следующим способом: в раствор 1,25 г 5-фенил- Δ- -5-пирролин-2-трет. бутилкарбоксилата (S) в 50 см3 этанола вводят 0,02 г окиси платины. Суспензию перемешивают в течение 3 ч при температуре около 20oC в атмосфере водорода (130 кПа). Катализатор отфильтровывают, а фильтрат концентрируют досуха при пониженном давлении и 45oC. Остаток очищают хроматографией на двуокиси кремния (элюент: дихлорметан-метанол (97,5-2,5 об.). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 1,2 г 5-фенилтрет.бутилпролината (2S, 5R) в виде масла, используемого при последующем синтезе.
2-Фенил -Δ- 5-пирролин-2-трет. бутилкарбоксилат (S) может быть получен следующим образом: в раствор 3,65 г 2-трет.бутоксикарбониламино-5-оксо-5-фенилтрет. бутилпентаноата (S) в 50 см3 дихлорметана добавляют при температуре около 20oC 1,4 см3 иодотриметилсилана. Реакционную смесь перемешивают в течение 20 ч при температуре около 20oC, затем добавляют 50 см3 насыщенного водного раствор кислого карбоната натрия. Органическую фазу отделяют декантированием, промывают в 20 см3 дистиллированной воды, высушивают над сульфатом магния и концентрируют при пониженном давлении. Таким образом, получают 2,0 г 5-фенил -Δ- 5-пирролин-2-трет.бутилкарбоксилата (S) в виде масла, используемого при последующем синтезе.
Пример 99. Действуют по способу, описанному в примере 2, но исходя из 1,93 г 1-(2-амино-ацетил) 5-фенилтрет. бутилпролината (2RS, 5SR) и 3,61 г 3-изоцианатбензилбензоата в 50 см3 тетрагидрофурана. После обработки получают 1 г 1-(2-(3-(3-бензилоксикарбонил-фенил)уреидо)ацетил) 5-фенилтрет.бутилпролината-(2RS, 5SR), плавящегося при 75oC.
3-Изоцианатбензилбензоат может быть получен, как описано в примере 64, A, но исходя из 27 г хлоргидрата 3-аминобензил бензоата в 200 см3 толуола, 14,4 см3 триэтиламина, 2 г угля и 12,5 см3 хлорформиата трихлорметила в 200 см3 толуола. После обработки получают 27 г 3-изоцианатбензилбензоата в виде масла, используемого при последующем синтезе.
3-аминобензилбензоат может быть получен по известному способу (H.A. SHONLE et coll., J.Am. Chem. Soc., 43, 361, 1921).
Пример 100. В раствор 1,7 г 1-(2-(3-(3-бензилоксикарбонил-фенил) уреидо)-ацетил 5-фенилэтилпролината-(2RS, 5SR) в 100 см3 этанола добавляют 0,5 г 5% палладия на угле. Суспензию перемешивают в течение 20 ч при температуре около 25oC в атмосфере водорода (130 кПа). Катализатор отфильтровывают, а фильтрат концентрируют досуха при пониженном давлении. Остаток рекристаллизуют в 80 см3 этанола. Таким образом, получают 0,7 г 3-(2-(2-этоксикарбонил-5- фенил-1-пирролидинил) 2-оксо-этил)уреидо) бензойной-(2RS, 5SR) кислоты, плавящейся при 240oC.
1-(2-/3-(3-Бензилоксикарбонил-фенил)уреидо/ацетил 5-фенил-этилпролинат-(2RS, 5SP) может быть получен, как описано в примере 41, A, но исходя из 2 г 5-фенил этилпролината-(2RS, 5SR), 2,99 г 2-(3-(3-бензилоксикарбонил- фенил)уреидо)уксусной кислоты и 2,07 г N,N'-дициклогексилкарбодиимида в 180 см3 тетрагидрофурана. После обработки получают 1,95 г 1-(2-(3-(3-бензилоксикарбонил-фенил)уреидо)ацетил 5-фенилэтилпролината-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
2-(3-(3-Бензилоксикарбонил-фенил)уреидо)уксусная кислота может быть получена, как описано в примере 1, A, но исходя из 3,97 г глицина и 14,62 г карбоната калия, растворенного в 90 см3 воды, и 13,4 г 3-изоцианатбензилбензоата, в растворенного в 80 см3 1,4-диоксана. После обработки получают 13 г 2-(3-(3-бензилоксикарбонил-фенил)уреидо) уксусной кислоты, используемой при последующем синтезе.
5-Фенилэтилпролинат- (2RS, 5SR) может быть получен следующим способом: в раствор 5,5 г 5-фенилпролина-(2RS, 5SR) в 50 см3 этанола прикапывают 0,5 см3 концентрированной серной кислоты. Затем реакционную смесь перемешивают при температуре около 80oC и концентрируют в течение 5 ч, затем охлаждают до температуры около 20oC и концентрируют при пониженном давлении. Остаток извлекают 50 см3 воды, доводят до pH около 9 добавлением нормального водного раствора гидроокиси натрия и экстрагируют три раза по 100 см3 этилацетата. Объединенные органические экстракты промывают в 50 см3 насыщенного водного раствора хлорида натрия, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении. Остаток подвергают хроматографии на двуокиси кремния (элюент: циклогексан-этилацетат (90-10 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении. Таким образом, получают 2,16 г 5-фенилэтилпролината-(2RS, 5SR) в виде масла, используемого при последующем синтезе.
Пример 101. Действуют по способу, описанному в примере 100, но исходя из 10,2 г 1-(2-(3-(3-бензилоксикарбонил-фенил)уреидо/ацетил) 5-фенилтрет.бутилпролината-(2RS, 5SR) и 1 г 5% палладия на угле в 300 см3 этанола. После обработки получают 7,1 г 3-(3-(2-(2-трет. бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо)бензойной (2RS, 5SR) кислоты, плавящейся при 236oC.
Пример 102. Действуют по способу, описанному в примере 100, но исходя из 0,8 г (2-(3-(3-бензилоксикарбонил-фенил)уреидо)ацетил)-5- (3-метил-фенил) трет. бутилпролината-(2RS, 5SR), растворенного в 50 см3 этилацетата, и 0,1 г 5% палладия на угле. После обработка получают 0,45 г 3-(3-(2-(2-трет.бутоксикарбнил-5-(3-метил-фенил)-1- пирролидинил) 2-оксо-этил)уреидо) бензойной-(2RS, 5SR) кислоты, плавящейся при 208oC.
1-(2-(3-(3-Бензилоксикарбонил-фенил)уреидо)ацетил)-5-(3-метил- фенил) трет. бутилпролинат-(2RS, 5SR) может быть получен, как описано в примере 41, A, но исходя из 0,4 г 5-(3-метил-фенил)-трет. бутилпролината-(2RS, 5SR), 0,5 г 2-(3-(3-бензилоксикарбонилфенил) уреидо)уксусной кислоты и 0,31 г N,N'-дициклогексилкарбодиимида в 20 см3 ацетонитрила. После обработки получают 0,8 г 1-(2-(3-(3- бензилоксикарбонил-фенил)уреидо/ацетил -5-(3-метилфенил) трет. бутилпролината-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
5-(3-Метил-фенил) трет.бутилпролинат-(2RS, 5SR) может быть получен, как в примере 2, A, но исходя из 1 г 1-трет.бутоксикарбонил- 5-(3-метил-фенил) трет. бутилпролината-(2RS, 5SR) и 0,4 см3 иодотриметилсилана в 40 см3 хлороформа. После обработки, получают 0,4 г 5-(3-метил-фенил) трет.бутилпролината-(2RS, 5SR) в виде масла, используемого при последующем синтезе.
1-Трет. бутоксикарбонил-5-(3-метил-фенил) трет.бутил-пролинат (2RS, 5SR) может быть получен, как описано в примере 90, A, но исходя из 2,4 г однозамещенного фосфата натрия и 7,63 г амальгамы с 6% натрия (в ртути) в растворе 2,5 г смеси двух эпимеров в положении 4 1-трет.бутоксикарбонил-5-(3-метил-фенил)-4-фенилсульфонил трет.бутилпролината (2RS, 5RS) в смеси 20 см3 метанола 40 см3 тетрагидрофурана. После обработки получают 1 г 1-трет.бутоксикарбонил-5-(3-метил-фенил) трет.бутилпролината-(2RS, 5SR) в виде масла, используемого при последующем синтезе.
1-Трет. бутоксикарбонил-5-(3-метил-фенил) 4-фенилсульфонилтрет. бутилпролинат (2RS, 5RS) может быть получен следующим способом: в раствор 2,01 г смеси двух эпимеров в положении 4 5-(3-метил-фенил) 4-фенилсульфонил трет. бутилпролината-(2RS, 5RS) в 50 см3 дихлорметана, добавляют последовательно 0,42 г кислого карбоната натрия, затем раствор 1,09 г бикарбоната дитрет-бутила в 20 см3 дихлорметана. Реакционную смесь перемешивают в течение 72 ч при температуре около 20oC, затем добавляют 50 см3 воды. Органическую фазу отделяют декантированием, высушивают над сульфатом магния и концентрируют досуха при пониженном давлении. Таким образом, получают 2,5 г 1-трет.бутоксикарбонил-5- (3-метил-фенил)-4-фенилсульфонилтрет. бутилпролината-(2RS, 5R), смесь двух эпимеров в 4) в виде масла, используемого при последующем синтезе.
5-(3-Метил-фенил)-4-фенилсульфонил трет. бутилпролинат-(2RS, 5RS) может быть получен, как описано в примере 89, A, но исходя из 4,7 г N-(метаметилбензилиден)-трет.бутилглицината, 5 г ацетата серебра, 3,4 г фенилвинилсульфона и 2,8 см3 триэтиламина. После обработки получают 6,1 г 5-(3-метил-фенил) 4-фенилсульфонилтрет. бутилпролината- (2RS, 5RS), смесь двух эпимеров в 4, плавящуюся при 139oC.
N-(метаметилбензилиден) трет. бутилглицинат может быть получен, как описано в примере 89, Б, но на основе 2,4 см3 метатолуальдегида, 3,35 г хлоргидрата трет-бутилглицината, 2,8 см3 триэтиламина и 3 г молекулярного сита  в 30 см3 дихлорметана, после обработки, получают 4,7 г N-(метаметилбензилиден) -трет-бутилглицината в виде масла, используемого при последующем синтезе.
в 30 см3 дихлорметана, после обработки, получают 4,7 г N-(метаметилбензилиден) -трет-бутилглицината в виде масла, используемого при последующем синтезе.
Пример 103. Действуют по способу, описанному в примере 9, но исходя из 1,2 г смеси 1-(2-(3-(3-этоксикарбонил-фенил)уреидо)-ацетил) 3-фенилтрет.бутилпролината-(2RS, 3SR) и -(2RS, 3RS) 85/15 по весу, в растворенной в 30 см3 тетрагидрофурана, и 2,4 см3 нормального раствора гидроокиси натрия, разбавленной в 15 см3 воды. После обработки получают смесь 0,7 г 3-(3-(2- (2-трет. бутоксикарбонил-3-фенил-1-пирролидинил) 2-оксо-этил)уреидо) бензойной кислоты (2RS, 3SR) и -(2RS, 3RS) 85/15 по весу, плавящуюся при 225oC.
1-(2-(3-(3-Этоксикарбонилфенил)уреидо)ацетил) 3-фенил- трет. бутилпролинат-(2RS, 3SR) может быть получен, как описано в примере 41, A, но исходя из 1 г смеси 3-фенилтрет.бутилпролината (2RS, 3SR) и -(2RS, 3RS) 85/15 по весу, 1,1 г 2-(3-3- этоксикарбонилфенил)уреидо) уксусной кислоты и 0,83 г N, N'- дициклогексилкарбодиимида в 30 см3 тетрагидрофурана. После обработки получают смесь 1,2 г 1-(2-(3-(3-этоксикарбонил-фенил) уреидо)ацетил) 3-фенилтрет. бутилпролината-(2RS, 3SR) и -(2RS, 3RS) 85/15 вес., плавящуюся при 154oC.
3-Фенилтрет. бутилпролинат-(2RS, 3SR) и 3-фенилтрет.бутилпролинат -(2RS, 3RS) могут быть получены, как описано в примере 1, Б, но исходя из 10 г смеси 3-фенилпролина (2RS, 5SP) и -(2RS, 5RS), изобутилена и 3 см3 серной кислоты в 200 см3 хлороформа. После обработки и хроматографии на двуокиси кремния (элюент: циклогексан-этилацетат (50 - 50 об.)) получают 1 г смеси 3-фенилтрет.бутилпролината-(2RS, 3SR) и -(2RS, 2RS) 85/15 вес. в виде масла, используемой при последующем синтезе, и 2,5 г смеси 3-фенилтрет.бутилпролината-(2RS, 3RS) и -(2RS, 3SR) 85.15 вес. в виде масла, используемого при последующем синтезе.
3-Фенилпролин-(2RS, 5SR) и -(2RS, 5RS) могут быть получены по известным методам (Y.N BELOKON et coll., J. Chem. Soc., Perkin. Trans., 1, 2075, 1988 и J. RIVIER., G.R. MARSHALL Peptides, Chemistry, Structure and Biology, Proceedings of the Eleventh American Peptide Symposium. July 9 - 14 1989, La Jolla, California, USA, ESCOM, Leiden, 1990).
Пример 104. Действуют по способу, описанному в примере 9, но исходя из 2,5 г смеси 1-(2-(3-(3-этоксикарбонил-фенил)уреидо)ацетил) 3-фенилтрет.бутилпролината-(2RS, 3RS) и -(2RS, 3SR) 80/20 вес. растворенной в 30 см3 тетрагидрофурана и 5 см3 нормального раствора гидроокиси натрия, разбавленного в 15 см3 воды. После обработки получают смесь 1 г 3-(3-(2-(2-трет. бутоксикарбонил-3-фенил-1-пирролидинил)2-оксо-этил) уреидо)бензойной- (2RS, 3RS) и -(2RS, 3SR) кислот (90-10 вес.), плавящуюся при 200oC.
1-(2-(3-(3-Этоксикарбонил-фенил)уреидо)ацетил) 3-фенил трет-бутилпропинат-(2RS, 3RS) может быть получен, как описано в примере 41, А, но исходя из 2,5 г смеси 3-фенилтрет.бутилпропината-(2RS, 3RS) и -(2RS, 3SR)(90/10 вес. ), 2,7 г 2-(3-(3-этоксикарбонилфенил)уреидо)уксусной кислоты и 2,1 г N, N'-дициклогексилкарбодиимида в 50 см3 тетрагидрофурана. После обработки получают смесь 1,2 г 1-(2-(3-(3-этоксикаронил-фенил)уреидо)ацетил) 3-фенилтрет. бутилпропината- (2RS, 3RS) и -(2RS, 3SR) 80/20 вес., плавящуюся при 141oC.
Пример 105. Действуют по способу, описанному в примере 9, но исходя из 2,1 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо)ацетил)-4-фенил- трет.бутилпролината-(2S, 4S), растворенного в 30 см3 метанола, и 0,5 г гидроокиси калия, растворенной в 15 см3 воды. После обработки и кристаллизации в смеси окись диизопропилацетат изопропила (90-10 об.), получают 0,65 г 3-(3-(2-(2-трет. бутоксикарбонил-4-фенил-1-пирролидинил) 2-оксо-этил)уреидо)бензойной-(2S, 4S) кислоты, плавящейся при 144oC.
1-(2-(3-(3-Метоксикарбонил-фенил)уреидо)ацетил) 4-фенил-трет. бутилпролинат-(2S, 4S) может быть получен, как описано в примере 41, А, но исходя из 1,2 г 4-фенилтрет.бутилпролината-(2S, 4S), 1,2 г 2-(3-(3-метоксикарбонил-фенил) уреидо)уксусной кислоты и 1 г N, N'-дициклогексилкарбодиимида в 30 см3 ацетонитрила. После обработки получают 2,1 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо)-ацетил)4- фенилтрет. бутилпролината-(2S, 4S) в виде воздушной массы, используемой при последующем синтезе.
4-фенилтрет. бутилпролинат-(2S, 4S) может быть получен, как описано в примере 1, Б, но исходя из 3,4 г 4-фенилпролина-(2S, 4S), изобутилена и 1,5 см3 серной кислоты в 150 см3 хлороформа. После обработки получают 1,2 г 4-фенил-трет. бутилпролината-(2S, 4S) в виде воздушной массы, используемой при последующем синтезе.
4-Фенилпролин-(2S, 4S) может быть получен по известным методам (J.K THOTTATHIL et J.L. MONIOT Tetrahedron Lett., 27, 151, 1986 и DR. KRONENTHAL et coll., Tetrahedron Lett, 31. 1241, 1990).
Пример 106. Действуют по способу, аналогичному описанному в примере 9, но исходя из 1,8 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо) ацетил) 4-фенилтрет. бутилпролината-(2S, 4S), растворенного в 30 см3 метанола, и 0,21 г гидроокиси калия, растворенной в 15 см3 воды. После обработки и кристаллизации в смеси окись диизипропил-ацетат изопропила (90-10 об.) получают 0,55 г 3-(3-(2-(2-трет. бутоксикарбонил-4-фенил-1-пирролидинил) 2-оксо-этил)уреидо) бензойной -(2S, 4R) кислоты, плавящейся при 140oC.
1-(2-(3-(3-Метоксикарбонил-фенил)уреидо)ацетил) 4-фенил-трет. бутилпролинат-(2S, 4R) может быть получен как описано в примере 41. А, но исходя из 1 г 4-фенилтрет. бутилпролината-(2S, 4R), 1 г 2-(3-(3-метоксикарбонил-фенил)уреидо)- уксусной кислоты и 0,85 г N, N'-дициклогексилкарбодиимида в 30 см3 ацетонитрила. После обработки получают 1,8 г 1-(2-(3-(3-метоксикарбонил-фенил)уреидо)ацетил 4-фенилтрет, бутилпролината-(2S, 4R) в виде воздушной массы, используемой при последующем синтезе.
4-Фенилтрет. бутилпролинат-(2S, 4R) может быть получен, как описано в примере 1, Б, но исходя из 4,4 г 4-фенилпролина-(2S, 4R), изобутилена и 1,5 см3 серной кислоты в 150 см3 хлороформа. После обработки получают 1 г 4-фенилтрет.бутилпролината (2S, 4R), плавящегося при 62oC.
4-фенилпролин-(2S, 4R) может быть получен по известным методикам (J.K. THOTTATHIL et J.L.MONIOT Tetrahedron Lett., 27, 151, 1986 и D.R.KRONENTHAL et coll.,Tetrahedron Lett, 31, 1241, 1990).
Пример 107. Действуют по способу, аналогичному описанному в примере 9, но на основе 0,55 г 1-(2-(3-(3-бензилоксикарбонил-фенил)уреидо)ацетил) 5-(2-пиридил)трет. бутилпролината-(2RS, 5SR),растворенного в 20 см3 метанола, и 10 см3 0,1 N водного раствора гидроокиси калия. После обработки получают 0,17 г 3-(3- (2-(2-трет.бутоксикарбонил 5-(2-пиридин) 1-пирролидинил) 2-оксоэтил)уреидо)бензойной -(2RS, 5SR) кислоты, плавящейся при 174oC.
1-(2-(3-(3-Бензилоксикарбонил-фенил)уреидо)ацетил)-5-(2-пиридилтрет. бутилпролинат-(2RS, 5SR) может быть получен, как описано в примере 2, но исходя из 0,5 г 1-(2-амино-ацетил)-5-(2-пиридин)трет-бутилпролината-(2RS, 5SR) и 0,45 г 3-изоцианатбензилбензоата в 25 см3 тетрагидрофурана. После обработки получают 0,7 г 1-(2-(3-(3-бензилоксикарбонил-фенил)уреидо) ацетил)-5-(2-пиридинл)-трет. бутилпролината-(2RS, 5SR) в виде воздушной массы, используемой при последующем синтезе.
1-(2-Амино-ацетил)-5-(2-пиридин)трет.бутилпролинат-(2RS, 5SR) может быть получен, как описано в примере 2, А, но исходя из 0,7 г 1-(2-трет.бутоксикарбониламино-ацетил)-5-(2-пиридил)трет. бутилпролината) -(2RS, 5SR) и 0,25 см3 иодитриметилсилана в 30 см3 хлороформа. После обработки получают 0,5 г 1-(2-амино-ацетил)-5-(2-пиридил)трет.бутилпролината-(2RS, 5SR) в виде масла, используемого при последующем синтезе.
1-(2-Трет. бутоксикарбониламино-ацетил)-5-(2-пиридил)-трет.бутилпролинат -(2RS, 5SR) может быть получен, как описано в примере 90, А, но исходя из 4,8 г однозамещенного фосфата натрия и 15,3 г амальгамы с 6% натрия (в ртути) в растворе 5,5 г смеси двух эпимеров в положении 4 1-(2-трет-бутоксикарбониламино-ацетил)-4-фенилсульфонил-5- (2-пиридилтрет.бутилпролината-(2RS, 5RS) в смеси 20 см3 метанола и 60 см3 тетрагидрофурана. После обработки получают 0,7 г 1-(2-трет. бутоксикарбониламино-ацетил)-5-(2-пиридил) трет. бутилпролината (2RS, 5SR) в виде масла, используемого при последующем синтезе.
1-(2-Трет. бутоксикарбониламино-ацетил)-4-фенилсульфонил-5-(2-пиридил) трет. бутилпролинат-(2RS, 5RS) может быть получен следующим образом: в раствор 2,7 г 2 -трет. бутоксикарбониламино-уксусной кислоты в смеси 2,2 см3 триэтиламина и 100 см3 дихлорметана, выдерживаемого при температуре около 0oC, добавляют 1,5 см3 этилового эфира хлоругольной кислоты. Реакционную среду перемешивают в течение 30 мин при температуре около 0oC, затем добавляют раствор 6 г 4-фенилсульфонил-5-(2-пиридинл)трет.бутилпролината-(2RS, 5RS) в 50 см3 дихлорметана. Реакционную смесь перемешивают в течение 20 ч при температуре около 20oC, затем добавляют 100 см3 воды. Органическую фазу отделяют декантированием, промывают в 25 см3 воды, высушивают над сульфатом магния и концентрируют при пониженном давлении. Остаток очищают хроматографией на двуокиси кремния (элюент: циклогексин-этилацетат (50/50 об.). Фракции, содержащие целевой продукт, объединяют и концентрируют при пониженном давлении. Таким образом, получают 5.6 г 1-(2-трет.бутоксикарбониламино-ацетил)-4-фенилсульфонил-5-(2-пиридил) трет.бутилпролината-(2RS, 5RS), смесь двух эпимеров в 4) в виде масла, используемого при последующем синтезе.
4-Фенилсульфонил-5-(2-пиридил_ -трет. бутилпролина (2RS, 5RS) может быть получен, как описано в примере 89, А, но исходя из 4,4 г N-/(2-пиридил)метилен-трет. бутилглицината, 5 г ацетата серебра, 3,4 г фенилвинилсульфона и 2,8 см3 триэтиламина в 200 см3 ацетонитрила. После обработки получают 6 г 4-фенилсульфонил-5-(2-пиридил)трет. бутилпролината-(2RS, 5R), смесь двух эпимеров в 4) в виде масла, используемого при последующем синтезе.
N-/(2-пиридил)метилен/-трет.бутилглицинат может быть получен, как описано в примере 89, Б, но исходя из 1,4 см3 2-пиридилкарбоксальдегида, 3,35 г хлоргидрата трет. бутилглицината, 2,8 см3 триэтиламина и 3 г молекулярного сита 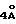 в 30 см3 дихлорметана. После обработки получают 4,8 г N-(2-пиридил)метилен)-трет-бутилглицината в виде масла, используемого при последующем синтезе.
в 30 см3 дихлорметана. После обработки получают 4,8 г N-(2-пиридил)метилен)-трет-бутилглицината в виде масла, используемого при последующем синтезе.
Пример 108. В раствор 0,65 г 2-(3-(3-(2-(4-трет.бутоксикарбонил- 2-(2-фтор-фенил)-3-тиазолидинил-(2R, 4R) 2-оксо-этил)уреидо)фенил) пропионовой кислоты (форма А) в 6 см3 метанола добавляют 1,3 г оксонаP, растворенного в 8 см3 дистиллированной воды. Реакционную среду перемешивают в течение 12 ч при температуре около 25oC, затем концентрируют при пониженном давлении и 40oC. Полученный продукт очищают хроматографией на двуокиси кремния (элюент: метиленхлорид-метанол (90/10 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом получают 0,4 г 2-(3-(3-(2-(4- трет.бутоксикарбонил-2-(2-фтор-фенил) 1-оксид-3-тиазолидинил-(1RS, 2R, 4R) / 2-оксо-этил)уреидо)фенил) пропионовой кислоты (форма А) в виде аморфного продукта.
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн, J в Гц), при 120oC наблюдается смесь двух диастереоизомеров в пропорции 85-15, характеристические химические сдвиги при 120oC: 1,32 (d, J = 7,5, 3H, -CH3), 1,50 и 1,58 (2S, 9H в совокупности, -C(CH3)3) наибольшего изомера, затем наименьшего изомера 2,85 (t, J = 12,5, 0,85H, 1H в S-CH2 для наибольшего изомера) 3,4 - 4,4 (m, 6,15H, -CH-COO, другой H в - S-CH2 для наибольшего изомера, -S-CH2 наименьшего изомера и N -COCH2-N), 4,8 (t, J = 8, 0,15H, S-CH-N наименьшего изомера), 4,97 (dd, J = 12,5 и 5,0 0,85H, S-CH-N наибольшего изомера), 6,3-6,45 (m, 1H, -NHCO-), 6,5-6,6 (2S, 0,15H и 0,85H, S-CH-N наименьшего и наибольшего изомеров), 6,8 (bd, J = 8, 1H, в позиции 4 в CO-NH-Ph-), 7,0-7,6 (m, 6H, ароматические), 7,77 и 8,10 (t, J = 8, 0,15H и 0,85 H, в позиции 6 в S-CH-Ph - наименьшего и наибольшего изомеров), 8,84 и 8,96 (2S, 0,85H и 0,15H, ArNHCO- в наименьшем и наибольшем изомерах).
Инфракрасный спектр: (KBr), характеристические полосы в см-1: 3390, 3100-3000, 2975, 2930, 2875, 2750-2300 (широкая полоса), 1735, 1670, 1615, 1595, 1555, 1490, 1460, 1400, 1395, 1370, 1150, 760, 700.
Пример 109. Действуют по способу, описанному в примере 41, но исходя из 1,85 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(1-нафтил)-3- тиазолидинил) 2-оксо-этил)уреидо)триметилсилилэтилбензоата (2R, 4R) и 5,82 см3 1M раствора фторида тетрабутиламмония. Таким образом, получают 0,47 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(1-нафтил) 3-тиазолидинил) 2-оксо-этил)уреидо)бензойной-(2R, 4R) кислоты в виде твердого вещества бежевого цвета, плавящегося при 210oC. /α/
Протонный ЯМР (200 МГц, DMCO D6 плюс несколько капель CD3COOD, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, характеристические химические сдвиги при 120oC: 1,53 (s, 9H, -C(CH3)3), 3,25 (dd, J = 12 и 7, 1H, 1H в S-CH2), 3,49 (dd, J = 12 и 6,5, 1H, другой H в S-CH2), 3,7 (bd, J = 17,5 1H в N-COCH2-N), 4,08 (d, J = 17,5 1H, другой H в N-COCH2-N), 5,02 (dd, J = 7,0 и 6,5, 1H, N-CH-COO), 7,08 (S, 1H, S-CH-N), 7,15-8,15 (m, 11H, ароматические).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3390, 3100-3000, 2975, 2930, 1735, 1655, 1595, 1555, 1510, 1485, 1400, 1150, 785, 770.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(1-нафтил)-3-тиазолидинил)-2- оксо-этил)уреидо) 2-триметилсилил-этилбензоат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 34, но исходя из 2,4 г 3-(2-амино-ацетил)-2-(1-нафтил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) и 2,1 г 3-изоцианат-2-триметилсилил- этилбензоата. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,85 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(1-нафтил)-3-тиазолидинил) 2-оксоэтил) уреидо) 2-триметилсилилбензоата-(4R) (форма А) в виде бежевой пасты, используемой при последующем синтезе.
3-(2-Амино-ацетил)-2-(1-нафтил)-4-тиазолидинтрет. бутилкарбоксилат-(2R, 4R) может быть получен методом, аналогичным описанному в примере 34, А, но исходя из 3,65 г 3-(2-трет. бутоксикарбониламиноацетил)- 2-(1-нафтил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) и 1,13 см3 иодтриметилсилана. Таким образом, получают 2,43 г 3-(2-амино-ацетил)-2-(2-нафтил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в виде желтого твердого продукта, используемого при последующем синтезе.
3-(2-Трет. бутоксикарбониламидо-ацетил)-2-(1-нафтил)-4- тиазолидинтрет. бутилкарбоксилат-(2R, 4R) может быть получен способом, аналогичным описанному в примере 34, Б, но исходя из 7,54 г 2-(1-нафтил)-4-тиазолидинтрет. бутилкарбоксилата-(2RS, 4R), 4,23 и 2-трет.бутоксикарбониламиноуксусной кислоты и 4,98 г N,N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении. Таким образом, получают 13,9 г 3-(2-трет.бутоксикарбониламино-ацетил)-2-(1-нафтил)-4-тиазолидинтрет. бутилкарбоксилата-(2R, 4R) в виде оранжевого масла, используемого при последующем синтезе.
2-(1-Нафтил)-4- тиазолидинтрет.бутилкарбоксилат-(2RS, 4R) может быть получен способом, аналогичным описанному в примере 34, В, но исходя из 20,0 г 2-(1-нафтил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, растворенной в 300 см3 хлороформа, 5,8 см3 концентрированной серной кислоты и избытка изобутилена. Полученный сырой продукт очищают хроматографией на двуокими кремния (элюент: этилацетатциклогексан (30/70 об. )). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 10,15 г 2-(1-нафтил)-4- тиазолидинтрет.бутилкарбоксилата-(2RS, 4R) в виде оранжевого масла, смеси изомеров (2R, 4R) и (2S, 4R), используемого при последующем синтезировании.
2-(1-Нафтил)-4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 20,0 г L-цистеина и 28,5 г 1-нафтилкарбоксальдегида. Таким образом, получают 36,3 г 2-(1-нафтил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 208oC, используемой при последующем синтезе.
Пример 110. Действуют по способу, описанному в примере 41, но исходя из 0,12 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(5-метил-2-тиенил) 3-тиазолидинил) 2-оксо-этил) уреидо) 2-триметилсилил-этилбензоата-(4R) (форма А) и 0,4 см3 1M раствора фторида тетрабутиламония. Таким образом, 0,08 г 3-(3-(2-(4-трет. бутоксикарбонил-2-(5-метил-2-тиенил) 3-тиазолидинил) 2-(оксо-этил) уреидо) бензойной-(4R) кислоты (форма А) в виде оранжевого твердого тела, плавящегося при 85 - 90oC.
Протонный ЯМР (300 МГц, DMCO D6 плюс несколько капель CD3COOD, δ в ч/млн, J в Гц), 2 ротамера при комнатной температуре, характеристические химические сдвиги при 120oC: 1,5 (s, 9H, -C(CH3)3, 2,46 (S, 3H, -CH3), 3,4 и 3,52 (2dd, J = 12 и 6,5, 1H каждый, - S-CH2), 3,96 (d, J = 17,5 1H, 1H в N-COOH2-N), 4,05 (d, J = 17,5, 1H, другой H в N-COCH2-N), 4,96 (t, J = 6,5, 1H, OOC-CH-N), 6,65 (S, 1H, S-CH-N), 6,65 (d, J = 4 1H, H в позиции 4 в тиениле), 7,12 (d, J = 4, 1H, H в позиции 3 в тиениле), 7,55 и 7,62 (2dd, J = 8 и 2. Каждый 1H, H в позиции 4 и 6 в CO-NH-Ph-), 8,02 (t, J = 2, 1H, H в позиции 2 в CO-NH-Ph-).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3390, 3100-3000, 2975, 2930, 2830, 2750-2300 (широкая полоса), 1750, 1695, 1650, 1615, 1595, 1560, 1490, 1425, 1395, 1370, 1150, 800, 785, 755.
3-(3-(2-(4-Трет. бутоксикарбонил-2-(5-метил-2-тиенил) 3-тиазолидинил) 2-оксо-этил)уреидо) 2-триметилсилил-этилбензонат (4R) (форма A) может быть получен способом, аналогичным описанному в примере 41, A, но исходя из 0,38 г 2-(5-метил-2-тиенил) 4-тиазолидинтрет.бутилкарбоксилата-(2RS, 4R), 0,45 г 2-(3-(3-(2-триметилсилил-этоксикарбонил) фенил)уреидо) уксусной кислоты и 0,30 г N, N'-дициклогексилкарбодиимида. Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетатциклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 0,12 г 3-(3-(2-(4-трет.бутоксикарбонил-2-(5-метил-2-тиенил)-3-тиазолидин) 2-оксо-этил)уреидо)-2-триметилсилил-этилбензоата-(4R) (форма A) в виде желто-оранжевого тела, используемого при последующем синтезе.
2-(5-Метил-2-тиенил)-4-тиазолидинтрет. бутилкарбоксилат-(2RS, 4R) может быть получен способом, аналогичным описанному в примере 48, Б, но исходя из 1,0 г 3-трет.бутоксикарбонил-2-(5-метил-2-тиенил) 4-триазолидинтрет.бутилкарбоксилата-(4R) (форма А) и 0,38 см3 иодотриметилсилана. Таким образом, получают 0,38 г 2-(5-метил-2-тиенил) 4-тиазолидинтрет.бутилкарбоксилата-(2RS, 4R) в виде масла желтого цвета, используемого при последующем синтезе.
3-Трет.бутоксикарбонил-2-(5-метил-2-тиенил)-4-тиазолидинтрет. бутоксикарбоксилат-(4R) (форма А) может быть получен способом, аналогичным описанному в примере 48, В, но исходя из 2,97 г 3-трет.бутоксикарбонил-2-(5-метил-2-тиенил)-4-тиазолидинкарбоновой-(4R) кислоты (форма А), 1,72 г паратолуолсульфонилхлорида и 0,67 г трет. бутанола. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (30/70 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,0 г 3-трет.бутоксикарбонил-2-(5-метил-2-тиенил)-4-триазолидинтрет. бутилкарбоксилата-(4R) (форма А) в виде оранжевого масла, используемого при последующем синтезе.
3-Трет. бутоксикарбонил-2-(5-метил-2-тиенил)-4-тиазолидинкарбоновая-(4R) кислота (форма А) может быть получена способом, аналогичным описанному в примере 48, Г, но исходя из 2,7 г 2-(5-метил-2-тиенил) -4-тиазолидинкарбоновой-(2RS, 4R) кислоты, 11,9 см3 водного 1N раствора гидроокиси натрия и 2,6 г дикарбоната дитрет.бутила. Таким образом, получают 3,0 г 3-трет. бутоксикарбонил-2-(5-метил-2-тиенил)-4-тиазолидинкарбоновой-(4R) кислоты (форма А) в виде желтого продукта, используемого при последующем синтезе.
2-(5-метил-2-тиенил)4-тиазолидинкарбоновая-(2RS, 4R) кислота может быть получена, как в примере 34, Г, но исходя из 10,0 г L-цистеина и 11,3 г (5-метил-2-тиени л) карбоксальдегида. Таким образом, получают 7,86 г 2-(5-метил-2-тиенил)-4-тиазолидинкарбоновой-(2RS, 4R) кислоты, плавящейся при 178oC, используемой при последующем синтезе.
Пример 111. Действует по способу, описанному в примере 84, но исходя из 0,88 г 2-(3-(3-(2-(2-трет.бутоксикарбонил-5-фенил-1-пиролидинил (2S, 5R))-2-оксо-этил)уреидо)фенил) бензилпропионата (форма В), растворенного в 50 см3 этилацетата, и 0,22 г 10%-ного палладия на угле. После обработки получают 0,5 г 2-(3-(3-(2-(2-трет. бутоксикарбонил-5-фенил-1-пирролидинил-(2S, 5R)) 2-оксо-этил)уреидо)фенил) пропионовой кислоты (форма В), плавящейся при 120oC, /α/
2-(3-(3-(2-(2-Трет. бутоксикарбонил-5-фенил-1-пиролидинил-2S, 5R)) 2-оксо-этил)уреидо)фенил)бензилпропионат (форма В) может быть получена по способу, аналогичному описанному в примере 2, но исходя из 0,79 г 1(2-амино-ацетил)-5-фенилтрет. бутилпролината-(2S, 5R) и 0,8 г 2-(3-изоцианато-фенил)бензилпропионата (форма В) в 40 см3 тетрагидрофурана. После обработки получают 0,9 г 2-(3-(3-(2-(2-трет-бутоксикарбонил-5-фенил-1-пиролидинил- (2S, 5R))-2-оксо-этил)уреидо)фенил) бензилпропионата, плавящегося при 75oC.
1-(2-Амино-ацетил) 5-фенил трет-бутилпролинат-(2S, 5R) может быть получен по способу, аналогичному описанному в примере 2А, но исходя из 1,22 г 1-(2-трет.бутоксикарбонил-аминоацетил) 5-фенилтрет. бутилпролината-(2S, 5R), 0,45 см3 иодотриметилсилана, растворенного в 30 см3 безводного хлороформа. Таким образом, получают 0,79 г 1-2(амино-ацетил)-5-фенил-трет.бутилпролината-(2S, 5R) в виде масла, используемого при последующем синтезе.
1-(2-Трет.бутоксикарбонил-амино-ацетил) 5-фенил-трет. бутилпролинат-(2S, 5R) может быть получен, как описано в примере 2Б, но исходя из раствора, содержащего 8 г 5-фенилтретбутилпролината- (2-S, 5R), 5,7 г 2-трет.бутоксикарбониламиноуксусной кислоты и 6,7 г N, N'-дициклогексилкарбодиимида в 75 см3 безводного ацетонитрила. Таким образом, получают 10,5 г 1-(2-трет.бутоксикарбониламино-ацетил) 5-фенил-трет.бутилпролината-(2S, 5R), плавящегося при 136oC, /α/
Пример 112. Действуют по способу, описанному в примере 2, но исходя из 2,5 г 1-(2-амино-ацетил)-5-фенилтрет. бутилпролината (2S, 5R), 5 г 1-(3-изоцианат-фенил)этансульфоната тетрабутиламмония (RS) в 60 см3 тетрагидрофурана. После обработки получают 0,23 г 1-(3-(3-(2-(2-трет.бутоксикарбониол-5-фенил-1-пирролидинил) 2-оксоэтил)уреидо)фенил)этансульфоната калия-(2S, 5R) (смесь обоих эпимеров в положении 1) в виде аморфного твердого продукта бежевого цвета, /α/
1-(3-Изоцианат-фенил) этансульфонат тетрабутиламмония (RS) может быть получен, как описано в примере 21, но исходя из 4,5 г 1-(3-амино-фенил) этансульфоната тетрабутиламмония (RS) в 40 см6 толуола, 0,2 г угля и 1,21 см3 хлорформиата трифторметила. После обработки получают 5 г 1-(3-изоцианатфенил) этан-сульфоната тетрабулиламмония (RS) в виде масла, используемого при последующем синтезе.
Пример 113. Действуют по способу, аналогичному описанному в примере 2, но исходя из 2,6 г 1-(2-амино-ацетил) 5-фенил-трет.бутил-пролината (2S, 5R), 4,8 г (3-изоцианат-фенилметансульфоната тетрабутиламмония в 60 см3 тетрагидрофурана. После обработки получают 0,21 г (1-(3-(3-(2-(2-трет.бутоксикарбонил-5-фенил-1-пирролидинил) 2-оксо-этил)уреидо)фенил) метансульфоната калия-(2S, 5R) в виде твердого продукта бежевого цвета, /α/
(3-Изоцианат-фенил)метан - сульфонат тетрабутиламмония может быть получен, как описано в примере 21, но исходя из (3-амино-фенил)метансульфоната тетрабутиламмония-(RS) в 80 см3 толуола, 0,49 г угля и 2,85 г хлорформиата трихлорметила. После обработки получают 11 г (3-изоцианат-фенил)метансульфоната тетрабутиламмония в виде масла, используемого при последующем синтезе.
Пример 114. В колбу, содержащую 1,38 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2,3-дифтор-фенил) 3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил)бензилпропионата (форма B), 0,85 г формиата аммония и 1,38 г 10%-ого палладия на угле, медленно добавляют в инертной атмосфере 20 см3 метанола. Реакционную среду нагревают с обратным холодильником в течение 1 ч, затем охлаждают до температуры около 25oC. Затем отфильтровывают катализатор, а фильтрат концентрируют досуха при пониженном давлении и 40oC. Полученный остаток растворяют в 20 см3 0,1 N водного раствора гидроокиси натрия и промывают два раза в 10 см3 диэтилового эфира. Водную фазу доводят до pH 2, добавляя 1 N водный раствор серной кислоты. Выпавший в осадок продукт отфильтровывают, промывают 2 раза в 10 см3 воды и высушивают на воздухе. Таким образом, получают 1,0 г 2-(3-(3-(2-(4-трет.бутокси-карбонил-2-(2,3-дифтор-фенил) 3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил)пропионовой кислоты (форма B), плавящейся при 130oC, /α/
2-(3-(3-(2-(4-трет-бутоксикарбонил-2-(2,3-дифтор-фенил) 3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил)бензилпропионат (форма B) может быть получен по способу, аналогичному описанному в примере 34, но исходя из 2,5 г 3-(2-аминоацетил) 2-(2,3-дифтор-фенил)-4-тиазолидинтрет. бутилкарбоксилата (2R, 4R) и 1,96 г 2-(3-изоцианат-фенил) бензилпропионата (форма B). Сырой продукт очищают хроматографией на двуокиси кремния (элюент: этилацетат-циклогексан (40/60 об.)). Фракции, содержащие продукт, объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 1,8 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2- (2,3-дифтор-фенил) 3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил бензилпроионата (форма B) в виде аморфного продукта, используемого при последующем синтезе.
Пример 115. Раствор 2,1 г (3-(3-(2-(2,5-дифенил-1-пирролидинил)2-оксо-этил)уреидо)фенил) метиленаминоксиацетата трет.бутила-(цис), растворенного в 10 см3 трифторуксусной кислоты, перемешивают в течение двух часов при температуре около 20oC, затем реакционную среду концентрируют досуха при пониженном давлении и температуре около 40oC. Остаток кристаллизуют в 50 см3 воды, фильтруют, промывают три раза в 15 см3 воды и высушивают при температуре около 20oC. Полученный продукт очищают хроматографией на двуокиси кремния (элюент: дихлорметан-метанол (95/5 об.)). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и температуре около 35oC. Таким образом, получают после перекристаллизации в ацетонитриле 1 г (3-(3-(2-(2,5-дифенил-1-пирролидинил) 2-оксо-этил)уреидо)фенил)метиленаминоксиуксусной-(цис) кислоты, плавящейся при 194oC.
(3-(3-(2-(2,5-Дифенил-1-пирролидинил)-2-оксо-этил)уреидо)фенил) метиленаминоксиацетат трет-бутила-(цис) может быть получен способом, аналогичным описанному в примере 3, но исходя из 4,2 г 1-(2-амино-ацетил) 2,5-дифенилпирролидина-(цис), 1,8 г N, N-диимидазол-карбонила и 2,5 г (3-амино-фенил)метиленаминоксиацетата трет.бутила в 60 см3 1,2-дихлорэтана. После обработки получают 2,1 г (3-(3-(2-(2,5-дифенил-1-пирролидинил) 2-оксо-этил)уреидо)фенил) метиленаминоксиацетата трет-бутила-(цис) в виде аморфного продукта бежевого цвета.
(3-амино-фенил)метиленаминоксиацетат трет-бутила может быть получен следующим способом: в раствор 7,5 г (3-нитро-фенил)метиленаминоксиацетата трет. бутила в 100 см3 этанола добавляют 0,75 г окиси платины. Суспензию перемешивают в течение 1 ч при температуре около 20oC в атмосфере водорода (130 кПа). Катализатор отфильтровывают, а фильтрат концентрируют досуха при пониженном давлении и 45oC. Остаток растворяют в 50 см3 этилацетата, а органическую фазу промывают в 50 см3 воды, высушивают над сульфатом магния и концентрируют при пониженном давлении. Таким образом, получают 6,2 г (3-аминофенил)метиленаминоксиацетата трет. бутила в виде масла, используемого при последующем синтезе.
(3-Нитро-фенил)метиленаминоксиацетат трет. бутила может быть получен следующим способом: раствор 4,3 г 60% гидрида натрия в вазелине обезжиривают, промывая два раза в 50 см3 гексана. Затем добавляют 100 см3 тетрагидрофурана, затем суспензию охлаждают до температуры около 5oC и прикапывают раствор 4,98 г 3-нитро-бензальдоксима в 20 см3 тетрагидрофурана. Реакционную смесь перемешивают в течение 1 ч при температуре около 25oC, добавляют раствор 6 г трет. бутилбромацетата в 5 см3 тетрагидрофурана и перемешивают в течение 1 ч при температуре около 25oC. Затем реакционную среду выливают в смесь 200 см3 воды и 100 см3 этилацетата. Органическую фазу отделяют декантированием, высушивают над сульфатом магния и концентрируют при пониженном давлении и температуре около 40oC. Таким образом, получают 7,5 г (3-нитро-фенил)метиленаминоксиацетата трет.бутила, плавящегося при 120oC.
3-Нитро-бензальдоксим может быть получен по известной методике (T.M.Oprishko et coll. . Izv. Vyssh Uchebn Zaved. Khim Khim. Tekhnol., 31, 53 (1988)).
Пример 116. Действуют, как описано в примере 108, но исходя из 0,5 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил) пропионовой кислоты (форма B) и 1,0 г оксонаP. Полученный сырой продукт очищают хроматографией на двуокиси кремния (элюент: метиленхлоридметанол (90/10 об.)), собирая фракции по 10 см3. Фракции 15-19 объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 0,2 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2- (2-фтор-фенил) 1-оксид-3-тиазолидинил-(1RS, 2R, 4R)) 2-оксо-этил)уреидо)фенил) пропионовой кислоты (форма B) в виде аморфного продукта. Фракции 9-14 объединяют и концентрируют досуха при пониженном давлении и 40oC. Таким образом, получают 0,1 г смеси 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил) 1-оксид-3-тиазолидинил-(1RS, 2R, 4R) 2-оксо-этил)уреидо)фенил) пропионовой кислоты (форма B) и 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил) 1-диоксид-3-тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил)пропионовой кислоты (форма B).
Протонный ЯМР (200 МГц, ДМСО D6, δ в ч/млн, J в Гц), при 120oC наблюдается смесь двух диастероизомеров в пропорции 88-12, характеристические химические сдвиги при 120oC: 1,32 (d, J = 7,5, 3H, -CH3), 1,50 и 1,58 (2s, 9H, в совокупности, -C(CH3)3) наибольшего изомера, затем наименьшего изомера), 2,85 (t, J = 12,5, 0,88H, 1H, d - S-CH2 наибольшего изомера), 3,4 - 4,4 (m, 6,12H, -CH-COO, другой H в -S-CH2 наибольшего изомера, -S-CH2 наименьшего изомера и N-COOH2-N), 4,8 (t, J = 8, 0,12H, S-CH-N наименьшего изомера), 4,96 (dd, J = 12,5 и 5,0, 0,88H, S-CH-N наибольшего изомера), 6,3 - 6,45 (m, 1H, -NHCO-), 6,5-6,6 (2S, 0,12H и 0,88H, S-CH-N наименьшего и наибольшего изомера), 6,8 (bd, J = 8, 1H, в позиции 4 в CO-NH-Ph), 7,0-7,6 (m, 6H, ароматические), 7,77 и 8,10 (t, J = 8, 0,12H и 0,88H, в позиции 6 в S-CH-Ph- для наименьшего и для наибольшего изомеров), 8,84 и 8,96 (2s, 0,88H и 0,12H, ArNHCO - для наибольшего и для наименьшего изомеров).
Инфракрасный спектр (KBr), характеристические полосы в см-1: 3380, 2975, 2930, 2650-2250 (широкая полоса), 1735, 1650, 1610, 1595, 1560, 1490, 1455, 1370, 1230, 1150, 1050, 760, 700.
2-(3-(3-(2-(4-трет. бутоксикарбонил-2-(2-фтор-фенил) 1-диоксид-3-тиазолидинил-(2R-, 4R) 2-оксо-этил)уреидо)фенил) пропионовую кислоты (форма B) выделяют из смеси 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил)-1-оксид-3- тиазолидинил-(1RS, 2R, 4R)) 2-оксо-этил)уреидо)фенил) пропионовой кислоты (форма B) и 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил)1-диоксид-3- тиазолидинил-(2R, 4R)) 2-оксо-этил)уреидо)фенил пропионовой кислоты (форма B) путем хроматографии на двуокиси кремния (элюент: этилацетат-метанол (95/5 об. ). Фракции, содержащие целевой продукт, объединяют и концентрируют досуха при пониженном давлении и 30oC. Таким образом, получают 0,02 г 2-(3-(3-(2-(4-трет.бутоксикарбонил-2-(2-фтор-фенил) 1-диоксид-3-тиазолидинил-(2R, 4R) 2-оксо-этил)уреидо)фенил)пропионовой кислоты (форма B) в виде аморфного вещества.
Протонный ЯМР (200 МГц, DMCO D6, δ в ч/млн): 11,3 (d, 3H, -CH3), 1,5 (s, 9H, -CH3)3), 3,6 (m, 2H, CH2- SO2), 4,0-4,3 (m, 3H, Ar -CH-COO и N-COCH2-N), 4,8 (bt, 1H, N-CH-COO), 6,4 (bt, 1H, -N-HCO-), 6,5 (S, 1H, S -CH-N), 6,8 (bd, 1H, в позиции 4 в CO-NH-Ph-), 7,1-7,8 (m, 6H, ароматические), 8,1 (bt, 1H, в позиции 6 в S -CH-Ph-), 8,8 (bs, 1H, Ar -NHCO-).
Инфракрасный спектр (KBr, характеристические полосы в см-1; 3380, 2975, 2930, 2650-2250 (широкая полоса), 1735, 1650, 1610, 1595, 1560, 1490, 1455, 1370, 1345, 1230, 1150, 760, 700, 550.
Пример 117. Работая согласно примеру 3, получают 5[3[3[2(2-трет.бутоксикарбонил-5-фенил-1-пирролидинил)-2-оксо-этил]фенил] тетразол-(2S, 5R).
Вращательная способность α
Изобретение относится также к фармацетвическим композициям, которые содержат в качестве активного начала по меньшей мере одно соединение формулы I, в свободной форме или в форме аддитивной соли с фармацевтически приемлемой кислотой, в сочетании с другим фармацевтически совместимым веществом, который может быть инертным или физиологически активным. Эти композиции могут применять оральным, парентеральным, ректальным или топическим способом.
В качестве твердых композиций для орального введения могут применяться таблетки, пилюли, порошки (желатиновые капсулы, облатки) или гранулы.
В этих композициях действующее начало, описанное в изобретении, смешено с одним или несколькими инертными разбавителями, такими как крахмал, целлюлоза, сахароза, лактоза или двуокись кремния, в потоке аргона. Эти композиции также могут содержать вещества, не являющиеся разбавителями, например, один или несколько смазывающих веществ, таких как стеарат магния или тальк, краситель, предохранительную оболочку (дражже) или покрытие.
В качестве жидкой композиции для орального способа введения можно применять фармацевтически приемлемые растворы, суспензии, эмульсии, сиропы и эликсиры, содержащие инертные разбавители, такие как вода, этанол, глиценрол, растительные масла или жидкий парафин. Эти композиции могут содержать вещества, не являющиеся разбавителями, например, смачиватели, подслащивающие, желатинирующие, ароматизирующие агенты или стабилизаторы.
Стерильные композиции для парентерального введения могут находится предпочтительно в виде водных или безводных растворов, суспензий или эмульсий. В качестве растворителя или носителя, можно использовать воду, пропиленгликоль, полиэтиленгликоль, растительные масла, в частности, оливковое, органические сложные эфиры для впрыскивания, например, олеат этила или другие соответствующие органические растворители. Эти композиции могут также содержать дополнительные вещества, в частности, смачиватели, изотонизаторы, эмульгаторы, диспергаторы, стабилизаторы. Стерилизация может осуществляться несколькими способами, например, асептической фильтрацией, вводя в композицию стерилизующие вещества, облучением или нагреванием. Они могут также приготавливаться в виде стерильных твердых композиций, которые непосредственно перед применением растворяют в воде или в любой другой стерильной среде, пригодной для впрыскивания.
Композиции для ректального введения представлены в виде свечей или ректальных капсул, которые содержат, кроме активного продукта, эксципиенты, такие, как масло какао, полусинтетические глицериды или полиэтиленгликоли.
Композиции для топического (местного) применения могут выпускаться, например, в виде кремов, лосьонов, глазных капель, лекарств для слизистой оболочки горла и рта, капель в нос или аэрозолей.
В терапии человека описанные в изобретении соединения применяются, в частности, для лечения и профилактики расстройств, связанных с ХЦК и гастрином на уровне центральной нервной системы и желудочно-кишечного тракта. Эти соединения могут применяться при лечении и профилактике психозов, состояний беспокойства, болезни Паркинсона, запоздалого дискинезиса, синдрома легковозбудимой оболочки кишки, острого панкреатита, язв, расстройств моторики кишечника, некоторых видов новообразований (опухолей), восприимчивых в ХЦК, расстройств памяти, в качестве анальгетических средств, средств для повышения анальгетической активного наркотического и ненаркотического анальгетических медицинских препаратов, и в качестве средств для регулирования аппетита.
Дозы зависят от желаемого эффекта, продолжительности лечения и способа введения, как правило, 0,05 - 1 г в день при оральном введении для взрослых, с единичной дозой 10-500 мг активного вещества.
Как правило, врач должен определять соответствующую позологическую (ежедневную) дозу в зависимости от возраста, веса и других индивидуальных особенностей пациента.
Пример А. По обычной методике изготавливают желатиновые капсулы с дозой активного вещества 50 мг, имеющие следующую композицию, мг:
3-(3-(2-(2-Трет. бутоксикарбонил-5-фенил-1-пирролидинил)-2-оксо- этил)уреидо)-3-фенил-бензойная-(2R, 5SR) кислота - 50
Целлюлоза - 18
Лактоза - 55
Колоидная двуокись кремния - 1
Натриевый карбоксиметилкрахмал - 10
Тальк - 10
Стеарат магния - 1
Пример Б. По обычной методике изготавливают таблетки с дозой активного вещества 50 мг, имеющие следующую композицию, мг:
3-((3-(2-(2-Трет. бутоксикарбонил-5-фенил-1-пирролидинил)-2-оксо- этил)-уреидо)-бензойная-(2R, 5S) кислота - 50
Целлюлоза - 40
Лактоза - 104
Поливидон - 10 г
Натриевый карбоксиметилкрахмал - 23
Тальк - 10
Коллоидная двуокись кремния - 2
Стеарат магния - 2
Смесь гидроксиметилцеллюлозы, глицерина, окиси титана (72-3,5-24,5) для 1 таблетки в оболочке на - 245
Пример В. Приготавливают раствор для впрыскивания, содержащий 10 мг активного вещества и имеющий следующую композицию, мг:
3-((3-(2-(4-Трет. бутоксикарбонил-2-фенил-3-тиазолидинил)-2- оксо-этил)уреидо)-фенилуксусная-(2R, 4R) кислота - 10
Бензойная кислота - 80
Бензиловый спирт - 0,06
Бензоат натрия - 80
95% этанол - 0,4 см3
Гидроокись натрия - 24
Пропиленгликоль - 1,6 см3
Вода - До 4 см3
Были проведены биологические исследования новых соединений для определения их сродства к рецепторам холецистокинина и гастрина с использованием известной методики (A.Saito et coll., J.Neuro. Chem., 37 483-490, 1981).
Исследования проводились на мембранных препаратах поджелудочной железы и коры головного мозга мыши в присутствии исследуемых соединений и рецепторов ССК.
Результаты выражены в минимальной концентрации исследованных соединений, ингибирующей на 50% связь холецистокинина со своим рецептором, и указаны в таблице.



Изобретение относится к производным пирролидина формулы (I), в которой либо R означает метилен-, этиленрадикал, >SO, >SO2 группы или атом серы; R1 означает пиридинил, фурил, тиенил, при необходимости замещенной одним или несколькими алкильными группами, нафтил, индолил или фенил, при необходимости замещенный одним или несколькими заместителями, выбранными из атомов галогена, алкил-, алкокси-, гидрокси- и диалкиламиногруппы; R5 означает атом водорода; либо R означает метилен, R1 - атом водорода и R5 означает фенил; либо R означает группу > CHR6, R1 и R5 означают атом водорода; R2 означает алкоксикарбонил, циклоалкил-алкилокси-карбонил- и др., R3 означает индолил- или фениламинорадикал, фенильное ядро которого замещено одним или несколькими заместителями, выбранными из ряда, содержащего атом галогена, алкил-, алкокси-, алкилтио-группу и др.; R4 означает атом водорода и алкилрадикал; R6 означает фенил-радикал в виде рицемической смеси или энантиомеров, а также их соли. Получают соединения общей формулы (I) либо путем взаимодействия активного производимого карбаминовой кислоты, полученного воздействием N,N - диимидазолкарбонила на производное общей формулы (II), с анилином, либо путем взаимодействия соединения общей формулы (II) с фенилизоцианатом, либо путем взаимодействия соединения общей формулы (IV) с кислотой HOOC-CH (R4)-NH-CO-R3 или с реакционноспособным производным этой кислоты. Способ разделения энантиомеров соединений формулы (I) заключающийся в том, что проводят хроматографию по хиральной фазе. Фармацевтическая композиция, обладающая активностью к рецепторам холецистокинина и гастрина, содержащая активное начало и фармацевтические добавки и соединение формулы (I) в качестве активного начала в эффективном количестве. 8 с. и 4 з.п - лы, 1 табл.  ;
;
\ \\1 1. Производные пирролидина общей формулы I \\\6 $$$ \\\1 в которой либо R метилен-, этиленрадикал, $$$группы или сера; \\\4 R<Mv>1<D> - пиридилрадикал, фурилрадикал, тиенилрадикал при необходимости замещенный одной или несколькими алкильными группами, нафтилрадикал, индолилрадикал или фенилрадикал, при необходимости замещенный одним или несколькими заместителями, выбранными из атомов галогена, алкил-, алкокси-, гидрокси- и диалкиламиногруппы; \ \ \ 4 R<Mv>5<D> - водород, \\\2 либо R - метиленрадикал; \\\4 R<Mv>1<D> - водород; \\\4 R<Mv>5<D> - фенилрадикал; \\\2 либо R - группа $$$ \ \ \ 4 R<Mv>1<D> и R<Mv>5<D> каждый - водород; \\\4 R<Mv>2<D> - алкоксикарбонил-, циклоалкил-алкилокси-карбонил-, -CONR<Mv>9<D>R<Mv>10<D> или фенилрадикал, при необходимости замещенный одним или несколькими заместителями, выбранным из алкил-, алкокси- или гидроксигруппы; \\\4 R<Mv>3<D> - индолил- или фениламинорадикал, фенильное ядро которого при необходимости замещено одним или несколькими заместителями, выбранными из ряда, содержащего атом галогена, алкил-, алкокси-, алкилтио-, трифторметил-, карбокси-, алкоксикарбонил-, гидрокси-, ацил-, гидроксиимино-алкил-, тетразолил-5, моно- или полигидроксиалкилгруппу, группу -алк-COOX, -акл'-COOX, -O-алк-COOX, -CH=CH-COOX, -алк-SO<Mv>3<D>H в форме соли, -S-алк-COOX, -CX= NO-алк-COOX; \\\4 R<Mv>4<D> - водород или алкилрадикал; \\\4 R<Mv>6<D> - фенилрадикал; \\\4 R<Mv>9<D> - водород или алкил; \\\4 R<Mv>10<D> - алкил или фенилрадикал, \\\ 4 или R<Mv>9<D> - R<Mv>10<D> образуют с атомом азота, с которым они связаны, 1,2,3,4-тетрагидрозинолил или пиперидинилрадикал, при необходимости замещенный одним или несколькими радикалами алкила; \\\4 X - алкилрадикал, водород или фенилалкилрадикал; \\\4 алк - алкилрадикал; \\\4 алк' - гидроксиалкилрадикал, \\\1 причем все перечисленные алкильные и алкоксиальные радикалы и алкильные и алкоксильные части содержат 1 - 4 атома углерода в прямой или разветвленной цепи, ацильные радикалы или ацильные части содержат 2 - 4 атома углерода и циклоалкильные радикалы и циклоалкильные части содержат 3 - 6 атомов углерода, \\\1 в виде рацемической смеси или энантиомеров при наличии по крайней мере одного асимметричного центра, а также их соли. \\\2 2. Соединение формулы I по п. 1, где R - метиленрадикал, сера или радикал $$$ R<Mv>1<D> - при необходимости замещенный фенилрадикал, R<Mv>2<D> - фенил- или алкоксикарбонилрадикал, R<Mv>4<D> и R<Mv>5<D> - водород, R<Mv>3<D> фениламинрадикал, фенильное ядро которого замещено карбокси-, алк-COOH, -S-алк-COOH, гидроксиалкил -алк'-COOH или -алк-SOR<Mv>3<D>-радикал. \\\2 3. Соединения формулы I по п.1 следующие: \\\2 1-{2-{3-[3-(1-гидроксиэтил-(RS))фенил] -уреидо} -ацетил}-5-фенил- пролинат трет-бутила-(2RS, 5SR), \\\2 2-{ 3-{3-[2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил(2S,5R)) 2-оксо-этил] уреидо} фенил} пропионовая кислота (форма B), \\\2 {3-{3-[2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил-2-оксо- этил] уреидо} фенилтио} уксусная-(2RS, 5SR) кислота, \\\2 3-{3-[4-трет-бутоксикарбонил-2-(2-фтор-3-триазолидинил)- 2-оксо-этил] уреидо} фенилуксусная-(2R,4R) кислота, \\\2 2-{3-{ 3-[2-(2-трет-бутоксикарбонил-2-(2-фтор-фенил)-3-тиазолидинил-(2R,4R))-2-оксо-этил] уреидо} фенил} пропионовая (2R,4R) кислота (форма B), \\\2 1-{3-{3-[2-(4-трет-бутоксикарбонил-2-фенил-3-тиазолидин- (2R, 4R))-2-оксо-этил]уреидо} фенил} этансульфонат калия (RS), смесь форм A и B, \\\2 1-{3-{3-[2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил-(2RS, 5SR) -2-оксо-этил]уреидо} фенил} этансульфонат калия-(RS), \\\2 3-{3-[2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил)-2-оксо- этил]уреидо}1-фенилметансульфонат калия-(2S,5R), \ \ \2 3-{3-[2-(2-трет-бутоксикарбонил-5-фенил-1-пирролидинил)-2-оксо- этил] уреидо} бензойная-(2S, 5R) кислота, \\\2 3-{3-[2-(2-трет-бутоксикарбонил-5-(2-фтор-фенил)-1-пирролидинил)-2-оксо- этил] уреидо} бензойная-(2RS, 5SR) кислота, \ \ \ 2 3-{3-[2-(2,5-дифенил-1-пирролидинил)-2-оксо-этил]уреидо}-бензойная -(цис) кислота, \\\2 3-{2-[2(2-гидрокси-фенил)5-фенил-1-пирролидинил] -2-оксоуреидо} фенилуксусная-(2RS, 5SR) кислота, \\\2 3-{3-[2-(4-трет-бутоксикарбонил-2-фенил-3-тиазолидинил)-2-оксо- этил]уреидо}фенилуксусная-(2R, 4R) кислота, \\\2 3-{3-[2-(4-трет-бутоксикарбонил-2-фенил-3-тиазолидинил)-2-оксо- этил]уреидо}бензойная-(2R,4R) кислота, \\\2 2-{3-{3-[2-(4-трет-бутоксикарбонил-2-(2-фтор-фенил)-1-оксид- 3-тиазолидинил-(1RS, 2R, 4R))-2-оксо-этил] уреидо} фенил} пропионовая кислота (форма A), \\\2 3-{{3-[4-трет-бутоксикарбонил-2-(2,3-дифтор-фенил)-3-тиазолидинил] - 2-оксо-этил] уреидо} фенилуксусная-(2R, 4R) кислота, \\\2 1-{2-{3-[3-(1-гидроксиимино-этил)-фенил-(E)] уреидо-ацетил} -5-фенил-пролинат трет-бутила-(2RS,5SR). \\\2 4. Способ получения соединения общей формулы I по одному из пп.1 - 3, где R - метилен-, этиленгруппа, группа $$$ или сера, а R<Mv>3<D> - фениламинорадикал, фенильное ядро которого при необходимости замещено одним или несколькими заместителями по пп.1 - 3, или его соли, отличающийся тем, что активное производное карбаминовой кислоты, полученное при необходимости in situ, путем воздействия N,N-диимидазолкарбонила на производное общей формулы II \\\6 $$$ \ \ \ 1 где R имеет указанные значения; \\\4 R<Mv>1<D>, R<Mv>2<D>, R<Mv>4<D> и R<Mv>5<D> имеют значения по пп.1 - 3, \\\1 подвергают взаимодействию с анилином, фенильное ядро которого при необходимости замещено одним или несколькими заместителями, указанными при характеристике радикала R<Mv>3<D>, с последующим выделением целевого продукта в свободном виде или в виде соли. \\\2 5. Способ по п.4, отличающийся тем, что реакцию проводят в инертном органическом растворителе при температуре от 20<198>C до температуры кипения растворителя. \\\2 6. Способ получения соединений общей формулы I по пп. 1 - 3, где R - метилен, этилен, радикал $$$ или сера, а R<Mv>3<D> - фениламинорадикал, фенильное ядро которого может быть замещено одним или несколькими заместителями, выбранными из ряда, содержащего атом галогена, алкил-, алкоксиалкилтио-, трифторметил-, ацил-, алкоксикарбонилгруппу и алк-SO<Mv>3<D>H в виде соли, а также их солей, отличающийся тем, что соединение общей формулы II \ \ \6 $$$ \\\1 где R, R<Mv>1<D>, R<Mv>2<D>, R<Mv>4<D> и R<Mv>5<D> имеют указанные значения, \\\1 подвергают взаимодействию с фенилизоцианатом, фенильное ядро которого может быть замещено одним или несколькими заместителями, выбранными из ряда, содержащего атом галогена, алкил-, алкокси-, алкилтио-, трифторметил-, ацил-, алкоксикарбонилгруппу и -алк-SO<Mv>3<D>H в виде соли, с последующим выделением целевого продукта и при необходимости превращением его в соль. \\\2 7. Способ по п.6, отличающийся тем, что взаимодействие проводят в инертном органическом растворителе при температуре от 10<198>C до температуры кипения растворителя. \\\2 8. Способ получения соединений общей формулы I по пп.1 - 3, где R - метилен, этилен, радикал $$$ или сера, а R<Mv>3<D> - фениламинорадикал, фенильное ядро которого замещено группой, выбранной из ряда, содержащего карбоксигруппу, -алк-COOH, -O-алк-COOH, -алк'-COOH, -CH=CH-COOH, -S-алк-COOH или CX=N-O-алк-COOH, а также их солей, отличающийся тем, что проводят гидролиз или гидрогенолиз соответствующего сложного эфира с последующим выделением целевого продукта и при необходимости превращением его в соль. \\\2 9. Способ получения соединений общей формулы I по пп.1 - 3, где R - метилен, этилен, радикал $$$ или сера, а R<Mv>3<D> - индолил- или фениламинорадикал, фенильное ядро которого при необходимости замещено одним или несколькими заместителями, выбранными из группы, содержащей атом галогена, алкил-, алкоксиалкилтио-, трифторметил-, ацетил-, алкоксикарбонилгруппу, группу -алк-COOX или -алк'-COOH, где X не является водородом, а также их солей, отличающийся тем, что проводят взаимодействие соединения общей формулы (IV) \\\6 $$$ \\\1 где R, R<Mv>1<D>, R<Mv>2<D> и R<Mv>5<D> определены выше, \\\1 с кислотой общей формулы \\\6 $$$ \ \ \ 1 где R<Mv>3<D> и R<Mv>4<D> имеют указанные значения; \\\1 или с реакционноспособным производным этой кислоты с последующим выделением целевого продукта и при необходимости превращением его в соль. \\\2 10. Фармацевтическая композиция, обладающая активностью к рецепторам холецистокинина и гастрина, содержащая активное начало и фармацевтические добавки, отличающаяся тем, что в качестве активного начала она содержит по крайней мере одно соединение по пп.1 - 3 в эффективном количестве. \\\2 11. Способ разделения энантиомеров соединений формулы I по одному из пп.1 - 3, отличающийся тем, что проводят хромотографию по хиральной фазе, имеющей структуру \\\6 $$$ \ \\1 где G<Mv>1<D> - группа 3,5-динитробензол. \\\2 12. Хиральная фаза для разделения энантиомеров соединений общей формулы I по пп.1 - 3, представляющая собой модифицированную двуокись кремния, имеющую структурные фрагменты, охарактеризованные в п.11.
| US 4499102 A, A 61 K 31/40, 1985 | |||
| Прием выполнения способа бензалирования целлюлозы по авторскому свидетельству № 38631 | 1938 |
|
SU59966A2 |

