Данное изобретение относится к чистому (-) энантиомеру [[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил)] фенил гидразоно] пропандинитрилу формулы I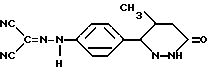
Изобретение относится также к солям, композициям и процессу получения этого энантиомера, а также и к новым промежуточным соединениям этого процесса.
Соединение, соответствующее данному изобретению, является пригодным для использования в качестве кардиотонического средства, антигипертензивного и сосудорасширяющего средства для лечения застойной сердечной недостаточности.
Рацемическая смесь [[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил)фенил] гидразино] пропандинитрила (1) с точкой плавления 258-263oC была описана ранее в патентной заявке Великобритании 2228004. Было показано, что соединение (1) является сильнодействующим при лечении застойной сердечной недостаточности и обладает значительной способностью в связывании кальция с тропонином. Наши дальнейшие исследования в настоящее время неожиданно выявили, что кардиотоническая активность преимущественно относится к оптически активному (-) энантиомеру этого соединения. Кроме того, было установлено, что растворимость в воде (-) энантиомера более, чем в 30 раз выше по сравнению с рацематом. Было найдено также, что биологическая доступность (-) энантиомера является более высокой по сравнению с рацематом. Следовательно, чистый (-) энантиомер является особенно пригодным по сравнению с рацемическим соединением для использования в качестве лекарственного средства для лечения застойной сердечной недостаточности.
(+) и (-) энантиомеры [[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил)фенил] гидразоно]пропандинитрила (1) могут быть разделены пропусканием рацемического соединения через хиральную фазу хроматографической колонки. Однако этот метод является трудоемким, если требуются большие количества вещества.
Другой возможностью получения чистых энантиомеров соединения (1) является использование соответствующих оптически активных энантиомеров 6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3-(2H)она в качестве промежуточного соединения. Рацемический 6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3 (2H)он формулы (II)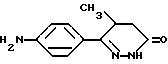
может быть синтезирован методами, известными из литературы J.Med.Chem. 17, 273-281 (1974). Однако разделение рацемического соединения (II) оказалось очень трудным из-за наличия в молекуле слабо основной 4-аминогруппы. Соли 6-(4-амино-фенил)-5-метил- 4,5-дигидропиридазин-3(2H)она с оптически активными кислотами легко гидролизуется при кристаллизации обратно в соединение (II) и в разрешающее соединение, что мешает процедуре разделения или делает ее вообще невозможной.
Разделение чистых энантиомеров соединения (II) на хиральной БЭЖХ-колонке было описано в заявке EP 208518. Однако этот метод неприменим в промышленном масштабе. Энантиоселективный семиступенчатый синтез (-)-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)она, начинающийся с (+)-2-хлорпропионовой кислоты, также был описан в литературе (J.Org.Chem., 56, 1963 (1991)). Общий выход в этом методе составляет всего 12% с образованием (-)-6-(4-аминофенил)-5- метил-4,5-дигидропиридазин-3(2H)она с оптической чистотой 97,2%.
В настоящее время установлено, что хорошее энантиомерное разделение соединения (II) может быть осуществлено при использовании избытка L- или D-винной кислоты, предпочтительно от 2 до 3 эквивалентов по отношению к соединению (II) в 2-пропаноле. Кислые соли (-)-6-(4-аминофенил)-5-метил-4,5-дигидропиридпзин-3(2H)она с D-винной кислотой 2-пропанол сольвата (IIIb) или соответствующего (+)-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)она с L-винной кислотой 2-пропанол сольвата (IIIа) кристаллизуются с хорошим выходом и практически являются оптически чистыми.
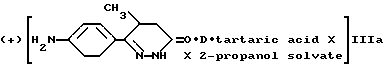
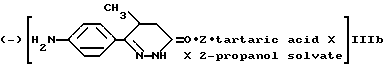
Далее, было установлено, что незначительный компонент в частично обогащенной энантиомерной смеси может быть выкристаллизован в виде рацемического соединения (II) из диоксана, оставляя основной компонент в растворе. Таким образом, соли (IIIa) или (IIIb), полученные в упомянутой выше кристаллизации, были отфильтрованы и с помощью карбоната калия было выделено свободное основание, а продукт был обработан диоксаном. Таким образом, оба энантиомера соединения (1) получают таким методом двухфазной кристаллизации с высокой оптической чистотой свыше 99%. Выход в этом процессе является очень высоким потому, что рацемическое соединение (I) получается из диоксана в кристаллическом состоянии и может быть рециркулировано. Оба разделенных соединения L- и D-винной кислоты могут быть поочередно использованы в вышеназванном процессе, но предпочтительной является Z-винная кислота из-за ее большой дешевизны.
В основном оптически чистые (-) и (+) энантиомеры соединения (I) могут быть получены затем из соответствующих в основном оптически чистых (-) и (+) энантиомеров соединения (II) соответственно с использованием обычного процесса, опубликованного в патентной заявке Великобритании 2228004 - с высокой оптической чистотой и с относительно высокими количественными выходами. Процесс, опубликованный в заявке Великобритании 2228004 для получения соединения (I), включает обработку соединения формулы (II) нитритом натрия и малононитрилом в кислых средах. Термин "в основном оптически чистый" означает здесь оптическую чистоту более 90%, предпочтительно более 95% и более предпочтительно - свыше 99%.
Соли энантиомеров соединения (I) могут быть получены известными методами. Фармацевтически приемлемые соли используются в качестве активных фармацевтических препаратов, однако предпочтительными являются соли с щелочными или щелочноземельными металлами.
Растворимость представлена в таблице 1.
Кардиотоническое действие.
Кардиотоническое действие (-) и (+) энантиомеров [[4-(1,4,5,6- тетрагидро-4-метил-6-оксо-3-пиридазинил)фенил] гидразоно]пропандинитрила (I) изучалось на выделенной, электрически напряженной, правой вентрикулярной мышце морской свинки. Опыты проводились в обычной ванне раствора Pyrode, как опубликовано Otani и др., Japan, J. Pharmacol. 45, 425, 1987.
Результаты представлены в таблице 2. Они показывают, что (-) энантиомер был в 47 раз более эффективным, чем (+) энантиомер.
Биологическая пригодность.
Чертеж иллюстрирует концентрацию, отвечающую суммарному содержанию [[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил) фенил]гидразоно]пропандинитрила в плазме собаки после орального приема единичной дозы рацемата (1мг/кг) и (-)-энантиомера (0,5 мг/кг). Кривая A относится к (-)-энантиомеру, а кривая B - к рацемическому [[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3- пиридазинил)фенил] гидразоно] пропандинитрилу. Чертеж показывает, что при использовании (-)-энантиомера вместо рацемата требуется менее, чем половина дозы для достижения того же уровня концентрации лекарственного вещества плазмы.
Фармацевтически активное соединение в соответствии с данным изобретением готовится в дозированных формах с использованием известных в данной области правил. Оно принимается млекопитающими, т.е. людьми, как таковое или в комбинации с подходящими фармацевтическими наполнителями в виде таблеток, драже, капсул, суппозиториев, эмульсий, суспензий или растворов. Состав, соответствующий изобретению, содержит терапевтически эффективное количество фармацевтически активного соединения изобретения. Содержания активного компонента в композиции составляют от 0,5 до 100% на вес. Обычно соединение в композиции составляют от 0,5 до 100% на вес. Обычно, соединение изобретения может приниматься в виде оральных доз низкого значения - от 1 до 50 мг в день. Выбор подходящих ингредиентов для композиции является обычным для специалистов. Очевидно, что для этого могут быть использованы применяемые обычно в этой области технологии соответствующие носители, растворители, гельобразующие ингредиенты, образующие дисперсию, антиоксиданты, подкрашивающие вещества, увлажняющие соединения и другие ингредиенты. Значение LD50 для (-) энантиомера, вводимого внутривенно крысам, составляло 57 мг/кг.
Композиции готовят с учетом цели применения лекарственного средства, обычно удовлетворительные результаты дает использование непокрытых таблеток. Иногда целесообразно использовать покрытые таблетки, т.е. так называемые энтеротаблетки для обеспечения того, что лекарственное средство достигает нужной части желудочно-кишечного тракта. Могут использоваться также драже и капсулы.
Пример 1.
Разделение рацемического 6-(4-аминофенил)-5-метил-4,5- дигидропиридазин-3(2H)она L-винной кислотой.
±-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3 (2H)он (203 г, 1 моль) растворялся в 2-пропаноле (40 дм3) при нагревании. К этому раствору постепенно добавляли раствор L-винной кислоты (300 г, 2 моля). Смесь перемешивалась при нагревании до получения прозрачного раствора. Раствор медленно охлаждали при перемешивании до комнатной температуры. После перемешивания раствора в течение ночи при 20oC был отфильтрован кристаллический продукт (IIIb). Влажная соль растворялась в воде (1,5 дм3) и при перемешивании добавлялся раствор карбоната калия (190 г K2CO3 в 0,75 дм3 воды). Свободное основание отфильтровывалось, промывалось водой и сушилось. Продукт (104,6 г) растворяли в диоксане (0,6 дм3) при нагревании и давали ему охлаждаться до комнатной температуры. Рацемический 6-(4-амитнофенил)-5-метил-4,5-дигидропиридазин-3(2H)он отфильтровывался (74,6 г), а фильтрат выпаривался до сухого состояния в вакууме, образуя (-)-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин- 3(2H)он в виде кристаллического твердого вещества (23,8 г) с оптической чистотой 99,5%, т. пл. 207-210oC, [a]
2-пропанольный раствор, содержащий (+)-энантиомер вместе с рацематом соединения (I) выпаривали в вакууме до сухого состояния. На остаток действовали раствором карбоната калия, как описано выше с получением смеси (+)-энантиомера и рацемата (87,3 г), которую растворяли при нагревании в диоксане (0,48 дм3). После охлаждения рацемат отфильтровывали (48,0 г), а фильтрат упаривали до сухого состояния в вакууме с получением (+)-6-(4-аминофенил)-3-метил-4,5-дигидропиридазин-3(2H)она в виде твердого кристаллического вещества (26,1 г) с оптической чистотой 99,5%, т. пл. 206-209oC, [a]
Пример 2.
(+)-[[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил) фенил] гидразоно]-пропандинитрил.
Озаглавленное соединение было получено как описано в патентной заявке Великобритании 2228004 из (+)-8-(4-амино-фенил)-5- метил-4,5-дигидропиридазин-3(2H)она. Выход 98%, т. пл. 210-214oC, [a]
Пример 3.
(-)-[[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил) фенил]гидразоно]-пропандинитрил.
Озаглавленное соединение было получено как описано выше из (-)-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)она. Выход 97%, т. пл. 210-214oC, [a]
Пример 4.
Получение чистой диастереоизомерной соли (IIIa).
508 мг (2,5 ммоль) чистого (+)-6-(4-аминофенил)-5- метил-4,5-дигидроппиридазин-3(2H)она, полученного в примере 1, растворялось в 100 мл 2-пропанола. Было добавлено 750 мг (5,0 ммоль) D-винной кислоты и смесь нагревалась до кипения. После охлаждения было получено 800 мг кристаллического (+)-6-(4-аминофенил)-5-метил- 4,5-дигидропиридазин-3(2H)она D-тартрата моно 2-пропанол сольвата, т. пл. 97-105oC.
Пример 5.
Получение чистой диастереоизомерной соли (IIIb).
Был повторен вышеприведенный процесс с использованием (-)-6- (4-амино-фенил)-5-метил-4,5-дигидропиридазин-3(2H)она и L-винной кислоты. Т. пл. 98-106oC.
Пример 6.
Получение (-)-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин- 3(2H)она разделением соответствующего рацемата L-винной кислотой.
±-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)он (203 г, 1 моль) растворялся при нагревании в 2-пропаноле (10 дм3). К этому раствору постепенно добавлялась (L)-винная кислота (300 г, 2 моль). Смесь перемешивалась при нагревании до тех пор, пока раствор не становился прозрачным, и медленно охлаждалась в течение 3 часов до 50oC, после чего перемешивалась далее при 50oC в течение ночи. Кристаллический продукт отфильтровывался и далее повторялась методика, описанная в примере 1. Выход (-)-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)она составил 30,3 г (97,4% от теоретического). Оптическая чистота была 99,7%. Всего было регенерировано 140,6 г рацемата.
Оптическая чистота соединений определялась методом высокоэффективной жидкостной хроматографии. Измерительный прибор представлял собой ступенчатый насос Waters 600 E с фотодиодной детекторной матрицей Waters 991 и инжектором Waters 700 Satellite Wisp (millipore Co), управляемый NEC Powermate SX Plus компьютером. Энантиомеры 6-(4-амино фенил)-5-метил-4,5-дигидропиридазин-3(2H)она были разделены с использованием хиральной колонки целлюлозного типа (Chiraeel=OJ, 4,6 x 250 мм, Daicel Chemical Industries LTD). Подвижная фаза состояла из 97% 2-пропанола и 3% гексана. Скорость пропускания была 0,3 мл/мин. Энантиомеры [[4-(1,4,5,6-тетрагидро-4-метил-6- оксо-3-пиридазинил)фенил] гидразино] пропандинитрила были разделены с использованием β--циклодекстриновой колонки (Cyclobond eb; 4,6 x 250 мм, advance Separation Technologies Inc. ). Подвижная фаза состояла из 41% метанола в воде, pH которой с помощью 1%-ного триэтиламмоний ацетата в качестве буфера была доведена до 4,0. Скорость прохождения составляла 0,3 мл/мин.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ [[4-(1,4,5,6-ТЕТРАГИДРО-4-МЕТИЛ-6-ОКСО-3-ПИРИДАЗИНИЛ)ФЕНИЛ]-ГИДРАЗОНО] ПРОПАНДИНИТРИЛА ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ИШЕМИИ МИОКАРДА, СПОСОБ ЛЕЧЕНИЯ ИШЕМИИ МИОКАРДА | 1993 |
|
RU2146142C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 1990 |
|
RU2048467C1 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ С ЩЕЛОЧНЫМИ ИЛИ ЩЕЛОЧНО-ЗЕМЕЛЬНЫМИ МЕТАЛЛАМИ | 1992 |
|
RU2068844C1 |
| СПОСОБ ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНЕЙ МОТОНЕЙРОНОВ | 2015 |
|
RU2706001C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННОГО ИМИДАЗОЛА ИЛИ ЕГО НЕТОКСИЧНОЙ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ КИСЛОТНО-АДДИТИВНОЙ СОЛИ | 1990 |
|
RU2021263C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3,4-ДИГИДРОКСИ-5-НИТРОБЕНЗАЛЬДЕГИДА | 1995 |
|
RU2130449C1 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ БЕНЗАЗЕПИНА, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2129123C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3,4-ДИГИДРОКСИ-5-НИТРОБЕНЗАЛЬДЕГИДА | 1993 |
|
RU2098405C1 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1992 |
|
RU2120440C1 |
| Способ получения замещенного имидазола или его нетоксичной фармацевтически приемлемой кислотно-аддитивной соли | 1991 |
|
SU1836355A3 |


Изобретение относится к чистому (-)-энанитомеру [[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил фенил] гидразоно] пропандинитрила, его фармацевтически приемлемым солям, фармацевтической композиции, обладающей кардиотоническим действием, которая содержит эффективное количество указанного энантиомера или его смеси фармацевтически приемлемым носителем. Предложен также способ получения промежуточного оптически чистого (-)-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3(2Н)-она путем обработки смеси исходных энантиомеров избытком L -винной кислоты, предпочтительно 2-3 эквивалентами, в пропаноле при нагревании с последующем охлаждением полученного раствора до комнатной температуры, отделением полученного кристаллического продукта, переводом его в свободное основание подщелачиванием, растворением свободного основания в диоксане при нагревании, охлаждением полученного раствора до комнатной температуры с последующим разделением полученной смеси на осадок, содержащий рацемический 6-(4-аминофенил)-5-метил-4,5-дигидропиридин-3(2Н)он, и фильтрат и выделением из фильтрата оптически чистого (-)-энантиомера названного промежуточного соединения. Для получения (-)-энантиомера целевого соединения, имеющего оптическую чистоту более 90%, промежуточное (-)-энантиомерное соединение, полученное вышеописанным образом, обрабатывают нитратом натрия и малонитрилом. 4 с. и 1 з.п.ф-лы, 1 ил., 2 табл.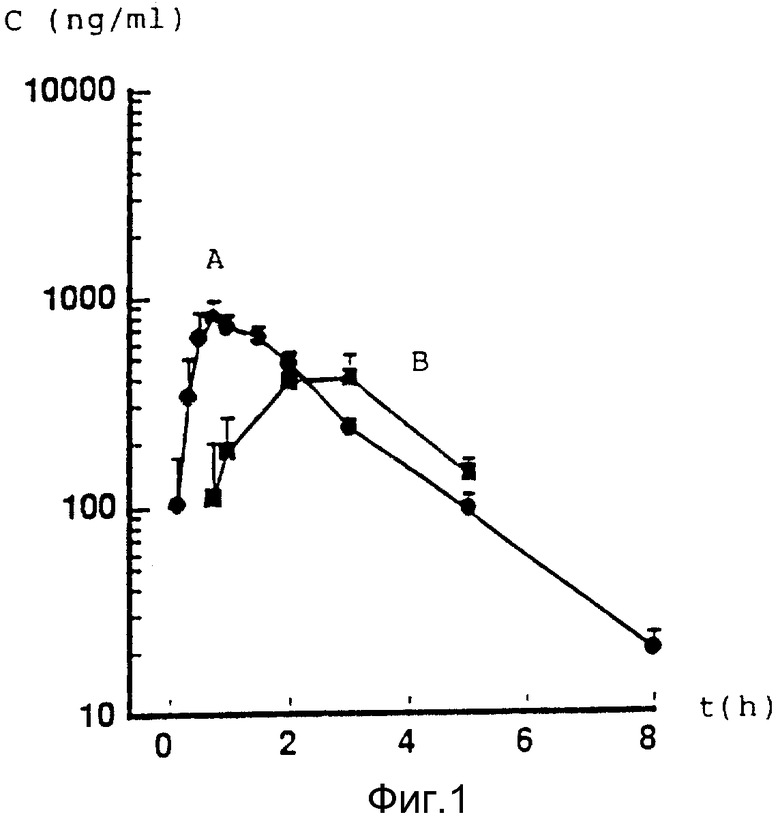
\ \\1 1. (-)-[[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил)фенил] гидразоно] пропандинитрил и его фармацевтически приемлемые соли. \\\2 2. Фармацевтическая композиция, обладающая кардиотоническим действием, содержащая эффективное количество [[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил)фенил] гидразоно] пропандинитрила и его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым носителем, отличающаяся тем, что содержит указанное соединение в (-)-энантиомерной форме. \\\2 3. Способ получения оптически чистого (-)-6-(4-аминофенил)-5-метил-4,5- дигидропиридазин-3(2H)-она, отличающийся тем, что рацемическую смесь исходных энантиомеров приводят в контакт с избытком L-винной кислоты в 2-пропаноле при нагревании с последующим охлаждением полученного раствора до комнатной температуры, отделением полученного кристаллического продукта и переводом его в свободное основание путем подщелачивания, растворением свободного основания в диоксане при нагревании, охлаждением полученного раствора до комнатной температуры с последующим разделением полученной смеси на осадок, содержащий рацемический 6-(аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)-он, и фильтрат и выделением из фильтрата оптически чистого (-)-6-(4-аминофенил)-5-метил-4,5-дигидропиридазин-3(2H)-она. \\\2 4. Способ по п.3, отличающийся тем, что L-винную кислоту берут в количестве от 2 до 3 эквивалентов на эквивалент рацемической смеси. \\\2 5. Способ получения (-)-[[4-(1,4,5,6-тетрагидро-4-метил-6-оксо-3-пиридазинил)-фенил] гидразоно] пропандинитрила, имеющего оптическую чистоту более 90%, отличающийся тем, что (-)-6-(4-аминофенил)-5-метил-4,5- дигидропиридазин-3(2H)-он, имеющий оптическую чистоту более 90% и полученный в соответствии с пп.3 и 4, подвергают обработке нитритом натрия и малононитрилом. \\\9 Приоритет по пунктам: \\\4 03.01.91 - по пп.1 и 2; \\ \4 05.09.91 - по пп.3 - 5.
| GB, N 2228004, A1 кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, N 0259835 A, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, N 0208518 B, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, N 4521415 A, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, N 4843072 A, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, N 4914093 A, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
