Изобретение относится к поверхностям теплообменников в реакторах и в трубчатых теплообменниках установок для превращения углеводородов и других органических соединений в связи с проблемой коксования этих обменных поверхностей.
Например, для изготовления этилена и других низких олефинов углеводороды или смеси углеводородов подвергают термическому крекированию в нагреваемых снаружи реакторах из металлических материалов, и полученные при этом горячие крекинг-продукты после выхода из крекинговых печей охлаждают в теплообменных аппаратах, омываемых снаружи водой под давлением в качестве охлаждающего агента.
Крекинговые печи изготавливают предпочтительно из высокотермостойких сталей, содержащих хром и никель, трубчатые теплообменники - предпочтительно из низколегированных сталей или сталей для котлостроения. Такие аппараты используют также для изготовления других органических продуктов, так, например, при получении винилхлорида путем пиролиза 1,2-дихлорэтана.
Эффективность работы таких аппаратов, изготовленных из металлических материалов, в значительной степени зависит от того, в каком объеме образуются во время работы обогащенные углеродом отложения на внутренних поверхностях, которые не только ухудшают переход тепла, но также могут уменьшить свободное поперечное сечение используемых труб, важное также для поддержания пропускаемого количества. Это имеет место в используемых в настоящее время аппаратах, причем для зависимости количества отложившегося коксообразного продукта m от времени реакции t типичной является характеристика, представленная кривой A на фиг. 1.
После определенного времени работы отложения, образованные на сторонах аппарата, контактирующих с органическими соединениями, достигают такой величины (фиг. 1, допустимая толщина слоя кокса S), что обусловленные ими снижения производительности вынуждают к остановке производства и к дорогостоящим работам по очистке. Удаление коксообразных отложений осуществляют большей частью путем продувания смесью чистого водяного пара и воздуха до обнажения металлической поверхности и обеспечения нужного теплового потока.
Несмотря на основательное удаление отложившегося кокса, уже через относительно короткое время работы (например, через 20 - 60 дней) вновь образовавшиеся отложения снова вынуждают к остановке производства и удалению кокса. Так как используемые процедуры удаления кокса, связанные с применением окислителей, одновременно вызывают изменение поверхности материалов, то такие процессы удаления кокса всегда связаны с повышением каталитической активности поверхности материалов, которая вызывает нежелательное образование кокса на поверхности. С возрастанием числа процедур удаления кокса, которым подвергается соответствующая поверхность теплообменника, эта каталитическая активность повышается, и время работы между двумя удалениями кокса постоянно снижается. Это в одинаковой степени нежелательно как с технической, так и с экономической точки зрения, потому что таким образом не только создаются помехи для, по возможности, более длительных стационарных рабочих состояний, но также снижается эффективное использование установки и все чаще расходуются средства на очистные работы. Поэтому издавна стараются найти решения, которые препятствовали бы быстрому коксованию внутренних поверхностей такой аппаратуры. Для достижения этой цели было предложено, среди прочего, предотвращение образования каталитически активных центров или его блокирование на внутренних поверхностях труб соответствующих аппаратов путем образования оказывающих пассивирующее действие окисных слоев (например, патент США 3919073), покрытие внутренних стенок труб тонкими слоями низколегированных или не содержащих никеля сталями (Выкладное описание к патенту ФРГ 3247568), получение покрытий или диффузионных слоев из хрома (Brown, S.M. и Albright L. F: ACS Symp. Ser.  (1976), 296), алюминия (Frech K.J; Hopotock F.H. и Hutchings D.A.: ACS Symp. Ser.
(1976), 296), алюминия (Frech K.J; Hopotock F.H. и Hutchings D.A.: ACS Symp. Ser.  (1976) 197) или кремния (Brown D.E., Clark J. T. K. , Foster A.J., McCarobI J.J. и Sims M.L.: ASC Symp. Ser. New York, 1982, 202, 23; Bach J., Zychlinski W., Zimmermann J., Kopinke F.-D. и Anders K: Chem. Techn. (Leipzig)
(1976) 197) или кремния (Brown D.E., Clark J. T. K. , Foster A.J., McCarobI J.J. и Sims M.L.: ASC Symp. Ser. New York, 1982, 202, 23; Bach J., Zychlinski W., Zimmermann J., Kopinke F.-D. и Anders K: Chem. Techn. (Leipzig)  (1990) 146; Ansari A.A., Sauders S.R.J., Bennett M. Y. , Tuson А.Т., Ayres C.F. и Steen W.M.: Materials Science and Engineering
(1990) 146; Ansari A.A., Sauders S.R.J., Bennett M. Y. , Tuson А.Т., Ayres C.F. и Steen W.M.: Materials Science and Engineering  (1987) 135) и добавка газо- или парообразных веществ из серосодержащих (например, Boene К.: Oilgas J.
(1987) 135) и добавка газо- или парообразных веществ из серосодержащих (например, Boene К.: Oilgas J.  (1983) 93), фосфоросодержащих (Josh К. К. и Kunzru D. : Jnd. Engng. Chem. Res.
(1983) 93), фосфоросодержащих (Josh К. К. и Kunzru D. : Jnd. Engng. Chem. Res.  (1988) 559; US 4835332; US 4842716; US 4900426) и азотсодержащих соединений (Egiasarov J.Y., Cores B. Ch. и Potapova L.L., Neftechimiya [Erdolchem]
(1988) 559; US 4835332; US 4842716; US 4900426) и азотсодержащих соединений (Egiasarov J.Y., Cores B. Ch. и Potapova L.L., Neftechimiya [Erdolchem]  (1985) 627) к используемому продукту.
(1985) 627) к используемому продукту.
Из патентов США 4835332, 4842716 и 4900426 известно, что образование подобных коксу отложений на внутренних поверхностях реакторов может быть снижено с помощью добавки органических соединений фосфора, причем фосфороорганические соединения (включая тиофосфорорганику) могут применяться как таковые, так и в качестве составляющих частей специальных компаундов. Добавка органических фосфорных соединений всегда связана с образованием более или менее легколетучих фосфинов, которые не только токсичны, но также могут привести к отравлению катализатора в подключенных процессах. Добавка фосфорорганики эффективна лишь в ограниченных объемах.
О действии соединений серы на образование кокса имеются противоречивые высказывания (ср. CS-A 180861 и Froment G. K. в: Reviews in Chem. Eng. 6(4)293(1990)).
Тем не менее, серные соединения до настоящего времени часто применяют в промышленной практике в том случае, если превращениям нужно подвергнуть свободные от серы углеводороды. Для большинства имеющихся в промышленности фракций углеводородов (нефть, керосин, газойль) добавка соединений серы едва ли оказывает влияние на коксообразование. Они имеют специальные соединения серы в качестве составных частей смеси, тем не менее при пиролизе таких фракций углеводородов наблюдается более или менее ярко выраженное образование коксообразных отложений.
Нанесение оксидных защитных слоев, как это предлагается в EP-A 0110486, хотя и дает улучшения, однако не может рассматриваться в качестве удовлетворяющего решения.
Другое улучшение получается с помощью покрытия на основе силиконового масла, которое затем, для получения защитного слоя, термически разрушается при совершенно определенных условиях (Chem. Techn. (Leipzig)  (1990), 146). Способ так же, как и получение поверхностных покрытий SiO2, индуцированных с помощью лазера, является относительно дорогим, а полученные при этом слои при смене температур в диапазоне между 750 и 1100oC (температура наружной стенки трубы) нестабильны. Это относится также к тем пассивным слоям, которые получаются согласно описанным Britich Petroleum Co Ltd кремниевым покрытиям (ACS Symp. Ser., New York, 1982, 202, 23 - 43; ср. к этому: Chem. Techn. (Leipzig)
(1990), 146). Способ так же, как и получение поверхностных покрытий SiO2, индуцированных с помощью лазера, является относительно дорогим, а полученные при этом слои при смене температур в диапазоне между 750 и 1100oC (температура наружной стенки трубы) нестабильны. Это относится также к тем пассивным слоям, которые получаются согласно описанным Britich Petroleum Co Ltd кремниевым покрытиям (ACS Symp. Ser., New York, 1982, 202, 23 - 43; ср. к этому: Chem. Techn. (Leipzig)  (1990) 146 ff).
(1990) 146 ff).
Наконец, следует еще сослаться на попытку применения труб из сталей, внутреннюю поверхность которых покрывают тонким слоем низколегированной или не содержащей никеля сталью (ДЕ-А 3247568). Оказалось, что при таком покрытии издержки и результат находятся в неприемлемом соотношении.
Если исходить из снижения коксообразования с помощью добавки фосфор- и/или серосодержащих веществ к используемым продуктам пиролиза, то для всех описанных до этого предложений решения общим является то, что они практически могут осуществляться лишь в новых установках или при проведении новых труб, но не в находящихся в эксплуатации установках.
Поэтому в основе изобретения лежит задача предложить новые улучшенные поверхности теплообменников, а также способ снижения коксообразования, с помощью которого можно таким образом обрабатывать соответствующие аппараты (оборудование) полностью оснащенной установки как перед первым пуском в производство, так и после каждого удаления кокса.
Согласно изобретению поверхность теплообменника в реакторах и/или теплообменниках установок для превращения углеводородов и других органических соединений в газовой фазе при высоких температурах отличается тем, что контактирующие с органическими веществами металлические поверхности обрабатывают продуктом, содержащим смесь кремния и серы, и сухим инертным по отношению к продукту, содержащему кремний и серу, потоком газа при температуре от 300 до 1000oC в течение 0,5 - 12 часов.
При этом продукт, содержащий кремний и серу, выбирают из: (1) одного или большего числа летучих соединений, содержащих кремний и серу; (2) из смеси летучих соединений, содержащих кремний, и смеси летучих соединений, содержащих серу; и (3) смеси летучих соединений, содержащих кремний и серу, и летучих соединений, содержащих кремний и/или летучих соединений, содержащих серу. Причем атомное соотношение кремния и серы в (1, 2) или (3) вариантах составляет соответственно от 5:1 до 1:1. Особенно предпочтительными соединениями являются при этом триметилсилилмеркаптан, диметилсульфид, диметилдисульфид и бис-триметилсилилсульфид и их смеси.
Если поверхность теплообменника, обработанная согласно изобретению, является металлической внутренней поверхностью трубы трубчатого реактора, то температура обработки составляет 800 - 1000oC. Если поверхность теплообменника, обработанная согласно изобретению, является металлической внутренней поверхностью трубы подключенного к трубчатому реактору теплообменника, то температура обработки составляет 300 - 750oC. В подключенном теплообменнике могут, разумеется, возникнуть местами еще более высокие температуры. Так, например, температура на отражательной пластине на входе в теплообменник в определенных случаях может составлять также более 800oC, например 875oC. Обычно же она лежит, однако, в указанном выше диапазоне.
Время обработки составляет, как указано выше, в общем случае 0,5 - 12 часов. При времени обработки менее 0,5 часов получающийся эффект слишком мал, чем это имело бы место при длительном воздействии. Время более 12 часов возможно, в общем же, однако, неэкономично.
Неожиданным образом в основе изобретения лежат сведения о том, что к началу каждого пуска крекинговой печи, реакторные трубы которой или новые, или внутренняя поверхность их труб освобождена от уже отложившихся богатых углеродом продуктов, можно значительно снизить постоянно наблюдаемое увеличение коксообразования, если перед первым пуском в производство крекинговой печи и/или после каждого повторного пуска крекинговой печи после предшествующего удаления кокса с помощью пара/воздуха внутренние поверхности труб, которые после пуска в производство контактируют с крекинговыми продуктами, подвергнуть соответствующей обработке при высокой температуре летучими соединениями, содержащими кремний и серу. Это получается целесообразно таким образом, что смесь из соединений, содержащих кремний и серу, и сухого инертного газа-носителя, который поглощает соединения, согласно изобретению направляется через трубы крекинговой печи и примыкающего к ней трубчатого теплообменника в таком составе, что не только имеющиеся первоначально и обусловливающие каталитическое коксообразование каталитически активные центры с помощью химических реакций превращаются в каталитически пассивные поверхностные соединения, но также имеет место концентрация содержащихся в соединениях согласно изобретению элементов кремния и серы в форме реактивных специй в поверхности металлических материалов. Если превращение каталитически активных центров на внутренней поверхности труб осуществлено при образовании каталитически инактивных поверхностных соединений и одновременно внедрение специй, содержащих кремний и серу, в поверхность материала достигла достаточной степени, то крекинговую печь вместе с трубчатым теплообменником можно снова пустить в работу. Так как слои внутренней поверхности труб, в частности, обогащены кремнием, а каталитически активные центры дезактивированы термически стабильными и каталитически инактивными специями кремний-сера, то новое коксование наступает лишь с большим замедлением во времени и на очень низком уровне (схематическое изображение см. на фиг. 1, кривая B). Благодаря этой сравнительно простой дополнительной обработке перед первым пуском в работу уже смонтированной крекинговой печи или после ее обычной очистки для удаления кокса с помощью водяной смеси пар/воздух, данное изобретение позволяет значительно увеличить производственные циклы крекинговых печей. Важным при этом является то, что на крекинговых печах и трубчатых теплообменниках не нужно предпринимать никаких конструктивных изменений и что способ применим также для уже работающих установок. Дорогостоящие покрытия изготовленных предварительно труб, которые во время монтажа нужно сваривать при частичном повреждении защитных слоев, причем желаемый эффект опять-таки частично уничтожается, отпадают. Кроме того, исключается нанесение плотных покрытий, которые могут снизить теплопередачу.
Оказалось эффективным через систему печи направлять смесь из инертного и сухого газа-носителя, как например головной продукт из деметанизатора установки разложения крекинг-газа или также азот, и соединений согласно изобретению при обычной температуре работы крекинг-печи, т.е. при температуре стенок труб выше 800oC, и трубчатого теплообменника (TLE), т.е. при 400 - 500oC, причем молярное соотношение соединений, содержащих кремний и серу, для газа-носителя предусмотрено в интервале 0,0005 - 0,03, а продолжительность обработки в зависимости от концентрации соединений, содержащих кремний и серу, - между 30 минутами и 12 часами. Наряду с соединениями, содержащими одновременно кремний и серу, можно использовать также смеси соединений, содержащих кремний и серу. Атомное соотношение кремния к сере может лежать между 5:1 и 1: 1; предпочтителен диапазон от 1:1 до 2:1. Давление направляемой смеси может соответствовать обычному давлению в системе крекинговой печи, т.е., например, составлять 0,5 - 20 бар; предпочтителен диапазон 1 - 2 бара. В качестве газа-носителя можно применять также другой, инертный по отношению к системе газ.
Далее изобретение более подробно поясняется на основе нескольких сравнительных примеров и примеров выполнения согласно изобретению. Фиг. 2-10 описывают зависимости скоростей коксования на предварительно активированных образцах из хромо-никелевой стали от времени испытаний при пиролизе n-гептана, отчасти после термических предварительных обработок согласно изобретению.
Фиг. 1 представляет зависимость количества отложившегося коксообразного продукта от времени реакции t в аппаратах согласно уровню техники.
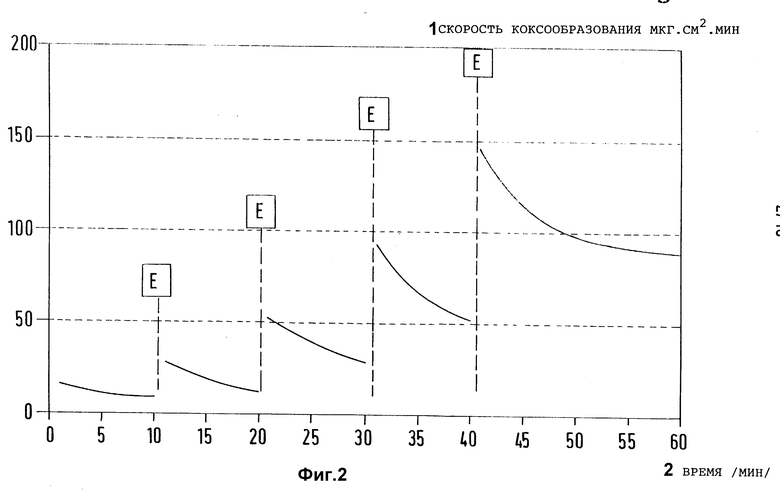
Фиг. 2 представляет зависимость скорости коксообразования на предварительно активированном (E - удаление кокса воздухом), но не обработанном согласно изобретению образце из хромо-никелевой стали X8CrNiTi 18 10 от времени испытаний при пиролизе чистого n-гептана (Ттр. = 715oC, τ = 1 с, N2 в качестве разбавителя).
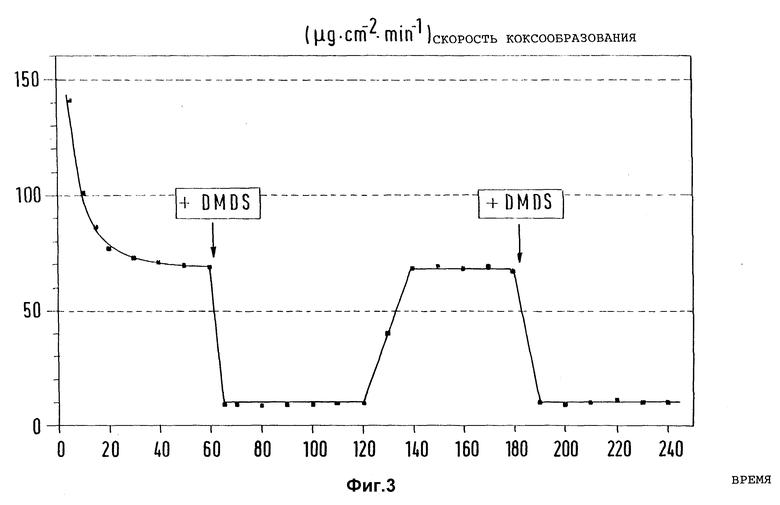
Фиг. 3 представляет влияние 85 ppm диметилдисульфида (DMDS) в качестве добавки к n-гептану на скорость коксообразования в предварительно активированном, но не обработанном предварительно согласно изобретению образце из стали X8CrNiTi 18 10 от времени испытаний при пиролизе n-гептана (Tтр = 715oC, τ = 1 с, N2 в качестве разбавителя).
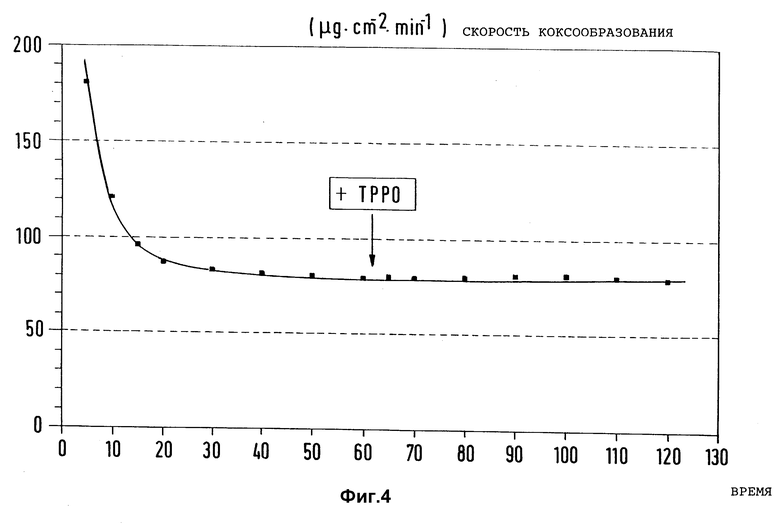
Фиг. 4 представляет влияние 1000 ppm трифенилфосфиноксида (ТРРО) вместо диметилдисульфида в качестве добавки к n-гептану на скорость коксообразования на предварительно активированном, но не обработанном согласно изобретению образце из стали X8CrNiTi 18 10 от времени испытаний при пиролизе n-гептана (Tтр = 715oC, τ = 1 с, N2 в качестве разбавителя).
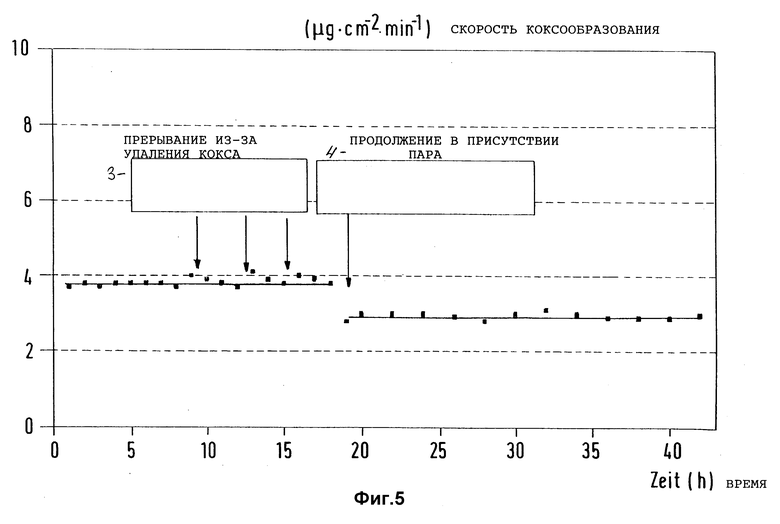
Фиг. 5 представляет зависимость скорости коксообразования на предварительно активированном и уже многократно подвергавшемся удалению кокса и предварительно обработанном термически при 880oC триметилсилилметилмеркаптаном согласно изобретению образце из стали X8CrNiTi 18 10 от времени испытаний при пиролизе n-гептана и многократном прерывании реакции пиролиза с целью сжигания отложившегося кокса с помощью воздуха (Tтр = 715oC, τ = 1 c; N2) или водяной пар в качестве разбавителя).
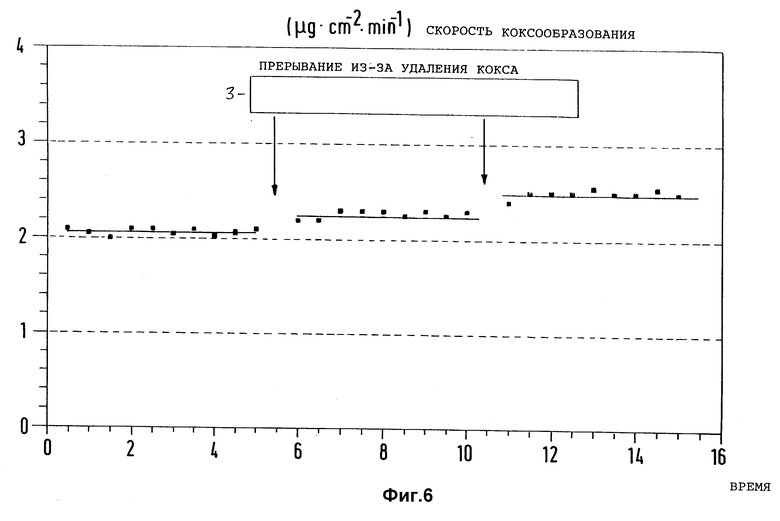
Фиг. 6 представляет зависимость скорости коксообразования от времени испытаний на предварительно обработанном согласно изобретению образце из неиспользованном, предварительно активированном инколое 800 при пиролизе n-гептана и многократном прерывании реакции пиролиза с целью обжигания отложившегося кокса с помощью воздуха (Ттр = 750oC, τ = 0,6 с; водяной пар в качестве разбавителя).
Фиг. 7 представляет влияние использованного для предварительной термической обработки образцов из стали X8CrNiTi 18 10 газа-носителя на скорость коксообразования при пиролизе n-гептана (Tтр = 715oC, τ = 1 с, N2 в качестве газа-разбавителя).
Фиг. 8 представляет влияние температуры при предварительной обработке согласно изобретению образцов из X8CrNiTi 18 10 на зависимость скорости коксообразования от времени испытаний при пиролизе n-гептана (Tтр = 715oC, τ = 1 с, N2 в качестве разбавителя).
Фиг. 9 представляет влияние времени предварительной обработки на зависимость скорости коксообразования от времени испытаний при пиролизе n-гептана (Ттр = 715oC, τ = 1 с, N2 в качестве газа-разбавителя).
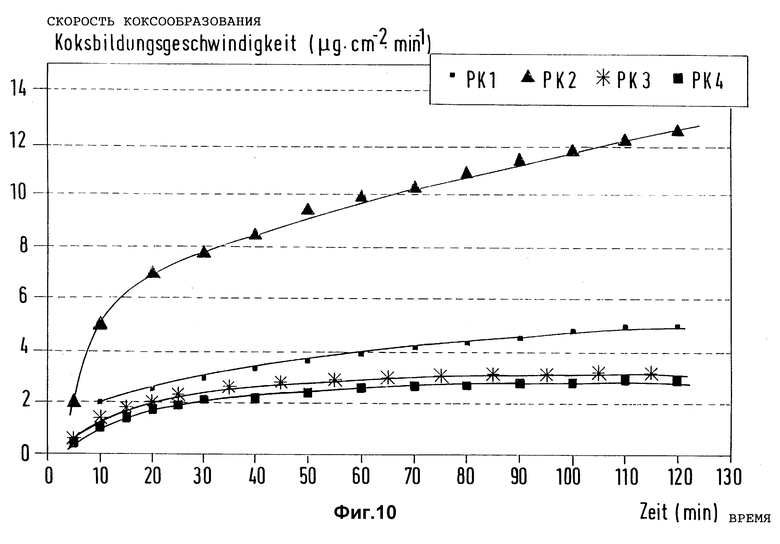
Фиг. 10 представляет зависимость скорости коксообразования на по-разному предварительно обработанных образцах из стали X8CrNiTi 18 10 от времени испытаний при пиролизе n-гептана (Tтр = 715oC, τ = 1 с, N2 в качестве разбавителя).
Пример 1 (для сравнения).
Скорости осаждающихся при пиролизе углеводородов на металлических материалах твердых коксообразных отложений можно замерить в специальных, расположенных вертикально лабораторных реакторах с электрическим подогревом, если внутри этих реакторов подвесить соответствующие образцы на тонкой платиновой или кварцевой проволоке и соединить с термовесами (ср. с Kopinke F.D., Bach G, Zimmermann G.: J. Anal. Appl. Pyrolysis 27 (1993) 45).
В такой аппаратуре из кварцевого стекла для пиролиза (dвн. = 20 мм, Vтр. = 13 мл), к которой присоединен нагреваемый отдельно сегмент трубы из кварцевого стекла такого же диаметра, в котором могут создаваться температуры газового пространства, соответствующие тем, которые применяют в технических трубчатых теплообменниках для охлаждения пиролизных газов, подвергли пиролизу n-гептан в качестве модели углеводорода при температурах между 715 и 800oC при условиях, которые привели к получению соотношения масс этилен: пропилен в пиролизном газе между 2,0 и 2,7. Если проводить пиролиз с азотом в качестве разбавителя 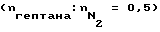 и в присутствии образцов материалов, на которых в целях провоцирования повышенного каталитического коксообразования путем пиролиза многократно осаждается кокс, и которые затем освобождаются от кокса путем его сжигания, можно затем замерить абсолютные значения скоростей коксообразования r, которые предпочтительно лежат в диапазоне между r = 50 и 300 мг/см2, причем величина замеренных скоростей коксообразования является интегральной замеренной величиной, которая при определенной степени крекинга и определенных условиях крекинга является характерной для замеренных в каждом случае образцов, однако в значительной степени зависит также от того, сколько циклов коксование/удаление кокса испытал соответствующий образец. Типичным примером для зависимости скорости коксообразования на образце из хромоникелевой стали X8CrNiTi 18 10 от времени реакции при пиролизе n-гептана при 780oC воспроизводит фиг. 2 для пяти следующих друг за другом циклов коксование/удаление кокса.
и в присутствии образцов материалов, на которых в целях провоцирования повышенного каталитического коксообразования путем пиролиза многократно осаждается кокс, и которые затем освобождаются от кокса путем его сжигания, можно затем замерить абсолютные значения скоростей коксообразования r, которые предпочтительно лежат в диапазоне между r = 50 и 300 мг/см2, причем величина замеренных скоростей коксообразования является интегральной замеренной величиной, которая при определенной степени крекинга и определенных условиях крекинга является характерной для замеренных в каждом случае образцов, однако в значительной степени зависит также от того, сколько циклов коксование/удаление кокса испытал соответствующий образец. Типичным примером для зависимости скорости коксообразования на образце из хромоникелевой стали X8CrNiTi 18 10 от времени реакции при пиролизе n-гептана при 780oC воспроизводит фиг. 2 для пяти следующих друг за другом циклов коксование/удаление кокса.
Пример 2 (для сравнения).
В такой же аппаратуре и при аналогичных внешних условиях, как и в примере 1, вначале определили характеристику скорости коксообразования на предварительно активированном образце из стали X8CrNiTi 18 10 при пиролизе n-гептана при температуре 715oC и времени испытаний 60 мин. Затем n-гептан, как используемый продукт для пиролиза, был заменен порцией n-гептана, которая содержала 85 ppm диметилдисульфида, соединения, известного как ингибитор коксообразования и имеющего промышленное применение.
Фиг. 3 информирует о характеристике замеренных при этом скоростей коксообразования на используемом образце в зависимости от времени испытаний, причем указанные загружаемые продукты многократно сменяются. Замеренные разницы в скоростях коксообразования подтверждают ингибирующее действие диметилдисульфида на коксообразование на металлических поверхностях материалов.
Пример 3 (для сравнения).
В такой же аппаратуре и при таких же условиях, как и в примере 1, проследили действие известного фосфорсодержащего ингибитора (US 4900426) вместо диметилдисульфида на скорость коксообразования при 715oC. На фиг. 4 сопоставлены результаты исследований.
Видно, что добавка 1000 ррм трифенилфосфиноксида (содержание P нормировано по содержанию S в соединении, использованном в примере 2) к n-гептану при примененных условиях пиролиза не оказывает заметного влияния на его коксообразование.
Пример 4 (пример выполнения согласно изобретению).
В такой же аппаратуре, как описано в примере 1, многократно предварительно активированный образец стали X8CrNiTi 18 10 в течение 60 мин обрабатывали 3 л/ч газового потока (объемная скорость V = 25 мл/(мл•мин)) 0,005 молярного триметилсилилметилмеркаптана в 3 л сухой эквимолярной смеси водорода и метана при 880oC. Реактор в течение 5 мин промывали азотом при 715oC. Затем n-гептан подвергли пиролизу в присутствии азота 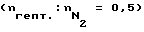 при 750oC, как описано в примере 1, и определили скорость коксообразования на обработанном образце в зависимости от времени реакции (фиг. 5). Скорость коксообразования r = 4 мкг/(см2•мин) в течение более 18 часов испытаний осталась примерно постоянной. Прерывая произвольно испытания, поверхность образца подвергли очистке после 8, 12 и 15 часов испытаний, в каждом случае путем обжигания кокса с помощью воздуха. Снижения пассивности поверхности из-за этого не возникло. После 18 часов испытаний применяемый в качестве разбавителя азот заменили водяным паром и испытания продолжили еще в течение 24 часов. Скорость коксообразования снизилась при этом до значений 3 мкг/(cм2•мин) и осталась в течение указанного промежутка времени примерно постоянной.
при 750oC, как описано в примере 1, и определили скорость коксообразования на обработанном образце в зависимости от времени реакции (фиг. 5). Скорость коксообразования r = 4 мкг/(см2•мин) в течение более 18 часов испытаний осталась примерно постоянной. Прерывая произвольно испытания, поверхность образца подвергли очистке после 8, 12 и 15 часов испытаний, в каждом случае путем обжигания кокса с помощью воздуха. Снижения пассивности поверхности из-за этого не возникло. После 18 часов испытаний применяемый в качестве разбавителя азот заменили водяным паром и испытания продолжили еще в течение 24 часов. Скорость коксообразования снизилась при этом до значений 3 мкг/(cм2•мин) и осталась в течение указанного промежутка времени примерно постоянной.
Пример 5 (пример выполнения согласно изобретению).
В такой же аппаратуре, как и описанная в примере 1, образцы из неиспользованного сплава инколой 800, как приведено в примере 4, подвергли предварительной обработке при приведенных там условиях, а затем проследили за скоростью коксообразования при пиролизе n-гептана при 750oC. При этом пиролиз выполнялся в присутствии водяного пара вместо азота в качестве разбавителя. На фиг. 6 замеренные значения скорости коксообразования нанесены по времени испытания, причем пиролиз многократно прерывали, и образцы подвергали удалению кокса с помощью воздуха. Результаты показали, что скорость коксообразования в течение всего времени испытания имела низкие значения около 2,5 мкг/(см2•мин).
Пример 6 (пример выполнения согласно изобретению).
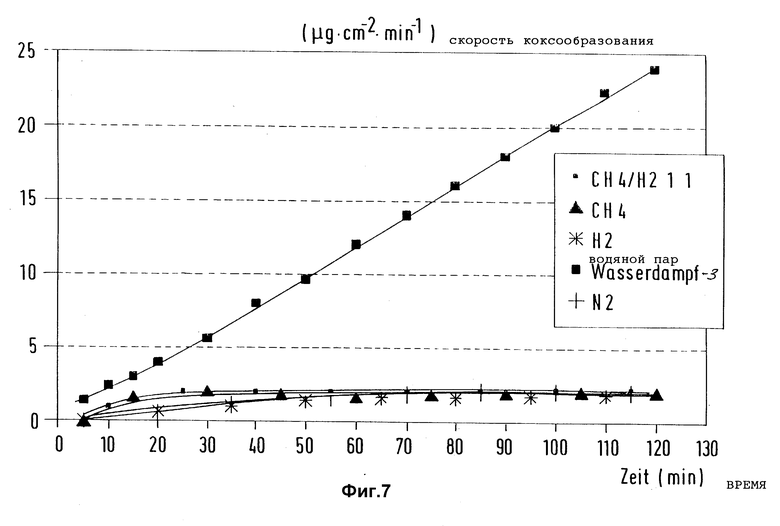
В такой же аппаратуре, как описано в примере 1, и при таких же условиях, как описано в примере 4, исследовали влияние применяемого газа-носителя на скорость коксообразования при пиролизе n-гептана. Вместо смеси водорода и метана в соотношении 1:1 использовали водород, метан, азот и водяной пар. Варианты использованного для предварительной обработки газа-носителя показали, что водород непригоден для длительного подавления коксообраэования на материалах, предварительно обработанных триметилсилилметилмеркаптаном. После замера сравнительно низких начальных значений (r = 1,7 мкг/(cм2•мин)) скорость коксообразования непрерывно росла и после времени испытания 120 мин достигла уже снова значений r = 25 мкг/(см2•мин).
На фиг. 7 представлены замеренные после соответствующих предварительных обработок на поверхности образцов при пиролизе n-гептана скорости коксообразования в зависимости от времени испытаний.
Пример 7 (пример выполнения согласно изобретению).
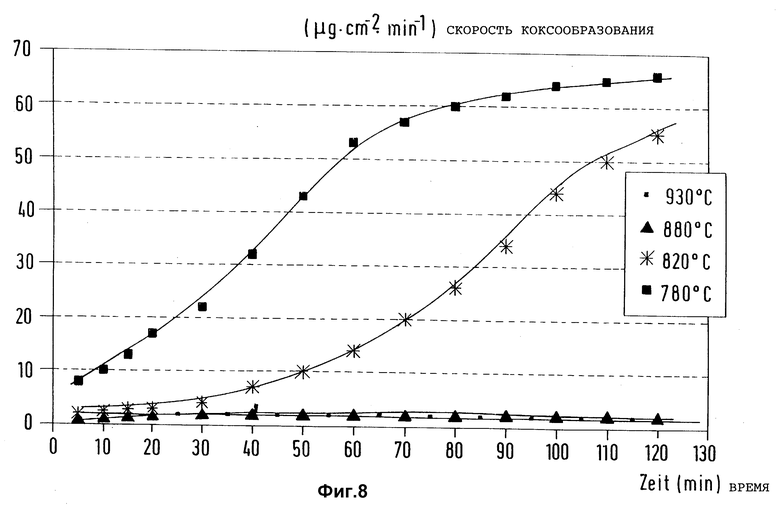
В описанной в примере 1 аппаратуре предварительно активированные образцы из стали X8CrNiTi 18 10 обработали эквимолярным потоком газа из водорода и метана при различных температурах в течение 60 мин при скорости потока 3 л/ч, к потоку при этом добавили 0,005 моль триметилсилилметилмеркаптана. После обработки и промывки реактора азотом на образцах замерили скорости коксообразования при пиролизе n-гептана в присутствии азота при 715oC 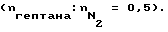
На фиг. 8 замеренные скорости коксообразования в зависимости от времени реакции на образцах, обработанных триметилсилилметилмеркаптаном при четырех различных температурах, представлены для сравнения. Видно, что обработка согласно изобретению поверхностей материалов перед пиролизом углеводородов зависит от температуры предварительной обработки. Температуры предварительной обработки выше 880oC способствуют подавлению коксообразования в течение длительного времени.
Пример 8 (пример выполнения согласно изобретению).
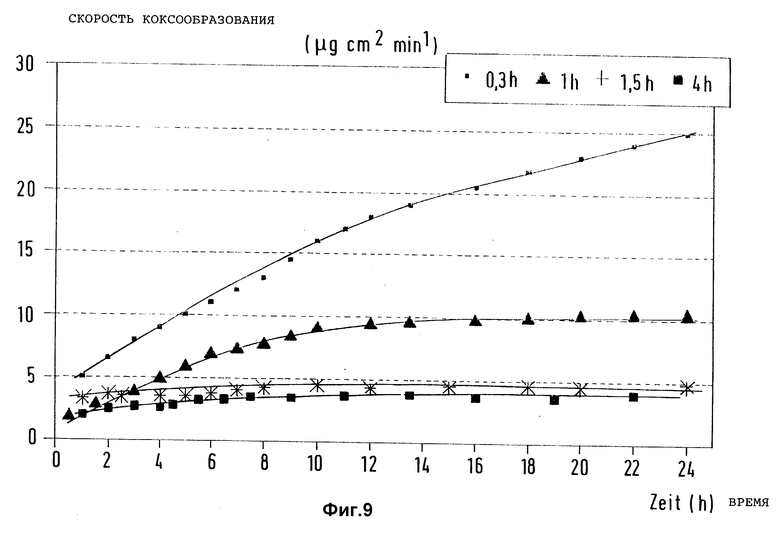
В такой же аппаратуре, как описано в примере 1, при условиях, аналогичных описанным в примере 7, предварительно активированные образцы из стали X8CrNiTi 18 10 подвергли предварительной обработке эквимолярной смесью из водорода и метана, содержащей триметилсилилметилмеркаптан, при 900oC в течение разного времени. Замеренные затем на этих образцах при пиролизе n-гептана в азоте при 715oC в зависимости от времени испытаний скорости коксообразования представлены для сравнения на фиг. 9 для четырех образцов.
Изменения продолжительности предварительной обработки показывают, что при времени предварительной обработки больше 1 час коксообразование в течение длительного времени испытания в равной степени эффективно подавляется.
Пример 9 (пример выполнения согласно изобретению).
В такой же аппаратуре, как описано в примере 1, и при условиях, одинаковых с описанными в примере 4, исследовали влияние вида и состава соединений, содержащих кремний и серу, при предварительной обработке предварительно активированного образца с помощью состоящего из соответственно 50 молярных процентов водорода и метана газа-носителя на скорость коксообразования при пиролизе n-гептана в азоте в качестве разбавителя.
Образцы, полученные при температуре предварительной обработки 880oC, продолжительности предварительной обработки 60 мин и при доле 0,005 моль соединений, содержащих кремний и серу, или сумме соединений, содержащих кремний и серу, в 3 л/ч эквимолярной смеси водород/метан друг за другом подвергли воздействию газовых реактивных фаз, возникающих при пиролизе, и исследовали скорости коксообразования на этих образцах в зависимости от времени реакции.
В табл. 1 представлены скорости коксообразования, полученные на образцах, предварительно обработанных различными соединениями кремния и серы, в зависимости от времени испытаний.
Видно, что цель предварительной обработки согласно изобретению не ограничивается применением соединений, содержащих одновременно кремний и серу; она достигается тем не менее также в том случае, если соединения, содержащие кремний или серу, применяют в смеси. Предварительная обработка согласно изобретению обеспечивается при этом в широком диапазоне атомных соотношений кремния и серы. Особенно предпочтительно соотношение Si:S = 2:1 - 1:1.
Пример 10 (пример выполнения согласно изобретению).
В такой же аппаратуре, как описано в примере 1, и при условиях, аналогичных условиям примера 4, определяли влияние содержания триметилсилилметилмеркаптана в применяемой для предварительной обработки эквимолярной смеси из водорода и метана на скорость коксообразования на образцах из стали X8CrNiTi 18 10. К примененной для предварительной обработки смеси водород-метан (3 л/ч) добавляли 0,002, 0,005, 0,01 и 0,02 моль триметилсилилметилмеркаптана и проводили предварительную обработку в каждом случае 3 л приведенного выше кондиционированного газа-носителя в течение 60 мин при 880oC.
Замеренные на предварительно обработанных в зависимости от содержания триметилсилилметилмеркаптана в смеси водород-метан образцах при пиролизе n-гептана в потоке азота при 715oC скорости коксообразования сопоставлены в табл. 2.
Результаты не показывают значительной зависимости замеренных скоростей коксообразования от содержания триметилсилилметилмеркаптана в используемой для предварительной обработки смеси водород-метан.
Пример 11 (изобретение и варианты для сравнения).
В лабораторной установке для пиролиза согласно примеру 1 предварительно обработали четыре образца из стали X8CrNiTi 18 10 в каждом случае 3 л газового потока, содержащего водород и метан в эквимолярных соотношениях, в течение промежутка времени 60 мин при 880oC, к которому (потоку) в каждом случае было добавлено 0,005 моль тетраметилсилана (образец РК1) или диметилсульфида (образец РК2) или смеси из тетраметилсилана и диметилсульфида в соотношении 1: 1 (образец РК3) или триметилсилилметилмеркаптана (образец РК4). Таким образом, лишь образцы РК3 и РК4 были обработаны согласно изобретению. Все четыре образца были затем подвергнуты друг за другом воздействию реактивной газовой фазы, которая возникает при пиролизе n-гептана в потоке азота при 715oC (время выдержки 1 с), и были замерены скорости коксообразования на этих образцах в зависимости от продолжительности опытов по пиролизу. Результаты графически представлены на фиг. 10. Сравнение показывает, что только на образцах 3 и 4, предварительно обработанных согласно изобретению, в течение длительного времени испытаний сохранились типичные для всех образцов низкие скорости коксообразования. Из полученных данных следует заключить, что предварительная обработка согласно изобретению дает ощутимое увеличение рабочего времени по сравнению со способами без предварительной обработки или с соединением, содержащим лишь кремний или серу.
В табл. 1 дано влияние соотношения кремния и серы в используемом для предварительной обработки предварительно активированных образцов из стали X8CrNiTi 18 10 (880oC, 60 мин) инертном газе (общее содержание суммы Si - S : 0,005 моля) на скорость коксообразования r при пиролизе n-гептана в потоке азота.
В табл. 1 обозначены: 1 - атомное соотношение Si:S, 2 - время испытания (мин); соединения Si, S, использованные для предварительной обработки: a) триметилсилилметилмеркаптан, b) смесь из тетраметилсилана и диметилсульфида в соотношении 1:1, c) бис-триметилсилилсульфид, d) смесь из тетраметилсилана и диметилсульфида в соотношении 2:1, e) смесь из триметилсилана и диметилсудьфида в соотношении 3:1, f) смесь из тетраметилсилана и диметилсульфида в соотношении 4: 1, g) смесь из тетраметилсилана и диметилсульфида в соотношении 5:1.
В табл. 2 дана зависимость скорости коксообразования r от содержания триметилсилилметилмеркаптана в инертном газе для термической предварительной обработки образцов из X8CrNiTi 18 10 при пиролизе n-гептана в потоке азота.
В табл. 2 обозначены: 1 - содержание триметилсилилметилмеркаптана в инертном газе, 2 - время испытаний (мин).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ НЕПРЕРЫВНОГО ПРЕОБРАЗОВАНИЯ ЭНЕРГИИ В ГАЗОТУРБИННОЙ УСТАНОВКЕ И ГАЗОТУРБИННАЯ УСТАНОВКА ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1991 |
|
RU2085754C1 |
| СПОСОБ ОБЕЗУГЛЕРОЖИВАНИЯ СТАЛЬНОГО РАСПЛАВА | 1996 |
|
RU2139355C1 |
| Способ получения из остатков переработки нефти алифатического типа углеродсодержащего материала, используемого в качестве спекающегося компонента в угольной шихте для получения кокса и алифатического масла | 1972 |
|
SU1087077A3 |
| СВАРИВАЕМАЯ ВЫСОКОПРОЧНАЯ КОНСТРУКЦИОННАЯ СТАЛЬ ДЛЯ ИЗГОТОВЛЕНИЯ БЕСШОВНЫХ КОРРОЗИОННО-СТОЙКИХ ТРУБ И ЕМКОСТЕЙ И СПОСОБ ИХ ИЗГОТОВЛЕНИЯ | 1992 |
|
RU2102521C1 |
| КОМПОЗИЦИИ ДЛЯ ПОДАВЛЕНИЯ КОКСООБРАЗОВАНИЯ В ПЕЧАХ ДЛЯ ПРОВЕДЕНИЯ ТЕРМИЧЕСКОГО КРЕКИНГА | 2001 |
|
RU2258731C2 |
| СПОСОБ ХИМИЧЕСКОЙ ОБРАБОТКИ ВНУТРЕННЕЙ ПОВЕРХНОСТИ РЕАКТОРА ДЛЯ ПИРОЛИЗА УГЛЕВОДОРОДОВ | 2013 |
|
RU2566244C2 |
| СПОСОБ ПАССИВАЦИИ ВНУТРЕННЕЙ ПОВЕРХНОСТИ РЕАКТОРА, ПОДВЕРГАЕМОГО ЗАКОКСОВЫВАНИЮ, И РЕАКТОР | 1992 |
|
RU2079569C1 |
| Способ снижения коксообразования в реакторах пиролиза углеводородов | 2018 |
|
RU2679610C1 |
| СПОСОБ ПОДГОТОВКИ ГАЗА ДЛЯ ТРАНСПОРТИРОВКИ ПО ТРУБАМ И КОМПРЕССОРНАЯ СТАНЦИЯ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1993 |
|
RU2104397C1 |
| Препятствующее коксообразованию оборудование, способ его изготовления и его применение | 2021 |
|
RU2800956C1 |
Изобретение относится к поверхности теплообменника в реакторах и/или теплообменниках установок для превращения углеводородов и других органических соединений при высокой температуре в газовой фазе, а также к способу изготовления каталитически дезактивированной металлической поверхности в химических реакторах и/или теплообменниках. Согласно изобретению контактирующие с органическими материалами металлические поверхности обрабатываюся смесью, состоящей из продукта, содержащего кремний и серу, и сухого инертного по отношению к продукту, содержащему кремний и серу, газового потока, при 300-1000oC в течение 0,5-12 ч. Предварительная обработка поверхностей согласно изобретению увеличивает время работы оборудования до момента необходимости удаления коксовых отложений. 2 с. и 10 з.п.ф-лы, 10 ил., 2 табл.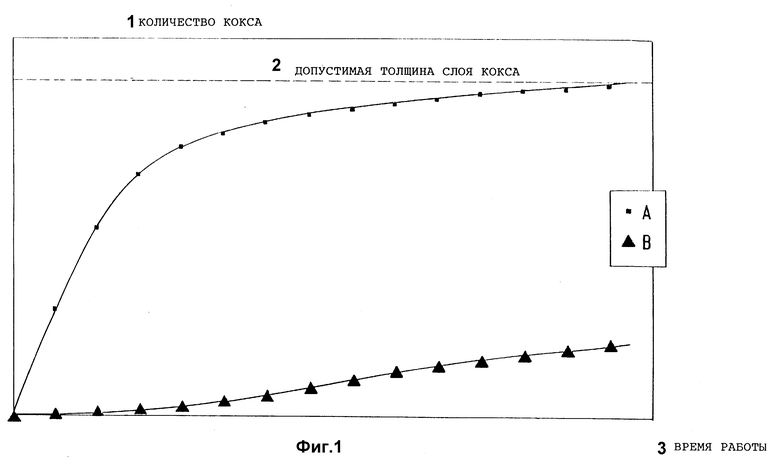
| Буровой раствор | 1985 |
|
SU1328363A1 |
| Способ защиты труб теплообменных аппаратов от органических загрязнений | 1987 |
|
SU1528785A1 |
| US 4410418 A, 18.10.83 | |||
| EP 0269332 A1, 01.06.88 | |||
| DE 3247568 A1, 30.06.83 | |||
| US 4116812 A, 26.09.78 | |||
| US 4280898 A, 28.07.81. | |||
