Настоящее изобретение относится к новым пептидам и их производным, имеющим точную химическую структуру. Изобретение также адресуется к методам их получения и применения. Гипоталамический гормон, стимулирующий выделение лютеинизирующего гормона (LHRH), действует на переднюю долю гипофиза, стимулируя секрецию лютеинизирующего гормона (LH) и фолликулостимулирующего гормона (FSH). Антагонистический аналог LHRH действует на переднюю долю гипофиза быстро, длительно, может быть безопасно и обратимо использован для контрацепции или селективного подавления секреции гонадотропных гормонов. При таком применении LHRH антагонисты преобладают над агонистами. К настоящему моменту предсказано и синтезировано более двух тысяч аналогов LHRH, среди которых аналог "Nal-Arg", который проявил также сильную активность в отношении стимуляции выделения гистамина (HRA). Он вызывал проходящий отек мордочки и конечностей у крыс при применении в дозах в 50-100 раз превышающих терапевтическую дозу. Результаты клинического испытания продемонстрировали системные воздействия, связанные с гистамином. Другие антагонисты ГСЛГ, содержащие DArg6 или DLys6, показали сходные побочные действия, их ED50 для HRA были ниже 1 мкг/мл. Настоящее изобретение представляет новые антагонисты, которые обладают очень высокой антиовуляторной активностью (АОА) и очень низкой активностью в отношении стимуляции выделения гистамина HRA и незначительными побочными эффектами. Содержание и примеры этого изобретения включают следующее.
Методология прогноза структуры этого изобретения основана на топологическом сходстве молекулы исходного соединения (NAc - D2Nal', DpClPhe2, D3Pal3, Ser4, Tyr5, DArg6, Leu7, Arg8, Pro9, DAla10)NH2 (II) и нейропептида, субстанция P, которая является характерной черной модификации как щелочной, так и липофильной области молекулы исходного соединения для получения новых антагонистов, обладающих как высокой АОА, так и низкой ГСА. Термин модификация здесь означает построение или замену аминокислот на участке Tyr5 - DArg6 - Arg8 в C-конце и ароматических кислот в N-конце (II). Более точно, создание структуры состоит во введении подходящей щелочной группы и замещений на природно несвойственные аминокислоты в положении 2, 3, 5, 6, 8 в (II).
Нижеследующее также входит в методы и примеры этого изобретения.
1. Замещение D3Pal, которая является ароматической аминокислотой, имеющей подходящую щелочность, вместо DArg8 в (II) с получением аналога (III): (NAc-2DNal', DpClPhe2, D3Pal3, Ser4, Tyr5, D3Pal6, Ser7, Arg8, Pro9, DAla10)NH2
2. Замещение Tyr5 на Arg5 в (III) для получения (IV): (NAc-D2Nal', DpClPhe2, D3Pal3, Ser4, Arg5, D3Pal6, Leu7, Arg8, Pro9, DAla10)NH2
3. Замещение D3Pal3 в (IV) на DPhe3 или его производные DXCH2Phe, чтобы получить (V): (NAc-D2Nal', DpClPhe2, DPhe3, Ser4, Arg5, D3Pal6, Leu7, Arg8, Pro9, DAla10)NH2 или его аналогов (DXCH2Phe3)
4. Замещения DPhe3 и его производными D3Pal3 в (III), чтобы получить (V'): (NAc-D2Nal', DpClPhe2, DPhe3, Ser4, Tyr5, D3Pal6, Leu7, Arg8, Pro9, DAla10)NH2 или его аналогов (DXCH2Phe3)
Синтезированы серии новых антагонистов LHRH с формулой (NAc-D2Nal', AA2, AA3, Ser4, AA5, AA6. Leu7, AA8, DAla10)NH2, где AA являются природными или неприродными аминокислотами, которые представлены D- или L-ArAla. Более конкретно показано на схеме А в конце текста.
Антагонисты LHRH, полученные при использовании вышеописанного метода, как пептидные лекарственные препараты, могут использоваться при лечении заболеваний репродуктивной эндокринной системы, таких как эндометриоз, преждевременное половое созревание детей, и для лечения рака простаты и рака молочных желез, так же как и в качестве мужских и женских контрацептивов для регулирования рождаемости, или использоваться для диагностики и лечения бесплодия и т. п. Такой пептидный лекарственный препарат может быть приготовлен в виде обычных инъекционных форм, инъецируемых капсул или других лекарственных форм для практического применения.
Дальнейшее описание этого изобретения состоит в следующем.
При естественном пути выделения гистамина в организме очень важную роль играет нейропептидная субстанция II, ее ED50 для HRA равна 5-15 мкМ. Химическая структура SP представляет собой (Arg', Pro2, Lys3, Pro4, Gln5, Gln6, Phe7, Phe8, Gly9, Leu10, Met11)NH2.
Изучение взаимосвязи между ее структурой и HRA показало, что Arg1-Pro2-Lys3 на N-конце молекулы SP существенно для ее HRA, так как при удалении этих трех аминокислот из молекулы полностью исчезает ее HRA. Для сравнения, при удалении одной, двух или трех аминокислот на C-конце HRA сохранялась на уровне одной четвертой от HRA первоначальной. Кроме того, удаление Phe8 и Phe7 снижает HRA на 4% и 0,5% таковой СП. Далее удаление Gln5,6 не вызывает значительного изменения HRA. Вышеприведенные данные означают, что липофильная область вокруг Phe7,8 определяет уровень HRA, эта область участвует в связывании молекулы с рецептором тучной клетки.
Как упомянуто ранее, аналоги (D2Nal', DArg6)LHRH показали очень высокую HPA, их молекулярное строение имеет топологическое сходство с SP: DArg6 - Leu7 - Arg8 в первом, по-видимому, соответствуют Arg'- Pro2 - Lys3 в последнем, оба состоят из пары сильно щелочных аминокислотных остатков, между которыми только один нейтральный аминокислотный остаток, т.е. оба аналога LHPH - 2DNal', DArg и SP содержат два сильно щелочных аминокислотных остатка, которые располагаются один по отношению к другому в позициях 1,3. С другой стороны, разброс остатков ароматических аминокислот в первом, как считают, соответствует области Phe7,8 в СП, с точки зрения определения величины HRA.
Создание структуры этого изобретения состоит из двух аспектов: один состоит в модификации участка Tyr5 - DArg6 - Arg8 на C-конце, и другим является тонкий подбор ароматических кислот после оптимизации модификации щелочного участка на C-конце. (NAc - D2Nal', DpClPhe2, D3Pal3, Tyr5, DArg6, Leu7, Arg8, Pro9, DAla10)NH2 (II) использован как исходное вещество, которое показало AOA 100% при дозе 0,5 мкг в кукурузном масле, 5% при 0,25 мкг.
В первую очередь, DArg6 в (II) мог быть замещен слабыми основными или нейтральными ароматическими кислотами, такими как D3Pal, D6Qal тетрагидротриптофан, метилтриптофан. (NAc - D2Nal', DpClPhe2, D3Pal3,6, DAla10)LHRH (III) было получено когда D3Arg6 в (II) был замещен на D3Pal6. (III) показал AOA 100% при дозе 3 мкг, 83% при 1 мкг (в кукурузном масле), и его ED50 по HRA была равна 9,8 мкг/мл, что значительно лучше, чем у аналога "Nal-Arg", что ED50 по HRA была меньше, чем 1 мкг/мл. По-видимому, для того чтобы получить высокую AOA, щелочность всей молекулы должна быть равной или близкой таковой пары аргинина. Поскольку позиция 5, как и позиция 6, не участвует в связывании с рецептором, в положение 5 может быть помещен широкий круг аминокислот, включая аргинин. Были созданы серии новых аналогов. Например, при замещении Tyr5 на Arg5 в (III) получены (NAc - DNal', DpClPhe2, D3Pal3,6, Arg5, DAla10)LHRH (IV). Оба (IV) и (II) содержали два аргининовых остатка, но расстояние между двумя аргининами в (IV), где их геометрическое взаиморасположение стало 1,4, т. е. между этими двумя аргининами были две другие аминокислоты, было больше, чем в (II). Поэтому, вероятно, HRA будет снижена и, с другой стороны, из-за наличия двух аргининов AOA не должна быть ниже, чем у (II). Результаты биологического исследования (IV) показали, что ED50 по HRA была равна 3,5 мкг/мл, тогда как AOA составила 60% при 0,12 мкг (кукурузное масло), 85% при 0,25 мкг, 100% при 0,5 мкг. Это было первым случаем для антагонистов LHRH, когда достигнутая ED50 для AOA была равна или менее 0,125 мкг.
Поэтому дальнейшее создание структуры базировалось на структуре (IV).
В молекуле (IV) присутствуют четыре остатка со щелочными ионами, D3Pal3,6 и Arg5,8, тогда как (II) содержит только три щелочных остатка. Поэтому приемлемо заменить один D3Pal нейтральной аминокислотой; с другой стороны, (IV) проявил очень высокую гидрофильность, и снижение гидрофильности замещением DPal в (IV) на гидрофобную аминокислоту, вероятно, будет благоприятным для задержки лекарства в организме и увеличения продолжительности эффекта. Затем были созданы новые серии аналогов путем замещения вместо DBPal3. (V) проявил 100% AOA при дозе 1 мкг (в физиологическом растворе), равную активности исходного вещества (IV), тогда как HRA снижена наполовину: ED50 по HRA была 7,4 мкг/мл.
Дополнительное замещение DClPhe2 на DPhe снизило липофильность этого участка молекулы и снизило HRA.
Arg5 - D3Pal6 - Leu7 - Arg8 на C-конце (IV), по-видимому, играют главную роль в пусковом механизме секреции гистамина. D3Pal соединяет в одной молекуле свойства ароматического строения, щелочность и гидрофильность, он также стереоприемлем в антагонистах LHRH для связывания с рецептором. Таким образом, создание новых серий неприродных аминокислот, используя подобные свойства, как (V) D3Pal, может привести к получению лучших антагонистов, чем (IV) или (V).
Модификация природных липофильных ароматических аминокислот, например фенилаланина, например, по методу, описанному ниже в разделе Синтез неприродных аминокислот, приводит к созданию серий новых аминокислот, которые соединяют свойства ароматических соединений, гидрофильность и щелочные свойства в одной молекуле и могут выражаться формулой
-R1R2NCH2C6H4CH2 CH(NH)CO2H
(VI), где
R1 и R2 могут быть одинаковыми или разными, могут быть представлены цепью или циклом, могут также содержать гетероатом. При изменении R1 и R2 могут быть получены серии аминокислот, которые постоянно системно измененные щелочность, гидрофильность и стереостроение. Введение этих аминокислот в положения 5, 6, 8 соединения (IV) позволило получить три серии новых антагонистов LHRH. Результаты биологического исследования показали, что каждая серия дала по крайней мере один новый антагонист, проявляющий 100% AOA при дозе 1 мкг, подобно (IV), в то же время была значительно снижена. Примером был (VII): (NAc - D2Nal', DpClPhe2, D3Pal3, Ser4, Mop5, D3Pal6, Leu7, Arg8, Pro9, DAla10)NH2,
который проявил 100% AOA при 1 мкг, ED50 при ГСА 14,7 мг/мл, явно лучше, чем у (V). При замещении Arg8 в (IV) на (VI) степень снижения HRA положительно коррелирует с длиной P в (VI), так что ED50 по HRA могла бы быть выше 200 мкг/мл, такие соединения могут легко растворяться в водном растворе и, как ожидается, будут использоваться в клинике без осложнений со стороны лекарственных форм. Результаты показали, что Arg8 или Lys8 не существенны для антагонистов LHRH с высокой активностью. Соответствующий щелочной центр в положении 8 может обеспечить высокую AOA, между тем, активность по индуцированию выработки гистамина тучными клетками заметно снижалась, когда встраивалась группа с основными свойствами, упомянутая выше, создающая значительное стереопрепятствие.
Это изобретение, объединяя модификации как на конце N, так и на конце C, приводит к созданию лучших антагонистов LHRH.
Процесс синтеза иллюстрируется следующим.
1. Синтез новых неприродных аминокислот
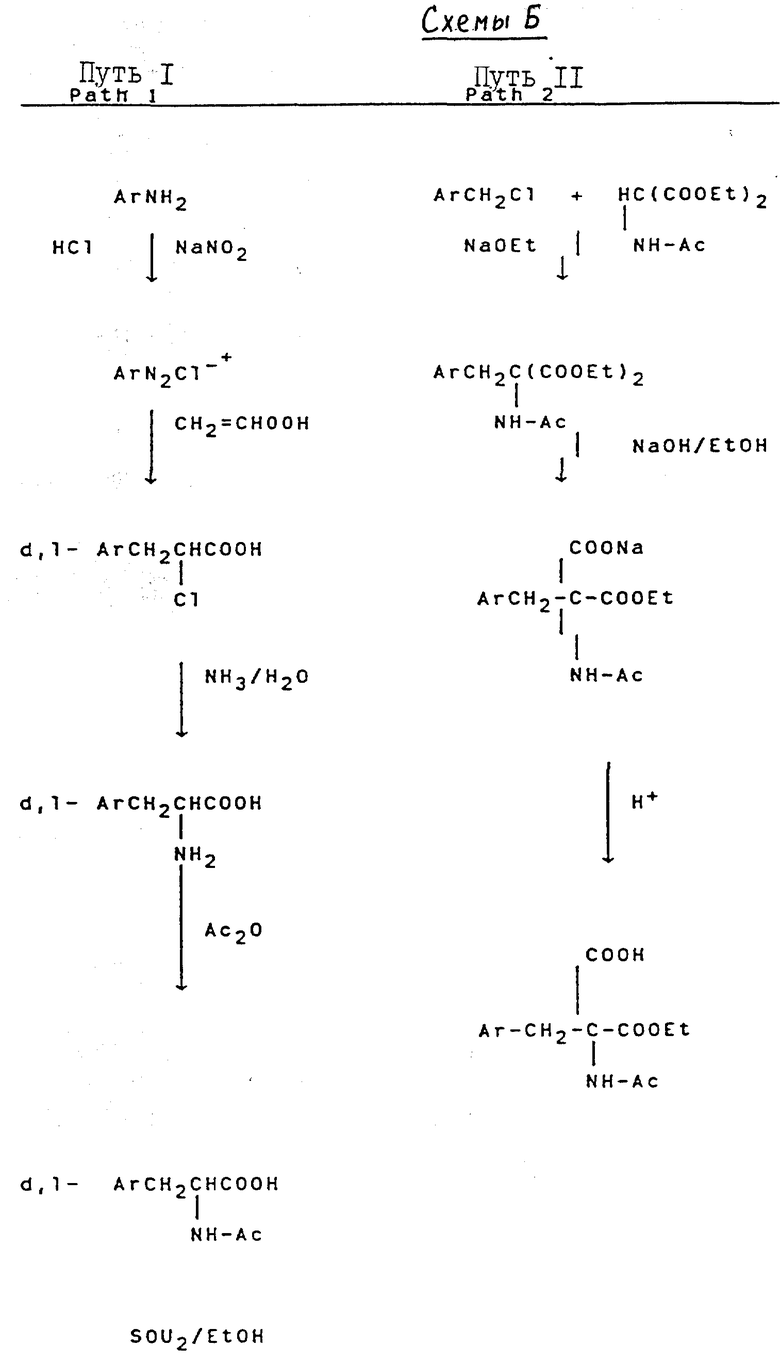
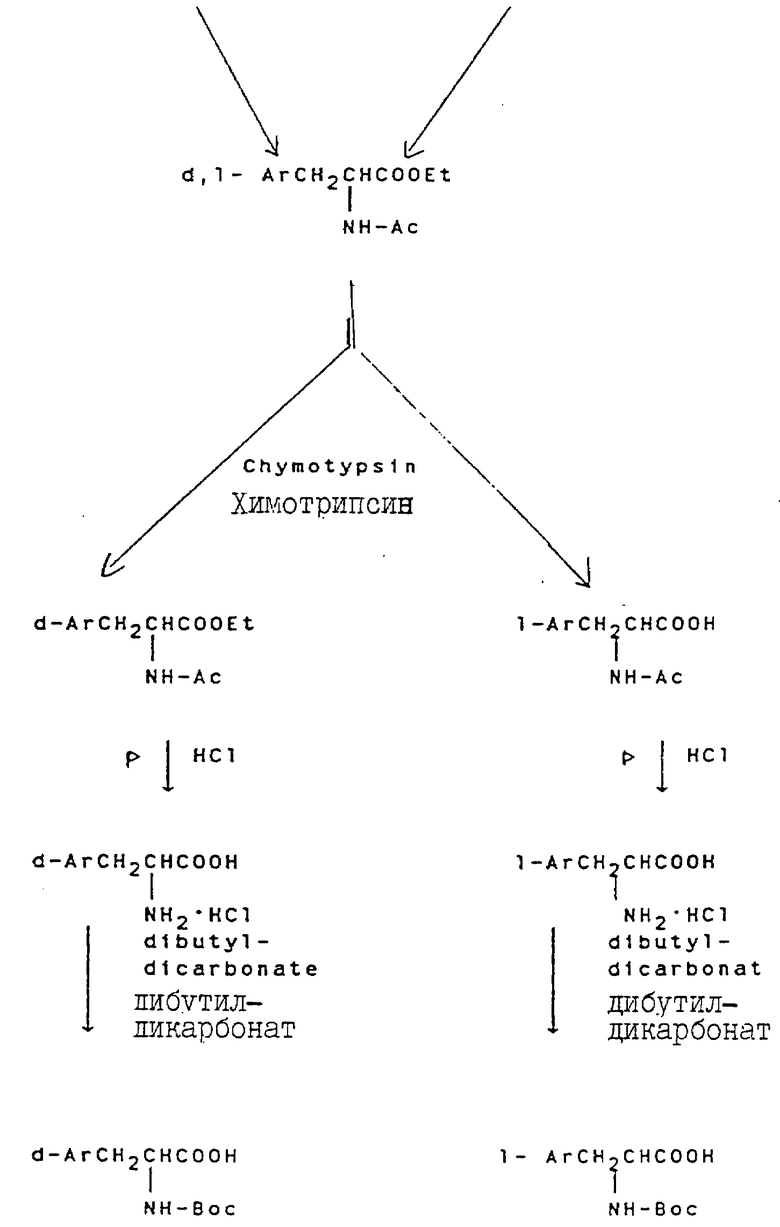
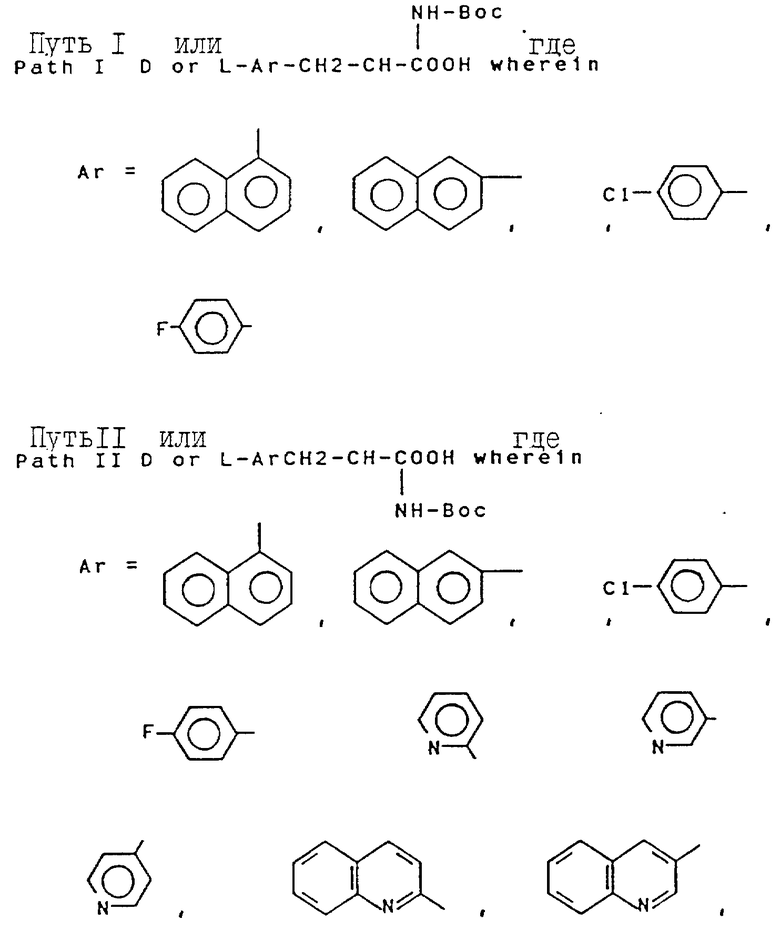
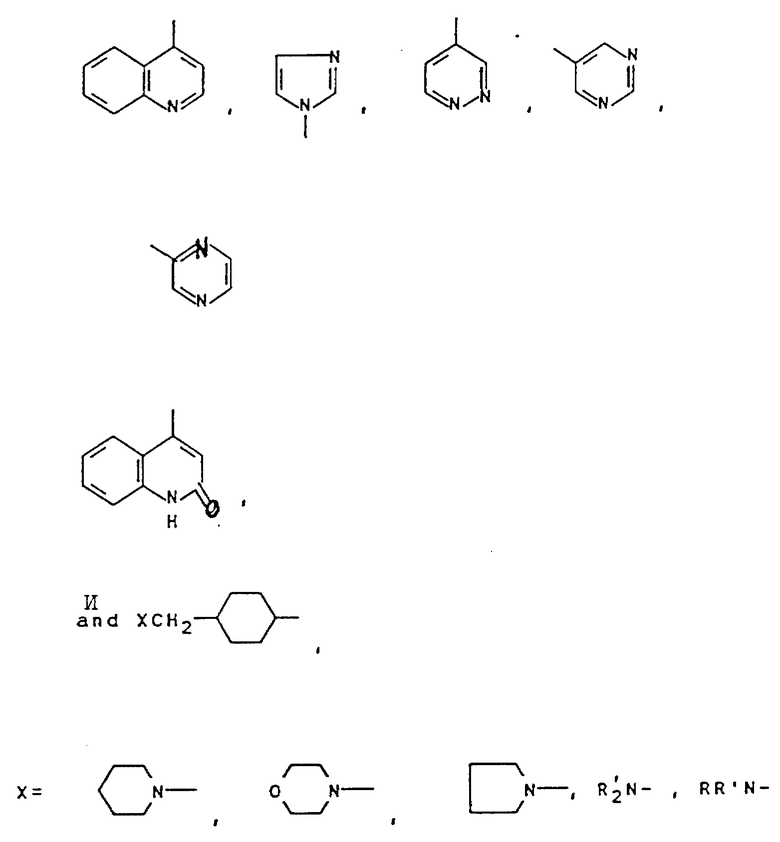
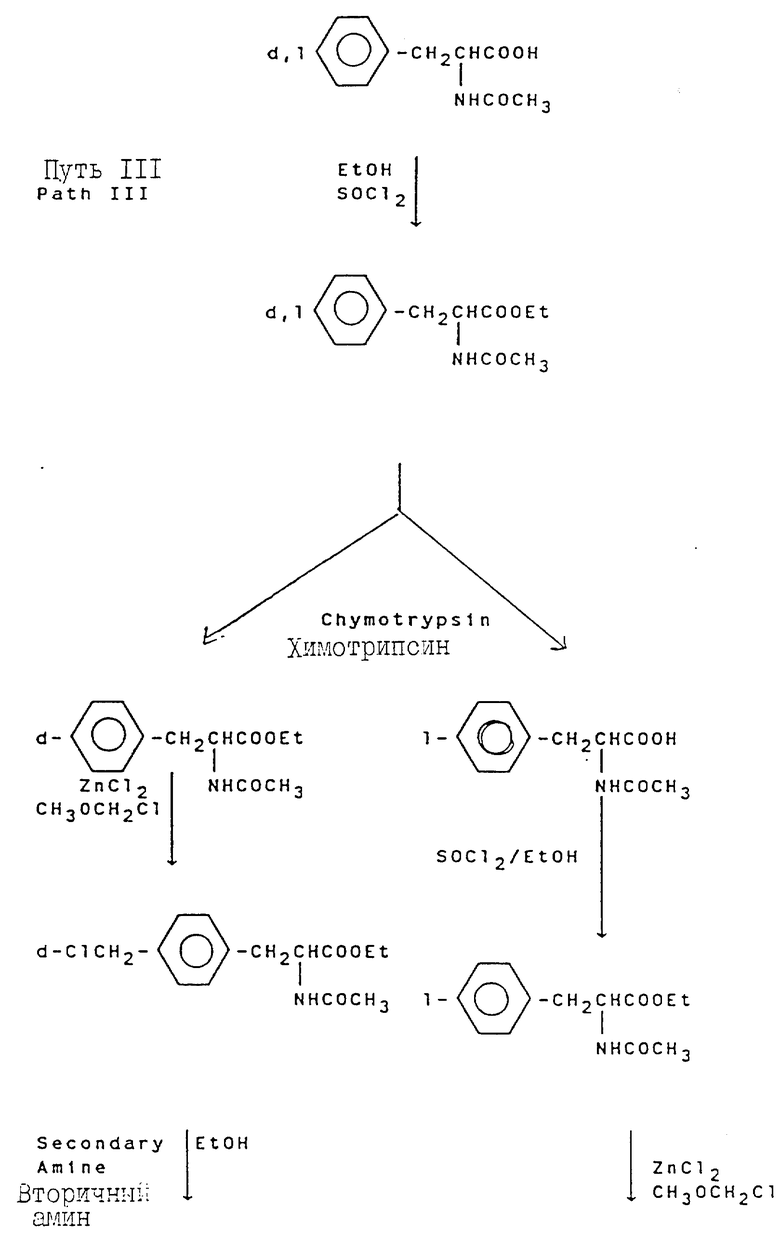
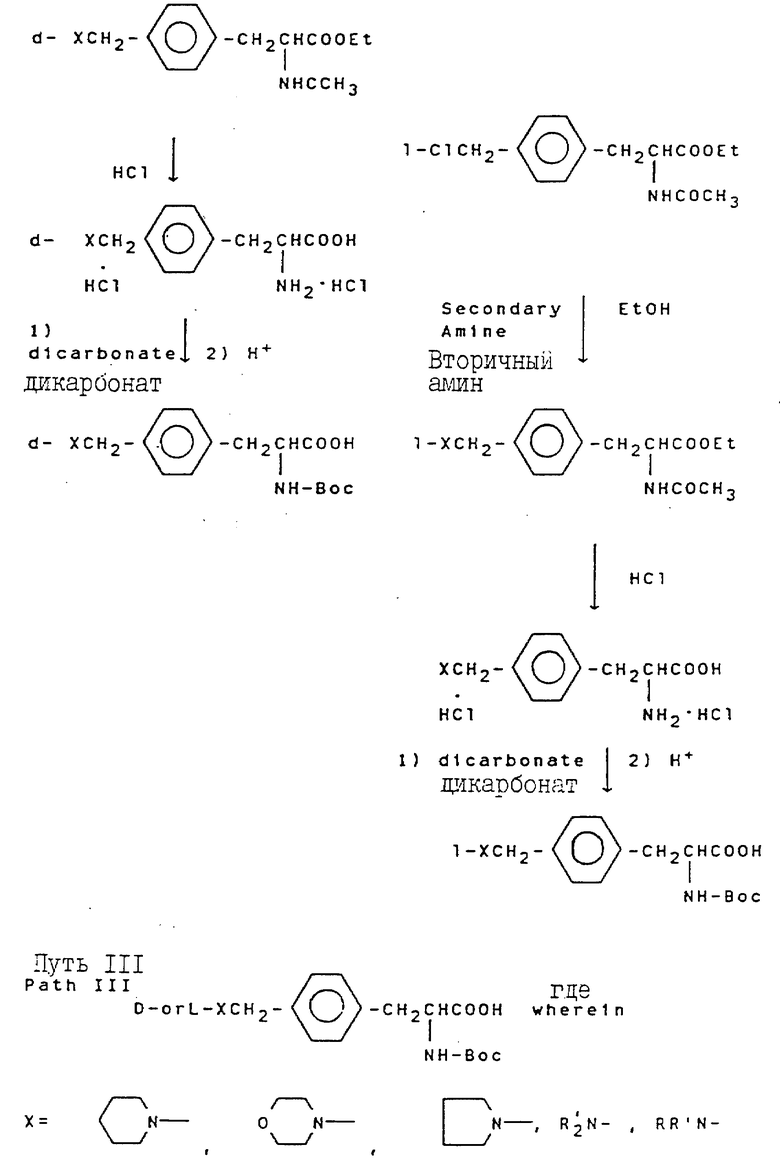
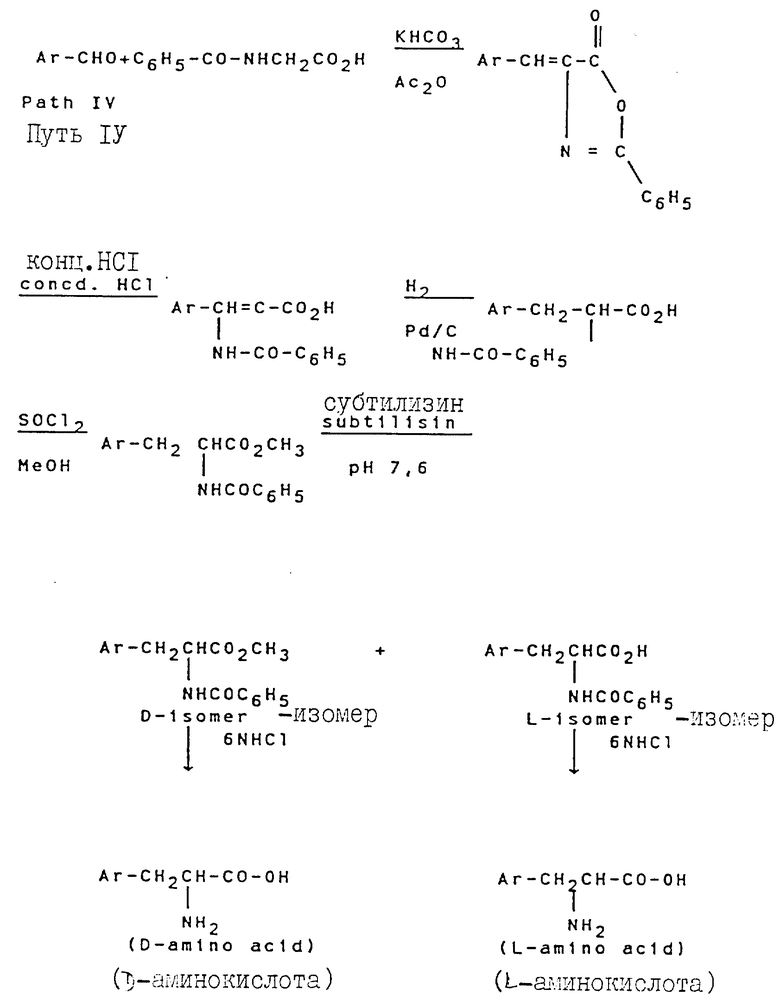
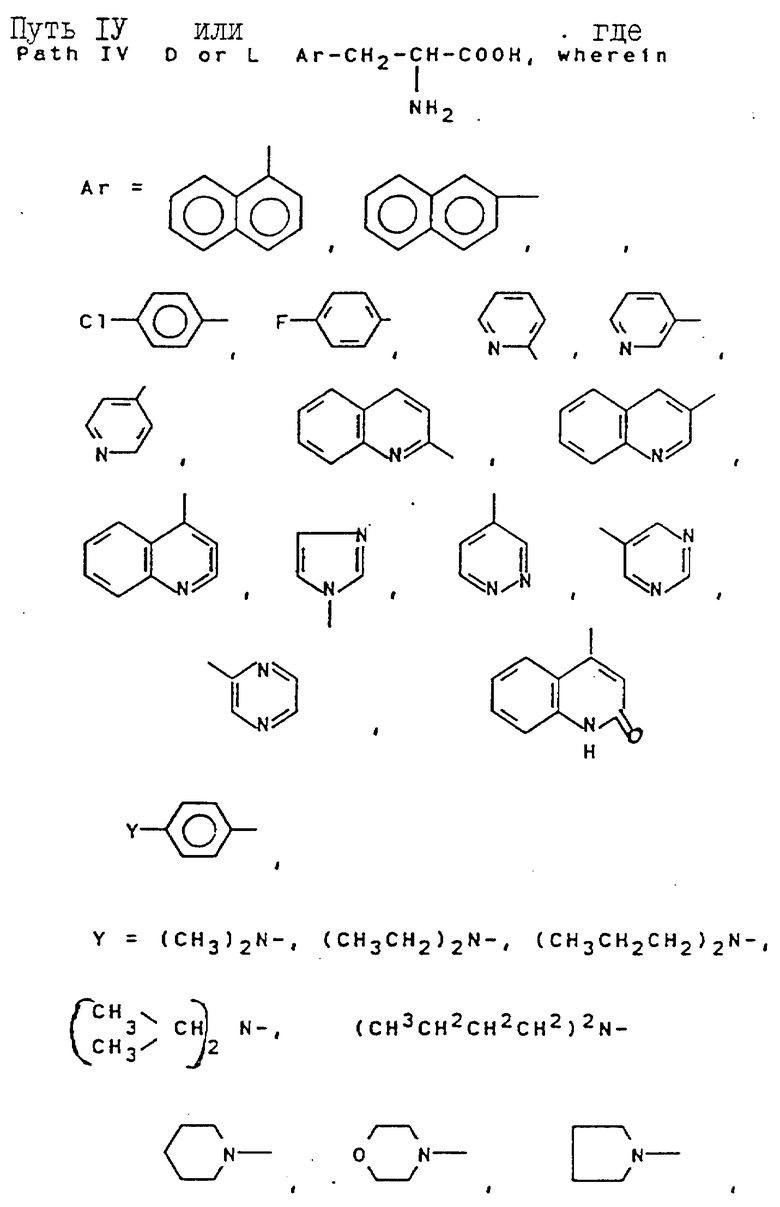
Свыше 60 серий и несерийных D- или L-аминокислот созданы и синтезированы четырьмя путями синтезами, представленными на схеме ниже (см. схемы Б в конце текста). Строение этих неприродных аминокислот показано в виде основных структурных формул, перечисленных в той же схеме. Некоторые их этих аминокислот обладают щелочностью, гидрофильностью и ароматическими свойствами соответственно, тогда как другие сочетают их все в одной молекуле.
2. Синтез пептида
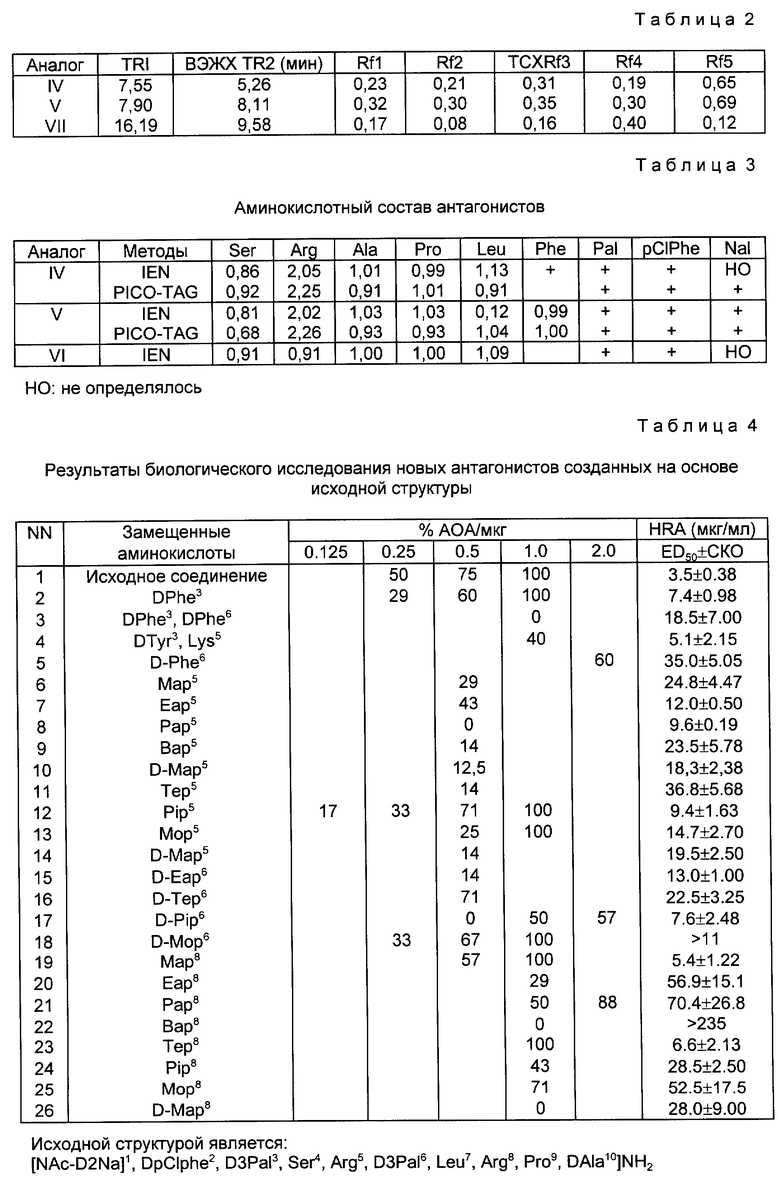
Синтез начинается с C-конца пептида на бензгидриламин-гидрохлоридной смоле (BHA-смоле) с использованием твердофазного пептидного синтеза, предложенного Меррифилд. Это трехфазный процесс, включающий прикрепление, взаимодействие и отщепление. Дихлорметан (DCM) является основным растворителем, используемым для отмывания перед каждой следующей стадией реакции, в то же время спирт изопропанол (IPA) и N, N-диметилформамид (DMF) также использовались, где необходимо. Реакция связывания осуществляется при катализе избытком дициклогексилкарбодиимида (DCC) при добавлении достаточного количества 1-гидроксибензотриазола (HOBT). Степень реакции связывания определялась по нингидриновому методу Кейзеса. Вторая реакция связывания должна проводиться, если она дает положительный результат в пробе Кейзеса. Пептидная цепь отщеплялась от смолы с использованием безводного фтористого водорода (HF) в присутствии анизола после завершения всех реакций, требующих связывания со смолой, в это же время удаляются все временные защищающие группы. После отмывания этилацетатом или эфиром сырые продукты LHRH-антагонистов получают путем экстракции водным раствором уксусной кислоты с последующей лиофилизацией. Выход свыше 50%.
3. Очистка пептида
(1) Пептид очищают с помощью гельпроникающей хроматографии или распределительной хроматографии на двуокиси кремния в колонке высокой 60 - 100 см при контроле с помощью УФ/ТСХ. Очистку LHRH-антагонистов проводили однократно и продукт получали после лиофилизации основных фракций. Выход составляет 50 - 90% и чистота может быть свыше 90%.
(2) Пептид затем очищали на аппарате для высокоэффективной жидкостной хроматографии (ВЭЖХ) Уотерса, используя колонку с обращением фазы C18 (7,8 x 300 мм) (m Bondapak 84176). Выход на этой стадии составляет 20 - 50%, при этом чистота не менее 99%.
4. Анализ чистоты пептида
(1) Анализ TCX
Он проводится на пластиковой пластинке, покрытой силикагелем 60F254, длиной 5 - 10 см. Во всех случаях выявлялось одиночное пятно при работе с пятью разными системами растворителей.
(2) Анализ ВЭЖХ
Во всех случаях обнаружен единичный пик при элюировании двумя видами систем растворителей, соответственно, при использовании аппарата Уотерса для ВЭЖХ с аналитической колонкой ( m Bondapak 27324) при УФ-детекции ( λ = 210). Величина образца составляла 10 - 200 мкг.
5. Аминокислотный анализ пептида
В соответствии с методом PICO-TAG, разработанным фирмой Уотерс, 50 мкг образца, который был высушен под вакуумом в течение 2 часов, точно отвешивают на чашечных весах с разрешением 10-5 г. После растворения в воде 10 мкг образца добавляют в реакционную пробирку, в которую, согласно руководству, добавлена соляная кислота 1:1 (содержащая 1% фенола). Реакция продолжается 20 - 24 ч при 105oC. Реакция продолжается 22 - 24 ч при 105oC в герметически закрытом контейнере, который наполнен азотом и откачен под вакуумом, чтобы удалить кислород из реакционной пробирки. Изотиоцианат фенола добавляется, чтобы получить аминогруппы после выпаривания избыточных количеств соляной кислоты. Затем он анализировался с помощью жидкостного хроматографа, снабженного аминокислотной аналитической колонкой PICO-TAG с УФ-детектором ( λ = 254 нм). Содержание каждой аминокислоты и относительное молярное соотношение рассчитывали, чтобы выявить аминокислотный состав образца, основываясь на сравнении составной части каждой аминокислоты с таковой H-стандартного образца Уотерса. Классический метод с ионообменным нингидриновым производным (IEN) был также использован как контрольный, который дал те же самые результаты. Но, чтобы получить удовлетворительные результаты, для этого метода необходимо в десять раз больше образца.
6. Оценка биологической активности
Использован метод антиовуляции крыс Корбина. В этом эксперименте использованы здоровые взрослые самки крыс SD (вес 200 - 500 г). Всех животных содержали при 22 - 24oC и при режиме 14 ч/10 ч (свет/темнота). Они получали стандартное питание и воду по потребности. В этом эксперименте могли быть использованы крысы, у которых выявлено по крайней мере два последовательных четырехдневных цикла течки при исследовании вагинальных мазков. Крысам вводили пептиды (антагонисты LHRH) в разных дозах в физрастворе в середине стадии протечки.
Крыс забивали на следующий день, их яйцеводы с двух сторон исследовали под препаровальной лупой, чтобы определить число созревших яйцеклеток. Крысы были разделены на несколько групп по получаемым дозам, каждая группа состояла из около 10 животных, и контрольная группа, животным которой вводили равное количество физраствора, состояла из 9 - 10 крыс. Антиовуляторная активность (AOA) показана в следующем равенстве: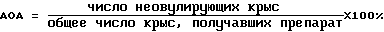
7. Оценка активности по стимуляции выделения гистамина
(1) Определение секреции гистамина (HRT)
В этом эксперименте использовали здоровых взрослых самцов крыс SD (вес 200 - 500 г), содержавшихся в вышеназванных условиях. После анестезии CO2 перитонеальную полость промывали 50 мл среды PIPAS AC, содержащей 20 единиц гепарина. После последующего центрифугирования при 200хg в течение 8 мин при 4oC клетки снова отмывали и окончательно ресуспендировали до концентрации от 8 до 24•105 общего числа лейкоцитов/мл PIPAS AC. Эта суспензия содержит примерно 5 - 10% тучных клеток. Отмытые клетки использовались сразу же после отбора и предварительно нагревались в течение 5 мин при 37oC, перед тем как 0,3 мл пробы пипеткой помещались в полистироловые пробирки, содержащие 0,3 мл разведенного пептида. Смеси инкубировали в течение 15 мин при 37oC и реакцию останавливали центрифугированием при 400хg в течение 15 мин при 4oC. Супернатанты исследовали на содержание гистамина с помощью метода флюорометрического определения, проводимого вручную, после последовательной экстракции н-бутанолом и н-гептаном. Содержание гистамина может быть установлено по стандартной гистаминовой кривой (см. ниже). Процесс секреции гистамина может быть рассчитан по следующему равенству ,
,
где
E является показанием флуорометрии для экспериментального образца; B - показание флуорометрии для образцов, содержащих только клетки и буфер; и C - показание флуорометрии "полного содержания" (клетки, обработанные HClO4).
Стандартная кривая может быть получена путем нанесения на график значений оптической плотности по показаниям флюориметра при 350 нм/450 нм (активация/флюоресценция) для концентрацией серий разведений раствора точно взвешенного количества гидрохлорида гистамина. Относительный параметр p стандартной гистаминовой кривой может быть 0,9998 и самая низкая определяемая концентрация гистамина равна 0,5 нг/мл.
Значение ED50 для пептида может быть получено из кривых зависимости ответной реакции от дозы путем нанесения на график уровня секреции гистамина в сравнении с концентрацией пептида в полулогарифмическом масштабе.
Все образцы пептидов должны быть испытаны с тучными клетками как минимум от 3 разных крыс.
(2) Кожная проба на анафилактоидную активность (CAT)
В этом эксперименте использованы здоровые взрослые крысы-самки (вес 250 г). Крысам вводили внутривенно раствор синего Еванса (1 мл 0,05%). Сразу же после этого внутрикожно вводили раствор пептида (5, 0,5 и 0,05 мг/мл соответственно) или физраствор (контроль) в обритый сектор на спине животных. Через 30 мин после инъекции крыс забивали и кожу спины отгибали. Диаметр поражений измеряли в миллиметрах в двух перпендикулярных направлениях с помощью циркуля с нониусом. Диаметр в контроле обычно меньше 5,5 мм.
Количество синего Еванса, проникающего в кожу из кровеносных сосудов, может быть тоже определено спектрофотометрически. Кожа, соответствующая области поражения, вырезалась и погружалась в смесь ацетона/физраствора (7:3, об/об) на ночь. После центрифугирования на следующий день содержание синего Еванса в супернатанте определялось на спектрофотометре (УФ-260) при 610 нм с раствором сравнения ацетон/физраствор (7:3). Каждый пептид испытывался как минимум на 3 разных крысах.
С помощью метода, описанного выше, создано и синтезировано множество новых антагонистов LRRH. Кратко, антагонисты с новой структурой были получены путем одиночных или множественных замещений разными природными и неприродными аминокислотами, перечисленными в предыдущих разделах.
Некоторые примеры новых антагонистов LHRH, полученных таким путем, показаны в таблице 1.
Применение этого изобретения
1. После завершения доклинических фармакологического и токсикологического изучения мы сможем применить эти новые антагонисты, которые обладают высокой терапевтической эффективностью и слабым побочным действием, в клинике так, чтобы разработать новое лекарство для лечения эндометриоза и других нарушений в репродуктивной эндокринной системе, включая преждевременное половое созревание у детей, рак простаты и рак молочных желез. Так как они подавляют секрецию гонадотропина путем конкурентного с эндогенным LHRH связывания с рецептором и действуют быстро, обратимо и безопасно, они могут, кроме того, разрабатываться как новый тип контрацептивов для мужчин и женщин. Сверх того, они могут быть также использованы при лечении бесплодия и для селективного и обратимого устранения функции гипофиза по секреции гонадотропина.
Являясь разновидностью лекарств пептидной природы, маловероятно, что антагонисты LHRH будут применяться перорально. Но они могут быть легко получены в виде лиофилизированного порошка, легко растворимого в физрастворе для инъекций внутривенно, подкожно или внутримышечно.
Кроме того, изучаются длительно действующие системы подачи лекарственного вещества, такие как биодеградируемые, инъецируемые капсулы. Капсулы могут имплантироваться подкожно с помощью специального шприца и должны абсорбироваться тканью после выделения всего количества пептида и не требуют хирургического удаления. Длительно действующие системы доставки особенно полезны для долговременного применения аналогов LHRH в клинике.
Следующее представляет собой анализы результатов из примеров (используя три аналога IV, V, VII как типичные примеры).
(1) Чистота
Тонкослойная хроматография (TCX)
На каждой из хроматограмм, полученных с пятью разными системами растворителей, присутствовало только одно пятно.
Высокоэффективная жидкостная хроматография (ВЭЖХ):
На каждой из хроматограмм при элюировании двумя разными системами растворителей присутствует только один пик.
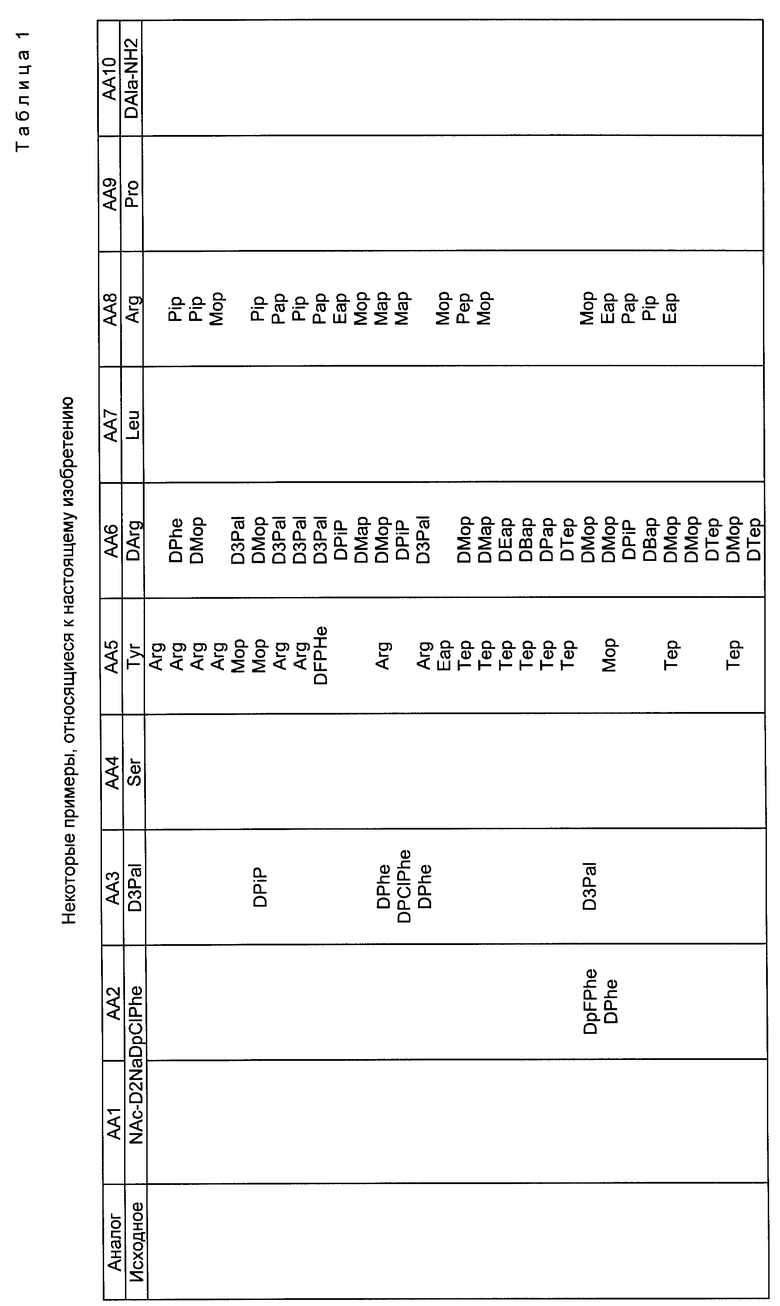
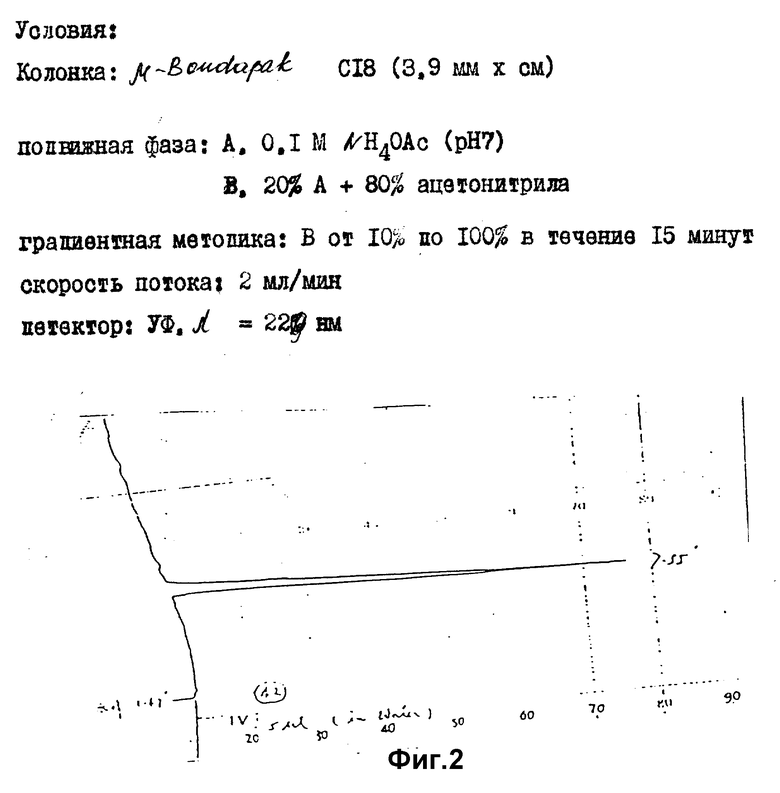
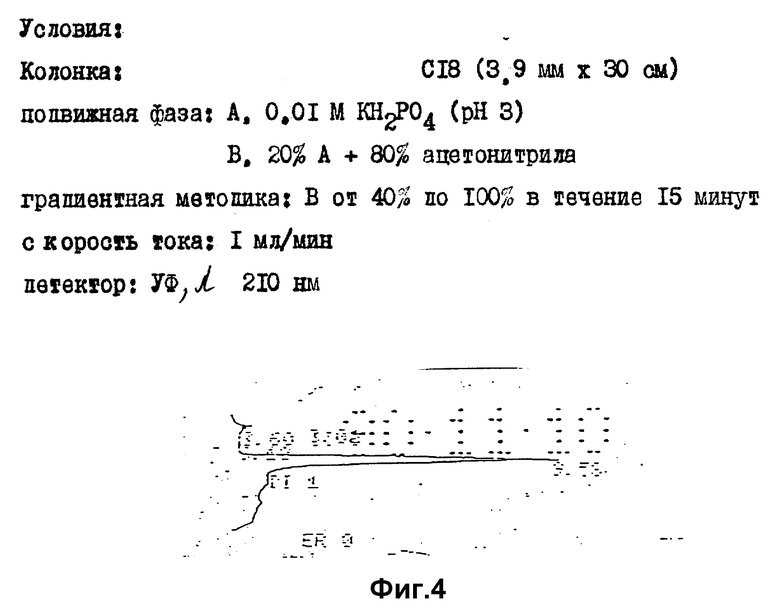
Значения Rf и времени удерживания (TP) представлены в таблице 2, хроматограммы проиллюстрированы фиг. 1 - 4.
Раствор A + 80% ацетонитрила
Система растворителей 2
Раствор A - 0,01 M KH2PO4 водный раствор (pH 3)
Раствор B - 20% раствор A + 80% ацетонитрила
Системы растворов для TCX
1. n BuOH/EtOAс/HOAс/H2 (5:5:1:1)
2. n BuOH/n BuOH/HOAс/H2O (2:8:2:3)
3. n BuOH/HOAc/H2O (4:1:5), восходящая фаза
4. n BuOH/HOAc/H2O (4:1:2)
5. n BuOH/EtOH/HOAc/H2O (1:1:1:1)
(2) Аминокислотный анализ
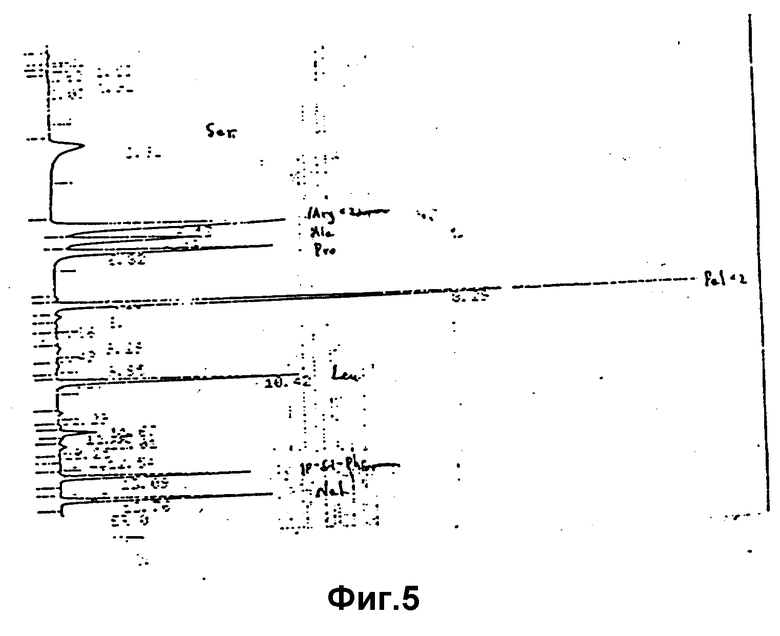
Анализ проводился по методу классической IEN и новой PICO-TAG, результаты представлены в таблице 3 и на фиг. 5, 6.
(3) Результаты биологических исследований
Результаты биологических исследований, включая антиовуляторную активность при разных дозах и ED50 по активности стимуляции выделения гистамина in vitro, показаны в таблице 4, в которой перечислены 26 антагонистов в качестве примеров.
Как показано и описано выше, антагонисты ГСЛГ, созданные и синтезированные в соответствии с этим изобретением, обнаруживают очень хорошие свойства. Они чисты по результатам анализа ТСХ или ВЭЖХ. Их структура правильна, т.е. она такая же, как запланирована. Их противозачаточная активность высока: они могут подавлять овуляцию у крыс при подкожной инъекции в позе от 0,1 до 2,0 мкг в середине стадии предтечки. Их побочное действие, связанное с гистамином низко: ED50 этих препаратов по активности стимуляции выделения гистамина in vitro (эффективная доза для тучных клеток крыс, стимулирующая выделение 50% гистамина) находится в пределах 5 - 300 мкг/мл; поражение, вызываемое ими при кожной анафилактоидной пробе у крыс мало настолько, что удовлетворяет клиническим требованиям. Они очень хорошо растворяются в воде, все биоисследования проводились нами с растворами препаратов в физрастворе, так что их легко получить в виде инъекционных лекарственных форм для клинического применения. Их также легко приготовить в виде длительно действующих систем доставки, среди которых инъецируемые микрокапсулы наиболее удобны для длительного подавления гонадотропина и половых гормонов. Поэтому они могут быть использованы как эффективные, с обратным действием и безопасные контрацептивы как для мужчин, так и для женщин. Они могут быть также использованы для лечения различных заболеваний, связанных с расстройствами репродуктивной эндокринной сферы, таких как гормонозависимый рак простаты, рак грудных желез, эндометриоз, преждевременное половое созревание у детей. Они также полезны при лечении бесплодия. Новые антагонисты LHRH могут также использоваться при основополагающих физиологических и фармакологических исследованиях репродуктивной сферы, таких как при изучении функции гипофиза, по действию половых гормонов или гонадотропинов или LHRH на сексуальное поведение и т.д.
Аббревиатуры
В тексте этого патентного заявочного документа были использованы следующие аббревиатуры
Ala аланин
AOA антиовуляторная активность
Arg аргинин
Bap дибутиламинометилфенилаланин
BOC т-бутилоксикарбонил
BuOAc бутилацетат
CAT кожная анафилактоидная проба
DCl дициклогексилкарбодиимид
DCM дихлорметан
D2Nal D -β- (2-нафтил)аланин
D3Pal D -β- (3-пиридил)аланин
DpClPhe p-хлор-D-фенилаланин
DpFPhe p-фтор-D-фенилаланин
D6Qal D -β- (6-хинолил)аланин
DMF N,N-диметилформамид
EAP диэтиламинометилфенилаланин
ED50 эффективная доза для 50% ответа
EtoAc этилацетат
FSH фолликулостимулирующий гормон
Glu глютаминовая кислота
Gly глицин
His гистидин
HOBT L-гидроксибензолтриазол
ВЭЖТ высокоразрешающая жидкостная хроматография нингидриновое производное
HRA активность по стимуляции выделения гистамина
HRT определение выделения гистамина
ISN ионообменная хроматография c
IPA изопропиловый спирт
LH лютеинизирующий гормон
LHRH гормон, стимулирующий выделение лютеинизирующего гормона
Leu лейцин
Lys лизин
Map диметиламинометилаланин
Met метионин
Mop морфолинометилфенилаланин
nBuOH н-бутиловый спирт
NS нормальный физиологический раствор
Pep дипропиламинометилфенилаланин
Phe фенилаланин
Pip пиперидинометилфенилаланин
Pipes пиперазин-N,N'-бис(2-этансульфоновая кислота)
Pro пролин
Rf степень смещения
SE стандартная ошибка
Ser серин
TFA трифторуксусная кислота
ТСХ тонкослойная хроматография
TR время удержания
Trp триптофан
Tyr тирозин
Tep тетрагидроперролилметилфенилаланинH
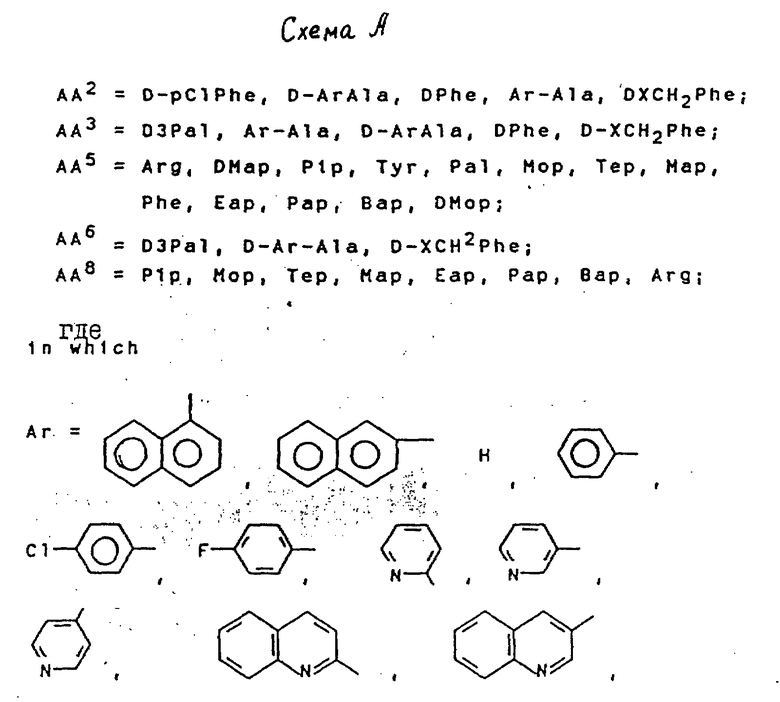

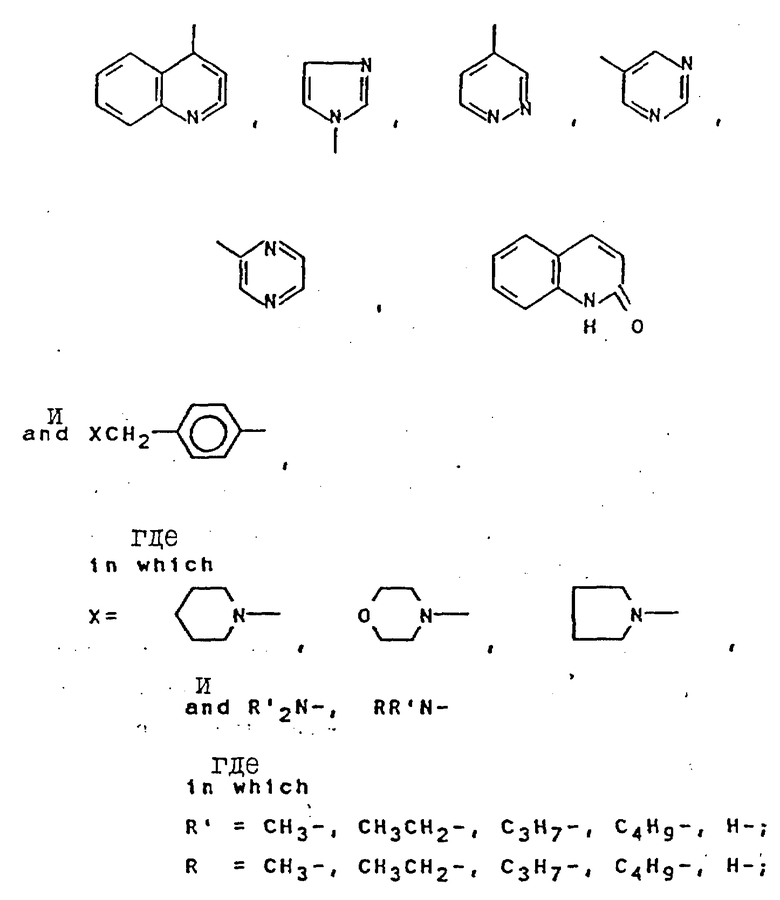


Сущность изобретения: новые антагонисты LHRH формулы [NАc - D2Nal1 - DpClphe2 - D3pal3 - Ser4 - Mop5 - D3pal6 - Leu7 - Ary8 - pro9 - DАla10]NH2 или [NАc - D2Nal1 - Dphe2 - D3pal3 - ser4 - Mop5 - D3pal3 - Leu7 - Arg8 - pro9 - DАla10] NH2 или [Nac - D2Nal1 - DpClphe2 - D3pal3 - Ser4 - Arg5 - D3pal6 - Leu7 - pap8 - pro9 - DАla10]NH2. Новые антагонисты LHRH формулы [NАc - D2Nal1 - AA2 - AA3 - Ser4 - AA5 - AA6 - Leu7 - AA8 - pro9 - DАla10] NH2, где DNal : DArAla, где 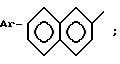 AA2 - DArAl, где
AA2 - DArAl, где 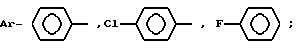 AA3: DArAla, где
AA3: DArAla, где 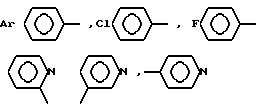 или
или 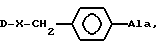 где
где 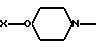 AA5 - Arg, Tyr, или DArAla, где
AA5 - Arg, Tyr, или DArAla, где 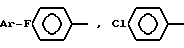 или
или 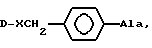 где
где 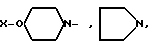 -N(R1R2), R1 и R2 - CH3, CH3CH2, C3H7, C4H9; AA6 - DАrphe, где
-N(R1R2), R1 и R2 - CH3, CH3CH2, C3H7, C4H9; AA6 - DАrphe, где 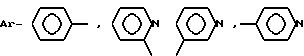 или
или 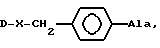 где
где 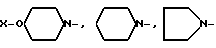 или NR1R2, где R1 и R2 - CH3, CH3CH2, AA8 - Arg или DArAla, где
или NR1R2, где R1 и R2 - CH3, CH3CH2, AA8 - Arg или DArAla, где 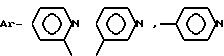 или
или 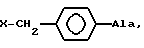 где
где 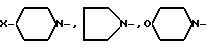 или NR1R2, где R1, R2 - CH3, CH3CH2, C3H7, C4H9 как новые пептиды могут быть использованы при лечении заболеваний эндокринной системы, для лечения рака, а также в качестве мужских и женских контрацептивов. Изобретение включает также способ получения антагонистов LHRH , заключающийся в том, что С-концевую N -защищенную аминокислоту присоединяют к бензгидриламиновой смоле, отщепляют защитную группу и продолжают наращивание пептидной цепи в последовательности, соответствующей пептиду формулы IV в условиях твердофазного синтеза, и от полученного пептидилполимера отщепляют пептид. Получают пептиды с ценными свойствами, с активностью по стимуляции выделения гистамина сниженной до такого уровня, чтобы удовлетворять клиническим требованиям. 3 с.п.ф-лы, 6 ил., 4 табл.
или NR1R2, где R1, R2 - CH3, CH3CH2, C3H7, C4H9 как новые пептиды могут быть использованы при лечении заболеваний эндокринной системы, для лечения рака, а также в качестве мужских и женских контрацептивов. Изобретение включает также способ получения антагонистов LHRH , заключающийся в том, что С-концевую N -защищенную аминокислоту присоединяют к бензгидриламиновой смоле, отщепляют защитную группу и продолжают наращивание пептидной цепи в последовательности, соответствующей пептиду формулы IV в условиях твердофазного синтеза, и от полученного пептидилполимера отщепляют пептид. Получают пептиды с ценными свойствами, с активностью по стимуляции выделения гистамина сниженной до такого уровня, чтобы удовлетворять клиническим требованиям. 3 с.п.ф-лы, 6 ил., 4 табл.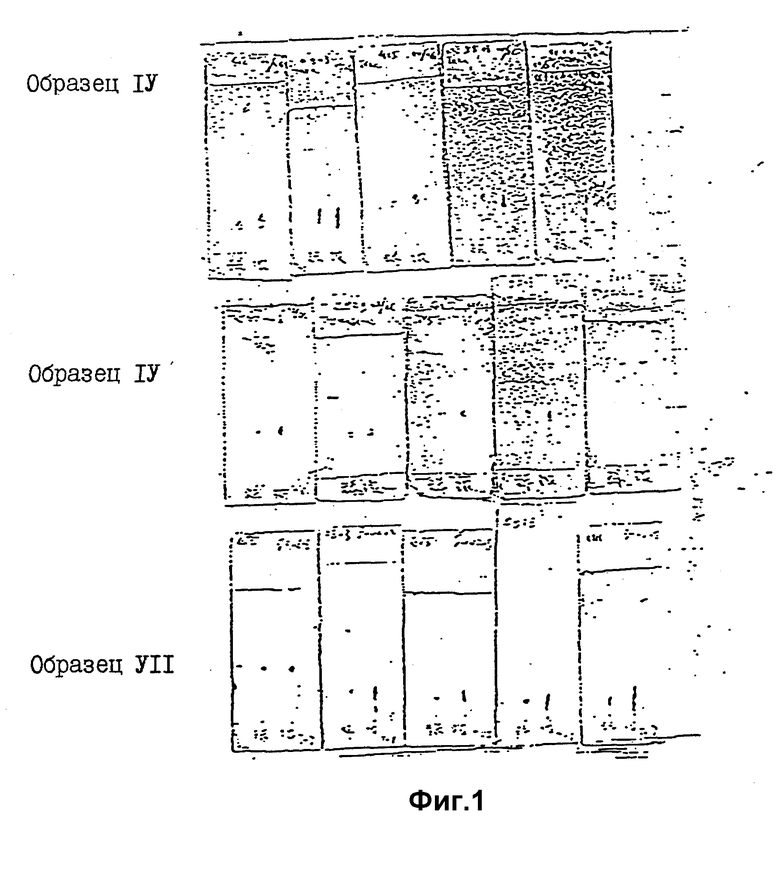
[NAc-D2Nal1-DpClPhe2-D3Pal3-Ser4-Mop5-D3Pal6-Leu7-Arg8-Pro9-DAla10] -NH2, I
или
[NAc-D2Nal1-Dphe2-D3Pal3-Ser4-Mop5-D3Pal3-Leu7-Arg8-Pro9-DAla10] NH2, II,
или
[NAc-D2Nal1-DpClphe2-D3Pal3-Ser4-Arg5-D3Pal6-Leu7-Pap8-Pro9-DAla10] NH2, III
2. Аналоги антагонистов LHRH формулы IV
[NAc-D2Nal1-AA2-AA3-Ser4-AA5-AA6-Leu7-AA8-Pro9-DAla10]NH2, DNal группа формулы
DArAla,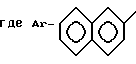
AA2 группа формулы
DArAla,
в которой Ar - группа 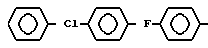
AA3 - группа формулы
DArAla,
где Ar - 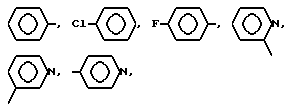
или группа формулы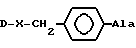
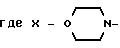
AA5 - Arg, Tyr, группа формулы
DArAla,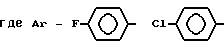
или группа формулы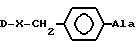
-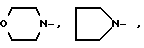 -NR1R2,
-NR1R2,
где R1, R2 - одинаковы и означают CH3, CH3CH2, C3H7, C4H9;
AA6 - группа формулы
DArPhe,
где Ar-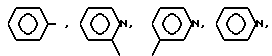
или группа формулы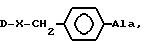
где X-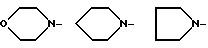
или группа
-NR1R2,
где R1, R2 - одинаковы и означают CH3, CH3CH2;
AA8 - Arg или группа формулы
DArAla,
где Ar-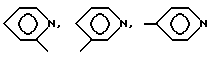
или группа формулы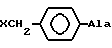
где X-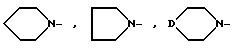
или группа формулы
-NR1R2,
где R1, R2 - одинаковы и означают CH3, CH3CH2, C3H7, C4H9.
| УСТРОЙСТВО ДЛЯ РЕГУЛИРОВКИ ТОЧЕЧНЫХ ПУТЕВЫХДАТЧИКОВ | 0 |
|
SU277829A1 |

