Введение.
Проблема целенаправленной локальной доставки фармакологических агентов непосредственно в зону патологического процесса (независимо от его этиологии) является одной из наиболее актуальных в современной фармакологии. Это связано с тем, что получение достаточно высоких концентраций лекарственного препарата в очаге поражения в течение необходимого промежутка времени считается одним из ключевых условий обеспечения эффективности терапии.
При стандартных методах лечения это обычно достигается системным введением больших концентраций препарата, что, однако, обладает рядом ограничений. Наиболее существенными из них являются побочные эффекты, связанные с токсичностью многих соединений в больших концентрациях (например цитостатиков), а также высокая скорость выведения из кровеносного русла и тканей.
Эти трудности могут быть успешно разрешены при направленной доставке фармакологического агента с использованием биоспецифических методов, в частности основанных на лигандрецепторных взаимодействиях. Одним из таких подходов является создание препаратов, состоящих из биоорганического носителя и лиганда с фармакологической активностью. На этом принципе, в чтности, основана система доставки цитостатика к раковым клеткам, являющаяся ближайшим аналогом изобретения (EP 398305, 1990, A 61 K 47/48).
Теоретическое обоснование изобретения.
Мы предлагаем собственную реализацию принципа направленного транспорта, которая заключается в связывании in vivo молекул-носителей фармакоактивных соединений через специфические рецепторы с форменными элементами крови, принимающими учтие в патологическом процессе и способными вследствие этого доставить фармакологический препарат непосредственно в зону патологического поражения.
В этом случае появляется принципиальная возможность минимизировать общее количество препарата, поступающего в организм, с одновременным достижением его максимальной концентрации в течение короткого времени в зоне поражения, что, наряду с обеспечением и усилением терапевтического эффекта, приведет к значимому снижению как общей токсичности, так и системной инактивации применяемого фармакологического соединения.
Для выполнения поставленной задачи нами синтезированы оригинальные производные пентапептида цистеинил-аргинилглицил-аспартил-цистена (в дальнейшем CRGDC), которые принадлежат к группе (RGD)-пептидов - соединений, содержащих последовательность, аргинин-глицин-аспарагиновая кислота (RGD).
Аминокислотная RGD-последовательность, согласно современным представлениям, является ключевым фрагментом комплементарного взаимодействия адгезивных белков (фибриногена, фибронектина, фактора Виллебранда, витронектина) с полифункциональным гликопротеидным комплексом 2b/3a (интегрином), обеспечивающим межклеточные взаимодействия и связь наружной поверхности клеточной мембраны с некоторыми биологически активными растворимыми соединениями [1]. Доказана вовлеченность RGD - содержащих белков, а также чувствительного к ним рецепторного комплекса - интегрина 2b/3a в процессы канцерогенеза, патогенез ряда гематологических и сердечно-сосудистых заболеваний [2,3].
Рецепторы 2b/3a, ответственные за связывание с клеточной поверхностью фиброногена и фибронектина, обнаружены в большом количестве в плазматических мембранах тромбоцитов, которые играют важную роль в формировании метастатических очагов опухолевого роста, образуя клеточные комплексы с мигрирующими в кровеносном русле злокачественными клетками [4]. Также хорошо известна роль тромбоцитов в локальном поражении сосудистой стенки в процессе образования морфологического субстрата атеросклероза - атеросклеротической бляшки [5].
Это дает основание предполагать, что фармакологический агент, присоединенный к RGD-пептиду при введении в кровеносное русло будет связываться с рецепторами 2b/3a тромбоцитов и адресно транспортироваться клетками в места повышенной адгезии тромбоцитов. Такими местами могут быть учтки атеросклеротического поражения коронарных сосудов, метастатические зоны опухолевого роста и другие точки скопления тромбоцитов.
В соответствии с изложенными принципами нами были выбраны лиганды для синтеза двух оригинальных производных RGD-пептидов.
Первый из них - 2,4-диоксо-5-фторпиримидин или 5-фторурацил является антиметаболическим препаратом. Противоопухолевая активность 5-фторурацила определяется его превращением в раковых клетках в 5-фтор-2-дезоксиуридин-5'-монофосфат, который является конкурентным ингибитором активности тимидинсинтетазы, принимающей учтие в синтезе ДНК [6]. 5-фторурацил показан как цитостатический препарат при лечении неоперабельных форм, рецидивов и метастазов рака желудка и толстого кишечника, при опухолях поджелудочной железы. Обычно вводится одномоментно внутривенно из расчета 10-15 мг на килограмм веса, в результате чего его концентрация в крови достигает 1-1,5 ммоль/л.
Второй лиганд - нитрозо-тиол, содержащий группу NO, являющуюся активной чтью коронарорасширяющих фармакологических соединений, относящихся к классу органических нитратов [7].
Для доставки этих биологически активных лигандов в места патологического поражения с использованием в качестве клеток носителей тромбоцитов нами синтезированы оригинальные соединения, представляющие собой гибридные молекулы, состоящие из описанных лигандов и пептидов, содержащих RGD- последовательность: карбоксиметил-5-фторурацил-ацетамидометил-цистеинил-аргинил-глицил-аспартил- ацетамидометил-цистеин (в дальнейшем обозначен как (CMF и GRGDC) и ацетиларгинилглицил-аспартил-S-нитрозоцистенин (Ac-RGDC-SNO).
Синтез оригинальных соединений
Материалы и методы
Для синтеза вышеописанных соединений были использованы аминокислоты и их производные фирмы "Reanal" (Венгрия).
Температуру плавления полученных соединений определяли на приборе "Boctins" фирмы "Nagema" (Германия). УФ-спектры снимали на спектрофотометре "Beckman" 24/25 (США). Индивидуальность полученных соединений контролировали с помощью тонкослойной хроматографии и электрофореза. Тонкослойную хроматографию проводили на пластинках "Silnfol" фирмы "Kavalier" (Чехословакия) (S), а также на пластинках фирмы "Merck" (Германия) (m). Использовались системы: (1) 1%-ный аммиак - вторбутиловый спирт 1:3 (2) трет-бутиловый спирт - уксусная кислота - вода 4:1:1; (3) бензол-этанол-этилацетат 3:1:1; (4) метанол - хлороформ 1:7.
Электрофорез проводили на приборе "Laboratorium Flszeresek" (Венгрия) в 2%-ной уксусной кислоте на бумаге FN-12 производства "Filtrak" (Германия) с напряжением на электродах 1500 В в течение 30 мин. Электрофоретическую подвижность определяли по отношению к глицину (EGly). Для всех синтезированных соединений данные элементного анализа соответствовали вычисленным значениям.
Препаративную высокоэффективную жидкостную хроматографию проводили на хроматографе "Waters" 600 Е (США).
По данным аналитической высокоэффективной жидкостной хроматографии чистота конечных продуктов составляла более 95%.
Аминокислотный анализ проводили на аминокислотном анализаторе "Microteohna" T 339 M (Чехословакия). Пептиды гидролизовали в 6 н. HCl в течение 20 ч при температуре 110oC. Данные аминокислотного анализа для всех конечных продуктов соответствовали расчетным значениям.
Масс-спектры снимали на времяпролетном масс-рефлектроне с источником "Электроспрей" (ФИНЭПХФ РАН).
Используемые сокращения:
5-FU - 5-фторурацил;
CMFU - карбоксиметил-5-фторурацил;
HONp - пара-нитрофенол;
ДМФА - диметилформамид;
Синтез CMFU
0,100 г (0,77 ммоль) 5-FU, 0,130 г (1,38 ммоль) хлоруксусной кислоты и 0,12 г (2,15 ммоль) KOH 30 мин кипятили в 10 мл воды с обратным холодильником. Охлажденный раствор наносили на колонку (2,5 х 10 см) со смолой "Dowex" 2 х 8 ("Serva", Германия) в Cl- форме, промывали колонку водой до полного выхода 5-FU, затем смывали продукт 30%-ной уксусной кислотой. Растворитель упаривали, остаток перекристаллизовывали из смеси этанола с серным эфиром. Выход - 0,040 г (27% от теоретического). m/z : 189 (MH+); Т.пл. - 190-200oC; Rf = 0,14 (1,S); O(2,S); O(3,S).
Синтез CMFU-ONp.
0,80 г (0,43 ммоль) CMFu суспендировали в пиридине, при перемешивании и охлаждении на ледяной бане добавляли 0,200 г (0,85 ммоль) трифторацетата л-нитрофенола. Через 1 ч убирали охлаждение и перемешивали при комнатной температуре до полного растворения осадка. Упаривали пиридин, остаток растирали с серным эфиром. Выход - 0,250 г (80% от теоретического). Т.пл. 195oC, Rf = 0,87 (2, S); 0,65 (3, S); 0,53 (4, S)
Синтез H-Cys(Acm)-Arg-Gly-Asp-Cys(Acm)-OH
Синтез проводили твердофазным методом на полимере Меррифильда при использовании автоматического пептидного синтезатора NPS -4000 (Neosistem Laboratoires, Франция). В работе использовали следующие производные аминокислот: Boc-Gly-OH; Boc-Arg(Mts)-OH); Boc-Asp(OBzl)OH; Boc-Cys(Acm)OH. Полноту прохождения реакции образования пептидной связи контролировали с помощью нингидринового теста (8). Трифторуксусную кислоту и растворители готовили в соответствии с требованиями для твердофазного пептидного синтеза, триэтиламин перегоняли над щелочью, а затем над нингидрином.
Все аминокислотные остатки были присоединены в соответствии с протоколом стандартного цикла, состоящего из стадий деблокирования, нейтрализации и конденсации.
Деблокирование
1) Промывка 20 мл хлористого метилена в течение 1 мин;
2) Двукратная обработка 20 мл 65% раствора трифторуксусной кислоты в хлористом метилене в течение 1 и 15 мин;
3) Двукратная промывка 20 мл хлористого метилена без перемешивания;
4) Двукратная промывка 20 мл хлористого метилена по 1 мине.
Нейтрализация
1) Промывка 20 мл ДМФА - 1 мин;
2) Двукратная обработка 20 мл 5% раствора триэтиламина в ДМФА в течение 1 и 2 мин;
2) Трехкратная промывка 20 мл ДМФА.
Конденсация
В 3 мл ДМФА растворяли 3 эквивалента (1,5 ммоль) защищенного производного аминокислоты и 1,5 ммоль 1-гидроксибензотриазола, при охлаждении до 0oC и перемешивании добавляли 1,5 ммоль N,N-диизопропилкарбодиимида, через 30 мин перемешивания, раствор вносили в реактор.
Продолжительность конденсации - 1 - 2 ч. По окончании конденсации:
1) Дважды промывали пептидилполимер 20 мл ДМФА по 1 мине;
2) Промывали 20 мл хлористого метилен 1 мин;
3) Выполняли тест на полноту прохождения реакции.
Отщепление пептида от полимерного носителя.
К 1 г пептидил-полимера добавляли 1 мл тиоанизола и 0,5 мл этандитиола перемешивали на магнитной мешалке 10 мин, затем при охлаждении до 0oC приливали 10 мл трифторуксусной кислоты, перемешивали 10 мин. По каплям добавляли 1 мл трифторметансульфокислоты, снимали охлаждение и перемешивали 0,5 ч при комнатной температуре. Разбавляли реакционную смесь 250 мл эфира и отфильтровывали осажденный на полимере пептид на фильтре Шотта. Затем смывали пептид с полимера трифторуксусной кислотой и высаживали 250 мл серного эфира. Выпавший продукт отфильтровывали, промывали эфиром и сушили в вакууме. Затем пептид обессоливали на колонке с сефадексом G-15(1,9 х 100 см) в 50% уксусной кислоте. Очистку препарата проводили с помощью ВЭЖХ на колонке Delta Pak C18 (19 х 300 мм, 300 А, 15 мкм); элюент - 0,1%-ная трифторуксусная кислота - ацетонитрил, скорость потока - 15 мл/мин, детекция при 230 нм. После очистки пептид лиофилизовали.
Синтез CMFU-Cys(Alm)-Arg-Gly-Asp-Cys(Acm)-OH (CMFU-CRG-DC).
0,12 г (0,17 ммоль) H-Cys(Acm)-Arg-Gly-Asp-Cys(Acm)-OH растворяли в смеси ДМФА - H2O, добавляли 0,7 г (2,26 ммоль) CMFu-ONp и доводили pH до 8 с помощью диизопропилэтиламина. Через сутки реакционную смесь нейтрализовали 1 н. H2SO4, растворитель упаривали и продукт закристаллизовывали в этаноле. Осадок отфильтровывали и промывали этанолом. Остаток растворяли в 0,2 н. уксусной кислоте и очищали гельфильтрацией на колонке с сефадексом G-15("Pharmacia", Швеция). Окончательную очистку проводили с помощью ВЭЖХ, на колонне Delta Pak C18 (19 х 300 мм, 300 А, 15 мкм); элюент - 0,1%-ная трифторукскусная кислота - ацетонитрил, скорость потока - 15 мл/мин, детекция при 230 нм. После очистки пептид лиофилизовали. EGly = 0,82.
Синтез Ac-Arg-Gly-Asp-Cys(SNO)-OH (Ac-RGDC-SNO).
К раствору 20 мл (0,0355 ммоль) Ac-Arg-Cly-Asp-Cys(Acm)-OH в 1 мл 30%-ной уксусной кислоты при перемешивании добавляли 11,3 мг (0,0355 ммоль) ацетата ртути. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли 0,05 мл (0,71 моль) 2-меркаптоэтанола и перемешивали еще 1 ч. Осадок солей ртути отфильтровывали, к фильтрату добавляли 46,5 мг нитрита натрия (раствор окрашивается в малиновый цвет) и перемешивали 0,5 ч при комнатной температура. Контроль за полнотой прохождения реакции осуществляли с помощью ВЭЖХ pH раствора доводили до 8 добавлением бикарбоната натрия и продукт очищали с помощью ВЭЖХ. После очистки пептид лиофилизировали.
В УФ спектре препарата присутствуют два характеристических максимума поглощения - при 337 нм и 545 нм.
Исследование биологической активности синтезированных соединений.
Материалы и методы
Исследования на культуре клеток.
Клетки линии Colo выращивали в питательной среде DMEM в течение 48-72 ч до достижения содержания общего белка 1 - 1,5 мг/мл, после чего в инкубационную среду вводили H3-тимидин в концентрации 10 мкКи/мл (удельная активность 80 Ки/ммоль), а также CMFU-CRGDC в концентрациях; O (контрольная группа), 10, 100, 500 и 1000 мкмоль/л. После 24-чой инкубации из культурных сосудов отбирали аликвоты в объеме 50 мкл и фильтровали из через микропористые фильтры "Whatman GF/B" с диаметром пор 1 мкм, с последующей промывкой 10-кратным избытком ледяного физиологического раствора (pH 7,35) для удаления несвязавшегося H3-тимидина. После высушивания фильтров радиоактивность контрольной и опытных проб измеряли на сцинтилляционном счетчике "Бета-2" с использованием стандартного толуольного сцинтиллятора. Для стандартизации процедуры в параллельно отобранных аликвотах определяли содержание общего белка по методу Лоури с предварительной солюбилилизацией клеточных структур. Результат эксперимента выражали в виде удельного включения радиоактивности на единицу массы белка (d.p.m./мгк).
Исследования агрегации тромбоцитов
Кровь для исследования брали из локтевой вены доноров в силиконизированные пробирки, содержащие 3,8% раствор цитрата натрия (1 мл на 9 мл крови). Богатую тромбоцитами плазму получали центрифугированием при 160 g в течение 10 мин. Бедную тромбоцитами плазму получали центрифугированием при 1600 g в течение 15 мин. Исследование влияния препаратов на АДФ-индуцированную агрегацию проводили в течение 2 ч с момента забора крови по нефелометрическому методу Born (9) с использованием агрегометра ФРМ-1. Все экспериментальные данные получены на пороговых концентрациях АДФ, которые предварительно устанавливались для данного образца и находились в настоящем исследовании в диапазоне от 0,5 до 2,5 мкмоль/л.
Стабильность образца проверяли по идентичности агрегатограмм при использовании пороговой концентрации АДФ. Исследуемые препараты растворяли в физиологическом растворе (pH 7,35) и вводили в богатую тромбоцитами плазму, находящуюся в кювете агрегометра в необходимых концентрациях. Время преинкубации препарата при 37oC с биологическим образцом составляло 3 мин при постоянном перемешивании (скорость магнитной мешалки - 400 об/мин). Количественно влияние препарата на АДФ-индуцированную агрегацию тромбоцитов человека оценивалось по величине вторичной агрегации. отнесенной к контрольной пробе.
Результаты экспериментов.
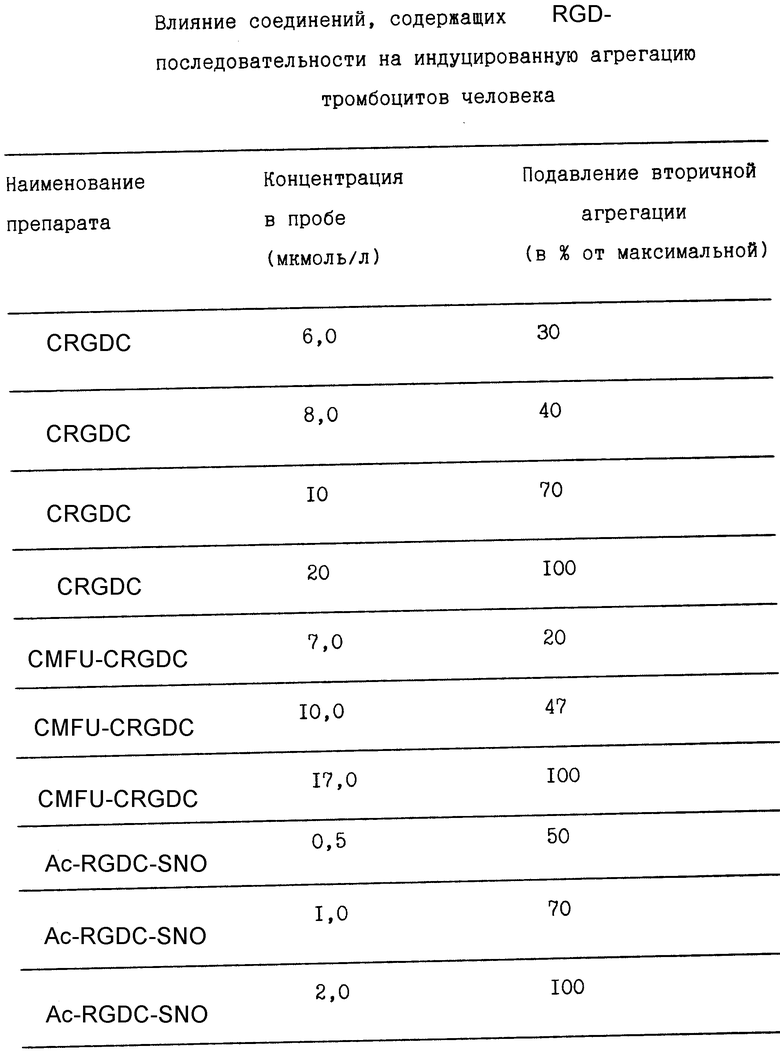
Возможность сохранения специфической активности лиганда в составе синтезированной гибридной молекулы продемонстрирована в исследованиях по влиянию CMFU-CRGDC на включение H3-тимидина в ДНК клеточной культуры рака толстого кишечника (линия Colo). Выбор именно этой линии культивируемых раковых клеток обоснован тем, что по клиническим данным одним из основных показаний для клинического применения 5-фторуарацила является рак прямой кишки и толстого кишечника.
Концентрации гибридного соединения 5-фторурацила с RGD-пептидом в инкубационной среде подобраны в соответствии с терапевтическими дозами лекарственных препаратов 5-фтор-урацила.
Инкубация культивируемых клеток CMFU-CRGDC приводит к достоверному снижению скорости синтеза в них ДНК, что является прямым свидетельством антипролиферативной активности исследуемого соединения.
Как видно из фиг. 1, имеет место эффективное дозозависимое подавление CMFU-CRGDC включения H3-тимидина клетками линии Colo.
На основании результатов описанных экспериментов можно сделать однозначный вывод о сохранении цитостатической активности 5-фторурацила в составе гибридной молекулы.
Второй задачей изучения синтезированных соединений была необходимость получения экспериментальных доказательств функциональной активности RGD-фрагмента молекулы, ответственного за специфическое связывание с 2b/3a рецепторами, так как именно это свойство гибридной молекулы позволяет реализовать идею направленного транспорта фармакологического агента с помощью тромбоцитов.
Сохранение способности синтезированных гибридных молекул связываться с клетками-носителями продемонстрирована в экспериментах по исследованию влияния CMFU-CRGDC, AC-RGDC-SNO на АДФ.
- индуцированную агрегацию тромбоцитов человека.
Для этого были испытаны три RGD-содержащих препарата:
1. Ацетамидометилцистеинил-аргинил-глицил-аспартил-ацетамидо- метил-цистеин (CRGSC), в качестве контрольного соединения.
2. Карбоксиметил-5-фторурацил-ацетамидометилцистеинил-аргинил- глицил-аспартил-ацетамидометилцистеин (CMFU-CRGDC).
3. Ацетиларгинил-глицил-аспартил-S-нитрозоцистеин (AC-RGDC-SNO).
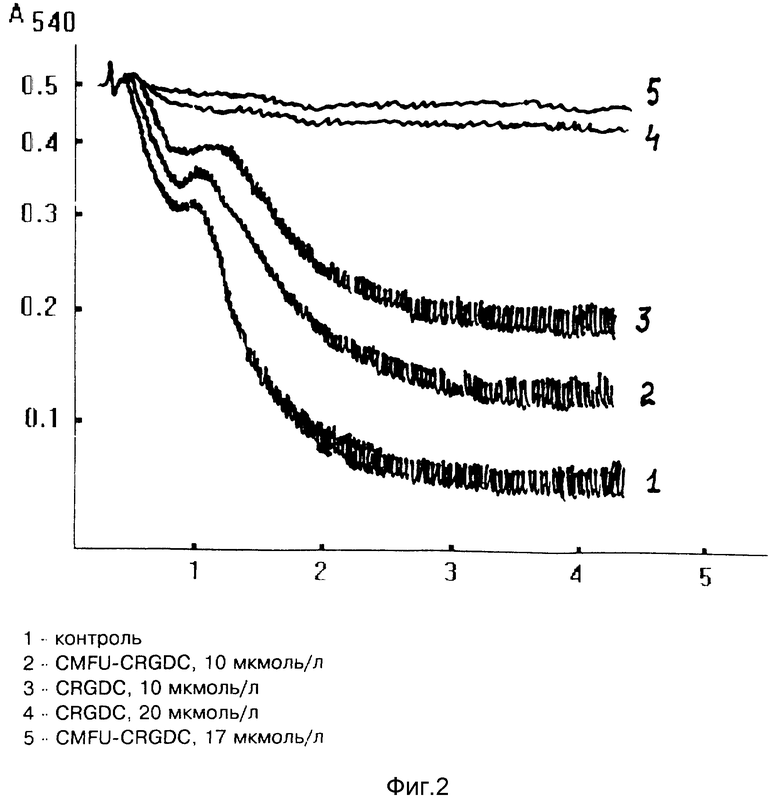
На фиг. 2 представлены сравнительные данные по влиянию CRGD и CMFU-CRGDC на АДФ-индуцированную агрегацию тромбоцитов человека. Оба препарата дозозависимо подавляли вторичную агрегацию как по скорости агрегации (т.е. изменению оптической плотности за мин. ), так и по амплитуде (т.е. по величине приращения оптической плотности за время, в течение которого процесс вторичной агрегации полностью заканчивается в контрольной пробе). Следует также отметить, что исследованные соединения оказывают ингибирующее влияние и на первичную агрегацию, хотя и в меньшей степени.
Количество экспериментов, выполненных с каждым из препаратов, равнялось пяти.
В таблице приведены количественные данные влияния исследованных соединений на вторичную агрегацию тромбоцитов.
Результаты сравнительного исследования влияния CMFU-CRGDC, Ac-RGDC-SNO и CRGDC на агрегацию тромбоцитов показывают, что RGD-пептидная последовательность, связанная с фармакоактивными лигандами сохраняет способность эффективно связываться с тромбоцитами.
Это свидетельствует о наличии принципиальной возможности использования указанных соединений для направленного транспорта активных лигандов в очаг патологического поражения, поскольку известно, что тромбоциты способы аффинно поглощать RGD-содержащие молекулы с депонированием их в альфагранулах и последующим высвобождением в месте активации тромбоцитов при осуществлении рилизинг-реакции.
Последнее, в чтности, может иметь место при ассоциации тромбоцитов с циркулирующими в кровеносном русле соматическими злокачественными клетками, сопровождающейся усилением адгезивных свойств последних и образованием метастатических очагов опухолевого роста. Предполагается что цитостатик (в данном случае 5-фторурацил), содержащий RGD-пептид, после введения в кровеносное русло будет связываться с 2b/3a рецепторами тромбоцитов. При последующем образовании комплекса циркулирующих опухолевых клеток с тромбоцитами и в результате инициации рилизинг-реакции цитостатик будет высвобождаться из тромбоцитов и воздействовать на ассоциированные с ними опухолевые клетки непосредственно в кровеносных сосудах, т.е. до их фиксации в тканях и органах. Следовательно, данные соединения можно рассматривать как потенциальные антиметастатические лекарственные препараты.
В том же случае, когда в кровеносное русло будет вводиться соединение с RGD-последовательностью, содержащее нитрогруппу, оно, связываясь в тромбоцитами, будет направленно транспортироваться в места обструкции сосудов, в чаcтности коронарных, вызванной атеросклеротическим повреждением интимы. В этих учтках эндотелия обычно происходит усиленная адгезия и агрегация тромбоцитов. Поэтому тромбоциты, нагруженные RGD-пептидами, несущими NO-группу, будут способствовать быстрой концентрации нитратов в зоне атеросклеротического сужения коронарной артерии.
Подобная схема направленного транспорта лекарственных препаратов, содержащих RGD-последовательнсоть, может реализовываться и при другой патологии, где имеет место локальная активация тромбоцитов. Терапевтический эффект в каждом конкретном случае будет зависеть от химической природы фармакоактивного лиганда, соединенного с RGD-последовательность аминокислотных остатков.
Источники информации
1. Zammaron C. , Ginsberg M.H., Plow F.T., J. Biol. Chem., 1991, 266: 16193
2. Clark E.A., Brigge J.S., Mol. Cell. Biol., 1993. 13: 1863.
3. Badimon L., Chesebro J.H., Badimon J.J., Circulation 1992, 86: 11174
4. Karpatkin S., Pearlstein F., Ann. Jntern Med. 1981. 85: 646
5. Ross R., Nature. 1993, 362: 801
6. Машковский М.Д., Лекарственные средства, 1987, 2: 455, М., Медицина,
7. Bromn B., G., Ciranlation, 1981, 64: 1089
8. Sarin V.K., Kent S.B.H., Engelhard M., Merrifield R.B., Anal Biochem. , 1981, 117: 147
9. Born G.V., Nature, 1962. 194: 927.
| название | год | авторы | номер документа |
|---|---|---|---|
| RGD-ПОДОБНЫЕ ПЕПТИДЫ | 2009 |
|
RU2396271C1 |
| ГЕТЕРОМЕРНЫЕ ПЕПТИДЫ НА ОСНОВЕ ИМИДАЗО[4,5-е]БЕНЗО[1,2-с;3,4-с']ДИФУРОКСАНА, ИНГИБИРУЮЩИЕ АГРЕГАЦИЮ ТРОМБОЦИТОВ | 2014 |
|
RU2550223C1 |
| БИОДЕГРАДИРУЕМЫЕ АРГИНИНСОДЕРЖАЩИЕ ПОЛИМЕРЫ | 2014 |
|
RU2549908C1 |
| ПЕПТИД АЗЕМИОПСИН, ИЗБИРАТЕЛЬНО ВЗАИМОДЕЙСТВУЮЩИЙ С НИКОТИНОВЫМИ ХОЛИНОРЕЦЕПТОРАМИ МЫШЕЧНОГО ТИПА И ПРИГОДНЫЙ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ МЫШЕЧНОГО РЕЛАКСАНТА В МЕДИЦИНЕ И КОСМЕТОЛОГИИ | 2011 |
|
RU2473559C1 |
| ПЕПТИДЫ И ИХ ПРОИЗВОДНЫЕ, ВЗАИМОДЕЙСТВУЮЩИЕ С НИКОТИНОВЫМ АЦЕТИЛХОЛИНОВЫМ РЕЦЕПТОРОМ И ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КОСМЕТОЛОГИИ ПРОТИВ МИМИЧЕСКИХ И ВОЗРАСТНЫХ МОРЩИН | 2013 |
|
RU2524428C1 |
| Производные триазола, проявляющие антиагрегационную активность | 2021 |
|
RU2770405C1 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДОМИМЕТИКИ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2519736C2 |
| МОДУЛЬНЫЙ МОЛЕКУЛЯРНЫЙ КОНЪЮГАТ ДЛЯ НАПРАВЛЕННОЙ ДОСТАВКИ ГЕНЕТИЧЕСКИХ КОНСТРУКЦИЙ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2012 |
|
RU2529034C2 |
| Средство с антиагрегационной активностью и способ его получения | 2021 |
|
RU2778686C1 |
| КОНЪЮГАТЫ RGD-(БАКТЕРИО)ХЛОРОФИЛЛ ДЛЯ ФОТОДИНАМИЧЕСКОЙ ТЕРАПИИ И ВИЗУАЛИЗАЦИИ НЕКРОТИЧЕСКИХ ОПУХОЛЕЙ | 2009 |
|
RU2518296C2 |
Изобретение относится к медицине. Предложен способ направленного транспорта лекарств, который заключается в связывании in vivo молекул-носителей фармакоактивных соединений с форменными элементами крови, принимающими участие в патологическом процессе. Для связывания синтезированы оригинальные производные фармакологических агентов и пептида, содержащего RGD. Фармакологический агент, присоединенный к RGD-пептиду при введении в кровеносное русло, способен связываться с рецепторами 2b/3a тромбоцитов и адресно транспортироваться клетками в места повышенной адгезии тромбоцитов. Такими местами могут быть участки атеросклеротического поражения корональных сосудов, метастические зоны опухолевого роста и другие точки скопления тромбоцитов. Данный способ позволяет получить высокие концентрации препаратов в очаге поражения в течение необходимого промежутка времени. 2 ил.,1 табл.
Способ направленного транспорта фармакологических препаратов в очаги патологического процесса, где имеет место тромбоцитарная адгезия и агрегация, отличающийся тем, что их связывают со специфическими рецепторами (2b/3a) тромбоцитов путем введения в состав молекул препаратов пептида с аминокислотной последовательностью аргинин-глицин-аспарагиновая кислота (RGD).
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| RU 94038047 A1, 10.06.96 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| НАКОПИТЕЛЬ-РАЗМАТЫВАТЕЛЬ ПРОВОЛОКИ | 0 |
|
SU398305A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Замок такелажный | 1972 |
|
SU459624A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ получения сывороточного альбумина | 1971 |
|
SU467536A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Экономайзер | 0 |
|
SU94A1 |
